

Abstract
The availability of long-reads is revolutionizing studies of structural variants (SVs). However, because SVs vary across individuals and are discovered through imprecise read technologies and methods, they can be difficult to compare. Addressing this, we present Jasmine and Iris (https://github.com/mkirsche/Jasmine), for fast and accurate SV refinement, comparison, and population analysis. Using an SV proximity graph, Jasmine outperforms six widely-used comparison methods, including reducing the rate of Mendelian discordance in trio datasets by more than five-fold, and reveals a set of high-confidence de novo SVs confirmed by multiple technologies. We also present a unified callset of 122,813 SVs and 82,379 indels from 31 samples of diverse ancestry sequenced with long reads. We genotype these variants in 1,317 samples from the 1000 Genomes Project and GTEx with DNA and RNA sequencing data and assess their widespread impact on gene expression, including within medically relevant genes.
Introduction
Structural variants (SVs) are defined as large-scale genomic mutations affecting more than 50 basepairs, and include insertions, deletions, duplications, inversions, and translocations 1,2. Such variants are responsible for more divergent basepairs across human genomes than any other class of variation 3, and have been associated with many major diseases and phenotypes, including cancer 4,5 and autism 6. They have also been shown to have phenotypic effects in other species, such as altered growth under stress in yeast 7. However, much of the impact of structural variants remains unknown because of the inability of SVs in complex regions to be accurately identified by short reads which make up the majority of existing genomic sequencing data 8,9. In a similar manner, indels larger than 30bp in length, while not typically considered to be SVs under the 50bp threshold, have been shown to be similarly associated with changes in phenotypes 1 and also suffer from an inability to be mapped and resolved in short-read genomic data 10–12. Therefore, while the main focus of our analysis is on SV calling, we also demonstrate how our methods can be applied to indels which affect at least 30bp as well. Throughout this manuscript, we use “SVs” to refer to variants affecting at least 50 basepairs, but use “SVs and indels” to refer collectively to all variants affecting 30 or more basepairs.
In recent years, the emergence of long-read genomic sequencing technologies 13–16 and the development of specialized software for alignment 17–19 and variant calling 18,20 have enabled the characterization of complex structural variants which were difficult or impossible to study from short reads alone 8. For this reason, many population variant inference studies include long-read sequencing data for multiple individuals instead of or in addition to short-read data 21–23.
Because there are multiple sequencing technologies, aligners, and SV callers that could be used, SV-processing pipelines for population-scale studies are frequently optimized for the particular dataset being analyzed 7,23, making it difficult to compare SVs called in different studies or to accurately screen newly sequenced samples for known variants. In addition, existing tools for comparing SV callsets from different samples have issues such as collapsing multiple variants in the same individual, including variants of different types, and producing callsets that vary substantially when the order of the input samples is changed. As the cost of long-read sequencing continues to fall and the number of population-scale SV studies continues to rise, there is an increasingly apparent need for methods which can accurately compare variants across a range of datasets.
To address this need, we introduce an optimized software pipeline for accurately detecting SVs and comparing these variant calls across large numbers of individuals (Figure 1). This pipeline enhances existing methods for alignment 17 and variant calling 18 with new methods for refining the sequences and breakpoints of SV calls, and for comparing variant calls between different individuals to achieve a unified callset. Using a combination of simulated and real datasets, we show that this pipeline produces more accurate SV calls than several widely used methods across a variety of metrics. First, by applying our methods to a HiFi dataset from the HG002 Genome-In-A-Bottle (GIAB) Ashkenazim trio, we illustrate that our approach achieves a five-fold reduction in the number of Mendelian discordant variants, while identifying multiple high-confidence de novo variants in the child supported by three independent sequencing platforms. We also analyze this trio to identify signatures of variants specifically derived from each particular technology. This enables us to establish recommended variant calling parameters for different sequencing technologies which minimize Mendelian discordance as well as false merges. We next show that Jasmine improves SV merging and addresses the major issues that other methods encounter when scaling up to large cohorts. We call variants with our pipeline from publicly available long-read data for 31 samples, and generate a panel of long-read SV and indel calls which can be used for screening further samples. Finally, we genotype this variant panel in 444 high-coverage short-read samples from the 1000 Genomes Project 24 along with 873 samples from GTEx 25 and discover thousands of previously undetected SV associations with gene expression. Many of these SVs have CAVIAR posterior probabilities of causality that exceed those of previously reported SNPs, indicating likely functional relevance, including within medically relevant genes.
Figure 1: SV Inference Pipeline.
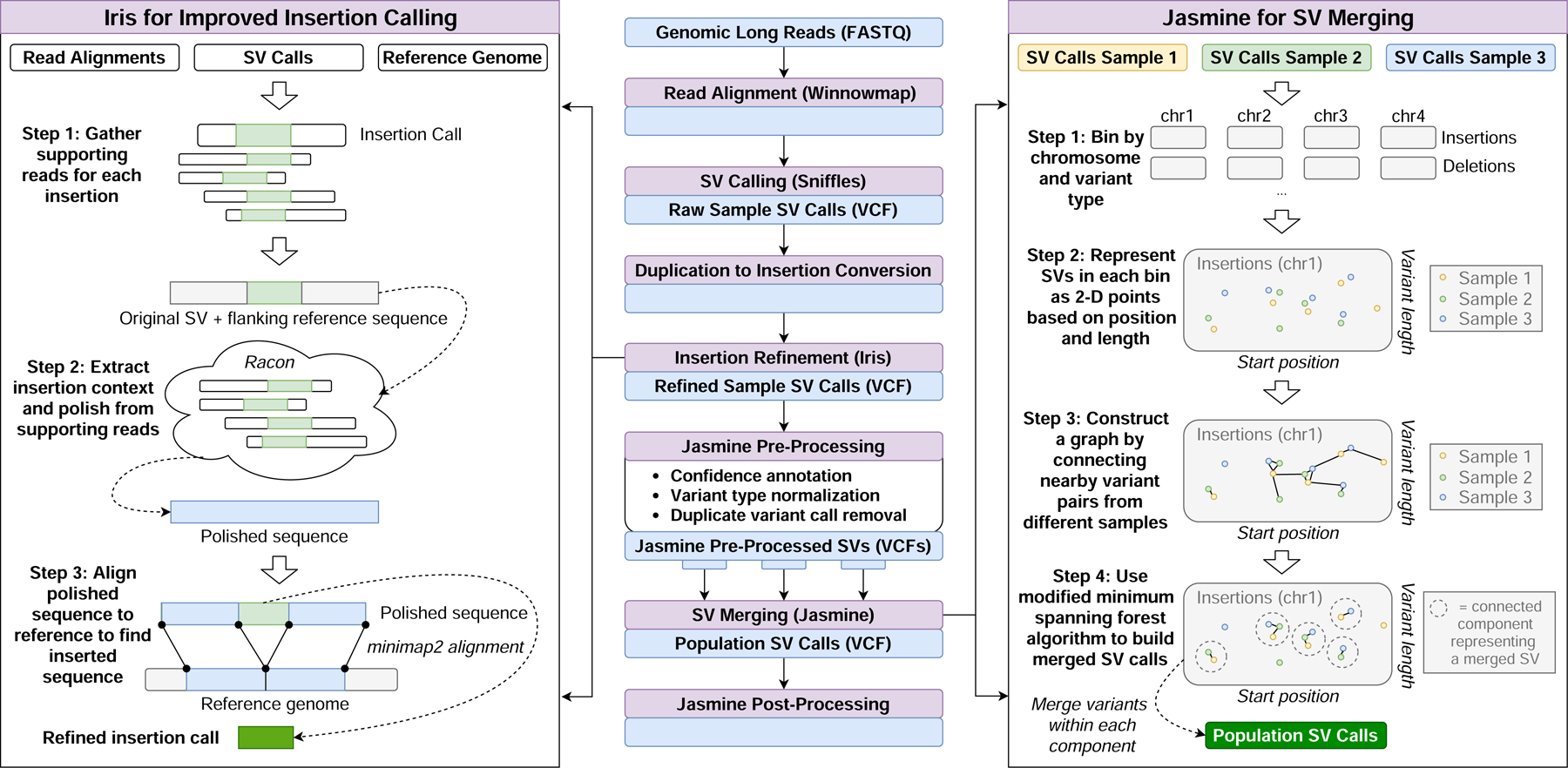
This pipeline produces population-level SV calls from FASTQ files using a number of existing methods as well as two novel methods, Iris and Jasmine. Iris uses consensus methods to improve the accuracy of the breakpoints and sequence of insertion SVs. Jasmine uses a graph of SV proximity and a constrained minimum spanning forest algorithm to compare and combine variants across multiple individuals.
Results
Optimized SV refinement, comparison, and population analysis with Iris and Jasmine
Addressing the need for accurate SV refinement, comparison, and population analysis we introduce two methods, Iris and Jasmine. The first method, Iris, refines variant calls by using Racon 26 to polish the variant sequence from reads supporting the alternate allele and realigning this polished sequence to the reference with minimap2 19.The second method, Jasmine, compares and merges calls in different individuals corresponding to the same variant. Jasmine represents variants as points in space based on their breakpoints and lengths and constructs a graph of SV proximity, where edges represent pairs of SVs with a small Euclidean distance between them. Jasmine then treats the comparison/merging problem as one of finding a minimal-weight acyclic subgraph of the proximity graph which satisfies constraints such as user-specified distance thresholds and the avoidance of intrasample merging. Jasmine solves this problem with a constrained version of Kruskal’s algorithm for minimum spanning trees 27, and avoids the high time and memory overhead of computing and storing the entire graph by using a KD-Tree 28 to dynamically locate nearby variant pairs and implicitly detect low-weight edges. This optimization is key to Jasmine’s performance, as it enables it to implicitly consider the entire SV proximity graph and prioritize merges which encompass edges of globally-minimal weight. This is in contrast to prior methods, which often perform sub-optimal merging because they utilize heuristics to consider smaller subgraphs of the variant proximity graph and potentially disregard minimum-weight edges which would be included in the optimal merging. Both Iris and Jasmine are available as stand-alone software packages and are available within bioconda as well as within Galaxy 29.
Reduced Mendelian Discordance in an Ashkenazim Trio
A common application of SV and other variant inference methods is the identification of de novo variants, or variants which are present in an individual but neither of their parents. Such variants have been associated with autism 30 and cancer 31, and de novo variant analysis is frequently used as a starting point for identifying the cause of genetic diseases or other phenotypes of interest 32. However, because of shortcomings in SV inference and comparison methods, identifying de novo SVs and indels remains a difficult problem. For example, one widely used pipeline consisting of ngmlr, sniffles 18, and SURVIVOR 7 gives thousands of candidate de novo SVs when applied to high-accuracy HiFi sequencing data from the HG002 Ashkenazim trio (Figure 2a). Because the number of de novo SVs is typically estimated to be less than ten per generation on average 33, almost all of these variant calls are either false positives in the child, false negatives in one or both parents, or errors in merging the callsets. Collectively, we refer to these false outcomes as Mendelian discordant variants.
Figure 2. Mendelian Discordance in the HG002 Ashkenazim Trio.
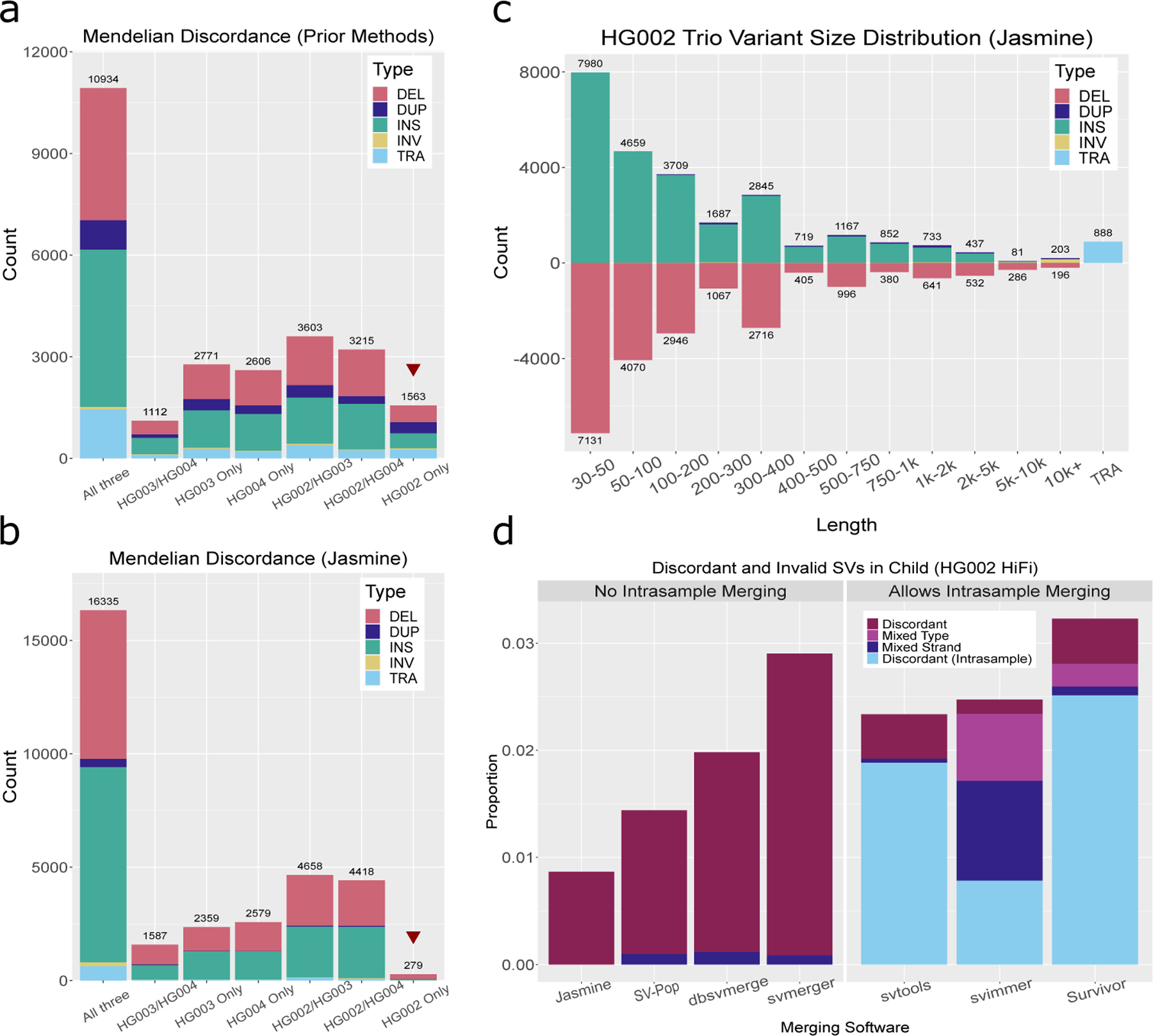
We called SVs from HiFi data for the Ashkenazim trio consisting of HG002 (son - 46,XY) and parents HG003 (46,XY), and HG004 (46,XX) using several prior methods as well as our pipeline. a.) The number of SVs called in each subset of individuals when using prior methods: ngmlr for alignment, Sniffles for SV calling, and SURVIVOR for consolidating SVs between samples. b.) The number of SVs called in each subset of individuals when using our optimized pipeline. c.) The distribution of variant types and lengths in the HG002 trio with our pipeline. d.) The rate of discordance when comparing SVs between individuals with Jasmine as well as six existing methods for population inference. Jasmine reduces the discordance rate while at the same time addressing issues present in other methods such as merging variants of different types, variants with the same type but corresponding to unique breakpoint adjacencies (mixed strand), or variants within the same sample.
To address the large number of discordant variants, our optimized pipeline offers a number of improvements which reduce the rate of Mendelian discordance by more than a factor of five with <1% (279/32,215 = 0.009) of merged SVs being discordant (Figure 2b). At the same time, our pipeline enabled the discovery of 10–20% more SVs than existing methods, with a size distribution and indel balance similar to prior work (Figure 2c, Supplementary Figure 1). The methodological improvements include double thresholding (see Methods: Double Thresholding) which mitigates threshold effects in variant detection (Supplementary Figure 2), improved variant calling parameters (Supplementary Figure 3), and using Jasmine for SV merging. Furthermore, we compared Jasmine to six existing methods for SV comparison between samples (Figure 2d; Supplementary Figure 4): dbsvmerge 34, SURVIVOR 7, svpop 35, svtools 36, sv-merger 23, and svimmer 37. For each software, we merged the unfiltered callset from each of the three samples, and after merging filtered the variants based on the read support, length, and breakpoint precision of the corresponding input SV calls. We found that Jasmine achieves the lowest rate of discordance and correctly avoids merging variants of different types or variants from the same sample. This is largely due to its ability to detect and merge the closest pair of variants among all variant pairs, which is in contrast to other methods that use heuristics to reduce the number of mergeable pairs beforehand, leading to suboptimal merging. In addition, Jasmine avoids merging mismatched variants corresponding to partial inversions or translocations, which is particularly important when resolving complex nested SVs (Supplementary Figure 5). The resulting reduction in Mendelian discordant variants is an important step towards the rapid identification of de novo variants, as it is typically necessary to screen all discordant variants manually when searching for true de novo variants.
We also evaluated the discordance rate among SVs overlapping tandem repeats (TRs), and found that the discordance of SVs overlapping tandem repeats was similar to the overall rate (195/22,626 = 0.0086 overlapping TRs; 84/9,589 = 0.0088 outside TRs). However, manual inspection revealed a large number of discordant variants where the true SV was within a tandem repeat, but disrupted alignment and variant calling resulted in an SV call just outside of the repeat region. We investigated discordance among SVs near TRs and found that there was a higher discordance rate for SVs within 500bp of tandem repeats (252/26,300 = 0.0096 within 500bp of TRs; 27/5,915 = 0.0046 at least 500bp outside TRs). Because the discordance is so much lower in regions at least 500bp away from TRs (<0.5%), we refer to these regions as non-TR regions.
SV Analysis Across Sequencing Technologies
Improved methods for comparing multiple SV callsets also enable the comparison of variants identified in a single individual from different sequencing technologies. We evaluated three different technologies applied to HG002: Pacific Biosciences Continuous Long Reads (CLR), Pacific Biosciences High-Fidelity (HiFi) circular consensus sequencing and Oxford Nanopore long reads (ONT) basecalled with Guppy 4.2.2. Variants were called separately from each technology, and the resulting callsets were merged with Jasmine. The three callsets were largely in agreement, with 18,778 out of 28,348 SVs being supported by all three technologies (Figure 3a and 3b; Supplementary Figure 6). The set of technology-concordant variants, shown in Figure 3c, shows that insertion and deletion calls are largely balanced, with a slight enrichment of insertions, shown in previous studies to be caused by missing sequence in the human reference genome 22, as well as a tendency for deletions to be more deleterious 38. There is also an increased number of variants around sizes of 300bp and 6–7kbp (Supplementary Figure 7), corresponding to SINE and LINE elements respectively.
Figure 3. SV Inference across Sequencing Technologies in HG002.
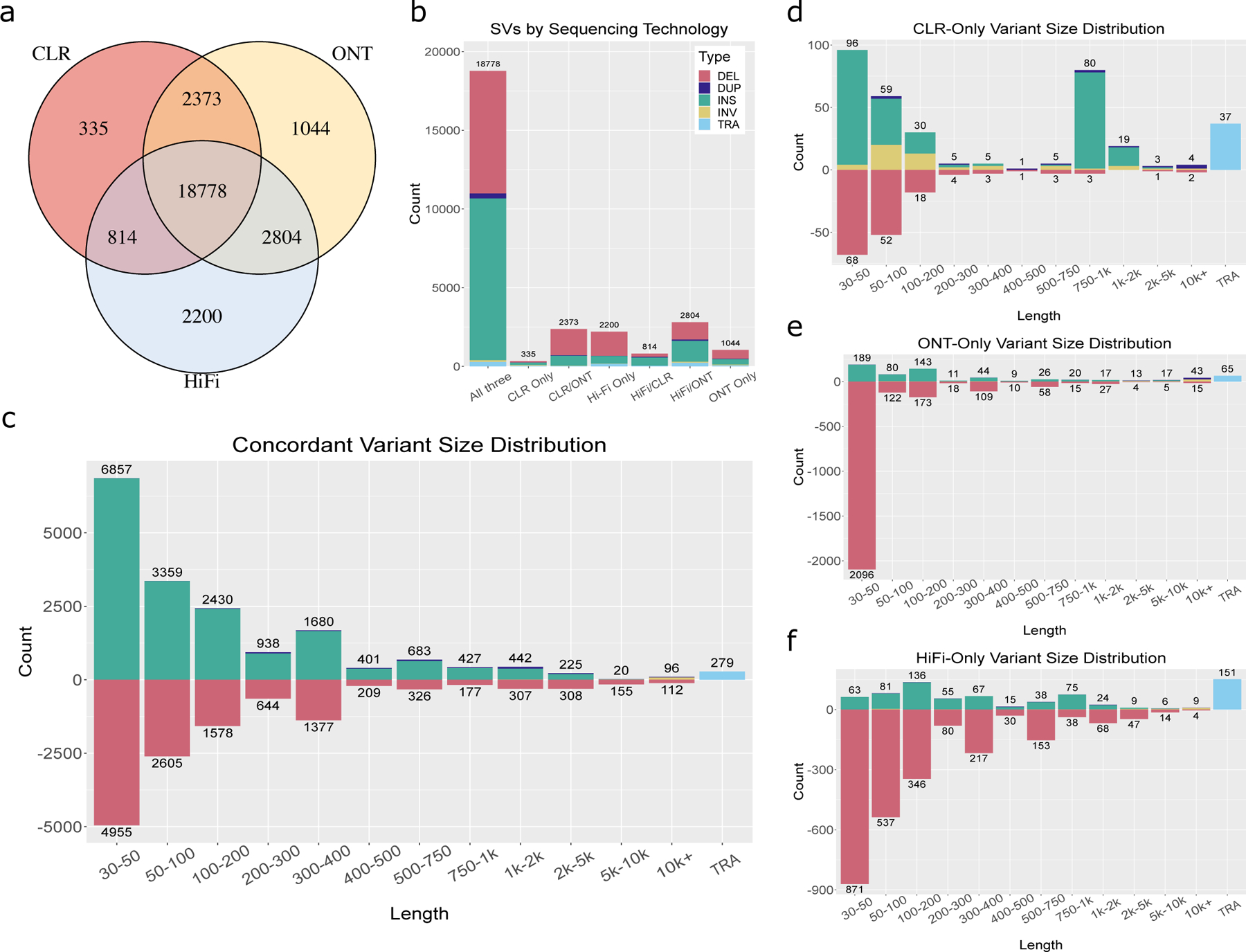
We called SVs in HG002 separately from Pacbio CLR data, Oxford Nanopore data, and Pacbio HiFi CCS data, and used Jasmine to compare the variants discovered by each of them. a.) The number of SVs discovered by each subset of technologies. b.) The SV type distribution within each subset of technologies. c.) The distribution of types and lengths among variants for which all of the technologies agree. d-f.) The type and length distributions for variants unique to CLR, ONT, and HiFi respectively.
We also examined variants that were identified only by a single technology, as these may reveal systematic biases in variant calling caused by each technology’s error model, particularly in CLR and ONT, which have higher rates of sequencing error. Of the 499 variants identified exclusively in CLR data (Figure 3d), there were 244 insertions and 155 deletions, with an excess of insertions in the size range 750 to 1000, corresponding to a known error characteristic of CLR sequencing 18. Of the 3,329 ONT-only variant calls (Figure 3e), there were 539 insertions and 2,652 deletions, with an enrichment of small deletions less than 50 basepairs in length. In addition, we found that many of the variants, particularly deletions, unique to ONT or HiFi are in centromeric regions or satellite repeats (Supplementary Figures 8-9). We also called and merged SVs separately for each technology across the HG002 trio and measured the discordance among the SVs discovered by the individual technologies. We found that ONT and HiFi data result in similar discordance rates (279/32,215 = .0087 in HiFi; 295/34,062 = .0087 in ONT), while CLR-derived calls have a higher rate of discordance (310/19,206 = 0.0161).
De Novo Variant Discovery
We next leveraged our methods, as well as data from all three technologies listed above, to screen the HG002 trio for de novo SVs and indels. We called variants from each of the three technologies in HG002 as well as both parents, for a total of nine callsets. We merged these nine callsets with Jasmine and filtered out any variants which were present in one or more of the six parent callsets. Of the remaining variants, we stratified them by which technologies supported their presence in the child and found that there were 16 which were supported by all three technologies (Figure 4a), with an additional 35 that were supported by HiFi and at least one other technology, a 43-fold reduction in candidates compared to evaluating HiFi data alone with prior methods (Supplementary Figure 10).
Figure 4. De Novo Variant Discovery in HG002.
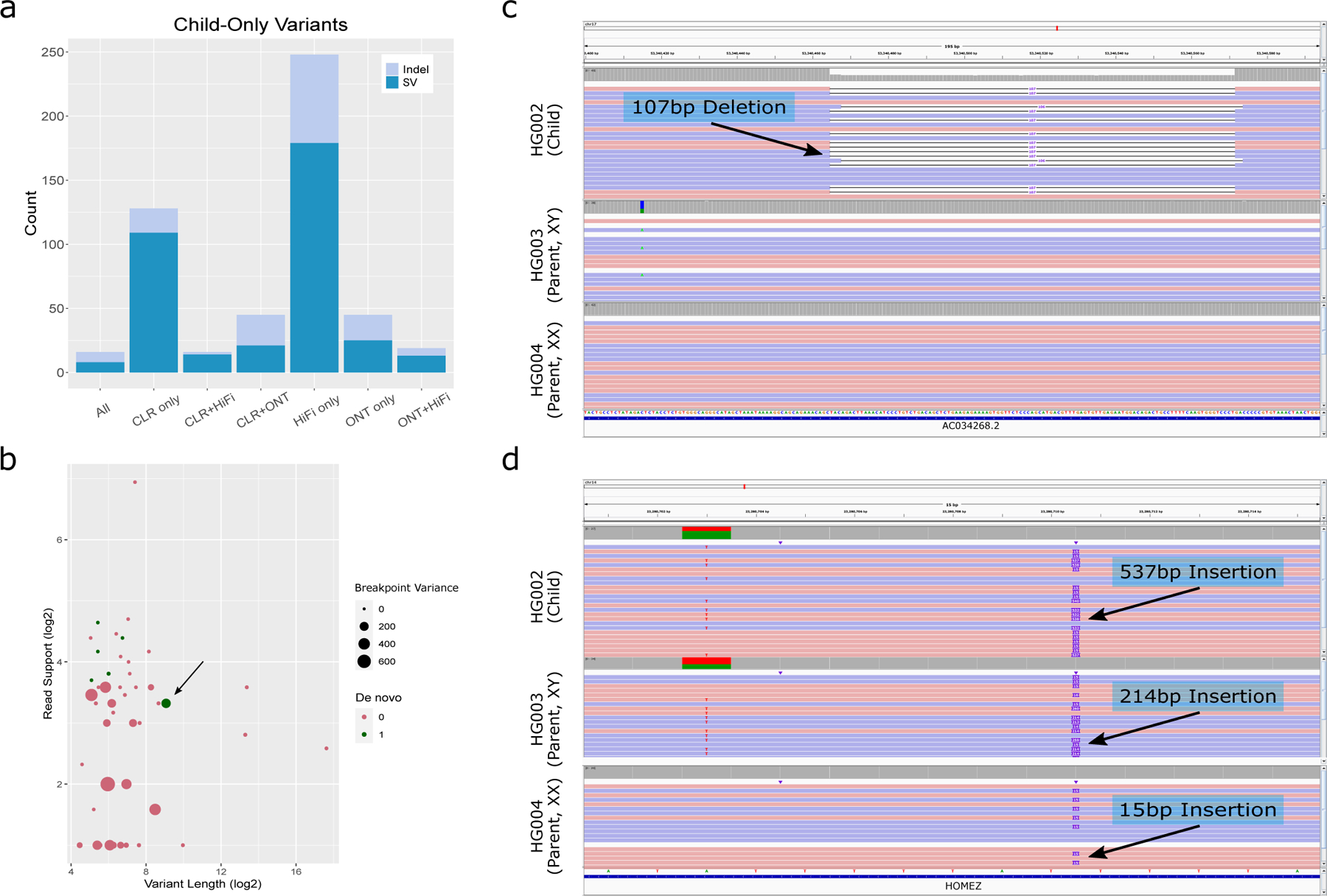
We called variants in each of HG002, HG003, and HG004 from three different sequencing technologies - CLR, ONT, and HiFi - to identify potential de novo variants that were called in none of the six parent callsets but one or more of the HG002 callsets. a.) The number of SVs and indels which are absent in all six parent callsets whose presence in HG002 is supported by each subset of technologies. While we manually inspected all SVs supported by HiFi and at least one other technology, both of the examples in (c) and (d) were supported by all three technologies. b.) All variants supported by HiFi and at least one other technology in HG002 that are absent in all parent callsets. The potential de novo variants we identified are highlighted in green, with the microsatellite repeat expansion denoted by an arrow. While filters based on length, read support, and breakpoint standard deviation could be used to filter out many false de novo candidates, the microsatellite repeat expansion is an example of a higher-confidence de novo SV which would be incorrectly filtered out. c.) A potential de novo 107bp deletion in HG002 at chr17:53340465. d.) A potential de novo microsatellite repeat expansion in HG002 at chr14:23280711.
Upon manual inspection, six of these were high-confidence de novo variants (Figure 4b), while the remaining candidates were in noisy regions that displayed split read alignments, but we could not be certain whether the alignments were correct (Supplementary Figure 11). One of the high-confident candidates, a 107bp deletion at chr17:53340465 (Figure 4c), was previously identified as a de novo SV in an effort to characterize the variants in HG002 39. Another example, a 537bp insertion at chr14:23280711, consists of a microsatellite repeat expansion on the paternal haplotype, a known class of mutations often caused by replication slippage 40 (Figure 4d). These and other examples (Supplementary Figures 11-13) show that our approach can correctly identify known de novo SVs as well as identify potential de novo variants which were previously undiscovered, and that these variants are supported by multiple independent sequencing technologies. This ability coupled with the reduced number of discordance demonstrates a major step towards automated de novo variant detection.
Population SV Inference
As the cost of long-read sequencing has continued to decrease in recent years, long-read studies including large cohorts have become more prevalent 23,34. As this trend is expected to continue 41, it is particularly important for SV inference methods to be able to scale to many samples. To compare Jasmine to existing approaches, we called SVs and indels in 31 publicly available long-read samples (Supplemental Table 2) and observed the results of merging these callsets with each method. We attempted to run all six prior methods, although sv-merger did not terminate after 72 hours, and so was excluded from this analysis. All other methods produced a population-level callset within a few hours with 24 threads on a modern 4GHz server with 192GB of RAM, but the callsets produced by existing approaches suffer from a number of issues. In addition to the invalid merges mentioned above (Figure 2d), several of the existing methods use algorithms which give different merging results, and consequently different numbers of total variant calls, based on the input order of the sample callsets (Figure 5a). This problem only worsens as the number of samples grows and the number of possible sample orderings increases exponentially. Conversely, Jasmine’s algorithm, which merges variant pairs in increasing order of their breakpoint distances irrespective of the input order, produces identical results after any permutation of input files. Jasmine additionally offers the lowest median breakpoint range within merged variants (Figure 5b, Supplementary Figure 14) and avoids merging variants from the same sample. Finally, there is an abundance of low-confidence likely false positive variant calls in samples sequenced with CLR (Supplementary Figures 15-16), and methods which use a constant breakpoint distance threshold incorrectly merge these calls with high-confidence variant calls in other samples to obtain an unreasonable trimodal allele frequency distribution (Supplementary Figures 17-18).
Figure 5. Population-Scale Inference from Public Datasets.
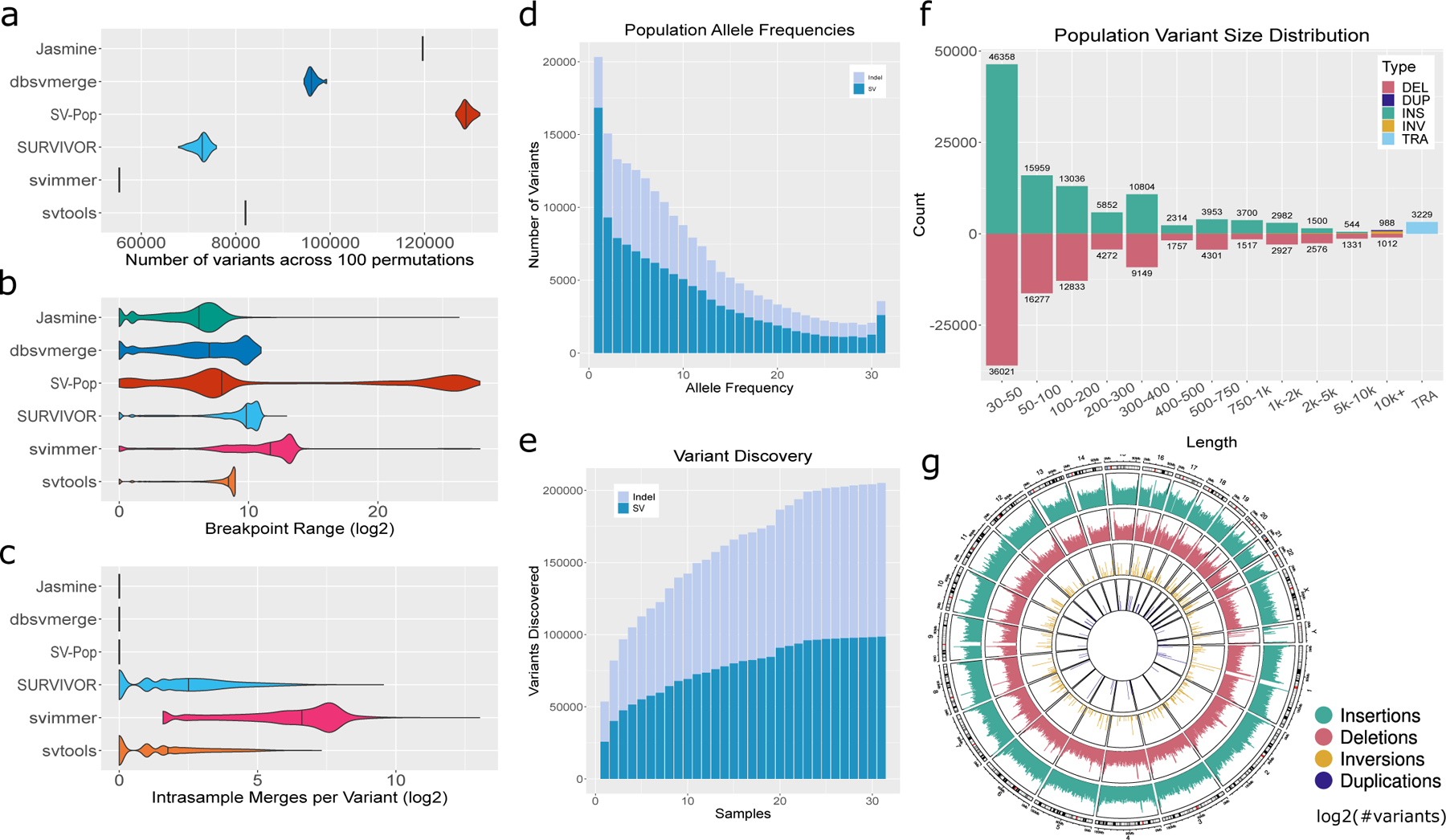
We called SVs and indels with our pipeline in a cohort of 31 samples from diverse ancestries and sequencing technologies and used Jasmine as well as five prior methods to combine the individual samples’ SVs into a population-scale callset. a.) The number of variants obtained with each merging software across 100 runs with the list of input VCFs randomly shuffled each time. b.) The distribution of the range of breakpoints of variant calls merged into single variants by each software, excluding unmerged variants. c.) The number of intrasample merges within single merged variants, defined as the number of variants minus the number of unique samples, for each software. d.) The allele frequency distribution of variants merged by Jasmine. e.) The number of variants discovered by Jasmine as the number of samples increases. f.) The distribution of variant types and lengths in the cohort when using Jasmine. g.) The number of SVs in the cohort in 1Mbp bins across the human genome.
Using our SV inference pipeline, we created a panel of long-read 122,813 SVs and 82,379 indels from these 31 samples. The datasets we used include individuals from a wide range of ancestral backgrounds, as well as sequencing data from multiple technologies. Variants were called in each sample separately and merged with Jasmine to create a unified callset. The allele frequency distribution is monotonically decreasing as expected, except an excess of variants at 100% corresponding to errors and/or minor alleles in the reference 22 (Figure 5d). The cumulative number of variants increases with the number of samples, but at a decreasing rate (Figure 5e). The indels are approximately balanced (Figure 5f), with a slight bias towards insertions, and there are spikes in the size distribution around 300bp and 6–7kbp for SINE and LINE elements (Supplementary Figure 19). There is also an enrichment of SVs in the centromeres and telomeres (Figure 5g; Supplementary Figure 20), likely due to a combination of missing reference sequence, repetitive sequence which is difficult to align to, and greater recombination rates 22. We also filtered our callset by the non-TR regions defined above (>500bp away from tandem repeats), and found that 22,132 SVs and 13,615 indels are contained in these regions.
Measuring Effects of SVs on Gene Expression
Previous expression quantitative trait loci (eQTL) studies have shown that SVs often have large effects on gene expression and that they are causal at 3.5–6.8% of eQTLs 3,42. To investigate this with our enhanced catalog of SVs, we first used Paragraph 43 to genotype each SV in 444 individuals from the 1000 Genomes Project (1KGP) for which gene expression data is publicly available 44, after removing SVs that were inconsistent with population genetics expectations based on the Hardy-Weinberg equilibrium (Supplementary Figure 21a). Following the prior studies, we mapped SV-eQTLs by pairing common (MAF ≥ 0.05) SVs to genes within 1 Mbp using gene expression data in lymphoblastic cell lines from the GEUVADIS consortium 44. Each SV-gene pair was considered independently. We then fit a linear model to measure the effect sizes of these SVs on gene expression, and found that 5,456 pairs passed a significance threshold with 10% FDR (matching previous studies of this dataset 44), which is substantially higher than the 478 pairs that we observe among short-read SVs using the same FDR. These associations occur for both deletions and insertions, and both have approximately the same effect size distribution (Supplementary Figure 21b). These data suggest that many of the SVs that are only visible through genotyping long-read-based variant calls have large effects on gene expression and thus are potentially functionally relevant.
In order to evaluate which SVs are likely to have causal effects on their associated genes, we used the fine-mapping tool CAVIAR 45 to measure the posterior probability that any given SV is causal compared to nearby SNPs within a 1 Mbp window, taking into account possible linkage disequilibrium (LD) between variants. We found that SVs had high posterior scores (>0.1) at 68 genes out of 1,863 genes examined (3.65%). Additionally, when compared to existing databases of SNP-eQTLs from the GTEx project 3,25, SVs had a higher CAVIAR posterior than reported SNPs for 53.5% of genes with an SV-eQTL (Supplementary Figure 21c). This shows that previously undetected SVs are likely causal at a large number of sites where the effects on gene expression were reported as SNP-eQTLs instead. Inspecting all SV-gene pairs with a CAVIAR posterior greater than that of any previously reported SNP-eQTL for that gene (and greater than 0.2 overall), we identified several potentially functional SVs in high linkage disequilibrium (LD) with reported SNPs (Supplementary Figures 22-23). Several of our top candidates have been reported by other studies as SV-eQTLs, which serves to validate our overall approach and increase confidence in our discoveries.
To further demonstrate the application of merging variants with Jasmine for SV-eQTL discovery, we next genotyped and analyzed the long-read reference SV set in the GTEx dataset 3,25. The GTEx dataset contains short-read WGS from over 800 individuals with matched RNA-seq data in up to 49 non-diseased tissues. We first genotyped 26,377 common SVs detected in the reference SV set with Paragraph43 within the NHGRI AnVIL Terra platform 46 in 873 GTEx individuals. Here we focused on common SVs with minor allele frequency of at least 0.05 that pass conservative Hardy-Weinberg filtering at genome-wide significant p-value. Using this approach we discovered over two-fold more variants per individual than previous efforts by the GTEx consortium 3 in identifying SVs exclusively using the short read data (Figure 6a).
Figure 6. Functional impact of SVs from Jasmine.
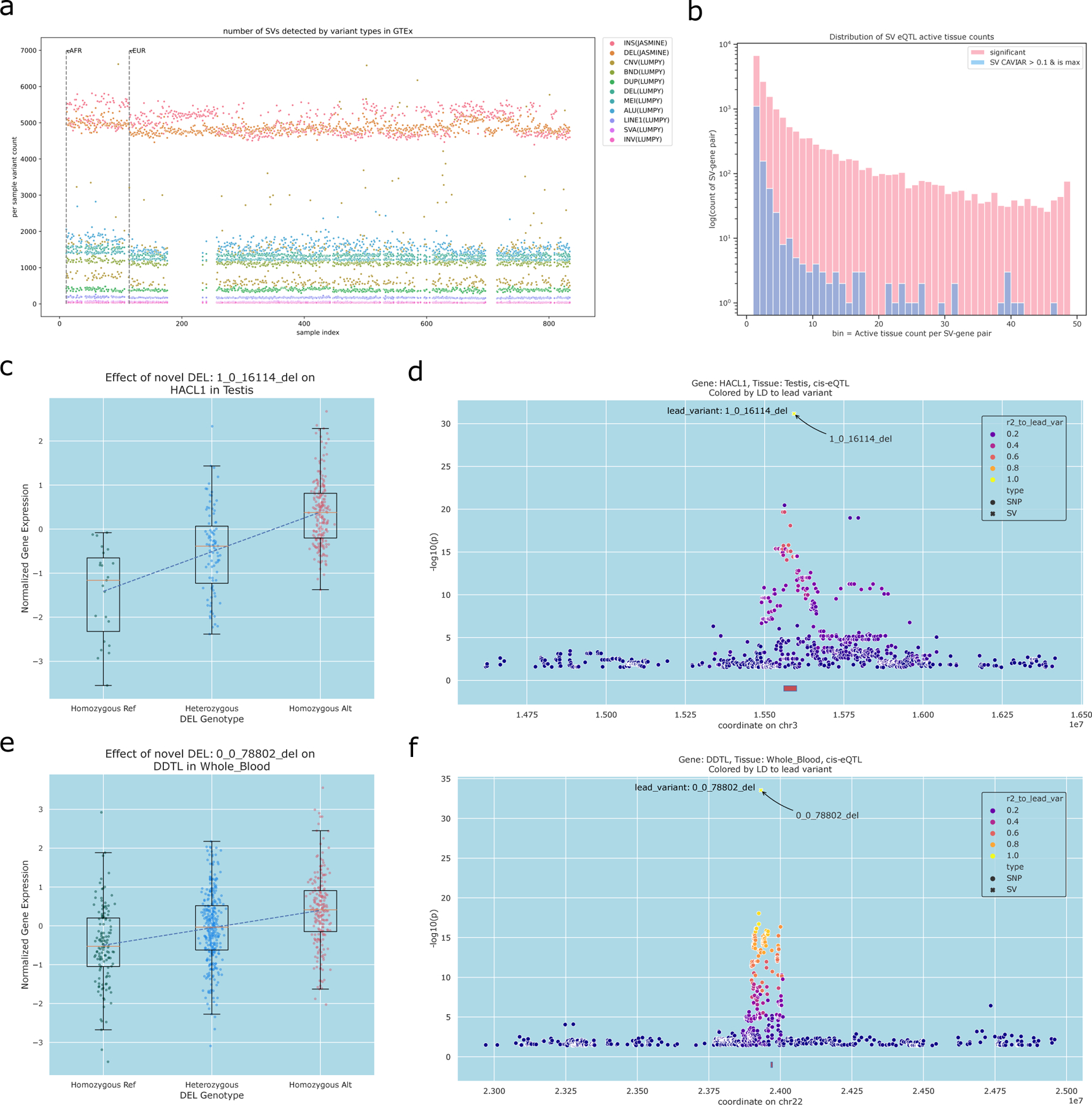
We used Paragraph to genotype SVs and indels from the cohort of 31 samples in 873 samples from the GTEx Consortium which have RNA-seq data in multiple tissues. We used 48 tissues in our analysis with sufficient samples. a.) Number of variants detected per sample for genotyped SVs and indels (Jasmine) versus SVs reported in the GTEx SV dataset after HWE filtering. Note short read-based SV calls are not available for all samples so some samples only display the counts using Jasmine. b.) Distribution of the number of tissues an SV-gene pair is found as a significant eQTLs (FDR correction at 5%). We further plot the distribution for SV-gene pairs with significant eQTLs where the SV has the maximum CAVIAR score compared to all flanking SNPs. c.) Genotype and gene expression distribution in GTEx samples with expression in testis for the HACL1-associated deletion (n=318). d.) Manhattan plot for SNPs and the novel SV near HACL1, with the log10 p-value measured by a generalized linear model accounting for GTEx covariates. The annotated variant is the top variant, 1_0_16114_del, and points are colored by LD to this variant. For c,d, we used 318 individuals with both SV calls and RNA seq in testis tissue. e.) Genotype and gene expression distribution in GTEx samples with expression in whole blood for DDTL-associated deletion (n=666). f.) Manhattan plot for SNPs and the novel SV near DDTL, with the log10 p-value measured by a generalized linear model accounting for GTEx covariates. The annotated variant is the top variant, 0_0_078802_del, and points are colored by LD to this variant. For e,f, we used 666 individuals with both SV calls and RNA seq in whole blood samples. Examples c and e were selected based on a two-sided t-test to assess nominal p-value of a variant gene pair after gene-level Bonferroni multiple hypothesis testing corrections at FDR 5%. Boxplots describes the 1st to 3rd quartile of the expression z-score distribution and the whiskers describes 1st quartile - 1.5 * IQR and 3rd quartile + 1.5 * IQR centered on the mean expression value of each genotype group.
We subsequently obtained gene expression measurements and technical covariates from GTEx for these individuals from 48 tissues (those with at least 70 individuals) and computed eQTLs using the same cis-eQTL calling framework as previously described in GTEx v8 25. As GTEx contains more individuals than GEUVADIS and provides gene expression measurements across dozens of tissue types, we used a 5% FDR rate, which is even more conservative than previous studies 47. At 5% FDR, we identified 111,291 significant eGenes across 48 tissues, including 11,046 SVs affecting the same genes in multiple tissues (Figure 6b). Among the eGenes, we intersected the SV-only eGenes with previously reported SNP-based eGenes, and conservatively estimated the new number of cases where an SV-eQTL is the top variant to be 10,436, which is over 2,000 more examples than previously reported even when using the stricter threshold 47. We next repeated the CAVIAR analysis on gene expression as with the 1000 Genomes dataset but scaled the analysis to all tissues. Overall, we find 5,580 SV-eQTLs where an SV has the highest CAVIAR score for the eGene, including 750 SVs affecting genes in two or more tissues (Figure 6b). The median proportion of significant eGenes with SV as lead causal variant within each tissue is 5.7%, and across all tissues, a SV is the top CAVIAR predicted causal variant in approximately 5% of the cases, consistent with our estimate from the 1000 Genome-Geuvadis SV-QTL dataset of 3.5–6.8%. We evaluated the SV eGenes with SV length <100,000 bp across all tissues available for enrichment and found a highly significant 9.5-fold enrichment (p-value=8.5e-10, Fisher Exact Test) for coding SVs to have high CAVIAR posteriors.
One notable example of an SV-eQTL identified using our Jasmine-Paragrah pipeline in GTEx is a deletion of 168bp within chromosome 3 in an intron of HACL1 (2-Hydroxyacyl-CoA Lyase 1), a gene associated with multiple metabolic diseases48. The deletion is not previously reported by GTEx or other major databases of variants but is strongly supported by the long read sequencing and genotyping results. Based on the GTEx expression data, we identified it as an eQTL in testis tissues with a log2 allelic fold change of 1.11 (Figure 6c). We also computed the t-statistic as the beta effect size divided by the variance of beta and found that both the p-value and t-statistic values are substantially stronger for the deletion than any flanking SNPs (Figure 6d, Supplementary Figure 24). The deletion is more common in the population than a non-deletion, indicating the reference genome itself carries a minor allele insertion variant. Consequently the direction of effect for the deletion is opposite the top SNP, as they are in linkage disequilibrium with an r2 of −0.6. Overall, the stronger CAVIAR score, p-value, and t-statistic suggests the SV is more likely than the flanking SNPs to be causal and the top SNP is effectively a marker for the SV. Another example of an SV-eQTL we discovered using our approach is a 37kbp deletion on chromosome 22 near the gene DDTL (D-Dopachrome Tautomerase Like), a paralog of the gene DDT, which has been associated with the chronic autoimmune disease Discoid Lupus Erythematosus 49. The deletion was previously reported by the 1000 Genomes Consortium, although previous reports did not report it as an SV-eQTL. Within Whole Blood, a log2 allelic fold change of 1.46 is observed, and as with HACL1, the p-value, t-statistic, and CAVIAR posterior are strongest for the SV compared to flanking SNPs (Figure 6e, f, Supplementary Figure 25). Interestingly, we find the SV-eQTL is putatively causal with CAVIAR posterior >0.9 for 36 tissues, and the tissue log p-value distribution is significantly higher (p-value= 1.1e-8, one-sided Wilcoxon rank sum test) than the top SNP associations in the same tissues (Supplementary Figure 26). A third significant SV-eQTL is a 60bp insertion on chromosome X that is an SV-eQTL of the gene ASMTL (Acetylserotonin O-Methyltransferase Like), a gene associated with Melanotic Neurilemmoma and other rare tumor types 50, in GTEx heart left ventricle tissue (Supplementary Figure 27). Overall, our eQTL and causal SV-QTL analysis broadly agrees with our analysis with 1000 Genome Project and previous GTEx analysis 3,47, although our Jasmine-Paragraph workflow enables us to genotype and analyze more SVs than previous approaches. Consequently, with our more accurate and complete SV catalog, we are able to discover substantially more significant and putatively causal eQTLs than in any previous analysis.
Discussion
Here we introduced Iris and Jasmine. Iris improves the sequence fidelity of SVs by computing the consensus of the reads that span each SV. Jasmine is a fast and accurate method for population-level structural variant comparison and analysis. It improves upon existing methods and achieves highly accurate results by merging pairs of variants in increasing order of their breakpoint distance, while maintaining favorable scaling qualities (Supplementary Figure 28) through the use of a KD-tree to efficiently locate nearby variant pairs. Jasmine also separately processes the SV calls by chromosome and SV type and strand to enable built-in parallelization, while many alternative methods incorrectly combine SVs of different types. By combining Jasmine with additional novel methods and carefully optimizing existing methods, we produced an SV-calling pipeline that reduces the rate of Mendelian discordance by more than a factor of five over prior pipelines, while at the same time being applicable to large cross-technology cohorts and resolving a number of issues encountered when using other methods. Finally, by calling SVs and indels in 31 publicly available long-read samples with our pipeline we developed and released a database of human structural variants. By genotyping these variants in 444 short-read samples from the 1000 Genomes Project and 873 samples from GTEx, we cataloged thousands of novel eQTLs across the human genome, including in medically relevant genes, and including 750 variants affecting multiple tissues.
While Jasmine offers highly accurate population SV analysis, we remain limited by the sequencing data that is available. A major challenge we faced when applying our methods to a cohort consisting of samples from multiple sequencing technologies was the additional noise in the samples sequenced with high-error CLR reads (Supplementary Figures 16 and 29). While we mitigated this noise through computational means, we expect that even more accurate SV calls could be obtained by using HiFi or ONT sequencing for all samples. We also found that the rate of discordance among SVs within 500bp of tandem repeats, while less than 1%, was more than double the discordance rate of SVs outside these regions. Other methods have mitigated this by separately processing and normalizing the breakpoints of these variants 23, and integrating these or similar modules with Jasmine’s merging algorithm could significantly improve SV analysis. In addition, there were systematic anomalies in the SV calls in highly repetitive regions such as the centromere and satellite repeats (Supplementary Figures 30-32) and an overall excess of variants that are found in all samples. There has recently been work to improve the reference genome to more accurately reflect these regions 51, and this reference has been shown to substantially improve long-read alignment and SV calling including improved indel balance, a reduction in uniform SVs, and SV calls in previously inaccessible regions of the genome 52. As tools for aligning to and calling variants in these regions continue to mature, we expect the accuracy of these calls to even further improve. Finally, while we have detected a large number of SVs in the 31 samples we studied, there is still much to be discovered. As the costs of long-read genome sequencing continue to decrease, we expect to apply these methods to even larger populations, as well to other species, to deepen our understanding of the role of SVs in disease, development, and evolution.
Methods
Refined Variant Breakpoints and Sequences with Iris
Many existing long-read SV callers identify variants from read alignments based on signatures such as an extended gap in the alignment or a segment of the read which aligns to a distant region of the genome 18,20. In the widely used variant caller sniffles 18, a variant is called when multiple reads show similar signatures that cluster together based on their type, span, and location. However, when reporting the variant’s breakpoints and sequence, the alignment from a single representative read (chosen arbitrarily) is used to infer this information. This is particularly problematic for insertions, where the novel sequence being inserted is taken directly from the single read. Since some read technologies, such as CLR and ONT have error rates of 5% or higher, it is expected that the sequence reported will have a sequence with a similar or higher rate of divergence from the true insertion sequence (Supplementary Figure 33). When comparing across samples, especially those sequenced with different technologies with different error models, this may cause the same variant in both individuals to be falsely identified as two separate variants.
Addressing this, we introduce Iris, a method for refining the breakpoints and novel sequence of SV calls by aggregating information from multiple reads which support each variant call (Figure 1). Iris refines each variant call separately, but supports the processing of multiple variants in parallel. In the case of an insertion variant call, Iris starts with an initial sequence consisting of the variant sequence plus flanking sequence from the reference genome (default 1kb on each side of the variant). Then, it gathers all of the reads which support the variant’s presence - indicated by the RNAMES field in the output of sniffles - and aligns those reads to the initial sequence with minimap2 19. These alignments are used as input to the polishing software racon 26, which polishes the initial sequence. Finally, the polished sequence is aligned to the reference with minimap2 and the CIGAR string is parsed to extract the insertion in the polished sequence relative to the reference which most closely resembles the original insertion call. If such an insertion is found, the variant call is refined to reflect the updated sequence and breakpoints. Iris also supports the refinement of deletion breakpoints, which is of particular interest when the sequencing technology being used has a biased error model in favor of either insertions and deletions. These are handled similarly to insertions, with the initial sequence instead consisting of the concatenation of the reference sequences immediately before and after the deleted region. Iris is available as a standalone tool at https://github.com/mkirsche/Iris.
Simulation Results:
To test the performance of Iris on simulated data, we simulated 400 indels with uniformly random lengths - 200 with length [50, 200] and 200 with length [900, 1100] - in a 5 Mbp segment of chr1 (chr1:20000000–24999999). Then, we used SURVIVOR 7 with a read error and length model trained on HG002 Oxford Nanopore reads to simulate 30x coverage of long reads. We aligned these reads back to the unmodified segment of chromosome 1 with winnowmap2 17 and called SVs with sniffles 18. From the insertion SV calls, we measured the similarity scores of the reported sequences to the ground truth using the formula: Similarity(S, T) = (1 - EditDistance(S, T) / max(length(S), length(T)). We also refined these variant calls with Iris and measured the similarity score of the updated insertion sequences (Supplementary Figure 34a). The average sequence similarity score increased from 94.7% to 98.6%, demonstrating that Iris refinement significantly improves insertion sequence accuracy.
Real Results in HG002:
While this simulated experiment demonstrated that Iris is able to improve sequence accuracy in simulation conditions, we wanted to ensure that it also improves the novel sequences of true genomic variants, where the novel sequences are typically more repetitive and the alignments noisier than when the insertions are random basepairs. To do this, we used the cell line HG002, which was sequenced with multiple technologies, notably including both ONT and HiFi. While the ONT reads have a high error rate around 8%, the HiFi reads have approximately 99.5% accuracy 15, so even novel insertion sequences taken from only a single HiFi read are expected to be highly accurate. Therefore, we used winnowmap and sniffles for variant calling as in the simulated experiment, but used the HiFi SV calls’ sequences in place of a ground truth. For each ONT SV call, we matched it with the nearest HiFi call if it was within 1 kbp, they shared at least 50% sequence identity, and no other ONT call had already matched with it. This resulted in 13,467 matched ONT calls before and 14,401 after refinement, with 12,978 having a matching HiFi call both before and after refinement. Among these, 9,522 (73.37%) had been changed by Iris. The average sequence identity among these 9,522 SVs increased from 91.6% before Iris to 96.2% after Iris, and the distributions of sequence accuracy scores are shown in Supplementary Figure 34b.
We also investigated the impact of Iris refinement on Mendelian discordance in the HiFi-derived SV and indel calls for the HG002 trio. To measure this effect, we called and merged variants in this trio with our SV calling pipeline but with Iris refinement disabled and compared the discordance to the results from the full pipeline shown in Supplementary Figure 35. Without refinement the discordance was 484/47,561 = 1.02%, while the discordance with our full pipeline was 404/47,326 = 0.85%.
Comparing Variant Calls at Population Scale with Jasmine
In order to perform SV inference at population scale and identify variants associated with diseases or phenotypes, it is important to identify when multiple individuals share the same (or functionally identical) variants. However, the same variant call can manifest differently in unique samples because of sequencing error or samples being processed with different sequencing technologies, levels of coverage, or upstream alignment and variant calling software. These differences, along with the increasing availability of long-read sequencing data for many individuals, highlight the need for careful variant comparison when analyzing SVs in multiple samples.
We refer to the problem of consolidating multiple variant callsets into a single set of variants as the “SV merging problem”. This is because the problem consists of identifying variant calls in separate samples which correspond to the same variant and merging them into a single call which is annotated with the samples in which it is present. A number of methods already exist for SV merging, but each has major issues such as invalid merges, results which vary significantly based on the order of input samples, or high levels of Mendelian discordance when evaluated on trio datasets.
Jasmine Methods:
We introduce Jasmine, a novel method which solves the SV merging problem. Jasmine takes as input a list of VCF files consisting of the variant callsets for each individual, and produces a single VCF file in which each variant is annotated with a list of samples in which it is present (as well as the IDs of the input calls which correspond to that variant).
Jasmine first separates the variants by their chromosome (or chromosome pair in the case of translocations), variant type, and strand. Each of these groups is processed independently with an option for parallelization because no two variants in different groups could be representations of the same variant. When processing a group of variants, Jasmine represents each variant as a 2-D point in space representing the start position and length of the variant. When represented this way, variants which are closer together along the genome (and are therefore more likely to represent the same variant) have a smaller Euclidean distance between them. Consequently, each pair of variants can be assigned a quantitative distance which reflects how dissimilar they are.
After projecting these variants into 2-D Euclidean space, Jasmine implicitly builds a variant proximity graph, or a graph in which nodes are individual variants and each pair of variants has an edge between them with a weight corresponding to the Euclidean distance between them. Then, the SV merging can be framed as selecting a set of edges (merges) making up a forest which is a subgraph of the variant proximity graph, and which minimizes the sum of edge weights chosen subject to a few constraints:
No intra-sample merging: No connected component of the forest contains multiple variants from the same individual because they have already been identified as different variants. Note that Jasmine enables this constraint to be disabled with the command line flag --allow_intrasample, which is useful if a single VCF has callsets from multiple SV discovery methods within a single individual.
Distance threshold: No chosen edge has a weight greater than the user-chosen distance threshold (default max(100bp, 50% of variant length))
Maximality: To prevent the trivial solution of no edges, we require that given a set of chosen edges, no additional edges can be added to the solution while still satisfying the other constraints.
Jasmine seeks to solve this optimization problem with a greedy algorithm similar in design to Kruskal’s algorithm for finding a minimum spanning tree. In this algorithm, the set of chosen edges is initially empty, and each edge is considered in order of non-decreasing edge weight. If adding the edge to the solution would violate any of the above constraints given the previously added edges, it is ignored; otherwise, it is added to the solution. When the edges being considered start to exceed the distance threshold, the algorithm terminates.
One issue with this algorithm is that in order to sort the edges by weight, they may need to be loaded into memory. This is problematic because some population datasets, with tens to hundreds of thousands of SVs per sample, include millions of variants, with the number of edges potentially scaling quadratically with the variant count. This is prohibitive even with existing datasets, and will only be more of a problem as even larger datasets are produced. Therefore, Jasmine instead stores the edges implicitly, making use of a KD-tree to quickly find the next smallest edge in the variant proximity graph.
To avoid storing the entire graph in memory, Jasmine maintains a list of a small number of nearest neighbors (initially 4) for each node, which are computed by forming a KD tree with all of the variant points, a data structure which supports k-nearest neighbor queries with a logarithmic runtime with respect to the number of variants. Then, the edge to the single nearest neighbor of each variant is stored in a minimum heap, and it is guaranteed that the first entry removed from this heap will be the edge with the smallest weight in the entire graph. When an edge is processed, the node for which it was the minimum-weight incident edge has its next nearest neighbor added to the heap based on the next entry in its nearest neighbor list. If the list of nearest neighbors for a node becomes empty, the KD-tree is queried for a set of twice as many nearest neighbors, and the list is refilled. In this manner, the next smallest edge in the graph will always be the edge removed from the heap, and the data structures Jasmine uses help to maintain this property without requiring a prohibitively large amount of time or memory. The pseudocode for this algorithm can be found in Supplemental Note 1.
Jasmine Distance Threshold:
When merging variants, it is important to determine for a given variant pair whether or not the two variants are sufficiently close together in terms of their breakpoints to be considered the same variant. In Jasmine, this is based on a distance threshold - if the distance between them (according to the chosen distance metric) is above the threshold they will be considered two different variants, while if their distance is less than or equal to the threshold they will be a candidate for merging. Jasmine offers a number of classes of distance thresholds, including constant thresholds, thresholds which vary based on a fixed proportion of each variant’s size, or even per-variant distance thresholds. By default, the distance threshold for Jasmine is max(100bp, 50% of variant length). We measured the difference in merging when using different values for the min_dist parameter, which is 100 by default, (Supplementary Figure 36), and found that while larger values for this parameter offer lower Mendelian discordance, these more lenient thresholds perform poorly in a cross-technology cohort setting because of false merges, and 100bp offers a good balance in performance across use cases.
Building an SV Inference Pipeline
Our SV inference pipeline is implemented in Snakemake, and supports multithreaded as well as multi-node execution. It takes as input a list of FASTQ files for each sample being studied as well as a reference genome, and produces as its final output a VCF file containing population-level SV calls. It is highly customizable, supporting unique configurations for alignment and variant calling on a per-sample or per-sequencing-technology level. It also enables the user to specify the alignment software to use - ngmlr, winnowmap, and minimap2 - and separately sets recommended default parameters for samples sequenced with each specific technology. On each sample we processed, the pipeline took about a day to run on a single Intel Cascade Lake 6248R compute node with 48 cores and 192GB RAM at 3.0GHz. The Snakemake files to run the pipeline are included in the Jasmine repository: https://github.com/mkirsche/Jasmine/tree/master/pipeline.
Evaluating Mendelian Discordance
When performing de novo variant analysis, we are particularly interested in Mendelian discordant variants, or variants which are called as present in the child of a trio but neither parent. This includes genuine de novo variants, but in practice most of these calls are actually false de novo variants caused by errors in variant calling or merging. Accordingly, one major goal of trio SV inference is to reduce the number of discordant variants while retaining any true de novo variants in that set.
To measure Mendelian discordance, we called variants in the Ashkenazim individual HG002 as well as their parents HG003 (46,XY) and HG004 (46,XX). We merged these three callsets with Jasmine (or other merging software when comparing them to Jasmine), and counted the number of variants which were identified in HG002 but not merged with any variants from either parent. We then filtered these variants by confidence by requiring that they be supported by at least min(10, 25% of average coverage) of the reads and have a length of at least 30. In addition, we filtered out any variants which were not marked with the PRECISE INFO field by the sniffles variant calling. The discordance rate was calculated as the quotient of the number of discordant variants over the total number of variants in the merged and filtered trio callset.
Optimized Sniffles Variant Calling Parameters
As shown in Supplementary Figure 3, we used Mendelian discordance to measure the effects of different variant calling parameters in HiFi data for HG002. We varied the max_dist parameter when running Sniffles for variant calling and measured the number of variants and discordance for each trio callset; based on these results we used max_dist=50 for calling variants from HiFi data.
Similar to the HiFi analysis, we used Mendelian discordance to measure the effects of different variant calling parameters in CLR data for HG002. We varied the max_dist parameter when running Sniffles for variant calling and measured the number of variants and discordance for each trio callset. Supplementary Figure 37 shows the effect of this parameter on these metrics, and based on these results we used max_dist=50 for calling variants from CLR data.
Next, to optimize variant calling parameters in ONT data from HG002, we repeated the experiment used for HiFi and CLR data, varying the max_dist variant calling parameter in Sniffles and measuring the number of variants and discordance for each trio callset. These results are shown in Supplementary Figure 38, and based on them we used max_dist=50 for calling variants from ONT data. While this doesn’t give the lowest discordance rate, all settings examined yielded less than 1% discordance, so we used a value of 50 to enable a high degree of variant discovery and consistency across technologies.
Double Thresholding
To reduce the impact of threshold effects on variant calling, our pipeline uses two different variant calling thresholds: a highly specific, strict high-confidence threshold and a highly sensitive, more lenient low-confidence threshold. To be a high-confident call, a variant must be at least 30bp long supported by a number of reads greater than or equal min(10, 25% of average coverage over that sample); otherwise a variant is called with low confidence if it is at least 20bp long and supported by at least two reads. All of the variants that meet either threshold are used as input to Jasmine’s cross-sample merging, and any low-confidence variants that do not get merged with any high-confidence variants are discarded. This allows variants which are close to the strict threshold to be properly detected in all of the samples in which they are present (Supplementary Figures 39-41).
When evaluating the impact of double thresholding, we consider the SV and indel calls in the HG002 trio which were identified as being present in HG002 and group them into one of four categories:
Discordant: Variants which were present only in HG002, regardless of whether we used double thresholding or only a single stricter threshold
Not discordant: Variants which were present in HG002 as well as one or both parents, regardless of whether we used double thresholding or only a single stricter threshold
Rescued from absence: Variants which were present in HG002 as well as one or both parents, but the call in HG002 had low enough length or read support that it would have been missed in that sample if just the stricter threshold were used.
Rescued from discordance: Variants which were present in HG002 as well as one or both parents, but the call in the parents had low enough length or read support that it would have been called only in HG002, and therefore discordant, if just the stricter threshold were used.
Associating Structural Variants to Genes
To obtain genotypes for SV-gene association, we called SVs in 31 long-read samples with our inference pipeline and merged them into a unified cohort-level callset with Jasmine. We then genotyped these SVs in the 1000 Genomes Collection with Paragraph after filtering out translocations and other variants which Paragraph cannot genotype, for a total of 189,581 genotyped variants across 444 individuals (Supplementary Figure 42). Following previous studies 43, we then used the Hardy-Weinberg Equilibrium (HWE) test to filter out variants not consistent with population genetic expectations, removing variants found to be significant with p < 0.0001 using an exact test of HWE 53. After filtering with HWE and additionally removing any variants that were left uncalled in 50% or more of the samples, we were left with 138,715 variants across the 444 individuals (Supplementary Figure 43).
We examined common cis-SV-eQTLs by associating our SV genotypes to gene expression data in the same cell lines collected by the GEUVADIS consortium 44. We first paired each gene with every structural variant that has a MAF ≥0.05 and resides within a window of 1 Mbp from the gene’s TSS. We then tested whether the distribution of normalized (zero-mean, unit variance) gene expression is different for those individuals with or without the variant by using a Wilcoxon rank-sum test for each variant-gene pair with a p-value cutoff reflecting a Benjamini-Hochberg multiple testing correction with an FDR of 0.1. For genes with multiple SVs tested, each individual SV-gene pair was considered independently. After identifying a set of significantly-associated SV-eQTLs, we fit a linear model between each variant genotype (where reference is encoded as 0 and the alternate allele is encoded as 1 if heterozygous and 2 if homozygous) and gene expression in order to determine the effect size (β) and the R2 of the association. We then analyzed the relationship between the effect size and various features of the SV or gene.
Comparing SVs and SNP-eQTLs with Fine Mapping:
We used the dataset of SNP-eQTLs from the GTEx project for all tissues 3 as a set of known SNP-eQTLs which we could use as a benchmark to compare the effects of SVs to SNPs on genes for which both may be associated. We examined the set of genes for which there were both associated SNP-eQTLs in GTEx (which were also significantly associated in our data) and significantly-associated SVs from our callset within a 1MB window. We then collected a set of 1,000 most-closely associated variants (SNP or SV) to each gene within the 1MB window and computed the Z-score from a linear regression as well as the linkage disequilibrium between each pair of variants. We used these values as input to the fine-mapping program CAVIAR 45 in order to predict which variants within the set are causal. We used CAVIAR’s posterior probability as a measure of how likely a particular variant was to be causal.
Measuring Enrichment of SVs based on CAVIAR Scores:
We examined the relationship between CAVIAR’s posterior probability for each SV’s most highly associated gene and various variant features, such as the distance to various regulatory elements (Supplementary Figure 44). We used the bedtools closest function to compute the distance between each SV and the nearest ENCODE candidate cis-regulatory element from the UCSC genome browser 54 (Supplementary Figure 44a). Using the Ensembl Regulatory Build 55, we performed a similar distance calculation to measure the distance between each variant and the nearest Ensembl Regulatory Element (Supplementary Figure 44b). We also found that higher CAVIAR posteriors are associated with other regulatory elements, distance to the associated gene (as previously reported in 3), as well as to FunSeq high occupancy of transcription factor (HOT) regions 56 (Supplementary Figures 44-45).
We also examined the relationship between CAVIAR posterior probability and various conservation scores, as well as other sequence features such as GC content. To compute conservation scores, inspired by previous works 57, we used pyBigWig to extract regions covered by the SV and computed the mean of the top 10 scores of individual bases within that region. For insertion variants, we extracted the flanking reference sequence - 75 basepairs in each direction - to assess the conservedness of the affected context. We calculated CADD scores 58, LINSIGHT scores 59, and PhastCons 60 in a similar fashion. Based on these prediction scores, we do not observe signs of enrichment of extreme pathogenicity or conservation among SVs with high CAVIAR posteriors (Supplementary Figures 46-47). We also do not observe a pattern among the GC percentage for SVs with high CAVIAR posteriors (Supplementary Figure 47a). However, larger-scale studies are needed to make definitive conclusions, as the number of SVs we observed with high CAVIAR posterior are limited.
Validating 1000 Genomes eQTL calls in GTEx lymphocyte tissue:
We implemented a WDL workflow in AnVIL Terra platform 46 to rapidly genotype the previously mentioned novel variants using paragraph. The environment is based off of the original docker containers provided by https://github.com/Illumina/paragraph/blob/master/doc/Installation.md. The latest version 2.4a can be found on a docker image in “bni1/paragraph:2.4a”. The workflow is available at https://portal.firecloud.org/?return=terra#methods/run_paragraph/run_paragraph/23. E-QTL calling was performed using the OLS module in statsmodel with GTEx expression and covariates publicly available on GTEx portal. We also performed fine mapping using CAVIAR with default parameters. Preprocessing of the data was performed using the aforementioned scripts.
Among the SV-eQTLs in the 1000 Genomes data is an intronic 3,143bp insertion in NCF4, upstream of the associated gene CSF2RB (Supplementary Figure 21e). These two genes have previously been shown to be linked to Crohn’s disease 61. We found that a SNP which was reported in the GTEx SNP-eQTL dataset to be associated with CSF2RB expression is in high LD with the insertion (r2=0.75), but the insertion is more strongly associated with gene expression than the reported SNP (Supplementary Figure 21f). To ensure that our finding is replicable, we proceeded to genotype this variant in 873 GTEx individuals using Paragraph 43 within the NHGRI AnVIL Terra platform, and found a similar alternate allele frequency of 0.796 in GTEx compared to 0.814 in 1KGP. We then analyzed GTEx publicly available expression measurements and expression covariates of the matched tissue, EBV-transformed lymphocytes, to evaluate the candidate SV-eQTL, and found the SV is an eQTL with p-value of 3.95e-8, which is even more significant than in 1KGP. The SV-eQTL measured in GTEx is in high LD (r2=0.79) with the reported SNP-eQTL, and has a more significant p-value than the reported top SNP association (p=1.6e-6). We similarly validated using GTEx data two additional strongly supported SV-eQTLs in LRGUK and CAMKMT that were detected using our cohort-level Jasmine SV calls. We found both SV-eQTLs to be more significant than the SNP-eQTLs reported by GTEx (Supplementary Figures 22-23).
GTEx SV-eQTL analysis:
We used the WDL-based Paragraph workflow described above in AnVIL Terra platform to rapidly genotype the SV variants in the GTEx v8 dataset. For this analysis, we cloned the GTEx data within AnVIL (https://anvil.terra.bio/#workspaces/anvil-datastorage/AnVIL_GTEx_V8_hg38). To reduce the effect of genotyping error, we filtered the variants by whether they significantly deviated from Hardy Weinberg Equilibrium at a genome-wide significance threshold. For eQTL analysis, we filtered for common variants with MAF>0.05. eQTL calling was performed using the OLS module in statsmodel with GTEx expression and covariates publicly available on GTEx portal. Gene-level eQTL p-values are obtained by Bonferroni correcting the minimal eQTL p-value associated with a gene by a factor of the number of eQTLs for that gene. Subsequently, the gene-level p-values are corrected for multiple testing using Benjamini-Hochberg method at a FDR rate <5%, yielding 111,291 significant eGenes across 48 tissues. We performed fine mapping with CAVIAR, using the top SV eQTL signal with the 1000 strongest SNP eQTLs for a gene. Preprocessing of the data was performed using the aforementioned scripts.
Data Availability
The sequencing data used in this study is available from the publications listed in Supplemental Table 1 and Supplemental Table 2. All variant calls and associations are available at http://data.schatz-lab.org/jasmine/.
Supplementary Material
Acknowledgements
We thank Fritz Sedlazeck and Michael Alonge for helpful discussions. This work was supported, in part, by National Science Foundation grants DBI-1350041 (MCS), IOS-1732253 (MCS) and IOS-1758800 (MCS) and National Institutes of Health grants NCI U01CA253481 (MCS), NCI U24CA231877 (MCS), NHGRI U41-HG006620 (MCS), NHGRI U24-HG010263 (MCS) and NIGMS R35GM139580 (AB). This work was also supported in part by the Mark Foundation for Cancer Research award 19–033-ASP (MCS) and a Microsoft Research Fellows award (AB). Part of this research project was conducted using computational resources at the Maryland Advanced Research Computing Center (MARCC). We also thank the investigators and the patient donors from the Human Pangenome Reference Consortium, GTEx, and 1000 Genomes for making their data available.
Footnotes
Competing Interests
S.A. has become an employee at Oxford Nanopore. R.S. has become an employee at Illumina.
Code Availability
The Jasmine and Iris code and documentation are available open-source at https://github.com/mkirsche/Jasmine and https://github.com/mkirsche/Iris. The versions used in the manuscript are archived in zenodo for Jasmine62 and Iris63. These methods are also available in bioconda and Galaxy to simplify use on the command line or within the Galaxy graphical user interface. The versions of all software packages used in the manuscript are described in Supplemental Table 3.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Peer review information:
Nature Methods thanks the anonymous reviewers for their contribution to the peer review of this work.
References
- 1.Alonge M et al. Major Impacts of Widespread Structural Variation on Gene Expression and Crop Improvement in Tomato. Cell 182, 145–161.e23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alkan C, Coe BP & Eichler EE Genome structural variation discovery and genotyping. Nature Reviews Genetics vol. 12 363–376 Preprint at 10.1038/nrg2958 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiang C et al. The impact of structural variation on human gene expression. Nature Genetics vol. 49 692–699 Preprint at 10.1038/ng.3834 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aganezov S et al. Comprehensive analysis of structural variants in breast cancer genomes using single-molecule sequencing. Genome Res 30, 1258–1273 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nattestad M et al. Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line. Genome Res 28, 1126–1135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brandler WM et al. Paternally inherited cis-regulatory structural variants are associated with autism. Science 360, 327–331 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeffares DC et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun 8, 14061 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sedlazeck FJ, Lee H, Darby CA & Schatz MC Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet 19, 329–346 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Mahmoud M et al. Structural variant calling: the long and the short of it. Genome Biol. 20, 246 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zook JM et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol 32, 246–251 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Sirén J et al. Pangenomics enables genotyping of known structural variants in 5202 diverse genomes. Science 374, abg8871 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Narzisi G et al. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat. Methods 11, 1033–1036 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korlach J et al. Real-Time DNA Sequencing from Single Polymerase Molecules. Methods in Enzymology 431–455 Preprint at 10.1016/s0076-6879(10)72001-2 (2010). [DOI] [PubMed]
- 14.Jain M, Olsen HE, Paten B & Akeson M The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol 17, 239 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wenger AM et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol 37, 1155–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodwin S, McPherson JD & McCombie WR Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet 17, 333–351 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jain C, Rhie A, Hansen NF, Koren S & Phillippy AM Long-read mapping to repetitive reference sequences using Winnowmap2. Nat. Methods (2022) doi: 10.1038/s41592-022-01457-8. [DOI] [PMC free article] [PubMed]
- 18.Sedlazeck FJ et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang T et al. Long-read-based human genomic structural variation detection with cuteSV. Genome Biol 21, 189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaisson MJP et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun 10, 1784 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Audano PA et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 176, 663–675.e19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beyter D et al. Long-read sequencing of 3,622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nat. Genet (2021) doi: 10.1038/s41588-021-00865-4. [DOI] [PubMed]
- 24.Byrska-Bishop M et al. High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185, 3426–3440.e19 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Consortium GTEx. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vaser R, Sović I, Nagarajan N & Šikić M Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27, 737–746 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kruskal JB On the shortest spanning subtree of a graph and the traveling salesman problem. Proceedings of the American Mathematical Society vol. 7 48–48 Preprint at 10.1090/s0002-9939-1956-0078686-7 (1956). [DOI] [Google Scholar]
- 28.Bentley JL Multidimensional binary search trees used for associative searching. Communications of the ACM vol. 18 509–517 Preprint at 10.1145/361002.361007 (1975). [DOI] [Google Scholar]
- 29.Jalili V et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2020 update. Nucleic Acids Res 48, W395–W402 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iossifov I et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Renaux-Petel M et al. Contribution of de novo and mosaic mutations to Li-Fraumeni syndrome. J. Med. Genet 55, 173–180 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Veltman JA & Brunner HG De novo mutations in human genetic disease. Nature Reviews Genetics vol. 13 565–575 Preprint at 10.1038/nrg3241 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Belyeu JR et al. De novo structural mutation rates and gamete-of-origin biases revealed through genome sequencing of 2,396 families. Am. J. Hum. Genet 108, 597–607 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi J et al. Structural variant selection for high-altitude adaptation using single-molecule long-read sequencing. bioRxiv 2021.03.27.436702 (2021) doi: 10.1101/2021.03.27.436702. [DOI]
- 35.Ebert P et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larson DE et al. svtools: population-scale analysis of structural variation. Bioinformatics 35, 4782–4787 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eggertsson HP et al. GraphTyper2 enables population-scale genotyping of structural variation using pangenome graphs. Nat. Commun 10, 1–8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooper GM et al. A copy number variation morbidity map of developmental delay. Nat. Genet 43, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zook JM et al. A robust benchmark for detection of germline large deletions and insertions. Nat. Biotechnol 38, 1347–1355 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ellegren H Microsatellites: simple sequences with complex evolution. Nature Reviews Genetics vol. 5 435–445 Preprint at 10.1038/nrg1348 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Ranallo-Benavidez TR et al. Optimized sample selection for cost-efficient long-read population sequencing. Genome Res (2021) doi: 10.1101/gr.264879.120. [DOI] [PMC free article] [PubMed]
- 42.Consortium T 1000 G. P. & The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature vol. 526 68–74 Preprint at 10.1038/nature15393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen S et al. Paragraph: a graph-based structural variant genotyper for short-read sequence data. Genome Biol 20, 291 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lappalainen T et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hormozdiari F, Kostem E, Kang EY, Pasaniuc B & Eskin E Identifying causal variants at loci with multiple signals of association. Genetics 198, 497–508 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schatz MC et al. Inverting the model of genomics data sharing with the NHGRI Genomic Data Science Analysis, Visualization, and Informatics Lab-space. Cell Genom 2, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scott AJ, Chiang C & Hall IM Structural variants are a major source of gene expression differences in humans and often affect multiple nearby genes. Genome Res (2021) doi: 10.1101/gr.275488.121. [DOI] [PMC free article] [PubMed]
- 48.Mezzar S et al. Phytol-induced pathology in 2-hydroxyacyl-CoA lyase (HACL1) deficient mice. Evidence for a second non-HACL1-related lyase. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 972–990 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Caltabiano R et al. Macrophage Migration Inhibitory Factor (MIF) and Its Homologue d-Dopachrome Tautomerase (DDT) Inversely Correlate with Inflammation in Discoid Lupus Erythematosus. Molecules 26, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Torres-Mora J et al. Malignant melanotic schwannian tumor: a clinicopathologic, immunohistochemical, and gene expression profiling study of 40 cases, with a proposal for the reclassification of ‘melanotic schwannoma’. Am. J. Surg. Pathol 38, 94–105 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Nurk S et al. The complete sequence of a human genome. Science 376, 44–53 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aganezov S et al. A complete reference genome improves analysis of human genetic variation. Science 376, eabl3533 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wigginton JE, Cutler DJ & Abecasis GR A note on exact tests of Hardy-Weinberg equilibrium. Am. J. Hum. Genet 76, 887–893 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Navarro Gonzalez J et al. The UCSC Genome Browser database: 2021 update. Nucleic Acids Res 49, D1046–D1057 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zerbino DR, Wilder SP, Johnson N, Juettemann T & Flicek PR The ensembl regulatory build. Genome Biol 16, 56 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fu Y et al. FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol 15, 480 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abel HJ et al. Mapping and characterization of structural variation in 17,795 human genomes. Nature 583, 83–89 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rentzsch P, Witten D, Cooper GM, Shendure J & Kircher M CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47, D886–D894 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang Y-F, Gulko B & Siepel A Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat. Genet 49, 618–624 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hubisz MJ, Pollard KS & Siepel A PHAST and RPHAST: phylogenetic analysis with space/time models. Brief. Bioinform 12, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chuang L-S et al. A Frameshift in CSF2RB Predominant Among Ashkenazi Jews Increases Risk for Crohn’s Disease and Reduces Monocyte Signaling via GMCSF. Gastroenterology 151, 710 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kirsche M Jasmine: Population-scale structural variant merging. Jasmine software release v1.1.0 from https://github.com/mkirsche/Jasmine.Zenodo. doi: 10.5281/zenodo.5586905 [DOI]
- 63.Kirsche M Iris: Structural variant breakpoint and sequence refinement. Iris software release v1.0.4 from https://github.com/mkirsche/Iris.Zenodo. doi: 10.5281/zenodo.5586965 [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data used in this study is available from the publications listed in Supplemental Table 1 and Supplemental Table 2. All variant calls and associations are available at http://data.schatz-lab.org/jasmine/.