

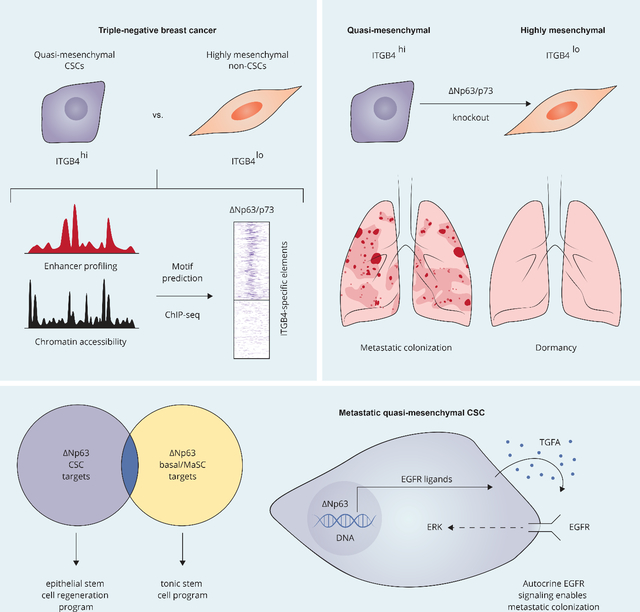
Summary
Cancer stem cells (CSCs) may serve as the cellular seeds of tumor recurrence and metastasis and can be generated via epithelial-mesenchymal transitions (EMTs). Isolating pure populations of CSCs is difficult because EMT programs generate multiple alternative cell states and phenotypic plasticity permits frequent interconversions between these states. Here, we used cell-surface expression of integrin β4 (ITGB4) to isolate highly enriched populations of human breast CSCs and identified the gene regulatory network operating in ITGB4+ CSCs. Specifically, we identified ΔNp63 and p73, the latter of which transactivates ΔNp63, as centrally important transcriptional regulators of quasi-mesenchymal CSCs that reside in an intermediate EMT state. We found that the transcriptional program controlled by ΔNp63 in CSCs is largely distinct from the one that it orchestrates in normal basal mammary stem cells and, instead, more closely resembles a regenerative epithelial stem cell response to wounding. Moreover, quasi-mesenchymal CSCs repurpose this program to drive metastatic colonization via autocrine EGFR signaling.
eTOC Blurb
Quasi-mesenchymal cancer stem cells (CSCs) are known to be highly metastatic. Lambert et al. demonstrate that ΔNp63 and p73 are key regulators of the quasi-mesenchymal CSC state. They show that these transcription factors control a regenerate epithelial stem cell program that drives metastatic colonization.
Graphical Abstract
Introduction
Cancer stem cells (CSCs) have been identified in numerous solid malignancies 1–4 and are thought to serve as the cellular seeds of both post-therapeutic recurrence at sites of primary tumor formation and, following dissemination to distant anatomical sites, of metastatic colonies 4–8. The epigenetic nature of the controls governing the formation of these cells is indicated by the bi-directional interconversions between CSCs and non-CSCs within tumor cell populations 9–12.
Variations of the cell-biological program termed epithelial-mesenchymal transition (EMT), drive epithelial cells into a range of more mesenchymal phenotypic states 13. We and others demonstrated that activation of the EMT program can lead to substantial increases in stem cells within carcinomas 14,15 and in normal epithelial tissues 16. However, demonstration of the precise relationship between the EMT and the stem cell state has been complicated by several factors, among them the various manifestations of the EMT program 17, which can generate cells residing in a number of intermediate, partially mesenchymal cell states 13,18–21. Ongoing phenotypic plasticity that enables extensive interconversions between the various intermediate quasi-mesenchymal states 13,19,22,23, further complicates attempts to isolate and characterize pure populations of CSCs.
Recent studies indicate that CSCs reside within an intermediate, quasi-mesenchymal phenotypic state 24–28. Indeed, quasi-mesenchymal carcinoma cells have been shown to initiate metastases with high efficiency 29, whereas more fully mesenchymal cells are limited in their colonization capacity and must proceed through some form of a mesenchymal-epithelial transition (MET) in order to form macrometastatic lesions 30–33. However, it remains unclear precisely how quasi-mesenchymal carcinoma cells differ from their highly mesenchymal counterparts and the underlying mechanisms that enable quasi-mesenchymal CSCs to initiate metastatic colonization are similarly obscure.
Because of the challenges in isolating pure populations of CSCs, the epigenetic configuration and underlying gene regulatory network of metastatic CSCs residing in a quasi-mesenchymal state(s) have yet to be resolved, including the critical regulators that establish and maintain their transcriptional network. In earlier work, the combination of CD44high/CD24low cell-surface markers was used to isolate mesenchymal carcinoma cells that exhibit stem cell activity 14,15, but this mesenchymal cell population is now known to harbor mixtures of cells residing at various points along the EMT spectrum. Recent work from our laboratory described an alternative, improved strategy for isolating populations of CSCs based on the cell-surface expression of integrin β4 (ITGB4) 28. In particular, ITGB4hi triple-negative human breast cancer cells were found to be highly enriched for CSCs and, at the same time, to display cellular and molecular features indicating their residence in an intermediate, quasi-mesenchymal cell state.
Here, we employed the SUM159 ITGB4hi human breast cancer cell line as a representative model for quasi-mesenchymal CSCs. This allowed us to define the detailed transcriptional network that supports the CSC state, to assess how this CSC network relates to the corresponding one operating in normal mammary stem cells, and determine how this epigenetic program enables metastatic colonization.
Results
p63 and p73 are transcriptional drivers of quasi-mesenchymal metastatic CSCs
Integrin-β4 (ITGB4; also termed CD104) is a cell-surface protein that can be used to isolate quasi-mesenchymal cancer stem cells (CSCs) 28. In the SUM159 human breast cancer line, ITGB4 can be used as a marker to separate two phenotypically stable cell populations – ITGB4lo and ITGB4hi – that exhibit little, if any, interconversion. ITGB4lo cells were found to be highly mesenchymal and weakly tumorigenic, whereas ITGB4hi cells were shown to reside in a quasi-mesenchymal state and to readily form tumors upon implantation at limiting dilutions 28. These properties, especially their limited phenotypic plasticity, made these cell populations a tractable experimental model to interrogate the underlying regulatory programs that distinguish quasi-mesenchymal CSCs from more mesenchymal carcinoma cells.
We first mapped the enhancer landscape and measured chromatin accessibility in both cell populations, using H3K27ac ChIP-seq and ATAC-seq, respectively (Figure 1A). The overall abundance and genomic distributions of this histone mark were similar between the ITGB4lo and ITGB4hi cells (Supplementary Figure 1A). Nonetheless, we identified 751 H3K27ac peaks that were significantly enriched in ITGB4hi cells and 561 significantly enriched in ITGB4lo cells (Supplementary Figure 1B). In order to identify sites of open, accessible chromatin, we employed ATAC-seq analysis 34, which revealed 5206 peaks that were uniquely present in the ITGB4hi cells and 4832 that were unique to the ITGB4lo cells (Supplementary Figure 1C).
Figure 1. p63 and p73 are transcriptional drivers of quasi-mesenchymal metastatic CSCs.
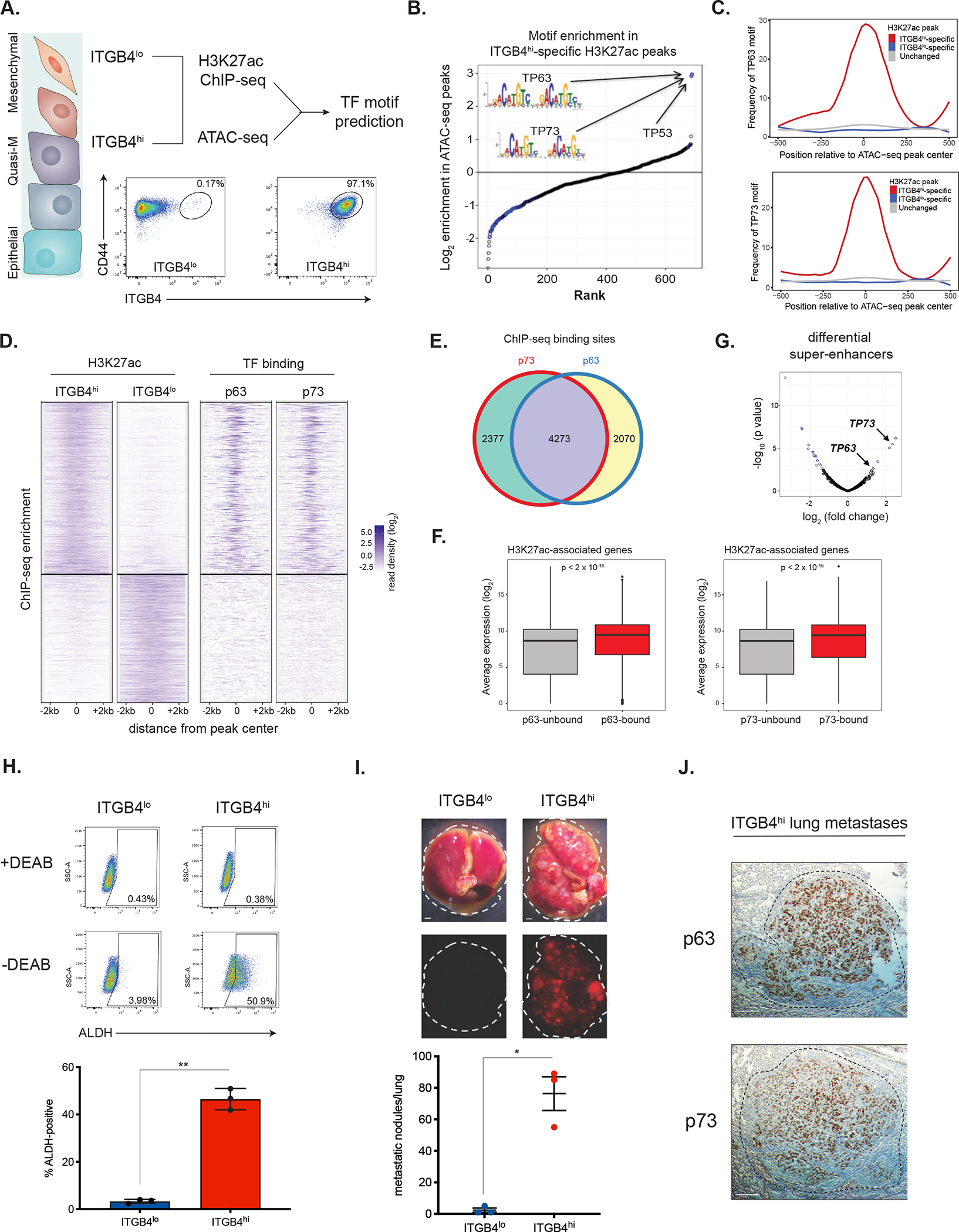
(A) Experimental approach used in this study.
(B) Motif enrichment scores across 547 open chromatin regions specific to ITGB4hi cells.
(C) Enrichment of TP63 and TP73 motifs in the indicated H3K27ac regions.
(D) Clustergram heatmap showing ITGB4hi- and ITGB4lo-specific H3K27ac ChIP-seq peaks (left) and p63/p73 binding at these sites in ITGB4hi cells (right).
(E) Venn diagram comparing p73 and p63 binding sites defined by ChIP-seq.
(F) Average gene expression of H3K27ac-associated genes bound or unbound by p63 (left) or p73 (right) in ITGB4hi cells.
(G) Differential super-enhancers (SEs) in the ITGB4hi vs. ITGB4lo cells. Statistically significant SEs are shown in blue.
(H) ALDH activity in ITGB4lo and ITGB4hi cells. Representative flow cytometry plots (above) and quantification (below). Data are ± SEM (n = 3), ** p ≤ 0.01.
(I) Metastatic colonization of ITGB4lo and ITGB4hi cells. Representative lung images (top) and quantification (below). Data are ± SEM (n = 3), * p ≤ 0.05. Scale bars, 1 mm.
(J) Immunohistochemistry (IHC) staining for p63 and p73 in representative ITGB4hi lung metastases. Scale bars, 100 μm.
See also Figure S1.
Integrating the enhancer landscape and chromatin accessibility datasets allowed us to identify potential master regulators of the transcriptional program operating in the quasi-mesenchymal ITGB4hi cells (Figure 1A). In particular, we focused on a unique set of 547 open chromatin regions, each encompassed within a larger DNA region of H3K27ac-positivity present specifically in the ITGB4hi cells. We predicted transcription factor (TF) binding events within these regions and then calculated the enrichment of these predicted binding sites relative to all genomic regions marked with H3K27ac. The three most significantly enriched motifs all belonged to members of the p53 family of transcription factors (p53, p63, and p73), which share similar DNA recognition motifs. These motifs were enriched nearly 4-fold above the next most-enriched TF motif and were enriched exclusively in ITGB4hi-specific H3K27ac peaks (Figure 1B and 1C).
Since the SUM159 cells used in these analyses harbor a mutation in the DNA-binding domain of TP53 that is thought to impair its ability to bind DNA 35, we focused our subsequent experiments on the actions of p63 and p73. Of note, p63 is known to function as a key regulator of adult stem cells in stratified epithelial tissues 36, including those in the mammary gland, where it is expressed by basal epithelial cells 37.
Transcript and protein levels of p63 and p73 were higher in ITGB4hi cells relative to ITGB4lo cells (Supplementary Figure 1D and 1E). Most ITGB4hi cells stained positive for both markers in their nuclei, while ITGB4lo cells had a smaller fraction of cells that were p73-positive and essentially lacked detectable p63 (Supplementary Figure 1F). Therefore, the majority of cells in the ITGB4hi compartment associated with CSC activity co-expressed p63 and p73.
We next performed ChIP-seq analysis for p63 and p73 in the ITGB4hi cells. Consistent with the motif enrichment analysis, there was a marked enrichment of both p63 and p73 binding at enhancers and promoters in ITGB4hi cells (Figure 1D), with 49% of ITGB4hi-specific H3K27ac peaks being bound by p63 and 57% bound by p73 (Supplementary Figure 1G); a majority of these sites (64.3%) were bound by both TFs (Figure 1E). p73 was bound to distinct loci in ITGB4lo cells compared to ITGB4hi cells (Supplementary Figure 1H). There was no enrichment of p63 or p73 at H3K27ac peaks that were specific to the ITGB4lo cells or in the regions that were shared in common between the two cell populations (Supplementary Figure 1G). In ITGB4hi cells, p63 and p73 bound primarily to intergenic enhancer regions and, to a lesser extent, proximal and distal promoters (Supplementary Figure 1I). The genes associated with p63 and p73 binding in ITGB4hi CSCs were expressed at significantly higher levels in these cells relative to other H3K27ac associated-genes not bound by these factors (Figure 1F), suggesting that p63/p73 were functioning primarily as transcriptional activators.
Super-enhancers (SEs) have been implicated in the control of cell-type specific transcriptional programs 38–40. Using the H3K27ac ChIP-seq data, we mapped SEs that were activated in ITGB4lo and ITGB4hi cells (see Methods). The TP73 locus contained one of the few SEs specifically activated in ITGB4hi cells and the TP63 locus was marked by a strong SE as well (Figure 1G). Moreover, we also found that p63 was bound, among other sites, to the enhancer site of its own encoding gene as well as to the gene encoding p73; a similar pattern of behavior applied to the p73 gene, with p73 binding at the TP63 and TP73 loci being particularly strong (Supplementary Figure 1J). Hence, p63 and p73 appeared to function in auto-regulatory positive-feedback loops, mirroring patterns of autoregulation that are characteristic of other cell type-specific master transcription factors (Supplementary Figure 1K). Collectively, these results implicated p63/p73 in the regulation of the chromatin landscape and the resulting transcriptional program of ITGB4hi quasi-mesenchymal CSCs.
Aldehyde dehydrogenase (ALDH) activity has been proposed as a marker of CSCs that reside closer toward the epithelial end of the EMT spectrum 41,42. Indeed, we found that the ITGB4hi cells contained ~14-fold more ALDH-positive cells than ITGB4lo cells (Figure 1H). We sorted ITGB4hi cells on the basis of ALDH activity and, in freshly prepared protein lysates, found higher levels of p63 and the EMT-TF Snail in ALDH+ cells compared to the ALDH− population (Supplementary Figure 1L). However, extensive plasticity among the ALDH− and ALDH+ subpopulations in ITGB4hi cells precluded further functional comparisons.
Since CSCs have been implicated in metastatic progression, we tested the ability of ITGB4hi and ITGB4lo cells to spawn lung metastases after tail-vein injection, which gauges the ability of cells to complete the later stages of the invasion-metastasis cascade, including survival in the circulation, extravasation, and the subsequent proliferative expansion of metastatic colonies. Only ITGB4hi cells were able to readily form macrometastatic colonies (Figure 1I). The metastases formed by the ITGB4hi cells stained strongly positive for both p63 and p73 (Figure 1J). Hence, ITGB4hi cells represented a population of quasi-mesenchymal CSCs with a strongly elevated metastatic colonization ability and possessed a unique chromatin landscape that we speculated was coordinated in large part by the actions of the TF(s) p63 and/or p73.
ΔNp63 and p73 are Required to Maintain CSCs in a Quasi-mesenchymal Cell State
The TP63 and TP73 genes each encode two main alternative isoforms: a full-length version carrying an intact N-terminal transactivation domain (TA) and a truncated form lacking the N-terminal TA domain (ΔN). First, we determined which specific isoforms of p63 and p73 were expressed in the ITGB4hi cells, since the different isoforms are known to exert different functional effects 43. Using isoform-specific qPCR44, we confirmed that ΔNp63 was the major isoform expressed by ITGB4hi CSCs (Supplementary Figure 2A), similar to the known role of this isoform in the stem cells of normal stratified epithelial tissues 36. In contrast, the ITGB4hi cells expressed two predominant p73 isoforms that could be readily resolved by western blot analysis and, we suspected, represented the expression of the TA and ΔN versions of p73 (Supplementary Figure 1E). This use of CRISPR/Cas9 isoform-specific gene knockouts supported this notion (Supplementary Figure 2B).
Compared to a non-targeting (NT) control, knockout of ΔNp63, of p73, or both (Supplementary Figure 2C) led to a clear shift of the quasi-mesenchymal ITGB4hi cells toward the ITGB4lo highly mesenchymal state (Figure 2A). Knockout of the larger p73 isoform using sgRNAs directed at exon 2 or 3, or complete knockout of both p73 isoforms using sgRNAs targeting the shared exon 4 had similar effects on ITGB4 status (Supplementary Figure 2D). Hence, in all subsequent experiments we proceeded with the p73 sgRNA that targeted the exon shared by both isoforms of this protein.
Figure 2. Loss of ΔNp63 and p73 impairs the post-extravasation proliferation of CSCs.
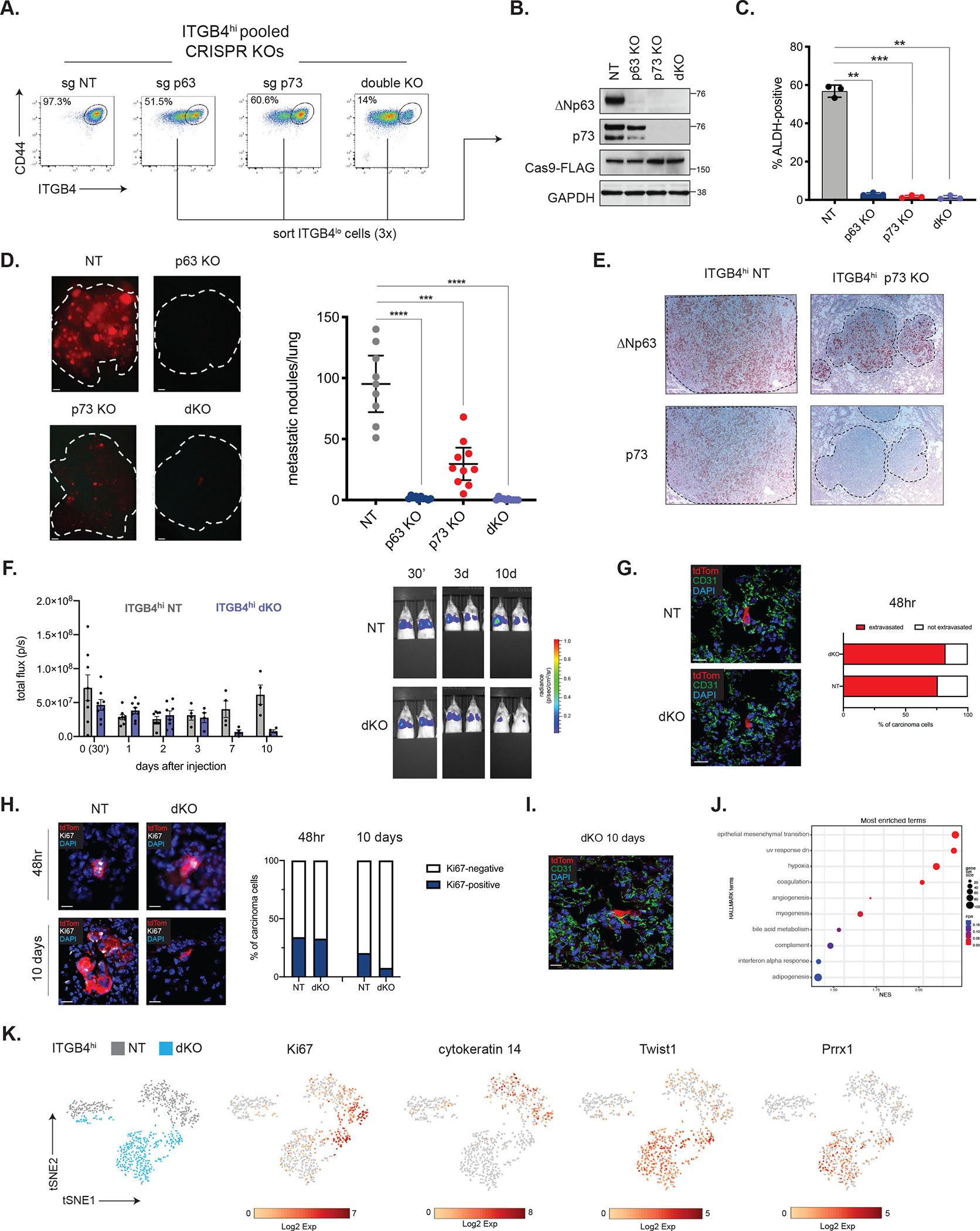
(A) Representative flow cytometry plots in the indicated ITGB4hi CRISPR-KO cells.
(B) Western blot analysis of the indicated proteins in control or KO cells that were successively sorted for loss of ITGB4 expression.
(C) Quantification of ALDH-positive cells in ITGB4hi control (NT) or KO cells. Data are ± SEM (n = 3), ** p ≤ 0.01, *** p ≤ 0.001.
(D) Metastatic colonization of ITGB4hi control or KO cells. Representative lung images (left) and quantification (right). Data are ± SEM (n = 10), *** p ≤ 0.001, **** p ≤ 0.0001. Scale bars, 1 mm.
(E) IHC staining for ΔNp63 and p73 in metastatic lung lesions. Scale bars, 100 μm.
(F) Quantification of luminescence after injection of ITGB4hi NT or dKO cells (n=8, days 0–2, n=4, days 3–10) and representative IVIS images (right).
(G) Representative IF staining for tdTom and CD31 in lung cryosections and quantification of extravasated ITGB4hi NT (n=29 cells from 2 mice) and dKO cells (n=28 cells from 2 mice) 48 hours after tail-vein injection (right). Scale bars, 20 μm.
(H) Representative IF staining for tdTom and Ki67 in lung cryosections and quantification of Ki67-positive cells 48 hours or 10 days after injection of ITGB4hi NT or dKO cells. 48hr: ITGB4hi NT (n=41 cells from 2 mice), dKO (n=79 cells from 2 mice). 10 days: ITGB4hi NT (n=166 cells from 2 mice), dKO (n=26 cells from 2 mice). Scale bars, 20 μm.
(I) Representative IF staining for tdTom and CD31 in lung cryosections 10 days after tail-vein injection of ITGB4hi dKO cells. Scale bar, 20 μm.
(J) GSEA of single-cell RNA-seq data comparing ITGB4hi NT and dKO cells isolated from the lungs of mice 3 days after tail-vein injection. Positive enrichment indicates higher expression in ITGB4hi dKO cells.
(K) t-SNE plot of tdTom-positive ITGB4hi NT and dKO cells isolated from the lungs.
Corresponding plots show expression of genes of interest.
Since the CRISPR/Cas9 knockout cells were generated in a pooled fashion, we suspected that these cell populations harbored a mix of cells that had undergone various degrees of gene knockout. Indeed, when we sorted the knockout populations on the basis of ITGB4 expression, we found that the cells that persisted in the ITGB4hi state retained expression of ΔNp63 and/or p73 (Supplementary Figure 2E), indicating that these cells had escaped CRISPR-mediated knockout. We therefore sorted for the ITGB4lo fraction to enrich for cells with knockout of ΔNp63 and/or p73 in both the single- and double-knockout (dKO) cell lines (Figure 2B) and used these sorted cells for all subsequent experiments.
Consistent with the binding of ΔNp63 and p73 to their respective enhancers, we found that knockout of either TF led to a corresponding decrease in the expression of the other, although this effect was far more pronounced with knockout of p73, which led to undetectable ΔNp63 protein expression (Figure 2B). This suggested that, at least in the ITGB4hi cells, p73 lies upstream of ΔNp63, acting as a potent activator of ΔNp63 transcription.
Since ITGB4 is a known transcriptional target of ΔNp63 and p73 45,46, we undertook to determine if the shift toward the ITGB4lo cell-surface phenotype observed following knockout of ΔNp63/p73 was truly indicative of a change in cell state rather than simply a loss of expression of a direct transcriptional target. In fact, ITGB4hi cells that had suffered knockout of either ΔNp63 and/or p73 became highly mesenchymal in nature, as gauged by their morphology (Supplementary Figure 2F), the high-level expression of canonical EMT markers, and increased expression of the EMT-inducing transcription factor Twist (Supplementary Figure 2G). Moreover, the ALDH+ population was significantly decreased in the knockout lines, mirroring the abundance of this population in the highly mesenchymal ITGB4lo state (Figure 2C and Supplementary Figure 2H). Notably, loss of ΔNp63 and/or p73 had no effect on cell proliferation in 2D culture (Supplementary Figure 2I) and did not compromise the ability of these cells to grow as tumorspheres (Supplementary Figure 2J). Altogether, these observations further substantiated the notion that ΔNp63/p73 are essential to maintain residence of these breast carcinoma cells in a quasi-mesenchymal state and that, in their absence, CSCs spontaneously lapse into a highly mesenchymal phenotypic state with associated loss of stemness.
ΔNp63 and p73 are Required for the Metastatic Ability of Cancer Stem Cells
We proceeded to test the relative abilities of the various cell populations to colonize the lungs in experimental metastasis assays. Whereas control ITGB4hi NT cells were highly efficient in generating metastatic colonies, cells lacking ΔNp63 and/or p73 were severely compromised in their ability to form metastases (Figure 2D).
Cells that ostensibly suffered knockout of p73 could still form an appreciable number of macrometastases. However, these metastases invariably stained positive for ΔNp63 (Figure 2E), suggesting that the growths derived from cells that had somehow re-activated ΔNp63 even in the absence of p73, which remained undetectable by IHC in the metastatic lesions (Figure 2E). These data indicated that while p73 could act upstream of ΔNp63, it was the downstream actions of ΔNp63 that appeared be the critical governor of ongoing residence of cells in the CSC state, enabling metastatic outgrowth in the lung.
We next determined with greater precision which of the multiple steps of metastasis formation depended upon ΔNp63/p73 in the experimental metastasis assay used above. We used IVIS bioluminescent imaging to track ITGB4hi NT and dKO cells immediately after tail-vein injection (30 minutes post-injection) and followed their fate for 10 days thereafter. The abundance of NT and dKO cells in the lung was comparable between days 1 and 3, with a reduction in luminescence in the dKO group only being evident at day 7 (Figure 2F). These results were supported using flow cytometry to estimate the number of tdTom+ cells in the lungs at day 2 and day 10 (Supplementary Figure 3A and 3B).
Immunofluorescent staining 2 days after injection revealed the majority of both NT (75.9%) and dKO (82.1%) cells had successfully extravasated (Figure 2G). These results echoed the in vitro behavior of these cells, as cells with KO of ΔNp63 and/or p73 exhibited comparable, or even slightly enhanced, rates of invasion in vitro through endothelial cells layers (Supplementary Figure 3C). These data demonstrated that dKO cells seeded the lung parenchyma with comparable efficiency as the control NT cells, and focused our attention instead on the post-extravasation behavior of such cells.
We assessed the proliferative potential of NT control or dKO cells after seeding the lung by quantifying the number of Ki67-positive tdTom+ cancer cells at 2- and 10-days post-injection. Two days after injection, the numbers of Ki67-positive cells were similar in NT and dKO groups (34.1% vs. 32.9%, respectively), but by 10 days only 7.7% of dKO cells stained positive for Ki67 whereas 20.5% of NT cells were positive (Figure 2H). We did not detect significant levels of apoptosis, as assessed by cleaved caspase 3 staining, in either group at days 2 or 10 (Supplementary Figure 3D). These observations indicated that the ability of carcinoma cells to sustain proliferation in the lung after extravasation, a process that lies at the heart of metastatic colonization, was compromised in CSCs lacking ΔNp63/p73. Importantly, this pronounced metastatic defect could not be attributed simply to the loss of surface ITGB4 expression in the KO cells because overexpression of ITGB4 in parental ITGB4lo cells did not significantly alter their colonization potential (Supplementary Figure 3E).
We noted that occasional dKO cells were capable of entering a dormant state, becoming closely affiliated with and spreading along the vascular capillaries of the lung (Figure 2I), a behavior that has previously been associated with metastatic dormancy 47. These cells could persist within the lung for extended periods of time, being detectable even 5 weeks after injection (Supplementary Figure 3E). Single-cell RNA-sequencing of NT and dKO cells isolated from the lungs 3 days after injection revealed the hallmark EMT gene set to be the top positively enriched gene set in the dKO cells (Figure 2J). Thus, in the parenchymal microenvironment of the lung, dKO cells maintained their highly mesenchymal nature, as evidenced by strong expression of the Twist1 and Prrx1 EMT-TFs together with the absence of cytokeratin 14 expression (Figure 2K). Taken together, these results supported the notion that CSCs lacking ΔNp63/p73 remain trapped in highly mesenchymal cell state in which cell proliferation within the lung parenchyma cannot be actively maintained and results, instead, in a small fraction of these carcinoma cells entering into metastatic dormancy.
Wishing to extend and refine these findings, we used NAMEC8 cells, another model of triple-negative breast cancer 28,48. These cells, which reside in a mesenchymal state, derive from genetically defined, experimentally immortalized human mammary epithelial cells 49. We proceeded to transform the NAMEC8 cells with oncogenic Ras and further engineered them to express a doxycycline-inducible version of ΔNp63. We then sorted the ITGB4lo fraction of NAMEC8 cells using FACS (Supplementary Figure 3G) and induced ΔNp63 expression with doxycycline treatment.
Induced expression of ΔNp63 in ITGB4lo NAMEC8 cells led to a clear shift toward the ITGB4hi state compared to control treated cells (Supplementary Figure 3H). Importantly, while control-treated ITGB4lo NAMEC8 cells were unable to generate macroscopic metastases, expression of ΔNp63, which we maintained in vivo, endowed these cells with a strongly enhanced capacity for metastatic colonization (Supplementary Figure 3I). These observations using genetically defined human breast cancer cells and experimentally induced ΔNp63 expression directly reinforced the notion that ΔNp63 is a key determinant of residence in the quasi-mesenchymal phenotypic state, which in turn is associated with strong increases in stemness as indicated by the greatly enhanced ability of cells to launch metastatic colony formation.
ΔNp63/p73 are required for maintenance of the CSC enhancer landscape and transcriptome
We next examined how loss of these TFs impacted the chromatin landscape and gene regulatory program of ITGB4hi CSCs. Enhancer mapping in control and KO cells using H3K27ac ChIP-seq showed that KO of ΔNp63, p73 or both, led to a collapse of the ITGB4hi CSC-specific enhancer landscape with a corresponding activation of ITGB4lo-specific enhancer elements (Figure 3A and 3B). This resulted in large-scale changes to the transcriptome with 483 genes coordinately up-regulated and 358 genes down-regulated across the three knockout cell lines (Figure 3C). Consistent with our previous observations, gene set enrichment analysis (GSEA) of RNA-seq data revealed that the top gene signatures positively enriched in the ΔNp63/p73 knockout lines were associated with activation of a strong EMT program (Figure 3D). Interestingly, however, the other gene sets negatively enriched upon ΔNp63/p73 KO were associated with various inflammatory signatures (Figure 3D). Hence, in addition to influencing mesenchymal polarization and stemness, ΔNp63/p73 appeared to control the expression of a number of inflammation-associated genes in ITGB4hi CSCs.
Figure 3. ΔNp63 regulates a distinct transcriptional program in CSCs and normal basal cells.
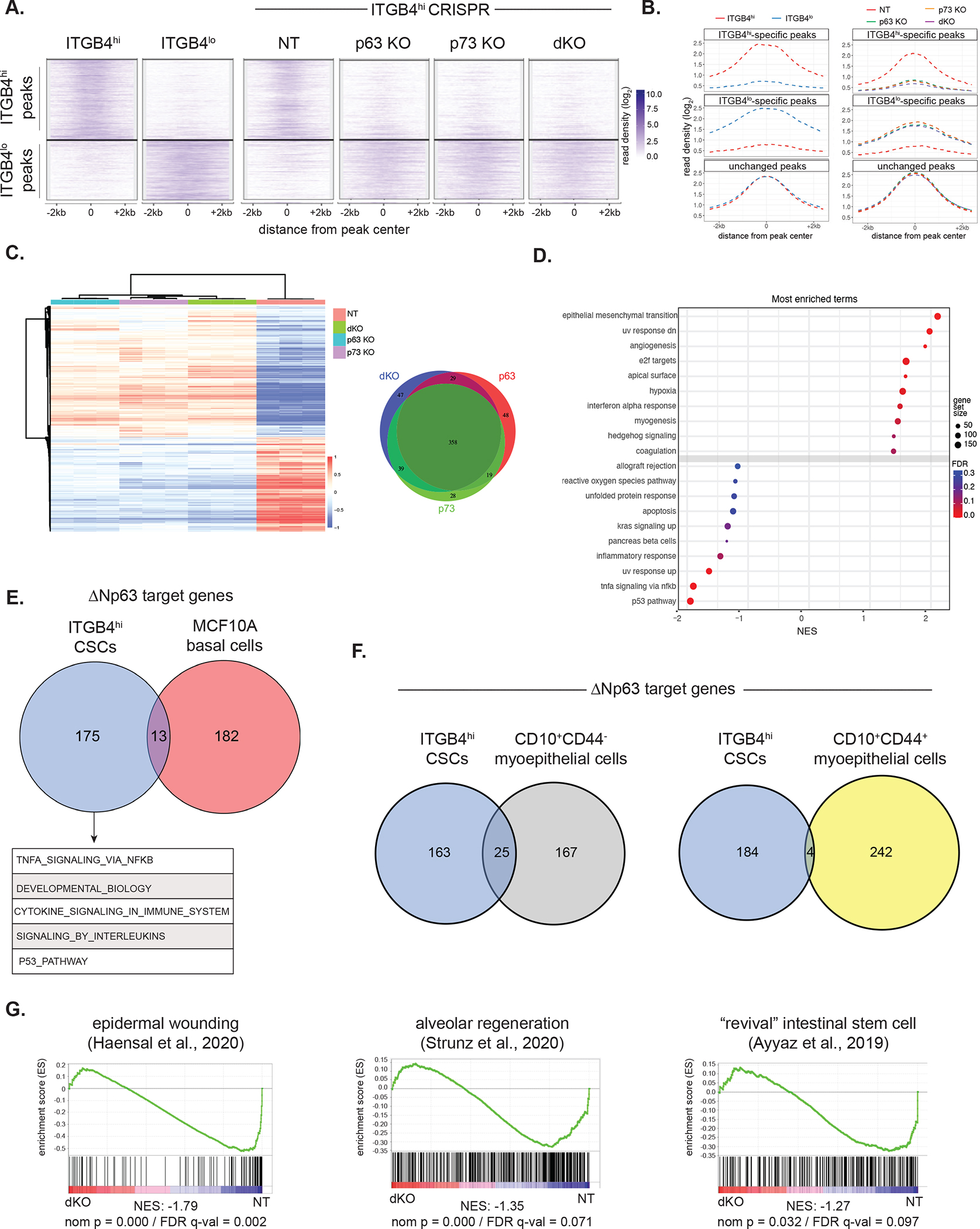
(A) Clustergram heatmap showing ITGB4hi- and ITGB4lo-specific H3K27ac ChIP-seq peaks (left) in the indicated ITGB4hi CRISPR KO cells (right).
(B) Metagene plot showing H3K27ac ChIP-seq reads in parental ITGB4lo and ITGB4hi cells (left) and ITGB4hi cells with the indicated CRSIPR KOs (right).
(C) Heatmap of the top differentially expressed genes (log2 fold-change ±1, p ≤ 0.01) in control (NT) ITGB4hi cells compared to the indicated KO lines (left). Venn diagram of genes downregulated across the three KO cell lines (right).
(D) GSEA based on RNA-seq data comparing ITGB4hi NT and dKO cells. Positive enrichment indicates higher expression in ITGB4hi dKO cells.
(E) Venn diagram showing ΔNp63 target genes in ITGB4hi cells and MCF10A cells 54. Top gene ontology (GO) terms of the ITGB4hi-specific ΔNp63 targets are shown below.
(F) Venn diagram showing ΔNp63 target genes in ITGB4hi cells and two subpopulations of CD10+ myoepithelial/basal 55.
(G) GSEA of RNA-seq data comparing ITGB4hi NT and dKO cells using the indicated epithelial wounding signatures.
See also Figure S4.
ΔNp63 regulates a distinct transcriptional program in CSCs and normal basal cells
Of great interest is the biological origin of CSCs: do carcinoma cells assemble a stem-cell program de novo that is unique to neoplastic cells or do they simply exploit and adapt with minor revisions a stem-cell program operative in the corresponding normal stem cell compartment? The observations above suggested that quasi-mesenchymal CSCs might rely on a transcriptional program that is closely related to that of normal epithelial stem cells since, as mentioned previously, ΔNp63 is also a key transcriptional regulator of untransformed epithelial stem cells, including those in the mammary gland 37.
On a global level, through use of the Epigenome Roadmap data 50, we found that the enhancers activated in quasi-mesenchymal ITGB4hi CSCs overlapped with H3K27ac regions associated with normal keratinocytes, mammary epithelial cells, and esophageal cells (Supplementary Figure 4A and 4B), in line with the known role of ΔNp63 in these cell types. In contrast, the enhancers that were activated in the highly mesenchymal ITGB4lo non-CSCs overlapped with those found in prototypical mesenchymal cell types, such fibroblasts, mesenchymal stem cells, and osteoblasts (Supplementary Figure 4A and 4B). Hence, CSCs residing in a quasi-mesenchymal cell state, retain at least some of the enhancers actively operating in the corresponding normal tissue, whereas carcinoma cells that have progressed toward more extreme mesenchymal states shed these enhancers and instead activate enhancers associated with bona fide mesenchymal cells. Moreover, KO of ΔNp63 and/or p73 in ITGB4hi cells led to reduced expression of a number of genes that have been implicated in basal mammary stem cells, including SLUG 16, PROCR 51, DLL1 52, and BCL11B 53 (Supplementary Figure 4C), suggesting, that certain parallels exist between the ΔNp63-driven transcriptional programs operating in quasi-mesenchymal CSCs and in normal basal stem cell.
Nonetheless, we wished to make a more precise assessment of the target genes controlled by ΔNp63 in normal and neoplastic mammary stem cells. Thus, we compared the genomic binding locations of ΔNp63 in ITGB4hi cells with its previously mapped binding locations in non-neoplastic, immortalized MCF10A human mammary basal cells 54. Although MCF10A cells may represent an imperfect basal cell model, this allowed us to define high-confidence ΔNp63 target genes in both cell types by employing two criteria: (1) ΔNp63 ChIP-seq peaks located near a gene (−11kb to +5.1kb of a TSS) and (2) gene downregulation upon loss of ΔNp63 expression (FDR <0.05 and log2 fold-change >1.5).
We found that ΔNp63 controlled an almost entirely distinct set of target genes in the quasi-mesenchymal CSC population when compared to those bound in the untransformed MCF10A mammary epithelial cells (Figure 3E). ITGB4hi-specific ΔNp63-bound target genes in the breast cancer cells were enriched for pathways corresponding to TNF-α signaling, developmental biology, and cytokine signaling (Figure 3E). Using previously published gene expression data from murine mammary epithelial cells 37, we confirmed that a representative selection of ITGB4hi-specific ΔNp63 targets from the top scoring gene sets were not more highly expressed in basal cells compared to luminal cells (Supplementary Figure 4D).
This suggested that many of the genes regulated by ΔNp63 in CSCs are not targets of this TF in normal basal cells. Further support for this notion was provided by an additional comparison of ΔNp63 targets in ITGB4hi cells with previously defined ΔNp63 targets in CD10+ myoepithelial/basal cells isolated from normal human breast tissues 55. Here, too, we observed minimal overlap of ΔNp63 target genes between ITGB4hi CSCs and either the CD10+CD44− and CD10+CD44+ subpopulations (Figure 3F). Of note, CD10+CD44+ cells were shown to have high ALDH activity and potential progenitor activity 55; still there were few shared ΔNp63 target genes between this subpopulation of myoepithelial/basal cells and ITGB4hi CSCs. Altogether, this indicated that although quasi-mesenchymal ITGB4hi CSCs exhibit some resemblance to their normal stem cell counterparts in terms of the overall enhancer landscape, the specific transcriptional circuits controlled by ΔNp63 in normal mammary stem cells and CSCs are actually quite distinct.
We were intrigued by the various inflammatory and cytokine gene signatures that were prominent features of the ΔNp63-controlled transcriptional program in ITGB4hi carcinoma cells, suggesting to us that CSCs might adopt a transcriptional program associated with wounding and regeneration. Indeed, there was strong overlap between the genes downregulated in ITGB4hi cells upon ΔNp63/p73 KO and transcriptional signatures associated with wounding and regeneration across various epithelial tissues. These signatures included those associated with the response of epidermal cells to skin wounding 56, with a transitional cell state linked with alveolar regeneration following lung injury 57, and with a damage-induced “revival” stem cell population in the intestine 58 (Figure 3G). In experimental models of epidermal wound healing, spatially distinct cellular regions emerge that include a migratory “leading edge” adjacent to the wound as well as a lagging “proliferative hub” of activated stem cells 59. Interestingly, ITGB4hi CSCs manifested hybrid transcriptional features of both the leading edge and proliferative hub that were dependent on ΔNp63 expression (Supplementary Figure 4D). Hence, the transcriptional program orchestrated by ΔNp63 in CSCs closely resembles a regenerative epithelial response to tissue damage in contrast to the tonic stem cell program expressed by basal mammary epithelial cells.
ΔNp63 is sufficient to activate the CSC transcriptional circuit
Given the apparent involvement of ΔNp63/p73 in maintaining residence in the CSC state, we next sought to determine if ΔNp63 and/or p73 was sufficient to establish the CSC gene regulatory network in non-CSCs. Accordingly, we forced expression of ΔNp63, TAp73, or both TFs, in ITGB4hi ΔNp63/p73 dKO cells using CRISPR-resistant expression vectors to do so (Figure 4A).
Figure 4. ΔNp63 is sufficient to activate the CSC transcriptional circuit.
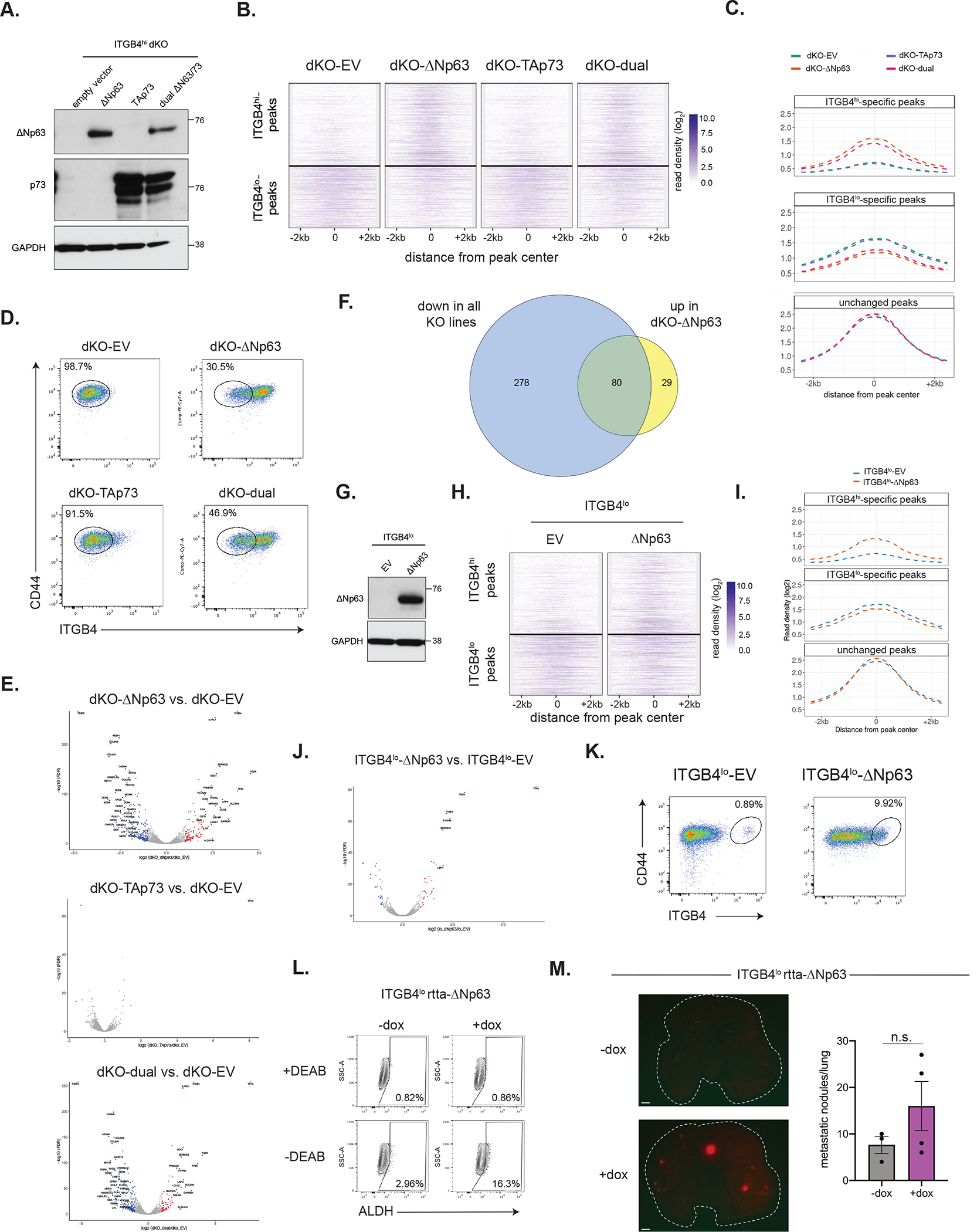
(A) Western blot analysis of ΔNp63 and p73 in ITGB4hi cells ectopically expressing the indicated vectors.
(B) Clustergram heatmap showing ITGB4hi- and ITGB4lo-specific H3K27ac ChIP-seq peaks in the indicated ITGB4hi dKO overexpression cells.
(C) Metagene plot showing H3K27ac ChIP-seq peaks in the indicated ITGB4hi dKO overexpression cells.
(D) Flow cytometry analysis of the indicated ITGB4hi dKO overexpression cells.
(E) Volcano plots showing genes differentially expressed between ITGB4hi dKO cells expressing the indicated overexpression vectors compared to cells expressing an empty vector (EV). Genes with significant expression changes are highlighted in blue (down) or red (up).
(F) Venn diagram showing the genes downregulated in all ITGB4hi KO lines and those upregulated upon re-expression of ΔNp63 in dKO cells. (log2 fold-change ±1, p ≤ 0.01).
(G) Western blot analysis of ΔNp63 and p73 in ITGB4lo cells ectopically expressing the indicated vectors.
(H) Clustergram heatmap showing ITGB4hi- and ITGB4lo-specific H3K27ac ChIP-seq peaks in control or ΔNp63-expressing ITGB4lo cells.
(I) Metagene plot showing H3K27ac ChIP-seq peaks in control or ΔNp63-expressing ITGB4lo cells.
(J) Volcano plot showing genes differentially expressed between ITGB4lo ΔNp63 and control EV cells.
(K) Flow cytometry analysis of control or ΔNp63-expressing ITGB4lo cells.
(L) ALDH activity in ITGB4lo cells expressing a doxycycline-inducible ΔNp63 expression vector.
(M) Representative lung images after injection of ITGB4lo cells expressing a doxycycline-inducible ΔNp63 expression vector (left) and quantification of macrometastatic lesions (right).
Data are ± SEM (n = 3 in −dox group and n=4 in +dox group). Scale bars, 1 mm.
Forced expression of ΔNp63 alone, but not TAp73, was sufficient to re-establish many of the ITGB4hi enhancer elements that were lost after ΔNp63/p73 dKO (Figure 4B and 4C). Moreover, forced expression of ΔNp63 on its own could switch cells to the ITGB4hi state and led to marked transcriptional changes, whereas expression of TAp73 essentially had no effect on the transcriptome of the dKO cells (Figure 4D and 4E). This demonstrated that ΔNp63, although it apparently lacks a transactivation domain, can nonetheless drive the transcriptional activation of CSC target genes. Of the 109 genes that became significantly transcriptionally activated upon expression of ΔNp63 in dKO cells, 73.4% of these were genes coordinately downregulated in the three knockout lines (Figure 4F). Also, concomitantly induced expression of ΔNp63 and TAp73 in dKO cells induced changes that were comparable to those resulting from ΔNp63 expression alone, indicating that the observed transcriptional effects of dual ΔNp63/p73 overexpression were mediated almost entirely by ΔNp63.
Forced expression of ΔNp63 in ITGB4lo cells had similar, albeit less dramatic effects on the enhancer and transcriptional network as well as conversion to the ITGB4hi state (Figures 4G–K). Notably, ITGB4lo cells expressing a doxycycline-inducible version of ΔNp63 harbored a higher percentage of ALDH+ cells (Figure 4L) and showed some ability to form macrometastatic lesions in the lung, although this result was not statistically significant (Figure 4M). This suggested that efficient transition of ITGB4lo cells to the quasi-mesenchymal state might be enhanced by other transcription factors in addition to ΔNp63.
Altogether, these observations provided direct evidence that the ΔNp63 isoform, not p73, is the proximal TF primarily responsible for enforcing activation of the CSC transcriptional circuit and strengthened the candidacy of ΔNp63 as a centrally acting master regulator of residence in the quasi-mesenchymal CSC state.
ITGB4hi CSCs rely on autocrine stimulation of the EGFR pathway to drive metastatic colonization
We found that ITGB4hi cells with KO of ΔNp63/p73 also displayed a strong negative enrichment for a gene signature associated with EGF response (Figure 5A), which was notable because EGFR signaling is a key mediator of the re-epithelialization phase of wound healing response 60. Indeed, relative to the more mesenchymal ITGB4lo cells, ITGB4hi cells displayed substantially elevated phosphorylation of the EGFR and downstream ERK phosphorylation, which was lost upon knockout of ΔNp63 and/or p73 (Figure 5B). Consistent with these findings, we also found that forced expression of ΔNp63 in ITGB4lo NAMEC8R cells was able to activate EGFR signaling (Supplementary Figure 5A). Both ITGB4lo and ITGB4hi dKO cells displayed surface levels of EGFR comparable to those seen in ITGB4hi CSCs (Supplementary Figure 5B), implying that the differential activation of the EGFR in these two cell types could reflect differences in the availability of the cognate ligands rather than in receptor protein levels.
Figure 5. ITGB4hi CSCs are dependent on EGFR signaling for metastatic colonization.
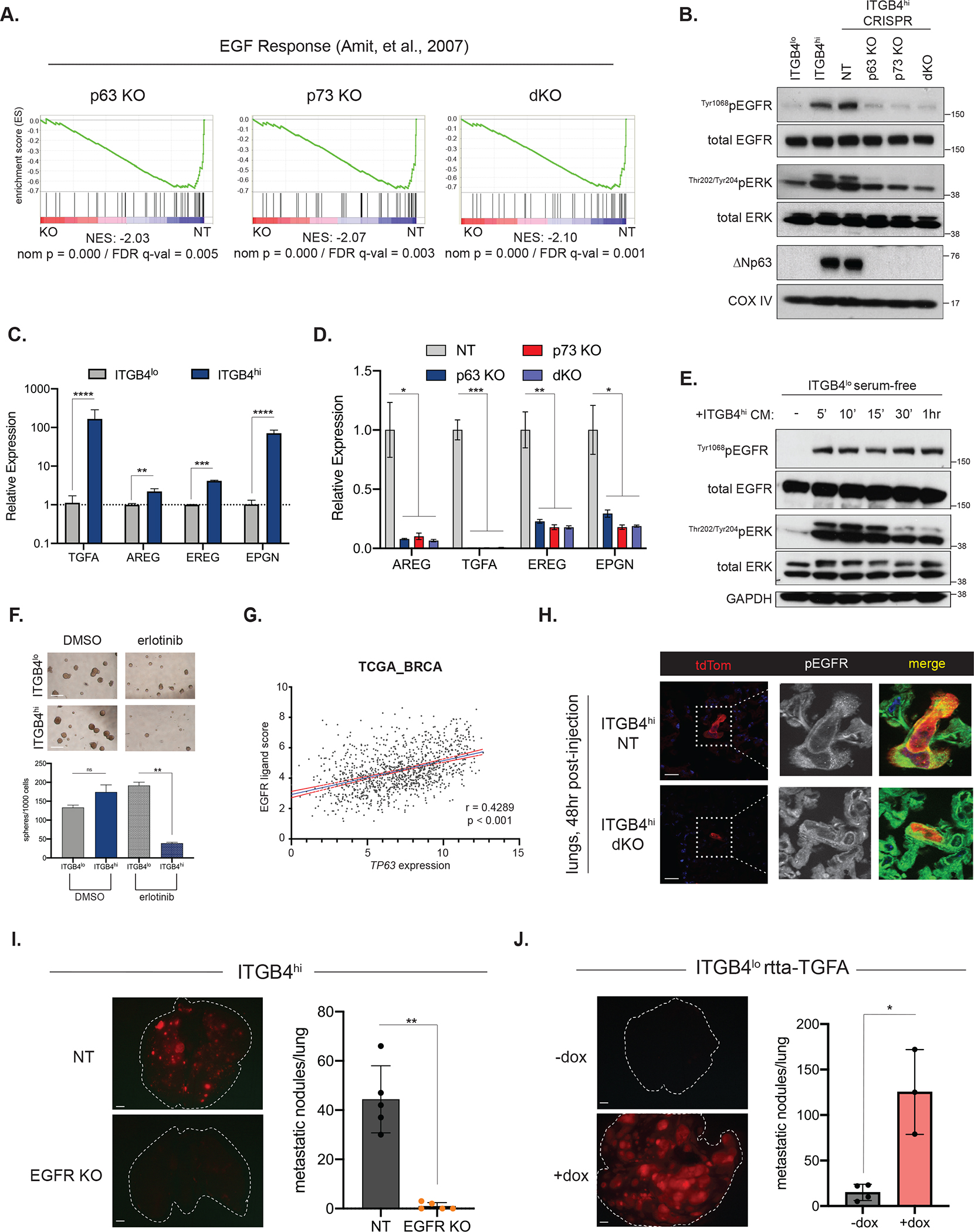
(A) GSEA of RNA-seq data comparing ITGB4hi NT and the indicated KO lines using an EGF response signature.
(B) Western blot analysis of EGFR and ERK activation in the indicated parental or KO cell lines.
(C) Representative qRT-PCR analysis of the indicated EGFR ligands in the ITGB4lo and ITGB4hi cells. Data are ± SEM, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001.
(D) Relative expression of the indicated EGFR ligands (from RNA-seq data) in control ITGB4hi cells (NT) or KO cell lines. Data are ± SEM (n=3), * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001,
(E) Western blot analysis of EGFR and ERK activation in serum-starved ITGB4lo cells treated with media conditioned from ITGB4hi cells. Negative control (−)cells were maintained in serum-free media.
(F) ITGB4lo or ITGB4hi tumorsphere growth after treatment with DMSO or erlotinib (1 μM). Representative images (left) and quantification (right). Data are ± SEM (n = 3), ** p ≤ 0.01. Scale bars, 100 μm.
(G) Correlation of TP63 expression with a 4 gene EGFR ligand score (TGFA, EREG, AREG, EPGN) in breast cancer specimens from the TCGA database.
(H) Representative IF staining for tdTom and pEGFR in lung cryosections prepared 48 hours after tail-vein injection of the indicated cell lines. Scale bars, 20 μm.
(I) Metastatic colonization of ITGB4hi control or EGFR KO cells. Representative lung images (left) and quantification (right). Data are ± SEM (n = 5), ** p ≤ 0.01. Scale bars, 1 mm.
(J) Metastatic colonization of ITGB4lo cells expressing a doxycycline-inducible TGFA expression vector. Representative lung images (left) and quantification (right). Data are ± SEM (n = 4 in −dox group and n=3 in +dox group), * p ≤ 0.05. Scale bars, 1 mm.
See also Figure S5.
In fact, we found that ITGB4hi cells expressed significantly elevated transcript levels of a number of EGFR ligands, including TGFA, AREG, EREG, and EPGN (Figure 5C). Moreover, knockout of ΔNp63 and/or p73 led to a strong reduction in the expression of these ligands (Figure 5D), suggesting that stimulation of EGFR signaling in ITGB4hi CSCs was maintained through autocrine secretion of one or more EGFR ligands. In further support of this idea, the highly mesenchymal ITGB4lo cells could be induced to activate EGFR and downstream ERK signaling by treatment with media conditioned from ITGB4hi cells (Figure 5E).
To test the functional importance of EGFR signaling to cells residing in the quasi-mesenchymal state, we treated ITGB4lo and ITGB4hi cells with erlotinib, a pharmacologic inhibitor of EGFR. Although we observed minimal effects when used in standard 2D monolayer culture (Supplementary Figure 5C), erlotinib treatment in tumorsphere culture strongly reduced the number of ITGB4hi-derived spheres but not the number of ITGB4lo spheres (Figure 5F). Treatment with erlotinib did not alter ITGB4 surface levels or ΔNp63 expression (Supplementary Figures 5D and 5E).
Consistent with these observations, we found a significant correlation between the expression of TP63 and a combined EGFR ligand score (comprising TGFA, AREG, EREG and EPGN) in patient samples from the breast TCGA dataset (Figure 5G). In addition, we analyzed single-cell RNA-sequencing data of circulating tumor cells isolated from breast cancer patients 61 and found expression of these ligands by a subset of circulating tumor cells (Supplementary Figure 5F). These observations suggested that the connection between ΔNp63 and the expression of EGFR ligands may be extrapolated to cases of human breast cancer.
Encouraged by these results, we next evaluated whether ITGB4hi CSCs also depended on EGFR signaling for metastatic colonization. In accord with the in vitro experiments described above, we noted that ITGB4hi NT control cells showed activation of EGFR in the lungs of mice 48 hours after tail-injection, as evidenced by numerous phospho-EGFR-positive membrane puncta, while dKO cells showed a strong reduction is such phospho-EGFR puncta (Figure 5H).
We then established ITGB4hi cells with knockout of EGFR (Supplementary Figure 5G) in order to determine its essentiality in the ΔNp63-positive CSCs. EGFR KO had no effect on cell viability in vitro, cell morphology, ΔNp63 or p73 protein expression, ALDH positivity, or ITGB4 cell-surface levels (Supplementary Figure 5H–5K). However, EGFR KO completely ablated the ability of ITGB4hi CSCs to colonize the lung (Figure 5I). Conversely, we found that overexpression of a single EGFR ligand, TGFA (Supplementary Figure 5L), strongly enabled metastatic colonization by the highly mesenchymal ITGB4lo cells (Figure 5J), indicating that autocrine EGFR signaling functionally distinguishes metastatic quasi-mesenchymal CSCs from carcinoma cells residing in a highly mesenchymal state, which are non-metastatic and instead may enter into a dormant state.
Discussion
Activation of an EMT program in carcinoma cells has been linked to both metastatic progression 8,13 and the formation of CSCs 24. However, EMT program generates a number of intermediate, quasi-mesenchymal cell types with differing phenotypes and functions 13,19,23. Multiple lines of evidence indicate that CSCs with high metastatic potential reside in an intermediate, quasi-mesenchymal state 18,26,29, but such cells have proven difficult to isolate and study due to the extensive phenotypic plasticity that is characteristic of the various cell types generated by an EMT program. Here, we took advantage of a ITGB4hi CSC population, which resides stably in a quasi-mesenchymal state, allowing us to undertake an in-depth analysis of how the chromatin configuration and enhancer landscape of these cells differs from those of the highly mesenchymal ITGB4lo cells.
As we discovered in a completely unbiased search for TFs that control the quasi-mesenchymal CSC state, our analysis pointed clearly to p63 and p73 as key regulators that orchestrate the central transcriptional program of quasi-mesenchymal breast CSCs. The wide-ranging functions of these TFs in cancer have been debated for many years and a loose dichotomy has emerged with the N-terminal truncated isoforms (ΔN) generally having oncogenic functions while the full-length isoforms (TA) tend to be tumor-suppressive 43,62. Our data indicated that p63, specifically the ΔNp63 isoform, is the TF proximally responsible for coordination of the CSC transcriptional program; the effects resulting from loss of p73 appear to be primarily due to its ability to transactivate the gene encoding ΔNp63. We were not able to distinguish unique roles for the different p73 isoforms, but we found that both isoforms seemed to be expressed in ITGB4hi CSCs. Although ΔNp63 has previously been implicated as a key factor in both normal and neoplastic mammary stem cells 37,63, our work adds to our understanding of how ΔNp63 functions in CSCs in two notable ways.
First, we found that a major function – perhaps the primary function – of ΔNp63 is to maintain CSCs in a quasi-mesenchymal cell state. Loss of either ΔNp63, p73, or both was sufficient to disrupt the CSC transcriptional circuit and caused carcinoma cells to lapse into a highly mesenchymal state that was associated with a loss of stem cell activity and an inability to generate lung metastases. Second, although these findings seemed, on the surface, to suggest a strong parallel between the normal mammary stem cell program and the one operating in breast CSCs, we were surprised to find that ΔNp63 regulated an almost completely distinct set of target genes in CSCs compared to normal mammary basal stem cells. This indicated that ΔNp63 controls normal and neoplastic epithelial stem cells in mechanistically different ways.
Indeed, our data suggested that the CSC program more closely approximates a regenerative epithelial response to wounding that is associated with various inflammatory mediators, cytokines, and activation of EGFR signaling. This is consistent with other observations that have linked inflammatory cytokines to CSCs 64–66 as well as recent reports that have found similarities in the epithelial transcriptional circuits activated in the wounding response and during metastatic progression 67–69. One intriguing possibility is that CSCs preferentially co-opt a transcriptional circuit normally reserved for activated stem cells that proliferate rapidly in response to tissue damage, in contrast to the program that operates in a tonic fashion and controls quiescent stem cells and tissue homeostasis 70.
The biochemical mechanisms responsible for ΔNp63 target selection in breast CSCs remain unknown, but conceivably could involve interactions with components of the SWI/SNF complex 69 or NF-kB pathway 71, similar to previous reports in head and neck squamous cell carcinoma. Additionally, we found that ectopic expression of ΔNp63 resulted in only limited conversion of ITGB4lo cells to the quasi-mesenchymal state, suggesting that other collaborating transcription factors may be required in order for highly mesenchymal carcinoma cells to efficiently transition to the CSC state.
Our study also provides one explanation for the high metastatic competence of quasi-mesenchymal carcinoma cells. We found that ΔNp63 was necessary for autocrine stimulation of EGFR, which sustained proliferation after extravasation into the lung parenchyma. Clusters of circulating tumor cells (CTCs) that disseminate in aggregate are also known to initiate metastases with high efficiency 72 and the EGFR ligand epigen (EPGN) is a key signal that promotes the metastatic growth of CTC clusters 73. Hence, the determinants of successful metastatic colonization may be quite similar for quasi-mesenchymal CSCs, which likely exhibit a single-cell mode of dissemination, and CTC clusters, which disseminate collectively. In fact, both seem to rely on a common mechanism for colonization that is centered on stimulation of EGFR.
It is also notable that highly mesenchymal carcinoma cells, which display strong PDGFR signaling but weak EGFR signaling 48, are compromised in their ability to generate macrometastatic colonies, suggesting potential qualitative or quantitative differences in these two mitogenic pathways, at least with respect to metastatic colonization. Indeed, we found that simply activating autocrine EGFR signaling in highly mesenchymal carcinoma cells, achieved via expression of TGFA, enabled metastatic colonization.
ΔNp63 has been found to promote metastasis by altering the stromal microenvironment 74,75, but we suspect such stromal interactions may come into play at later phases of metastatic colonization, long after the initial requirement for proliferation that is mediated by EGFR signaling.
Through a detailed analysis of chromatin access and enhancer activation we have elucidated the underlying transcriptional network that distinguishes highly metastatic quasi-mesenchymal CSCs from more extreme mesenchymal cells that lack stem cell activity and have limited colonization potential. Our findings revealed that the transcriptional circuits operative in breast CSCs are substantially different from those associated with normal mammary stem cells, provide a mechanistic explanation for the high metastatic competence of quasi-mesenchymal cells, and suggest potential dependencies and vulnerabilities for specifically targeting cells residing in the CSC state.
Limitations of the Study
This study used a limited number of breast cancer cell lines to provide an in-depth characterization of the nature and function of the gene regulatory circuit controlled by ΔNp63 and p73 in quasi-mesenchymal CSCs. However, the specific transcriptional program controlled by these factors could vary within and across cancer types. Also, because we focused on human cancer cells, our metastasis models involved immunocompromised mice and therefore excluded potentially relevant interactions between CSCs and components of the immune system.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Robert Weinberg (weinberg@wi.mit.edu).
Materials Availability
Plasmids and cell lines generated for this study are available upon request from the lead contact.
Data and Code Availability
Sequencing data have been deposited at GEO and are publicly available as of the date of publication. The accession number for the ChIP-seq and ATAC-seq reported in this paper is GSE172407. The accession numbers for the RNA-seq and single-cell RNA-seq reported in this paper are GSE171629, GSE171626 and GSE171628.
No original code was generated for this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
Isolation of SUM159 ITGB4lo and ITGB4hi cells was described previously 28. Both lines were cultured in F12 supplemented with 5% inactivated fetal bovine serum (IFS), hydrocortisone (1 μg/mL), insulin (5 μg/mL) and 5% penicillin/streptomycin. 293T cells used for viral production were maintained in DMEM supplemented with 10% IFS and 5% penicillin/streptomycin. NAMEC8 cells were cultured as previously described 48. Cells lines were confirmed to be mycoplasma-free.
Mice and Animal Procedures
A colony of NOD scid gamma (NOD.Cg-Prkdscid Il2rgtm1 Wjl/SzJ, originally from Jackson Laboratory) mice was maintained in-house and used for all xenograft experiments. Animals were housed in the Whitehead Institute animal facility under standard conditions, including 12-hour light/dark cycle, temperature of 18–23°C and humidity of 40–60%. Mice were housed in sterilized cages, with food (irradiated chow) and water available ad libitum. All procedures and experiments described here were approved by the Massachusetts Institute of Technology Committee on Animal Care (CAC). Male NSG mice, aged 6–10 weeks, were used for metastatic colonization assays.
METHOD DETAILS
Plasmids and Cloning
Single-guide RNAs were designed using software from the Broad Institute, Genetic Perturbation Platform (https://portals.broadinstitute.org/gppx/crispick/public) and cloned into lenti-CRISPR-V2-puro (Addgene plasmid # 52961), a gift from Feng Zhang 79 or lenti-CRSIPR-V2-GFP vector (Addgene plasmid # 82416), a gift from David Feldser 80, as previously described 79. Cells were labeled with a Luciferase-tdTomato-encoding lentiviral plasmid 77. For rescue experiments, a plasmid encoding ΔNp63alpha (Addgene plasmid # 26979), a gift from David Sidransky 81, was used to subclone ΔNp63 into the Gateway entry vector pENTR1A (Thermo Fisher); an entry vector (pENTR223) encoding TAp73 was obtained from the PlasmID Repository at Harvard Medical School (Clone ID: HsCD00399879). Silent mutations of the PAM sites were made in the entry vectors using the Q5 Site-directed Mutagenesis kit according to the manufacturer’s protocols (New England Biolabs) and subsequently recombined into the Gateway destination vector pLenti6/V5-DEST (Thermo Fisher). Doxycycline-induced vectors for ΔNp63 (FLAG/blasticidin) and TGFA (EGFP) were designed and manufactured by VectorBuilder and co-transduced with pLenti-CMV-rtta3-hygro (Addgene plasmid #26730). All plasmids were verified by sequencing.
Lentiviral Production and Generation of Cell Lines
Lentivirus was produced by co-transfection of 293T cells with the viral vector and the packaging plasmids pMD2.G (Addgene plasmid # 12259) and psPax2 (Addgene plasmid # 12260) as previously described 82. Cells were infected in the presence of 2 μg/mL polybrene (Millipore Sigma) and subsequently selected with puromycin (2 μg/mL), blasticidin (10 μg/mL) or hygromycin (250 μg/mL). Cells infected with viral constructs bearing a fluorescent tag were selected by cell sorting.
Fluorescence-Activated Cell Sorting (FACS)
Single-cell suspensions of ~ 1 × 106 cells were washed with ice-cold FACS buffer (PBS− + 2% IFS) and resuspended in 100 μL FACS buffer for staining. Cells were stained with antibodies for 30 minutes at 4°C and washed one time prior to analysis for sorting. Cell were analyzed on a BD Fortessa or LSRII instrument and sorted on a BD FACSAria, both running FACSDiva software (BD Biosciences). FlowJo (FlowJo, LLC) software was used for the analysis of FACS data. The following antibodies were used for flow cytometry: CD44-PeCy7 (1:500, Thermo Fisher), ITGB4-eFluor660 (1:50, Thermo Fisher), and EGFR (1:100, BD Biosciences). ALDH activity was assessed using the ALDEFLUOR kit (STEMCELL Technologies) according to the manufacturer’s protocol (5 μL/test of ALDEFLUOR reagent and DEAB), with cells incubated for 30 minutes at 37°C prior to analysis.
Quantitative RT-PCR
RNA was extracted with TRIzol (Thermo Fisher) and purified using the RNeasy Plus Kit (Qiagen). cDNA was synthesized with the High-capacity cDNA Reverse Transcription Kit (Thermo Fisher) using oligo-dT primers (Thermo Fisher) and an RNase inhibitor (New England Biolabs). Quantitative real-time PCR was performed with the SYBR Green Mastermix (Roche) using a LightCycler 480 II (Roche).
Western Blotting
Cells were washed in ice-cold PBS− and lysed on ice with RIPA (Millipore Sigma) containing protease (Roche) and phosphatase inhibitors (Roche). Protein lysates were cleared by centrifugation at 14,000g for 15 minutes and resolved on NuPAGE Novex 4–12% Bis-Tris gels as described by the manufacturer (Thermo Fisher). Proteins were transferred to PVDF membranes and blocked with TBS-T containing 5% non-fat dry milk for 30 minutes prior to incubation with primary antibodies overnight at 4°C. Blots were washed in TBS-T, incubated with HRP-conjugated secondary antibodies (Cell Signaling Technologies) and detected using Western Lightning Plus-ECL chemiluminescence substrate (PerkinElmer). Condition media was collected from cells grown in serum-starved media for 24hr and was cleared by centrifugation. The following antibodies were used: anti-p63α (CST 13109, 1:1000), anti-ΔNp63α (CST 67825, 1:1000) anti-p73 (CST 14620, 1:1000), anti-GAPDH (CST 5174, 1:10000), anti-FLAG (Millipore Sigma F1804, 1:1000), anti-E-cadherin (CST 3195, 1:1000), anti-N-cadherin (CST 13116, 1:1000), anti-fibronectin (BD 610078, 1:1000), anti-Slug (CST 9585, 1:1000), anti-Snail (CST 3879, 1:1000), anti-Twist (Abcam ab50887, 1:1000), anti-Zeb1 (CST 3396, 1:1000), anti-phosphoEGFRTyr1068 (CST 3777, 1:1000), anti-EGFR (CST 4267, 1:1000), anti-phospho-ERKThr202/Tyr204 (CST 4370, 1:1000), anti-ERK (CST 4695, 1:1000), anti-COX IV (CST 4850, 1:5000), and anti-beta-tubulin (CST 2128, 1:5000).
Immunostaining
For cellular immunofluorescence, cells were cultured in tissue culture-treated 8-well chamber slides (Thermo Fisher). Cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton-X, and blocked in PBS- with 3% horse serum (Vector Laboratories). Cells were incubated overnight at 4°C with the following primary antibodies: anti-p63α (CST 13109, 1:800), anti-ΔNp63 (CST 67825, 1:400) anti-p73 (CST 14620, 1:250) and secondary staining was performed using the TSA amplification kit according the manufacturer’s protocol (PerkinElmer). Fixed paraffin-embedded tissue sections were deparaffinized with Histo-Clear II (National Diagnostics) and rehydrated through a descending alcohol series. Antigen retrieval was performed with a homemade or commercial (Dako) citrate buffer (pH 6.1). For immunohistochemistry, incubatation with HRP-conjugated secondary antibodies was performed using the VECTASTAIN Elite ABC Kit (Vector Laboratories) and detected with ImmPACT DAB peroxidase substrate kit (Vector Laboratories). Slides were counterstained with hematoxylin QS (Vector Laboratories). For immunofluorescence, tissue sections were incubated with secondary antibodies conjugated to Alexa Fluor-488, -555, or -633 (Biotium, 1:400) and slides were mounted with Prolong anti-fade with NucBlue (Thermo Fisher). For visualization and staining of carcinoma cells following tail-vein injection, the lungs were perfused and embedded in O.C.T. compound (Sakura), and snap frozen in liquid nitrogen as previously described 76. Cryosections (20 μM) were thawed overnight, fixed in 4% paraformaldehyde, and stained as described above for cellular immunofluorescence with the following primary antibodies: anti-CD31-Alexa Fluor657 (BioLegend 102416, 1:100), anti-Ki67 (CST 9129, 1:400), anti-cleaved-caspase 3 (CST 9661, 1:400), anti-phospho-EGFRTyr1068 (CST 3777, 1:1000). Images were acquired on a Zeiss inverted fluorescent microscope and analyzed with the ZEN imaging software (Zeiss).
Metastasis Assays
For lateral tail-vein injections, male NSG mice were injected with 5 × 105 cells resuspended in 100 μl PBS− and lungs were analyzed for visible macroscopic metastases formation 4–6 weeks after injection using a Leica MZ12 fluorescence dissection microscope. Lungs were fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned (5 μM), and stained with H&E. Tumor cell extravasation was assessed on cryosections prepared 48 hours after tail-vein injections. Tdtom+ cells were scored as extravasated if they were completely outside a capillary vessel and/or if there was no overlap between their nuclei and CD31+ vessels. To examine tdTom+ carcinoma cells in the lungs with flow cytometry, lungs were minced and incubated in DMEM with 1.5 mg/ml collagenase A and 0.01% DNase for 30 minutes at 37°C and dissociated using a gentleMACS tissue dissociator (Miltenyi Biotec). RBCs were lysed by resuspension in RBS Lysis Buffer (BioLegend) and cells were passed through a cell strainer (BD Biosciences) to obtain a single-cell suspension prior to flow cytometric analysis. For metastasis experiments involving cells carrying doxycycline-inducible constructs, expression was induced in vitro for 5–7 days prior to tail-vein injection and mice were maintained on a doxycycline-containing diet (625 mg/kg, Teklad/Envigo) for the duration of the experiment.
Bioluminescent Imaging
Mice were imaged using an IVIS Spectrum imaging system following intraperitoneal injection with D-luciferin (165 mg/kg, PerkinElmer) and the data analyzed using Living Image software (PerkinElmer).
ChIP-seq
ChIP-seq was performed as described 83,84. Briefly, cells were crosslinked with 1% formaldehyde in PBS for 8 minutes and quenched in 1mM glycine. Cells were washed sequentially in lysis buffer 1 (140 mM NaCl, 1 mM EDTA, 50 mM HEPES, 10% glycerol, 0.5% NP-40, 0.25% Triton-X-100), lysis buffer 2 (10 mM TRIS [pH 8.0], 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA) and sonication buffer (10 mM TRIS [pH 8.0], 1 mM EDTA, 0.1% SDS) containing 1x protease inhibitor (Thermo Fisher 78430) and 1mM sodium butyrate on ice for 10 minutes. Cells were sonicated for 5 minutes total (Covaris E220) and supernatants collected. Sheared chromatin was diluted 1:1 in dilution buffer (300 mM NaCl, 2 mM EDTA, 50 mM TRIS [pH 8.0], 1.5% Triton-X, 0.1% SDS) and incubated with anti-H3K27ac (Abcam, ab4729), anti-p63 (CST 13109) or anti-p73 (Bethyl A300-126a) conjugated Protein G Dynabeads (Thermo Fisher) overnight (8–16 hours, with rotation) at 4°C, and then washed two times with wash buffer 1 (20 mM TRIS [pH 8.0], 150 mM NaCl, 2 mM EDTA, 1% Triton-X, 0.1% SDS, 0.05% DOC), two times with high-salt wash buffer (20 mM TRIS [pH 8.0], 500 mM NaCl, 2 mM EDTA, 1% Triton-X, 0.1% SDS, 0.05% DOC), one time with LiCl buffer (250 mM LiCl, 10 mM TRIS 8.0, 1mM EDTA, 0.5% NP-40) and one time with TE-NaCl buffer (10 mM Tris [pH 8.0], 1 mM EDTA, 50 mM NaCl). Samples were eluted from beads for 1 hr at 65°C in elution buffer (50 mM TRIS [pH8.0], 10 mM EDTA, 1% SDS) and the supernatant reverse-crosslinked at 65°C for 8 hours. Samples were diluted 1:1 with TE buffer (50 mM TRIS [pH 8.0], 1 mM EDTA) and treated with RNase A (0.2 mg/ml) for 2 hours at 37°C and then Proteinase K (0.2 mg/ml) for 2 hours at 55°C. DNA was isolated by phenol-chloroform extraction and ethanol precipitation. DNA libraries were prepared using Swift Biosciences ChIP kit (Cat# 21024) according to the manufacturer’s instructions using primers and amplification instructions from KAPA Biosystems HIFI library amp kit (Cat# KK2611) and were sequenced on an Illumina HiSeq.
Enhancer calling
Enhancers and were called and quantified from H3K27ac ChIP-seq data as done previously 84. Briefly, peaks were called using MACS v1.4 78 using a p-value cutoff of 1e-9. Enhancers were then formed from these peaks by merging them together if they were within 12.5kb of each other. In order to compare between multiple sample replicates, we created a merged enhancer map by merging together any overlapping enhancers between the ITGB4hi and ITGB4lo replicates. We then calculated a score for each enhancer as before 84, by calculating the read coverage in each enhancer (with input subtraction) and fitting to a negative binomial distribution. The scores were divided by the point at which the fit distribution was at 0.975, such that those scores below 1 were called “typical enhancers” and those scores above 1 were called “super-enhancers”. This produced an enhancer score for each enhancer in each replicate. These were then quantile normalized. The enhancer scores for replicates of the same state were averaged together to produce an enhancer score for each cell state.
Differential enhancers
Differential enhancers were determined using the merged enhancer map of all replicates. We then counted the number of unique reads from the IP sample overlapping each enhancer to use in a differential analysis. The R package DESeq2 85 was used to determine enhancers with differential read coverage in the ITGB4hi vs. ITGB4lo state, using default parameters. We called differential super-enhancers by filtering the differential enhancers to those enhancers with an enhancer score of at least 1 in at least one replicate. A new adjusted p-value (FDR) was calculated using the Benjamini & Hochberg method on only these super-enhancers. Those super-enhancers with a FDR<0.05 and a log2 fold-change of at least 1 were called differential, and were further filtered for those with an enhancer score of at least 1 in the differential cell state.
Differential H3K27ac peaks
We created a merged map of H3K27ac peaks by merging the peaks from all the replicates from the ITGB4hi and ITGB4lo states. The number of reads overlapping each peak from each replicate (IP only) were then counted. We created a score for each peak by normalizing the number of reads in each peak using the function varianceStabilizingTransformation from DESeq2. Only reads with a score of at least the median in at least one sample were used for downstream analysis. We then performed a differential analysis using DESeq2 on the un-normalized counts to find peaks differential between ITGB4hi and ITGB4lo states. We used an FDR cutoff of 0.05 and a log2 fold-change cutoff of 1 to call differential peaks.
Enhancer linking to genes
Enhancers were linked to genes using the Pearson correlation of the H3K27ac ChIP-seq signal at the enhancers with all promoters (+5kb to −1kb) within 500kb of the enhancer. Promoter reads were normalized using and the varianceStabilizingTransformation function from DESeq2 and the quantile normalized enhancer score was used for enhancers. This was done across a set of breast cancer cell lines and patient samples. Only positive correlations were kept. Then, promoters were filtered for those either with a correlation of at least 0.5 and p<0.05 or within 10kb of the enhancer. Finally, only those promoters with a correlation within 0.1 of the best correlated promoters to that enhancer were linked to the gene.
p63/p73 ChIP-seq analysis
p63/p73 ChIP-seq peaks were called using MACS2 and IDR with pseudo-replicates as described above in the ATAC-seq peak calling section. An IDR threshold of 0.01 was used. p63/p73 peaks were linked to genes by defining a gene regulatory region for each gene. This was defined as the basal regulatory region (−5kb to +1kb) around the TSS of each gene, plus up to 10kb beyond the basal regulatory region, up to the next gene’s basal regulatory region. If a p63/p73 peak was located in the gene regulatory region of the gene, it was said to bind that gene. For the clustergram heatmap, we used 100bp windows in the 5kb around the center of each of the H3K27ac peaks in the merged map. The number of reads per million (RPM) mapped reads in each window was calculated for each IP replicate for H3K27ac and p63/p73. The replicates of H3K27ac in ITGB4hi or ITGB4lo were averaged together. The heatmap shows the log2(RPM+0.1).
Expression of p63/p73 bound genes
Genes were considered bound by p63 or p73 if a ChIP-seq peak for the TF was present in the gene regulatory region of the gene as defined above. A Student’s t-test was used to compare the expression of p63/p73 bound vs. unbound genes, only considering genes with an H3K27ac enhancer linked to the gene.
Roadmap enhancer analysis
Enhancers based on H3K27ac for all cell types in the Epigenome Roadmap 50 were compared to the H3K27ac enhancers from this study: those activated in ITGB4hi cells, activated in the ITGB4lo cells, or unchanged. The fraction of the enhancers from this study overlapping each cell type was calculated.
ATAC-seq
ATAC-seq was performed as described 86. Briefly, cells were treated in culture medium with 200 U/ml DNase (Worthington cat# LS002007) at room temperature for 30 minutes. Cells were pelleted and washed twice with cold PBS. Fifty thousand cells were centrifuged, washed with 50 μl of cold PBS and resuspended in cold lysis buffer. Cells were pelleted for 10 minutes at 500×g, 4°C and placed on ice. Cells were combined with transposition mix (25 μl TD [2x reaction buffer, 2.5 μl TDE1, Tn5 Transposase, 22.5 μl nuclease-free H2O]) from Illumina Tagment DNA TDE1 Enzyme and Buffer Kit (Cat# 20034198) and incubated at 37°C for 30 minutes. Immediately following transposition, DNA was purified using a Qiagen MinElute Reaction Cleanup Kit according to the manufacturer’s protocol. DNA was amplified by PCR using the NEBNext High-Fidelity 2x PCR Master Mix using 1 cycle of 5 minutes 72°C, 30 seconds 98°C. Amplified DNA was purified using Qiagen MinElute Reaction Cleanup Kit and sequenced on an Illumina HiSeq.
ATAC-seq analysis
We first trimmed off an adapter sequence in the ATAC-seq reads and then aligned them to hg19 using Bowtie2. Duplicate reads were then removed before peak calling. Peak calling was performed using MACS2 78 and IDR (https://sites.google.com/site/anshulkundaje/projects/idr). Aligned reads from each replicate of a given state were combined. Pseudo-replicates from these combined reads were then used for peaking calling with MACS2. IDR was then performed on the peak calls from the pseudo-replicates and an IDR cutoff of 0.1 was used. These comprised the ATAC-seq peaks from each cell state.
Association of ATAC-seq with H3K27ac
ATAC-seq peaks were associated with differential H3K27ac by checking for overlaps of the ATAC-seq peaks and H3K27ac peaks. We used a merged map of ATAC-seq peaks from both cell states and the merged map of H3K27ac peaks from both cell states. Those ATAC-seq peaks overlapping H3K27ac peaks stronger in ITGB4hi or ITGB4lo (FDR<0.05 and log2 fold-change > 1) were labeled as ATAC-seq peaks with differential H3K27ac in either ITGB4hi or ITGB4lo. Background sets of ATAC-seq peaks for each state were determined using overlaps with H3K27ac peaks that were not differential for each state (either p-value>0.1 and log2 fold-change < 0.5, or log2 fold-change < 0).
Predicted TF binding sites by motifs and ATAC-seq
We determined predicted TF binding sites by associating a set of TF sequence motifs with the merged set of ATAC-seq peaks. Motifs were drawn from the following databases: motifs based on ENCODE data 87 (http://compbio.mit.edu/encode-motifs), JASPAR 88, uniPROBE 89, and high-throughput SELEX 90. Motifs mapping to the same TF were ranked according to the database that they originally came from (some sources of motifs contained motifs from multiple original databases): 1) uniPROBE, 2) JASPAR, 3) Jolma, 4) Transfac, 5) ENCODE. For each TF, motifs were chosen only from the database with the highest-ranking motif(s), creating the “Curated motif set.” Next, FIMO 91 was used to find instances of the motifs in the genome, with a p-value cutoff of 10−4, and using only the top 500k locations per motif.
Enrichment of predicted TF binding sites in differential H3K27ac
To find the TFs for which binding sites were associated with differential H3K27ac from ITGB4hi, we determined which ATAC-seq peaks with differential H3K27ac in ITGB4hi had a predicted binding site compared to which ATAC-seq peaks from the background set for ITGB4hi had a predicted binding site. We then used a Fisher’s exact test to determine enrichment and adjusted the p-values using the Benjamini & Hochberg method to produce FDRs. If a TF had multiple motifs in the database, the one with the greatest log-odds ratio of enrichment was used. Motifs with an FDR<0.01 were considered enriched.
RNA-sequencing
Total RNA was extracted with TRIzol (Thermo Fisher) and purified using the RNeasy Plus Kit (Qiagen). Libraries were prepared for RNA-Seq using KAPA mRNA HyperPrep Kit (Roche), according to manufacturer’s directions. Briefly, total RNA was enriched for polyadenylated sequences using oligo-dT magnetic bead capture. The enriched mRNA fraction was then fragmented, and first-strand cDNA generated using random primers. Strand specificity is achieved during second-strand cDNA synthesis by replacing dTTP with dUTP. The resulting cDNA was A-Tailed and ligated with indexed adapters. Finally, the library was amplified using KAPA qPCR library quant kit (Roche) as per manufacturers protocol. Libraries were then pooled at equimolar concentration, for each lane, using qPCR concentrations. The pooled libraries were denatured using the Illumina protocol. The denatured libraries were sequenced on an Illumina HiSeq 2500 (40-nt long single end). Basecalls were performed using Illumina offline basecaller (OLB) and then demultiplexed.
Reads were mapped with STAR 92 to hg38 version of human genome using the “sjdbOverhang” parameter set to 39, and the annotation file from ENSEMBL GRCh38.90. At least 22 and 32 million of reads were uniquely mapped reads for the knockout and forced overexpression experiments, respectively. The samples were stranded, and counts for the 2nd read strand aligned with RNA were used for calculating the number of counts per gene. If none of the samples within comparisons had at least one read assigned to a gene, this gene was considered to be not expressed and removed from further analysis. Normalization and differential expression were done with DESeq2 (https://pubmed.ncbi.nlm.nih.gov/25516281/), which taking batch effect into design consideration. Genes with at least two-fold differences plus adjusted p-values no more than 0.01 were considered to be differentially expressed. To create the heatmap, the differentially expressed genes were used for hierarchical clustering with Cluster 3.0 93 based on uncentered correlation and average linkage.
Single-cell RNA-sequencing
Three days after tail-vein injection of ITGB4hi NT or dKO cells, mice were euthanized and the lungs dissociated as described above. Flow cytometry was used to isolate live tdTom+ carcinoma cells, which were immediately processed for single-cell RNA-sequencing. Cells were processed using the 10X Genomics Chromium Controller with the Single Cell 3ʹ v3 Reagent Kit, according to manufacturer’s directions. Briefly, a target number of 1250 cells per library was roughly achieved by loading 1.6 times the target number of cells in suspension. The Chromium Controller processes individual cells so that each cell is marked with its unique barcode during reverse transcription. The end result is a single library representing one cell suspension, containing data for each individual cell. Libraries were amplified using KAPA qPCR library quant kit (Roche) as per manufacturers protocol. Libraries were then pooled at equimolar concentration, for each lane, using qPCR concentrations. The pooled libraries were denatured using the Illumina protocol. The denatured libraries were loaded onto an HiSeq 2500 (Illumina) and sequenced for 60×60 cycles. Fastq files were generated and demultiplexed with the bcl2fastq Conversion Software (Illumina).
Alignments, counts matrix and loupe browser files were generated using cellrangerv4 (10X Genomics) using mixed species reference genome (refdata-gex-GRCh38_and_mm10-2020-A) and human only reference genome (refdata-gex-GRCh38-2020-A). Reference genomes were downloaded from 10x Genomics website (https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest).
Further downstream analysis was done using Seurat Bioconductor package (Lambert_scRNASeq_Analysis.R, https://github.com/sumeetg23/DifferentialExpression/blob/master/Lambert_scRNASeq_Analysis.R) to ensure any contaminating mouse cells were eliminated from the analysis. Specifically, from dual species analysis, the cell barcodes classified as human were identified from the gem_classification.csv file. We used the list of human cell barcodes to select for cells in the analysis done with human only reference genome, using cellranger reanalyze option. Read count matrix for human only dataset was then normalized and cluster specific markers for each cluster were identified using FindAllMarkers function (parameters - min.pct = 0.25, logfc.threshold = 0.25). The list of markers was further filtered for a fold-change of 1.5 and sorted by p-value to identify the top markers.
Gene Set Enrichment Analysis
Protein coding genes ranked by fold changes were used as input for GseaPreranked tool from the Broad Institute 94. Hallmark annotation h.all.v7.2.symbols.gmt was downloaded from the Broad Institute. EGF response 95 was from the GSEA result using the curated gene sets c2.all.v6.2.symbols.gmt as input. For epidermal wounding gene set, we downloaded the differentially expressed genes between basal cells from the unwounded (UW) and (wounded) WO mouse samples (Supplemental Table 4 from Haensel et al. 56. To create the gene set of alveolar regeneration (Strunz et al. 57), we downloaded Krt8_ADI_all genes.xlsx (http://146.107.176.18:3838/Bleo_webtool_v2/) selected mouse genes with at least two-fold up in Krt8+ ADI cells plus adjusted p-value less than 0.001. To get the gene set from revival intestinal stem cell (Ayyaz et al. 58), we used the human homologs of genes from the revival stem cell signature ayyaz_ganesh.xls (downloaded from Ganesh et al. 68, Supplementary Table 1). We analyzed the gene signature regulating wound healing in mouse tail epidermis (Aragona et al. 59) using the limma package 96. We fit a linear model to their GSE93638 microarray data, and used empirical bayes statistics for differential expression. Genes with adjusted p-value less than 0.01 and at least two-fold differences were selected to be in the gene sets. Since our ranked genes came from human cancer cells, all the genes from mouse gene sets were converted to human symbols using orthologs from ENSEMBL before running GSEA analysis.
Cell Proliferation Assay
Cell proliferation was measured using the CellTiter 96 Aqueous Cell Proliferation Assay according to the manufacturer’s protocol (Promega).
Tumor Sphere Culture
Tumorspheres were grown in Mammocult medium (STEMCELL Technologies) containing 1% methylcellulose and supplemented with 4 μg/mL heparin, 0.48 μg/mL hydrocortisone and penicillin/streptomycin. Between 500–1000 cells were seeded in 1mL of sphere media in 24-well ultra-low attachment plates (Corning). For erlotinib treatment experiments, a single dose of erlotinib (1μM) of DMSO was added at the time of cell seeding. Spheres were quantified after 7–10 days of growth.
Transendothelial Migration Assays
The transendothelial migration assay was performed using QCM Transendothelial Migration Assay according to the manufacturer’s instructions (Millipore Sigma).
TCGA and CTC Analysis
Normalized RNA-seq gene expression data for primary breast cancer profiles as part of The Cancer Genome Atlas (TCGA) were obtained using Firehose (http://firebrowse.org/?cohort=BRCA). EGFR ligand score was calculated as geometric mean of EREG, TGFA, AREG and EPGN genes. Correlation analysis was performed using PRISM 9 software. Gene expression data of circulating tumor cells from breast cancer patients was from the GSE111065 dataset 61.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are presented as the mean ± standard error of the mean (SEM) and statistical significance was assessed using two-tailed Student’s t-test unless specified otherwise. p < 0.05 was considered significant. Statistical analyses were performed using Prism software and detailed statistical information (including n of cell or animals) can be found in the figure legends.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| p63α (immunoblotting, ChIP, IHC/IF) | Cell Signaling Technology | Cat#13109 |
| ΔNp63α (immunoblotting, IHC/IF) | Cell Signaling Technology | Cat#67825 |
| p73 (immunoblotting, IHC/IF) | Cell Signaling Technology | Cat#14620 |
| p73 (ChIP) | Bethyl Laboratories | Cat#A300-126a |
| H3K27ac (ChIP) | Abcam | Cat#ab4729 |
| E-cadherin | Cell Signaling Technology | Cat#3195 |
| N-cadherin | Cell Signaling Technology | Cat#13116 |
| Fibronectin | BD Biosciences | Cat#610078 |
| Zeb1 | Cell Signaling Technology | Cat#3396 |
| Snail | Cell Signaling Technology | Cat#3879 |
| Slug | Cell Signaling Technology | Cat#9585 |
| Twist1 | Abcam | Cat#ab50887 |
| phospho-ERK1/2 (T202/Y204) | Cell Signaling Technology | Cat#4370 |
| ERK1/2 | Cell Signaling Technology | Cat#4695 |
| phospho-EGFR (Y1068) | Cell Signaling Technology | Cat#3777 |
| EGFR | Cell Signaling Technology | Cat#4267 |
| FLAG | Millipore Sigma | Cat#F1804 |
| GAPDH | Cell Signaling Technology | Cat#2118 |
| COX IV | Cell Signaling Technology | Cat#4850 |
| β-tubulin | Cell Signaling Technology | Cat#2128 |
| CD44-PeCy7 | Thermo Fisher Scientific | Cat#25-0441-81 |
| ITGB4 (CD104)-e660 | Thermo Fisher Scientific | Cat#50-1049-82 |
| EGFR-BV786 | BD Biosciences | Cat#742606 |
| Ki67 | Cell Signaling Technology | Cat#9129 |
| CD31-Alexa Fluor 647 | BioLegend | Cat#102416 |
| Cleaved caspase-3 | Cell Signaling Technology | Cat#9661 |
| Bacterial and virus strains | ||
| Stbl3™ E. coli | Thermo Fisher Scientific | Cat#C737303 |
| DH5α E. coli | Thermo Fisher Scientific | Cat#18265017 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Millipore Sigma | Cat#D6171 |
| Ham’s F-12 | Thermo Fisher Scientific | Cat#21700075 |
| MammoCult | STEMCELL Technologies | Cat#05620 |
| Polybrene | Millipore Sigma | Cat#TR-1003-G |
| X-tremeGENE 9 | Roche | Cat#19128100 |
| TRIzol | Thermo Fisher Scientific | Cat#15596018 |
| Oligo-dT | Thermo Fisher Scientific | Cat#SO132 |
| RNase inhibitor | Thermo Fisher Scientific | Cat#N8080119 |
| RIPA | Millipore Sigma | Cat#R0278 |
| Complete, EDTA-free protease inhibitor cocktail | Roche | Cat#11873580001 |
| PhosSTOP phosphatase inhibitor cocktail | Roche | Cat#04906845 |
| Protease inhibitor cocktail | Thermo Fisher Scientific | Cat#78430 |
| Protein G Dynabeads | Thermo Fisher Scientific | Cat#10004D |
| NuPAGE MOPS SDS running buffer | Thermo Fisher Scientific | Cat#NP001 |
| NuPAGE transfer buffer | Thermo Fisher Scientific | Cat#NP0006-1 |
| Enhanced Plus-ECL solution | PerkinElmer | Cat#NEL104001EA |
| 10% neutral buffered formalin | VWR | Cat# 89370-094 |
| 16% formaldehyde | Thermo Fisher Scientific | Cat#28908 |
| Histo-Clear II | National Diagnostics | Cat#HS-202 |
| Target retrieval solution | Agilent Dako | Cat#S1699 |
| Horse serum | Vector Laboratories | Cat#S-2000 |
| Hematoxylin QS | Vector Laboratories | Cat#H3404 |
| ProLong glass antifade with NucBlue | Thermo Fisher Scientific | Cat#P36981 |
| Tissue-Tek O.C.T compound | Sakura | Cat#4583 |
| heparin | STEMCELL Technologies | Cat#07980 |
| hydrocortisone | Millipore Sigma | Cat# H0396 |
| DMSO | Millipore Sigma | Cat#D2650 |
| erlotinib | Selleckchem | Cat#S1023 |
| Collagenase | Millipore Sigma | Cat#11088793001 |
| DNase | Worthington Biochemical | Cat#LS002007 |
| RBC lysis buffer | BioLegend | Cat#420301 |
| D-luciferin | PerkinElmer | Cat#122799 |
| Critical commercial assays | ||
| DNA ligation kit, mighty mix | Takara | Cat#6023 |
| Gateway LR clonase II | Thermo Fisher Scientific | Cat#11791020 |
| High-fidelity 2x PCR master mix | New England Biolabs | Cat#M0541 |
| Q5 site-directed mutagenesis kit | New England Biolabs | Cat#E0554S |
| Gel DNA recovery kit | Zymo Research | Cat#D4002 |
| Zyppy plasmid minprep kit | Zymo Research | Cat#D4036 |
| RNeasy mini kit | Qiagen | Cat#74104 |
| MiniElute reaction cleanup kit | Qiagen | Cat#28206 |
| High-capacity cDNA reverse transcription kit | Thermo Fisher Scientific | Cat#4368814 |
| SYBR Green I master | Roche | Cat#14571520 |
| ALDEFLUOR kit | STEMCELL Technologies | Cat#01700 |
| VECTASTAIN Elite ABC peroxidase kit | Vector Laboratories | Cat#PK-6101 |
| ImmPACT DAB peroxidase substrate kit | Vector Laboratories | Cat#SK-4100 |
| TSA Plus cyanine 3 system | PerkinElmer | Cat#NEL744001KT |
| CellTiter 96 AQueous cell proliferation assay | Promega | Cat#G3582 |
| QCM transendothelial migration assay | Millipore Sigma | Cat#ECM558 |
| 2S Plus DNA Library kit | Swift Biosciences | Cat#20024 |
| KAPA HiFi amplification kit | Roche | Cat#KK2611 |
| Illumina Tagment DNA TDE1 Enzyme and Buffer | Illumina | Cat# 20034198 |
| Deposited data | ||
| H3K27ac ChIP-seq | This paper | GSE172407 |
| ATAC-seq | This paper | GSE172407 |
| p63/p73 ChIP-seq | This paper | GSE172407 |
| RNA-sequencing of NT and p63/p73 KO lines | This paper | GSE171629 |
| RNA-sequencing of control and p63/p73 OE lines | This paper | GSE171626 |
| Single-cell RNA-sequencing of NT vs. dKO cells isolated from lungs | This paper | GSE171628 |
| Experimental models: cell lines | ||
| Human 293T | Shibue, et al. 76 | N/A |
| Human SUM159 ITGB4lo | Bierie et al. 28 | N/A |
| Human SUM159 ITGB4hi | Bierie et al. 28 | N/A |
| Human NAMEC8 | Tam et al., 48 | N/A |
| Human SUM159 ITGB4lo with ITGB4OE | Bierie et al. 28 | N/A |
| Experimental models: organisms/strains | ||
| Mouse NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) | The Jackson Laboratory | Cat#005557 |
| Oligonucleotides | ||
| See Table S1 | ||
| Recombinant DNA | ||
| lenti-CRISPR-V2-puro | Addgene | #52961 |
| lenti-CRISPR-V2-GFP | Addgene | #82416 |
| Luciferase-tdTomato | Frose et al. 77 | |
| ΔNp63alpha-FLAG | Addgene | #26979 |
| pWZL-HRAS (G12V)-blast | Tam et al. 48 | |
| pLenti-CMV-rtta3-hygro | Addgene | #26730 |
| doxycycline-inducible ΔNp63-FLAG | VectorBuilder | |
| doxycycline-inducible TGFA | VectorBuilder | |
| pENTR223-TAp73 | Harvard PlasmID Database | Clone ID: HsCD003999879 |
| pENTR1A | Thermo Fisher Scientific | Cat#A10462 |
| pLenti6/V5-DEST | Thermo Fisher Scientific | Cat#V49610 |
| pMD2.G | Addgene | #12259 |
| psPax2 | Addgene | #12260 |
| Software and algorithms | ||
| FlowJo | BD Biosciences | https://www.flowjo.com/ |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Living Image | PerkinElmer | https://www.perkinelmer.com/product/spectrum-200-living-image-v4series-1-128113 |
| Zen | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| Cell Ranger | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Loupe Browser | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/visualization/latest/what-is-loupe-cell-browser |
| MACS2 | Zhang et al. 78 | https://pypi.org/project/MACS2/ |
Highlights.
ΔNp63 and p73 maintain residence in the quasi-mesenchymal breast CSC state
ΔNp63 and p73 are required for the post-extravasation proliferation of CSCs
ΔNp63 controls distinct transcriptional programs in CSCs and normal basal SCs
Quasi-mesenchymal CSCs use autocrine EGFR signaling to enable metastatic colonization
Acknowledgements
We thank Drs. Richard Goldsby, Christian Fritz, Tsukasa Shibue, George Bell and all members of the Weinberg laboratory for critical discussions and feedback. We thank Caitlin Rausch for preparation of the graphical abstract. We also thank the core facilities at the Whitehead Institute (Flow Cytometry, Genome Technology, Bioinformatics and Research Computing) and the Swanson Biotechnology Center at the MIT Koch Institute (Histology, Preclinical Imaging). This research was supported by the MIT Stem Cell Initiative, the Breast Cancer Research Foundation, the Advanced Medical Research Foundation, the Ludwig Center at MIT, and grants R01-CA078461 (to R.A.W.) and R35-CA220487 (to R.A.W.) from the National Institutes of Health/National Cancer Institute Program. A.W.L. was supported by an American Cancer Society – New England Division – Ellison Foundation Postdoctoral Fellowship (PF-15-131-01-CSM) and a postdoctoral fellowship from the Ludwig Center at MIT. Y.Z. was supported by a Susan G. Komen Postdoctoral Fellowship (PDF15301255). R.A.W. is an American Cancer Society Research Professor and a Daniel K. Ludwig Cancer Research Professor.
Footnotes
Declarations of Interests
R.A.W. has a consulting agreement with Verastem Inc., and held shares of this company. C.F., Y.C. M.W.C., B.W., M.E., D.F., J.C., E.R.O. and M.G.G. were or are employees of Syros Pharmaceuticals.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, and Clarke MF (2003). Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 100, 3983–3988. 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beck B, and Blanpain C (2013). Unravelling cancer stem cell potential. Nat Rev Cancer 13, 727–738. 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- 3.Batlle E, and Clevers H (2017). Cancer stem cells revisited. Nat Med 23, 1124–1134. 10.1038/nm.4409. [DOI] [PubMed] [Google Scholar]
- 4.Lytle NK, Barber AG, and Reya T (2018). Stem cell fate in cancer growth, progression and therapy resistance. Nat Rev Cancer 18, 669–680. 10.1038/s41568-018-0056-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pattabiraman DR, and Weinberg RA (2014). Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov 13, 497–512. 10.1038/nrd4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oskarsson T, Batlle E, and Massague J (2014). Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell 14, 306–321. 10.1016/j.stem.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Celia-Terrassa T, and Kang Y (2016). Distinctive properties of metastasis-initiating cells. Genes Dev 30, 892–908. 10.1101/gad.277681.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lambert AW, Pattabiraman DR, and Weinberg RA (2017). Emerging Biological Principles of Metastasis. Cell 168, 670–691. 10.1016/j.cell.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta PB, Chaffer CL, and Weinberg RA (2009). Cancer stem cells: mirage or reality? Nat Med 15, 1010–1012. 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 10.Meacham CE, and Morrison SJ (2013). Tumour heterogeneity and cancer cell plasticity. Nature 501, 328–337. 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, et al. (2011). Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A 108, 7950–7955. 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta PB, Pastushenko I, Skibinski A, Blanpain C, and Kuperwasser C (2019). Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 24, 65–78. 10.1016/j.stem.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nieto MA, Huang RY, Jackson RA, and Thiery JP (2016). Emt: 2016. Cell 166, 21–45. 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 14.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. (2008). The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715. 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, and Puisieux A (2008). Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 3, e2888. 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer-Hardi U, Bell G, et al. (2012). Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148, 1015–1028. 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lambert AW, and Weinberg RA (2021). Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer. 10.1038/s41568-021-00332-6. [DOI] [PubMed] [Google Scholar]
- 18.Jolly MK, Boareto M, Huang B, Jia D, Lu M, Ben-Jacob E, Onuchic JN, and Levine H (2015). Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front Oncol 5, 155. 10.3389/fonc.2015.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, and Weinberg RA (2018). Epithelial-to-mesenchymal transition in cancer: complexity and opportunities. Front Med 12, 361–373. 10.1007/s11684-018-0656-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams ED, Gao D, Redfern A, and Thompson EW (2019). Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat Rev Cancer 19, 716–732. 10.1038/s41568-019-0213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS, et al. (2017). Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 171, 1611–1624 e1624. 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aiello NM, and Kang Y (2019). Context-dependent EMT programs in cancer metastasis. J Exp Med 216, 1016–1026. 10.1084/jem.20181827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pastushenko I, and Blanpain C (2019). EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol 29, 212–226. 10.1016/j.tcb.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Shibue T, and Weinberg RA (2017). EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 14, 611–629. 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kroger C, Afeyan A, Mraz J, Eaton EN, Reinhardt F, Khodor YL, Thiru P, Bierie B, Ye X, Burge CB, and Weinberg RA (2019). Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc Natl Acad Sci U S A 116, 7353–7362. 10.1073/pnas.1812876116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grosse-Wilde A, Fouquier d’Herouel A, McIntosh E, Ertaylan G, Skupin A, Kuestner RE, del Sol A, Walters KA, and Huang S (2015). Stemness of the hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLoS One 10, e0126522. 10.1371/journal.pone.0126522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jolly MK, Jia D, Boareto M, Mani SA, Pienta KJ, Ben-Jacob E, and Levine H (2015). Coupling the modules of EMT and stemness: A tunable ‘stemness window’ model. Oncotarget 6, 25161–25174. 10.18632/oncotarget.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bierie B, Pierce SE, Kroeger C, Stover DG, Pattabiraman DR, Thiru P, Liu Donaher J, Reinhardt F, Chaffer CL, Keckesova Z, and Weinberg RA (2017). Integrin-beta4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc Natl Acad Sci U S A 114, E2337–E2346. 10.1073/pnas.1618298114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, Van Keymeulen A, Brown D, Moers V, Lemaire S, et al. (2018). Identification of the tumour transition states occurring during EMT. Nature 556, 463–468. 10.1038/s41586-018-0040-3. [DOI] [PubMed] [Google Scholar]
- 30.Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A, and Nieto MA (2012). Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 22, 709–724. 10.1016/j.ccr.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 31.Celia-Terrassa T, Meca-Cortes O, Mateo F, Martinez de Paz A, Rubio N, Arnal-Estape A, Ell BJ, Bermudo R, Diaz A, Guerra-Rebollo M, et al. (2012). Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest 122, 1849–1868. 10.1172/JCI59218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beerling E, Seinstra D, de Wit E, Kester L, van der Velden D, Maynard C, Schafer R, van Diest P, Voest E, van Oudenaarden A, et al. (2016). Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Rep 14, 2281–2288. 10.1016/j.celrep.2016.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Del Pozo Martin Y, Park D, Ramachandran A, Ombrato L, Calvo F, Chakravarty P, Spencer-Dene B, Derzsi S, Hill CS, Sahai E, and Malanchi I (2015). Mesenchymal Cancer Cell-Stroma Crosstalk Promotes Niche Activation, Epithelial Reversion, and Metastatic Colonization. Cell Rep 13, 2456–2469. 10.1016/j.celrep.2015.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wasielewski M, Elstrodt F, Klijn JG, Berns EM, and Schutte M (2006). Thirteen new p53 gene mutants identified among 41 human breast cancer cell lines. Breast Cancer Res Treat 99, 97–101. 10.1007/s10549-006-9186-z. [DOI] [PubMed] [Google Scholar]
- 36.Melino G, Memmi EM, Pelicci PG, and Bernassola F (2015). Maintaining epithelial stemness with p63. Sci Signal 8, re9. 10.1126/scisignal.aaa1033. [DOI] [PubMed] [Google Scholar]
- 37.Chakrabarti R, Wei Y, Hwang J, Hang X, Andres Blanco M, Choudhury A, Tiede B, Romano RA, DeCoste C, Mercatali L, et al. (2014). DeltaNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat Cell Biol 16, 1004–1015, 1001–1013. 10.1038/ncb3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al. (2013). Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24, 777–790. 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD, et al. (2014). Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports 2, 78–91. 10.1016/j.stemcr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brooks MD, Burness ML, and Wicha MS (2015). Therapeutic Implications of Cellular Heterogeneity and Plasticity in Breast Cancer. Cell Stem Cell 17, 260–271. 10.1016/j.stem.2015.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Botchkarev VA, and Flores ER (2014). p53/p63/p73 in the epidermis in health and disease. Cold Spring Harb Perspect Med 4. 10.1101/cshperspect.a015248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang H, Bierie B, Li AG, Pathania S, Toomire K, Dimitrov SD, Liu B, Gelman R, Giobbie-Hurder A, Feunteun J, et al. (2016). BRCA1/FANCD2/BRG1-Driven DNA Repair Stabilizes the Differentiation State of Human Mammary Epithelial Cells. Mol Cell 63, 277–292. 10.1016/j.molcel.2016.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carroll DK, Carroll JS, Leong CO, Cheng F, Brown M, Mills AA, Brugge JS, and Ellisen LW (2006). p63 regulates an adhesion programme and cell survival in epithelial cells. Nat Cell Biol 8, 551–561. 10.1038/ncb1420. [DOI] [PubMed] [Google Scholar]
- 46.Xie N, Vikhreva P, Annicchiarico-Petruzzelli M, Amelio I, Barlev N, Knight RA, and Melino G (2018). Integrin-beta4 is a novel transcriptional target of TAp73. Cell Cycle 17, 589–594. 10.1080/15384101.2017.1403684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, Almeida D, Koller A, Hajjar KA, Stainier DY, et al. (2013). The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 15, 807–817. 10.1038/ncb2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, Wu ZJ, Krall JA, Bierie B, Guo W, et al. (2013). Protein kinase C alpha is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 24, 347–364. 10.1016/j.ccr.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, and Weinberg RA (2001). Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev 15, 50–65. 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang D, Cai C, Dong X, Yu QC, Zhang XO, Yang L, and Zeng YA (2015). Identification of multipotent mammary stem cells by protein C receptor expression. Nature 517, 81–84. 10.1038/nature13851. [DOI] [PubMed] [Google Scholar]
- 52.Chakrabarti R, Celia-Terrassa T, Kumar S, Hang X, Wei Y, Choudhury A, Hwang J, Peng J, Nixon B, Grady JJ, et al. (2018). Notch ligand Dll1 mediates cross-talk between mammary stem cells and the macrophageal niche. Science 360. 10.1126/science.aan4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cai S, Kalisky T, Sahoo D, Dalerba P, Feng W, Lin Y, Qian D, Kong A, Yu J, Wang F, et al. (2017). A Quiescent Bcl11b High Stem Cell Population Is Required for Maintenance of the Mammary Gland. Cell Stem Cell 20, 247–260 e245. 10.1016/j.stem.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karsli Uzunbas G, Ahmed F, and Sammons MA (2019). Control of p53-dependent transcription and enhancer activity by the p53 family member p63. J Biol Chem 294, 10720–10736. 10.1074/jbc.RA119.007965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ding L, Su Y, Fassl A, Hinohara K, Qiu X, Harper NW, Huh SJ, Bloushtain-Qimron N, Jovanovic B, Ekram M, et al. (2019). Perturbed myoepithelial cell differentiation in BRCA mutation carriers and in ductal carcinoma in situ. Nat Commun 10, 4182. 10.1038/s41467-019-12125-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haensel D, Jin S, Sun P, Cinco R, Dragan M, Nguyen Q, Cang Z, Gong Y, Vu R, MacLean AL, et al. (2020). Defining Epidermal Basal Cell States during Skin Homeostasis and Wound Healing Using Single-Cell Transcriptomics. Cell Rep 30, 3932–3947 e3936. 10.1016/j.celrep.2020.02.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Strunz M, Simon LM, Ansari M, Kathiriya JJ, Angelidis I, Mayr CH, Tsidiridis G, Lange M, Mattner LF, Yee M, et al. (2020). Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat Commun 11, 3559. 10.1038/s41467-020-17358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ayyaz A, Kumar S, Sangiorgi B, Ghoshal B, Gosio J, Ouladan S, Fink M, Barutcu S, Trcka D, Shen J, et al. (2019). Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 569, 121–125. 10.1038/s41586-019-1154-y. [DOI] [PubMed] [Google Scholar]
- 59.Aragona M, Dekoninck S, Rulands S, Lenglez S, Mascre G, Simons BD, and Blanpain C (2017). Defining stem cell dynamics and migration during wound healing in mouse skin epidermis. Nat Commun 8, 14684. 10.1038/ncomms14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arnoux V, Nassour M, L’Helgoualc’h A, Hipskind RA, and Savagner P (2008). Erk5 controls Slug expression and keratinocyte activation during wound healing. Mol Biol Cell 19, 4738–4749. 10.1091/mbc.E07-10-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gkountela S, Castro-Giner F, Szczerba BM, Vetter M, Landin J, Scherrer R, Krol I, Scheidmann MC, Beisel C, Stirnimann CU, et al. (2019). Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 176, 98–112 e114. 10.1016/j.cell.2018.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dotsch V, Bernassola F, Coutandin D, Candi E, and Melino G (2010). p63 and p73, the ancestors of p53. Cold Spring Harb Perspect Biol 2, a004887. 10.1101/cshperspect.a004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Memmi EM, Sanarico AG, Giacobbe A, Peschiaroli A, Frezza V, Cicalese A, Pisati F, Tosoni D, Zhou H, Tonon G, et al. (2015). p63 Sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc Natl Acad Sci U S A 112, 3499–3504. 10.1073/pnas.1500762112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li HJ, Reinhardt F, Herschman HR, and Weinberg RA (2012). Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov 2, 840–855. 10.1158/2159-8290.CD-12-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, et al. (2013). Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152, 25–38. 10.1016/j.cell.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 66.Korkaya H, Liu S, and Wicha MS (2011). Regulation of cancer stem cells by cytokine networks: attacking cancer’s inflammatory roots. Clin Cancer Res 17, 6125–6129. 10.1158/1078-0432.CCR-10-2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ge Y, Gomez NC, Adam RC, Nikolova M, Yang H, Verma A, Lu CP, Polak L, Yuan S, Elemento O, and Fuchs E (2017). Stem Cell Lineage Infidelity Drives Wound Repair and Cancer. Cell 169, 636–650 e614. 10.1016/j.cell.2017.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ganesh K, Basnet H, Kaygusuz Y, Laughney AM, He L, Sharma R, O’Rourke KP, Reuter VP, Huang YH, Turkekul M, et al. (2020). L1CAM defines the regenerative origin of metastasis-initiating cells in colorectal cancer. Nat Cancer 1, 28–45. 10.1038/s43018-019-0006-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saladi SV, Ross K, Karaayvaz M, Tata PR, Mou H, Rajagopal J, Ramaswamy S, and Ellisen LW (2017). ACTL6A Is Co-Amplified with p63 in Squamous Cell Carcinoma to Drive YAP Activation, Regenerative Proliferation, and Poor Prognosis. Cancer Cell 31, 35–49. 10.1016/j.ccell.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mascre G, Dekoninck S, Drogat B, Youssef KK, Brohee S, Sotiropoulou PA, Simons BD, and Blanpain C (2012). Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 489, 257–262. 10.1038/nature11393. [DOI] [PubMed] [Google Scholar]
- 71.Yang X, Lu H, Yan B, Romano RA, Bian Y, Friedman J, Duggal P, Allen C, Chuang R, Ehsanian R, et al. (2011). DeltaNp63 versatilely regulates a Broad NF-kappaB gene program and promotes squamous epithelial proliferation, migration, and inflammation. Cancer Res 71, 3688–3700. 10.1158/0008-5472.CAN-10-3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, et al. (2014). Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122. 10.1016/j.cell.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wrenn ED, Yamamoto A, Moore BM, Huang Y, McBirney M, Thomas AJ, Greenwood E, Rabena YF, Rahbar H, Partridge SC, and Cheung KJ (2020). Regulation of Collective Metastasis by Nanolumenal Signaling. Cell 183, 395–410 e319. 10.1016/j.cell.2020.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kumar S, Wilkes DW, Samuel N, Blanco MA, Nayak A, Alicea-Torres K, Gluck C, Sinha S, Gabrilovich D, and Chakrabarti R (2018). DeltaNp63-driven recruitment of myeloid-derived suppressor cells promotes metastasis in triple-negative breast cancer. J Clin Invest 128, 5095–5109. 10.1172/JCI99673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Somerville TD, Biffi G, Dassler-Plenker J, Hur SK, He XY, Vance KE, Miyabayashi K, Xu Y, Maia-Silva D, Klingbeil O, et al. (2020). Squamous trans-differentiation of pancreatic cancer cells promotes stromal inflammation. Elife 9. 10.7554/eLife.53381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shibue T, Brooks MW, Inan MF, Reinhardt F, and Weinberg RA (2012). The outgrowth of micrometastases is enabled by the formation of filopodium-like protrusions. Cancer Discov 2, 706–721. 10.1158/2159-8290.CD-11-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Frose J, Chen MB, Hebron KE, Reinhardt F, Hajal C, Zijlstra A, Kamm RD, and Weinberg RA (2018). Epithelial-Mesenchymal Transition Induces Podocalyxin to Promote Extravasation via Ezrin Signaling. Cell Rep 24, 962–972. 10.1016/j.celrep.2018.06.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Walter DM, Venancio OS, Buza EL, Tobias JW, Deshpande C, Gudiel AA, Kim-Kiselak C, Cicchini M, Yates TJ, and Feldser DM (2017). Systematic In Vivo Inactivation of Chromatin-Regulating Enzymes Identifies Setd2 as a Potent Tumor Suppressor in Lung Adenocarcinoma. Cancer Res 77, 1719–1729. 10.1158/0008-5472.CAN-16-2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chatterjee A, Upadhyay S, Chang X, Nagpal JK, Trink B, and Sidransky D (2008). U-box-type ubiquitin E4 ligase, UFD2a attenuates cisplatin mediated degradation of DeltaNp63alpha. Cell Cycle 7, 1231–1237. 10.4161/cc.7.9.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep 9, 1163–1170. 10.1016/j.celrep.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 84.McKeown MR, Corces MR, Eaton ML, Fiore C, Lee E, Lopez JT, Chen MW, Smith D, Chan SM, Koenig JL, et al. (2017). Superenhancer Analysis Defines Novel Epigenomic Subtypes of Non-APL AML, Including an RARalpha Dependency Targetable by SY-1425, a Potent and Selective RARalpha Agonist. Cancer Discov 7, 1136–1153. 10.1158/2159-8290.CD-17-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–21 29 29. 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kheradpour P, and Kellis M (2014). Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res 42, 2976–2987. 10.1093/nar/gkt1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mathelier A, Fornes O, Arenillas DJ, Chen CY, Denay G, Lee J, Shi W, Shyr C, Tan G, Worsley-Hunt R, et al. (2016). JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 44, D110–115. 10.1093/nar/gkv1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Newburger DE, and Bulyk ML (2009). UniPROBE: an online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res 37, D77–82. 10.1093/nar/gkn660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, Morgunova E, Enge M, Taipale M, Wei G, et al. (2013). DNA-binding specificities of human transcription factors. Cell 152, 327–339. 10.1016/j.cell.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 91.Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.de Hoon MJ, Imoto S, Nolan J, and Miyano S (2004). Open source clustering software. Bioinformatics 20, 1453–1454. 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 94.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Amit I, Citri A, Shay T, Lu Y, Katz M, Zhang F, Tarcic G, Siwak D, Lahad J, Jacob-Hirsch J, et al. (2007). A module of negative feedback regulators defines growth factor signaling. Nat Genet 39, 503–512. 10.1038/ng1987. [DOI] [PubMed] [Google Scholar]
- 96.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data have been deposited at GEO and are publicly available as of the date of publication. The accession number for the ChIP-seq and ATAC-seq reported in this paper is GSE172407. The accession numbers for the RNA-seq and single-cell RNA-seq reported in this paper are GSE171629, GSE171626 and GSE171628.
No original code was generated for this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.