

Abstract
Recent advances in machine learning (ML) have enabled the development of next-generation predictive models for complex computational biology problems, thereby spurring the use of interpretable machine learning (IML) to unveil biological insights. However, guidelines for using IML in computational biology are generally underdeveloped. We provide an overview of IML methods and evaluation techniques and discuss common pitfalls encountered when applying IML methods to computational biology problems. We also highlight open questions, especially in the era of large language models, and call for collaboration between IML and computational biology researchers.
Introduction
Machine learning (ML) has significantly shaped the landscape of computational biology, with the integration of high-throughput data acquisition and burgeoning computational power leading to the creation of potent prediction models. More recently, the advancements inspired by large language models (LLMs) – a term which conventionally refers to models that can perform a wide variety of natural language processing tasks, but is increasingly encompassing transformer-based, pretrained large-scale models in other domains such as computational biology – have further enhanced our ability to model and analyze biomolecular sequences and gene expression data. With the rapid development of these new models, there is a growing need for techniques that can yield interpretation or comprehension of model behavior, enabling researchers to verify that the proposed model reflects actual biological mechanisms. Yet, the complexity of these prediction models often renders them challenging to interpret. For instance, merely presenting the multitude of weights from deep learning models such as convolutional neural networks or transformer models to a user will not yield immediate comprehension or interpretation.
To address this challenge, researchers have embraced techniques from the field of Interpretable Machine Learning (IML), which aims to elucidate why a prediction model has arrived at a particular score or classification, either by deriving an explanation post-training or by building interpretable mechanisms into the model architecture [1, 2]. The application of IML methods has surged in prominence within computational biology research across a wide range of biological tasks [3–7]:
These techniques have been extensively employed to uncover critical sequence patterns and interpret sequence variants in DNA, RNA, and protein sequence-based tasks, such as the prediction of gene expression, epigenetic modifications, 3D structures, and protein-DNA/RNA interactions [8–13].
IML methods have also exhibited promising results in identifying key biomarkers in phenotype predictions based on gene expression, metagenomics, and genetic variations, often referred to as one-dimensional tabular data. The tasks commonly associated with tabular data involve the prediction of cellular phenotypes, such as cell states and cellular response, as well as clinical phenotypes, such as complex diseases [14–17].
Additionally, IML has demonstrated its potential to capture distinctive features in biomedical imaging tasks [18–20].
Despite the growing significance of IML in computational biology, guidance on the application and evaluation of IML methods within complex biological settings is scarce, leading to ad-hoc applications of popular IML techniques and inconsistent, and potentially unreliable, interpretations of IML outputs. In this Perspective, we first provide an overview of classical IML methods and evaluation techniques. We then expand upon prior discussions on the pitfalls of applying ML techniques to the sciences [21–23] by highlighting potential issues concerning the current use of IML methods in computational biology applications. We specifically focus on pitfalls in the selection of IML methods, interpretation of IML outputs, and improper evaluation of findings from IML methods. Each pitfall is also delineated using illustrative examples from the literature, which include applications of IML methods to the newest transformer-based models and other biologically-informed networks. Finally, in light of recent advancements in LLMs, we close by highlighting IML techniques that have been developed to explain these new models and presenting multiple opportunities for how they can be adapted for biological problems. These directions for future work represent ripe opportunities for collaboration between the IML and computational biology communities toward developing better practices and methodologies.
IML Methods and Evaluations in Computational Biology
Methods
We first provide an overview of the two primary IML approaches employed to explain prediction models in computational biology applications, as shown in Fig. 1. Both approaches are featured in many of the examples cited in subsequent sections. For an additional discussion of each approach and an overview of other IML methods that are less commonly used in computational biology applications, we refer the readers to Azodi et al. [3], Chen et al. [24], and Rauker et al. [25].
Figure 1:

The two main IML approaches used to explain prediction models are post-hoc explanations and by-design explanations. Each approach has its canonical workflows and popular types of IML methods: post-hoc explanations are model-agnostic and are applied after a model is trained while by-design explanations are typically built into or inherent to the model architecture.
Post-hoc Explanations
The most widely used IML methods are post-hoc explanations, which are flexible and typically model-agnostic due to their application after the design and training of a prediction model. Feature importance methods are commonly used in computational biology applications [6, 26]. These methods assign each input feature, such as a pixel in a cellular image or a DNA sequence feature, an importance value based on its contribution to the model prediction. Consequently, a large magnitude feature importance score would imply a significant contribution. These importance values can be calculated in one of two ways: (1) gradient-based methods (e.g., DeepLIFT [27], Integrated Gradients [28], GradCAM [29]), and (2) perturbation-based methods (e.g., in silico mutagenesis [30], SHAP/DeepExplainer [31], LIME [32], Fourier-based attributions [33]).
By-design Methods
Interpretable by-design models are, as the name suggests, models that are naturally interpretable [34]. For instance, a linear model is deemed interpretable because one can easily inspect the coefficient weights to ascertain the importance of each feature to the prediction outcome. Similarly, decision trees are interpretable as one can examine the splits in the tree. Other interpretable by-design models include logistic regression, decision rules, and generalized additive models (GAMs) [35]. While the aforementioned models are conventional by-design models in the IML literature, new by-design IML approaches that leverage recent advancements and superior performance of deep neural networks are emerging in computational biology. These methods construct biologically-informed neural networks or incorporate attention mechanisms.
Biologically-informed neural networks are model architectures that encode domain knowledge. The process of architecture design is application-specific and constitutes an open problem that is beyond the scope of this work. Examples of biologically-informed neural networks include DCell [14], which represent the hierarchical cell subsystems capturing interwoven intracellular components and cellular processes in the neural network design, P-NET [36], which leverages the organization of biological pathways, and KPNN [37], which integrates biological networks, such as gene regulatory network and protein signaling pathways, into the network architecture. In these examples, the hidden nodes in the neural network correspond to biological entities such as genes and pathways. The relative importance of the biological entities is often interpreted using a self-defined measure, such as the Relative Local Improvement in Predictive Power score defined in DCell [14] and the node weight-based importance score defined in KPNN [37]. Post-hoc explanations can still be applied to the by-design neural networks, for instance, as in P-NET [36], to explain the model prediction using importance scores with respect to a certain biologically meaningful hidden layer. In addition, PAUSE [38] demonstrates one way to bridge post-hoc and by-design methods, where a biologically-constrained autoencoder model is explained using a post-hoc game theoretic approach.
Attention is a technique that has become a popular addition to neural networks that handle sequence-based inputs, where the network incorporates and learns a set of weights indicating the amount of attention the model is assigning to specific parts of the input. Most notably, transformers are encoder-decoder models that have capitalized on attention mechanisms through a variation called self-attention and move beyond the need for recurrent units [39]. Attention weights, which do not incorporate domain knowledge, are automatically learned as part of the training process and have been shown empirically to assist the network in focusing on the correct parts of the input sequence. The weights have often been considered as an explanation [16, 40]. More recently in transformer-based models, for example, Enformer [8] utilizes attention scores to identify the potential enhancers regulating gene expression. The recent Geneformer [17] inspects the attention weights from different layers to probe how the model encodes the gene regulatory network hierarchy. However, the validity and reliability of such an approach to explain the model’s reasoning remains debatable [41–44]. In the later section on “Opportunities for IML Developments in the Age of LLMs”, we review more recent IML approaches designed for transformers, and in particular the direction of mechanistic interpretability [45, 46].
Evaluation Techniques
To algorithmically assess the quality of explanations generated by IML methods, several concepts have been proposed, which are generally agnostic to the type of IML method that is applied. We focus on two metrics, depicted in Fig. 2, that frequently appear in the IML literature and are being increasingly adopted by computational biology publications. We provide a brief background on these evaluation measures and summarize existing evaluations of popular IML methods along both evaluation metrics.
Figure 2:
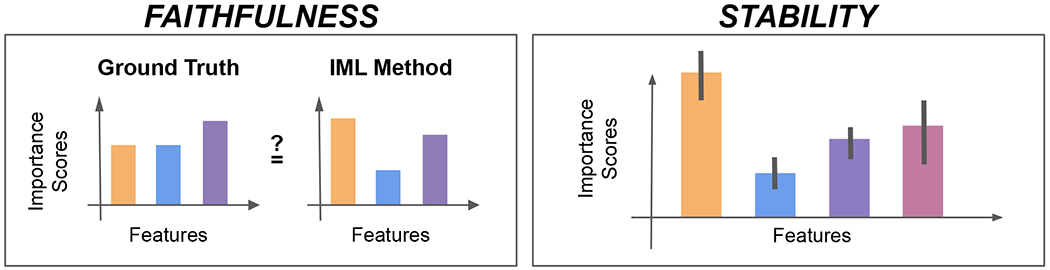
How do we assess explanations, which attribute importance scores to features of an input, generated by an IML method? IML methods are typically evaluated for the faithfulness of their computed feature importance scores as compared to a known ground truth mechanism and the stability of computed feature importance scores (e.g., as denoted by error bars) across varied input data.
Faithfulness (or fidelity) is the most common metric used to evaluate explanations generated by IML methods. This metric captures the degree to which an explanation reflects the ground truth mechanisms of the underlying ML model [47, 48]. While several benchmarking efforts have been conducted to evaluate the faithfulness of IML methods across multiple datasets and approaches to generating ground truth mechanisms [49–53], there is no method which generally outperforms other methods across the board, pointing to general unreliability of existing methods. We note that these evaluation approaches heavily relied on the use of synthetic data to encode variations of ground truth logic in the data, which may not be feasible in many computational biology contexts, where synthetic data fails to encapsulate the complexities of real biological processes. Consequently, it might be more suitable to identify and test IML methods against real data for which the ground truth mechanism is known, examples of which we discuss further in Pitfall #1.
Stability is a measure of consistency that can complement an evaluation of faithfulness. It primarily answers the question: “how consistent are the explanations for similar inputs?” This evaluation was proposed in response to the observation that feature importance often varies substantially when small perturbations are applied to an input [47, 54]. Many popular methods, including SHAP [31] and LIME [32], have been empirically shown to cause unstableness and again, there is no single method that is most stable across multiple real-world datasets [53]. These findings motivate our later discussion in Pitfall #3 about the importance of stability evaluations for clearer biological interpretations of results derived from IML methods.
Pitfalls of IML Usage in Computational Biology
As the computational biology community increasingly adopts IML methods as an important tool to understand ML model behaviors, we identify three pitfalls that should be avoided when using IML methods, depicted in Fig. 3. We describe each pitfall with illustrative examples and discuss how prior work has addressed them.
Figure 3:

An overview of three common pitfalls of IML interpretation in biological contexts and how to avoid these pitfalls. 1) Only considering one IML method. Consideration of multiple IML methods can inform the downstream interpretation of the outputs. 2) IML output disconnected from biological interpretation. Oftentimes, a post-processing step is necessary to enable interpretation of the IML output, particularly when the method is applied to sequence or pixel-level data. 3) Cherry-picked presentation of results. Many prior works do not present a complete picture of the extent to which the IML output reflects known biological mechanisms.
Pitfall #1: Only considering one IML method
While one might naturally apply a well-known IML method as a first step of model interpretation, it is important to note that different methods often produce different interpretations of the same prediction due to differences in their underlying assumptions and algorithms [49–51]. In fact, these underlying differences can lead to disagreements between IML method outputs (e.g., when the top-k most important features output by methods differs), a phenomenon that has been increasingly characterized across ML contexts [55]. Disagreement among IML methods has been reported in computational biology applications. DeepLIFT [27] demonstrates that not all IML methods can correctly identify key motifs for cooperative binding of transcription factors (TFs). Assessments on transcriptomics data show that different IML methods identify varying top genes for tissue type classification [56]. Additionally, even for a specific IML method, different hyperparameters (e.g., the baseline input in DeepLIFT [27] and Integrated Gradients [28]), or multiple runs of the same workflow, may lead to variance in the derived importance scores. Therefore, relying on a single run of one IML method may result in biased feature importance.
To obtain a more comprehensive view of the model’s behavior, we recommend employing multiple IML methods with diverse sets of hyperparameters and comparing their results. For example, Enformer is a transformer-based neural network that predicts gene expression from DNA sequences [8]. The authors computed feature importance scores using a diverse set of methods, including attention scores, input gradients, and perturbation-based scores, to understand important regulatory elements for gene expression. To identify the distinctive features for the classification of Alzheimer’s disease pathologies, Tang et al. [57] examined the differences in feature maps captured by Guided Grad-CAM [29] and the feature occlusion method [58], focusing on the specific features each method reveals under different conditions. Furthermore, KPNN [37], which represents a by-design, biological network with a partially connected neural network, proposed several design modifications to enhance the robustness of feature importance and assessed the effectiveness using the simulation data with known ground truth. These applications illustrate how incorporating multiple IML methods can foster a more reliable and comprehensive assessment of model behavior and feature importance.
However, if applying multiple IML methods leads to conflicting conclusions, it is important to develop evaluation mechanisms to assess the faithfulness of each IML method (i.e., comparing the generated explanation to real data or expert knowledge). This practice is increasingly common and has been applied to problems where there is some prior knowledge about the underlying mechanisms, such as TF binding motifs. Additionally, when the ground truth is significantly lacking, experimental validation will be crucial to verify the predictions made by IML methods. Incorporating “human-in-the-loop” and “lab-in-the-loop” approaches can further enhance interpretation and ensure the findings are biologically relevant and actionable.
Pitfall #2: IML output disconnected from biological interpretation
Although IML methods can identify features that are highly predictive of the output labels, they may not directly provide a biological interpretation of the resulting importance scores. For example, while gradient-based methods assign nucleotide-level feature importance in DNA sequence-based prediction tasks, post-processing steps are necessary to summarize the genome-wide importance scores and reveal the important sequence patterns. Similarly, for cell imaging-based classification tasks, the set of pixels that are highlighted by the feature attribution methods need to be converted to human-interpretable features.
Post-hoc explanation methods typically compute importance scores for the input features. The post-processing techniques to uncover the biological interpretation of these importance scores are highly domain-specific: For sequence-based tasks, various methods can extract meaningful insights from importance scores. For instance, TF-MoDISco [59] summarizes the nucleotide-level importance scores and performs de novo motif discovery to reveal the important sequence patterns. Additionally, statistical enrichment analysis can detect the known sequence patterns that frequently occur in regions with high importance. In the context of metagenomic sequence analysis, IDMIL [60] uses local sequence alignment search tool to identify the microbiome species with high-attention sequence fragments based on metagenomic data. In gene expression-related tasks, importance scores are typically assigned to individual genes using the feature importance attribution methods, such as DeepLIFT and SHAP. Subsequently, Gene Ontology (GO) enrichment analysis is commonly leveraged to identify the key functions of the top-ranked genes based on the computed importance scores [15, 61]. For cellular imaging analysis, the important regions highlighted by IML methods need to be translated into human interpretable features [62]. Moreover, when imaging data are transformed into a vector representation, generative models can visualize phenotype changes associated with important features in the latent space [19, 63]. Likewise, generative models can generate counterfactual images, enabling human experts to better understand the underlying reasoning processes [20].
In contrast, by-design explanation methods inherently embed feature importance within the network, utilizing components such as neurons, hidden layers, or attention matrices. Unveiling this information often requires subsequent post-processing steps. Recent methods, particularly in the context of single-cell RNA-seq, have incorporated prior knowledge (e.g., known gene functions, regulatory relationships, and biological pathways) into the network architecture to improve the intrinsic interpretability of neural networks [14, 37, 38]. For these biologically informed models, it is important to interpret the learned weights of various biologically relevant components and establish meaningful connections to prior knowledge. For instance, DCell [14], which embeds the GO hierarchical structures into the network architecture to perform phenotype predictions, further identifies the mechanisms of the genotype-phenotype connections by evaluating how subsystems mimic different logic gates.
Interpreting 2D attention matrices in attention-based models as pairwise dependencies between distinct loci may initially appear straightforward; however, these attention-based models typically comprise multiple layers, each containing multiple attention heads. Each attention head within a layer may learn a distinct aspect of pairwise attention, making it challenging to summarize and extract the biologically meaningful features or pairwise interactions. The existing works have attempted to tackle this challenge with a diverse set of approaches. Enformer, which predicts gene expression levels from DNA sequences through a combination of convolutional neural network (CNN) and transformer layers, summarizes the attention weights by averaging the attention matrices across all heads and layers [8]. C.Origami, a multi-modal transformer-based neural network that predicts chromatin organization from DNA sequences and chromatin features, inspects the layer-specific attention scores by averaging all the attention heads within the same layer [64]. Nucleotide Transformer, a pre-trained model based on DNA sequences, employs the BERTology method to evaluate attention scores associated with key genomic elements learned by individual attention heads [65]. Besides the methods discussed above, attention rollout and attention flow are other techniques to process the raw attention matrices [66].
After examining the application of post-processing techniques for both post-hoc and by-design methods, it is evident that the post-processing step is essential for converting the raw IML output into meaningful biological interpretations. Moreover, the selection of these techniques is closely linked to the specific characteristics of the data type and model architecture.
Pitfall #3: Cherry-picked presentation of results
Evaluations of faithfulness have been increasingly conducted to comprehend the quality of IML outputs across various inputs of interest, but are often presented cherry-picked manner. Firstly, evaluations that present only selective examples where the IML output aligns with previously identified biological mechanisms may be misleading, overlooking the remaining samples that could suggest different underlying behaviors. Additionally, evaluations may also selectively highlight certain conclusions drawn from the examples, leading to an incomplete understanding of the overall findings. Oftentimes, this pitfall occurs not because researchers are intentionally trying to present misleading IML interpretations but rather because they try to select the “best” examples to showcase in a paper.
We now provide concrete examples of cherry-picking in various computational biology applications. For example, in DNA/RNA/protein sequence-based prediction tasks, cherry-picking involves presenting results that only showcase local regions where the subsequences with high importance scores are consistent with the existing annotations, or partially showcase only the convolutional kernels that match the known sequence patterns. In phenotype prediction tasks based on tabular data, cherry-picked results include those that only report the biomarkers assigned high importance scores, which overlap with previously known biomarkers. Such practices are problematic and may lead to a biased interpretation of the underlying mechanisms.
To present a more robust evaluation, we recommend conducting a quantitative analysis of the faithfulness of the importance score to prior knowledge across the entire dataset and summarizing the overall feature importance. It is crucial not to overlook the non-trivial feature importance attributions that may appear inconsistent with prior knowledge. For instance, for BPNet, [9] conducted a comprehensive genome-wide scanning to identify the motifs for each TF. Importantly, the authors reported all 51 identified motifs, assessing the sequence properties to decide whether to include them in the final representative set, providing sufficient justifications for their choices. The final representative set was compared with the previously known transcription factor binding motifs. CITRUS [16] embeds cancer somatic variations and predicts gene expression through an attention-based architecture. To evaluate the consistency between the high-attention genes and the known cancer driver genes, the authors split the gene set into more and less attended groups and performed statistical tests to confirm that cancer drivers are indeed enriched in the high-attention genes, supported by small p-values. IAIA-BL [18] is an imaging-based workflow for classifying breast lesions. An interpretability metric, named activation precision, is defined to assess the concordance between important regions identified by Grad-CAM [29] and human expert annotations. In summary, the examples presented above showcase the recommended practice of assessing feature importance across the entire dataset and employing quantitative metrics for evaluation.
For each IML method, we also recommend including a measure of stability. This can be computed in several ways. For example, UnitedNet [67] assesses the robustness of SHAP in determining feature relevance in single-cell multi-modality data across various hyperparameters and models trained on distinct subsets of data. Washburn et al. [68] calculated the average gradient value attributed to each DNA base pair across 10 iterations of five-fold cross-validation. C. Origami [64] conducted a comparison among three different methods, including Gradient-weighted Regional Activation Mapping (GRAM), attention scores, and perturbation impact scores. Notably, they evaluated feature importance scores for GRAM with varied window sizes and random seeds, revealing its relatively lower robustness compared with the other methods. Stability can also be measured by verifying whether the most important features identified in one dataset are consistent when applying the same workflow to an independent dataset. All the methods discussed above are effective ways of evaluating the stability of the feature importance, thereby assessing the faithfulness of interpretation and enhancing confidence in biological discoveries.
Opportunities for IML Developments in the Age of LLMs
Besides establishing better practices to avoid the pitfalls of IML usage, there are multiple opportunities to develop novel IML techniques for new model architectures and biological applications. Despite the rapid development in predictive modeling for biological applications, particularly with recent advancements in large language models (LLMs), specialized techniques to interpret these models lag behind. Concretely, we observe that the state-of-the-art transformer-based models, such as Enformer [8] and Geneformer [17], still utilize the classic IML methods, such as attention, for explanations. However, the validity and reliability of this explanatory approach remain open to debate, as we addressed in the section “IML Methods and Evaluations in Computational Biology”. We highlight challenges and associated opportunities for developing and applying IML techniques to pre-trained LLMs:
What is the right choice of tokenization for biological applications? Interpretations of explanations can be greatly impacted by the choice of tokenization scheme, the process that splits an input sequence into smaller units that are then encoded by the LLM. For example, for DNA sequences, commonly used tokenization techniques include single-nucleotide tokenization [69], fixed k-mer tokenization [65], and byte pair encoding tokenization [70], which could lead to hypotheses and explanations at different resolutions. There remain opportunities to develop schemes to handle other types of biological data and enable existing tokenization schemes to better represent the underlying biology of the input sequences by incorporating prior knowledge.
How can LLM-specific IML methods be adapted to biological contexts? Recent techniques proposed for transformers are still in their infancy and may not be directly applicable in their current forms; we discuss two prominent approaches. Mechanistic interpretability techniques aim to translate complex transformer models into human-interpretable algorithms (e.g., circuits [45] and human-readable programs [46]). While applications of existing techniques have been limited to relatively simple functions, there are opportunities to apply and enhance mechanistic interpretability methods for more complex, biological contexts. Prompting LLMs is another common approach to explain LLM-generated output using natural language (e.g., via chain-of-thought [71]). Natural language explanations may not be directly applicable to computational biology, which often focuses on predictive rather than generative outputs, but future LLMs that are pre-trained on both natural language and biological corpora may allow computational biologists to leverage these explanations. We note that prompting-based explanations provide no guarantees with regard to the faithfulness to model internals [72], but we might consider this to be a feature, rather than a bug, when using these methods to identify novel, testable hypotheses in biological applications.
How do we develop IML techniques to handle multi-modal applications? There is a recent boom in the integration and modeling of multi-modal data, enabling the understanding of cellular mechanisms from a more comprehensive point of view: Bichrom incorporates DNA sequence with epigenetic signals to predict TF binding [73], and Pathomic Fusion leverages both cancer histology images and genomic features for survival prediction [74]. Matilda [75] explores the integrative modeling of single-cell multi-omic data. While some of these prior works have considered IML methods for their proposed models, their applications of IML methods are limited to unimodal explanations. However, many biological data discussed above, such as sequence and epigenomic features, exhibit high levels of correlation, which can present challenges when assigning accurate importance scores and drawing meaningful biological conclusions. An open research question is defining evaluation techniques to check whether explanations properly attribute importance scores to each modality and building on early work from the ML literature for understanding multi-modal models [76].
What types of novel visualization tools can best facilitate interpretation? Novel IML techniques for LLMs or multi-modal data need to be accompanied by open-source visualization approaches and evaluation platforms. These visualization methods should be tailored to the various data types and applications that are common in computational biology [77]. For example, DNABERT-Viz [78], an attention-weight visualization module designed for DNA sequences, allows users to explore the important genomic regions and sequence motifs. While DNABERT-Viz serves as a good starting point for the development of visualization tools specific to analyzing the attention weights for DNA sequences, a suite of tools across different data types is necessary to enable a more standardized IML workflow across computational biology applications.
Finally, there are still areas within computational biology, such as genetic perturbation studies, sequence comparisons, cellular structure and function modeling, and bioimage analysis, where the application of ML methods is prevalent, but the adoption of IML methods remains relatively limited. Therefore, there are significant opportunities in these areas to consider applying existing methods or developing novel domain-specific IML methods (e.g., by integrating prior biological knowledge into neural network architectures) to improve the interpretability of the analyses.
Conclusion
As IML methods continue to gain traction in computational biology applications, the need for a standardized guideline detailing best practices for evaluating IML methods has become more apparent. In this article, we provided an overview of common IML methods and evaluation metrics, discussed three pitfalls of current evaluation practices when applying IML methods to computational biology applications, and highlighted the importance of engaging additional validations, including incorporating “human-in-the-loop” and “lab-in-the-loop” approaches, to assess IML predictions and enhance their reliability. Nonetheless, we believe that these recommendations signify only the start of a set of contributions toward solidifying the foundations of IML usage and evaluation in computational biology.
There is a persistent demand for concerted efforts within the IML and computational biology communities to continuously improve the ways in which IML methods and evaluations can be tailored to suit diverse biological applications. This is particularly timely given the expected upcoming wave of LLM applications to molecular and cellular datasets. Through such collaborations, we aspire to facilitate the formulation of new IML problems in ways that are likely to significantly promote hypothesis generation and new discoveries across a broad spectrum of biological and biomedical contexts.
Acknowledgements
This work was supported in part by the National Institutes of Health Common Fund 4D Nucleome Program grant UM1HG011593 (J.M.), National Institutes of Health Common Fund Cellular Senescence Network Program grant UH3CA268202 (J.M.), National Institutes of Health grants R01HG007352 (J.M.), R01HG012303 (J.M.), and U24HG012070 (J.M.), and National Science Foundation grants IIS1705121 (A.T.), IIS1838017 (A.T.), IIS2046613 (A.T.), and IIS2112471 (A.T.). J.M. was additionally supported by a Guggenheim Fellowship from the John Simon Guggenheim Memorial Foundation, a Google Research Collabs Award and a Single-Cell Biology Data Insights award from the Chan Zuckerberg Initiative. A.T. was additionally supported by funding from Meta, Morgan Stanley and Amazon. Any opinions, findings and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of any of these funding agencies.
Competing Interests
A.T. received gift research grants from Meta, Morgan Stanley, and Amazon. J.M. received gift research grant from Google Research. A.T. works part-time for Amplify Partners. The other authors declare no competing interests.
References
- [1].Miller T Explanation in artificial intelligence: Insights from the social sciences. Artificial Intelligence 267, 1–38 (2019). [Google Scholar]
- [2].Doshi-Velez F & Kim B Towards a rigorous science of interpretable machine learning. arXiv preprint arXiv:1702.08608 (2017). [Google Scholar]
- [3].Azodi CB, Tang J & Shiu S-H Opening the black box: Interpretable machine learning for geneticists. Trends in Genetics 36, 442–455 (2020). [DOI] [PubMed] [Google Scholar]
- [4].Eraslan G, Avsec Ž, Gagneur J & Theis FJ Deep learning: new computational modelling techniques for genomics. Nature Reviews Genetics 20, 389–403 (2019). This paper gives an extensive review of the application of deep learning models in genomics. [DOI] [PubMed] [Google Scholar]
- [5].Talukder A, Barham C, Li X & Hu H Interpretation of deep learning in genomics and epigenomics. Briefings in Bioinformatics 22, bbaa177 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Novakovsky G, Dexter N, Libbrecht MW, Wasserman WW & Mostafavi S Obtaining genetics insights from deep learning via explainable artificial intelligence. Nature Reviews Genetics 24, 125–137 (2023). This paper provides a comprehensive review for the commonly applied IML methods in biology through the examples from regulatory genomics. [DOI] [PubMed] [Google Scholar]
- [7].Klauschen F et al. Toward explainable artificial intelligence for precision pathology. Annual Review of Pathology: Mechanisms of Disease 19, 541–570 (2024). [DOI] [PubMed] [Google Scholar]
- [8].Avsec Ž et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nature Methods 18, 1196–1203 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Avsec Ž et al. Base-resolution models of transcription-factor binding reveal soft motif syntax. Nature Genetics 53, 354–366 (2021). This paper is a representative example of applying post-hoc explanation methods and connecting the feature importance scores with biological interpretations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schwessinger R et al. DeepC: predicting 3D genome folding using megabase-scale transfer learning. Nature Methods 17, 1118–1124 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Karimi M, Wu D, Wang Z & Shen Y DeepAffinity: interpretable deep learning of compound–protein affinity through unified recurrent and convolutional neural networks. Bioinformatics 35, 3329–3338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Vig J et al. BERTology meets biology: interpreting attention in protein language models. arXiv preprint arXiv:2006.15222 (2020). [Google Scholar]
- [13].Taujale R et al. Mapping the glycosyltransferase fold landscape using interpretable deep learning. Nature Communications 12, 5656 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ma J et al. Using deep learning to model the hierarchical structure and function of a cell. Nature Methods 15, 290–298 (2018). This paper illustrates a biologically informed neural network that incorporates the hierarchical cell subsystems into the neural network architecture. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tasaki S, Gaiteri C, Mostafavi S & Wang Y Deep learning decodes the principles of differential gene expression. Nature Machine Intelligence 2, 376–386 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tao Y et al. Interpretable deep learning for chromatin-informed inference of transcriptional programs driven by somatic alterations across cancers. Nucleic Acids Research 50, 10869–10881 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- [17].Theodoris CV et al. Transfer learning enables predictions in network biology. Nature 618, 616–624 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Barnett AJ et al. A case-based interpretable deep learning model for classification of mass lesions in digital mammography. Nature Machine Intelligence 3, 1061–1070 (2021). [Google Scholar]
- [19].Zaritsky A et al. Interpretable deep learning uncovers cellular properties in label-free live cell images that are predictive of highly metastatic melanoma. Cell Systems 12, 733–747 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].DeGrave AJ, Cai ZR, Janizek JD, Daneshjou R & Lee S-I Auditing the inference processes of medical-image classifiers by leveraging generative AI and the expertise of physicians. Nature Biomedical Engineering 1–13 (2023). [DOI] [PubMed] [Google Scholar]
- [21].Heil BJ et al. Reproducibility standards for machine learning in the life sciences. Nature Methods 18, 1132–1135 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Whalen S, Schreiber J, Noble WS & Pollard KS Navigating the pitfalls of applying machine learning in genomics. Nature Reviews Genetics 23, 169–181 (2022). [DOI] [PubMed] [Google Scholar]
- [23].Sapoval N et al. Current progress and open challenges for applying deep learning across the biosciences. Nature Communications 13, 1728 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen V, Li J, Kim JS, Plumb G & Talwalkar A Interpretable machine learning: Moving from mythos to diagnostics. Communications of the ACM 65, 43–50 (2022). This paper describes the disconnect between IML techniques and downstream use cases and outlines paths forward to bridge the disconnect. [Google Scholar]
- [25].Räuker T, Ho A, Casper S & Hadfield-Menell D Toward transparent AI: A survey on interpreting the inner structures of deep neural networks. In 2023 IEEE Conference on Secure and Trustworthy Machine Learning (SaTML), 464–483 (IEEE, 2023). [Google Scholar]
- [26].Yang M & Ma J Machine learning methods for exploring sequence determinants of 3D genome organization. Journal of Molecular Biology 167666 (2022). [DOI] [PubMed] [Google Scholar]
- [27].Shrikumar A, Greenside P & Kundaje A Learning important features through propagating activation differences. In International Conference on Machine Learning, 3145–3153 (2017). [Google Scholar]
- [28].Sundararajan M, Taly A & Yan Q Axiomatic attribution for deep networks. In International Conference on Machine Learning, 3319–3328 (2017). [Google Scholar]
- [29].Selvaraju RR et al. Grad-CAM: Visual explanations from deep networks via gradient-based localization. In Proceedings of the IEEE International Conference on Computer Vision, 618–626 (2017). [Google Scholar]
- [30].Nair S, Shrikumar A, Schreiber J & Kundaje A fastISM: performant in silico saturation mutagenesis for convolutional neural networks. Bioinformatics 38, 2397–2403 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lundberg SM & Lee S-I A unified approach to interpreting model predictions. In Proceedings of the 31st International Conference on Neural Information Processing Systems, 4768–4777 (2017). [Google Scholar]
- [32].Ribeiro MT, Singh S & Guestrin C “why should i trust you?” explaining the predictions of any classifier. In Proceedings of the 22nd ACM SIGKDD, 1135–1144 (2016). [Google Scholar]
- [33].Tseng A, Shrikumar A & Kundaje A Fourier-transform-based attribution priors improve the interpretability and stability of deep learning models for genomics. In Advances in Neural Information Processing Systems, vol. 33, 1913–1923 (2020). [Google Scholar]
- [34].Rudin C Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nature Machine Intelligence 206–215 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hastie T & Tibshirani R Generalized additive models: some applications. Journal of the American Statistical Association 82, 371–386 (1987). [Google Scholar]
- [36].Elmarakeby HA et al. Biologically informed deep neural network for prostate cancer discovery. Nature 598, 348–352 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fortelny N & Bock C Knowledge-primed neural networks enable biologically interpretable deep learning on single-cell sequencing data. Genome Biology 21, 1–36 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Janizek JD et al. PAUSE: principled feature attribution for unsupervised gene expression analysis. Genome Biology 24, 81 (2023). This paper proposes an approach to combining the post-hoc and by-design explanation methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Vaswani A et al. Attention is all you need. In Advances in Neural Information Processing Systems, 5998–6008 (2017). [Google Scholar]
- [40].Karbalayghareh A, Sahin M & Leslie CS Chromatin interaction–aware gene regulatory modeling with graph attention networks. Genome Research 32, 930–944 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Serrano S & Smith NA Is attention interpretable? In Proceedings of the 57th Annual Meeting of the Association for Computational Linguistics (2019). [Google Scholar]
- [42].Jain S & Wallace BC Attention is not explanation. In Proceedings of NAACL-HLT, 3543–3556 (2019). [Google Scholar]
- [43].Wiegreffe S & Pinter Y Attention is not not explanation. In Proceedings of EMNLP-IJCNLP, 11–20 (2019). [Google Scholar]
- [44].Bai B et al. Why attentions may not be interpretable? In Proceedings of the 27th ACM SIGKDD Conference on Knowledge Discovery & Data Mining, 25–34 (2021). [Google Scholar]
- [45].Conmy A, Mavor-Parker AN, Lynch A, Heimersheim S & Garriga-Alonso A Towards automated circuit discovery for mechanistic interpretability. In Thirty-seventh Conference on Neural Information Processing Systems (2023). [Google Scholar]
- [46].Friedman D, Wettig A & Chen D Learning transformer programs. In Advances in Neural Information Processing Systems, vol. 36 (2023). [Google Scholar]
- [47].Alvarez Melis D & Jaakkola T Towards robust interpretability with self-explaining neural networks. In Advances in Neural Information Processing Systems, vol. 31 (2018). [Google Scholar]
- [48].Jacovi A & Goldberg Y Towards faithfully interpretable NLP systems: How should we define and evaluate faithfulness? arXiv preprint arXiv:2004.03685 (2020). [Google Scholar]
- [49].Yang M & Kim B Benchmarking attribution methods with relative feature importance. arXiv preprint arXiv:1907.09701 (2019). [Google Scholar]
- [50].Adebayo J, Muelly M, Liccardi I & Kim B Debugging tests for model explanations. arXiv preprint arXiv:2011.05429 (2020). [Google Scholar]
- [51].Kim JS, Plumb G & Talwalkar A Sanity simulations for saliency methods. In Proceedings of the 39th International Conference on Machine Learning, 11173–11200 (2022). [Google Scholar]
- [52].Zhou Y, Booth S, Ribeiro MT & Shah J Do feature attribution methods correctly attribute features? In Proceedings of the AAAI Conference on Artificial Intelligence, vol. 36, 9623–9633 (2022). [Google Scholar]
- [53].Agarwal C et al. Openxai: Towards a transparent evaluation of model explanations. In Advances in Neural Information Processing Systems, vol. 35 (2022). [Google Scholar]
- [54].Ghorbani A, Abid A & Zou J Interpretation of neural networks is fragile. In Proceedings of the AAAI Conference on Artificial Intelligence, vol. 33, 3681–3688 (2019). [Google Scholar]
- [55].Krishna S et al. The disagreement problem in explainable machine learning: A practitioner’s perspective. arXiv preprint arXiv:2202.01602 (2022). [Google Scholar]
- [56].Zhao Y, Shao J & Asmann YW Assessment and optimization of explainable machine learning models applied to transcriptomic data. Genomics, Proteomics and Bioinformatics 20, 899–911 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tang Z et al. Interpretable classification of Alzheimer’s disease pathologies with a convolutional neural network pipeline. Nature Communications 10, 2173 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zeiler MD & Fergus R Visualizing and understanding convolutional networks. In ECCV 2014, 818–833 (2014). [Google Scholar]
- [59].Shrikumar A et al. Technical note on transcription factor motif discovery from importance scores (TF-MoDISco) version 0.5. 6.5. arXiv preprint arXiv:1811.00416 (2018). [Google Scholar]
- [60].Rahman MA & Rangwala H IDMIL: an alignment-free interpretable Deep Multiple Instance Learning (MIL) for predicting disease from whole-metagenomic data. Bioinformatics 36, i39–i47 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wang L et al. An interpretable deep-learning architecture of capsule networks for identifying cell-type gene expression programs from single-cell RNA-sequencing data. Nature Machine Intelligence 2, 693–703 (2020). [Google Scholar]
- [62].Nagao Y, Sakamoto M, Chinen T, Okada Y & Takao D Robust classification of cell cycle phase and biological feature extraction by image-based deep learning. Molecular Biology of the Cell 31, 1346–1354 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Lafarge MW et al. Capturing single-cell phenotypic variation via unsupervised representation learning. In International Conference on Medical Imaging with Deep Learning, 315–325 (2019). [PMC free article] [PubMed] [Google Scholar]
- [64].Tan J et al. Cell-type-specific prediction of 3D chromatin organization enables high-throughput in silico genetic screening. Nature Biotechnology 41, 1140–1150 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Dalla-Torre H et al. The nucleotide transformer: Building and evaluating robust foundation models for human genomics. bioRxiv 2023–01 (2023). [Google Scholar]
- [66].Abnar S & Zuidema W Quantifying attention flow in transformers. arXiv preprint arXiv:2005.00928 (2020). [Google Scholar]
- [67].Tang X et al. Explainable multi-task learning for multi-modality biological data analysis. Nature Communications 14, 2546 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Washburn JD et al. Evolutionarily informed deep learning methods for predicting relative transcript abundance from DNA sequence. Proceedings of the National Academy of Sciences 116, 5542–5549 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Nguyen E et al. HyenaDNA: Long-range genomic sequence modeling at single nucleotide resolution. Advances in neural information processing systems 36 (2024). [Google Scholar]
- [70].Zhou Z et al. DNABERT-2: Efficient foundation model and benchmark for multi-species genome. arXiv preprint arXiv:2306.15006 (2023). [Google Scholar]
- [71].Wei J et al. Chain-of-thought prompting elicits reasoning in large language models. Advances in Neural Information Processing Systems 35, 24824–24837 (2022). [Google Scholar]
- [72].Liu K, Casper S, Hadfield-Menell D & Andreas J Cognitive dissonance: Why do language model outputs disagree with internal representations of truthfulness? arXiv preprint arXiv:2312.03729 (2023). [Google Scholar]
- [73].Srivastava D, Aydin B, Mazzoni EO & Mahony S An interpretable bimodal neural network characterizes the sequence and preexisting chromatin predictors of induced transcription factor binding. Genome Biology 22, 1–25 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Chen RJ et al. Pathomic fusion: an integrated framework for fusing histopathology and genomic features for cancer diagnosis and prognosis. IEEE Transactions on Medical Imaging (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Liu C, Huang H & Yang P Multi-task learning from multimodal single-cell omics with Matilda. Nucleic Acids Research 51, e45–e45 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Liang PP et al. MultiViz: Towards visualizing and understanding multimodal models. In The Eleventh International Conference on Learning Representations (2023). [Google Scholar]
- [77].Valeri JA et al. BioAutoMATED: An end-to-end automated machine learning tool for explanation and design of biological sequences. Cell Systems 14, 525–542 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Ji Y, Zhou Z, Liu H & Davuluri RV DNABERT: pre-trained bidirectional encoder representations from transformers model for dna-language in genome. Bioinformatics 37, 2112–2120 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]