

Abstract
Pancreatic ductal adenocarcinoma (PDAC) remains one of the deadliest cancers. Significant efforts have largely defined major genetic factors driving PDAC pathogenesis and progression. Pancreatic tumors are characterized by a complex microenvironment that orchestrates metabolic alterations and supports a milieu of interactions among various cell types within this niche. In this review, we highlight the foundational studies that have driven our understanding of these processes. We further discuss the recent technological advances that continue to expand our understanding of PDAC complexity. We posit that the clinical translation of these research endeavors will enhance the dismal survival of this recalcitrant disease.
eTOC
Enabled by technological development on various fronts, our understanding of the complexity in the biology of pancreatic ductal adenocarcinoma deepens, laying the foundation for promising translation of the biological insights into clinically actionable advances.
Introduction:
Pancreatic ductal adenocarcinoma (PDAC) has surpassed breast cancer to become the third leading cause of cancer-related death in the United States1,2. More concerningly, PDAC is projected to overtake colorectal cancer before 2040, moving only behind lung cancer as a leading cause of cancer-related mortality. The factors driving the lethality of PDAC are numerous, centered on an inability to detect the disease until late in progression, often after distant metastasis3. Moreover, outside of the minority (10–15%) of cases ascribed to germline mutations or known risk factors, such as mucinous cystic lesions and chronic pancreatitis, there is no single attributable risk factor for most patients4. Further complicating diagnosis, localized disease is largely asymptomatic or accompanied by ill-defined symptoms, there is a paucity of diagnostic biomarkers for earlier stage tumors, and the difficult to access anatomical location of the pancreas prevents routine office-based screening. The steadily increasing incidence of disease5, driven by a combination of factors, including the obesity epidemic and greater life expectancy, underscores the urgent need to identify and deploy new therapeutic strategies to achieve benefit for most patients.
As most patients present with advanced, unresectable disease, only about 12% are predicted to survive beyond 5 years. While these numbers are dismal at first glance, some reasons for optimism exist. PDAC survival statistics have doubled over the past decade through a combination of improved clinical care and understanding of the biology of pancreatic cancer6,7. This progress coincides with a newfound appreciation for the complexity of PDAC, which occurs not only across patients but also can vary dramatically within individual tumors. Growing understanding of these variances in genetics, treatment response, and aspects of tumor composition are clarifying the scope of the problem that needs to be solved to accelerate the progress in extending patient survival.
In this review, we will briefly introduce the current treatment paradigms for PDAC, and then expand upon the biological underpinnings driving its pathogenesis and progression. We will also discuss the complexity of the pancreatic tumor microenvironment (TME) and interactions among the numerous cell types within this niche. The translational implications of research advances, such as novel therapeutic strategies and technologies, will be highlighted within pertinent sections. Finally, we will discuss how the clinical trial landscape of PDAC has evolved over the past decade, including lessons learned from pivotal trials that were unsuccessful in prolonging survival and the adoption of innovative trial designs to improve outcomes in this recalcitrant disease.
Treatment Strategies for Pancreatic Ductal Adenocarcinoma
Chemotherapy and surgery are the primary treatment options for PDAC. However, only 15–20% of patients are eligible for surgery at diagnosis3. Most PDAC patients present with distant metastasis at diagnosis, where removal of the primary lesion through a major surgical procedure is unlikely to impact the outcome. Second, the pancreas is close to critical vasculature. Even in patients without clinical evidence of extra-pancreatic spread, significant involvement or occlusion of these vessels by the primary tumor can prevent eligibility for surgery. Finally, partial removal of the pancreas, often along with a portion of the duodenum and the creation of de novo anastomoses, is a major surgery with lasting impacts on digestion and systemic metabolism. Accordingly, patients must be in robust health to withstand the operation and recover to benefit from the procedure.
The clinical care for most patients, regardless of surgical eligibility, includes systemic chemotherapy. In the past decade, two new combination regimens have emerged as the first-line therapy in patients with advanced PDAC. The first is a combination of 5-fluorouracil (5-FU), leucovorin, irinotecan and oxaliplatin, which carries the acronym FOLFIRINOX. The second is the combination of gemcitabine and an albumin nanoparticle conjugate of paclitaxel (nab-paclitaxel)8,9. Patients progressing on first-line regimens can be offered the possibility of switching to the other regimen, or a second-line regimen that includes a liposomal formulation of irinotecan in combination with 5-FU10, provided they have not received either of these agents previously. Overall, there are no universal standards for second-line regimens and beyond in PDAC, and the course of therapy is often determined by the patient’s performance status, presence of “actionable” targets, and the availability of appropriate clinical trials.
Unfortunately, even in cases where surgical resection can be performed, nearly 3 in 4 of patients will develop recurrence within two years, suggesting patients undergoing resection also harbor micro-metastatic disease11. Supporting this, pre-clinical studies have demonstrated that disseminated PDAC cells can be observed in circulation before localized invasion is readily visualized in primary tumors12. Accordingly, to enhance the possibility of eliminating PDAC cells that have escaped the primary tumor, prior to or during resection, patients who undergo surgery are typically treated with adjuvant chemotherapy in the post-operative setting. Based on the patient’s performance status, this can be either a modification of the original FOLFIRINOX regimen, or one containing gemcitabine, such as gemcitabine and cisplatin or gemcitabine and nab-paclitaxel13–15. Finally, one of the practices that has become increasingly adopted in the context of PDAC therapy over the past decade, albeit not uniformly around the globe, is that of systemic pre-operative (neoadjuvant) therapy16. Neoadjuvant therapy has the potential to shrink “borderline” resectable PDAC and enable the tumor to undergo resection. Indeed, patients whose PDAC exhibits a major or complete pathological response to neoadjuvant therapy demonstrate significantly improved overall survival17, although the biological underpinnings of this response remain to be discerned. In recently completed randomized clinical trials18, neoadjuvant chemoradiation in conjunction with adjuvant chemotherapy shows significantly improved five-year overall survival rates compared to patients who received upfront surgery with adjuvant therapy (20.5% versus 6.5%).
While the aforementioned data describe advances in standard of care therapy of PDAC, they also highlight the significant challenges with achieving cures in this recalcitrant disease. There is reasonable evidence that earlier diagnosis and surgical resection of PDAC provides the best opportunity for long-term survival. In retrospective analyses of a national cancer surveillance database in the United States, a remarkable 80% of patients who present with the lowest stages of PDAC (Stage IA or IB disease) were alive at five years, compared to less than 10% of patients with advanced disease19. Unfortunately, patients with such low stage tumors are typically diagnosed incidentally and comprise no more than 3–5% of cases. This has led to a burgeoning interest at enabling earlier diagnosis of PDAC in cohorts that are higher than average risk for this disease20. This approach is effective in individuals or families who undergo prospective surveillance for PDAC due to an inherited lifetime risk, such as a germline pathogenic mutation4. In one large multi-center international trial performed in high-risk cohorts, almost three-fourths of patients who were diagnosed with PDAC on active surveillance had Stage I disease, and an equal proportion were still alive at five years, reiterating that earlier detection can translate into improved outcomes21.
While surgery and chemotherapy remain the mainstay of PDAC clinical management, there have been several compelling developments with the application of targeted therapies in this disease, and these will be discussed in the context of selected genomic drivers in the section below.
Precursor Lesions of Pancreatic Cancer: An Opportunity for Early Detection
Among the histological subtypes of cancer arising in the pancreas, PDAC is both the most frequent exocrine neoplasm and the most frequent cancer overall. PDAC accounts for greater than 90% of pancreas cancers and has a distinct molecular profile and natural history from pancreatic neuroendocrine tumors, the second most common neoplasm in this organ22. Early molecular profiling coupled with histology allowed for the development of a putative pathological progression model; analogous to the adenoma-carcinoma model in the colon23 (Fig. 1). Here, intraductal precursors are postulated to undergo a progression from low to high-grade lesions, acquiring increasing cytological atypia and genetic aberrations, culminating in invasive adenocarcinomas. At least two pathways to invasive cancer in the pancreas have been recognized, each with somewhat distinct frequency, natural history, and genetics. Pancreatic Intraepithelial Neoplasia (PanINs) are the most frequently observed precursor lesions and are, by definition, microscopic lesions that cannot be observed on abdominal imaging scans24. In contrast, cystic mucinous neoplasms of the pancreas, of which intraductal papillary mucinous neoplasms (IPMNs) are the most common histological subtype, are macroscopic precursors that arise within the main pancreatic duct or one of its branches. These can be observed and followed for progression by imaging techniques25. Approximately 85–90% of PDAC cases are ascribed to originate on the backdrop of PanIN lesions, the remaining 10–15% in cystic precursors20. Recent phylogenetic analyses of precursor lesions from the pancreata of patients with established cancer have confirmed the stepwise progression model and accumulation of genetic alterations in the transition from PanINs to PDAC postulated using more rudimentary techniques26. Further, modeling studies performed on the mutational data have estimated that the timeline of multistep progression to invasive neoplasia in the pancreas likely spans multiple years and provides a theoretically wide window of opportunity for early detection27.
Figure 1: Initiation and Progression of Pancreatic Cancer.
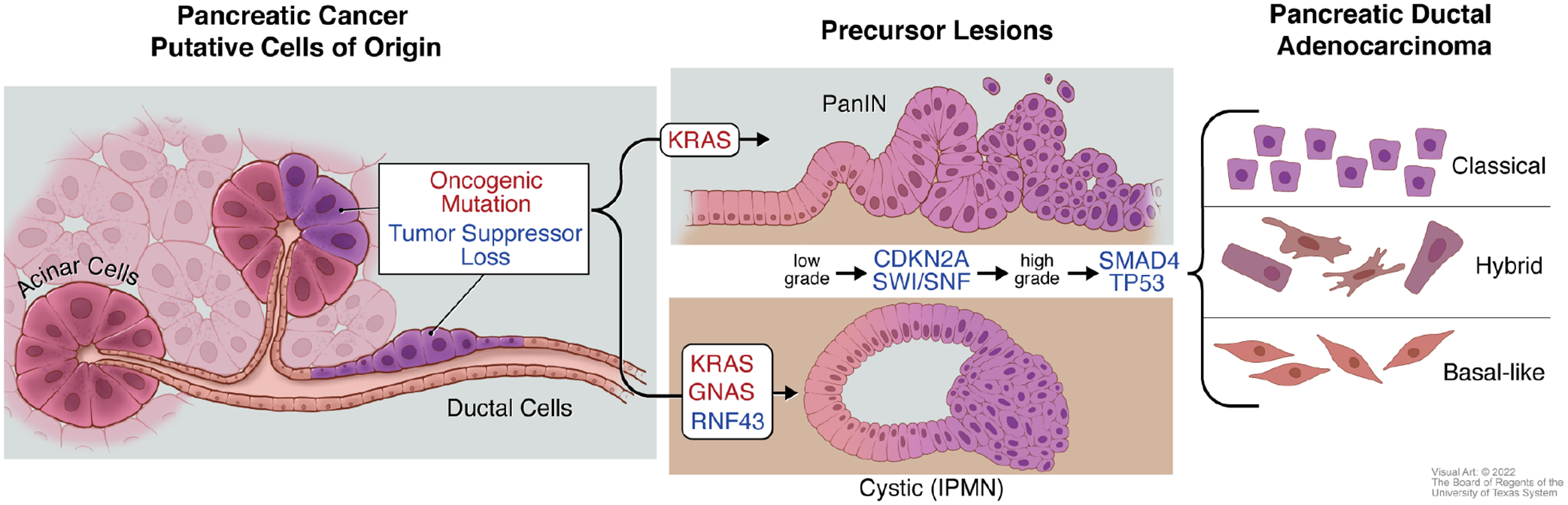
Pancreatic ductal adenocarcinoma (PDAC) forms from the exocrine tissue of the pancreas. Acinar and ductal cells have both shown the potential to serve as cells of origin for PDAC upon acquisition of oncogenic mutations and/or loss of tumor suppressor function. Activating mutations in the oncogene KRAS are found in the two most commonly observed PDAC precursor lesions: pancreatic intraepithelial neoplasia (PanINs) and cystic lesions termed intraductal papillary mucinous neoplasm (IPMNs). In addition to KRAS mutations, activating mutations in the gene encoding for the G-protein alpha subunit Gαs (GNAS) and loss of function of the tumor suppressor gene RING-type E3 ubiquitin ligase (RNF43) are associated with IPMNs. As these precursor lesions progress from low grade to high grade lesions, loss of the tumor suppressor CDKN2A or components of the SWI/SNF chromatin-remodeling complexes are observed. Further deletion or inactivating mutations of tumor suppressor genes SMAD4 or TP53 accompany the advancement of precursor lesions to PDAC. PDAC has also been classified into several RNA-based transcriptomic subtypes. “Classic” and “Basal-like” have emerged as two consensus groups, with a third “Hybrid” capturing those with overlapping features.
The plausibility of multistep progression of PanIN and IPMN has also been validated using genetically engineered mouse models (GEMMs). For example, conditional expression of oncogenic mutant Kras in the pancreas, using Cre-recombinase driven by pancreatic transcription factor promoters activated during embryonic development, leads to development of murine PanIN lesions histologically reminiscent of human disease28. Only a fraction of these mice will go on to develop invasive cancer, and those which do show a concurrent loss of a tumor suppressor gene. However, combined expression of oncogenic Kras coupled with the deletion or loss of function of Trp53, Cdkn2a, or Smad4 in the pancreas of mice readily leads to invasive PDAC29–32. Early studies in these models demonstrated that the combined haploinsufficiency of Smad4, coupled with oncogenic KRAS expression, leads to the development of cystic neoplasms, linking Kras mutations to other putative PDAC precursor lesions33. More recent IPMN models have been generated that genocopy recurrent driver alterations observed in cognate human lesions (i.e. Kras, Gnas, Rnf43) (Fig 1)34–36, and these models demonstrate the comparable stepwise progression observed in patient derived IPMNs.
Importantly, the understanding that PDAC arises through a multistep series of precursor lesions, with a prolonged timeline for progression to invasive neoplasia, has generated substantial interest in discovery of circulating biomarkers for early detection, especially in high-risk cohorts37. The carbohydrate antigen 19–9 (CA19–9), also known as sialyl-Lewisa, is currently the most widely used diagnostic biomarker for PDAC, although it has a suboptimal sensitivity in early-stage disease. Nonetheless, CA19–9 elevations can be observed in plasma samples obtained in the pre-diagnostic setting, suggesting a potential window of opportunity for early detection38. While an in-depth discussion of ongoing biomarker research in PDAC is beyond the scope of this review, a multitude of circulating tumor-derived analytes (proteins, autoantibodies, metabolites, DNA, non-coding RNAs and extracellular vesicles, among others) have been assessed in the context of PDAC early detection39–41. For patients with cystic lesions, aspirated cyst fluid (obtained using endoscopic ultrasound guided biopsy) is an attractive proximal biospecimen for assessment of analytes42. There is burgeoning enthusiasm for the potential of “multi-cancer early detection” (MCED) tests, which can interrogate for the presence of multiple cancer types, typically using aberrant tumor-derived DNA signals (mutation, methylation or fragments) in circulation43. One cautionary note worth reiterating with any biomarker endeavors in PDAC is the potential for false positives in the average-risk population, mandating exceptionally superlative performance characteristics (high specificity and high sensitivity). Therefore, current guidelines advise against screening the general population and instead emphasize biomarker studies be focused on well-defined high-risk cohorts44.
Genomic Drivers of Pancreatic Cancer
In contrast to the absence of a single attributable cause in most patients, the somatic genetic drivers of pancreatic cancer are well characterized (Fig. 1). Mutations in the v-Kiras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) are found in over 90% of patient samples45. KRAS mutations are observed in most low-grade PanINs and IPMNs, suggesting KRAS is the initiating genetic event driving exocrine neoplasia46,47. In addition to KRAS mutations, the genomic landscape of PDAC is also dominated by loss of function alterations of TP53, SMAD4 (also known as DPC4), and CDKN2A45,48–50. The temporal appearance of these alterations during multistep progression suggests that TP53 and SMAD4 typically occur late, usually in the setting of high-grade lesions or cancer in both PanINs and IPMNs23,51.
Ras has pleiotropic cell autonomous and non-cell autonomous effects in PDAC. Each of the other major drivers adds one or more unique facets the phenotype, natural history and therapeutic vulnerability of PDAC. For example, CDKN2A is often lost by homozygous deletions at the chromosomal 9p21 locus, which is associated with codeletions of a neighboring interferon cluster in half of cases. Interferon cluster loss creates a “cold” tumor immune milieu and resistance to immunotherapy52, a phenotype that is likely to be generalized across other solid cancers with chromosomal 9p21 deletions53. SMAD4 loss has a profound deleterious effect on prognosis54, and is associated with higher rates of metastatic dissemination during neoadjuvant therapy and terminal disease55,56. TP53 is the most commonly mutated tumor suppressor gene in PDAC, and altered p53 function has a multitude of tumor-promoting effects on neoplastic cells, including increased genomic instability30, reprogrammed cellular metabolism57,58, and enhanced metastatic propensity59. PDAC mouse models have suggested TP53 mutations promote a patterned loss of genomic integrity as opposed to a chaotic process60. Finally, it is important to note that “hotspot” TP53 missense mutations might have a distinct impact on the pathophysiology of cancer cells from that of truncating mutations or homozygous deletions which lead to absence of functional protein59,61, underscoring an additional layer of nuance when considering the role of tumor suppressor genes in PDAC.
Given the near ubiquitous presence in pancreatic tumorigenesis, it was postulated that PDAC might be “addicted” to oncogenic Ras signaling. Providing an early clue toward the complex heterogeneity of PDAC, RNAi-mediated gene silencing of oncogenic KRAS revealed a significant portion of PDAC cell lines demonstrate no reliance on KRAS expression for viability62. Further insights into the requirement of KRAS signaling in pancreatic tumor maintenance was made through the development of GEMMs with inducible and reversable expression of mutant Kras in the pancreas. Withdrawal of oncogenic Kras resulted in regression of invasive neoplasia, including regression of metastatic disease63–65. Notably, in such an inducible system, PanINs can be observed to re-differentiate back to acinar cells in the absence of tumor suppressor loss66. However, withdrawal of mutant Ras signaling in preclinical models that include loss of tumor suppressor function leads to tumors that can re-occur in a Ras-independent fashion67. While “Ras-escaper” cells have orthogonal targetable vulnerabilities68, the ability of PDAC to survive Ras inhibition highlights a likely challenge of newly developed small molecule inhibitors of mutant KRAS.
The advent of allele-specific (targeting KRASG12C and KRASG12D)69,70, and a broader cache of allele agnostic (“pan-RAS”) small molecule inhibitors71, has generated considerable enthusiasm for their application in KRAS-mutated cancers, including PDAC. Covalent inhibitors of the KRASG12C allele were the first to be evaluated in the clinic72,73, and two agents have recently been approved by the US FDA for use in KRASG12C-mutant non-small cell lung cancer. However, the KRASG12C allele occurs in only 1.6% of PDAC cases, whereas the KRASG12D allele is present in greater than 40%, followed in frequency by KRASG12V and KRASG12R mutations74. In the first published clinical series of KRASG12C inhibitor monotherapy in advanced PDAC, responses occurred in a minority of patients (~21%) and were generally transient75. This is not surprising, as correlative studies conducted in other KRASG12C-mutant tumor types treated with these inhibitors have demonstrated the emergence of pleiotropic resistance through both cell autonomous and non-cell autonomous mechanisms76–78. Recently, data from pre-clinical PDAC models has shown promising responses to a small molecule inhibitor targeting KRASG12D mutations common in PDAC79. However, given the observed resistance to KRASG12C inhibitors and observations from KRAS extinction models, it is probable these drugs will face similar challenges as they enter the clinic. Accordingly, significant activity is currently focused on identifying combinations that would lead to more sustained responses in patients80.
While much of the excitement on therapeutic targeting of the erstwhile “undruggable” Ras oncoprotein in PDAC has focused on small molecule inhibitors, additional modalities of direct KRAS inhibition have been developed. For example, a directly binding protein “degrader” that forms a ternary complex with mutant Ras and E3 ubiquitin ligase is currently being evaluated in the clinic81. Similarly, genetic extinction of mutant KRASG12D allele using exosome delivered synthetic siRNAs is currently undergoing early phase trials in advanced PDAC82. Finally, adoptive T cell therapy using autologous T cells expressing an engineered T cell receptor targeting HLA-restricted mutant KRAS neoantigen has shown promising early results and is currently being expanded to other epitopes and HLA contexts83.
The most frequently encountered driver mutations in PDAC were identified prior to the advent of next generation sequencing (NGS) by targeted PCR, immunohistochemistry, or through their association with hereditary syndromes. The widespread adoption of NGS has greatly expanded the genomic landscape of PDAC and identified a large compendium of less frequently altered genes (10% or lower) that play a role in selected tumor subsets45,48–50. These less-frequent mutations largely cluster around central themes, including DNA maintenance and DNA damage response, epigenetic modifying proteins, and axon guidance pathway components. However, despite its aggressive nature, the overall mutational load of PDAC is less than many common solid tumors, such as lung cancers and melanoma84.
Approximately 5–7% of patients with PDAC harbor a germline mutation in one of the Fanconi anemia family of genes (BRCA1, BRCA2, and PALB2) encoding for proteins engaged in homologous recombination repair85. It is postulated that somatic loss of the remaining wild-type allele in the tumor cells would lead to a homologous repair deficient (HRD) phenotype, rendering such tumors exquisitely sensitive to agents that induce double strand DNA breaks (e.g., platinum agents like cisplatin and oxaliplatin), or to inhibitors of the DNA repair enzyme PARP, which would result in synthetic lethality given the pre-existing compromise in DNA repair function86. Indeed, a pivotal clinical trial in patients with PDAC and germline BRCA1/2 mutations established the benefit of administering an orally bioavailable PARP inhibitor on improved progression free survival (PFS). This led to the approval of targeted therapy in this setting87. Unfortunately, longer follow up data did not demonstrate a similar improvement in overall survival (OS) compared to patients who received placebo alone following platinum-based therapy88. Nonetheless, PARP inhibitors continue to be embraced in the clinic, largely due to the impact of a “chemo-free” regimen on the quality of life for patients.
More recent studies have demonstrated that, in addition to patients with germline mutations, those harboring de novo somatic mutations of BRCA1/2 and PALB2 also show benefit from PARP inhibitors, provided there is bi-allelic loss and established homologous repair deficiency in the tumors89. Ongoing trials are also PARP inhibitors in additional scenarios, such as in the postoperative (adjuvant) setting and in combination with immunotherapy90. Mutations of ATM are the second most frequent known cause of germline mutations in PDAC, after BRCA2, conferring a comparable lifetime increased risk of PDAC in mutation carriers91. While the ATM protein is a serine/threonine kinase that is recruited to sites of DNA double strand breaks and is involved in DNA repair, bi-allelic mutations of ATM have not been shown to result in susceptibility to PARP inhibitor monotherapy like BRCA1/292,93.
Chromatin remodeling and SWI/SNF complex genes are one of the most frequently altered class of genes in PDAC45,48–50, and can occur via somatic deletions of the associated chromosomal loci94. Perturbation of gene function in GEMMs has strongly suggested a tumor suppressor role for many of these altered genes. For example, deletion of KDM6A results in accelerated tumorigenesis and acquisition of an aggressive transcriptional subtype95–97. Similarly, deletion of SWI/SNF factors Arid1a or Brg1 results in the acceleration of pancreatic tumorigenesis on the backdrop of cystic neoplasms resembling human IPMNs98–100. In contrast to prototypal tumor suppressor genes like TP53 and CDKN2A, chromatin regulatory factors may have a more context-specific and biphasic function in PDAC, with studies in autochthonous models suggesting both tumor suppressor and oncogenic functions during distinct stages of tumorigenesis101,102. Given the cumulative frequency of PDAC cases that exhibit alterations in this class of genes, there is significant promise of leveraging therapeutic vulnerabilities that arise within these patients97,103.
Finally, there are also so-called “alternative drivers” found in the 8–10% of cases that are KRAS wild type104,105. These are enriched in patients who have younger onset PDAC (<50 years) and can manifest with variant histological features. The alternative drivers (e.g., ALK, TRK, RET, NRG1, BRAF, EGFR) activate the MAP kinase signaling pathway, reiterating the primacy of this downstream Ras effector to PDAC pathogenesis. Importantly, many alternative drivers are activated by fusions, rather than point mutations. Given the availability of inhibitors approved in a tumor-agnostic manner for such “actionable” targets, every attempt should be made to identify the presence of these drivers, particularly in younger onset, KRAS-wild type tumors106–109.
Pancreatic Cancer Cell-of-Origin: An Unsettled Debate
Given the ductal histological appearance of PDAC, it was widely assumed that precursor lesions arise from mutations in the normal pancreatic ductal epithelial cells (Fig. 1). However, numerous studies have demonstrated that murine acinar cells can de-differentiate to a progenitor-like state with a ductal appearance. This has been termed acinar-to-ductal-metaplasia (ADM)110,111, and lineage tracing studies in mice confirmed that acinar cells can repopulate damaged tissue112. The early demonstration of the oncogenic transformation of the pancreas in mice utilized embryonic Cre-drivers that enable recombination in the entire exocrine pancreas28–31. Further studies using Cre-drivers specific to adult acinar cells in mice demonstrated ADM and PanIN formation by expression of oncogenic KRAS113–117; however, these models did not eliminate the potential of ductal transformation.
The first direct comparison using adult acinar vs. ductal expression of oncogenic KRAS demonstrated that KRAS promotes acinar cells to generate neoplastic lesions at a frequency over a hundred-fold higher than ductal cells118. This study also supported key aspects of the ADM to PanIN tumorigenesis model. Deletion of the ductal progenitor gene Sox9 prevented KRAS-driven transformation, while Sox9 overexpression accelerated PanIN development. Further studies have demonstrated the heterozygous or homozygous deletion of factors associated with terminal acinar differentiation such as Ptf1a, Mist1, and Nr5a2 accelerate KRAS-driven tumorigenesis119–122. Conversely, stabilized expression of PTF1A and MIST1 inhibit PanIN formation in the context of mutant KRAS expression122,123.
Subsequent studies have demonstrated that, while the plasticity of acinar cells allows them to be more readily transformed by oncogenic KRAS, their ability to serve as a sole cell-of-origin for neoplasia has proven context dependent. Combining oncogenic KRAS expression in the adult-ductal lineage with Pten loss leads to the formation of IPMNs124, and the deletion of Trp53 leads to the rapid development of invasive carcinoma125–127. Further, the induction of pancreatic damage by pancreatic ductal ligation is sufficient to promote KRAS-mediated ductal-lineage tumorigenesis128. This was not observed in the context of damage through cerulein-induced pancreatitis118, which drives inflammation through damage of acinar cells. Taken together, these observations directly link the ability of oncogenic KRAS to promote tumorigenesis to lineage-specific regenerative responses.
Overall, the accumulation of evidence in murine models has shown that, in the right context, either acinar or ductal cells can serve as a progenitor population for PDAC (Fig. 1). These studies have largely leaned on lineage tracing approaches that are only possible in mouse models, and conclusively connecting observations from the different cells of origin to human disease remains a work in progress. However, recent advances in our understanding of potential subtypes of patient disease, discussed in detail in following section, have begun to allow interpretation of the cell-of-origin models derived in mice to human disease127, as well as potential phenotypes129.
Transcriptional Subtypes of Human Pancreatic Cancer
Approaches to comprehensively interrogate the transcriptional landscape, as opposed to DNA mutations, has identified several PDAC subtypes across patients (Fig. 1). Collisson, Sadanandam and colleagues led one of the first major efforts to subtype pancreatic cancers, utilizing a combination of patient microarray datasets and PDAC cell-lines. They identified three subtypes, termed “classical”, “quasimesenchymal”, and “exocrine-like”130. Importantly, these subtypes could predict drug response in cell culture models, and patient outcomes were significantly different across their subtypes. Similarly, using an RNA-sequencing approach to assess PDAC patient tumors and low passage cell lines, Bailey and colleagues identified four subtypes of PDAC, which they termed “squamous”, “pancreatic progenitor”, “immunogenic”, and “aberrantly differentiated endocrine exocrine” (ADEX) type tumors50.
These efforts to subtype patient samples were confounded by the characteristic histology of PDAC, in which cancer cells frequently dwarfed by the immune and stromal cells populations. To ensure the genomic and transcriptomic signatures represented those of the cancer cells, samples were screened for high levels of epithelial content, resulting in a drop out of a significant number of samples49,50. To address this shortcoming, Moffit and colleagues devised a “virtual dissection strategy” to computationally subtract the transcriptional background imposed by stromal and immune cells from bulk-RNA sequencing signatures131. Using this approach, they identified two subtypes of PDAC tumors, termed “classical” and “basal-like”, in addition to two stromal subtypes termed “normal stroma” and “activated stroma”.
Taking an alternative approach to the constraints imposed by the low percentage of the PDAC cells within the TME, several efforts have been made using laser capture microdissection (LCM) on PDAC sections to collect epithelial or stromal enriched areas of tissues132,133. Using this approach, Puleo et al. identified five subtypes, termed “pure basal like”, “stroma activated”, “desmoplastic”, “pure classical”, and “immune classical”133. They also noted overlaps with their subtypes, the Moffitt subtypes, and most of the Bailey subtypes. However, they suggested that their findings demonstrate that the Bailey ADEX subtype is likely contamination of pancreatic acinar cells.
Similarly, Mauer et al., found isolated epithelial tissues supported the Moffitt “classical” or “basal-like” subtypes, while they found no evidence of the Bailey “ADEX” subtype or Collisson “exocrine-like” subtypes in epithelial enriched tumor samples132. The lack of “ADEX” or “exocrine-like” subtype in these two studies fits with an observation from the Cancer Genome Atlas (TGCA). Namely, samples “ADEX” or “exocrine-like” signatures are associated with low PDAC cellularity45, suggesting these signals derive from stromal/immune/non-cancerous elements of tissues. Finally, Maurer stromal areas of PDAC tumors revealed two prominent subtypes, which they postulated represented “ECM-rich” or “Immune-rich” phenotypes132.
Following these studies, another major sequencing effort was made by Chan-Seng-Yu and colleagues. Here, the authors performed LCM enrichment of epithelial tissue from a large cohort of advanced PDAC patients combined with whole-genome sequencing and transcriptomic sequencing134. They identified five different subtypes of PDAC tumors: “basal-like-A”, “basal-like-B”, “hybrid”, “classical-A” and “classical-B”. They reported best overlap with the Moffitt subtypes, and their resolution allowed for subclassification of both, while their hybrid subtype did not consistently fall into previously reported classes. Importantly, correlating their subtypes with single-cell RNA sequencing data, they demonstrated that different subtypes can co-exist within individual tumors.
Overall, as our understanding of transcriptional subtypes of PDAC has evolved, consensus has best coalesced around two major subtypes, classical and basal-like, with some populations showing mixed features135 (Fig. 1). There is potential to differentiate these into further subclasses, and multiple types have potential to co-exist within induvial tumors134. The cell state of the classical and basal-like subtypes is potentially plastic and several candidate regulators such as TP63136, which can be induced by KDM6A loss97, can promote more aggressive subtypes. Further, the transcriptional landscape of PDAC cells can also be influenced by microenvironmental factors137. However, single cell sequencing, as discussed in detail later, has showcased how much more of the transcriptional states of PDAC cells remains to be uncovered.
A related question is how to exploit this concept therapeutically: the recent COMPASS trial provided proof of principle validation of subtype-based treatment of patients. Patients with classical subtype of PDAC had superior responses to first line 5-flurouracil-basd regimens than those with “basal-like” cancers138. This initial observation is now being further validated with a multi-center trial that is also examining whether contemporaneously established ex vivo organoid cultures from patient biopsies can provide phenotypic data on a broader range of drug sensitivities.
The Pancreatic Cancer Tumor Microenvironment
One of the most fascinating and challenging features of pancreatic cancer is the tumor microenvironment (TME). The pancreatic TME is composed of numerous populations of fibroblasts, dense extracellular matrix, a poorly formed vascular system, and a diverse and largely suppressive populations of immune cells. The neoplastic cells in PDAC often constitute a minority of the overall cellularity within the tumor. Indeed, the aggressive biology, resistance to therapy, and heterogeneity of the disease is driven nearly as much by the non-cancerous components of the tumor as the PDAC cells. However, the oncogenic KRAS signaling in the PDAC cells initiates and coordinates the development of this growth supportive niche139, and the vast network of interactions between and among these populations is only beginning to be understood.
Cancer Associated Fibroblasts
A significant effort to understand the pancreatic TME has focused on understanding stromal fibroblast populations (Fig. 2). Fibroblasts represent the most abundant cell type in the TME and are largely responsible for the deposition of the extracellular matrix driving the characteristic physiological properties of PDAC140. However, the extent of the diversity of these cancer-associated fibroblasts (CAFs), and their specific progenitor populations remain unknown and is a major area of ongoing investigation.
Figure 2: Cancer-associated Fibroblast Origin and Heterogeneity.
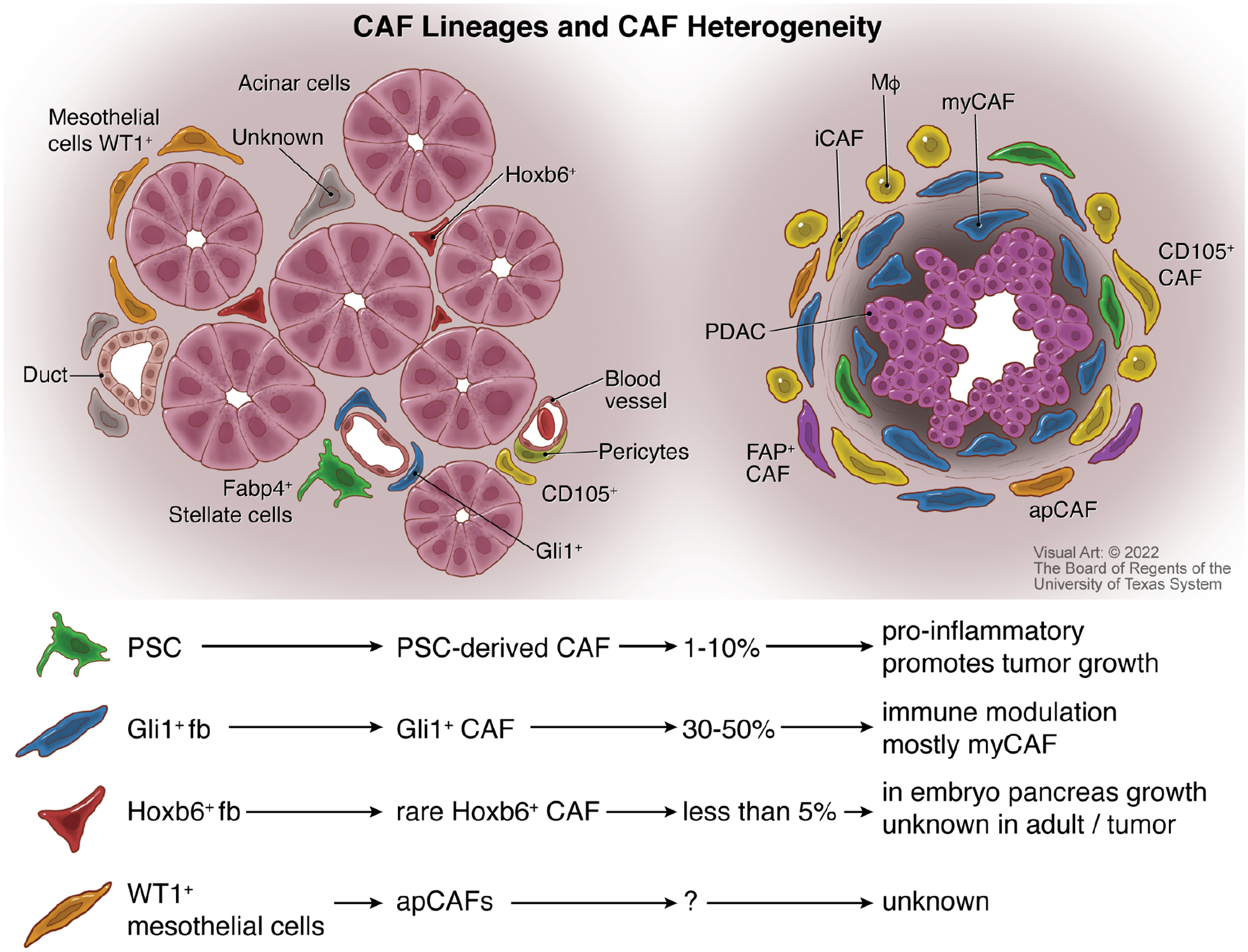
Several types of fibroblasts are present in both the normal pancreas (left) and PDAC (right). Normal pancreatic fibroblast populations include pancreatic stellate cells (PSCs), Gli1+ fibroblasts, Hoxb6+ fibroblasts, Fabp4+ fibroblasts, CD105+ fibroblasts, pericytes, and WT1+ mesothelial cells that encase the organ. Within PDAC, cancer-associated fibroblast (CAFs) populations include myofibroblastic CAFs (myCAFs) associated with collagen deposition, inflammatory CAFs (iCAFs) that associate with macrophages (MΦ), antigen-presenting CAFs (apCAFs), CD105+ CAFs, and fibroblast activation protein containing (FAP+) fibroblasts. Lineage tracing has demonstrated that PSC-derived CAFs make up less than 10% of the overall number of CAFs and have roles in promoting tumor growth and inflammation. Gli1+ CAFs can account for nearly half of all the fibroblasts in pancreatic cancer, with roles in depositing matrix and modulating immune response. Hoxb6-derived CAFs are infrequent and have no known function akin to their role in embryonic pancreas growth. The number of mesothelial-derived CAFs is unknown, as is their function outside of a potential antigen presentation role.
The normal pancreas contains heterogeneous fibroblasts populations (Fig. 2) including pancreatic stellate cells —similar to liver stellate cells and containing vitamin A-rich lipid droplets141— perivascular fibroblasts, and other parenchymal fibroblasts. Another potential origin of CAFs is from bone marrow derived progenitors. While stellate cells were long believed to be the only source of stromal fibroblasts, recent lineage tracing studies targeting Fabp4+ stellate cells revealed that they give rise to a minority of CAFs142,143. While not abundant, stellate cell derived CAFs have a tumor promoting role supported by unique gene expression and metabolic profiles. Conversely, lineage tracing analysis of perivascular fibroblasts, expressing the transcription factor Gli1, showed this population expands to form nearly half of pre-CAFs in PanINs and CAFs in cancer; in contrast, parenchymal fibroblasts, expressing Hoxb6 do not expand during carcinogenesis144. Finally, mesothelial cells, expressing Wilms Tumor 1 (Wt1) also contribute to a sub-population of CAFs145. Thus, among resident pancreatic fibroblasts, different populations have vastly different abilities to expand during carcinogenesis. A recent study took a step further back in tracing the origins of CAFs in PDAC. First, a combination of dual labeling approaches and bone marrow transplants showed the contribution of bone marrow progenitors and cancer epithelial cells to the CAF population is minimal146. Conversely, tracing the embryonic origin of tissue resident fibroblasts through the splanchnic mesenchyme expressing Isl Lim homeodomain 1 (Isl1) labeled most tissue resident fibroblasts and pancreatic CAFs146. A key area of further research is whether different cellular origins account for the phenotypic and functional differences of CAFs, as addressed below.
The heterogeneity of CAFs within PDAC tumors is an actively evolving area, in part due to the advent of single cell technologies (Fig. 2). Early efforts to identify and understand different CAF populations identified a myofibroblast-like population (myCAF) and an inflammatory fibroblast population (iCAF)147. MyCAFs are localized close to the neoplastic cells and thought to be responsible for deposition of extracellular matrix, whereas iCAFs tend to occupy spaces between islands of the PDAC cells and release IL6 and other anti-inflammatory cytokines. Interestingly, mechanistic investigation into myCAF and iCAF biology in vitro suggested potential plasticity between the two populations148, dependent on extracellular signaling molecules present.
Beyond myCAF and iCAFs, several additional populations of CAFs have been characterized (Fig. 2). One population expresses several components of the antigen presentation machinery typically found on myeloid populations. Given this feature, these have been termed apCAFs, although whether those CAFs prime a functional immune response is unclear149. Of note, the transcriptional profiles of apCAFs overlap with mesothelial cells150, and based on lineage tracing studies in mice have been postulated to derive from a mesothelial origin145.
Other classifications of CAFs are based on their expression of specific factors. For example, CAFs expressing Fibroblast Activation Protein (FAP) also express high levels of CXCL12 and inhibit tumor T cell infiltration. Depletion of these fibroblasts, or targeting of CXCL12, sensitizes PDAC to immune checkpoint blockade in preclinical models151. As another example, single-cell mass cytometric analysis has identified that the cell surface marker CD105 can be used to identify two functionally distinct and non-interconvertible populations of CAFs, namely, tumor permissive CD105+ CAFs, and tumor suppressive CD105− CAFs152. Recently, fibroblasts expressing leucine-rich-repeat-containing protein 15 (LRRC15) were identified in mouse PDAC but not in the normal pancreas, and these fibroblasts are postulated to promote tumor growth and inhibit anti-tumor immune responses153.
In addition to heterogeneity among CAF populations, fibroblast status throughout tumorigenesis and progression can be influenced by genetic mutations present in the PDAC cells154,155, suggesting the overall characteristics of the TME might be specific to each patient. Further complexity of PDAC TME has been observed spatially, as CAF populations and their organization of tumor architecture can vary across different regions of tumors creating “sub-tumor microenvironments” within patients156. Interestingly, the specific nature of the sub-TME in an individual tumor did not predict outcome, but higher sub-TME heterogeneity was a poor prognostic sign, possibly because heterogeneous systems are inherently more resilient. CAF biology in pancreatic cancer is an area of active investigation. The functional role of specific CAF populations is not fully clear; whether CAFs exist as distinct, stable populations, or if their status is plastic is not known, and, finally, whether it would be beneficial to eliminate or, conversely, reprogram, CAFs is an area of debate.
Extracellular Matrix
CAFs are largely responsible for deposition of the dense extracellular matrix found in PDAC, largely consisting of collagen and hyaluronic acid. These matrix molecules create a physical barrier, presence of which is debated as a hinderance to chemotherapy delivery140,157. Early studies suggested that targeting the sonic hedgehog pathway, important for epithelial-fibroblast paracrine signaling, would improve chemotherapy by reducing this matrix barrier158. However, clinical trials showed a worsened survival with combined hedgehog inhibition and chemotherapy. Subsequent studies suggested depletion of stromal fibroblasts through hedgehog inhibition, epithelial sonic hedgehog deletion, stromal ablation, or inhibition of lysyl oxidase like-2 increases tumor access to the blood supply and facilitates metastasis, having deleterious consequences on natural history of the disease159–162.
Collagen is the most abundant matrix molecule found in PDAC163. The majority of collagen is type I collagen164, largely generated by CAFs147,165. Targeted deletion of type I collagen from SMA-expressing fibroblasts increases immune suppressive myeloid infiltration, accelerating tumor progression166. Beyond physiological heterotrimeric collagen I produced by fibroblasts, PDAC cells produce “tonic” (low) levels of a distinct oncogenic homo-trimeric collagen that binds to α3β1 integrin, thereby mediating immune suppression and reprogramming the tumor-associated microbiome167. Collagen also acts as a signaling molecule, activating discoidin domain receptor signaling that shape multiple roles in PDAC pathology168–171. Accordingly, the role of collagens in the pancreatic TME extends beyond a structural role, and the potential of targeting collagen-mediated crosstalk is only beginning to be understood.
Hyaluronic acid is a glycosaminoglycan polymer, thought to be released by predominantly by CAFs, that avidly retains water and promotes the characteristically high interstitial fluid pressure found in pancreatic tumors140. Targeting hyaluronic acid in the TME was hoped to be a more effective way to improve penetration of chemotherapy, releasing pressure on collapsed blood vessels while leaving cancer restraining fibrosis intact. Initial preclinical data in mouse models suggested that use of hyaluronidase combined with chemotherapy showed great promise140,157. Nonetheless, randomized clinical trials combining a pegylated hyaluronic acid degrading enzyme, PEG-hyaluronidase, with standard-of-care chemotherapy for advanced PDAC did not show an increase in overall survival regardless of chemotherapy backbone or selection of patients with high levels of hyaluronic acid by immunostaining172,173.
Alternative methods to target stromal matrix have also demonstrated preclinical promise. Focal adhesion kinase (FAK), hyperactivated in pancreatic cancer cells, is an important driver of fibrosis and immune suppression in the TME174. FAK inhibition was shown to markedly reduced fibrosis and decreased the infiltration of tumor-promoting immune cells in mice. Further, combined treatment of FAK inhibition with immune checkpoint blockade demonstrated tumor regression in an aggressive mouse model of PDAC, suggesting this approach will translate to the clinic. More recently, FAK inhibition-mediated reprogramming of the stroma was shown to overcome radiation resistance and sensitize the tumors to immunotherapy175. Taken together, it has become clear that targeting the non-cellular component of the TME represents a challenging prospect. However, there is optimism that new approaches are poised to overcome the shortcomings of previous approaches to target the PDAC tumor matrix.
Innate Immune Cells
Myeloid cell populations make up the largest immune cell population in pancreatic tumors (Fig. 3). This has prompted investigation into the roles of various myeloid populations in PDAC biology, particularly related to immune suppression. Oncogenic KRAS signaling drives expression of granulocyte colony stimulating factor (GM-CSF) by PDAC cells, promoting recruitment of immunosuppressive myeloid-derived suppressor cells (MDSCs)176,177. Tumor-associated macrophages (TAMs) arising from both tissue resident macrophages or circulating monocytes appear to play important but potentially different roles in shaping the TME, and suppression of the adaptive immune response178. Importantly, targeting MDSCs and/or TAMs improves adaptive immune responses in mouse models of pancreatic cancer and can potentiate checkpoint inhibition approaches176,177,179,180 (Fig. 3). Among these, novel agonists developed in PDAC models to target the CD11b/CD18 receptor present on most tumor-infiltrating myeloid subsets have shown promising pre-clinical data, currently being tested in the clinic181,182. A different approach uses CD40 agonists to reprogram macrophages to a less immunosuppressive status, leading to CD8 T cell infiltration that would drive the conversion from a “cold” to a “hot tumor”183. Two distinct CD40 agonists have recently been evaluated in clinical trials in the advanced and neoadjuvant settings in PDAC. Although the response was modest, extensive tissue and blood based correlative studies confirmed CD40 agonism enhanced T cell infiltration in PDAC184,185.
Figure 3: Immune Interactions in Pancreatic Cancer.
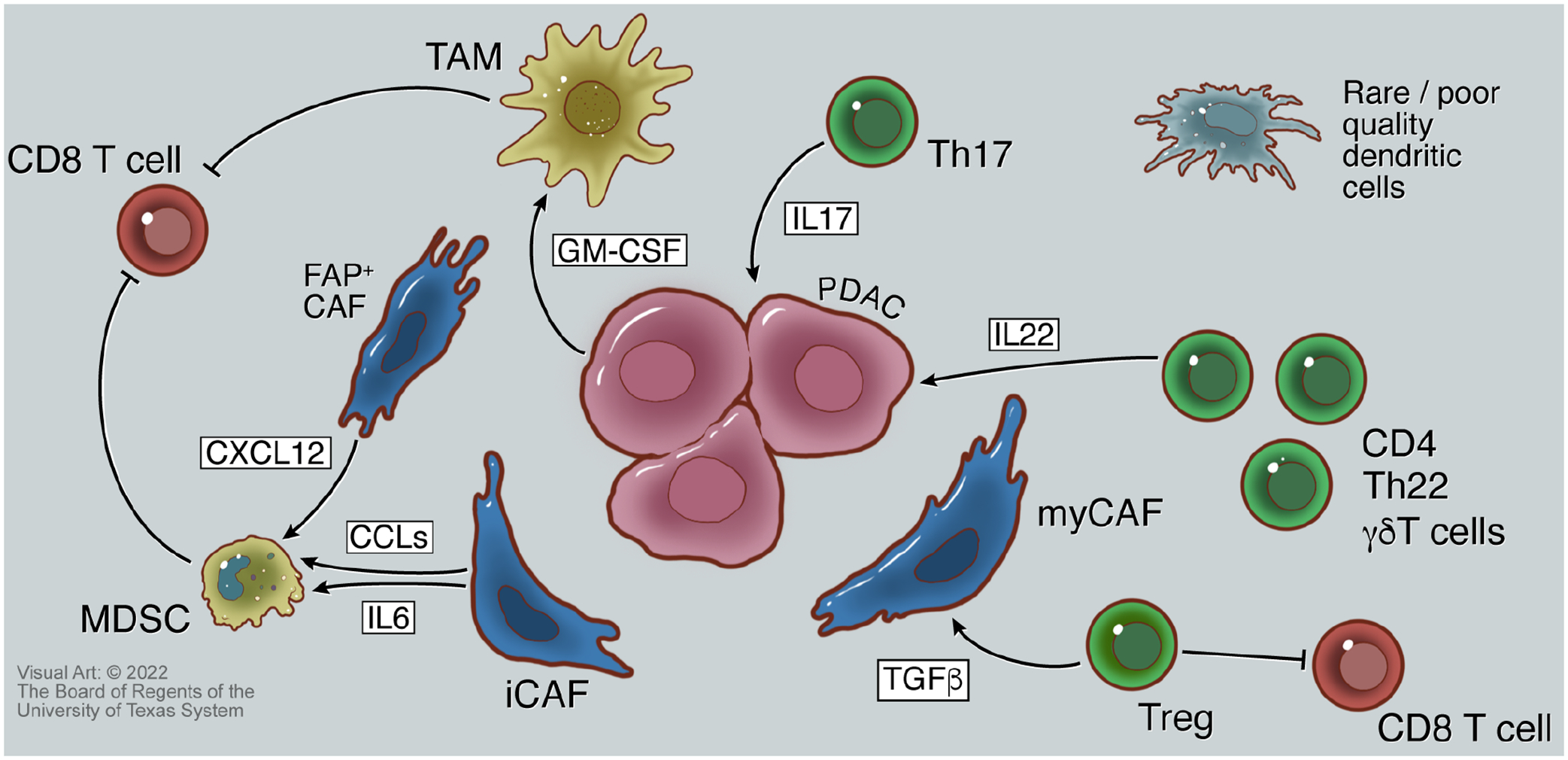
A network of interactions between fibroblasts, cancer cells, and immune cells create an immune-suppressive tumor microenvironment. Monocyte-derived suppressor cells (MDSCs) that inhibit CD8 cytotoxic T cells are polarized by chemokine ligands (CCLs) and interleukin 6 (IL6) from inflammatory cancer-associated fibroblasts (iCAFs), and by CXCL12 from fibroblast activation protein containing (FAP+) fibroblasts. Granulocyte macrophage colony-stimulating factor (GM-CSF) released by neoplastic cells also polarizes tumor-associated macrophages that inhibit CD8 T cells. Further, T regulatory cells (Tregs) inhibit CD8 T cells, and supply TGF-β that allows for myofibroblastic cancer-associated fibroblast (myCAF) activation. IL17 and IL22 released by Th17 and Th22 cells, respectively, provide pro-growth signals to cells to PDAC cells via binding to cognate cell surface receptors. Dendritic cells are sparse in pancreatic cancers, and those present are of low quality, further contributing to the lack of an effective anti-tumor adaptive immune response to PDAC.
Beyond immune-suppression by myeloid cells, a major barrier to mounting an anti-cancer adaptive immune response to PDAC appears to be a lack of adequate antigenpresentation. Dendritic cells (DCs) are found in low numbers in PDAC, as compared to other tumor types, and the DCs present are of poor quality for antigen presentation186 (Fig. 3). These mechanistic data lend additional weight to efforts to increase DC licensing as part of enhancing PDAC patient response to checkpoint inhibition186,187.
Adaptive Immune Cells
One of the most notable features of the PDAC TME is the lack of infiltration by cytotoxic CD8 T cells. Other T cell populations within the TME have all been noted to play protumorigenic roles, including Th17, Th22, CD4, and γδ T cells188–191 (Fig. 3). Interestingly, even though CD4 T cell ablation enabled CD8 mediated regression of PDAC tumors in mice, depletion of FoxP3+ T regulatory cells (Tregs) accelerated tumorigenesis due to compensatory myeloid infiltration192. This shift in immune infiltration follows a loss of myCAFs upon Treg depletion, as Tregs proved a significant source of TGF-beta.
This is only one example of the complicated interplay between the immune and stromal compartments in PDAC, illustrating why efforts to enable a sustained immune response have proven difficult in this complex TME. Currently, several combinatorial innovative approaches to overcome these barriers are being tested in the clinic, and some of these principles are revisited in the last section of the review.
Pancreatic Cancer Metabolism: Adaptations Present Therapeutic Opportunities
The physical barriers imposed within PDAC tumors function to limit access to blood-derived nutrients and oxygen, leading to dysregulate nutrient profiles and promote regional hypoxia. Direct measurement of the metabolites in human PDAC tumors revealed classes of metabolites are in low supply for cells within the tumors, and others are increased in abundance193. Additionally, analysis of the nutrients available within murine tumor interstitial fluid demonstrates a starkly different profile within pancreatic tumors vs. serum levels194. PDAC cells exhibit numerous adaptations to not only survive but thrive under the nutrient constraints imposed by the TME. These center around two themes: oncogenic KRAS mediated reprogramming of cellular metabolism to mediate survival and enable proliferation, and adaptations to enhance nutrient acquisition and/or scavenging pathways (Figure 4a).
Figure 4: Altered Metabolism and Metabolic Interactions of Pancreatic Tumors.
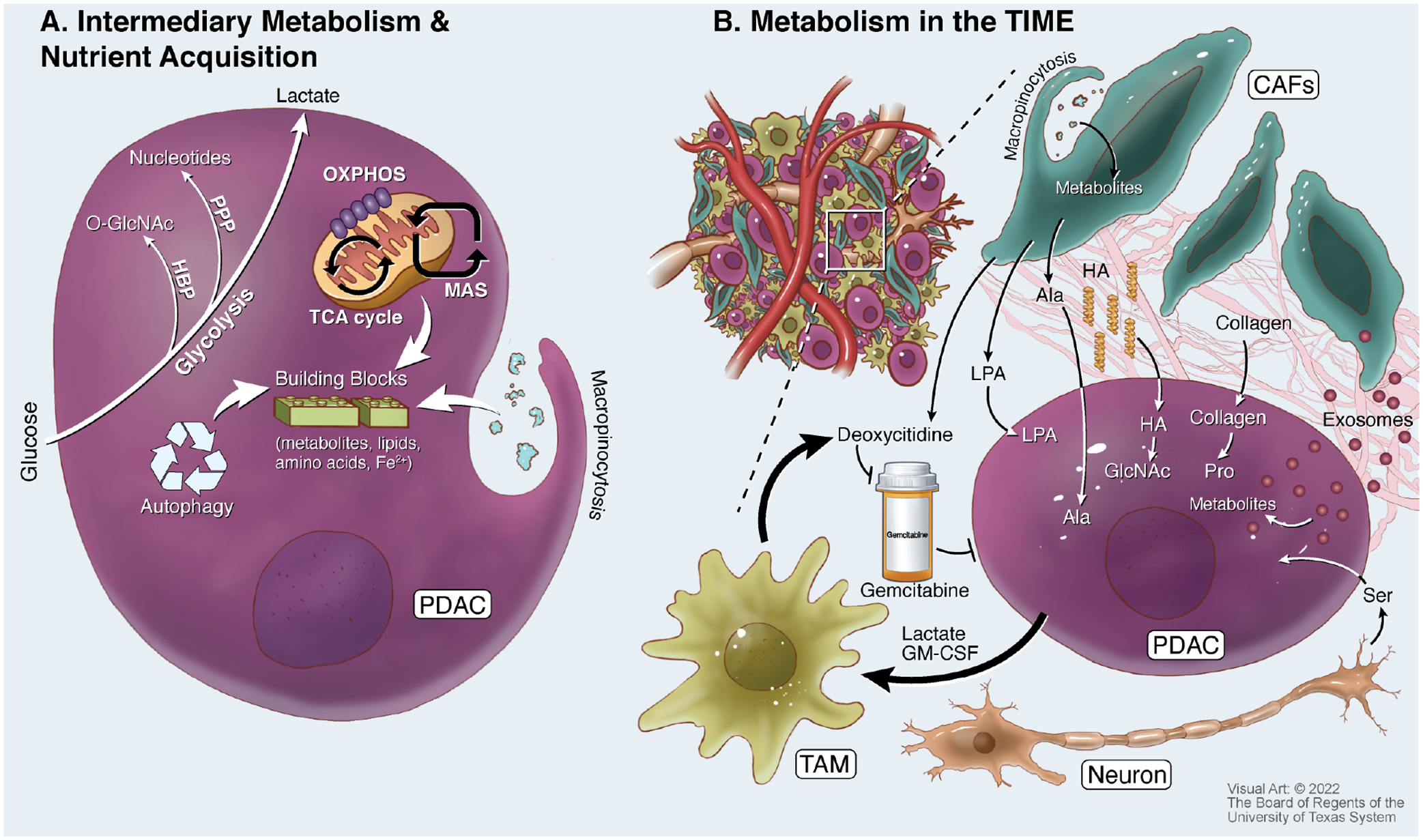
A. Pancreatic ductal adenocarcinoma (PDAC) cells have increased glucose consumption to fuel glycolysis and anabolic metabolism from glycolytic intermediates. These include upregulation of the hexosamine biosynthetic pathway (HBP) to produce O-GlcNAc needed for protein glycosylation, and the pentose phosphate pathway (PPP) to produce nucleotides. PDAC cells also take advantage of a re-wired malate-aspartate shuttle (MAS) to fuel mitochondrial metabolism and oxidative phosphorylation (OXPHOS), and intermediates from the tricarboxylic acid (TCA) cycle provide synthetic metabolic building blocks. In addition to creating new biosynthetic material, PDAC cells recycle proteins and organelles through autophagy, and obtain extracellular materials through a non-specific fluid uptake process known as macropinocytosis. B. Many metabolic interactions shape the tumor microenvironment (TME). Cancer-associated fibroblasts (CAFs) provide alanine (Ala), lysophosphatidic acid (LPA), and exosomes loaded with metabolites to PDAC cells. The nutrient support of CAFs is at least in part fueled by macropinocytotic uptake of extracellular materials. PDAC cells metabolize extracellular matrix deposited in the TME to obtain hyaluronic acid (HA) to fuel GlcNAc pools and collagen to support proline (Pro) pools. Tumor-associated macrophages (TAMs) are polarized by signals including lactate and granulocyte macrophage colony-stimulating factor (GM-CSF) from PDAC cells. TAMs release deoxycytidine, also released by CAFs, that directly competes with the anti-metabolite chemotherapy gemcitabine. In addition, neurons in PDAC tumors can share serine (Ser), which is used to support mRNA translation.
Intermediary Metabolism and Potential Targets in PDAC
The metabolic reprogramming of central carbon metabolism in PDAC occurs in both glucose and glutamine metabolism. Mutant KRAS drives upregulated glycolysis, not only increasing production of lactate, but also providing key carbon inputs for synthesis of molecules required for proliferation. Examples include the non-oxidative pentose phosphate pathway, which enables nucleotide biosynthesis, and the hexosamine biosynthesis pathway, which generates GlcNAc required for posttranslational modifications64,195 (Figure 4a). Directly targeting glycolysis through lactate dehydrogenase inhibition has shown mixed results depending on p53 status58, and this approach can be potentially overcome through compensatory upregulation of oxidative phosphorylation196. Further, combining inhibition of glycolysis with blockade of KRAS effector signaling pathways has shown efficacy in culture and xenograft models197, suggesting utility for paired oncogenic metabolism and signaling combination treatments.
Oncogenic Ras also drives glutamine anaplerosis to provide a significant source of carbon for TCA cycle, biosynthesis of glutathione necessary to detoxify reactive oxygen species (ROS), and enables a non-canonical malate-aspartate shuttle to generate NADPH for redox balance198 (Figure 4a). Given the importance of glutamine metabolism in pancreatic cancer, glutaminase (GLS), which drives the hydrolysis of glutamine to glutamate and ammonia, has drawn significant interest as a drug target. However, despite showing potent growth inhibitory effects in short-term PDAC cell proliferation assays, GLS inhibition was found to be ineffective in pancreatic tumor models through remodeling of compensatory metabolic pathways199. Further, PDAC can engage the biosynthesis of glutamine through glutamate ammonia ligase in response to glutamine withdrawal, further demonstrating metabolic plasticity by pancreatic cancer cells200.
However, there appears to be promise in targeting glutamine metabolism using inhibitors with less substrate specificity. 6-Diazo-5-oxo-l-norleucine (DON) was identified as a potential anti-cancer glutamine antagonist more than a half century ago201, but its lack of substrate specificity provided too much off-target toxicity202. However, recent advances have optimized DON as a pro-drug with pharmacokinetic properties that increase tumor targeting203, including an orally available analog with promising pre-clinical results in murine PDAC models204. Interestingly, recent studies have shown that the use of DON or analogs in murine PDAC models also appear to have immunomodulatory effects204,205, highlighting the potential of targeting metabolism in combination with immunotherapy.
Nutrient Recycling and Scavenging
Beyond alterations in central carbon metabolism, pancreatic cancer cells exhibit dramatic changes in organelle programming and function. One of the best studied examples of this centers on the increased biogenesis of lysosomes and constitutive engagement of the autophagy “self-eating” recycling program206 (Figure 4a). Autophagy is critical for cancer cell growth and tumorigenesis in mouse models of PDAC207–209, and genetic inhibition of critical autophagy proteins in mice leads to regression of autochthonous PDAC tumors210. Mitophagy, the selective engulfment and degradation of mitochondria through autophagy, has also been shown to be important in PDAC tumorigenesis in mice and organoid models211. Importantly, clinical trials have shown that hydroxychloroquine, which inhibits autophagic flux by preventing lysosomal acidification, enhances PDAC patient response to chemotherapy212,213.
In addition to organelle maintenance, autophagy plays a critical role in maintaining iron homeostasis in PDAC cells214. Iron is a necessary co-factor for proteins in metabolism and other critical cellular processes215; however, free intracellular iron must be stored within the protein ferritin to prevent generation of free radicals. In PDAC cells, ferritin is trafficked to autophagosomes via nuclear receptor coactivator 4 (NCOA4), and free iron is subsequently released by autophagy through a process termed “ferritinophagy”214. Increased expression of NCOA4 accelerates tumorigenesis in mice, and genetic ablation leads to delayed tumor growth that is eventually overcome through development of compensatory iron acquisition pathways216. Importantly, MEK-inhibition in PDAC increased both lysosome biosynthesis and ferritinophagy, necessary to support increased iron-sulfur cluster proteins for respiration217 (Figure 4a). Labile iron can also potentiate cell death via the lipid oxidation process known as “ferroptosis”218, which is currently an active and exciting area of research in PDAC219–223.
Autophagy has also been shown to play important roles in resistance to targeted therapies. Inhibition of the mutant KRAS-MEK-MAPK signaling cascade using ERK inhibitors drives increased autophagic flux in PDAC cells to facilitate survival224,225. Combined pharmacological targeting of MEK or ERK with hydroxychloroquine potently inhibits xenograft tumor growth and has shown efficacy in clinical treatment of PDAC, with additional trials on-going. Beyond direct consequences on intercellular metabolism, autophagy also facilitate tumor survival by influencing cellular immunoreactivity. Major histocompatibility complex I (MHC-I) down-regulation is a major mechanism by which cancer cells escape detection by the immune system226. Indeed, MHC-I expression is low at the PDAC cell surface, instead found predominantly localized within autophagosomes and lysosomes227. Genetic or pharmacological inhibition of autophagy in mice bearing PDAC tumors promotes enhanced cell-surface MHC-I expression and antigen presentation, leading to an increased response to immune checkpoint blockade therapy. The autophagy-MHC-I relationship provides mechanistic insight to functional genomics studies in mice, which have also linked autophagy and immune evasion in PDAC228.
Autophagy drives degradation/recycling of components already within cancer cells; however, deregulated activation of nutrient uptake processes also fuel pancreatic cancer metabolism. For example, macropinocytosis is a non-substrate specific fluid phase uptake process engaged by oncogenic KRAS signaling229. Numerous studies have now demonstrated that PDAC cells utilize macropinocytosis to obtain amino acids through the uptake and degradation of albumin and collagen230–232 (Figure 4a). Macropinocytosis has also been shown to utilize necrotic cell debris as a fuel source, which can provide sugars, fatty acids, nucleotides, and amino acids to fuel cancer cell metabolism233,234. Macropinocytosis was initially thought to be a ubiquitous feature of oncogenic KRAS mutations231. However, only a subset of PDAC cell lines with mutant KRAS demonstrate a constitutive macropinocytosis program, whereas others engage this process only when challenged with glutamine deprivation235.
Collectively, these studies highlight the heterogeneity of metabolic features and behaviors utilized by PDAC cells extend beyond simple re-wiring of anabolic and catabolic pathways. These combine to enable PDAC cells to survive nutrient challenging conditions in the primary tumor and provide metabolic flexibility to enable metastasis. However, the unique metabolic features of PDAC cells also present numerous therapeutic opportunities to eliminate cancer cells, while sparing normal tissues.
Metabolic Heterogeneity in Pancreatic Cancer
As discussed above, the metabolic milieu in pancreatic tumors is deregulated, with some nutrients being enriched and others depleted. The differential metabolism of these regions can be readily observed through imaging techniques236,237, including spatial heterogeneity of glycolysis. Recognition of regional metabolic heterogeneity within pancreatic tumors has inspired efforts to classify PDAC cells in a manner reminiscent of the transcriptional subtypes. PDAC cell lines classified by bioenergetic preferences and response to metabolic inhibitors revealed “glycolytic”, “lipogenic”, and “slow proliferating” metabolic subtypes238. These states also correspond to transcriptional subtypes. The glycolytic metabolic subtype aligns with a “quasimesenchymal” transcriptional subtype and lipogenic metabolism state with a “classical” signature130. More recently, metabolic characterization of clonal lines isolated from tumors revealed that two distinct populations can co-exist within the same tumor239. Accordingly, the levels of intratumoral metabolic heterogeneity are only beginning to be appreciated.
The disparity between nutrient availability imposed by the pancreatic TME, the bloodstream and other routes of dissemination, and secondary organs colonized in metastasis allow for varied metabolic states for PDAC cells194. Glutamine deprivation promotes an epithelial-to-mesenchymal transition (EMT) through the upregulation of the master EMT regulator Slug240, enabling PDAC metastasis to the lung. Comparison of the epigenetic and metabolic state of primary vs. metastatic PDAC patient tumors revealed the upregulation of anabolic glucose metabolism with a concurrent dependence on the oxidative pentose phosphate pathway during the evolution of metastasis241. This re-wired metabolic pathway revealed a dependence on a unique pentose conversion pathway, centered on the upregulated activity of phosphogluconate dehydrogenase (PGD)242. High glycolytic flux is required to maintain upregulated PGD metabolism, and this in maintained in part through suppression of thioredoxin-interacting protein (TXNIP) transcription243. Targeting TXNIP can reverse the metabolic and epigenetic programming identified to be unique in metastatic PDAC.
Several other links between PDAC metabolism and cell state have also been reported. PDAC cells that survive withdrawal of oncogenic KRAS signaling exhibit a targetable dependence on mitochondrial metabolism68. Similarly, pancreatic tumor-initiating cells have been found to preferentially utilize mitochondrial metabolism, as compared to glycolytic metabolism preferred by bulk tumor cells244. Differential activation of the integrated stress response can also be used to define sub-populations within individual tumors, which drives differential sensitivity to metabolic inhibitors239.
Given the breadth of metabolic diversity that can exist within tumors, there is growing interest in developing techniques to simultaneously inhibit multiple pathways to combat heterogeneity239,245. For example, extracellular asparagine from serum and produced by a constitutively active integrated stress response in subpopulations of PDAC cells can be targeted using asparaginase, potently sensitizing PDAC tumors to mitochondrial inhibition in pre-clinical models239,246. Further, metabolic inhibition approaches combined with RAS pathway inhibitors have shown strong pre-clinical data and are currently in clinical trials197,224,225. These signaling/metabolic inhibitor combinations have even more potential to grow as mutant KRAS inhibitors are established in the clinic.
Metabolic Interactions in the Pancreatic Tumor Immune Microenvironment
As previously discussed, the neoplastic epithelial cells within PDAC tumors are often vastly outnumbered by the stromal and immune cell populations. The varied localization, motility, and function of these different cell populations translate to differential nutrient requirements and can lead to tumor-supporting metabolic interactions. These symbiotic interactions fuel PDAC metabolism, promote resistance to chemotherapy, and drive mechanisms of immune suppression (Figure 4b).
Stromal Metabolic Crosstalk
Pancreatic CAFs are in close proximity to PDAC cells147. CAFs provide signals that promote epigenetic and metabolic re-wiring of cancer cell metabolism247. Examination of metabolic exchange between pancreatic CAFs and cancer cells revealed that alanine is selectively released by fibroblasts and consumed by PDAC cells248. The exchange of alanine and other non-essential amino acids from fibroblasts to promote PDAC metabolism is linked to both autophagy and macropinocytosis in the fibroblasts248,249 (Figure 4b). Further, the exchange of metabolites such as pyruvate can be used by PDAC cells to maintain redox balance250.
Multiple mechanisms have been identified that promote exchange of amino acids from fibroblasts to cancer cells. Stellate cell-derived exosomes have been shown to carry numerous metabolite and protein cargo to pancreatic cancer cells, including amino acids251,252. Differential expression of solute carrier proteins (SLCs) can also function to promote a one-way fibroblast-SLC1A4 / PDAC-SLC38A2 exchange axis, enabling the exchange of alanine253.
Fibroblasts have also been shown to engage in reciprocal reprogramming to drive metabolite exchange axes. Activation of CAFs from quiescent stellate cells engages a metabolic program supporting the secretion of several lipid species. Among these lysophosphatidylcholines are hydrolyzed by autotaxin released from cancer cells to provide the potent signaling molecule lysophosphatidic acid, which promotes PDAC proliferation and migration254 (Figure 4b). Similarly, PDAC program CAFs to engage a BCAT1-driven metabolic program, leading to CAF production of branched-chain α-ketoacid precursors reciprocally exchanged to fuel PDAC metabolism255.
In addition to direct metabolic exchange, extracellular matrix produced by stromal fibroblasts and PDAC cells can serve as a fuel for PDAC metabolism. As previously noted, PDAC cells can utilize macropinocytosis to obtain proline from CAF produced collagen230. PDAC cells can also obtain GlcNAC through N-acetylglucosamine kinase salvage from matrix components256, including hyaluronic acid257 (Figure 4b).
Tumor-Associated Macrophage Metabolic Crosstalk
Myeloid cells, including TAMs, provide another potential source of metabolic support to fuel PDAC metabolism. TAMs and other anti-inflammatory macrophages release numerous pyrimidine species that are avidly consumed by PDAC cells258. Beyond a potential role in supporting PDAC metabolism, the deoxycytidine released among these pyrimidines inhibits the efficacy of the standard of care chemotherapy gemcitabine, and depletion of myeloid cells in mice sensitizes PDAC tumors to gemcitabine treatment. Deoxycytidine can also be released from CAFs259, and both TAM and CAF-released deoxycytidine act to inhibit the efficacy of gemcitabine through molecular competition at deoxycytidine kinase258,259 (Figure 4b).
The programming of TAMs is mediated by oncogenic-KRAS reprogramming of PDAC cells. Lactate produced from upregulated glycolysis in PDAC cells combines with GM-CSF, released in a KRAS-dependent fashion, to stimulate macrophage polarization260. The polarization of TAMs occurs through a PI3K-AKT pathway, activating ACLY signaling and the expression of the arginine catabolic enzyme Arginase 1 (Arg1). Arginase-mediated arginine depletion by myeloid cells is a metabolic mediator of immune suppression through limitation of arginine to cytotoxic T cells, which impairs their activity261. In addition, anti-inflammatory macrophages and pancreatic TAMs utilize a predominantly mitochondrial bioenergetic metabolic programming, fueled by fatty acid metabolism258. Accordingly, there are multiple avenues macrophage metabolism can be targeted to relieve immune suppression in pancreatic cancer, and ongoing studies in this area aim to exploit these metabolic vulnerabilities to facilitate anti-tumor immunity.
Other Metabolic Interactions in PDAC
Neural invasion is a well-recognized feature of pancreatic tumors, with the severity negatively correlating to prognosis262. Neurons can also provide serine that functions to support mRNA translation in PDAC263 (Figure 4b). Beyond host cells supporting the neoplastic cells within the tumors, bacterial and fungal pathogens can infiltrate into PDAC264–266. Among these, gammaproteobacterial PDAC have been shown to directly detoxify gemcitabine through expression of cytidine deaminase267.
Heterogeneity between PDAC cells also supports multiple axes of metabolic symbiosis. For example, lactate produced by cancer cells in hypoxic regions can be used to fuel PDAC cells in well oxygenated areas195. Additionally, asparagine produced by subpopulations of PDAC cells that have a constitutively active integrated stress response support the mitochondrial metabolism of other PDAC cells during limited respiration239, providing a barrier to metabolic targeting.
Taken together, these studies demonstrate that targeting metabolism is not as simple as defining metabolic vulnerabilities of the cancer cells, and the metabolic state of PDAC cells is heterogenous within individual tumors. However, our increased understanding of the symbiotic support mechanisms of nutrients within the pancreatic TME have already identified several therapeutic opportunities. These targets will only expand as our understanding of the PDAC metabolic crosstalk network is further defined.
New Technologies Driving New Insights
The advancement of single-cell analysis technologies and high-dimensional spatial analysis techniques have rapidly broadened our understanding of PDAC. A common thread emerging from these cutting-edge studies is that the levels of heterogeneity that can exist within PDAC tumors extend far beyond what has previously been described. This underappreciated level of complexity spans populations of stromal cells, immune cells, and distinct populations of cancer cells that co-exist within individual tumors. Here, we will highlight studies that illustrate examples of several new technologies, and how they can be combined to bring new insights to clinical therapy.
Mass cytometry
Mass cytometry by time-of-flight (CyTOF) combines the use of heavy metal conjugated antibodies with flow-cytometry coupled to a mass spectrometer. This broadly expands the number of antigens that can be run simultaneously for single cell analysis, allowing high-dimensional profiling to better define cell types in the TME. For example, stromal heterogeneity in PDAC is widely recognized, but the relationships between different lineages and a consensus of markers to identify fibroblast behaviors within tumor remains less defined. The use of mass cytometry comparing millions of cells isolated from 19 murine PDAC tumors against library of 40+ known fibroblast and mesenchymal cell markers was integral to the discovery of CD105 as a key indicator of tumor promoting/tumor suppressive fibroblast behavior152. The extended number of unique markers that can be used in parallel with mass cytometry has also proven to be a powerful tool for immunophenotyping. Illustrating this, a 36 marker CyTOF characterization of peripheral blood from PDAC patients revealed neoadjuvant FOLFIRINOX promotes an increase in effector T cells and concurrent decrease in suppressive immune populations268. The data gained through this approach suggests neoadjuvant treated patients would potentially respond better to adjuvant immunotherapy.
Single-cell RNA Sequencing
The advancement of single-cell RNA sequencing (scRNA-seq) technology has completely re-shaped our view of the pancreatic TME. scRNA-seq data confirmed the existence of multiple CAF populations within human and murine pancreatic tumors and allowed the discovery of apCAFs149. Integration of multiple scRNA-seq datasets combined with lineage tracing studies demonstrated that ap-CAFs can arise from a mesothelial origin and regulate the expansion of regulatory T cells and immune suppression145. scRNA-seq datasets from human and murine tumors have identified multiple unique neoplastic epithelial populations within individual tumors149,269, which represent multiple PDAC subtypes within the same tumor270. In fact, PDAC cell lines and patient-derived organoid cultures demonstrate multiple, distinct, sub-populations by scRNA-seq271, highlighting epithelial cell heterogeneity even in ex vivo models. Further, patient-derived organoid transcriptional landscape and sub-population proportions shift during passaging137,271, suggesting that representation of the in vivo biology of the original tumor is not stable in culture. However, addition of contextual factors lacking in such ex vivo culture models can potentially rescue the heterogeneity of the original tumor models and the retain the ability to predict response to therapy137. Additional iterations of single cell profiling approaches – such as single cell T-cell receptor sequencing (scTCR-seq) of tumor infiltrating lymphocytes – have now been developed and have provided insights into antigenic targets of immune response in this disease272.
Spatial Analysis Techniques
CyTOF and scRNA-seq of isolated cell populations from PDAC tissue has provided an unprecedented depth of knowledge on the complexity and heterogeneity of the TME. However, the spatial organization of cell populations also has a central role in shaping their relationships and phenotypes. Fortunately, advances in multiplexed immunohistochemical staining and imaging technology have enabled the expansion of markers that can be used in spatial assessment of cancer cell number, function and evolutionary trajectories273,274. Illustrating the utility of this approach, multiplexed myeloid, lymphoid, and functional antibody panels were able to spatially define the relationship between myeloid and T cell populations in PDAC patient tissue275. Of note, while many differences in immune infiltration and behavior were found between different patients, this ability to spatially assess these data allowed the observation of widespread heterogeneity in individual patients. The extrapolation of spatial profiling technologies into the threedimensional (3D) space has the potential to further expand our understanding into the heterogeneity of PDAC and its constituent elements276.
Synergistic Applications of New Techniques
Parallel use of spatial and isolated single-cell analysis techniques has proven adept at identification of interactions between cell types in the TME. For example, combined scRNA-seq, multiplex IHC, and CyTOF immune profiling demonstrated an inverse correlation between suppressive myeloid populations and CD8 effector T cell infiltrations277. These further identified that CD8 T cells show indications of systemic exhaustion, and a network analysis of potential checkpoint receptor-ligand pairing suggested TIGIT as a key node of immune-suppression. In line with these observations, subsequent studies using murine PDAC models have further implicated the CD155/TIGIT axis as a potential target to move forward to clinical trials to enhance immunotherapy278. Similarly, the combined use of spatial profiling with single-nucleus RNA sequencing has enabled the interrogation of the response of PDAC to neoadjuvant therapy and identified a treatment-refractory “neural-like” progenitor cell type that escapes standard of care regimens279. Recent advances in spatial profiling technologies have also allowed the newer platforms to approach true single cell, and even subcellular, resolution280. Given the increasingly facile extension of these technologies to archival samples and limited biospecimens typically obtained from clinical trials, there is strong optimism that the correlative studies enabled by these platforms will lead to the insights needed to develop lasting and durable approaches to treat PDAC.
Clinical Trials: Setting Expectations for Future Success
Increased understanding of the biology of PDAC has led to a plethora of clinical trials to improve treatment; however, most have fallen short of expectations. In fact, PDAC suffers from one of the highest rates of Phase 3 failures amongst common solid cancers. This has led to calls to revamp how drug development is furthered for this disease, and to redefine the thresholds needed for agents to move forward to larger phase trials281,282.
The indelible lessons we continue to learn are that the past is prologue and that the underlying biology remains one of the most pivotal arbitrators of meaningful success in the clinic. For example, it is now evident that simply combining two “off the shelf” immune checkpoint inhibitors to enhance effector T cell function – a strategy that has worked in several other solid cancers – has minimal to no impact in a disease where a multiplicity of immune defects co-exist283,284. In a recent preclinical study, dual therapy against CD8 T cell checkpoints using a 41-BB agonist and LAG-3 inhibitor by itself was ineffectual, and yet when administered with a myeloid cell targeted CXCR1/2 antagonist, the triple regimen led to sustained responses in biologically relevant models285. Not surprisingly, therefore, the first clinical hints of success at deploying immunotherapy in PDAC have thus come from regimens that combine chemotherapy with targeting of one or more additional nodes within the dysfunctional TME (myeloid cells, Tregs) in combination with immune checkpoint inhibitors286,287. This has the dual benefit of tumor shrinkage and acts as an in situ antigen release mechanism to allow for an immune response.
Many of the recent promising clinical signals in PDAC have arisen through rigorous preclinical analyses in syngeneic and autochthonous models that predated the trial and informed its design288. While preclinical models remain far from perfect, they do offer valuable insights into potential mechanisms of failure when trials are unsuccessful. One notable example of an ultimately unsuccessful but informative clinical paradigm came by way of a series of pre-clinical studies proposing the efficacy of systemic therapy would improve through efforts to penetrate the dense fibrosis and high interstitial pressure promoted by the CAFs in pancreatic tumors140,158. Subsequently, attempts to inactivate CAFs through sonic hedgehog inhibition proved ineffective in patients289, and preclinical studies that were conducted after the fact pinpointed reasons for this failure159,290.
Even rigorously conducted preclinical studies are not predictive of success upon subsequent clinical translation if they deviate significantly from the preclinical “recipe”. An unfortunate example of this pertains to the trials of hyaluronic acid targeted therapies in patients with advanced PADC, which used dosages that were three orders of magnitude lower than what garnered improved survival in animal models291. Moving forward, it is imperative that clinical trials are only implemented when preceded by rigorous and efficacious preclinical studies in biologically relevant models. Further, trials should recapitulate methodologies and read outs that generated success in model systems. Finally, trials should be designed in a manner where we “learn from every patient” through the implementation of a multitude of tissue and blood-based correlative analyses. In fact, partly driven by the challenging trajectory of “conventional” drug development in PDAC, there has been an evolving shift from large, empirical trials towards those that are focused on fewer patient numbers, but which feature in-depth correlative studies that provide insights into mechanisms of response and resistance184,292,293.
In many instances, these “team science” efforts are funded by philanthropic foundations such as Stand Up to Cancer (SU2C), the Lustgarten Foundation for Pancreatic Cancer Research, the Parker Institute for Cancer Immunotherapy (PICI), and Break Through Cancer, amongst others. Pairing the most promising early phase trials with larger, multi-institutional registration efforts that feature an innovative adaptive design platform, such as PanCAN’s Precision Promise294, has the potential to completely transform the path to accelerated therapeutic approval in PDAC.
Summary:
Significant progress has been made in understanding the biology of pancreatic cancer over the past several decades, unfortunately, these have not translated into a breakthrough in clinical care for the majority of patients. We have presented several themes within this review that we believe can serve as “Hallmarks of Pancreatic Cancer Therapy” (Figure 5). These center on genomic alterations, metabolism, the tumor microenvironment, immunotherapy, and innovative clinical trial design. Importantly, approaches that encompass as many of these features as possible present the highest likelihood of success. Examples of these can be found across the spectrum of investigational compounds and clinically deployed approaches to target the genetic and immunological features of PDAC295,296, cancer metabolism297, and chemoresistance298 There are also other exciting avenues of new therapeutic development, such as cancer vaccines and antibody-drug conjugates, that we were not able to cover within the scope of this review. Overall, we are optimistic that these collective efforts will soon transform PDAC from recalcitrant to a manageable disease.
Figure 5: Hallmarks of Pancreatic Cancer Therapy.
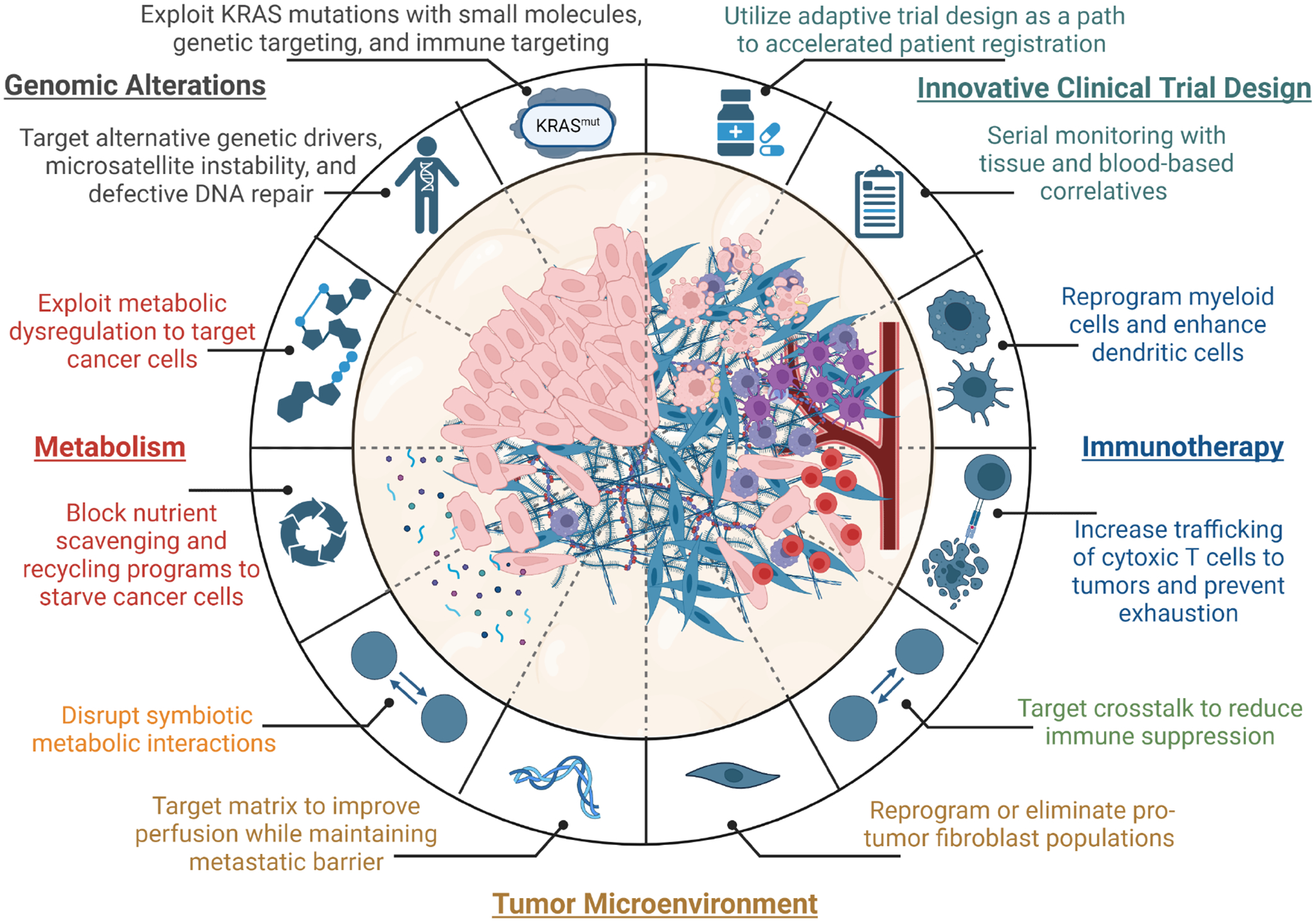
Five major themes have emerged as priorities to improve pancreatic cancer treatment. Genomic Alterations: Numerous approaches to directly target mutant RAS are entering clinical care and sequencing can identify alternative genetic drivers that can be exploited to effectively treat smaller subsets of patients. Metabolism: The rewired metabolism of pancreatic cancer cells presents opportunities to selectively target neoplastic cells. Scavenging, recycling, and metabolic crosstalk programs engaged to deal with nutrient dysregulation in pancreatic tumors can be blocked to starve cancer cells and prevent therapy resistance. Tumor Microenvironment: The characteristic pancreatic tumor microenvironment can be remodeled to increase drug perfusion by targeting stromal fibroblasts or the matrix directly, however, these strategies must not remove the barrier to cancer cell migration. Immunotherapy: Disruption of interactions between cell populations and reprogramming of myeloid cells in pancreatic tumors can relieve immune suppression. Decreases in immune suppression will likely need to be coupled with efforts to increase antigen presentation, increase cytotoxic T cell infiltration into tumors, and prevent their exhaustion. Innovative Clinical Trial Design: Clinical studies testing need to prioritize approaches most likely to succeed, continuously reassess patients on the trials using molecular correlatives, and quickly transition patients who are not responding onto different treatments. Importantly, these priorities are not mutually exclusive, and best probability of success lies in methods that will address multiple themes described above.
Acknowledgements:
While we strived to be as inclusive as possible within the confines of a broad overview of the field, we apologize to the authors of the numerous important studies we were unable to include due to brevity. C.J.H. was supported by a Sky Foundation Research Grant, a V Scholar Award (V2021-026), a UCI Anti-Cancer Challenge Pilot Award, a University of California Pancreatic Cancer Consortium Pilot Award, R00CA241357, and the Chao Family Comprehensive Cancer Center support grant P30CA062203; C.A.L. by R37CA237421, R01CA244931, and R01CA248160; MPM by R01CA271510, R01CA264843, and R01CA260752 and by the Pancreatic Cancer Action Network; C.A.L. and M.P.M. were also supported by the UMCCC Core Grant (P30CA046592) and U01CA274154. A.M. is supported by the MD Anderson Pancreatic Cancer Moon Shot Program, the Sheikh Khalifa Bin Zayed Al-Nahyan Foundation and NIH (U01CA200468, U24CA274274, R01CA220236). A.M., C.A.L and M.P.M were also supported by U54CA274371 TBEL consortium award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest:
C.A.L. has received consulting fees from Astellas Pharmaceuticals, Odyssey Therapeutics, and T-Knife Therapeutics, and is an inventor on patents pertaining to Kras regulated metabolic pathways, redox control pathways in pancreatic cancer, and targeting the GOT1-pathway as a therapeutic approach (US Patent No: 2015126580-A1, 05/07/2015; US Patent No: 20190136238, 05/09/2019; International Patent No: WO2013177426-A2, 04/23/2015). A.M. receives royalties for a pancreatic cancer biomarker test from Cosmos Wisdom Biotechnology, and this financial relationship is managed and monitored by the UTMDACC Conflict of Interest Committee. A.M. is also listed as an inventor on a patent that has been licensed by Johns Hopkins University to ThriveEarlier Detection. A.M. serves as a consultant for Freenome and Tezcat Biotechnology.
References:
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, and Matrisian LM (2014). Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 74, 2913–2921. 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Wehner MR, Matrisian LM, and Nead KT (2021). Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw Open 4, e214708. 10.1001/jamanetworkopen.2021.4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, Neale RE, Tempero M, Tuveson DA, Hruban RH, and Neoptolemos JP (2016). Pancreatic cancer. Nat Rev Dis Primers 2, 16022. 10.1038/nrdp.2016.22. [DOI] [PubMed] [Google Scholar]
- 4.Petersen GM (2016). Familial pancreatic cancer. Semin Oncol 43, 548–553. 10.1053/j.seminoncol.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cronin KA, Scott S, Firth AU, Sung H, Henley SJ, Sherman RL, Siegel RL, Anderson RN, Kohler BA, Benard VB, et al. (2022). Annual report to the nation on the status of cancer, part 1: National cancer statistics. Cancer. 10.1002/cncr.34479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jemal A, Siegel R, Xu J, and Ward E (2010). Cancer statistics, 2010. CA Cancer J Clin 60, 277–300. 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 7.Siegel RL, Miller KD, Wagle NS, and Jemal A (2023). Cancer statistics, 2023. CA Cancer J Clin 73, 17–48. 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- 8.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, et al. (2011). FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364, 1817–1825. 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 9.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et al. (2013). Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369, 1691–1703. 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang-Gillam A, Li CP, Bodoky G, Dean A, Shan YS, Jameson G, Macarulla T, Lee KH, Cunningham D, Blanc JF, et al. (2016). Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 387, 545–557. 10.1016/S0140-6736(15)00986-1. [DOI] [PubMed] [Google Scholar]
- 11.Groot VP, Rezaee N, Wu W, Cameron JL, Fishman EK, Hruban RH, Weiss MJ, Zheng L, Wolfgang CL, and He J (2018). Patterns, Timing, and Predictors of Recurrence Following Pancreatectomy for Pancreatic Ductal Adenocarcinoma. Ann Surg 267, 936–945. 10.1097/SLA.0000000000002234. [DOI] [PubMed] [Google Scholar]
- 12.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, et al. (2012). EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361. 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neoptolemos JP, Palmer DH, Ghaneh P, Psarelli EE, Valle JW, Halloran CM, Faluyi O, O’Reilly DA, Cunningham D, Wadsley J, et al. (2017). Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): a multicentre, open-label, randomised, phase 3 trial. Lancet 389, 1011–1024. 10.1016/S0140-6736(16)32409-6. [DOI] [PubMed] [Google Scholar]
- 14.Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, Chone L, Francois E, Artru P, Biagi JJ, et al. (2018). FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N Engl J Med 379, 2395–2406. 10.1056/NEJMoa1809775. [DOI] [PubMed] [Google Scholar]
- 15.Tempero MA, Pelzer U, O’Reilly EM, Winter J, Oh DY, Li CP, Tortora G, Chang HM, Lopez CD, Bekaii-Saab T, et al. (2022). Adjuvant nab-Paclitaxel + Gemcitabine in Resected Pancreatic Ductal Adenocarcinoma: Results From a Randomized, Open-Label, Phase III Trial. J Clin Oncol, JCO2201134. 10.1200/JCO.22.01134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mavros MN, Moris D, Karanicolas PJ, Katz MHG, O’Reilly EM, and Pawlik TM (2021). Clinical Trials of Systemic Chemotherapy for Resectable Pancreatic Cancer: A Review. JAMA Surg 156, 663–672. 10.1001/jamasurg.2021.0149. [DOI] [PubMed] [Google Scholar]
- 17.Cloyd JM, Wang H, Egger ME, Tzeng CD, Prakash LR, Maitra A, Varadhachary GR, Shroff R, Javle M, Fogelman D, et al. (2017). Association of Clinical Factors With a Major Pathologic Response Following Preoperative Therapy for Pancreatic Ductal Adenocarcinoma. JAMA Surg 152, 1048–1056. 10.1001/jamasurg.2017.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Versteijne E, van Dam JL, Suker M, Janssen QP, Groothuis K, Akkermans-Vogelaar JM, Besselink MG, Bonsing BA, Buijsen J, Busch OR, et al. (2022). Neoadjuvant Chemoradiotherapy Versus Upfront Surgery for Resectable and Borderline Resectable Pancreatic Cancer: Long-Term Results of the Dutch Randomized PREOPANC Trial. J Clin Oncol 40, 1220–1230. 10.1200/JCO.21.02233. [DOI] [PubMed] [Google Scholar]
- 19.Blackford AL, Canto MI, Klein AP, Hruban RH, and Goggins M (2020). Recent Trends in the Incidence and Survival of Stage 1A Pancreatic Cancer: A Surveillance, Epidemiology, and End Results Analysis. J Natl Cancer Inst 112, 1162–1169. 10.1093/jnci/djaa004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singhi AD, Koay EJ, Chari ST, and Maitra A (2019). Early Detection of Pancreatic Cancer: Opportunities and Challenges. Gastroenterology 156, 2024–2040. 10.1053/j.gastro.2019.01.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dbouk M, Katona BW, Brand RE, Chak A, Syngal S, Farrell JJ, Kastrinos F, Stoffel EM, Blackford AL, Rustgi AK, et al. (2022). The Multicenter Cancer of Pancreas Screening Study: Impact on Stage and Survival. J Clin Oncol 40, 3257–3266. 10.1200/JCO.22.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Wilde RF, Edil BH, Hruban RH, and Maitra A (2012). Well-differentiated pancreatic neuroendocrine tumors: from genetics to therapy. Nat Rev Gastroenterol Hepatol 9, 199–208. 10.1038/nrgastro.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maitra A, Adsay NV, Argani P, Iacobuzio-Donahue C, De Marzo A, Cameron JL, Yeo CJ, and Hruban RH (2003). Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol 16, 902–912. 10.1097/01.MP.0000086072.56290.FB. [DOI] [PubMed] [Google Scholar]
- 24.Overbeek KA, Cahen DL, Canto MI, and Bruno MJ (2016). Surveillance for neoplasia in the pancreas. Best Pract Res Clin Gastroenterol 30, 971–986. 10.1016/j.bpg.2016.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matthaei H, Schulick RD, Hruban RH, and Maitra A (2011). Cystic precursors to invasive pancreatic cancer. Nat Rev Gastroenterol Hepatol 8, 141–150. 10.1038/nrgastro.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Makohon-Moore AP, Matsukuma K, Zhang M, Reiter JG, Gerold JM, Jiao Y, Sikkema L, Attiyeh MA, Yachida S, Sandone C, et al. (2018). Precancerous neoplastic cells can move through the pancreatic ductal system. Nature 561, 201–205. 10.1038/s41586-018-0481-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, et al. (2010). Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117. 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. (2003). Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4, 437–450. 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 29.Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D, and DePinho RA (2006). Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 20, 3130–3146. 10.1101/gad.1478706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, and Tuveson DA (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 31.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, and DePinho RA (2003). Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 17, 3112–3126. 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qiu W, Sahin F, Iacobuzio-Donahue CA, Garcia-Carracedo D, Wang WM, Kuo CY, Chen D, Arking DE, Lowy AM, Hruban RH, et al. (2011). Disruption of p16 and activation of Kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget 2, 862–873. 10.18632/oncotarget.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Izeradjene K, Combs C, Best M, Gopinathan A, Wagner A, Grady WM, Deng CX, Hruban RH, Adsay NV, Tuveson DA, and Hingorani SR (2007). Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 11, 229–243. 10.1016/j.ccr.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 34.Ideno N, Yamaguchi H, Ghosh B, Gupta S, Okumura T, Steffen DJ, Fisher CG, Wood LD, Singhi AD, Nakamura M, et al. (2018). GNAS(R201C) Induces Pancreatic Cystic Neoplasms in Mice That Express Activated KRAS by Inhibiting YAP1 Signaling. Gastroenterology 155, 1593–1607 e1512. 10.1053/j.gastro.2018.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patra KC, Kato Y, Mizukami Y, Widholz S, Boukhali M, Revenco I, Grossman EA, Ji F, Sadreyev RI, Liss AS, et al. (2018). Mutant GNAS drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat Cell Biol 20, 811–822. 10.1038/s41556-018-0122-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hosein AN, Dangol G, Okumura T, Roszik J, Rajapakshe K, Siemann M, Zaid M, Ghosh B, Monberg M, Guerrero PA, et al. (2022). Loss of Rnf43 Accelerates Kras-Mediated Neoplasia and Remodels the Tumor Immune Microenvironment in Pancreatic Adenocarcinoma. Gastroenterology 162, 1303–1318 e1318. 10.1053/j.gastro.2021.12.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Kaur S, Huang Y, Fahrmann JF, Rinaudo JA, Hanash SM, Batra SK, Singhi AD, Brand RE, Maitra A, and Haab BB (2020). Biomarkers and Strategy to Detect Preinvasive and Early Pancreatic Cancer: State of the Field and the Impact of the EDRN. Cancer Epidemiol Biomarkers Prev 29, 2513–2523. 10.1158/1055-9965.EPI-20-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fahrmann JF, Schmidt CM, Mao X, Irajizad E, Loftus M, Zhang J, Patel N, Vykoukal J, Dennison JB, Long JP, et al. (2021). Lead-Time Trajectory of CA19–9 as an Anchor Marker for Pancreatic Cancer Early Detection. Gastroenterology 160, 1373–1383 e1376. 10.1053/j.gastro.2020.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chari ST, Kelly K, Hollingsworth MA, Thayer SP, Ahlquist DA, Andersen DK, Batra SK, Brentnall TA, Canto M, Cleeter DF, et al. (2015). Early detection of sporadic pancreatic cancer: summative review. Pancreas 44, 693–712. 10.1097/MPA.0000000000000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singhi AD, and Wood LD (2021). Early detection of pancreatic cancer using DNA-based molecular approaches. Nat Rev Gastroenterol Hepatol 18, 457–468. 10.1038/s41575-021-00470-0. [DOI] [PubMed] [Google Scholar]
- 41.Pereira SP, Oldfield L, Ney A, Hart PA, Keane MG, Pandol SJ, Li D, Greenhalf W, Jeon CY, Koay EJ, et al. (2020). Early detection of pancreatic cancer. Lancet Gastroenterol Hepatol 5, 698–710. 10.1016/S2468-1253(19)30416-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paniccia A, Polanco PM, Boone BA, Wald AI, McGrath K, Brand RE, Khalid A, Kubiliun N, O’Broin-Lennon AM, Park WG, et al. (2023). Prospective, Multi-Institutional, Real-Time Next-Generation Sequencing of Pancreatic Cyst Fluid Reveals Diverse Genomic Alterations That Improve the Clinical Management of Pancreatic Cysts. Gastroenterology 164, 117–133 e117. 10.1053/j.gastro.2022.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loomans-Kropp HA, Umar A, Minasian LM, and Pinsky PF (2022). Multi-Cancer Early Detection Tests: Current Progress and Future Perspectives. Cancer Epidemiol Biomarkers Prev 31, 512–514. 10.1158/1055-9965.EPI-21-1387. [DOI] [PubMed] [Google Scholar]
- 44.Force USPST, Owens DK, Davidson KW, Krist AH, Barry MJ, Cabana M, Caughey AB, Curry SJ, Doubeni CA, Epling JW Jr., et al. (2019). Screening for Pancreatic Cancer: US Preventive Services Task Force Reaffirmation Recommendation Statement. JAMA 322, 438–444. 10.1001/jama.2019.10232. [DOI] [PubMed] [Google Scholar]
- 45.Cancer Genome Atlas Research Network. Electronic address, a.a.d.h.e., and Cancer Genome Atlas Research, N. (2017). Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 32, 185–203 e113. 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, Hruban RH, Maitra A, Kinzler K, Vogelstein B, and Goggins M (2012). Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 142, 730–733 e739. 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujikura K, Hosoda W, Felsenstein M, Song Q, Reiter JG, Zheng L, Beleva Guthrie V, Rincon N, Dal Molin M, Dudley J, et al. (2021). Multiregion whole-exome sequencing of intraductal papillary mucinous neoplasms reveals frequent somatic KLF4 mutations predominantly in low-grade regions. Gut 70, 928–939. 10.1136/gutjnl-2020-321217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. (2008). Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801–1806. 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al. (2015). Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518, 495–501. 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 51.Sohn TA, Yeo CJ, Cameron JL, Iacobuzio-Donahue CA, Hruban RH, and Lillemoe KD (2001). Intraductal papillary mucinous neoplasms of the pancreas: an increasingly recognized clinicopathologic entity. Ann Surg 234, 313–321; discussion 321–312. 10.1097/00000658-200109000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barriga FM, Tsanov KM, Ho Y-J, Sohail N, Zhang A, Baslan T, Wuest AN, Del Priore I, Meškauskaitė B, Livshits G, et al. (2022). MACHETE identifies interferon-encompassing chromosome 9p21.3 deletions as mediators of immune evasion and metastasis. Nature Cancer. 10.1038/s43018-022-00443-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Han G, Yang G, Hao D, Lu Y, Thein K, Simpson BS, Chen J, Sun R, Alhalabi O, Wang R, et al. (2021). 9p21 loss confers a cold tumor immune microenvironment and primary resistance to immune checkpoint therapy. Nat Commun 12, 5606. 10.1038/s41467-021-25894-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blackford A, Serrano OK, Wolfgang CL, Parmigiani G, Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Eshleman JR, et al. (2009). SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res 15, 4674–4679. 10.1158/1078-0432.CCR-09-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, Vilardell F, Wang Z, Keller JW, Banerjee P, et al. (2009). DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol 27, 1806–1813. 10.1200/JCO.2008.17.7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ecker BL, Court CM, Janssen QP, Tao AJ, D’Angelica MI, Drebin JA, Gonen M, O’Reilly EM, Jarnagin WR, Wei AC, and David, M.R.C.f.P.C.R.G.a.M.S.K.C.C. (2022). Alterations in Somatic Driver Genes Are Associated with Response to Neoadjuvant FOLFIRINOX in Patients with Localized Pancreatic Ductal Adenocarcinoma. J Am Coll Surg 235, 342–349. 10.1097/XCS.0000000000000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morris J.P.t., Yashinskie JJ, Koche R, Chandwani R, Tian S, Chen CC, Baslan T, Marinkovic ZS, Sanchez-Rivera FJ, Leach SD, et al. (2019). alpha-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 573, 595–599. 10.1038/s41586-019-1577-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rajeshkumar NV, Dutta P, Yabuuchi S, de Wilde RF, Martinez GV, Le A, Kamphorst JJ, Rabinowitz JD, Jain SK, Hidalgo M, et al. (2015). Therapeutic Targeting of the Warburg Effect in Pancreatic Cancer Relies on an Absence of p53 Function. Cancer Res 75, 3355–3364. 10.1158/0008-5472.CAN-15-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weissmueller S, Manchado E, Saborowski M, Morris J.P.t., Wagenblast E, Davis CA, Moon SH, Pfister NT, Tschaharganeh DF, Kitzing T, et al. (2014). Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell 157, 382–394. 10.1016/j.cell.2014.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baslan T, Morris J.P.t., Zhao Z, Reyes J, Ho YJ, Tsanov KM, Bermeo J, Tian S, Zhang S, Askan G, et al. (2022). Ordered and deterministic cancer genome evolution after p53 loss. Nature 608, 795–802. 10.1038/s41586-022-05082-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Escobar-Hoyos LF, Penson A, Kannan R, Cho H, Pan CH, Singh RK, Apken LH, Hobbs GA, Luo R, Lecomte N, et al. (2020). Altered RNA Splicing by Mutant p53 Activates Oncogenic RAS Signaling in Pancreatic Cancer. Cancer Cell 38, 198–211 e198. 10.1016/j.ccell.2020.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, and Settleman J (2009). A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 15, 489–500. 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, Rakshit S, Flannagan KS, Adsay NV, and Pasca di Magliano M (2012). Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 122, 639–653. 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ying HQ, Kimmelman AC, Lyssiotis CA, Hua SJ, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang HL, Coloff JL, et al. (2012). Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 149, 656–670. 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Collins MA, Brisset JC, Zhang Y, Bednar F, Pierre J, Heist KA, Galban CJ, Galban S, and di Magliano MP (2012). Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS One 7, e49707. 10.1371/journal.pone.0049707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Collins MA, Yan W, Sebolt-Leopold JS, and Pasca di Magliano M (2014). MAPK signaling is required for dedifferentiation of acinar cells and development of pancreatic intraepithelial neoplasia in mice. Gastroenterology 146, 822–834 e827. 10.1053/j.gastro.2013.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, et al. (2014). Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 158, 185–197. 10.1016/j.cell.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, et al. (2014). Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632. 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hallin J, Bowcut V, Calinisan A, Briere DM, Hargis L, Engstrom LD, Laguer J, Medwid J, Vanderpool D, Lifset E, et al. (2022). Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nat Med 28, 2171–2182. 10.1038/s41591-022-02007-7. [DOI] [PubMed] [Google Scholar]
- 70.Kim D, Xue JY, and Lito P (2020). Targeting KRAS(G12C): From Inhibitory Mechanism to Modulation of Antitumor Effects in Patients. Cell 183, 850–859. 10.1016/j.cell.2020.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hofmann MH, Gerlach D, Misale S, Petronczki M, and Kraut N (2022). Expanding the Reach of Precision Oncology by Drugging All KRAS Mutants. Cancer Discov 12, 924–937. 10.1158/2159-8290.CD-21-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, Falchook GS, Price TJ, Sacher A, Denlinger CS, et al. (2020). KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med 383, 1207–1217. 10.1056/NEJMoa1917239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Janne PA, Riely GJ, Gadgeel SM, Heist RS, Ou SI, Pacheco JM, Johnson ML, Sabari JK, Leventakos K, Yau E, et al. (2022). Adagrasib in Non-Small-Cell Lung Cancer Harboring a KRAS(G12C) Mutation. N Engl J Med 387, 120–131. 10.1056/NEJMoa2204619. [DOI] [PubMed] [Google Scholar]
- 74.Waters AM, and Der CJ (2018). KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb Perspect Med 8. 10.1101/cshperspect.a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Strickler JH, Satake H, George TJ, Yaeger R, Hollebecque A, Garrido-Laguna I, Schuler M, Burns TF, Coveler AL, Falchook GS, et al. (2022). Sotorasib in KRAS p.G12C-Mutated Advanced Pancreatic Cancer. N Engl J Med. 10.1056/NEJMoa2208470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tsai YS, Woodcock MG, Azam SH, Thorne LB, Kanchi KL, Parker JS, Vincent BG, and Pecot CV (2022). Rapid idiosyncratic mechanisms of clinical resistance to KRAS G12C inhibition. J Clin Invest 132. 10.1172/JCI155523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, Michel AG, Syed MU, Fella KA, Sakhi M, et al. (2021). Clinical Acquired Resistance to KRAS(G12C) Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov 11, 1913–1922. 10.1158/2159-8290.CD-21-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, Johnson ML, Heist RS, Patil T, Riely GJ, et al. (2021). Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N Engl J Med 384, 2382–2393. 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kemp SB, Cheng N, Markosyan N, Sor R, Kim IK, Hallin J, Shoush J, Quinones L, Brown NV, Bassett JB, et al. (2022). Efficacy of a small molecule inhibitor of KrasG12D in immunocompetent models of pancreatic cancer. Cancer Discov. 10.1158/2159-8290.CD-22-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yaeger R, Weiss J, Pelster MS, Spira AI, Barve M, Ou SI, Leal TA, Bekaii-Saab TS, Paweletz CP, Heavey GA, et al. (2022). Adagrasib with or without Cetuximab in Colorectal Cancer with Mutated KRAS G12C. N Engl J Med. 10.1056/NEJMoa2212419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nagashima T, Inamura K, Nishizono Y, Suzuki A, Tanaka H, Yoshinari T, and Yamanaka Y (2022). ASP3082, a First-in-class novel KRAS G12D degrader, exhibits remarkable anti-tumor activity in KRAS G12D mutated cancer models. European Journal of Cancer 174, S30. 10.1016/S0959-8049(22)00881-4. [DOI] [Google Scholar]
- 82.Mendt M, Kamerkar S, Sugimoto H, McAndrews KM, Wu CC, Gagea M, Yang S, Blanko EVR, Peng Q, Ma X, et al. (2018). Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 3. 10.1172/jci.insight.99263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leidner R, Sanjuan Silva N, Huang H, Sprott D, Zheng C, Shih YP, Leung A, Payne R, Sutcliffe K, Cramer J, et al. (2022). Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N Engl J Med 386, 2112–2119. 10.1056/NEJMoa2119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, Schrock A, Campbell B, Shlien A, Chmielecki J, et al. (2017). Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 9, 34. 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhen DB, Rabe KG, Gallinger S, Syngal S, Schwartz AG, Goggins MG, Hruban RH, Cote ML, McWilliams RR, Roberts NJ, et al. (2015). BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genet Med 17, 569–577. 10.1038/gim.2014.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lord CJ, and Ashworth A (2017). PARP inhibitors: Synthetic lethality in the clinic. Science 355, 1152–1158. 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park JO, Hochhauser D, Arnold D, Oh DY, et al. (2019). Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N Engl J Med 381, 317–327. 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kindler HL, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park JO, Hochhauser D, Arnold D, Oh DY, et al. (2022). Overall Survival Results From the POLO Trial: A Phase III Study of Active Maintenance Olaparib Versus Placebo for Germline BRCA-Mutated Metastatic Pancreatic Cancer. J Clin Oncol, JCO2101604. 10.1200/JCO.21.01604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Momtaz P, O’Connor CA, Chou JF, Capanu M, Park W, Bandlamudi C, Berger MF, Kelsen DP, Suehnholz SP, Chakravarty D, et al. (2021). Pancreas cancer and BRCA: A critical subset of patients with improving therapeutic outcomes. Cancer 127, 4393–4402. 10.1002/cncr.33812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brown TJ, and Reiss KA (2021). PARP Inhibitors in Pancreatic Cancer. Cancer J 27, 465–475. 10.1097/PPO.0000000000000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hsu FC, Roberts NJ, Childs E, Porter N, Rabe KG, Borgida A, Ukaegbu C, Goggins MG, Hruban RH, Zogopoulos G, et al. (2021). Risk of Pancreatic Cancer Among Individuals With Pathogenic Variants in the ATM Gene. JAMA Oncol 7, 1664–1668. 10.1001/jamaoncol.2021.3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Park W, O’Connor CA, Bandlamudi C, Forman D, Chou JF, Umeda S, Reyngold M, Varghese AM, Keane F, Balogun F, et al. (2022). Clinico-genomic Characterization of ATM and HRD in Pancreas Cancer: Application for Practice. Clin Cancer Res 28, 4782–4792. 10.1158/1078-0432.CCR-22-1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Javle M, Shacham-Shmueli E, Xiao L, Varadhachary G, Halpern N, Fogelman D, Boursi B, Uruba S, Margalit O, Wolff RA, and Golan T (2021). Olaparib Monotherapy for Previously Treated Pancreatic Cancer With DNA Damage Repair Genetic Alterations Other Than Germline BRCA Variants: Findings From 2 Phase 2 Nonrandomized Clinical Trials. JAMA Oncol 7, 693–699. 10.1001/jamaoncol.2021.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shain AH, Giacomini CP, Matsukuma K, Karikari CA, Bashyam MD, Hidalgo M, Maitra A, and Pollack JR (2012). Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc Natl Acad Sci U S A 109, E252–259. 10.1073/pnas.1114817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kalisz M, Bernardo E, Beucher A, Maestro MA, Del Pozo N, Millan I, Haeberle L, Schlensog M, Safi SA, Knoefel WT, et al. (2020). HNF1A recruits KDM6A to activate differentiated acinar cell programs that suppress pancreatic cancer. EMBO J 39, e102808. 10.15252/embj.2019102808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yi Z, Wei S, Jin L, Jeyarajan S, Yang J, Gu Y, Kim HS, Schechter S, Lu S, Paulsen MT, et al. (2022). KDM6A Regulates Cell Plasticity and Pancreatic Cancer Progression by Noncanonical Activin Pathway. Cell Mol Gastroenterol Hepatol 13, 643–667. 10.1016/j.jcmgh.2021.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Andricovich J, Perkail S, Kai Y, Casasanta N, Peng W, and Tzatsos A (2018). Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell 33, 512–526 e518. 10.1016/j.ccell.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Livshits G, Alonso-Curbelo D, Morris J.P.t., Koche R, Saborowski M, Wilkinson JE, and Lowe SW (2018). Arid1a restrains Kras-dependent changes in acinar cell identity. Elife 7. 10.7554/eLife.35216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang SC, Nassour I, Xiao S, Zhang S, Luo X, Lee J, Li L, Sun X, Nguyen LH, Chuang JC, et al. (2019). SWI/SNF component ARID1A restrains pancreatic neoplasia formation. Gut 68, 1259–1270. 10.1136/gutjnl-2017-315490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.von Figura G, Fukuda A, Roy N, Liku ME, Morris Iv JP, Kim GE, Russ HA, Firpo MA, Mulvihill SJ, Dawson DW, et al. (2014). The chromatin regulator Brg1 suppresses formation of intraductal papillary mucinous neoplasm and pancreatic ductal adenocarcinoma. Nat Cell Biol 16, 255–267. 10.1038/ncb2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Roy N, Malik S, Villanueva KE, Urano A, Lu X, Von Figura G, Seeley ES, Dawson DW, Collisson EA, and Hebrok M (2015). Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev 29, 658–671. 10.1101/gad.256628.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ferri-Borgogno S, Barui S, McGee AM, Griffiths T, Singh PK, Piett CG, Ghosh B, Bhattacharyya S, Singhi A, Pradhan K, et al. (2020). Paradoxical Role of AT-rich Interactive Domain 1A in Restraining Pancreatic Carcinogenesis. Cancers (Basel) 12. 10.3390/cancers12092695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tomihara H, Carbone F, Perelli L, Huang JK, Soeung M, Rose JL, Robinson FS, Lissanu Deribe Y, Feng N, Takeda M, et al. (2021). Loss of ARID1A Promotes Epithelial-Mesenchymal Transition and Sensitizes Pancreatic Tumors to Proteotoxic Stress. Cancer Res 81, 332–343. 10.1158/0008-5472.CAN-19-3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Singhi AD, George B, Greenbowe JR, Chung J, Suh J, Maitra A, Klempner SJ, Hendifar A, Milind JM, Golan T, et al. (2019). Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology 156, 2242–2253 e2244. 10.1053/j.gastro.2019.02.037. [DOI] [PubMed] [Google Scholar]
- 105.Aguirre AJ, Nowak JA, Camarda ND, Moffitt RA, Ghazani AA, Hazar-Rethinam M, Raghavan S, Kim J, Brais LK, Ragon D, et al. (2018). Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov 8, 1096–1111. 10.1158/2159-8290.CD-18-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Heining C, Horak P, Uhrig S, Codo PL, Klink B, Hutter B, Frohlich M, Bonekamp D, Richter D, Steiger K, et al. (2018). NRG1 Fusions in KRAS Wild-Type Pancreatic Cancer. Cancer Discov 8, 1087–1095. 10.1158/2159-8290.CD-18-0036. [DOI] [PubMed] [Google Scholar]
- 107.Singhi AD, Ali SM, Lacy J, Hendifar A, Nguyen K, Koo J, Chung JH, Greenbowe J, Ross JS, Nikiforova MN, et al. (2017). Identification of Targetable ALK Rearrangements in Pancreatic Ductal Adenocarcinoma. J Natl Compr Canc Netw 15, 555–562. 10.6004/jnccn.2017.0058. [DOI] [PubMed] [Google Scholar]
- 108.Topham JT, Tsang ES, Karasinska JM, Metcalfe A, Ali H, Kalloger SE, Csizmok V, Williamson LM, Titmuss E, Nielsen K, et al. (2022). Integrative analysis of KRAS wildtype metastatic pancreatic ductal adenocarcinoma reveals mutation and expression-based similarities to cholangiocarcinoma. Nat Commun 13, 5941. 10.1038/s41467-022-33718-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Philip PA, Azar I, Xiu J, Hall MJ, Hendifar AE, Lou E, Hwang JJ, Gong J, Feldman R, Ellis M, et al. (2022). Molecular Characterization of KRAS Wild-type Tumors in Patients with Pancreatic Adenocarcinoma. Clin Cancer Res 28, 2704–2714. 10.1158/1078-0432.CCR-21-3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Means AL, Meszoely IM, Suzuki K, Miyamoto Y, Rustgi AK, Coffey RJ Jr., Wright CV, Stoffers DA, and Leach SD (2005). Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development 132, 3767–3776. 10.1242/dev.01925. [DOI] [PubMed] [Google Scholar]
- 111.Miyamoto Y, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, Sriuranpong V, Iso T, Meszoely IM, Wolfe MS, et al. (2003). Notch mediates TGF alphainduced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 3, 565–576. 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 112.Desai BM, Oliver-Krasinski J, De Leon DD, Farzad C, Hong N, Leach SD, and Stoffers DA (2007). Preexisting pancreatic acinar cells contribute to acinar cell, but not islet beta cell, regeneration. J Clin Invest 117, 971–977. 10.1172/JCI29988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, Feldmann G, Stoffers DA, Konieczny SF, Leach SD, and Maitra A (2008). Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A 105, 18913–18918. 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP, and Barbacid M (2007). Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11, 291–302. 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 115.Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, Vasile E, DePinho RA, and Jacks T (2009). Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 16, 379–389. 10.1016/j.ccr.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.De La OJ, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB, and Murtaugh LC (2008). Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A 105, 18907–18912. 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Carriere C, Seeley ES, Goetze T, Longnecker DS, and Korc M (2007). The Nestin progenitor lineage is the compartment of origin for pancreatic intraepithelial neoplasia. Proc Natl Acad Sci U S A 104, 4437–4442. 10.1073/pnas.0701117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris J.P.t., Pan FC, Akiyama H, Wright CV, Jensen K, et al. (2012). Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 22, 737–750. 10.1016/j.ccr.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.von Figura G, Morris J.P.t., Wright CV, and Hebrok M (2014). Nr5a2 maintains acinar cell differentiation and constrains oncogenic Kras-mediated pancreatic neoplastic initiation. Gut 63, 656–664. 10.1136/gutjnl-2012-304287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Flandez M, Cendrowski J, Canamero M, Salas A, del Pozo N, Schoonjans K, and Real FX (2014). Nr5a2 heterozygosity sensitises to, and cooperates with, inflammation in KRas(G12V)-driven pancreatic tumourigenesis. Gut 63, 647–655. 10.1136/gutjnl-2012-304381. [DOI] [PubMed] [Google Scholar]
- 121.Shi G, Zhu L, Sun Y, Bettencourt R, Damsz B, Hruban RH, and Konieczny SF (2009). Loss of the acinar-restricted transcription factor Mist1 accelerates Kras-induced pancreatic intraepithelial neoplasia. Gastroenterology 136, 1368–1378. 10.1053/j.gastro.2008.12.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Krah NM, De La OJ, Swift GH, Hoang CQ, Willet SG, Chen Pan F, Cash GM, Bronner MP, Wright CV, MacDonald RJ, and Murtaugh LC (2015). The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. Elife 4. 10.7554/eLife.07125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shi G, DiRenzo D, Qu C, Barney D, Miley D, and Konieczny SF (2013). Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene 32, 1950–1958. 10.1038/onc.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kopp JL, Dubois CL, Schaeffer DF, Samani A, Taghizadeh F, Cowan RW, Rhim AD, Stiles BL, Valasek M, and Sander M (2018). Loss of Pten and Activation of Kras Synergistically Induce Formation of Intraductal Papillary Mucinous Neoplasia From Pancreatic Ductal Cells in Mice. Gastroenterology 154, 1509–1523 e1505. 10.1053/j.gastro.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lee AYL, Dubois CL, Sarai K, Zarei S, Schaeffer DF, Sander M, and Kopp JL (2019). Cell of origin affects tumour development and phenotype in pancreatic ductal adenocarcinoma. Gut 68, 487–498. 10.1136/gutjnl-2017-314426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bailey JM, Hendley AM, Lafaro KJ, Pruski MA, Jones NC, Alsina J, Younes M, Maitra A, McAllister F, Iacobuzio-Donahue CA, and Leach SD (2016). p53 mutations cooperate with oncogenic Kras to promote adenocarcinoma from pancreatic ductal cells. Oncogene 35, 4282–4288. 10.1038/onc.2015.441. [DOI] [PubMed] [Google Scholar]
- 127.Flowers BM, Xu H, Mulligan AS, Hanson KJ, Seoane JA, Vogel H, Curtis C, Wood LD, and Attardi LD (2021). Cell of Origin Influences Pancreatic Cancer Subtype. Cancer Discov 11, 660–677. 10.1158/2159-8290.CD-20-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shi C, Pan FC, Kim JN, Washington MK, Padmanabhan C, Meyer CT, Kopp JL, Sander M, Gannon M, Beauchamp RD, et al. (2019). Differential Cell Susceptibilities to Kras(G12D) in the Setting of Obstructive Chronic Pancreatitis. Cell Mol Gastroenterol Hepatol 8, 579–594. 10.1016/j.jcmgh.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Espinet E, Gu Z, Imbusch CD, Giese NA, Buscher M, Safavi M, Weisenburger S, Klein C, Vogel V, Falcone M, et al. (2021). Aggressive PDACs Show Hypomethylation of Repetitive Elements and the Execution of an Intrinsic IFN Program Linked to a Ductal Cell of Origin. Cancer Discov 11, 638–659. 10.1158/2159-8290.CD-20-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, et al. (2011). Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 17, 500–503. 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH, et al. (2015). Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 47, 1168–1178. 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Maurer C, Holmstrom SR, He J, Laise P, Su T, Ahmed A, Hibshoosh H, Chabot JA, Oberstein PE, Sepulveda AR, et al. (2019). Experimental microdissection enables functional harmonisation of pancreatic cancer subtypes. Gut 68, 1034–1043. 10.1136/gutjnl-2018-317706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Puleo F, Nicolle R, Blum Y, Cros J, Marisa L, Demetter P, Quertinmont E, Svrcek M, Elarouci N, Iovanna J, et al. (2018). Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 155, 1999–2013 e1993. 10.1053/j.gastro.2018.08.033. [DOI] [PubMed] [Google Scholar]
- 134.Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K, Figueroa EF, O’Kane GM, Connor AA, Denroche RE, Grant RC, McLeod J, et al. (2020). Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet 52, 231–240. 10.1038/s41588-019-0566-9. [DOI] [PubMed] [Google Scholar]
- 135.Collisson EA, Bailey P, Chang DK, and Biankin AV (2019). Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol 16, 207–220. 10.1038/s41575-019-0109-y. [DOI] [PubMed] [Google Scholar]
- 136.Somerville TDD, Xu Y, Miyabayashi K, Tiriac H, Cleary CR, Maia-Silva D, Milazzo JP, Tuveson DA, and Vakoc CR (2018). TP63-Mediated Enhancer Reprogramming Drives the Squamous Subtype of Pancreatic Ductal Adenocarcinoma. Cell Rep 25, 1741–1755 e1747. 10.1016/j.celrep.2018.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Raghavan S, Winter PS, Navia AW, Williams HL, DenAdel A, Lowder KE, Galvez-Reyes J, Kalekar RL, Mulugeta N, Kapner KS, et al. (2021). Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 184, 6119–6137 e6126. 10.1016/j.cell.2021.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Aung KL, Fischer SE, Denroche RE, Jang GH, Dodd A, Creighton S, Southwood B, Liang SB, Chadwick D, Zhang A, et al. (2018). Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin Cancer Res 24, 1344–1354. 10.1158/1078-0432.CCR-17-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Velez-Delgado A, Donahue KL, Brown ZKL, Du W, Irizarry-Negron V, Menjivar RE, Lasse Opsahl EL, Steele NG, The S, Lazarus J, et al. (2022). Extrinsic KRAS signaling shapes the pancreatic microenvironment through fibroblast reprogramming. Cell Mol Gastroenterol Hepatol. 10.1016/j.jcmgh.2022.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, and Hingorani SR (2012). Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 21, 418–429. 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Apte MV, Park S, Phillips PA, Santucci N, Goldstein D, Kumar RK, Ramm GA, Buchler M, Friess H, McCarroll JA, et al. (2004). Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas 29, 179–187. [DOI] [PubMed] [Google Scholar]
- 142.Helms EJ, Berry MW, Chaw RC, DuFort CC, Sun D, Onate MK, Oon C, Bhattacharyya S, Sanford-Crane H, Horton W, et al. (2022). Mesenchymal Lineage Heterogeneity Underlies Nonredundant Functions of Pancreatic Cancer-Associated Fibroblasts. Cancer Discov 12, 484–501. 10.1158/2159-8290.CD-21-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Helms EJ, Berry MW, Chaw RC, DuFort CC, Sun D, Onate MK, Oon C, Bhattacharyya S, Sanford-Crane H, Horton W, et al. (2021). Mesenchymal Lineage Heterogeneity Underlies Nonredundant Functions of Pancreatic Cancer-Associated Fibroblasts. Cancer Discov. 10.1158/2159-8290.CD-21-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Garcia PE, Adoumie M, Kim EC, Zhang Y, Scales MK, El-Tawil YS, Shaikh AZ, Wen HJ, Bednar F, Allen BL, et al. (2020). Differential Contribution of Pancreatic Fibroblast Subsets to the Pancreatic Cancer Stroma. Cell Mol Gastroenterol Hepatol 10, 581–599. 10.1016/j.jcmgh.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Huang H, Wang Z, Zhang Y, Pradhan RN, Ganguly D, Chandra R, Murimwa G, Wright S, Gu X, Maddipati R, et al. (2022). Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell 40, 656–673 e657. 10.1016/j.ccell.2022.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Han L, Wu Y, Fang K, Sweeney S, Roesner UK, Parrish M, Patel K, Walter T, Piermattei J, Trimboli A, et al. (2023). The splanchnic mesenchyme is the tissue of origin for pancreatic fibroblasts during homeostasis and tumorigenesis. Nat Commun 14, 1. 10.1038/s41467-022-34464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA, Lee EJ, et al. (2017). Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 214, 579–596. 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, Preall J, and Tuveson DA (2019). IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov 9, 282–301. 10.1158/2159-8290.CD-18-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. (2019). Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov 9, 1102–1123. 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dominguez CX, Muller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, Breart B, Foreman O, Bainbridge TW, Castiglioni A, et al. (2020). Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov 10, 232–253. 10.1158/2159-8290.CD-19-0644. [DOI] [PubMed] [Google Scholar]
- 151.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. (2013). Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 110, 20212–20217. 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hutton C, Heider F, Blanco-Gomez A, Banyard A, Kononov A, Zhang X, Karim S, Paulus-Hock V, Watt D, Steele N, et al. (2021). Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell 39, 1227–1244 e1220. 10.1016/j.ccell.2021.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Krishnamurty AT, Shyer JA, Thai M, Gandham V, Buechler MB, Yang YA, Pradhan RN, Wang AW, Sanchez PL, Qu Y, et al. (2022). LRRC15(+) myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature. 10.1038/s41586-022-05272-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Shaashua L, Ben-Shmuel A, Pevsner-Fischer M, Friedman G, Levi-Galibov O, Nandakumar S, Barki D, Nevo R, Brown LE, Zhang W, et al. (2022). BRCA mutational status shapes the stromal microenvironment of pancreatic cancer linking clusterin expression in cancer associated fibroblasts with HSF1 signaling. Nat Commun 13, 6513. 10.1038/s41467-022-34081-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Vennin C, Melenec P, Rouet R, Nobis M, Cazet AS, Murphy KJ, Herrmann D, Reed DA, Lucas MC, Warren SC, et al. (2019). CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat Commun 10, 3637. 10.1038/s41467-019-10968-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Grunwald BT, Devisme A, Andrieux G, Vyas F, Aliar K, McCloskey CW, Macklin A, Jang GH, Denroche R, Romero JM, et al. (2021). Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 184, 5577–5592 e5518. 10.1016/j.cell.2021.09.022. [DOI] [PubMed] [Google Scholar]
- 157.Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, et al. (2013). Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 62, 112–U153. 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. (2009). Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461. 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, et al. (2014). Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747. 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. (2014). Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734. 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lee JJ, Perera RM, Wang H, Wu DC, Liu XS, Han S, Fitamant J, Jones PD, Ghanta KS, Kawano S, et al. (2014). Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U S A 111, E3091–3100. 10.1073/pnas.1411679111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jiang H, Torphy RJ, Steiger K, Hongo H, Ritchie AJ, Kriegsmann M, Horst D, Umetsu SE, Joseph NM, McGregor K, et al. (2020). Pancreatic ductal adenocarcinoma progression is restrained by stromal matrix. J Clin Invest 130, 4704–4709. 10.1172/JCI136760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Perez VM, Kearney JF, and Yeh JJ (2021). The PDAC Extracellular Matrix: A Review of the ECM Protein Composition, Tumor Cell Interaction, and Therapeutic Strategies. Front Oncol 11, 751311. 10.3389/fonc.2021.751311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Imamura T, Iguchi H, Manabe T, Ohshio G, Yoshimura T, Wang ZH, Suwa H, Ishigami S, and Imamura M (1995). Quantitative analysis of collagen and collagen subtypes I, III, and V in human pancreatic cancer, tumor-associated chronic pancreatitis, and alcoholic chronic pancreatitis. Pancreas 11, 357–364. 10.1097/00006676-199511000-00007. [DOI] [PubMed] [Google Scholar]
- 165.Hosein AN, Brekken RA, and Maitra A (2020). Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol 17, 487–505. 10.1038/s41575-020-0300-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chen Y, Kim J, Yang S, Wang H, Wu CJ, Sugimoto H, LeBleu VS, and Kalluri R (2021). Type I collagen deletion in alphaSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 39, 548–565 e546. 10.1016/j.ccell.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chen Y, Yang S, Tavormina J, Tampe D, Zeisberg M, Wang H, Mahadevan KK, Wu CJ, Sugimoto H, Chang CC, et al. (2022). Oncogenic collagen I homotrimers from cancer cells bind to alpha3beta1 integrin and impact tumor microbiome and immunity to promote pancreatic cancer. Cancer Cell 40, 818–834 e819. 10.1016/j.ccell.2022.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Aguilera KY, Huang H, Du W, Hagopian MM, Wang Z, Hinz S, Hwang TH, Wang H, Fleming JB, Castrillon DH, et al. (2017). Inhibition of Discoidin Domain Receptor 1 Reduces Collagen-mediated Tumorigenicity in Pancreatic Ductal Adenocarcinoma. Mol Cancer Ther 16, 2473–2485. 10.1158/1535-7163.MCT-16-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ruggeri JM, Franco-Barraza J, Sohail A, Zhang Y, Long D, Pasca di Magliano M, Cukierman E, Fridman R, and Crawford HC (2020). Discoidin Domain Receptor 1 (DDR1) Is Necessary for Tissue Homeostasis in Pancreatic Injury and Pathogenesis of Pancreatic Ductal Adenocarcinoma. Am J Pathol 190, 1735–1751. 10.1016/j.ajpath.2020.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Su H, Yang F, Fu R, Trinh B, Sun N, Liu J, Kumar A, Baglieri J, Siruno J, Le M, et al. (2022). Collagenolysis-dependent DDR1 signalling dictates pancreatic cancer outcome. Nature 610, 366–372. 10.1038/s41586-022-05169-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Aguilera KY, Rivera LB, Hur H, Carbon JG, Toombs JE, Goldstein CD, Dellinger MT, Castrillon DH, and Brekken RA (2014). Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma. Cancer Res 74, 1032–1044. 10.1158/0008-5472.CAN-13-2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ramanathan RK, McDonough SL, Philip PA, Hingorani SR, Lacy J, Kortmansky JS, Thumar J, Chiorean EG, Shields AF, Behl D, et al. (2019). Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients With Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J Clin Oncol 37, 1062–1069. 10.1200/JCO.18.01295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Van Cutsem E, Tempero MA, Sigal D, Oh DY, Fazio N, Macarulla T, Hitre E, Hammel P, Hendifar AE, Bates SE, et al. (2020). Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J Clin Oncol 38, 3185–3194. 10.1200/JCO.20.00590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, et al. (2016). Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 22, 851–860. 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lander VE, Belle JI, Kingston NL, Herndon JM, Hogg GD, Liu X, Kang LI, Knolhoff BL, Bogner SJ, Baer JM, et al. (2022). Stromal reprogramming by FAK inhibition overcomes radiation resistance to allow for immune priming and response to checkpoint blockade. Cancer Discov. 10.1158/2159-8290.CD-22-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, and Vonderheide RH (2012). Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21, 822–835. 10.1016/j.ccr.2012.04.025 S1535–6108(12)00167–5 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, and Bar-Sagi D (2012). Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 21, 836–847. 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, Cullinan DR, Luo J, Bearden AR, Lavine KJ, et al. (2017). Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 47, 597. 10.1016/j.immuni.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo JQ, Wang-Gillam A, Goedegebuure SP, Linehan DC, and DeNardo DG (2014). CSF1/CSF1R Blockade Reprograms Tumor-Infiltrating Macrophages and Improves Response to T-cell Checkpoint Immunotherapy in Pancreatic Cancer Models. Cancer Res 74, 5057–5069. 10.1158/0008-5472.Can-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Candido JB, Morton JP, Bailey P, Campbell AD, Karim SA, Jamieson T, Lapienyte L, Gopinathan A, Clark W, McGhee EJ, et al. (2018). CSF1R(+) Macrophages Sustain Pancreatic Tumor Growth through T Cell Suppression and Maintenance of Key Gene Programs that Define the Squamous Subtype. Cell Rep 23, 1448–1460. 10.1016/j.celrep.2018.03.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Panni RZ, Herndon JM, Zuo C, Hegde S, Hogg GD, Knolhoff BL, Breden MA, Li X, Krisnawan VE, Khan SQ, et al. (2019). Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci Transl Med 11. 10.1126/scitranslmed.aau9240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.DeNardo DG, Galkin A, Dupont J, Zhou L, and Bendell J (2021). GB1275, a first-in-class CD11b modulator: rationale for immunotherapeutic combinations in solid tumors. J Immunother Cancer 9. 10.1136/jitc-2021-003005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Vonderheide RH (2020). CD40 Agonist Antibodies in Cancer Immunotherapy. Annu Rev Med 71, 47–58. 10.1146/annurev-med-062518-045435. [DOI] [PubMed] [Google Scholar]
- 184.Padron LJ, Maurer DM, O’Hara MH, O’Reilly EM, Wolff RA, Wainberg ZA, Ko AH, Fisher G, Rahma O, Lyman JP, et al. (2022). Sotigalimab and/or nivolumab with chemotherapy in first-line metastatic pancreatic cancer: clinical and immunologic analyses from the randomized phase 2 PRINCE trial. Nat Med 28, 1167–1177. 10.1038/s41591-022-01829-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Byrne KT, Betts CB, Mick R, Sivagnanam S, Bajor DL, Laheru DA, Chiorean EG, O’Hara MH, Liudahl SM, Newcomb C, et al. (2021). Neoadjuvant Selicrelumab, an Agonist CD40 Antibody, Induces Changes in the Tumor Microenvironment in Patients with Resectable Pancreatic Cancer. Clin Cancer Res 27, 4574–4586. 10.1158/1078-0432.CCR-21-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hegde S, Krisnawan VE, Herzog BH, Zuo C, Breden MA, Knolhoff BL, Hogg GD, Tang JP, Baer JM, Mpoy C, et al. (2020). Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell 37, 289–307 e289. 10.1016/j.ccell.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Byrne KT, and Vonderheide RH (2016). CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Rep 15, 2719–2732. 10.1016/j.celrep.2016.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Daley D, Zambirinis CP, Seifert L, Akkad N, Mohan N, Werba G, Barilla R, Torres-Hernandez A, Hundeyin M, Mani VRK, et al. (2016). gammadelta T Cells Support Pancreatic Oncogenesis by Restraining alphabeta T Cell Activation. Cell 166, 1485–1499 e1415. 10.1016/j.cell.2016.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.McAllister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, Rattigan Y, Roeser JC, Lankapalli RH, Zhang H, et al. (2014). Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell 25, 621–637. 10.1016/j.ccr.2014.03.014 S1535–6108(14)00122–6 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Perusina Lanfranca M, Zhang Y, Girgis A, Kasselman S, Lazarus J, Kryczek I, Delrosario L, Rhim A, Koneva L, Sartor M, et al. (2020). Interleukin 22 Signaling Regulates Acinar Cell Plasticity to Promote Pancreatic Tumor Development in Mice. Gastroenterology 158, 1417–1432 e1411. 10.1053/j.gastro.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhang Y, Yan W, Mathew E, Bednar F, Wan S, Collins MA, Evans RA, Welling TH, Vonderheide RH, and di Magliano MP (2014). CD4+ T lymphocyte ablation prevents pancreatic carcinogenesis in mice. Cancer Immunol Res 2, 423–435. 10.1158/2326-6066.CIR-14-0016-T 2326–6066.CIR-14–0016-T [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhang Y, Lazarus J, Steele NG, Yan W, Lee HJ, Nwosu ZC, Halbrook CJ, Menjivar RE, Kemp SB, Sirihorachai VR, et al. (2020). Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov 10, 422–439. 10.1158/2159-8290.CD-19-0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar-Sagi D, et al. (2015). Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75, 544–553. 10.1158/0008-5472.CAN-14-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Vander Heiden MG, and Muir A (2019). Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8. 10.7554/eLife.44235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Guillaumond F, Leca J, Olivares O, Lavaut MN, Vidal N, Berthezene P, Dusetti NJ, Loncle C, Calvo E, Turrini O, et al. (2013). Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A 110, 3919–3924. 10.1073/pnas.1219555110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Boudreau A, Purkey HE, Hitz A, Robarge K, Peterson D, Labadie S, Kwong M, Hong R, Gao M, Del Nagro C, et al. (2016). Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat Chem Biol 12, 779–786. 10.1038/nchembio.2143. [DOI] [PubMed] [Google Scholar]
- 197.Yan L, Tu B, Yao J, Gong J, Carugo A, Bristow CA, Wang Q, Zhu C, Dai B, Kang Y, et al. (2021). Targeting Glucose Metabolism Sensitizes Pancreatic Cancer to MEK Inhibition. Cancer Res 81, 4054–4065. 10.1158/0008-5472.CAN-20-3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105. 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Biancur DE, Paulo JA, Malachowska B, Del Rey MQ, Sousa CM, Wang XX, Sohn ASW, Chu GC, Gygi SP, Harper JW, et al. (2017). Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nature Communications 8. ARTN 15965 10.1038/ncomms15965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bott AJ, Shen J, Tonelli C, Zhan L, Sivaram N, Jiang YP, Yu X, Bhatt V, Chiles E, Zhong H, et al. (2019). Glutamine Anabolism Plays a Critical Role in Pancreatic Cancer by Coupling Carbon and Nitrogen Metabolism. Cell Rep 29, 1287–1298 e1286. 10.1016/j.celrep.2019.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Magill GB, Myers WP, Reilly HC, Putnam RC, Magill JW, Sykes MP, Escher GC, Karnofsky DA, and Burchenal JH (1957). Pharmacological and initial therapeutic observations on 6-diazo-5-oxo-1-norleucine (DON) in human neoplastic disease. Cancer 10, 1138–1150. 10.1002/1097-0142(195711/12)10:6<1138::aid-cncr2820100608>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 202.Lynch G, Kemeny N, and Casper E (1982). Phase II evaluation of DON (6-diazo-5-oxo-L-norleucine) in patients with advanced colorectal carcinoma. Am J Clin Oncol 5, 541–543. [PubMed] [Google Scholar]
- 203.Rais R, Jancarik A, Tenora L, Nedelcovych M, Alt J, Englert J, Rojas C, Le A, Elgogary A, Tan J, et al. (2016). Discovery of 6-Diazo-5-oxo-l-norleucine (DON) Prodrugs with Enhanced CSF Delivery in Monkeys: A Potential Treatment for Glioblastoma. J Med Chem 59, 8621–8633. 10.1021/acs.jmedchem.6b01069. [DOI] [PubMed] [Google Scholar]
- 204.Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, Arwood ML, Bettencourt IA, Patel CH, Wen J, et al. (2019). Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021. 10.1126/science.aav2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sharma NS, Gupta VK, Garrido VT, Hadad R, Durden BC, Kesh K, Giri B, Ferrantella A, Dudeja V, Saluja A, and Banerjee S (2020). Targeting tumor-intrinsic hexosamine biosynthesis sensitizes pancreatic cancer to anti-PD1 therapy. J Clin Invest 130, 451–465. 10.1172/JCI127515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al. (2015). Transcriptional control of autophagyly-sosome function drives pancreatic cancer metabolism. Nature 524, 361–365. 10.1038/nature14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, et al. (2011). Pancreatic cancers require autophagy for tumor growth. Genes Dev 25, 717–729. 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, et al. (2013). p53 status determines the role of autophagy in pancreatic tumour development. Nature 504, 296–300. 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 209.Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, Von Hoff DD, Maitra A, and Kimmelman AC (2014). Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov 4, 905–913. 10.1158/2159-8290.CD-14-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, Deng J, Hai J, Yang S, Wong KK, and Kimmelman AC (2018). Autophagy Sustains Pancreatic Cancer Growth through Both Cell-Autonomous and Nonautonomous Mechanisms. Cancer Discov 8, 276–287. 10.1158/2159-8290.CD-17-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Humpton TJ, Alagesan B, DeNicola GM, Lu D, Yordanov GN, Leonhardt CS, Yao MA, Alagesan P, Zaatari MN, Park Y, et al. (2019). Oncogenic KRAS Induces NIX-Mediated Mitophagy to Promote Pancreatic Cancer. Cancer Discov 9, 1268–1287. 10.1158/2159-8290.CD-18-1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Boone BA, Bahary N, Zureikat AH, Moser AJ, Normolle DP, Wu WC, Singhi AD, Bao P, Bartlett DL, Liotta LA, et al. (2015). Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann Surg Oncol 22, 4402–4410. 10.1245/s10434-015-4566-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Zeh HJ, Bahary N, Boone BA, Singhi AD, Miller-Ocuin JL, Normolle DP, Zureikat AH, Hogg ME, Bartlett DL, Lee KK, et al. (2020). A Randomized Phase II Preoperative Study of Autophagy Inhibition with High-Dose Hydroxychloroquine and Gemcitabine/Nab-Paclitaxel in Pancreatic Cancer Patients. Clin Cancer Res 26, 3126–3134. 10.1158/1078-0432.CCR-19-4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Mancias JD, Wang X, Gygi SP, Harper JW, and Kimmelman AC (2014). Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109. 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Pantopoulos K, Porwal SK, Tartakoff A, and Devireddy L (2012). Mechanisms of mammalian iron homeostasis. Biochemistry 51, 5705–5724. 10.1021/bi300752r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Santana-Codina N, Del Rey MQ, Kapner KS, Zhang H, Gikandi A, Malcolm C, Poupault C, Kuljanin M, John KM, Biancur DE, et al. (2022). NCOA4-Mediated Ferritinophagy Is a Pancreatic Cancer Dependency via Maintenance of Iron Bioavailability for Iron-Sulfur Cluster Proteins. Cancer Discov 12, 2180–2197. 10.1158/2159-8290.CD-22-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ravichandran M, Hu J, Cai C, Ward NP, Venida A, Foakes C, Kuljanin M, Yang A, Hennessey CJ, Yang Y, et al. (2022). Coordinated Transcriptional and Catabolic Programs Support Iron-Dependent Adaptation to RAS-MAPK Pathway Inhibition in Pancreatic Cancer. Cancer Discov 12, 2198–2219. 10.1158/2159-8290.CD-22-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, Sagalovskiy IR, Ma A, Kapilian J, Firl CEM, et al. (2020). Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89. 10.1126/science.aaw9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Kremer DM, Nelson BS, Lin L, Yarosz EL, Halbrook CJ, Kerk SA, Sajjakulnukit P, Myers A, Thurston G, Hou SW, et al. (2021). GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat Commun 12, 4860. 10.1038/s41467-021-24859-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Sharbeen G, McCarroll JA, Akerman A, Kopecky C, Youkhana J, Kokkinos J, Holst J, Boyer C, Erkan M, Goldstein D, et al. (2021). Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma Determine Response to SLC7A11 Inhibition. Cancer Res 81, 3461–3479. 10.1158/0008-5472.CAN-20-2496. [DOI] [PubMed] [Google Scholar]
- 222.Dai E, Han L, Liu J, Xie Y, Zeh HJ, Kang R, Bai L, and Tang D (2020). Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun 11, 6339. 10.1038/s41467-020-20154-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Kshattry S, Saha A, Gries P, Tiziani S, Stone E, Georgiou G, and DiGiovanni J (2019). Enzyme-mediated depletion of l-cyst(e)ine synergizes with thioredoxin reductase inhibition for suppression of pancreatic tumor growth. NPJ Precis Oncol 3, 16. 10.1038/s41698-019-0088-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM, George SD, et al. (2019). Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med 25, 628–640. 10.1038/s41591-019-0368-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT, Yap JT, et al. (2019). Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med 25, 620–627. 10.1038/s41591-019-0367-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, and van Hall T (2016). The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol 39, 44–51. 10.1016/j.coi.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, Sohn ASW, Mukhopadhyay S, Lin EY, Parker SJ, et al. (2020). Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581, 100–105. 10.1038/s41586-020-2229-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Zhu XG, Chudnovskiy A, Baudrier L, Prizer B, Liu Y, Ostendorf BN, Yamaguchi N, Arab A, Tavora B, Timson R, et al. (2021). Functional Genomics In Vivo Reveal Metabolic Dependencies of Pancreatic Cancer Cells. Cell Metab 33, 211–221 e216. 10.1016/j.cmet.2020.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Bar-Sagi D, and Feramisco JR (1986). Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science 233, 1061–1068. 10.1126/science.3090687. [DOI] [PubMed] [Google Scholar]
- 230.Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, Lac S, Roques J, Lavaut MN, Berthezene P, et al. (2017). Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun 8, 16031. 10.1038/ncomms16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, et al. (2016). Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab 23, 517–528. 10.1016/j.cmet.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Jayashankar V, and Edinger AL (2020). Macropinocytosis confers resistance to therapies targeting cancer anabolism. Nat Commun 11, 1121. 10.1038/s41467-020-14928-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kim SM, Nguyen TT, Ravi A, Kubiniok P, Finicle BT, Jayashankar V, Malacrida L, Hou J, Robertson J, Gao D, et al. (2018). PTEN Deficiency and AMPK Activation Promote Nutrient Scavenging and Anabolism in Prostate Cancer Cells. Cancer Discov 8, 866–883. 10.1158/2159-8290.CD-17-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Lee SW, Zhang Y, Jung M, Cruz N, Alas B, and Commisso C (2019). EGFR-Pak Signaling Selectively Regulates Glutamine Deprivation-Induced Macropinocytosis. Dev Cell 50, 381–392 e385. 10.1016/j.devcel.2019.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Yamamoto K, Brender JR, Seki T, Kishimoto S, Oshima N, Choudhuri R, Adler SS, Jagoda EM, Saito K, Devasahayam N, et al. (2020). Molecular Imaging of the Tumor Microenvironment Reveals the Relationship between Tumor Oxygenation, Glucose Uptake, and Glycolysis in Pancreatic Ductal Adenocarcinoma. Cancer Res 80, 2087–2093. 10.1158/0008-5472.CAN-19-0928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Kishimoto S, Brender JR, Crooks DR, Matsumoto S, Seki T, Oshima N, Merkle H, Lin P, Reed G, Chen AP, et al. (2019). Imaging of glucose metabolism by 13C-MRI distinguishes pancreatic cancer subtypes in mice. Elife 8. 10.7554/eLife.46312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Daemen A, Peterson D, Sahu N, McCord R, Du X, Liu B, Kowanetz K, Hong R, Moffat J, Gao M, et al. (2015). Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci U S A 112, E4410–4417. 10.1073/pnas.1501605112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Halbrook CJ, Thurston G, Boyer S, Anaraki C, Jimenez JA, McCarthy A, Steele NG, Kerk SA, Hong HS, Lin L, et al. (2022). Differential integrated stress response and asparagine production drive symbiosis and therapy resistance of pancreatic adenocarcinoma cells. Nat Cancer 3, 1386–1403. 10.1038/s43018-022-00463-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Recouvreux MV, Moldenhauer MR, Galenkamp KMO, Jung M, James B, Zhang Y, Lowy A, Bagchi A, and Commisso C (2020). Glutamine depletion regulates Slug to promote EMT and metastasis in pancreatic cancer. J Exp Med 217. 10.1084/jem.20200388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, Word AE, Carrer A, Salz TH, Natsume S, et al. (2017). Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet 49, 367–376. 10.1038/ng.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Bechard ME, Word AE, Tran AV, Liu X, Locasale JW, and McDonald OG (2018). Pentose conversions support the tumorigenesis of pancreatic cancer distant metastases. Oncogene 37, 5248–5256. 10.1038/s41388-018-0346-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Bechard ME, Smalling R, Hayashi A, Zhong Y, Word AE, Campbell SL, Tran AV, Weiss VL, Iacobuzio-Donahue C, Wellen KE, and McDonald OG (2020). Pancreatic cancers suppress negative feedback of glucose transport to reprogram chromatin for metastasis. Nat Commun 11, 4055. 10.1038/s41467-020-17839-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, Barneda D, Sellers K, Campos-Olivas R, Grana O, et al. (2015). MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab 22, 590–605. 10.1016/j.cmet.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 245.Elgogary A, Xu QG, Poore B, Alt J, Zimmermann SC, Zhao L, Fu J, Chen BW, Xia SY, Liu YF, et al. (2016). Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proceedings of the National Academy of Sciences of the United States of America 113, E5328–E5336. 10.1073/pnas.1611406113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Krall AS, Mullen PJ, Surjono F, Momcilovic M, Schmid EW, Halbrook CJ, Thambundit A, Mittelman SD, Lyssiotis CA, Shackelford DB, et al. (2021). Asparagine couples mitochondrial respiration to ATF4 activity and tumor growth. Cell Metab 33, 1013–1026 e1016. 10.1016/j.cmet.2021.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Sherman MH, Yu RT, Tseng TW, Sousa CM, Liu S, Truitt ML, He N, Ding N, Liddle C, Atkins AR, et al. (2017). Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc Natl Acad Sci U S A 114, 1129–1134. 10.1073/pnas.1620164114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. 10.1038/nature19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Zhang Y, Recouvreux MV, Jung M, Galenkamp KMO, Li Y, Zagnitko O, Scott DA, Lowy AM, and Commisso C (2021). Macropinocytosis in Cancer-Associated Fibroblasts Is Dependent on CaMKK2/ARHGEF2 Signaling and Functions to Support Tumor and Stromal Cell Fitness. Cancer Discov 11, 1808–1825. 10.1158/2159-8290.CD-20-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Kerk SA, Lin L, Myers AL, Sutton DJ, Andren A, Sajjakulnukit P, Zhang L, Zhang Y, Jimenez JA, Nelson BS, et al. (2022). Metabolic requirement for GOT2 in pancreatic cancer depends on environmental context. Elife 11. 10.7554/eLife.73245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, Marini JC, Tudawe T, Seviour EG, San Lucas FA, et al. (2016). Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 5, e10250. 10.7554/eLife.10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Raghavan KS, Francescone R, Franco-Barraza J, Gardiner JC, Vendramini-Costa DB, Luong T, Pourmandi N, Andren A, Kurimchak A, Ogier C, et al. (2022). NetrinG1(+) cancer-associated fibroblasts generate unique extracellular vesicles that support the survival of pancreatic cancer cells under nutritional stress. Cancer Res Commun 2, 1017–1036. 10.1158/2767-9764.crc-21-0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Parker SJ, Amendola CR, Hollinshead KER, Yu Q, Yamamoto K, Encarnacion-Rosado J, Rose RE, LaRue MM, Sohn ASW, Biancur DE, et al. (2020). Selective Alanine Transporter Utilization Creates a Targetable Metabolic Niche in Pancreatic Cancer. Cancer Discov 10, 1018–1037. 10.1158/2159-8290.CD-19-0959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, Tumanov S, Allen-Petersen BL, Link J, Kendsersky ND, et al. (2019). A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer Discov 9, 617–627. 10.1158/2159-8290.CD-18-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Zhu Z, Achreja A, Meurs N, Animasahun O, Owen S, Mittal A, Parikh P, Lo TW, Franco-Barraza J, Shi J, et al. (2020). Tumour-reprogrammed stromal BCAT1 fuels branched-chain ketoacid dependency in stromal-rich PDAC tumours. Nat Metab 2, 775–792. 10.1038/s4225-5020-0226-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Campbell S, Mesaros C, Izzo L, Affronti H, Noji M, Schaffer BE, Tsang T, Sun K, Trefely S, Kruijning S, et al. (2021). Glutamine deprivation triggers NAGK-dependent hexosamine salvage. Elife 10. 10.7554/eLife.62644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Kim PK, Halbrook CJ, Kerk SA, Radyk M, Wisner S, Kremer DM, Sajjakulnukit P, Andren A, Hou SW, Trivedi A, et al. (2021). Hyaluronic acid fuels pancreatic cancer cell growth. Elife 10. 10.7554/eLife.62645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee HJ, Thurston G, Zhang Y, Lazarus J, Sajjakulnukit P, et al. (2019). Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab 29, 1390–1399 e1396. 10.1016/j.cmet.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Dalin S, Sullivan MR, Lau AN, Grauman-Boss B, Mueller HS, Kreidl E, Fenoglio S, Luengo A, Lees JA, Vander Heiden MG, et al. (2019). Deoxycytidine Release from Pancreatic Stellate Cells Promotes Gemcitabine Resistance. Cancer Res 79, 5723–5733. 10.1158/0008-5472.CAN-19-0960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Boyer S, Lee HJ, Steele N, Zhang L, Sajjakulnukit P, Andren A, Ward MH, Singh R, Basrur V, Zhang Y, et al. (2022). Multiomic characterization of pancreatic cancer-associated macrophage polarization reveals deregulated metabolic programs driven by the GM-CSF-PI3K pathway. Elife 11. 10.7554/eLife.73796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Van de Velde LA, Subramanian C, Smith AM, Barron L, Qualls JE, Neale G, Alfonso-Pecchio A, Jackowski S, Rock CO, Wynn TA, and Murray PJ (2017). T Cells Encountering Myeloid Cells Programmed for Amino Acid-dependent Immunosuppression Use Rictor/mTORC2 Protein for Proliferative Checkpoint Decisions. J Biol Chem 292, 15–30. 10.1074/jbc.M116.766238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Liebl F, Demir IE, Mayer K, Schuster T, D’Haese JG, Becker K, Langer R, Bergmann F, Wang K, Rosenberg R, et al. (2014). The impact of neural invasion severity in gastrointestinal malignancies: a clinicopathological study. Ann Surg 260, 900–907; discussion 907–908. 10.1097/SLA.0000000000000968. [DOI] [PubMed] [Google Scholar]
- 263.Banh RS, Biancur DE, Yamamoto K, Sohn ASW, Walters B, Kuljanin M, Gikandi A, Wang H, Mancias JD, Schneider RJ, et al. (2020). Neurons Release Serine to Support mRNA Translation in Pancreatic Cancer. Cell 183, 1202–1218 e1225. 10.1016/j.cell.2020.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, Mohan N, Aykut B, Usyk M, Torres LE, et al. (2018). The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov 8, 403–416. 10.1158/2159-8290.CD-17-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI, Shadaloey SA, Wu D, Preiss P, Verma N, et al. (2019). The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 574, 264–267. 10.1038/s41586-019-1608-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, Quesada P, Sahin I, Chandra V, San Lucas A, et al. (2019). Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 178, 795–806 e712. 10.1016/j.cell.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, Gavert N, Zwang Y, Cooper ZA, Shee K, et al. (2017). Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 357, 1156–1160. 10.1126/science.aah5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Peng H, James CA, Cullinan DR, Hogg GD, Mudd JL, Zuo C, Takchi R, Caldwell KE, Liu J, DeNardo DG, et al. (2021). Neoadjuvant FOLFIRINOX Therapy Is Associated with Increased Effector T Cells and Reduced Suppressor Cells in Patients with Pancreatic Cancer. Clin Cancer Res 27, 6761–6771. 10.1158/1078-0432.CCR-21-0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Hosein AN, Huang H, Wang Z, Parmar K, Du W, Huang J, Maitra A, Olson E, Verma U, and Brekken RA (2019). Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 5. 10.1172/jci.insight.129212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Lee JJ, Bernard V, Semaan A, Monberg ME, Huang J, Stephens BM, Lin D, Rajapakshe KI, Weston BR, Bhutani MS, et al. (2021). Elucidation of Tumor-Stromal Heterogeneity and the Ligand-Receptor Interactome by Single-Cell Transcriptomics in Real-world Pancreatic Cancer Biopsies. Clin Cancer Res 27, 5912–5921. 10.1158/1078-0432.CCR-20-3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Monberg ME, Geiger H, Lee JJ, Sharma R, Semaan A, Bernard V, Wong J, Wang F, Liang S, Swartzlander DB, et al. (2022). Occult polyclonality of preclinical pancreatic cancer models drives in vitro evolution. Nat Commun 13, 3652. 10.1038/s41467-022-31376-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Schalck A, Sakellariou-Thompson D, Forget MA, Sei E, Hughes TG, Reuben A, Bai S, Hu M, Kumar T, Hurd MW, et al. (2022). Single-Cell Sequencing Reveals Trajectory of Tumor-Infiltrating Lymphocyte States in Pancreatic Cancer. Cancer Discov 12, 2330–2349. 10.1158/2159-8290.CD-21-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Hickey JW, Neumann EK, Radtke AJ, Camarillo JM, Beuschel RT, Albanese A, McDonough E, Hatler J, Wiblin AE, Fisher J, et al. (2022). Spatial mapping of protein composition and tissue organization: a primer for multiplexed antibody-based imaging. Nat Methods 19, 284–295. 10.1038/s41592-021-01316-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Seferbekova Z, Lomakin A, Yates LR, and Gerstung M (2022). Spatial biology of cancer evolution. Nat Rev Genet. 10.1038/s41576-022-00553-x. [DOI] [PubMed] [Google Scholar]
- 275.Liudahl SM, Betts CB, Sivagnanam S, Morales-Oyarvide V, da Silva A, Yuan C, Hwang S, Grossblatt-Wait A, Leis KR, Larson W, et al. (2021). Leukocyte Heterogeneity in Pancreatic Ductal Adenocarcinoma: Phenotypic and Spatial Features Associated with Clinical Outcome. Cancer Discov 11, 2014–2031. 10.1158/2159-8290.CD-20-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Kiemen AL, Braxton AM, Grahn MP, Han KS, Babu JM, Reichel R, Jiang AC, Kim B, Hsu J, Amoa F, et al. (2022). CODA: quantitative 3D reconstruction of large tissues at cellular resolution. Nat Methods 19, 1490–1499. 10.1038/s41592-022-01650-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Steele NG, Carpenter ES, Kemp SB, Sirihorachai VR, The S, Delrosario L, Lazarus J, Amir ED, Gunchick V, Espinoza C, et al. (2020). Multimodal Mapping of the Tumor and Peripheral Blood Immune Landscape in Human Pancreatic Cancer. Nat Cancer 1, 1097–1112. 10.1038/s43018-020-00121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Freed-Pastor WA, Lambert LJ, Ely ZA, Pattada NB, Bhutkar A, Eng G, Mercer KL, Garcia AP, Lin L, Rideout WM 3rd, et al. (2021). The CD155/TIGIT axis promotes and maintains immune evasion in neoantigen-expressing pancreatic cancer. Cancer Cell 39, 1342–1360 e1314. 10.1016/j.ccell.2021.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Hwang WL, Jagadeesh KA, Guo JA, Hoffman HI, Yadollahpour P, Reeves JW, Mohan R, Drokhlyansky E, Van Wittenberghe N, Ashenberg O, et al. (2022). Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat Genet 54, 1178–1191. 10.1038/s41588-022-01134-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.He S, Bhatt R, Brown C, Brown EA, Buhr DL, Chantranuvatana K, Danaher P, Dunaway D, Garrison RG, Geiss G, et al. (2022). High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat Biotechnol 40, 1794–1806. 10.1038/s41587-022-01483-z. [DOI] [PubMed] [Google Scholar]
- 281.Rahib L, Fleshman JM, Matrisian LM, and Berlin JD (2016). Evaluation of Pancreatic Cancer Clinical Trials and Benchmarks for Clinically Meaningful Future Trials: A Systematic Review. JAMA Oncol 2, 1209–1216. 10.1001/jamaoncol.2016.0585. [DOI] [PubMed] [Google Scholar]
- 282.Thota R, Maitra A, and Berlin JD (2017). Preclinical Rationale for the Phase III Trials in Metastatic Pancreatic Cancer: Is Wishful Thinking Clouding Successful Drug Development for Pancreatic Cancer? Pancreas 46, 143–150. 10.1097/MPA.0000000000000753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.O’Reilly EM, Oh DY, Dhani N, Renouf DJ, Lee MA, Sun W, Fisher G, Hezel A, Chang SC, Vlahovic G, et al. (2019). Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol 5, 1431–1438. 10.1001/jamaoncol.2019.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. (2012). Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366, 2455–2465. 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Gulhati P, Schalck A, Jiang S, Shang X, Wu CJ, Hou P, Ruiz SH, Soto LS, Parra E, Ying H, et al. (2022). Targeting T cell checkpoints 41BB and LAG3 and myeloid cell CXCR1/CXCR2 results in antitumor immunity and durable response in pancreatic cancer. Nat Cancer. 10.1038/s43018-022-00500-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.O’Hara MH, O’Reilly EM, Varadhachary G, Wolff RA, Wainberg ZA, Ko AH, Fisher G, Rahma O, Lyman JP, Cabanski CR, et al. (2021). CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: an open-label, multicentre, phase 1b study. Lancet Oncol 22, 118–131. 10.1016/S1470-2045(20)30532-5. [DOI] [PubMed] [Google Scholar]
- 287.Bockorny B, Macarulla T, Semenisty V, Borazanci E, Feliu J, Ponz-Sarvise M, Abad DG, Oberstein P, Alistar A, Munoz A, et al. (2021). Motixafortide and Pembrolizumab Combined to Nanoliposomal Irinotecan, Fluorouracil, and Folinic Acid in Metastatic Pancreatic Cancer: The COMBAT/KEYNOTE-202 Trial. Clin Cancer Res 27, 5020–5027. 10.1158/1078-0432.CCR-21-0929. [DOI] [PubMed] [Google Scholar]
- 288.Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, Clendenin C, Stanger BZ, Furth EE, Wherry EJ, and Vonderheide RH (2015). Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol Res 3, 399–411. 10.1158/2326-6066.CIR-14-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Catenacci DVT, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR, Marsh R, Wallace J, Kozloff M, Rajdev L, et al. (2015). Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. Journal of Clinical Oncology 33, 4284–+. 10.1200/Jco.2015.62.8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. (2015). Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 28, 831–833. 10.1016/j.ccell.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 291.Hingorani SR (2022). Epithelial and stromal co-evolution and complicity in pancreatic cancer. Nat Rev Cancer. 10.1038/s41568-022-00530-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Balachandran VP, Rojas LA, Sethna Z, Soares K, Derhovanessian E, Mueller F, Yadav M, Basturk O, Gonen M, Wei A.C. c., et al. (2022). Phase I trial of adjuvant autogene cevumeran, an individualized mRNA neoantigen vaccine, for pancreatic ductal adenocarcinoma. Journal of Clinical Oncology 40, 2516–2516. 10.1200/JCO.2022.40.16_suppl.2516. [DOI] [Google Scholar]
- 293.Knox JJ, Jaffee EM, O’Kane GM, Plenker D, Zhang A, Ramotar S, Dodd A, Prince RM, Laheru D, Yu KH, et al. (2022). PASS-01: Pancreatic adenocarcinoma signature stratification for treatment–01. Journal of Clinical Oncology 40, TPS635–TPS635. 10.1200/JCO.2022.40.4_suppl.TPS635. [DOI] [Google Scholar]
- 294.Picozzi VJ, Duliege A-M, Collisson EA, Maitra A, Hidalgo M, Hendifar AE, Beatty GL, Doss S, Matrisian LM, Herena PS, et al. (2022). Precision Promise (PrP): An adaptive, multi-arm registration trial in metastatic pancreatic ductal adenocarcinoma (PDAC). Journal of Clinical Oncology 40, TPS4188–TPS4188. 10.1200/JCO.2022.40.16_suppl.TPS4188. [DOI] [Google Scholar]
- 295.Falcomata C, Barthel S, Schneider G, Rad R, Schmidt-Supprian M, and Saur D (2023). Context-Specific Determinants of the Immunosuppressive Tumor Microenvironment in Pancreatic Cancer. Cancer Discov, OF1–OF20. 10.1158/2159-8290.CD-22-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Hosein AN, Dougan SK, Aguirre AJ, and Maitra A (2022). Translational advances in pancreatic ductal adenocarcinoma therapy. Nat Cancer 3, 272–286. 10.1038/s43018-022-00349-2. [DOI] [PubMed] [Google Scholar]
- 297.Lemberg KM, Gori SS, Tsukamoto T, Rais R, and Slusher BS (2022). Clinical development of metabolic inhibitors for oncology. J Clin Invest 132. 10.1172/JCI148550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Beutel AK, and Halbrook CJ (2022). Barriers and Opportunities for Gemcitabine in Pancreatic Cancer Therapy. Am J Physiol Cell Physiol. 10.1152/ajpcell.00331.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]