

Abstract
Fracture risk is increased in patients with type 2 diabetes mellitus (T2DM). In addition, these patients sustain fractures despite having higher levels of areal bone mineral density, as measured by dual-energy X-ray absorptiometry, than individuals without T2DM. Thus, additional factors such as alterations in bone quality could have important roles in mediating skeletal fragility in patients with T2DM. Although the pathogenesis of increased fracture risk in T2DM is multifactorial, impairments in bone material properties and increases in cortical porosity have emerged as two key skeletal abnormalities that contribute to skeletal fragility in patients with T2DM. In addition, indices of bone formation are uniformly reduced in patients with T2DM, with evidence from mouse studies published over the past few years linking this abnormality to accelerated skeletal ageing, specifically cellular senescence. In this Review, we highlight the latest advances in our understanding of the mechanisms of skeletal fragility in patients with T2DM and suggest potential novel therapeutic approaches to address this problem.
Type 2 diabetes mellitus (T2DM) is increasing in incidence and prevalence around the world1. Although considerable attention has been appropriately focused on the well-recognized complications of T2DM, including retinopathy, nephropathy, neuropathy and vascular disease, data are now accumulating that warrant skeletal fragility being added to the list of known diabetic complications. Fracture risk is clearly increased in patients with T2DM2. However, numerous studies have demonstrated that areal bone mineral density (BMD) as measured by dual-energy X-ray absorptiometry (DXA) is preserved, or even elevated, in patients with T2DM relative to individuals without T2DM2–4. In addition to elevated fracture risk, patients with T2DM have increased morbidity following a fracture compared with patients with a fracture but without T2DM5. Further complicating the issue is the observation that the most widely used fracture risk assessment tool (FRAX)6 underestimates fracture risk in patients with T2DM4. This observation indicates that additional factors beyond BMD and the risk factors for fracture included in FRAX (prior fragility fracture, parental history of hip fracture, smoking, glucocorticoid use, rheumatoid arthritis and alcohol consumption) probably contribute to increased skeletal fragility in patients with T2DM.
The topic of diabetic skeletal fragility in type 1 diabetes mellitus and T2DM was extensively reviewed in Nature Reviews Endocrinology in 2017 by Napoli and colleagues2. As such, the goal of this article is to provide an update with new information that has been published since that review. Furthermore, we focus on specific areas emerging as key to the pathogenesis of skeletal fragility in these patients and potential therapeutic approaches to manage increased fracture risk in patients with T2DM. In addition, we summarize the evidence that skeletal fragility should now be included in the list of well-recognized diabetic complications, given shared mechanisms across the different complications of T2DM. For this Review, we selected published papers based on the authors’ knowledge of the literature as well as PubMed searches using ‘diabetes’ and ‘bone’ as search keywords, focusing largely on papers published since the review by Napoli and colleagues2.
Epidemiology of fracture risk in T2DM
Evidence for increased fracture risk in T2DM.
The clinical importance of fragility fractures in patients with T2DM has considerably increased worldwide, as increasing life expectancy in people with T2DM has led to rapid growth in the number of ageing patients with T2DM7. In 2008, we reported a population-based study of 700 residents with T2DM in Olmsted County, Minnesota, USA, with 23,236 person-years of follow-up, who experienced 1,369 fractures8. Fracture risk was elevated (standardized incidence ratio (SIR) 1.3, 95% CI 1.2–1.4) compared with residents without T2DM. Moreover, fractures were increased among patients with neuropathy (HR 1.3, 95% CI 1.1–1.6) and those on insulin (HR 1.3, 95% CI 1.1–1.5), but the risk was reduced among users of biguanides (HR 0.7, 95% CI 0.6–0.96)8. These findings are consistent with a meta-analysis published in 2020 that reported data from 17,571,738 participants with 319,652 hip fractures and 2,978,487 participants with 181,228 non-vertebral fractures9. Patients with T2DM had an elevated risk of fracture at the hip (relative risk (RR) 1.33, 95% CI 1.19–1.49) and non-vertebral sites (RR 1.19, 95% CI 1.11–1.28) compared with participants without T2DM. In addition, a long duration of T2DM and insulin use were independently associated with an increased risk of hip fracture9.
Out of all fracture sites, the risk of hip fractures has been most consistently increased in T2DM3,10,11. Interestingly, a large-scale, retrospective, longitudinal, nationwide population-based cohort study of 5,761,785 individuals in the Republic of Korea confirmed that the hazard ratios of hip fractures were slightly increased in people with prediabetes compared with people without T2DM (HR 1.032, 95% CI 1.009–1.056)12. Further progressive increases in hip fracture risk were observed in those with newly diagnosed T2DM (meaning individuals diagnosed at the time of the initial clinic visit for the study and without a prior history of T2DM) (HR 1.168, 95% CI 1.113–1.225), T2DM of less than 5 years’ duration (HR 1.543, 95% CI 1.495–1.592) and T2DM of greater than 5 years’ duration (HR 2.105, 95% CI 2.054–2.157). The secular trend of risk of hip fracture increasing according to the duration of T2DM was consistent in both sexes and all age groups. Moreover, a meta-analysis evaluating fractures in patients with diabetes mellitus showed an increased risk of total, hip, upper arm and ankle fractures: patients who had type 1 diabetes mellitus had a greater risk than those with T2DM13. Another meta-analysis focusing on peripheral fractures found an increased risk of ankle fractures (RR 1.30, 95% CI 1.15–1.48) but a decreased risk of wrist fractures (RR 0.85, 95% CI 0.77–0.95) in patients with T2DM14.
The most recent meta-analysis from 2020 indicated a decrease in prevalence (OR 0.84, 95% CI 0.70–0.95) but an increase in incidence (OR 1.35, 95% CI 1.27–1.44)5 of vertebral fracture in patients with T2DM compared with control individuals without diabetes mellitus. Furthermore, individuals with both T2DM and vertebral fracture have increased mortality compared with individuals without T2DM or vertebral fracture (HR 2.11, 95% CI 1.72–2.59) or with vertebral fracture alone (HR 1.84, 95% CI 1.49–2.28), and a borderline increased risk compared with individuals with T2DM alone (HR 1.23, 95% CI 0.99–1.52). In addition, a paper published in 2020 reported an Italian nationwide study of 59,950 women of whom 5.2% had diabetes mellitus (predominantly T2DM) and noted an association between diabetes mellitus and any fracture (OR 1.3, 95% CI 1.1–1.4, and OR 1.3, 95% CI 1.2–1.5, for vertebral or hip fractures and non-vertebral, non-hip fractures, respectively)15. Interestingly, the prevalence of vertebral or hip fracture was higher in participants with diabetes mellitus but without obesity (OR 1.9, 95% CI 1.7–2.1) than in patients with obesity and diabetes mellitus (OR 1.5, 95% CI 1.3–1.8), suggesting that obesity might be partially protective against vertebral or hip fractures in T2DM15.
However, data from the Osteoporotic Fractures in Men Study, which enrolled men aged 65 years and older with T2DM (n = 875) and men without diabetes mellitus (n = 4,679), showed that the prevalence (7.0% versus 7.7%, respectively) and incidence (4.4% versus 4.5%, respectively) of vertebral fracture were not higher in men with T2DM than in men without diabetes mellitus16. The risk of prevalent (OR, 1.05, 95% CI 0.78–1.40) or incident (OR, 1.28, 95% CI 0.81–2.00) vertebral fracture was also not higher in men with T2DM than in men without diabetes mellitus in models adjusted for age, clinic site, race, BMI and BMD. Nevertheless, the results revealed a trend for an increased risk of incident vertebral fractures by 30% in BMD-adjusted models16. Collectively, these findings raise the possibility that increased vertebral fracture risk might be more consistently present in older women with T2DM than in older men with T2DM.
The impact of glycaemic control on fracture risk in T2DM.
The association between glycaemic control and fracture risk is best described as a J-shaped relationship, as observational studies have found that both poor glycaemic control and very strict glycaemic control are associated with increased fracture risk17–20 This relationship is probably best explained by hypoglycaemia resulting from very strict glycaemic control21–23. For example, in a study published in 2020, 4,706 Japanese patients with T2DM (2,755 men and 1,951 postmenopausal women; mean age 66 years) were followed prospectively (median of 5.3 years; follow-up rate 97.6%) and stratified by severe hypoglycaemia status and glycaemic control24. Fractures occurred in 662 participants (249 men and 413 women). FIGURE 1 shows the age-adjusted and sex-adjusted incidence rates (expressed per 1,000 person-years) based on multiple or single episodes of hypoglycaemia as well as HbA1c measurements. Having multiple episodes of hypoglycaemia was associated with a marked increase in fracture risk, and even a history of a single episode of hypoglycaemia led to an increase in fracture risk. In the absence of hypoglycaemia, fracture risk was similar in those with HbA1c values of <7%, 7% to <8%, and 8% to <9%, but increased statistically significantly in patients with HbA1c values of ≥9%24.
Fig. 1 |. Age-adjusted and sex-adjusted incidence of fractures based on the number of hypoglycaemic episodes and baseline HbA1c in a cohort of Japanese patients with T2DM.
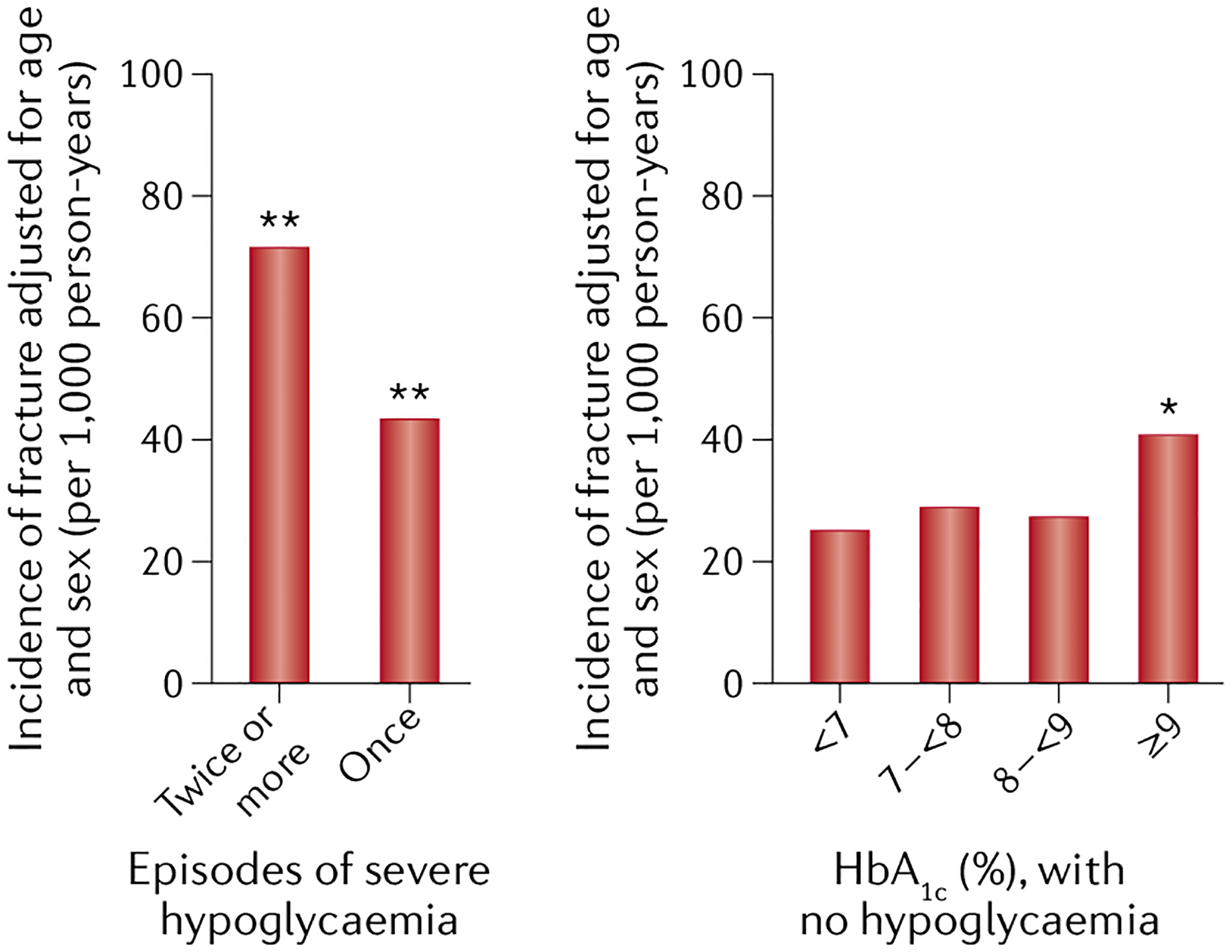
Multiple episodes of hypoglycaemia are associated with a marked increase in fracture risk, and even a history of a single episode of hypoglycaemia results in an increase in fracture risk. In the absence of hypoglycaemia, fracture risk is increased significantly in patients with HbA1c values ≥9%. *P < 0.01, **P < 0.001 versus the reference group (HbA1c <7% without severe hypoglycaemia). Adapted with permission from Komorita et al. 202024.
Another study published in 2020 confirmed the relationship between poor glycaemic control and increased fracture risk. Here, Chinese individuals aged ≥60 years with T2DM were identified from electronic health records in Hong Kong between 2008 and 2012 and observed for incident hip fractures25. A total of 83,282 participants were included, with a mean age of 71.3 ± 7.5 years, duration of diabetes mellitus of 11.7 ± 7.7 years, baseline HbA1c of 7.33 ± 1.23%, and median follow-up of 6.8 years. A mean HbA1c of ≥8.0% was associated with a 25% increase in incident hip fractures compared with a mean HbA1c of <7.0%. In addition, all HbA1c variability indices were independent predictors of incident hip fractures, with an adjusted hazard ratio of up to 1.29 (all P < 0.001) and persisted as independent predictors across groups of different intensities of glycaemic control. Thus, both poor glycaemic control and HbA1c variability across the spectrum of glycaemic control are independent positive predictors of hip fractures in patients with T2DM25.
Pathogenesis of skeletal fragility in T2DM
Clinical studies on skeletal fragility pathogenesis in T2DM.
Despite the increase in fracture risk, most studies have found that patients with T2DM have preserved, or even increased, BMD compared with control individuals without diabetes mellitus. For example, in a subset (n = 6,384) of postmenopausal women from the Women’s Health Initiative, women with T2DM consistently had higher spine and hip BMD values than women without diabetes mellitus both at baseline and over 9 years of follow-up (TABLE 1)26. A meta-analysis that included studies of women and men with T2DM showed similar findings, with high Z-scores (standard deviations from the mean) of 0.41 at the spine and 0.27 at the hip in patients with T2DM3. These findings have been subsequently confirmed and extended by perhaps the most comprehensive meta-analysis to date on the association between BMD and T2DM, which included 15 observational studies (3,437 patients with T2DM and 19,139 control individuals)27. This analysis showed that BMD in patients with T2DM was significantly higher than in participants without diabetes mellitus, with pooled mean differences of 0.04 g/cm2 (95% CI 0.02–0.05) at the femoral neck, 0.06 g/cm2 (95% CI 0.04–0.08) at the hip and 0.06 g/cm2 (95% CI 0.04–0.07) at the spine. These differences were equivalent to about a 25–50% higher standard deviation for BMD in patients with T2DM than in control individuals, depending on the skeletal site. The differences between patients with T2DM and participants without diabetes mellitus for forearm BMD were not significant, perhaps due to reduced statistical power as forearm BMD was included in fewer studies.
Table 1 |.
BMD values in women with and without T2DM
| Timepoint | Spine BMD (g/cm2) | Hip BMD (g/cm2) | ||
|---|---|---|---|---|
| Women with T2DM (n) | Women without T2DM (n) | Women with T2DM (n) | Women without T2DM (n) | |
| Baseline | 1.04 ± 0.19 (472) | 0.97 ± 0.17 (5,922) | 0.90 ± 0.16 (469) | 0.84 ± 0.14 (5,915) |
| Year 3 | 1.06 ± 0.20 (331) | 0.99 ± 0.17 (4,839) | 0.89 ± 0.16 (331) | 0.84 ± 0.13 (4,831) |
| Year 6 | 1.07 ± 0.21 (253) | 1.00 ± 0.18 (4,203) | 0.87 ± 0.16 (261) | 0.84 ± 0.13 (4,262) |
| Year 9 | 1.12 ± 0.24 (91) | 1.02 ± 0.18 (1,608) | 0.88 ± 0.17 (92) | 0.82 ± 0.13 (1,606) |
Because T2DM is associated with obesity, this meta-analysis27 also evaluated the effects of BMI and found that in general, the association between T2DM and BMD remained despite correction for BMI. The authors postulated that additional factors, including altered adipokine levels (for example, increased leptin and reduced adiponectin) and hyperinsulinaemia might potentially have mediated the effects of obesity on BMD in T2DM27.
Along with increased BMD, several studies have noted reduced bone turnover in patients with T2DM28–30. As shown in FIG. 2 from a previous study from our group29, patients with T2DM have reduced markers of bone formation (serum levels of procollagen type 1 amino-terminal propeptide (P1NP); FIG. 2a) and resorption (carboxy-terminal telopeptide of type 1 collagen (CTx); FIG. 2b). The mechanisms underlying the reduced bone turnover in patients with T2DM are unclear. However, one study30 found that bone formation and resorption markers were not reduced in participants with obesity who were insulin sensitive, but were reduced in participants with obesity who were insulin resistant and in patients with T2DM, indicating that insulin resistance contributed to the reduced bone turnover in T2DM. In addition, these investigators found that visceral adipose tissue (assessed by CT) was negatively correlated with both serum levels of P1NP (FIG. 2c) and serum levels of CTx (FIG. 2d). Thus, both insulin resistance and increased visceral adipose tissue contribute to the reduced bone formation and resorption indices in patients with T2DM, although there are probably also other factors involved.
Fig. 2 |. Bone turnover is reduced in patients with T2DM.

Serum levels of procollagen type 1 amino-terminal propeptide (P1NP) (part a) and of carboxy-terminal telopeptide of type 1 collagen (CTx) (part b) in a cohort of patients with type 2 diabetes mellitus (T2DM) compared with a cohort without diabetes mellitus, demonstrating the reduction in bone turnover observed across studies in patients with T2DM29. Correlation between visceral adipose tissue area (assessed by CT) and serum levels of P1NP (part c) and of CTx (part d) in a combined cohort of people without diabetes mellitus (including those who were lean and those with obesity) and patients with T2DM. Parts c and d adapted with permission from Tonks et al.30.
Importantly, BMD still predicts fracture risk in patients with T2DM; however, the FRAX algorithm generally underestimates fracture risk in patients with T2DM4. Although this underestimation is related, at least in part, to an increased risk of falls31, additional factors are probably also involved, leading to the search for indices of ‘bone quality’ that might be impaired in patients with T2DM. The spine trabecular bone score, which is derived from the texture of the spine DXA image, is reduced in patients with T2DM and predicts fracture risk independent of BMD32. In addition, a number of studies have utilized high-resolution peripheral quantitative CT (HR-pQCT) to evaluate the effects of T2DM on bone microarchitecture in the peripheral skeleton (radius and tibia). These studies have generally found preserved, or even improved, trabecular bone microarchitecture in patients with T2DM compared with control individuals without diabetes mellitus33–38. However some35,36,38–40, but not all29,37,41, studies have found increased cortical porosity in patients with T2DM, which independently predicts fracture risk, at least in postmenopausal women without diabetes mellitus42,43.
Impairments in bone material properties and increased cortical porosity in T2DM.
Another aspect of bone quality that might be impaired in patients with T2DM is the material property of bone. Using in vivo microindentation, our group initially reported that postmenopausal women with longstanding (>10 years) T2DM had a reduced bone material strength index (BMSi) compared with age-matched control women who did not have diabetes mellitus29. Other groups subsequently reported similar findings44,45. Of note, one study found reductions in BMSi in Black patients with T2DM, but not in white patients with T2DM as compared with race-matched subjects without T2DM46. However, in a larger cohort of patients with T2DM, we were unable to find a statistically significant difference in BMSi between patients with T2DM and control participants37.
These data suggest that although patients with T2DM might have alterations in bone quality, specifically increased cortical porosity and impaired bone material properties (that is, BMSi), heterogeneity could exist within these patients leading to the conflicting findings noted in this section. We reasoned that these skeletal abnormalities could be related to underlying diabetic complications, which might vary from cohort to cohort. Thus, we performed a comprehensive assessment of diabetic complications in a fairly large (n = 171) cohort of men ≥50 years of age and postmenopausal women with T2DM, as well as 108 control individuals without diabetes mellitus. This assessment included evaluation of urine microalbuminuria, retinopathy, detailed neuropathy testing and vascular testing37. The vascular testing included transcutaneous oxygen tension (a non-invasive measure of microvascular blood flow assessed on the dorsum of the foot) and the ankle brachial index (a measure of macrovascular blood flow). These measures, as well as skin advanced glycation end products (AGEs) assessed non-invasively using skin autofluorescence47 were then related to bone microarchitectural parameters by HR-pQCT and BMSi. AGEs are the products of irreversible, non-enzymatic reactions between proteins and sugars48,49. The long-lived and slowly turned-over proteins in bone, most notably collagens50, are particularly susceptible to these modifications, which negatively affect bone material properties.
In terms of the HR-pQCT parameters, we found that the patients with T2DM had statistically significantly higher bone volume fraction and trabecular thickness at the tibia but not the radius than the control participants, even following adjustment for age, sex and BMI37. Overall, cortical porosity was not statistically significantly different between the groups, but we found that patients with T2DM and clinically relevant microvascular disease (defined as a transcutaneous oxygen tension of ≤40 mm Hg51,52) had increased cortical porosity (+21%, P = 0.031) at the distal tibia compared with the control participants without diabetes mellitus37. As noted already, in this cohort, BMSi did not differ significantly between the groups, but skin AGEs were significantly higher (+17%, P < 0.001) in the T2DM patients than in the control participants. Interestingly, significant negative correlations were observed between BMSi and skin AGEs in both the T2DM patients (r = −0.30, P < 0.001) and the control participants (r = −0.23, P < 0.001)37.
Based on these data and previous work29,44–46, we have proposed a working model for the pathogenesis of skeletal abnormalities and increased fracture risk in patients with T2DM (FIG. 3)37. These patients generally have preserved, or even increased, BMD and trabecular bone volume fraction, which is probably related to obesity and/or hyperinsulinaemia (see section on “Mechanisms of skeletal fragility in T2DM”)53,54. However, these beneficial trabecular bone changes are negated by impaired bone quality, specifically increased accumulation of AGEs that probably contributes to impaired bone material properties, and microvascular disease that could be responsible for increased cortical porosity. The low bone turnover associated with T2DM28–30 might also contribute to reduced bone quality due to impaired microfracture repair. Collectively, the impaired bone material properties and increased cortical porosity, along with impaired microfracture repair secondary to the low bone turnover lead to skeletal fragility, which, in the setting of peripheral neuropathy and an increased propensity for falls31, results in the increased fracture risk observed in these patients despite a relative preservation of BMD. Of note, weight loss, which can be crucial in the management of T2DM, might also increase fracture risk. For example, in the Look AHEAD trial, an intensive lifestyle intervention (≥7% weight loss) in individuals with T2DM and obesity or overweight increased the risk of fragility fractures (composite of hip, pelvis or upper arm and/or shoulder) (HR 1.39, 95% CI 1.02–1.89)55. Clearly, additional human and animal studies are needed to further test and refine this model, but the model (FIG. 3) does provide a potentially useful framework for such studies.
Fig. 3 |. A working model for the pathogenesis of skeletal fragility and increased fracture risk in patients with T2DM.
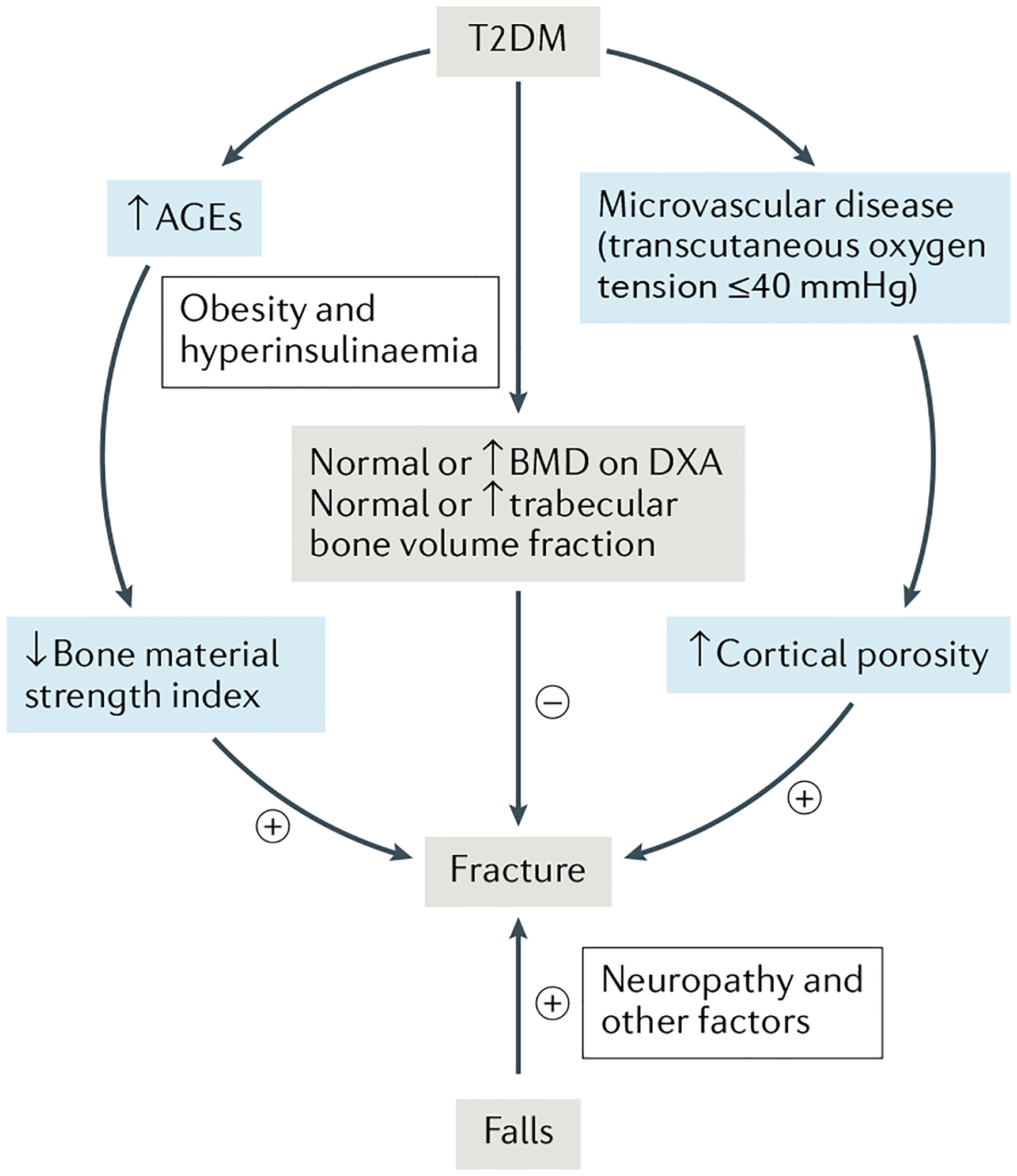
Patients with type 2 diabetes mellitus (T2DM) generally have preserved or increased bone mineral density (BMD) as measured by dual-energy X-ray absorptiometry (DXA) and trabecular bone fraction as measured by high-resolution peripheral quantitative CT (HR-pQCT), which is probably related to obesity and/or hyperinsulinaemia. However, these patients have impaired bone quality, including increased accumulation of advanced glycation end products (AGEs) in bone that leads to impaired bone material properties and microvascular disease that contributes to increased cortical porosity. Patients with T2DM also have an increased propensity for falls, which further contributes to fracture risk. +, increases fracture risk; −, decreases fracture risk. FIGURE 3 reproduced with permission from Samakkarnthai et al.37.
Mechanisms of skeletal fragility in T2DM
Overview of potential mechanisms.
A complex network of hormones, neurotransmitters and other factors are necessary to regulate bone metabolism and energy homeostasis. Mounting evidence has established that the skeleton not only regulates itself via remodelling of bone, but also has far-reaching systemic influences, for example, on whole-body energy metabolism56–58. Conversely, basic mechanisms central to energy metabolism have important roles in regulating skeletal homeostasis59,60. Precisely how these remarkable functions go awry in T2DM is still unclear and the complexity of the process is becoming increasingly apparent. As reviewed previously2,61, several multifactorial cellular and molecular mechanisms probably interact to either protect against or contribute to skeletal fragility in T2DM at various stages of the natural course of the disease. These mechanisms include, but are not limited to, effects of hyperinsulinaemia, obesity and increased bone marrow adiposity, long-term hyperglycaemia and accumulation of AGEs, inflammation, reactive oxygen species, oxidative stress, accumulation of senescent cells and the presence of diabetic complications such as microvascular disease2,62.
In the early stages of T2DM, hyperinsulinaemia is probably responsible for the preserved, or improved, BMD and trabecular bone microarchitectural parameters observed in patients with T2DM35,54,63, as insulin is osteoanabolic53,64. The benefit of hyperinsulinaemia to bone is independent of adipose tissue mass65. This early stage advantage in trabecular bone microarchitecture is not maintained at the later stages of T2DM63, when hyperinsulinaemia subsides due to β-cell failure.
The increased BMI and percentage body adiposity that is common in patients with T2DM might have beneficial effects on the skeleton due to the protective effects of greater mechanical loading. These effects are perhaps either potentiated or offset by the complex actions of adipokines (for example, adiponectin and leptin) and by the pathophysiological effects of adipose-derived pro-inflammatory cytokines61. The skeletal effects of obesity could also be dependent on adipose distribution, as depots associated with insulin resistance and the metabolic syndrome, such as visceral adipose tissue and bone marrow adipose tissue (BMAT), have been linked to increased chronic inflammation60,61. Examples of pro-inflammatory cytokines associated with increased visceral adipose tissue mass include TNF and IL-6 (REF.66). Along with other factors, these cytokines promote bone resorption by stimulating production of the osteoclastogenic cytokine RANKL67, which could have a causal role in the development of cortical porosity in uncontrolled T2DM.
Obesity in the setting of T2DM is also associated with increased BMAT in both rodents68 and humans69; however, the functional roles of this adipose depot remain incompletely understood. Although some evidence suggests that BMAT is negatively correlated with BMD in postmenopausal women with T2DM and obesity, and that women with poorly controlled diabetes mellitus have increased levels of BMAT70,71, the mechanism underlying these observations are unknown. Clearly, a better understanding of the roles of BMAT in potentially mediating skeletal fragility in T2DM is needed. Some evidence from the past few years suggests that high levels of BMAT-derived lipids both locally in the bone marrow and in the circulation generate reactive oxygen species and oxidative stress that are detrimental to the functions of stem and progenitor cells as well as to cells of the osteoblast lineage60. Furthermore, studies in rodents72 and humans73,74 have found that levels of sclerostin, a potent soluble antagonist of the bone-anabolic canonical WNT–β-catenin signalling pathway75, are increased in T2DM and correlate with BMAT accumulation76. Taken together, these mechanisms might, at least in part, explain the low bone formation rates observed in T2DM.
The role of cellular senescence in mediating skeletal fragility in T2DM.
Diverse forms of age-related stress or metabolic insults, including DNA breaks, reactive oxygen species, proteotoxic aggregates and inflammation, converge to cause a cell to enter an essentially irreversible permanent growth arrest, termed senescence77. The senescence programme is activated by cyclin-dependent kinase inhibitors, most notably p16Ink4a and p21Cip1, that antagonize the actions of cyclin-dependent kinases to halt cell proliferation and malignant transformation78,79. Senescent cells develop an altered gene expression profile that includes upregulation of senescent cell anti-apoptotic pathways80 and a senescence-associated secretory phenotype (SASP), typically consisting of pro-inflammatory cytokines, chemokines and matrix remodelling proteins81,82. Accumulation of senescent cells increases with ageing, obesity and T2DM77,83,84. In these contexts, the accumulation of senescent cells is presumably due to metabolic dysfunction, inefficient immune system removal and resistance to apoptosis85,86. The biological relevance and consequences of senescent cells are becoming evident in several models of ageing and disease77, including models of T2DM.
Obesity and T2DM are associated with the premature increased burden of senescent cells in adipose tissue87,88, pancreatic β-cells89, liver90, brain91 and bone62, which contributes to metabolic dysfunction and several accelerated ageing phenotypes77. Mechanistically, the SASP of senescent cells, characterized by increased levels of pro-inflammatory cytokines (for example, IL-6, TNF and activin A), contributes to insulin resistance in T2DM by disrupting insulin signalling, attracting macrophages that exacerbate inflammation and spreading senescence to neighbouring cells and tissues88,92–95. In addition, the consequences of T2DM (for example, glucotoxicity and lipotoxicity) can amplify the accumulation of senescent cells in multiple tissues, resulting in a pathogenic positive feedback loop83. This systemic increase in senescent cell burden contributes to the development of metabolic dysfunction and several other diabetic complications, such as cardiovascular disease, neurodegeneration, renal disease and hepatic steatosis77. These effects are exacerbated in patients with poorly controlled T2DM.
With regard to skeletal fragility, work from our group published in 2020 demonstrated accelerated osteocyte senescence and poor bone quality in an inducible obese mouse model of T2DM62. Importantly, obesity was induced in these mice during adulthood, after skeletal maturity. After 3 months of established disease, these mice display several detrimental alterations in bone quality that closely mirror those in bones from humans with T2DM, including defective cortical bone microarchitecture, reduced biomechanical strength and impaired bone material properties62. In addition, bone histomorphometry revealed lower bone formation rates in mice with T2DM than in non-diabetic mice62, again consistent with data in patients with T2DM96.
A combination of multiple key characteristics (as encouraged by a consensus article97) was used to identify senescent osteocytes in mice with T2DM. First, mRNA expression of the key mediators of senescence, p16Ink4a and p21Cip1, were found to be statistically significantly elevated in osteocyte-enriched bones of T2DM mice (FIG. 4a). Further, as shown in FIG. 4b, the senescence-associated distension of satellites assay98 (that is, large-scale unravelling of peri-centromeric satellite DNA characteristic of senescent cells), revealed that the percentage of senescent osteocytes was statistically significantly higher in bone cortices of T2DM mice than in control mice (FIG. 4c). We also measured telomere-associated foci (TAF), a specific, robust marker of senescence, in osteocytes (FIG. 4d). TAF are sites of DNA damage (γH2AX), which colocalize with telomeres97. As quantified (FIG. 4e), the percentage of TAF+ osteocytes increased in bone cortices of T2DM mice. Finally, quantitative PCR with reverse transcription was used to measure a panel of established SASP genes81,82,99 in the osteocyte-enriched samples of T2DM mice62. Interestingly, this analysis revealed a unique pro-inflammatory SASP composed predominantly of upregulated levels of matrix metalloproteinases (MMPs; that is, Mmp3, Mmp9, Mmp12 and Mmp13) (FIG. 4f). In addition, the expression of Nfkb1 (which encodes NF-κB), a downstream target of the RAGE pathway48,100 that is activated by AGEs48, was also statistically significantly elevated in osteocyte-enriched bones of T2DM mice (FIG. 4f). Thus, elevated levels of MMPs and NF-κB seem to be part of the SASP signature of senescent osteocytes unique to T2DM62. These findings establish that T2DM, in the context of obesity, causes the premature accumulation of senescent osteocytes during young adulthood, at least in a mouse model. Other bone-resident cell types, however, might also become senescent and additional key SASP factors specific to T2DM are likely to exist.
Fig. 4 |. Accelerated osteocyte senescence in T2DM.

mRNA expression of p16Ink4a and p21Cip1 in osteocyte-enriched bones (part a). Senescence-associated distension of satellites (SADS)+ osteocytes in control mice versus mice with type 2 diabetes mellitus (T2DM) (parts b, c). Arrows in part b indicate SADS (unravelling of peri-centromeric satellite DNA) in the osteocyte nucleus. Telomere-associated foci (TAF)+ osteocytes in control mice versus mice with T2DM (parts d, e). Arrows in part d indicate TAFs (yellow) and sites of DNA damage (γH2AX, green), which colocalize with telomeres (red) in the osteocyte nucleus. mRNA expression of senescence-associated secretory phenotype (SASP) markers in control mice versus mice with T2DM (part f). Collectively, these data demonstrate that T2DM, at least in a mouse model, is associated with accelerated osteocyte senescence and the secretion of a SASP that might have deleterious skeletal effects. Data are means ± standard error of the mean. *P < 0.05; **P < 0.01; ***P < 0.001. Adapted with permission from Eckhardt et al.62.
Role of AGEs.
As noted above, the production and accumulation of AGEs is an important complication of hyperglycaemia observed in patients with T2DM. Based on rodent and cadaver studies, AGEs lead to impairments in the ability of collagen to dissipate energy and a reduction in the capacity of organic and mineralized matrix to creep (deform under mechanical stress), leading to bone fracture at low levels of strain101. In addition to their direct effects on bone material properties through collagen modification, AGEs also bind to and can signal through a transmembrane protein termed receptor for AGE (RAGE, the protein product of the Ager gene) in diverse cell types throughout the body, including in the osteoclastic and osteoblastic cell lineages in bone102,103. Indeed, Nε-(1-carboxymethyl)-l-lysine is a dominant RAGE ligand in bone and has been found to accumulate in the bone and serum of mice with T2DM62,104,105.
Of note, almost all of the data regarding the accumulation of AGEs in bone are currently derived from rodent studies, as studying this issue in humans is a challenge. However, one study has quantified serum levels of AGEs (pentosidine and total AGEs) as well as AGEs from proximal femur specimens from participants with and without T2DM106. Serum levels of pentosidine or total AGEs did not differ between groups, but cortical bone levels of AGEs tended to be higher in the participants with T2DM than in those without T2DM (+21.3%, P = 0.08), whereas trabecular bone levels of AGEs were similar between the groups. Cortical or trabecular bone levels of AGEs were only weakly correlated (r = 0.28–0.39) with serum levels of AGEs. Additional human studies are needed to evaluate the accumulation of AGEs in bone in patients with T2DM and the relationship of these to AGEs in serum, urine or skin. In terms of fracture risk, a study found an association between urinary levels of pentosidine and vertebral fracture risk in patients with T2DM107. Collectively, these data demonstrate that AGEs accumulate in patients with T2DM and provide the basis for future studies of accumulated AGEs in the context of T2DM and its effects on bone material properties.
A number of intracellular signalling pathways are potentially activated through RAGE signalling (including PKC, JAK–STAT, PI3K and MAPK), many of which converge on the activation of NF-κB signalling, thereby generating an inflammatory response that contributes to T2DM48,100. This observation suggests that specific blockade of RAGE signalling might alleviate the increased inflammatory response seen in bone in patients with T2DM. Indeed, it has been demonstrated that the soluble RAGE molecule (sRAGE) inhibits HMGB1-induced inflammation108. In addition, sRAGE slowed the rate of alveolar bone loss in a diabetic model of periodontal disease109. Small-molecule inhibitors have also been shown to inhibit RAGE signalling and have anti-inflammatory effects in Alzheimer disease, neuroinflammation and cancer110,111. Some of these RAGE inhibitors have also been shown to prevent bone loss through RAGE-dependent inhibition of osteoblast and/or osteocyte apoptosis112,113. Whether blockade of RAGE signalling in bone improves bone material properties in the context of T2DM is a subject of ongoing research.
Links between the vasculature and bone.
In addition to the accumulation of AGEs and senescent cells in bone, another potential contributor to skeletal fragility in T2DM is macrovascular and microvascular disease. Histological examination of bone remodelling units, the clusters of cells responsible for the active processes of bone resorption and formation, has revealed the presence of a capillary system that provides the bone remodelling unit access to the bloodstream, nutrients and other cells involved in bone homeostasis114. The crosstalk between the vasculature and bone remodelling compartments is essential for proper bone development, normal functioning and repair following bone injury115. Disruptions in this interface can lead to impaired bone homeostasis, as is often seen in diseases such as T2DM115. This crosstalk occurs between vascular endothelial cells and other neighbouring cells that assist in the control of bone metabolism. Mesenchymal stem cells for example, have intrinsic osteogenic capacity and promote vascularization through communication with vascular endothelial cells, which occurs via pro-angiogenic factors such as vascular endothelial growth factors, insulin-like growth factors, platelet-derived growth factors and fibroblast growth factors116,117. Crosstalk of vascular endothelial cells also exists with other cell types, such as periosteum-derived progenitors118, adipose-derived progenitors119, stromal cells120, macrophages121 and pericytes122. The interactions between endothelial cells and these auxiliary cells promote angiogenesis and bone mineralization, leading to normal and healthy bone metabolism.
The appearance of vascular disease in patients with T2DM is a common diabetic complication, due to hyperglycaemia damaging blood vessels123. Another common feature in diabetic bone disease is increased cortical porosity, which has been found in patients with T2DM in several studies35,36,39,40. In our 2020 study37, we found that our cohort of patients with T2DM and clinically relevant microvascular disease (defined as exhibiting a lowered oxygen tension, as described previously) had increased cortical porosity at the distal tibia. The causal link between lowered oxygen tension and cortical porosity in T2DM is unknown, but impaired crosstalk between the bone vasculature (for example, vascular endothelial cells) and osteogenic cell precursors could be a potential mechanism. In this model, the decreased vascularization, and therefore the decreased number of vascular endothelial cells, would lead to reduced signalling for the recruitment and differentiation of osteogenic precursor cells. This reduced signalling would inhibit the restoration of bone formation at sites of intracortical remodelling, leading to the appearance of a cortical pore that cannot be filled. However, further studies are clearly needed to fully understand the link between the vasculature and cortical porosity.
FIGURE 5 builds on FIG. 3 and presents a working model of the cellular and molecular changes leading to impaired bone quality and skeletal fragility in patients with T2DM, focusing specifically on AGEs, cellular senescence and microvascular disease. Multiple key mechanisms and effects, stemming from the accumulation of AGEs and senescent cells, converge to cause accelerated ageing of several tissues, including bone. Over long periods, these mechanisms (and probably additional mechanisms) contribute to established skeletal complications in T2DM, such as low bone turnover and abnormal collagen and mineralization, which leads to impaired bone material properties. In addition, the microvascular disease associated with T2DM probably contributes to compromised cortical bone microarchitecture, ultimately increasing fracture risk124. Given that these mechanisms are linked and overlap, interventions that target one could in theory ameliorate others and have beneficial influences on multiple systems and physiological functions in T2DM. Therefore, basic mechanisms central to energy metabolism and accelerated ageing, such as cellular senescence and the RAGE pathway, are potential therapeutic targets for interventions that could alleviate or partially treat T2DM and its complications, including skeletal fragility.
Fig. 5 |. Emerging pathophysiological mechanisms at the nexus of complications related to T2DM, including skeletal fragility.

Microvascular disease, including impaired vascular–bone crosstalk contributes to increased cortical porosity and reduced bone quality. Accumulation of advanced glycation end products (AGEs) and cellular senescence activate the senescence-associated secretory phenotype (SASP), which contributes to reduced bone formation and further impairments in bone quality. Coloured circles represent AGEs and coloured triangles represent the SASP. CML, Nε-(carboxymethyl)lysine; MMPs, matrix metalloproteinases. Adapted with permission from Eckhardt et al. 202062.
Treatment of skeletal fragility in T2DM
Currently, limited evidence exists for therapeutic interventions to prevent or treat skeletal fragility in patients with T2DM. However, similar to individuals without T2DM, lifestyle measures, including appropriate physical activity, smoking cessation, avoidance of alcohol abuse and assuring adequate dietary calcium and vitamin D intake, should be implemented as the mainstay of osteoporosis prevention and treatment in patients with T2DM125. As noted already, evidence from the Look AHEAD trial shows that intensive lifestyle intervention leading to considerable weight loss in individuals with T2DM and obesity or overweight increases the risk of fragility fractures55. Thus, a fracture prevention programme, especially weight-bearing exercise, to balance the potential negative effects of weight loss in patients with T2DM should be considered.
Patients with T2DM have an increased risk of falls126,127, so risk factors for falls, including visual impairment due to diabetic retinopathy, peripheral neuropathy, poor balance, sarcopenia, cardiovascular diseases, sequalae of stroke (neurological and cognitive impairment) and hypoglycaemic events, should be included in the evaluation of fracture risk in patients with T2DM23,127. Fall risk should be rigorously assessed and, where appropriate, preventive measures instituted. A systematic review demonstrated that fall prevention programmes in patients with T2DM128 and peripheral neuropathy129 improved balance without the risk of adverse effects in older adults (≥60 years of age) with T2DM.
Impact of diabetes mellitus medications on fracture risk.
As discussed already, maintenance of good glycaemic control and avoidance of hypoglycaemia should reduce fracture risk. Although no prospective trials have been performed on the effects of diabetes mellitus medications on bone fragility, evidence from epidemiological studies and adverse effects surveillance in clinical trials in diabetes mellitus have provided important insights into the potentially beneficial or adverse effects of diabetes mellitus medications on fracture risk (TABLE 2)2,61. Insulin use has also been associated with an increased risk of fractures130; however, whether insulin use is a marker of the severity and/or duration of the disease or the occurrence of hypoglycaemic events that precipitate falls is uncertain. The choice of insulin formulation might affect fracture risk, as long-standing therapy with insulin glargine was associated with a lower fracture risk than treatment with NPH insulin131; whether this difference was related to a reduced frequency of hypoglycaemia with insulin glargine remains to be determined. However, another study found no differences between types of insulin and risk of fractures132.
Table 2 |.
Effects of hypoglycaemic agents on fracture risk in T2DM
| Medication | Risk of fracture | Remarks |
|---|---|---|
| Metformin | No effect154, decrease8,155–157 | Sub-analysis of the TECOS study (testing the cardiovascular safety of sitagliptin) showed that patients treated with metformin have a reduced fracture risk157 |
| Thiazolidinedione | Increase135,156 | Sub-analysis of data from the ACCORD trial indicated that risk of fractures decreases with time after thiazolidinedione discontinuation158 |
| Sulfonylurea | Decrease155, no effect154, increase156 | Sulfonylureas seem not to directly increase fracture risk in patients with T2DM; however, fracture risk might increase due to increased risk of falls secondary to hypoglycaemia156,159 |
| Glucagon-like peptide 1 agonist | No effect160, decrease134 | Liraglutide and lixisenatide were associated with decreased fracture events134 |
| Dipeptidyl peptidase 4 inhibitor | No effect160, decrease161 | Results of the cardiovascular outcome trials with saxagliptin (SAVOR-TIMI 54)162 and sitagliptin (TECOS)157 show the safety (but also lack of benefit) of dipeptidyl peptidase 4 inhibitor treatment regarding the risk of fractures |
| Sodium–glucose cotransporter 2 inhibitor | No effect137,160, increase136,163 | Canagliflozin increases the risk of hip fractures136; dapagliflozin increases fractures in patients with T2DM who have moderate renal failure163 |
| Insulin | Increase132,156 | Insulin use might increase fracture risk due to hypoglycaemia-induced falls156 |
T2DM, type 2 diabetes mellitus.
Medications with a neutral or favourable effect on bone metabolism, such as metformin and incretin-based treatments, should be the preferred treatment in patients with T2DM at increased fracture risk based on BMD testing and/or FRAX scores133. Interestingly, the latest meta-analysis of GLP1 receptor antagonists, which included 38 studies with 39,795 patients (241 incident fractures, 107 in the GLP1 receptor antagonist group and 134 in the control group), found that when compared with placebo and other antidiabetic drugs, liraglutide and lixisenatide were associated with a statistically significant reduction in the risk of fractures and the beneficial effects were dependent on the duration of treatment134. By contrast, use of thiazolidinediones should be avoided as these drugs are associated with an increase in fracture risk135. SGLT2 inhibitors, specifically canagliflozin, should also probably be used with caution in patients with T2DM as this drug has also been associated with increased fracture risk136 However, the most recent meta-analysis (from 2019) which included 27 eligible randomized controlled trials with 20,895 participants with T2DM, with an average study duration of 64.22 weeks, did not find an increased risk of fracture in patients with T2DM treated with SGLT2 inhibitors137. More long-term follow-up data are needed to clarify the impact of SGLT2 inhibitors on fracture risk.
In addition, concomitant medications related to comorbidities should also be carefully considered for their possible effects on fracture risk in patients with T2DM; note that much of the data cited in this section has come from studies largely in patients without diabetes mellitus. In terms of antihypertensive medications, in the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack trial, thiazide diuretic users had a lower risk of fracture in adjusted analyses (HR 0.79, 95% CI 0.63–0.98) than users of calcium channel blockers or angiotensin-converting enzyme inhibitors138. In a large Swedish cohort study published in 2020, loop diuretics were associated with an increased risk of hip fracture (HR 1.23, 95% CI 1.11–1.35). No statistically significant associations were found between the risk of hip fracture and current exposure to β-blockers, calcium channel blockers, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers or aldosterone receptor blockers139. An updated meta-analysis of randomized controlled trials revealed no significant effect of statin treatment on the risk of fractures (RR 1.00, 95% CI 0.87–1.15)140. However, antidepressants and drugs for treatment of diabetic neuropathy should be prescribed cautiously because meta-analyses of observational studies have shown increased risk of fractures among users of selective serotonin reuptake inhibitors141 and tricyclic antidepressants142 compared with participants not using these medications.
Impact of osteoporosis medications on fracture risk in T2DM.
Studies evaluating the antifracture efficacy of current osteoporosis drugs in patients with T2DM have not been widely published. As osteoporosis in T2DM is a condition with a low bone turnover, antiresorptive medications, which further reduce bone turnover, might not be the preferred option28–30. However, a meta-analysis evaluating osteoporosis drugs, including bisphosphonates (which are potent antiresorptive drugs), found an efficacy similar to that achieved in patients with osteoporosis but without diabetes mellitus143. Nonetheless, randomized control trials are needed to assess the antifracture efficacy of osteoporosis drugs in patients with T2DM. In the absence of further evidence, bisphosphonates remain the first choice for osteoporosis treatment in patients with T2DM. However, the possibility that the RANKL antagonist, denosumab could have favourable effects on glucose metabolism could make that a more attractive antiresorptive option for patients with T2DM than bisphosphonates. Thus, although a post hoc analysis of the FREEDOM trial showed no overall effect of denosumab on fasting glucose levels in postmenopausal women with diabetes mellitus or prediabetes, a statistically significant decrease in fasting serum levels of glucose was observed in women with diabetes mellitus not using antidiabetic medications144. Interestingly, as, in animal studies, denosumab induced human β-cell proliferation both in vitro and in vivo145, the possibility of using it in the future as a treatment for T2DM itself has been raised146. Indeed, in 2020, our group reported that denosumab-treated patients showed a statistically significant improvement in HbA1c of a magnitude similar to that seen with commonly used antidiabetic medications, relative to no treatment or bisphosphonate use147.
Finally, post hoc analysis of patients with T2DM from the Abaloparatide Comparator Trial In Vertebral Endpoints (ACTIVE), a phase III, double-blind, randomized, placebo-controlled and active-controlled trial, showed that in women with postmenopausal osteoporosis and T2DM, abaloparatide treatment led to statistically significant improvements in BMD compared with placebo, similar to the improvements in the overall ACTIVE population148.
Future directions.
Future directions for treatment of T2DM-related skeletal fragility will be driven by mechanistic insights into the factors that cause compromised bone quality (for example, RAGE inhibitors and senolytic drugs). Based on the pathogenesis of T2DM-related skeletal fragility, RAGE inhibitors are potential agents that might help alleviate fracture risk in patients with T2DM. Several studies have demonstrated beneficial effects with RAGE inhibitors in various age-related pathologies, including T2DM, cardiovascular disease, neurodegeneration and cancer149. Moreover, downregulation of RAGE signalling has been shown to protect against disease-induced bone and muscle loss150,151. Notably, in bone, genetic RAGE deficiency protects against ovariectomy-induced bone loss in mice150. However, studies have shown that short-term pharmacological RAGE inhibition does not protect against early ageing-induced bone loss152. Further studies of RAGE inhibitors are needed to evaluate their role in preventing or treating T2DM-related skeletal fragility. As described already, senescent cells accumulate in the bone microenvironment in a T2DM mouse model62 and these cells could lead to a reduction in bone formation and a relative increase in bone resorption153. Intermittent senolytic therapy reduces senescent cell burden, which simultaneously suppresses bone resorption with either an increase (in cortical bone) or maintenance (in trabecular bone) of bone formation, leading to higher levels of bone formation relative to bone resorption153. However, further research is needed to better understand whether the clearance of the senescent cells would be beneficial for skeletal fragility in the setting of T2DM.
Conclusions
The evidence is now clear that skeletal fragility should be included among known, established diabetic complications such as retinopathy, nephropathy, neuropathy and vascular disease. Patients with T2DM tend to sustain fragility fractures despite higher levels of BMD than individuals who do not have diabetes mellitus, with impaired bone material properties seeming to most consistently contribute to skeletal frailty in patients with T2DM. Alterations in bone material properties are related, at least in part, to AGE accumulation. In addition, patients with T2DM also have increased cortical porosity, which is linked to microvascular disease. The underlying cellular and molecular mechanisms leading to skeletal fragility in T2DM are complex, but probably involve AGE effects not only on bone material properties, but also on bone cell function through RAGE signalling. Additionally, premature accumulation of senescent cells could possibly lead to an accelerated skeletal ageing phenotype in T2DM, as seems to be present in other tissues. Although standard osteoporosis drugs remain the mainstay in preventing fractures in patients with T2DM, future research should focus on targeting the underlying mechanisms (for example, RAGE signalling and senescence) that mediate the skeletal fragility in patients with T2DM.
Key points.
Fracture risk is increased in patients with type 2 diabetes mellitus (T2Dm) despite normal, or even increased, bone mineral density.
Clinical studies have revealed that the two most consistent alterations in bone quality in patients with T2Dm are impaired bone material properties and increased cortical porosity.
These abnormalities seem to be linked, at least in part, to accumulation of advanced glycation end products (leading to impaired bone material properties) and microvascular disease (leading to increased cortical porosity).
evidence from the past few years also indicates that T2Dm, at least in mice, is associated with accelerated skeletal ageing and increased accumulation of senescent cells, in bone as well as in other tissues.
Current strategies for fracture prevention in patients with T2Dm include minimizing exposure to diabetes mellitus drugs that increase fracture risk and use of osteoporosis medications shown to be effective in patients without diabetes mellitus.
Further studies are needed to evaluate the efficacy of osteoporosis medications specifically in patients with T2Dm and to develop new drugs targeting the mechanisms potentially driving skeletal fragility in patients with T2Dm.
Acknowledgements
The authors acknowledge the support of NIH grants AG062413 (S.K., J.N.F.), AG004875 (S.K., D.G.M.), AR027065 (S.K.), AR070241 (J.N.F.), AG065868 (J.N.F., S.K.), AG063707 (D.G.M.) and AR068275 (D.G.M.).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Zimmet PZ Diabetes and its drivers: the largest epidemic in human history? Clin. Diabetes Endocrinol 3, 1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Napoli N et al. Mechanisms of diabetes mellitus-induced bone fragility. Nat. Rev. Endocrinol 13, 208–219 (2017). [DOI] [PubMed] [Google Scholar]; A comprehensive review of the mechanisms of bone fragility in T2DM; the current review aims largely to update new information since this publication.
- 3.Vestergaard P Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes–a meta-analysis. Osteoporos. Int 18, 427–444 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Schwartz AV et al. Association of BMD and FRAX score with risk of fracture in older adults with type 2 diabetes. JAMA 305, 2184–2192 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; A key paper demonstrating that patients with diabetes mellitus experience fractures at higher levels of BMD than individuals without diabetes mellitus.
- 5.Koromani F et al. Vertebral fractures in individuals with type 2 diabetes: more than skeletal complications alone. Diabetes Care 43, 137–144 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanis JA, Johnell O, Oden A, Johansson H & McCloskey E FRAX™ and the assessment of fracture probability in men and women from the UK. Osteoporos. Int 19, 385–397 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ioacara S et al. Improvements in life expectancy in adult type 2 diabetes patients in the last six decades. Diabetes Res. Clin. Pract 92, 400–404 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Melton LJ 3rd, Leibson CL, Achenbach SJ, Therneau TM & Khosla S Fracture risk in type 2 diabetes: update of a population-based study. J. Bone Miner. Res 23, 1334–1342 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vilaca T et al. The risk of hip and non-vertebral fractures in type 1 and type 2 diabetes: a systematic review and meta-analysis update. Bone 137, 115457 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Janghorbani M, Van Dam RM, Willett WC & Hu FB Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. Am. J. Epidemiol 166, 495–505 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Fan Y, Wei F, Lang Y & Liu Y Diabetes mellitus and risk of hip fractures: a meta-analysis. Osteoporos. Int 27, 219–228 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Park HY, Han K, Kim Y, Kim YH & Sur YJ The risk of hip fractures in individuals over 50 years old with prediabetes and type 2 diabetes – a longitudinal nationwide population-based study. Bone 142, 115691 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Wang H, Ba Y, Xing Q & Du JL Diabetes mellitus and the risk of fractures at specific sites: a meta-analysis. BMJ Open 9, e024067 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vilaca T, Walsh J & Eastell R Discordant pattern of peripheral fractures in diabetes: a meta-analysis on the risk of wrist and ankle fractures. Osteoporos. Int 30, 135–143 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Adami G et al. Risk of fragility fractures in obesity and diabetes: a retrospective analysis on a nation-wide cohort. Osteoporos. Int 31, 2113–2122 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Napoli N et al. Vertebral fracture risk in diabetic elderly men: the MrOS study. J. Bone Miner. Res 33, 63–69 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oei L et al. High bone mineral density and fracture risk in type 2 diabetes as skeletal complications of inadequate glucose control: the Rotterdam study. Diabetes Care 36, 1619–1628 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneider AL et al. Diabetes and risk of fracture-related hospitalization: the Atherosclerosis Risk in Communities study. Diabetes Care 36, 1153–1158 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li CI et al. Glycated hemoglobin level and risk of hip fracture in older people with type 2 diabetes: a competing risk analysis of Taiwan diabetes cohort study. J. Bone Miner. Res 30, 1338–1346 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Conway BN, Long DM, Figaro MK & May ME Glycemic control and fracture risk in elderly patients with diabetes. Diabetes Res. Clin. Pract 115, 47–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ntouva A et al. Hypoglycaemia is associated with increased risk of fractures in patients with type 2 diabetes mellitus: a cohort study. Eur. J. Endocrinol 180, 51–58 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Hung YC et al. Severe hypoglycemia and hip fracture in patients with type 2 diabetes: a nationwide population-based cohort study. Osteoporos. Int 28, 2053–2060 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Johnston SS, Conner C, Aagren M, Ruiz K & Bouchard J Association between hypoglycaemic events and fall-related fractures in Medicare-covered patients with type 2 diabetes. Diabetes Obes. Metab 14, 634–643 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Komorita Y et al. Both hypo- and hyperglycaemia are associated with increased fracture risk in Japanese people with type 2 diabetes: the Fukuoka Diabetes Registry. Diabet. Med 37, 838–847 (2020). [DOI] [PubMed] [Google Scholar]; An observational study demonstrating that recurrent hypoglycaemia as well as poorly controlled T2DM are associated with an increase in fracture risk.
- 25.Lui DTW et al. HbA1c variability, in addition to mean HbA1c, predicts incident hip fractures in Chinese people with type 2 diabetes. Osteoporos. Int 31, 1955–1964 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Bonds DE et al. Risk of fracture in women with type 2 diabetes: the Women’s Health Initiative Observational study. J. Clin. Endocrinol. Metab 91, 3404–3410 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Ma L et al. Association between bone mineral density and type 2 diabetes mellitus: a meta-analysis of observational studies. Eur. J. Epidemiol 27, 319–332 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Starup-Linde J & Vestergaard P Biochemical bone turnover markers in diabetes mellitus – a systematic review. Bone 82, 69–78 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Farr JN et al. In vivo assessment of bone quality in postmenopausal women with type 2 diabetes. J. Bone Miner. Res 29, 787–795 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; The first in vivo demonstration that bone material properties are impaired in patients with T2DM.
- 30.Tonks KT, White CP, Center JR, Samocha-Bonet D & Greenfield JR Bone turnover is suppressed in insulin resistance, independent of adiposity. J. Clin. Endocrinol. Metab 102, 1112–1121 (2017). [DOI] [PubMed] [Google Scholar]; This study demonstrates that the reduced bone turnover in patients with T2DM is linked to insulin resistance and increases in visceral adipose tissue.
- 31.Timar B et al. The impact of diabetic neuropathy on balance and on the risk of falls in patients with type 2 diabetes mellitus: a cross-sectional study. PLoS ONE 11, e0154654 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leslie WD, Aubry-Rozier B, Lamy O & Hans D TBS (trabecular bone score) and diabetes-related fracture risk. J. Clin. Endocrinol. Metab 98, 602–609 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Patsch JM et al. Increased cortical porosity in type 2 diabetic postmenopausal women with fragility fractures. J. Bone Miner. Res 28, 313–324 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heilmeier U et al. Cortical bone laminar analysis reveals increased midcortical and periosteal porosity in type 2 diabetic postmenopausal women with history of fragility fractures compared to fracture-free diabetics. Osteoporos. Int 27, 2791–2802 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burghardt AJ et al. High-resolution peripheral quantitative computed tomographic imaging of cortical and trabecular bone microarchitecture in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab 95, 5045–5055 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu EW et al. Defects in cortical microarchitecture among African-American women with type 2 diabetes. Osteoporos. Int 26, 673–679 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samakkarnthai P et al. Determinants of bone material strength and cortical porosity in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab 105, e3718–e372 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that, in patients with T2DM, impaired bone material properties are associated with accumulation of AGEs, whereas increased cortical porosity is associated with microvascular disease.
- 38.Samelson EJ et al. Diabetes and deficits in cortical bone density, microarchitecture, and bone size: Framingham HR-pQCT study. J. Bone Miner. Res 33, 54–62 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paccou J et al. Bone microarchitecture in men and women with diabetes: the importance of cortical porosity. Calcif. Tissue Int 98, 465–473 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Shanbhogue VV et al. Compromised cortical bone compartment in type 2 diabetes mellitus patients with microvascular disease. Eur. J. Endocrinol 174, 115–124 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Shu A et al. Bone structure and turnover in type 2 diabetes mellitus. Osteoporos. Int 23, 635–641 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bala Y et al. Cortical porosity identifies women with osteopenia at increased risk for forearm fractures. J. Bone Miner. Res 29, 1356–1362 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bala Y et al. Risedronate slows or partly reverses cortical and trabecular microarchitectural deterioration in postmenopausal women. J. Bone Miner. Res 29, 380–388 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Furst JR et al. Advanced glycation endproducts and bone material strength in type 2 diabetes. J. Clin. Endocrinol. Metab 101, 2502–2510 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nilsson AG et al. Type 2 diabetes mellitus is associated with better bone microarchitecture but lower bone material strength and poorer physical function in elderly women: a population-based study. J. Bone Miner. Res 32, 1062–1071 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Dawson-Hughes B, Bouxsein M & Shea K Bone material strength in normoglycemic and hyperglycemic black and white older adults. Osteoporos. Int 30, 2429–2435 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meerwaldt R et al. Simple non-invasive assessment of advanced glycation endproduct accumulation. Diabetologia 47, 1324–1330 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Litwinoff E, Hurtado Del Pozo C, Ramasamy R & Schmidt AM Emerging targets for therapeutic development in diabetes and its complications: the RAGE signaling pathway. Clin. Pharmacol. Ther 98, 135–144 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fournet M, Bonté F & Desmoulière A Glycation damage: a possible hub for major pathophysiological disorders and aging. Aging Dis 9, 880–900 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Byun K et al. Advanced glycation end-products produced systemically and by macrophages: a common contributor to inflammation and degenerative diseases. Pharmacol. Ther 177, 44–55 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Desai CS, Blumenthal RS & Greenland P Screening low-risk individuals for coronary artery disease. Curr. Atheroscler. Rep 16, 402 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Bacharach JM, Rooke TW, Osmundson PJ & Gloviczki P Predictive value of transcutaneous oxygen pressure and amputation success by use of supine and elevation measurements. J. Vasc. Surg 15, 558–563 (1992). [PubMed] [Google Scholar]
- 53.Thrailkill KM, Lumpkin CK Jr., Bunn RC, Kemp SF & Fowlkes JL Is insulin an anabolic agent in bone? Dissecting the diabetic bone for clues. Am. J. Physiol. Endocrinol. Metab 289, E735–745 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stolk RP et al. Hyperinsulinemia and bone mineral density in an elderly population: the Rotterdam study. Bone 18, 545–549 (1996). [DOI] [PubMed] [Google Scholar]
- 55.Johnson KC et al. The effect of intentional weight loss on fracture risk in persons with diabetes: results from the look AHEAD randomized clinical trial. J. Bone Miner. Res 32, 2278–2287 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DiGirolamo DJ, Clemens TL & Kousteni S The skeleton as an endocrine organ. Nat. Rev. Rheumatol 8, 674–683 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Liu JM, Rosen CJ, Ducy P, Kousteni S & Karsenty G Regulation of glucose handling by the skeleton: insights from mouse and human studies. Diabetes 65, 3225–3232 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dirckx N, Moorer MC, Clemens TL & Riddle RC The role of osteoblasts in energy homeostasis. Nat. Rev. Endocrinol 15, 651–665 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lecka-Czernik B & Rosen CJ Energy excess, glucose utilization, and skeletal remodeling: new insights. J. Bone Miner. Res 30, 1356–1361 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Rendina-Ruedy E & Rosen CJ Lipids in the bone marrow: an evolving perspective. Cell Metab 31, 219–231 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shanbhogue VV, Mitchell DM, Rosen CJ & Bouxsein ML Type 2 diabetes and the skeleton: new insights into sweet bones. Lancet Diabetes Endocrinol 4, 159–173 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Eckhardt BA et al. Accelerated osteocyte senescence and skeletal fragility in mice with type 2 diabetes. JCI Insight 5, e135236 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrates that at least in a mouse model, the combination of obesity and hyperglycaemia is associated with increased cellular senescence in bone.
- 63.Starr JF et al. Robust trabecular microstructure in type 2 diabetes revealed by individual trabecula segmentation analysis of HR-pQCT Images. J. Bone Miner. Res 33, 1665–1675 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ogata N et al. Insulin receptor substrate-1 in osteoblast is indispensable for maintaining bone turnover. J. Clin. Invest 105, 935–943 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abrahamsen B, Rohold A, Henriksen JE & Beck-Nielsen H Correlations between insulin sensitivity and bone mineral density in non-diabetic men. Diabet. Med 17, 124–129 (2000). [DOI] [PubMed] [Google Scholar]
- 66.Fontana L, Eagon JC, Trujillo ME, Scherer PE & Klein S Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 56, 1010–1013 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Martin TJ & Sims NA RANKL/OPG; critical role in bone physiology. Rev. Endocr. Metab. Disord 16, 131–139 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Krings A et al. Bone marrow fat has brown adipose tissue characteristics, which are attenuated with aging and diabetes. Bone 50, 546–552 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sheu Y et al. Vertebral bone marrow fat, bone mineral density and diabetes: the osteoporotic fractures in men (MrOS) study. Bone 97, 299–305 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Baum T et al. Does vertebral bone marrow fat content correlate with abdominal adipose tissue, lumbar spine bone mineral density, and blood biomarkers in women with type 2 diabetes mellitus? J. Magn. Reson. Imaging 35, 117–124 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fazeli PK et al. Marrow fat and bone–new perspectives. J. Clin. Endocrinol. Metab 98, 935–945 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nuche-Berenguer B et al. Exendin-4 exerts osteogenic actions in insulin-resistant and type 2 diabetic states. Regul. Pept 159, 61–66 (2010). [DOI] [PubMed] [Google Scholar]
- 73.Gennari L et al. Circulating sclerostin levels and bone turnover in type 1 and type 2 diabetes. J. Clin. Endocrinol. Metab 97, 1737–1744 (2012). [DOI] [PubMed] [Google Scholar]
- 74.Piccoli A et al. Sclerostin regulation, microarchitecture, and advanced glycation end-products in the bone of elderly women with type 2 diabetes. J. Bone Miner. Res 35, 2415–2422 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baron R & Kneissel M WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med 19, 179–192 (2013). [DOI] [PubMed] [Google Scholar]
- 76.Ma YH et al. Circulating sclerostin associated with vertebral bone marrow fat in older men but not women. J. Clin. Endocrinol. Metab 99, E2584–2590 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Khosla S, Farr JN, Tchkonia T & Kirkland JL The role of cellular senescence in ageing and endocrine disease. Nat. Rev. Endocrinol 16, 263–275 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alcorta DA et al. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl Acad. Sci. USA 93, 13742–13747 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Beauséjour CM et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 22, 4212–4222 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wissler Gerdes EO, Zhu Y, Tchkonia T & Kirkland JL Discovery, development, and future application of senolytics: theories and predictions. FEBS J 287, 2418–2427 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Coppé JP, Desprez PY, Krtolica A & Campisi J The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol 5, 99–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Acosta JC et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol 15, 978–990 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Palmer AK et al. Cellular senescence in type 2 diabetes: a therapeutic opportunity. Diabetes 64, 2289–2298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Palmer AK, Gustafson B, Kirkland JL & Smith U Cellular senescence: at the nexus between ageing and diabetes. Diabetologia 62, 1835–1841 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Prata L, Ovsyannikova IG, Tchkonia T & Kirkland JL Senescent cell clearance by the immune system: emerging therapeutic opportunities. Semin. Immunol 40, 101275 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang E Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res 55, 2284–2292 (1995). [PubMed] [Google Scholar]
- 87.Tchkonia T et al. Fat tissue, aging, and cellular senescence. Aging Cell 9, 667–684 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Palmer AK et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 18, e12950 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aguayo-Mazzucato C et al. Acceleration of β cell aging determines diabetes and senolysis improves disease outcomes. Cell Metab 30, 129–142.e124 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ogrodnik M et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun 8, 15691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ogrodnik M et al. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab 29, 1061–1077.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xu M et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. eLife 4, e12997 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zaragosi LE et al. Activin A plays a critical role in proliferation and differentiation of human adipose progenitors. Diabetes 59, 2513–2521 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nelson G et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11, 345–349 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.da Silva PFL et al. The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell 18, e12848 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Manavalan JS et al. Circulating osteogenic precursor cells in type 2 diabetes mellitus. J. Clin. Endocrinol. Metab 97, 3240–3250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gorgoulis V et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019). [DOI] [PubMed] [Google Scholar]
- 98.Swanson EC, Manning B, Zhang H & Lawrence JB Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J. Cell Biol 203, 929–942 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Coppé JP et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6, 2853–2868 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ramasamy R, Shekhtman A & Schmidt AM The multiple faces of RAGE–opportunities for therapeutic intervention in aging and chronic disease. Expert. Opin. Ther. Targets 20, 431–446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Poundarik AA et al. A direct role of collagen glycation in bone fracture. J. Mech. Behav. Biomed. Mater 52, 120–130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhou Z et al. Regulation of osteoclast function and bone mass by RAGE. J. Exp. Med 203, 1067–1080 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Franke S et al. Advanced glycation end products affect growth and function of osteoblasts. Clin. Exp. Rheumatol 29, 650–660 (2011). [PubMed] [Google Scholar]
- 104.Ahmed N & Thornalley PJ Quantitative screening of protein biomarkers of early glycation, advanced glycation, oxidation and nitrosation in cellular and extracellular proteins by tandem mass spectrometry multiple reaction monitoring. Biochem. Soc. Trans 31, 1417–1422 (2003). [DOI] [PubMed] [Google Scholar]
- 105.O’Grady KL et al. Development and application of mass spectroscopy assays for Nε-(1-carboxymethyl)-L-lysine and pentosidine in renal failure and diabetes. J. Appl. Lab. Med 5, 558–568 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Karim L et al. Bone microarchitecture, biomechanical properties, and advanced glycation end-products in the proximal femur of adults with type 2 diabetes. Bone 114, 32–39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schwartz AV et al. Pentosidine and increased fracture risk in older adults with type 2 diabetes. J. Clin. Endocrinol. Metab 94, 2380–2386 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang J, Wang H, Shi J & Ding Y Effects of bone marrow MSCs transfected with sRAGE on the intervention of HMGB1 induced immuno-inflammatory reaction. Int. J. Clin. Exp. Pathol 8, 12028–12040 (2015). [PMC free article] [PubMed] [Google Scholar]
- 109.Lalla E et al. Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J. Clin. Invest 105, 1117–1124 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Walker D, Lue LF, Paul G, Patel A & Sabbagh MN Receptor for advanced glycation endproduct modulators: a new therapeutic target in Alzheimer’s disease. Expert. Opin. Investig. Drugs 24, 393–399 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Galasko D et al. Clinical trial of an inhibitor of RAGE-Aβ interactions in Alzheimer disease. Neurology 82, 1536–1542 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mao YX et al. RAGE-dependent mitochondria pathway: a novel target of silibinin against apoptosis of osteoblastic cells induced by advanced glycation end products. Cell Death Dis 9, 674 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen H et al. Advanced glycation end products induced IL-6 and VEGF-A production and apoptosis in osteocyte-like MLO-Y4 cells by activating RAGE and ERK1/2, P38 and STAT3 signalling pathways. Int. Immunopharmacol 52, 143–149 (2017). [DOI] [PubMed] [Google Scholar]
- 114.Lafage-Proust MH et al. Assessment of bone vascularization and its role in bone remodeling. Bonekey Rep 4, 662 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Andersen TL et al. A physical mechanism for coupling bone resorption and formation in adult human bone. Am. J. Pathol 174, 239–247 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rehman J, Li J, Orschell CM & March KL Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation 107, 1164–1169 (2003). [DOI] [PubMed] [Google Scholar]
- 117.Guo P et al. Platelet-derived growth factor-B enhances glioma angiogenesis by stimulating vascular endothelial growth factor expression in tumor endothelia and by promoting pericyte recruitment. Am. J. Pathol 162, 1083–1093 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Colnot C Skeletal cell fate decisions within periosteum and bone marrow during bone regeneration. J. Bone Miner. Res 24, 274–282 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liao YH et al. Osteogenic differentiation of adipose-derived stem cells and calvarial defect repair using baculovirus-mediated co-expression of BMP-2 and miR-148b. Biomaterials 35, 4901–4910 (2014). [DOI] [PubMed] [Google Scholar]
- 120.Divya MS et al. Umbilical cord blood-derived mesenchymal stem cells consist of a unique population of progenitors co-expressing mesenchymal stem cell and neuronal markers capable of instantaneous neuronal differentiation. Stem Cell Res. Ther 3, 57 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wynn TA & Vannella KM Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44, 450–462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Doherty MJ et al. Vascular pericytes express osteogenic potential in vitro and in vivo. J. Bone Miner. Res 13, 828–838 (1998). [DOI] [PubMed] [Google Scholar]
- 123.Rask-Madsen C & King GL Vascular complications of diabetes: mechanisms of injury and protective factors. Cell Metab 17, 20–33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Farr JN & Khosla S Determinants of bone strength and quality in diabetes mellitus in humans. Bone 82, 28–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cosman F et al. Clinician’s guide to prevention and treatment of osteoporosis. Osteoporos. Int 25, 2359–2381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Roman de Mettelinge T, Cambier D, Calders P, Van Den Noortgate N & Delbaere K Understanding the relationship between type 2 diabetes mellitus and falls in older adults: a prospective cohort study. PLoS ONE 8, e67055 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schwartz AV et al. Older women with diabetes have a higher risk of falls: a prospective study. Diabetes Care 25, 1749–1754 (2002). [DOI] [PubMed] [Google Scholar]
- 128.Chapman A, Meyer C, Renehan E, Hill KD & Browning CJ Exercise interventions for the improvement of falls-related outcomes among older adults with diabetes mellitus: a systematic review and meta-analyses. J. Diabetes Complicat 31, 631–645 (2017). [DOI] [PubMed] [Google Scholar]
- 129.Gu Y & Dennis SM Are falls prevention programs effective at reducing the risk factors for falls in people with type-2 diabetes mellitus and peripheral neuropathy: a systematic review with narrative synthesis. J. Diabetes Complicat 31, 504–516 (2017). [DOI] [PubMed] [Google Scholar]
- 130.Napoli N et al. Fracture risk in diabetic elderly men: the MrOS study. Diabetologia 57, 2057–2065 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pscherer S, Kostev K, Dippel FW & Rathmann W Fracture risk in patients with type 2 diabetes under different antidiabetic treatment regimens: a retrospective database analysis in primary care. Diabetes Metab. Syndr. Obes 9, 17–23 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Losada-Grande E et al. Insulin use and excess fracture risk in patients with type 2 diabetes: a propensity-matched cohort analysis. Sci. Rep 7, 3781 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Palermo A et al. Oral anti-diabetic drugs and fracture risk, cut to the bone: safe or dangerous? A narrative review. Osteoporos. Int 26, 2073–2089 (2015). [DOI] [PubMed] [Google Scholar]
- 134.Cheng L et al. Glucagon-like peptide-1 receptor agonists and risk of bone fracture in patients with type 2 diabetes: a meta-analysis of randomized controlled trials. Diabetes Metab. Res. Rev 35, e3168 (2019). [DOI] [PubMed] [Google Scholar]
- 135.Zhu ZN, Jiang YF & Ding T Risk of fracture with thiazolidinediones: an updated meta-analysis of randomized clinical trials. Bone 68, 115–123 (2014). [DOI] [PubMed] [Google Scholar]
- 136.Watts NB et al. Effects of canagliflozin on fracture risk in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab 101, 157–166 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li X et al. Effects of SGLT2 inhibitors on fractures and bone mineral density in type 2 diabetes: an updated meta-analysis. Diabetes Metab. Res. Rev 35, e3170 (2019). [DOI] [PubMed] [Google Scholar]
- 138.Barzilay JI et al. The impact of antihypertensive medications on bone mineral density and fracture risk. Curr. Cardiol. Rep 19, 76 (2017). [DOI] [PubMed] [Google Scholar]
- 139.Bokrantz T et al. Antihypertensive drug classes and the risk of hip fracture: results from the Swedish primary care cardiovascular database. J. Hypertens. 38, 167–175 (2020). [DOI] [PubMed] [Google Scholar]
- 140.Shi R, Mei Z, Zhang Z & Zhu Z Effects of statins on relative risk of fractures for older adults: an updated systematic review with meta-analysis. J. Am. Med. Dir. Assoc 20, 1566–1578.e3 (2019). [DOI] [PubMed] [Google Scholar]
- 141.Wu Q, Bencaz AF, Hentz JG & Crowell MD Selective serotonin reuptake inhibitor treatment and risk of fractures: a meta-analysis of cohort and case-control studies. Osteoporos. Int 23, 365–375 (2012). [DOI] [PubMed] [Google Scholar]
- 142.Wu Q, Qu W, Crowell MD, Hentz JG & Frey KA Tricyclic antidepressant use and risk of fractures: a meta-analysis of cohort and case-control studies. J. Bone Miner. Res 28, 753–763 (2013). [DOI] [PubMed] [Google Scholar]
- 143.Anagnostis P et al. Efficacy of anti-osteoporotic medications in patients with type 1 and 2 diabetes mellitus: a systematic review. Endocrine 60, 373–383 (2018). [DOI] [PubMed] [Google Scholar]
- 144.Napoli N et al. Effect of denosumab on fasting glucose in women with diabetes or prediabetes from the FREEDOM trial. Diabetes Metab. Res. Rev 34, e2991 (2018). [DOI] [PubMed] [Google Scholar]
- 145.Kondegowda NG et al. Osteoprotegerin and denosumab stimulate human beta cell proliferation through inhibition of the receptor activator of NF-κB ligand pathway. Cell Metab 22, 77–85 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Schmitz F, Roscioni S & Lickert H Repurposing an osteoporosis drug for β cell regeneration in diabetic patients. Cell Metab 22, 58–59 (2015). [DOI] [PubMed] [Google Scholar]
- 147.Weivoda MM et al. Identification of osteoclast-osteoblast coupling factors in humans reveals links between bone and energy metabolism. Nat. Commun 11, 87 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dhaliwal R et al. Abaloparatide in postmenopausal women with osteoporosis and type 2 diabetes: a post hoc analysis of the ACTIVE study. JBMR 4, e10346 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schmidt AM Soluble RAGEs - prospects for treating & tracking metabolic and inflammatory disease. Vasc. Pharmacol 72, 1–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ding KH et al. Disordered osteoclast formation in RAGE-deficient mouse establishes an essential role for RAGE in diabetes related bone loss. Biochem. Biophys. Res. Commun 340, 1091–1097 (2006). [DOI] [PubMed] [Google Scholar]
- 151.Egawa T et al. Potential involvement of dietary advanced glycation end products in impairment of skeletal muscle growth and muscle contractile function in mice. Br. J. Nutr 117, 21–29 (2017). [DOI] [PubMed] [Google Scholar]
- 152.Davis HM et al. Short-term pharmacologic RAGE inhibition differentially affects bone and skeletal muscle in middle-aged mice. Bone 124, 89–102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Farr JN et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med 23, 1072–1079 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; A study demonstrating that reducing the burden of senescent cells ameliorates age-related bone loss in mice, which raises the possibility that a similar approach might be useful in alleviating the skeletal fragility associated with T2DM.
- 154.Monami M et al. Bone fractures and hypoglycemic treatment in type 2 diabetic patients: a case-control study. Diabetes Care 31, 199–203 (2008). [DOI] [PubMed] [Google Scholar]
- 155.Vestergaard P, Rejnmark L & Mosekilde L Relative fracture risk in patients with diabetes mellitus, and the impact of insulin and oral antidiabetic medication on relative fracture risk. Diabetologia 48, 1292–1299 (2005). [DOI] [PubMed] [Google Scholar]
- 156.Hidayat K, Du X, Wu MJ & Shi BM The use of metformin, insulin, sulphonylureas, and thiazolidinediones and the risk of fracture: systematic review and meta-analysis of observational studies. Obes. Rev 20, 1494–1503 (2019). [DOI] [PubMed] [Google Scholar]
- 157.Josse RG et al. Sitagliptin and risk of fractures in type 2 diabetes: results from the TECOS trial. Diabetes Obes. Metab 19, 78–86 (2017). [DOI] [PubMed] [Google Scholar]
- 158.Schwartz AV et al. Effects of TZD use and discontinuation on fracture rates in ACCORD bone study. J. Clin. Endocrinol. Metab 100, 4059–4066 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lapane KL, Jesdale BM, Dubé CE, Pimentel CB & Rajpathak SN Sulfonylureas and risk of falls and fractures among nursing home residents with type 2 diabetes mellitus. Diabetes Res. Clin. Pract 109, 411–419 (2015). [DOI] [PubMed] [Google Scholar]
- 160.Hidayat K, Du X & Shi BM Risk of fracture with dipeptidyl peptidase-4 inhibitors, glucagon-like peptide-1 receptor agonists, or sodium-glucose cotransporter-2 inhibitors in real-world use: systematic review and meta-analysis of observational studies. Osteoporos. Int 30, 1923–1940 (2019). [DOI] [PubMed] [Google Scholar]
- 161.Monami M, Dicembrini I, Antenore A & Mannucci E Dipeptidyl peptidase-4 inhibitors and bone fractures: a meta-analysis of randomized clinical trials. Diabetes Care 34, 2474–2476 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mosenzon O et al. Incidence of fractures in patients with type 2 diabetes in the SAVOR-TIMI 53 trial. Diabetes Care 38, 2142–2150 (2015). [DOI] [PubMed] [Google Scholar]
- 163.Kohan DE, Fioretto P, Tang W & List JF Long-term study of patients with type 2 diabetes and moderate renal impairment shows that dapagliflozin reduces weight and blood pressure but does not improve glycemic control. Kidney Int 85, 962–971 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]