

Abstract
Renal fibrosis is a devastating consequence of progressive chronic kidney disease, representing a major public health challenge worldwide. The underlying mechanisms in the pathogenesis of renal fibrosis remain unclear, and effective treatments are still lacking. Renal tubular epithelial cells (RTECs) maintain kidney function, and their dysfunction has emerged as a critical contributor to renal fibrosis. Cellular quality control comprises several components, including telomere homeostasis, ubiquitin-proteasome system (UPS), autophagy, mitochondrial homeostasis (mitophagy and mitochondrial metabolism), endoplasmic reticulum (ER, unfolded protein response), and lysosomes. Failures in the cellular quality control of RTECs, including DNA, protein, and organelle damage, exert profibrotic functions by leading to senescence, defective autophagy, ER stress, mitochondrial and lysosomal dysfunction, apoptosis, fibroblast activation, and immune cell recruitment. In this review, we summarize recent advances in understanding the role of quality control components and intercellular crosstalk networks in RTECs, within the context of renal fibrosis.
Keywords: Renal tubular epithelial cells, Quality control, Renal fibrosis, Telomere homeostasis, Autophagy, Mitochondria
Graphical abstract
Renal fibrosis is a devastating consequence of progressive chronic kidney disease, and poses a major public health challenge worldwide. Renal tubular epithelial cells (RTECs) play an essential role in maintaining kidney function, and their cellular quality control imbalance has emerged as a critical contributor to renal fibrosis.
Highlights
-
•
Quality control mechanisms within RTECs are multi-tiered, involving DNA-, protein-, and organelle-level.
-
•
Dysfunctional RTECs quality control components drive the progression of renal fibrosis.
-
•
The crosstalk of RTEC/(myo)fibroblast and RTEC/immune cell promote progressive renal fibrosis.
1. Introduction
Fibrosis is a devastating disease with a pathological feature of the progressive accumulation of excessive extracellular matrix (ECM) owing to the dysregulated repair progress in a wide range of organs systems, such as kidney, liver, lung, heart, and skin [1]. Renal fibrosis is a paramount pathological hallmark for the evolution of virtually all types of chronic kidney diseases (CKDs), which can progress to end-stage renal disease. The pathology of renal fibrosis is principally identified by irreversible nephron loss, tubular atrophy, renal interstitial fibrosis, and glomerulosclerosis [2].
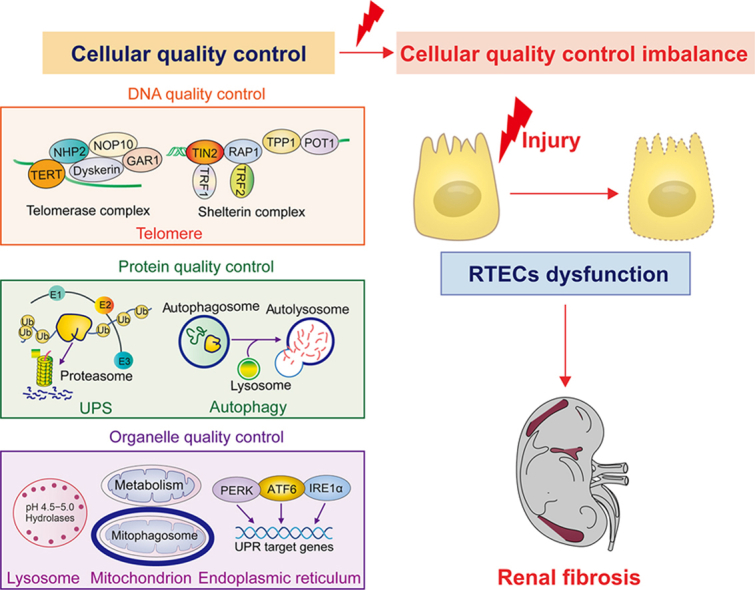
Multiple noxious stimulations, such as ischemia, toxins, proteinuria, metabolic disorders, hypoxia, and senescence, are the leading causes of kidney injury [3]. Renal tubules are essential components of the kidney. From a histopathological view, chronic tubular injury leads to the formation of the fibrogenic niche, which accelerates fibroblast activation, tubular atrophy, inflammatory cell infiltration, and vascular rarefaction during fibrosis [4]. Currently, no safe and effective treatments for renal fibrosis are available. Renal tubular epithelial cells (RTECs) exert an active role in responding to injury, and injured RTECs are significant drivers of fibrotic kidney disease progression. Cellular quality control is an anthropomorphic decision-making process that discriminates between normal and malfunctioning proteins or subcellular structures [5]. Intracellular quality control involves a great diversity of self-regulating activities, such as maintaining the balance between functional protein synthesis and dysfunctional protein breakdown, which play essential roles in biological homeostasis [6]. Accumulating evidence suggests that imbalance in cellular quality control is closely related to various diseases, including fibrosis, cancer, neurodegenerative disorders, aging, and other aging-related diseases. RTECs can maintain homeostasis at DNA, protein, and organelle levels in response to exogenous or endogenous insults (Fig. 1). In this review, we summarize the cellular quality control and biology of dysfunctional RTECs in renal fibrosis and discuss the potential implications of quality control in RTECs for the future diagnosis and treatment of renal fibrotic diseases.
Fig. 1.

Cellular quality control in renal tubular epithelial cells (RTECs). Maintenance of telomere homeostasis (DNA), ubiquitin-proteasome system (UPS) and autophagy (protein), endoplasmic reticulum (ER), mitochondrion, and lysosome (organelle) are essential for cellular homeostasis. TIN2: TRF1 interacting nuclear factor 2; RAP1: repressor/activator protein 1; TPP1: telomere protection protein 1; POT1: protection of telomeres 1; TRF1: telomeric repeat-binding factor 1; TRF2: telomeric repeat-binding factor 2; TERT: telomerase reverse transcriptase; NHP2: H/ACA ribonucleoprotein complex subunit 2; NOP10: H/ACA ribonucleoprotein complex subunit 3; GAR1: H/ACA ribonucleoprotein complex subunit 1; E1: ubiquitin-activating enzyme; E2: ubiquitin-conjugating enzyme; E3: ubiquitin ligase; Ub: ubiquitin; LC3: microtubule-associated protein 1 light chain 3; p62: sequestosome 1; PERK: protein kinase RNA-like ER kinase; ATF6: activating transcription factor 6; IRE1α: inositol-requiring protein 1α; UPR: unfolded protein response; PINK1: phosphatase and tensin homolog on chromosome ten (PTEN)-induced putative kinase 1; TFEB: transcription factor EB; TFEC: transcription factor EC.
2. RTECs as critical contributors of renal fibrosis
The role of RTECs in the pathogenesis of renal fibrosis has recently received attention. Following kidney injury, damaged RTECs undergo a series of changes. On the one hand, RTECs undergo dedifferentiation into mesenchymal states, known as epithelial-to-mesenchymal transition (EMT). In living organisms, EMT is associated with multiple biological and pathological processes, such as embryo development, tissue regeneration, fibrosis, and cancer progression [7]. During this process, RTECs remodel cell cytoskeleton and undergo distinct changes in signaling and transcriptional regulatory networks [8]. Numerous genetic drivers and regulators are involved in the activation and maintenance of EMT, including zinc finger E-box binding homeobox 1/2 (ZEB1/2), zinc finger protein SNAI1/2 (Snail1/2), and twist-related protein 1/2 (Twist1/2). E-cadherin is a common epithelial marker; and N-cadherin, vimentin, and matrix metalloproteinases are known as mesenchymal marks. EMT translational factors can suppress the expression of E-cadherin and activate mesenchymal marks, all of which promote RTECs’ loss of tight junctions and apical polarity and thus provide these cells with mesenchymal characteristics [9]. Furthermore, EMT significantly induces the upregulation of a large amount of profibrotic cytokines, such as connective tissue growth factor (CTGF), fibronectin, and collagen I, thereby accelerating interstitial ECM deposition [10,11]. Multiple signaling pathways such as transforming growth factor-β (TGF-β), Wnt/β-catenin, sonic hedgehog (SHH), Hedgehog, Notch, and Hippo serve as the regulatory networks of EMT [12,13]. TGF-β can induce EMT in a significant way and elicit small mothers against decapentaplegic (Smad)-dependent classical pathway and non-Smad pathway responses in renal fibrosis. On the other hand, damaged RTECs transform into a secretory phenotype and subsequently release a series of proinflammatory mediators, such as toll-like receptors and inflammasomes. In turn, this phenomenon exacerbates the inflammation response in the interstitium [14]. After renal insult, RTECs are stimulated to enter the cell cycle but are arrested at G1/S and G2/M checkpoints in response to maladaptive DNA repair [15]. The increased G2/M-arrested RTECs can produce TGF-β and CTGF, thereby inducing renal fibrosis [16]. Thus, damaged RTECs can exacerbate renal inflammation and fibrosis through these mechanisms.
3. DNA quality control in RTECs
Telomere homeostasis is the core of DNA quality control in RTECs, and telomere dysfunction in RTECs can drive renal fibrosis (Fig. 2).
Fig. 2.

DNA quality control in fibrotic renal tubular epithelial cells (RTECs). Telomere dysfunction can be induced by irreparable DNA damage and telomere shortening. Persistent damage and telomere shortening inhibit DNA damage repair in fibrotic RTECs, trigger persistent DNA damage responses (DDRs), and induce cell cycle arrest and cellular senescence, all of which ultimately lead to renal fibrosis. TIN2: TRF1 interacting nuclear factor 2; RAP1: repressor/activator protein 1; TRF1: telomeric repeat-binding factor 1; TRF2: telomeric repeat-binding factor 2; TPP1: telomere protection protein 1; POT1: protection of telomeres 1; TERT: telomerase reverse transcriptase; NHP2: H/ACA ribonucleoprotein complex subunit 2; NOP10: H/ACA ribonucleoprotein complex subunit 3; GAR1: H/ACA ribonucleoprotein complex subunit 1; TERC: telomerase RNA component; p16: cyclin dependent kinase inhibitor 2A; p21: cyclin-dependent kinase inhibitor 1A; SASP: senescence-associated secretory phenotype.
3.1. Telomere homeostasis
Telomeres are formed by satellite DNA repeats sequences and a specialized six-protein shelterin complex that prevents linear chromosome ends from triggering DNA damage response (DDR) and contributes to the maintenance of genome integrity and stability [17]. Telomere dysfunction typically manifests in three primary ways: telomere shortening, telomere structure collapse, and displacement of shelterin complexes from telomeres. Progressive telomere shortening followed by each successive cell division disrupts the protective cap, activates persistent DDR, and ultimately results in growth arrest and cell apoptosis [18,19]. Telomere function is closely related to shelterin complex, which involves six core telomeric proteins: telomeric repeat-binding factor 1 (TRF1), telomeric repeat-binding factor 2 (TRF2), TRF1 interacting nuclear factor 2 (TIN2), repressor/activator protein 1 (RAP1), telomere protection protein 1 (TPP1), and protection of telomeres 1 (POT1) [20]. Telomerase is a specialized enzyme that can elongate telomeres and maintain telomere DNA repeat sequences. It is mainly composed of a telomerase reverse transcriptase catalytic subunit called telomerase reverse transcriptase (TERT) and a telomerase RNA subunit that encodes the template sequence called telomerase RNA component (TERC) [21]. Telomerase complex also contains additional proteins such as H/ACA ribonucleoprotein complex subunit 1, H/ACA ribonucleoprotein complex subunit 2, H/ACA ribonucleoprotein complex subunit 3, and dyskerin. Telomerase plays a crucial role in the glomerular renewal of adult mice kidneys due to the activation and clonal expansion of podocyte progenitor cells medicating by TERT [22]. Genome instability, telomeric dysfunction, and DNA damage have been implicated in the damage and senescence of RTECs in the kidney [23]. MiR-155 deficiency distinctly alleviates cisplatin-induced acute kidney injury (AKI) by suppressing genome instability and telomeric dysfunction in RTECs [24].
3.2. Telomere shortening and senescence
Telomere shortening has emerged as a hallmark and causal factor for cellular senescence. The length of telomeres tends to shorten with successive DNA replication in proliferating tissues [25]. When telomeres reach a critically short length, the induction of DDR suppresses cell proliferation by activating cell cycle inhibitors such as cyclin-dependent kinase inhibitor 1A and 2A (p21 and p16). Moreover, irreparable DNA damage can trigger degradation or loss in terminal portions of telomeres, thereby causing telomere shortening and arrested proliferation in nonproliferating tissues [26]. The main characteristics of cellular senescence include an irreversible arrest of proliferation and variations of gene expression, chromatin reorganization, and cell morphology. RTECs can exhibit a senescence-like phenotype during abnormal kidney repair [27]. Cells undergoing senescence maintain a high metabolic activity and produce a variety of pro-inflammatory and proteolytic factor cytokines, an ability called senescence-associated secretory phenotype [28]. Accelerated telomere shortening is the result of multiple factors (oxidative damage, replicative stress, and physiological stress) and is also strongly linked to kidney aging, AKI, diabetic nephropathy (DN) and aging-related diseases such as renal fibrosis [29,30]. The number of sensitive DDR marker histone H2A variant H2AX is significantly increased in the tubules of kidney tissue from patients with CKD and RTECs of mice after cisplatin-induced AKI [31]. Telomere length is positively associated with high levels of reactive oxygen species (ROS), and elevated ROS production can lead to cellular senescence [32]. RTECs are central sites of renal senescence [33,34]. RTECs undergoing senescence promote maladaptive kidney repair, leading to the progression of renal fibrosis [35,36]. Urinary decoy receptor 2 has been proposed as a potential biomarker for tubulointerstitial damage in patients with DN. Its knockdown significantly attenuates renal fibrosis via inhibiting the cellular senescence of RTECs [37,38].
4. Protein quality control in RTECs
Protein homeostasis, or proteostasis, is controlled by a complicated regulatory network of elements that comprises molecular chaperones, proteases, and their regulators [39]. These elements work together to maintain the balance of protein synthesis, folding, conformational maintenance, and degradation in living organisms. Deficient protein degradation can affect protein homeostasis. The cellular protein quality control system acts as a proteostasis protector by modulating the proper folding of new proteins [40]. Eukaryotic cells, including RTECs, are composed of two main proteolytic systems, including ubiquitin-proteasome system (UPS) and autophagy, both of which constitute an interconnected quality control network (Fig. 3). Persistent mechanical or environmental stresses can pose ongoing threats to the accurate folding and integrity of the cellular protein network in the kidney, ultimately leading to renal fibrosis, DN, and renal senescence.
Fig. 3.

Protein quality control in renal tubular epithelial cells (RTECs). Misfolded and damaged protein can be degraded by ubiquitin-proteasome system (UPS) and autophagy system. The collaboration between UPS and autophagy is critical for protein homeostasis in RTECs. The majority of aberrant proteins are targeted to 26S proteasome for degradation in UPS. When UPS is inhibited, the autophagy system can be activated to remove incremental aberrant proteins. Ub: ubiquitin; E1: ubiquitin-activating enzyme; E2: ubiquitin-conjugating enzyme; E3: ubiquitin ligase; p62: sequestosome 1; LC3: microtubule-associated protein 1 light chain 3.
4.1. UPS
UPS is a highly specific cellular response to misfolded, aggregated, and damaged proteins. Approximately 70%–80% of proteolysis in cells is performed via UPS, which primarily degrades individual and unfolded polypeptides. These polypeptides can typically access the narrow axial channel of the proteasome [5,41]. The initial step is adenosine triphosphate (ATP)-dependent ubiquitination through the cooperative action of three enzymes, namely, ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3) [42]. In particular, E1 is responsible for initiating enzyme in the ubiquitination cascade because it hydrolyses ATP and adenylates the C-terminal glycine residue of ubiquitin to yield ubiquitin adenylates. The critical step of this system is the conjugation of ubiquitin to substrates. Activated ubiquitin can be transferred to E2 and forming an E2–ubiquitin complex. E3 then recruits substrates and accelerates the transfer of ubiquitin from E2 to the targeted protein [43]. The following step is the recognition and degradation of ubiquitin-tagged substrate by the 26S proteasome complex. A functional 26S proteasome is assembled by a barrel-shaped 20S core particle (CP) and one or two 19S regulatory particles (RPs) [44]. RPs directly control the processing and entry of ubiquitinated cargo into CP, in which substrates can be hydrolyzed and broken down into short peptides [45]. UPS is closely linked to the progress of kidney diseases. The deubiquitinating enzyme ubiquitin C-terminal hydrolase L1 (UCH-L1) modulates protein degradation through UPS in the kidney, and UCH-L1-deficient mice show fibrinoid necrosis [46]. Ubiquitin-like protein human leukocyte antigen F locus adjacent transcript 10 (FAT10) can facilitate renal fibrosis by stabilizing ubiquitin-specific protease 7 (USP7) to agitate checkpoint kinase 1-mediated G2/M arrest in RTECs [47]. TGF-β signaling is primarily mediated by intracellular Smad signal transducer proteins, and numerous steps of TGF-β signaling cascade are regulated through UPS [48]. The activation of TGF-β/Smads signaling in RTECs can directly promote renal fibrosis [49,50]. Von Hippel-Lindau (VHL) is an E3 ubiquitin ligase, and VHL-recruiting proteolytic targeting chimeras can alleviate renal fibrosis via the degradation of mothers against decapentaplegic homolog 3 (SMAD3) by UPS [51]. Kindlin-2 is implicated in the regulation of TGF-β, and Hippo signaling pathway. Kindlin-2 in RTECs suppresses Hippo signaling and facilitates renal fibrosis by enhancing the degradation of Mps one binder kinase activator 1 (MOB1) [52].
4.2. Autophagy
Macroautophagy, often referred to as autophagy, is a major intracellular quality control process that is essential for maintaining cellular and tissue homeostasis. During autophagy, potentially harmful cytosolic entities, such as protein aggregates, damaged mitochondria, and invading bacteria, are enclosed in double-membrane vesicles named autophagosomes, which then transport them to lysosomes for degradation [53]. Autophagosome biogenesis is mainly composed of several sequential steps, such as initiation, isolation membrane nucleation, elongation, fusion, and degradation. From a mechanistical perspective, UNC-51-like kinase 1 or 2 (ULK1 or ULK2) serine threonine kinase complex involving ULK1, ULK2, RB1-inducible coiled-coil protein 1 (RB1CC1), autophagy-related (ATG) protein 13 and 101, can initiate the formation of autophagosome and phosphorylate class III phosphatidylinositol 3-kinase (PI3K-III) complex. The PI3K complex involving ATG14, Beclin 1, vacuolar protein sorting 34 and 15 can generate phosphatidylinositol 3-phosphate (PI3P) to participate in autophagosome nucleation [54,55]. In addition, ATG9A vesicles deliver components to form phagophore membrane. WD repeat domain phosphoinositide-interacting proteins and their binding partners ATG2A and ATG2B are recruited to work as PI3P effectors in autophagy [56]. The elongation and closure of phagophore are primarily regulated by two ubiquitin-like protein conjugation systems, namely, ATG12–ATG5–ATG16 complex and phosphatidylethanolamine (PE) conjugated-ATG8 family proteins (LC3 and gamma-aminobutyric acid receptor–associated protein) [57]. ATG12–ATG–ATG16 complex facilitates the conjugation between ATG8 family proteins and PE, all of which can convert into autophagosome with the accumulation of cytosolic contents [58]. Finally, autophagosomes fuse with lysosomes to produce autolysosomes and then degrade their autophagosomal contents through lumenal acidification and lysosomal hydrolases activation.
RTECs are highly dependent on autophagy to respond to stressors for homeostasis maintenance. A clinical study found that in human diabetic kidneys, RTECs exhibited an accumulation of autophagosomes and a reduction in autophagic clearance [59]. Charged multivesicular body protein 2A (CHMP2A)-mediated phagophore closure plays a crucial role in the formation of functional autolysosome, and phosphatase and tensin homolog on chromosome ten (PTEN) can restrain the maladaptive repair of RTECs by restoring CHMP2A-mediated phagosome closure [60]. ATG5-mediated autophagy in RTECs can ameliorate renal fibrosis by inhibiting G2/M cell cycle arrest [61]. In addition, AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signaling pathway participate in the regulation of autophagy [62]. Glucagon-like peptide-1 and ginsenoside Rb1 (a natural autophagy regulator) attenuates autophagy and renal fibrosis by mediating the AMPK/mTOR pathway in RTECs [63,64]. AMPK exists as a highly conserved heterotrimeric complex containing α, β, and γ subunits. The mechanism of AMPK activation is achieved through the phosphorylation of a conserved threonine residue, phospho-AMPKα. Liver kinase B1 (LKB1) and calcium/calmodulin-dependent protein kinase kinase (CaMKK) are the two major upstream kinases for AMPK activation. LKB1-mediated AMPK activation is induced by the increase of the adenosine monophosphate (AMP)/ATP ratio, and its CaMKK-mediated activation is dependent on intracellular Ca2+ ion concentration [65,66]. LKB1 dysfunction is directly involved in renal fibrosis. Piericidin analogue S14, an activator of LKB1, can attenuate RTEC senescence and renal fibrosis by promoting LKB1/AMPK-mediated autophagy [67]. Vitamin D receptor promotes the activation of Ca2+-CaMKK2-AMPK pathway to restore the defective autophagy of RTECs, eventually alleviating streptozotocin-induced DN [68]. In addition, brahma-related gene 1 accelerates RTEC senescence and drives renal fibrosis via the inhibition of Wnt/β-catenin-mediated autophagy [69].
5. Organelle quality control in RTECs
The organelle quality control in RTECs mainly includes ER, mitochondria, and lysosome quality control system (Fig. 4).
Fig. 4.

Organelle quality control in fibrotic renal tubular epithelial cells (RTECs). The core elements of organelle quality control network in RTECs include the homeostasis of endoplasmic reticulum (ER), mitochondria, and lysosome. Upon ER stress, RTECs exert complementary adaptive mechanisms, namely, unfolded protein response (UPR), to deal with protein-folding alterations. Upon irreversible ER stress, UPR further promotes RTEC apoptosis. An increased UPR can be found in fibrotic RTECs. In addition, lysosome dysfunction suppresses normal mitophagy, and mitochondrial damage results in defective fatty acid β-oxidation in RTECs, both of which promote renal fibrosis. PERK: protein kinase RNA-like ER kinase; eIF2α: factor 2 subunit-α; ATF4: activating transcription factor 4; ATF6: activating transcription factor 6; S1P: site-1 protease; S2P: site-2 protease; ATF6f: fragment of ATF6; IRE1α: inositol-requiring protein 1α; XBP-1: X-box binding protein 1; XBP-1s: spliced form of XBP1; JNK: c-Jun N-terminal kinase; CHOP: CCAAT/enhancer-binding protein-homologous protein; TCA: tricarboxylic acid; ATP: adenosine triphosphate; LMP: lysosomal membrane permeabilization; PINK1: phosphatase and tensin homolog on chromosome ten (PTEN)-induced putative kinase 1; Ub: ubiquitin; LC3: microtubule-associated protein 1 light chain 3.
5.1. ER quality control
Cells response to ER stress by activating a complementary adaptive mechanism known as the unfolded protein response (UPR). The aim of the UPR is to restore ER homeostasis by eliminating misfolded proteins. This mechanism involves three signal transducers, namely, inositol-requiring protein 1α (IRE1α), protein kinase RNA-like ER kinase (PERK), and activates transcription factor 6 (ATF6). ER stress triggers the dimerization and autotransphosphorylation of IRE1α, which can accelerate the noncanonical splicing of X-box binding protein 1 (XBP1) mRNA via RNase activity [70]. XBP1 is an unfolded protein response effector, and IRE1α-dependent XBP1 splicing produces an active XBP1 transcription factor to regulate the expression of various genes that are strongly implicated in protein folding, ER protein quality control, ER-associated degradation (ERAD), and phospholipid synthesis regulation [71]. Activated PERK promotes the dimerization and phosphorylation of eukaryotic translation initiation factor 2 subunit-α (eIF2α), which can inhibit protein translation and attenuate the overload of unfolded and misfolded proteins in the ER lumen [72]. ATF6 is a transmembrane protein embedded in the ER that encompasses a cytosolic domain encoding basic Leu zipper transcription factor. During ER stress, ATF6 is dissociated and transported to the Golgi apparatus, in which the cytoplasmic domain is proteolytically processed by Golgi-resident site-1 protease and site-2 protease, thus generating an active cytosolic domain fragment of ATF6 (ATF6f) [73]. ATF6f, which migrates to the nucleus, can serve as a transcription factor to directly regulate ERAD and pro-apoptotic pathway, is involved in the activation of XBP1, and further contributes to the inhibition of ER stress [74]. The structural and functional characteristics of RTECs are strongly linked to ER proteostasis activity. Various stressors, including oxidative stress, glycative stress, and hypoxia, can lead to the dysregulation of UPR in RTECs. In addition, sustained UPR triggers the apoptosis of RTECs [75,76]. AKI promotes the activation of UPR, which generally manifests as the up-regulation of p-PERK, cleaved ATF6, and p-IRE1. Nevertheless, ER stress master regulator XBP1-deficient RTECs contribute to fibrosis progression via promoting DDR, cell cycle G2/M arrest, and TGF-β secretion [77]. CCAAT/enhancer-binding protein-homologous protein (CHOP) is a characteristic UPR target gene that can induce caspase-dependent apoptosis activation by regulating the balance between pro- and anti-apoptotic proteins that attach to B-cell lymphoma-2 (Bcl-2) family [78]. Proteinuria overload induces RTEC apoptosis through the PERK-CHOP-dependent pathway [79]. ATF6 is a significant regulator of CHOP during UPR, and cleaved ATF6 triggers the early activation of pro-apoptotic CHOP [80,81]. Meanwhile, 3-hydroxy-3-methylglutaryl reductase degradation protein (HRD1) is known as an ERAD-associated E3 ubiquitin ligase and EIF2α is one of the substrate of HRD1. HRD1 overexpression can inhibit RTECs apoptosis by mediating eIF2α ubiquitylation and degradation [82].
5.2. Mitochondrial quality control
5.2.1. Mitophagy
Mitophagy is a crucial mechanism for mitochondrial quality control and aims to degrade damaged mitochondria to maintain cellular homeostasis [83]. This process involves the formation of autophagosomes around the mitochondria, which is facilitated by ubiquitin molecules and mitochondrial outer membrane receptors that are conjugated to proteins [84]. PTEN-induced putative kinase 1 (PINK1)-Parkin-mediated and receptor-mediated mitophagy plays renoprotective roles in several kidney diseases. In PINK1-Parkin-dependent mitophagy, the loss of mitochondrial membrane potential (MMP) recruits Parkin to depolarized mitochondria, thereby motivating the clearance of damaged mitochondria via autophagy [85]. PTEN-PINK1 triggers Parkin E3 ubiquitin ligase activity by phosphorylating serine 65 upon mitochondrial depolarization, consequently creating a PINK1-Parkin-pSer65-Ub feedforward loop to initiate mitophagy [86,87]. Receptor-mediated mitophagy pathways are mediated by several receptors on the outer mitochondrial membrane (FUN14 domain-containing 1, NIX/Bcl-2 interacting protein 3, FK506-binding protein 8, and Bcl-2-like protein 13) or inner mitochondrial membrane (prohibitin-2). These receptors directly bind to LC3 and form mitophagosome [88]. Optineurin is a well-recognized autophagy receptor that can initiate PINK1-Parkin-mediated mitophagy in a nonclassical path. Rather than ULK1 or ULK2, it uses TANK binding kinase 1 that directly binds and phosphorylates the PI3K complex, thereby triggering mitophagy [89]. Mitophagy has emerged as the effective protective regulator of RTECs during renal injury and fibrosis. In ischemia-reperfusion injury (IRI) or contrast-induced AKI, the activation of PINK1-Parkin-mediated mitophagy can protect RTECs by suppressing mitochondrial damage, ROS production, and the NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome-induced inflammatory response [90,91]. The accumulation of damaged and dysfunctional mitochondria can accelerate the cell apoptosis and premature senescence of RTECs during renal injury and fibrosis. The mitochondria-targeted antioxidant mitoQ abolishes suppressive PINK1-Parkin-mediated mitophagy and RTEC apoptosis in DN [92]. Hypoxia-inducible factor 1-α (HIF-1α) knockout in RTECs prominently inhibits hypoxia and reoxygenation-induced mitophagy and promotes cell apoptosis and ROS production; the activation of HIF-1α-Bcl-2 interacting protein 3 (BNIP3)-mediated mitophagy exerts a protective role by reversing these changes during AKI [93]. Inhibition of NLRP3 inflammasome can trigger HIF-1α-BNIP3-mediated mitophagy to alleviate RTEC apoptosis and renal injury [94]. AMPK and sirtuin-1 (Sirt1) are involved in PINK1-Parkin-mediated mitophagy activation. AMPK agonist metformin alleviates renal oxidative stress and fibrosis via activating p-AMPK-Pink1-Parkin-mediated mitophagy [95]. Quercetin, a natural flavonoid compound, can effectively ameliorate RTEC senescence and renal fibrosis via activating Sirt1-PINK1/Parkin-mediated mitophagy [96]. The upregulation of optineurin prevents RTEC senescence during DN progression by enhancing mitophagy [97]. Nevertheless, the up-regulation of tumor necrosis factor alpha-induced protein 8-like 1 in RTECs aggravates renal fibrosis and DN via destabilizing PHB2-mediated mitophagy [98].
5.2.2. Mitochondrial metabolism
Metabolism refers to the set of life-sustaining chemical reactions that take place in living organisms and is critical for maintaining life. Dysregulation of cellular metabolism has been implicated in kidney dysfunction [99]. As a highly metabolic organ, the kidney requires abundant energy; RTECs have numerous mitochondria, which are the crucial regulators for kidney energy metabolism [100]. Fatty acid uptake, oxidation, lipogenesis, and lipolysis serve as the most important factors of intracellular lipid metabolism. Following persistent injuries, the dysregulation of lipid metabolism in RTECs can result in lipid accumulation and lipotoxicity [101]. A shift occurs from glycolysis to fatty acid β-oxidation (FAO) during proximal tubule development; proximal RTECs require high levels of baseline energy consumption, primarily relying on mitochondrial respiration and FAO as an energy source [102]. FAO inhibition can induce metabolic reprogramming in RTECs, thus promoting cell apoptosis, dedifferentiation, and renal fibrosis [103]. Therefore, defective FAO in RTECs has emerged as a critical contributor for the development of CKD. Fatty acid transporter 2 in RTECs induces lipid metabolic reprogramming through abnormal fatty acid uptake and defective FAO, ultimately leading to renal fibrosis [104]. Nevertheless, the overexpression of carnitine palmitoyl transferase 1A in RTECs attenuates renal fibrosis via restoring mitochondrial homeostasis and FAO [105]. Sodium-glucose cotransporter 2 inhibitors exert renoprotective effects by restraining aberrant glycolytic metabolism and mitochondrial ROS formation in hyperglycemia-induced proximal RTECs [106]. Nuclear farnesoid X receptor inhibits cisplatin-induced lipid accumulation and increases FAO in RTECs by activating peroxisome proliferator-activated receptor-g [107]. Sirtuins, including Sirt3 and Sirt5, have emerged as critical regulators of FAO in RTECs. Sirt3 can regulate FAO via deacetylating LKB1 and activating AMPK, and Sirt5 can modulate the balance between mitochondrial and peroxisomal FAO to protect against AKI [108,109].
5.3. Lysosome quality control
Lysosomes were discovered in the 1950s by Christian de Duve and have since been considered as central catabolic membrane-enclosed organelle in cells [110]. Lysosomes are composed of a series of hydrolytic enzymes, including proteases, nucleases, lipases, sulfates, and phosphatases. These enzymes in lysosomes decompose various biological macromolecules, such as proteins, lipids, carbohydrates, polysaccharides, nucleic acids, and cell organelles [111]. In addition to their role in degradation, lysosomes are important for various biological processes, including cell metabolism and cellular material circulation, ultimately contributing to the maintenance of intracellular homeostasis [112]. Lysosomal enzyme leakage, abnormal lysosomal intracellular localization, lysosomal pH elevation, and lysosomal membrane permeabilization (LMP) are the main underlying mechanisms of impaired lysosome function [113]. The overloading of urinary proteins induces LMP and lysosomal dysfunction via oxidative stress in RTECs, which further leads to RTEC injury and apoptosis [114]. An indolizidine alkaloid called swainsonine also triggers lysosomal function in RTECs, suppresses autophagy degradation, and eventually promotes RTEC apoptosis [115]. Lysosome dysfunction and depletion during progressive injury can damage autophagic activity, further inducing mitochondria dysfunction in kidney injury, fibrosis, and DN diseases. Lysosome biogenesis is highly regulated by microphthalmia/transcription factor E protein family, which contains transcription factor EB (TFEB), transcription factor EC, transcription factor binding to IGHM enhancer 3, and melanocyte inducing transcription factor [116,117]. In IRI-induced AKI, the nuclear translocation and accumulated expression of TFEB in RTECs can further activate coordinated lysosomal expression and regulation (CLEAR)-network genes to promote lysosomal biogenesis [118]. In addition, high-fat diet (HFD) feeding causes an accumulation of phospholipids and thus leads to the progression of lipotoxicity; the TFEB-mediated lysosomal exocytosis in RTECs suppresses HFD-induced lipotoxicity in the kidney [119,120]. Lysosome depletion results in the autophagic flux blockage of RTECs, and SMAD3 accelerates lysosome depletion by inhibiting TFEB-dependent damaged lysosome clearance and lysosome biogenesis in the RTECs, thereby promoting the development of progressive renal fibrosis in DN [121].
6. RTEC participation in renal fibrosis crosstalk networks
The tubulointerstitium is mainly composed of multiple cell types, including RTECs, fibroblasts, and immune and endothelial cells. Renal fibrosis is a complex biological process resulting from the combined effects of these cell components. Dysfunctional RTECs undergo crosstalk with surrounding cells and promote the progression of renal fibrosis (Fig. 5).
Fig. 5.

Crosstalk of renal tubular epithelial cells (RTECs) with effector cells in renal fibrosis. Injured RTECs produce various cytokines to promote the crosstalk among RTECs, fibroblasts/myofibroblasts, and immune cells, accelerate epithelial-to-mesenchymal transition (EMT) and apoptosis program, and further contribute to excessive extracellular matrix (ECM) deposition and promote renal fibrosis. TGF-β: transforming growth factor-β; CTGF: connective tissue growth factor.
6.1. Crosstalk between RTECs and fibroblasts/myofibroblasts
In renal fibrosis, RTECs usually act as the initial reactor, and fibroblasts and myofibroblasts are the principal effectors and final executors, respectively. Myofibroblasts mainly originate from resident fibroblasts, pericytes, and circulating bone marrow-derived fibrocytes [122]. In addition to EMT, fibroblast–myofibroblast differentiation (FMD) is an important cellular phenotype during the development and deterioration of renal fibrosis. Notch1 is a member of the Notch family; an injury can stimulate the activation of Notch1 signaling pathway in RTECs and fibroblasts. The activated Notch1 aggravates EMT and FMD responses by activating TGF-β/Smad2/3 signaling pathway, leading to renal fibrosis [123]. Injured RTECs undergoing EMT and endothelial cells undergoing endothelial-to-mesenchymal transition are also important sources of myofibroblasts during renal fibrosis [124]. Upon maladaptive kidney injury, RTECs produce a great variety of growth factors and cytokines, including TGF-β, platelet-derived growth factor (PDGF), hedgehog, and Wnt ligands [125]. These cytokines can accelerate the regeneration of damaged RTECs and promote the differentiation of fibroblasts into myofibroblasts. Activated myofibroblasts then undergo rapid proliferation and produce a large amount of ECM components, such as fibronectin and collagen [126]. The consecutive and excessive deposition of ECM ultimately results in progressive fibrotic kidney disease. ECM deposition is influenced by the rate of ECM production and degradation. The degradation of ECM is modulated by proteases, such as matrix metalloproteinases [127]. Several regulatory cytokines produced by RTECs are involved in the initiation of fibrosis. For instance, RTECs undergoing autophagy can produce fibroblast growth factor 2 (FGF2) as a paracrine factor to induce fibroblast activation and renal fibrosis [128]. RTEC-derived exosomes act as new couriers for cell-to-cell communication during renal fibrosis. Injured RTECs produce miR-150-containing exosomes to initiate the activation and proliferation of fibroblasts, ultimately contributing to renal fibrosis [129]. Activated myofibroblasts also exert profibrotic effects on RTECs. The accumulation collagen I aggrandizes the production of TGF-β or the destabilization of E-cadherin adhesion complex to evoke the EMT activation of RTECs [130,131]. Growing evidence revealed the distinct role of interleukin 6 (IL-6) family members in the pathogenesis of renal fibrosis. Leukemia inhibitory factor (LIF), a member of the IL-6 family, works in alliance with SHH signaling to create a vicious cycle between fibroblasts and RTECs, increasing the expression of LIF and further contributing to profibrotic responses by activating extracellular signal-regulated kinase, the signal transducer and activator of transcription 3 signaling pathway [132,133].
6.2. Crosstalk between RTECs and immune cells
Inflammation has been recognized as a critical accelerant in initiating renal fibrosis after injury. Immune cells such as T cells, B cells, neutrophils, natural killer (NK) cells, dendritic cells, monocytes, and tissue-resident macrophages, comprise the pivotal contributors and regulators of inflammation in the fibrotic niche [134]. Injured RTECs produce numerous cytokines, including chemokines and proinflammatory cytokines, to recruit and activate surrounding immune cells. In turn, these immune cells further secrete cytokines to drive the EMT of RTECs [135].
Human fibrotic kidneys have a significant increase in T cells expressing cluster of differentiation 3 and 4 (CD3 and CD4), CD4+ T cells are pivotal players in renal fibrosis [136,137]. The recruitment and activation of T cells occur prior to the infiltration of macrophages into the damage kidneys [138]. Injured RTECs are renal nonprofessional antigen-presenting cells (APCs) playing a key regulatory role in the pathogenesis of renal fibrosis. RTECs functioning as APCs can induce antigen-specific CD4+ T cell proliferation, activation, and cytokine production [139]. MHC class II (MHCII) molecules are expressed on APCs in the immune system and play a leading part in the initiation of an effective immune response through the presentation of antigens to CD4+ T cells [140]. Rather than MHC-dependent cell-to-cell contact, RTECs can also directly induce the proliferation of CD4+ and CD8+ T cells in the kidney via an CD155, αVb3-integrin, and vitronectin-mediated mechanism [141]. In addition, MHCII is expressed in RTECs and renal cortical tubules, where its expression can be significantly up-regulated during renal fibrosis. The increased MHCII-expressing renal cortical tubules can serve as APCs to promote CD4+ T cell activation, thereby promoting renal fibrosis [142]. In turn, CD4+ T cells can act directly on RTECs, which promote the secretion of TGF-β1, PDGF, FGF2, and CTGF, thus aggravating renal fibrosis [143].
Damaged RTECs yield an endogenous toll-like receptor 2 ligand high-mobility group box protein 1, which further triggers the production of CC chemokine receptor 5 to recruit NK cells. In turn, infiltrated NK cells stimulate RTECs to secrete chemokine C-X-C motif ligand 1 and 2 (CXCL1 and CXCL2) via activating CD137L signaling, thereby promoting the accumulation of neutrophils in the damage kidney [144,145]. Moreover, NK cells can produce direct cytotoxic effects on RTECs to facilitate cell apoptosis [146]. Monocytes are produced in bone marrow and released to blood circulation. Injured RTECs secrete chemokine C–C motif ligand 2 (CCL2) and chemokine C-X3-C motif ligand 1 (CX3CL1) to recruit and accumulate monocytes [147]. In addition to resident macrophages, the majority of macrophages in the damaged kidney are derived from bone marrow monocytes. The recruited circulating monocytes can differentiate into pro-inflammatory M1 macrophages, and this process can be accelerated by matrix metallopeptidase 9 produced by injured RTECs in response to the early renal injury [148,149]. In addition, maladaptive RTECs can produce colony stimulating factor 1 to promote resident macrophages differentiate into anti-inflammatory M2 phenotype throughout kidney repair after injury. The increase in M2 macrophages expressing CD206 and CD163 is also closely related to fibrosis in human kidney [150,151]. M1 and M2 macrophages can induce the EMT activation of RTECs [[152], [153], [154]]. In turn, RTECs that undergo EMT generate a large amount of exosomes, which can serve as a crucial mediator of M1 macrophages activation and further promote renal fibrosis [155]. Circular RNA actin-related protein 2 in macrophages can trigger macrophage-induced EMT program and renal fibrosis by activating NLRP3 inflammasome [156]. Therefore, RTEC-to-macrophage communications form a feedback loop to aggravate the progression of renal fibrosis by leading to exaggerated inflammation and EMT.
7. Summary and future perspectives
RTECs have been identified as significant drivers of renal fibrosis following injury. Disordered quality control within RTECs and their crosstalk with other cells are essential pathogenic factors and therapeutic targets for renal fibrosis. A deep understanding of the quality control processes within RTECs and their interactions with other cells can offer valuable insights and open up new avenues for the development of novel therapeutics.
CRediT author statement
Yini Bao: Funding acquisition, Conceptualization, Methodology, Writing - Original draft preparation; Qiyuan Shan: Writing - Reviewing and Editing; Keda Lu: Writing - Reviewing and Editing; Qiao Yang: Formal analysis; Ying Liang: Formal analysis; Haodan Kuang: Visualization; Lu Wang: Visualization; Min Hao: Data curation; Mengyun Peng: Data curation; Shuosheng Zhang: Data curation; Gang Cao: Funding acquisition, Supervision, Project administration.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (Grant No.: 82204625), the Natural Science Foundation of Zhejiang Province (Grant Nos.: LQ23H280013 and LZ22H280001), the Chinese Medicine Research Program of Zhejiang Province (Program Nos.: 2021ZZ009, 2023ZR009, and 2021ZQ023), and the Youth Natural Science Program of Zhejiang Chinese Medical University (Program No.: 2021RCZXZK14). We appreciate the great help from the Pharmaceutical Research Center and Medical Research Center, Academy of Chinese Medical Sciences, Zhejiang Chinese Medical University.
References
- 1.Zhao M., Wang L., Wang M., et al. Targeting fibrosis, mechanisms and cilinical trials. Signal Transduct. Target. Ther. 2022;7 doi: 10.1038/s41392-022-01070-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan H., Xu J., Xu Z., et al. Defining therapeutic targets for renal fibrosis: Exploiting the biology of pathogenesis. Biomed. Pharmacother. 2021;143 doi: 10.1016/j.biopha.2021.112115. [DOI] [PubMed] [Google Scholar]
- 3.Huang R., Fu P., Ma L. Kidney fibrosis: From mechanisms to therapeutic medicines. Signal Transduct. Target. Ther. 2023;8 doi: 10.1038/s41392-023-01379-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li L., Fu H., Liu Y. The fibrogenic niche in kidney fibrosis: Components and mechanisms. Nat. Rev. Nephrol. 2022;18:545–557. doi: 10.1038/s41581-022-00590-z. [DOI] [PubMed] [Google Scholar]
- 5.Pohl C., Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019;366:818–822. doi: 10.1126/science.aax3769. [DOI] [PubMed] [Google Scholar]
- 6.Hurtley S., Alderton G. Quality control in cell biology. Science. 2019;366:816–817. doi: 10.1126/science.aba0302. [DOI] [PubMed] [Google Scholar]
- 7.Nieto M.A., Huang R.Y., Jackson R.A., et al. Emt: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 8.Yang J., Antin P., Berx G., et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020;21:341–352. doi: 10.1038/s41580-020-0237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brabletz S., Schuhwerk H., Brabletz T., et al. Dynamic EMT: A multi-tool for tumor progression. EMBO J. 2021;40 doi: 10.15252/embj.2021108647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katsuno Y., Derynck R. Epithelial plasticity, epithelial-mesenchymal transition, and the TGF-β family. Dev. Cell. 2021;56:726–746. doi: 10.1016/j.devcel.2021.02.028. [DOI] [PubMed] [Google Scholar]
- 11.Rayego-Mateos S., Campillo S., Rodrigues-Diez R.R., et al. Interplay between extracellular matrix components and cellular and molecular mechanisms in kidney fibrosis. Clin. Sci. (Lond.) 2021;135:1999–2029. doi: 10.1042/CS20201016. [DOI] [PubMed] [Google Scholar]
- 12.Huang Y., Hong W., Wei X. The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. J. Hematol. Oncol. 2022;15 doi: 10.1186/s13045-022-01347-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deshmukh A.P., Vasaikar S.V., Tomczak K., et al. Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc. Natl. Acad. Sci. U. S. A. 2021;118 doi: 10.1073/pnas.2102050118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu B., Tang T., Lv L., et al. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int. 2018;93:568–579. doi: 10.1016/j.kint.2017.09.033. [DOI] [PubMed] [Google Scholar]
- 15.Liu B., Tang T., Lv L. How tubular epithelial cell injury contributes to renal fibrosis. Adv. Exp. Med. Biol. 2019;1165:233–252. doi: 10.1007/978-981-13-8871-2_11. [DOI] [PubMed] [Google Scholar]
- 16.Lee K., Gusella G.L., He J.C. Epithelial proliferation and cell cycle dysregulation in kidney injury and disease. Kidney Int. 2021;100:67–78. doi: 10.1016/j.kint.2021.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith E.M., Pendlebury D.F., Nandakumar J. Structural biology of telomeres and telomerase. Cell. Mol. Life Sci. 2020;77:61–79. doi: 10.1007/s00018-019-03369-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roger L., Tomas F., Gire V. Mechanisms and regulation of cellular senescence. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms222313173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herranz N., Gil J. Mechanisms and functions of cellular senescence. J. Clin. Invest. 2018;128:1238–1246. doi: 10.1172/JCI95148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghilain C., Gilson E., Giraud-Panis M.J. Multifunctionality of the telomere-capping shelterin complex explained by variations in its protein composition. Cells. 2021;10 doi: 10.3390/cells10071753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schrumpfová P.P., Fajkus J. Composition and function of telomerase-a polymerase associated with the origin of eukaryotes. Biomolecules. 2020;10 doi: 10.3390/biom10101425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montandon M., Hamidouche T., Yart L., et al. Telomerase is required for glomerular renewal in kidneys of adult mice. NPJ Regen. Med. 2022;7 doi: 10.1038/s41536-022-00212-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uddin M.J., Farjana M., Moni A., et al. Prospective pharmacological potential of resveratrol in delaying kidney aging. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22158258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin Q., Zhao Y., Ni W., et al. miR-155 deficiency protects renal tubular epithelial cells from telomeric and genomic DNA damage in cisplatin-induced acute kidney injury. Theranostics. 2022;12:4753–4766. doi: 10.7150/thno.72456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aguado J., d’Adda di Fagagna F., Wolvetang E. Telomere transcription in ageing. Ageing Res. Rev. 2020;62 doi: 10.1016/j.arr.2020.101115. [DOI] [PubMed] [Google Scholar]
- 26.Rossiello F., Jurk D., Passos J.F., et al. Telomere dysfunction in ageing and age-related diseases. Nat. Cell Biol. 2022;24:135–147. doi: 10.1038/s41556-022-00842-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andrade L., Rodrigues C.E., Gomes S.A., et al. Acute kidney injury as a condition of renal senescence. Cell Transplant. 2018;27:739–753. doi: 10.1177/0963689717743512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumari R., Jat P. Mechanisms of cellular senescence: Cell cycle arrest and senescence associated secretory phenotype. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.645593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin J., Epel E. Stress and telomere shortening: Insights from cellular mechanisms. Ageing Res. Rev. 2022;73 doi: 10.1016/j.arr.2021.101507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang M. Telomere shortening promotes kidney fibrosis. Nat. Rev. Nephrol. 2021;17 doi: 10.1038/s41581-021-00432-4. [DOI] [PubMed] [Google Scholar]
- 31.Kishi S., Brooks C.R., Taguchi K., et al. Proximal tubule ATR regulates DNA repair to prevent maladaptive renal injury responses. J. Clin. Invest. 2019;129:4797–4816. doi: 10.1172/JCI122313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berby B., Bichara C., Rives-Feraille A., et al. Oxidative stress is associated with telomere interaction impairment and chromatin condensation defects in spermatozoa of infertile males. Antioxidants. 2021;10 doi: 10.3390/antiox10040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen X.J., Kim S.R., Jiang K., et al. Renovascular disease induces senescence in renal scattered tubular-like cells and impairs their reparative potency. Hypertension. 2021;77:507–518. doi: 10.1161/HYPERTENSIONAHA.120.16218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu J., Zhou L., Liu Y. Cellular senescence in kidney fibrosis: Pathologic significance and therapeutic strategies. Front. Pharmacol. 2020;11 doi: 10.3389/fphar.2020.601325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J., Li Y., Zhang X., et al. Cellular senescence of renal tubular epithelial cells in renal fibrosis. Front. Endocrinol. 2023;14 doi: 10.3389/fendo.2023.1085605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou B., Wan Y., Chen R., et al. The emerging role of cellular senescence in renal diseases. J. Cell Mol. Med. 2020;24:2087–2097. doi: 10.1111/jcmm.14952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen J., Zhang W., Chen K., et al. Urinary DcR2 is a novel biomarker for tubulointerstitial injury in patients with diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2017;313:F273–F281. doi: 10.1152/ajprenal.00689.2016. [DOI] [PubMed] [Google Scholar]
- 38.Chen J., Chen K., Xiao F., et al. Decoy receptor 2 mediation of the senescent phenotype of tubular cells by interacting with peroxiredoxin 1 presents a novel mechanism of renal fibrosis in diabetic nephropathy. Kidney Int. 2020;98:645–662. doi: 10.1016/j.kint.2020.03.026. [DOI] [PubMed] [Google Scholar]
- 39.Jayaraj G.G., Hipp M.S., Hartl F.U. Functional modules of the proteostasis network. Cold Spring Harb. Perspect. Biol. 2020;12 doi: 10.1101/cshperspect.a033951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dubnikov T., Ben-Gedalya T., Cohen E. Protein quality control in health and disease. Cold Spring Harb. Perspect. Biol. 2017;9 doi: 10.1101/cshperspect.a023523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collins G.A., Goldberg A.L. The logic of the 26S proteasome. Cell. 2017;169:792–806. doi: 10.1016/j.cell.2017.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Celebi G., Kesim H., Ozer E., et al. The effect of dysfunctional ubiquitin enzymes in the pathogenesis of most common diseases. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21176335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kwon Y.T., Ciechanover A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem. Sci. 2017;42:873–886. doi: 10.1016/j.tibs.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 44.Bard J.A.M., Goodall E.A., Greene E.R., et al. Structure and function of the 26S proteasome. Annu. Rev. Biochem. 2018;87:697–724. doi: 10.1146/annurev-biochem-062917-011931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pla-Prats C., Thomä N.H. Quality control of protein complex assembly by the ubiquitin-proteasome system. Trends Cell Biol. 2022;32:696–706. doi: 10.1016/j.tcb.2022.02.005. [DOI] [PubMed] [Google Scholar]
- 46.Radón V., Czesla M., Reichelt J., et al. Ubiquitin C-Terminal Hydrolase L1 is required forregulated protein degradation through theubiquitin proteasome system in kidney. Kidney Int. 2018;93:110–127. doi: 10.1016/j.kint.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 47.Shao Y., Zhang W., Du D., et al. Ubiquitin-like protein FAT10 promotes renal fibrosis by stabilizing USP7 to prolong CHK1-mediated G2/M arrest in renal tubular epithelial cells. Aging. 2022;14:7527–7546. doi: 10.18632/aging.204301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peng D., Fu M., Wang M., et al. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol. Cancer. 2022;21 doi: 10.1186/s12943-022-01569-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fukasawa H. The role of the ubiquitin-proteasome system in kidney diseases. Clin. Exp. Nephrol. 2012;16:507–517. doi: 10.1007/s10157-012-0643-1. [DOI] [PubMed] [Google Scholar]
- 50.Chen L., Yang T., Lu D., et al. Central role of dysregulation of TGF-β/Smad in CKD progression and potential targets of its treatment. Biomed. Pharmacother. 2018;101:670–681. doi: 10.1016/j.biopha.2018.02.090. [DOI] [PubMed] [Google Scholar]
- 51.Yang J., Ruan Y., Wang D., et al. VHL-recruiting PROTAC attenuates renal fibrosis and preserves renal function via simultaneous degradation of Smad3 and stabilization of HIF-2α. Cell Biosci. 2022;12 doi: 10.1186/s13578-022-00936-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song J., Wang T., Chi X., et al. Kindlin-2 inhibits the hippo signaling pathway by promoting degradation of MOB1. Cell Rep. 2019;29:3664–3677.e5. doi: 10.1016/j.celrep.2019.11.035. [DOI] [PubMed] [Google Scholar]
- 53.Galluzzi L., Green D.R. Autophagy-independent functions of the autophagy machinery. Cell. 2019;177:1682–1699. doi: 10.1016/j.cell.2019.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mercer T.J., Ohashi Y., Boeing S., et al. Phosphoproteomic identification of ULK substrates reveals VPS15-dependent ULK/VPS34 interplay in the regulation of autophagy. EMBO J. 2021;40 doi: 10.15252/embj.2020105985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nishimura T., Tooze S.A. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 2020;6 doi: 10.1038/s41421-020-0161-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li L., Tong M., Fu Y., et al. Lipids and membrane-associated proteins in autophagy. Protein Cell. 2021;12:520–544. doi: 10.1007/s13238-020-00793-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klionsky D.J., Petroni G., Amaravadi R.K., et al. Autophagy in major human diseases. EMBO J. 2021;40 doi: 10.15252/embj.2021108863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saha S., Panigrahi D.P., Patil S., et al. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018;104:485–495. doi: 10.1016/j.biopha.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 59.Ma Z., Li L., Livingston M.J., et al. p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease. J. Clin. Invest. 2020;130:5011–5026. doi: 10.1172/JCI135536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang H., Wang Y., Wang X., et al. PTEN alleviates maladaptive repair of renal tubular epithelial cells by restoring CHMP2A-mediated phagosome closure. Cell Death Dis. 2021;12 doi: 10.1038/s41419-021-04372-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peng X., Wang Y., Li H., et al. ATG5-mediated autophagy suppresses NF-κB signaling to limit epithelial inflammatory response to kidney injury. Cell Death Dis. 2019;10 doi: 10.1038/s41419-019-1483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y., Liu Z., Shu S., et al. AMPK/mTOR signaling in autophagy regulation during cisplatin-induced acute kidney injury. Front. Physiol. 2020;11 doi: 10.3389/fphys.2020.619730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang S., Lin C., Zhuo X., et al. Glucagon-like peptide-1 alleviates diabetic kidney disease through activation of autophagy by regulating AMP-activated protein kinase-mammalian target of rapamycin pathway. Am. J. Physiol. Endocrinol. Metab. 2020;319:E1019–E1030. doi: 10.1152/ajpendo.00195.2019. [DOI] [PubMed] [Google Scholar]
- 64.Liu X., Chen J., Sun N., et al. Ginsenoside Rb1 ameliorates autophagy via the AMPK/mTOR pathway in renal tubular epithelial cells in vitro and in vivo. Int. J. Biol. Macromol. 2020;163:996–1009. doi: 10.1016/j.ijbiomac.2020.07.060. [DOI] [PubMed] [Google Scholar]
- 65.MacDonald A.F., Bettaieb A., Donohoe D.R., et al. Concurrent regulation of LKB1 and CaMKK2 in the activation of AMPK in castrate-resistant prostate cancer by a well-defined polyherbal mixture with anticancer properties. BMC Complement. Altern. Med. 2018;18 doi: 10.1186/s12906-018-2255-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liang S., Wu Y., Li D., et al. Autophagy and renal fibrosis. Aging Dis. 2022;13:712–731. doi: 10.14336/AD.2021.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu C., Wang X., Wang X., et al. A new LKB1 activator, piericidin analogue S14, retards renal fibrosis through promoting autophagy and mitochondrial homeostasis in renal tubular epithelial cells. Theranostics. 2022;12:7158–7179. doi: 10.7150/thno.78376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li A., Yi B., Han H., et al. Vitamin D-VDR (vitamin D receptor) regulates defective autophagy in renal tubular epithelial cell in streptozotocin-induced diabetic mice via the AMPK pathway. Autophagy. 2022;18:877–890. doi: 10.1080/15548627.2021.1962681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gong W., Luo C., Peng F., et al. Brahma-related gene-1 promotes tubular senescence and renal fibrosis through Wnt/β-catenin/autophagy axis. Clin. Sci. (Lond.) 2021;135:1873–1895. doi: 10.1042/CS20210447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Papaioannou A., Centonze F., Metais A., et al. Stress-induced tyrosine phosphorylation of RtcB modulates IRE1 activity and signaling outputs. Life Sci. Alliance. 2022;5 doi: 10.26508/lsa.202201379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Preissler S., Ron D. Early events in the endoplasmic reticulum unfolded protein response. Cold Spring Harb. Perspect. Biol. 2019;11 doi: 10.1101/cshperspect.a033894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hetz C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 73.Wiseman R.L., Mesgarzadeh J.S., Hendershot L.M. Reshaping endoplasmic reticulum quality control through the unfolded protein response. Mol. Cell. 2022;82:1477–1491. doi: 10.1016/j.molcel.2022.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Khanna M., Agrawal N., Chandra R., et al. Targeting unfolded protein response: A new horizon for disease control. Expert Rev. Mol. Med. 2021;23 doi: 10.1017/erm.2021.2. [DOI] [PubMed] [Google Scholar]
- 75.Inagi R., Ishimoto Y., Nangaku M. Proteostasis in endoplasmic reticulum: New mechanisms in kidney disease. Nat. Rev. Nephrol. 2014;10:369–378. doi: 10.1038/nrneph.2014.67. [DOI] [PubMed] [Google Scholar]
- 76.Inagi R. Endoplasmic reticulum stress in the kidney as a novel mediator of kidney injury. Nephron Exp. Nephrol. 2009;112:e1–e9. doi: 10.1159/000210573. [DOI] [PubMed] [Google Scholar]
- 77.Chen J., Wu C.H., Jheng J.R., et al. The down-regulation of XBP1, an unfolded protein response effector, promotes acute kidney injury to chronic kidney disease transition. J. Biomed. Sci. 2022;29 doi: 10.1186/s12929-022-00828-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ju Y., Su Y., Chen Q., et al. Protective effects of Astragaloside IV on endoplasmic reticulum stress-induced renal tubular epithelial cells apoptosis in type 2 diabetic nephropathy rats. Biomed. Pharmacother. 2019;109:84–92. doi: 10.1016/j.biopha.2018.10.041. [DOI] [PubMed] [Google Scholar]
- 79.Wu X., He Y., Jing Y., et al. Albumin overload induces apoptosis in renal tubular epithelial cells through a CHOP-dependent pathway. OMICS. 2010;14:61–73. doi: 10.1089/omi.2009.0073. [DOI] [PubMed] [Google Scholar]
- 80.Yang H., Niemeijer M., van de Water B., et al. ATF6 is a critical determinant of CHOP dynamics during the unfolded protein response. iScience. 2020;23 doi: 10.1016/j.isci.2020.100860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Limonta P., Moretti R.M., Marzagalli M., et al. Role of endoplasmic reticulum stress in the anticancer activity of natural compounds. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20040961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang Y., Sun Y., Cao Y., et al. HRD1 prevents apoptosis in renal tubular epithelial cells by mediating eIF2α ubiquitylation and degradation. Cell Death Dis. 2017;8 doi: 10.1038/s41419-017-0002-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang X., Pan W., Xu G., et al. Mitophagy: A crucial modulator in the pathogenesis of chronic diseases. Clin. Chim. Acta. 2020;502:245–254. doi: 10.1016/j.cca.2019.11.008. [DOI] [PubMed] [Google Scholar]
- 84.Onishi M., Yamano K., Sato M., et al. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021;40 doi: 10.15252/embj.2020104705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang N., Zhu P., Huang R., et al. PINK1: The guard of mitochondria. Life Sci. 2020;259 doi: 10.1016/j.lfs.2020.118247. [DOI] [PubMed] [Google Scholar]
- 86.Eldeeb M.A., Thomas R.A., Ragheb M.A., et al. Mitochondrial quality control in health and in Parkinson’s disease. Physiol. Rev. 2022;102:1721–1755. doi: 10.1152/physrev.00041.2021. [DOI] [PubMed] [Google Scholar]
- 87.Killackey S.A., Philpott D.J., Girardin S.E. Mitophagy pathways in health and disease. J. Cell Biol. 2020;219 doi: 10.1083/jcb.202004029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lampert M.A., Orogo A.M., Najor R.H., et al. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy. 2019;15:1182–1198. doi: 10.1080/15548627.2019.1580095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nguyen T.N., Sawa-Makarska J., Khuu G., et al. Unconventional initiation of PINK1/Parkin mitophagy by Optineurin. Mol. Cell. 2023;83:1693–1709.e9. doi: 10.1016/j.molcel.2023.04.021. [DOI] [PubMed] [Google Scholar]
- 90.Tang C., Han H., Yan M., et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy. 2018;14:880–897. doi: 10.1080/15548627.2017.1405880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lin Q., Li S., Jiang N., et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019;26 doi: 10.1016/j.redox.2019.101254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xiao L., Xu X., Zhang F., et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017;11:297–311. doi: 10.1016/j.redox.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fu Z., Wang Z., Xu L., et al. HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 2020;36 doi: 10.1016/j.redox.2020.101671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lin Q., Li S., Jiang N., et al. Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1A and BNIP3-mediated mitophagy. Autophagy. 2021;17:2975–2990. doi: 10.1080/15548627.2020.1848971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Han Y., Tang S., Liu Y., et al. AMPK agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and streptozotocin induced diabetic mice. Cell Death Dis. 2021;12 doi: 10.1038/s41419-021-04184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu T., Yang Q., Zhang X., et al. Quercetin alleviates kidney fibrosis by reducing renal tubular epithelial cell senescence through the SIRT1/PINK1/mitophagy axis. Life Sci. 2020;257 doi: 10.1016/j.lfs.2020.118116. [DOI] [PubMed] [Google Scholar]
- 97.Chen K., Dai H., Yuan J., et al. Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis. 2018;9 doi: 10.1038/s41419-017-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu L., Bai F., Song H., et al. Upregulation of TIPE1 in tubular epithelial cell aggravates diabetic nephropathy by disrupting PHB2 mediated mitophagy. Redox Biol. 2022;50 doi: 10.1016/j.redox.2022.102260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Votava J.A., Reese S.R., Deck K.M., et al. Dysregulation of the sensory and regulatory pathways controlling cellular iron metabolism in unilateral obstructive nephropathy. Am. J. Physiol. Ren. Physiol. 2022;322:F89–F103. doi: 10.1152/ajprenal.00537.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Clark A.J., Parikh S.M. Mitochondrial metabolism in acute kidney injury. Semin. Nephrol. 2020;40:101–113. doi: 10.1016/j.semnephrol.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wahl P., Ducasa G.M., Fornoni A. Systemic and renal lipids in kidney disease development and progression. Am. J. Physiol. Ren. Physiol. 2016;310:F433–F445. doi: 10.1152/ajprenal.00375.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang G., Heijs B., Kostidis S., et al. Spatial dynamic metabolomics identifies metabolic cell fate trajectories in human kidney differentiation. Cell Stem Cell. 2022;29:1580–1593.e7. doi: 10.1016/j.stem.2022.10.008. [DOI] [PubMed] [Google Scholar]
- 103.Kang H.M., Ahn S.H., Choi P., et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015;21:37–46. doi: 10.1038/nm.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen Y., Yan Q., Lv M., et al. Involvement of FATP2-mediated tubular lipid metabolic reprogramming in renal fibrogenesis. Cell Death Dis. 2020;11 doi: 10.1038/s41419-020-03199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Miguel V., Tituaña J., Herrero J.I., et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J. Clin. Invest. 2021;131 doi: 10.1172/JCI140695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shirakawa K., Sano M. Sodium-glucose co-transporter 2 inhibitors correct metabolic maladaptation of proximal tubular epithelial cells in high-glucose conditions. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21207676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xu S., Jia P., Fang Y., et al. Nuclear farnesoid X receptor attenuates acute kidney injury through fatty acid oxidation. Kidney Int. 2022;101:987–1002. doi: 10.1016/j.kint.2022.01.029. [DOI] [PubMed] [Google Scholar]
- 108.Li M., Li C., Ye Z., et al. Sirt3 modulates fatty acid oxidation and attenuates cisplatin-induced AKI in mice. J. Cell Mol. Med. 2020;24:5109–5121. doi: 10.1111/jcmm.15148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chiba T., Peasley K.D., Cargill K.R., et al. Sirtuin 5 regulates proximal tubule fatty acid oxidation to protect against AKI. J. Am. Soc. Nephrol. 2019;30:2384–2398. doi: 10.1681/ASN.2019020163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bonam S.R., Wang F., Muller S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019;18:923–948. doi: 10.1038/s41573-019-0036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ballabio A., Bonifacino J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020;21:101–118. doi: 10.1038/s41580-019-0185-4. [DOI] [PubMed] [Google Scholar]
- 112.Wu M., Zhang M., Zhang Y., et al. Relationship between lysosomal dyshomeostasis and progression of diabetic kidney disease. Cell Death Dis. 2021;12 doi: 10.1038/s41419-021-04271-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang Z., Yue P., Lu T., et al. Role of lysosomes in physiological activities, diseases, and therapy. J. Hematol. Oncol. 2021;14 doi: 10.1186/s13045-021-01087-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu W., Xu B., Ye L., et al. Urinary proteins induce lysosomal membrane permeabilization and lysosomal dysfunction in renal tubular epithelial cells, Am. J. Physiol. Renal Physiol. 2015;308:F639–F649. doi: 10.1152/ajprenal.00383.2014. [DOI] [PubMed] [Google Scholar]
- 115.Wang S., Guo R., Su Y., et al. Swainsonine promotes apoptosis by impairing lysosomal function and inhibiting autophagic degradation in rat primary renal tubular epithelial cells. Chem. Biol. Interact. 2021;336 doi: 10.1016/j.cbi.2020.109319. [DOI] [PubMed] [Google Scholar]
- 116.Perera R.M., Di Malta C., Ballabio A. MiT/TFE family of transcription factors, lysosomes, and cancer. Annu. Rev. Cancer Biol. 2019;3:203–222. doi: 10.1146/annurev-cancerbio-030518-055835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kim S., Song H.S., Yu J., et al. MiT family transcriptional factors in immune cell functions. Mol. Cells. 2021;44:342–355. doi: 10.14348/molcells.2021.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Li Z., Liu Z., Luo M., et al. The pathological role of damaged organelles in renal tubular epithelial cells in the progression of acute kidney injury. Cell Death Dis. 2022;8 doi: 10.1038/s41420-022-01034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yamamoto T., Takabatake Y., Takahashi A., et al. High-fat diet-induced lysosomal dysfunction and impaired autophagic flux contribute to lipotoxicity in the kidney. J. Am. Soc. Nephrol. 2017;28:1534–1551. doi: 10.1681/ASN.2016070731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nakamura J., Yamamoto T., Takabatake Y., et al. TFEB-mediated lysosomal exocytosis alleviates high-fat diet-induced lipotoxicity in the kidney. JCI Insight. 2023;8 doi: 10.1172/jci.insight.162498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yang C., Chen X., Li Z., et al. SMAD3 promotes autophagy dysregulation by triggering lysosome depletion in tubular epithelial cells in diabetic nephropathy. Autophagy. 2021;17:2325–2344. doi: 10.1080/15548627.2020.1824694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mack M., Yanagita M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 2015;87:297–307. doi: 10.1038/ki.2014.287. [DOI] [PubMed] [Google Scholar]
- 123.Hong W., Zhang G., Lu H., et al. Epithelial and interstitial Notch1 activity contributes to the myofibroblastic phenotype and fibrosis. Cell Commun. Signal. 2019;17 doi: 10.1186/s12964-019-0455-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.LeBleu V.S., Taduri G., O’Connell J., et al. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013;19:1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gewin L., Zent R., Pozzi A. Progression of chronic kidney disease: Too much cellular talk causes damage. Kidney Int. 2017;91:552–560. doi: 10.1016/j.kint.2016.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Plikus M.V., Wang X., Sinha S., et al. Fibroblasts: Origins, definitions, and functions in health and disease. Cell. 2021;184:3852–3872. doi: 10.1016/j.cell.2021.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhao X., Chen J., Sun H., et al. New insights into fibrosis from the ECM degradation perspective: The macrophage-MMP-ECM interaction. Cell Biosci. 2022;12 doi: 10.1186/s13578-022-00856-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Livingston M.J., Shu S., Fan Y., et al. Tubular cells produce FGF2 via autophagy after acute kidney injury leading to fibroblast activation and renal fibrosis. Autophagy. 2023;19:256–277. doi: 10.1080/15548627.2022.2072054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Guan H., Peng R., Mao L., et al. Injured tubular epithelial cells activate fibroblasts to promote kidney fibrosis through miR-150-containing exosomes. Exp. Cell Res. 2020;392 doi: 10.1016/j.yexcr.2020.112007. [DOI] [PubMed] [Google Scholar]
- 130.Li M., Luan F., Zhao Y., et al. Epithelial-mesenchymal transition: An emerging target in tissue fibrosis. Exp. Biol. Med. (Maywood) 2016;241:1–13. doi: 10.1177/1535370215597194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Imamichi Y., Menke A. Signaling pathways involved in collagen-induced disruption of the E-cadherin complex during epithelial-mesenchymal transition. Cells Tissues Organs. 2007;185:180–190. doi: 10.1159/000101319. [DOI] [PubMed] [Google Scholar]
- 132.Xu S., Yang X., Chen Q., et al. Leukemia inhibitory factor is a therapeutic target for renal interstitial fibrosis. EBioMedicine. 2022;86 doi: 10.1016/j.ebiom.2022.104312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shen A., Lv L. Tubule epithelial cells and fibroblasts communication: Vicious cycle of renal fibrosis. EBioMedicine. 2022;86 doi: 10.1016/j.ebiom.2022.104360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Huang E., Peng N., Xiao F., et al. The roles of immune cells in the pathogenesis of fibrosis. Int. J. Mol. Sci. 2020;21:5203. doi: 10.3390/ijms21155203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Qi R., Yang C. Renal tubular epithelial cells: The neglected mediator of tubulointerstitial fibrosis after injury. Cell Death Dis. 2018;9:1126. doi: 10.1038/s41419-018-1157-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.do Valle Duraes F., Lafont A., Beibel M., et al. Immune cell landscaping reveals a protective role for regulatory T cells during kidney injury and fibrosis. JCI Insight. 2020;5 doi: 10.1172/jci.insight.130651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tapmeier T.T., Fearn A., Brown K., et al. Pivotal role of CD4+ T cells in renal fibrosis following ureteric obstruction. Kidney Int. 2010;78:351–362. doi: 10.1038/ki.2010.177. [DOI] [PubMed] [Google Scholar]
- 138.Wang J., Tian J., Sun J., et al. Two identified subsets of CD8 T cells in obstructed kidneys play different roles in inflammation and fibrosis. Aging. 2020;12:17528–17540. doi: 10.18632/aging.103764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Breda P.C., Wiech T., Meyer-Schwesinger C., et al. Renal proximal tubular epithelial cells exert immunomodulatory function by driving inflammatory CD4+ T cell responses. Am. J. Physiol. Ren. Physiol. 2019;317:F77–F89. doi: 10.1152/ajprenal.00427.2018. [DOI] [PubMed] [Google Scholar]
- 140.Jurewicz M.M., Stern L.J. Class II MHC antigen processing in immune tolerance and inflammation. Immunogenetics. 2019;71:171–187. doi: 10.1007/s00251-018-1095-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sadasivam M., Jie C., Hamad A.R.A. Renal tubular epithelial cells are constitutive non-cognate stimulators of resident T cells. Cell Rep. 2023;42 doi: 10.1016/j.celrep.2023.113210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhou Y., Luo Z., Liao C., et al. MHC class II in renal tubules plays an essential role in renal fibrosis. Cell. Mol. Immunol. 2021;18:2530–2540. doi: 10.1038/s41423-021-00763-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nikolic-Paterson D.J. CD4+ T cells: a potential player in renal fibrosis. Kidney Int. 2010;78:333–335. doi: 10.1038/ki.2010.182. [DOI] [PubMed] [Google Scholar]
- 144.Cantoni C., Granata S., Bruschi M., et al. Recent advances in the role of natural killer cells in acute kidney injury. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.01484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kim H.J., Lee J.S., Kim A., et al. TLR2 signaling in tubular epithelial cells regulates NK cell recruitment in kidney ischemia-reperfusion injury. J. Immunol. 2013;191:2657–2664. doi: 10.4049/jimmunol.1300358. [DOI] [PubMed] [Google Scholar]
- 146.Turner J.E., Rickassel C., Healy H., et al. Natural killer cells in kidney health and disease. Front. Immunol. 2019;10:587. doi: 10.3389/fimmu.2019.00587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.von Vietinghoff S., Kurts C. Regulation and function of CX3CR1 and its ligand CX3CL1 in kidney disease. Cell Tissue Res. 2021;385:335–344. doi: 10.1007/s00441-021-03473-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Meng X., Jin J., Lan H. Driving role of macrophages in transition from acute kidney injury to chronic kidney disease. Chin. Med. J. 2022;135:757–766. doi: 10.1097/CM9.0000000000002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Masola V., Zaza G., Bellin G., et al. Heparanase regulates the M1 polarization of renal macrophages and their crosstalk with renal epithelial tubular cells after ischemia/reperfusion injury. FASEB J. 2018;32:742–756. doi: 10.1096/fj.201700597R. [DOI] [PubMed] [Google Scholar]
- 150.Cao Q., Harris D.C.H., Wang Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology (Bethesda) 2015;30:183–194. doi: 10.1152/physiol.00046.2014. [DOI] [PubMed] [Google Scholar]
- 151.Wang X., Chen J., Xu J., et al. The role of macrophages in kidney fibrosis. Front. Physiol. 2021;12 doi: 10.3389/fphys.2021.705838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yu C.C., Chien C.T., Chang T.C. M2 macrophage polarization modulates epithelial-mesenchymal transition in cisplatin-induced tubulointerstitial fibrosis. Biomedicine (Taipei) 2016;6 doi: 10.7603/s40681-016-0005-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tan T.K., Zheng G., Hsu T.T., et al. Matrix metalloproteinase-9 of tubular and macrophage origin contributes to the pathogenesis of renal fibrosis via macrophage recruitment through osteopontin cleavage. Lab. Invest. 2013;93:434–449. doi: 10.1038/labinvest.2013.3. [DOI] [PubMed] [Google Scholar]
- 154.Jiang W., Xu C., Du C., et al. Tubular epithelial cell-to-macrophage communication forms a negative feedback loop via extracellular vesicle transfer to promote renal inflammation and apoptosis in diabetic nephropathy. Theranostics. 2022;12:324–339. doi: 10.7150/thno.63735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lu Y., Zhang R., Gu X., et al. Exosomes from tubular epithelial cells undergoing epithelial-to-mesenchymal transition promote renal fibrosis by M1 macrophage activation. FASEB Bioadv. 2023;5:101–113. doi: 10.1096/fba.2022-00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Fu H., Gu Y., Tan J., et al. CircACTR2 in macrophages promotes renal fibrosis by activating macrophage inflammation and epithelial-mesenchymal transition of renal tubular epithelial cells. Cell. Mol. Life Sci. 2022;79 doi: 10.1007/s00018-022-04247-9. [DOI] [PMC free article] [PubMed] [Google Scholar]