

Abstract
Purpose
BRAF V600E mutation is seen in 5% to 8% of patients with metastatic colorectal cancer (CRC) and is associated with poor prognosis. Vemurafenib, an oral BRAF V600 inhibitor, has pronounced activity in patients with metastatic melanoma, but its activity in patients with BRAF V600E–positive metastatic CRC was unknown.
Patients and Methods
In this multi-institutional, open-label study, patients with metastatic CRC with BRAF V600 mutations were recruited to an expansion cohort at the previously determined maximum-tolerated dose of 960 mg orally twice a day.
Results
Twenty-one patients were enrolled, of whom 20 had received at least one prior metastatic chemotherapy regimen. Grade 3 toxicities included keratoacanthomas, rash, fatigue, and arthralgia. Of the 21 patients treated, one patient had a confirmed partial response (5%; 95% CI, 1% to 24%) and seven other patients had stable disease by RECIST criteria. Median progression-free survival was 2.1 months. Patterns of concurrent mutations, microsatellite instability status, CpG island methylation status, PTEN loss, EGFR expression, and copy number alterations were not associated with clinical benefit. In contrast to prior expectations, concurrent KRAS and NRAS mutations were detected at low allele frequency in a subset of the patients' tumors (median, 0.21% allele frequency) and were apparent mechanisms of acquired resistance in vemurafenib-sensitive patient-derived xenograft models.
Conclusion
In marked contrast to the results seen in patients with BRAF V600E–mutant melanoma, single-agent vemurafenib did not show meaningful clinical activity in patients with BRAF V600E mutant CRC. Combination strategies are now under development and may be informed by the presence of intratumor heterogeneity of KRAS and NRAS mutations.
INTRODUCTION
BRAF mutations are present in between 5% and 15% of colorectal cancer (CRC). Classically, these mutations are common in the sessile serrated adenoma pathway of adenoma to carcinoma progression and are associated with unique molecular features, including microsatellite instability, hypermethylation, and minimal chromosomal instability.1–3 BRAF mutations are present at higher frequency in right-sided tumors and are more likely to present with poorly differentiated and node-positive tumors. BRAF mutations frequently coexist with high microsatellite instability and provide modest prognostic information for recurrence.4,5 Patients with metastatic BRAF-mutated CRC have a poor prognosis, with a median survival of approximately 12 months despite the utilization of combination chemotherapy.6
The mutation of a valine to glutamate in codon 600 in the BRAF kinase domain (V600E) results in a constitutively active protein. The V600E mutation accounts for approximately 95% of the activating mutations in BRAF in CRC. This mutation results in activation of the MAPK pathway, as highlighted by the near mutual exclusivity with activating mutations in KRAS and NRAS. The unique conformation induced by V600E mutations is inhibited by vemurafenib, a kinase inhibitor designed to have increased affinity for the mutated form of BRAF. Treatment with vemurafenib has demonstrated a high response rate and prolonged progression-free survival in BRAF mutant melanoma, leading to US Food and Drug Administration approval for this indication.7 Preclinical studies in BRAF mutant CRC cell line models suggested activity with vemurafenib, with growth arrest and inhibition of MAPK signaling, and provided a strong rationale for clinical evaluation.8
However, the activity of vemurafenib in patients with CRC with BRAF V600 mutations had not been defined. Therefore, we conducted a multisite expansion cohort on the phase I trial of vemurafenib after establishment of the recommended phase II dose in melanoma.7 To explore the biology and potential predictive markers of response to therapy, we subjected patient material for correlative studies based on the emerging data on the distinct biology of BRAF mutant CRC.
PATIENTS AND METHODS
Eligibility Criteria
For the CRC expansion cohort, eligible patients had metastatic CRC with centrally confirmed V600E-positive BRAF, evaluable or measurable disease by RECIST version 1.0, and Eastern Cooperative Oncology Group performance status of 0 or 1. Patients also had to be ≥ 18 years old with adequate hematopoietic, hepatic, and kidney function and a life expectancy ≥ 3 months. Key exclusion criteria included chemotherapy within 2 weeks, prior therapy with targeted agents against BRAF or MEK, uncontrolled CNS metastases, and mean QTc ≥ 500 milliseconds. Given the risk of squamous cell carcinoma (SCC) with BRAF inhibition, patients were required to be free of pre-existing nonmelanoma skin lesions including keratoacanthoma, actinic keratosis, and squamous carcinoma in situ. The protocol (ClinicalTrials.gov identifier: NCT00405587) was approved by the institutional review boards of all participating institutions, and written informed consent was obtained for all patients before performing study-related procedures.
Drug Administration and Study Design
Vemurafenib was provided in microprecipitated bulk powder formulation as 240-mg film-coated tablets, dosed at the previously determined maximum-tolerated dose of 960 mg orally twice a day, and administered continuously in 28-day cycles.9 Patients underwent history and physical examination, CBC, chemistries, coagulation parameters, urinalysis, carcinoembryonic antigen, ECG, and tumor measurements within 3 weeks before start of therapy. While on study, patients underwent evaluations, including brief history and physical examination, vital signs, urinalysis, CBC, and chemistries, on days 8, 15, and 29 during cycle 1 and on day 1 of each subsequent cycle. ECGs were performed at baseline and before cycle 2. Adverse events were classified according to the Common Terminology Criteria of Adverse Events (version 3.0). Response was assessed radiographically after every two cycles or more frequently at the treating physician's discretion. Given the risk of SCC with BRAF inhibition, dermatologic, head, neck, and chest (with chest computed tomography) monitoring for SCC were performed at baseline and periodically thereafter.
Pharmacokinetic Studies
Pharmacokinetic samples were obtained on days 1 and 15 (before dose and 0.5, 1, 2, 4, and 8 hours after dose), day 16 (before dose), and day 29 (before dose). During continued dosing, pharmacokinetic samples were obtained before dose once every 4 weeks. Vemurafenib plasma concentration was evaluated by noncompartmental methods. Pharmacokinetic samples were also collected from the parallel expansion cohort in melanoma, as previously reported.9
Correlative Studies
DNA was extracted from macrodissected tumor on formalin-fixed paraffin-embedded (FFPE) slides, including hematoxylin and eosin–stained reference slide according to manufacturer methods (Qiagen DNeasy, Qiagen, Hilden, Germany; and Epicentre, Madison, WI).10 To determine copy number and hotspot gene mutations, the tumor was subjected to OncoScan FFPE Express V2 (Affymetrix, Santa Clara, CA) and Sequenom MassARRAY (Sequenom, San Diego, CA), both of which were optimized for use of FFPE samples with a sensitivity of 10% mutant alleles. The mutations detected are included in hotspot regions in KRAS, NRAS, PIK3CA, and AKT, among others (Data Supplement). Copy number alterations were determined by segmentation analysis produced by Nexus Copy Number (BioDiscovery, El Segundo, CA). PTEN and EGFR protein expression was determined using immunohistochemistry as previously described (PTEN clone 6H2.1, Dako, Carpinteria, CA; EGFR clone 31G7, Zymed, San Francisco, CA).11,12 Methylation analysis was performed to classify patients based on the degree of tumor methylation using bisulfite polymerase chain reaction (PCR) followed by pyrosequencing for a previously established six-marker CpG island methylator phenotype (CIMP) panel (MINT1, MINT2, MINT31, hMLH1, p16, and p14).13,14
To assess low-frequency clones, DNA was subjected to emulsion PCR (BEAMing, Sysmex Inostics, Baltimore, MD), a methodology to perform single-molecule PCRs on magnetic beads in water-in-oil emulsions, followed by allele-specific hybridization and flow cytometry–based allele quantification with sensitivity of 0.15% on FFPE tissue specimens (Data Supplement).15 A complementary droplet digital PCR was applied to a second cohort of 19 BRAF mutant tumors from patients not treated on the protocol but obtained from the Royal Melbourne Hospital (Parkville, Victoria, Australia). Analyzes were performed with a droplet digital PCR KRAS screening multiplex kit (BioRad QX100, BioRad, Hercules, CA) according to the manufacturer's instructions, with sensitivity to 0.15% mutant allele frequency for FFPE samples and 0.05% for fresh tissue. Patient-derived xenografts were derived from core biopsies and implanted in NOD-SCID-γ mice (Jackson Labs, Bar Harbor, ME). Established tumors were treated with vemurafenib analog PLX4720. Tumor sequencing of the resistant xenograft tumors for 409 exons was performed by Ion AmpliSeq Comprehensive Cancer Panel on the Ion Torrent PGM (Life Technologies, Carlsbad, CA).
RESULTS
Patient Characteristics and Treatment
Twenty-one patients with BRAF V600E mutation–positive metastatic CRC (20 colon cancers and one rectal cancer) were enrolled at multiple institutions in the United States and Australia. Of the 21 patients, 11 were men and 10 were women, with a median age of 65 years (range, 38 to 91 years; Table 1). Baseline Eastern Cooperative Oncology Group performance status was 0 and 1 in 11 and 10 patients, respectively. Results from local standard KRAS testing (codons 12 and 13) were available for 16 of the 21 patients, with all tumors characterized as wild type. All patients received prior therapy, and all except one patient received at least one prior chemotherapy regimen in the metastatic setting; 14 patients received at least two lines. Consistent with the known chemotherapy-refractory nature of the disease, only three of the 20 previously treated patients achieved a partial response to prior chemotherapy treatments.16
Table 1.
Baseline Demographic and Clinical Characteristics
| Characteristic | No. of Patients | % |
|---|---|---|
| Sex | ||
| Male | 11 | 52 |
| Female | 10 | 48 |
| Age, years | ||
| Median | 65 | |
| Range | 38-91 | |
| ECOG performance status | ||
| 0 | 11 | 52 |
| 1 | 10 | 48 |
| Race | ||
| White | 19 | 91 |
| Asian | 2 | 9 |
| Previous therapies | ||
| Surgery | 21 | 100 |
| Chemotherapy | 20 | 95 |
| Radiation | 2 | 10 |
| No. of previous therapies for metastatic disease | ||
| 0 | 1 | 5 |
| 1 | 1 | 5 |
| 2 | 4 | 19 |
| ≥ 3 | 15 | 71 |
Abbreviation: ECOG, Eastern Cooperative Oncology Group.
Safety
The most common toxicities of any grade while on treatment were fatigue, hyperglycemia, proteinuria, and diarrhea (Table 2). There were no grade 4 toxicities, and the most common grade 3 toxicity was SCC of the skin (a Common Terminology Criteria for Adverse Events term that was inclusive of keratoacanthomas). Overall, 15 of 21 patients experienced at least one grade 3 AE while on study, with seven patients experiencing severe adverse events attributed to the therapy. There were no toxicity-related discontinuations or deaths, but 52% of patients (11 patients) had AEs leading to dose interruption across all cycles. The most common causes of dose interruption included rash (n = 4), elevated liver function tests (n = 2), and diarrhea (n = 2). Furthermore, 19% of patients (four patients) required one or more dose reductions secondary to drug-related toxicities of rash, fatigue, nausea, or anorexia.
Table 2.
Summary of All Adverse Events While Receiving Treatment in ≥ 25% of Patients, Irrespective of Causality
| Adverse Event | No. of Patients (%) |
|
|---|---|---|
| Grade ≥ 3 | All Grades | |
| Squamous cell carcinoma of skin* | 5 (23.8) | 5 (23.8) |
| Hyperbilirubinemia | 3 (14.3) | 7 (33.3) |
| Rash | 2 (9.5) | 7 (33.3) |
| Hyponatremia | 2 (9.5) | 6 (28.8) |
| Fatigue | 1 (4.8) | 13 (61.9) |
| Diarrhea | 1 (4.8) | 8 (38.1) |
| Arthralgia | 1 (4.8) | 8 (38.1) |
| Vomiting | 1 (4.8) | 7 (33.3) |
| Lymphopenia | 1 (4.8) | 6 (28.8) |
| Proteinuria | 0 | 9 (42.9) |
| Photosensitivity reaction | 0 | 7 (33.3) |
| Hyperglycemia | 0 | 7 (33.3) |
| Nausea | 0 | 7 (33.3) |
| Hypoalbuminemia | 0 | 6 (28.6) |
| Pyrexia | 0 | 6 (28.6) |
| Headache | 0 | 6 (28.6) |
Cutaneous squamous cell carcinomas were reported as grade 3 adverse events, and a majority were managed by excision. One patient required treatment discontinuation for numerous lesions unable to be adequately managed by excision.
Patients were monitored closely and underwent scheduled skin exams by dermatologists during therapy. Five patients developed new cutaneous SCCs while on treatment (Appendix Fig A1, online only). In addition, two patients developed seborrheic dermatitis, and one patient each developed a skin papilloma and actinic keratosis. There were no SCCs at other monitored sites including the head, neck, and lung. Other cutaneous adverse events included photosensitivity in 33.3% (seven patients), rash in 19% (four patients), dry skin in 24% (five patients), pruritus in 14% (three patients), and hyperkeratosis in 10% (two patients).
Given the reported potential for vemurafenib to induce QT prolongation, ECGs were evaluated at baseline and the beginning of cycle 2, with a mean increase in QTc of 12.5 milliseconds (standard deviation, 17.8 milliseconds). Three patients experienced an increase greater than 30 milliseconds and less than 45 milliseconds.
Pharmacokinetic studies were available on all patients. When compared with melanoma patients in the concurrently enrolled cohort, there was no statistically significant difference in vemurafenib concentration after the first dose (mean ± standard deviation: maximum concentration, 5.5 ± 2.3 μg/mL; area under the curve from 0 to 8 hours, 31.5 ± 15.1 μg/mL·h; Appendix Fig A2, online only). Thirteen patients were available for day 15 sampling, with a maximum concentration of 38.6 ± 15.9 μg/mL and area under the curve from 0 to 8 hours of 262.4 ± 101.4 μg/mL·h.
Antitumor Activity
Nineteen of the 21 patients in this trial had postbaseline scans. There were no complete responses. One patient had a confirmed partial response, which was durable for 21 weeks. Seven other patients had stable disease as best response for at least 8 weeks (range, 8 to 50 weeks; Fig 2A). Ten patients had some degree of tumor shrinkage on the first planned computed tomography scan. Three patients had pretreatment and follow-up [18F]fluorodeoxyglucose positron emission tomography; two of these patients had a metabolic response with greater than 25% reduction in standard uptake value (Appendix Fig A3, online only). The median progression-free survival was 2.1 months (range, 0.4 to 11.6 months), with two patients free from progression for more than 6 months. Median overall survival time was 7.7 months (range, 1.4 to 13.1 months).
Fig 2.

Correlation of best overall response rate with molecular characterization. Gray boxes represent samples unable to be analyzed for the specified assay. Numbers in the copy number (CN) alteration boxes represent the number of unique segments with CN alterations. Percentages in the RAS clone box represent mutant allele frequency. (*) Not evaluable for response.
Fig 1.
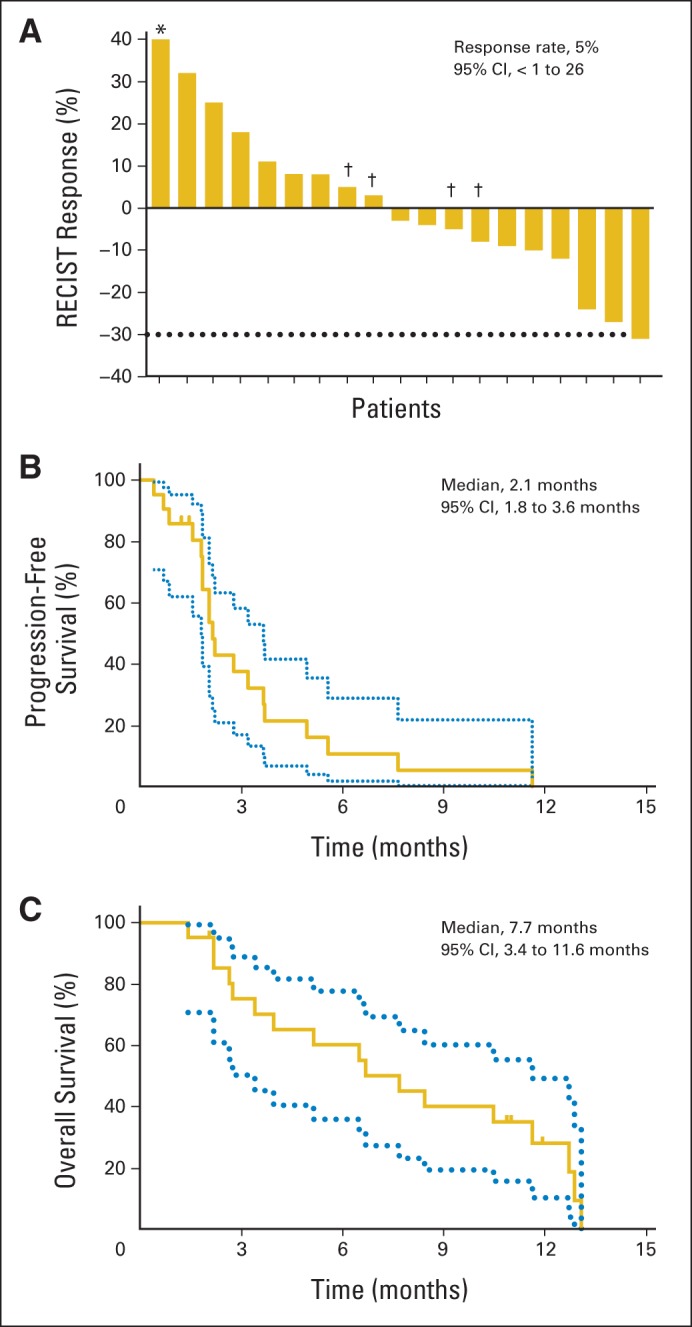
(A) Waterfall plot of best RECIST response to vemurafenib in evaluable patients. (*) Fifty-nine percent growth from baseline. (†) Patients with notable mixed responses. (B) Progression-free survival. (C) Overall survival.
Correlative Biomarkers
To evaluate the lower than expected overall response rate (compared with that seen in melanoma) and heterogeneous response to therapy in some patients, baseline tumor tissue from requested archival FFPE slides was analyzed for molecular and protein biomarkers in exploratory analyses. Prior preclinical studies had suggested that activation of the PI3K pathway may be associated with resistance to BRAF inhibition based on correlation to sensitivity in a panel of BRAF mutant CRC cell lines.8,17 The PI3K pathway was activated by complete loss of PTEN protein staining or mutations in PIK3CA or AKT in four of 14 evaluable tumors, with no association with response to therapy (Appendix Fig A4, online only). Because EGFR has been implicated in feedback activation of the MAPK pathway after BRAF inhibition, total expression of EGFR in the tumor was evaluated as a potential predictive biomarker. When dichotomized based on any expression versus no expression, there was no correlation with tumor response (Appendix Fig A4B).
Hypermethylation is a nearly universal feature of tumors with BRAF mutations, and analysis of a previously established six-gene CIMP panel confirmed this finding.18 The pattern of methylation in these markers has been previously used to subclassify CIMP-high tumors but did not correlate with clinical activity in this cohort. Complete loss of hMLH1 by protein expression was seen in patients with methylation of hMLH1 in the promoter region and defined a subset of patients (four of 15 patients) with a high level of microsatellite instability. Tumors were further annotated by the number of unique segments with copy number alterations. The expected inverse correlation between higher levels of copy number alteration and microsatellite instability was seen, but neither correlated with radiographic response.
Prior studies have demonstrated the presence of rare KRAS or NRAS clones that can lead to resistance in patients treated with EGFR monoclonal antibodies. To explore whether a similar finding is seen in BRAF-mutated CRC, tumors were subjected to a high-sensitivity emulsion PCR methodology to evaluate for the presence of rare KRAS or NRAS mutant clones in the pretreatment tissue. Low-frequency KRAS or NRAS mutations were seen in nine of 16 evaluable tumors (56%), with a median allele frequency of 0.21% (interquartile range, 0.18% to 0.42%; Fig 3A). These mutations were not detected by standard-of-care clinical sequencing or mass spectroscopy genotyping with a sensitivity of 10%. There was no correlation of the presence of low-frequency alleles with radiographic response or duration of treatment. To confirm this finding, a second cohort of primary BRAF V600E mutant CRC tumors was evaluated for low-frequency KRAS mutations by an independent digital PCR methodology. Thirteen (68%) of the 19 tumors evaluated demonstrated detectable KRAS mutations at a median allele frequency of 0.31% (interquartile range, 0.24% to 0.61%). Postprogression tissue was not available from patients who initially developed a response to directly determine the clinical relevance of these low-frequency mutations. Instead, we treated a vemurafenib-sensitive BRAF V600E patient-derived xenograft with vemurafenib until progression and explored mechanisms of resistance (Fig 3B). In both cases, the implanted tumor had low-frequency KRAS clones present at 0.07% and 0.08% by droplet digital PCR. Tumor regression was seen before development of resistance within 8 weeks. Two separate resistant tumors were sequenced, confirming persistent presence of BRAF V600E, with KRAS G12D or G12R mutations present at an allele frequency of greater than 40%.
Fig 3.
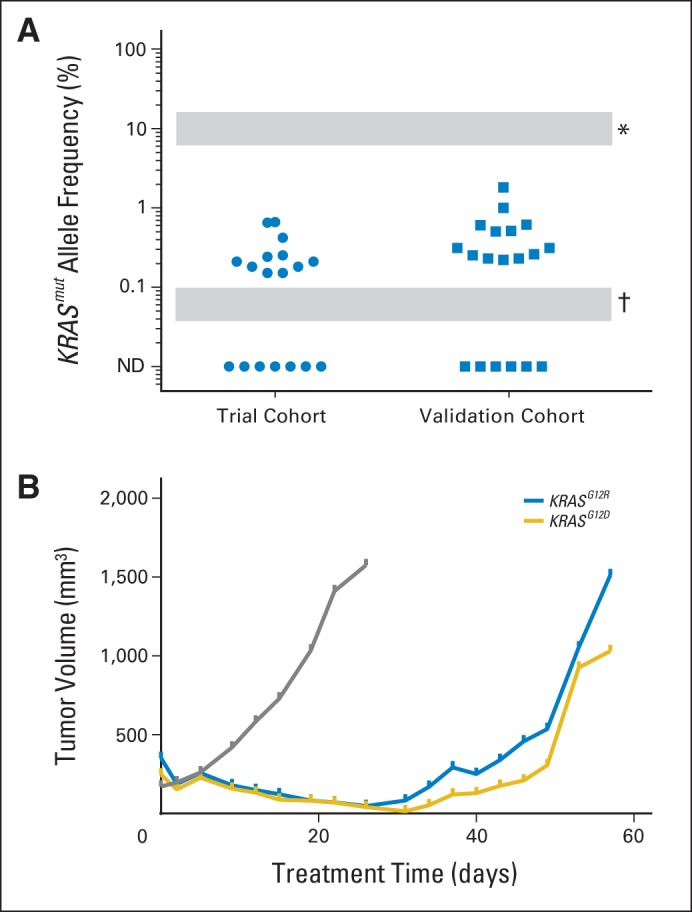
(A) Rare KRAS-mutant (mut) clones were detected in a validated cohort of BRAF V600E–mutant colorectal cancer tumors by droplet digital polymerase chain reaction (ddPCR). (*) Lower limit of detection for clinical sequencing is approximately 5% to 20% depending on assays used. (†) Lower limit of detection for ddPCR. (B) A patient-derived xenograft model developed resistance to vemurafenib analog PLX4720 provided through ad lib chow at a dose of 417 mg/kg. Gray represents control arm; blue and gold represent PLX4720 treatment arm with acquisition of KRAS G12R and G12D mutations, respectively.
DISCUSSION
CRCs with mutations in BRAF V600E represent a unique CRC subtype with poor prognosis and limited response to standard-of-care therapies. In contrast to melanoma, the tumors have limited response to single-agent BRAF inhibition. This discrepancy is not attributed to differences in dose-limiting adverse events or pharmacokinetics but is tumor specific and suggests either differences in oncogene dependency or pharmacodynamic failure as a result of feedback mechanisms. The presence of the mutation in early sessile serrated adenomas and the ability of the BRAF V600E mutation to recapitulate tumor development in a genetically engineered mouse model of CRC suggest that this mutation is a driver mutation important for carcinoma initiation.19,20 The population of enrolled patients with BRAF mutant CRC reflected the anticipated molecular phenotype, with high levels of DNA methylation, few copy number alterations, and enrichment for microsatellite instability. Our results demonstrate no meaningful differences in pharmacokinetics between patients with CRC and melanoma, with similar adverse event profiles and administered dose-intensity.21 Although vemurafenib is a substrate for cellular drug exporters such as P-glycoprotein, which is upregulated in CRC relative to melanoma, a similar low response rate (one responder) with the non–P-glycoprotein substrate BRAF inhibitor dabrafenib has been reported in patients with CRC.22
Preclinical work has suggested that the MAPK pathway is insufficiently inhibited with single-agent BRAF inhibition in CRC, in contrast to what is considered adequate inhibition in melanoma, irrespective of the BRAF inhibitor used.17,23,24 This alone may be sufficient to limit clinical activity in CRC. In addition, in the clinical setting in melanoma, there seems to be a threshold of inhibition as measured by phospho-Erk expression required to induce response.25 Because this study did not incorporate on-study biopsies, direct assessment of the ability of vemurafenib to sufficiently inhibit the MAPK pathway is not possible. Preclinical studies since this clinical trial was first reported have added considerably to our understanding of MAPK inhibition and early reactivation of the pathway in CRC, with perhaps the most compelling reactivation being through feedback EGFR reactivation.23,24 It has been proposed that EGFR expression levels may predict for this reactivation; however, no correlation was seen between total EGFR expression in the primary tumor and clinical activity in our study, suggesting that baseline EGFR expression may be a poor correlate of subsequent EGFR pathway reactivation after BRAF inhibition. The combination of EGFR and vemurafenib is being explored in several clinical trials, with some promising early results with panitumumab and an ongoing randomized study of cetuximab and irinotecan, with or without vemurafenib (ClinicalTrials.gov identifier: NCT02164916).26 In contrast, prior preclinical studies suggested that PI3K pathway activation may be associated with resistance to BRAF inhibition. However, neither PTEN protein loss nor PIK3CA mutation was correlated with resistance in our study, consistent with the limited additional clinical activity seen with the addition of a PI3K inhibitor to BRAF and EGFR inhibition.27
The finding of KRAS and NRAS mutations with low allele frequency in the BRAF mutant tumors is unexpected. With rare exception, BRAF V600E and KRAS/NRAS mutations are mutually exclusive when assessed by standard sensitivity sequencing.28–30 Acquisition of NRAS mutations has been described as a mechanism of resistance to BRAF inhibition in melanoma, and our patient-derived xenograft models suggest that a similar mechanism may be present in BRAF-sensitive CRCs.31 Because low allele frequency KRAS/NRAS mutations have been associated with resistance to cetuximab and current clinical trials are combining BRAF and EGFR inhibition, there is potential for these low allele frequency KRAS/NRAS mutations to be important drivers for resistance to the combination. The allele frequency observed in the patient-derived xenograft models is consistent with BRAF and KRAS mutations coexisting in the same tumor clones, suggesting that strategies to address both mutations will be needed. Evaluation of KRAS/NRAS mutations should be incorporated into future clinical trials of BRAF inhibition, including assessment at the time of progression. Other described mechanisms of acquired resistance in CRC to BRAF inhibition in preclinical models and patients include EGFR, KRAS, and MET amplification.32
The contrast of activity from BRAF inhibition in melanoma and CRC has been an important cautionary tale for the trend toward tumor-agnostic, oncogene-defined basket clinical trials. Histology can be a surrogate for potentially important differences in biology. BRAF-mutated CRC and melanoma differ in the tissue lineage (endoderm v ectoderm, respectively) and concurrent epigenetic features (nearly universal CIMP positive v CIMP negative, respectively), among other factors. BRAF inhibition has demonstrated some single-agent activity in non–small-cell lung cancer and thyroid cancer, among others, although the magnitude of the benefit remains to be defined.33,34 In each setting, alternate mechanisms of resistance to BRAF inhibition have been proposed, reiterating the concept that tumor histology is still a major determinant of sensitivity to targeted therapies. Further studies should leverage these similarities and differences in BRAF inhibition sensitivity to optimize future combination strategies.
Supplementary Material
Acknowledgment
We thank Richard Lee for project management.
Appendix
Fig A1.

Development of keratoacanthoma while on therapy. (A) Before initiation of therapy and (B) after 8 weeks of treatment. (C and D) Development of multiple keratoacanthomas requiring treatment discontinuation.
Fig A2.
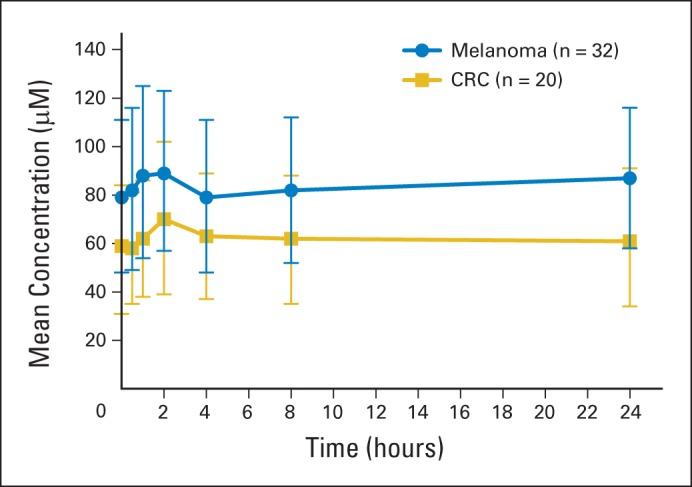
Mean steady-state concentration-time profile of patients with metastatic colorectal cancer (CRC) compared with patients with melanoma treated with vemurafenib 960 mg twice per day.
Fig A3.
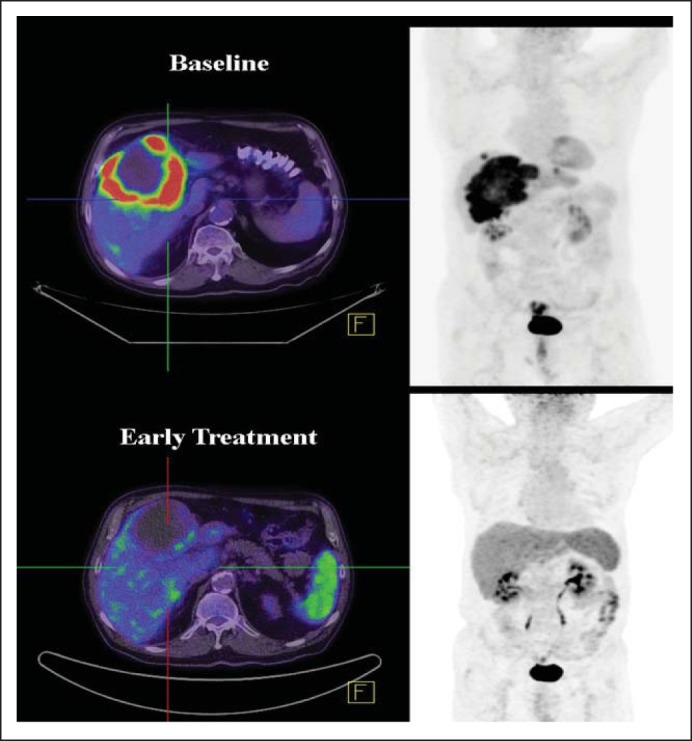
Response by positron emission tomography to vemurafenib. (Top) Baseline scan. (Bottom) After one cycle of vemurafenib.
Fig A4.
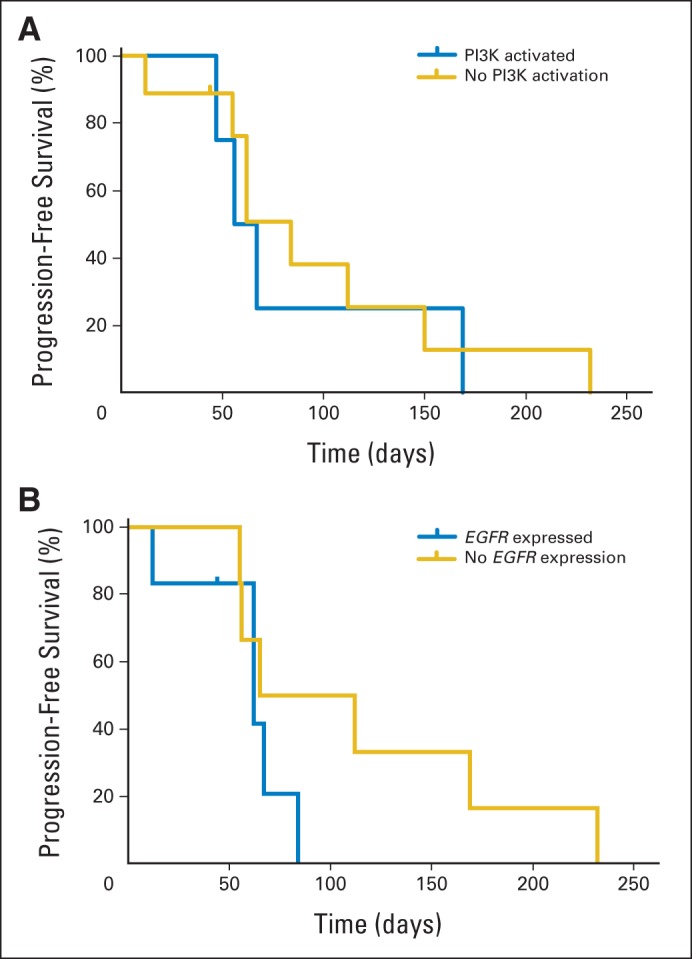
(A) Progression-free survival (PFS) for patients with or without identified alterations resulting in PI3K pathway activation (as denoted in Fig 2). (B) PFS for patients with tumors with intact or absent EGFR expression by immunohistochemistry.
Footnotes
Supported by Plexxikon and Roche. The correlative work was supported in part by the National Institutes of Health Cancer Center Support Grant No. CA16670 to The University of Texas MD Anderson Cancer Center. The authors acknowledge support from the National Cancer Institute under Awards No. R01CA172670 (S.K., G.P.), R01 CA160398 (G.P.), and R01CA184843 (S.K.).
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
Clinical trial information: NCT00405587.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Disclosures provided by the authors are available with this article at www.jco.org.
AUTHOR CONTRIBUTIONS
Conception and design: Scott Kopetz, Jayesh Desai, Peter J. O'Dwyer, Filip Janku, Keith B. Nolop, Suman Bhattacharya, Leonard Saltz
Financial support: Scott Kopetz
Provision of study materials or patients: Scott Kopetz, Jayesh Desai, Emily Chan, Leonard Saltz
Collection and assembly of data: Scott Kopetz, Emily Chan, Joel Randolph Hecht, Peter J. O'Dwyer, Dipen Maru, Van Morris, Arvind Dasari, Woonbook Chung, Peter Gibbs, Brian James, Garth Powis, Keith B. Nolop, Suman Bhattacharya
Data analysis and interpretation: Scott Kopetz, Jayesh Desai, Emily Chan, Joel Randolph Hecht, Peter J. O'Dwyer, Dipen Maru, Jean-Pierre J. Issa, Peter Gibbs, Brian James, Garth Powis, Keith B. Nolop, Suman Bhattacharya, Leonard Saltz
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Scott Kopetz
Consulting or Advisory Role: Amgen, Roche, GlaxoSmithKline, Janssen, Bristol-Myers Squibb, Agendia, Merrimack, Sysmex, Bayer, Taiho Pharmaceutical, Sanofi, Array BioPharma
Research Funding: Roche, Amgen, GlaxoSmithKline, Sanofi, Sysmex, Biocartis, Guardant Health, Agendia
Jayesh Desai
Consulting or Advisory Role: Bayer, Novartis, Merck Serono, Bionomics, Vegenics
Research Funding: Novartis, GlaxoSmithKline, Roche
Emily Chan
Consulting or Advisory Role: Amgen, Lilly, Castle Biosciences, Bayer, Taiho Pharmaceutical
Research Funding: Millennium, Aduro Biotech, Halozyme, Amgen, Merrimack, Bristol-Myers Squibb, Karyopharm Therapeutics, Boehringer Ingelheim, Taiho Pharmaceutical, Dekkun Corporation
Joel Randolph Hecht
Consulting or Advisory Role: Amgen, Sanofi, Genentech
Research Funding: GlaxoSmithKline, Novartis, OncoMed, Immunomedics, Amgen, Celgene, Pfizer, Gilead Sciences
Peter J. O'Dwyer
Consulting or Advisory Role: Genentech, Bristol-Myers Squibb
Research Funding: Genentech, Genentech, GlaxoSmithKline, Novartis, Pfizer, Mirati, BBI, Bayer, Celgene, Merck, Millennium, Bristol-Myers Squibb
Dipen Maru
Consulting or Advisory Role: Bristol-Myers Squibb, Houston, TX - Advisory Board Member
Van Morris
No relationship to disclose
Filip Janku
Consulting or Advisory Role: Trovagene, Deciphera
Research Funding: Biocartis, Trovagene, BioMed Valley Discoveries, Novartis, Astellas Pharma, Plexxikon
Arvind Dasari
No relationship to disclose
Woonbook Chung
No relationship to disclose
Jean-Pierre J. Issa
Stock or Other Ownership: Exelixis
Honoraria: GlaxoSmithKline, Janssen, Teva
Consulting or Advisory Role: Astex Pharmaceuticals, GlaxoSmithKline
Research Funding: Astex Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Biomarkers of Cancer
Travel, Accommodations, Expenses: Astex Pharmaceuticals, Janssen
Peter Gibbs
Honoraria: Roche, Amgen, Bayer, Merck, Sirtex Medical, Alchemia
Research Funding: Roche (Inst), Pfizer (Inst)
Brian James
Employment: Biogen Idec (I)
Stock or Other Ownership: Biogen Idec (I), Amgen (I)
Garth Powis
Stock or Other Ownership: Oncothyreon, PHusis Therapeutics
Patents, Royalties, Other Intellectual Property:
Keith B. Nolop
Employment: Kite Pharma
Leadership: Kite Pharma
Stock or Other Ownership: Kite Pharma
Consulting or Advisory Role: Nuevolution
Suman Bhattacharya
Employment: Medivation
Leadership: Medivation
Stock or Other Ownership: Medivation
Leonard Saltz
Consulting or Advisory Role: Boehringer Ingelheim, Sun Pharma, Roche/Genentech, Pfizer, Bayer, Lilly, McNeil PPC (I), Abbvie
Research Funding: Taiho Pharmaceutical
REFERENCES
- 1.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 2.Michaloglou C, Vredeveld LC, Mooi WJ, et al. BRAF(E600) in benign and malignant human tumours. Oncogene. 2008;27:877–895. doi: 10.1038/sj.onc.1210704. [DOI] [PubMed] [Google Scholar]
- 3.Joneson T, Bar-Sagi D. Ras effectors and their role in mitogenesis and oncogenesis. J Mol Med (Berl) 1997;75:587–593. doi: 10.1007/s001090050143. [DOI] [PubMed] [Google Scholar]
- 4.Sinicrope FA, Shi Q, Smyrk TC, et al. Molecular markers identify subtypes of stage III colon cancer associated with patient outcomes. Gastroenterology. 2015;148:88–99. doi: 10.1053/j.gastro.2014.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst. 2013;105:1151–1156. doi: 10.1093/jnci/djt173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer. 2011;117:4623–4632. doi: 10.1002/cncr.26086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang H, Higgins B, Kolinsky K, et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res. 2012;72:779–789. doi: 10.1158/0008-5472.CAN-11-2941. [DOI] [PubMed] [Google Scholar]
- 9.Abrams JS, Mooney MM, Zwiebel JA, et al. Implementation of timeline reforms speeds initiation of National Cancer Institute-sponsored trials. J Natl Cancer Inst. 2013;105:954–959. doi: 10.1093/jnci/djt137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morikawa T, Shima K, Kuchiba A, et al. No evidence for interference of H&E staining in DNA testing: Usefulness of DNA extraction from H&E-stained archival tissue sections. Am J Clin Pathol. 2012;138:122–129. doi: 10.1309/AJCP28LAOOKSZSVW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang X, Varella-Garcia M, Xavier AC, et al. Epidermal growth factor receptor abnormalities in the pathogenesis and progression of lung adenocarcinomas. Cancer Prev Res (Phila) 2008;1:192–200. doi: 10.1158/1940-6207.CAPR-08-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shroff S, Overman MJ, Rashid A, et al. The expression of PTEN is associated with improved prognosis in patients with ampullary adenocarcinoma after pancreaticoduodenectomy. Arch Pathol Lab Med. 2013;137:1619–1626. doi: 10.5858/arpa.2012-0418-OA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahn JB, Chung WB, Maeda O, et al. DNA methylation predicts recurrence from resected stage III proximal colon cancer. Cancer. 2011;117:1847–1854. doi: 10.1002/cncr.25737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colella S, Shen L, Baggerly KA, et al. Sensitive and quantitative universal pyrosequencing methylation analysis of CpG sites. Biotechniques. 2003;35:146–150. doi: 10.2144/03351md01. [DOI] [PubMed] [Google Scholar]
- 15.Dressman D, Yan H, Traverso G, et al. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U S A. 2003;100:8817–8822. doi: 10.1073/pnas.1133470100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morris V, Overman MJ, Jiang ZQ, et al. Progression-free survival remains poor over sequential lines of systemic therapy in patients with BRAF-mutated colorectal cancer. Clin Colorectal Cancer. 2014;13:164–171. doi: 10.1016/j.clcc.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mao M, Tian F, Mariadason JM, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res. 2013;19:657–667. doi: 10.1158/1078-0432.CCR-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen L, Toyota M, Kondo Y, et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci U S A. 2007;104:18654–18659. doi: 10.1073/pnas.0704652104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beach R, Chan AO, Wu TT, et al. BRAF mutations in aberrant crypt foci and hyperplastic polyposis. Am J Pathol. 2005;166:1069–1075. doi: 10.1016/S0002-9440(10)62327-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coffee EM, Faber AC, Roper J, et al. Concomitant BRAF and PI3K/mTOR blockade is required for effective treatment of BRAF(V600E) colorectal cancer. Clin Cancer Res. 2013;19:2688–2698. doi: 10.1158/1078-0432.CCR-12-2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falchook GS, Long GV, Kurzrock R, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: A phase 1 dose-escalation trial. Lancet. 2012;379:1893–1901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corcoran RB, Ebi H, Turke AB, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 25.Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yaeger R, Cercek A, O'Reilly EM, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21:1313–1320. doi: 10.1158/1078-0432.CCR-14-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Geel R, Elez E, Bendell JC, et al. Phase I study of the selective BRAFV600 inhibitor encorafenib (LGX818) combined with cetuximab and with or without the α-specific PI3K inhibitor BYL719 in patients with advanced BRAF-mutant colorectal cancer. J Clin Oncol. 2014;(suppl):32. abstr 3514. [Google Scholar]
- 28.Rajagopalan H, Bardelli A, Lengauer C, et al. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 29.Tie J, Gibbs P, Lipton L, et al. Optimizing targeted therapeutic development: Analysis of a colorectal cancer patient population with the BRAF(V600E) mutation. Int J Cancer. 2011;128:2075–2084. doi: 10.1002/ijc.25555. [DOI] [PubMed] [Google Scholar]
- 30.Fransen K, Klintenas M, Osterstrom A, et al. Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas. Carcinogenesis. 2004;25:527–533. doi: 10.1093/carcin/bgh049. [DOI] [PubMed] [Google Scholar]
- 31.Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahronian LG, Sennott EM, Van Allen EM, et al. Clinical acquired resistance to RAF inhibitor combinations in BRAF-mutant colorectal cancer through MAPK pathway alterations. Cancer Discov. 2015;5:358–367. doi: 10.1158/2159-8290.CD-14-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dadu R, Shah K, Busaidy NL, et al. Efficacy and tolerability of vemurafenib in patients with BRAF(V600E)-positive papillary thyroid cancer: M.D. Anderson Cancer Center off label experience. J Clin Endocrinol Metab. 2015;100:E77–E81. doi: 10.1210/jc.2014-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peters S, Michielin O, Zimmermann S. Dramatic response induced by vemurafenib in a BRAF V600E-mutated lung adenocarcinoma. J Clin Oncol. 2013;31:e341–e344. doi: 10.1200/JCO.2012.47.6143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.