

Abstract
Puerperal sepsis is a leading cause of maternal mortality worldwide. Streptococcus pyogenes (Group A Streptococcus; GAS) is a major etiologic agent of severe postpartum sepsis yet little is known regarding the pathogenesis of these infections. Tissue macrophages provide innate defense against GAS and their actions are highly regulated. The intracellular second messenger cAMP can negatively regulate macrophage actions against GAS. Because leukotriene (LT) B4 has been shown to suppress intracellular cAMP in macrophages, we hypothesized that it could enhance innate defenses against GAS. We assessed the capacity of LTB4 to modulate anti-streptococcal actions of human macrophages, including placental and decidual macrophages and used a novel intrauterine infection model of GAS in mice lacking the 5-lipoxygenase (5LO) enzyme to determine the role of endogenous LTs in host defense against this pathogen. Animals lacking 5LO were significantly more vulnerable to intrauterine GAS infection than wild-type mice and showed enhanced dissemination of bacteria out of the uterus and a more robust inflammatory response compared to wild-type mice. Additionally, LTB4 reduced intracellular cAMP levels via the BLT1 receptor and was a potent stimulant of macrophage phagocytosis and NADPH oxidase-dependent intracellular killing of GAS. Importantly, interference was observed between the macrophage immunomodulatory actions of LTB4 and the cAMP-inducing lipid prostaglandin E2, suggesting that interplay between pro- and anti-inflammatory compounds may be important in vivo. This work underscores the potential for pharmacological targeting of lipid mediator signaling cascades in the treatment of invasive GAS infections.
Introduction
Worldwide, more than 500,000 women die per year as a complication of childbirth (1). One of the principal causes of maternal death is puerperal sepsis, which causes more than 75,000 maternal deaths per year globally (2). The gram-positive bacterium Streptococcus pyogenes, also known as a group A Streptococcus (GAS), is the most common cause of severe postpartum sepsis (3). Puerperal GAS is an important cause of maternal death in resource-poor settings, and it is a reemerging problem in industrialized countries (3). For example, the rate of maternal deaths from sepsis increased in the United Kingdom between 2000–2008, primarily because of increases in postpartum GAS (4). Significant gaps exist in understanding fundamental mechanisms of GAS pathogenesis and host defense in the female reproductive tract that lead to intrauterine infection and sepsis.
Pregnancy is associated with major shifts in immune surveillance (5) as the maternal immune system must be “detuned” to accommodate the immunologically-distinct fetus (6). Despite this, the mother’s immune system must be able to detect and respond to potentially pathogenic organisms. However, some pathogens have evolved mechanisms to evade host defense, apparently taking advantage of the immunological shifts associated with pregnancy. For example, certain Gram positive bacteria are adept at causing pregnancy-related infections, including Listeria monocytogenes, Streptococcus pneumoniae, Group A Streptococcus, Group B Streptococcus, and the clostridia (3, 7, 8). The incidence of invasive GAS is more than 25 times higher for postpartum vs. nonpregnant women but the reasons for this are not defined (7). It is also unclear why only a subset of women exposed to GAS develops overt disease. Bacterial virulence factors are important determinants of GAS infection, but these alone do not predict the development and clinical severity of infection (9). Greater attention is needed to the basic mechanisms of disease pathogenesis, with an emphasis on host-microbial interactions.
Tissue macrophages are critical cellular immune barriers against invasive GAS infection (10, 11), although this has not been examined in the reproductive tract. Macrophage phagocytosis and intracellular killing of tissue-invasive microbes are highly regulated processes that present an important first line of defense against potential pathogens. The host factors that regulate the capacity of macrophages to respond to GAS have not been well characterized. Defining the mechanisms governing antimicrobial functions of tissue macrophages provides an opportunity to develop innovate measures for preventing or treating invasive bacterial infections in susceptible hosts. Recently, host-derived lipid mediators have been found to regulate the capacity for macrophages to respond to GAS (12) but this has not been explored in the context of reproductive immunology.
Eicosanoids are immunoregulatory lipids that are oxygenated metabolites of the cell membrane constituent molecule arachidonic acid. They are potent modulators of innate immunity, including macrophage behaviors (13–17). For example, recent studies suggest that the cyclooxygenase-derived PG compound, PGE2, suppresses macrophage phagocytosis and intracellular killing of GAS through acutely activating the synthesis of the intracellular second messenger molecule cAMP (18). In general, receptor-dependent increases in cAMP suppress macrophage immune responses, possibly an evolved mechanism for limiting the amplitude and duration of inflammatory responses (19). In contrast, the 5-lipoxygenase (5LO)-derived eicosanoid leukotriene (LT) B4 has been reported to stimulate macrophage phagocytosis and intracellular killing of several pathogens in association with suppression of intracellular cAMP production (17, 20). The ability of LTB4 to enhance host elimination of streptococci has been most clearly defined in a model of lung infection caused by S. pneumoniae (21–24). In addition, the administration of aerosolized LTB4 to mice with preexisting pneumococcal pneumonia increased mononuclear phagocyte/macrophage accumulation in the lungs, NADPHox subunit expression in pulmonary macrophages, and pulmonary bacterial clearance (25).
Leukotriene B4 is synthesized from arachidonic acid when an acute calcium (Ca2+) flux is induced in cells following activation by inflammatory mediators or pathogens (26, 27). There are two G protein coupled receptors for LTB4 that inhibit adenylate cyclase. The BLT1 receptor (high affinity) is expressed primarily in leukocytes, including granulocytes, eosinophils, macrophages and differentiated T cells, while the BLT2 receptor (low affinity) is expressed in many cell types (28–32).
It is notable that both PGE2 and LTB4 are found in the gravid uterus (33–35) and both have immunoregulatory actions that might play a role in defining a postpartum woman’s vulnerability of invasive bacterial infection. Because PGE2 stimulates intracellular cAMP to suppress macrophage immune defenses against GAS (18), we hypothesized that LTB4 would stimulate macrophage actions against this organism because its actions on cAMP are opposite to those of PGE2. We further speculated that mice unable to generate LTB4 because they lack the gene encoding 5LO would be susceptible to intrauterine GAS infection. Both in vivo and in vitro models of infection were used to assess the role of LTs as immunoregulators of innate defense against GAS.
Material and Methods
Reagents
RPMI 1640 culture medium, antibiotic solution (penicillin and streptomycin) including an antimycotic (amphotericin) was from Invitrogen (Carlsbad, CA). Charcoal-stripped and dextran-treated FBS was from HyClone Laboratories (Waltham, MA). Pertussis toxin (PTX), cytochalasin D from Zygosporium mansonii and apocynin were obtained from EMD Chemicals-Calbiochem (Gibbstown, NJ). FITC, trypan blue, deoxyribonuclease, hyaluronidase, collagenase, Percoll®, citrate buffer, fucoidan, saponin, PMA, non-enzymatic cell dissociation solution, NaHCO3 buffer, and sodium bicarbonate were from Sigma-Aldrich (St. Louis, MO). LTB4, PGE2, 12-HHT, U75302 and A23187 were from Cayman Chemicals (Ann Arbor, MI). Todd Hewitt Broth, Tryptic Soy Agar, and yeast extract were from BD Biosciences (San Jose, CA). Sheep blood was from Remel (Lenexa, CA). Ficcoll-Hypaque was from GE Healthcare (Piscataway, NJ). Lysis buffer for erythrocytes was purchased from eBioscience (San Diego, CA). FLUO-4 NW used for intracellular calcium assays and 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (Carboxy H2DCFDA) for reactive oxygen species measure were provided from Molecular Probes (Grand Island, NY). ELISA kits to measure intracellular cAMP were from Enzo LifeSciences (Farmingdale, NY). Magnetic MACS® CD14 microbeads (human) were from Miltenyi Biotec (Auburn, CA). Required dilutions of all compounds were prepared immediately before use, and equivalent quantities of vehicle were added to the appropriate controls. Compounds requiring reconstitution were dissolved in DMSO (Sigma).
Animals
Age-matched, six-to eight-week-old, female, non-pregnant, C57BL/6 wild-type or 5LO deficient mice (5LO−/−) mice on a C57BL/6 background) were purchased from Jackson Laboratory (Bar Harbor, ME). Animals were treated according to National Institutes of Health guidelines for the use of experimental animals with approval of the University of Michigan Committee for the Use and Care of Animals.
Bacteria
The GAS M1T1 strain 5448 was generously provided by Dr. Malak Kotb of the University of Cincinnati and aliquots grown in THY. Estimates of bacterial concentrations in inocula were made by comparing the absorbance of light at 600 nm (OD600) and using a standard curve of CFU/ml vs OD600. The actual CFU/ml concentrations were confirmed for all bacterial suspensions used in these studies by culturing serial 10-fold dilutions on TSA blood plates. For phagocytosis experiments, heat-killed (HK) FITC-labeled GAS was used. These were generated as previously described for S. pneumoniae (36).
Intrauterine Infection
GAS cultures were grown 18 h, shaking at 200 rpm, at 37°C in 10 mL of THY; the bacteria were then centrifuged 3,000 rpm for 10 min and the pellet was washed twice in sterile 1x PBS, and resuspended in 1 mL total volume. Dilutions were prepared in 1x PBS to obtain the correct approximate inoculum of CFUs per mouse. The actual inoculum was determined by plating serial 10-fold dilutions of the PBS suspension onto TSA blood plates. The infection was performed in anesthetized animals. A low 2.0 cm midline abdominal incision exposed the right uterine horn, the uterine horn was ligated at the cervical junction to prevent inoculum loss, and 20 μL of prepared culture was directly injected into the horn (37, 38). For survival experiments, mice were monitored every 2 h on weekdays and 2×/day on weekends for 1 week after inoculation.
Quantitation of Bacterial Loads
After 24 h of infection, mice were euthanized and bacterial load was determined in the blood, spleen and uterus. The blood was serially diluted in sterile 1x PBS and cultured on TSA blood at 37°C for 18 h. The spleen and uterus were homogenized with a handheld tissue tearer in 1mL sterile 1x PBS, serially diluted in PBS, and then cultured on TSA blood at 37°C for 18 h. Colonies were counted as previously described (37).
Human Subjects
Following appropriate informed consent, human decidual tissue was obtained from healthy adult women aged 18–44 years undergoing elective surgical termination of pregnancy during the first trimester. Human placental tissue was obtained from healthy women undergoing caesarean section at the term of pregnancy. Human peripheral blood was obtained from healthy adults who were not smokers, not taking nonsteroidal antiinflamatory drugs (NSAIDs), not taking oral or inhaled steroids and not pregnant. Blood was collected by venous puncture into heparinized collection tube. These studies were reviewed and approved by the University of Michigan Institutional Review Board.
Cell Culture
THP-1 monocytic cells were obtained from the American Type Culture Collection (ATCC, TIB-202; Manassas, VA) and cultured in RPMI+/+. Cells were passaged every 2–4 days and were only used through the 10th passage, at which time a new culture was started. THP-1 cells were differentiated into macrophage-like cells by culturing with 100 nM of PMA in RPMI +/+ for 18 h at 37°C with 5% CO2. Cells were detached from the flask with non-enzymatic cell dissociation solution, scraped after 5 min of incubation at 37°C with 5% CO2. The protocol was adapted as described before (39, 40). PBMCs, PMs, DMs and peritoneal macrophages were cultured RPMI+/+ for 18 h at 37°C with 5% CO2 at a density of 2 × 105 cells/well in 6, 96 or 384 well-plate dishes (Costar, Corning, NY) depending on the experiment.
Isolation and Culture of Placental and Decidual Macrophages
The procedure for decidual macrophage (DM) and placental macrophage (PM) culture was similar to that used for THP-1 cells above. Isolation was adapted from a previously described protocol (38, 41). Briefly, the tissue was collected in 50 mL conical tubes containing 15 mL of sterile PBS and washed three times with 50 mL of PBS at 1,500 rpm for 10 min. The tissue was weighed and minced into small pieces with autoclaved scissors and weighed again to determine final grams collected. After this, the fragments were placed into 50 mL conical tubes with digestion solution containing deoxyribonuclease, collagenase and hyaluronidase at 10 mL per gram of tissue. Cells were filtered through 280 μm autoclaved metal sieve, followed by 180 and 80 μm autoclaved nylon screens. Cells were centrifuged again and resuspended in 25% Percoll® diluted in cold RPMI+/+ and overlayed onto 50% Percoll® plus 2 mL of PBS on top of the density gradient. CD14+ cells were performed using magnetic MACS® large cell separation column system (Miltenyi Biotec) according to the manufacturer’s instructions and adapted as previously reported (38).
Human Peripheral Blood Monocyte Isolation
Human peripheral blood mononuclear cells (PBMC) were isolated from heparinized blood by Ficoll-Hypaque gradient density centrifugation by 20 min at 1,700 rpm, 22°C without active breaking. The PBMCs were collected and transferred to new tubes and washed with PBS at 1,400 rpm × 10 min at room temperature. The pellets were broken and pooled together in one tube and washed again. The erythrocytes were lysed after the addition of 10 mL of lysis buffer at 22°C × 10 min. The cells were washed again and the final solution was resuspended in 50 mL of PBS and total cells were counted. CD14+ cells were negatively selected using magnetic MACS® small cell separation column system (Miltenyi Biotec) according to the manufacturer’s instructions.
Isolation and Culture of Peritoneal Macrophages
Resident peritoneal macrophages (RPM) were collected from uninfected mice. Mice were euthanized by CO2 inhalation and peritoneal cavities were washed twice with 5 mL of cold RPMI+/−. Collected fluid was placed in conical tubes on ice and centrifuged at 1,500 rpm for 10 min. Cell counts were performed immediately and 2 × 105 cells/well were added to culture in 384 well plates (Costar) followed by incubation at 37°C, 5% CO2 for 2 hr. After this period, the media was changed to RPMI-I+/+, and adherent cells were cultivated by 18 h incubation at 37°C, 5% CO2 as published before (42, 43)
Fluorometric Phagocytosis Assay with FITC-labeled Group A Streptococcus
The phagocytosis of unopsonized FITC-labeled HK GAS was assessed using PMA treated THP-1 cells, PBMCs, DMs, PMs and (RPM). All cell types were plated onto 384-well tissue culture-treated plates with 2 × 105 cells per well and the phagocytosis was performed as previously reported (20, 38, 44). After an overnight rest, media was removed and RPMI+/− containing vehicle or different stimuli were added as indicated in the figure legends. Specific treatments included LTB4 (15 min pre-treatment 1–1,000 nM), PGE2 (15 min pre-treatment 1 μM), 12-HHT (15 min pre-treatment 0.001–1 μM), U75302 (15 min pre-treatment 10 μM) and PTX (18 h pre-treatment 3 μg/mL). After appropriate incubations, GAS were added at multiplicity of infection (MOI) of 150:1 and incubated at 37°C with 5% CO2 for 3 h. Extracellular bacterial fluorescence was quenched with trypan blue (500 μg/mL in 0.09 M citrate buffer) for 15 min in the dark before the fluorescence was determined on a microplate flourometer (485ex/535em), SPECTRAMax GEMINI EM; Molecular Devices, Sunnyvale, CA. A minimum of eight replicates were done for each condition in each experiment. Fluorometric data were expressed in arbitrary relative fluorescence emitted from intracellular phagocytosed bacteria (RFUi) and was determined by subtracting the fluorescence from unquenched, extracellular bacteria (RFUex) from the total fluorescence of the experimental well (RFUtotal). The RFUex was determined using the fluorescence of cells exposed for 30 min to the phagocytosis inhibitor cytochalasin D (20 μg/mL). Thus, the PI = RFUi = RFUtotal − RFUex. (44, 45).
Bacterial Killing Assay
PMA treated THP-1 cells were adjusted to a suspension of 2 × 105 cells per well in 100 μL of RPMI−/− in replicates of 8 per condition in a 96 well plate and was centrifuged at 500 rpm for 30 sec to settle all cells into a monolayer. Cells adhered for 1 h before starting with the treatments. One set of replicate conditions within each plate was treated with cytochalasin D for 30 min. Cells were pretreated with the NADPH oxidase inhibitor apocynin 100 μM and/or LTB4 10 nM for 30 and 15 min respectively before infecting cells with a 10:1 MOI of live GAS. The plates were centrifuged again to synchronize bacterial contact with the monolayer and incubated for 30 min. After this time, all media was removed and plates washed gently 3 times with RPMI+/− to remove nonphagocytosed bacteria. Then 100 μL of 0.5% saponin in PBS was added and incubated for 10 min. Serial dilutions were made and the lysates were plated onto TSA blood plates; CFUs were counted 24 h later (adapted from (14, 22)).
Measurement of Intracellular Reactive Oxygen Intermediates
Reactive oxygen intermediate (ROI) generation was performed using 1.2 × 105 PMA treated THP-1 cells per well using 384-well plate, according to our previously reported protocol (14, 22, 46). Medium was replaced the following morning, and cells were rested for 2 days. The cells were incubated with the ROI-sensitive fluorophore 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (Carboxy-H2DCFDA) at 10 μM for 30 min followed by two washes with Hank’s Balance Salt Solution (HBSS). Then LTB4 (10 nM), live unopsonized GAS 10:1 or both were added and incubated for 30 min. The fluorescence intensity was measured at an excitation wavelength of 493 nm and emission wavelength of 522 nm using the fluorescent plate reader (above).
Measurement of Intracellular cAMP
For the intracellular cAMP measurement, 3 × 106 PMA treated THP-1 per well were used. The medium was changed to RPMI+/− and cells were incubated for 0, 15, 60, 300, 600 and 900 sec with 10 nM of LTB4 for some experimentsor 60 sec with 10 nM of LTB4 plus 900 sec followed by 1 μM of PGE2 for others. Culture supernatants were aspirated and the cells were lysed by incubation for 10 min with 0.1 M HCl at room temperature, followed by disruption using a cell scraper (20). Intracellular cAMP levels were determined by ELISA according to the manufacturer (Enzo Lifesciences).
Measurement of LTB4 and Cytokines
Uteri were removed 24 h post-infection to measure LTB4 and cytokines. Briefly, tissue was homogenized (Tissuemiser, Fisher Scientific, Suwanee, GA) in 1 ml of PBS, centrifuged and stored at −80°C until assayed. The OD of samples was determined at 405 nm on a microplate reader (VERSAMAX, Molecular Devices, Sunnyvale, CA), and concentrations of eicosanoids were calculated based on a standard curve. The sensitivity for LTB4 was 5.63 pg/ml. A specific enzyme immunoassay was used to quantify LTB4 (Enzo Lifesciences.) according to the manufacturer’s instructions. The tissue was recovered and stored at −80 °C until assays were performed to determine the levels of IL-17A, IL-6, IL-1β, TNF-α, IL-10, IFN-γ and MCP-1 by ELISA (R&D Duoset, R&D Systems). ELISAs were performed by the University of Michigan Cancer Center Cellular Immunology Core (14).
Analysis of Calcium Mobilization
PMA treated THP-1 cells were used at a density of 4 × 105 cells per well using 96-well, white bottom plates (BD Biosciences). The cells were incubated at 37°C plus 5% CO2 for 1 h to rest, and then the media was removed from the adherent cell culture, in order to eliminate sources of baseline fluorescence, particularly esterase activity. A volume of 100 μL of the dye loading solution was added to each well immediately. The plate was incubated for 30–45 min at 37°C, and after this, LTB4 (10 nM) and our positive control A23187 (1 μM) were added in the specific wells at the moment of the reading. Fluorescence was measured with excitation at 494 nm and emission at 516 nm. The assay was performed using a Fluo4-NM Calcium Kit (Invitrogen) according to the manufacturer’s instructions and adapted according to previous works (47, 48).
Statistical Analyses
Data are presented as the mean ± SEM and were analyzed with the Prism 5.0 statistical program (GraphPad Software). Comparisons among three or more experimental groups were performed with one-way ANOVA followed by a Bonferroni correction and between two experimental groups a Student t test was used, unless otherwise stated. Differences were considered significant for values of P < 0.05. Animal survival experiments were evaluated for differences using a Mantel-Cox log-rank test.
Results
5LO−/− mice have enhanced susceptibility to intrauterine GAS infection
Leukotrienes, including LTB4, are important endogenous regulators of immune defense against bacterial pathogens (21, 25, 49, 50), suggesting that they might be involved in supporting host defense against GAS. To test this, wild-type C57BL/6 mice or 5LO−/− (5LO−/−) mice were subjected to intrauterine GAS infections and monitored for survival, uterine bacterial clearance, and bacterial dissemination. As illustrated in Fig. 1A, 5LO−/− mice were significantly more susceptible to overwhelming infection caused by GAS than wild-type mice. It was found that 80% of the wild-type mice survived after seven days, while only 30% of 5LO−/− mice survived (P<0.0001). The enhanced susceptibility of 5LO−/− mice to intrauterine GAS infection was correlated with an impaired ability of these genetically deficient mice to contain the infection within the uterus (Fig. 1B). 24 h after infection, the number of bacteria that disseminated to the spleen in 5LO−/− mice was 1.48 times greater than was observed for wild-type mice. Levels of LTB4 were measured in uterine homogenates from wild-type and 5LO−/− mice following GAS infection. As shown in figure 1C, intrauterine infection with GAS significantly increased LTB4 production in the wild-type uterus. However, uterine tissues obtained from infected 5LO−/− mice did not reflect an increase in LTB4 generation compared to uninfected tissues. Interestingly, as evidenced in figure 1D, inflammatory mediator generation, including IL-1β, IL-6, IL-17 and MCP-1, was markedly increased in the uterine tissues of infected 5LO−/− mice compared with wild-type animals.
Figure 1. Leukotrienes are important determinants of survival and containment of intrauterine GAS infection in mice.

(A) Wild-type C57BL/6 or 5LO−/− mice(n = 10 per group) were inoculated with 104 CFU of GAS in the right uterine horn on day 0 and survival was recorded over time. Vehicle (PBS)-treated mice did not die (n = 10; data not shown). Data are expressed as mean ± SEM followed by Log-rank (Mantel-Cox) Test; ***P < 0.001. (B) Nonpregnant mice C57BL/6 or 5LO−/− (n = 10 per group) were inoculated with 104 CFU of GAS in the right uterine horn and then euthanized 24 h after infection. Bacterial loads from blood, spleen and uterus were determined as described in Materials and Methods. Data shown are from one representative experiment from two independent experiments with similar results. Data are expressed as mean ± SEM followed by unpaired Student t test; *P < 0.05. (C) After 24 h infection, enzyme immunoassay quantification of LTB4 concentrations were performed on uterine homogenates obtained from mice that had received either an intrauterine PBS injection (uninfected) or GAS. Data are presented as the mean ± SEM followed by one-way ANOVA and are representative of two independent experiments with similar results (n = 10 mice per group). ***P<0.001 compared with wild-type uninfected control. (D) Inflammatory mediators in uterine homogenates of C57BL/6 or 5LO−/− mice 24 h after inoculation with 104 CFU of GAS. 24 h after intrauterine inoculation with GAS cytokines and chemokines were measured by ELISA.***P < 0.001 by Student t test (n = 10 mice per group from 2 combined experiments with similar results). IL, interleukin; MCP, monocyte chemotactic protein-1.
LTB4 enhances the phagocytosis of unopsonized GAS
The importance of LTs in supporting clearance of GAS in vivo and the role of macrophages in host defense against GAS (10, 11) suggested that LTB4 might regulate macrophage interactions with GAS in vitro. Thus, primary human macrophages, mouse macrophages or THP-1 cells were pretreated with various concentrations of LTB4 (1–100 nM) that were found to enhance macrophage phagocytosis in other model systems (20, 50, 51). As outlined in figures 2A-E, LTB4 significantly increased the phagocytosis of unopsonized GAS by THP-1 cells, human peripheral blood monocytes, human DMs, human PMs, and mouse peritoneal macrophages. Because LTB4’s actions on THP-1 cells were similar to primary cells, we used this human macrophage-like cell line for mechanistic studies to build a model of how LTB4 regulates phagocyte function during GAS infection.
Figure 2. LTB4 increases leukocyte phagocytosis of GAS.

Macrophages were exposed for 15 min to LTB4 before challenge with FITC-labeled HK GAS and phagocytosis was determined as described in Materials and Methods. Data shown are from (A) PMA-treated human THP-1 cells, (B) human peripheral blood monocytes, (C) human decidual macrophages, (D) human placental macrophages, and (E) C57BL/6 mouse peritoneal macrophages. All data are expressed as a percentage of the phagocytic activity of untreated cells. *P < 0.05, **P<0.01 vs untreated as determined by one-way ANOVA for A-D and t-test for E.
The high-affinity, Gαi-coupled BLT1 receptor for LTB4 modulates phagocytosis in THP-1 cells
There are two major G protein-coupled receptors for LTB4, the high-affinity BLT1 receptor and the low-affinity BLT2 receptor (28). The BLT1 receptor has been implicated in macrophage immunoregulation by LTB4 (52) through a PTX-sensitive suppression of intracellular cAMP (20). We confirmed that THP-1 cells express the BLT1 receptor (39) by Western immunoblot analysis (not shown) and demonstrated that the LTB4-mediated enhancement of GAS phagocytosis by THP-1 cells was completely blocked by the BLT1 receptor antagonist U75302 (Fig. 3A). Further, 12-HHT, an agonist for the BLT2 receptor that is more potent than LTB4 (28), had no significant effect on THP-1 internalization of unopsonized bacteria (Fig. 3B).
Figure 3. LTB4 enhances GAS phagocytosis in THP-1 cells via the BLT-1 receptor.
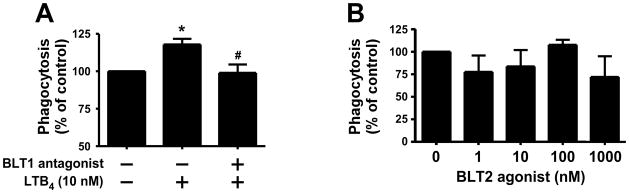
(A) PMA-treated THP-1 cells were incubated with the BLT1 receptor antagonist U75302 (10 μM) or LTB4 (10 nM) for 15 min before challenge with FITC-labeled HK GAS as described in Materials and Methods. Data shown are mean ± SEM of four independent experiments. (B) PMA-treated THP-1 cells were incubated with 0.001–1 μM of BLT2 agonist (12-HHT) for 15 min before challenge with HK FITC-GAS at 150:1. Phagocytosis was quantified after 3h by fluorometry as detailed in Materials and Methods. *P<0.05 vs untreated and #P<0.05 vs LTB4 as determined by one-way ANOVA.
The capacity of LTB4-BLT1 signaling to enhance GAS phagocytosis suggested that LTB4 reduced cAMP in these cells. We measured intracellular cAMP in macrophages after exposure to LTB4. Notably, cAMP levels fell acutely. Within 15 sec of exposure the intracellular levels were undetectable (Fig. 4A). Phagocytosis experiments with PTX (Fig. 4B) confirmed that the capacity for LTB4 to stimulate THP-1 cell ingestion of GAS was Gαi-dependent. Because the BLT1 receptor can signal through a Gαq-dependent release of intracellular calcium (53), we tested whether LTB4 treatment could increase Ca2+ in THP-1 cells. As opposed to the positive control compound, the calcium ionophore A23187, LTB4 failed to significantly alter basal calcium levels (Fig. 4C), consistent with results in lung macrophages (20). This was verified over a range of LTB4 concentrations and incubation times (data not shown).
Figure 4. Enhancement of phagocytosis by LTB4 depends on Gαi-receptor activation.

(A) PMA-treated THP-1 cells were cultured for 0, 15, 60, 300, 600 and 900 sec with LTB4 (10 nM) and intracellular cAMP was measure by ELISA. Mean +/− SEM data from five independent experiments were combined with n = 3 per each condition per experiment. ***P<0.001 (B) PMA-treated THP-1 cells were cultured overnight with 3 μg/mL of PTX and the 18 h later the cells were incubated for 15 min with LTB4 (10 nM) before GAS phagocytosis was quantified as detailed in Materials and Methods. *P<0.05 vs untreated cells. (C) FLUO-4NM-loaded, PMA-treated THP-1 cells were exposed to LTB4 (10 nM) and A23187 (1μM) and changes in intracellular Ca2+ were measured as described in Materials and Methods. Data are expressed as a percentage of relative to untreated cells. *P<0.05 vs untreated cells. .
LTB4 enhances bacterial killing of GAS
Along with phagocytosis, intracellular microbicidal activity is an important step in the control of infection. Previous work in other model systems has revealed that PGE2 can suppress the capacity of macrophages to kill bacteria (13, 18, 54) while endogenous and exogenous LTs enhance this action (20, 55). We determined the importance of LTB4 to macrophage killing of GAS using THP-1 cells. As noted in figure 5A, the number of intracellular bacteria present in THP-1 cells following 30 min of incubation was significantly reduced by treating cells with LTB4 (10 nM). Reactive oxygen intermediates (ROIs) generated by NADPHox are important for macrophage elimination of GAS (56) and it is known that LTB4 enhances NADPHox activation and ROI generation in macrophages (25, 55, 57–59). The extent to which the LTB4 stimulation of ROI production enhanced macrophage GAS killing was evaluated using the selective NADPHox inhibitor apocynin (60) Treatment with apocynin prevented the acute killing of intracellular bacteria during the 30 min of incubation of GAS with THP-1 cells (Fig. 5A). In addition, LTB4 failed to reduce the number of intracellular bacteria in macrophages pretreated with apocynin, suggesting that LTB4’s actions depend upon NAPDHox activation. We then tested whether LTB4 enhanced intracellular ROI generation in THP-1 cells infected with GAS (Fig. 5B). Notably, live GAS suppressed cellular ROI concentrations, an effect not associated with cell death (data not shown). However, LTB4 alone significantly increased ROI generation and this overcame the suppressive action of GAS (Fig. 5B).
Figure 5. LTB4 enhances macrophage killing of GAS through activation of reactive oxygen species production.
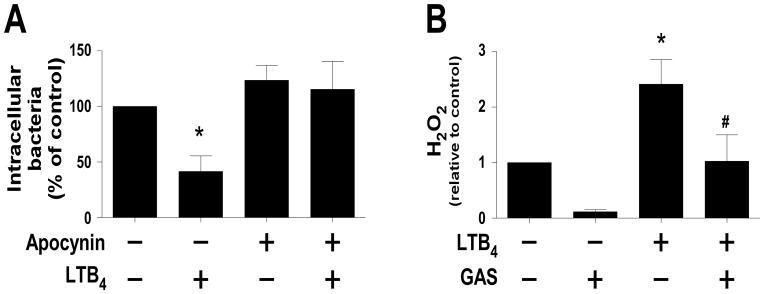
(A) PMA-treated THP-1 cells were exposed to apocynin (100 μM) for 30 min and/or LTB4 (10 nM) for 15 min prior to inoculation with live unopsonized GAS (10 bacteria per cell). Phagocytosis was determined as detailed in Materials and Methods, after 30 min THP-1 cells were washed to remove extracellular bacteria and lysed to release intracellular bacteria, which were enumerated on solid agar. Data are expressed as the percent of bacterial CFU relative to the untreated control and are presented as the mean ± SEM of five independent experiments. *P< 0.05 compared to untreated control as determined by one-way ANOVA. (B) PMA-treated THP-1 cells were incubated with H2DCFDA (10 μM) for 30 min followed by LTB4 (10 nM) and/or live unopsonized GAS (10 bacteria per cell) for another 30 min and H2O2 production was measured as described. Data are expressed relative to untreated cells and display the mean ± SEM of eight independent experiments. *P< 0.05 compared to untreated cells; #P<0.05 compared to LTB4 as determined by one-way ANOVA.
The cAMP-reducing actions of LTB4 antagonize PGE2 actions on GAS phagocytosis
If the actions of LTB4 are mediated through suppression of intracellular cAMP, then it can be hypothesized that this lipid would antagonize the capacity for PGE2 (which stimulates cAMP) to suppress macrophage host defense functions. Indeed, PGE2 raised intracellular cAMP concentrations and inhibited the phagocytosis of GAS by THP-1 cells (Fig. 6A and Fig. 6 B). As noted in Fig. 6A, PGE2 increased cAMP, LTB4 reduced it, and the effect of a combination of both lipids was intermediate (Fig. 6A). In addition (Fig. 6B), 10 μM of PGE2 significantly inhibited GAS phagocytosis by 56.5 ± 3.0% while 100 nM of LTB4 enhanced it by 44.4 ± 7.3%. However, when cells were exposed to both compounds, the net effect was a lack of significant change in phagocytic activity relative to untreated cells (Fig. 6B).
Figure 6. Effects of exogenous PGE2 and LTB4 on phagocytosis and cAMP production in macrophages.
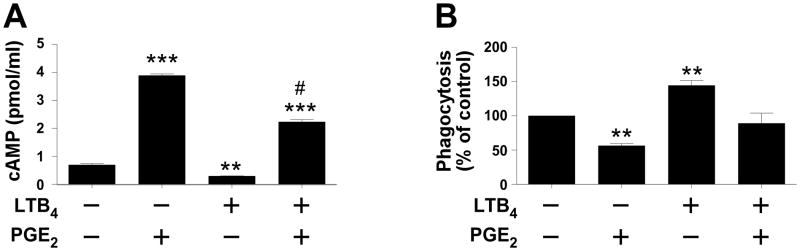
(A) PMA-treated THP-1 cells were exposed for 1 min to LTB4 (10 nM) or vehicle followed by PGE2 (1 μM) for 3 min. Intracellular cAMP was determined as described in the text. Data are expressed as the mean ± SEM from seven independent experiments. ***P<0.001; **P<0.01 compared to untreated cells; #P<0.001 vs. PGE2 treatment. (B) PMA-treated THP-1 cells were cultured with LTB4 (100 nM) and PGE2 (10 μM) for 15 min before challenge with HK FITC-GAS. Phagocytosis was quantified after 3 h as detailed in Materials and Methods. Data are expressed as the mean ± SEM relative to untreated cells for four independent experiments. ** P<0.01 compared to untreated and #P<0.0001 compared to PGE2 as determined by one-way ANOVA.
Discussion
Streptococcus pyogenes is a leading cause of severe postpartum sepsis worldwide (1, 61). A better understanding of why pregnancy and the postpartum state increase the risk for GAS is needed to develop better preventive or therapeutic strategies. Herein we report the novel finding that LTB4 is an endogenous lipid mediator that augments innate immune defenses against invasive GAS infection. Macrophages are important sentinels of host defense against GAS (10, 11). These studies suggest that LTB4, a major product of the 5LO metabolic pathway, enhances the phagocytosis and bacterial killing of unopsonized GAS by macrophages, including primary human macrophages from the female reproductive tract. The mechanism for these actions involves signalling via the Gαi –coupled BLT1 receptor with an acute suppression of cAMP.
Earlier studies established the importance of LTs as immunostimulatory mediators during infection with non-GAS pathogens (21, 25, 42, 49, 62, 63). In fact, the capacity of LTB4 to regulate host-streptococcal interactions has been shown in a model of lung infection caused by S. pneumoniae (21–24). In addition, the administration of aerosolized LTB4 to mice with pre-existing pneumococcal pneumonia increased mononuclear phagocyte/macrophage accumulation in the lungs, NADPHox subunit expression in pulmonary macrophages, and pulmonary bacterial clearance (25). While these data provided an important rationale for our hypothesis, there was little known about the role of LTB4 in GAS infections or reproductive tract infections.
A significant finding includes the critical importance of the enzyme 5LO in supporting immune containment of intrauterine S. pyogenes infection. Unlike wild-type mice, the mice lacking the 5LO enzyme did not produce meaningful quantities of LTB4 following infection, they were significantly more susceptible to death from GAS endometritis, they were less capable of preventing dissemination of bacteria from the uterus to distant sites, and they displayed a significantly enhanced inflammatory response to infection. These findings suggest that an overwhelming inflammatory response, due to poor innate immune containment of a local GAS infection, played a role in the mortality of this infection in 5LO−/− mice. Establishing a causal relationship between the exaggerated inflammatory response to GAS in 5LO−/− mice and their mortality will require further study.
It is important that LTB4 has been shown to augment macrophage immune defenses against several pathogens in vivo (49, 51, 55, 62, 63) and to amplify the ability of leukocytes do phagocytose and kill microbes (43, 49, 51, 62–65). Our results support the generalizability of the previous reports and add evidence that one causal mechanism by which LTB4-BLT1 coupling regulates macrophage behaviors is by reducing cAMP production through a PTX-sensitive signaling cascade. How the acute lowering of basal cAMP levels impair host defense against GAS requires more exploration but both phagocytosis and ROI-dependent bacterial killing are certainly involved. cAMP is a critical intracellular immunoregulatory molecule that can rapidly dampen macrophage antimicrobial responses (19). The lipid mediator PGE2, for example, has been shown to suppress macrophage-GAS interactions via this second messenger (18). Our results confirm that report and extend them to demonstrate that a significant interaction is possible when LTB4 and PGE2 costimulate a macrophage at the time of GAS infection (summarized in Fig. 7). Such crosstalk between these lipid mediators has been shown in other systems (17), adding generalizability to this finding.
Figure 7. Summary of immunoregulation of macrophages by LTB4 during Streptococcus pyogenes infection.

Ligation of BLT1 by LTB4 results in the activation of Gαi protein, with a subsequent decrease in intracellular cAMP levels. This, in turn, limits the activation of protein kinase A (PKA), an endogenous suppressor of both phagocytosis and NADPHox-dependent reactive oxygen intermediate (ROI) generation. These actions oppose those of the endogenous lipid mediator prostaglandin E2 (PGE2), which stimulates cAMP production via EP2 and/or EP4 receptors in macrophages. Compounds used in our studies to modulate signaling are shown. AC, adenylate cyclase; APY, apocynin; PTX, pertussis toxin.
This work has limitations, which underscore areas requiring investigation in future studies. For example, 5LO−/− mice not only failed to generate increased LTB4 during infection, but they also lacked the capacity to produce the cysteinyl LTs, such as LTC4, D4, and E4. The extent to which such compounds affect survival or bacterial clearance during infection will require further study. Preliminary studies in our lab suggest that exogenously-added cysteinyl LTs also have positive immunomodulatory actions on macrophage-GAS interactions (data not shown) and their contribution to the in vivo phenotype of infected mice remains to be determined. Another limitation was our use of a human macrophage-like cell line to build a model of LTB4 immunoregulation. We used this THP-1 cell line because it demonstrated similar responsiveness to LTB4 in the regulation of GAS phagocytosis as several other primary macrophage types, including human and mouse macrophages. A longer term plan is to validate our complete model in primary cells.
The mouse model of intrauterine infection that we have newly developed for this study involved nonpregnant mice that were also not immediately postpartum. It is possible that different results would be obtained if experiments were conducted in such animals, particularly given the hormonal and immunological differences associated with pregnancy and the puerperium (6, 66). Experiments to assess the impact of pregnancy and the postpartum state on this infection are planned.
In summary, this study newly establishes a role for LTs in positively regulating innate immune defenses against the puerperal sepsis pathogen GAS. These findings confirm those in other model systems and suggest that exploiting this endogenous signaling system pharmacologically might be a robust mechanism for improving therapeutic approaches against this infection. For example, amplifying the synthesis and signaling LTB4 while limiting the production and responsiveness to PGE2 might significantly enhance the efficacy of existing medical treatments of severe GAS infections. Future studies to assess such approaches are needed. The global problem of increasing antimicrobial resistance further highlights the ongoing need for new treatment modalities for life-threatening infections. Reducing maternal mortality requires ongoing studies of fundamental mechanisms of disease with a goal of identifying novel targets for treatment and prevention.
Acknowledgments
The authors wish to thank Jill Mogle, RN for technical assistance.
Funding
This work was supported by CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior), grant number 2652110, FAPESP (Fundação de Amparo a Pesquisa do Estado de São Paulo), Brazil (EMS, LHF); an Investigators in the Pathogenesis of Infectious Disease grant from the Burroughs Wellcome Fund (DMA), a Scientific Exchange Fellowship from the International Society for Infectious Diseases (EMS), and a National Institutes of Health grant, T32HL007749 (KLM) and HL103777-03 (CHS).
Abbreviations used in this paper
- AM
alveolar macrophage
- Anti-anti
antibiotic (penicillin and streptomycin) antimycotic (amphotericin) 100X. Ca+2, calcium
- CFU
colony forming unit
- COX
cyclooxygenase. DM, decidual macrophage
- GAS
Group A Streptococcus
- GPCR
G protein coupled receptor. HK, heat killed. LT, leukotriene
- MOI
multiple of infection
- NADPHox
NADPH-oxidase. PM, placental macrophage
- PKA
protein kinase A
- PTX
pertussis toxin. ROI, reactive oxygen intermediate
- RPMI+/+
RPMI 1640 supplemented with 1% antibiotic-antimycotic and 10% charcoal/dextran treated FBS
- RPMI+/−
RPMI 1640 supplemented with 1% antibiotic-antimycotic in the absence of serum
- THY
Todd Hewitt Broth plus 5% of yeast
- TSA-blood
Tryptic soy agar plus 5% sheep blood
- PBS
Phosphate buffered saline
- PMA
phorbol myristate acetate
- VASP
vasodilator-stimulated phosphoprotein
- 12-HHT-12(S)-hydroxyheptadeca-5Z
8E, 10E-trienoic acid
Footnotes
Disclosures
The authors have no conflict of interest.
References
- 1.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 2.van Dillen J, Zwart J, Schutte J, van Roosmalen J. Maternal sepsis: epidemiology, etiology and outcome. Curr Opin Infect Dis. 2010;23:249–254. doi: 10.1097/QCO.0b013e328339257c. [DOI] [PubMed] [Google Scholar]
- 3.Sriskandan S. Severe peripartum sepsis. J R Coll Physicians Edinb. 2011;41:339–346. doi: 10.4997/JRCPE.2011.411. [DOI] [PubMed] [Google Scholar]
- 4.Cantwell R, Clutton-Brock T, Cooper G, Dawson A, Drife J, Garrod D, Harper A, Hulbert D, Lucas S, McClure J, Millward-Sadler H, Neilson J, Nelson-Piercy C, Norman J, O’Herlihy C, Oates M, Shakespeare J, de Swiet M, Williamson C, Beale V, Knight M, Lennox C, Miller A, Parmar D, Rogers J, Springett A. Saving Mothers’ Lives: Reviewing maternal deaths to make motherhood safer: 2006–2008. The Eighth Report of the Confidential Enquiries into Maternal Deaths in the United Kingdom. BJOG. 2011;118(Suppl 1):1–203. doi: 10.1111/j.1471-0528.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 5.Mor G, Cardenas I. The immune system in pregnancy: a unique complexity. American journal of reproductive immunology. 2010;63:425–433. doi: 10.1111/j.1600-0897.2010.00836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mason KL, Aronoff DM. Postpartum group A Streptococcus sepsis and maternal immunology. Am J Reprod Immunol. 2012;67:91–100. doi: 10.1111/j.1600-0897.2011.01083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deutscher M, Lewis M, Zell ER, Taylor TH, Jr, Van Beneden C, Schrag S. Incidence and severity of invasive Streptococcus pneumoniae, group A Streptococcus, and group B Streptococcus infections among pregnant and postpartum women. Clin Infect Dis. 2011;53:114–123. doi: 10.1093/cid/cir325. [DOI] [PubMed] [Google Scholar]
- 8.Goulet V, Hebert M, Hedberg C, Laurent E, Vaillant V, De Valk H, Desenclos JC. Incidence of listeriosis and related mortality among groups at risk of acquiring listeriosis. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2012;54:652–660. doi: 10.1093/cid/cir902. [DOI] [PubMed] [Google Scholar]
- 9.Davis SM, Clark EA, Nelson LT, Silver RM. The association of innate immune response gene polymorphisms and puerperal group A streptococcal sepsis. Am J Obstet Gynecol. 2010;202:308, e301–308. doi: 10.1016/j.ajog.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Goldmann O, Rohde M, Chhatwal GS, Medina E. Role of macrophages in host resistance to group A streptococci. Infect Immun. 2004;72:2956–2963. doi: 10.1128/IAI.72.5.2956-2963.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mishalian I, Ordan M, Peled A, Maly A, Eichenbaum MB, Ravins M, Aychek T, Jung S, Hanski E. Recruited macrophages control dissemination of group A Streptococcus from infected soft tissues. J Immunol. 2011;187:6022–6031. doi: 10.4049/jimmunol.1101385. [DOI] [PubMed] [Google Scholar]
- 12.Goldmann O, Hertzen E, Hecht A, Schmidt H, Lehne S, Norrby-Teglund A, Medina E. Inducible Cyclooxygenase Released Prostaglandin E2 Modulates the Severity of Infection Caused by Streptococcus pyogenes. J Immunol. 2010 doi: 10.4049/jimmunol.1000838. [DOI] [PubMed] [Google Scholar]
- 13.Serezani CH, Chung J, Ballinger MN, Moore BB, Aronoff DM, Peters-Golden M. Prostaglandin E2 suppresses bacterial killing in alveolar macrophages by inhibiting NADPH oxidase. Am J Respir Cell Mol Biol. 2007;37:562–570. doi: 10.1165/rcmb.2007-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aronoff DM, I, Bergin L, Lewis C, Goel D, O’Brien E, Peters-Golden M, Mancuso P. E-prostanoid 2 receptor signaling suppresses lung innate immunity against Streptococcus pneumoniae. Prostaglandins Other Lipid Mediat. 2012;98:23–30. doi: 10.1016/j.prostaglandins.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bourdonnay E, Serezani CH, Aronoff DM, Peters-Golden M. Regulation of alveolar macrophage p40phox: hierarchy of activating kinases and their inhibition by PGE2. J Leukoc Biol. 2012;92:219–231. doi: 10.1189/jlb.1211590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serezani CH, Lewis C, Jancar S, Peters-Golden M. Leukotriene B4 amplifies NF-kappaB activation in mouse macrophages by reducing SOCS1 inhibition of MyD88 expression. J Clin Invest. 2011;121:671–682. doi: 10.1172/JCI43302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee SP, Serezani CH, Medeiros AI, Ballinger MN, Peters-Golden M. Crosstalk between prostaglandin E2 and leukotriene B4 regulates phagocytosis in alveolar macrophages via combinatorial effects on cyclic AMP. J Immunol. 2009;182:530–537. doi: 10.4049/jimmunol.182.1.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldmann O, Hertzen E, Hecht A, Schmidt H, Lehne S, Norrby-Teglund A, Medina E. Inducible cyclooxygenase released prostaglandin E2 modulates the severity of infection caused by Streptococcus pyogenes. J Immunol. 2010;185:2372–2381. doi: 10.4049/jimmunol.1000838. [DOI] [PubMed] [Google Scholar]
- 19.Serezani CH, Ballinger MN, Aronoff DM, Peters-Golden M. Cyclic AMP: master regulator of innate immune cell function. Am J Respir Cell Mol Biol. 2008;39:127–132. doi: 10.1165/rcmb.2008-0091TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peres CM, Aronoff DM, Serezani CH, Flamand N, Faccioli LH, Peters-Golden M. Specific leukotriene receptors couple to distinct G proteins to effect stimulation of alveolar macrophage host defense functions. J Immunol. 2007;179:5454–5461. doi: 10.4049/jimmunol.179.8.5454. [DOI] [PubMed] [Google Scholar]
- 21.Mancuso P, Peters-Golden M, Goel D, Goldberg J, Brock TG, Greenwald-Yarnell M, Myers MG., Jr Disruption of leptin receptor-STAT3 signaling enhances leukotriene production and pulmonary host defense against pneumococcal pneumonia. J Immunol. 2011;186:1081–1090. doi: 10.4049/jimmunol.1001470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aronoff DM, Lewis C, Serezani CH, Eaton KA, Goel D, Phipps JC, Peters-Golden M, Mancuso P. E-prostanoid 3 receptor deletion improves pulmonary host defense and protects mice from death in severe Streptococcus pneumoniae infection. J Immunol. 2009;183:2642–2649. doi: 10.4049/jimmunol.0900129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mancuso P, Huffnagle GB, Olszewski MA, Phipps J, Peters-Golden M. Leptin corrects host defense defects after acute starvation in murine pneumococcal pneumonia. Am J Respir Crit Care Med. 2006;173:212–218. doi: 10.1164/rccm.200506-909OC. [DOI] [PubMed] [Google Scholar]
- 24.Schultz MJ, Wijnholds J, Peppelenbosch MP, Vervoordeldonk MJ, Speelman P, van Deventer SJ, Borst P, van der Poll T. Mice lacking the multidrug resistance protein 1 are resistant to Streptococcus pneumoniae-induced pneumonia. J Immunol. 2001;166:4059–4064. doi: 10.4049/jimmunol.166.6.4059. [DOI] [PubMed] [Google Scholar]
- 25.Mancuso P, Lewis C, Serezani CH, Goel D, Peters-Golden M. Intrapulmonary administration of leukotriene B4 enhances pulmonary host defense against pneumococcal pneumonia. Infect Immun. 2010;78:2264–2271. doi: 10.1128/IAI.01323-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haeggstrom JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev. 2011;111:5866–5898. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- 27.Peters-Golden M, Henderson WR., Jr Leukotrienes. N Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 28.Yokomizo T. Leukotriene B4 receptors: novel roles in immunological regulations. Adv Enzyme Regul. 2011;51:59–64. doi: 10.1016/j.advenzreg.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 29.Iizuka Y, Yokomizo T, Terawaki K, Komine M, Tamaki K, Shimizu T. Characterization of a mouse second leukotriene B4 receptor, mBLT2: BLT2-dependent ERK activation and cell migration of primary mouse keratinocytes. J Biol Chem. 2005;280:24816–24823. doi: 10.1074/jbc.M413257200. [DOI] [PubMed] [Google Scholar]
- 30.Kamohara M, Takasaki J, Matsumoto M, Saito T, Ohishi T, Ishii H, Furuichi K. Molecular cloning and characterization of another leukotriene B4 receptor. J Biol Chem. 2000;275:27000–27004. doi: 10.1074/jbc.C000382200. [DOI] [PubMed] [Google Scholar]
- 31.Wang S, Gustafson E, Pang L, Qiao X, Behan J, Maguire M, Bayne M, Laz T. A novel hepatointestinal leukotriene B4 receptor. Cloning and functional characterization. J Biol Chem. 2000;275:40686–40694. doi: 10.1074/jbc.M004512200. [DOI] [PubMed] [Google Scholar]
- 32.Yokomizo T, Kato K, Hagiya H, Izumi T, Shimizu T. Hydroxyeicosanoids bind to and activate the low affinity leukotriene B4 receptor, BLT2. J Biol Chem. 2001;276:12454–12459. doi: 10.1074/jbc.M011361200. [DOI] [PubMed] [Google Scholar]
- 33.Pasetto N, Zicari A, Piccione E, Lenti L, Pontieri G, Ticconi C. Influence of labor and oxytocin on in vitro leukotriene release by human fetal membranes and uterine decidua at term gestation. Am J Obstet Gynecol. 1992;166:1500–1506. doi: 10.1016/0002-9378(92)91626-l. [DOI] [PubMed] [Google Scholar]
- 34.Brown NL, Slater DM, Alvi SA, Elder MG, Sullivan MH, Bennett PR. Expression of 5-lipoxygenase and 5-lipoxygenase-activating protein in human fetal membranes throughout pregnancy and at term. Mol Hum Reprod. 1999;5:668–674. doi: 10.1093/molehr/5.7.668. [DOI] [PubMed] [Google Scholar]
- 35.Nagamatsu T, Schust DJ. The immunomodulatory roles of macrophages at the maternal-fetal interface. Reprod Sci. 2010;17:209–218. doi: 10.1177/1933719109349962. [DOI] [PubMed] [Google Scholar]
- 36.Arredouani M, Yang Z, Ning Y, Qin G, Soininen R, Tryggvason K, Kobzik L. The scavenger receptor MARCO is required for lung defense against pneumococcal pneumonia and inhaled particles. J Exp Med. 2004;200:267–272. doi: 10.1084/jem.20040731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aronoff DM, Hao Y, Chung J, Coleman N, Lewis C, Peres CM, Serezani CH, Chen GH, Flamand N, Brock TG, Peters-Golden M. Misoprostol impairs female reproductive tract innate immunity against Clostridium sordellii. J Immunol. 2008;180:8222–8230. doi: 10.4049/jimmunol.180.12.8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thelen T, Hao Y, Medeiros AI, Curtis JL, Serezani CH, Kobzik L, Harris LH, Aronoff DM. The class A scavenger receptor, macrophage receptor with collagenous structure, is the major phagocytic receptor for Clostridium sordellii expressed by human decidual macrophages. J Immunol. 2010;185:4328–4335. doi: 10.4049/jimmunol.1000989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hashidate T, Murakami N, Nakagawa M, Ichikawa M, Kurokawa M, Shimizu T, Nakamura M. AML1 enhances the expression of leukotriene B4 type-1 receptor in leukocytes. FASEB J. 2010;24:3500–3510. doi: 10.1096/fj.10-156844. [DOI] [PubMed] [Google Scholar]
- 40.Daigneault M, Preston JA, Marriott HM, Whyte MK, Dockrell DH. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS ONE. 2010;5:e8668. doi: 10.1371/journal.pone.0008668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vince GS, Starkey PM, Jackson MC, Sargent IL, Redman CW. Flow cytometric characterisation of cell populations in human pregnancy decidua and isolation of decidual macrophages. J Immunol Methods. 1990;132:181–189. doi: 10.1016/0022-1759(90)90028-t. [DOI] [PubMed] [Google Scholar]
- 42.Secatto A, Rodrigues LC, Serezani CH, Ramos SG, Dias-Baruffi M, Faccioli LH, Medeiros AI. 5-Lipoxygenase deficiency impairs innate and adaptive immune responses during fungal infection. PLoS ONE. 2012;7:e31701. doi: 10.1371/journal.pone.0031701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bailie MB, Standiford TJ, Laichalk LL, Coffey MJ, Strieter R, Peters-Golden M. Leukotriene-deficient mice manifest enhanced lethality from Klebsiella pneumonia in association with decreased alveolar macrophage phagocytic and bactericidal activities. J Immunol. 1996;157:5221–5224. [PubMed] [Google Scholar]
- 44.Aronoff DM, Canetti C, Peters-Golden M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J Immunol. 2004;173:559–565. doi: 10.4049/jimmunol.173.1.559. [DOI] [PubMed] [Google Scholar]
- 45.Thiele L, Rothen-Rutishauser B, Jilek S, Wunderli-Allenspach H, Merkle HP, Walter E. Evaluation of particle uptake in human blood monocyte-derived cells in vitro. Does phagocytosis activity of dendritic cells measure up with macrophages? J Control Release. 2001;76:59–71. doi: 10.1016/s0168-3659(01)00412-6. [DOI] [PubMed] [Google Scholar]
- 46.McGuire KA, Barlan AU, Griffin TM, Wiethoff CM. Adenovirus type 5 rupture of lysosomes leads to cathepsin B-dependent mitochondrial stress and production of reactive oxygen species. J Virol. 2011;85:10806–10813. doi: 10.1128/JVI.00675-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanson BJ. Multiplexing Fluo-4 NW and a GeneBLAzer transcriptional assay for high-throughput screening of G-protein-coupled receptors. J Biomol Screen. 2006;11:644–651. doi: 10.1177/1087057106289982. [DOI] [PubMed] [Google Scholar]
- 48.Xi Q, Wang S, Ye Z, Liu J, Yu X, Zhu Z, Su S, Bai J, Li C. Adenovirus-delivered microRNA targeting the vitamin D receptor reduces intracellular Ca(2)(+) concentrations by regulating the expression of Ca(2)(+)-transport proteins in renal epithelial cells. BJU Int. 2011;107:1314–1319. doi: 10.1111/j.1464-410X.2010.09444.x. [DOI] [PubMed] [Google Scholar]
- 49.Peres CM, de Paula L, Medeiros AI, Sorgi CA, Soares EG, Carlos D, Peters-Golden M, Silva CL, Faccioli LH. Inhibition of leukotriene biosynthesis abrogates the host control of Mycobacterium tuberculosis. Microbes and infection/Institut Pasteur. 2007;9:483–489. doi: 10.1016/j.micinf.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mancuso P, Nana-Sinkam P, Peters-Golden M. Leukotriene B4 augments neutrophil phagocytosis of Klebsiella pneumoniae. Infect Immun. 2001;69:2011–2016. doi: 10.1128/IAI.69.4.2011-2016.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mancuso P, Standiford TJ, Marshall T, Peters-Golden M. 5-Lipoxygenase reaction products modulate alveolar macrophage phagocytosis of Klebsiella pneumoniae. Infect Immun. 1998;66:5140–5146. doi: 10.1128/iai.66.11.5140-5146.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Serezani CH, Aronoff DM, Sitrin RG, Peters-Golden M. FcgammaRI ligation leads to a complex with BLT1 in lipid rafts that enhances rat lung macrophage antimicrobial functions. Blood. 2009;114:3316–3324. doi: 10.1182/blood-2009-01-199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Canetti C, Aronoff DM, Choe M, Flamand N, Wettlaufer S, Toews GB, Chen GH, Peters-Golden M. Differential regulation by leukotrienes and calcium of Fc gamma receptor-induced phagocytosis and Syk activation in dendritic cells versus macrophages. J Leukoc Biol. 2006;79:1234–1241. doi: 10.1189/jlb.0705374. [DOI] [PubMed] [Google Scholar]
- 54.Medeiros AI, Serezani CH, Lee SP, Peters-Golden M. Efferocytosis impairs pulmonary macrophage and lung antibacterial function via PGE2/EP2 signaling. J Exp Med. 2009 doi: 10.1084/jem.20082058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Serezani CH, Aronoff DM, Jancar S, Mancuso P, Peters-Golden M. Leukotrienes enhance the bactericidal activity of alveolar macrophages against Klebsiella pneumoniae through the activation of NADPH oxidase. Blood. 2005;106:1067–1075. doi: 10.1182/blood-2004-08-3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goldmann O, von Kockritz-Blickwede M, Holtje C, Chhatwal GS, Geffers R, Medina E. Transcriptome analysis of murine macrophages in response to infection with Streptococcus pyogenes reveals an unusual activation program. Infect Immun. 2007;75:4148–4157. doi: 10.1128/IAI.00181-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Serezani CH, Aronoff DM, Jancar S, Peters-Golden M. Leukotriene B4 mediates p47phox phosphorylation and membrane translocation in polyunsaturated fatty acid-stimulated neutrophils. J Leukoc Biol. 2005;78:976–984. doi: 10.1189/jlb.1004587. [DOI] [PubMed] [Google Scholar]
- 58.Yun MR, Park HM, Seo KW, Lee SJ, Im DS, Kim CD. 5-Lipoxygenase plays an essential role in 4-HNE-enhanced ROS production in murine macrophages via activation of NADPH oxidase. Free Radic Res. 2010;44:742–750. doi: 10.3109/10715761003758122. [DOI] [PubMed] [Google Scholar]
- 59.Morato-Marques M, Campos MR, Kane S, Rangel AP, Lewis C, Ballinger MN, Kim SH, Peters-Golden M, Jancar S, Serezani CH. Leukotrienes target F-actin/cofilin-1 to enhance alveolar macrophage anti-fungal activity. J Biol Chem. 2011;286:28902–28913. doi: 10.1074/jbc.M111.235309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vejrazka M, Micek R, Stipek S. Apocynin inhibits NADPH oxidase in phagocytes but stimulates ROS production in non-phagocytic cells. Biochim Biophys Acta. 2005;1722:143–147. doi: 10.1016/j.bbagen.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 61.Aronoff DM, Mulla ZD. Postpartum invasive group A streptococcal disease in the modern era. Infect Dis Obstet Gynecol. 2008;2008:796892. doi: 10.1155/2008/796892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Medeiros AI, Sa-Nunes A, Turato WM, Secatto A, Frantz FG, Sorgi CA, Serezani CH, Deepe GS, Jr, Faccioli LH. Leukotrienes are potent adjuvant during fungal infection: effects on memory T cells. J Immunol. 2008;181:8544–8551. doi: 10.4049/jimmunol.181.12.8544. [DOI] [PubMed] [Google Scholar]
- 63.Medeiros AI, Sa-Nunes A, Soares EG, Peres CM, Silva CL, Faccioli LH. Blockade of endogenous leukotrienes exacerbates pulmonary histoplasmosis. Infect Immun. 2004;72:1637–1644. doi: 10.1128/IAI.72.3.1637-1644.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mancuso P, Peters-Golden M. Modulation of alveolar macrophage phagocytosis by leukotrienes is Fc receptor-mediated and protein kinase C-dependent. Am J Respir Cell Mol Biol. 2000;23:727–733. doi: 10.1165/ajrcmb.23.6.4246. [DOI] [PubMed] [Google Scholar]
- 65.Serezani CH, Perrela JH, Russo M, Peters-Golden M, Jancar S. Leukotrienes are essential for the control of Leishmania amazonensis infection and contribute to strain variation in susceptibility. J Immunol. 2006;177:3201–3208. doi: 10.4049/jimmunol.177.5.3201. [DOI] [PubMed] [Google Scholar]
- 66.Robinson DP, Klein SL. Pregnancy and pregnancy-associated hormones alter immune responses and disease pathogenesis. Horm Behav. 2012 doi: 10.1016/j.yhbeh.2012.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]