

Abstract
RNA-binding proteins (RBPs) are effectors and regulators of posttranscriptional gene regulation (PTGR). RBPs regulate stability, maturation, and turnover of all RNAs, often binding thousands of targets at many sites. The importance of RBPs is underscored by their dysregulation or mutations causing a variety of developmental and neurological diseases. This chapter globally discusses human RBPs and provides a brief introduction to their identification and RNA targets. We review RBPs based on common structural RNA-binding domains, study their evolutionary conservation and expression, and summarize disease associations of different RBP classes.
Keywords: RNA-binding domains, overview; RNA-binding proteins, tissue specificity; RNA-binding proteins, abundance; RNA-binding proteins, genetic diseases
1 Principles of Posttranscriptional Gene Regulation
RNA is an essential constituent of all living organisms and central to decoding the genetic information of every cell. Recent advances in RNA sequencing technologies have facilitated the discovery of novel transcripts and we will soon know the precise composition of most cellular transcriptomes. While functional annotation for many RNAs is still in progress, the major classes of RNAs have now been described (Table 1.1). The most abundant RNAs, constituting 90 % of cellular RNAs by copy number, are shared by all organisms and required for protein synthesis: rRNAs, tRNAs, and mRNAs (Table 1.1). The remaining 10 % are noncod-ing RNAs (ncRNAs) that mainly serve as guides or molecular scaffolds in a variety of processes including RNA splicing, RNA modification, and RNA silencing. The structure, length, and composition of these RNAs and their ribonucleoprotein particles (RNP) are distinct and allow their integration into diverse functions and layers of regulation to control target RNAs and their many functions.
Table 1.1.
Functional description of the main RNA classes in humans and their length distribution
| RNA class | Size (nt) | Biological role (additional reviews on function and biogenesis) |
|---|---|---|
| Messenger RNA (mRNA) | ~200–100,000 | Encodes the information for protein-coding genes, translated by ribosomes (Dreyfuss et al. 2002; Glisovic et al. 2008; Müller-McNicoll and Neugebauer 2013) |
| Transfer RNA (tRNA) | ~70–95 | RNA adaptor molecule, transports amino acids to ribosome and recognizes specific triplet codons on mRNA (Suzuki et al. 2011; Maraia and Lamichhane 2011; Simos and Hurt 1999) |
| Ribosomal RNA (rRNA) | 121–5,072 | Structural component of ribosomes (Boisvert et al. 2007; Ciganda and Williams 2011; Granneman and Baserga 2004) |
| Small nuclear RNA (snRNA) | ~70–190 | snRNAs U1, U2, U4, U5, U6, U11, U12, U4atac, and U6atac are core components of the spliceosome; U7 snRNA functions in 3′end maturation of histone RNAs (Kiss 2004; Matera et al. 2007) |
| Small nucleolar RNA (snoRNA) and small Cajal-body-specific RNA (scaRNA) | ~50–450 | Guide chemical modifications (methylation and pseudouridylation) of rRNAs, snRNAs, and snoRNAs (Filipowicz and Pogacić 2002; Kiss et al. 2006; Matera et al. 2007) |
| microRNA (miRNA) and small interfering RNA (siRNA) | 21–22 | Associate with AGO proteins, guide them to target sequences predominantly in the 3′UTRs of mRNAs, induce degradation and translational repression (Bartel 2009; Kim et al. 2009) |
| piwi-interacting RNA (piRNA) | ~28–32 | Associates with PIWI proteins; PIWI RNP complexes induce ribonucleolytic cleavage and epigenetic silencing of transposable elements (Kim et al. 2009; Siomi et al. 2011) |
| Long intervening noncoding RNA (lincRNA), 7SK RNA | >200 | Recruits chromatin modifiers and remodeling complexes, modulates transcription by recruitment of protein cofactors to transcription starts sites and enhancers, functions as molecular scaffolds for nuclear RBPs (Batista and Chang 2013; Ulitsky and Bartel 2013); 7SK RNA regulates transcription elongation (Peterlin et al. 2011) |
| Ribonuclease P/(RNase P) and mitochondrial RNA-processing endonuclease (MRP RNase) | ~260–340 | Ribonucleolytic RNP complexes that carry out processing of precursor tRNAs, rRNAs, snRNAs, and other noncoding RNAs (Xiao et al. 2002; Jarrous 2002; Ellis and Brown 2009; Esakova and Krasilnikov 2010) |
| Y RNA | ~80–110 | Small noncoding RNAs that form an RNP complex with TROVE2 (Ro60) protein and act as RNA chaperones, have a role in DNA replication and immune response (Hall et al. 2013; Köhn et al. 2013) |
| Signal recognition particle RNA (7SL/SRP RNA) | ~300 | RNA of the signal recognition particle; the complex recognizes signal sequences of newly synthesized peptides and targets them to the endoplasmatic reticulum (Akopian et al. 2013) |
| Vault-associated RNA (vtRNA) | ~80–120 | Small noncoding RNAs, part of the vault RNP complex, involved in drug resistance, downregulate mRNA targets through posttranscriptional gene silencing (Berger et al. 2008) |
| Telomerase RNA (telRNA) | ~450 | RNA component of the telomerase complex TERC, which acts as reverse transcriptase and elongates telomerase repeats, TERC is structurally related to box H/ACA snoRNAs (Egan and Collins 2012) |
Additional reviews on biogenesis pathways and RBP components interacting with each class of RNA are referenced
Posttranscriptional gene regulation (PTGR) is a term that refers to the cellular processes that control gene expression at the level of RNA; it encompasses RNA maturation, modification, transport, and degradation. Consequently, every RNA molecule independent of its ultimate function is at some level subject to PTGR. RNA- binding proteins (RBPs) are central players of PTGR, as they directly bind to RNAs to form RNPs. In many cases, the RNP is the most basic unit, comprising a complex of obligate RNA and protein partners (e.g., snRNPs, snoRNPs, RNase P, ribosome subunits), which elicits its respective function. However, many other types of RNAs, particularly mRNAs and tRNAs, only transiently associate with RBPs, whose functions are necessary for their proper maturation, localization, and turnover (Dreyfuss et al. 2002; Granneman and Baserga 2004; Phizicky and Hopper 2010; Müller-McNicoll and Neugebauer 2013). Indeed most mRNA-binding proteins (mRBPs) have thousands of targets they regulate (Ascano et al. 2011). Hence the proper assembly and function of RNA-protein complexes are critical for development and maintenance of all cells and organisms. For a large fraction of RBPs, we are only starting to understand the complexity of their basic molecular roles, modes of recognition, and global targets.
In this chapter we review the current state of knowledge of the protein components involved in PTGR in humans. We discuss common patterns found among RBPs, based on targets, evolutionary conservation, shared structural domains, and cell-type-specific or ubiquitous expression. We then examine various classes of RBPs commonly implicated in human disease.
2 Human RBPs
2.1 Experimental and Bioinformatic Approaches Leading Towards a Census of RBPs
A complete catalogue of the proteins involved in PTGR is an important goal. Historically, different strategies have been employed towards the identification of RBPs (Ascano et al. 2013). Common approaches used RNA pull-down assays to recover associated proteins in cell lysates, followed by their mass spectrometric identification, or candidate proteins were recombinantly expressed and interrogated for their RNA-binding properties in vitro. These RNA-centric approaches identified the interactome of subsets of RNAs but did not capture the whole RBP proteome, and were not suitably of high throughput.
The first genome-wide approaches for the identification of proteins involved in PTGR utilized predictive methods and searched for the presence of protein domains conferring RNA binding. Early studies on the protein components of heteronuclear RNPs (hnRNPs) led to the identification of the first conserved, canonical RNA- binding domain (RBD) within RBPs (Burd and Dreyfuss 1994). Following these initial discoveries, and facilitated by advances in genome sequencing and the acquisition of protein structures, more precise classification of structural and functional protein domains followed rapidly (Henikoff et al. 1997). Computational prediction algorithms that use probability matrices from multiple sequence alignments enabled the detection of structural domains in uncharacterized protein sequences across organisms. The results of these predictions are publically available in a number of databases such as Interpro, Pfam, SCOP, SMART, or CDD (Murzin et al. 1995; Apweiler et al. 2001; Marchler-Bauer et al. 2003; Letunic et al. 2009; Finn et al. 2010). Among these domain classifications, at least 600 can be found with annotation referring to involvement in RNA-related processes.
Predicting the number of RBPs encoded in various genomes has remained a challenge. RBPs were defined by the presence of one or more canonical RBDs, such as RRM, KH, CSD, zinc fingers, and PUF domains (Lunde et al. 2007). Selecting these predominantly mRNA-binding RBDs, the number of RBPs was initially estimated to ~400–500 in human and mouse (McKee et al. 2005; Galante et al. 2009; Cook et al. 2011), ~300 in D. melanogaster (Lasko 2000; Gamberi et al. 2006), ~250–500 in C. elegans (Lasko 2000; Lee and Schedl 2006; Tamburino et al. 2013), and ~500 RBPs in S. cerevisiae (Hogan et al. 2008). Inclusion of RNA-processing domains involved in RNA metabolism of every known type of RNA leads to numbers near ~700 RBPs in humans (Anantharaman et al. 2002).
Other predictive approaches such as the Kyoto Encyclopedia of Genes and Genomes database (KEGG) (Kanehisa and Goto 2000) and the Gene Ontology project (GO) (Ashburner et al. 2000) integrate domain annotations, protein homologies, and searches of scientific literature statements. These estimate the number of human proteins with RNA-related functions to ~1,800 proteins (Fig. 1.1). However, these methods are often not reliable due to false classifications of proteins, leading to a large number false positives and false negatives.
Fig. 1.1.
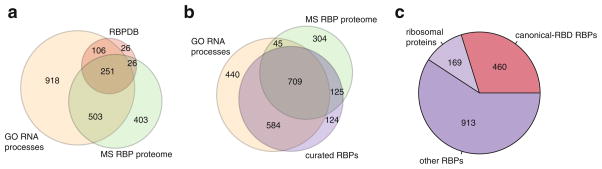
Different approaches to define the catalogue of human RBPs. (a) Venn diagram showing the overlap of proteins with RNA-related Gene Ontology (GO) categories (Ashburner et al. 2000) (orange), the human RNA-binding proteome identified by RNA-cross-linking and mass spectrometry studies (MS RBP proteome, green) (Baltz et al. 2012; Castello et al. 2012; Kwon et al. 2013), and the RBPDB database of human RBPs with canonical RBDs (Cook et al. 2011) (red). (b) Venn diagram showing the overlaps of GO RBPs (orange), MS RBP proteome (green), and the curated RBP list based on analysis of RNA-binding domains and experimental evidence of RNA binding found in the literature (violet). (c) Composition of RBPs in the curated RBP list: Canonical-RBD RBPs (containing canonical RBDs (Lunde et al. 2007; Cook et al. 2011), red), ribosomal proteins (bright violet), other RBPs (dark violet)
In parallel, experimental proteome-wide methods were employed to identify the number of known and novel RBPs such as the development of protein microarrays, which allowed increased throughput for probing the RNA-binding capabilities of a fraction of the proteome in vitro, using RNA probes of defined sequence (Scherrer et al. 2010; Tsvetanova et al. 2010; Siprashvili et al. 2012). In an attempt to comprehensively identify existing and novel RBPs in human at large scale with a singular approach, cross-linking-based methods were recently introduced. In these methods, RBPs were covalently cross-linked to endogenous RNAs using in vivo UV cross-linking, followed by polyA selection of mRNAs, and subsequent identification of interacting proteins by mass spectrometry. These approaches identified ~800 mRBPs in human HEK293 and HeLa cell lines, respectively (Baltz et al. 2012; Castello et al. 2012), 555 in mouse embryonic stem cells (mESCs) (Kwon et al. 2013), and 200 mRBPs in yeast (Mitchell et al. 2013). Together, 1,100 of known and putative human mRBPs were experimentally defined and, assuming homologous function between mouse and human proteins, an additional ~80 proteins may be added (Fig. 1.1a, b). A significant portion of these (64 %) overlapped with known GO-classified RBPs (Fig. 1.1a, b). Many of the residual mRBP candidates did not contain previously described RBDs and require further experimental validation, while other known and expressed RBPs were missed due to the sensitivity of the experiments. However, in comparison to earlier predictive counts of the number of mRBPs (Cook et al. 2011), this approach expanded the mRBP proteome from ~400 to ~1,200 proteins and may, with increasing sensitivity, represent the most suitable method to identify novel RBPs in proteome-wide experiments in different cell types.
Here, we describe our attempt in generation of a curated and comprehensive list of RBPs involved in PTGR processes to guide us in their study of molecular and cellular function and definition of all RNA-related processes.
2.2 Generation of a Curated List of Human RBPs
Our approach selects RBDs involved in RNA-related processes as defined by Pfam (Finn et al. 2010) and searches the human genome for any protein-coding gene that contains at least one of the selected domains, but remains overall unbiased to the putative function of the gene and its RNA targets. Arriving at a list of 2,130 candidates, we added known RBPs from literature searches with unclassified RBDs and additionally screened proteins defined as RBPs by GO (Ashburner et al. 2000) and proteome-wide mass spectrometry datasets (Baltz et al. 2012; Castello et al. 2012; Kwon et al. 2013) for literature-based evidence of their involvement in PTGR.
The list of RBPs was finalized according to the following main criteria: (1) the proteins possessed defined RNA-binding or RNA-enzymatic domains, (2) the proteins were experimentally shown to be part of RNP complexes and thereby involved in RNA metabolic pathways, or (3) they possessed high sequence identity to homologs and paralogs involved in PTGR. Some of the candidate RBPs identified in the recent cross-linking-mass spectrometry studies were not considered as RBPs, if their RNA-binding activity could not be confirmed independently in other published datasets or their domain structure, family members, and homologs were not indicative of an RBP. We furthermore disregarded proteins containing putative RBDs if they showed strong evidence for exclusive roles in RNA-unrelated pathways, such as the majority of C2H2 zinc finger transcription factors of which only a small subset are RNA-binding, e.g., TF3A binding to 5S rRNA (Brown 2005). We included proteins, which are components of well-defined, large RBP complexes, such as the ribosome or the spliceosome, as it is difficult to establish with certainty which proteins interact with RNA directly or indirectly in these large RNPs. This approach is supported by recent proteome-wide cross-linking studies, and the RNA-binding properties of scaffold proteins CNOT1 and TDRD3 have for example emerged through this process (Thomson and Lasko 2005; Siomi et al. 2010; Baltz et al. 2012; Castello et al. 2012; Kwon et al. 2013). Through this curation process we reduced the union of ~3,700 proteins, derived from domain annotation, mass spectrometry datasets, literature search, and GO annotation, to arrive at a final of 1,542 proteins (Fig. 1.1b). The resulting curated list of RBPs contains proteins interacting with all RNA classes. A comparison to the conventionally named “canonical RBPs” (Lunde et al. 2007; Cook et al. 2011) shows that canonical RBPs only represent one-third of RBPs in this set and the majority of RBPs in our set would not be considered using currently available datasets (Fig. 1.1c). The following sections discuss abundance, evolution, expression, and RBPs in human disease based on this curated set of RBPs.
3 Quantitative Aspects of Proteins in RNA Metabolism
The dynamics of complex assembly and composition of RNPs, their targets, and protein cofactors are extremely sensitive to the quantitative relationship between the abundance of RBPs and their targets (Dreyfuss et al. 2002; Müller-McNicoll and Neugebauer 2013). RBPs are in constant competition for binding to frequently occurring short and degenerated RNA sequence elements and thus the cellular compartment concentration of RNA and RBPs will affect the equilibrium of dynamic RNP formation and disassembly. Processes such as pre-mRNA splicing and alternative polyadenylation, where the choice of alternative splice sites or 3′UTR lengths is dependent on the abundance of splicing enhancers, silencers, or U1 snRNP (Smith and Valcarcel 2000; Kaida et al. 2010; Berg et al. 2012; Kornblihtt et al. 2013), emphasize the importance of determining precise RBP levels.
Approximately 7 % of all protein-coding genes are committed to PTGR, but their contribution to the pool of expressed proteins in cells is much higher. We analyzed expression level of ubiquitous RBPs based on RNA-seq data in HEK293 cells (Teplova et al. 2013) (Fig. 1.2). In this cell line, RBPs represented 9 % (1,364 genes) of the ~16,300 expressed genes (expressed with RPKM > 1), but their corresponding transcripts represent more than 25 % of total cellular mRNA, including 7 % mRBPs and 14 % ribosomal proteins (RBP categorization discussed in next section), stressing the abundance of mRNA metabolism and the central role of protein translation (Fig. 1.2). In contrast, transcription factors and cytoskeletal proteins were not overrepresented in the transcriptome of HEK293 cells. In summary, about a quarter of the transcriptome is committed to RNA metabolism, highlighting its fundamental role in the cell.
Fig. 1.2.

Analysis of RBP abundance. Curated RBPs are subclassified into ribosomal proteins (orange), mRBPs (bright red), and other RBPs (dark red). Percentages of RBPs are compared to a set of GO-defined transcription factors (TFs, yellow), a set of GO-defined cytoskeletal proteins (CSK proteins, green) (Ashburner et al. 2000), and all other expressed genes (grey). (a) Count of expressed genes with RPKM > 1 in RNA-seq data from HEK293 cells (Teplova et al. 2013). (b) Relative abundance of each gene group is given by the summation of expression levels of genes in each category
4 Paralogous RBP Families and their Targets
Determining the evolutionary relationships and the conservation of gene families has been critical for understanding gene function and emphasized the utility of model organisms for the study of fundamental biological processes (Henikoff et al. 1997). To account for redundancies among RBPs it is therefore beneficial to consider the RBP family as the smallest functional unit. Grouping the 1,542 RBPs into paralogous gene families with at least 20 % homology gives 1,113 RBP families with one or two members on average. The large number of families reflects a high diversity of RBPs in human.
Here, we categorized RBPs and RBP families based on their reported natural targets and examined their distribution and evolutionary relationships among different classes. Although RBPs often show some degree of interaction with a range of target RNAs in vivo, most of them are committed to one subtype of RNA (Hafner et al. 2010; Wang et al. 2012; Ascano et al. 2012; Hussain et al. 2013; Lovci et al. 2013; Wang et al. 2013) (Table 1.1). Some exceptions remain, such as RNA nucleases, and RBPs acting at the interface of two different RNA classes, such as spliceo-somal proteins, XPO5, or EEF1A, recognizing snRNA/mRNAs, pre-miRNAs/mRNAs, and tRNAs/mRNAs, respectively (Liu et al. 2002; Lund 2004; Mickleburgh et al. 2006; Bennasser et al. 2011; Dever and Green 2012). For the RBPs with multiple targets, we either classified them as diverse in target preference or counted the proteins towards the predominant group of targets based on available literature. The resulting distribution of RBPs and RBP families across all RNA targets in human and their conserved homologs in yeast is shown in Fig. 1.3a–c.
Fig. 1.3.

Number of RBPs involved in different RNA pathways. Curated RBPs are categorized into the following groups: (1) Ribosomal proteins and RBP-interacting proteins (e.g., TUDOR proteins, RBP transport proteins) (dark blue), (2) mRNA-binding proteins (orange), tRNA-binding proteins (red), rRNA-binding proteins (dark green), snRNA-binding proteins (bright green), snoRNA-binding proteins (yellow), ncRNA-binding proteins (ncRNAs defined as miRNA, piRNA, MRP, 7SL, XIST, lincRNAs, telRNA, etc.) (light grey), RNA/DNA-hybrid-interacting proteins (violet), RBPs interacting unselectively with a range of RNA targets (light blue), RBPs with unknown RNA targets (marine blue). Distribution into the listed categories of the (a) 1,542 curated human RBPs, (b) 1,113 human paralogous RBP families, and (c) conserved paralogous RBP families in S. cerevisiae and their average conservation score (orange box)
Our analysis shows that mRBPs form the largest group among RBPs comprising 45 % of all human RBPs (~700 proteins). mRBPs frequently represent families of RBPs with more than two members. Ribosomal proteins constitute the next larger group of RBPs with ~170 proteins of the cytosolic and mitochondrial ribosomes. The next smaller groups of RBPs are committed to tRNA (~150) and rRNA (~120) biogenesis pathways, followed by proteins involved in snRNA, snoRNA, and other ncRNA pathways (Fig. 1.3a, b).
Of the ~1,100 human RBP families, ~550 have homologs in yeast with on average 30 % homology. Different clades of RBPs display varied degrees of conservation. Cytosolic ribosomal proteins are the most conserved with ~57 % homology, while proteins associating with mRNAs or ncRNAs are least conserved, 27 % and 20 %, respectively, and also have the least number of conserved homologs, 45 % and 21 %, respectively (Fig. 1.3c). Nevertheless, despite the gene expansions within protein families at later evolutionary stages (Venter 2001; Van de Peer et al. 2009), the relative ratios of paralogous RBPs families invested in the different RNA pathways remain approximately the same across evolution as seen for the distribution between human and yeast RBP families (Fig. 1.3b, c). This breadth of PTGR factors agrees with an earlier analysis of 32 RBP domain classes of canonical RBDs (including RRM, KH, dsrm, DEAD, PUF, Piwi, PAZ, zinc finger, LSM) showing that the large diversity of RBPs found in contemporary metazoans was already established in the last common ancestor (LCA) of animals, and which possessed an estimated total number of 88 RRM, 15 KH, 49 DEAD box, 9 dsrm, and 38 other RBD proteins (Kerner et al. 2011). Thus the complexity of PTGR was present at the earliest stages of evolution, reflecting that RNA metabolism lies at the heart of eukaryotic gene regulation.
Visualization of the evolutionary relationships of RBP families facilitates systems biology approaches to dissect their regulatory roles. Phylogenetic trees give an intuitive graphic representation of the conservation of proteins, highlight closely related homologs, and thereby provide a glimpse into function of uncharacterized RBPs if function has been already established for a relative. Phylogenetic comparison of the predominantly mRNA-binding KH-domain-containing proteins and the proteins of the small subunit of the cytosolic ribosome illustrates the differences in their evolutionary trajectory (Fig. 1.4).
Fig. 1.4.

Phylogenetic trees of (a) KH-containing proteins and (b) ribosomal proteins of the small subunit. Branch lengths are scaled to the sequence identity of the proteins. S. cerevisiae proteins are marked in red, human proteins in black, homologous families with conserved members in S. cerevisiae highlighted in yellow
KH proteins experienced multiple gene expansions, as noted earlier for mRBPs, and evolved new RBP families at the later metazoan stages, thereby expanding and diversifying components involved in various regulatory pathways, such as mRNA splicing, translational regulation, and transport. KH protein families contain between one and four members in human, and possess generally one distantly related homolog in yeast (Fig. 1.4a). Multiple family members often have redundant biological functions and RNA target spectra. For example, members of the FMR1 family (FMR1, FXR1, FXR2) or the IGF2BP1 family (IGF2BP1, 2, and 3) show >90 % identical RNA-binding specificities (Hafner et al. 2010; Ascano et al. 2012).
In contrast, cytosolic ribosomal proteins display an unusually high conservation, not too surprising, given that the process of protein translation is conserved to such a high degree between prokaryotes and all clades of eukaryotes that functional details of translation determined in bacteria are almost identical to higher systems (Wool et al. 1995; Melnikov et al. 2012; Dever and Green 2012). The ~90 human ribosomal cytosolic proteins are highly similar in structure and function between yeast and human and show late divergence in evolution, as illustrated for the phylogenetic tree of small ribosomal subunit proteins (Fig. 1.4b). With on average 57 % protein identity, all human cytosolic ribosomal proteins have direct one-to-one, or due to a whole-genome duplication in yeast, one-to-two or two-two matching homologs (Wool 1979; Wool et al. 1995; Anger et al. 2013). In contrast, the majority (80 %) of the ~80 human mitochondrial ribosomal proteins (Matthews et al. 1982) have no homologs in yeast, and the few that are conserved have comparatively low homology (22 % identity), reflecting that mitochondrial ribosomes, acquired through eubacterial endosymbiosis, rapidly evolved independently across species and that major remodeling events happened later in evolution (Cavdar Koc et al. 2001; O’Brien 2003).
5 Structural Analysis of RNA-Binding Domains
Analysis of structural features in proteins and the grouping of proteins into domain classes can help to understand their biological function (Henikoff et al. 1997; Anantharaman et al. 2002). Structural domains can predict how RBPs recognize and bind RNAs. It can also uncover redundancies to other RBPs in target recognition, as well as highlight families of RBPs that remain to be characterized. The size of a domain class mirrors its diversity and evolutionary adaptation to biological pathways.
Structure-guided searches can be valuable to place proteins into biological pathways and, for instance, DICER1 and DROSHA were identified as the endonucle-ases responsible for double-stranded RNA (dsRNA) processing in microRNA (miRNA) maturation based on known structure and substrate preferences of the dsRNA-processing bacterial and yeast RNase III enzymes (Hammond et al. 2000) (Bernstein et al. 2001; Lee et al. 2003). Similarly, the structural similarity of AGO proteins and the germline-specific PIWI proteins sparked the search for PIWI- interacting small RNAs with similar features as miRNAs, now known as piRNAs (Girard et al. 2006; Grivna 2006; Aravin et al. 2006).
For a review of the structural features of RBPs, we analyzed characteristic domain combinations of RBD classes (Fig. 1.5a) and will give here a brief overview over the abundant RBD classes and their modes of RNA-binding, natural targets, and the processes they are involved in. For excluded classes, we refer to a number of excellent review articles (Burd and Dreyfuss 1994; Sommerville 1999; Aravind and Koonin 2001a; Aravind and Koonin 2001b; Anantharaman et al. 2002; Arcus 2002; Szymczyna et al. 2003; Kim and Bowie 2003; Maraia and Bayfield 2006; Lunde et al. 2007; Rajkowitsch et al. 2007; Glisovic et al. 2008; Curry et al. 2009; Mihailovich et al. 2010; Zhang et al. 2010). To give additional insight into the structural properties of RBPs, we distinguished between RBDs with only RNA-binding properties (nonenzymatic RBDs), and RBDs that also contain enzymatic functions (enzymatic RBDs), such as RNA helicases, ATPases, polymerases, editing enzymes, and nucleases.
Fig. 1.5.

(a) Analysis of structural patterns of the most abundant RNA-binding domains (with ≥ 9 members) in humans. Shown are the counts for the number of genes containing the listed RNA-binding domains (with ≥ 9 members) named by its Pfam abbreviation (Finn et al. 2010). RNA-binding domains are categorized into those binding RNA without additional enzymatic activity (RBD) (black) and those with additional enzymatic activity (enzRBD) (red). The RBD category was broadly defined to include protein-protein interacting domains known to interact with RBPs, such as those found in TUDOR family proteins (TUDOR) or ribosomal proteins. The following structural patterns are counted: (1) singular occurrence of an RNA-binding domain (RBD—dark blue, enzRBD—yellow), (2) single RBD repeated (RBD—marine blue), (3) multiple RBDs (RBD—light blue, enzRBD—red), (4) combinations of RBDs and enzRBDs (green), and (5) combination of at least one RBD/enzRBD with at least one other, non-RNA-related protein domain (grey). (b) Scheme of domain structure organization of representative RBPs, categorized into the domain combination classes listed in (a)
5.1 Modes of RNA Interaction by RBPs and their Domain Organization
Prototypical single-stranded RNA (ssRNA)-binding domains interact with their targets in a nucleobase-sequence-specific manner typically binding between 4 and 8 nucleotides (Singh and Valcarcel 2005; Lunde et al. 2007; Glisovic et al. 2008). Specificity is introduced mainly by hydrogen bonding and van der Waals interactions of the nucleobases with the protein side chains or the carbonyl and amide groups of the main chain (Auweter et al. 2006), often leaving the RNA phosphate backbone exposed to the solvent. Additional base stacking interactions with aromatic amino acids or positively charged residues in cationic π interactions serve to increase affinity. Double-stranded RNA (dsRNA)-binding proteins achieve specificity through recognition of shape of RNA secondary structure, such as stem–loops (Masliah et al. 2013). Non-sequence-specific RBDs generally interact with the negatively charged phosphate backbone, leaving the bases exposed to the solvent. To achieve specificity, these RBPs can interact with cofactors recruiting them to specific targets, as has been observed for many RNA helicases (Rocak and Linder 2004; Auweter et al. 2006).
Many RBDs (but also DNA-binding domains) derive from a few common superfamily folds, such as the oligonucleotidyl transferase fold and the oligosaccharide-binding fold (OB-fold). Oligonucleotidyl transferase fold proteins include enzymatic RBPs such as TUTases, polyA polymerases, RNA ligases, tRNA CCA-adding enzymes, and immune-stimulatory 2′, 5′-oligoadenylate synthases (Kuchta et al. 2009). RBDs of the OB-fold superfamily are the S1, PAZ, and CSD domains (Murzin 1993; Arcus 2002; Lunde et al. 2007). However, RBDs largely diversified throughout evolution and most RBD classes have only one member, while only 4 % of all RBD classes found in human have more than eight members. Members of the 26 most abundant RBD classes (with 9 and more members) constitute a third of the 1,542 curated RBPs (Fig. 1.5a); most of them are mRBPs. Some of the highly studied RBDs (Lunde et al. 2007), such as the PUF (two proteins), S1, CSD, and PIWI domains (eight members each), define smaller RBD classes in humans. Particularly, ribosomal structural components and proteins involved in processes related to ribosome maturation are unique and thus cannot be classified into large families of related structural organization (Korobeinikova et al. 2012).
More than half of all RBPs contain only one RBD; mRBPs, however, form a notable exception and often have multiple RBDs, either one repeated RBD or multiple RBDs in combination. This modular design allows flexibility and versatility for target recognition and, as RBDs usually recognize relatively short stretches of RNA, increases the affinity and specificity for RNA targets by extending the RNA recognition element (RRE) of the protein (Lunde et al. 2007). A few RBDs are exclusively found in combination with other conserved protein domains, such as RBM1CTR in hnRNPs or the PAZ domain found in the Argonaute proteins and DICER1.
5.2 Abundant Nonenzymatic RBDs
5.2.1 RNA-Recognition Motif
The ssRNA-recognition motif (RRM) is the most frequently found RNA-binding domain in eukaryotes, and has 226 members in humans. The ~90 amino-acid-long domain adopts a βαββαβ topology and is composed of two RNP consensus motifs that recognize 4–6 nucleotides (nts) by stacking interactions of the bases with three conserved aromatic amino acids in the β-sheets (Auweter et al. 2006; Lunde et al. 2007; Cléry et al. 2008). Its small size and modular organization yield flexibility to adaptive change and allowed the RRM domain to vastly expand during evolution (Anantharaman et al. 2002).
Deviations from this canonical binding mode, including N- and C-terminal extensions of the domain, as well as usage of the linker regions and other regions outside of the β-sheet, have been characterized and allow for recognition of up to 8 nt (Maris et al. 2005; Auweter et al. 2006; Lunde et al. 2007; Cléry et al. 2008; Muto and Yokoyama 2012).
Sixty-one RRM proteins only comprise a single isolated RRM domain; examples include the polyA-binding protein CPEB family and the nuclear cap-binding protein NCBP2 (CBP20) (Fig. 1.5b). Sixty RBPs occur with several repeated RRMs; among them are the PTBP and the ELAVL families, regulators of mRNA splicing, stability, and localization (Sawicka et al. 2008; Simone and Keene 2013). Another 68 RRM proteins are found in combination with other RBDs; prominent examples in this group include the IGF2BP1 and FUS families, involved in translational regulation, mRNA transport, and splicing (Tan and Manley 2009; Bell et al. 2013) (Fig. 1.5b). Hence, RRM proteins are found in a variety of biological pathways, the majority of which involve mRNA-related processes, such as regulation of mRNA stability, splicing, translation, and transport. RRM domains are generally a diagnostic indicator for ssRNA binding. In a few cases, however, protein-binding partners have been shown to occlude interaction of the RRM domain with RNA, as seen for the RRM domain of the exon junction complex protein RBM8A (Y14), which binds to the protein cofactor MAGOH (Maris et al. 2005; Glisovic et al. 2008).
5.2.2 K-Homology Domain
The heterogeneous nuclear RNP K-homology (KH) domain binds to ssRNA and ssDNA, and has 39 members in humans. KH domains are ~70 amino acids long and characterized by a hydrophobic core domain with an (I/L/V)IGXXGXX(I/L/V) consensus sequence in the center. Structurally, all KH domains form a three-stranded β-sheet packed against three α-helices and belong either to the eukaryotic type I, of βααβα topology, or the prokaryotic type II, of αββααβ topology (Grishin 2001; Lunde et al. 2007; Valverde et al. 2008). KH domains typically recognize 4-nt ssRNA sequences through electrostatic interactions. Signal transduction and activation of RNA (STAR) proteins, such as SAM68 and QKI, contain just a single KH domain sandwiched between two short signaling motifs, which modulate the protein activity through posttranslational modifications in response to intracellular signaling pathways (Lasko 2003; Chénard and Richard 2008). However, most KH proteins contain combinations of RBDs including the IGF2BP1 family, with four KH and two RRM motifs (Bell et al. 2013), and the brain-specific NOVA splicing family with three repeated KH domains (Li et al. 2007) (Fig. 1.5b). The most extreme example of multiplication of RBDs is found in HDLBP, conserved from yeast to humans, which has 14 repeated KH domains in human (Fig. 1.5b). Analogous to RRM proteins, KH domain proteins predominantly interact with mRNAs and are found in posttranscriptional processes, such as mRNA splicing (PCBP and NOVA family), transport, and translation (IGF2BP1, FMR1, and the MEX family). The two, highly conserved, KH domain proteins PNO1 and RPS3 represent an exception in their target specificity and interact with rRNA during ribosome biogenesis (Vanrobays et al. 2004; Anger et al. 2013).
5.2.3 Double-Stranded RNA-Binding Motif
The dsRNA-binding motif (dsrm) has 21 members and 4 dsrm-like members in humans. Dsrm domains are ~70 amino acids long and adopt an αβββα topology, in which the two α-helices are packed against the three β-sheets. This facilitates nonspecific, shape-dependent contacts with the RNA backbone along the minor and major grooves of A-form dsRNA helix, as well as base contacts along the minor groove and the apical loop (Chang and Ramos 2005; Lunde et al. 2007; Masliah et al. 2013). Dsrm domains are rarely found alone; 24 of the 25 of human dsrm proteins contain multiple dsrm domains or other enzymatic and nonenzymatic RBDs that modulate their function. While the best known dsrm-containing proteins are the Staufen family (STAU1, STAU2) of mRNA stability and transport regulators (Miki et al. 2005; Park and Maquat 2013), this domain type is not confined to mRBPs; instead, most dsrm proteins interact with a range of RNA substrates and are commonly found in RNA enzymes. Members include the adenine-to-inosine RNA-editing ADAR family, processing stem–loops or double strands in mRNAs, viral RNAs, and miRNA precursors (Savva et al. 2012), the two miRNA-processing endonucleases DROSHA and DICER1 (Kim et al. 2009; Wilson and Doudna 2013), as well as the interferon-inducible protein kinase EIF2AK2 (PKR), which, upon binding dsRNA, activates its kinase domain (Saunders and Barber 2003; Raven and Koromilas 2008) (Fig. 1.5b).
5.2.4 CCCH and CCHC Zinc Fingers
The two ssRNA-binding zinc fingers (zf), zf-CCCH and zf-CCHC, form rigid structures by coordination of a Zn2+ ion with three cysteine (C) and one histidine (H) residues. In humans, 45 genes contain the zf-CCCH (C-×8-C-×5-C-×3-H type) and 21 contain the zf-CCHC (C-×2-C-×4-H-×4-C) motif, also known as zinc knuckle. Zf proteins form sequence-specific interaction with RNAs through hydrogen bonding and van der Waals interactions of the protein backbone (Lunde et al. 2007; Kaymak et al. 2010), and use stacking interactions of aromatic side chains with the bases to increase RNA-binding affinity. In contrast to other ssRNA-binding domains, the rigidity and shape of the protein structure are the key determinant for specificity of zinc-finger proteins to their target RNAs. The domains generally occur in repeats or in combination with other RBDs. While for most of the CCHC and CCCH zf proteins the molecular function remains unclear, characterized zf proteins are predominantly involved in regulation of mRNA-related processes. Classic examples of zf-CCCH proteins are the AU-rich-binding ZFP36 (TTP) proteins, which participate in rapid degradation of mRNAs transcribed after immune stimulation (Sandler and Stoecklin 2008; Brooks and Blackshear 2013), and the muscleblind (MBNL1,2,3) family, which regulates alternative splicing during muscle differentiation (Pascual et al. 2006; Cooper et al. 2009). Characterized zf-CCHC proteins include the CPSF4 mRNA polyadenylation and cleavage factor (Colgan and Manley 1997; Shatkin and Manley 2000; Proudfoot and O’Sullivan 2002; Proudfoot 2004) (Fig. 1.5b), and the ZCCHC7 (AIR1) protein, member of the nuclear polyadenylation TRAMP complex, required for the degradation of aberrant nuclear ncRNAs (Anderson and Wang 2009). Another prominent member in this class is the LIN28 family (Fig. 1.5b), which posttran-scriptionally maintains pluripotency in early embryonic development by inhibiting maturation of miRNA let-7 family precursors and increasing stability and translation of mRNA targets (Thornton and Gregory 2012; Wilbert et al. 2012; Cho et al. 2012; Hafner et al. 2013).
5.2.5 LSM Domain
The LSM domain is found in 19 proteins in humans. First discovered in Sm proteins, it later was re-named LSM (“like-Sm”) to include proteins outside of its founding members. The LSM fold is a bipartite domain that stretches along a region of ~65 amino acids and folds into an N-terminal α-helix followed by a twisted five- stranded β-strand (Wilusz and Wilusz 2005; Tharun 2009). The two motifs, motif I, ~22 residues long, and motif II, ~16 residues long, are connected by a variable linker region. LSM proteins form hexa- and heptameric-ring-shaped complexes around RNA. Binding to short, internal polyU- and polyA-rich stretches, they generally associate with snRNAs and some snoRNAs to form stable snRNP complexes (Achsel et al. 2001; Khusial et al. 2005). These RNP complexes commonly act as RNA-RNA and RNA-protein chaperones (Wilusz and Wilusz 2005; Tharun 2009).
The originally characterized Sm proteins (SNRPB, SNRBPD1, SNRBPD2, SNRBPD3, SNRBPE, SNRBPF, SNRBPG) form the core protein complex around the snRNAs of the major and minor spliceosome (U1, U2, U4, U5, U11, U12, U4atac) (Tharun 2009). Other combinations of LSM-fold proteins and their RNA components lead to functional complexes (Beggs 2005; Wilusz and Wilusz 2005; Tharun 2009), such as the nuclear LSM2-8 protein complex (LSM2, LSM3, LSM4, LSM5, LSM6, LSM7, LSM8), which forms around U6 and U6atac snRNA and participates in mRNA splicing. Association of the same LSM proteins around the C/D box U8 snoRNA results in an RNP complex functioning in rRNA maturation (Pannone et al. 2001; Tomasevic and Peculis 2002). In addition, binding of LSM2-8 to nuclear polyadenylated mRNAs promotes mRNA decapping (Kufel et al. 2004). Other LSM complexes include the U7 snRNP complex (SNRPB, LSM10, SNRPD3, LSM11, SNRPE, SNRPF, SNRPG), which is essential for histone 3′end processing (Pillai et al. 2001), and the cytoplasmic LSM1-7 complex (LSM1, LSM2, LSM3, LSM4, LSM5, LSM6, LSM7), which localizes to P bodies and facilitates mRNA decapping after deadenylation (Tharun et al. 2000; Coller and Parker 2004; Parker and Song 2004; Parker and Sheth 2007). We can expect the identification of novel functions and target specificities in PTGR by as yet uncharacterized variations in the composition of LSM complexes (Wilusz and Wilusz 2005).
5.2.6 PIWI and PAZ Domain
The combination of PIWI, PAZ, and MID domains characterizes the Argonaute RBP family, a clade wirh four AGO and four PIWI protein members in humans (Peters and Meister 2007; Kim et al. 2009). Proteins of this clade bind miRNAs, siRNAs, and piRNAs by anchoring the 5′ phosphate in the MID-domain pocket and the 3′end in the PAZ domain, while the PIWI domain interacts with the RNA backbone (Song 2004; Song and Joshua-Tor 2006; Wang et al. 2008b; Tian et al. 2011; Simon et al. 2011; Schirle and MacRae 2012). The PAZ RBD is also a structural component of the miRNA-processing endonuclease DICER1 (Zhang et al. 2004).
The 110 amino-acid-long PAZ domain consists of a β-barrel followed by an αβ-domain, and is structurally related to OB folds, S1, and LSM domains (Lunde et al. 2007). Forming a clamp-like structure, the PAZ domain selectively binds the two-nucleotide overhangs of small RNA duplexes at the 3′end, thereby acting as an anchor to position small RNAs for cleavage (Jinek and Doudna 2009). The PIWI domain is structurally similar to the RNase H endonuclease domain; however, in mammals, only PIWI proteins (Siomi et al. 2011) and AGO2 (Meister et al. 2004; Liu 2004) display nuclease activity, while in other AGO proteins subtle changes in the active site or the N-terminal regulatory domain prevent catalytic activity (Hauptmann et al. 2013; Faehnle et al. 2013; Nakanishi et al. 2013). AGO proteins initiate, guided by miRNAs and siRNAs, posttranscriptional silencing of mRNAs (Hutvagner and Simard 2008), and PIWI proteins, guided by piRNAs, silence transposons at the posttranscriptional and epigenetic levels (Kim et al. 2009; Siomi et al. 2011). Given the variability of possible guide RNA sequences, Argonaute proteins are tremendously versatile and by using different endogenously expressed guide RNA sequences they can form hundreds of distinct RNP complexes in vivo. Capable of targeting virtually any given cytosolic RNA sequence in a specific manner, they are used extensively as a tool in biotechnological applications (Dorsett and Tuschl 2004).
5.2.7 PUF Repeat
In humans, PUF repeats are only found in the two members of the Pumilio family; however, the structure and RNA-recognition mechanism of this domain are highly conserved and probably the best understood among all RBDs. PUF domains are ~40 amino acids long and consist of three α-helices that pack together into a half-ring structure. Each PUF domain recognizes only one nucleotide, but multiple repeats additively increase the number of bases recognized, and Pumilio proteins contain multiple PUF repeats that recognize highly sequence-specifically stretches within mRNAs (Wang et al. 2001). The extremely high specificity is achieved by hydrogen bonding interactions of two residues per repeat, while aromatic side chains wrap the bases into a tight fit. Human Pumilio proteins (PUM1, PUM2) contain eight PUF repeats, which together recognize the sequence UGUANAUA frequently located within the 3′UTRs of its targets to regulate mRNA stability and translation (Wickens et al. 2002; Wang et al. 2002; Hafner et al. 2010). Compared to other RBDs recognizing short and often degenerated RNA sequences, the RRE of Pumilio repeats is highly predictive for identifying Pumilio protein targets. Indeed, predictions of conserved RREs within 3′UTRs of mRNAs mainly identified, next to miRNA, Pumilio protein-binding sites (Xie et al. 2005), highlighting the exceptionally high information content of the Pumilio RRE. The high molecular specificity of the interaction has allowed engineering of RNA-binding specificity of Pumilio proteins to recognize different sequences (Cheong and Hall 2006).
5.3 Predominant Enzymatic RNA-Binding Domains
5.3.1 DExD/H helicases
DExD/H helicases, comprising DEAD and DEAH box helicases, are ATP-dependent enzymes that are involved in RNA-protein remodeling in the cell. They form the second largest class of RBPs comprising 73 members in humans, of which 62 interact specifically with RNA, and the remaining with DNA. The majority of the human RNA-binding DexD/H helicases, 42 members, belong to the DEAD box class, while the others are DEAH and DExH Ski-like helicases (named after its founding member Ski2p) (la Cruz et al. 1999). DExD/H RNA helicases belong to the SF2 helicase superfamily and contain NTPase characteristic Walker A and B motifs; their seven helicase signature motifs extend over ~400 amino acids (Tanner and Linder 2001; Rocak and Linder 2004; Pyle 2008; Jankowsky and Fairman-Williams 2010; Fairman-Williams et al. 2010). The helicases are differentiated by their catalytic core residues Asp-Glu-Ala-Asp for DEAD box helicases, and Asp-Glu-Ala/x- His for the related DEAH box and Ski2-like helicases. The enzymatic core arranges into two discrete domains connected by a linker that forms a cleft, in which an ATP can bind (Tanner and Linder 2001), whose hydrolysis provides the energy for unwinding RNA secondary structures or reorganizing RNPs in either a directional (DEAH helicases) or a bidirectional (DEAD helicases) manner. DExD/H RNA helicases generally lack substrate specificity, or even affinity, towards RNA and DNA. This allows them to promiscuously unwind and remodel a broad range of targets, but also requires their association with cofactors that give specificity and affinity for their targets (Rocak and Linder 2004; Jankowsky and Fairman-Williams 2010). While most members of DExD/H helicases are involved in mRNA-related processes, in particular splicing, they play essential roles in diverse PTGR pathways such as transcriptional regulation, rRNA and tRNA maturation, viral defense, miRNA RISC loading, translation initiation, RNA export, and degradation (Rocak and Linder 2004; Fukuda et al. 2007; Pyle 2008; Jankowsky 2011; Linder and Jankowsky 2011; Martin et al. 2013; Schmidt and Butler 2013; Fullam and Schröder 2013). Next to RNA-RNA and RNA-protein remodeling, DExD/H helicases are also important in RNA-protein complex disassembly and facilitate removal of protein interactors from their targets during RNA export (Linder and Jankowsky 2011).
Paralogs within one RBP family can function in highly diverse roles and pathways, but even one helicase can assume a variety of different biological functions depending on its associated cofactors. For instance, EIF4A1, the first DEAD box helicase for which remodeling and unwinding was mechanistically characterized, forms the EIF4F translation initiation complex, together with the cap-binding protein EIF4E and the scaffolding protein EIF4G (Gingras et al. 1999; Andreou and Klostermeier 2013). Complexed with EIF4H or EIF4B cofactors, EIF4A1 unwinds secondary structures in the 5′UTR, allowing binding of the 43S ribosome complex for AUG start codon scanning. In contrast, although structurally very similar (65 %), the family member EIF4A3 is a core component of the exon junction complex (EJC), in which it acts as an RNA clamp to assist correct positioning of the EJC 20–24 nt upstream of mRNA exon-exon junctions (Linder and Fuller-Pace 2013).
5.3.2 EF-Tu GTP-Binding Domain
The EF-Tu GTP-binding domain (GTP_EFTU), named after its prokaryotic founding member EF-Tu, is a highly conserved domain across all kingdoms of life, and shared by 21 genes in humans. The domain is typically found in GTP-binding translation elongation factors, which are composed of three structural domains, the GTP- binding domain, and two β-barrel nucleotide-binding domains, D2 and D3, which bind to aminoacylated tRNAs (Nissen et al. 1995; Wang et al. 1997; Negrutskii and El’skaya 1998). Eukaryotic EF-1α (human ortholog EEF1A1) has also been shown to interact with higher molecular weight G/U-rich RNAs and rRNAs at a tRNA- independent binding site (Negrutskii and El’skaya 1998). Translation elongation factors are essential for protein synthesis; they bind aminoacyl-tRNAs in a GTP- dependent manner and direct them to the A-site of the ribosome where, upon codon recognition by the tRNA, GTP is hydrolyzed and the factor released (Dever and Green 2012). Furthermore, the GTP_EFTU domains are not only found in combination with D2 and D3 in various translation initiation and release factors, but also alone in GTPases involved in mRNA splicing (EFTUD2) (Fabrizio et al. 1997) and rRNA biogenesis (GNL3 family) (Du et al. 2006; Kressler et al. 2010). The biological role for a majority of the human GTP_EFTU-containing proteins has not been characterized. Particularly, the translation-independent roles of EF1-α and family members, which have been found to directly interact with stem–loops in β-actin (ACTB) and metallothionein 1 (human orthologs MT1 family) mRNAs, are not well understood (Liu et al. 2002; Mickleburgh et al. 2006). More than half of the GTP_EFTU-containing proteins were isolated from direct mRNA-cross-linking experiments by Castello et al. (Castello et al. 2012), supporting a wider role for these proteins in PTGR.
6 Tissue Specificity of RNA-Binding Proteins
Given the evolutionary conservation of RBPs and their involvement in general RNA metabolism, one anticipates predominantly ubiquitous expression for the majority of RBPs. Most tissue-specific proteins evolved recently and have ancestral proteins with low conservation, or no homologs in lower eukaryotes (Winter et al. 2004). Hence, tissue-specific RBPs evolved at later stages of metazoan evolution, the most extreme example of which is represented by the vertebrate-specific secreted RNA ribonucleases A (RNase A) family comprising eight nucleolytic and five inactive pseudo-nuclease members (Rosenberg 2011). Among the active members of this class are angiogenin (ANG), which promotes blood vessel growth, cleaves tRNAs under oxidative stress and is predominantly expressed and secreted from the liver, as well as the EDN and ECP RNases (RNASE2 and RNASE3), which play a role in innate immune response and are expressed in bone marrow eosinophiles (Yamasaki et al. 2009; Rosenberg 2011; Ivanov et al. 2011) (Fig. 1.6a).
Fig. 1.6.

Tissue specificity of RBPs based on their mRNA expression profiles in a human microar-ray tissue atlas (Dezso et al. 2008). (a) Plot of maximum expression intensity versus their tissue specificity score. The threshold for tissue specificity is set at 1 (dashed line), to include the brain- specific ELAVL and NOVA splicing factor families and the germline-specific PIWI family. Protein-coding genes are marked as grey-filled circles and RBPs as red-filled circles. Tissue-specific RBPs are marked based on the tissue with their highest expression: testis (orange triangle), muscle/heart (dark blue triangle), liver/pancreas (dark green inverted triangle), lymphocytes/bone marrow (light blue triangle), brain (dark red triangle). In (b) the number of tissue-specific RBPs is compared to all expressed RBPs and in (c) the number of tissue-specific proteins is compared to total expressed proteins
In order to obtain an overview of the number of tissue-specific RBPs, we examined mRNA expression levels across tissues, which provide a reliable estimate of protein abundance in the cell (Guo et al. 2010). Current RNA-seq resources do not provide a comprehensive collection of tissues profiled in one study and thus we used a publically available microarray tissue atlas comprising 31 different human tissues to examine the tissue specificity of ~17,000 profiled genes (Dezso et al. 2008). We devised a metric measuring tissue specificity by calculating the deviation for each gene from a uniform expression across all tissues. Our analysis likely underestimates tissue specificity, particularly of low-expressed genes, due to tissue heterogeneity, which adds additional noise to the true expression profiles. In addition, mRNA splicing or alternative polyadenylation events, which can give rise to cell-type- specific protein isoforms with different biological functions (Black 2000; Kornblihtt et al. 2013) (Matlin et al. 2005; Sandberg et al. 2008; Mayr and Bartel 2009; Hogg and Goff 2010), cannot be captured by microarray analysis. Nevertheless, a few general themes contrasting RBPs from other protein-coding genes emerge from the present analysis.
About 90 % of the curated RBPs (~1,400) were profiled in the study. While RBPs showed similar average expression levels as compared to other protein-coding genes, they displayed markedly lower variation of expression across tissues. Only ~2 % of RBPs showed very high, tissue-specific expression on the mRNA level compared to 8 % among other protein-coding genes (Dezso et al. 2008). Even when setting the criteria for tissue-specific expression more lenient to include known RBPs with enrichment in selected tissues, only about ~6 % of RBPs were classified as tissue specific, while at the same threshold 20 % of non-RNA-binding proteins displayed tissue-specific expression (Fig. 1.6b, c). Tissue-specific RBPs were predominantly located to the germline, and to a lesser degree to brain, bone marrow, liver, and muscle, where they play distinct biological roles in RNA metabolic pathways (Fig. 1.6a).
RNA metabolism in the germline is uniquely specialized and many of the exclusively expressed RBPs are involved in the piRNA-induced silencing pathway, comprising piRNA biogenesis as well as their effector components such as PIWI (PIWIL1,2,3,4) and TUDOR proteins, MOV10L1, Maelstrom, and PLD6 and DDX4 (VASA) (Siomi et al. 2010; Siomi et al. 2011). Other germline-specific RBPs are the DAZL family (DAZ1,2,3, DAZL, BOLL) and the nanos zinc fingers (NANOS2, NANOS3), both of which are regulators of mRNA translation (Parisi and Lin 2000; Brook et al. 2009; Barckmann and Simonelig 2013). Loss of function of these genes commonly results in arrest of germline development and leads to infertility (Yen 2004; Klattenhoff and Theurkauf 2008; Kim et al. 2009; Brook et al. 2009; Thomson and Lin 2009; Siomi et al. 2011; Massart et al. 2012; Ishizu et al. 2012; Lasko 2013).
The brain shows higher levels of alternative splicing, polyadenylation, and editing compared to other tissues, which are required for neuronal development, plasticity, and memory functions (Xu et al. 2002; Yeo et al. 2005; Wang et al. 2008a; Grabowski 2011; Miura et al. 2013). The higher level of posttranscriptional regulation points towards the brain’s demand to rapidly adapt to evolutionary pressure and PTGR can be considered a means for accelerating evolution of protein diversity (Xing and Lee 2006). Hence, in particular splicing factors are overrepre-sented among the brain-specific RBPs such as the NOVA family, PTB2, and the neuronal members of the RBFOX and ELAVL families (Li et al. 2007). Dysregulation of these proteins results in defects in neurological function and brain development.
We examined paralogous RBP protein families in further detail and defined three categories for tissue specificity in RBP families: (1) highly tissue-specific RBP families, (2) ubiquitous RBP families containing tissue-specific members that evolved independently to meet cell-type-specific needs, and (3) ubiquitous families wherein the individual members display a gradient of expression in different tissues and cell types.
About 2 % of RBP families fall into the first category and display a high degree of tissue specificity in all of their members, expressed in only one or two tissues. Among them are the DAZL, NOVA, RNase A, and PIWI family.
RBP families in the second category (~3 %) have ubiquitously expressed members and one or two tissue-specific paralogs. Members generally have highly overlapping functions, with the tissue-specific paralogs carrying out similar functions to the ubiquitous paralog. A number of germline-restricted helicases, essential for gametogenesis, belong to this category, including DDX4 and DDX3Y, the RNA export factor DDX25, and the piRNA pathway MOV10L1 helicase, all of which have ubiquitously expressed paralogs (DDX3X paralog of DDX4 and DDX3Y, DDX19A and DDX19B paralog of DDX25, MOV10 paralog of MOV10L) (Abdelhaleem 2005; Tsai-Morris et al. 2010; Frost et al. 2010; Zheng et al. 2010; Lasko 2013). Other RBPs include the AU-rich element- binding, nucleocytoplasmic shuttling ELAVL mRBP family (ELAVL1,2,3,4, also known as HuR,B,C,D) (Brennan and Steitz 2001; Simone and Keene 2013). Tissue-specific paralogs likely fulfill a specific demand in PTGR by potentially regulating an increased abundance or number of RNA targets in this tissue. Several mRBP families, such as the ELAVL family, possess identical RNA-binding specificities (Ascano et al. 2011). Whether ubiquitous paralogs are able to functionally compensate for loss of function of their tissue-specific counterparts has not been evaluated systematically in vivo. However, despite their close homology, overexpression of ELAVL4 but not ELAVL1 in cultured neural crest cells is able to induce neurite outgrowth (Wakamatsu and Weston 1997), suggesting that, differences in expression levels alone may not fully account for the different biological functions of paralogs.
About 25 % of paralogous families are ubiquitously expressed but their members display some tissue-specific variation in expression. For these proteins, different expression levels in different tissues can reflect a higher demand or sensitivity for the expressed RBP due to different metabolic activities in different cell types. As expected, dysregulation of the RBPs shows the strongest phenotype in the tissues with highest expression. For instance loss of FMR1, expressed ubiquitously in every tissue but found elevated in brain and gonads (Wang et al. 2008a), leads to mental retardation and autism, macroorchidism, and ovarian insufficiency (Ascano et al. 2012). The variations in RBP expression leading to tissue-specific phenotypes may be considered in the selection of suitable model systems for the investigation of their biological function.
The residual 70 % of paralogous RBP families are expressed ubiquitously with marginal variation in expression. Dysregulation of ubiquitous RBPs impacts PTGR in multiple tissues; however, the severity of the phenotype in different tissues varies. Thus, counterintuitively, loss of abundantly and ubiquitously expressed SMN1 leads to spinal muscular atrophy (SMA), and mutation or loss of TARDBP/TDP43 is associated with amyotrophic lateral sclerosis (ALS) (Ule 2008; Lagier-Tourenne et al. 2010). These examples indicate that tissue-specific defects caused by dysregu-lation of an RBP do not strictly correlate with levels of expression and that differences in expression of RNA targets, protein partners, or additional biological roles of the protein must also be taken into consideration. To understand tissue-specific phenotypes of ubiquitous RBPs it may be necessary to also consider the tissue-specific expression levels of RNA targets or protein cofactors.
7 RBPs in Human Disease
Efforts to understand the role and the pathomechanisms of RBPs in disease have focused predominantly on mRBPs (Wang and Cooper 2007; Lukong et al. 2008; Ule 2008; Cooper et al. 2009; Hanson et al. 2011; Kapeli and Yeo 2012; Castello et al. 2013; Ramaswami et al. 2013). Disease associations of RBPs targeting other classes of RNA such as tRNAs, rRNAs, snoRNAs, snRNAs, and others have been less extensively covered (Scheper et al. 2007a, b; Antonellis and Green 2008; Perron and Provost 2009; Narla and Ebert 2010; Wapinski and Chang 2011; Esteller 2011; Batista and Chang 2013). Here we will give a brief overview of RBPs identified as Mendelian disease factors, categorized by targets, and will focus on their characteristic disease phenotypes (Table 1.2).
Table 1.2.
Overview of RBPs with identified genetic disease-causing mutations collected in the OMIM database (Hamosh et al. 2005), categorized into their main RNA target groups Mitochondrially localized proteins are indicated with (mt). Proteins within the same RBP family are written in one line; if family members are also involved in disease they are highlighted in bold; if family members are involved in diseases other than the listed category they are highlighted in bold brown and found elsewhere in a separate category in the table
| RBP class | disease category | RBP family | disease | genetic mutation | reference |
|---|---|---|---|---|---|
| mRNA-binding | Cancer | EWSR1, FUS, TAF15 | Ewing sarcoma, soft tissue tumors | Gene fusion | (May et al. 1993; Ichikawa et al. 1994; Panagopoulos et al. 1994; Gill et al. 1995; Panagopoulos et al. 1999) |
| TPR | Gastric, thyroid carcinoma, sarcoma | Gene fusion | (Dean et al. 1987; Gonzatti-Haces et al. 1988) | ||
| Muscular/cardiac disease | CNBP, ZCCHC13 | Myotonic dystrophy | RNA repeat expansion sequesters RBPs | (Liquori et al. 2001) | |
| MBNL1, MBNL2, MBNL3 | Myotonic dystrophy | Sequestered RBP in repeat expansion | (Miller 2000; Mankodi et al. 2001; Fardaei 2002) | ||
| CELF1, CELF2-6 | Myotonic dystrophy | Sequestered RBP in repeat expansion | (Timchenko et al. 1996; Roberts et al. 1997) | ||
| MATR3, RBM20 | Cardio-/distal myopathy | Missense mutation | (Senderek et al. 2009; Brauch et al. 2009) | ||
| NOL3 | Myoclonus | Missense mutation | (Russell et al. 2012) | ||
| PABPN1, PABN1L | Muscular dystrophy | Polyalanine expansion leading to protein aggregation | (Brais et al. 1998) | ||
| Neurological disease | AFF1, AFF2, AFF3, AFF4 | Mental retardation | Deletion, loss-of-function through repeat expansion in mRNA | (Knight et al. 1993; Stettner et al. 2011) | |
| ATXN1, ATXN1L | Spinocerebellar ataxia | Polyglutamine expansion leading to protein aggregation | (Orr et al. 1993; Banfi et al. 1994; Servadio et al. 1995) | ||
| ATXN2, ATXN2L | Spinocerebellar ataxia, susceptibility to late-onset Parkinson disease, susceptibility to amyotrophic lateral sclerosis (ALS) | Polyglutamine expansion leading to protein aggregation | (Pulst et al. 1996; Cancel et al. 1997; Gwinn-Hardy et al. 2000; Elden et al. 2010) | ||
| DYNC1H1, DNAH1-11, DNAH17, DYNC2H1 | Charcot-Marie-Tooth disease, mental retardation, spinal muscular atrophy (SMA) | Missense mutation | (Vissers et al. 2010; Weedon et al. 2011; Harms et al. 2012) | ||
| UBA1, UBA6-7 | Spinal muscular atrophy (SMA) | Missense mutation | (Ramser et al. 2008) | ||
| EIF2B1 | Leukoencephalopathy with vanishing white matter | Missense mutation | (van der Knaap et al. 2002) | ||
| EIF2B2 | Leukoencephalopathy with vanishing white matter | Missense, nonsense mutation | (Leegwater et al. 2001) | ||
| EIF2B3 | Leukoencephalopathy with vanishing white matter | Missense mutation | (van der Knaap et al. 2002) | ||
| EIF2B4 | Leukoencephalopathy with vanishing white matter | Missense mutation | (van der Knaap et al. 2002) | ||
| EIF2B5 | Leukoencephalopathy with vanishing white matter | Missense mutation | (Leegwater et al. 2001; van der Knaap et al. 2002; Fogli et al. 2002) | ||
| EIF4G1, EIF4G2, EIF4G3 | Parkinson disease | Missense mutation | (Chartier-Harlin et al. 2011) | ||
| FMR1, FXR1, FXR2 | Fragile X mental retardation syndrome (FXS), fragile X tremor/ataxia syndrome (FXTAS), premature ovarian failure | Deletion, repeat expansion leading to protein loss-of-function (FXS) or RNA-gain-of-function (FXTAS) sequesters RBPs | (Kremer et al. 1991; Devys et al. 1992; Gedeon et al. 1992; Wöhrle et al. 1992; Murray et al. 1998; Hagerman et al. 2001) | ||
|
|
Amyotrophic lateral sclerosis (ALS) | Missense mutation leading to prion-like protein aggregation | (Kwiatkowski et al. 2009; Vance et al. 2009) | ||
| HNRNPA2B1, HNRNPA0, HNRNPAB, HNRNPA1L2, HNRNPA1, HNRNPA3, HNRNPD, HNRNPDL | Amyotrophic lateral sclerosis (ALS) | Missense mutation leading to prion-like protein aggregation | (Kim et al. 2013) | ||
| SETX | Amyotrophic lateral sclerosis (ALS) | Missense, nonsense mutation, deletion | (Moreira et al. 2004) | ||
| TARDBP | Amyotrophic lateral sclerosis (ALS) | Missense mutation leading to protein aggregation | (Sreedharan et al. 2008) | ||
| IGHMBP2 | Distal spinal muscular atrophy (DSMA1) | Missense mutation | (Grohmann et al. 2001) | ||
| LRPPRC | Leigh syndrome | Missense mutation | (Mootha et al. 2003) | ||
| MECP2 | Rett syndrome, X-linked mental retardation | Missense, nonsense mutation, frameshift, deletion | (Amir et al. 1999; Wan et al. 1999; Cheadle et al. 2000; Huppke et al. 2000) | ||
| MTPAP, TUT1 | Spastic ataxia | Missense mutation | (Crosby et al. 2010) | ||
| PARK7 | Parkinson disease | Missense mutation | (Bonifati et al. 2003) | ||
| PQBP1 | Renpenning syndrome 1 | Frameshift | (Kalscheuer et al. 2003) | ||
|
|
Dystonia | Frameshift, missense mutation | (Camargos et al. 2008; Seibler et al. 2008) | ||
| RANBP2, RGPD1-6, RGPD8 | Acute, infection-induced susceptibility to encephalopathy | Missense mutation | (Neilson et al. 2009) | ||
| RBM28 | Alopecia, neurologic defects, endocrinopathy syndrome | Missense mutation | (Nousbeck et al. 2008) | ||
| UPF3A, UPF3B | Mental retardation | Frameshift, missense, nonsense mutation | (Tarpey et al. 2007) | ||
| TIA1, TIAL1 | Welander distal myopathy | Missense mutation | (Hackman et al. 2012) | ||
| RBFOX1, RBFOX2, RBFOX3 | Mental retardation, epilepsy | Deletion, breakpoint | (Bhalla et al. 2004; Martin et al. 2007) | ||
| Neurological/developmental disease | GLE1 | Lethal congenital contracture syndrome | Splice site mutation, missense mutation | (Nousiainen et al. 2008) | |
| Developmental disease | BICC1 | Susceptibility to renal dysplasia | Missense, nonsense mutation | (Kraus et al. 2012) | |
| EIF2AK1, EIF2AK2, EIF2AK3 | Wolcott-Rallison syndrome, multiple epiphyseal dysplasia | Missense, nonsense mutation, splice site mutation | (Delépine et al. 2000; Brickwood et al. 2003; Durocher et al. 2006) | ||
| FTO | Growth, developmental delay | Missense mutation | (Boissel et al. 2009) | ||
| MKRN1, MKRN2, MKRN3 | Central precocious puberty 2 | Missense mutation, insertion | (Abreu et al. 2013) | ||
| NR0B1, NR0B2 | Congenital adrenal hypoplasia | Deletion, missense mutation | (Muscatelli et al. 1994; Yanase et al. 1996) | ||
| RBM5, RBM6, RBM10 | TARP syndrome | Frameshift, missense mutation | (Johnston et al. 2010) | ||
| SF3B4 | Acrofacial dysostosis | Missense, nonsense mutation, frameshift | (Bernier et al. 2012; Czeschik et al. 2013) | ||
| SKIV2L | Trichohepatonenteric syndrome 2 | Missense, nonsense mutation | (Fabre et al. 2012) | ||
| Infertility | BOLL, DAZ1-4, DAZL | Azoospermia | Deletion | (Reijo et al. 1995) | |
| Metabolic disease | AUH, ECH1, ECHS1, ECHDC2, ECHDC3 | 3-methylglutaconic aciduria | Nonsense mutation, frameshift, splice site mutation | (IJlst et al. 2002; Ly et al. 2003) | |
| C12ORF65 (mt) | Combined oxidative phosphorylation deficiency, spastic paraplegia-55 (SPG55) | Frameshift, missense, nonsense mutation | (Antonicka et al. 2010; Shimazaki et al. 2012) | ||
| GFM1 (mt) | Combined oxidative phosphorylation deficiency | Missense, nonsense mutation | (Coenen et al. 2004; Valente et al. 2007) | ||
| TSFM (mt) | Combined oxidative phosphorylation deficiency | Missense mutation | (Smeitink et al. 2006) | ||
| EEFSEC, TUFM (mt) | Combined oxidative phosphorylation deficiency | Missense mutation | (Valente et al. 2007) | ||
| SECISBP2, SECISBP2L | Abnormal thyroid metabolism | Missense mutation | (Dumitrescu et al. 2005) | ||
| Hematologic disease | FIP1L1 | Spontaneous hypereosinophilic syndrome | Deletion leading to gene fusion | (Cools et al. 2003; Griffin et al. 2003) | |
| U2AF1, U2AF1L4 | Myelodysplastic syndrome | Missense mutation | (Graubert et al. 2012) | ||
| Ophthalmologic disease | TDRD7 | Cataract | Frameshift | (Lachke et al. 2011) | |
| Immunological/skin disease | ADAD1, ADAD2, ADAT, ADAR, ADARB1, ADARB2 | Aicardi-Goutieres syndrome (AGS), dyschromatosis symmetrica hereditaria 1 (DSH1) | Missense, nonsense mutation | (Miyamura et al. 2003; Rice et al. 2012) | |
| tRNA-binding | Cancer/metabolic disease | ELAC2 | Prostate cancer, combined oxidative phosphorylation deficiency | Missense, nonsense mutation, frameshift | (Tavtigian et al. 2001; Haack et al. 2013) |
| Muscular/metabolic/hematologic disease | PUS1 | Myopathy, lactic acidosis and sideroblastic anemia 1 | Missense, nonsense mutation | (Bykhovskaya et al. 2004; Fernandez-Vizarra et al. 2007) | |
| YARS2 (mt) | Myopathy, lactic acidosis and sideroblastic anemia 2 | Missense mutation | (Riley et al. 2010) | ||
| Neurological disease |
|
Charcot-Marie Tooth disease | Missense mutation | (Latour et al. 2010; Lin et al. 2011) | |
| ADAT3 | Mental retardation | Missense mutation | (Alazami et al. 2013) | ||
| AIMP1 | Hypomyelinating leukodystrophy | Frameshift | (Feinstein et al. 2010) | ||
| YARS | Charcot-Marie Tooth disease | Missense mutation, frameshift | (Jordanova et al. 2006) | ||
| KARS | Charcot-Marie Tooth disease, deafness | Missense mutation, frameshift | (McLaughlin et al. 2010; Santos-Cortez et al. 2013) | ||
| GARS | Charcot-Marie Tooth disease | Missense mutation | (Antonellis et al. 2003) | ||
| CLP1 | Microencephalopathy, motor sensory defects | Missense mutation | (Karaca et al. 2014) | ||
| DARS | Hypomyelination with brainstem and spinal cord involvement | Missense mutation | (Taft et al. 2013) | ||
| DARS2 (mt) | Leukoencephalopathy | Frameshift, missense, nonsense mutation, splice site mutation | (Scheper et al. 2007) | ||
| NSUN2 | Mental retardation | Nonsense, missense mutation, splice site mutation | (Abbasi-Moheb et al. 2012; Khan et al. 2012) | ||
| FTSJ1 | Mental retardation | Frameshift, splice site mutation | (Freude et al. 2004; Ramser et al. 2004) | ||
| RARS, RARS2 (mt) | Pontocerebellar hypoplasia | Splice site mutation, missense mutation | (Edvardson et al. 2007; Rankin et al. 2010) | ||
| SEPSECS | Pontocerebellar hypoplasia | Missense mutation | (Agamy et al. 2010) | ||
| TSEN2 | Pontocerebellar hypoplasia | Missense mutation | (Budde et al. 2008) | ||
| TSEN34 | Pontocerebellar hypoplasia | Missense mutation | (Budde et al. 2008) | ||
| TSEN54 | Pontocerebellar hypoplasia | Missense, nonsense mutation, deletion | (Budde et al. 2008; Cassandrini et al. 2010) | ||
| Developmental disease |
|
Mandibulofacial dysostosis with microcephaly | Splice site mutation, nonsense, missense mutation, frameshift | (Lines et al. 2012; Bernier et al. 2012; Gordon et al. 2012) | |
| Metabolic disease |
|
Spinocerebellar ataxia | Missense mutation | (Hekman et al. 2012) | |
|
|
Combined oxidative phosphorylation deficiency | Missense mutation | (Götz et al. 2011) | ||
| MTFMT (mt) | Combined oxidative phosphorylation deficiency | Missense, nonsense mutation, deletion | (Neeve et al. 2013; Haack et al. 2014) | ||
| MTO1 (mt) | Combined oxidative phosphorylation deficiency | Frameshift, missense mutation | (Ghezzi et al. 2012) | ||
| EARS2 (mt) | Combined oxidative phosphorylation deficiency | Missense mutation, insertion | (Steenweg et al. 2012; Talim et al. 2013) | ||
| FARS2 (mt) | Combined oxidative phosphorylation deficiency | Missense mutation | (Shamseldin et al. 2012; Elo et al. 2012) | ||
| SARS2 (mt) | Hyperuricemia, pulmonary hypertension, renal failure, and alkalosis | Missense mutation | (Belostotsky et al. 2011) | ||
| TRMU (mt) | Liver failure, deafness | Missense mutation | (Guan et al. 2006; Zeharia et al. 2009) | ||
| Opthalmologic disease/hearing loss | HARS, HARS2 (mt) | Usher syndrome, Perrault syndrome | Missense mutation | (Pierce et al. 2011; Puffenberger et al. 2012) | |
| LARS2 (mt) | Perrault syndrome | Deletion | (Pierce et al. 2013) | ||
| rRNA-binding | Developmental disease | EMG1 | Bowen-Conradi syndrome | Missense mutation | (Armistead et al. 2009) |
| MURC, PTRF, PRKCDBP, SDPR | Lipodystrophy, muscular dystrophy | Frameshift | (Hayashi et al. 2009; Shastry et al. 2010) | ||
| TCOF1 | Treacher Collins syndrome 1 | Missense, nonsense mutation, deletion, insertion | (Caluseriu et al. 2013; Marszalek et al. 2003; Splendore et al. 2002; Wise et al. 1997) | ||
| Developmental/hematologic disease | SBDS | Shwachman-Diamond syndrome | Frameshift, missense, nonsense mutation | (Boocock et al. 2003; Nakashima et al. 2004) | |
| Opthalmologic disease | WDR36 | Open angle glaucoma | Missense mutation | (Monemi et al. 2005) | |
| snRNA-binding | Neurological disease | SMN1, SMN2 | Spinal muscular atrophy (SMA) | Missense, nonsense mutation, frameshift, deletion | (Lefebvre et al. 1995; Cobben et al. 1995; Parsons et al. 1996; Hahnen et al. 1997; Gambardella et al. 1998; Sossi et al. 2001) |
| Skin disease | SART3 | Porokeratosis | Missense mutation | (Zhang et al. 2005) | |
| SNRPE | Hypotrichosis | Missense mutation | (Pasternack et al. 2013) | ||
| USB1 | Poikiloderma with neutropenia | Deletion, splice site mutation, frameshift | (Volpi et al. 2010; Tanaka et al. 2010) | ||
| Opthalmologic disease | SNRNP200, ASCC3 | Retinitis pigmentosa | Missense mutation | (Zhao et al. 2009) | |
| PRPF3 | Retinitis pigmentosa | Missense mutation | (Chakarova et al. 2002) | ||
| PRPF31 | Retinitis pigmentosa | Splice site mutation/deletion, missense mutation | (Vithana et al. 2001) | ||
| PRPF6 | Retinitis pigmentosa | Missense mutation | (Tanackovic et al. 2011) | ||
| PRPF8 | Retinitis pigmentosa | Missense mutation | (McKie et al. 2001) | ||
| RP9 | retinitis pigmentosa | Missense mutation | (Keen et al. 2002) | ||
| snoRNA-binding | Neurological disease | NOP56 | Spinocerebellar ataxia | RNA repeat expansion sequesters RBPs | (Kobayashi et al. 2011) |
| Hematologic/neurodevelopmental/developmental disorder | DKC1 | Dyskeratosis congenita | Missense mutation, deletion, intron insertion, splice site mutation | (Heiss et al. 1998; Knight et al. 1999; Vulliamy et al. 1999; Knight et al. 2001; Kanegane et al. 2005; Pearson et al. 2008) | |
| NHP2 | Dyskeratosis congenita | Missense mutation | (Vulliamy et al. 2008) | ||
| NOP10 | Dyskeratosis congenita | Missense mutation | (Walne et al. 2007) | ||
| cytosolic ribosomal proteins | Neurological disease | RPL10, RPL10L | Autism | Missense mutation | (Klauck et al. 2006) |
| Hematologic disease | RPL11 | Diamond-Blackfan anemia | Frameshift, deletion, splice site mutation, nonsense mutation | (Gazda et al. 2008) | |
| RPL35A | Diamond-Blackfan anemia | Missense, nonsense mutation, deletion | (Farrar et al. 2008) | ||
| RPL5 | Diamond-Blackfan anemia | Missense, nonsense mutation, frameshift, splice site mutation | (Gazda et al. 2008) | ||
| RPS10 | Diamond-Blackfan anemia | Missense, nonsense mutation, frameshift | (Doherty et al. 2010) | ||
| RPS17, RPS17L | Diamond-Blackfan anemia | Missense, frameshift | (Cmejla et al. 2007; Gazda et al. 2008) | ||
| RPS19 | Diamond-Blackfan anemia | Missense, nonsense mutation, frameshift | (Draptchinskaia et al. 1999; Matsson et al. 1999) | ||
| RPS24 | Diamond-Blackfan anemia | Nonsense mutation, frameshift | (Gazda et al. 2006) | ||
| RPS26 | Diamond-Blackfan anemia | Missense mutation, splice site mutation, frameshift | (Doherty et al. 2010) | ||
| RPS7 | Diamond-Blackfan anemia | Splice site mutation | (Gazda et al. 2008) | ||
| mitochondrial (mt) ribosomal proteins | Metabolic disease | MRPL3 | Combined oxidative phosphorylation deficiency | Missense mutation | (Galmiche et al. 2011) |
| MRPS16 | Combined oxidative phosphorylation deficiency | Nonsense mutation | (Miller et al. 2004) | ||
| MRPS22 | Combined oxidative phosphorylation deficiency | Missense mutation | (Saada et al. 2007) | ||
| lincRNA-binding | Cancer | BRCA1 | Breast, ovarian, pancreatic cancer | Missense, nonsense mutation, deletion, frameshift | (Castilla et al. 1994; Simard et al. 1994; Al-Sukhni et al. 2008) |
| Neurological disease | DNMT1 | Cerebellar ataxia, neuropathy | Missense mutation | (Klein et al. 2011; Winkelmann et al. 2012) | |
| Developmental disorder | EZH1, EZH2 | Weaver syndrome 2 | Missense mutation, frameshift | (Gibson et al. 2012) | |
| miRNA-binding | Cancer | DICER1 | Pleuropulmonary blastoma, goiter with testicular tumours, embryonal habdomyosarcoma | Missense, nonsense mutation, frameshift | (Hill et al. 2009; Rio Frio et al. 2011; Foulkes et al. 2011) |
| XPO5 | Colecteral cancer | Frameshift, insertion | (Melo et al. 2010) | ||
| Cancer/developmental disorder |

|
Colorectal cancer, Loeys-Dietz syndrome, juvenile polyposis, pancreatic cancer | Missense, nonsense mutation, frameshift | (Schutte et al. 1996; Howe et al. 1998; Broderick et al. 2007; van de Laar et al. 2011; Regalado et al. 2011) | |
|
|
Colecteral cancer | Frameshift | (Melo et al. 2010) | ||
| Neurological disease | SNIP1 | Psychomotor retardation, epilepsy, craniofacial dymorphism | Missense mutation | (Puffenberger et al. 2012) | |
| Pulmonary disease |

|
Pulmonary hypertension, aortic valve disease (AOVD2) | Missense, nonsense mutation | (Nasim et al. 2011; Drake et al. 2011; Tan et al. 2012) | |
| telRNA-binding | Developmental/cardiovascular/pulmonary disease | TERT | Coronary artery disease, dyskeratosis congenita, aplastic anemia | Missense mutation, frameshift | (Yamaguchi et al. 2005; Armanios et al. 2007; Tsakiri et al. 2007; Marrone et al. 2007) |
| Developmental disorder | WRAP53 | Dyskeratosis congenita | Missense mutation | (Zhong et al. 2011) | |
| 7SL-RNA-binding | Hematologic disease | SRP72 | Bone marow failure | Missense mutation | (Kirwan et al. 2012) |
| Developmental disorder | LARP7, SSB | Alazami syndrome | Base insertion | (Alazami et al. 2012) | |
| immune-stimulatory RNA-binding | Autoimmune disease | IFIH1, DDX58, DHX58 | Aicardi-Goutieres syndrome (AGS) | Missense mutation | (Rice et al. 2014) |
| Metabolic disease | OAS1, OAS2, OAS3, OASL | Susceptibility to diabetes mellitus | Missense mutation | (Tessier et al. 2006) | |
| Cancer | RNASEL | Prostate cancer | Nonsense, missense mutation | (Carpten et al. 2002; Casey et al. 2002) | |
| RNA/DNA-hybrid-binding | Autoimmune/neurological disease | RNASEH2A | Aicardi-Goutieres syndrome (AGS) | Missense mutation | (Crow et al. 2006; Rice et al. 2013) |
| RNASEH2B | Aicardi-Goutieres syndrome (AGS) | Missense mutation | (Crow et al. 2006) | ||
| RNASEH2C | Aicardi-Goutieres syndrome (AGS) | Missense mutation | (Crow et al. 2006) | ||
| SAMHD1 | Aicardi-Goutieres syndrome (AGS), Chilbain lupus 2 | Missense mutation | (Rice et al. 2009; Ravenscroft et al. 2011) | ||
| DNMT3A, DNMT3B, DNMT3L | Immunodeficiency-centromeric instability-facial anomalies syndrome 1 | Missense mutation, deletion | (Xu et al. 1999) | ||
| Neurological disease | APTX | Ataxia | Deletion, splice site mutation, missense mutation | (Amouri et al. 2004; Criscuolo et al. 2005) | |
| diverse targets | Neurological disease | ANG, RNASE1-4, RNASE6-8 | Amyotrophic lateral sclerosis (ALS) | Missense mutation | (Greenway et al. 2006) |
| EXOSC3 | Pontocerebellar hypoplasia, spinal motor neuron degeneration | Missense mutation, deletion | (Wan et al. 2012) | ||
| RNASET2 | Leukoencephalopathy, cancer | Missense mutation, deletion, splice site mutation | (Henneke et al. 2009) | ||
| Developmental disorder | DIS3, DIS3L, DIS3L2 | Perlman syndrome | Deletion, splice site mutation, missense mutation | (Astuti et al. 2012) | |
| Metabolic disease/deafness | PNPT1 | Combined oxidative phosphorylation deficiency | Missense mutation | (Ameln et al. 2012; Vedrenne et al. 2012) | |
| unknown targets | Cardiac disease | CALR3, CALR, CANX, CLGN | Cardiomyopathy | Missense mutation | (Chiu et al. 2007) |
| Developmental disorder | ASCC1 | Barrett esophagus | Missense mutation | (Orloff et al. 2011) | |
| Pulmonary disease | DNAAF2 | Ciliary dyskinesia | Insertion, nonsense mutation | (Omran et al. 2008) |
Despite the interdependence of RNA regulatory pathways, many of the observed disease phenotypes are specific for RBPs binding to particular classes of RNAs, and symptoms of diseases with unknown molecular mechanisms could point towards putative dysregulated RNA metabolic pathways. The majority of RBPs have not been associated with diseases yet, but the recurring patterns occurring for the known proteins may support, facilitated by the growing genomic and transcriptomic data from patients, the interpretation of the dysregulated pathways involving new candidates in disease.
7.1 Diseases Involving mRNA-Binding Proteins
Most of the ~150 RBPs currently listed in the OMIM database (Hamosh et al. 2005) are mRNA-binding proteins with phenotypes characteristically showing neurological and neuromuscular dysfunctions due to dysregulation of splicing, translation, localization, or protein aggregation (Lukong et al. 2008; Ule 2008; Cooper et al. 2009; Hanson et al. 2011; Kapeli and Yeo 2012). Family members tend to have overlapping phenotypes reflecting functional redundancies; for instance the paralogs RBM20 and MATR3 are both involved in myopathies due to dysregulated splicing of their targets (Senderek et al. 2009; Guo et al. 2012).
RNA gain of function leading to altered mRBP-binding patterns is a common pathomechanism (Cooper et al. 2009; Echeverria and Cooper 2012; Nelson et al. 2013). In these disorders, repeat expansions in introns or UTRs of mRNAs often lead to sequestration of mRBPs in the nucleus, thus causing dysregulation of their respective targets. Such diseases comprise FXTAS (fragile X-associated tremor/ataxia syndrome), caused by trinucleotide repeat expansions in the 5′UTR of the FMR1 mRNA (Hagerman 2013), and the myotonic dystrophies DM1 and DM2, caused by repeat expansions in the 3′UTR of DMPK and the intron of CNBP (ZNF9), sequestering the mRBPs CELF1 and MBNL1 and their paralogs (Echeverria and Cooper 2012).
Mutations leading to prion-like aggregation of cytoplasmic or shuttling mRBPs and the dysregulation in assembly and disassembly processes of cytoplasmic RNP granules have been found to be the underlying cause in a range of neurodegenerative disorders. Cytoplasmic inclusion of TARDBP/TDP43 and FUS has been found in amyotrophic lateral sclerosis, a motor neuron disease leading to muscle atrophy (Anthony and Gallo 2010; Lagier-Tourenne et al. 2010; Lee et al. 2012; Ling et al. 2013). Accumulation and inefficient removal of these RNA-protein granules lead to cellular stress predominantly affecting neuronal cells (Liu-Yesucevitz et al. 2011; Li et al. 2013; Buchan et al. 2013; Ramaswami et al. 2013).
7.2 Mitochondrial RBPs in Disease
mRBPs involved in mitochondrial metabolism, such as translation elongation factors GFM1 and TSFM, typically cause deficiencies in oxidative phosphorylation that manifest themselves on a physiological level as neurological and muscular myopathies (Smeitink et al. 2006). Analogously, defects in mitochondrially localized tRNA- and rRNA-binding proteins and subunits of the mitochondrial ribosome also cause deficiencies in oxidative phosphorylation (Smits et al. 2010; Yao and Fox 2013) (Table 1.2).
7.3 Diseases Involving snRNA-Binding Proteins
Mutation or loss of function of snRBPs involved in snRNP complex formation or snRNA biogenesis causes defects in proper mRNA splicing and affects mRNA splicing patterns. Overall, only a few snRBPs have been linked to disease.
The most studied snRBP is the SMN protein, the loss of function of which causes spinal muscular atrophy (SMA). SMN proteins form a multimeric complex with Gemin proteins, which carries out the assembly of snRNPs and other RNP complexes in the cytoplasm (Paushkin et al. 2002; Battle et al. 2006). Autosomal recessive loss of function of the SMN1 gene is the molecular cause for SMA, affecting 1 in 6,000 births (Gubitz et al. 2004). The pathomechanism of SMA exemplifies a striking example of how a single point mutation within a cryptic splice-enhancer site can lead to dramatically altered protein levels that result in global defects in mRNA splicing. Two copies, SMN1 and SMN2, are found in the human genome encoding identical forms of the mature protein. Transcription from the SMN1 locus gives full-length mRNA. The SMN2 gene, however, harbors a single, silent nucleotide mutation in exon 7 leading to frequent exon skipping and resulting in only a small fraction of functional protein being produced, with the majority of transcripts giving rise to truncated, inactive protein. In the disease, deletion of the SMN1 gene results in the SMN2 locus being the only source for full-length mRNAs and protein. The very low amounts of SMN protein produced lead to highly skewed ratios of snRNPs, resulting in global aberrant splicing patterns (Zhang et al. 2008; Cooper et al. 2009). While snRNP assembly defects are detected in all tissues upon SMN1 deletion, the physiological phenotype manifests itself mainly in motor neurons (Glisovic et al. 2008; Cooper et al. 2009; Liu-Yesucevitz et al. 2011).
Mutations in other snRNP complexes have also been shown to cause disease. Loss of function of snRBPs of the U2, U12, and U4/U6-U5 snRNP complexes specifically cause retinitis pigmentosa, a retinal degeneration leading to blindness caused by incorrect splicing of mRNAs encoding for photoreceptors (Daiger et al. 2013). Why mutations in components of the general splicing machinery display highly tissue-specific phenotypes in the eye remains unclear (Wang and Cooper 2007; Singh and Cooper 2012).
7.4 Diseases Involving tRNA Metabolism of tRNA-Binding Proteins
Disease-causing mutations in tRNA-binding proteins are found in the tRNA maturation and aminoacylation pathways and show predominantly neurological phenotypes (Scheper et al. 2007a, b) (Table 1.2). Mutations in a number of cytoplasmic tRNA synthetases cause Charcot-Marie-Tooth disease, affecting the peripheral nervous system and leading to muscular atrophy (Antonellis and Green 2008; Yao and Fox 2013). Mutations in TSEN tRNA-splicing endonucleases lead to pontocerebellar hypoplasia, a sometimes fatal underdevelopment of the cerebellum, causing intellectual disability and impairing muscle control and motor skills (Budde et al. 2008). Dysfunctions of cytoplasmic tRNA aminoacyl synthetases lead to inefficient translation and molecularly link tRNAs with mRNA-binding proteins (Scheper et al. 2007a, b), explaining the overlapping range of symptoms observed for these classes of RBPs in neurological diseases.
7.5 Diseases Involving rRNA-Biogenesis Factors and Ribosomal Proteins
Loss of function of rRNA biogenesis factors and ribosomal proteins is generally embryonically lethal and only few diseases, classified as ribosomopathies, are known for these RBPs (Ruggero and Pandolfi 2003; Narla and Ebert 2010). Ribosomopathies commonly show growth retardation, organ malformation, and frequently bone marrow failure (Liu 2006). Examples of ribosomopathies include mutations in the SBDS rRNA biogenesis gene that cause Shwachman-Bodian syndrome, displaying a deficit in neutrophils and other blood cell types (Boocock et al. 2003), as well as a number of ribosomal proteins predominantly belonging to the small ribosomal subunit that cause Diamond-Blackfan anemia, an impairment of red blood cell formation (Narla and Ebert 2010).
7.6 Diseases Involving snoRNA-Binding Proteins
snoRNA-binding proteins are involved in maturation pathways of rRNAs, snRNAs, and the H/ACA-snRNA-like telomerase RNA. Dysregulation of snoRNPs leads to deficiencies in nucleotide modifications, pseudouridylation, or methylation of their targets (Bachellerie et al. 2002; Filipowicz and Pogacić 2002). As a consequence, snoRNP disease phenotypes overlap with those observed for defects in rRNA biogenesis, ribosomal proteins, as well as proteins involved in the telomerase assembly pathway, such as the protein components TERT and WRAP53 of the telomerase complex. Defects in snoRNA biogenesis manifest themselves in the severe developmental disorder dyskeratosis congenita, leading to bone marrow failure, growth retardation, neurological defects, and premature aging (Filipowicz and Pogacić 2002; Smogorzewska and de Lange 2004).
7.7 Diseases Involving microRNA Pathway Components
Mutations in miRNA-binding proteins are involved in different cancers and developmental disorders and have been well characterized (Merritt et al. 2008; Perron and Provost 2009; Kaneko et al. 2011). For example, mutations and loss of function or reduced levels of DICER1, TARBP2, and XPO5 have been found in pleuropul-monary blastomas and ovarian and other types of cancers (Zhang et al. 2006; Melo et al. 2009; Hill et al. 2009; Melo et al. 2010). In addition, changes in miRNA profiles are observed in many more human diseases due to changes in the cellular metabolism, and may prove valuable diagnostic tools, analogous to mRNA expression profiles (Esquela-Kerscher and Slack 2006; Kloosterman and Plasterk 2006; Calin and Croce 2006; Li and Kowdley 2012).
7.8 Immune Stimulatory and Stress-Related Diseases Caused by RBPs
In recent years it has become evident that nucleic acids play a central role in autoimmune and cellular stress-related diseases. Mutations or loss of function in three RNA/DNA nucleases, SAMHD1, the RNase H2 complex, and TREX1, leads to development of the autoimmune disease Aicardi-Goutieres syndrome (AGS), a neurodevelopmental disorder causing white matter abnormalities and cerebral atrophy. Symptomatically, AGS overlaps with the autoimmune disorder systemic lupus erythematosus (SLE) (Crow et al. 2006b). In both cases, a failure to remove accumulating nucleic acids is central to disease development and activates the innate immune system by type I interferon signaling (Atianand and Fitzgerald 2013; Rabe 2013).
The triphosphatase SAMHD1 possesses 3′–5′ exonuclease activity for ssRNA, ssDNA, and DNA/RNA hybrids. Its antiviral and autoimmune-suppressive function has been attributed to its role in the removal of nucleotides and nucleic acids in the cell (Beloglazova et al. 2013). The heterotrimeric RNase H2 complex endonucleolytically cleaves DNA/RNA hybrids and is thought to be required for the removal of Okazaki fragments during DNA replication (Cerritelli and Crouch 2009; Rabe 2013). Mutations in all three subunits of the RNase H2 complex (RNASEH2A, RNASEH2B, RNASEH2C) have been found to cause AGS (Crow et al. 2006b; Rabe 2013). The 3′–5′ DNA exonuclease TREX1 is implicated in degradation of ssDNA fragments during replication and antiviral defense (Rabe 2013). Mutations in TREX1 are found in AGS patients (Crow et al. 2006a) and TREX1 knockout mice accumulate endogenous retroelements; the accumulating nucleic acids are thought to trigger a subsequent interferon response (Stetson et al. 2008). Interestingly, loss of function of the dsRNA-editing ADAR enzyme has also been shown to cause AGS by an as yet unknown mechanism (Rice et al. 2012).
Many autoantibodies against other RBPs and even RNA have been detected in paraneoplastic syndromes and autoimmune diseases (DeHoratius et al. 1975; Hendrick et al. 1981; Pettersson et al. 1984; Gold et al. 1988; Gelpi et al. 1992; Sakai et al. 1994; Buckanovich et al. 1996). It is thought that dysregulation of RNA clearance mechanisms triggers innate immune responses and leads to apoptosis and release of RBP-RNA complexes into circulation. There these granules mobilize the immune system to develop autoantibodies against self-RNA-protein complexes (Gaipl et al. 2005; Muñoz et al. 2010). The Ro60 complex, consisting of the TROVE2 (Ro60) protein and Y RNAs, was among the first identified targets of autoimmune antibodies in SLE patients was the Ro-RNP particle (Lerner et al. 1981; Hendrick et al. 1981). The Ro60 complex plays a regulatory role in DNA replication and stress response, removing misfolded RNAs, and mice lacking Ro60 develop lupus-like syndromes (Chen and Wolin 2004; Sim and Wolin 2011; Hall et al. 2013). Cleavage of tRNAs and Y RNAs accompanies cellular stress response and apoptosis (Phizicky and Hopper 2010; Nawrot et al. 2011; Hall et al. 2013; Köhn et al. 2013) and these stress-induced small RNA fragments may also act as immune-stimulatory RNAs. Autoantibodies against RBPs associating with these RNAs have been found in serum of SLE patients, stressing the importance of efficient clearance of circulating RNP granules in autoimmune diseases.
Dysfunctional nucleic acid clearance has not only been associated with autoimmune diseases but also with neurological defects of the peripheral nervous system, as seen in loss of function of the RNA exosome component EXOSC3 and RNASET2 (Henneke et al. 2009; Wan et al. 2012), which also acts as a tumor suppressor (Monti et al. 2008). Given the autoimmune-stimulatory role of nucleic acids it is surprising that loss of these general RNA turnover factors more closely resembles loss of mRBPs or tRNA-binding proteins. It will be interesting to understand in mechanistic detail whether the defects caused by these general factors are due to accumulation of RNA or protein aggregates inside the cell, or due to impaired RNA biogenesis pathways.
8 Conclusion
The central role of PTGR in cellular metabolism can be appreciated by considering the large number of proteins interacting with RNA. Over 1,500 of the 21,000 unique human proteins are directly contributing to PTGR. RBPs form many distinct families with few members and human RBPs can be grouped into ~1,100 paralogous families related by 20 % identity.
The complexity of PTGR was established early evolutionary time scales. The lowest common ancestor of metazoans had a set of ~200 RBPs (Kerner et al. 2011), and of the ~1,100 human RBP families ~600 families have homologs in yeast. As a comparison, a census of human and S. cerevisiae transcription factors containing transcription factor domains estimated ~1,400 and ~150 genes in human and yeast, respectively (Costanzo et al. 2000; Lee et al. 2002; Vaquerizas et al. 2009). The difference reflects that transcription factors underwent large expansions in the eumetazoan lineage (Larroux et al. 2008; Degnan et al. 2009), suggesting that the rapidly evolved complexity of transcriptional regulation was required for the development of multiple cell types in eukaryotes.
Consistent with their high degree of conservation, most RBPs (98 %) do not display highly tissue-specific expression, but they are abundant and make up to 25 % of the total cellular protein content. Interestingly, dysregulation of ubiquitous and general components in PTGR often shows highly tissue-specific phenotypes; for instance defects involving mRNA- and tRNA-binding proteins are most frequently associated with neurological diseases, especially of the peripheral nervous system.
Given that the common RBD folds have been characterized and the majority of RBPs do not fall into large families, novel RBPs are most probably singular or have recently evolved RNA-binding activity independent of their family. This makes RBP prediction highly challenging and leaves experimental approaches as the most suitable strategy for their identification. Novel genome-wide experimental methods such as covalent RNA-protein cross-linking coupled with high-throughput sequencing to identify RNA target sites, or combined with mass spectrometric approaches to identify proteome-wide RBPs cross-linked to RNAs, have substantially advanced the efforts towards the elucidation of posttranscriptional regulatory networks. With the increasing sensitivity of experimental approaches, the number of RBPs is likely to grow. Even at present though, the precise biological functions and RNA targets for the majority of known human RBPs have not been characterized and some processes, such as noncoding RNA maturation pathways, control of RNA transport, sensing of intracellular RNA, and mechanisms of RNA/RNP clearance, remain poorly understood. Thus, the main challenges in the field are the characterization of these processes and the mechanisms leading to human disease.
References
- Abbasi-Moheb L, Mertel S, Gonsior M, et al. Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am J Hum Genet. 2012;90:847–855. doi: 10.1016/j.ajhg.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abreu AP, Dauber A, Macedo DB, et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N Engl J Med. 2013;368:2467–2475. doi: 10.1056/NEJMoa1302160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelhaleem M. RNA, helicases: regulators of differentiation. Clinical Biochem. 2005;38:499–503. doi: 10.1016/j.clinbiochem.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Achsel T, Stark H, Luhrmann R. The Sm domain is an ancient RNA-binding motif with oligo(U) specificity. Proc Natl Acad Sci U S A. 2001;98:3685–3689. doi: 10.1073/pnas.071033998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agamy O, Ben Zeev B, Lev D, et al. Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am J Hum Genet. 2010;87:538–544. doi: 10.1016/j.ajhg.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akopian D, Shen K, Zhang X, Shan S-O. Signal recognition particle: an essential protein-targeting machine. Annu Rev Biochem. 2013;82:693–721. doi: 10.1146/annurev-biochem-072711-164732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alazami AM, Al-Owain M, Alzahrani F, et al. Loss of function mutation in LARP7, chaperone of 7SK ncRNA, causes a syndrome of facial dysmorphism, intellectual disability, and primordial dwarfism. Hum Mutat. 2012;33:1429–1434. doi: 10.1002/humu.22175. [DOI] [PubMed] [Google Scholar]
- Alazami AM, Hijazi H, Al-Dosari MS, et al. Mutation in ADAT3, encoding adenosine deaminase acting on transfer RNA, causes intellectual disability and strabismus. J Med Genet. 2013;50:425–430. doi: 10.1136/jmedgenet-2012-101378. [DOI] [PubMed] [Google Scholar]
- Al-Sukhni W, Rothenmund H, Borgida AE, et al. Germline BRCA1 mutations predispose to pancreatic adenocarcinoma. Hum Genet. 2008;124:271–278. doi: 10.1007/s00439-008-0554-0. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Amouri R, Moreira M-C, Zouari M, et al. Aprataxin gene mutations in Tunisian families. Neurology. 2004;63:928–929. doi: 10.1212/01.wnl.0000137044.06573.46. [DOI] [PubMed] [Google Scholar]
- Anantharaman V, Koonin EV, Aravind L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002;30:1427–1464. doi: 10.1093/nar/30.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JT, Wang X. Nuclear RNA surveillance: no sign of substrates tailing off. Crit Rev Biochem Mol Biol. 2009;44:16–24. doi: 10.1080/10409230802640218. [DOI] [PubMed] [Google Scholar]
- Andreou AZ, Klostermeier D. The DEAD-box helicase eIF4A: paradigm or the odd one out? RNA Biol. 2013;10:19–32. doi: 10.4161/rna.21966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anger AM, Armache J-P, Berninghausen O, et al. Structures of the human and Drosophila 80S ribosome. Nature. 2013;497:80–85. doi: 10.1038/nature12104. [DOI] [PubMed] [Google Scholar]
- Anthony K, Gallo JM. Aberrant RNA processing events in neurological disorders. Brain Res. 2010;1338:67–77. doi: 10.1016/j.brainres.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Antonellis A, Green ED. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu Rev Genomics Hum Genet. 2008;9:87–107. doi: 10.1146/annurev.genom.9.081307.164204. [DOI] [PubMed] [Google Scholar]
- Antonellis A, Ellsworth RE, Sambuughin N, et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonicka H, Ostergaard E, Sasarman F, et al. Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am J Hum Genet. 2010;87:115–122. doi: 10.1016/j.ajhg.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler R, Attwood TK, Bairoch A, et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 2001;29:37–40. doi: 10.1093/nar/29.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin A, Gaidatzis D, Pfeffer S, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442:203–207. doi: 10.1038/nature04916. [DOI] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. A natural classification of ribonucleases. Meth Enzymol. 2001a;341:3–28. doi: 10.1016/s0076-6879(01)41142-6. [DOI] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. THUMP–a predicted RNA-binding domain shared by 4-thiouridine, pseudouridine synthases and RNA methylases. Trends Biochem Sci. 2001b;26:215–217. doi: 10.1016/s0968-0004(01)01826-6. [DOI] [PubMed] [Google Scholar]
- Arcus V. OB-fold domains: a snapshot of the evolution of sequence, structure and function. Curr Opin Struct Biol. 2002;12:794–801. doi: 10.1016/S0959-440X(02)00392-5. [DOI] [PubMed] [Google Scholar]
- Armanios MY, Chen JJ-L, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- Armistead J, Khatkar S, Meyer B, et al. Mutation of a gene essential for ribosome biogenesis, EMG1, causes Bowen-Conradi syndrome. Am J Hum Genet. 2009;84:728–739. doi: 10.1016/j.ajhg.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano M, Hafner M, Cekan P, et al. Identification of RNA-protein interaction networks using PAR-CLIP. WIREs RNA. 2011;3:159–177. doi: 10.1002/wrna.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano M, Mukherjee N, Bandaru P, et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492:382–386. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano M, Gerstberger S, Tuschl T. Multi-disciplinary methods to define RNA-protein interactions and regulatory networks. Curr Opin Genet Dev. 2013;23:20–28. doi: 10.1016/j.gde.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astuti D, Morris MR, Cooper WN, et al. Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat Genet. 2012;44:277–284. doi: 10.1038/ng.1071. [DOI] [PubMed] [Google Scholar]
- Atianand MK, Fitzgerald KA. Molecular basis of DNA recognition in the immune system. J Immunol. 2013;190:1911–1918. doi: 10.4049/jimmunol.1203162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auweter SD, Oberstrass FC, Allain FHT. Sequence-specific binding of single-stranded RNA: is there a code for recognition? Nucleic Acids Res. 2006;34:4943–4959. doi: 10.1093/nar/gkl620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachellerie JP, Cavaille J, Huttenhofer A. The expanding snoRNA world. Biochimie. 2002;84:775–790. doi: 10.1016/s0300-9084(02)01402-5. [DOI] [PubMed] [Google Scholar]
- Baltz AG, Munschauer M, Schwanhausser B, et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 2012;46:674–690. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- Banfi S, Servadio A, Chung MY, et al. Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat Genet. 1994;7:513–520. doi: 10.1038/ng0894-513. [DOI] [PubMed] [Google Scholar]
- Barckmann B, Simonelig M. Control of maternal mRNA stability in germ cells and early embryos. Biochim Biophys Acta. 2013;1829:714–724. doi: 10.1016/j.bbagrm.2012.12.011. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle DJ, KASIM M, Yong J, et al. The SMN complex: an assembly machine for RNPs. Cold Spring Harbor Symp Quant Biol. 2006;71:313–320. doi: 10.1101/sqb.2006.71.001. [DOI] [PubMed] [Google Scholar]
- Beggs JD. Lsm proteins and RNA processing. Biochem Soc Trans. 2005;33:433–438. doi: 10.1042/BST0330433. [DOI] [PubMed] [Google Scholar]
- Bell JL, Wächter K, Mühleck B, et al. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression? Cell Mol Life Sci. 2013;70:2657–2675. doi: 10.1007/s00018-012-1186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloglazova N, Flick R, Tchigvintsev A, et al. Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J Biol Chem. 2013;288:8101–8110. doi: 10.1074/jbc.M112.431148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belostotsky R, Ben-Shalom E, Rinat C, et al. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet. 2011;88:193–200. doi: 10.1016/j.ajhg.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennasser Y, Chable-Bessia C, Triboulet R, et al. Competition for XPO5 binding between Dicer mRNA, pre-miRNA and viral RNA regulates human Dicer levels. Nat Struct Mol Biol. 2011;18:323–327. doi: 10.1038/nsmb.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg MG, Singh LN, Younis I, et al. U1 snRNP determines mRNA length and regulates isoform expression. Cell. 2012;150:53–64. doi: 10.1016/j.cell.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger W, Steiner E, Grusch M, et al. Vaults and the major vault protein: Novel roles in signal pathway regulation and immunity. Cell Mol Life Sci. 2008;66:43–61. doi: 10.1007/s00018-008-8364-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier FP, Caluseriu O, Ng S, et al. Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am J Hum Genet. 2012;90:925–933. doi: 10.1016/j.ajhg.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- Bhalla K, Phillips HA, Crawford J, et al. The de novo chromosome 16 translocations of two patients with abnormal phenotypes (mental retardation and epilepsy) disrupt the A2BP1 gene. J Hum Genet. 2004;49:308–311. doi: 10.1007/s10038-004-0145-4. [DOI] [PubMed] [Google Scholar]
- Black DL. Protein diversity from alternative splicing: a challenge for bioinformatics and post-genome biology. Cell. 2000;103:367–370. doi: 10.1016/s0092-8674(00)00128-8. [DOI] [PubMed] [Google Scholar]
- Boissel S, Reish O, Proulx K, et al. Loss-of-function mutation in the dioxygenase-encoding FTO gene causes severe growth retardation and multiple malformations. Am J Hum Genet. 2009;85:106–111. doi: 10.1016/j.ajhg.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert F-M, van Koningsbruggen S, Navascués J, Lamond AI. The multifunctional nucleolus. Nat Rev Mol Cell Biol. 2007;8:574–585. doi: 10.1038/nrm2184. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Boocock GRB, Morrison JA, Popovic M, et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003;33:97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- Brais B, Bouchard J-P, Xie Y-G, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998;18:164–167. doi: 10.1038/ng0298-164. [DOI] [PubMed] [Google Scholar]
- Brauch KM, Karst ML, Herron KJ, et al. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol. 2009;54:930–941. doi: 10.1016/j.jacc.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CM, Steitz JA. HuR and mRNA stability. Cell Mol Life Sci. 2001;58:266–277. doi: 10.1007/PL00000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickwood S, Bonthron DT, Al-Gazali LI, et al. Wolcott-Rallison syndrome: pathogenic insights into neonatal diabetes from new mutation and expression studies of EIF2AK3. J Med Genet. 2003;40:685–689. doi: 10.1136/jmg.40.9.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick P, Carvajal-Carmona L, Pittman AM, et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet. 2007;39:1315–1317. doi: 10.1038/ng.2007.18. [DOI] [PubMed] [Google Scholar]
- Brook M, Smith JWS, Gray NK. The DAZL and PABP families: RNA-binding proteins with interrelated roles in translational control in oocytes. Reproduction. 2009;137:595–617. doi: 10.1530/REP-08-0524. [DOI] [PubMed] [Google Scholar]
- Brooks SA, Blackshear PJ. Tristetraprolin (TTP): Interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochim Biophys Acta. 2013;1829:666–679. doi: 10.1016/j.bbagrm.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RS. Zinc finger proteins: getting a grip on RNA. Curr Opin Struct Biol. 2005;15:94–98. doi: 10.1016/j.sbi.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Buchan JR, Kolaitis R-M, Taylor JP, Parker R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell. 2013;153:1461–1474. doi: 10.1016/j.cell.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckanovich RJ, Yang YY, Darnell RB. The onconeural antigen Nova-1 is a neuron-specific RNA-binding protein, the activity of which is inhibited by paraneoplastic antibodies. J Neurosci. 1996;16:1114–1122. doi: 10.1523/JNEUROSCI.16-03-01114.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde BS, Namavar Y, Barth PG, et al. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet. 2008;40:1113–1118. doi: 10.1038/ng.204. [DOI] [PubMed] [Google Scholar]
- Burd CG, Dreyfuss G. Conserved structures and diversity of functions of RNA-binding proteins. Science. 1994;265:615–621. doi: 10.1126/science.8036511. [DOI] [PubMed] [Google Scholar]
- Bykhovskaya Y, Casas K, Mengesha E, et al. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA) Am J Hum Genet. 2004;74:1303–1308. doi: 10.1086/421530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- Caluseriu O, Lowry BR, McLeod R, et al. The hutterite variant of Treacher Collins syndrome: a 28-year-old story solved. Am J Med Genet. 2013;161A:2855–2859. doi: 10.1002/ajmg.a.36172. [DOI] [PubMed] [Google Scholar]
- Camargos S, Scholz S, Simón-Sánchez J, et al. DYT16, a novel young-onset dystonia-parkinsonism disorder: identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol. 2008;7:207–215. doi: 10.1016/S1474-4422(08)70022-X. [DOI] [PubMed] [Google Scholar]
- Cancel G, Dürr A, Didierjean O, et al. Molecular and clinical correlations in spinocerebellar ataxia 2: a study of 32 families. Hum Mol Genet. 1997;6:709–715. doi: 10.1093/hmg/6.5.709. [DOI] [PubMed] [Google Scholar]
- Carpten J, Nupponen N, Isaacs S, et al. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat Genet. 2002;30:181–184. doi: 10.1038/ng823. [DOI] [PubMed] [Google Scholar]
- Casey G, Neville PJ, Plummer SJ, et al. RNASEL Arg462Gln variant is implicated in up to 13 % of prostate cancer cases. Nat Genet. 2002;32:581–583. doi: 10.1038/ng1021. [DOI] [PubMed] [Google Scholar]
- Cassandrini D, Biancheri R, Tessa A, et al. Pontocerebellar hypoplasia: clinical, pathologic, and genetic studies. Neurology. 2010;75:1459–1464. doi: 10.1212/WNL.0b013e3181f88173. [DOI] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Castello A, Fischer B, Hentze MW, Preiss T. RNA-binding proteins in Mendelian disease. Trends Genet. 2013;29:318–327. doi: 10.1016/j.tig.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Castilla LH, Couch FJ, Erdos MR, et al. Mutations in the BRCA1 gene in families with early-onset breast and ovarian cancer. Nat Genet. 1994;8:387–391. doi: 10.1038/ng1294-387. [DOI] [PubMed] [Google Scholar]
- Cavdar Koc E, Burkhart W, Blackburn K, et al. The small subunit of the mammalian mitochondrial ribosome. Identification of the full complement of ribosomal proteins present. J Biol Chem. 2001;276:19363–19374. doi: 10.1074/jbc.M100727200. [DOI] [PubMed] [Google Scholar]
- Cerritelli SM, Crouch RJ. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 2009;276:1494–1505. doi: 10.1111/j.1742-4658.2009.06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakarova CF, Hims MM, Bolz H, et al. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:87–92. doi: 10.1093/hmg/11.1.87. [DOI] [PubMed] [Google Scholar]
- Chang K-Y, Ramos A. The double-stranded RNA-binding motif, a versatile macromolecular docking platform. FEBS J. 2005;272:2109–2117. doi: 10.1111/j.1742-4658.2005.04652.x. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin M-C, Dachsel JC, Vilariño-Güell C, et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet. 2011;89:398–406. doi: 10.1016/j.ajhg.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheadle JP, Gill H, Fleming N, et al. Long-read sequence analysis of the MECP2 gene in Rett syndrome patients: correlation of disease severity with mutation type and location. Hum Mol Genet. 2000;9:1119–1129. doi: 10.1093/hmg/9.7.1119. [DOI] [PubMed] [Google Scholar]
- Chen X, Wolin SL. The Ro 60 kDa autoantigen: insights into cellular function and role in autoimmunity. J Mol Med. 2004;82:232–239. doi: 10.1007/s00109-004-0529-0. [DOI] [PubMed] [Google Scholar]
- Chénard CA, Richard S. New implications for the QUAKING RNA binding protein in human disease. J Neurosci Res. 2008;86:233–242. doi: 10.1002/jnr.21485. [DOI] [PubMed] [Google Scholar]
- Cheong C-G, Hall TMT. Engineering RNA sequence specificity of Pumilio repeats. Proc Natl Acad Sci U S A. 2006;103:13635–13639. doi: 10.1073/pnas.0606294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu C, Tebo M, Ingles J, et al. Genetic screening of calcium regulation genes in familial hypertrophiccardiomyopathy. J Mol Cell Cardiol. 2007;43:337–343. doi: 10.1016/j.yjmcc.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Cho J, Chang H, Kwon SC, et al. LIN28A is a suppressor of ER-associated translation in embryonic stem cells. Cell. 2012;151:765–777. doi: 10.1016/j.cell.2012.10.019. [DOI] [PubMed] [Google Scholar]
- Ciganda M, Williams N. Eukaryotic 5S rRNA biogenesis. WIREs RNA. 2011;2:523–533. doi: 10.1002/wrna.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cléry A, Blatter M, Allain FHT. RNA recognition motifs: boring? Not quite. Curr Opin Struct Biol. 2008;18:290–298. doi: 10.1016/j.sbi.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Cmejla R, Cmejlova J, Handrkova H, et al. Ribosomal protein S17 gene (RPS17) is mutated in Diamond-Blackfan anemia. Hum Mutat. 2007;28:1178–1182. doi: 10.1002/humu.20608. [DOI] [PubMed] [Google Scholar]
- Cobben JM, van der Steege G, Grootscholten P, et al. Deletions of the survival motor neuron gene in unaffected siblings of patients with spinal muscular atrophy. Am J Hum Genet. 1995;57:805–808. [PMC free article] [PubMed] [Google Scholar]
- Coenen MJH, Antonicka H, Ugalde C, et al. Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N Engl J Med. 2004;351:2080–2086. doi: 10.1056/NEJMoa041878. [DOI] [PubMed] [Google Scholar]
- Colgan DF, Manley JL. Mechanism and regulation of mRNA polyadenylation. Genes Dev. 1997;11:2755–2766. doi: 10.1101/gad.11.21.2755. [DOI] [PubMed] [Google Scholar]
- Coller J, Parker R. Eukaryotic mRNA decapping. Annu Rev Biochem. 2004;73:861–890. doi: 10.1146/annurev.biochem.73.011303.074032. [DOI] [PubMed] [Google Scholar]
- Cook KB, Kazan H, Zuberi K, et al. RBPDB: a database of RNA-binding specificities. Nucleic Acids Res. 2011;39:D301–D308. doi: 10.1093/nar/gkq1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med. 2003;348:1201–1214. doi: 10.1056/NEJMoa025217. [DOI] [PubMed] [Google Scholar]
- Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo MC, Hogan JD, Cusick ME, et al. The yeast proteome database (YPD) and Caenorhabditis elegans proteome database (WormPD): comprehensive resources for the organization and comparison of model organism protein information. Nucleic Acids Res. 2000;28:73–76. doi: 10.1093/nar/28.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscuolo C, Mancini P, Menchise V, et al. Very late onset in ataxia oculomotor apraxia type I. Ann Neurol. 2005;57:777. doi: 10.1002/ana.20463. [DOI] [PubMed] [Google Scholar]
- Crosby AH, Patel H, Chioza BA, et al. Defective mitochondrial mRNA maturation is associated with spastic ataxia. Am J Hum Genet. 2010;87:655–660. doi: 10.1016/j.ajhg.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Hayward BE, Parmar R, et al. Mutations in the gene encoding the 3 “-5” DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006a;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Leitch A, Hayward BE, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006b;38:910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- Curry S, Kotik-Kogan O, Conte MR, Brick P. Getting to the end of RNA: structural analysis of protein recognition of 5′ and 3′ termini. Biochim Biophys Acta. 2009;1789:653–666. doi: 10.1016/j.bbagrm.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Czeschik JC, Voigt C, Alanay Y, et al. Clinical and mutation data in 12 patients with the clinical diagnosis of Nager syndrome. Hum Genet. 2013;132:885–898. doi: 10.1007/s00439-013-1295-2. [DOI] [PubMed] [Google Scholar]
- Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84:132–141. doi: 10.1111/cge.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz J, Kressler D, Linder P. Unwinding RNA in Saccharomyces cerevisiae: DEAD-box proteins and related families. Trends Biochem Sci. 1999;24:192–198. doi: 10.1016/s0968-0004(99)01376-6. [DOI] [PubMed] [Google Scholar]
- Dean M, Park M, Vande Woude GF. Characterization of the rearranged tpr-met oncogene breakpoint. Mol Cell Biol. 1987;7:921–924. doi: 10.1128/mcb.7.2.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan BM, Vervoort M, Larroux C, Richards GS. Early evolution of metazoan transcription factors. Curr Opin Genet Dev. 2009;19:591–599. doi: 10.1016/j.gde.2009.09.008. [DOI] [PubMed] [Google Scholar]
- DeHoratius RJ, Pillarisetty R, Messner RP, Talal N. Anti-nucleic acid antibodies in systemic lupus erythematosus patients and their families. Incidence and correlation with lymphocyto-toxic antibodies. J Clin Invest. 1975;56:1149–1154. doi: 10.1172/JCI108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delépine M, Nicolino M, Barrett T, et al. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet. 2000;25:406–409. doi: 10.1038/78085. [DOI] [PubMed] [Google Scholar]
- Dever TE, Green R. The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harbor Pers Biol. 2012;4:a013706. doi: 10.1101/cshperspect.a013706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devys D, Biancalana V, Rousseau F, et al. Analysis of full fragile X mutations in fetal tissues and monozygotic twins indicate that abnormal methylation and somatic heterogeneity are established early in development. Am J Med Genet. 1992;43:208–216. doi: 10.1002/ajmg.1320430134. [DOI] [PubMed] [Google Scholar]
- Dezso Z, Nikolsky Y, Sviridov E, et al. A comprehensive functional analysis of tissue specificity of human gene expression. BMC Biol. 2008;6:49. doi: 10.1186/1741-7007-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty L, Sheen MR, Vlachos A, et al. Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2010;86:222–228. doi: 10.1016/j.ajhg.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett Y, Tuschl T. siRNAs: applications in functional genomics and potential as therapeutics. Nat Rev Drug Discov. 2004;3:318–329. doi: 10.1038/nrd1345. [DOI] [PubMed] [Google Scholar]
- Drake KM, Zygmunt D, Mavrakis L, et al. Altered MicroRNA processing in heritable pulmonary arterial hypertension: an important role for Smad-8. Am J Respir Crit Care Med. 2011;184:1400–1408. doi: 10.1164/rccm.201106-1130OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draptchinskaia N, Gustavsson P, Andersson B, et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet. 1999;21:169–175. doi: 10.1038/5951. [DOI] [PubMed] [Google Scholar]
- Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- Du X, Rao MRKS, Chen XQ, et al. The homologous putative GTPases Grn1p from fission yeast and the human GNL3L are required for growth and play a role in processing of nucleolar pre-rRNA. Mol Biol Cell. 2006;17:460–474. doi: 10.1091/mbc.E05-09-0848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu AM, Liao X-H, Abdullah MSY, et al. Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat Genet. 2005;37:1247–1252. doi: 10.1038/ng1654. [DOI] [PubMed] [Google Scholar]
- Durocher F, Faure R, Labrie Y, et al. A novel mutation in the EIF2AK3 gene with variable expressivity in two patients with Wolcott-Rallison syndrome. Clin Genet. 2006;70:34–38. doi: 10.1111/j.1399-0004.2006.00632.x. [DOI] [PubMed] [Google Scholar]
- Echeverria GV, Cooper TA. RNA-binding proteins in microsatellite expansion disorders: Mediators of RNA toxicity. Brain Res. 2012;1462:100–111. doi: 10.1016/j.brainres.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Shaag A, Kolesnikova O, et al. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan ED, Collins K. Biogenesis of telomerase ribonucleoproteins. RNA. 2012;18:1747–1759. doi: 10.1261/rna.034629.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim H-J, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JC, Brown JW. The RNase P family. RNA Biol. 2009;6:362–369. doi: 10.4161/rna.6.4.9241. [DOI] [PubMed] [Google Scholar]
- Elo JM, Yadavalli SS, Euro L, et al. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum Mol Genet. 2012;21:4521–4529. doi: 10.1093/hmg/dds294. [DOI] [PubMed] [Google Scholar]
- Esakova O, Krasilnikov AS. Of proteins and RNA: The RNase P/MRP family. RNA. 2010;16:1725–1747. doi: 10.1261/rna.2214510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- Fabre A, Charroux B, Martinez-Vinson C, et al. SKIV2L mutations cause syndromic diarrhea, or trichohepatoenteric syndrome. Am J Hum Genet. 2012;90:689–692. doi: 10.1016/j.ajhg.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Laggerbauer B, Lauber J, et al. An evolutionarily conserved U5 snRNP-specific protein is a GTP-binding factor closely related to the ribosomal translocase EF-2. EMBO J. 1997;16:4092–4106. doi: 10.1093/emboj/16.13.4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faehnle CR, Elkayam E, Haase AD, et al. The making of a slicer: activation of human argonaute-1. Cell Rep. 2013;3:1901–1909. doi: 10.1016/j.celrep.2013.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairman-Williams ME, Guenther U-P, Jankowsky E. SF1 and SF2 helicases: family matters. Curr Opin Struct Biol. 2010;20:313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fardaei M. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum Mol Genet. 2002;11:805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- Farrar JE, Nater M, Caywood E, et al. Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood. 2008;112:1582–1592. doi: 10.1182/blood-2008-02-140012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein M, Markus B, Noyman I, et al. Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am J Hum Genet. 2010;87:820–828. doi: 10.1016/j.ajhg.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Vizarra E, Berardinelli A, Valente L, et al. Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA) J Med Genet. 2007;44:173–180. doi: 10.1136/jmg.2006.045252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipowicz W, Pogacić V. Biogenesis of small nucleolar ribonucleoproteins. Curr Opin Cell Biol. 2002;14:319–327. doi: 10.1016/s0955-0674(02)00334-4. [DOI] [PubMed] [Google Scholar]
- Finn RD, Mistry J, Tate J, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogli A, Wong K, Eymard-Pierre E, et al. Cree leukoencephalopathy and CACH/VWM disease are allelic at the EIF2B5 locus. Ann Neurol. 2002;52:506–510. doi: 10.1002/ana.10339. [DOI] [PubMed] [Google Scholar]
- Foulkes WD, Bahubeshi A, Hamel N, et al. Extending the phenotypes associated with DICER1 mutations. Hum Mutat. 2011;32:1381–1384. doi: 10.1002/humu.21600. [DOI] [PubMed] [Google Scholar]
- Freude K, Hoffmann K, Jensen L-R, et al. Mutations in the FTSJ1 gene coding for a novel S-adenosylmethionine-binding protein cause nonsyndromic X-linked mental retardation. Am J Hum Genet. 2004;75:305–309. doi: 10.1086/422507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost RJA, Hamra FK, Richardson JA, et al. MOV10L1 is necessary for protection of spermatocytes against retrotransposons by Piwi-interacting RNAs. Proc Natl Acad Sci U S A. 2010;107:11847–11852. doi: 10.1073/pnas.1007158107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T, Yamagata K, Fujiyama S, et al. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nat Cell Biol. 2007;9:604–611. doi: 10.1038/ncb1577. [DOI] [PubMed] [Google Scholar]
- Fullam A, Schröder M. DExD/H-box RNA helicases as mediators of anti-viral innate immunity and essential host factors for viral replication. Biochim Biophys Acta. 2013;1829:854–865. doi: 10.1016/j.bbagrm.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaipl US, Voll RE, Sheriff A, et al. Impaired clearance of dying cells in systemic lupus erythematosus. Autoimmun Rev. 2005;4:189–194. doi: 10.1016/j.autrev.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Galante PAF, Sandhu D, de Sousa Abreu R, et al. A comprehensive in silico expression analysis of RNA binding proteins in normal and tumor tissue: Identification of potential players in tumor formation. RNA Biol. 2009;6:426–433. doi: 10.4161/rna.6.4.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galmiche L, Serre V, Beinat M, et al. Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum Mutat. 2011;32:1225–1231. doi: 10.1002/humu.21562. [DOI] [PubMed] [Google Scholar]
- Gambardella A, Mazzei R, Toscano A, et al. Spinal muscular atrophy due to an isolated deletion of exon 8 of the telomeric survival motor neuron gene. Ann Neurol. 1998;44:836–839. doi: 10.1002/ana.410440522. [DOI] [PubMed] [Google Scholar]
- Gamberi C, Johnstone O, Lasko P. Drosophila RNA binding proteins. Int Rev Cytol. 2006;248:43–139. doi: 10.1016/S0074-7696(06)48002-5. [DOI] [PubMed] [Google Scholar]
- Gazda HT, Grabowska A, Merida-Long LB, et al. Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2006;79:1110–1118. doi: 10.1086/510020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazda HT, Sheen MR, Vlachos A, et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am J Hum Genet. 2008;83:769–780. doi: 10.1016/j.ajhg.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedeon AK, Baker E, Robinson H, et al. Fragile X syndrome without CCG amplification has an FMR1 deletion. Nat Genet. 1992;1:341–344. doi: 10.1038/ng0892-341. [DOI] [PubMed] [Google Scholar]
- Gelpi C, Sontheimer EJ, Rodriguez-Sanchez JL. Autoantibodies against a serine tRNA-protein complex implicated in cotranslational selenocysteine insertion. Proc Natl Acad Sci U S A. 1992;89:9739–9743. doi: 10.1073/pnas.89.20.9739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi D, Baruffini E, Haack TB, et al. Mutations of the mitochondrial-tRNA modifier MTO1 cause hypertrophic cardiomyopathy and lactic acidosis. Am J Hum Genet. 2012;90:1079–1087. doi: 10.1016/j.ajhg.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson WT, Hood RL, Zhan SH, et al. Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet. 2012;90:110–118. doi: 10.1016/j.ajhg.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill S, McManus AP, Crew AJ, et al. Fusion of the EWS gene to a DNA segment from 9q22-31 in a human myxoid chondrosarcoma. Genes Chromosome Cancer. 1995;12:307–310. doi: 10.1002/gcc.2870120412. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: Effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442:199–202. doi: 10.1038/nature04917. [DOI] [PubMed] [Google Scholar]
- Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582:1977–1986. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold HA, Craft J, Hardin JA, et al. Antibodies in human serum that precipitate ribonuclease P. Proc Natl Acad Sci U S A. 1988;85:5483–5487. doi: 10.1073/pnas.85.15.5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzatti-Haces M, Seth A, Park M, et al. Characterization of the TPR-MET oncogene p65 and the MET protooncogene p140 protein-tyrosine kinases. Proc Natl Acad Sci U S A. 1988;85:21–25. doi: 10.1073/pnas.85.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CT, Petit F, Oufadem M, et al. EFTUD2 haploinsufficiency leads to syndromic oesophageal atresia. J Med Genet. 2012;49:737–746. doi: 10.1136/jmedgenet-2012-101173. [DOI] [PubMed] [Google Scholar]
- Götz A, Tyynismaa H, Euro L, et al. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88:635–642. doi: 10.1016/j.ajhg.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski P. Alternative splicing takes shape during neuronal development. Curr Opin Genet Dev. 2011;21:388–394. doi: 10.1016/j.gde.2011.03.005. [DOI] [PubMed] [Google Scholar]
- Granneman S, Baserga SJ. Ribosome biogenesis: of knobs and RNA processing. Exp Cell Res. 2004;296:43–50. doi: 10.1016/j.yexcr.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Graubert TA, Shen D, Ding L, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2012;44:53–57. doi: 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenway MJ, Andersen PM, Russ C, et al. ANG mutations segregate with familial and “sporadic” amyotrophic lateral sclerosis. Nat Genet. 2006;38:411–413. doi: 10.1038/ng1742. [DOI] [PubMed] [Google Scholar]
- Griffin JH, Leung J, Bruner RJ, et al. Discovery of a fusion kinase in EOL-1 cells and idiopathic hypereosinophilic syndrome. Proc Natl Acad Sci U S A. 2003;100:7830–7835. doi: 10.1073/pnas.0932698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishin NV. KH domain: one motif, two folds. Nucleic Acids Res. 2001;29:638–643. doi: 10.1093/nar/29.3.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivna ST. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev. 2006;20:1709–1714. doi: 10.1101/gad.1434406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohmann K, Schuelke M, Diers A, et al. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat Genet. 2001;29:75–77. doi: 10.1038/ng703. [DOI] [PubMed] [Google Scholar]
- Guan M-X, Yan Q, Li X, et al. Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am J Hum Genet. 2006;79:291–302. doi: 10.1086/506389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubitz AK, Feng W, Dreyfuss G. The SMN complex. Exp Cell Res. 2004;296:51–56. doi: 10.1016/j.yexcr.2004.03.022. [DOI] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Schafer S, Greaser ML, et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med. 2012;18:766–773. doi: 10.1038/nm.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn-Hardy K, Chen JY, Liu HC, et al. Spinocerebellar ataxia type 2 with parkinsonism in ethnic Chinese. Neurology. 2000;55:800–805. doi: 10.1212/wnl.55.6.800. [DOI] [PubMed] [Google Scholar]
- Haack TB, Gorza M, Danhauser K, et al. Phenotypic spectrum of eleven patients and five novel MTFMT mutations identified by exome sequencing and candidate gene screening. Mol Genet Metab. 2014;111:342–352. doi: 10.1016/j.ymgme.2013.12.010. [DOI] [PubMed] [Google Scholar]
- Haack TB, Kopajtich R, Freisinger P, et al. ELAC2 Mutations Cause a Mitochondrial RNA Processing Defect Associated with Hypertrophic Cardiomyopathy. Am J Hum Genet. 2013;93:211–223. doi: 10.1016/j.ajhg.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackman P, Sarparanta J, Lehtinen S, et al. Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1. Ann Neurol. 2012;73:500–509. doi: 10.1002/ana.23831. [DOI] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, et al. Transcriptome-wide identification of RNA-binding protein and MicroRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Max KEA, Bandaru P, et al. Identification of mRNAs bound and regulated by human LIN28 proteins and molecular requirements for RNA recognition. RNA. 2013;19:613–626. doi: 10.1261/rna.036491.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman P. Fragile X-associated tremor/ataxia syndrome (FXTAS): pathology and mechanisms. Acta Neuropathol. 2013;126:1–19. doi: 10.1007/s00401-013-1138-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Leehey M, Heinrichs W, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–130. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- Hahnen E, Schönling J, Rudnik-Schöneborn S, et al. Missense mutations in exon 6 of the survival motor neuron gene in patients with spinal muscular atrophy (SMA) Hum Mol Genet. 1997;6:821–825. doi: 10.1093/hmg/6.5.821. [DOI] [PubMed] [Google Scholar]
- Hall AE, Turnbull C, Dalmay T. Y RNAs: recent developments. BioMol Concepts. 2013;4:103–110. doi: 10.1515/bmc-2012-0050. [DOI] [PubMed] [Google Scholar]
- Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000;404:293–296. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger JS, et al. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson KA, Kim SH, Tibbetts RS. RNA-binding proteins in neurodegenerative disease: TDP-43 and beyond. WIREs RNA. 2011;3:265–285. doi: 10.1002/wrna.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms MB, Ori-McKenney KM, Scoto M, et al. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology. 2012;78:1714–1720. doi: 10.1212/WNL.0b013e3182556c05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauptmann J, Dueck A, Harlander S, et al. Turning catalytically inactive human Argonaute proteins into active slicer enzymes. Nat Struct Mol Biol. 2013;20:814–817. doi: 10.1038/nsmb.2577. [DOI] [PubMed] [Google Scholar]
- Hayashi YK, Matsuda C, Ogawa M, et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest. 2009;119:2623–2633. doi: 10.1172/JCI38660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiss NS, Knight SW, Vulliamy TJ, et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet. 1998;19:32–38. doi: 10.1038/ng0598-32. [DOI] [PubMed] [Google Scholar]
- Hekman KE, Yu G-Y, Brown CD, et al. A conserved eEF2 coding variant in SCA26 leads to loss of translational fidelity and increased susceptibility to proteostatic insult. Hum Mol Genet. 2012;21:5472–5483. doi: 10.1093/hmg/dds392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrick JP, Wolin SL, Rinke J, et al. Ro small cytoplasmic ribonucleoproteins are a subclass of La ribonucleoproteins: further characterization of the Ro and La small ribonucleoproteins from uninfected mammalian cells. Mol Cell Biol. 1981;1:1138–1149. doi: 10.1128/mcb.1.12.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Greene EA, Pietrokovski S, et al. Gene families: the taxonomy of protein paralogs and chimeras. Science. 1997;278:609–614. doi: 10.1126/science.278.5338.609. [DOI] [PubMed] [Google Scholar]
- Henneke M, Diekmann S, Ohlenbusch A, et al. RNASET2-deficient cystic leukoencephalopathy resembles congenital cytomegalovirus brain infection. Nat Genet. 2009;41:773–775. doi: 10.1038/ng.398. [DOI] [PubMed] [Google Scholar]
- Hill DA, Ivanovich J, Priest JR, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325:965. doi: 10.1126/science.1174334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan DJ, Riordan DP, Gerber AP, et al. Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol. 2008;6:e255. doi: 10.1371/journal.pbio.0060255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg JR, Goff SP. Upf1 senses 3′UTR length to potentiate mRNA decay. Cell. 2010;143:379–389. doi: 10.1016/j.cell.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe JR, Roth S, Ringold JC, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086–1088. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- Huppke P, Laccone F, Krämer N, et al. Rett syndrome: analysis of MECP2 and clinical characterization of 31 patients. Hum Mol Genet. 2000;9:1369–1375. doi: 10.1093/hmg/9.9.1369. [DOI] [PubMed] [Google Scholar]
- Hussain S, Sajini AA, Blanco S, et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 2013;4:255–261. doi: 10.1016/j.celrep.2013.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutvagner G, Simard MJ. Argonaute proteins: key players in RNA silencing. Nat Rev Mol Cell Biol. 2008;9:22–32. doi: 10.1038/nrm2321. [DOI] [PubMed] [Google Scholar]
- Ichikawa H, Shimizu K, Hayashi Y, Ohki M. An RNA-binding protein gene, TLS/FUS, is fused to ERG in human myeloid leukemia with t(16;21) chromosomal translocation. Cancer Res. 1994;54:2865–2868. [PubMed] [Google Scholar]
- IJlst L, Loupatty FJ, IRuiter JPN, et al. 3-Methylglutaconic aciduria type I is caused by mutations in AUH. Am J Hum Genet. 2002;71:1463–1466. doi: 10.1086/344712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizu H, Siomi H, Siomi MC. Biology of PIWI-interacting RNAs: new insights into biogenesis and function inside and outside of germlines. Genes Dev. 2012;26:2361–2373. doi: 10.1101/gad.203786.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov P, Emara MM, Villen J, et al. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol Cell. 2011;43:613–623. doi: 10.1016/j.molcel.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky E. RNA helicases at work: binding and rearranging. Trends Biochem Sci. 2011;36:19–29. doi: 10.1016/j.tibs.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky E, Fairman-Williams ME. An introduction to RNA helicases: superfamilies, families, and major themes. In: Jankowsky E, editor. RNA helicases. Vol. 19. RSC Biomolecular Sciences; London: 2010. pp. 1–31. [Google Scholar]
- Jarrous N. Human ribonuclease P: subunits, function, and intranuclear localization. RNA. 2002;8:1–7. doi: 10.1017/s1355838202011184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Doudna JA. A three-dimensional view of the molecular machinery of RNA interference. Nature. 2009;457:405–412. doi: 10.1038/nature07755. [DOI] [PubMed] [Google Scholar]
- Johnston JJ, Teer JK, Cherukuri PF, et al. Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am J Hum Genet. 2010;86:743–748. doi: 10.1016/j.ajhg.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordanova A, Irobi J, Thomas FP, et al. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet. 2006;38:197–202. doi: 10.1038/ng1727. [DOI] [PubMed] [Google Scholar]
- Kaida D, Berg MG, Younis I, et al. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664–668. doi: 10.1038/nature09479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalscheuer VM, Freude K, Musante L, et al. Mutations in the polyglutamine binding protein 1 gene cause X-linked mental retardation. Nat Genet. 2003;35:313–315. doi: 10.1038/ng1264. [DOI] [PubMed] [Google Scholar]
- Kanegane H, Kasahara Y, Okamura J, et al. Identification of DKC1 gene mutations in Japanese patients with X-linked dyskeratosis congenita. Br J Haematol. 2005;129:432–434. doi: 10.1111/j.1365-2141.2005.05473.x. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko H, Dridi S, Tarallo V, et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011;471:325–330. doi: 10.1038/nature09830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapeli K, Yeo GW. Genome-wide approaches to dissect the roles of RNA binding proteins in translational control: implications for neurological diseases. Front Neurosci. 2012;6:144. doi: 10.3389/fnins.2012.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca E, Weitzer S, Pehlivan D, et al. Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell. 2014;157:636–650. doi: 10.1016/j.cell.2014.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaymak E, Wee LM, Ryder SP. Structure and function of nematode RNA-binding proteins. Curr Opin Struct Biol. 2010;20:305–312. doi: 10.1016/j.sbi.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen TJ, Hims MM, McKie AB, et al. Mutations in a protein target of the Pim-1 kinase associated with the RP9 form of autosomal dominant retinitis pigmentosa. Eur J Hum Genet. 2002;10:245–249. doi: 10.1038/sj.ejhg.5200797. [DOI] [PubMed] [Google Scholar]
- Kerner P, Degnan SM, Marchand L, et al. Evolution of RNA-binding proteins in animals: insights from genome-wide analysis in the sponge Amphimedon queenslandica. Mol Biol Evol. 2011;28:2289–2303. doi: 10.1093/molbev/msr046. [DOI] [PubMed] [Google Scholar]
- Khan MA, Rafiq MA, Noor A, et al. Mutation in NSUN2, which encodes an RNA methyl-transferase, causes autosomal-recessive intellectual disability. Am J Hum Genet. 2012;90:856–863. doi: 10.1016/j.ajhg.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khusial P, Plaag R, Zieve GW. LSm proteins form heptameric rings that bind to RNA via repeating motifs. Trends Biochem Sci. 2005;30:522–528. doi: 10.1016/j.tibs.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Kim CA, Bowie JU. SAM domains: uniform structure, diversity of function. Trends Biochem Sci. 2003;28:625–628. doi: 10.1016/j.tibs.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Kim NC, Wang Y-D, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirwan M, Walne AJ, Plagnol V, et al. Exome sequencing identifies autosomal-dominant SRP72 mutations associated with familial aplasia and myelodysplasia. Am J Hum Genet. 2012;90:888–892. doi: 10.1016/j.ajhg.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T. Biogenesis of small nuclear RNPs. J Cell Sci. 2004;117:5949–5951. doi: 10.1242/jcs.01487. [DOI] [PubMed] [Google Scholar]
- Kiss T, Fayet E, Jády BE, et al. Biogenesis and intranuclear trafficking of human box C/D and H/ACA RNPs. Cold Spring Harbor Symp Quant Biol. 2006;71:407–417. doi: 10.1101/sqb.2006.71.025. [DOI] [PubMed] [Google Scholar]
- Klattenhoff C, Theurkauf W. Biogenesis and germline functions of piRNAs. Development. 2008;135:3–9. doi: 10.1242/dev.006486. [DOI] [PubMed] [Google Scholar]
- Klauck SM, Felder B, Kolb-Kokocinski A, et al. Mutations in the ribosomal protein gene RPL10 suggest a novel modulating disease mechanism for autism. Mol Psychiatry. 2006;11:1073–1084. doi: 10.1038/sj.mp.4001883. [DOI] [PubMed] [Google Scholar]
- Klein CJ, Botuyan M-V, Wu Y, et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet. 2011;43:595–600. doi: 10.1038/ng.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman WP, Plasterk RHA. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441–450. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Knight SJL, Flannery AV, Hirst MC, et al. Trinucleotide repeat amplification and hypermethylation of a CpG island in FRAXE mental retardation. Cell. 1993;74:127–134. doi: 10.1016/0092-8674(93)90300-F. [DOI] [PubMed] [Google Scholar]
- Knight SW, Heiss NS, Vulliamy TJ, et al. X-linked dyskeratosis congenita is predominantly caused by missense mutations in the DKC1 gene. Am J Hum Genet. 1999;65:50–58. doi: 10.1086/302446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight SW, Vulliamy TJ, Morgan B, et al. Identification of novel DKC1 mutations in patients with dyskeratosis congenita: implications for pathophysiology and diagnosis. Hum Genet. 2001;108:299–303. doi: 10.1007/s004390100494. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Abe K, Matsuura T, et al. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet. 2011;89:121–130. doi: 10.1016/j.ajhg.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhn M, Pazaitis N, Hüttelmaier S. Why YRNAs? About versatile RNAs and their functions. Biomolecules. 2013;3:143–156. doi: 10.3390/biom3010143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblihtt AR, Schor IE, Allo M, et al. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol. 2013;14:153–165. doi: 10.1038/nrm3525. [DOI] [PubMed] [Google Scholar]
- Korobeinikova AV, Garber MB, Gongadze GM. Ribosomal proteins: structure, function, and evolution. Biochem (Mosc) 2012;77:562–574. doi: 10.1134/S0006297912060028. [DOI] [PubMed] [Google Scholar]
- Kraus MR-C, Clauin S, Pfister Y, et al. Two mutations in human BICC1 resulting in Wnt pathway hyperactivity associated with cystic renal dysplasia. Hum Mutat. 2012;33:86–90. doi: 10.1002/humu.21610. [DOI] [PubMed] [Google Scholar]
- Kremer EJ, Pritchard M, Lynch M, et al. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science. 1991;252:1711–1714. doi: 10.1126/science.1675488. [DOI] [PubMed] [Google Scholar]
- Kressler D, Hurt E, Bassler J. Driving ribosome assembly. Biochim Biophys Acta. 2010;1803:673–683. doi: 10.1016/j.bbamcr.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Kuchta K, Knizewski L, Wyrwicz LS, et al. Comprehensive classification of nucleotidyl-transferase fold proteins: identification of novel families and their representatives in human. Nucleic Acids Res. 2009;37:7701–7714. doi: 10.1093/nar/gkp854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufel J, Bousquet-Antonelli C, Beggs JD, Tollervey D. Nuclear pre-mRNA decapping and 5′ degradation in yeast require the Lsm2-8p complex. Mol Cell Biol. 2004;24:9646–9657. doi: 10.1128/MCB.24.21.9646-9657.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Kwon SC, Yi H, Eichelbaum K, et al. The RNA-binding protein repertoire of embryonic stem cells. Nat Struct Mol Biol. 2013;20:1122–1130. doi: 10.1038/nsmb.2638. [DOI] [PubMed] [Google Scholar]
- Lachke SA, Alkuraya FS, Kneeland SC, et al. Mutations in the RNA granule component TDRD7 cause cataract and glaucoma. Science. 2011;331:1571–1576. doi: 10.1126/science.1195970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19:R46–R64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larroux C, Luke GN, Koopman P, et al. Genesis and expansion of metazoan transcription factor gene classes. Mol Biol Evol. 2008;25:980–996. doi: 10.1093/molbev/msn047. [DOI] [PubMed] [Google Scholar]
- Lasko P. The Drosophila melanogaster genome: translation factors and RNA binding proteins. J Cell Biol. 2000;150:F51–F56. doi: 10.1083/jcb.150.2.f51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasko P. Gene regulation at the RNA layer: RNA binding proteins in intercellular signaling networks. Sci STKE. 2003;2003:RE6. doi: 10.1126/stke.2003.179.re6. [DOI] [PubMed] [Google Scholar]
- Lasko P. The DEAD-box helicase Vasa: evidence for a multiplicity of functions in RNA processes and developmental biology. Biochim Biophys Acta. 2013;1829:810–816. doi: 10.1016/j.bbagrm.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Latour P, Thauvin-Robinet C, Baudelet-Méry C, et al. A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet. 2010;86:77–82. doi: 10.1016/j.ajhg.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M-H, Schedl T. RNA-binding proteins. WormBook. 2006:1–13. doi: 10.1895/wormbook.1.79.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han JJ, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- Lee EB, Lee VMY, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci. 2012;13:38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leegwater PA, Vermeulen G, Könst AA, et al. Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat Genet. 2001;29:383–388. doi: 10.1038/ng764. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Lerner MR, Boyle JA, Hardin JA, Steitz JA. Two novel classes of small ribonucleoproteins detected by antibodies associated with lupus erythematosus. Science. 1981;211:400–402. doi: 10.1126/science.6164096. [DOI] [PubMed] [Google Scholar]
- Letunic I, Doerks T, Bork P. SMART 6: recent updates and new developments. Nucleic Acids Res. 2009;37:D229–D232. doi: 10.1093/nar/gkn808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kowdley KV. MicroRNAs in common human diseases. Genom Proteom Bioinform. 2012;10:246–253. doi: 10.1016/j.gpb.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Lee J-A, Black DL. Neuronal regulation of alternative pre-mRNA splicing. Nat Rev Neurosci. 2007;8:819–831. doi: 10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. 2013;201:361–372. doi: 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K-P, Soong B-W, Yang C-C, et al. The mutational spectrum in a cohort of Charcot-Marie-Tooth disease type 2 among the Han Chinese in Taiwan. PLoS One. 2011;6:e29393. doi: 10.1371/journal.pone.0029393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder P, Fuller-Pace FV. Looking back on the birth of DEAD-box RNA helicases. Biochim Biophys Acta. 2013;1829:750–755. doi: 10.1016/j.bbagrm.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Linder P, Jankowsky E. From unwinding to clamping - the DEAD box RNA helicase family. Nat Rev Mol Cell Biol. 2011;12:505–516. doi: 10.1038/nrm3154. [DOI] [PubMed] [Google Scholar]
- Lines MA, Huang L, Schwartzentruber J, et al. Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly. Am J Hum Genet. 2012;90:369–377. doi: 10.1016/j.ajhg.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S-C, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- Liu J. Argonaute2 Is the catalytic engine of Mammalian RNAi. Science. 2004;305:1437–1441. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- Liu JM. Ribosomes and marrow failure: coincidental association or molecular paradigm? Blood. 2006;107:4583–4588. doi: 10.1182/blood-2005-12-4831. [DOI] [PubMed] [Google Scholar]
- Liu G, Grant WM, Persky D, et al. Interactions of elongation factor 1alpha with F-actin and beta-actin mRNA: implications for anchoring mRNA in cell protrusions. Mol Biol Cell. 2002;13:579–592. doi: 10.1091/mbc.01-03-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bassell GJ, Gitler AD, et al. Local RNA translation at the synapse and in disease. J Neurosci. 2011;31:16086–16093. doi: 10.1523/JNEUROSCI.4105-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovci MT, Ghanem D, Marr H, et al. Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat Struct Mol Biol. 2013;20:1434–1442. doi: 10.1038/nsmb.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukong KE, Chang KW, Khandjian EW, Richard S. RNA-binding proteins in human genetic disease. Trends Genet. 2008;24:416–425. doi: 10.1016/j.tig.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Lund E. Nuclear export of MicroRNA precursors. Science. 2004;303:95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol. 2007;8:479–490. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly TBN, Peters V, Gibson KM, et al. Mutations in the AUH gene cause 3-methylglutaconic aciduria type I. Hum Mutat. 2003;21:401–407. doi: 10.1002/humu.10202. [DOI] [PubMed] [Google Scholar]
- Mankodi A, Urbinati CR, Yuan QP, et al. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet. 2001;10:2165–2170. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- Maraia RJ, Bayfield MA. The La protein-RNA complex surfaces. Mol Cell. 2006;21:149–152. doi: 10.1016/j.molcel.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Maraia RJ, Lamichhane TN. 3′ processing of eukaryotic precursor tRNAs. WIREs RNA. 2011;2:362–375. doi: 10.1002/wrna.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, DeWeese-Scott C, et al. CDD: a curated Entrez database of conserved domain alignments. Nucleic Acids Res. 2003;31:383–387. doi: 10.1093/nar/gkg087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maris C, Dominguez C, Allain FHT. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005;272:2118–2131. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- Marrone A, Walne A, Tamary H, et al. Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood. 2007;110:4198–4205. doi: 10.1182/blood-2006-12-062851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalek B, Wisniewski SA, Wojcicki P, et al. Novel mutation in the 5′ splice site of exon 4 of the TCOF1 gene in the patient with Treacher Collins syndrome. Am J Med Genet. 2003;123A:169–171. doi: 10.1002/ajmg.a.20312. [DOI] [PubMed] [Google Scholar]
- Martin CL, Duvall JA, Ilkin Y, et al. Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:869–876. doi: 10.1002/ajmg.b.30530. [DOI] [PubMed] [Google Scholar]
- Martin R, Straub AU, Doebele C, Bohnsack MT. DExD/H-box RNA helicases in ribosome biogenesis. RNA Biol. 2013;10:4–18. doi: 10.4161/rna.21879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah G, Barraud P, Allain FHT. RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell Mol Life Sci. 2013;70:1875–1895. doi: 10.1007/s00018-012-1119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massart A, Lissens W, Tournaye H, Stouffs K. Genetic causes of spermatogenic failure. Asian J Androl. 2012;14:40–48. doi: 10.1038/aja.2011.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera AG, Terns RM, Terns MP. Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat Rev Mol Cell Biol. 2007;8:209–220. doi: 10.1038/nrm2124. [DOI] [PubMed] [Google Scholar]
- Matlin AJ, Clark F, Smith CWJ. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005;6:386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- Matsson H, Klar J, Draptchinskaia N, et al. Truncating ribosomal protein S19 mutations and variable clinical expression in Diamond-Blackfan anemia. Hum Genet. 1999;105:496–500. doi: 10.1007/s004399900165. [DOI] [PubMed] [Google Scholar]
- Matthews DE, Hessler RA, Denslow ND, et al. Protein composition of the bovine mitochondrial ribosome. J Biol Chem. 1982;257:8788–8794. [PubMed] [Google Scholar]
- May WA, Gishizky ML, Lessnick SL, et al. Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proc Natl Acad Sci U S A. 1993;90:5752–5756. doi: 10.1073/pnas.90.12.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–684. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AE, Minet E, Stern C, et al. A genome-wide in situ hybridization map of RNA-binding proteins reveals anatomically restricted expression in the developing mouse brain. BMC Dev Biol. 2005;5:14. doi: 10.1186/1471-213X-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKie AB, McHale JC, Keen TJ, et al. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13) Hum Mol Genet. 2001;10:1555–1562. doi: 10.1093/hmg/10.15.1555. [DOI] [PubMed] [Google Scholar]
- McLaughlin HM, Sakaguchi R, Liu C, et al. Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet. 2010;87:560–566. doi: 10.1016/j.ajhg.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G, Landthaler M, Patkaniowska A, et al. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell. 2004;15:185–197. doi: 10.1016/j.molcel.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Melnikov S, Ben-Shem A, Garreau de Loubresse N, et al. One core, two shells: bacterial and eukaryotic ribosomes. Nat Struct Mol Biol. 2012;19:560–567. doi: 10.1038/nsmb.2313. [DOI] [PubMed] [Google Scholar]
- Melo SA, Ropero S, Moutinho C, et al. A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat Genet. 2009;41:365–370. doi: 10.1038/ng.317. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Melo SA, Moutinho C, Ropero S, et al. A genetic defect in exportin-5 traps precursor micrornas in the nucleus of cancer cells. Cancer Cell. 2010;18:303–315. doi: 10.1016/j.ccr.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Merritt WM, Lin YG, Han LY, et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med. 2008;359:2641–2650. doi: 10.1056/NEJMoa0803785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickleburgh I, Chabanon H, Nury D, et al. Elongation factor 1alpha binds to the region of the metallothionein-1 mRNA implicated in perinuclear localization–importance of an internal stem-loop. RNA. 2006;12:1397–1407. doi: 10.1261/rna.2730106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihailovich M, Militti C, Gabaldón T, Gebauer F. Eukaryotic cold shock domain proteins: highly versatile regulators of gene expression. Bioessays. 2010;32:109–118. doi: 10.1002/bies.200900122. [DOI] [PubMed] [Google Scholar]
- Miki T, Takano K, Yoneda Y. The role of mammalian Staufen on mRNA traffic: a view from its nucleocytoplasmic shuttling function. Cell Struct Funct. 2005;30:51–56. doi: 10.1247/csf.30.51. [DOI] [PubMed] [Google Scholar]
- Miller JW. Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C, Saada A, Shaul N, et al. Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann Neurol. 2004;56:734–738. doi: 10.1002/ana.20282. [DOI] [PubMed] [Google Scholar]
- Mitchell SF, Jain S, She M, Parker R. Global analysis of yeast mRNPs. Nat Struct Mol Biol. 2013;20:127–133. doi: 10.1038/nsmb.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura P, Shenker S, Andreu-Agullo C, et al. Widespread and extensive lengthening of 3′ UTRs in the mammalian brain. Genome Res. 2013;23:812–825. doi: 10.1101/gr.146886.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamura Y, Suzuki T, Kono M, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet. 2003;73:693–699. doi: 10.1086/378209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monemi S, Spaeth G, DaSilva A, et al. Identification of a novel adult-onset primary open-angle glaucoma (POAG) gene on 5q22.1. Hum Mol Genet. 2005;14:725–733. doi: 10.1093/hmg/ddi068. [DOI] [PubMed] [Google Scholar]
- Monti L, Rodolfo M, Rudolfo M, et al. RNASET2 as a tumor antagonizing gene in a melanoma cancer model. Oncol Res. 2008;17:69–74. doi: 10.3727/096504008784523658. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lepage P, Miller K, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci U S A. 2003;100:605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira M-C, Klur S, Watanabe M, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. 2004;36:225–227. doi: 10.1038/ng1303. [DOI] [PubMed] [Google Scholar]
- Müller-McNicoll M, Neugebauer KM. How cells get the message: dynamic assembly and function of mRNA-protein complexes. Nat Rev Genet. 2013;14:275–287. doi: 10.1038/nrg3434. [DOI] [PubMed] [Google Scholar]
- Muñoz LE, Lauber K, Schiller M, et al. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. 2010;6:280–289. doi: 10.1038/nrrheum.2010.46. [DOI] [PubMed] [Google Scholar]
- Murray A, Webb J, Grimley S, et al. Studies of FRAXA and FRAXE in women with premature ovarian failure. J Med Genet. 1998;35:637–640. doi: 10.1136/jmg.35.8.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin AG. OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J. 1993;12:861–867. doi: 10.1002/j.1460-2075.1993.tb05726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Muscatelli F, Strom TM, Walker AP, et al. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature. 1994;372:672–676. doi: 10.1038/372672a0. [DOI] [PubMed] [Google Scholar]
- Muto Y, Yokoyama S. Structural insight into RNA recognition motifs: versatile molecular Lego building blocks for biological systems. WIREs RNA. 2012;3:229–246. doi: 10.1002/wrna.1107. [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Ascano M, Gogakos T, et al. Eukaryote-specific insertion elements control human ARGONAUTE slicer activity. Cell Rep. 2013;3:1893–1900. doi: 10.1016/j.celrep.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima E, Mabuchi A, Makita Y, et al. Novel SBDS mutations caused by gene conversion in Japanese patients with Shwachman-Diamond syndrome. Hum Genet. 2004;114:345–348. doi: 10.1007/s00439-004-1081-2. [DOI] [PubMed] [Google Scholar]
- Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115:3196–3205. doi: 10.1182/blood-2009-10-178129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasim MT, Ogo T, Ahmed M, et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat. 2011;32:1385–1389. doi: 10.1002/humu.21605. [DOI] [PubMed] [Google Scholar]
- Nawrot B, Sochacka E, Düchler M. tRNA structural and functional changes induced by oxidative stress. Cell Mol Life Sci. 2011;68:4023–4032. doi: 10.1007/s00018-011-0773-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neeve VCM, Pyle A, Boczonadi V, et al. Clinical and functional characterisation of the combined respiratory chain defect in two sisters due to autosomal recessive mutations in MTFMT. Mitochondrion. 2013;13:743–748. doi: 10.1016/j.mito.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrutskii BS, El’skaya AV. Eukaryotic translation elongation factor 1 alpha: structure, expression, functions, and possible role in aminoacyl-tRNA channeling. Prog Nucleic Acid Res Mol Biol. 1998;60:47–78. doi: 10.1016/s0079-6603(08)60889-2. [DOI] [PubMed] [Google Scholar]
- Neilson DE, Adams MD, Orr CMD, et al. Infection-triggered familial or recurrent cases of acute necrotizing encephalopathy caused by mutations in a component of the nuclear pore, RANBP2. Am J Hum Genet. 2009;84:44–51. doi: 10.1016/j.ajhg.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DL, Orr HT, Warren ST. The unstable repeats—three evolving faces of neurological disease. Neuron. 2013;77:825–843. doi: 10.1016/j.neuron.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen P, Kjeldgaard M, Thirup S, et al. Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog. Science. 1995;270:1464–1472. doi: 10.1126/science.270.5241.1464. [DOI] [PubMed] [Google Scholar]
- Nousbeck J, Spiegel R, Ishida-Yamamoto A, et al. Alopecia, neurological defects, and endocrinopathy syndrome caused by decreased expression of RBM28, a nucleolar protein associated with ribosome biogenesis. Am J Hum Genet. 2008;82:1114–1121. doi: 10.1016/j.ajhg.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nousiainen HO, Kestila M, Pakkasjarvi N, et al. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat Genet. 2008;40:155–157. doi: 10.1038/ng.2007.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien TW. Properties of human mitochondrial ribosomes. IUBMB Life. 2003;55:505–513. doi: 10.1080/15216540310001626610. [DOI] [PubMed] [Google Scholar]
- Omran H, Kobayashi D, Olbrich H, et al. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orloff M, Peterson C, He X, et al. Germline mutations in MSR1, ASCC1, and CTHRC1 in patients with Barrett esophagus and esophageal adenocarcinoma. JAMA. 2011;306:410–419. doi: 10.1001/jama.2011.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HT, Chung MY, Banfi S, et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4:221–226. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- Panagopoulos I, Aman P, Fioretos T, et al. Fusion of the FUS gene with ERG in acute myeloid leukemia with t(16;21)(p11;q22) Genes Chromosome Cancer. 1994;11:256–262. doi: 10.1002/gcc.2870110408. [DOI] [PubMed] [Google Scholar]
- Panagopoulos I, Mencinger M, Dietrich CU, et al. Fusion of the RBP56 and CHN genes in extraskeletal myxoid chondrosarcomas with translocation t(9;17)(q22;q11) Oncogene. 1999;18:7594–7598. doi: 10.1038/sj.onc.1203155. [DOI] [PubMed] [Google Scholar]
- Pannone BK, Kim SD, Noe DA, Wolin SL. Multiple functional interactions between components of the Lsm2-Lsm8 complex, U6 snRNA, and the yeast La protein. Genetics. 2001;158:187–196. doi: 10.1093/genetics/158.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, Lin HF. Translational repression: A duet of Nanos and Pumilio. Curr Biol. 2000;10:R81–R83. doi: 10.1016/s0960-9822(00)00283-9. [DOI] [PubMed] [Google Scholar]
- Park E, Maquat LE. Staufen-mediated mRNA decay. WIREs RNA. 2013;4:423–435. doi: 10.1002/wrna.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R, Sheth U. P bodies and the control of mRNA translation and degradation. Mol Cell. 2007;25:635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Parker R, Song H. The enzymes and control of eukaryotic mRNA turnover. Nat Struct Mol Biol. 2004;11:121–127. doi: 10.1038/nsmb724. [DOI] [PubMed] [Google Scholar]
- Parsons DW, McAndrew PE, Monani UR, et al. An 11 base pair duplication in exon 6 of the SMN gene produces a type I spinal muscular atrophy (SMA) phenotype: further evidence for SMN as the primary SMA-determining gene. Hum Mol Genet. 1996;5:1727–1732. doi: 10.1093/hmg/5.11.1727. [DOI] [PubMed] [Google Scholar]
- Pascual M, Vicente M, Monferrer L, Artero R. The Muscleblind family of proteins: an emerging class of regulators of developmentally programmed alternative splicing. Differentiation. 2006;74:65–80. doi: 10.1111/j.1432-0436.2006.00060.x. [DOI] [PubMed] [Google Scholar]
- Pasternack SM, Refke M, Paknia E, et al. Mutations in SNRPE, which encodes a core protein of the spliceosome, cause autosomal-dominant hypotrichosis simplex. Am J Hum Genet. 2013;92:81–87. doi: 10.1016/j.ajhg.2012.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paushkin S, Gubitz AK, Massenet S, Dreyfuss G. The SMN complex, an assemblyosome of ribonucleoproteins. Curr Opin Cell Biol. 2002;14:305–312. doi: 10.1016/s0955-0674(02)00332-0. [DOI] [PubMed] [Google Scholar]
- Pearson T, Curtis F, Al-Eyadhy A, et al. An intronic mutation in DKC1 in an infant with Høyeraal-Hreidarsson syndrome. Am J Med Genet. 2008;146A:2159–2161. doi: 10.1002/ajmg.a.32412. [DOI] [PubMed] [Google Scholar]
- Perron MP, Provost P. Protein components of the microRNA pathway and human diseases. Methods Mol Biol. 2009;487:369–385. doi: 10.1007/978-1-60327-547-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterlin BM, Brogie JE, Price DH. 7SK snRNA: a noncoding RNA that plays a major role in regulating eukaryotic transcription. WIREs RNA. 2011;3:92–103. doi: 10.1002/wrna.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters L, Meister G. Argonaute proteins: mediators of RNA silencing. Mol Cell. 2007;26:611–623. doi: 10.1016/j.molcel.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Pettersson I, Hinterberger M, Mimori T, et al. The structure of mammalian small nuclear ribonucleoproteins. Identification of multiple protein components reactive with anti-(U1) ribonucleoprotein and anti-Sm autoantibodies. J Biol Chem. 1984;259:5907–5914. [PubMed] [Google Scholar]
- Phizicky EM, Hopper AK. tRNA biology charges to the front. Genes Dev. 2010;24:1832–1860. doi: 10.1101/gad.1956510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce SB, Chisholm KM, Lynch ED, et al. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci U S A. 2011;108:6543–6548. doi: 10.1073/pnas.1103471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce SB, Gersak K, Michaelson-Cohen R, et al. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am J Hum Genet. 2013;92:614–620. doi: 10.1016/j.ajhg.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai RS, Will CL, Luhrmann R, et al. Purified U7 snRNPs lack the Sm proteins D1 and D2 but contain Lsm10, a new 14 kDa Sm D1-like protein. EMBO J. 2001;20:5470–5479. doi: 10.1093/emboj/20.19.5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot N. New perspectives on connecting messenger RNA 3′ end formation to transcription. Curr Opin Cell Biol. 2004;16:272–278. doi: 10.1016/j.ceb.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Proudfoot N, O’Sullivan J. Polyadenylation: a tail of two complexes. Curr Biol. 2002;12:R855–R857. doi: 10.1016/s0960-9822(02)01353-2. [DOI] [PubMed] [Google Scholar]
- Puffenberger EG, Jinks RN, Sougnez C, et al. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS One. 2012;7:e28936. doi: 10.1371/journal.pone.0028936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulst SM, Nechiporuk A, Nechiporuk T, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet. 1996;14:269–276. doi: 10.1038/ng1196-269. [DOI] [PubMed] [Google Scholar]
- Pyle AM. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu Rev Biophys. 2008;37:317–336. doi: 10.1146/annurev.biophys.37.032807.125908. [DOI] [PubMed] [Google Scholar]
- Rabe B. Aicardi-Goutières syndrome: clues from the RNase H2 knock-out mouse. J Mol Med. 2013;91:1235–1240. doi: 10.1007/s00109-013-1061-x. [DOI] [PubMed] [Google Scholar]
- Rajkowitsch L, Chen D, Stampfl S, et al. RNA chaperones, RNA annealers and RNA helicases. RNA Biol. 2007;4:118–130. doi: 10.4161/rna.4.3.5445. [DOI] [PubMed] [Google Scholar]
- Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013;154:727–736. doi: 10.1016/j.cell.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramser J, Ahearn ME, Lenski C, et al. Rare missense and synonymous variants in UBE1 are associated with X-linked infantile spinal muscular atrophy. Am J Hum Genet. 2008;82:188–193. doi: 10.1016/j.ajhg.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramser J, Winnepenninckx B, Lenski C, et al. A splice site mutation in the methyltransferase gene FTSJ1 in Xp11.23 is associated with non-syndromic mental retardation in a large Belgian family (MRX9) J Med Genet. 2004;41:679–683. doi: 10.1136/jmg.2004.019000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin J, Brown R, Dobyns WB, et al. Pontocerebellar hypoplasia type 6: A British case with PEHO-like features. Am J Med Genet. 2010;152A:2079–2084. doi: 10.1002/ajmg.a.33531. [DOI] [PubMed] [Google Scholar]
- Raven JF, Koromilas AE. PERK and PKR: old kinases learn new tricks. Cell Cycle. 2008;7:1146–1150. doi: 10.4161/cc.7.9.5811. [DOI] [PubMed] [Google Scholar]
- Ravenscroft JC, Suri M, Rice GI, et al. Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am J Med Genet. 2011;155A:235–237. doi: 10.1002/ajmg.a.33778. [DOI] [PubMed] [Google Scholar]
- Regalado ES, Guo D-C, Villamizar C, et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res. 2011;109:680–686. doi: 10.1161/CIRCRESAHA.111.248161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijo R, Lee TY, Salo P, et al. Diverse spermatogenic defects in humans caused by Y chromosome deletions encompassing a novel RNA-binding protein gene. Nat Genet. 1995;10:383–393. doi: 10.1038/ng0895-383. [DOI] [PubMed] [Google Scholar]
- Rice GI, Bond J, Asipu A, et al. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41:829–832. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, del Toro DY, Jenkinson EM, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet. 2014;46:503–509. doi: 10.1038/ng.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GMA, et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Reijns MAM, Coffin SR, et al. Synonymous mutations in RNASEH2A create cryptic splice sites impairing RNase H2 enzyme function in Aicardi-Goutières syndrome. Hum Mutat. 2013;34:1066–1070. doi: 10.1002/humu.22336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley LG, Cooper S, Hickey P, et al. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia–MLASA syndrome. Am J Hum Genet. 2010;87:52–59. doi: 10.1016/j.ajhg.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio Frio T, Bahubeshi A, Kanellopoulou C, et al. DICER1 mutations in familial multinodular goiter with and without ovarian Sertoli-Leydig cell tumors. JAMA. 2011;305:68–77. doi: 10.1001/jama.2010.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R, Timchenko NA, Miller JW, et al. Altered phosphorylation and intracellular distribution of a (CUG)n triplet repeat RNA-binding protein in patients with myotonic dystrophy and in myotonin protein kinase knockout mice. Proc Natl Acad Sci U S A. 1997;94:13221–13226. doi: 10.1073/pnas.94.24.13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocak S, Linder P. DEAD-box proteins: the driving forces behind RNA metabolism. Nat Rev Mol Cell Biol. 2004;5:232–241. doi: 10.1038/nrm1335. [DOI] [PubMed] [Google Scholar]
- Rosenberg HF. Vertebrate secretory (RNase A) ribonucleases and host defense. Nucleic Acids Mol Biol. 2011;26:35–53. [Google Scholar]
- Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3:179–192. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- Russell JF, Steckley JL, Coppola G, et al. Familial cortical myoclonus with a mutation in NOL3. Ann Neurol. 2012;72:175–183. doi: 10.1002/ana.23666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saada A, Shaag A, Arnon S, et al. Antenatal mitochondrial disease caused by mitochondrial ribosomalprotein (MRPS22) mutation. J Med Genet. 2007;44:784–786. doi: 10.1136/jmg.2007.053116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K, Gofuku M, Kitagawa Y, et al. A hippocampal protein associated with paraneoplastic neurologic syndrome and small cell lung carcinoma. Biochem Biophys Res Commun. 1994;199:1200–1208. doi: 10.1006/bbrc.1994.1358. [DOI] [PubMed] [Google Scholar]
- Sandberg R, Neilson JR, Sarma A, et al. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer MicroRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler H, Stoecklin G. Control of mRNA decay by phosphorylation of tristetraprolin. Biochem Soc Trans. 2008;36:491. doi: 10.1042/BST0360491. [DOI] [PubMed] [Google Scholar]
- Santos-Cortez RLP, Lee K, Azeem Z, et al. Mutations in KARS, encoding lysyl-tRNA synthetase, cause autosomal-recessive nonsyndromic hearing impairment DFNB89. Am J Hum Genet. 2013;93:132–140. doi: 10.1016/j.ajhg.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders LR, Barber GN. The dsRNA binding protein family: critical roles, diverse cellular functions. FASEB J. 2003;17:961–983. doi: 10.1096/fj.02-0958rev. [DOI] [PubMed] [Google Scholar]
- Savva YA, Rieder LE, Reenan RA. The ADAR protein family. Genome Biol. 2012;13:252. doi: 10.1186/gb-2012-13-12-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicka K, Bushell M, Spriggs KA, Willis AE. Polypyrimidinetract-binding protein: a multifunctional RNA-binding protein. Biochem Soc Trans. 2008;36:641–647. doi: 10.1042/BST0360641. [DOI] [PubMed] [Google Scholar]
- Scheper GC, van der Knaap MS, Proud CG. Translation matters: protein synthesis defects in inherited disease. Nat Rev Genet. 2007a;8:711–723. doi: 10.1038/nrg2142. [DOI] [PubMed] [Google Scholar]
- Scheper GC, van der Klok T, van Andel RJ, et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007b;39:534–539. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- Scherrer T, Mittal N, Janga SC, Gerber AP. A screen for RNA-binding proteins in yeast indicates dual functions for many enzymes. PLoS One. 2010;5:e15499. doi: 10.1371/journal.pone.0015499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirle NT, MacRae IJ. The crystal structure of human argonaute2. Science. 2012;336:1037–1040. doi: 10.1126/science.1221551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K, Butler JS. Nuclear RNA surveillance: role of TRAMP in controlling exosome specificity. WIREs RNA. 2013;4:217–231. doi: 10.1002/wrna.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutte M, Hruban RH, Hedrick L, et al. DPC4 gene in various tumor types. Cancer Res. 1996;56:2527–2530. [PubMed] [Google Scholar]
- Seibler P, Djarmati A, Langpap B, et al. A heterozygous frameshift mutation in PRKRA (DYT16) associated with generalised dystonia in a German patient. Lancet Neurol. 2008;7:380–381. doi: 10.1016/S1474-4422(08)70075-9. [DOI] [PubMed] [Google Scholar]
- Senderek J, Garvey SM, Krieger M, et al. Autosomal-dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am J Hum Genet. 2009;84:511–518. doi: 10.1016/j.ajhg.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servadio A, Koshy B, Armstrong D, et al. Expression analysis of the ataxin-1 protein in tissues from normal and spinocerebellar ataxia type 1 individuals. Nat Genet. 1995;10:94–98. doi: 10.1038/ng0595-94. [DOI] [PubMed] [Google Scholar]
- Shamseldin HE, Alshammari M, Al-Sheddi T, et al. Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J Med Genet. 2012;49:234–241. doi: 10.1136/jmedgenet-2012-100836. [DOI] [PubMed] [Google Scholar]
- Shastry S, Delgado MR, Dirik E, et al. Congenital generalized lipodystrophy, type 4 (CGL4) associated with myopathy due to novel PTRF mutations. Am J Med Genet. 2010;152A:2245–2253. doi: 10.1002/ajmg.a.33578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatkin AJ, Manley JL. The ends of the affair: capping and polyadenylation. Nat Struct Biol. 2000;7:838–842. doi: 10.1038/79583. [DOI] [PubMed] [Google Scholar]
- Shimazaki H, Takiyama Y, Ishiura H, et al. A homozygous mutation of C12orf65 causes spastic paraplegia with optic atrophy and neuropathy (SPG55) J Med Genet. 2012;49:777–784. doi: 10.1136/jmedgenet-2012-101212. [DOI] [PubMed] [Google Scholar]
- Sim S, Wolin SL. Emerging roles for the Ro 60-kDa autoantigen in noncoding RNA metabolism. WIREs RNA. 2011;2:686–699. doi: 10.1002/wrna.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard J, Tonin P, Durocher F, et al. Common origins of BRCA1 mutations in Canadian breast and ovarian cancer families. Nat Genet. 1994;8:392–398. doi: 10.1038/ng1294-392. [DOI] [PubMed] [Google Scholar]
- Simon B, Kirkpatrick JP, Eckhardt S, et al. Recognition of 2“-O-methylated 3-”end of piRNA by the PAZ domain of a Piwi protein. Structure. 2011;19:172–180. doi: 10.1016/j.str.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Simone LE, Keene JD. Mechanisms coordinating ELAV/Hu mRNA regulons. Curr Opin Genet Dev. 2013;23:35–43. doi: 10.1016/j.gde.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simos G, Hurt E. Transfer RNA biogenesis: A visa to leave the nucleus. Curr Biol. 1999;9:R238–R241. doi: 10.1016/s0960-9822(99)80152-3. [DOI] [PubMed] [Google Scholar]
- Singh RK, Cooper TA. Pre-mRNA splicing in disease and therapeutics. Trends Mol Med. 2012;18:472–482. doi: 10.1016/j.molmed.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Valcarcel J. Building specificity with nonspecific RNA-binding proteins. Nat Struct Mol Biol. 2005;12:645–653. doi: 10.1038/nsmb961. [DOI] [PubMed] [Google Scholar]
- Siomi MC, Mannen T, Siomi H. How does the royal family of tudor rule the PIWI-interacting RNA pathway? Genes Dev. 2010;24:636–646. doi: 10.1101/gad.1899210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi MC, Sato K, Pezic D, Aravin AA. PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol. 2011;12:246–258. doi: 10.1038/nrm3089. [DOI] [PubMed] [Google Scholar]
- Siprashvili Z, Webster DE, Kretz M, et al. Identification of proteins binding coding and non-coding human RNAs using protein microarrays. BMC Genomics. 2012;13:633. doi: 10.1186/1471-2164-13-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeitink JAM, Elpeleg O, Antonicka H, et al. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am J Hum Genet. 2006;79:869–877. doi: 10.1086/508434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CW, Valcarcel J. Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem Sci. 2000;25:381–388. doi: 10.1016/s0968-0004(00)01604-2. [DOI] [PubMed] [Google Scholar]
- Smits P, Smeitink J, van den Heuvel L. Mitochondrial translation and beyond: processes implicated in combined oxidative phosphorylation deficiencies. J Biomed Biotech. 2010;2010:737385. doi: 10.1155/2010/737385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, de Lange T. Regulation of telomerase by telomeric proteins. Annu Rev Biochem. 2004;73:177–208. doi: 10.1146/annurev.biochem.73.071403.160049. [DOI] [PubMed] [Google Scholar]
- Sommerville J. Activities of cold-shock domain proteins in translation control. Bioessays. 1999;21:319–325. doi: 10.1002/(SICI)1521-1878(199904)21:4<319::AID-BIES8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Song JJ. Crystal structure of argonaute and its implications for RISC slicer activity. Science. 2004;305:1434–1437. doi: 10.1126/science.1102514. [DOI] [PubMed] [Google Scholar]
- Song JJ, Joshua-Tor L. Argonaute and RNA–getting into the groove. Curr Opin Struct Biol. 2006;16:5–11. doi: 10.1016/j.sbi.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Sossi V, Giuli A, Vitali T, et al. Premature termination mutations in exon 3 of the SMN1 gene are associated with exon skipping and a relatively mild SMA phenotype. Eur J Hum Genet. 2001;9:113–120. doi: 10.1038/sj.ejhg.5200599. [DOI] [PubMed] [Google Scholar]
- Splendore A, Passos-Bueno MR, Jabs EW, et al. TCOF1 mutations excluded from a role in other first and second branchial arch-related disorders. Am J Med Genet. 2002;111:324–327. doi: 10.1002/ajmg.10567. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenweg ME, Ghezzi D, Haack T, et al. Leukoencephalopathy with thalamus and brainstem involvement and high lactate “LTBL” caused by EARS2 mutations. Brain. 2012;135:1387–1394. doi: 10.1093/brain/aws070. [DOI] [PubMed] [Google Scholar]
- Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stettner GM, Shoukier M, Höger C, et al. Familial intellectual disability and autistic behavior caused by a small FMR2 gene deletion. Am J Med Genet. 2011;155:2003–2007. doi: 10.1002/ajmg.a.34122. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Nagao A, Suzuki T. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu Rev Genet. 2011;45:299–329. doi: 10.1146/annurev-genet-110410-132531. [DOI] [PubMed] [Google Scholar]
- Szymczyna BR, Bowman J, McCracken S, et al. Structure and function of the PWI motif: a novel nucleic acid-binding domain that facilitates pre-mRNA processing. Genes Dev. 2003;17:461–475. doi: 10.1101/gad.1060403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talim B, Pyle A, Griffin H, et al. Multisystem fatal infantile disease caused by a novel homozygous EARS2 mutation. Brain. 2013;136:e228. doi: 10.1093/brain/aws197. [DOI] [PubMed] [Google Scholar]
- Tamburino AM, Ryder SP, Walhout AJM. A compendium of caenorhabditis elegans RNA binding proteins predicts extensive regulation at multiple levels. G3 (Bethesda) 2013;3:297–304. doi: 10.1534/g3.112.004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AY, Manley JL. The TET family of proteins: functions and roles in disease. J Mol Cell Biol. 2009;1:82–92. doi: 10.1093/jmcb/mjp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HL, Glen E, Töpf A, et al. Nonsynonymous variants in the SMAD6 gene predispose to congenital cardiovascular malformation. Hum Mutat. 2012;33:720–727. doi: 10.1002/humu.22030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanackovic G, Ransijn A, Ayuso C, et al. A missense mutation in PRPF6 causes impairment of pre-mRNA splicing and autosomal-dominant retinitis pigmentosa. Am J Hum Genet. 2011;88:643–649. doi: 10.1016/j.ajhg.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Morice-Picard F, Lacombe D, et al. Identification of a homozygous deletion mutation in C16orf57 in a family with Clericuzio-type poikiloderma with neutropenia. Am J Med Genet. 2010;152A:1347–1348. doi: 10.1002/ajmg.a.33455. [DOI] [PubMed] [Google Scholar]
- Tanner NK, Linder P. DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol Cell. 2001;8:251–262. doi: 10.1016/s1097-2765(01)00329-x. [DOI] [PubMed] [Google Scholar]
- Tarpey PS, Raymond FL, Nguyen LS, et al. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat Genet. 2007;39:1127–1133. doi: 10.1038/ng2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Simard J, Teng DH, et al. A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet. 2001;27:172–180. doi: 10.1038/84808. [DOI] [PubMed] [Google Scholar]
- Teplova M, Hafner M, Teplov D, et al. Structure-function studies of STAR family Quaking proteins bound to their in vivo RNA target sites. Genes Dev. 2013;27:928–940. doi: 10.1101/gad.216531.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier M-C, Qu H-Q, Fréchette R, et al. Type 1 diabetes and the OAS gene cluster: association with splicing polymorphism or haplotype? J Med Genet. 2006;43:129–132. doi: 10.1136/jmg.2005.035212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharun S. Roles of eukaryotic Lsm proteins in the regulation of mRNA function. Int Rev Cell Mol Biol. 2009;272:149–189. doi: 10.1016/S1937-6448(08)01604-3. [DOI] [PubMed] [Google Scholar]
- Tharun S, He W, Mayes AE, et al. Yeast Sm-like proteins function in mRNA decapping and decay. Nature. 2000;404:515–518. doi: 10.1038/35006676. [DOI] [PubMed] [Google Scholar]
- Thomson T, Lasko P. Tudor and its domains: germ cell formation from a Tudor perspective. Cell Res. 2005;15:281–291. doi: 10.1038/sj.cr.7290297. [DOI] [PubMed] [Google Scholar]
- Thomson T, Lin H. The biogenesis and function of PIWI proteins and piRNAs: progress and prospect. Annu Rev Cell Dev Biol. 2009;25:355–376. doi: 10.1146/annurev.cellbio.24.110707.175327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton JE, Gregory RI. How does Lin28 let-7 control development and disease? Trends Cell Biol. 2012;22:474–482. doi: 10.1016/j.tcb.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Simanshu DK, Ma J-B, Patel DJ. Structural basis for piRNA 2“-O-methylated 3-”end recognition by Piwi PAZ (Piwi/Argonaute/Zwille) domains. Proc Natl Acad Sci U S A. 2011;108:903–910. doi: 10.1073/pnas.1017762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timchenko LT, Timchenko NA, Caskey CT, Roberts R. Novel proteins with binding specificity for DNA CTG repeats and RNA CUG repeats: implications for myotonic dystrophy. Hum Mol Genet. 1996;5:115–121. doi: 10.1093/hmg/5.1.115. [DOI] [PubMed] [Google Scholar]
- Tomasevic N, Peculis BA. Xenopus LSm proteins bind U8 snoRNA via an internal evolutionarily conserved octamer sequence. Mol Cell Biol. 2002;22:4101–4112. doi: 10.1128/MCB.22.12.4101-4112.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taft RJ, Vanderver A, Leventer RJ, et al. Mutations in DARS cause hypomyelination with brain stem and spinal cord involvement and leg spasticity. Am J Hum Genet. 2013;92:774–780. doi: 10.1016/j.ajhg.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai-Morris CH, Sheng Y, Gutti RK, et al. Gonadotropin-regulated testicular RNA helicase (GRTH/DDX25): a multifunctional protein essential for spermatogenesis. J Androl. 2010;31:45–52. doi: 10.2164/jandrol.109.008219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007;104:7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetanova NG, Klass DM, Salzman J, Brown PO. Proteome-wide search reveals unexpected RNA-binding proteins in Saccharomyces cerevisiae. PLoS One. 2010;5(9) doi: 10.1371/journal.pone.0012671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J. Ribonucleoprotein complexes in neurologic diseases. Curr Opin Neurobiol. 2008;18:516–523. doi: 10.1016/j.conb.2008.09.018. [DOI] [PubMed] [Google Scholar]
- Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente L, Tiranti V, Marsano RM, et al. Infantile encephalopathy and defective mitochondrial DNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EFTu. Am J Hum Genet. 2007;80:44–58. doi: 10.1086/510559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde R, Edwards L, Regan L. Structure and function of KH domains. FEBS J. 2008;275:2712–2726. doi: 10.1111/j.1742-4658.2008.06411.x. [DOI] [PubMed] [Google Scholar]
- van de Laar IMBH, Oldenburg RA, Pals G, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43:121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- Van de Peer Y, Maere S, Meyer A. The evolutionary significance of ancient genome duplications. Nat Rev Genet. 2009;10:725–732. doi: 10.1038/nrg2600. [DOI] [PubMed] [Google Scholar]
- van der Knaap MS, Leegwater PAJ, Könst AAM, et al. Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann Neurol. 2002;51:264–270. doi: 10.1002/ana.10112. [DOI] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanrobays E, Gélugne J-P, Caizergues-Ferrer M, Lafontaine DLJ. Dim2p, a KH-domain protein required for small ribosomal subunit synthesis. RNA. 2004;10:645–656. doi: 10.1261/rna.5162204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nat Rev Genet. 2009;10:252–263. doi: 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- Vedrenne V, Gowher A, de Lonlay P, et al. Mutation in PNPT1, which encodes a polyribonucleotide nucleotidyltransferase, impairs RNA import into mitochondria and causes respiratory-chain deficiency. Am J Hum Genet. 2012;91:912–918. doi: 10.1016/j.ajhg.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter JC. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Vissers LELM, de Ligt J, Gilissen C, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- Vithana EN, Abu-Safieh L, Allen MJ, et al. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11) Mol Cell. 2001;8:375–381. doi: 10.1016/s1097-2765(01)00305-7. [DOI] [PubMed] [Google Scholar]
- Volpi L, Roversi G, Colombo EA, et al. Targeted next-generation sequencing appoints c16orf57 as clericuzio-type poikiloderma with neutropenia gene. Am J Hum Genet. 2010;86:72–76. doi: 10.1016/j.ajhg.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Ameln S, Wang G, Boulouiz R, et al. A mutation in PNPT1, encoding mitochondrial-RNA-import protein PNPase, causes hereditary hearing loss. Am J Hum Genet. 2012;91:919–927. doi: 10.1016/j.ajhg.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulliamy TJ, Knight SW, Heiss NS, et al. Dyskeratosis congenita caused by a 3′ deletion: germline and somatic mosaicism in a female carrier. Blood. 1999;94:1254–1260. [PubMed] [Google Scholar]
- Vulliamy T, Beswick R, Kirwan M, et al. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc Natl Acad Sci U S A. 2008;105:8073–8078. doi: 10.1073/pnas.0800042105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamatsu Y, Weston JA. Sequential expression and role of Hu RNA-binding proteins during neurogenesis. Development. 1997;124:3449–3460. doi: 10.1242/dev.124.17.3449. [DOI] [PubMed] [Google Scholar]
- Walne AJ, Vulliamy T, Marrone A, et al. Genetic heterogeneity in autosomal recessive dys-keratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet. 2007;16:1619–1629. doi: 10.1093/hmg/ddm111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan M, Lee SS, Zhang X, et al. Rett syndrome and beyond: recurrent spontaneous and familialMECP2mutationsatCpGhotspots. Am J Hum Genet. 1999;65:1520–1529. doi: 10.1086/302690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, Yourshaw M, Mamsa H, et al. Mutations in the RNA exosome component gene EXOSC3 cause pontocerebellar hypoplasia and spinal motor neuron degeneration. Nat Genet. 2012;44:704–708. doi: 10.1038/ng.2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G-S, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- Wang Y, Jiang Y, Meyering-Voss M, et al. Crystal structure of the EF-Tu. EF-Ts complex from Thermus thermophilus. Nat Struct Biol. 1997;4:650–656. doi: 10.1038/nsb0897-650. [DOI] [PubMed] [Google Scholar]
- Wang XQ, Zamore PD, Hall TMT. Crystal structure of a Pumilio homology domain. Mol Cell. 2001;7:855–865. doi: 10.1016/s1097-2765(01)00229-5. [DOI] [PubMed] [Google Scholar]
- Wang X, McLachlan J, Zamore PD, Hall TMT. Modular recognition of RNA by a human pumilio-homology domain. Cell. 2002;110:501–512. doi: 10.1016/s0092-8674(02)00873-5. [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008a;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sheng G, Juranek S, et al. Structure of the guide-strand-containing argonaute silencing complex. Nature. 2008b;456:209–213. doi: 10.1038/nature07315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Cody NAL, Jog S, et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150:710–724. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, et al. N(6)-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2013;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21:354–361. doi: 10.1016/j.tcb.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Weedon MN, Hastings R, Caswell R, et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet. 2011;89:308–312. doi: 10.1016/j.ajhg.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickens M, Bernstein DS, Kimble J, Parker R. A PUF family portrait: 3′UTR regulation as a way of life. Trends Genet. 2002;18:150–157. doi: 10.1016/s0168-9525(01)02616-6. [DOI] [PubMed] [Google Scholar]
- Wilbert ML, Huelga SC, Kapeli K, et al. LIN28 binds messenger RNAs at GGAGA motifs and regulates splicing factor abundance. Mol Cell. 2012;48:195–206. doi: 10.1016/j.molcel.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RC, Doudna JA. Molecular Mechanisms of RNA Interference. Annu Rev Biophys. 2013;42:217–239. doi: 10.1146/annurev-biophys-083012-130404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz CJ, Wilusz J. Eukaryotic Lsm proteins: lessons from bacteria. Nat Struct Mol Biol. 2005;12:1031–1036. doi: 10.1038/nsmb1037. [DOI] [PubMed] [Google Scholar]
- Winkelmann J, Lin L, Schormair B, et al. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum Mol Genet. 2012;21:2205–2210. doi: 10.1093/hmg/dds035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter EE, Goodstadt L, Ponting CP. Elevated rates of protein secretion, evolution, and disease among tissue-specific genes. Genome Res. 2004;14:54–61. doi: 10.1101/gr.1924004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise CA, Chiang LC, Paznekas WA, et al. TCOF1 gene encodes a putative nucleolar phosphoprotein that exhibits mutations in Treacher Collins Syndrome throughout its coding region. Proc Natl Acad Sci U S A. 1997;94:3110–3115. doi: 10.1073/pnas.94.7.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wöhrle D, Kotzot D, Hirst MC, et al. A microdeletion of less than 250 kb, including the proximal part of the FMR-I gene and the fragile-X site, in a male with the clinical phenotype of fragile-X syndrome. Am J Hum Genet. 1992;51:299–306. [PMC free article] [PubMed] [Google Scholar]
- Wool IG. The structure and function of eukaryotic ribosomes. Annu Rev Biochem. 1979;48:719–754. doi: 10.1146/annurev.bi.48.070179.003443. [DOI] [PubMed] [Google Scholar]
- Wool IG, Chan YL, Glück A. Structure and evolution of mammalian ribosomal proteins. Biochem Cell Biol. 1995;73:933–947. doi: 10.1139/o95-101. [DOI] [PubMed] [Google Scholar]
- Xiao S, Scott F, Fierke CA, Engelke DR. Eukaryotic ribonuclease P: a plurality of ribonucleoprotein enzymes. Annu Rev Biochem. 2002;71:165–189. doi: 10.1146/annurev.biochem.71.110601.135352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas EJ, et al. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, Lee C. Alternative splicing and RNA selection pressure–evolutionary consequences for eukaryotic genomes. Nat Rev Genet. 2006;7:499–509. doi: 10.1038/nrg1896. [DOI] [PubMed] [Google Scholar]
- Xu GL, Bestor TH, Bourc’his D, et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402:187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- Xu Q, Modrek B, Lee C. Genome-wide detection of tissue-specific alternative splicing in the human transcriptome. Nucleic Acids Res. 2002;30:3754–3766. doi: 10.1093/nar/gkf492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Calado RT, Ly H, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005;352:1413–1424. doi: 10.1056/NEJMoa042980. [DOI] [PubMed] [Google Scholar]
- Yamasaki S, Ivanov P, Hu G-F, Anderson P. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J Cell Biol. 2009;185:35–42. doi: 10.1083/jcb.200811106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanase T, Takayanagi R, Oba K, et al. New mutations of DAX-1 genes in two Japanese patients with X-linked congenital adrenal hypoplasia and hypogonadotropic hypogonadism. J Clin Endocrinol Metabol. 1996;81:530–535. doi: 10.1210/jcem.81.2.8636263. [DOI] [PubMed] [Google Scholar]
- Yao P, Fox PL. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol Med. 2013;5:332–343. doi: 10.1002/emmm.201100626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen PH. Putative biological functions of the DAZ family. Int J Androl. 2004;27:125–129. doi: 10.1111/j.1365-2605.2004.00469.x. [DOI] [PubMed] [Google Scholar]
- Yeo GW, Van Nostrand E, Holste D, et al. Identification and analysis of alternative splicing events conserved in human and mouse. Proc Natl Acad Sci U S A. 2005;102:2850–2855. doi: 10.1073/pnas.0409742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeharia A, Shaag A, Pappo O, et al. Acute infantile liver failure due to mutations in the TRMU gene. Am J Hum Genet. 2009;85:401–407. doi: 10.1016/j.ajhg.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Kolb FA, Jaskiewicz L, et al. Single processing center models for human dicer and bacterial RNase III. Cell. 2004;118:57–68. doi: 10.1016/j.cell.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Zhang ZH, Niu ZM, Yuan WT, et al. A mutation in SART3 gene in a Chinese pedigree with disseminated superficial actinic porokeratosis. Br J Dermatol. 2005;152:658–663. doi: 10.1111/j.1365-2133.2005.06443.x. [DOI] [PubMed] [Google Scholar]
- Zhang L, Huang J, Yang N, et al. microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci U S A. 2006;103:9136–9141. doi: 10.1073/pnas.0508889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lotti F, Dittmar K, et al. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Theler D, Kaminska KH, et al. The YTH domain is a novel RNA binding domain. J Biol Chem. 2010;285:14701–14710. doi: 10.1074/jbc.M110.104711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Bellur DL, Lu S, et al. Autosomal-dominant retinitis pigmentosa caused by a mutation in SNRNP200, a gene required for unwinding of U4/U6 snRNAs. Am J Hum Genet. 2009;85:617–627. doi: 10.1016/j.ajhg.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K, Xiol J, Reuter M, et al. Mouse MOV10L1 associates with Piwi proteins and is an essential component of the Piwi-interacting RNA (piRNA) pathway. Proc Natl Acad Sci U S A. 2010;107:11841–11846. doi: 10.1073/pnas.1003953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong F, Savage SA, Shkreli M, et al. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev. 2011;25:11–16. doi: 10.1101/gad.2006411. [DOI] [PMC free article] [PubMed] [Google Scholar]