

Introduction
Osteoarthritis (OA) is the single most common painful joint condition; knee OA alone currently affects >250 million people globally, and it is one of the fastest increasing health conditions worldwide1,2. The individual impact of OA includes pain and loss of both mobility and independence, 25% of patients being unable to carry out normal activities of daily life3,4. OA’s societal burden is enormous, with current annual direct healthcare costs of knee OA alone estimated at up to $15 billion in the USA5. This figure is dwarfed by indirect costs of work absenteeism, early retirement, and loss of productivity associated with OA and associated medication use6. OA remains one of the major unresolved medical conditions, with no registered therapies that halt structural damage, and symptom-modifying interventions having only moderate long-term effect at best7.
It is clear that developing new, safe, effective OA treatments is an international healthcare and socioeconomic priority. A key underpinning requirement for therapeutic advancement in OA, as it is for all diseases, is knowledge of the cellular and molecular pathophysiology8. There has been an extraordinary increase in understanding of human-relevant OA bio-molecular mechanisms over the last 15 years9–11. This has been associated with the recognition of OA as a joint-wide disease affecting and involving molecular and mechanical cross-talk between multiple tissues, and these with systemic processes and pathways. The complexity and breadth of new knowledge in OA pathophysiology, means the task of summarizing the key pathways is immense and crosses diverse mechano-biological domains. In the current review, we have therefore taken the approach to ask individuals with expertise in six different aspects of OA pathogenesis (cartilage matrix degradation, inflammation, fibrosis, failed cartilage repair, bone remodelling, and ageing), to provide a brief narrative review of what they consider the key disease mechanisms in their domain, with a lens to focus on those that may that offer the most promise for therapeutic targeting. The essays were written independently to avoid unintended collusion bias and are presented below, followed by a brief conclusion written after collation of the individual sections. We hope this approach will not only provide a different, interesting and more approachable review on a daunting topic but also allow identification of pathways and mechanisms that cross multiple aspects of OA and contribute to the changing crosstalk between joint tissues as disease progresses.
Targeting cartilage degradation to treat OA – Linda Troeberg
Degradation of the cartilage extracellular matrix (ECM) is appreciated to be an important feature of OA pathogenesis that, together with bone remodelling, leads to progressive joint damage and structural failure. Breakdown of type II collagen and aggrecan are thought to be most important, as these are the two most abundant cartilage matrix biomolecules and their loss reduces tensile strength and resistance to compression. A large body of evidence supports the conclusion that matrix metalloproteinases (MMPs) mediate type II collagen degradation, while related metalloproteinases, the adamalysins with thrombospondin motifs (ADAMTSs) are responsible for the degradation of aggrecan (Figure 1).
Figure 1: MMPs and ADAMTSs metalloproteases cleave type II collagen and aggrecan in the OA cartilage extracellular matrix.
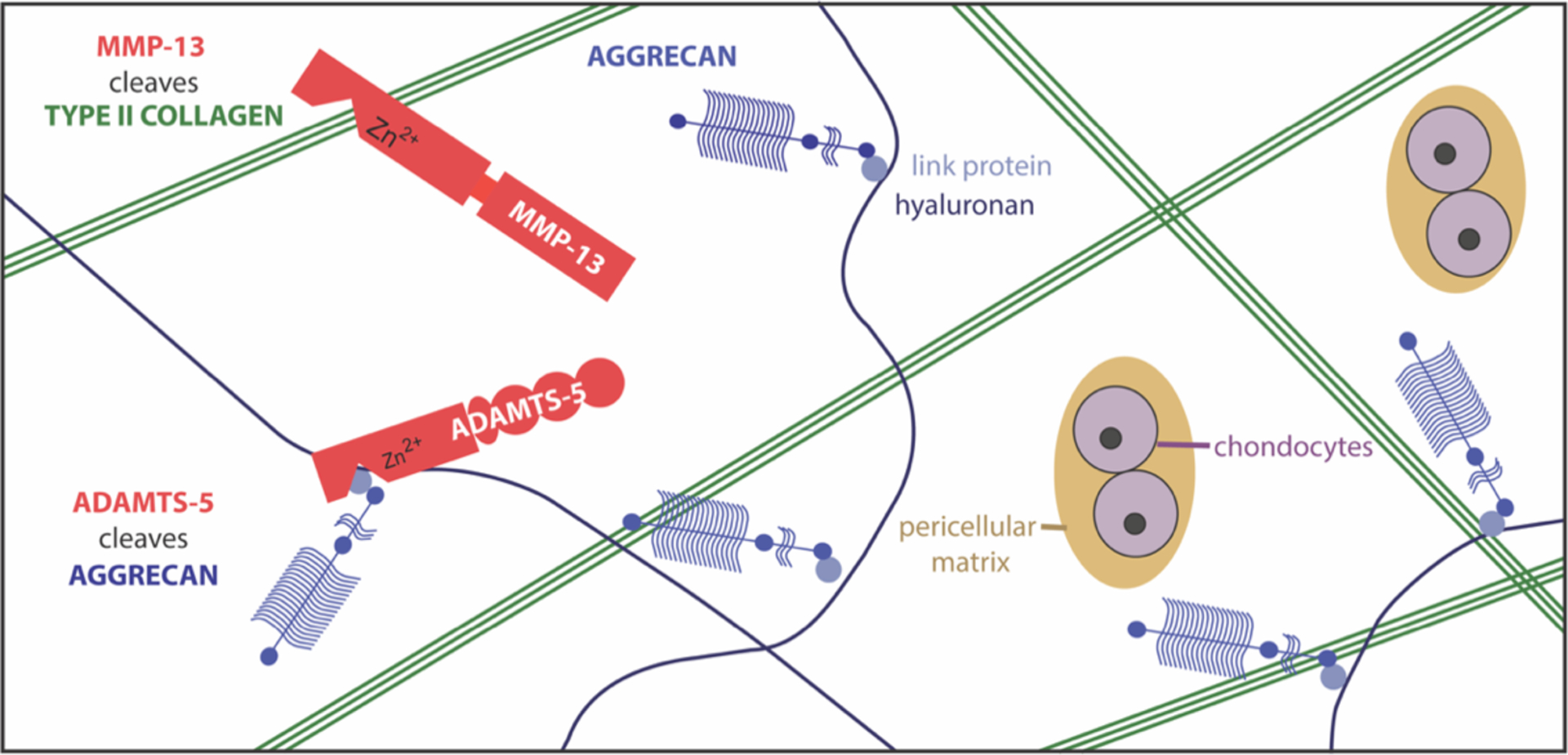
Chondrocytes secrete metalloproteases that degrade the cartilage extracellular matrix in OA. Studies on transgenic mice suggest that MMP13 is the key collagenase in cartilage, while ADAMTS5 is the main ‘aggrecanase’.
Type II collagen is a very stable molecule whose triple helical structure can only be cleaved by a handful of proteases, including cathepsin K and 4 collagenolytic MMPs (i.e. MMP1, 8, 13 and 14). Collagen degradation occurs progressively in osteoarthritic cartilage12,13, and can be blocked by metalloprotease inhibitors in vitro14,15, suggesting that the collagenolytic MMPs play a central role in this catabolic process. Two key papers support the assertion that MMP13 is a key collagenase in OA. Firstly, transgenic mice overexpressing MMP-13 in cartilage exhibited increased collagen degradation by 5 months of age, along with increased cartilage erosion and joint pathology16. Secondly, Mmp13-null mice developed significantly less cartilage erosion 8 weeks after surgical induction of OA17. Expression of MMP13 is increased in human and murine OA cartilage, and is highly inducible in vitro by inflammatory cytokines. The catalytic domains of MMPs are structurally homologous, so it has historically been difficult to design inhibitors that effectively target a single MMP without undesirable side-effects. This is thought to be the reason that metalloproteinase inhibitors failed as cancer therapies18, despite clear evidence showing the important roles of MMPs in cancer metastasis and progression. MMP13, however, is unusual among MMPs in that it has deep pockets in its active site, so attempts have been made to design MMP13 inhibitors as potential OA therapies - these fared well in pre-clinical models19, but have not progressed further at present, most likely due to lingering concerns about lack of specificity and consequent toxicity.
The sequence of events in early OA is difficult to ascertain, but in vitro studies indicate that collagen breakdown starts relatively late in the pathogenesis of OA, while breakdown of aggrecan occurs earlier20,21. Importantly, aggrecan loss in these models is reversible, while collagen loss is not. For many years, MMPs were thought to be responsible for the pathological degradation of both collagen and aggrecan in OA cartilage, but this view was challenged by Sandy et al.22, who showed that aggrecan fragments released into the synovial fluid of OA patients had been cleaved at the Glu373~Ala374 bond, which is not targeted by MMPs. This sparked considerable interest in identifying the ‘aggrecanases’ or enzyme(s) responsible for pathological breakdown of aggrecan, as targets for development of OA therapies. The first ‘aggrecanase’ was purified from IL-1-stimulated bovine cartilage by Tortorella et al.23, and named A Disintegrin And Metalloproteinase with Thrombospondin motifs 4 (ADAMTS4) based on its homology to ADAMTS1. Another aggrecanase, ADAMTS5, was cloned shortly afterwards24,25, and subsequent studies indicated this is the main murine aggrecanase, since mice lacking Adamts5 were protected against aggrecan degradation and cartilage damage in 2 pre-clinical models of OA26,27. ADAMTS5 may also be the primary human aggrecanase28, although ADAMTS4 may also play a role.
Aggrecanase inhibitors have been designed by several groups, with some of these showing promising efficacy in pre-clinical models29. For example, Galapagos and Servier developed an ADAMTS5 catalytic domain inhibitor, GLPG1972/S201086, with good selectivity for ADAMTS5 and efficacy in preclinical rat and mouse OA models30,31. However, this inhibitor failed to meet its primary outcome (reduction in cartilage loss over 1 year by qMRI) or secondary outcomes, including pain and structural progression in a clinical trial (https://clinicaltrials.gov, NCT03595618). Some groups have taken the approach of designing inhibitors that target the non-catalytic domains of ADAMTSs, to reduce the potential for cross-reactivity and off-target inhibition of homeostatic MMPs and related metalloproteases such as ADAMs. For example, Santamaria et al.32 recently generated small molecule exosite inhibitors of ADAMTS5, and Merck generated a cross-domain bi-specific nanobody with good efficacy in a murine OA model33. However, a word of caution was raised by GlaxoSmithKline34, who found that their antibody against ADAMTS5 caused cardiac abnormalities in cynomolgus monkeys, which they suggest may relate to expression of ADAMTS5 in cardiovascular tissue. ADAMTS5 also has homeostatic roles in other tissues (reviewed by Santamaria35), suggesting further challenges for inhibitor design.
Targeting inflammation to treat OA - Christopher B. Little
Historically osteoarthritis (OA) was considered a non-inflammatory “degenerative-joint-disease”, and alternative names such as osteoarthrosis were proposed. However, just as the concept of passive “wear-and-tear” OA cartilage loss has been replaced with an understanding of a dynamic balance between bio-cellular repair and destruction (see sections by Troeberg and Vincent), the presence of synovial inflammation is now a well-recognized and consistent finding in OA patients and pre-clinical animal models10,36–38. OA synovium, even in early-stage disease, displays focal hyperplasia and hypertrophy of synovial lining cells, subintimal accumulation of inflammatory cells (macrophages, lymphocytes, plasma cells) and increased vascularity39,40, along with progressive fibrosis of the joint capsule (see section by Kapoor). In the OA knee, the infrapatellar fat pad as part of the functional synovial unit also has increased inflammatory cells and fibrotic changes, although notably with some unique characteristics compared with other synovial tissues41–43.
Synovial inflammation in OA is associated not only with symptoms but structural disease severity and progression in patients44–48. Beyond simply being a secondary response to late-stage joint tissue breakdown, synovial inflammatory mediators are more elevated acutely after OA-inducing joint injury49 and in early compared with late OA39,46,50,51. Importantly, synovitis/joint-effusion is associated not only with faster progression of established disease, but also more incident OA44 and increased risk of post-traumatic OA following joint injury52. In light of this, it seems clear that the “itis” in OA is indeed appropriate, not only from the perspective of correctly describing the presence of synovial inflammation but also its potential pathophysiological role in initiation and progression of structural and symptomatic disease.
Activation of the innate inflammatory/immune response in OA has been well-described and may be triggered by mechanical injury directly, as has been proposed for articular cartilage53. It is characterized by the influx of blood-derived monocytes and macrophages, which may contribute directly to increases in cytokines, growth factors and pathologically-relevant enzymes (e.g. IL1, IL6, TNF, TGFβ, MMP1, MMP13, ADAMTS4, ADAMTS5)54–56. Lymphocytes, particularly CD4 and CD8 T-cells are also increased in OA synovium even in early disease stages, these cells producing cytokines, chemokines and enzymes implicated in disease progression (e.g. IL8, IL17, TNF, CCL2, MMP1, MMP3, MMP9)39,41,43,57–59. Stromal cells in the synovium and other joint tissues (e.g. injured cruciate ligament) also increase synthesis of cytokines and chemokines60–62, and OA chondrocytes themselves increase expression of pro-catabolic cytokines (e.g. IL8, IL12, IL17) that may act in an autocrine or paracrine manner to promote cartilage degradation63. Finally, systemic inflammation, particularly circulating cytokines (e.g. IL6, TNF) and activated monocytes (associated for example, with obesity/metabolic-syndrome), further contribute to the pro-inflammatory milieux and complex cellular cross-talk that may initiate, perpetuate and exacerbate joint-wide OA structural pathology and pain (Figure 2)54,64–67.
Figure 2:
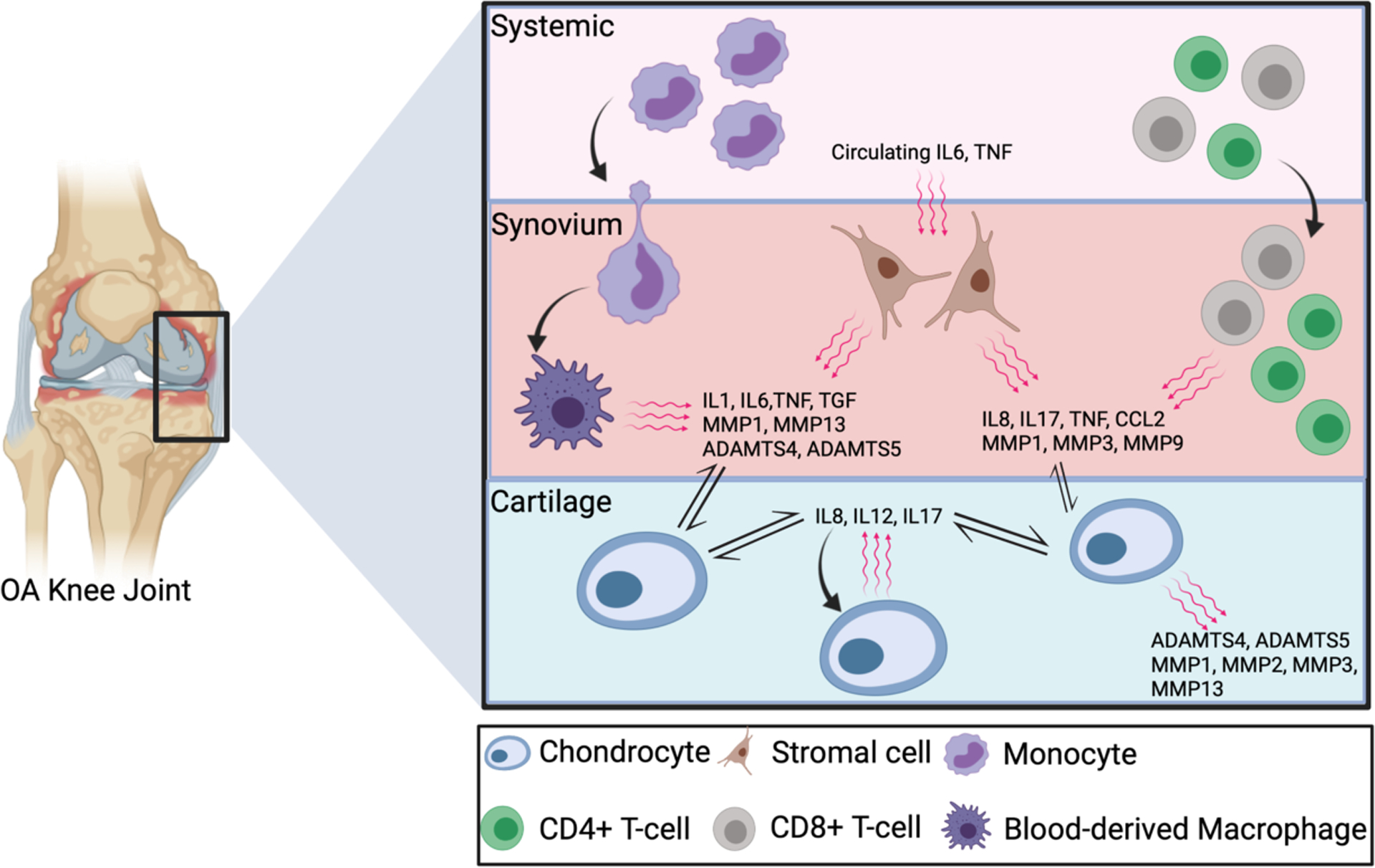
Schematic image depicting the key inflammatory cells and soluble mediators and pathways implicated in osteoarthritis pathogenesis.
The discussion above provides a glimpse of the burgeoning evidence for up-regulation of a multitude of inflammatory pathways locally and systemically in OA, involving innate and adaptive immune cells, and numerous cytokines, chemokines and growth factors. Dysregulation does not equal causality however, so is there data supporting the therapeutic potential of targeting “inflammation” in OA, and which if any of the pathways may hold the most promise? Notwithstanding that samples are predominantly from late-stage disease, unbiased genome-wide mRNA expression and network analyses of different human joint tissues have identified highly-relevant/hub genes and/or inflammatory processes in OA54,57,60,63,68. While there are, not surprisingly, some differences between joint-tissue compartments and even cells with a given tissue, the commonly identified dysregulated inflammatory pathways in OA include: IL1, IL6, IL8, IL12, IL17, TNF, CCL2, M1/M2 macrophage polarization and Th1 and Th17 CD4 T-cells. There is supporting evidence from pre-clinical tissue culture and/or animal models, that inhibiting or ablating any of the above identified inflammatory pathways can modify onset, progression and/or severity of various aspects of joint-wide structural and/or symptomatic OA10,38.
Despite the above evidence, clinical trials targeting some of these pathways in OA patients have been disappointing69,70. Does this mean inflammation is not as important an OA-therapeutic target as it appeared? In answering this, it is noteworthy that variable outcomes are reported in different OA disease models and model systems e.g. IL1 in mono-idodacetate-, meniscal destabilization-, meniscectomy- and collagenase-induced OA71–74; IL6 in post-traumatic and age-associated OA75,76. This pre-clinical data suggests that the specific inflammatory pathways involved and therefore usefully therapeutically targeted, may differ depending on the disease model i.e. it is disease-phenotype-dependent. This is consistent with human OA patient data showing differences in inflammatory dysregulation e.g. in hip versus knee OA77, in knee OA in males versus females78, and the cytokines that correlate with different aspects of knee OA pain46,79. Even within a given OA population, distinct inflammatory cell/cytokine patient clusters can be identified e.g. in those presenting for knee replacement80.
This OA inflammatory heterogeneity may, at least in part explain the poor outcomes from clinical trials70. Just as in recognized inflammatory arthropathies81,82, a more nuanced approach to anti-inflammatory therapy in OA may be needed for example selecting patients with a more inflammatory clinical phenotype, or potentially using biomarker analysis to identify particular inflammatory molecular endotypes within OA sub-populations. This is supported by serendipitous data from a large trial of IL1β inhibition for myocardial infarction in patients with elevated C-reactive protein, that demonstrated a significant reduction of incident or worsening OA symptoms and rates of total knee and hip replacement83. While not designed with OA-relevant structure and symptom outcomes, this study strongly suggests that targeting the right inflammatory pathways in the right patients at the right time may make significant inroads to successfully treating OA (Table 1). As with many of the potential OA therapeutic approaches based on targeting pathophysiologic pathways, developing biomarkers to identify different patient cohorts is a key research imperative.
Table 1.
Evidence in favour of targeting inflammation in osteoarthritis
|
Targeting synovial fibrosis to treat OA – Mohit Kapoor
The synovial membrane is a thin membrane that surrounds articular joints and comprises two main layers; a cellular intima and underlying collagen I-rich sub-intima84,85. The synovium is required to maintain joint integrity, lubrication and homeostasis. While the majority of OA research has focused on mechanisms associated with articular cartilage degeneration, it is now believed that changes in the synovium may play an active role in driving OA pathogenesis. During OA, synovium presents with different synoviopathies, including inflammatory (see preceding section), hyperplastic, fibrotic and detritus-rich forms86.
Synovial fibrosis is characterized by excessive ECM deposition and contributes to the joint stiffness and pain associated with OA. Underlying endogenous mechanisms associated with synovial fibrosis are not well characterized and several critical questions remain to be answered: (1) Why and how does synovial fibrosis occur?; (2) Which cell types are responsible for the initiation and progression of synovial fibrosis?; (3) How can we control fibrosis to reduce structural and symptomatic OA?
Fibrosis is speculated to occur due to uncontrolled tissue repair responses, prolonged inflammatory insults, and cross talk between a variety of endogenous pro-fibrotic molecular and cellular mechanisms. The synovium consists of cells including, but not limited to, fibroblast-like synoviocytes (FLS) and macrophages. FLS are the key cell type of the synovium that is responsible for maintaining homeostatic functions and promoting inflammatory and fibrogenic responses (reviewed in87). FLS respond to a wide array of stimuli in the OA joint microenvironment resulting in increased proliferation, migratory capacity and acquiring a myofibroblast like phenotype (Figure 3), all contributing towards increased ECM deposition and fibrogenic responses in the synovium. Some of the key triggers associated with FLS activation include transforming growth factor-beta (TGFβ), cartilage wear products, Wnt/beta-catenin signaling pathway, and hypoxia inducible factor-1 alpha88–94, among others. TGFβ is a major pro-fibrotic mediator known to activate FLS and induce their transition to highly contractile myofibroblast-like cells that are believed to be involved in excessive ECM accumulation in the synovium95. Targeting TGFβ and its signaling to achieve anti-fibrotic effects has proved to be complex due to its homeostatic roles in other joint tissues such as the articular cartilage96,97.
Figure 3:
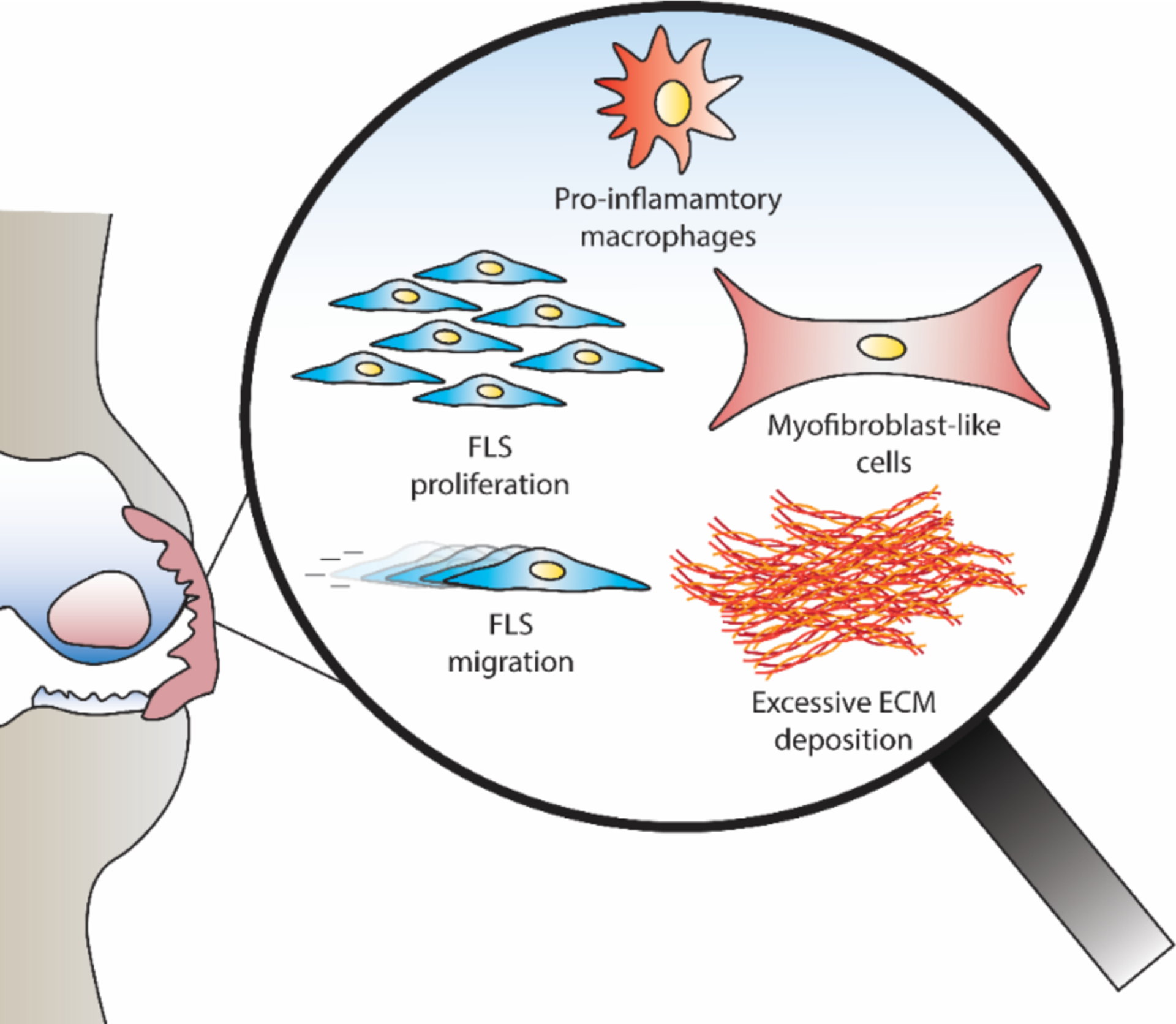
Fibroblast like synoviocytes (FLS) and macrophages are key cell types present in the synovium. FLS exhibit increased proliferation and migration, and also acquire a myofibroblast like phenotype, resulting in the excessive ECM deposition in the synovium during osteoarthritis.
At this point, clinical evidence to support efficacy of anti-fibrotic therapies to minimize the degree of joint destruction during OA requires further investigation. One could speculate that controlling inflammation during early stages of OA initiation and development could indirectly minimize the pro-fibrotic events and associated pathological mechanisms. Another potential therapeutic modality may include the simultaneous targeting of inflammation and fibrosis using a combination of anti-inflammatory and anti-fibrotic agent(s). In this context, Pirfenidone, an anti-inflammatory and anti-fibrotic drug currently approved for the treatment of idiopathic pulmonary fibrosis98, has been shown to attenuate synovial fibrosis and delay the progression of OA in a preclinical model99. Future clinical trials would help determine the therapeutic efficacy of such drugs and agents in reducing fibrosis and minimizing the degree of joint destruction during OA.
The Wnt family of proteins are also involved in OA pathogenesis100–102 and have drawn significant attention in the OA field. For instance, a phase II study of Lorecivivint (SM04690) an inhibitor of intranuclear kinases CDC-like kinase 2 (CLK2) and dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) that modulates the Wnt pathway, shows initial efficacy in improving pain, function and joint space narrowing in patients with unilateral moderate to severe symptomatic knee OA103, with phase III trials currently underway104. Preclinical studies using SM04690, shows cartilage protective effects in vivo105. It would therefore be of interest to investigate the potential of SM04690 to reduce synovial inflammation and fibrosis in preclinical animal models and in clinical trials. In this context, intra-articular injection with XAV-939, a small-molecule inhibitor of Wnt/β-catenin signaling, reduces the degree of synovitis and cartilage degeneration in a mouse model of knee OA in vivo, and reduces proliferation and collagen synthesis of FLS treated with XAV-939 in vitro90; however, it remains to be determined if XAV-939-induced cartilage protective effects are driven by reductions in synovitis or vice versa.
Research on synovium as a key driver of OA pathogenesis is garnering significant attention in the OA field. To better understand the contribution of synovium and to devise adequate therapeutic strategies to control processes such as synovial fibrosis and inflammation in joint destruction, it is essential to identify and understand the roles of individual synovial cell types and subpopulations that are involved in the initiation and progression of OA. The emergence of single cell sequencing, high throughput omics technologies and advanced bioinformatics provides an excellent opportunity to deep dive into the role of the synovium in OA pathogenesis. Applying these technologies to investigations using pre-clinical animal models and well characterized human OA synovial samples will allow for the identification of putative therapeutic targets that may limit pathological processes in OA, including synovial fibrosis.
Targeting regeneration of cartilage to treat osteoarthritis - Tonia L. Vincent
OA textbooks frequently describe OA as a disease determined by the balance between catabolic and anabolic pathways activated within the tissue. Historically this was based on the observation that some chondrocytes appeared to display an exuberant synthetic response in OA tissue when measuring uptake of radiolabelled sulfate (indicative of synthesis of sulfated proteoglycans), whilst other chondrocytes were in regions of the matrix completely devoid of proteoglycan106. Later evidence was based on transcriptomic analyses where evidence of new matrix synthesis was often upregulated alongside catabolic enzymes and other inflammatory molecules107,108. The textbooks shied away from describing the anabolic response as evidence for regenerative activities, as the pervasive view had been that articular cartilage was incapable of repairing itself.
Mounting evidence in the past 10 years indicates that this paradigm is incorrect. Not only is there evidence from careful prospective arthroscopy studies that many focal cartilage lesions heal spontaneously (reviewed in109), but also that established OA can repair if the hostile mechanical environment of the joint is corrected e.g. by joint distraction, using an external frame attached above and below the joint, or by high tibial osteotomy110,111. Such studies demonstrate MRI-proven regeneration of cartilage-like tissue even where the erosion was down to the underlying bone112. In the case of joint distraction, which is typically in situ for 6 weeks, this tissue appears to be maintained up to 2 years after removal of the frame and is associated with a sustained clinical benefit over longer periods113. Whether the tissue that is produced is true hyaline cartilage with newly synthesized type II collagen or fibrocartilage (type I collagen rich) is unclear. This fact may also be irrelevant so long as it shows resilience over time with associated symptom improvement. Several studies point to minimal type II collagen incorporation after skeletal maturity, which does not appear to change with OA114,115.
Molecular mechanisms that underly regenerative activities in articular cartilage are being revealed. These fall into two broad areas: the identity, control and activity of progenitor cells in the joint that mediate cartilage repair, and tissue factors that signal the injury and activate the tissue repair response. Meachim famously described two distinct repair responses in articular cartilage; one which was ‘intrinsic’ to the cartilage, mediated by cells that resided within the substance of the tissue, and a second which was mediated by cells migrating from the underlying bone marrow (especially where the osteochondral junction had been breached), which he called ‘extrinsic’ repair. Intrinsic repair was thought to be mechanically superior, producing excellent integrated hyaline cartilage compared with the fibrocartilaginous response elicited by extrinsic bone marrow derived cells. He concluded that, as extrinsic repair was rapid, you needed to suppress this to enable intrinsic repair to occur (reviewed in116). Several groups since this time have described pluripotent progenitor cells that can be expanded in vitro from cells derived from the articular cartilage117–119. Repair cells have also been identified in the synovium, periosteum and synovial fluid taken from human OA joints120–122. Such cells may be quite distinct from classical mesenchymal stem cells derived from the bone marrow. For instance, synovial derived cells are marked by being GDF5 positive, arising from those cells that originated from the joint interzone during development123. Collectively these results are consistent with Meachim’s idea that intrinsic repair cells are distinct from those derived from the bone marrow. It also raises the possibility that orthopedic procedures such as Pridie drilling may be encouraging extrinsic repair by stimulating bone marrow derived MSCs, and this may not be in the long term interests of the tissue.
The tissue injury signals likely originate from the cartilage matrix itself. The pericellular matrix, a region immediately surrounding individual chondrocytes in the tissue is rich in the proteoglycan perlecan, upon whose heparan sulfate chains are attached a number of heparin binding growth factors124. Four such growth factors were identified by proteomic analysis, including FGF2, CCN2 bound to latent TGFβ, hepatoma derived growth factor and CCN1125,126. These are released immediately in response to mechanical injury of the tissue by a mechanism that involves a localized increase in sodium concentration as water is squeezed out of the compressed tissue127. This is sufficient to displace the growth factors from their pericellular matrix binding sites and allow their binding to high affinity cell surface receptors (Figure 4). In osteoarthritis, when proteolytic activity causes loss of the negatively charged aggrecan from the tissue, the sodium is no longer held in the tissue and mechanical compression is unable to generate the concentration of sodium required to release growth factors127. These results indicate that proteolytic loss of aggrecan in OA suppresses intrinsic repair just at the time it is most needed. FGF2 and TGFβ are the best described of these molecules and are known chondroprotective and chondrogenic molecules in preclinical and in vitro studies128–130. They are also implicated in repair responses in other tissues such as the skin131.
Figure 4. Balance of pro-regenerative and mechano-inflammatory responses in articular cartilage with abnormal mechanical load.
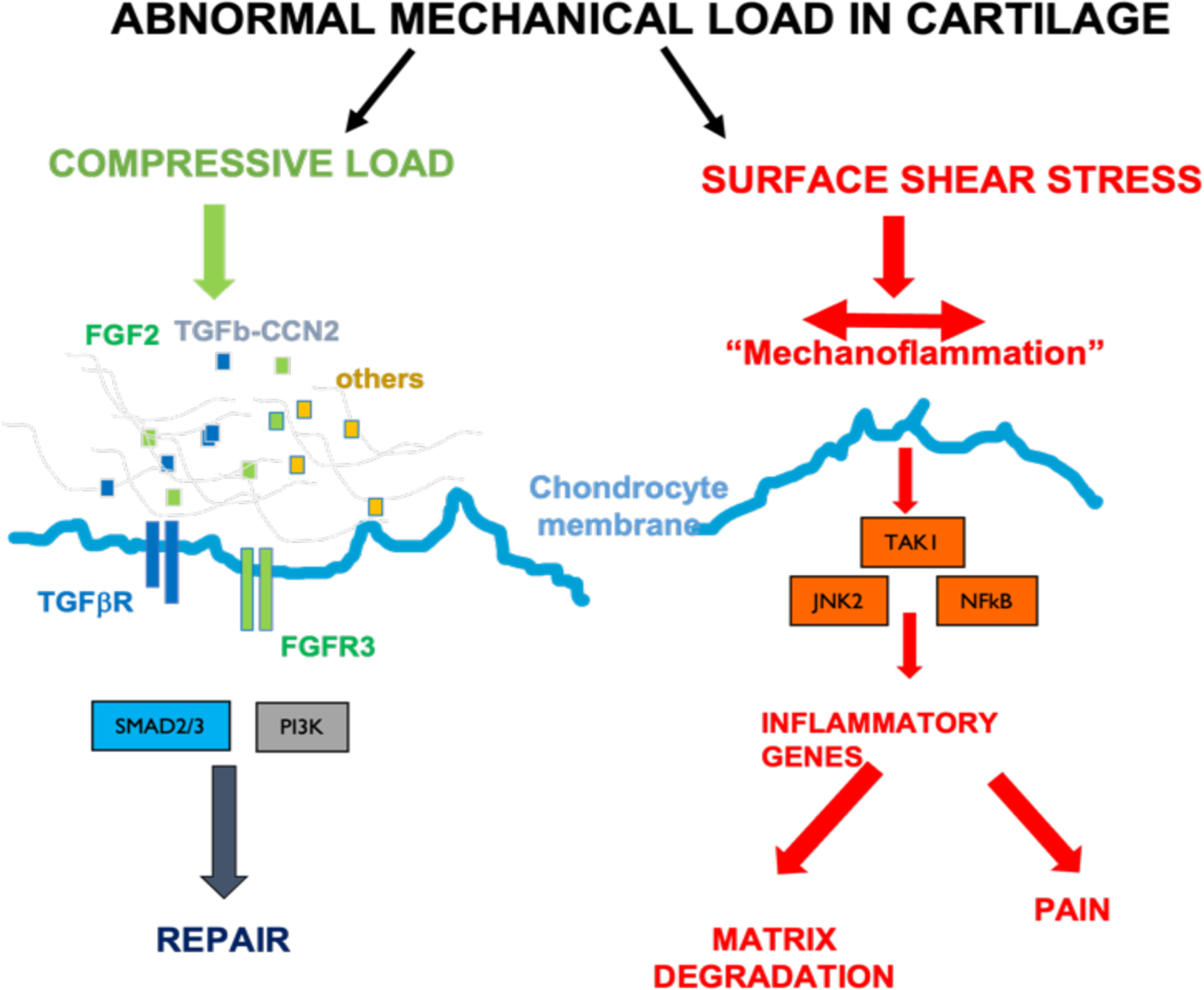
Compressive load leads to sodium dependent release of pericellular matrix growth factors, which drive repair and chondroprotection through a variety of intracellular signalling pathways. Surface shear stress (perpendicular to compressive load) leads to activation of TGFβ-activated kinase 1 (TAK1) dependent inflammatory signalling and results in nerve growth factor regulation (driving pain) and matrix degradation.
The clinical relevance of TGFβ and FGF family members in cartilage repair is strongly supported by agnostic evidence arising from recent genome wide association studies in OA. To date, polymorphic variants associated with expression of eight members of the TGFβ family (TGFβ1, TGFβ2, LTBP1, LTBP3, GDF5, SMAD3, ACVR1, BMP5) and two members of the FGF family (FGF18, FGFR3) have been documented132–136. Where described, these are hypomorphic variants associated with increased OA, thus confirming their chondroprotective role in human OA. Other growth factor families also emerge, such as the Wnts (DOT1L; WNT9a, WNT1, WNT10a) and TGFα, a ligand for the epidermal growth factor receptor (EGFR). Very few recognizable ‘inflammatory’ genes are identified in these analyses raising the possibility that OA could be viewed primarily as a disease of failed repair (Table 2).
Table 2.
Evidence in favour of targeting cartilage regeneration in osteoarthritis
|
So will this change our approach to disease modification in OA? Evidence to support this concept is already emerging. To date, the only successful structure modifying pharmacological trial is that using intraarticular injections of sprifermin, a truncated form of FGF18. In this extended 3 year trial, (the study was originally 2 years137), there was evidence of a delay in cartilage loss in the sprifermin group and increased cartilage thickness measured in the affected and unaffected regions of the joint138. Although not reaching its primary endpoint for symptoms, a recent post hoc analysis, considering a ‘subgroup at risk’ of progression (defined by lower joint space width and higher pain at baseline), was able to demonstrate both structural and symptomatic improvement over the study period139. Collectively these data appear to represent a striking U-turn for molecular pathogenesis and target discovery in OA.
Targeting bone remodeling to treat OA – Tamara Alliston
In the healthy joint, subchondral bone provides mechanical and vascular support to overlying avascular cartilage140. Given this vital role in joint structure, function, and shape, it is not surprising that subchondral bone is thought to be both a target and a driver of osteoarthritis progression.
Human imaging studies demonstrate that changes in the subchondral bone compartment both precede and predict degradative changes in overlaying cartilage; with the effects of OA apparent on the thin subchondral bone plate, subchondral trabecular bone, and the surrounding bone marrow. First, subchondral bone loss early in OA, due to increased bone remodeling by osteoclasts and osteoblasts, is followed by radiographic detection of sclerosis, or thickening, of the subchondral bone plate and trabecular bone141 142. Second, machine learning analysis of magnetic resonance imaging (MRI) in the Osteoarthritis Initiative identify changes in subchondral bone shape as one of the earliest known predictors of OA, as well as joint pain143. Third, the appearance of bone marrow lesions (BML) in clinical MRI is associated with joint pain and increased cartilage loss144. Histologically, BMLs are associated with greater cartilage degeneration, increased marrow vasculature, fibrosis, and edema, and increased osteoid deposition and osteocyte density145,146. BMLs appear to be a response to subchondral bone microdamage, resulting from traumatic injury or mechanical insufficiency of subchondral bone. Therefore, the bony sclerosis, changes in joint shape, and bone marrow lesions in OA subchondral bone are diagnostically and clinically significant because they can be detected early in OA and can predict OA progression and joint pain.
These changes in subchondral bone motivate bone-targeting therapies to prevent or treat OA, some of which have been tested clinically, but still with limited success. In an effort to abrogate the hyperactive subchondral bone remodeling that occurs early in OA, osteoclast-inhibitory bisphosphonates have been evaluated in clinical trials for OA. Bisphosphonates may indeed be therapeutically beneficial in a subset of non-overweight individuals with early stage OA147, even though this clinical benefit was not observed in a meta-analysis of randomized control trials148. In clinical trials, cathepsin K inhibitors, which suppress bone remodeling, prevent changes in subchondral bone and cartilage, but were ineffective for treating OA pain149. Other bone-targeting agents with potential to impact OA progression, including estrogen, PTH, TGFβ antagonists, and calcitonin, show benefits in pre-clinical studies, but have yet to be tested in randomized clinical trials, or to show reproducible clinical benefits in diverse human cohorts140.
Discrepancies between the success of pre-clinical and clinical studies still limit the clinical application of bone-targeting agents to treat OA. A more precise stratification of OA subtypes, perhaps with the help of new genetic, serum, and imaging biomarkers, could improve the identification of patients who would benefit from bone-targeting therapies. Another possibility is that bone-targeting therapies for OA are still missing a critical cellular target – osteocytes.
The cellular mechanisms by which changes in subchondral bone propel cartilage degeneration have largely been attributed to osteoblasts and osteoclasts. However, the contribution of osteocytes, the most abundant bone cell type, in OA has been overlooked until recently140. Over the past ten years, the dynamic role of bone-embedded osteocytes in bone homeostasis has become more clear. Osteocytes couple mechanical demands to bone resorption and deposition by osteoclasts and osteoblasts through mechanosensitive secretion of Rank Lignad (RANKL;TNFSf11) and Sclerostin, respectively150–152. Furthermore, through the process of perilacunar/canalicular remodeling (PLR), osteocytes directly resorb their local ECM) by secreting acid and proteases such as MMP13 and cathepsin K, and then later deposit new ECM. PLR maintains systemic mineral homeostasis, bone quality, and the intricate lacunocanalicular network (LCN), which enables osteocytes to communicate with one another and the vascular supply153–155. Since cartilage relies on subchondral bone for mechanical and vascular support, understanding the impact of OA on osteocytes, and vice versa, became a critical question.
Several lines of evidence support a causal role for osteocytes in the progression of OA. Relative to non-OA cadaveric controls, subchondral bone from human OA surgical retrieval specimens shows several hallmarks of deregulated osteocyte function, including LCN degeneration, collagen disorganization, and heterogeneous mineralization156. Furthermore, osteocyte-intrinsic defects in genetically modified mice were sufficient to exacerbate cartilage degeneration and mimic several features of human OA subchondral bone. Specifically, mice with an osteocyte-targeted ablation of the PLR enzyme MMP13 exhibit cartilage degeneration, accompanied by sclerotic subchondral bone with degenerated LCN, disorganized collagen, and heterogeneous mineralization156. Similar results are observed upon osteocyte-intrinsic inhibition of TGFβ signaling through targeted ablation of the TGFβ type II receptor (TβRIIocy−/−)157. Recently, several genes in the osteocyte transcriptome were shown to have significant associations with OA in a human GWAS study, including MEPE, TSKU, SEMA3F, SEMA3G and SEMA7A, which are expressed in osteocytes but not in chondrocytes158. Thus data from human clinical and genetic studies, as well as from mouse models with osteocyte-intrinsic mutations, support a causal role of osteocyte dysfunction in OA.
While the mechanisms by which osteocytes affect cartilage remain to be determined, the importance of their participation in subchondral bone and cartilage homeostasis, and joint disease, is clear. Computational modeling predicts that degeneration of the osteocyte LCN in aged or TβRIIocy−/− mouse bone, relative to young or control bone, is sufficient to compromise bone mechanosensitivity and solute transport159. Either of these mechanisms could compromise cartilage integrity. Uncoupling bone remodeling from mechanical stimuli could contribute to subchondral bone sclerosis. LCN degeneration could interfere with the ability of bone vasculature to support cartilage. Interestingly, the downregulation of osteocytic TGFβ signaling and MMP13 is a common feature in human OA subchondral bone, the TβRIIocy−/− mouse model, aging mouse bone, and wild type mouse bone following meniscal ligamentous injury156,157,159 (Table 3). Determining whether the relationship between OA and osteocytic TGFβ signaling and MMP13 are correlative or causal in aging will require further investigation. Either way, these observations highlight the need to consider the joint-compartment-specific effects of each factor. For example, agents that suppress MMP13 may protect cartilage from proteolytic degradation, while simultaneously interfering with osteocyte functions required for cartilage homeostasis. Unfortunately, diagnostic markers of osteocyte function, or osteocyte-specific therapies currently do not exist. Although the “osteocyte transcriptome“ identifies genes that are specific to osteocytes, relative to other skeletal cell types, more work is needed158. Continued efforts to understand osteocyte function and regulation, in the healthy skeleton and in aging and disease, are needed to develop new strategies to monitor and target subchondral bone to prevent or treat joint disease.
Table 3.
Osteocyte-intrinsic inhibition of MMP13 or TGFβ signaling is sufficient to mimic several hallmarks of human osteoarthritis
| Human OA | MMP13ocy−/− | TβRIIocy−/− | |
|---|---|---|---|
| Cartilage Degeneration | ✓ | ✓ | ✓ |
| Subchondral Sclerosis | ✓ | ✓ | ✓ |
| Thickened Subchondral Plate | ✓ | ✓ | ✓ |
| Collagen Disorganization | ✓ | ✓ | |
| Mineral Heterogeneity | ✓ | ✓ | |
| Degenerated Osteocyte LCN | ✓ | ✓ | ✓ |
| Impaired Mechanosensitivity | ✓ | ||
| Altered TGFβ signaling | ✓ | ✓ | |
| Altered MMP13 activity | ✓ | ✓ | ✓ |
Targeting aging and cell senescence to treat OA - Richard F. Loeser
There is no doubt that aging processes, both systemic and within joint tissues, contribute to the pathophysiology of OA. The prevalence of radiographic and symptomatic OA in all the commonly affected joints, including hands, hips, knees, and spine, increases with increasing age160. The prevalence of OA and the pain and loss of function associated with it make OA one of the leading causes of disability in older adults worldwide161. What is not clear is precisely how aging promotes the development of OA or if targeting aging processes would slow or halt OA progression. This essay will focus on cell senescence in OA and address the question of whether targeting senescent cells would be of therapeutic benefit.
Nine hallmarks of aging have been proposed that include genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, stem cell exhaustion, altered intercellular communication, and perhaps most importantly, cellular senescence162. Many, if not all these aging hallmarks have been investigated in the context of joint tissue aging, with the majority of the published work focused on articular cartilage and its resident cell, the chondrocyte163. A common denominator to the hallmarks of aging is cell senescence, as the other hallmarks can either lead to senescence or result from the senescent state.
The literature to date strongly supports cell senescence as a major factor contributing to age-related diseases including OA164,165. Cell senescence can be defined as a state of growth arrest that prevents further cell division and results in typical phenotypic changes162,164. Importantly, cell senescence is not just a phenomenon seen after replicating cells have stopped dividing due to telomere shortening. Senescent cells contribute to tissue development during embryogenesis, tissue repair during wound healing, and suppress tumor formation by preventing the propagation of damaged cells164,166. Cell senescence can result from multiple chronic stresses that result in an accumulation of cellular damage, many of which are relevant to factors thought to contribute to OA (Figure 5). DNA damage is a central mediator of cell senescence and has been shown to induce senescence in chondrocytes167. The OA joint has often been referred to as a “chronic wound” with irreparable damage, the type of environment that can promote cell senescence. Chronic signaling from inflammatory factors such as cytokines has been proposed to result in “stress-induced” senescence resulting from a feed forward loop168. This could be a very relevant mechanism for senescence in the joint.
Figure 5:
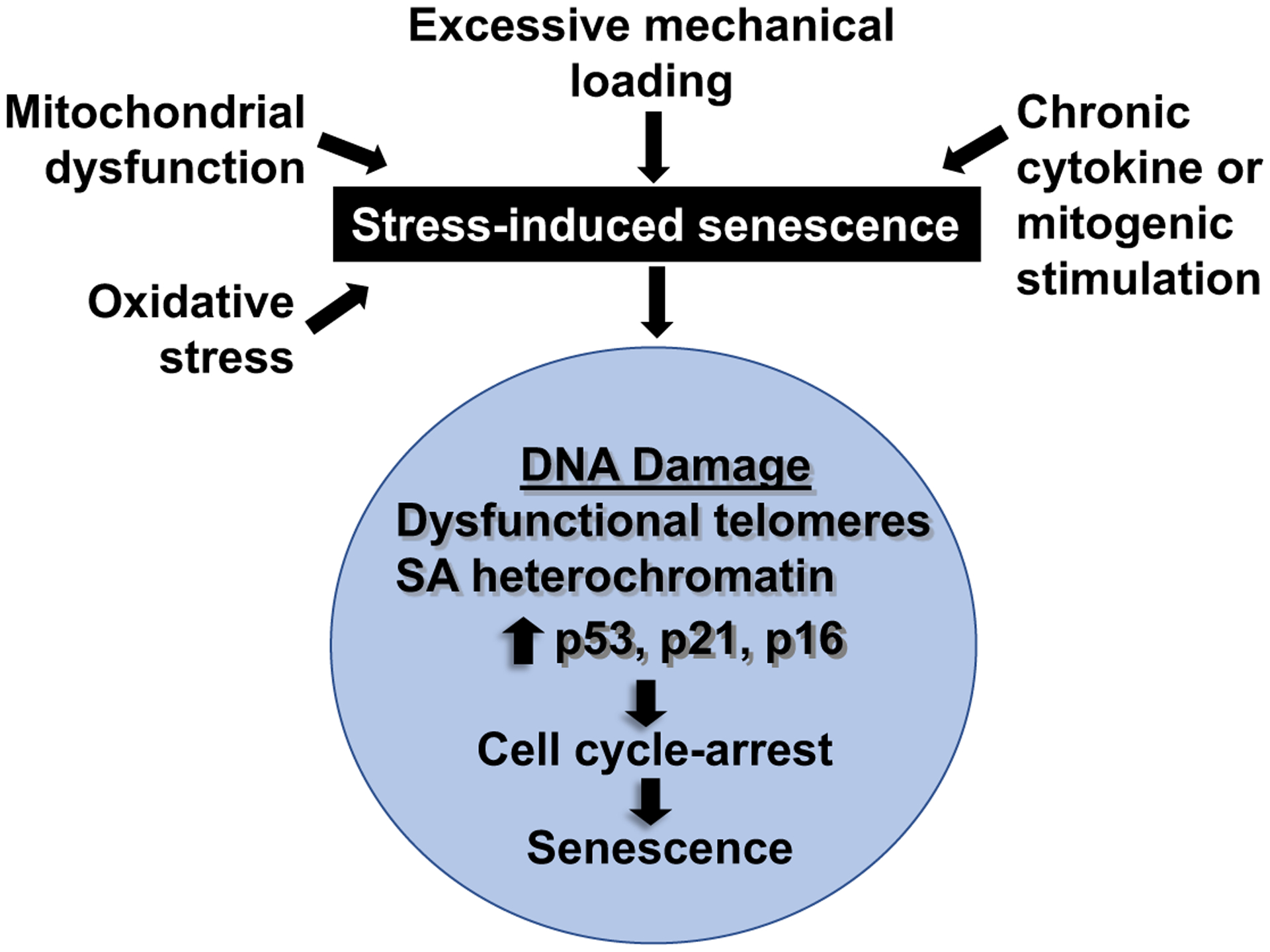
Factors that promote stress-induced senescence
A central mechanism by which senescence contributes to disease is through the production of inflammatory cytokines and matrix degrading enzymes, referred to as the senescence-associated secretory phenotype or SASP164. Many of the proinflammatory mediators and matrix degrading enzymes considered to be SASP factors (Table 4) are found in the OA joint54,169,170 and may directly contribute to the tissue changes seen in OA. Increased expression of p16INK4a, a cell cycle inhibitor, is considered one of the most reliable markers of cell senescence164. p16INK4a mRNA expression was found to be significantly increased with age in murine cartilage and in primary human chondrocytes from cadaveric tissue donors and this correlated with expression of the SASP transcripts IGFBP3, MMP1 and MMP13171. However, deletion of p16INK4a in chondrocytes of adult mice did not mitigate SASP expression and did not alter the severity of age-related OA, suggesting the effects of chondrocyte senescence on OA are most likely driven by the production of SASP factors and not by the loss of chondrocyte replicative function that occurs with increased p16INK4a.
Table 4.
Senescence-Associated Secretory Phenotype (SASP) Factors Most Relevant to OA
| Class | Component |
|---|---|
| Cytokines | IL1, IL6, IL7, IL13, IL15, IL17, OSM |
| Chemokines | IL8 (CXCL15), GRO (CXCL1), MCP1 (CCL2), MIP1α (CCL3), ENA78 (CCXL5) |
| Other inflammatory molecules | TGFβ, MIF |
| Growth factors, regulators | EGF, FGF2, HGF, VEGF, SDF1 (CXCL12), NGF, IGFBP2, IGFBP3, IGFBP4, IGFBP6, IGFBP7 |
| Proteases and regulators | MMP1, MMP3, MMP10, MMP12, MMP13, MMP14, TIMP1, TIMP2, PAI1 (SERPINE1), PAI2 (SERPINEB2), CTSB |
| Receptors and ligands | OPG (TNFRSF11B), sTNFRI (TNFRSF1B), sTNFRII (TNFRSF1A), FAS, uPAR (PLAUR), EGFR |
| Non-protein molecules | PGE2, nitric oxide, reactive oxygen species |
| Insoluble factors | fibronectins, collagens |
Adapted from Gorgoulis et al, Cell 2019; 179:813–827. Abbreviations: bFGF, basic fibroblast growth factor; EGF, epidermal growth factor; ENA, epithelial neutrophil-activating peptide; GRO, growth-related oncogene; HGF, hepatocyte growth factor; IGFBP, insulin-like growth factor binding protein; IL, interleukin; MCP, monocyte chemotactic protein; MIF, macrophage inhibitory factor; MIP, macrophage inflammatory protein; MMP, matrix metalloproteinase; NGF, nerve growth factor; OPG, osteoprotegerin; OSM, oncostatin M; PAI, plasminogen activator inhibitor; PGE2, prostaglandin E2; SDF, stromal cell-derived factor; TGF, transforming growth factor; sTNFR, soluble tumor necrosis factor receptor; TIMP, tissue inhibitor of metalloproteinases; uPAR, urokinase-type plasminogen activator receptor; VEGF, vascular endothelial growth factor.
It has been suggested that senescent progenitor cells may be present in aged cartilage and release inflammatory mediators, including IL8, to promote the SASP172. Transplantation of senescent cells into mouse knee joints was shown to promote OA-like changes173. NFκB is considered a key regulator of the SASP164 and a recent study found activation of NFκB signaling in mice promoted age-related OA and production of SASP factors174. Other important regulators of the SASP include C/EBPβ, STAT3, and GATA4, while the SASP may be inhibited by activity of FOXOs164,166. Importantly, all these mediators have also been implicated in OA pathogenesis175–179, providing further support for a strong connection between SASP regulation and the development of OA.
Perhaps the strongest evidence for a causal role of senescent joint tissue cells in OA comes from studies that have demonstrated reduced OA severity in the anterior cruciate ligament transection model of post-traumatic OA and in age-related OA in mice treated with small molecules called “senolytics” to selectively kill senescent cells or using a molecular approach to kill senescent cells expressing p16180,181. However, translation of this pre-clinical work to the treatment of human OA has not yet been realized. The senolytic compound UBX0101 that reduced OA severity in mice, did not achieve a significant reduction in WOMAC knee pain compared to a placebo when tested as an intra-articular therapy in a 12-week Phase 2 clinical study in humans (UNITY Biotechnology Announces 12-week data from UBX0101 Phase 2 Clinical Study in Patients with Painful Osteoarthritis of the Knee | Unity Biotechnology).
There are many possible reasons why a single injection of a senolytic drug would fail in a short-term trial with pain as the outcome. Clearly, further work is needed to: a) define an OA phenotype that may be more responsive to an intervention targeting senescent cells by discovering one or more biomarkers of joint tissue senescence; b) decide on the timing in the disease course of when such an intervention would be most useful; c) establish how many doses of the senolytic would be needed, and d) determine what outcome measures in early phase studies would best predict efficacy. Alternatives to killing senescent joint tissue cells with a senolytic also need to be developed such as “senomorphics” that target the production of SASP factors182. Although the link between aging and the development of OA is well established, and the underlying mechanisms are becoming clearer, the field is still not at the point where targeting a specific aging process to slow OA progression and improve symptoms is possible.
Conclusions
Molecular pathogenesis is a relatively new scientific discipline in OA. The scientific community has needed to overcome significant hurdles associated with working with matrix-rich pauci-cellular tissues, and to develop pre-clinical models of disease that are accepted as being clinically informative. In recent years, additional molecular insights have emerged from agnostic õmic studies such as genome wide association studies. Being a highly prevalent condition, such studies can be performed in very large numbers to elucidate common pathways associated with OA risk135. As demonstrated above, there has been a rapid expansion of cellular and molecular pathogenic understanding across multiple tissues of the OA joint. But how likely is it that this knowledge will deliver translational success?
Epidemiology, perhaps the oldest discipline in OA research, has much to teach us. It reminds us that mechanical strain remains a principal driver of OA development and progression183. It also teaches us that the disease is heterogeneous; having a variable course and symptoms184. Using all sources of data available, we should be able to improve our chances of success but as independently highlighted by the authors of the individual sections, there are key questions that need constant re-inforcement if we are to translate our ever more detailed understanding of OA pathophysiology to treatment and patient care.
Which of the pathways are targetable?
If targetable, do they deliver a clinically meaningful effect?
Does the target have benefits across all tissues of the joint or is it tissue-specific (see conflicting roles of MMP13 in bone and cartilage above)?
Do several targets need to be delivered in combination?
Will treatments work when the adverse mechanical environment of the joint is uncorrected?
Are the described processes active in all patients at all stages of disease, or will patient stratification be necessary?
We don’t have all the answers yet, but progress has been rapid, there is a recognized urgency across funders and patient groups, and as this review demonstrates, the scientific community is working collaboratively and imaginatively to combat this challenging disease.
Acknowledgements:
Dr Patrick Haubruck, Raymond Purves Research Laboratories for producing Figure 2 using biorender.com.
Funding:
The research of the authors related to the specific topics explored in this review were supported by funding from numerous sources:
TLV Centre for OA Pathogenesis Versus Arthritis (grant no. 20205 and 21621); TA; MK Tier 1 Canada Research Chair Award (#950-232237) and Tony and Shari Fell Platinum Chair in Arthritis Research; RFL National Institute on Aging RO1 AG044034; LT VA grants 21776 and 22194; CBL Australian National Health and Medical Research Council (NHMRC: Project Grant APP1045890), the Hillcrest Foundation through Perpetual Philanthropies, and Arthritis Australia.
Footnotes
Competing interests: The authors have no potential or apparent conflicts of interest with regard to this work. No benefits in any form have been or will be received from a commercial party related directly or indirectly to the subject of this manuscript.
Patient and Public Involvement: While papers and studies relating to patients are cited in the manuscript, patients/consumers were not involved in the design, conduct, or writing of this manuscript.
Data and materials availability:
All data associated with this study are present in the paper.
References
- 1.Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396(10258):1204–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cross M, Smith E, Hoy D, et al. The global burden of hip and knee osteoarthritis: estimates from the global burden of disease 2010 study. Ann Rheum Dis. 2014;73(7):1323–1330. [DOI] [PubMed] [Google Scholar]
- 3.Ackerman IN, Pratt C, Gorelik A, Liew D. Projected burden of osteoarthritis and rheumatoid arthritis in Australia: a population-level analysis. Arthritis Care Res (Hoboken). 2018;70(6):877–883. [DOI] [PubMed] [Google Scholar]
- 4.Hunter DJ, Nicolson PJA, Little CB, Robbins SR, Wang X, Bennell KL. Developing strategic priorities in osteoarthritis research: Proceedings and recommendations arising from the 2017 Australian Osteoarthritis Summit. BMC Musculoskelet Disord. 2019;20(1):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bedenbaugh AV, Bonafede M, Marchlewicz EH, Lee V, Tambiah J. Real-world health care resource utilization and costs among US patients with knee osteoarthritis compared with controls. Clinicoecon Outcomes Res. 2021;13:421–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huizinga JL, Stanley EE, Sullivan JK, et al. Societal cost of opioid use in symptomatic knee osteoarthritis patients in the United States. Arthritis Care Res. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bannuru RR, Osani MC, Vaysbrot EE, et al. OARSI guidelines for the non-surgical management of knee, hip, and polyarticular osteoarthritis. Osteoarthritis Cartilage. 2019;27(11):1578–1589. [DOI] [PubMed] [Google Scholar]
- 8.Cook D, Brown D, Alexander R, et al. Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. 2014;13(6):419–431. [DOI] [PubMed] [Google Scholar]
- 9.Little CB, Fosang AJ. Is cartilage matrix breakdown an appropriate therapeutic target in osteoarthritis--insights from studies of aggrecan and collagen proteolysis? Curr Drug Targets. 2010;11(5):561–575. [DOI] [PubMed] [Google Scholar]
- 10.Little CB, Hunter DJ. Post-traumatic osteoarthritis: from mouse models to clinical trials. Nat Rev Rheumatol. 2013;9(8):485–497. [DOI] [PubMed] [Google Scholar]
- 11.Soul J, Barter MJ, Little CB, Young DA. OATargets: a knowledge base of genes associated with osteoarthritis joint damage in animals. Ann Rheum Dis. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollander AP, Pidoux I, Reiner A, Rorabeck C, Bourne R, Poole AR. Damage to type II collagen in aging and osteoarthritis starts at the articular surface, originates around chondrocytes, and extends into the cartilage with progressive degeneration. J Clin Invest. 1995;96(6):2859–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lohmander LS, Atley LM, Pietka TA, Eyre DR. The release of crosslinked peptides from type II collagen into human synovial fluid is increased soon after joint injury and in osteoarthritis. Arthritis Rheum. 2003;48(11):3130–3139. [DOI] [PubMed] [Google Scholar]
- 14.Billinghurst RC, Dahlberg L, Ionescu M, et al. Enhanced cleavage of type II collagen by collagenases in osteoarthritic articular cartilage. J Clin Invest. 1997;99(7):1534–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim NH, Kashiwagi M, Visse R, et al. Reactive-site mutants of N-TIMP-3 that selectively inhibit ADAMTS-4 and ADAMTS-5: biological and structural implications. Biochem J. 2010;431(1):113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neuhold LA, Killar L, Zhao W, et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J Clin Invest. 2001;107(1):35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Little CB, Barai A, Burkhardt D, et al. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009;60(12):3723–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295(5564):2387–2392. [DOI] [PubMed] [Google Scholar]
- 19.Johnson AR, Pavlovsky AG, Ortwine DF, et al. Discovery and characterization of a novel inhibitor of matrix metalloprotease-13 that reduces cartilage damage in vivo without joint fibroplasia side effects. J Biol Chem. 2007;282(38):27781–27791. [DOI] [PubMed] [Google Scholar]
- 20.Karsdal MA, Madsen SH, Christiansen C, Henriksen K, Fosang AJ, Sondergaard BC. Cartilage degradation is fully reversible in the presence of aggrecanase but not matrix metalloproteinase activity. Arthritis Res Ther. 2008;10(3):R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pratta M, Yao W, Decicco C, et al. Aggrecan protects cartilage collagen from proteolytic cleavage. J Biol Chem. 2003;278:45539–45545. [DOI] [PubMed] [Google Scholar]
- 22.Sandy JD, Flannery CR, Neame PJ, Lohmander LS. The structure of aggrecan fragments in human synovial fluid. Evidence for the involvement in osteoarthritis of a novel proteinase which cleaves the Glu 373-Ala 374 bond of the interglobular domain. J Clin Invest. 1992;89:1512–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tortorella MD, Burn TC, Pratta MA, et al. Purification and cloning of aggrecanase-1: a member of the ADAMTS family of proteins. Science. 1999;284(5420):1664–1666. [DOI] [PubMed] [Google Scholar]
- 24.Abbaszade I, Liu RQ, Yang F, et al. Cloning and characterization of ADAMTS11, an aggrecanase from the ADAMTS family. J Biol Chem. 1999;274(33):23443–23450. [DOI] [PubMed] [Google Scholar]
- 25.Hurskainen TL, Hirohata S, Seldin MF, Apte SS. ADAM-TS5, ADAM-TS6, and ADAM-TS7, novel members of a new family of zinc metalloproteases. General features and genomic distribution of the ADAM-TS family. J Biol Chem. 1999;274(36):25555–25563. [DOI] [PubMed] [Google Scholar]
- 26.Glasson SS, Askew R, Sheppard B, et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005;434(7033):644–648. [DOI] [PubMed] [Google Scholar]
- 27.Stanton H, Rogerson FM, East CJ, et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature. 2005;434(7033):648–652. [DOI] [PubMed] [Google Scholar]
- 28.Ismail HM, Yamamoto K, Vincent TL, Nagase H, Troeberg L, Saklatvala J. Interleukin-1 acts via the JNK-2 signaling pathway to induce aggrecan degradation by human chondrocytes. Arthritis Rheumatol. 2015;67(7):1826–1836. [DOI] [PubMed] [Google Scholar]
- 29.Chockalingam PS, Sun W, Rivera-Bermudez MA, et al. Elevated aggrecanase activity in a rat model of joint injury is attenuated by an aggrecanase specific inhibitor. Osteoarthritis Cartilage. 2011;19(3):315–323. [DOI] [PubMed] [Google Scholar]
- 30.Brebion F, Gosmini R, Deprez P, et al. Discovery of GLPG1972/S201086, a potent, selective, and orally bioavailable ADAMTS-5 inhibitor for the treatment of osteoarthritis. J Med Chem. 2021;64(6):2937–2952. [DOI] [PubMed] [Google Scholar]
- 31.Clement-Lacroix P, Little CB, Smith MM, et al. Pharmacological characterization of GLPG1972/S201086, a potent and selective small molecule inhibitor of ADAMTS5. Osteoarthritis Cartilage. 2021. [DOI] [PubMed] [Google Scholar]
- 32.Santamaria S, Cuffaro D, Nuti E, et al. Exosite inhibition of ADAMTS-5 by a glycoconjugated arylsulfonamide. Sci Rep. 2021;11(1):949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brenneis C, Serruys B, Van Belle T, et al. Structural and symptomatic benefit of a half-live extended, systemically applied anti-ADAMTS-5 inhibitor (M6495). Osteoarthritis Cartilage. 2018;26:S299–S300. [Google Scholar]
- 34.Larkin J, Lohr T, Elefante L, et al. The highs and lows of translational drug development: antibody-mediated inhibition of ADAMTS-5 for osteoarthritis disease modification. Osteoarthrits Cartilage. 2014;22:S483–S484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santamaria S ADAMTS-5: A difficult teenager turning 20. Int J Exp Pathol. 2020;101(1–2):4–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Lange-Brokaar BJ, Ioan-Facsinay A, Yusuf E, et al. Evolution of synovitis in osteoarthritic knees and its association with clinical features. Osteoarthritis Cartilage. 2016;24(11):1867–1874. [DOI] [PubMed] [Google Scholar]
- 37.Wang X, Hunter DJ, Jin X, Ding C. The importance of synovial inflammation in osteoarthritis: current evidence from imaging assessments and clinical trials. Osteoarthritis Cartilage. 2018;26(2):165–174. [DOI] [PubMed] [Google Scholar]
- 38.Soul J, Barter MJ, Little CB, Young DA. OATargets: a knowledge base of genes associated with osteoarthritis joint damage in animals. Ann Rheum Dis. 2020;80(3):376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benito MJ, Veale DJ, FitzGerald O, van den Berg WB, Bresnihan B. Synovial tissue inflammation in early and late osteoarthritis. Ann Rheum Dis. 2005;64(9):1263–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith MD, Triantafillou S, Parker A, Youssef PP, Coleman M. Synovial membrane inflammation and cytokine production in patients with early osteoarthritis. J Rheumatol. 1997;24(2):365–371. [PubMed] [Google Scholar]
- 41.Klein-Wieringa IR, de Lange-Brokaar BJ, Yusuf E, et al. Inflammatory cells in patients with endstage knee osteoarthritis: a comparison between the synovium and the infrapatellar fat pad. J Rheumatol. 2016;43(4):771–778. [DOI] [PubMed] [Google Scholar]
- 42.Macchi V, Stocco E, Stecco C, et al. The infrapatellar fat pad and the synovial membrane: an anatomo-functional unit. J Anat. 2018;233(2):146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sae-Jung T, Leearamwat N, Chaiseema N, et al. The infrapatellar fat pad produces interleukin-6-secreting T cells in response to a proteoglycan aggrecan peptide and provides dominant soluble mediators different from that present in synovial fluid. Int J Rheum Dis. 2021;24(6):834–846. [DOI] [PubMed] [Google Scholar]
- 44.Atukorala I, Kwoh CK, Guermazi A, et al. Synovitis in knee osteoarthritis: a precursor of disease? Ann Rheum Dis. 2016;75(2):390–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ayral X, Pickering EH, Woodworth TG, Mackillop N, Dougados M. Synovitis: a potential predictive factor of structural progression of medial tibiofemoral knee osteoarthritis -- results of a 1 year longitudinal arthroscopic study in 422 patients. Osteoarthritis Cartilage. 2005;13(5):361–367. [DOI] [PubMed] [Google Scholar]
- 46.Li L, Li Z, Li Y, Hu X, Zhang Y, Fan P. Profiling of inflammatory mediators in the synovial fluid related to pain in knee osteoarthritis. BMC Musculoskelet Disord. 2020;21(1):99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nees TA, Rosshirt N, Zhang JA, et al. Synovial cytokines significantly correlate with osteoarthritis-related knee pain and disability: inflammatory mediators of potential clinical relevance. J Clin Med. 2019;8(9):1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neogi T, Guermazi A, Roemer F, et al. Association of joint inflammation with pain sensitization in knee osteoarthritis: the multicenter osteoarthritis study. Arthritis Rheumatol. 2016;68(3):654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lieberthal J, Sambamurthy N, Scanzello CR. Inflammation in joint injury and post-traumatic osteoarthritis. Osteoarthritis Cartilage. 2015;23(11):1825–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosshirt N, Trauth R, Platzer H, et al. Proinflammatory T cell polarization is already present in patients with early knee osteoarthritis. Arthritis Res Ther. 2021;23(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scanzello CR, Umoh E, Pessler F, et al. Local cytokine profiles in knee osteoarthritis: elevated synovial fluid interleukin-15 differentiates early from end-stage disease. Osteoarthritis Cartilage. 2009;17(8):1040–1048. [DOI] [PubMed] [Google Scholar]
- 52.MacFarlane LA, Yang H, Collins JE, et al. Association of Changes in Effusion-Synovitis With Progression of Cartilage Damage Over Eighteen Months in Patients With Osteoarthritis and Meniscal Tear. Arthritis Rheumatol. 2019;71(1):73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vincent TL. Mechanoflammation in osteoarthritis pathogenesis. Semin Arthritis Rheum. 2019;49:S36–38. [DOI] [PubMed] [Google Scholar]
- 54.Chou CH, Jain V, Gibson J, et al. Synovial cell cross-talk with cartilage plays a major role in the pathogenesis of osteoarthritis. Sci Rep. 2020;10(1):10868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Griffin TM, Scanzello CR. Innate inflammation and synovial macrophages in osteoarthritis pathophysiology. Clin Exp Rheumatol. 2019;37 Suppl 120(5):57–63. [PMC free article] [PubMed] [Google Scholar]
- 56.Menarim BC, Gillis KH, Oliver A, et al. Macrophage activation in the synovium of healthy and osteoarthritic equine joints. Front Vet Sci. 2020;7:568756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen Z, Ma Y, Li X, Deng Z, Zheng M, Zheng Q. The immune cell landscape in different anatomical structures of knee in osteoarthritis: a gene expression-based study. Biomed Res Int. 2020;2020:9647072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nees TA, Rosshirt N, Zhang JA, et al. T helper cell infiltration in osteoarthritis-related knee pain and disability. J Clin Med. 2020;9(8):2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Platzer H, Nees TA, Reiner T, et al. Impact of mononuclear cell infiltration on chondrodestructive MMP/ADAMTS production in osteoarthritic knee joints - an ex vivo study. J Clin Med. 2020;9(5):1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brophy RH, Tycksen ED, Sandell LJ, Rai MF. Changes in transcriptome-wide gene expression of anterior cruciate ligament tears based on time from injury. Am J Sports Med. 2016;44(8):2064–2075. [DOI] [PubMed] [Google Scholar]
- 61.Han D, Fang Y, Tan X, et al. The emerging role of fibroblast-like synoviocytes-mediated synovitis in osteoarthritis: An update. J Cell Mol Med. 2020;24(17):9518–9532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pap T, Dankbar B, Wehmeyer C, Korb-Pap A, Sherwood J. Synovial fibroblasts and articular tissue remodelling: Role and mechanisms. Semin Cell Dev Biol. 2020;101:140–145. [DOI] [PubMed] [Google Scholar]
- 63.Sandy JD, Chan DD, Trevino RL, Wimmer MA, Plaas A. Human genome-wide expression analysis reorients the study of inflammatory mediators and biomechanics in osteoarthritis. Osteoarthritis Cartilage. 2015;23(11):1939–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Visser HM, Mastbergen SC, Kozijn AE, et al. Metabolic dysregulation accelerates injury-induced joint degeneration, driven by local inflammation; an in vivo rat study. J Orthop Res. 2018;36(3):881–890. [DOI] [PubMed] [Google Scholar]
- 65.Pearson MJ, Herndler-Brandstetter D, Tariq MA, et al. IL-6 secretion in osteoarthritis patients is mediated by chondrocyte-synovial fibroblast cross-talk and is enhanced by obesity. Sci Rep. 2017;7(1):3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stannus O, Jones G, Cicuttini F, et al. Circulating levels of IL-6 and TNF-alpha are associated with knee radiographic osteoarthritis and knee cartilage loss in older adults. Osteoarthritis Cartilage. 2010;18(11):1441–1447. [DOI] [PubMed] [Google Scholar]
- 67.Stannus OP, Jones G, Blizzard L, Cicuttini FM, Ding C. Associations between serum levels of inflammatory markers and change in knee pain over 5 years in older adults: a prospective cohort study. Ann Rheum Dis. 2013;72(4):535–540. [DOI] [PubMed] [Google Scholar]
- 68.Li Z, Wang Q, Chen G, et al. Integration of gene expression profile data to screen and verify hub genes involved in osteoarthritis. Biomed Res Int. 2018;2018:9482726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Persson MSM, Sarmanova A, Doherty M, Zhang W. Conventional and biologic disease-modifying anti-rheumatic drugs for osteoarthritis: a meta-analysis of randomized controlled trials. Rheumatology (Oxford). 2018;57(10):1830–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oo WM, Little C, Duong V, Hunter DJ. The development of disease-modifying therapies for osteoarthritis (DMOADs): the evidence to date. Drug Des Devel Ther. 2021;15:2921–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Glasson SS. In vivo osteoarthritis target validation utilizing genetically-modified mice. Curr Drug Targets. 2007;8(2):367–376. [DOI] [PubMed] [Google Scholar]
- 72.Na HS, Park JS, Cho KH, et al. Interleukin-1-interleukin-17 signaling axis induces cartilage destruction and promotes experimental osteoarthritis. Front Immunol. 2020;11:730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nasi S, Ea HK, So A, Busso N. Revisiting the role of interleukin-1 pathway in osteoarthritis: interleukin-1α and −1β, and NLRP3 inflammasome are not involved in the pathological features of the murine menisectomy model of osteoarthritis. Front Pharmacol. 2017;8:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Dalen SC, Blom AB, Slöetjes AW, et al. Interleukin-1 is not involved in synovial inflammation and cartilage destruction in collagenase-induced osteoarthritis. Osteoarthritis Cartilage. 2017;25(3):385–396. [DOI] [PubMed] [Google Scholar]
- 75.de Hooge AS, van de Loo FA, Bennink MB, Arntz OJ, de Hooge P, van den Berg WB. Male IL-6 gene knock out mice developed more advanced osteoarthritis upon aging. Osteoarthritis Cartilage. 2005;13(1):66–73. [DOI] [PubMed] [Google Scholar]
- 76.Ryu JH, Yang S, Shin Y, Rhee J, Chun CH, Chun JS. Interleukin-6 plays an essential role in hypoxia-inducible factor 2alpha-induced experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum. 2011;63(9):2732–2743. [DOI] [PubMed] [Google Scholar]
- 77.Grieshaber-Bouyer R, Kammerer T, Rosshirt N, et al. Divergent mononuclear cell participation and cytokine release profiles define hip and knee osteoarthritis. J Clin Med. 2019;8(10):1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kriegova E, Manukyan G, Mikulkova Z, et al. Gender-related differences observed among immune cells in synovial fluid in knee osteoarthritis. Osteoarthritis Cartilage. 2018;26(9):1247–1256. [DOI] [PubMed] [Google Scholar]
- 79.Leung YY, Huebner JL, Haaland B, Wong SBS, Kraus VB. Synovial fluid pro-inflammatory profile differs according to the characteristics of knee pain. Osteoarthritis Cartilage. 2017;25(9):1420–1427. [DOI] [PubMed] [Google Scholar]
- 80.Labinsky H, Panipinto PM, Ly KA, et al. Multiparameter Analysis Identifies Heterogeneity in Knee Osteoarthritis Synovial Responses. Arthritis Rheumatol. 2020;72(4):598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Letarouilly JG, Salmon JH, Flipo RM. Factors affecting persistence with biologic treatments in patients with rheumatoid arthritis: a systematic literature review. Expert Opin Drug Saf. 2021;20(9):1087–1094. [DOI] [PubMed] [Google Scholar]
- 82.Noviani M, Feletar M, Nash P, Leung YY. Choosing the right treatment for patients with psoriatic arthritis. Ther Adv Musculoskelet Dis. 2020;12:1759720x20962623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schieker M, Conaghan PG, Mindeholm L, et al. Effects of interleukin-1β inhibition on incident hip and knee replacement : exploratory analyses from a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2020;173(7):509–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Singh JA, Arayssi T, Duray P, Schumacher HR. Immunohistochemistry of normal human knee synovium: a quantitative study. Ann Rheum Dis. 2004;63(7):785–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Smith MD. The normal synovium. The open rheumatology journal. 2011;5:100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oehler S, Neureiter D, Meyer-Scholten C, Aigner T. Subtyping of osteoarthritic synoviopathy. Clin Exp Rheumatol. 2002;20(5):633–640. [PubMed] [Google Scholar]
- 87.Maglaviceanu A, Wu B, Kapoor M. Fibroblast-like synoviocytes: role in synovial fibrosis associated with osteoarthritis. Wound Repair Regen. 2021;29(4):642–649. [DOI] [PubMed] [Google Scholar]
- 88.Estell EG, Silverstein AM, Stefani RM, et al. Cartilage wear particles induce an inflammatory response similar to cytokines in human fibroblast-like synoviocytes. J Orthop Res. 2019;37(9):1979–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kuo SJ, Liu SC, Huang YL, et al. TGF-beta1 enhances FOXO3 expression in human synovial fibroblasts by inhibiting miR-92a through AMPK and p38 pathways. Aging. 2019;11(12):4075–4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lietman C, Wu B, Lechner S, et al. Inhibition of Wnt/beta-catenin signaling ameliorates osteoarthritis in a murine model of experimental osteoarthritis. JCI Insight. 2018;3(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Remst DF, Blom AB, Vitters EL, et al. Gene expression analysis of murine and human osteoarthritis synovium reveals elevation of transforming growth factor beta-responsive genes in osteoarthritis-related fibrosis. Arthritis Rheumatol. 2014;66(3):647–656. [DOI] [PubMed] [Google Scholar]
- 92.Silverstein AM, Stefani RM, Sobczak E, et al. Toward understanding the role of cartilage particulates in synovial inflammation. Osteoarthritis Cartilage. 2017;25(8):1353–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vaamonde-Garcia C, Malaise O, Charlier E, et al. 15-Deoxy-Delta-12, 14-prostaglandin J2 acts cooperatively with prednisolone to reduce TGF-beta-induced pro-fibrotic pathways in human osteoarthritis fibroblasts. Biochem Pharmacol. 2019;165:66–78. [DOI] [PubMed] [Google Scholar]
- 94.Zhang L, Zhang L, Huang Z, et al. Increased HIF-1alpha in knee osteoarthritis aggravate synovial fibrosis via fibroblast-like synoviocyte pyroptosis. Oxid Med Cell Longev. 2019;2019:6326517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mattey DL, Dawes PT, Nixon NB, Slater H. Transforming growth factor beta 1 and interleukin 4 induced alpha smooth muscle actin expression and myofibroblast-like differentiation in human synovial fibroblasts in vitro: modulation by basic fibroblast growth factor. Ann Rheum Dis. 1997;56(7):426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Finnson KW, Chi Y, Bou-Gharios G, Leask A, Philip A. TGF-b signaling in cartilage homeostasis and osteoarthritis. Front Biosci. 2012;4:251–268. [DOI] [PubMed] [Google Scholar]
- 97.Remst DF, Blaney Davidson EN, van der Kraan PM. Unravelling osteoarthritis-related synovial fibrosis: a step closer to solving joint stiffness. Rheumatology (Oxford). 2015;54(11):1954–1963. [DOI] [PubMed] [Google Scholar]
- 98.Kim ES, Keating GM. Pirfenidone: a review of its use in idiopathic pulmonary fibrosis. Drugs. 2015;75(2):219–230. [DOI] [PubMed] [Google Scholar]
- 99.Wei Q, Kong N, Liu X, et al. Pirfenidone attenuates synovial fibrosis and postpones the progression of osteoarthritis by anti-fibrotic and anti-inflammatory properties in vivo and in vitro. J Transl Med. 2021;19(1):157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dell’Accio F, De Bari C, El Tawil NM, et al. Activation of WNT and BMP signaling in adult human articular cartilage following mechanical injury. Arthritis Res Ther. 2006;8(5):R139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dell’accio F, De Bari C, Eltawil NM, Vanhummelen P, Pitzalis C. Identification of the molecular response of articular cartilage to injury, by microarray screening: Wnt-16 expression and signaling after injury and in osteoarthritis. Arthritis Rheum. 2008;58(5):1410–1421. [DOI] [PubMed] [Google Scholar]
- 102.van den Bosch MH, Blom AB, Sloetjes AW, et al. Induction of canonical Wnt signaling by synovial overexpression of selected Wnts leads to protease activity and early osteoarthritis-like cartilage damage. Am J Pathol. 2015;185(7):1970–1980. [DOI] [PubMed] [Google Scholar]
- 103.Yazici Y, McAlindon TE, Gibofsky A, et al. Lorecivivint, a novel intraarticular CDC-like kinase 2 and dual-specificity tyrosine phosphorylation-regulated kinase 1A inhibitor and Wnt pathway modulator for the treatment of knee osteoarthritis: a phase II randomized trial. Arthritis Rheumatol. 2020;72(10):1694–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.A study utilizing patient-reported and radiographic outcomes and evaluating the safety and efficacy of lorecivivint (SM04690) for the treatment of moderately to severely symptomatic knee osteoarthritis (STRIDES-X-ray). Updated: October 8, 2021. https://clinicaltrials.gov/ct2/show/NCT03928184. Accessed October 22, 2021.
- 105.Deshmukh V, O’Green AL, Bossard C, et al. Modulation of the Wnt pathway through inhibition of CLK2 and DYRK1A by lorecivivint as a novel, potentially disease-modifying approach for knee osteoarthritis treatment. Osteoarthritis Cartilage. 2019;27(9):1347–1360. [DOI] [PubMed] [Google Scholar]
- 106.Collins DH, Meachim G. Sulphate (35SO4) fixation by human articular cartilage compared in the knee and shoulder joints. Ann Rheum Dis. 1961;20:117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Aigner T, Fundel K, Saas J, et al. Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 2006;54(11):3533–3544. [DOI] [PubMed] [Google Scholar]
- 108.Soul J, Dunn SL, Anand S, et al. Stratification of knee osteoarthritis: two major patient subgroups identified by genome-wide expression analysis of articular cartilage. Ann Rheum Dis. 2018;77(3):423–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dell’Accio F, Vincent TL. Joint surface defects: clinical course and cellular response in spontaneous and experimental lesions. Eur Cells Mater. 2010;20:210–217. [DOI] [PubMed] [Google Scholar]
- 110.Wiegant K, van Roermund PM, Intema F, et al. Sustained clinical and structural benefit after joint distraction in the treatment of severe knee osteoarthritis. Osteoarthritis Cartilage. 2013;21(11):1660–1667. [DOI] [PubMed] [Google Scholar]
- 111.Parker DA, Beatty KT, Giuffre B, Scholes CJ, Coolican MRJ. Articular cartilage changes in patients with osteoarthritis after osteotomy. Am J Sports Med. 2011;39(5):1039–1045. [DOI] [PubMed] [Google Scholar]
- 112.Jansen MP, Maschek S, van Heerwaarden RJ, et al. Changes in cartilage thickness and denuded bone area after knee joint distraction and high tibial osteotomy - post-hoc analyses of two randomized controlled trials. J Clin Med. 2021;10(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.van der Woude J-TAD, Wiegant K, Van Roermund PM, et al. Five-Year follow-up of knee joint distraction: clinical benefit and cartilaginous tissue repair in an open uncontrolled prospective study. Cartilage. 2017;8(3):263–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ariosa-Morejon Y, Santos A, Fischer R, et al. Age-dependent changes in protein incorporation into collagen-rich tissues of mice by in vivo pulsed SILAC labelling Elife. 2021;10:e66635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Heinemeier KM, Schjerling P, Heinemeier J, et al. Radiocarbon dating reveals minimal collagen turnover in both healthy and osteoarthritic human cartilage. Sci Transl Med. 2016;8(346):346ra390. [DOI] [PubMed] [Google Scholar]
- 116.Stockwell RA. Biology of cartilage cells. 1979. [Google Scholar]
- 117.Alsalameh S, Amin R, Gemba T, Lotz M. Identification of mesenchymal progenitor cells in normal and osteoarthritic human articular cartilage. Arthritis Rheum. 2004;50(5):1522–1532. [DOI] [PubMed] [Google Scholar]
- 118.Dowthwaite GP, Bishop JC, Redman SN, et al. The surface of articular cartilage contains a progenitor cell population. J Cell Sci. 2004;117(Pt 6):889–897. [DOI] [PubMed] [Google Scholar]
- 119.Fellows CR, Williams R, Davies IR, et al. Characterisation of a divergent progenitor cell sub-populations in human osteoarthritic cartilage: the role of telomere erosion and replicative senescence. Sci Rep. 2017;7:41421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.De Bari C, Dell’Accio F, Tylzanowski P, Luyten FP. Multipotent mesenchymal stem cells from adult human synovial membrane. Arthritis Rheum. 2001;44(8):1928–1942. [DOI] [PubMed] [Google Scholar]
- 121.De Bari C, Dell’Accio F, Vanlauwe J, et al. Mesenchymal multipotency of adult human periosteal cells demonstrated by single-cell lineage analysis. Arthritis Rheum. 2006;54(4):1209–1221. [DOI] [PubMed] [Google Scholar]
- 122.Jones EA, English A, Henshaw K, et al. Enumeration and phenotypic characterization of synovial fluid multipotential mesenchymal progenitor cells in inflammatory and degenerative arthritis. Arthritis Rheum. 2004;50(3):817–827. [DOI] [PubMed] [Google Scholar]
- 123.Roelofs AJ, Zupan J, Riemen AHK, et al. Joint morphogenetic cells in the adult mammalian synovium. Nat Commun. 2017;8:15040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Poole CA, Flint MH, Beaumont BW. Chondrons in cartilage: ultrastructural analysis of the pericellular microenvironment in adult human articular cartilages. J Orthop Res. 1987;5(4):509–522. [DOI] [PubMed] [Google Scholar]
- 125.Tang X, Muhammad H, McLean C, et al. Connective tissue growth factor contributes to joint homeostasis and osteoarthritis severity by controlling the matrix sequestration and activation of latent TGFβ. Ann Rheum Dis. 2018;77(9):1372–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vincent T, Hermansson M, Bolton M, Wait R, Saklatvala J. Basic FGF mediates an immediate response of articular cartilage to mechanical injury. Proc Natl Acad Sci USA. 2002;99(12):8259–8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Keppie SJ, Mansfield JC, Tang X, et al. Matrix-bound growth factors are released upon cartilage compression by an aggrecan-dependent sodium flux that is lost in osteoarthritis. Function (Oxf). 2021;2(5):zqab037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chia S-L, Sawaji Y, Burleigh A, et al. Fibroblast growth factor 2 is an intrinsic chondroprotective agent that suppresses ADAMTS-5 and delays cartilage degradation in murine osteoarthritis. Arthritis Rheum. 2009;60(7):2019–2027. [DOI] [PubMed] [Google Scholar]
- 129.Huang L, Yi L, Zhang C, et al. Synergistic effects of FGF-18 and TGF-β3 on the chondrogenesis of human adipose-derived mesenchymal stem cells in the pellet culture. Stem Cells Int. 2018;2018:7139485–7139410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Valverde-Franco G, Binette JS, Li W, et al. Defects in articular cartilage metabolism and early arthritis in fibroblast growth factor receptor 3 deficient mice. Hum Mol Genet. 2006;15(11):1783–1792. [DOI] [PubMed] [Google Scholar]
- 131.Moulin V Growth factors in skin wound healing. Eur J Cell Biol. 1995;68(1):1–7. [PubMed] [Google Scholar]
- 132.Styrkarsdottir U, Stefansson OA, Gunnarsdottir K, et al. GWAS of bone size yields twelve loci that also affect height, BMD, osteoarthritis or fractures. Nat Commun. 2019;10(1):2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Styrkarsdottir U, Lund SH, Thorleifsson G, et al. Meta-analysis of Icelandic and UK data sets identifies missense variants in SMO, IL11, COL11A1 and 13 more new loci associated with osteoarthritis. Nat Genet. 2018;50(12):1681–1687. [DOI] [PubMed] [Google Scholar]
- 134.Zengini E, Hatzikotoulas K, Tachmazidou I, et al. Genome-wide analyses using UK Biobank data provide insights into the genetic architecture of osteoarthritis. Nat Genet. 2018;50(4):549–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Boer CG, Hatzikotoulas K, Southam L, et al. Deciphering osteoarthritis genetics across 826,690 individuals from 9 populations. Cell. 2021;184(18):4784–4818 e4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Castano-Betancourt MC, Evans DS, Ramos YF, et al. Novel genetic variants for cartilage thickness and hip osteoarthritis. PLoS Genet. 2016;12(10):e1006260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lohmander LS, Hellot S, Dreher D, et al. Intraarticular sprifermin (recombinant human fibroblast growth factor 18) in knee osteoarthritis: a randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 2014;66(7):1820–1831. [DOI] [PubMed] [Google Scholar]
- 138.Hochberg MC, Guermazi A, Guehring H, et al. Effect of intra-articular sprifermin vs placebo on femorotibial foint cartilage thickness in patients with osteoarthritis: the FORWARD randomized clinical trial. JAMA. 2019;322(14):1360–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Guehring H, Moreau F, Daelken B, et al. The effects of sprifermin on symptoms and structure in a subgroup at risk of progression in the FORWARD knee osteoarthritis trial. Semin Arthritis Rheum. 2021;51(2):450–456. [DOI] [PubMed] [Google Scholar]
- 140.Zhu X, Chan YT, Yung PSH, Tuan RS, Jiang Y. Subchondral bone remodeling: a therapeutic target for osteoarthritis. Front Cell Dev Biol. 2020;8:607764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen Y, Hu Y, Yu YE, et al. Subchondral trabecular rod loss and plate thickening in the development of osteoarthritis. J Bone Miner Res. 2018;33(2):316–327. [DOI] [PubMed] [Google Scholar]
- 142.Goldring SR, Goldring MB. Changes in the osteochondral unit during osteoarthritis: structure, function and cartilage-bone crosstalk. Nat Rev Rheumatol. 2016;12(11):632–644. [DOI] [PubMed] [Google Scholar]
- 143.Kanthawang T, Bodden J, Joseph GB, et al. Obese and overweight individuals have greater knee synovial inflammation and associated structural and cartilage compositional degeneration: data from the osteoarthritis initiative. Skeletal Radiol. 2021;50(1):217–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Alliston T, Hernandez CJ, Findlay DM, Felson DT, Kennedy OD. Bone marrow lesions in osteoarthritis: What lies beneath. J Orthop Res. 2018;36(7):1818–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Muratovic D, Cicuttini F, Wluka A, et al. Bone marrow lesions detected by specific combination of MRI sequences are associated with severity of osteochondral degeneration. Arthritis Res Ther. 2016;18:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Muratovic D, Findlay DM, Cicuttini FM, Wluka AE, Lee YR, Kuliwaba JS. Bone matrix microdamage and vascular changes characterize bone marrow lesions in the subchondral bone of knee osteoarthritis. Bone. 2018;108:193–201. [DOI] [PubMed] [Google Scholar]
- 147.Hayes KN, Giannakeas V, Wong AKO. Bisphosphonate use is protective of radiographic knee osteoarthritis progression among those with low disease severity and being non-overweight: data from the osteoarthritis initiative. J Bone Miner Res. 2020;35(12):2318–2326. [DOI] [PubMed] [Google Scholar]
- 148.Vaysbrot EE, Osani MC, Musetti MC, McAlindon TE, Bannuru RR. Are bisphosphonates efficacious in knee osteoarthritis? A meta-analysis of randomized controlled trials. Osteoarthritis Cartilage. 2018;26(2):154–164. [DOI] [PubMed] [Google Scholar]
- 149.Conaghan PG, Bowes MA, Kingsbury SR, et al. Disease-modifying effects of a novel cathepsin K inhibitor in osteoarthritis: a randomized controlled trial. Ann Intern Med. 2020;172(2):86–95. [DOI] [PubMed] [Google Scholar]
- 150.Nakashima T, Hayashi M, Fukunaga T, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17(10):1231–1234. [DOI] [PubMed] [Google Scholar]
- 151.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17(10):1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Robling AG, Niziolek PJ, Baldridge LA, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283(9):5866–5875. [DOI] [PubMed] [Google Scholar]
- 153.Qing H, Ardeshirpour L, Pajevic PD, et al. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J Bone Miner Res. 2012;27(5):1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tang SY, Herber RP, Ho SP, Alliston T. Matrix metalloproteinase-13 is required for osteocytic perilacunar remodeling and maintains bone fracture resistance. J Bone Miner Res. 2012;27(9):1936–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26(2):229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mazur CM, Woo JJ, Yee CS, et al. Osteocyte dysfunction promotes osteoarthritis through MMP13-dependent suppression of subchondral bone homeostasis. Bone Res. 2019;7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bailey KN, Nguyen J, Yee CS, Dole NS, Dang A, Alliston T. Mechanosensitive control of articular cartilage and subchondral bone homeostasis in mice requires osteocytic transforming growth factor beta signaling. Arthritis Rheumatol. 2021;73(3):414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Youlten SE, Kemp JP, Logan JG, et al. Osteocyte transcriptome mapping identifies a molecular landscape controlling skeletal homeostasis and susceptibility to skeletal disease. Nat Commun. 2021;12(1):2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Schurman CA, Verbruggen SW, Alliston T. Disrupted osteocyte connectivity and pericellular fluid flow in bone with aging and defective TGF-beta signaling. Proc Natl Acad Sci U S A. 2021;118(25). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Anderson SA, Loeser RF. Why is osteoarthritis an age-related disease? Best practice & research. 2010;24(1):15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Safiri S, Kolahi AA, Cross M, et al. Prevalence, deaths, and disability-adjusted life years due to musculoskeletal disorders for 195 countries and territories 1990–2017. Arthritis Rheumatol. 2021;73(4):702–714. [DOI] [PubMed] [Google Scholar]
- 162.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12:412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gorgoulis V, Adams PD, Alimonti A, et al. Cellular senescence: defining a path forward. Cell. 2019;179(4):813–827. [DOI] [PubMed] [Google Scholar]
- 165.Jeon OH, David N, Campisi J, Elisseeff JH. Senescent cells and osteoarthritis: a painful connection. J Clin Invest. 2018;128(4):1229–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev. 2020;34(23–24):1565–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Copp ME, Flanders MC, Gagliardi R, et al. The combination of mitogenic stimulation and DNA damage induces chondrocyte senescence. Osteoarthritis Cartilage. 2021;29(3):402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chandrasekaran A, Idelchik M, Melendez JA. Redox control of senescence and age-related disease. Redox Biol. 2017;11:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2011;7(1):33–42. [DOI] [PubMed] [Google Scholar]
- 170.van den Bosch MHJ, van Lent P, van der Kraan PM. Identifying effector molecules, cells, and cytokines of innate immunity in OA. Osteoarthritis Cartilage. 2020;28(5):532–543. [DOI] [PubMed] [Google Scholar]
- 171.Diekman BO, Sessions GA, Collins JA, et al. Expression of p16(INK)(4a) is a biomarker of chondrocyte aging but does not cause osteoarthritis. Aging Cell. 2018:e12771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Liu W, Brodsky AS, Feng M, et al. Senescent tissue-resident mesenchymal stromal cells are an internal source of inflammation in human osteoarthritic cartilage. Front Cell Dev Biol. 2021;9:725071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Xu M, Bradley EW, Weivoda MM, et al. Transplanted senescent cells induce an osteoarthritis-like condition in mice. J Gerontol A Biol Sci Med Sci. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Catheline SE, Bell RD, Oluoch LS, et al. IKKβ–NF-κB signaling in adult chondrocytes promotes the onset of age-related osteoarthritis in mice. Sci Signal. 2021;14(701). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hirata M, Kugimiya F, Fukai A, et al. C/EBPbeta and RUNX2 cooperate to degrade cartilage with MMP-13 as the target and HIF-2alpha as the inducer in chondrocytes. Hum Mol Genet. 2012;21(5):1111–1123. [DOI] [PubMed] [Google Scholar]
- 176.Latourte A, Cherifi C, Maillet J, et al. Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann Rheum Dis. 2016;76(4):748–755. [DOI] [PubMed] [Google Scholar]
- 177.Kang D, Shin J, Cho Y, et al. Stress-activated miR-204 governs senescent phenotypes of chondrocytes to promote osteoarthritis development. Sci Transl Med. 2019;11(486). [DOI] [PubMed] [Google Scholar]
- 178.Matsuzaki T, Alvarez-Garcia O, Mokuda S, et al. FoxO transcription factors modulate autophagy and proteoglycan 4 in cartilage homeostasis and osteoarthritis. Sci Transl Med. 2018;10(428). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wang C, Shen J, Ying J, Xiao D, O’Keefe RJ. FoxO1 is a crucial mediator of TGF-β/TAK1 signaling and protects against osteoarthritis by maintaining articular cartilage homeostasis. Proc Natl Acad Sci U S A. 2020;117(48):30488–30497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jeon OH, Kim C, Laberge RM, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017;23(6):775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Faust HJ, Zhang H, Han J, et al. IL-17 and immunologically induced senescence regulate response to injury in osteoarthritis. J Clin Invest. 2020;130(10):5493–5507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Coryell PR, Diekman BO, Loeser RF. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat Rev Rheumatol. 2021;17(1):47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Felson DT. Osteoarthritis as a disease of mechanics. Osteoarthritis Cartilage. 2013;21(1):10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Leyland KM, Hart D, Javaid MK, et al. The natural history of radiographic knee osteoarthritis: A fourteen year population-based cohort study. Arthritis Rheum. 2012;64:2243–2251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data associated with this study are present in the paper.