


Abstract
The K homology (KH) module is a widespread RNA-binding motif that has been detected by sequence similarity searches in such proteins as heterogeneous nuclear ribonucleoprotein K (hnRNP K) and ribosomal protein S3. Analysis of spatial structures of KH domains in hnRNP K and S3 reveals that they are topologically dissimilar and thus belong to different protein folds. Thus KH motif proteins provide a rare example of protein domains that share significant sequence similarity in the motif regions but possess globally distinct structures. The two distinct topologies might have arisen from an ancestral KH motif protein by N- and C-terminal extensions, or one of the existing topologies may have evolved from the other by extension, displacement and deletion. C-terminal extension (deletion) requires β-sheet rearrangement through the insertion (removal) of a β-strand in a manner similar to that observed in serine protease inhibitors serpins. Current analysis offers a new look on how proteins can change fold in the course of evolution.
INTRODUCTION
Since the emergence of the first three-dimensional protein structures, it has been widely accepted that spatial structure is more conserved than protein sequence (1–6). Many examples of very close structural resemblance in the absence of detectable sequence similarity have been catalogued (7–12). The opposite situation remains obscure. We know very few proteins with statistically supported sequence similarity that fold into radically different structures (13–16). These rare cases are of exceptional interest since they have a profound impact on our understanding of the protein world. Practically, their existence indicates difficulties for homology modeling techniques that rely heavily on the assumption ‘similar sequences, similar structures’ and brings inconsistencies between sequence- and structure-based protein classification schemes. The most fundamental questions, however, concern evolution of protein structure, its relation to evolution of sequence and function, and mechanisms by which protein folds can change. These mechanisms remain largely unexplored both experimentally and theoretically.
A unique example of proteins with clear sequence similarity while having considerably different folds is presented here and it appears to be by far the most striking case of this kind. Sequence similarity between the two proteins described below has been detected and was widely known before the structures were solved, but the protein folds turned out to be topologically different.
K homology (KH) motif was first biochemically characterized in the major pre-mRNA-binding protein K (heterogeneous nuclear ribonucleoprotein K, hnRNP K) and described as a 45-amino acid repeat detected by sequence similarity in a number of RNA-binding proteins (17). Siomi et al. (17) note that similarity was particularly strong with ribosomal protein S3. The first KH domain of human hnRNP K and the KH domain of Halobacterium halobium S3 display 36% identity (54% similarity, z-score of 12.5, calculation through the entire alignment length of 39 residues), which is larger than that between the first and the second KH domains of hnRNP K (31% identity) (17). KH motifs can occur in multiple copies (15 in chicken vigilin) (18). The most conserved sequence with the consensus VIGXXGXXI maps to the middle of the motif (17,19,20). A single amino acid substitution (I304 to N) in this consensus sequence of FMR1 protein (21) affects its RNA-binding properties (22) and causes fragile X mental-retardation syndrome (23). There has been no question that significant sequence similarity in the KH motif reflects descent from a common ancestor (17,19,20). The conservation of KH motif in diverse organisms such as Bacteria, Archaea and Eukaryotes suggests that KH arose early in evolution.
MATERIALS AND METHODS
Sequence similarity searches against the non-redundant protein database (nr) maintained at the National Center for Biotechnology Information (NCBI; Bethesda, MD) were performed using the PSI-BLAST program (24,25). The BLOSUM62 matrix (26) was used for scoring, and 0.01 or 0.001 were used as E-value thresholds for inclusion in the profile calculation. Sequence analysis protocols were carried out using SEALS (27). Structure similarity searches against the protein data bank (PDB) (28,29) maintained at the Research Collaboratory for Structural Bioinformatics (RCSB) were performed using DALI (30–32), VAST (33,34) and CE (35) programs with default parameters. The Structural Classification of Proteins (SCOP) database (release 1.53, 11 410 PDB entries, July 1, 2000) (11,12) was used as a source of protein classification. Protein structures were visualized and superimposed using InsightII package (MSI) and the multiple structure-based alignment was built on the basis of the superpositions made in InsightII. Structure diagrams were rendered using Bobscript (36), a modified version of Molscript (37).
RESULTS AND DISCUSSION
Sequence similarity between KH domains of hnRNP K and ribosomal protein S3 described back in 1993 (17) can be detected by PSI-BLAST. Even the gapped BLAST program finds S3 when hnRNP K is used as a query and vice versa. For example, gapped BLAST aligns the first KH domain of human hnRNP K [NCBI database gene identification number (GI) 585911, residues 35–95] taken as a query with the ribosomal protein S3 from Deinococcus radiodurans (GI:7473848) detected in the nr protein sequence database (November 2000, 582 290 sequences; 183 345 511 letters). The alignment spans through 64 residues, which constitutes virtually the entire KH motif and displays 32% identity (47% similarity, score 31.4 bits, E-value 1.3). BLAST alignment of hnRNP K (GI:585911 residues 35–95) and H.halobium S3 (GI:133930) spans through 36 residues giving 38% identity (63% similarity, no gaps, score 31 bits, E-value 1.6). In this alignment, a nine-residue segment in the KH signature region is invariant between the two sequences: VIGKGGKNI (GI:585911 residues 57–65, GI:133930 residues 54–62). Conversely, when D.radiodurans S3 (GI:7473848 residues 63–126) is taken as a gapped BLAST query, the first KH domain of human hnRNP K (GI:585911) is found with a score of 34 bits (E-value 0.19). Additionally, the KH domain of GTPase ERA from Mycobacterium leprae is found with a score of 34.3 bits (E-value 0.16), 31% identity (50% similarity) in a 47-residue alignment.
When the KH motif was first described (17), no spatial structure for KH-containing proteins was determined. By now, we have several KH domain structures in hand (18,38–44), including those detected by sequence similarity in the original paper that identified the motif (17): hnRNP K and ribosomal protein S3. The structure of the C-terminal KH domain of human hnRNP K has been determined by NMR spectroscopy (Fig. 1B) (40) and the coordinates for S3 became available recently following the solution of the X-ray structure of the entire 30S ribosome subunit from Thermus thermophilus (Fig. 1E) (44). Was the prediction of structural similarity between hnRNP K and S3 based on sequence similarity in the KH motif region fulfilled? Yes and no. The conformations of residues in and around the KH consensus VIGXXGXXI are indeed very similar between the two structures (Figs 1B and E and 2A). Near the consensus, the protein chain is folded as two α-helices, A and B (Fig. 1), arranged at an angle of 100–120° to each other. A two-residue protruding turn connects the α-helices A and B (Figs 1B and E and 2A). The two largely invariant glycines separated by two variable residues in the turn (GXXG) serve as C- and N-caps of the two α-helices A and B. The side chains of residues around the consensus are conformationally similar (Fig. 1B and E) and are likely to bear the same functional role. The KH consensus sequence has been implied in direct contact with nucleic acids (17,19,21,22,45) and the recent crystal structure of nova-2 KH domain bound to a 20mer RNA hairpin (43) confirmed this hypothesis (Fig. 2B). The α-helix A, the following turn and the β-strand b (Fig. 1) are involved in extensive contacts with RNA.
Figure 1.

Structural comparison of KH domains. Ribbon diagrams of type I (maxi) KH domains (A and B), type II (mini) KH domains (D and B) and non-KH proteins (C and F) were drawn by Bobscript (36), a modified version of Molscript (37). The structures were superimposed and then separated for clarity. N- and C-termini are labeled. The spatially equivalent structural elements are colored correspondingly. N- and C-terminal extension in type II (mini) and type I (maxi) KH domains and their structural equivalencies in non-KH proteins of similar fold are colored in blue and green, respectively. α-Helices and β-strands are labeled in upper and lower case italic letters, respectively. Letter color matches the color of the secondary structure element. Side chains (Cα atoms for Gly) of residues conserved in KH domains are displayed. (A) Repeat 6 of vigilin [PDB (29) entry 1VIH, residues 7–76]; (B) C-terminal KH domain of hnRNP K (PDB entry 1KHM, residues A11–A89); (C) C-terminal domain of E.coli arginine repressor (PDB entry 1XXA, residues A92–A152, the first β-strand is not shown); (D) C-terminal domain of GTPase ERA (PDB entry 1EGA, residues A186–A283); (E) N-terminal domain of ribosomal protein S3 (PDB entry 1FJF, residues C24–C106); (F) C-terminal domain of E.coli GMP synthetase (PDB entry 1GPM, residues A416–A523); (G) structure-based sequence alignment of KH motif regions from structures shown in (A, B, D and E). The panel label, PDB entry name, starting and ending residue numbers are given for each protein. Color shading and labels of secondary structure elements correspond to those in A–F. Highly conserved residues in KH domains (KH signature) are boxed with black. Hydrophobic amino acids in buried sites are shown in bold letters. Blue and green shading points to the presence of N- and C-terminal extensions whose sequences are not shown. The residues with side chains (Cα atoms for Gly) displayed on ribbon diagrams are marked with a black dot below the alignment.
Figure 2.
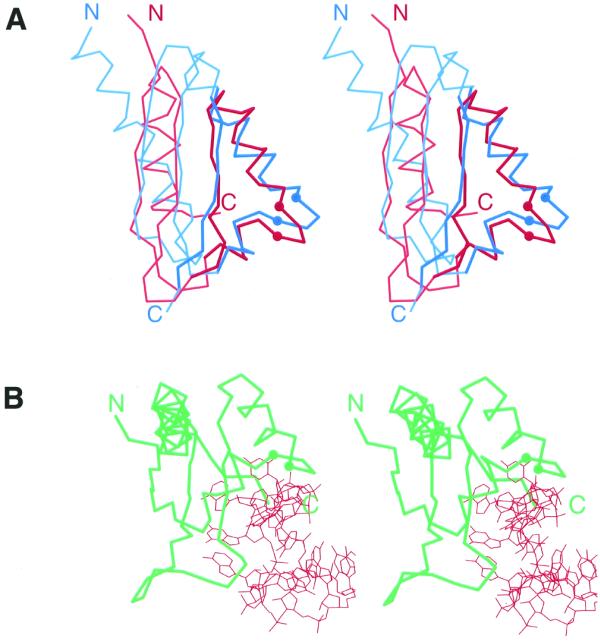
Stereo diagrams of KH domains. The Cα traces of proteins are shown and the Cα atoms of the two conserved glycines in KH motif signature region are displayed as balls. N- and C-termini are labeled. (A) Stereo diagram of superimposed Cα traces of type I (maxi) KH of vigilin (red, PDB entry 1VIH, residues 6–76) and type II (mini) KH of ribosomal protein S3 (blue, PDB entry 1FJF, residues C28–C108). Superposition was performed using Insight II package (MSI). The regions used in RMSD minimization are outlined in darker colors and thicker lines. The RMSD is 2.4 Å. (B) Stereo diagram of Nova-2 KH domain (green) bound to RNA (red), PDB entry 1EC6, residues A4–A90, RNA chain C.
Thus the local motif identified by the statistically supported sequence similarity is folded the same way in hnRNP K and S3 structures, and is likely to bind nucleic acids by the same mechanism. But are the global folds of the two proteins similar? The first spatial structure of a KH motif protein, the sixth KH domain of vigilin (Fig. 1A), revealed the presence of a compact domain. In addition to the motif sequence covering the βααβ unit (Fig. 1, a, A, B and b), the KH domain included a βα unit at the C-terminus that is inherently important for its structural integrity (18). Indeed, the β-strand c is the central element of the three-stranded anti-parallel β-sheet (Fig. 1A and B). The α-helix C (Fig. 1A and B) completes the hydrophobic core of the protein and the KH domain is unable to fold when this α-helix is deleted (18). The vigilin KH domain can be described as an α+β two-layer sandwich with α-β plate topology (9,10). This topology is also known as the ‘ferredoxin-like’ protein fold (11,12) (the last strand of the ferredoxin common fold is missing in the KH domain). An example of a protein with α-β plate topology that does not share sequence similarity with the KH domain, namely the C-terminal domain of the Escherichia coli arginine repressor (46), is illustrated in Figure 1C.
The structure of the vigilin domain leads to re-definition of the KH motif boundaries to cover the helix C (18), making the domain length equal to approximately 70 residues. However, several KH sequences lack the C helix. These include ribosomal protein S3, amongst others. The shorter KH sequences that match the original definition of the KH motif (17) were termed ‘mini-KH’, in contrast to typical ‘vigilin-like’ ‘maxi-KH’ domains (18). Surprisingly, the structure of the ribosomal protein S3 N-terminal domain (44) revealed that the β-sheet topology of the mini-KH domain is drastically different from the one established for maxi-KH (Fig. 1E). Indeed, not only the α-helix C, but also the central β-strand c, which seemed to be crucially important for the fold, is lacking in S3 structure (Fig. 1E). Alternatively, another β-strand (a′) and α-helix (A′) donated by the N-terminal part of the domain complete the hydrophobic core of the mini-KH. Such an arrangement results in architectural similarity between maxi- and mini-KH: both domains are built from a three-stranded β-sheet with three α-helices packed on one side of it (Figs 1 and 2A). The difference is topological: while in maxi-KH the β-sheet is anti-parallel, in mini-KH it is mixed. Parallel β-strands a and b that were included in the original definition of KH motif (17,19) form hydrogen bonds with each other in the S3 structure (Fig. 1E), but are separated by the β-strand c in maxi-KH (Fig. 1B). Another structure of a mini-KH domain-containing protein, GTPase ERA (41), displays significant topological similarity to S3 (Fig. 1D) and thus confirms that the structure of S3 is not an exception, but a template for mini-KH domains. The structures topologically similar to mini-KH domain are known among proteins that do not contain KH motif. For example, the C-terminal domain of E.coli GMP synthetase (47) is shown in Figure 1F.
Global structure similarity search programs such as DALI (30–32), VAST (33,34) and CE (35) find similarity significant within mini- and maxi-KH classes, but concur on the global structural differences between the two classes. For example, DALI finds the structures of two mini-KH domains similar: the KH domains of S3 (PDB entry 1FJF chain C) and GTPase ERA (1EGA chain A, C-terminal domain) are aligned with z-score of 4.1, root mean-squared deviation (RMSD) of 4.3 Å and 7% sequence identity in the alignment of 89 residues. DALI does not report similarity of these proteins to any of the maxi-KH domains implying that corresponding z-scores are <2.0.
The analysis presented forces us to return to the original definition of the KH motif boundaries that include only the βααβ unit shared between maxi- and mini-KH domains (Fig. 1G). In addition to this shared KH motif element, maxi- and mini-KH domains contain C- and N-terminal extensions, respectively. Therefore in terms of the overall domain size, the mini-KH domain is not smaller than the maxi-KH domain: both comprise approximately 70 residues. The mini-/maxi-KH terminology was originally meaningful. The mini-KH domain does not contain the C-terminal β-strand and α-helix (Fig. 1A and B, c and C) of maxi-KH that were included in the modified KH domain definition (18). Prior to mini-KH structure determination it was not known that sequence segments upstream of the N-terminal boundary set by maxi-KH would be part of the hydrophobic core of the mini-KH domain, thus the mini-KH domain appeared to be shorter than the maxi-KH domain. However, due to the lack of chain length differences between the two domains, as revealed by their crystal structures, mini/maxi terminology loses its meaning. We suggest naming the two topologically different KH domains KH type I for the KH domain with the C-terminal βα extension [maxi-KH, its structure was determined first (18)], and KH type II for the KH domain with N-terminal αβ extension (mini-KH).
It is clear that the type I and II KH domains belong to different protein folds (Fig. 1A, B, D and E). It is also clear that they share the same KH motif (Fig. 1G). What is the evolutionary connection between the two different KH domains with the same KH motif? The simplest, and well-documented, mechanism of topological changes in protein evolution (48–50), circular permutation, is not possible in this case since the order of secondary structural elements differs: a βα unit is present at the C-terminus of the type I KH, but an αβ unit starts type II KH. It is therefore likely that type I and II KH domains are not homologous throughout their entire length. Theoretically, four evolutionary scenarios are possible. First, local sequence, structural and functional similarities in the KH motif region were acquired independently by type I and II KH domains and thus are convergent. Second, the element of the local sequence similarity (minimally, sequence segment around the turn between the α-helices A and B; Fig. 1) was inserted in two different structural templates: type I and II KH domains. Third, the homology region covers the entire βααβ unit, which represents a ‘primordial’ KH domain. This domain was expanded by the C-terminal extension to form a type I KH domain fold or by the N-terminal extension to form a type II KH domain fold. Fourth, one of the two types represents the ancestral form and the other type evolved through N- or C-terminal extension, and displacement and deletion at the other end.
It appears that the third and fourth scenarios offer the simplest explanation to the available data. Indeed, insertions, deletions and terminal extensions are very common events in protein evolution (51,52). Also, it was argued, and largely accepted, that statistically significant similarity detected from the sequence alone (without consideration of spatial structure) reflects descent from the common ancestor, i.e. homology (16,53,54). Programs that are routinely used for sequence similarity searches, such as PSI-BLAST (24,25), are based on amino acid similarity matrices which are derived under evolutionary models (55) or computed from the aligned homologous sequences (26) and thus are intended to find homologs. Therefore, convergent origin of KH domains appears unlikely due to their highly significant sequence similarity (17–19). At present, it is hard to discriminate between the third and fourth scenarios. The third scenario might seem unrealistic, since it assumes the existence of a putative primordial βααβ domain, which might not be stable in the absence of the N- or C-terminal α-helix to pack against the β-sheet. However, it is likely that primordial proteins existed in tight contacts with RNA and might not be foldable in the absence of RNA molecules. It is also reasonable to assume that primordial proteins were significantly shorter than average present-day domains. The fourth scenario offers a physically realistic model that might pass through an intermediate protein containing both N- and C-terminal extensions before one of the extensions was eliminated. There is a chance that such a ‘hybrid’ protein still exists in nature. Thus to discover the KH motif-containing protein with topology αββααββα (a combination of both type I and II domains, four-stranded β-sheet with four α-helices on one side; Fig. 1A, B, D and E), would be an argument favoring the fourth scenario.
Interestingly, the C-terminal extension cC (Fig. 1A and B) in the type I KH domains required rearrangement of the β-sheet: hydrogen-bonding between β-strands a and b of the putative ‘primordial’ KH domain should have been broken to accommodate the central β-strand c. Typically, terminal extensions do not disrupt the β-sheet topology, but add up to the existing structural core, like the N-terminal extension a′A′ (Fig. 1D and E) in the type II KH domain. However, the KH domain is not the first example for which the rearrangement of β-sheet topology has been suggested. Serine protease inhibitors, serpins, are known to undergo the conformational change during which one of the β-strands is inserted between the two hydrogen-bonded parallel β-strands (56). P-loop ATPases that display statistically significant sequence similarity in Walker A and B motifs (57) are known to possess several distinct topologies that can be transformed to each other through the β-sheet rearrangement (58,59). β-Sheet rearrangement was postulated for the triabin that shares sequence similarity with lipocalins but possesses distinct topology (14).
In summary, analysis of available spatial structures revealed that there are two different KH domains that belong to different protein folds, but share a single KH motif. The KH motif is folded into a βααβ unit. In addition to the motif core, type II KH domains (e.g. ribosomal protein S3) include N-terminal extension αβ and type I KH domains (e.g. hnRNP K) contain C-terminal extension βα. A β-strand of this extension in type I KH is inserted into the β-sheet formed by the KH motif βααβ unit offering a clear example of a rare structural rearrangement. KH domains demonstrate how proteins can change fold in the course of evolution.
Acknowledgments
ACKNOWLEDGEMENTS
The author is grateful to Hong Zhang and Sara Cheek for critical reading of the manuscript and the two anonymous reviewers for constructive suggestions.
References
- 1.Chothia C. and Lesk,A.M. (1986) The relation between the divergence of sequence and structure in proteins. EMBO J., 5, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hubbard T.J. and Blundell,T.L. (1987) Comparison of solvent-inaccessible cores of homologous proteins: definitions useful for protein modelling. Protein Eng., 1, 159–171. [DOI] [PubMed] [Google Scholar]
- 3.Flores T.P., Orengo,C.A., Moss,D.S. and Thornton,J.M. (1993) Comparison of conformational characteristics in structurally similar protein pairs. Protein Sci., 2, 1811–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grishin N.V. (1997) Estimation of evolutionary distances from protein spatial structures. J. Mol. Evol., 45, 359–369. [DOI] [PubMed] [Google Scholar]
- 5.Doolittle R.F. (1981) Similar amino acid sequences: chance or common ancestry? Science, 214, 149–159. [DOI] [PubMed] [Google Scholar]
- 6.Holm L. and Sander,C. (1997) New structure—novel fold? Structure, 5, 165–171. [DOI] [PubMed] [Google Scholar]
- 7.Russell R.B., Saqi,M.A., Bates,P.A., Sayle,R.A. and Sternberg,M.J. (1998) Recognition of analogous and homologous protein folds—assessment of prediction success and associated alignment accuracy using empirical substitution matrices. Protein Eng., 11, 1–9. [DOI] [PubMed] [Google Scholar]
- 8.Russell R.B., Saqi,M.A., Sayle,R.A., Bates,P.A. and Sternberg,M.J. (1997) Recognition of analogous and homologous protein folds: analysis of sequence and structure conservation. J. Mol. Biol., 269, 423–439. [DOI] [PubMed] [Google Scholar]
- 9.Orengo C.A., Michie,A.D., Jones,S., Jones,D.T., Swindells,M.B. and Thornton,J.M. (1997) CATH—a hierarchic classification of protein domain structures. Structure, 5, 1093–1108. [DOI] [PubMed] [Google Scholar]
- 10.Pearl F.M., Lee,D., Bray,J.E., Sillitoe,I., Todd,A.E., Harrison,A.P., Thornton,J.M. and Orengo,C.A. (2000) Assigning genomic sequences to CATH. Nucleic Acids Res., 28, 277–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murzin A.G., Brenner,S.E., Hubbard,T. and Chothia,C. (1995) SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol., 247, 536–540. [DOI] [PubMed] [Google Scholar]
- 12.Lo Conte L., Ailey,B., Hubbard,T.J., Brenner,S.E., Murzin,A.G. and Chothia,C. (2000) SCOP: a structural classification of proteins database. Nucleic Acids Res., 28, 257–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matte A., Goldie,H., Sweet,R.M. and Delbaere,L.T. (1996) Crystal structure of Escherichia coli phosphoenolpyruvate carboxykinase: a new structural family with the P-loop nucleoside triphosphate hydrolase fold. J. Mol. Biol., 256, 126–143. [DOI] [PubMed] [Google Scholar]
- 14.Fuentes-Prior P., Noeske-Jungblut,C., Donner,P., Schleuning,W.D., Huber,R. and Bode,W. (1997) Structure of the thrombin complex with triabin, a lipocalin-like exosite-binding inhibitor derived from a triatomine bug. Proc. Natl Acad. Sci. USA, 94, 11845–11850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grishin N.V., Osterman,A.L., Brooks,H.B., Phillips,M.A. and Goldsmith,E.J. (1999) X-ray structure of ornithine decarboxylase from Trypanosoma brucei: the native structure and the structure in complex with alpha-difluoromethylornithine. Biochemistry, 38, 15174–15184. [DOI] [PubMed] [Google Scholar]
- 16.Murzin A.G. (1998) How far divergent evolution goes in proteins. Curr. Opin. Struct. Biol., 8, 380–387. [DOI] [PubMed] [Google Scholar]
- 17.Siomi H., Matunis,M.J., Michael,W.M. and Dreyfuss,G. (1993) The pre-mRNA binding K protein contains a novel evolutionarily conserved motif. Nucleic Acids Res., 21, 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Musco G., Stier,G., Joseph,C., Castiglione Morelli,M.A., Nilges,M., Gibson,T.J. and Pastore,A. (1996) Three-dimensional structure and stability of the KH domain: molecular insights into the fragile X syndrome. Cell, 85, 237–245. [DOI] [PubMed] [Google Scholar]
- 19.Gibson T.J., Thompson,J.D. and Heringa,J. (1993) The KH domain occurs in a diverse set of RNA-binding proteins that include the antiterminator NusA and is probably involved in binding to nucleic acid. FEBS Lett., 324, 361–366. [DOI] [PubMed] [Google Scholar]
- 20.Burd C.G. and Dreyfuss,G. (1994) Conserved structures and diversity of functions of RNA-binding proteins. Science, 265, 615–621. [DOI] [PubMed] [Google Scholar]
- 21.Gibson T.J., Rice,P.M., Thompson,J.D. and Heringa,J. (1993) KH domains within the FMR1 sequence suggest that fragile X syndrome stems from a defect in RNA metabolism. Trends Biochem. Sci., 18, 331–333. [DOI] [PubMed] [Google Scholar]
- 22.Siomi H., Choi,M., Siomi,M.C., Nussbaum,R.L. and Dreyfuss,G. (1994) Essential role for KH domains in RNA binding: impaired RNA binding by a mutation in the KH domain of FMR1 that causes fragile X syndrome. Cell, 77, 33–39. [DOI] [PubMed] [Google Scholar]
- 23.De Boulle K., Verkerk,A.J., Reyniers,E., Vits,L., Hendrickx,J., Van Roy,B., Van den Bos,F., de Graaff,E., Oostra,B.A. and Willems,P.J. (1993) A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nature Genet., 3, 31–35. [DOI] [PubMed] [Google Scholar]
- 24.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altschul S.F. and Koonin,E.V. (1998) Iterated profile searches with PSI-BLAST—a tool for discovery in protein databases. Trends Biochem. Sci., 23, 444–447. [DOI] [PubMed] [Google Scholar]
- 26.Henikoff S. and Henikoff,J.G. (1992) Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA, 89, 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker D.R. and Koonin,E.V. (1997) SEALS: a system for easy analysis of lots of sequences. Ismb, 5, 333–339. [PubMed] [Google Scholar]
- 28.Abola E.E., Sussman,J.L., Prilusky,J. and Manning,N.O. (1997) Protein Data Bank archives of three-dimensional macromolecular structures. Methods Enzymol., 277, 556–571. [DOI] [PubMed] [Google Scholar]
- 29.Berman H.M., Westbrook,J., Feng,Z., Gilliland,G., Bhat,T.N., Weissig,H., Shindyalov,I.N. and Bourne,P.E. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holm L. and Sander,C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol., 233, 123–138. [DOI] [PubMed] [Google Scholar]
- 31.Holm L. and Sander,C. (1996) The FSSP database: fold classification based on structure–structure alignment of proteins. Nucleic Acids Res., 24, 206–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holm L. and Sander,C. (1997) Dali/FSSP classification of three-dimensional protein folds. Nucleic Acids Res., 25, 231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibrat J.F., Madej,T. and Bryant,S.H. (1996) Surprising similarities in structure comparison. Curr. Opin. Struct. Biol., 6, 377–385. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y., Addess,K.J., Geer,L., Madej,T., Marchler-Bauer,A., Zimmerman,D. and Bryant,S.H. (2000) MMDB: 3D structure data in Entrez. Nucleic Acids Res., 28, 243–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shindyalov I.N. and Bourne,P.E. (1998) Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng., 11, 739–747. [DOI] [PubMed] [Google Scholar]
- 36.Esnouf R.M. (1997) An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model., 15, 133–138. [DOI] [PubMed] [Google Scholar]
- 37.Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- 38.Castiglone Morelli M.A., Stier,G., Gibson,T., Joseph,C., Musco,G., Pastore,A. and Trave,G. (1995) The KH module has an alpha beta fold. FEBS Lett., 358, 193–198. [DOI] [PubMed] [Google Scholar]
- 39.Musco G., Kharrat,A., Stier,G., Fraternali,F., Gibson,T.J., Nilges,M. and Pastore,A. (1997) The solution structure of the first KH domain of FMR1, the protein responsible for the fragile X syndrome. Nature Struct. Biol., 4, 712–716. [DOI] [PubMed] [Google Scholar]
- 40.Baber J.L., Libutti,D., Levens,D. and Tjandra,N. (1999) High precision solution structure of the C-terminal KH domain of heterogeneous nuclear ribonucleoprotein K, a c-myc transcription factor. J. Mol. Biol., 289, 949–962. [DOI] [PubMed] [Google Scholar]
- 41.Chen X., Court,D.L. and Ji,X. (1999) Crystal structure of ERA: a GTPase-dependent cell cycle regulator containing an RNA binding motif. Proc. Natl Acad. Sci. USA, 96, 8396–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lewis H.A., Chen,H., Edo,C., Buckanovich,R.J., Yang,Y.Y., Musunuru,K., Zhong,R., Darnell,R.B. and Burley,S.K. (1999) Crystal structures of Nova-1 and Nova-2 K-homology RNA-binding domains. Structure Fold Des., 7, 191–203. [DOI] [PubMed] [Google Scholar]
- 43.Lewis H.A., Musunuru,K., Jensen,K.B., Edo,C., Chen,H., Darnell,R.B. and Burley,S.K. (2000) Sequence-specific RNA binding by a Nova KH domain: implications for paraneoplastic disease and the fragile X syndrome. Cell, 100, 323–332. [DOI] [PubMed] [Google Scholar]
- 44.Wimberly B.T., Brodersen,D.E., Clemons,W.M.,Jr, Morgan-Warren,R.J., Carter,A.P., Vonrhein,C., Hartsch,T. and Ramakrishnan,V. (2000) Structure of the 30S ribosomal subunit. Nature, 407, 327–339. [DOI] [PubMed] [Google Scholar]
- 45.Baber J.L., Levens,D., Libutti,D. and Tjandra,N. (2000) Chemical shift mapped DNA-binding sites and 15N relaxation analysis of the C-terminal KH domain of heterogeneous nuclear ribonucleoprotein K. Biochemistry, 39, 6022–6032. [DOI] [PubMed] [Google Scholar]
- 46.Van Duyne G.D., Ghosh,G., Maas,W.K. and Sigler,P.B. (1996) Structure of the oligomerization and L-arginine binding domain of the arginine repressor of Escherichia coli. J. Mol. Biol., 256, 377–391. [DOI] [PubMed] [Google Scholar]
- 47.Tesmer J.J., Klem,T.J., Deras,M.L., Davisson,V.J. and Smith,J.L. (1996) The crystal structure of GMP synthetase reveals a novel catalytic triad and is a structural paradigm for two enzyme families. Nature Struct. Biol., 3, 74–86. [DOI] [PubMed] [Google Scholar]
- 48.Lindqvist Y. and Schneider,G. (1997) Circular permutations of natural protein sequences: structural evidence. Curr. Opin. Struct. Biol., 7, 422–427. [DOI] [PubMed] [Google Scholar]
- 49.Uliel S., Fliess,A., Amir,A. and Unger,R. (1999) A simple algorithm for detecting circular permutations in proteins. Bioinformatics, 15, 930–936. [DOI] [PubMed] [Google Scholar]
- 50.Jeltsch A. (1999) Circular permutations in the molecular evolution of DNA methyltransferases. J. Mol. Evol., 49, 161–164. [DOI] [PubMed] [Google Scholar]
- 51.Pascarella S. and Argos,P. (1992) Analysis of insertions/deletions in protein structures. J. Mol. Biol., 224, 461–471. [DOI] [PubMed] [Google Scholar]
- 52.Benner S.A., Cohen,M.A. and Gonnet,G.H. (1993) Empirical and structural models for insertions and deletions in the divergent evolution of proteins. J. Mol. Biol., 229, 1065–1082. [DOI] [PubMed] [Google Scholar]
- 53.Doolittle R.F. (1994) Convergent evolution: the need to be explicit. Trends Biochem. Sci., 19, 15–18. [DOI] [PubMed] [Google Scholar]
- 54.Aravind L. and Koonin,E.V. (1999) Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. J. Mol. Biol., 287, 1023–1040. [DOI] [PubMed] [Google Scholar]
- 55.Dayhoff M.O., Schwartz,R.M. and Orcutt,B.C. (1978) In Dayhoff,M.O. (ed.), Atlas of Protein Sequences and Structures. National Biomedical Research Foundation, Washington, DC, Vol. 5, Suppl. 3, pp. 345–352.
- 56.Whisstock J., Skinner,R. and Lesk,A.M. (1998) An atlas of serpin conformations. Trends Biochem. Sci., 23, 63–67. [DOI] [PubMed] [Google Scholar]
- 57.Walker J.E., Saraste,M., Runswick,M.J. and Gay,N.J. (1982) Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J., 1, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dreusicke D., Karplus,P.A. and Schulz,G.E. (1988) Refined structure of porcine cytosolic adenylate kinase at 2.1 Å resolution. J. Mol. Biol., 199, 359–371. [DOI] [PubMed] [Google Scholar]
- 59.Story R.M., Weber,I.T. and Steitz,T.A. (1992) The structure of the E. coli recA protein monomer and polymer. Nature, 355, 318–325. [DOI] [PubMed] [Google Scholar]