

SUMMARY
The last 50 years have witnessed extraordinary developments in understanding mechanisms of carcinogenesis, synthesized as the hallmarks of cancer. Despite this logical framework, our understanding of the molecular basis of systemic manifestations and the underlying causes of cancer-related death remains incomplete. Looking forward, elucidating how tumors interact with distant organs and how multifaceted environmental and physiological parameters impinge on tumors and their hosts will be crucial for advances in preventing and more effectively treating human cancers. In this perspective, we discuss complexities of cancer as a systemic disease, including tumor initiation and promotion, tumor micro- and immune macro-environments, aging, metabolism and obesity, cancer cachexia, circadian rhythms, nervous system interactions, tumor-related thrombosis, and the microbiome. Model systems incorporating human genetic variation will be essential to decipher the mechanistic basis of these phenomena and unravel gene-environment interactions, providing a modern synthesis of molecular oncology that is primed to prevent cancers and improve patient quality of life and cancer outcomes.
INTRODUCTION
Looking back 50 years, when Cell launched as a new journal, it is evident that the landscape of cancer research has changed dramatically. During this time, reductionist cell biology utilizing elegant and simple model systems has formed the mainstay of scientific discovery, producing extraordinary insights into the regulation of the cell cycle, apoptosis, cell motility, invasion, and immune dysregulation, leading to significant progress in the diagnosis and treatment of cancer; 5-year survival rates have increased from 35% in the 1950s to 69.7% by 2017 (SEER Cancer Statistics Review 1975–2018). Despite this progress, cancer remains a leading cause of death worldwide.
We are in the midst of a renaissance where knowledge about cancer biology and genetics is exploding, with >1,000 genes identified that are altered in tumors, either genetically by recurrent mutations, or epigenetically, resulting in changes in their regulation and expression.1 This milestone has facilitated ever more precise delineation of the molecular circuitry, cellular constitutions, and heterogeneous population dynamics of mutant cancer cells, as well as the mechanisms of tumor progression and metastatic dissemination in concert with ostensibly normal—but functionally corrupted—cells of distinctive origins. Such knowledge, which tends to be focused on the cancer cell itself and its local microenvironment, has enabled new generations of mechanism-guided therapeutic drugs and treatment regimens that have benefited some patients with certain forms of cancer. Frustratingly, few of these innovative new therapeutic strategies are broadly beneficial across the spectrum of human cancers and, with many avenues for tumors to evolve resistance, even fewer significantly prolong overall survival.
The multi-dimensional archaeology of cancer, combined with advancing technologies, creates a dizzyingly vast scope for “big data” down to the single-cell level. These technologies aim to characterize cancers in ever-increasing detail, shedding light on the neo-Darwinian development and progression of cancers from different cells of origin that face tissue-specific barriers requiring distinctive adaptations reflected in their multistep tumorigenesis. Appreciation of this sobering diversity and complexity has been growing for decades, looming as a potentially insurmountable challenge for the “war on cancer.” 24 years ago, in the millennium issue of Cell, an attempt to reconcile this daunting diversity with the growing knowledge about mechanisms of cancer was published.2 The hallmarks of cancer posited that virtually all tumors acquire a common set of qualitatively distinct functional capabilities that collectively enable cancer cells to proliferate expansively while orchestrating the formation of tumors that grow and often disseminate. The corollary concept was that a common pair of phenotypic characteristics—genomic instability and mutation, along with inflammation—facilitate their acquisition. Thus, the immense complexity of cancer pathogenesis could be distilled, it was argued, as different solutions to the same challenge, namely acquiring the same set of hallmark capabilities during tumorigenesis and malignant progression. This simple concept, refined in subsequent years,3-5 has resonated through cancer medicine to the present day, indicating that it has some utility as a conceptual organizing principle.
However, this modern synthesis of cancer biology does not fully consider the broader interactions of an evolving tumor with distant organs of the host, nor the impact of host pathophysiology, germline genetic diversity, and environmental exposures, on cancer initiation and evolution. The reassuring simplicity of the hallmarks is clearly insufficient to fully understand the multifarious manifestations of disease mechanisms. As such, there is a clear need to develop new therapeutic strategies that improve both the quality and length of life for patients suffering from cancer, tackling some of the most life-threatening conditions such as cancer cachexia, thrombosis, and paraneoplastic syndromes and, importantly, opportunities to intervene at the earliest stages of cancer initiation in new prevention strategies. An expanding repertoire of (multi-omic) research tools and refined model systems are now poised to address cancer as a systemic disease consequent to the complex interplay between the diversity of the host genome, chance events, and a legacy of human behavior resulting in complex environmental exposures.
Aging is the number one prognostic factor for most tumors, given that over 90% of patients diagnosed with cancer are over the age of 50. In addition to this major contributor to cancer complexity, and indeed a route for actionable preventive strategies, is the environment in which we live, estimated from epidemiological studies to influence the development of up to 80% of human cancers.6 The term “environment” encompasses a wide range of exogenous factors, including industrial pollutants in the air or in our diet, as well as occupational or medical exposures to toxic substances or certain types of radiation and pathogenic viral and bacterial infection. It also includes what have come to be known as “lifestyle factors”: diet, alcohol consumption, cigarette smoking, sun exposure, and sedentary behavior, all of which impact cancer risk. Remarkably, it is becoming increasingly clear that these disparate environmental agents increase cancer risk by affecting the same cancer hallmark characteristics described above—genomic mutations, altered metabolism, chromosome instability, and inflammation.
In this perspective, a culmination of four Cancer-Research-UK-sponsored Marshall Symposia, we contemplate—as illustrated in Figure 1—the new frontier that complements the hallmarks of cancer by embracing the complexity of human cancer pathogenesis, using technological advances to unravel the diverse interplay between an evolving tumor within an aging environment and distant organ systems in genetically diverse populations who have engaged in distinct behaviors and been exposed to heterogeneous environmental exposures. Importantly, we argue that over the next 50 years, we will need to acknowledge the limitations of inbred animal models in highly controlled environments to fully recapitulate such complexity. Addressing this challenge will require well-conceived studies in human subjects as a vehicle for advancing discovery, interfaced with increasingly sophisticated ex vivo and animal models of cancer complexities, in order to advance therapeutic intervention and early interception to more effectively prevent, diagnose and treat cancer and its systemic manifestations.
Figure 1. The “clouds of complexity” impinging on the frontiers of cancer biology and cancer medicine.
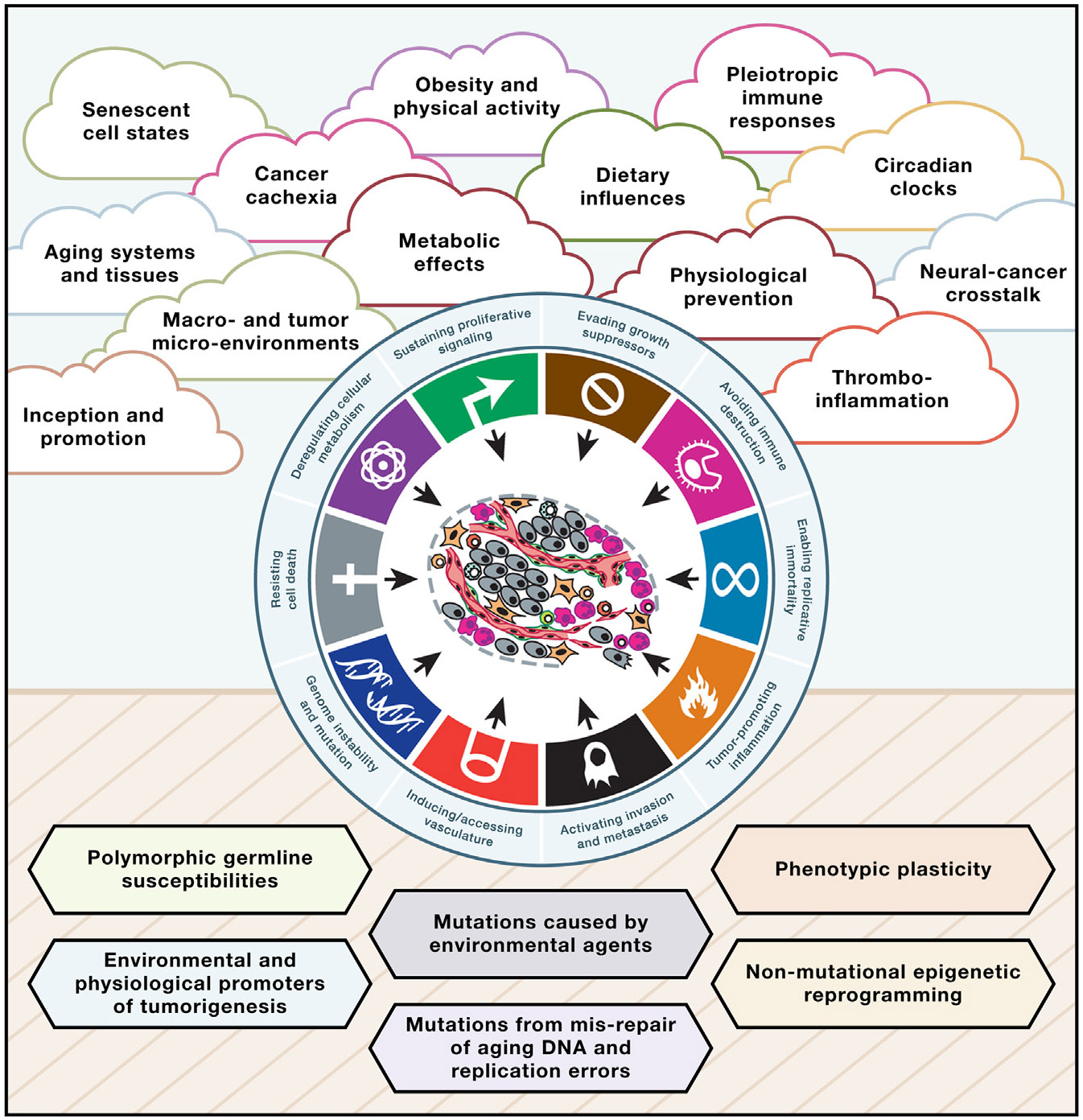
Although the hallmarks of cancer have provided an overarching conceptual rationale for the myriad manifestations encompassing cancer as a disease, below this simplicity lies a dizzying diversity in mechanistic effects and phenotypes, both inside tumors and system-wide in the affected individual. Thus, above the horizon are clouds of complexity that, we argue in this perspective, are important and incompletely understood. Below the horizon lie mechanistic effectors—the building blocks of cancer—governing the inception and progression of cancer, which are also incompletely understood. Elucidating both dimensions of cancer as a systemic disease will be instrumental for ground-breaking innovations in prevention and enduring treatment of human cancer.
Any review with a visionary goal to chart future directions for cancer research in the next 50 years might well consider other salient topics that are not highlighted in Figure 1. Prominent among these is the role of biological sex in cancer susceptibility, development, and therapeutic responses. Apart from the importance of sex hormones in the development of breast, ovarian, and prostate cancers, biological sex is associated with distinct incidences of a range of other cancer types, including bladder, kidney and esophagus,7,8 all of which are higher in males. Increasing evidence supports a role for sex-chromosome gene effects9 and crosstalk between sex hormones and other systems involved in inflammation and immunity,10,11 effects that have clear implications for cancer treatment and therapeutic responses. For example, understanding why lung cancer in never smokers is more common in females will require unraveling the role of the environment, genetics, and human behavior. This field is ripe for deeper analysis in the coming decades.
It would be remiss to discuss cancer complexity without mentioning how the ever-increasing pace of developing artificial intelligence (AI) algorithms may impact our ability to process the vast amounts of data being generated, and to understand how the emergent properties of interconnected networks of genes can help us to understand the progression of cancer from initiated cells to metastasis at the single-cell level. Ultimately, we anticipate a time when these new methods will be applied to multiple dimensions of cancer prevention and treatment, ranging from analysis of the clonal architecture of normal tissues, to illuminating signs of increased risk, and to the prediction of stem cell plasticity states that lead to drug resistance and poor patient survival. Even when these clouds of complexity are understood and AI is primed to improve outcomes for patients, the role of “bad luck” in cancer-initiating mutations may never be fully mitigated. Finally, a focus on cancer complexity should not detract from reductionist approaches to understanding cancer’s hallmarks, particularly with respect to the dynamic, evolving nature of the disease over space and time. Deep longitudinal sampling studies integrated with autopsy programs to decipher the co-evolution of the tumor within its microenvironment during the disease course and at distant metastatic sites, exemplified by studies such as renal, lung, and breast TRACERx, lung TRACERx EVO, and the UK national PEACE autopsy program, are primed to help with these endeavors to create “dynamic tumor atlases” across cancer subtypes. However, space considerations do not allow us to consider these important areas in the depth they deserve.
Here, we lay out a roadmap delineating a selection of overarching themes—clouds of complexity—looming on the horizon of cancer biology and medicine that, if mechanistically addressed, promise to improve patient outcomes and quality of life.
MULTISTEP TUMORIGENESIS: UNDERSTANDING TUMOR PROMOTION AND PROGRESSION
It has long been appreciated that cancers develop via pathways of stepwise tumorigenesis and malignant progression, and, more recently, during adaptive resistance to therapy. In part, these stepwise transitions and stages reflect the acquisition and refinement of hallmark capabilities, enabled in particular via the prominent phenotypic characteristics of genome instability and gene mutation, and tissue inflammation.2,4
Cancer genetics has taught us that mutations in specific regulatory genes—termed oncogenes, of which the RAS genes are prototypes—are essential drivers of this disease. In concert with loss-of-function mutations in tumor suppressor genes, these complementary genetic events can lead to the acquisition of the multiple cancer hallmark capabilities found in most human cancers. Indeed, this paradigm has been strongly supported by elegant mouse models of cancer in which germline or somatic genetic modifications can combine to unleash the complete set of phenotypes associated with human cancers, in the absence of any evident environmental carcinogenic factors. However, new knowledge has revealed that ostensibly “normal” tissues throughout the body harbor a multitude of oncogenic driver events comprising a patchwork of mutant cells that persist and can expand as asymptomatic clones during aging.12,13 This provocative result informs us that mutations in “driving” oncogenes may be necessary but not always sufficient for tumorigenesis (Figure 2). It is estimated that approximately 40% of the population will develop cancer in their lifetime, and given that a typical human has 30 trillion cells, these observations prompt the question: why is cancer so rare at the single-cell level?
Figure 2. The interplay between cancer driver mutations and environmental or endogenous tumor promoters in cancer risk.

Mutations are essential for cancers to develop and may arise as a result of spontaneous errors in normal DNA replication, during aging of quiescent cells, or by exposure to mutagens in the environment. Obesity, dietary factors, and inflammation may also contribute to mutation burden indirectly, for example, through generation of reactive oxygen species (ROS), but this is unlikely to make a major contribution to overall mutation numbers. Mutations may remain dormant in normal tissues for long periods, unless the tissue is repeatedly exposed to an inducer of inflammation or tissue wounding, causing selection of cells carrying specific mutations, leading to clonal outgrowth. The particular mutations selected depend on many factors including the tissue or cell of origin, the host genetic background, or the nature of the specific promoting factor. Known or suspected exogenous promoting factors are shown as examples, as are potential endogenous or lifestyle-associated promoting factors.
The historical concept that chemically induced skin carcinogenesis, mimicking natural environmental insults, involves “initiating” and “promoting” events14 has been conceptually refined by the realization that many normal tissues are replete with cells containing latent mutations in oncogenes and tumor suppressor genes.12,13 Certainly, some environmental carcinogens act synergistically as both mutagens and inducers of inflammation (typified by tobacco smoke) to mutationally initiate and promote cancer. This suggestion, first made in the 1940s, is one that has important implications for cancer prevention and unproven assumptions regarding the long-term safety of e-cigarettes.15 However, Riva and colleagues recently demonstrated that many known or suspected initiating carcinogens do not appear to cause mutations themselves, but rather may act by stimulating other hallmarks—including but not limited to chronic inflammation—that awaken and trigger clonal expansion of cells carrying latent oncogenic mutations.16 The concept that non-mutational factors—e.g., wound healing, chronic inflammation, and exposure to chemicals in the environment—can stimulate the growth and selection of cells containing such activating mutations in oncogenes or inactivating mutations in tumor suppressor genes has been documented in mouse cancer models,16-20 in human lung cancers linked to air pollution,21 and in mesothelioma through asbestos exposure,22 and is implicated in other human cancers, including those arising in the esophagus,23 pancreas,24 and colon.25
A logical and overlooked implication of these important insights is that we lack biological assays to assess the potential “tumor-promoting activity” of existing or new chemical matter introduced into the environment. We foresee the need to chart the entirety of multistage pathways of tumorigenesis across tissues, to clarify the mechanisms by which environmental or endogenous physiological promoters can trigger stem/progenitor-cell-like properties in pre-initiated cells harboring oncogenic mutations, acting through diverse inflammatory/wounding pathways across different tissues and cell types to alter clonal selection. The roster of suspects that may act as environmental tumor promoters includes, but is certainly not limited to, microplastics, glyphosate,26 per/polyfluoroalkyl substances (PFASs), hot liquids,27 and infectious agents such as H. pylori (Figure 2). Moreover, serious questions remain unanswered with respect to the long-term safety of e-cigarettes and the realistic possibility that human exposure to vaping substances could promote tumor initiation, independent of DNA mutagenesis, in much the same way as air pollution is thought to act. Endogenous or lifestyle factors, including diet, stress, sleep deprivation, sedentary behavior, and circadian rhythm disruptions, many of which can impact the microbiome, are also likely to play formative roles.
We envisage that defining encyclopedias of environmental (and physiological) promoter-induced inflammatory and other reactive responses that orchestrate tumorigenesis will facilitate the development of crucially important technical capabilities for early detection of incipient neoplasia that can distinguish lesions likely to progress to malignancy from those that will not.
Rigorously assaying for non-mutagenic tumor promoters will require improved technological capabilities, including single-cell RNA and DNA sequencing, as well as spatial transcriptomics/proteomics/metabolomics, to unveil heterotypic cellular interactions and actionable pathways suitable for molecular cancer prevention efforts. While the foundation of cancer may lie in mutant oncogenes and tumor suppressor genes, it is increasingly evident that tumor progression involves non-mutational epigenetic programming,4 as reflected in the dynamic heterogeneity apparent in many tumors, and as such, the epigenomes of both cancer cells and the diverse cells of the tumor microenvironment (TME) will necessarily require illumination at all stages. There is evidence for “epigenetic memory” of prior exposure to inflammation-inducing agents, which we envisage could subsequently contribute to tumorigenesis.28 While past exposure to mutagens can be identified by whole-genome sequencing of human cells and tissues that reveals mutational signatures of distinctive carcinogens,29 there is currently no technology for identifying past or current exposure to tumor promoters. The technical development of such tools will enable molecular cancer prevention efforts aimed to dampen inflammatory tumor promoter activities, further refined by deep appreciation of the broader macro-environmental complexities of the host, as elaborated in the topical sections that follow.
TUMORIGENESIS AND WOUND HEALING: TWO SIDES OF THE SAME COIN
While the complexity and uniqueness of each human cancer is undeniable, there are also remarkable commonalities across cancers. Some of these commonalities are general—all cancers engage the same cell cycle machinery and almost all exhibit p53 pathway inactivation, and promiscuous activation of both Myc and the Ras/phosphatidylinositol 3-kinase (PI3K) pathway. Furthermore, cancers of specific types or tissues of origin share signature tumor/stromal phenotypes, even when driven by different oncogenic mutations, and these differ profoundly from adenocarcinomas arising in other organs, even when driven by the same oncogenic mutations. Dvorak offered us the first clue as to the source of these organ-specific neoplastic constraints with his proposal that tumors are unresolved wounds,30 and there are clear parallels between the hallmarks of cancer and the hallmarks of wound healing.31 More lately this has been directly confirmed by showing that activation of the same core oncogenic mutations—KRasG12D and Myc—in adult tissues (lung and pancreas) directly and immediately drives formation of distinct adenocarcinomas whose tumor-stromal phenotypes perfectly match those of their spontaneous human pancreatic or lung adenocarcinoma counterparts.32,33 Thus, the principal determinant of cancer phenotypes is not their unique panoply of oncogenic mutations but their organ/tissue of origin. In contemporary parlance, the same oncogenic pathways are hacking into the unique endogenous wound repair programs of each target tissue/cell type.
This is a potent conceptual advance for two reasons. First, it speaks to the widely held presumption that cancer’s hallmarks (inflammation, immune suppression, promiscuous proliferation, suppression of apoptosis, etc.) are neomorphic traits selected through tumor evolution because they benefit tumor progression.2 However, if cancers are merely persistent hacks of normal regenerative programs, then most of cancer’s “hallmarks” actually evolved to optimize tissue repair, not neoplastic subterfuge. Second, wound repair has major components—an initial regenerative phase, which is followed by resolution, a discrete morphogenic program that reorganizes the inchoate tissue and reasserts homeostatic architecture, cellularity, and function. Emerging evidence indicates that, in wound healing, the switch from regeneration to resolution is triggered by a decrease in mitogenic signaling. Provocatively, it has long been known from genetic and pharmacological studies that blockade of oncogenic signaling triggers rapid regression of tumors (both cancer cells and stroma)—at least initially. This is typically attributed to “oncogene addiction,” a phenomenon lacking a coherent mechanistic explanation. However, the similarity between post-injury wound resolution, due to cessation of mitogenic signaling, and regression of tumors upon oncogene blockade suggests that a close examination of injury resolution could yield entirely novel strategies for cancer treatment.
TME: UNRAVELING AND TARGETING CELLULAR NICHES
The inflammation induced by cancer promoters and resultant multistep tumorigenesis discussed above are intertwined with stage-specific alterations in the complex multicellular TME and its associated extracellular milieu, which progressively coevolves with the cancer cells. Emerging evidence indicates that alterations in tissue integrity associated with age, tobacco exposure, or environmental pollutants among others (see aging and cancer: cellular fitness, microenvironment dynamics, and evolution over time section) can permit clonal expansions of normal somatic cells harboring oncogenic mutations. The TME is evidently involved in regulating disease progression and modulating the response to a broad range of cancer therapies.31,34 Within the TME, cellular niches provide unique habitats that influence tumor behavior, treatment response, and immune surveillance. Understanding the complex interactions within these spatial niches is essential for developing more effective cancer treatments. Moreover, there is potential that by identifying and targeting the key cell types, processes, or signaling pathways within particular niches, the effects of such therapeutic interventions can be amplified. We briefly summarize below knowledge about cellular niches within the TME and highlight questions that warrant investigation over the next decades.
Cellular niches in the TME consist of diverse cell populations, including cancer cells, immune cells, fibroblasts, vasculature, fat, nerve cells, and extracellular matrix (ECM) components. Each niche may contribute to tumor growth, invasion, and metastasis in distinct ways by supporting the specific features of cancer cells that inhabit them (Figure 3). For instance, cancer cells with high stemness potential reside within stem-cell-like niches,32 which support their self-renewal and resistance to conventional therapies, leading to disease relapse.33,35 Immune cell niches can either promote or suppress anti-tumor immune responses, thereby profoundly influencing treatment outcomes.36 The spatial heterogeneity of other TME components such as fibroblasts in the tumor stroma, can contribute to desmoplasia, angiogenesis, and immune modulation.37
Figure 3. Tumor micro- and macro-environments.

Representation of the complex tumor ecosystem. Interactions between diverse components of the tumor micro- and macro-environments are depicted, involving influences of the host, as well as external factors, on tumor growth and metastatic dissemination.
The importance of cancer niches can be particularly appreciated in the context of metastatic colonization, where metastatic cells must recreate a supportive environment upon seeding a new organ. Notably, the fact that metastases may erupt years after primary tumor resection, indicates that metastasiscompetent cells can reside in distant organs in a non-proliferating, dormant state. The existence of dormant niche components that sustain the quiescent but viable state of metastatic cells, with the propensity to enable outgrowth, is reported.38,39 However, the properties of these dormant niches and how they change over time to re-awaken cancer cells are still largely unknown. Certainly, tissue aging has implications for dormancy reversion.40 As we discuss in the following sections, aging as well as progressive systemic changes that are directly linked to metabolic and inflammatory perturbations caused by cancer-derived factors may contribute to evolving niches (Figure 3).
Significant technological progress has been made in characterizing cellular niches within the TME, revealing their dynamic and complex nature. Advanced imaging techniques, single-cell sequencing, spatial transcriptomics, multiplex high-resolution imaging techniques, and niche-labeling strategies are allowing researchers to identify distinct cellular populations and map their spatial organization within tumors.41 This has enabled, for instance, identification of a tissue-intrinsic regenerative program activated at early stages of metastatic niche initiation,41 the phenotypic states inherent to particular tumor niches linked to certain genetic abnormalities or between patients with primary or metastatic cancers,42,43 the tumor-stromal composition changes associated with cancer progression,44 as well as the features in the TME that can be correlated with clinical outcome or predict therapy responses.45-47 Based on such multi-omics profiles, new classifications of tumors are emerging.48 Although we expect that many tumors will ultimately be amenable to TME modulation in the future, it is conceivable that certain cancers will prove especially challenging, such as liver metastases in patients with microsatellite-stable colorectal cancers or glioblastoma (GBM). Notably, these are also organs subject to immune suppression under steady-state conditions, which likely contributes an additional barrier to mounting an effective immune response.
Looking forward, it will be important to determine the biology underlying TME spatial diversity; how hypoxia, nutrient availability, metabolites, and ECM composition influence the behavior of cellular niches. Understanding the intricate interplay between the distinct cellular niches within tumors is essential, for example, to discriminate between cancer-promoting and cancer-restrictive niches. Investigating the temporal evolution of these cellular niches will provide insights into how the TME evolves both during cancer progression and following therapeutic intervention. Longitudinal studies capturing the dynamics of cellular populations within niches over time, for example, from sequential patient biopsies combined with multimodal imaging strategies in preclinical models,49 will be key to unravel the spatiotemporal heterogeneity of tumors.
We envisage that charting the dynamic evolution of the TME and its heterogeneous niches will reveal new avenues to manipulate TME states to interfere with tumor growth and progression, metastatic outgrowth, and adaptive resistance to therapy. Understanding the key determinants of dormant niches that can maintain metastatic cells in a quiescent state for decades, and the subsequent alterations that lead to a switch toward metastatic outgrowth, will be an essential requirement for developing strategies to detect and eliminate dormant cells. Generating refined model systems to investigate these questions will be paramount to gaining mechanistic knowledge around metastatic dormancy and other niche phenotypes. Exploiting vulnerabilities within cellular niches holds great potential for the development of effective precision-based therapeutic strategies through the identification of specific markers or pathways that are essential for niche maintenance and tumor progression. Beyond the TME is the tumor “macro-environment” of the host, also depicted in Figure 3, which encompasses a number of the complexities discussed in the following sections.
THE IMMUNE MACRO-ENVIRONMENT: HOMEOSTATIC CONTROL AND HACKING SYSTEMS PHYSIOLOGY
The effects of tumor-induced perturbations of the immune system extend beyond the local tumor-immune environment, involving pronounced alterations to the systemic immune landscape during tumorigenesis. Paracrine molecules produced by cancer cells, immune cells, and non-immune stromal cells throughout tumor progression and released into the cardiovascular system have actions beyond the local TME (Figure 4). Several mechanisms leading to the release of immuno-modulatory molecules during cancer progression have been identified. First, the tumor itself can act in an autocrine manner on neighboring tumor and non-tumor cells, leading to the activation of both immune and non-immune cells and the release of additional molecules that add to the TME’s secretome.50 Second, cellular senescence programs that are commonly active in malignant and non-malignant cells can be exacerbated with chemotherapy agents, resulting in a secretory phenotype marked by high production of pro-inflammatory cytokines such as interleukin (IL)-6 and IL-8.51-54 Third, genetically unstable tumor cells activate DNA damage sensors that produce type-1 interferons or activate inflammasomes and IL-1β production, which can act peripherally on stromal cells and bone marrow progenitors.50,55-58 Fourthly, microbial elements influenced by patients’ diet and environment can lead to the activation of cells expressing pattern recognition receptors and the release of inflammatory molecules such as IL-6, IL-1β, and tumor necrosis factor (TNF).59-65 Lastly, stress-induced metabolites produced by genetically unstable tumor cells or surrounding stromal and immune cells can also be actively released into the circulatory system.66-69 These systemic cues are sensed by hematopoietic progenitors during the course of tumor development, leading to dysregulated myelopoiesis and expansion of aberrant myeloid cells that contribute to the dampening of local and systemic anti-tumor immunity, and to the shaping of a pro-tumorigenic TME (Figure 4).70,71
Figure 4. Local and systemic drivers of protumorigenic myeloid dysregulation.
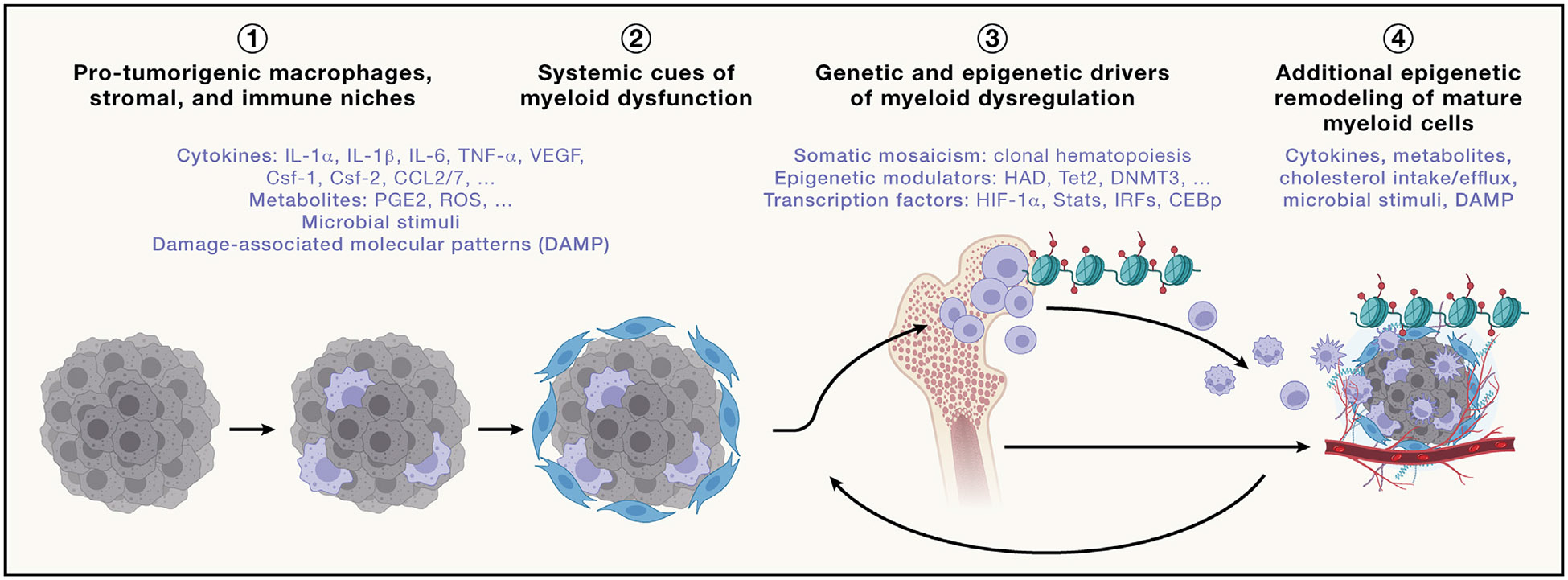
(1) Molecules produced during tumor initiation that contribute to the recruitment and activation of macrophages that play a key role in the initiation of the inflammatory cascade and the release of inflammatory molecules in the blood circulation.
(2) Release of inflammatory molecules in the blood circulation that drives the enhancement and dysregulation of myelopoiesis.
(3) Epigenetic remodeling of myeloid progenitors in the bone marrow that contributes to the induction of nodes of molecular suppressive programs along the myeloid lineage.
(4) Additional drivers produced in the local tumor microenvironment that contribute to further enhancement of myeloid suppression that further dampen antitumor immunity and promote tumor proliferation and growth. The list of molecules provided in each category is not exhaustive and is only provided as examples.
The co-option of developmental and regenerative programs by tumor cells further contributes to changes in the systemic immune environment, as exemplified by the cooperation between MYC and KRAS in driving an immunosuppressive microenvironment, marked by the exclusion of systemic T and B cell infiltrates in highly proliferative, invasive carcinomas. Deactivation of MYC triggers tumor regression that is accompanied by the rapid reversal of these immunological changes, involving a program that parallels the resolution phase of wound healing responses.72
Additionally, the characteristics of systemic immune responses in tumor-burdened hosts are heavily influenced by changes in physiological homeostatic controls, such as circadian control, endocrine response to stress and metabolism, and physical activity-induced changes in the circulatory system. Understanding how the systemic immune response changes during cancer progression, and how systemic immune functions are altered as the antigenic and phenotypic diversity of a tumor increases, and whether adaptive immunosuppression brought about by tumor evolution contributes to cancer-related death, are all important topics for future research.
These complexities suggest that the design of effective immune-based therapies will require a comprehensive understanding of the molecular underpinnings of the interplay between local and systemic immunity. Broadly, these analyses will comprise the identification of the systemic drivers of pathogenic myelopoiesis through deep and dynamic profiling of cancer patients’ secretome, as well as charting the chromatin and transcriptomic programs along the myeloid lineage, starting from early hematopoietic progenitors in the bone marrow to tumor-associated myeloid cells in order to identify initiating nodes of myeloid dysregulation. The results may guide the development of novel strategies to target mediators of systemic immune dysfunction. Concurrent assessments of systemic cancer patients’ secretomes and their clinical features, along with an understanding of tumor somatic evolution, could form the foundation for a therapeutic framework to guide patient immunestratification and immunotherapeutic decision-making.
Importantly, multistep tumorigenesis, malignant progression, and the evolving phenotypes of cancer-promoting micro- and systemic macro-environments are also modulated by a plethora of other complexities, as delineated below.
AGING AND CANCER: CELLULAR FITNESS, MICROENVIRONMENT DYNAMICS, AND EVOLUTION OVER TIME
Most cancers occur in people over the age of 60, and by 2050, it is projected that over 2 billion of the world’s population will be over 60 years old.73 The age-related rise in cancer can be attributed to various factors, including the accumulation of chronic genetic damage, epigenetic drift, alterations in tissue microenvironments (including increased senescent cells), and changes in adaptive and innate immunity.74 These factors can alter tissue homeostasis and thus fitness, enabling selection for proliferative expansion of mutant cells responsive to such altered landscapes. However, given the frequency with which they are observed in normal tissues, it can be surmised that mutation-driven clonal expansions rarely evolve to become life-threatening malignancies in aging tissue microenvironments.75 It is critical to view the association of cancers with old age through the lens of evolutionary biology—animals have evolved strategies to maintain tissue functions and avoid disease, maximizing reproductive success. These mechanisms wane in post-reproductive periods. Therefore, we need to appreciate how young/healthy tissue microenvironments maximize the somatic fitness of stem and progenitor cells, preventing the persistence or expansion of cells with potentially malignant mutations—youth is tumor suppressive. Unfortunately, aged tissues progressively lose their ability to limit cancer evolution.75,76 Notably, inherited progeria syndromes often (but not always) exhibit accelerated and augmented cancer incidence,77 which we envisage results from progeria-associated tissue dysfunction in the context of increased frequency of mutations.
Recent studies have revealed that aging is characterized by an increase in clonal expansions of somatic cells harboring oncogenic mutations in histologically normal tissue,78 which can vary widely in their pathogenic potential, as has been most clearly shown for clonal hematopoiesis, which is associated with increased risk for leukemias and solid cancers.79 A challenge will be to better understand how healthy tissues impede tissue-impairing clonal expansion and the phenotypic evolution of somatic cells that leads to cancer pathogenesis, and how environmental carcinogens and lifestyle factors in concert with natural aging affect these protective mechanisms (see multistep tumorigenesis: understanding tumor promotion and progression section). These expansions are associated with conditions that change tissue landscapes, including inflammatory bowel disease,80-82 sun exposure for the skin,83 alcohol and smoking for the esophagus,84 air pollution and smoking exposures for the lung,21 and obesity and smoking for clonal hematopoiesis.85 Although it is increasingly accepted that healthy lifestyles promote tissue repair/maintenance and cancer protection and that unhealthy lifestyles engender chronic inflammation, poor tissue repair, and increased risk of cancer and other diseases,86,87 we do not understand how healthy lifestyles impact the earliest stages of carcinogenesis, including such mutation-driven clonal expansions. The forthcoming knowledge may support the development of interventions that can block abnormal clonal expansions in aging tissues, with the aim of reducing cancer incidence as well as physiological aging. Understanding how non-mutagenic carcinogens (analogous to the tumor-promoting potential of pollutant particulate matter) impact this process and drive the expansion of cells with stem-like potential harboring oncogenic mutations will be instrumental. We also need to characterize suspected feedback loops whereby aging promotes particular mutation-driven clonal expansions, which then further contribute to tissue aging (which itself promotes oncogenesis). Analyzing clonal mutational patterns in tissue samples (e.g., blood and accessible epithelial tissues) from clinical trials of candidate anti-aging agents and from individuals of different ages, lifestyles (e.g., exercise), and diet could reveal strategies to limit the clonal evolution that can lead to aging-associated tissue decline and cancer.80,81
Aging also influences the ECM, a core constituent of the TME secreted by tumor-associated fibroblasts and other non-tumor cells.76 The ECM modulates tumor cell behavior through mechano-transduction. Aged fibroblasts release molecules that induce significant changes in tumor cells, impacting signaling pathways, reactive oxygen species (ROS) response, and metabolism,74,76 as well as emergence from proliferative dormancy.40 Indeed, it is possible that the regulatory processes that augment selection for somatic clones carrying cancer-associated mutations during aging overlap with those that awaken cells from the dormant state to instruct metastatic colonization (see the above discussion on dormant metastatic niches).40,88 While fibroblast senescence may be involved in both proliferative expansions, it is important to distinguish between senescence and aging, as senescence can occur across the lifespan, having both pro-tumor and anti-tumor effects, as described in the next section. Targeting actionable elements in the microenvironment of aging tissues, beyond senescence, has the potential to inhibit metastasis and overcome therapy resistance in elderly cancer patients. This pursuit will require sophisticated bioengineering techniques, AI-based pathology, and exploration of intersecting factors such as biological sex and stress with age. Specifically, spatial transcriptomics layered with sequencing and the 3D reconstruction and annotation of the TME using AI-based pathology techniques such as CODA,89 quantitative analysis of multiplex immunohistochemistry using techniques like AstroPath,89,90 as well as bioinformatics tools for gene behavior analysis to predict cell-cell communication will be critical to understand how the aging TME impacts the expansion of cells with stem-like potential leading to tumor initiation or the awakening of dormant metastatic cells.91 The ultimate goal is to identify agents that induce tissue landscapes to assume phenotypic states that limit all stages of cancer evolution, from early clonal expansions to metastatic outgrowths. Applications could range from early prevention (e.g., interventions that maintain more youthful landscapes) to therapies (e.g., adjuvant/neoadjuvant therapies that reduce metastatic outgrowth) that can be used to reduce cancer burden and mortality in a rapidly aging population. Among these, leveraging the expanding knowledge base and insights about the complexities connecting (partially age-related) cell senescence to cancer are likely to lay the groundwork, as discussed below.
SENESCENCE AND CANCER: CELL STATE TRANSITIONS AND THERAPEUTIC APPROACHES
Senescence is a stress response characterized by a stable cessation of proliferation, with changes in cellular morphology, gene expression, chromatin states, as well as increased secretion of cytokines. The unambiguous identification of senescent cells is complicated by a lack of gold standard markers of the senescent state. Moreover, there are several closely related cell states, such as quiescence, dormancy, diapause, and drug-tolerant-persister (DTP) cells, which share features with senescent cells. For example, the senescence marker senescence-associated β-galactosidase is also expressed in some DTPs. Moreover, while the dogma in the senescence field is that the proliferation arrest of senescent cells is irreversible (which is not the case for the related states mentioned above), there is growing evidence that senescence is also reversible.92 Multiple gene expression signatures can identify senescent cells, but there is significant variability in gene expression among senescent cancer cells derived from different tissues.93
In cancer cells, senescence can be triggered by genotoxic stress (resulting from chemotherapy or radiotherapy; referred to as “therapy-induced senescence”), oxidative stress, or hyperactivated mitogenic signaling. A proliferation arrest can be considered beneficial for cancer therapy in the short term. However, the persistence of senescent cancer cells can be unfavorable in the long term due to the creation of an inflamed microenvironment, thereby acting as a tumor promoter (see tumor promotion section). It is increasingly feasible, and potentially beneficial, to kill senescent cancer cells, fibroblasts, and collaterally affected normal cells, via so-called senolytic therapy, aiming to avoid undesired phenotypic effects of senescence in cancer. Indeed, evidence from mouse models suggests that several of the side effects of chemotherapeutic agents are caused by the induction of senescence in normal cells, such that the removal of senescent cells in chemotherapy-treated mice reduced bone marrow suppression and improved renal function.94 Aging studies have also shown an increase in senescent cells—principally fibroblasts—over time, and senolytic clearing of such senescent cells in animal models delays aging-associated disorders, including cancer (see aging section).95,96
Based on these considerations, a “one-two punch” approach to cancer therapy has been proposed, consisting of sequential treatment with a senescence-inducing therapy, followed by a senolytic therapy.92 Conceptually, sequential treatment should be highly synergistic, while reducing the toxicities of combination therapy. A practical complication in applying this approach is the heterogeneity of cancer cell senescence pathways, making it challenging to find senolytic drugs that act broadly. For instance, the BH3 mimetic drug navitoclax (ABT-263), which is used widely as a senolytic drug in aging research, is only senolytic in a fraction of senescent cancer cells. Recent data indicate that activation of death receptor signaling with agonistic antibodies has broader senolytic activity in cancer cells.97 An alternative approach to senolysis is the exploitation of the immune cells attracted to the senescent tumor mass by secreted cytokines that are components of the senescence-associated secretory program (SASP). In preclinical models of pancreatic cancer, induction of senescence resulted in recruitment of CD8+ T cells into tumors, resulting in sensitivity to checkpoint immunotherapy.98
Despite these initial advances, many questions remain to be answered. Successful exploitation of the “one-two punch” pro-senescence therapeutic approach will require an understanding of which combinations of pro-senescence plus senolytic drugs are most active, and of the context dependency of such drug pairs. Moreover, we need to better understand the complexity of the infiltrating immune cells in senescent tumors and how to exploit their presence therapeutically. There is, however, another layer of complexity, namely that maintaining senescent cells can be demonstrably beneficial in certain tumors, perhaps reflecting variabilities in their SASP4; this dichotomy of function requires further investigation in regard to fine-tuning therapeutic targeting of senescence. Finally, in the aging human brain, neuronal cells abundantly express senescence markers.99 It will therefore be crucial to understand whether a therapeutic window can be defined for aged patients with cancer in the context of senolytic therapy. A successful proof-of-concept clinical study using pro-senescence and senolytic therapies will act as a catalyst for this approach to cancer therapy. Intertwined with aging and senescence are the complex effects of metabolic variation, in cancer cells, in tumors, and in the host, as elaborated below.
METABOLISM, DIET, AND CANCER: SYSTEMIC EFFECTS AND THERAPEUTIC OPPORTUNITIES
The widespread dysregulation of metabolism and cellular energetics in cancer suggests that altered metabolism is not merely an ancillary consequence of tumorigenesis but is rather a selected requirement for tumor initiation and progression. While metabolic changes intrinsic to cancer cells support tumorigenesis, the complex metabolic crosstalk between the tumor and the host—both within the immediate TME and across distant organs—highlights our emerging appreciation of cancer as a systemic disease (Figure 5). Early during carcinogenesis, cancer cells are selected for metabolic reprogramming that maximizes the production and supply of nutrients from the microenvironment to satisfy the metabolic demands required for aberrant growth.100 However, the reciprocal relationship between tumor metabolism and systemic metabolism remains largely unexplored. We focus in this section on our increasing appreciation of these intertwining complexities and their potential to provide new approaches to prevent, detect, and treat cancer.
Figure 5. Cancer as a systemic disease.

Reciprocal relationships between cancer cell metabolism, the metabolism of heterotypic cells of the tumor microenvironment, and systemic metabolism. These metabolic crosstalks modulate cancer progression and could lead to cancer-associated cachexia. Detecting these metabolic alterations provides clinical opportunities for early detection and disease monitoring.
Effects of cancer on the body
Cancer cells subvert non-cancer cells (e.g., fibroblasts, adipocytes, immune cells, and neurons) in the TME to provide metabolic support and supplement nutrient-poor tumor environments,101,102 wherein avid nutrient uptake and metabolic waste production can suppress anti-cancer immune responses.103-105 In the macro-environment, tumors alter the host’s systemic metabolism both directly102 (by the tumor secreting particular signaling molecules or altering nutrient availability) or indirectly106 (e.g., via the immune system’s response to the tumor). The reprogramming of metabolism in distant organs, such as the liver107 and brain,108 alters whole-body metabolism to promote cancer-related systemic manifestations, including metastasis, resistance to anti-cancer therapy, cancer-associated cachexia (CAC), and death (Figure 5).109,110 Since these alterations in systemic metabolism start early, understanding them may guide and enable the design of strategies aimed to preserve host metabolism and restrict systemic manifestations of late-stage cancers.
A clear example of the metabolic effect of cancer on the body is the induction of the active catabolic state of CAC, a complex, debilitating wasting syndrome that limits fitness and the effectiveness of therapies, resulting in poor patient outcomes.111,112 CAC likely represents corruption of a normal and transient wound healing process, which cannot be resolved in the context of cancer.113 The mechanisms by which tumor progression and metastasis cause systemic changes that facilitate the loss of muscle and/or fat remains largely unknown but may reflect a cancer/host interplay that causes a systemic metabolic imbalance favoring the tumor at the expense of the host. Consequently, interventions that prevent or reverse CAC may also limit tumor growth.114 A deeper understanding of distinct metabolic, inflammatory, molecular, and neuro-endocrine drivers of CAC and their origins from the host or the evolving tumor may open new therapeutic avenues that extend beyond treating the causes and consequences of CAC to hampering the growth of primary and metastatic tumor.114
Obesity, physical activity, and cancer
Metabolic disorders as well as variations in healthy physiological states impacted by factors like exercise can influence the development and progression of cancers and are likely to involve interplay between the host’s genome, diet, physical state, physiological status, and microbiome.115 Approximately 4%–8% of all cancers are attributed directly to obesity,116 but this statistic is set to increase rapidly as obesity rates grow, particularly in low- and middle-income countries. Although strategies to reduce obesity and increase regular physical activity are obvious interventions with potential to prevent or treat cancer, the mechanistic link between obesity and cancer remains unclear. Outstanding questions include the following: is obesity per se or the accompanying metabolic perturbation responsible for increased cancer incidence? Does adipocyte expansion indiscriminately release factors that are pro-oncogenic? Do some individuals have a specific pathophysiologic response to obesity that makes them more susceptible to cancer? And, what is the role of the gut microbiome (discussed in more detail below)?
Regular physical activity has been implicated in the prevention and/or improved cancer-specific survival for several cancer types,117 ostensibly functioning not only by reducing body mass index but also by lowering levels of hormones, suppressing inflammation, improving immune function, and altering intermediary metabolism.118 The emergence of new technologies for identifying molecular effectors of physical activity at the organism level119,120 will provide unprecedented opportunities to define physiological responses to physical activity that can be leveraged for cancer prevention and control.
Cancer and host metabolism in detection and therapy
Metabolic interventions that impact the cancer cells, the TME, or systemic host metabolism—through limitation of nutrient support of cancers or by boosting anti-tumor-host responses—may each provide new therapeutic opportunities. However, targeting the metabolic alterations in cancer has proven to be challenging, due to many factors including potential toxicities of targeting pathways that are essential in normal cells and the difficulty of making selective drugs for highly abundant enzymes, as well as the metabolic flexibility and plasticity of cancer cells. New approaches to overcome these limitations will be needed, including strategies to influence the pathologic activities of metabolic enzymes while maintaining their function in normal organs.121
Cancers’ abilities to alter systemic and tissue-specific metabolism also suggest new opportunities for early disease detection through changes in the levels or types of circulating or excreted metabolites.122 Redox metabolism, urea cycle metabolism, sulfur metabolism (including bile acid production), aspects of one-carbon and nucleotide metabolism, and possibly even tricarboxylic acid (TCA) cycle function or glycolysis, could be monitored systematically using increasingly refined technologies to inform on the metabolism of the host, the tumor, and their interplay.
Dietary interventions are another promising avenue, most likely to be effective in augmenting cancer therapy.123,124 Fasting or fasting-mimicking diets (FMDs) can improve cancer outcomes for many organ-specific tumors in both rodents and humans,125,126 evidently functioning through the regulation of various regulatory pathways, including reduced insulin and leptin signaling, and attenuated inflammation (which are also downregulated by exercise).
Precision nutrition—selective alterations of specific dietary carbohydrates, amino acids, and lipids—offers another way to target cancer vulnerabilities. Reducing the intake of non-essential amino acids such as serine, glycine, and methionine can retard tumor growth and sensitize treatment-resistant cancer cells.127,128 Additionally, manipulating the balance of saturated and unsaturated lipids can directly affect membrane composition and thereby impair the survival of cancer cells.129 Moreover, low-carbohydrate diets that limit insulin production can increase the efficacy of PIK3CA-targeted therapies in certain cancers.130 While several of these dietary approaches are now being explored in clinical settings, further understanding of how diet affects normal physiology and anti-cancer responses is still emerging123,124,30 and can be anticipated to guide further applications of this strategy.
The success of such metabolic interventions depends on a deeper mechanistic understanding of the requirements imposed on cancer cells by their origin, genetics, and environment, coupled with the impact of therapy.123,131 We also need to understand the interplay and effects of metabolism and its physiological and pharmacological modulations on many of the systemic host factors that govern cancer progression, as discussed elsewhere in this review, including circadian rhythms, as discussed below.
CIRCADIAN RHYTHMS AND CANCER: CLOCKS AS CRITICAL REGULATORS OF PHYSIOLOGY AND DISEASE
A poorly understood aspect of tumor-host dynamics involves the potential temporal effects on cancer cell behavior and response to therapy. Circadian rhythms describe 24-h oscillations of light, temperature, and other aspects of the earthly environment, as well as daily fluctuations in gene expression and physiology driven by genetically encoded molecular oscillators (“circadian clocks”) present in virtually all mammalian cells. Disruption of circadian rhythms caused by nightshift work, travel across multiple time zones, and residence at the western edge of time zones, are all associated with increased cancer incidence.132 Recent work has uncovered mechanisms that may contribute to these associations.
Mammalian circadian clocks are based on a transcriptiontranslation feedback loop in which a heterodimer of the transcription factors CLOCK and BMAL1 drives the expression of its repressors, PERIODs (PER1–3) and CRYPTOCHROMEs (CRY1 and 2). PERs and CRYs accumulate and inactivate CLOCK-BMAL1 after which PERs and CRYs are tagged for proteasomal degradation, renewing the cycle. Thousands of genes exhibit daily oscillations of expression in every organ that has been examined, such that over half the genome is rhythmically expressed somewhere in the body.133 Several mechanisms contribute to transcriptional oscillations including transactivation by CLOCK-BMAL1, and to repression of diverse transcription factors by PERs and CRYs. Intriguingly, both PER2 and CRY2 have been shown to influence P53, the most frequently inactivated tumor suppressor in cancer.134,135 CRY2 also stimulates turnover of the oncoprotein c-MYC in rapidly proliferating cells,136 although its deletion does not appear to affect MYC in healthy spleens in vivo.137
Molecular connections between clocks and proteins that are well-known to influence cancer, such as P53 and c-MYC, encourage speculation that disruption of these pathways might explain enhanced cancer incidence associated with circadian abnormalities. However, recent work suggests alternative mechanisms, including disruption of anti-tumor immunity and of cellular stress responses.138,139 Genetic deletion of individual clock components has varied impacts on tumor growth in mouse models,132,140,141 so it seems unlikely that a particular mechanism based on disrupting a core clock component will universally explain increased cancer incidence caused by circadian disruption. Using transformed murine cells to initiate tumors results in striking differences in tumor growth depending on the time of day at which cells were implanted,139 implicating circadian regulation of anti-tumor-immune responses. Exposure of host mice to chronic circadian disruption exacerbated tumor growth and eliminated the impact of engraftment time, which suggests that impaired immune responses may contribute to increased tumor growth upon circadian disruption. Chronic circadian disruption also increased tumor burden in genetically engineered mouse models of KRAS-driven lung adenocarcinoma.138,142 Unbiased analysis of gene expression in lung tumors revealed that circadian disruption increases expression of heat shock factor 1 (HSF1), which has been linked to enhanced tumorigenesis in a variety of contexts.143 Additional research is needed to determine whether reduced immune surveillance and/or elevated HSF1 is required for enhanced tumorigenesis caused by chronic circadian disruption.
Multiple mechanisms likely contribute to enhanced tumorigenesis observed in people exposed to circadian disruption. Circadian disruption also influences cancer-associated phenotypes, including metastasis and cachexia.144,145 Circadian disruption acts as a tumor-promoting lifestyle factor through unknown mechanisms, likely including sleep deprivation and metabolic disturbances. Continuing research is needed to delineate the contributions of the mechanisms discussed here and the additional effects of circadian disruption on the etiology of diverse tumor types. Few clinical trials include multiple dosing times and “circadian logic” in their design, although there are clues that such regimens can affect chemotherapy outcomes,146 and the response to checkpoint blockade in melanoma, where emerging evidence from the MEMOIR study suggests that adaptive immune responses are less robust in the evenings.147 Recording the time of day at which biopsies are collected, and when treatments are delivered during clinical trials, would also markedly improve the potential to advance understanding in this area. Another systemic regulator of the interplay between the host and tumor phenotypes, likely intertwined with circadian rhythms, involves the nervous system, as considered below.
INFLUENCES OF THE NERVOUS SYSTEM: EMERGING ROLES OF NEURO-CANCER CROSSTALK
Neurons, glial cells, and nerves: A critical component of the cancer ecosystem
In recent years, both the central and peripheral nervous system have emerged as critical components of tumor-host interactions. Exciting discoveries have established neural innervation and signaling subversion as potential new factors of the tumor macro- and microenvironment, both in brain tumors and most other cancers, making it possible that neuronal-cancer interactions might eventually become another hallmark of cancer. Much as it governs physiological wound healing, tissue development, and organogenesis, as well as homeostasis and plasticity throughout life, the nervous system seems to play instrumental roles in the regulation of cancers.5,148 Specifically, synaptic and paracrine neuronal activity, but also cancer-intrinsic neural features, can govern cancer initiation, growth, dissemination, and treatment resistance; conversely, tumors can negatively affect the nervous system and even reprogram it, leading to detrimental feedback loops.5,148 Moreover, the nervous system influences cancer biology and cancer therapy response through dysregulation of the immune system, alterations to angiogenesis, and broader systemic effects.
A salient example is GBM (gliomas), wherein pathways of normal neurodevelopment are hijacked to interconnect cancer cells into a network, a syncytium involving cell-cell communications that foster tumor phenotypes.149 This cancer network receives direct input from bona fide glutamatergic (excitatory) neuron-glioma synapses that activate the network, driving tumor growth.150 The cell population is constantly stimulated by an autonomous rhythmic activity generated by pacemaker-like cancer cells within the network, recapitulating neurodevelopmental processes.151 GBM dissemination—perineural invasion—in the brain is governed by this cancer network via its neuron-glioma associations, as well as other co-opted neuronal mechanisms.152 Remarkably, in the human brain, GBM also remodels neural circuits, such as in regions of language representation, developing functional interconnectivity between the cancer and the normal brain, which is associated with decreased patient survival.153
Outside the brain, tumors secrete growth factors (e.g., nerve growth factor, NGF) that attract innervation by peripheral nerves and can even reprogram them. This innervation by sympathetic, parasympathetic, and sensory nerves has been demonstrated to drive tumor growth, invasion, metastasis, and therapeutic resistance in most cancer sites throughout the body, largely via paracrine secretion of neural factors. New therapeutic avenues that are based on these neuro-cancer interactions are currently being explored.148
Cancer pain
An important example of how tumors and cancer therapies can negatively impact a patient’s nervous system involves the sensation of pain. Debilitating bone pain associated with metastases in prostate, breast, and lung cancers can severely impact quality of life. Opioid analgesics remain the mainstay of cancer pain management, but the long-term consequences of tolerance, dependence, and hyperalgesia are problematic.154 The potent analgesic lidocaine has both pain-killing and anti-tumorigenic activity as well as effects on immune responses, consistent with a vital role for the neuro-immune cancer axis in approaches to treatment.155 Clinical trials of new approaches to pain treatment focused on blocking input into the central nervous system have promise, for example, using antibodies that neutralize immune-derived mediators such as NGF or TNF to alleviate cancer pain (NCT02609828). Similarly, channel-blocking small molecules are also under evaluation in phase 3 trials aimed at reducing cancer-associated pain.156 Pain control is an increasingly important dimension to managing and treating cancer, with encouraging promise.
The road to translation
Future research in translational cancer neuroscience will need to address three main points: (1) mapping the world of neuro-cancer interactions (e.g., synaptic, paracrine, and systemic), including the classes of neurotransmitters, neurotrophins, neuropeptides, and hormones that are relevant for distinct tumor entities and disease stages, combining molecular imaging, circulating biomarkers and morphological, neurophysiological, and molecular profiling of freshly resected tumor tissues, for which bespoke methodologies need to be developed; (2) developing informative biomarkers to monitor neuro-cancer interactions in a given patient; (3) establishing optimal combination therapy partners, e.g., immunotherapies, as part of the emerging field of neuro-immuno-oncology. Since more than 100 drugs are approved in neurology, psychiatry, and internal medicine that target neural signaling pathways, drug repurposing may prove productive, as exemplified by clinical trials targeting stimulatory neuron-cancer synapses of the AMPA receptor subtype with the antiepileptic drug perampanel,150,157 or targeting neural-like cancer cell networks with meclofenamate (EudraCT 2021-000708-39).149 Moreover, drug development aimed at early neurodevelopmental regulators usurped by cancer cells and at cancer-specific neural interactions present promising avenues. While interfering with normal neurotransmission can lead to side effects that need to be monitored, understanding the mediators and receptors specifically implicated in driving tumorigenesis should produce therapeutic targets based on more precise mechanistic insights.
In summary, the recent discovery of intimate neuro-cancer interfaces opens new horizons in our broader understanding of cancer, including the possibility that modulating cognitive or other neural states might impair crucial cancer phenotypes. Future research will determine whether this emerging knowledge can be leveraged for the development of new therapeutic strategies.
PARITY: LESSONS FOR TUMOR PROTECTION
The aforementioned examples of how the host can modulate tumor development and malignant progression have largely focused on tumor-promoting intersections. The converse consideration is the potential for human behavior and environmental exposures to protect against tumor initiation. Recent work has shed light on the role of sex hormones such as androgens and the endocrine system in such interactions and revealed mechanisms via which they govern anti-tumor immunity and T cell checkpoint responses. This crosstalk is consistent with knowledge about autoimmunity and infectious diseases, where, e.g., biological sex is associated with apparent differences in incidence and magnitude of vaccine-mediated immune responses. A clear example of this is parity and the duration of breastfeeding, which are well established to have positive health benefits for both mother and child. Pregnancy and breastfeeding have been associated with protection from breast and ovarian cancer, though the risk does rise in the short term, and a lower risk from all-cause mortality later in life.158 The risk of breast cancer is reduced by 4% for every 12 months of breastfeeding in addition to the 7% decrease in risk observed for each birth.159 Breastfeeding particularly reduces the risk of triple-negative breast cancers (TNBCs).160 The mechanisms behind breast cancer protection associated with pregnancy are thought to be related to the maturation of the breast epithelial cells, making them less susceptible to transformation, and to a reduction in circulating estrogen through amenorrhea.
Previous studies have established a positive prognostic effect from high levels of T cell infiltration in early-stage breast cancers, particularly in TNBC.161 For instance, a small subset of T cells with a tissue-resident phenotype were shown to be highly proliferative and cytotoxic, essential qualities for a durable anti-tumor response.162 Recently, using mouse models, the critical role of T cells with a resident memory (TRM) phenotype in immune protection from mammary tumor rechallenge has been reported, suggesting that TRMs may confer protection against breast cancer.163 Healthy, non-cancer affected breast tissue contains a multiplicity of immune cells, including myeloid, natural killer (NK), B, and T cells that do not express high levels of T cell-inhibitory checkpoint molecules.162,164 Given this epidemiological data, future work should seek evidence for an immunological mechanism that might explain the relationship between breastfeeding, pregnancy, and resultant long-term breast cancer protection. Gene expression patterns in the human breast significantly change years after pregnancy, notably with an upregulation of immune-related genes.165 Pregnancy itself causes dramatic changes in the gut and vaginal microbiome of the mother through the three trimesters.166 It is also well accepted that breast milk contains a rich microbiome that changes throughout the stages of lactation.167 In colostrum samples, Weisella, Leuconostoc, Staphylococcus, Streptococcus, and Lactococcus are the predominant species. In contrast, at 1 and 6 months, microbes that are typically found in the oral cavity (e.g., Veillonella, Leptotrichia, and Prevotella) are significantly increased in breast milk.168 Overall, breast milk can contain up to 600 different bacterial species, which could ultimately influence the immune repertoire of both mother and child.169
Research into the inflammatory and microbial changes during pregnancy and post-pregnancy, as well the composition of breast milk, has mainly focused on the benefits to the infant but has not yet deeply investigated how these effects may benefit the mother in the long term, as well as contribute to immune surveillance of the local breast microenvironment. Further research into sex hormones, their change at various reproductive stages, in both females and males, and how they alter anti-tumor immunity may help elucidate underlying molecular mechanisms governing cancer protection as well as progression, not only in hormone-responsive organs.
THROMBOINFLAMMATION AND CANCER
Thromboembolism is the obstruction of a blood vessel by the pathological formation of a clot. It is a leading cause of death in people with cancer and is associated with a poor prognosis that cannot be explained by cancer stage.170,171 Individuals with cancer have a 9-fold higher risk of thrombosis than the general population and cancer-associated thromboembolism is growing in incidence.172
An underlying predisposition to thrombosis relates to a dysregulated thromboinflammatory response that involves the interplay between coagulation and inflammatory mediators (Figure 6).173 Neutrophils, the most abundant myeloid leukocytes, promote thromboinflammation through the release of neutrophil-extracellular traps (NETs), a negatively charged combination of histones, proteolytic enzymes, and DNA that activates thrombosis.174 Several cancer types are also known to secrete tissue factor, the primary activator of coagulation in vivo. Higher plasma levels of tissue factor are associated with a worse cancer stage, poorer outcomes, and a risk of thrombosis.175
Figure 6. Thromboinflammation and cancer.

The interplay between the coagulation and innate immunity systems plays important roles throughout the development and growth of a tumor. Early interactions between neutrophils and tumor cells, including neutrophil-extracellular traps (NETs) produced by neutrophils, promote dormant tumor cell awakening, tumor survival and immune escape. Later aggregations between platelets and tumor cells promote tumor survival intravascularly and tumor spread. Activation of coagulation through tumor-specific mechanisms, such as the release of tissue factor, increases the risk of cancer-associated thrombosis.
Thromboinflammation potentially promotes tumor growth and spread. Tumor cell-platelet aggregates are hypothesized to promote tumor migration.176 An elevated platelet count is a marker of poor prognosis as well as a risk for thromboembolism. Murine models of GBM demonstrate that platelets bind to tumor cells through podoplanin and, importantly, elevated plasma levels of podoplanin are associated with a higher risk of thromboembolism and lower survival rates in patients.176,177
Studies in mice and humans have established associations between the number and type of intra-tumoral neutrophils and disease progression.178 The neutrophil phenotype is influenced by the environment and many types of cancer elicit potent immunosuppressive and pro-angiogenic functions of neutrophils that promote tumor growth and/or dissemination.179,180 Balancing this view, emerging evidence also reveals anti-tumoral activities of neutrophils and tumor-derived factors that act by promoting the education and activation of cytotoxic T cells.181,182
Beyond these functions, the release of NETs has been shown to promote intravascular tumor cell entrapment, which facilitates metastasis, by physically shielding tumors from the actions of cytotoxic T cells, or awakening dormant cancer cells by remodeling their local microenvironment (Figure 6).183 Conversely, NETs can also be cytotoxic and have been shown to kill tumor cells by the action of associated proteolytic enzymes and histones.
Managing thrombosis
Many factors can alter thrombotic risk during treatment of patients with cancer, including surgery, chemotherapy, infection, and intravenous inoculation of therapeutic drugs. A large-scale cohort study that serially measures multiple biomarkers in people with cancer receiving therapy is needed to help identify risk biomarkers that could be used to define a change in an individual’s risk over time. Future assay development should look for reliable global markers of thrombotic risk that would allow a more flexible and personalized approach. This leads us to the question: once we can identify the risk, can we do anything to mitigate it?
The use of low-dose anticoagulation (thromboprophylaxis) to prevent venous thromboembolism is effective in higher-risk cancer populations. However, there is insufficient evidence to support the use of thromboprophylaxis in all people with cancer due to the risk of bleeding caused by this treatment. Clinical scores, the most validated being the Khorana score, can identify some but not all at higher risk.184 Newer anticoagulants such as factor XI inhibitors, which aim to “uncouple” haemostasis from thrombosis are currently being trialed for treatment of cancer thrombosis (NCT05171075). Future randomized controlled trials should investigate their effectiveness in preventing thrombosis.
To improve outcomes for people with cancer, we need both to understand the role of thromboinflammation, and to develop methods to monitor thrombotic risk throughout cancer management so as to improve both prevention and treatment strategies.
The intersection of clonal hematopoiesis in inflammation, thrombosis, and tumor progression
Somatic mutations in hematopoietic stem cells are linked to increased NET formation and the promotion of thrombosis, both in the context of hematologic malignancies and clonal hematopoiesis (e.g., JAK2V617F in myeloproliferative neoplasms or in clonal hematopoiesis).185
Clonal hematopoiesis of indeterminate potential (CHIP) is a phenomenon of aging defined by the presence of leukemia-associated somatic mutations in the hematopoietic stem cells of individuals without apparent hematologic clonal disorders.85 These mutated hematopoietic stem cells give rise to altered immune progenies that influence host immunity.186 The most frequent CHIP mutations are recognized to promote myeloid-derived inflammation through increased production of inflammatory cytokines.
Furthermore, CHIP is linked to non-hematologic cancer in several ways. CHIP is associated with an increased risk of developing solid tumors, such as lung cancer, kidney cancer, lymphoma, and sarcoma.187 CHIP is observed in 20%–30% of patients with cancer overall, is shaped by anti-cancer therapies,188 and is associated with an increased risk of death through the progression of the primary non-hematologic tumors.189 However, little is known about the relationships between immune dysfunction in CHIP and anti-tumor immunity during tumor initiation, progression, and response to therapies.190 Future work delving into the tumor-immune interface within the context of CHIP are poised to unveil prognostic biomarkers and potentially anti-inflammatory interventions to attenuate disease initiation and progression.
MICROBIOMES AND CANCER: DISEASE PATHOGENESIS, THERAPY RESPONSE, AND THERAPEUTIC TARGETING
Another environmental complexity that transcends the host per se, in addition to those discussed above, involves the human microbiome, composed of microbes and their genomes.191 It is increasingly well established that the microbiome variably profoundly impacts disease pathogenesis and therapy outcomes in human malignancies,192 with polymorphic microbiomes recently highlighted as a hallmark-enabling phenotypic characteristic of cancer.4 There is increasing evidence for the impact of gut and other tissue microbiota on the pathogenesis of cancer as well as therapy responses, along with intriguing implications for intra-tumoral microbiota.
The role of gut microbes on pathogenesis and therapy response
Microbes within the gut have diverse effects on immunity and cancer via a number of different mechanisms. They can directly modulate cellular constituents promoting carcinogenesis, for example, in colorectal cancer,193 and influence systemic and anti-tumor immunity.194 The importance of the gut microbiota in shaping responses to immune checkpoint blockade was first shown in preclinical models59 and was quickly followed by landmark studies in human cohorts,61,62,195 now bolstered by a number of studies demonstrating prognostic associations of the gut microbiota with human cancers.192 Importantly, we are gaining more clarity into the taxa and functional characteristics of gut microbiota that are associated with response and resistance to immunotherapy, with data suggesting that the presence of unfavorable taxa in the gut is associated with systemic inflammation and impaired anti-tumor immunity along with increased risk for adverse events.196 Recent studies have also suggested that dietary intake can impact the gut microbiota and immunotherapy response and toxicity,197-199 thus providing a tractable angle for therapeutic intervention.
The role of intra-tumoral microbes on pathogenesis and therapy response
Nucleic acid sequencing and imaging analyses have revealed the presence of intra-tumoral microbes in various human cancers.200-204 Microbial composition and load have been associated in certain tumor types with treatment responses and patient survival.203,205 Advances in multi-omics approaches have provided mechanistic insight into the role of intra-tumoral microbes within the oral and colorectal TME.204 Initially, intra-tumoral microbiome studies focused on gastrointestinal cancers and microbe-accessible mucosal sites. However, in 2020, two landmark studies reported tumor-type-specific bacterial communities in many solid cancer types,206,207 suggesting diagnostic potential involving bacterial nucleic acids in cancer patients’ blood.207 One study reported that intra-tumoral bacteria were predominantly intracellular in non-gastrointestinal cancers.206 Similarly, cancer-type-specific intra-tumoral fungal communities have been reported.208,209 However, recent data reanalysis has raised important concerns about the prevalence of tumortype-specific microbiomes, in particular inside cancer cells highlighting microbial contamination in datasets, human DNA contamination of microbial references, and machine learning issues.210,211 Bacterial imaging reanalysis questioned bacterial localization within breast cancer epithelial cells,206 instead suggesting their presence in macrophages and interstitial stroma. Reproducing findings on intra-tumoral fungi in pancreatic cancers has also faced challenges.212-214 Thus, while there is compelling evidence for the presence of tumor-infecting microbes in specific cancer types, such as colorectal cancers,200,201,204,215 there is now controversy regarding the generality of a bona fide tumor-type-specific microbiome across human cancer types. Further research is needed to determine whether the presence of intra-tumoral bacterial and fungal communities is unique to specific cases or is a more widespread phenomenon, and in which types of cancer they are most prevalent, and functionally impactful.
Targeting gut and tumor microbes for cancer treatment, interception, and prevention
A growing body of evidence supports the modulation of microbiota as a strategy to enhance outcomes to cancer treatment,192 including fecal microbiota transplants216,217 or inoculation with well-defined, mechanism-guided microbial species.218 Other strategies include elimination of tumor-promoting species within the TME via targeted antibiotic approaches.219 As we move forward, it will be imperative to develop standardized approaches for assessing pathogenic (and beneficial) microbes acting at a distance from the gut and mucosal tissues they naturally populate, as well as within tumors. We must enhance our mechanistic understanding of how gut and intra-tumoral microbes directly and indirectly shape cancer pathogenesis, and thereby impact patient outcomes. A corollary opportunity is to leverage this information to identify microbial-based biomarkers for cancer prevention or prognostics, to aid in the design and testing of therapeutic strategies aimed to modulate microbes and thereby better treat cancer, and ultimately even to prevent or intercept malignant transformation.
FUTURE STRATEGIES AIMED TO LEVERAGE CANCER COMPLEXITIES
Experimental systems for modeling cancer and its complexity
Genetically engineered (and traditional cancer cell transplant) mouse models of human cancer have for 40 years illuminated mechanisms of multistep tumorigenesis and malignant progression, and increasingly, responses and adaptive resistance to therapies, the latter enabled by the possibility of assembling age- and disease-matched cohorts to conduct preclinical trials.220-222 However, the spectrum of cancer complexities discussed in this review cannot be fully addressed in such models, including, for example, assessing the impact of gene-environment (GxE) interactions. The study of large human cohorts, such as the UK Biobank and “All of Us,” has made us aware of the significant impact of confounding factors, including population structure, ethnic background, and environmental exposures, on the outcomes of genetic analyses and therapeutic targeting. This simple lesson, which is by its nature largely observational, has not yet been effectively embraced by bio-medical experimentalists. It is therefore not surprising that experiments in inbred animals, aimed to support the causal and mechanistic links between genotypes and phenotypes suggested by human genetics, can fail to translate into humans. Such genetically constrained experimental models have raised debate about the limitations of animal models for cancer research and have fueled the notion that humans are the best model to study human disease, despite the reality that performing experiments to fully address underlying mechanisms are very difficult if not impossible in humans.223 Looking forward, in order to address the challenges of cancer complexities and the translation of their illumination to better treatments for human cancers, we envisage increasing refinement and utilization of other model systems, both in genetically diverse mouse models and in ex vivo models involving human cells and tissues.
In one approach to modeling GxE effects in cancer, mouse genetic diversity can be embraced. Such diversity can be achieved through studies in genetic reference populations (GRP), such as the BXD and the Collaborative Cross GRPs224,225 or in outbred mice, such as the diversity outbred mice,226 that collectively capture ~90% of the genetic variation in laboratory mice, surpassing the genetic diversity present in humans. Such genetically diverse mice can be used not only to study genetic perturbations that are protective or predisposing to cancer development (e.g., generating F1 animals of a GRP crossed with a strain carrying an oncogene) as well as to study environmental modifiers ranging from dietary challenges, stressors, to pharmacological interventions.227 Such longitudinal population studies in mice have the prospect to (1) model the genetic diversity present in human populations, (2) study GxE interactions, (3) allow access to internal tumors and tissues for molecular characterization, and (4) map genes responsible for a given phenotype through quantitative trait locus (QTL) analysis. An unresolved technical challenge for the future will be to develop efficient means for genetic engineering of outbred cancer models to produce the remarkable diversity of organ-specific tumors carrying the same oncogenic mutations as cognate human cancers in cohorts amenable to preclinical trials. Certainly, the cancer field has much to gain by embracing genetic diversity to model tumor initiation and progression, and to use these models for preclinical trials of new cancer drugs and mechanism-guided combinations, advancing knowledge and its application to cancer medicine.
Beyond all the myriad variations of mouse models of human cancer, we envisage that ex vivo systems involving human cells and tissues will be increasingly refined and creatively applied to elucidate the mechanisms underlying cancer complexity and develop “precision/personalized” assays both to test therapeutic hypotheses and to guide treatment decisions for individual cancer patients. Once it became evident that tissue stem cells, alone or with stromal cells, could be propagated ex vivo as tissue-like structures in 3D matrix,228,229 organoid cultures have grown in use as a bridge between simplistic 2D cultures and the complexity of in vivo human biology. Organoids offer approaches to propagate tumors ex vivo in three dimensions, representing the pathological features of the parental tumor, therefore expanding the number of patient-derived model systems amenable to experimentation aiming to complement observations from mouse models. Organoids can be established from either adult tissue- or cancer-cell-specific stem cells as well as pluripotent stem cells (either induced or embryonic),230 although the latter have the additional challenge of faithfully recapitulating the biochemical signals and spatial organization of tissue/tumor development.
Notwithstanding the challenge of intratumor heterogeneity and tissue sampling bias where subclonal events may not be well represented in ex vivo propagated tumor models from patients, patient-derived organoids have been shown to maintain tumor clonal events,231 and their transcriptomic signatures can be similar to the tumors from which they were derived.232 Recent progress has been made in developing organoids to more effectively mirror the TME observed in human cancers, with lymphocyte/tumor organoid co-culture methods under development,233 as are means to create a functional vasculature. Another limitation of the organoid approach in modeling cancer is the potential lack of cancer-specific ECM and the physical microenvironment (stiffness, interstitial fluid pressure, vascularization, etc.). Currently most organoid culture approaches utilize Matrigel, collagen, or a mixture of the two. Leveraging the field of bioengineering, many synthetic materials are being developed that allow the generation of more controlled environments enabling the modulation of tissue stiffness.234 We envisage future development of “tumoroids” that more fully recapitulate all of these salient features of the bona fide TME.
Tumor-derived explants and organ-on-chip technologies offer additional solutions to these challenges, which may enhance cancer drug discovery and development by permitting appropriate and diverse human models of cancer with representative TMEs. Organ-on-chip technologies provide more elaborate models of disease, employing microfluidics to model tissue interfaces and to begin to recapitulate the complex TME, allowing the investigation of tumor/stromal interactions and anti-cancer drug testing235-237 Patient-derived tumor explants are initiated by culturing fragments or thick (vibratome) sections of fresh tumor tissues, permitting the short-term maintenance of the histopathological features of the parental tumors. Since tumor spatial morphology can be maintained, such approaches may offer the ability to combine ex vivo drug testing with biomarker analysis to identify predictors of drug response, and to decipher the impact of the TME on drug response and resistance.238
Patient-derived xenograft (PDX) mice also facilitate in vivo testing of drugs in the context of a human tumor in an immunodeficient background; partially “humanized” PDX mice bearing some features of the human immune system have further extended their use to evaluate immunotherapies,239 although further refinement will be needed to fully model the complexity and polymorphic diversity of human immune response to human tumors in mouse hosts. Moreover, PDX models are cumbersome, expensive, and, in many cases, unable to replicate the complexity and heterogeneity of human tumors and their microenvironments. Patient-derived tumor fragments (PDTFs) address some of these issues by allowing ex vivo testing of the impact of novel drugs in multiple tumor fragments obtained from patients.240,241 The PDTF platform allows perturbation of fresh or frozen tumor fragments retaining the microenvironment of the original tumor, albeit only briefly. Crucially, the PDTF response ex vivo to checkpoint inhibition was shown in one study to be highly concordant with the clinical response of the same patient to anti-programmed cell death protein 1 (anti-PD1) agents, illustrating the potential value of this platform.242 While still limited in some respects (such as short-term viability of the fragments and loss of innate immune cells and functional vasculature), we envisage that further improvements in this platform will significantly contribute to our understanding of responses (and resistance) to cancer therapeutics within the complexity and heterogeneity of human tumors.
Exploiting the other side of oncogene addiction
Studying resistance to targeted therapies over the past two decades has revealed that re-activation of the drug-inhibited signaling pathway by secondary mutations is a dominant mechanism of resistance. By inhibiting signals to which the cancer cell is addicted, we can steer the cancer cell to evolve into the direction of higher signaling. This begs the question whether we can also steer cancer cells in the opposite direction, of evolving toward a less malignant phenotype through reduced oncogenic signaling. In considering this concept, it is important to realize that cancer cells strive to optimize rather than maximize mitogenic signaling. Consistent with this, emerging evidence indicates that further stimulation of mitogenic signaling in cancer cells can be as toxic as the inhibition of these signals, by further activating cellular stress response pathways.243 Indeed, a combination of hyperactivation of oncogenic signaling and inhibition of the associated stress responses is very effective in animal models of colon cancer. Remarkably, resistance to this combination was associated with downregulation of oncogenic signaling and loss of oncogenic capacity.244 An obvious concern in such a therapeutic approach is the presence of many apparently normal cells in the aging soma that harbor oncogenic mutations (see tumor promotion section). Such cells may be stimulated to expand by drugs that further activate oncogenic signaling. However, in animal models of precancerous intestinal adenomas, a further increase in Wnt signaling by treatment with lithium chloride was associated with a reduction in adenoma formation rather than an increase, indicating that also in a pre-cancer state, there is a “goldilocks” scenario in which more oncogenic signal is not better.245 Consistently, epidemiological studies show that long-term treatment of bipolar disorder patients with lithium chloride is associated with a reduced risk of colorectal cancer.246 Although the risks and hurdles of this paradoxical approach must be addressed, steering cancer cells toward a less malignant phenotype through deliberate hyperactivation of oncogenic signaling holds promise for future cancer therapy.
TRANSLATING DISCOVERIES TO PATIENTS: NEXT GENERATION CLINICAL TRIALS
Many basic science discoveries inform us about the early steps of tumorigenesis. There is, therefore, an opportunity to develop clinical trials testing drugs in early-stage cancers and prevention platforms. Two distinct types of preoperative trials represent attractive designs for drug development and clinical translation. First, neoadjuvant clinical trials evaluate the efficacy of drugs administered with therapeutic intent several months before surgery. Those trials also allow for the investigation of the mechanisms of resistance.247 Second, window-of-opportunity trials are not designed to assess drug efficacy but are particularly appealing to explore the mechanisms of action and the pharmacodynamics of new drugs administered for a short duration (typically a few days) prior to the initiation of cancer therapy.248 Moreover, in a prevention setting, the categorization of studies as primary, secondary, and tertiary is an appealing framework that may prove useful for future drug development if endorsed by scientific societies.249 Primary chemoprevention studies describe the use of medicines to prevent cancer in high-risk individuals. Precise definitions of clinical, genomic, demographic, and environmental factors capable of highlighting at-risk patient populations will be required to enable such studies. This capability might be achieved through combined epidemiological and preclinical studies, facilitated by improving capabilities for clinical data collection, including digital AI-facilitated tumor pathology.21,250 Novel biological endpoints that capture treatment effects associated with impaired cancer development will be necessary as early indicators of treatment benefit. Secondary chemoprevention studies refer to evaluating potential interventions in patients with established pre-malignant conditions. There is a need for prospective randomized evaluation of promising agents from the preclinical space in this therapeutic setting for multiple cancer indications.251 Tertiary chemoprevention studies involve approaches to reduce the risk of cancer relapse, locally or at other organ sites. In this category, circulating tumor DNA will have an increasing role as a tool to non-invasively detect minimal residual disease (MRD) as a biomarker and an early endpoint of therapeutic efficacy, as this technology is capable of detecting and determining treatment response in micrometastatic disease that is not identifiable by clinical imaging.252-256
Since cancer is a disease driven by biological mechanisms, there is a need to establish new classification systems based on the biology of the disease, in addition to the current ones based on the organ of origin. Multi-omic classifications could dramatically accelerate the development of drugs targeting biological pathways through organ-agnostic clinical trials.257 The previous sections of this review have shown that cancer is a complex disease driven by multiple and interrelated biological mechanisms. These complexities should therefore be integrated into portraits that recapitulate the complete biology of each cancer patient. Trials testing the clinical utility of these multiplex, comprehensive molecular portraits of cancer are being developed,258 although at present validating the utility of individual components of the multiplex analyses on an extended number of patients is a challenge. Moreover, the future of cancer therapy is almost certainly one of combinatorial multi-targeting with distinctive classes of cancer drugs that disrupt tumor-driving mechanisms as well as systemic manifestations. Therefore, as knowledge progresses about molecular and cellular mechanisms driving the complexities of cancer and their pathogenic effects, these and other innovative clinical (and preclinical) therapeutic trial designs will be instrumental in translating the results to the benefit of cancer patients.
CONCLUDING REMARKS
Improving the quantity of life for cancer patients remains an overarching goal of cancer research. Looking forward, improving the quality and duration of life (“cancer without disease”), decreasing the burden of symptoms, and improving healthcare sustainability, affordability, and accessibility face monumental yet ultimately tractable challenges, enabled by embracing and better understanding the complex biology of the disease, locally and systemically, as elaborated herein.
Despite progress in our understanding of the disease, it is estimated that cancer incidence will increase by 20%–50% over the next 20 years, reflecting in part aging populations, increased obesity and the multi-faceted environmental factors impinging on the inception and malignant progression of cancer, a reality mandating ongoing investment and redoubled efforts to address its complexities. Furthermore, the incidence of early-onset cancers is increasing,259 for reasons that are unclear. An overarching issue related to the new era of addressing cancer’s complexities involves the systemic manifestations of the disease and the impact of environmental exposures and influences—from well-recognized and cryptic pollution to unhealthy diets, microbiota, and beyond—to the course of multistep tumorigenesis, malignant progression, and therapy resistance across the spectrum of human cancers. Successfully elucidating and addressing these complexities will benefit from the recruitment into the cancer research enterprise of scientists and clinical academics with expertise in areas outside of oncology such as data science, mathematical modeling, neuroscience, inflammation and autoimmunity, aging, metabolism, endocrinology, and muscle physiology. This expansion in expertise and capabilities will require new funding and research opportunities that cross disciplines and international borders and move beyond the traditional (largely governmental) funding mechanisms, analogous to the philanthropically-based CRUK/NCI Cancer Grand Challenges Program, specifically aimed at encouraging multi-disciplinary collaborations to address the plethora of questions delineated in this review. Furthermore, at a time when commitments to clinical academic training and career paths thereafter is under threat around the world, there is a pressing need to reinvigorate clinical academic MD/PhD training programs, along with post-MD research fellowships in lieu of formal PhDs, as well as innovative initiatives and funding mechanisms to “buy” protected time for physician-scientists to substantively engage in translational research.
Understandably, much effort has been focused on achieving “cures” for this disease; while an appropriate strategic goal, the reality of neo-Darwinian adaptation of cancer cells, enabled by genome instability and mutational evolution as well as epigenetic plasticity, suggest that all forms of cancer may never be completely curable. Optimistically, however, elucidating and targeting the mechanisms that enable cancer cells’ phenotypic plasticity and flexibility through tumorigenesis, malignant progression, and adaptive resistance to therapy may provide opportunities to transform cancer into a chronic, largely asymptomatic disease, increasing survival and improving the quality of life of patients with cancer. Connections between aging and cancer extend beyond the accumulation of mutations. Understanding how organ/tissue and tumor landscapes change with age and with diverse environmental exposures, and how such changes influence all stages of cancer evolution, from selection of initiated clones to metastasis, will be essential for winning more battles and maybe even wars on cancer.
Over the next two decades, as the world is faced with an aging population in which cancer will touch at least one in three and global incidence is set to rise, this review may help to provide a logical framework for initiatives aimed to address the clouds of complexity described herein, aiming to increasingly prevent the inception of cancer, to reduce the incidence of symptomatic cancer, as well as to diagnose cancers earlier, collectively alleviating suffering and prolonging the quality and quantity of life for those suffering from this daunting disease.
ACKNOWLEDGMENTS
CRUK thanks Alison Howe for her generosity in supporting the creation of the CRUK Marshall Symposium series and Lucy Stuart for coordinating the organization of this workshop. J.A. was supported by Ecole Polytechnique Fédérale de Lausanne (EPFL), and grants from the European Research Council (ERC-AdG-787702) and the Swiss National Science Foundation (SNSF 31003A_179435). A.B. is supported by National Cancer Institutes (NCI) grant R35CA210018; Barbara Bass Bakar Professorship of Cancer Genetics; Cancer Research UK/NCI PROMINENT Cancer Grand Challenge Award (Project Number: OT2CA278665 and CGCATF-2021/100006), D.B.-S. NIH/National Cancer Institute (NCI) grant R35CA210263. R.B. was supported by ERC-787925. J.D. was supported by R01AG067584. A.E. was supported by ERC818943, ISF860/18, and ICRF 837124. M.A.F. was supported by an Investigator Grant APP1194141 from the National Health & Medical Research Council of Australia (NHMRC). A.H. was supported by 1R01AI165661. J.A.J. was supported by the Swiss National Science Foundation Advanced Grant TMAG-3_209224, Ludwig Institute for Cancer Research, and the University of Lausanne. K.L. was supported by CA271500. I.M. was supported by ERC CoGH2020-725492; Francis Crick Institute/Wellcome Trust CC2051; Francis Crick Institute/UK Medical Research Council CC2051; Francis Crick Institute/Cancer Research UK CC2051. K.H.V. was supported by Cancer Research UK grant C596/A26855; Francis Crick Institute/Wellcome Trust CC2073; Francis Crick Institute/UK Medical Research Council CC2073; Francis Crick Institute/Cancer Research UK. E.W. was supported by the CRUK/NCI Cancer Grand Challenge CANCAN award, the Ludwig Institute for Cancer Research, NCI grants CA163591 and CA243547, and the Fifth Generation Foundation. F.W. was supported by the Deutsche Forschungsgemeinschaft (SFB 1389, UNITE Glioblastoma). D.H. was supported by the Ludwig Institute for Cancer Research.
Footnotes
DECLARATION OF INTERESTS
C.A. reports employment at Saga Diagnostics and reports options to own stock in the company. C.A. and C.S. are listed as inventors on a European patent application relating to assay technology to detect tumor recurrence (PCT/GB2017/053289). This patent has been licensed to commercial entities and, under their terms of employment, C.A. and C.S. are due a revenue share of any revenue generated from such license(s). C.A. and C.S. declare a patent application (PCT/US2017/028013) for methods to detect lung cancer. C.A. and C.S. are named inventors on a patent application to determine methods and systems for tumor monitoring (PCT/EP2022/077987). C.A. and C.S. are named inventors on a provisional patent protection related to a ctDNA detection algorithm.
J.A.J. has received honoraria for speaking at research symposia organized by Bristol Meyers Squibb and Glenmark Pharmaceuticals and previously served on the SAB of Pionyr Immunotherapeutics.
D.H. is a founder and member of the BoD and SAB of Opna Bio, and a member of the SABs of Pfizer, Cellestia Biotech, 4D Molecular Therapeutics, and AtG Therapeutics.
F.A. reports travel/accommodation/expenses from AstraZeneca, GlaxoSmithKline, Novartis, Pfizer, and Roche, and his institution has received research funding from AstraZeneca, Daiichi Sankyo, Lilly, Novartis, Pfizer, and Roche. C.M. Consultant/Advisory fees from Amgen, Astellas, AstraZeneca, Bayer, BeiGene, BMS, Celgene, Debiopharm, Genentech, Ipsen, Janssen, Lilly, MedImmune, MSD, Novartis, Pfizer, Roche, Sanofi, Orion. Principal/sub-Investigator of Clinical Trials for AbbVie, Aduro, Agios, Amgen, Argen-x, Astex, AstraZeneca, Aveo pharmaceuticals, Bayer, Beigene, Blueprint, BMS, Boeringer Ingelheim, Celgene, Chugai, Clovis, Daiichi Sankyo, Debiopharm, Eisai, Eos, Exelixis, Forma, Gamamabs, Genentech, Gortec, GSK, H3 biomedecine, Incyte, InnatePharma, Janssen, Kura Oncology, Kyowa, Lilly, Loxo, Lysarc, Lytix Biopharma, Medimmune, Menarini, Merus, MSD, Nanobiotix, Nektar Therapeutics, Novartis, Octimet, Oncoethix, Oncopeptides AB, Orion, Pfizer, Pharmamar, Pierre Fabre, Roche, Sanofi, Servier, Sierra Oncology, Taiho, Takeda, Tesaro, and Xencor.
R.B. is founder of Oncosence.
M.A.F. is founder and shareholder of Celesta Therapeutics.
A.H. is a paid consultant for Calida Therapeutics.
M.J.-H. has consulted for, and is a member of, the Achilles Therapeutics Scientific Advisory Board and Steering Committee, has received speaker honoraria from Pfizer, Astex Pharmaceuticals, Oslo Cancer Cluster, Bristol Myers Squibb, and is listed as a co-inventor on a European patent application relating to methods to detect lung cancer PCT/US2017/028013), this patent has been licensed to commercial entities and, under terms of employment, M.J.-H. is due a share of any revenue generated from such license(s).
J.W.L. advises Restoration Foodworks, Nanocare Technologies, and Cornerstone Pharmaceuticals.
S.L. received research funding to institution from Novartis, Bristol Myers Squibb, MSD, Puma Biotechnology, Eli Lilly, Nektar Therapeutics, AstraZeneca/Daiichi Sankyo, and Seattle Genetics. S.L. has acted as consultant (not compensated) to Seattle Genetics, Novartis, Bristol Myers Squibb, MSD, AstraZeneca/Daiichi Sankyo, Eli Lilly, Pfizer, Gilead Therapeutics, Nektar Therapeutics, PUMA Biotechnologies and Roche-Genentech. S.L. has acted as consultant (paid to institution) to Novartis, GlaxoSmithKline, Roche-Genentech, AstraZeneca/Daiichi Sankyo, Pfizer, Gilead Therapeutics, Seattle Genetics, MSD, Tallac Therapeutics, Eli Lilly, and Bristol Myers Squibb.
I.M. is a consultant of LIfT BioSciences.
K.M. has received honoraria from LEO Pharma, Pfizer and Bayer PLC. She has received research funding from LEO Pharma and Bayer PLC.
S.Q. is a founder, CSO, and holds stock options in Achilles Therapeutics.
C.S. acknowledges grants from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Pfizer, Roche-Ventana, Invitae (previously Archer Dx Inc—collaboration in minimal residual disease sequencing technologies), Ono Pharmaceutical, and Personalis. He is Chief Investigator for the AZ MeRmaiD 1 and 2 clinical trials and is the Steering Committee Chair. He is also Co-Chief Investigator of the NHS Galleri trial funded by GRAIL and a paid member of GRAIL’s Scientific Advisory Board. He receives consultant fees from Achilles Therapeutics (also SAB member), Bicycle Therapeutics (also a SAB member), Genentech, Medicxi, China Innovation Centre of Roche (CI-CoR) formerly Roche Innovation Centre—Shanghai, Metabomed (until July 2022), Relay Therapeutics, and the Sarah Cannon Research Institute. C.S. has received honoraria from Amgen, AstraZeneca, Bristol Myers Squibb, GlaxoSmithKline, Illumina, MSD, Novartis, Pfizer, and Roche-Ventana. C.S. has previously held stock options in Apogen Biotechnologies and GRAIL, and currently has stock options in Epic Bioscience, Bicycle Therapeutics, and has stock options and is co-founder of Achilles Therapeutics. Patents: C.S. declares a patent application (PCT/US2017/028013) for methods to lung cancer); targeting neoantigens (PCT/EP2016/059401); identifying patent response to immune checkpoint blockade (PCT/EP2016/071471), determining HLA LOH (PCT/GB2018/052004); predicting survival rates of patients with cancer (PCT/GB2020/050221); identifying patients who respond to cancer treatment (PCT/GB2018/051912); methods for lung cancer detection (US20190106751A1). C.S. is an inventor on a European patent application (PCT/GB2017/053289) relating to assay technology to detect tumor recurrence. This patent has been licensed to a commercial entity and under their terms of employment C.S. is due a revenue share of any revenue generated from such license(s).
K.H.V. is on the board of directors and shareholder of Bristol Myers Squibb and on the science advisory board (with stock options) of PMV Pharma, RAZE Therapeutics, Volastra Pharmaceuticals and Kovina Therapeutics. She is on the SAB of Ludwig Cancer and a co-founder and consultant of Faeth Therapeutics. She has been in receipt of research funding from Astex Pharmaceuticals and AstraZeneca and contributed to CRUK Cancer Research Technology filing of patent application WO/2017/144877.
J.W. is an inventor on a US patent application (PCT/US17/53.717) submitted by the University of Texas MD Anderson Cancer Center which covers methods to enhance immune checkpoint blockade responses by modulating the microbiome, reports compensation for speaker’s bureau and honoraria from Imedex, Dava Oncology, Omniprex, Illumina, Gilead, PeerView, Physician Education Resource, MedImmune, Exelixis and Bristol Myers Squibb, and has served as a consultant/advisory board member for Roche/Genentech, Novartis, AstraZeneca, GlaxoSmithKline, Bristol Myers Squibb, Micronoma, OSE therapeutics, Merck, and Everimmune. Dr. Wargo receives stock options from Micronoma and OSE therapeutics.
A.W. is on the board of Regain Therapeutics.
In memory of Professor Chris Marshall FRS 1949–2015.
REFERENCES
- 1.Wishart DS (2015). Is Cancer a Genetic Disease or a Metabolic Disease? EBiomedicine 2, 478–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, and Weinberg RA (2000). The hallmarks of cancer. Cell 100, 57–70. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D. (2022). Hallmarks of Cancer: New Dimensions. Cancer Discov. 12, 31–46. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, and Monje M (2023). Cancer hallmarks intersect with neuroscience in the tumor microenvironment. Cancer Cell 41, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brennan P, and Davey-Smith G (2022). Identifying Novel Causes of Cancers to Enhance Cancer Prevention: New Strategies Are Needed. J. Natl. Cancer Inst 114, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clocchiatti A, Cora E, Zhang Y, and Dotto GP (2016). Sexual dimorphism in cancer. Nat. Rev. Cancer 16, 330–339. [DOI] [PubMed] [Google Scholar]
- 8.Haupt S, Caramia F, Klein SL, Rubin JB, and Haupt Y (2021). Sex disparities matter in cancer development and therapy. Nat. Rev. Cancer 21, 393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Lan Z, Liao W, Horner JW, Xu X, Liu J, Yoshihama Y, Jiang S, Shim HS, Slotnik M, et al. (2023). Histone demethylase KDM5D upregulation drives sex differences in colon cancer. Nature 619, 632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Özdemir BC, and Dotto GP (2019). Sex Hormones and Anticancer Immunity. Clin. Cancer Res 25, 4603–4610. [DOI] [PubMed] [Google Scholar]
- 11.Vellano CP, White MG, Andrews MC, Chelvanambi M, Witt RG, Daniele JR, Titus M, McQuade JL, Conforti F, Burton EM, et al. (2022). Androgen receptor blockade promotes response to BRAF/MEK-targeted therapy. Nature 606, 797–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martincorena I, and Campbell PJ (2015). Somatic mutation in cancer and normal cells. Science 349, 1483–1489. [DOI] [PubMed] [Google Scholar]
- 13.Fowler JC, and Jones PH (2022). Somatic Mutation: What Shapes the Mutational Landscape of Normal Epithelia? Cancer Discov. 12, 1642–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berenblum I, and Shubik P (1947). The role of croton oil applications, associated with a single painting of a carcinogen, in tumour induction of the mouse’s skin. Br. J. Cancer 1, 379–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedewald WF, and Rous P (1944). THE INITIATING AND PROMOTING ELEMENTS IN TUMOR PRODUCTION: AN ANALYSIS OF THE EFFECTS OF TAR, BENZPYRENE, AND METHYLCHOLANTHRENE ON RABBIT SKIN. J. Exp. Med 80, 101–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riva L, Pandiri AR, Li YR, Droop A, Hewinson J, Quail MA, Iyer V, Shepherd R, Herbert RA, Campbell PJ, et al. (2020). The mutational signature profile of known and suspected human carcinogens in mice. Nat. Genet 52, 1189–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP, and Barbacid M (2007). Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 11, 291–302. [DOI] [PubMed] [Google Scholar]
- 18.Bailleul B, Surani MA, White S, Barton SC, Brown K, Blessing M, Jorcano J, and Balmain A (1990). Skin hyperkeratosis and papilloma formation in transgenic mice expressing a ras oncogene from a suprabasal keratin promoter. Cell 62, 697–708. [DOI] [PubMed] [Google Scholar]
- 19.Alonso-Curbelo D, Ho YJ, Burdziak C, Maag JLV, Morris JP, Chandwani R, Chen HA, Tsanov KM, Barriga FM, Luan W, et al. (2021). A gene-environment-induced epigenetic program initiates tumorigenesis. Nature 590, 642–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, Lamprecht TL, Shen D, Hundal J, Fulton RS, et al. (2015). Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 518, 552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hill W, Lim EL, Weeden CE, Lee C, Augustine M, Chen K, Kuan FC, Marongiu F, Evans EJ, Moore DA, et al. (2023). Lung adenocarcinoma promotion by air pollutants. Nature 616, 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kadariya Y, Menges CW, Talarchek J, Cai KQ, Klein-Szanto AJ, Pietrofesa RA, Christofidou-Solomidou M, Cheung M, Mossman BT, Shukla A, et al. (2016). Inflammation-Related IL1β/IL1R Signaling Promotes the Development of Asbestos-Induced Malignant Mesothelioma. Cancer Prev. Res. (Phila) 9, 406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moody S, Senkin S, Islam SMA, Wang J, Nasrollahzadeh D, Cortez Cardoso Penha R, Fitzgerald S, Bergstrom EN, Atkins J, He Y, et al. (2021). Mutational signatures in esophageal squamous cell carcinoma from eight countries with varying incidence. Nat. Genet 53, 1553–1563. [DOI] [PubMed] [Google Scholar]
- 24.Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andrén-Sandberg A, and Domellöf L (1993). Pancreatitis and the risk of pancreatic cancer. International pancreatitis study group. N. Engl. J. Med 328, 1433–1437. [DOI] [PubMed] [Google Scholar]
- 25.Collins RH Jr., Feldman M, and Fordtran JS (1987). Colon cancer, dysplasia, and surveillance in patients with ulcerative colitis. A critical review. N. Engl. J. Med 316, 1654–1658. [DOI] [PubMed] [Google Scholar]
- 26.George J, Prasad S, Mahmood Z, and Shukla Y (2010). Studies on glyphosate-induced carcinogenicity in mouse skin: a proteomic approach. J. Proteomics 73, 951–964. [DOI] [PubMed] [Google Scholar]
- 27.Luo H, and Ge H (2022). Hot Tea Consumption and Esophageal Cancer Risk: A Meta-Analysis of Observational Studies. Front. Nutr 9, 831567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naik S, Larsen SB, Gomez NC, Alaverdyan K, Sendoel A, Yuan S, Polak L, Kulukian A, Chai S, and Fuchs E (2017). Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 550, 475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. (2020). The repertoire of mutational signatures in human cancer. Nature 578, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanarek N, Petrova B, and Sabatini DM (2020). Dietary modifications for enhanced cancer therapy. Nature 579, 507–517. [DOI] [PubMed] [Google Scholar]
- 31.Bejarano L, Jordāo MJC, and Joyce JA (2021). Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 11, 933–959. [DOI] [PubMed] [Google Scholar]
- 32.Plaks V, Kong N, and Werb Z (2015). The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 16, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Debaugnies M, Rodríguez-Acebes S, Blondeau J, Parent MA, Zocco M, Song Y, de Maertelaer V, Moers V, Latil M, Dubois C, et al. (2023). RHOJ controls EMT-associated resistance to chemotherapy. Nature 616, 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Visser KE, and Joyce JA (2023). The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 41, 374–403. [DOI] [PubMed] [Google Scholar]
- 35.Cañellas-Socias A, Cortina C, Hernando-Momblona X, Palomo-Ponce S, Mulholland EJ, Turon G, Mateo L, Conti S, Roman O, Sevillano M et al. (2022). Metastatic recurrence in colorectal cancer arises from residual EMP1+ cells. Nature 611, 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, Sheng K, Niu Y, Goldstein A, Zhao N, et al. (2019). Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell Biol 21, 1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lavie D, Ben-Shmuel A, Erez N, and Scherz-Shouval R (2022). Cancer-associated fibroblasts in the single-cell era. Nat. Cancer 3, 793–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dai J, Cimino PJ, Gouin KH 3rd, Grzelak CA, Barrett A, Lim AR, Long A, Weaver S, Saldin LT, Uzamere A, et al. (2022). Astrocytic laminin-211 drives disseminated breast tumor cell dormancy in brain. Nat. Cancer 3, 25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Martino JS, Nobre AR, Mondal C, Taha I, Farias EF, Fertig EJ, Naba A, Aguirre-Ghiso JA, and Bravo-Cordero JJ (2022). A tumor-derived type III collagen-rich ECM niche regulates tumor cell dormancy. Nat. Cancer 3, 90–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fane ME, Chhabra Y, Alicea GM, Maranto DA, Douglass SM, Webster MR, Rebecca VW, Marino GE, Almeida F, Ecker BL, et al. (2022). Stromal changes in the aged lung induce an emergence from melanoma dormancy. Nature 606, 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ombrato L, Nolan E, Kurelac I, Mavousian A, Bridgeman VL, Heinze I, Chakravarty P, Horswell S, Gonzalez-Gualda E, Matacchione G, et al. (2019). Metastatic-niche labelling reveals parenchymal cells with stem features. Nature 572, 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, Brouland JP, Iacobuzio-Donahue CA, Brennan C, Tabar V, et al. (2020). Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 181, 1643–1660.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Álvarez-Prado ÁF, Maas RR, Soukup K, Klemm F, Kornete M, Krebs FS, Zoete V, Berezowska S, Brouland JP, Hottinger AF, et al. (2023). Immunogenomic analysis of human brain metastases reveals diverse immune landscapes across genetically distinct tumors. Cell Rep. Med 4, 100900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Risom T, Glass DR, Averbukh I, Liu CC, Baranski A, Kagel A, McCaffrey EF, Greenwald NF, Rivero-Gutiérrez B, Strand SH, et al. (2022). Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stroma. Cell 185, 299–310.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jackson HW, Fischer JR, Zanotelli VRT, Ali HR, Mechera R, Soysal SD, Moch H, Muenst S, Varga Z, Weber WP, et al. (2020). The single-cell pathology landscape of breast cancer. Nature 578, 615–620. [DOI] [PubMed] [Google Scholar]
- 46.Schürch CM, Bhate SS, Barlow GL, Phillips DJ, Noti L, Zlobec I, Chu P, Black S, Demeter J, McIlwain DR et al. (2020). Coordinated Cellular Neighborhoods Orchestrate Antitumoral Immunity at the Colorectal Cancer Invasive Front. Cell 183, 838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sammut SJ, Crispin-Ortuzar M, Chin SF, Provenzano E, Bardwell HA, Ma W, Cope W, Dariush A, Dawson SJ, Abraham JE, et al. (2022). Multi-omic machine learning predictor of breast cancer therapy response. Nature 601, 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Z, Zhao Y, Kong P, Liu Y, Huang J, Xu E, Wei W, Li G, Cheng X, Xue L, et al. (2023). Integrated multi-omics profiling yields a clinically relevant molecular classification for esophageal squamous cell carcinoma. Cancer Cell 41, 181–195.e9. [DOI] [PubMed] [Google Scholar]
- 49.Croci D, Santalla Méndez R, Temme S, Soukup K, Fournier N, Zomer A, Colotti R, Wischnewski V, Flögel U, van Heeswijk RB, et al. (2022). Multispectral fluorine-19 MRI enables longitudinal and noninvasive monitoring of tumor-associated macrophages. Sci. Transl. Med 14, eabo2952. [DOI] [PubMed] [Google Scholar]
- 50.Dmitrieva-Posocco O, Dzutsev A, Posocco DF, Hou V, Yuan W, Thovarai V, Mufazalov IA, Gunzer M, Shilovskiy IP, Khaitov MR, et al. (2019). Cell-Type-Specific Responses to Interleukin-1 Control Microbial Invasion and Tumor-Elicited Inflammation in Colorectal Cancer. Immunity 50, 166–180.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. (2017). Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, Dörr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA, et al. (2018). Senescence-associated reprogramming promotes cancer stemness. Nature 553, 96–100. [DOI] [PubMed] [Google Scholar]
- 53.Cai Z, Aguilera F, Ramdas B, Daulatabad SV, Srivastava R, Kotzin JJ, Carroll M, Wertheim G, Williams A, Janga SC, et al. (2020). Targeting Bim via a lncRNA Morrbid Regulates the Survival of Preleukemic and Leukemic Cells. Cell Rep. 31, 107816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biavasco R, Lettera E, Giannetti K, Gilioli D, Beretta S, Conti A, Scala S, Cesana D, Gallina P, Norelli M, et al. (2021). Oncogene-induced senescence in hematopoietic progenitors features myeloid restricted hematopoiesis, chronic inflammation and histiocytosis. Nat. Commun 12, 4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, Betz KS, Penz-Oesterreicher M, Bjorkdahl O, Fox JG, et al. (2008). Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 14, 408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al. (2014). STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. (2014). STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahn J, Xia T, Rabasa Capote A, Betancourt D, and Barber GN (2018). Extrinsic Phagocyte-Dependent STING Signaling Dictates the Immunogenicity of Dying Cells. Cancer Cell 33, 862–873.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP, et al. (2015). Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350, 1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, et al. (2015). Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, et al. (2018). Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, Luke JJ, and Gajewski TF (2018). The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xing C, Wang M, Ajibade AA, Tan P, Fu C, Chen L, Zhu M, Hao ZZ, Chu J, Yu X, et al. (2021). Microbiota regulate innate immune signaling and protective immunity against cancer. Cell Host Microbe 29, 959–974.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsay J-CJ, Wu BG, Sulaiman I, Gershner K, Schluger R, Li Y, Yie TA, Meyn P, Olsen E, Perez L, et al. (2021). Lower Airway Dysbiosis Affects Lung Cancer Progression. Cancer Discov. 11, 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Villemin C, Six A, Neville BA, Lawley TD, Robinson MJ, and Bakdash G (2023). The heightened importance of the microbiome in cancer immunotherapy. Trends Immunol. 44, 44–59. [DOI] [PubMed] [Google Scholar]
- 66.Zhao H, Teng D, Yang L, Xu X, Chen J, Jiang T, Feng AY, Zhang Y, Frederick DT, Gu L, et al. (2022). Myeloid-derived itaconate suppresses cytotoxic CD8+ T cells and promotes tumour growth. Nat. Metab 4, 1660–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Low V, Li Z, and Blenis J (2022). Metabolite activation of tumorigenic signaling pathways in the tumor microenvironment. Sci. Signal 15, eabj4220. [DOI] [PubMed] [Google Scholar]
- 68.Juarez D, and Fruman DA (2021). Targeting the Mevalonate Pathway in Cancer. Trends Cancer 7, 525–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen YJ, Li GN, Li XJ, Wei LX, Fu MJ, Cheng ZL, Yang Z, Zhu GQ, Wang XD, Zhang C, et al. (2023). Targeting IRG1 reverses the immunosuppressive function of tumor-associated macrophages and enhances cancer immunotherapy. Sci. Adv 9, eadg0654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Messmer MN, Netherby CS, Banik D, and Abrams SI (2015). Tumor-induced myeloid dysfunction and its implications for cancer immunotherapy. Cancer Immunol. Immunother 64, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gabrilovich DI, and Nagaraj S (2009). Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol 9, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, Littlewood TD, and Evan GI (2017). Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 171, 1301–1315.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rahib L, Wehner MR, Matrisian LM, and Nead KT (2021). Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 4, e214708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fane M, and Weeraratna AT (2020). How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 20, 89–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rozhok AI, and DeGregori J (2015). Toward an evolutionary model of cancer: Considering the mechanisms that govern the fate of somatic mutations. Proc. Natl. Acad. Sci. USA 112, 8914–8921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chhabra Y, and Weeraratna AT (2023). Fibroblasts in cancer: unity in heterogeneity. Cell 186, 1580–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hoeijmakers JHJ (2009). DNA damage, aging, and cancer. N. Engl. J. Med 361, 1475–1485. [DOI] [PubMed] [Google Scholar]
- 78.Marongiu F, and DeGregori J (2022). The sculpting of somatic mutational landscapes by evolutionary forces and their impacts on agingrelated disease. Mol. Oncol 16, 3238–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ahmad H, Jahn N, and Jaiswal S (2023). Clonal Hematopoiesis and Its Impact on Human Health. Annu. Rev. Med 74, 249–260. [DOI] [PubMed] [Google Scholar]
- 80.Olafsson S, McIntyre RE, Coorens T, Butler T, Jung H, Robinson PS, Lee-Six H, Sanders MA, Arestang K, Dawson C, et al. (2020). Somatic Evolution in Non-neoplastic IBD-Affected Colon. Cell 182, 672–684.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nanki K, Fujii M, Shimokawa M, Matano M, Nishikori S, Date S, Takano A, Toshimitsu K, Ohta Y, Takahashi S, et al. (2020). Somatic inflammatory gene mutations in human ulcerative colitis epithelium. Nature 577, 254–259. [DOI] [PubMed] [Google Scholar]
- 82.Kakiuchi N, Yoshida K, Uchino M, Kihara T, Akaki K, Inoue Y, Kawada K, Nagayama S, Yokoyama A, Yamamoto S, et al. (2020). Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature 577, 260–265. [DOI] [PubMed] [Google Scholar]
- 83.Fowler JC, King C, Bryant C, Hall MWJ, Sood R, Ong SH, Earp E, Fernandez-Antoran D, Koeppel J, Dentro SC, et al. (2021). Selection of Oncogenic Mutant Clones in Normal Human Skin Varies with Body Site. Cancer Discov. 11, 340–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, Shiozawa Y, Sato Y, Aoki K, Kim SK, et al. (2019). Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 565, 312–317. [DOI] [PubMed] [Google Scholar]
- 85.Jaiswal S, and Ebert BL (2019). Clonal hematopoiesis in human aging and disease. Science 366, eaan4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kerr J, Anderson C, and Lippman SM (2017). Physical activity, sedentary behaviour, diet, and cancer: an update and emerging new evidence. Lancet Oncol. 18, e457–e471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, Ferrucci L, Gilroy DW, Fasano A, Miller GW, et al. (2019). Chronic inflammation in the etiology of disease across the life span. Nat. Med 25, 1822–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Turrell FK, Orha R, Guppy NJ, Gillespie A, Guelbert M, Starling C, Haider S, and Isacke CM (2023). Age-associated microenvironmental changes highlight the role of PDGF-C in ER+ breast cancer metastatic relapse. Nat. Cancer 4, 468–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kiemen AL, Braxton AM, Grahn MP, Han KS, Babu JM, Reichel R, Jiang AC, Kim B, Hsu J, Amoa F, et al. (2022). CODA: quantitative 3D reconstruction of large tissues at cellular resolution. Nat. Methods 19, 1490–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Berry S, Giraldo NA, Green BF, Cottrell TR, Stein JE, Engle EL, Xu H, Ogurtsova A, Roberts C, Wang D, et al. (2021). Analysis of multispectral imaging with the AstroPath platform informs efficacy of PD-1 blockade. Science 372, eaba2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Drapela S, and Gomes AP (2023). The aging lung reawakens dormant tumor cells. Nat. Cancer 4, 442–443. [DOI] [PubMed] [Google Scholar]
- 92.Wang L, Lankhorst L, and Bernards R (2022). Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 22, 340–355. [DOI] [PubMed] [Google Scholar]
- 93.Jochems F, Thijssen B, De Conti G, Jansen R, Pogacar Z, Groot K, Wang L, Schepers A, Wang C, Jin H, et al. (2021). The Cancer SENESCopedia: A delineation of cancer cell senescence. Cell Rep. 36, 109441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, et al. (2017). Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 7, 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, and van Deursen JM (2011). Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. (2016). Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang L, Jin H, Jochems F, Wang S, Lieftink C, Martinez IM, De Conti G, Edwards F, de Oliveira RL, Schepers A, et al. (2022). cFLIP suppression and DR5 activation sensitize senescent cancer cells to senolysis. Nat. Cancer 3, 1284–1299. [DOI] [PubMed] [Google Scholar]
- 98.Ruscetti M, Morris JP 4th, Mezzadra R, Russell J, Leibold J, Romesser PB, Simon J, Kulick A, Ho Y-J, Fennell M, et al. (2020). Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 181, 424–441.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, and Elledge SJ (2015). The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349, aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li F, and Simon MC (2020). Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor Microenvironments. Dev. Cell 54, 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kimmelman AC, and White E (2017). Autophagy and Tumor Metabolism. Cell Metab. 25, 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Al-Zoughbi W, Al-Zhoughbi W, Huang J, Paramasivan GS, Till H, Pichler M, Guertl-Lackner B, and Hoefler G (2014). Tumor macroenvironment and metabolism. Semin. Oncol 41, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li X, Yang Y, Zhang B, Lin X, Fu X, An Y, Zou Y, Wang JX, Wang Z, and Yu T (2022). Lactate metabolism in human health and disease. Signal Transduct. Target. Ther 7, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Zhan, Yanxiang Guo J, et al. (2017). Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Du Z, Zhang H, Feng Y, Zhan D, Li S, Tu C, Liu J, and Wang J (2023). Tumour-derived small extracellular vesicles contribute to the tumour progression through reshaping the systemic immune macroenvironment. Br. J. Cancer 128, 1249–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Goldman O, Adler LN, Hajaj E, Croese T, Darzi N, Galai S, Tishler H, Ariav Y, Lavie D, Fellus-Alyagor L, et al. (2023). Early Infiltration of Innate Immune Cells to the Liver Depletes HNF4α and Promotes Extrahepatic Carcinogenesis. Cancer Discov. 13, 1616–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Burfeind KG, Zhu X, Norgard MA, Levasseur PR, Huisman C, Buenafe AC, Olson B, Michaelis KA, Torres ER, Jeng S, et al. (2020). Circulating myeloid cells invade the central nervous system to mediate cachexia during pancreatic cancer. eLife 9, e54095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Argilés JM, López-Soriano FJ, Stemmler B, and Busquets S (2023). Cancer-associated cachexia - understanding the tumour macroenvironment and microenvironment to improve management. Nat. Rev. Clin. Oncol 20, 250–264. [DOI] [PubMed] [Google Scholar]
- 110.Pryce BR, Wang DJ, Zimmers TA, Ostrowski MC, and Guttridge DC (2023). Cancer cachexia: involvement of an expanding macroenvironment. Cancer Cell 41, 581–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Martin L, Birdsell L, Macdonald N, Reiman T, Clandinin MT, McCargar LJ, Murphy R, Ghosh S, Sawyer MB, and Baracos VE (2013). Cancer cachexia in the age of obesity: skeletal muscle depletion is a powerful prognostic factor, independent of body mass index. J. Clin. Oncol 31, 1539–1547. [DOI] [PubMed] [Google Scholar]
- 112.Baracos VE, Martin L, Korc M, Guttridge DC, and Fearon KCH (2018). Cancer-associated cachexia. Nat. Rev. Dis. Primers 4, 17105. [DOI] [PubMed] [Google Scholar]
- 113.Ferrer M, Anthony TG, Ayres JS, Biffi G, Brown JC, Caan BJ, Cespedes Feliciano EM, Coll AP, Dunne RF, Goncalves MD, et al. (2023). Cachexia: A systemic consequence of progressive, unresolved disease. Cell 186, 1824–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Neyroud D, Laitano O, Dasgupta A, Lopez C, Schmitt RE, Schneider JZ, Hammers DW, Sweeney HL, Walter GA, Doles J, et al. (2023). Blocking muscle wasting via deletion of the muscle-specific E3 ligase MuRF1 impedes pancreatic tumor growth. Commun. Biol 6, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ternes D, Tsenkova M, Pozdeev VI, Meyers M, Koncina E, Atatri S, Schmitz M, Karta J, Schmoetten M, Heinken A, et al. (2022). The gut microbial metabolite formate exacerbates colorectal cancer progression. Nat. Metab 4, 458–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pati S, Irfan W, Jameel A, Ahmed S, and Shahid RK (2023). Obesity and Cancer: A Current Overview of Epidemiology, Pathogenesis, Outcomes, and Management. Cancers 15, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schmitz KH, Campbell AM, Stuiver MM, Pinto BM, Schwartz AL, Morris GS, Ligibel JA, Cheville A, Galvão DA, Alfano CM, et al. (2019). Exercise is medicine in oncology: engaging clinicians to help patients move through cancer. CA Cancer J. Clin 69, 468–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McTiernan A. (2008). Mechanisms linking physical activity with cancer. Nat. Rev. Cancer 8, 205–211. [DOI] [PubMed] [Google Scholar]
- 119.Moore TM, Lee S, Olsen T, Morselli M, Strumwasser AR, Lin AJ, Zhou Z, Abrishami A, Garcia SM, Bribiesca J, et al. (2023). Conserved multi-tissue transcriptomic adaptations to exercise training in humans and mice. Cell Rep. 42, 112499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wei W, Riley NM, Lyu X, Shen X, Guo J, Raun SH, Zhao M, Moya-Garzon MD, Basu H, Sheng-Hwa Tung A, et al. (2023). Organism-wide, cell-type-specific secretome mapping of exercise training in mice. Cell Metab. 35, 1261–1279.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liberti MV, Dai Z, Wardell SE, Baccile JA, Liu X, Gao X, Baldi R, Mehrmohamadi M, Johnson MO, Madhukar NS, et al. (2017). A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product. Cell Metab. 26, 648–659.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Altea-Manzano P, Cuadros AM, Broadfield LA, and Fendt SM (2020). Nutrient metabolism and cancer in the in vivo context: a metabolic game of give and take. EMBO Rep. 21, e50635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tajan M, and Vousden KH (2020). Dietary Approaches to Cancer Therapy. Cancer Cell 37, 767–785. [DOI] [PubMed] [Google Scholar]
- 124.Locasale JW (2022). Diet and Exercise in Cancer Metabolism. Cancer Discov. 12, 2249–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Di Biase S, Lee C, Brandhorst S, Manes B, Buono R, Cheng CW, Cacciottolo M, Martin-Montalvo A, de Cabo R, Wei M, et al. (2016). Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 30, 136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Valdemarin F, Caffa I, Persia A, Cremonini AL, Ferrando L, Tagliafico L, Tagliafico A, Guijarro A, Carbone F, Ministrini S, et al. (2021). Safety and Feasibility of Fasting-Mimicking Diet and Effects on Nutritional Status and Circulating Metabolic and Inflammatory Factors in Cancer Patients Undergoing Active Treatment. Cancers 13, 4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, van den Broek NJF, Mackay GM, Labuschagne CF, Gay D, Kruiswijk F, et al. (2017). Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376. [DOI] [PubMed] [Google Scholar]
- 128.Gao X, Sanderson SM, Dai Z, Reid MA, Cooper DE, Lu M, Richie JP, Ciccarella A, Calcagnotto A, Mikhael PG, et al. (2019). Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 572, 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lien EC, Westermark AM, Zhang Y, Yuan C, Li Z, Lau AN, Sapp KM, Wolpin BM, and Vander Heiden MG (2021). Low glycaemic diets alter lipid metabolism to influence tumour growth. Nature 599, 302–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, Amadiume SC, Goncalves MD, Hodakoski C, Lundquist MR, et al. (2018). Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Taylor SR, Falcone JN, Cantley LC, and Goncalves MD (2022). Developing dietary interventions as therapy for cancer. Nat. Rev. Cancer 22, 452–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pariollaud M, and Lamia KA (2020). Cancer in the Fourth Dimension: What Is the Impact of Circadian Disruption? Cancer Discov. 10, 1455–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhang R, Lahens NF, Ballance HI, Hughes ME, and Hogenesch JB (2014). A circadian gene expression atlas in mammals: implications for biology and medicine. Proc. Natl. Acad. Sci. USA 111, 16219–16224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gotoh T, Vila-Caballer M, Santos CS, Liu J, Yang J, and Finkielstein CV (2014). The circadian factor Period 2 modulates p53 stability and transcriptional activity in unstressed cells. Mol. Biol. Cell 25, 3081–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chan AB, Parico GCG, Fribourgh JL, Ibrahim LH, Bollong MJ, Partch CL, and Lamia KA (2021). CRY2 missense mutations suppress P53 and enhance cell growth. Proc. Natl. Acad. Sci. USA 118, e2101416118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Huber AL, Papp SJ, Chan AB, Henriksson E, Jordan SD, Kriebs A, Nguyen M, Wallace M, Li Z, Metallo CM, et al. (2016). CRY2 and FBXL3 Cooperatively Degrade c-MYC. Mol. Cell 64, 774–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu Z, Selby CP, Yang Y, Lindsey-Boltz LA, Cao X, Eynullazada K, and Sancar A (2020). Circadian regulation of c-MYC in mice. Proc. Natl. Acad. Sci. USA 117, 21609–21617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pariollaud M, Ibrahim LH, Irizarry E, Mello RM, Chan AB, Altman BJ, Shaw RJ, Bollong MJ, Wiseman RL, and Lamia KA (2022). Circadian disruption enhances HSF1 signaling and tumorigenesis in Kras-driven lung cancer. Sci. Adv 8, eabo1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang C, Barnoud C, Cenerenti M, Sun M, Caffa I, Kizil B, Bill R, Liu Y, Pick R, Garnier L, et al. (2023). Dendritic cells direct circadian anti-tumour immune responses. Nature 614, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chun SK, Fortin BM, Fellows RC, Habowski AN, Verlande A, Song WA, Mahieu AL, Lefebvre AEYT, Sterrenberg JN, Velez LM, et al. (2022). Disruption of the circadian clock drives Apc loss of heterozygosity to accelerate colorectal cancer. Sci. Adv 8, eabo2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Qu M, Zhang G, Qu H, Vu A, Wu R, Tsukamoto H, Jia Z, Huang W, Lenz HJ, Rich JN, et al. (2023). Circadian regulator BMAL1::CLOCK promotes cell proliferation in hepatocellular carcinoma by controlling apoptosis and cell cycle. Proc. Natl. Acad. Sci. USA 120, e2214829120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Papagiannakopoulos T, Bauer MR, Davidson SM, Heimann M, Subbaraj L, Bhutkar A, Bartlebaugh J, Vander Heiden MG, and Jacks T (2016). Circadian Rhythm Disruption Promotes Lung Tumorigenesis. Cell Metab. 24, 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Alasady MJ, and Mendillo ML (2020). The Multifaceted Role of HSF1 in Tumorigenesis. Adv. Exp. Med. Biol 1243, 69–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Diamantopoulou Z, Castro-Giner F, Schwab FD, Foerster C, Saini M, Budinjas S, Strittmatter K, Krol I, Seifert B, Heinzelmann-Schwarz V, et al. (2022). The metastatic spread of breast cancer accelerates during sleep. Nature 607, 156–162. [DOI] [PubMed] [Google Scholar]
- 145.Verlande A, Chun SK, Goodson MO, Fortin BM, Bae H, Jang C, and Masri S (2021). Glucagon regulates the stability of REV-ERBα to modulate hepatic glucose production in a model of lung cancer-associated cachexia. Sci. Adv 7, eabf3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Innominato PF, Ballesta A, Huang Q, Focan C, Chollet P, Karaboué A, Giacchetti S, Bouchahda M, Adam R, Garufi C, et al. (2020). Sex-dependent least toxic timing of irinotecan combined with chronomodulated chemotherapy for metastatic colorectal cancer: Randomized multicenter EORTC 05011 trial. Cancer Med. 9, 4148–4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Qian DC, Kleber T, Brammer B, Xu KM, Switchenko JM, Janopaul-Naylor JR, Zhong J, Yushak ML, Harvey RD, Paulos CM, et al. (2021). Effect of immunotherapy time-of-day infusion on overall survival among patients with advanced melanoma in the USA (MEMOIR): a propensity score-matched analysis of a single-centre, longitudinal study. Lancet Oncol. 22, 1777–1786. [DOI] [PubMed] [Google Scholar]
- 148.Winkler F, Venkatesh HS, Amit M, Batchelor T, Demir IE, Deneen B, Gutmann DH, Hervey-Jumper S, Kuner T, Mabbott D, et al. (2023). Cancer neuroscience: state of the field, emerging directions. Cell 186, 1689–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, Blaes J, Weil S, Horstmann H, Wiestler B, Syed M, et al. (2015). Brain tumour cells interconnect to a functional and resistant network. Nature 528, 93–98. [DOI] [PubMed] [Google Scholar]
- 150.Venkataramani V, Tanev DI, Strahle C, Studier-Fischer A, Fankhauser L, Kessler T, Körber C, Kardorff M, Ratliff M, Xie R, et al. (2019). Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 573, 532–538. [DOI] [PubMed] [Google Scholar]
- 151.Hausmann D, Hoffmann DC, Venkataramani V, Jung E, Horschitz S, Tetzlaff SK, Jabali A, Hai L, Kessler T, Azoŕin DD, et al. (2023). Autonomous rhythmic activity in glioma networks drives brain tumour growth. Nature 613, 179–186. [DOI] [PubMed] [Google Scholar]
- 152.Venkataramani V, Yang Y, Schubert MC, Reyhan E, Tetzlaff SK, Wißmann N, Botz M, Soyka SJ, Beretta CA, Pramatarov RL, et al. (2022). Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 185, 2899–2917.e31. [DOI] [PubMed] [Google Scholar]
- 153.Krishna S, Choudhury A, Keough MB, Seo K, Ni L, Kakaizada S, Lee A, Aabedi A, Popova G, Lipkin B, et al. (2023). Glioblastoma remodelling of human neural circuits decreases survival. Nature 617, 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Haroun R, Wood JN, and Sikandar S (2022). Mechanisms of cancer pain. Front. Pain Res. (Lausanne) 3, 1030899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zhang C, Xie C, and Lu Y (2021). Local Anesthetic Lidocaine and Cancer: Insight Into Tumor Progression and Recurrence. Front. Oncol. 11, 669746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mullard A. (2022). Vertex’s Nav1.8 inhibitor passes phase II pain point. Nat. Rev. Drug Discov 21, 327. [DOI] [PubMed] [Google Scholar]
- 157.Heuer S, Burghaus I, Gose M, Kessler T, Sahm F, Vollmuth P, Venkataramani V, Hoffmann D, Schlesner M, Ratliff M, et al. (2024). PerSurge (NOA-30) phase II trial of perampanel treatment around surgery in patients with progressive glioblastoma. BMC Cancer 24, 135. 10.1186/s12885-024-11846-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Britt KL, Cuzick J, and Phillips KA (2020). Key steps for effective breast cancer prevention. Nat. Rev. Cancer 20, 417–436. [DOI] [PubMed] [Google Scholar]
- 159.Collaborative Group on Hormonal Factors in Breast Cancer (2002). Breast cancer and breastfeeding: collaborative reanalysis of individual data from 47 epidemiological studies in 30 countries, including 50302 women with breast cancer and 96973 women without the disease. Lancet 360, 187–195. [DOI] [PubMed] [Google Scholar]
- 160.Islami F, Liu Y, Jemal A, Zhou J, Weiderpass E, Colditz G, Boffetta P, and Weiss M (2015). Breastfeeding and breast cancer risk by receptor status–a systematic review and meta-analysis. Ann. Oncol 26, 2398–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Loi S, Drubay D, Adams S, Pruneri G, Francis PA, Lacroix-Triki M, Joensuu H, Dieci MV, Badve S, Demaria S, et al. (2019). Tumor-Infiltrating Lymphocytes and Prognosis: A Pooled Individual Patient Analysis of Early-Stage Triple-Negative Breast Cancers. J. Clin. Oncol 37, 559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, Salgado R, Byrne DJ, Teo ZL, Dushyanthen S, et al. (2018). Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med 24, 986–993. [DOI] [PubMed] [Google Scholar]
- 163.Virassamy B, Caramia F, Savas P, Sant S, Wang J, Christo SN, Byrne A, Clarke K, Brown E, Teo ZL, et al. (2023). Intratumoral CD8+ T cells with a tissue-resident memory phenotype mediate local immunity and immune checkpoint responses in breast cancer. Cancer Cell 41, 585–601.e8. [DOI] [PubMed] [Google Scholar]
- 164.Pal B, Chen Y, Vaillant F, Capaldo BD, Joyce R, Song X, Bryant VL, Penington JS, Di Stefano L, Tubau Ribera N, et al. (2021). A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast. EMBO J. 40, e107333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Asztalos S, Gann PH, Hayes MK, Nonn L, Beam CA, Dai Y, Wiley EL, and Tonetti DA (2010). Gene expression patterns in the human breast after pregnancy. Cancer Prev. Res. (Phila) 3, 301–311. [DOI] [PubMed] [Google Scholar]
- 166.Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Bäckhed HK, Gonzalez A, Werner JJ, Angenent LT, Knight R, et al. (2012). Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 150, 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hunt KM, Foster JA, Forney LJ, Schütte UM, Beck DL, Abdo Z, Fox LK, Williams JE, McGuire MK, and McGuire MA (2011). Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS One 6, e21313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Cabrera-Rubio R, Collado MC, Laitinen K, Salminen S, Isolauri E, and Mira A (2012). The human milk microbiome changes over lactation and is shaped by maternal weight and mode of delivery. Am. J. Clin. Nutr 96, 544–551. [DOI] [PubMed] [Google Scholar]
- 169.Camacho-Morales A, Caba M, García-Juárez M, Caba-Flores MD, Viveros-Contreras R, and Martínez-Valenzuela C (2021). Breastfeeding Contributes to Physiological Immune Programming in the Newborn. Front. Pediatr 9, 744104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Khorana AA, Francis CW, Culakova E, Kuderer NM, and Lyman GH (2007). Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapy. J. Thromb. Haemost 5, 632–634. [DOI] [PubMed] [Google Scholar]
- 171.Khorana AA, Francis CW, Culakova E, Fisher RI, Kuderer NM, and Lyman GH (2006). Thromboembolism in hospitalized neutropenic cancer patients. J. Clin. Oncol 24, 484–490. [DOI] [PubMed] [Google Scholar]
- 172.Mulder FI, Horváth-Puhó E, van Es N, van Laarhoven HWM, Pedersen L, Moik F, Ay C, Büller HR, and Sørensen HT (2021). Venous thromboembolism in cancer patients: a population-based cohort study. Blood 137, 1959–1969. [DOI] [PubMed] [Google Scholar]
- 173.Engelmann B, and Massberg S (2013). Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol 13, 34–45. [DOI] [PubMed] [Google Scholar]
- 174.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, Wrobleski SK, Wakefield TW, Hartwig JH, and Wagner DD (2010). Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 107, 15880–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kasthuri RS, Taubman MB, and Mackman N (2009). Role of tissue factor in cancer. J. Clin. Oncol 27, 4834–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Anvari S, Osei E, and Maftoon N (2021). Interactions of platelets with circulating tumor cells contribute to cancer metastasis. Sci. Rep 11, 15477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tawil N, Bassawon R, Meehan B, Nehme A, Montermini L, Gayden T, De Jay N, Spinelli C, Chennakrishnaiah S, Choi D, et al. (2021). Glioblastoma cell populations with distinct oncogenic programs release podoplanin as procoagulant extracellular vesicles. Blood Adv. 5, 1682–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Xue R, Zhang Q, Cao Q, Kong R, Xiang X, Liu H, Feng M, Wang F, Cheng J, Li Z, et al. (2022). Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature 612, 141–147. [DOI] [PubMed] [Google Scholar]
- 179.Ballesteros I, Rubio-Ponce A, Genua M, Lusito E, Kwok I, Fernández-Calvo G, Khoyratty TE, van Grinsven E, González-Hernández S, Nicolás-Ávila JÁ, et al. (2020). Co-option of Neutrophil Fates by Tissue Environments. Cell 183, 1282–1297.e18. [DOI] [PubMed] [Google Scholar]
- 180.Maas RR, Soukup K, Fournier N, Massara M, Galland S, Kornete M, Wischnewski V, Lourenco J, Croci D, Álvarez-Prado ÁF, et al. (2023). The local microenvironment drives activation of neutrophils in human brain tumors. Cell 186, 4546–4566.e27. [DOI] [PubMed] [Google Scholar]
- 181.Ponzetta A, Carriero R, Carnevale S, Barbagallo M, Molgora M, Perucchini C, Magrini E, Gianni F, Kunderfranco P, Polentarutti N, et al. (2019). Neutrophils Driving Unconventional T Cells Mediate Resistance against Murine Sarcomas and Selected Human Tumors. Cell 178, 346–360.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Cui C, Chakraborty K, Tang XA, Zhou G, Schoenfelt KQ, Becker KM, Hoffman A, Chang YF, Blank A, Reardon CA, et al. (2021). Neutrophil elastase selectively kills cancer cells and attenuates tumorigenesis. Cell 184, 3163–3177.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Adrover JM, McDowell SAC, He XY, Quail DF, and Egeblad M (2023). NETworking with cancer: The bidirectional interplay between cancer and neutrophil extracellular traps. Cancer Cell 41, 505–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Khorana AA, Kuderer NM, Culakova E, Lyman GH, and Francis CW (2008). Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood 111, 4902–4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, Silver AJ, Adams D, Castellano CA, Schneider RK, et al. (2018). Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med 10, eaan8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Belizaire R, Wong WJ, Robinette ML, and Ebert BL (2023). Clonal haematopoiesis and dysregulation of the immune system. Nat. Rev. Immunol 23, 595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kar SP, Quiros PM, Gu M, Jiang T, Mitchell J, Langdon R, Iyer V, Barcena C, Vijayabaskar MS, Fabre MA, et al. (2022). Genomewide analyses of 200,453 individuals yield new insights into the causes and consequences of clonal hematopoiesis. Nat. Genet 54, 1155–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Bolton KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, Patel M, Berthon A, Syed A, Yabe M, et al. (2020). Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet 52, 1219–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Coombs CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P, Hyman DM, Solit DB, Robson ME, Baselga J, et al. (2017). Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 21, 374–382.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lee M, Li J, Li J, Fang S, Zhang J, Vo ATT, Han W, Zeng H, Isgandarova S, Martinez-Moczygemba M, et al. (2021). Tet2 Inactivation Enhances the Antitumor Activity of Tumor-Infiltrating Lymphocytes. Cancer Res. 81, 1965–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Budden KF, Shukla SD, Rehman SF, Bowerman KL, Keely S, Hugenholtz P, Armstrong-James DPH, Adcock IM, Chotirmall SH, Chung KF, et al. (2019). Functional effects of the microbiota in chronic respiratory disease. Lancet Respir. Med 7, 907–920. [DOI] [PubMed] [Google Scholar]
- 192.Sepich-Poore GD, Zitvogel L, Straussman R, Hasty J, Wargo JA, and Knight R (2021). The microbiome and human cancer. Science 371, eabc4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Wong SH, and Yu J (2019). Gut microbiota in colorectal cancer: mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol 16, 690–704. [DOI] [PubMed] [Google Scholar]
- 194.Baruch EN, Wang J, and Wargo JA (2021). Gut Microbiota and Antitumor Immunity: Potential Mechanisms for Clinical Effect. Cancer Immunol. Res 9, 365–370. [DOI] [PubMed] [Google Scholar]
- 195.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, et al. (2018). Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97. [DOI] [PubMed] [Google Scholar]
- 196.McCulloch JA, Davar D, Rodrigues RR, Badger JH, Fang JR, Cole AM, Balaji AK, Vetizou M, Prescott SM, Fernandes MR, et al. (2022). Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1. Nat. Med 28, 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Spencer CN, McQuade JL, Gopalakrishnan V, McCulloch JA, Vetizou M, Cogdill AP, Khan MAW, Zhang X, White MG, Peterson CB, et al. (2021). Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science 374, 1632–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lam KC, Araya RE, Huang A, Chen Q, Di Modica M, Rodrigues RR, Lopès A, Johnson SB, Schwarz B, Bohrnsen E, et al. (2021). Microbiota triggers STING-type I IFN-dependent monocyte reprogramming of the tumor microenvironment. Cell 184, 5338–5356.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Simpson RC, Shanahan ER, Batten M, Reijers ILM, Read M, Silva IP, Versluis JM, Ribeiro R, Angelatos AS, Tan J, et al. (2022). Diet-driven microbial ecology underpins associations between cancer immunotherapy outcomes and the gut microbiome. Nat. Med 28, 2344–2352. [DOI] [PubMed] [Google Scholar]
- 200.Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, et al. (2012). Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 22, 292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, Neuberg D, Huang K, Guevara F, Nelson T, et al. (2017). Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 358, 1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, Gavert N, Zwang Y, Cooper ZA, Shee K, et al. (2017). Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 357, 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, Quesada P, Sahin I, Chandra V, San Lucas A, et al. (2019). Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 178, 795–806.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Galeano Niño JL, Wu H, LaCourse KD, Kempchinsky AG, Baryiames A, Barber B, Futran N, Houlton J, Sather C, Sicinska E, et al. (2022). Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancer. Nature 611, 810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Serna G, Ruiz-Pace F, Hernando J, Alonso L, Fasani R, Landolfi S, Comas R, Jimenez J, Elez E, Bullman S, et al. (2020). Fusobacterium nucleatum persistence and risk of recurrence after preoperative treatment in locally advanced rectal cancer. Ann. Oncol 31, 1366–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, Rotter-Maskowitz A, Weiser R, Mallel G, Gigi E, et al. (2020). The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 368, 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, Kosciolek T, Janssen S, Metcalf J, Song SJ, et al. (2020). Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 579, 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 208.Narunsky-Haziza L, Sepich-Poore GD, Livyatan I, Asraf O, Martino C, Nejman D, Gavert N, Stajich JE, Amit G, González A, et al. (2022). Pan-cancer analyses reveal cancer-type-specific fungal ecologies and bacteriome interactions. Cell 185, 3789–3806.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Dohlman AB, Klug J, Mesko M, Gao IH, Lipkin SM, Shen X, and Iliev ID (2022). A pan-cancer mycobiome analysis reveals fungal involvement in gastrointestinal and lung tumors. Cell 185, 3807–3822.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gihawi A, Ge Y, Lu J, Puiu D, Xu A, Cooper CS, Brewer DS, Pertea M, and Salzberg SL (2023). Major data analysis errors invalidate cancer microbiome findings. mBio 14, e0160723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gihawi A, Cooper CS, and Brewer DS (2023). Caution regarding the specificities of pan-cancer microbial structure. Microb. Genom 9, mgen001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Aykut B, Pushalkar S, Chen R, Li Q, Abengozar R, Kim JI, Shadaloey SA, Wu D, Preiss P, Verma N, et al. (2019). The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 574, 264–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Fletcher AA, Kelly MS, Eckhoff AM, and Allen PJ (2023). Revisiting the intrinsic mycobiome in pancreatic cancer. Nature 620, E1–E6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Xu F, Saxena D, Pushalkar S, and Miller G (2023). Reply to: Revisiting the intrinsic mycobiome in pancreatic cancer. Nature 620, E7–E9. [DOI] [PubMed] [Google Scholar]
- 215.Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, Wu X, DeStefano Shields CE, Hechenbleikner EM, Huso DL, et al. (2018). Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 359, 592–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, Adler K, Dick-Necula D, Raskin S, Bloch N, et al. (2021). Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 371, 602–609. [DOI] [PubMed] [Google Scholar]
- 217.Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, Deblasio RN, Menna C, Ding Q, Pagliano O, et al. (2021). Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 371, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Dizman N, Meza L, Bergerot P, Alcantara M, Dorff T, Lyou Y, Frankel P, Cui Y, Mira V, Llamas M, et al. (2022). Nivolumab plus ipilimumab with or without live bacterial supplementation in metastatic renal cell carcinoma: a randomized phase 1 trial. Nat. Med 28, 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Holt RA (2023). Oncomicrobial vaccines: The potential for a Fusobacterium nucleatum vaccine to improve colorectal cancer outcomes. Cell Host Microbe 31, 141–145. [DOI] [PubMed] [Google Scholar]
- 220.Maertens O, McCurrach ME, Braun BS, De Raedt T, Epstein I, Huang TQ, Lauchle JO, Lee H, Wu J, Cripe TP, et al. (2017). A Collaborative Model for Accelerating the Discovery and Translation of Cancer Therapies. Cancer Res. 77, 5706–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Olson B, Li Y, Lin Y, Liu ET, and Patnaik A (2018). Mouse Models for Cancer Immunotherapy Research. Cancer Discov. 8, 1358–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Drapkin BJ, and Rudin CM (2021). Advances in Small-Cell Lung Cancer (SCLC) Translational Research. Cold Spring Harb. Perspect. Med 11, a038240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Nadeau JH, and Auwerx J (2019). The virtuous cycle of human genetics and mouse models in drug discovery. Nat. Rev. Drug Discov 18, 255–272. [DOI] [PubMed] [Google Scholar]
- 224.Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, Beavis WD, Belknap JK, Bennett B, Berrettini W, et al. (2004). The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat. Genet 36, 1133–1137. [DOI] [PubMed] [Google Scholar]
- 225.Ashbrook DG, Arends D, Prins P, Mulligan MK, Roy S, Williams EG, Lutz CM, Valenzuela A, Bohl CJ, Ingels JF, et al. (2021). A platform for experimental precision medicine: The extended BXD mouse family. Cell Syst. 12, 235–247.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Churchill GA, Gatti DM, Munger SC, and Svenson KL (2012). The Diversity Outbred mouse population. Mamm. Genome 23, 713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Halliwill KD, Quigley DA, Kang HC, Del Rosario R, Ginzinger D, and Balmain A (2016). Panx3 links body mass index and tumorigenesis in a genetically heterogeneous mouse model of carcinogen-induced cancer. Genome Med. 8, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ootani A, Li X, Sangiorgi E, Ho QT, Ueno H, Toda S, Sugihara H, Fujimoto K, Weissman IL, Capecchi MR, et al. (2009). Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat. Med 15, 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Pastuła A, Middelhoff M, Brandtner A, Tobiasch M, Höhl B, Nuber AH, Demir IE, Neupert S, Kollmann P, Mazzuoli-Weber G, et al. (2016). Three-Dimensional Gastrointestinal Organoid Culture in Combination with Nerves or Fibroblasts: A Method to Characterize the Gastrointestinal Stem Cell Niche. Stem Cells Int. 2016, 3710836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Yan HHN, Chan AS, Lai FP-L, and Leung SY (2023). Organoid cultures for cancer modeling. Cell Stem Cell 30, 917–937. [DOI] [PubMed] [Google Scholar]
- 231.Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K, Chua CW, Barlow LJ, Kandoth C, Williams AB, et al. (2018). Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 173, 515–528.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Li X, Francies HE, Secrier M, Perner J, Miremadi A, Galeano-Dalmau N, Barendt WJ, Letchford L, Leyden GM, Goffin EK, et al. (2018). Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat. Commun 9, 2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, Slagter M, van der Velden DL, Kaing S, Kelderman S, et al. (2018). Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 174, 1586–1598.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.LeSavage BL, Suhar RA, Broguiere N, Lutolf MP, and Heilshorn SC (2022). Next-generation cancer organoids. Nat. Mater 21, 143–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Ingber DE (2018). Developmentally inspired human “organs on chips.”. Development 145, dev156125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sontheimer-Phelps A, Hassell BA, and Ingber DE (2019). Modelling cancer in microfluidic human organs-on-chips. Nat. Rev. Cancer 19, 65–81. [DOI] [PubMed] [Google Scholar]
- 237.Ma C, Peng Y, Li H, and Chen W (2021). Organ-on-a-Chip: A New Paradigm for Drug Development. Trends Pharmacol. Sci 42, 119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Powley IR, Patel M, Miles G, Pringle H, Howells L, Thomas A, Kettleborough C, Bryans J, Hammonds T, MacFarlane M, et al. (2020). Patient-derived explants (PDEs) as a powerful preclinical platform for anti-cancer drug and biomarker discovery. Br. J. Cancer 122, 735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Chiorazzi M, Martinek J, Krasnick B, Zheng Y, Robbins KJ, Qu R, Kaufmann G, Skidmore Z, Juric M, Henze LA, et al. (2023). Autologous humanized PDX modeling for immuno-oncology recapitulates features of the human tumor microenvironment. J. Immunother. Cancer 11, e006921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Thommen DS (2022). Tumour avatars to model patients’ responses to immunotherapy. Nat. Rev. Cancer 22, 660. [DOI] [PubMed] [Google Scholar]
- 241.Roelofsen LM, Voabil P, de Bruijn M, Herzig P, Zippelius A, Schumacher TN, and Thommen DS (2023). Protocol for ex vivo culture of patient-derived tumor fragments. Star Protoc. 4, 102282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Voabil P, de Bruijn M, Roelofsen LM, Hendriks SH, Brokamp S, van den Braber M, Broeks A, Sanders J, Herzig P, Zippelius A, et al. (2021). An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat. Med 27, 1250–1261. [DOI] [PubMed] [Google Scholar]
- 243.Dias MH, and Bernards R (2021). Playing cancer at its own game: activating mitogenic signaling as a paradoxical intervention. Mol. Oncol 15, 1975–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Dias MH, Friskes A, Wang S, Fernandes Neto JM, van Gemert F, Mourragui S, Kuiken HJ, Mainardi S, Alvarez-Villanueva D, Lieftink C, et al. (2023). Paradoxical activation of oncogenic signaling as a cancer treatment strategy. Preprint at bioRxiv. 10.1101/2023.02.06.527335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.van Neerven SM, de Groot NE, Nijman LE, Scicluna BP, van Driel MS, Lecca MC, Warmerdam DO, Kakkar V, Moreno LF, Vieira Braga FA, et al. (2021). Apc-mutant cells act as supercompetitors in intestinal tumour initiation. Nature 594, 436–441. [DOI] [PubMed] [Google Scholar]
- 246.Ge W, and Jakobsson E (2019). Systems Biology Understanding of the Effects of Lithium on Cancer. Front. Oncol 9, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Bardia A, and Baselga J (2013). Neoadjuvant therapy as a platform for drug development and approval in breast cancer. Clin. Cancer Res 19, 6360–6370. [DOI] [PubMed] [Google Scholar]
- 248.Marron TU, Galsky MD, Taouli B, Fiel MI, Ward S, Kim E, Yankelevitz D, Doroshow D, Guttman-Yassky E, Ungar B, et al. (2022). Neoadjuvant clinical trials provide a window of opportunity for cancer drug discovery. Nat. Med 28, 626–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Reynolds AR, Moschetta M, Yohannes AR, Walcott F, Ashford M, Szucs Z, Sarbajna T, Hadfield J, Harrison E, Challis BG, et al. (2023). A View on Drug Development for Cancer Prevention. Cancer Discov. 13, 1058–1083. [DOI] [PubMed] [Google Scholar]
- 250.Inan OT, Tenaerts P, Prindiville SA, Reynolds HR, Dizon DS, Cooper-Arnold K, Turakhia M, Pletcher MJ, Preston KL, Krumholz HM, et al. (2020). Digitizing clinical trials. NPJ Digit. Med 3, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Alkhayyat M, Kumar P, Sanaka KO, and Thota PN (2021). Chemoprevention in Barrett’s esophagus and esophageal adenocarcinoma. Therap. Adv. Gastroenterol 14, 17562848211033730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Gale D, Heider K, Ruiz-Valdepenas A, Hackinger S, Perry M, Marsico G, Rundell V, Wulff J, Sharma G, Knock H, et al. (2022). Residual ctDNA after treatment predicts early relapse in patients with early-stage non-small cell lung cancer. Ann. Oncol 33, 500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Abbosh C, Frankell AM, Harrison T, Kisistok J, Garnett A, Johnson L, Veeriah S, Moreau M, Chesh A, Chaunzwa TL, et al. (2023). Tracking early lung cancer metastatic dissemination in TRACERx using ctDNA. Nature 616, 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Powles T, Assaf ZJ, Davarpanah N, Banchereau R, Szabados BE, Yuen KC, Grivas P, Hussain M, Oudard S, Gschwend JE, et al. (2021). ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature 595, 432–437. [DOI] [PubMed] [Google Scholar]
- 255.Lipsyc-Sharf M, de Bruin EC, Santos K, McEwen R, Stetson D, Patel A, Kirkner GJ, Hughes ME, Tolaney SM, Partridge AH, et al. (2022). Circulating Tumor DNA and Late Recurrence in High-Risk Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Breast cancer. J. Clin. Oncol 40, 2408–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Tie J, Cohen JD, Lahouel K, Lo SN, Wang Y, Kosmider S, Wong R, Shapiro J, Lee M, Harris S, et al. (2022). Circulating Tumor DNA Analysis Guiding Adjuvant Therapy in Stage II Colon Cancer. N. Engl. J. Med 386, 2261–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.André F. (2018). Developing Anticancer Drugs in Orphan Molecular Entities - A Paradigm under Construction. N. Engl. J. Med 378, 763–765. [DOI] [PubMed] [Google Scholar]
- 258.Andre F, Filleron T, Kamal M, Mosele F, Arnedos M, Dalenc F, Sablin MP, Campone M, Bonnefoi H, Lefeuvre-Plesse C, et al. (2022). Genomics to select treatment for patients with metastatic breast cancer. Nature 610, 343–348. [DOI] [PubMed] [Google Scholar]
- 259.Koh B, Tan DJH, Ng CH, Fu CE, Lim WH, Zeng RW, Yong JN, Koh JH, Syn N, Meng W, et al. (2023). Patterns in Cancer Incidence Among People Younger Than 50 Years in the US, 2010 to 2019. JAMA Netw. Open 6, e2328171. [DOI] [PMC free article] [PubMed] [Google Scholar]