

Abstract
Several current immunotherapy approaches target private neoantigens derived from mutations that are unique to individual patients’ tumors. However, immunotherapeutic agents can also be developed against public neoantigens derived from recurrent mutations in cancer driver genes. The latter approaches target proteins that are indispensable for tumor growth, and each therapeutic agent can be applied to numerous patients. Here we review the opportunities and challenges involved in the identification of suitable public neoantigen targets and the development of therapeutic agents targeting them.
The aim of cancer immunotherapy is to engage the immune system against targets that are present in cancer cells but not normal tissues. One such class of targets are mutation-associated neoantigens, which arise when somatic mutations generate altered peptides that are processed and presented by the major histocompatibility complex (MHC) on the cell surface1. Such MHC-presented mutant peptides can be recognized by the immune system as distinct from their wild-type counterparts by several mechanisms (Fig. 1a–c). For instance, the altered peptide sequence can allow the mutant peptide to be processed differently for presentation, cause differential mutant versus wild-type peptide binding to the MHC, alter contacts with a T cell receptor (TCR) or change peptide conformation and the overall structure of the peptide–MHC–TCR binding interface. Because they are present exclusively in cancer cells, mutation-associated neoantigens are attractive targets for precision immunotherapies including vaccines, antibodies and cellular therapeutics.
Fig. 1 |. Generation and immune recognition of public neoantigens.

a, Amino acid alterations can enable a mutant peptide to be processed differently for presentation or to bind to the HLA, whereas the wild-type peptide does not bind. b, Differential contact with the TCR by a mutant amino acid residue allows discrimination between the mutant and wild-type peptides within the HLA. c, The overall structure of the peptide–HLA–TCR binding interface is altered through altered conformation of mutant peptide binding, distinguishing the mutant peptide from the wild-type peptide. d, Genetic mutations produce changes in the amino acid sequences of proteins. These proteins with altered sequences can be degraded by the proteasome and the resulting peptides can be processed and presented by HLA on the cell surface. The genes listed are those for which public neoantigens have been identified with defined HLA restriction and strong evidence for endogenous presentation (Supplementary Table 1).
Cancer driver mutations, in oncogenes or select tumor suppressor genes, tend to be localized in genome hotspots that change protein function and are recurrent among patients2. Altered peptides produced from these mutations and presented by common human leukocyte antigen (HLA) alleles (the human MHC molecules) can therefore yield neoantigens that are shared among individuals whose tumors harbor the same genetic alterations and HLA, demarcating them as public (Fig. 1d). Conversely, private neoantigens are generated from passenger mutations or non-recurrent driver mutations that are observed in individual patients with cancer. The majority of driver gene mutations do not yield neoantigens that are presented by common HLAs, and the vast majority of presented neoantigens are private3–5. Thus, identifying the subset of patients for whom public neoantigens are relevant targets is a major challenge. Nevertheless, the ability provided by the expansion of standard-of-care next-generation sequencing to screen patients affords the opportunity for off-the-shelf precision immunotherapies against public neoantigens that would be broadly applicable to many patients and would have tremendous advantages in scalability relative to targeting private neoantigens (Fig. 2). Here we discuss the rationale and recent progress in discovering and targeting public neoantigens, as well as future prospects for developing relevant immunotherapies.
Fig. 2 |. Strategies to target public neoantigens.

Sequencing of tumor specimens from patients enables mutation identification and HLA typing. Patients can then be matched to appropriate therapies for targeting their identified public neoantigens. These therapies include vaccines, TCR-T cells, CAR-T cells and bispecific antibodies.
The rationale for targeting public neoantigens
Current immunotherapeutic approaches against specific neoantigens, such as vaccines and autologous cell transfer, typically rely on a multistep development process that is personalized to each patient and may target non-recurrent private as well as putative public neoantigens1. The logistical and financial realities of producing personalized therapies for each patient present a substantial obstacle to widespread accessibility6. The extended production timeline also increases the potential for disease progression before treatment can be initiated.
A common benefit of targeting public and private neoantigens is the cancer cell specificity conferred by the underlying genetic mutations, which would ensure minimal toxicity to normal tissue. However, targeting public neoantigens could circumvent many of the limitations, including the complex logistics, inherent in personalized immunotherapy approaches. In addition, because driver gene mutations drive tumorigenesis, their public neoantigens should be present in every cell within the cancer—something that is not necessarily true for private neoantigens7—with cancer cells unable to lose the antigen without a concomitant loss of fitness8.
One potential disadvantage of off-the-shelf public neoantigen-targeting therapies is that no patient is likely to have more than one or a few public neoantigens available to be targeted. In contrast, cancers generally have multiple private neoantigens, in principle facilitating combinatorial targeting and mitigating the risk of treatment resistance due to antigen loss during clonal evolution9.
The frequency of driver gene mutations (Fig. 3) indicates that targeting their public neoantigens could potentially benefit a large proportion of patients. Apart from oncogenes, some tumor suppressor genes also exhibit recurrent mutations, but tumor suppressor genes in general are unlikely sources for public neo-antigens as they are often inactivated by non-recurrent mutations or expressed at low levels because of nonsense-mediated decay of their messenger RNA transcripts. An exception is TP53. Among the several TP53 mutation hotspots, the most common is the one leading to the protein alteration p.Arg175His, which was recently shown to be targetable with a bispecific antibody in pre-clinical studies10.
Fig. 3 |. Frequency of recurrent mutations across cancer types.
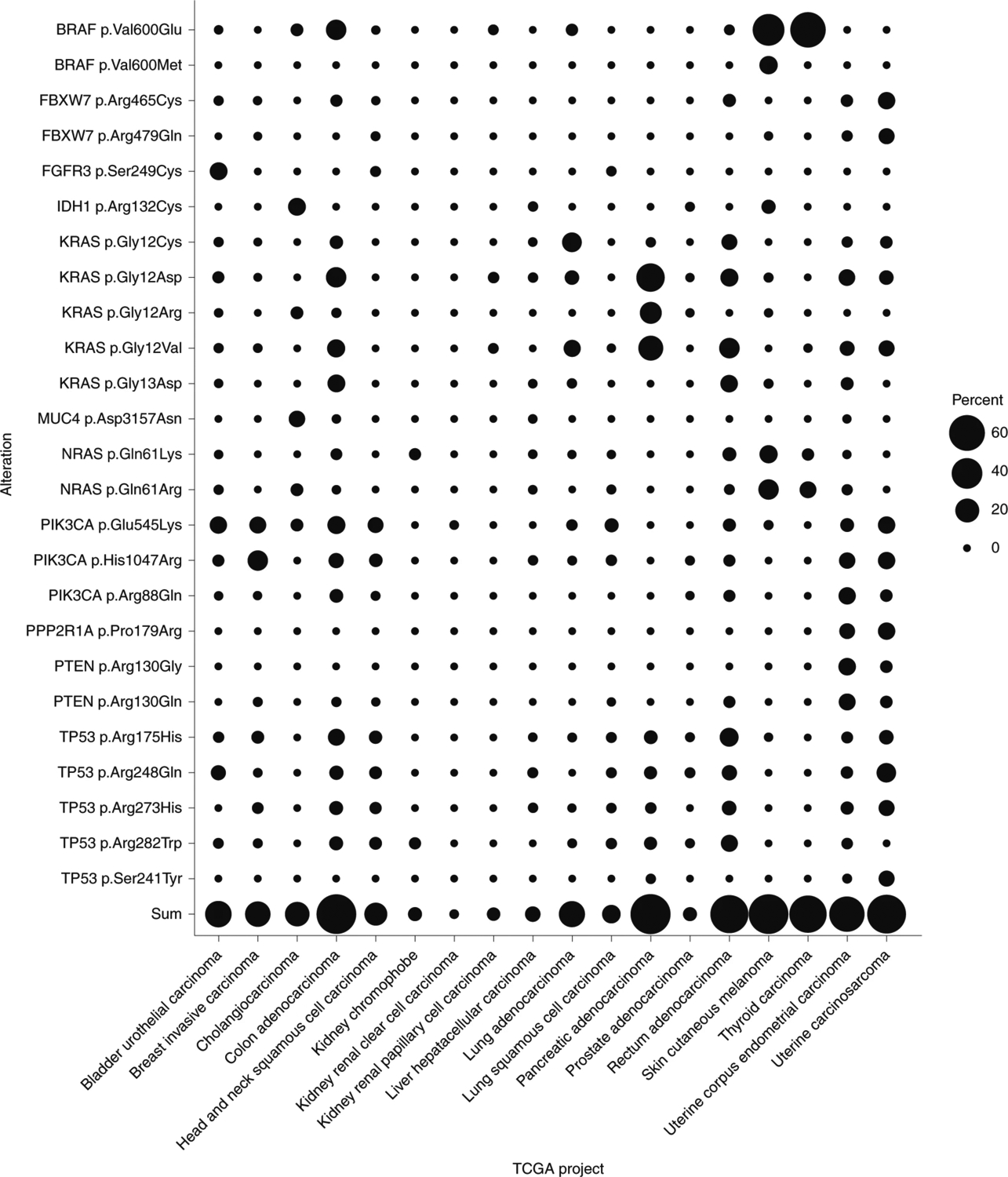
For the top 12 solid tumor organ sites projected to be responsible for most new cancer cases in the United States in 2020 (National Cancer Institute Surveillance, Epidemiology, and End Results Cancer Statistics Factsheets; https://seer.cancer.gov/statfacts/html/common.html), the corresponding The Cancer Genome Atlas (TCGA) projects were selected159. Shown are the frequencies of occurrence in each TCGA project of each substitution that accounts for at least 5% of cases in at least one TCGA project. Sum represents the summation of frequencies of all included mutations for a given TCGA project.
A broadly applicable public neoantigen-targeting therapy also requires the presence of common HLA alleles, many of which do occur at high frequencies (Table 1). The product of the frequency of a specific hotspot mutation and the frequency of a specific HLA allele yields the theoretical frequency of patients who could benefit from public neoantigen-based therapies. For example, when the most common HLA alleles (Table 1) and the most common driver gene mutations (Fig. 3) are considered, the fraction of patients with pancreatic cancer who could potentially benefit from targeting KRAS p.Gly12Asp public neoantigens is ~10%. Given that most driver gene mutations, such as those in PIK3CA, TP53 and KRAS, occur in a large number of tumor types, those shown to generate validated neoantigens with reasonably frequent HLA alleles would be common enough to enable the development of off-the-shelf immunotherapies. For the 12 most common solid tumor organ sites in the United States, ~1% of patients could in theory benefit from an immunotherapeutic approach targeting a KRAS p.Gly12Asp peptide in the context of an HLA-A*03 allele (Table 2). This number of patients is comparable to or higher than those who might benefit from other targeted therapies used in clinical practice or currently in development. Examples include larotrectinib for solid tumors with NTRK fusions (which may be present in up to 1% of all solid tumors) and avapritinib for PDGFRA exon 18 mutant gastrointestinal stromal tumors (which occur in a few thousand patients per year)11,12. If immunotherapeutic agents could be developed just to the top ten public neoantigens, at least one of these drugs could in theory be used to treat more than one million patients with cancer per year at the international level.
Table 1.
Frequencies of common HLA alleles
| HLA | Phenotype frequency in population (%) | ||||
|---|---|---|---|---|---|
| Asian | Black | Hispanic | White | Weighted average | |
| A*02:01 | 18 | 23 | 35 | 50 | 42 |
| C*07:01 | 8 | 23 | 20 | 31 | 26 |
| C*07:02 | 27 | 13 | 21 | 28 | 25 |
| A*01:01 | 10 | 9 | 13 | 31 | 24 |
| C*04:01 | 15 | 34 | 30 | 20 | 23 |
| A*03:01 | 5 | 16 | 15 | 27 | 22 |
| B*07:02 | 5 | 14 | 11 | 26 | 20 |
| B*08:01 | 3 | 8 | 9 | 23 | 17 |
| A*24:02 | 33 | 4 | 23 | 17 | 17 |
| C*06:02 | 13 | 17 | 11 | 18 | 16 |
Phenotype frequencies of the ten most common HLA alleles in the United States. The alleles are ranked by average phenotype frequency153,154, weighted according to ethnicity representation in the United States (United States Census Bureau QuickFacts; https://www.census.gov/quickfacts/fact/table/US/PST045219).
Table 2.
Top ten public neoantigens
| Gene | Protein alteration | HLA | Top cancer | HLA frequencya (%) | Mutation frequencyb (%) | Neoantigen frequencyc (%) | Incidence of cancers carrying neoantigend | Reference for neoantigen validation | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| All cancerse | Top cancerf | All cancerse | Top cancerf | All cancerse | Top cancerf | ||||||
| BRAF | p.Val600Glu | A*02 | Melanoma | 41.9 | 7.1 | 43.9 | 3.0 | 18.4 | 40,000 | 18,000 | 155 |
| KRAS | p.Gly12Asp | A*03 | Pancreas | 21.7 | 3.8 | 32.4 | 0.8 | 7.0 | 11,000 | 4,000 | 24 |
| KRAS | p.Gly12Val | A*03:01 | Lung | 21.7 | 3.3 | 6.7 | 0.7 | 1.5 | 10,000 | 3,000 | 40 |
| TP53 | p.Arg175His | A*02:01 | Colorectum | 41.9 | 1.6 | 6.5 | 0.7 | 2.7 | 9,000 | 4,000 | 10,42,46,156 |
| KRAS | p.Gly12Asp | A*11:01 | Pancreas | 10.8 | 3.8 | 32.4 | 0.4 | 3.5 | 6,000 | 2,000 | 45 |
| KRAS | p.Gly12Val | B*35 | Lung | 11.4 | 3.3 | 6.7 | 0.4 | 0.8 | 5,000 | 1,700 | 41 |
| HRAS/KRAS/NRAS g | p.Gln61Arg | A*01:01 | Melanoma | 23.6 | 1.6 | 12.4 | 0.4 | 2.9 | 5,000 | 2,900 | 24,157 |
| KRAS | p.Gly12Val | A*11:01 | Lung | 10.8 | 3.3 | 6.7 | 0.4 | 0.7 | 5,000 | 1,700 | 45 |
| BRAF | p.Val600Glu | B*27:05 | Melanoma | 4.8 | 7.1 | 43.9 | 0.3 | 2.1 | 5,000 | 2,100 | 158 |
| KRAS | p.Gly12Asp | C*08:02 | Pancreas | 7.5 | 3.8 | 32.4 | 0.3 | 2.4 | 4,000 | 1,400 | 79 |
This table lists public neoantigens identified with defined HLA restriction and with evidence for endogenous processing and presentation, ranked by the number of new patients annually in the United States for whom the public neoantigen applies. This analysis includes public neoantigens generated by substitution mutations recurrent in the top 12 most common solid tumor organ sites and identified with HLA class I restriction.
Calculated according to phenotype frequencies in the United States, weighted for proportional ethnicity representation153,154 (United States Census Bureau QuickFacts; https://www.census.gov/quickfacts/fact/table/US/PST045219).
Calculated according to The Cancer Genome Atlas mutation frequencies, weighted according to the 2020 projected incidence of the top 12 most common solid tumor organ sites in the United States159 (National Cancer Institute Surveillance, Epidemiology, and End Results Cancer Statistics Factsheets; https://seer.cancer.gov/statfacts/html/common.html).
Calculated by multiplying the HLA frequency by the mutation frequency.
Number of new patients annually whose tumors carry the neoantigen, calculated by multiplying the cancer incidence by the neoantigen frequency.
All of the top 12 most common solid tumor organ sites in the United States.
The cancer type with the greatest number of applicable new patients annually.
HRAS, KRAS and NRAS p.Gln61Arg mutations are considered together because the protein sequences flanking the mutation site are identical.
The actual number of patients who could benefit from public neoantigen-targeted therapies could be lower than calculated maximum estimates because the existence of recurrent mutations and common HLA alleles does not guarantee the existence of public neoantigens. For a mutated gene to produce a neoantigen, the resulting altered peptide must be expressed, processed by cellular machinery, presented stably by an HLA on the cell surface and show a distinct feature recognizable to the immune system. Thus, the paramount question is whether specific driver gene mutations generate neoantigens that are presented by common HLAs. The high frequency of hotspot mutations has been suggested to be partly due to the inefficient neoantigen presentation of their product peptides, thereby permitting cells harboring the mutation to evade elimination by the immune system13,14. Others have reported the lack of evidence supporting the depletion of mutations that generate neoantigens under negative selective pressure15. Evidence for a tumor’s ability to induce T cell tolerance to its own mutations further suggests that neither failure to present nor negative selection of peptides derived from driver mutations is universal16. Computational means have been employed to predict whether peptides containing hotspot mutations identified from sequencing data are likely to be presented. These efforts reveal that neoantigens generated from recurrent driver mutations are indeed predicted to exist (for example, for driver mutations in EGFR, KRAS, PIK3CA, TP53 and others)17–19. However, computational methods to assess HLA binding of specific epitopes can generate both false negative and false positive predictions1,20. Experimental validation is therefore critical for a more accurate estimate of the potential applicability of public neoantigen targeting to patients.
Experimental validation of public neoantigens
The most direct way to demonstrate the existence of public neoantigens is through mass spectrometry to detect HLA-complexed mutant peptides (pHLA), although the low sensitivity of detection may lead to false negatives21. Mass spectrometry experiments have been performed in primary tumor samples and in cell lines endogenously expressing the neoantigen or engineered to overexpress it22–24, providing unequivocal evidence that neoantigens can be processed and presented by specific HLAs. However, such experiments do not provide evidence that these antigens are presented by tumors that harbor the mutation in every patient with that particular HLA allele. To demonstrate such presentation in an individual patient, mass spectrometry would have to be performed on pHLA complexes purified from the neoplastic cells of the patient, which is technically challenging but possible21,25.
Another approach to identify or validate public neoantigens is to do so indirectly, through the existence of reactive T cells, as this suggests presentation of the putative neoantigen on the tumor cells20,26,27. Neoantigens validated by this approach are immunogenic by definition. Although the majority of characterized public neoantigens recognized by T cells are HLA I restricted, HLA II-restricted T cell responses have also been identified, such as against the common BRAF mutation that leads to a p.Val600Glu alteration28. Reactive T cells do not necessarily show that the neoantigen is actually processed and presented by tumor cells29. Specifically, if rather than expressing the relevant gene the target cells are experimentally loaded with exogenous peptide, endogenous processing is not taken into account30. More definitive evidence is therefore provided by detecting T cell reactivity against cells that express the driver gene and comparing that with isogenic cells engineered in a way, such as with knockout, that they do not express the gene. In vivo T cell clonal expansion also indicates presentation of a neoantigen that is sufficiently immunogenic to drive this expansion31,32.
Overall, each experimental approach provides a different level of substantiation that a putative public neoantigen is presented by cancer cells and is immunogenic. We propose an eight-point hierarchy for levels of evidence, ordered from the strongest to the weakest:
Generation of a clinical response in a randomized, controlled trial of a public neoantigen-targeted therapy. No public neoantigen has yet achieved this level of validation.
Documentation of a clinical response to a public neoantigen-targeted therapy in an uncontrolled trial33–35.
Experimental confirmation of neoantigen presentation by mass spectrometry of primary cancer cells from patients with the expected neoantigen23,36,37.
Identification of T cell reactivity against primary cancer cells in patients with the expected neoantigen28,38,39. This is less rigorous than mass spectrometry, because the absence of false positives due to cross-reactivity cannot be guaranteed, but it does provide evidence of immunogenicity rather than presentation alone.
Experimental confirmation of presentation in model cell lines endogenously expressing the public neoantigen of interest10,22–24,40–42.
Experimental confirmation of presentation in model cell lines engineered to overexpress the public neoantigen4,5,24,31,42–46.
Confirmation of presentation in peptide-loaded cell lines.
Computational prediction.
As our understanding of the roles of cross-presented public neoantigens and ways to exploit them increases, it will also be useful to integrate evidence for cross-presentation into the validation framework. Supplementary Table 1 lists the public neoantigens identified to date that meet at least level 6 evidence for validation and for which the HLA restriction has been defined. The top ten of these neoantigens have also been ranked according to the number of new patients with cancer per year in the United States for whom a targeted therapy would be applicable (Table 2). The ranking is based on the overall incidence, including early-stage cancers. Although immunotherapies targeting public neoantigens are currently most relevant for advanced cancers, the prospective safety profile of these precision therapies, owing to their specificity, may enable them to eventually be applicable even in early disease settings.
Future identification of public neoantigens
As noted above, validating a candidate neoantigen as a bona fide one is still challenging, and showing proper processing and presentation is a major bottleneck. For immunotherapeutic strategies that make use of T cell responses, such as vaccination or adoptive cell transfer, a second bottleneck lies in determining whether presented antigens are immunogenic. Indeed, the majority of neoantigens confirmed by mass spectrometry as presented by HLAs do not induce T cell responses21,25,47. The failure of T cells to recognize many neoantigens may be due to the similarity of the latter to their wild-type counterparts and the corresponding gaps in the functional TCR repertoire induced by central or peripheral tolerance mechanisms48. For immunotherapies such as engineered antibody-based constructs, immunogenicity is not a limiting factor, providing a major advantage to such modalities compared with those that are dependent on a natural immune response, including immune checkpoint inhibitors.
Several strategies could help to overcome these bottlenecks. First, more accurate prediction of the binding of candidate peptides to specific HLA alleles is essential. Traditional computational methods for predicting the presentation of neoantigens were developed using affinity data of peptide binding to HLA. However, more recent prediction tools using machine learning methods trained on expanded mass spectrometry data of endogenously presented peptides account for additional factors in the presentation process beyond affinity and have achieved increased accuracy in predicting neoantigen presentation for both HLA class I and class II49–52. The growing volume of publicly available mass spectrometry data should facilitate continued improvements in neoantigen prediction methods53–55. Similarly, using binding affinity alone to forecast immunogenicity may lead to poor predictions. Indeed, peptides predicted to have low affinity for HLA can still be immunogenic, as is the case for the TP53 p.Arg175His alteration46. In contrast, many neoantigens predicted to have reasonable binding affinity have not been identified in empirical searches of presentation or the ability to induce T cell responses3,5,25. Evidence suggests that features other than affinity, such as pHLA stability, greater difference between altered versus wild-type peptide binding to HLA, or peptide foreignness, could better predict immunogenicity and prioritize targets56–58. An advantage of some of the public neoantigen-targeted approaches described here, such as those using antibodies or cells directed to specific pHLA complexes, is that they do not require immunogenicity of the pHLA complex, but rather only its presence on the surface of the cancer cell.
Second, neoantigen discovery could be improved by screening with new innovations in recombinant HLA reagents. One traditional strategy is to measure T cell binding to soluble recombinant HLA molecules complexed with neoantigen peptides, as the existence of reactive T cells implies neoantigen presentation. Reagents with combinatorial fluorescent, isotope or DNA barcodes or nanoparticle functionalization have vastly expanded throughput and sensitivity. These methods also support simultaneous interrogation of multiple candidate neoantigens with a single precious clinical sample59–61. Fully formed HLA molecules in which peptides can be inserted at will have also been reported62–64. These new technologies should enable rapid testing of candidate neoantigens to all common HLA alleles. Positive results would not prove that a candidate neoantigen is properly processed and presented, but negative results would exclude neoantigen–HLA combinations from further testing in cell-based assays.
Third, advances in assays measuring functional T cell responses to neoantigen-presenting cells will continue to be important. For example, tandem minigene constructs that allow for unbiased and simultaneous screening of both HLA class I- and class II-restricted neoantigens from multiple mutation sites have aided the identification of candidate public neoantigens in numerous studies4,5,33,46.
Fourth, combinations of recurrent mutations (Fig. 3) and common HLA alleles (Table 1) could be screened (for example, by expressing the top 25 genes harboring hotspot mutations in cells expressing common HLA alleles, followed by mass spectrometry of purified pHLA complexes). Testing various cancer cell lines representing the most common cancer types and HLA types would help to mitigate differences in presentation capabilities. Though costly, this project is feasible and would have immediate clinical ramifications. Such experiments on cells engineered to express these mutations could be a starting point for interrogating variability between cancer types and heterogeneity among the tumors of individual patients.
Fifth, it is important to determine whether candidate neoantigens demonstrated to be processed and presented on the cell surface in an experimental system are presented on a particular individual’s cancer cells. Genetic or epigenetic differences in protein processing or presentation create interpatient and intratumor heterogeneity. For example, if cancer cells do not express HLA complex components, they cannot present public neoantigens. Inactivation of HLA genes or other genes involved in HLA class I and II antigen processing and presentation is known to occur in some patients, making tumors resistant to any immunotherapy relying on particular pHLA complexes, including immune checkpoint inhibitors9,33,65. Assessment of functional HLA molecules on cancer cells is likely to become the standard of care for assessing the value of selected immunotherapeutic approaches in the future. Because neoantigen cross-presentation can be clinically relevant, neoantigen presentation by cell types other than the cancer cells themselves should be measured in tumors66,67—an issue that is more pertinent to the development of vaccines against public neoantigens than for antibodies or cells that react with specific pHLA complexes.
At present, the approach used to judge an individual patient’s likelihood of responding to an immunotherapy targeting a public neoantigen is to evaluate the tumor DNA sequence and cell surface HLA expression. However, this does not definitively determine presentation of the public neoantigen by the patient’s tumor cells. In the future, it is hoped that mass spectrometry methods will have advanced sufficiently to be applied directly to patient samples. The identification of neoantigens in pHLA complexes by mass spectrometry currently requires hundreds of millions of cells24, but sensitivity for both HLA class I and class II alleles continues to be improved24,25,67–69. The application of such techniques to hundreds of thousands rather than millions of neoplastic cells would enable demonstration that an individual patient’s cancer cells do present the candidate public neoantigens. Documentation that T cells from the patient with cancer can react with candidate public neoantigens could provide additional evidence of presentation and would further suggest that stimulation of this immune response, particularly in combination with immune checkpoint inhibitors, could be therapeutic.
Although these emerging and aspirational advances are not essential for moving public neoantigen-targeting therapy candidates forward in the short term, they will be essential for expanding the number of patients public neoantigen-targeting therapies can routinely reach in the long term. Advancing technologies will also help to increase the number of validated public neoantigens, making them relevant to more patients. To take advantage of off-the-shelf therapies, companion diagnostics to identify suitable patients must similarly be available in an off-the-shelf fashion and at scale. Some of these approaches, such as adding HLA typing to next-generation sequencing panels or implementing HLA expression assays, should be straightforward to integrate into clinical workflows. Other assays, such as individualized mass spectrometry or T cell reactivity assays, would be more difficult to implement and standardize in a clinical setting. Ultimately, applying them to every patient may not be essential if a large proportion of patients with the mutation and HLA type can be shown to present the relevant public neoantigen and respond to treatment.
Therapeutic targeting of public neoantigens
The validation of public neoantigens opens opportunities to exploit them therapeutically. In the future, sequencing cancer driver genes from patients’ tumor DNA is likely to be routine, to determine the applicability of targeted therapy. The same DNA could be readily assessed for HLA type and the patient matched to an off-the-shelf therapy corresponding to that public neoantigen and HLA type combination.
Four general treatment modalities targeting public neoantigens have been explored to date (Fig. 2). The first is vaccination. The treatment of patients with cancer with vaccines engendering an immune response against public neoantigens derived from KRAS mutations has a near three-decade history34,41,70. Vaccination strategies against TP53 and BCR/ABL fusion public neoantigens have also been conducted70,71. Although such vaccines have succeeded in generating neoantigen-responsive T cells70, measures of clinical benefits have been modest and anecdotal, with no vaccinations yet demonstrating statistically significant patient benefit in randomized trials72. This suggests that the vaccines did not stimulate a sufficiently strong immune response to eliminate cancer cells in an immunosuppressive tumor environment. Better adjuvants, improved delivery formats and combinations with immune checkpoint inhibitors could improve efficacy. Much of the current intense effort to improve the efficacy of immune checkpoint inhibitors will also be applicable to vaccines, and in particular to vaccine–immune checkpoint combinations. Trials might also benefit from focusing on patients with tumors that have been validated to present the neoantigen target. One illustrative study designed a vaccine targeting public neoantigens derived from 20 recurrent mutations that were predicted to cover a median of 11% of nine types of common solid tumors34. Similar vaccines targeting multiple public neoantigens could simplify regulatory certification and production. For example, the need to prepare individual vaccines for each patient (as will always be the case for private neoantigens) could be alleviated by producing vaccines specific for each HLA type, with each one including the most common recurrent mutant peptides presented by that HLA.
The second immunotherapeutic modality that can be employed to target public neoantigens is the adoptive transfer of specifically reactive tumor-infiltrating lymphocytes (TILs) or cytotoxic T cells derived from autologous or allogeneic sources. One study of a patient with metastatic colorectal cancer reported tumor regression after adoptive transfer of autologous TILs exhibiting reactivity to a KRAS p.Gly12Asp neoantigen33. In a second study, adoptive transfer of autologous TILs exhibiting reactivity for a BRAF p.Val600Glu neoantigen was associated with melanoma regression35. Autologous T cells specific for a BCR/ABL translocation public neoantigen, transferred in three patients with acute lymphocytic leukemia, led to remission when combined with tyrosine kinase inhibitors73. This work suggests that adoptive cell transfer of T cells specific for public neoantigens can produce durable clinical responses. This suggestion was supported by another study in which adoptive transfer of T cells with reactivity to a TP53 p.Arg175His neoantigen was evaluated46. However, only some lesions responded in this study and an immune checkpoint inhibitor was used, complicating interpretation and raising the possibility of alternative explanations for the observed efficacy.
Adoptive transfer therapies using patient-specific, public neoantigen-reactive TILs are limited in their scalability, but they have heralded the third modality of public neoantigen targeting: validated TCRs cloned from the naturally occurring T cells. TCRs from TILs and peripheral T cells that recognize a variety of public neoantigens have been described37,38,42,43,74. Thus, it is theoretically possible to genetically engineer the relevant TCR into the T cells of other patients to furnish reactivity against a specific neoantigen. Transposon or CRISPR–Cas9 systems can streamline the rapid and safe engineering of TCRs into T cells75,76. Notably, trials in which patients receive autologous T cells engineered to express TCRs targeting KRAS p.Gly12Val or p.Gly12Asp altered peptides presented by HLA-A*11:01 are already underway72. Because these trials employ a murine TCR, it will be important to analyze whether host reactivity against the TCR may decrease the treatment efficacy or contribute to adverse effects45. Alternatively, soluble constructs directed by monoclonal TCR moieties could be used as an off-the-shelf therapy for matched public neoantigens. One example of this format is ImmTACs (immune mobilizing monoclonal T cell receptors against cancer), a class of bispecific molecules comprising a TCR component connected to an anti-CD3 antibody component, which together crosslink target cells to T cells, thereby activating them77,78. Improving pipelines to identify relevant TCR sequences from patients with cancer, healthy donors or engineered mice could accelerate the implementation of these TCR-based strategies44,79–82.
In contrast with the three modalities described above that share reliance on neoantigen-reactive TCRs, a fourth approach employs antibody moieties to drive specific activity (for example, through full-length antibodies, antibody–drug conjugates, bispecific antibodies or chimeric antigen receptor T cell (CAR-T) therapies)83,84. Two representative studies used phage display to select antibody fragments against KRAS, EGFR and CTNNB1 public neoantigens, but endogenous processing and presentation of these neoantigens remains to be validated85,86. A third study used yeast display to select antibody fragments against an NPM1 public neoantigen and demonstrated conversion into a CAR format39. In two other recent studies, bispecific antibodies directed to TP53 p.Arg175His, KRAS p.Gly12Val or RAS p.Gln61His/Leu/Arg public neoantigens were shown to be somewhat efficacious in both in vitro and in vivo models10,40. Such antibodies against pHLA molecules are sometimes referred to as TCR mimics and tend to have much higher affinity than natural TCRs87–90. Thus, they must be carefully screened to avoid cross-reactivity or binding of the HLA component independent of the presented peptide. As with engineered TCRs, negative selection against off-target peptides during directed evolution is an important method for avoiding cross-reactivity39,85,86. Synthetic reagents with less cross-reactivity than analogous natural receptors have been created in at least one instance91. Combinatorial library screening approaches can provide determinations of specificity on a large scale59,92,93. Structural analysis of a reagent in combination with its cognate pHLA can also help to predict cross-reactivity and guide rational designs to improve specificity10,94–98.
Comparisons and optimizations of therapeutic formats
Vaccine therapies provide practical advantages over cell-based therapies because they are easier to manufacture and more scalable. Neoantigen vaccine testing in patients has also proven to be generally safe99,100. In contrast, data on objective clinical responses to cancer vaccines are sparse, as illustrated by the lack of responses to vaccines against KRAS hotspot neoantigens101,102. The limited efficacy so far may be a result of inhibitory factors within the tumor microenvironment that ultimately provide barriers that are too high for vaccine-induced T cells to overcome. Innovating vaccine design, composition and delivery to maximize immune stimulation might bolster efficacy103–106. Rather than being used against advanced disease, vaccines may be more effective in early disease settings or adjuvant settings to prevent relapse103,107,108. The efficacy of an experimental vaccine against human papilloma virus antigens in premalignant lesions serves as a model, although clinical benefit awaits the results of an ongoing randomized phase 3 clinical trial109. As a more radical but potentially effective strategy, public neoantigen-targeting vaccines could be used prophylactically110–112. In theory, healthy individuals could ultimately be vaccinated against all validated public neoantigens matched to their HLA alleles.
Cell therapies with CARs or TCRs have been more potent than vaccines, offering hope that these platforms can be adapted for targeting public neoantigens113. Although they are more scalable than approaches that seek to grow natural neoantigen-specific T cells from individual patients, important challenges still persist in the manufacturing, scalability and safety of these therapies114. The personalized manufacturing component for autologous cell products limits their potential for widespread application of public neoantigen-targeted therapies. In contrast, universal T cells, in which allogeneic cells are used for therapy, would in theory overcome this hurdle115. The major challenges for the treatment of patients with an allogeneic product are graft-versus-host reactivity and rejection by the host’s immune system. Approaches to overcome these limitations include genetically ablating TCR genes (the source of graft reactivity against the host), genetically ablating HLA genes (the source of host recognition of transferred allogeneic cells), adoptive transfer into lympho-ablated patients, engineering cells with synthetic modules to make them resistant to rejection, or using alternative cell types such as natural killer cells116,117. Methods to program immune cells in patients in situ without an ex vivo manufacturing process could streamline delivery for any type of cellular therapy118,119.
Soluble antibody or TCR construct-based therapies, such as bispecific antibodies or ImmTACs, represent a bridge between the power of specifically redirecting T cells and the logistical advantages of non-cell-based therapy. Combining soluble or cell-based therapeutics with vaccines or with immune checkpoint inhibition therapies may enable targeting of a patient’s full public and private neoantigen repertoire to induce maximum responses while preserving T cell functionality. T cells can be genetically engineered to overcome immunosuppression and to function effectively in the hostile microenvironment of solid tumors, offering tremendous opportunities for future innovation120–123. As the efficacies of various engineered T cell and T cell-engaging therapies targeting public neoantigens evolve, each therapy’s unique and relative toxicities will also require attention to fine tune anti-tumor immunity while reducing treatment-associated morbidity and mortality.
Emerging insights from biology
Emerging knowledge of the biological differences between redirecting T cells with CARs, TCRs or bispecific antibodies will further illuminate the optimal properties for platforms targeting public neoantigens. The immunological synapses formed with TCRs, CARs and bispecific antibodies are distinct in their structure, affinity and binding kinetics124–126, leading to differing downstream signaling between CARs and TCRs127–129. A consequence of these differences is the distinct sensitivity to antigen density levels conferred by various therapeutic formats130–132—a clinically important point given that low antigen expression is a mechanism of resistance to and relapse following CAR-T therapies133. Peptide–HLA complexes, including those of public neoantigens, are typically present on the cell surface at copy numbers of less than 100 per cell10,24,40,134,135. TCRs can trigger a response to one or a few antigens136. CARs, with some exceptions, seem to require an antigen density threshold on the order of hundreds to thousands per cell and are less sensitive than TCRs when compared using synthetic constructs130,131,137–139. Bispecific antibodies generally require hundreds to thousands of antigens per cell140, but recent studies have also shown reactivities of special formats of bispecific antibodies against antigens at densities of single digits per cell10,40. Similarly, ImmTACs are active against cells with one to tens of copies per cell77. Within each format, optimization of architecture can potentially decrease neoantigen density requirements139. Beyond antigen density sensitivity, in-depth analyses relating cell and treatment characteristics with patient outcomes also hold potential for informing additional engineering efforts for different therapeutic platforms141–143.
A final issue is how best to approach cancers that carry HLA class II-restricted public neoantigens. Immunologic responses have been achieved using adoptive transfer of CD4+ T cells that recognize class II-restricted neoantigens144,145. Additionally, neoantigen vaccines that have demonstrated enhanced anti-tumor immunity have been associated predominantly with CD4+ T cell responses146–148. The precise mechanisms of these CD4+ T cells in mediating anti-cancer activity remain to be determined100,149,150, although augmenting activation of cytotoxic T cells and inducing tumoricidal inflammatory macrophages are two established functions of tumor antigen-specific CD4+ cells. Directly cytotoxic HLA class II-dependent CD4+ T cells have also been observed151. Further study of CD4+ T cell roles and of which cell types within the tumor microenvironment present class II-restricted public neoantigens is important for future progress.
Conclusions and outlook
Public neoantigens are unquestionably a compelling therapeutic target. Initial work has validated their existence and is translating that knowledge into therapies. Indeed, the first clinical trials of therapeutic agents targeting public neoantigens are underway. Progress in identifying more public neoantigens will be critical for expanding the applicability of these approaches to more patients. Moreover, the widespread adoption of cancer genome sequencing will facilitate the pairing of patients with therapies that target the public neoantigens in their tumors. In the future, an armamentarium of public neoantigen-targeted therapeutics could be developed to power precision immunotherapies against the most common genetic changes that drive cancer3,10,40,152.
Supplementary Material
Acknowledgements
We thank J. Cohen, M. Miller, S. Paul and K. Wright for insightful discussions, and E. Cook for assistance with Figs. 1 and 2. This work was supported by the Virginia and D. K. Ludwig Fund for Cancer Research, Lustgarten Foundation for Pancreatic Cancer Research, Commonwealth Fund, Burroughs Wellcome Career Award for Medical Scientists, Bloomberg~Kimmel Institute for Cancer Immunotherapy, Bloomberg Philanthropies, Mark Foundation for Cancer Research, NIH Cancer Center Support Grant P30 CA006973 and National Cancer Institute grant R37 CA230400. A.H.P., B.J.M., J.D. and S.R.D. were supported by NIH T32 grant GM136577. M.F.K. was supported by NIH T32 grant AR048522.
Footnotes
Reporting Summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Competing interests
The Johns Hopkins University has filed patent applications related to technologies described in this paper, on which A.H.P., M.S.H., E.H.-C.H., J.D., B.J.M., N.P., K.W.K., B.V., D.M.P. and S.Z. are listed as inventors: HLA-restricted epitopes encoded by somatically mutated genes (15/560,241, USPTO; 2016235251, European Patent Office); MANAbodies and Methods of Using (16/614,005, USPTO; 18802867.4, European Patent Office); MANAbodies Targeting Tumor Antigens and Methods of Using (63/059,638, USPTO; PCT/US2020/065617, World IP Organization). These applications include methods for identifying public neoantigens and the development of therapeutic agents that target these neoantigens. B.V., K.W.K. and N.P. are founders of Thrive Earlier Detection. K.W.K. and N.P. are consultants to Thrive Earlier Detection and were on its Board of Directors. B.V., K.W.K., N.P. and S.Z. own equity in Exact Sciences. B.V., K.W.K., N.P., S.Z. and D.M.P. are founders of, and serve or may serve as consultants to, ManaT Bio, and hold or may hold equity in ManaT Holdings, LLC. B.V., K.W.K., N.P. and S.Z. are founders of, hold equity in and serve as consultants to Personal Genome Diagnostics. S.Z. has a research agreement with BioMed Valley Discoveries. S.B.G. is a founder of and holds equity in AMS. K.W.K. and B.V. are consultants to Sysmex, Eisai and Cage Pharma and hold equity in Cage Pharma. B.V. is also a consultant to Catalio. K.W.K., B.V., S.Z. and N.P. are consultants to and hold equity in NeoPhore. N.P. is an advisor to and holds equity in Cage Pharma. C.B. is a consultant to DePuy Synthes and Bionaut Labs. The companies named above, as well as other companies, have licensed previously described technologies related to the work described in this paper from Johns Hopkins University. B.V., K.W.K., S.Z., N.P. and C.B. are inventors on some of these technologies. Licenses to these technologies are or will be associated with equity or royalty payments to the inventors, as well as to Johns Hopkins University. The terms of all of these arrangements are being managed by Johns Hopkins University in accordance with its conflict of interest policies. M.F.K. received personal fees from Bristol Myers Squibb and Celltrion. D.M.P. reports grant and patent royalties through his institution from Bristol Myers Squibb, a grant from Compugen, stock from Trieza Therapeutics and Dracen Pharmaceuticals and founder equity from Potenza; is a consultant for Aduro Biotech, Amgen, AstraZeneca (MedImmune/Amplimmune), Bayer, DNAtrix, Dynavax Technologies Corporation, Ervaxx, FLX Bio, Rock Springs Capital, Janssen, Merck, Tizona and Immunomic Therapeutics; is on the scientific advisory board of Five Prime Therapeutics, Catalio and WindMIL; and is on the board of directors for Dracen Pharmaceuticals.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s43018-021-00210-y.
Data availability
Data for The Cancer Genome Atlas mutation frequencies used in the analyses presented in Fig. 3 and Table 2 are available from the National Cancer Institute Genomics Data Commons (https://gdc.cancer.gov/). Data for the HLA frequencies used in the analyses presented in Tables 1 and 2 and Supplementary Table 1 are available from the Allele Frequency Net Database (http://www.allelefrequencies.net/) and National Marrow Donor Program (https://bioinformatics.bethematchclinical.org/hla-resources/haplotype-frequencies/high-resolution-hla-alleles-and-haplotypes-in-the-us-population/). Data for cancer incidence used in the analyses presented in Fig. 3 and Table 2 are available from the National Cancer Institute Surveillance, Epidemiology, and End Results Program (https://seer.cancer.gov/statfacts/html/common.html). Data for ethnicity representation in the United States used in the analyses presented in Tables 1 and 2 and Supplementary Table 1 are available from the United States Census Bureau (https://www.census.gov/quickfacts/fact/table/US/PST045219).
References
- 1.Schumacher TN, Scheper W & Kvistborg P Cancer neoantigens. Annu. Rev. Immunol 37, 173–200 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Vogelstein B et al. Cancer genome landscapes. Science 339, 1546–1558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leko V & Rosenberg SA Identifying and targeting human tumor antigens for T cell-based immunotherapy of solid tumors. Cancer Cell 38, 454–472 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deniger DC et al. T-cell responses to TP53 ‘hotspot’ mutations and unique neoantigens expressed by human ovarian cancers. Clin. Cancer Res 24, 5562–5573 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parkhurst MR et al. Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discov. 9, 1022–1035 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guedan S, Ruella M & June CH Emerging cellular therapies for cancer. Annu. Rev. Immunol 37, 145–171 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reiter JG et al. An analysis of genetic heterogeneity in untreated cancers. Nat. Rev. Cancer 19, 639–650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGranahan N & Swanton C Neoantigen quality, not quantity. Sci. Transl. Med 11, eaax7918 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Rosenthal R et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 567, 479–485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsiue EH-C et al. Targeting a neoantigen derived from a common TP53 mutation. Science 371, eabc8697 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heinrich MC et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): a multicentre, open-label, phase 1 trial. Lancet Oncol. 21, 935–946 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Hong DS et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 21, 531–540 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marty R et al. MHC-I genotype restricts the oncogenic mutational landscape. Cell 171, 1272–1283.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marty Pyke R et al. Evolutionary pressure against MHC class II binding cancer mutations. Cell 175, 416–428.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van den Eynden J, Jiménez-Sánchez A, Miller ML & Larsson E Lack of detectable neoantigen depletion signals in the untreated cancer genome. Nat. Genet 51, 1741–1748 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pardoll D Cancer and the immune system: basic concepts and targets for intervention. Semin. Oncol 42, 523–538 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castle JC, Uduman M, Pabla S, Stein RB & Buell JS Mutation-derived neoantigens for cancer immunotherapy. Front. Immunol 10, 1856 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Segal NH et al. Epitope landscape in breast and colorectal cancer. Cancer Res. 68, 889–892 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Thorsson V et al. The immune landscape of cancer. Immunity 48, 812–830. e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia-Garijo A, Fajardo CA & Gros A Determinants for neoantigen identification. Front. Immunol 10, 1392 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bassani-Sternberg M et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun 7, 13404 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chheda ZS et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J. Exp. Med 215, 141–157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narayan R et al. Acute myeloid leukemia immunopeptidome reveals HLA presentation of mutated nucleophosmin. PLoS ONE 14, e0219547 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Q et al. Direct detection and quantification of neoantigens. Cancer Immunol. Res 7, 1748–1754 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalaora S et al. Combined analysis of antigen presentation and T-cell recognition reveals restricted immune responses in melanoma. Cancer Discov. 8, 1366–1375 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arnaud M et al. Biotechnologies to tackle the challenge of neoantigen identification. Curr. Opin. Biotechnol 65, 52–59 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Gerber H-P, Sibener LV, Lee LJ & Gee MH Identification of antigenic targets. Trends Cancer 6, 299–318 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Sharkey MS, Lizée G, Gonzales MI, Patel S & Topalian SL CD4+ T-cell recognition of mutated B-RAF in melanoma patients harboring the V599E mutation. Cancer Res. 64, 1595–1599 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto TN, Kishton RJ & Restifo NP Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat. Med 25, 1488–1499 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Jaigirdar A, Rosenberg SA & Parkhurst M A high-avidity WT1-reactive T-cell receptor mediates recognition of peptide and processed antigen but not naturally occurring WT1-positive tumor cells. J. Immunother 39, 105–116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tran E et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 350, 1387–1390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith KN et al. Persistent mutant oncogene specific T cells in two patients benefitting from anti-PD-1. J. Immunother. Cancer 7, 40 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran E et al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med 375, 2255–2262 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen F et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J. Clin. Invest 129, 2056–2070 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Veatch JR et al. Tumor-infiltrating BRAFV600E-specific CD4+ T cells correlated with complete clinical response in melanoma. J. Clin. Invest 128, 1563–1568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clark RE et al. Direct evidence that leukemic cells present HLA-associated immunogenic peptides derived from the BCR–ABL b3a2 fusion protein. Blood 98, 2887–2893 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Van der Lee DI et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J. Clin. Invest 129, 774–785 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biernacki MA et al. CBFB–MYH11 fusion neoantigen enables T cell recognition and killing of acute myeloid leukemia. J. Clin. Invest 130, 5127–5141 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie G et al. CAR-T cells targeting a nucleophosmin neoepitope exhibit potent specific activity in mouse models of acute myeloid leukaemia. Nat. Biomed. Eng 10.1038/s41551-020-00625-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Douglass J et al. Bispecific antibodies targeting mutant RAS neoantigens. Sci. Immunol 6, eabd5515 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gjertsen MK, Bjorheim J, Saeterdal I, Myklebust J & Gaudernack G Cytotoxic CD4+ and CD8+ T lymphocytes, generated by mutant p21-ras (12VAL) peptide vaccination of a patient, recognize 12VAL-dependent nested epitopes present within the vaccine peptide and kill autologous tumour cells carrying this mutation. Int. J. Cancer 72, 784–790 (1997). [DOI] [PubMed] [Google Scholar]
- 42.Malekzadeh P et al. Antigen experienced T cells from peripheral blood recognize p53 neoantigens. Clin. Cancer Res 26, 1267–1276 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tubb VM et al. Isolation of T cell receptors targeting recurrent neoantigens in hematological malignancies. J. Immunother. Cancer 6, 70 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gros A et al. Recognition of human gastrointestinal cancer neoantigens by circulating PD-1+ lymphocytes. J. Clin. Invest 129, 4992–5004 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang QJ et al. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol. Res 4, 204–214 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lo W et al. Immunologic recognition of a shared p53 mutated neoantigen in a patient with metastatic colorectal cancer. Cancer Immunol. Res 7, 534–543 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strønen E et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 352, 1337–1341 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Calis JJA, de Boer RJ & Keşmir C Degenerate T-cell recognition of peptides on MHC molecules creates large holes in the T-cell repertoire. PLoS Comput. Biol 8, e1002412 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shao XM et al. High-throughput prediction of MHC class I and II neoantigens with MHCnuggets. Cancer Immunol. Res 8, 396–408 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bulik-Sullivan B et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol 37, 55–63 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Sarkizova S et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol 38, 199–209 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abelin JG et al. Defining HLA-II ligand processing and binding rules with mass spectrometry enhances cancer epitope prediction. Immunity 51, 766–779.e17 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Vita R et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 43, D405–D412 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vizcaíno JA et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shao W et al. The SysteMHC Atlas project. Nucleic Acids Res. 46, D1237–D1247 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hundal J et al. pVACtools: a computational toolkit to identify and visualize cancer neoantigens. Cancer Immunol. Res 8, 409–420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wells DK et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell 183, 818–834.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jappe EC et al. Thermostability profiling of MHC-bound peptides: a new dimension in immunopeptidomics and aid for immunotherapy design. Nat. Commun 11, 6305 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bentzen AK et al. T cell receptor fingerprinting enables in-depth characterization of the interactions governing recognition of peptide–MHC complexes. Nat. Biotechnol 36, 1191–1196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang S-Q et al. High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat. Biotechnol 36, 1156–1159 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peng S et al. Sensitive detection and analysis of neoantigen-specific T cell populations from tumors and blood. Cell Rep. 28, 2728–2738.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moritz A et al. High-throughput peptide–MHC complex generation and kinetic screenings of TCRs with peptide-receptive HLA-A*02:01 molecules. Sci. Immunol 4, eaav0860 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Saini SK et al. Empty peptide-receptive MHC class I molecules for efficient detection of antigen-specific T cells. Sci. Immunol 4, eaau9039 (2019). [DOI] [PubMed] [Google Scholar]
- 64.Overall SA et al. High throughput pMHC-I tetramer library production using chaperone-mediated peptide exchange. Nat. Commun 11, 1909 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zaretsky JM et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med 375, 819–829 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sánchez-Paulete AR et al. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann. Oncol 28, xii44–xii55 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Marino F et al. Biogenesis of HLA ligand presentation in immune cells upon activation reveals changes in peptide length preference. Front. Immunol 11, 1981 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Purcell AW, Ramarathinam SH & Ternette N Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc 14, 1687–1707 (2019). [DOI] [PubMed] [Google Scholar]
- 69.Klatt MG et al. Solving an MHC allele-specific bias in the reported immunopeptidome. JCI Insight 5, e141264 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cafri G et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Invest 130, 5976–5988 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bocchia M et al. Complete molecular response in CML after p210 BCR–ABL1-derived peptide vaccination. Nat. Rev. Clin. Oncol 7, 600–603 (2010). [DOI] [PubMed] [Google Scholar]
- 72.Chatani PD & Yang JC Mutated RAS: targeting the ‘untargetable’ with T cells. Clin. Cancer Res 26, 537–544 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Comoli P et al. BCR–ABL-specific T-cell therapy in Ph+ ALL patients on tyrosine-kinase inhibitors. Blood 129, 582–586 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Veatch JR et al. Endogenous CD4+ T cells recognize neoantigens in lung cancer patients, including recurrent oncogenic KRAS and ERBB2 (Her2) driver mutations. Cancer Immunol. Res 7, 910–922 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roth TL et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559, 405–409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Querques I et al. A highly soluble Sleeping Beauty transposase improves control of gene insertion. Nat. Biotechnol 37, 1502–1512 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liddy N et al. Monoclonal TCR-redirected tumor cell killing. Nat. Med 18, 980–987 (2012). [DOI] [PubMed] [Google Scholar]
- 78.Lowe KL et al. Novel TCR-based biologics: mobilising T cells to warm ‘cold’ tumours. Cancer Treat. Rev 77, 35–43 (2019). [DOI] [PubMed] [Google Scholar]
- 79.Lu Y-C et al. An efficient single-cell RNA-seq approach to identify neoantigen-specific T cell receptors. Mol. Ther 26, 379–389 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dijkstra KK et al. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 174, 1586–1598. e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Paria BC et al. Rapid identification and evaluation of neoantigen-reactive T-cell receptors from single cells. J. Immunother 10.1097/CJI.0000000000000342 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Spindler MJ et al. Massively parallel interrogation and mining of natively paired human TCRαβ repertoires. Nat. Biotechnol 38, 609–619 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Goebeler M-E & Bargou RC T cell-engaging therapies—BiTEs and beyond. Nat. Rev. Clin. Oncol 17, 418–434 (2020). [DOI] [PubMed] [Google Scholar]
- 84.MacKay M et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat. Biotechnol 38, 233–244 (2020). [DOI] [PubMed] [Google Scholar]
- 85.Skora AD et al. Generation of MANAbodies specific to HLA-restricted epitopes encoded by somatically mutated genes. Proc. Natl Acad. Sci. USA 112, 9967–9972 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Miller MS et al. An engineered antibody fragment targeting mutant β-catenin via major histocompatibility complex I neoantigen presentation. J. Biol. Chem 294, 19322–19334 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dao T et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat. Biotechnol 33, 1079–1086 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chang AY et al. A therapeutic T cell receptor mimic antibody targets tumor-associated PRAME peptide/HLA-I antigens. J. Clin. Invest 127, 2705–2718 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ahmed M et al. TCR-mimic bispecific antibodies targeting LMP2A show potent activity against EBV malignancies. JCI Insight 3, e97805 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Low L, Goh A, Koh J, Lim S & Wang C-I Targeting mutant p53-expressing tumours with a T cell receptor-like antibody specific for a wild-type antigen. Nat. Commun 10, 5382 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sharma P, Harris DT, Stone JD & Kranz DM T-cell receptors engineered de novo for peptide specificity can mediate optimal T-cell activity without self cross-reactivity. Cancer Immunol. Res 7, 2025–2035 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Riley TP et al. T cell receptor cross-reactivity expanded by dramatic peptide–MHC adaptability. Nat. Chem. Biol 14, 934–942 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gejman RS et al. Identification of the targets of T-cell receptor therapeutic agents and cells by use of a high-throughput genetic platform. Cancer Immunol. Res 8, 672–684 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ataie N et al. Structure of a TCR mimic antibody with target predicts pharmacogenetics. J. Mol. Biol 428, 194–205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hellman LM et al. Improving T cell receptor on-target specificity via structure-guided design. Mol. Ther 27, 300–313 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Holland CJ et al. Specificity of bispecific T cell receptors and antibodies targeting peptide–HLA. J. Clin. Invest 130, 2673–2688 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sim MJW et al. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc. Natl Acad. Sci. USA 117, 12826–12835 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wu D, Gallagher DT, Gowthaman R, Pierce BG & Mariuzza RA Structural basis for oligoclonal T cell recognition of a shared p53 cancer neoantigen. Nat. Commun 11, 2908 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hu Z et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat. Med 27, 515–525 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Blass E & Ott PA Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol 18, 215–229 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rosenberg SA, Yang JC & Restifo NP Cancer immunotherapy: moving beyond current vaccines. Nat. Med 10, 909–915 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Klebanoff CA, Acquavella N, Yu Z & Restifo NP Therapeutic cancer vaccines: are we there yet? Immunol. Rev 239, 27–44 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vormehr M, Türeci Ö & Sahin U Harnessing tumor mutations for truly individualized cancer vaccines. Annu. Rev. Med 70, 395–407 (2019). [DOI] [PubMed] [Google Scholar]
- 104.Mehta NK et al. Pharmacokinetic tuning of protein–antigen fusions enhances the immunogenicity of T-cell vaccines. Nat. Biomed. Eng 4, 636–648 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ott PA et al. A phase Ib trial of personalized neoantigen therapy plus anti-PD-1 in patients with advanced melanoma, non-small cell lung cancer, or bladder cancer. Cell 183, 347–362.e24 (2020). [DOI] [PubMed] [Google Scholar]
- 106.Sahin U et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 585, 107–112 (2020). [DOI] [PubMed] [Google Scholar]
- 107.Romero P et al. The Human Vaccines Project: a roadmap for cancer vaccine development. Sci. Transl. Med 8, 334ps9 (2016). [DOI] [PubMed] [Google Scholar]
- 108.Türeci Ö et al. Challenges towards the realization of individualized cancer vaccines. Nat. Biomed. Eng 2, 566–569 (2018). [DOI] [PubMed] [Google Scholar]
- 109.Van Poelgeest MIE et al. Vaccination against oncoproteins of HPV16 for noninvasive vulvar/vaginal lesions: lesion clearance is related to the strength of the T-cell response. Clin. Cancer Res 22, 2342–2350 (2016). [DOI] [PubMed] [Google Scholar]
- 110.Schumacher T et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 512, 324–327 (2014). [DOI] [PubMed] [Google Scholar]
- 111.Pan J et al. Immunoprevention of KRAS-driven lung adenocarcinoma by a multipeptide vaccine. Oncotarget 8, 82689–82699 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Morrison AH, Byrne KT & Vonderheide RH Immunotherapy and prevention of pancreatic cancer. Trends Cancer 4, 418–428 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Weber EW, Maus MV & Mackall CL The emerging landscape of immune cell therapies. Cell 181, 46–62 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rafiq S, Hackett CS & Brentjens RJ Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol 17, 147–167 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Depil S, Duchateau P, Grupp SA, Mufti G & Poirot L ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov 19, 185–199 (2020). [DOI] [PubMed] [Google Scholar]
- 116.Liu E et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med 382, 545–553 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mo F et al. Engineered off-the-shelf therapeutic T cells resist host immune rejection. Nat. Biotechnol 39, 56–63 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Smith TT et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol 12, 813–820 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Agarwal S et al. In vivo generation of CAR T cells selectively in human CD4+ lymphocytes. Mol. Ther 28, 1783–1794 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Roybal KT & Lim WA Synthetic immunology: hacking immune cells to expand their therapeutic capabilities. Annu. Rev. Immunol 35, 229–253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lynn RC et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yamamoto TN et al. T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J. Clin. Invest 129, 1551–1565 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Stadtmauer EA et al. CRISPR-engineered T cells in patients with refractory cancer. Science 367, eaba7365 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Davenport AJ et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA 115, E2068–E2076 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Roda-Navarro P & Álvarez-Vallina L Understanding the spatial topology of artificial immunological synapses assembled in T cell-redirecting strategies: a major issue in cancer immunotherapy. Front. Cell Dev. Biol 7, 370 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Skokos D et al. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Sci. Transl. Med 12, eaaw7888 (2020). [DOI] [PubMed] [Google Scholar]
- 127.Salter AI et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal 11, eaat6753 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ramello MC et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci. Signal 12, eaap9777 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu Y et al. Chimeric STAR receptors using TCR machinery mediate robust responses against solid tumors. Sci. Transl. Med 13, eabb5191 (2021). [DOI] [PubMed] [Google Scholar]
- 130.Harris DT et al. Comparison of T cell activities mediated by human TCRs and CARs that use the same recognition domains. J. Immunol 200, 1088–1100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gudipati V et al. Inefficient CAR-proximal signaling blunts antigen sensitivity. Nat. Immunol 21, 848–856 (2020). [DOI] [PubMed] [Google Scholar]
- 132.Wu L, Wei Q, Brzostek J & Gascoigne NRJ Signaling from T cell receptors (TCRs) and chimeric antigen receptors (CARs) on T cells. Cell. Mol. Immunol 17, 600–612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fry TJ et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med 24, 20–28 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bossi G, Buisson S, Oates J, Jakobsen BK & Hassan NJ ImmTAC-redirected tumour cell killing induces and potentiates antigen cross-presentation by dendritic cells. Cancer Immunol. Immunother 63, 437–448 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wu T et al. Quantification of epitope abundance reveals the effect of direct and cross-presentation on influenza CTL responses. Nat. Commun 10, 2846 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Huang J et al. A single peptide–major histocompatibility complex ligand triggers digital cytokine secretion in CD4+ T cells. Immunity 39, 846–857 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nerreter T et al. Super-resolution microscopy reveals ultra-low CD19 expression on myeloma cells that triggers elimination by CD19 CAR-T. Nat. Commun 10, 3137 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pillai V et al. CAR T-cell therapy is effective for CD19-dim B-lymphoblastic leukemia but is impacted by prior blinatumomab therapy. Blood Adv. 3, 3539–3549 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Majzner RG et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discov. 10, 702–723 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Stone JD, Aggen DH, Schietinger A, Schreiber H & Kranz DM A sensitivity scale for targeting T cells with chimeric antigen receptors (CARs) and bispecific T-cell engagers (BiTEs). Oncoimmunology 1, 863–873 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Deng Q et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med 26, 1878–1887 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nobles CL et al. CD19-targeting CAR T cell immunotherapy outcomes correlate with genomic modification by vector integration. J. Clin. Invest 130, 673–685 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sheih A et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat. Commun 11, 219 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tran E et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zacharakis N et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med 24, 724–730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Keskin DB et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hilf N et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 565, 240–245 (2019). [DOI] [PubMed] [Google Scholar]
- 148.Platten M et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 10.1038/s41586-021-03363-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Linnemann C et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med 21, 81–85 (2015). [DOI] [PubMed] [Google Scholar]
- 150.Germano G et al. CD4 T cell dependent rejection of beta 2 microglobulin null mismatch repair deficient tumors. Cancer Discov. 10.1158/2159-8290.CD-20-0987 (2021). [DOI] [PubMed] [Google Scholar]
- 151.Oh DY et al. Intratumoral CD4+ T cells mediate anti-tumor cytotoxicity in human bladder cancer. Cell 181, 1612–1625.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hwang MS et al. Targeting loss of heterozygosity for cancer-specific immunotherapy. Proc. Natl Acad. Sci. USA 118, e2022410118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gonzalez-Galarza FF et al. Allele Frequency Net Database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 48, D783–D788 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Maiers M, Gragert L & Klitz W High-resolution HLA alleles and haplo types in the United States population. Hum. Immunol 68, 779–788 (2007). [DOI] [PubMed] [Google Scholar]
- 155.Somasundaram R et al. Human leukocyte antigen-A2-restricted CTL responses to mutated BRAF peptides in melanoma patients. Cancer Res. 66, 3287–3293 (2006). [DOI] [PubMed] [Google Scholar]
- 156.Malekzadeh P et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Invest 129, 1109–1114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Linard B et al. A ras-mutated peptide targeted by CTL infiltrating a human melanoma lesion. J. Immunol 168, 4802–4808 (2002). [DOI] [PubMed] [Google Scholar]
- 158.Andersen MH et al. Immunogenicity of constitutively active V599EBRaf. Cancer Res. 64, 5456–5460 (2004). [DOI] [PubMed] [Google Scholar]
- 159.Cancer Genome Atlas Research Network et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet 45, 1113–1120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data for The Cancer Genome Atlas mutation frequencies used in the analyses presented in Fig. 3 and Table 2 are available from the National Cancer Institute Genomics Data Commons (https://gdc.cancer.gov/). Data for the HLA frequencies used in the analyses presented in Tables 1 and 2 and Supplementary Table 1 are available from the Allele Frequency Net Database (http://www.allelefrequencies.net/) and National Marrow Donor Program (https://bioinformatics.bethematchclinical.org/hla-resources/haplotype-frequencies/high-resolution-hla-alleles-and-haplotypes-in-the-us-population/). Data for cancer incidence used in the analyses presented in Fig. 3 and Table 2 are available from the National Cancer Institute Surveillance, Epidemiology, and End Results Program (https://seer.cancer.gov/statfacts/html/common.html). Data for ethnicity representation in the United States used in the analyses presented in Tables 1 and 2 and Supplementary Table 1 are available from the United States Census Bureau (https://www.census.gov/quickfacts/fact/table/US/PST045219).