

Abstract
Investigations into seizure initiation, in recent years, have focused almost entirely upon alterations of interneuronal function, chloride homeostasis, and extracellular potassium levels. In contrast, little attention has been directed toward a possible role of dendritic plateau potentials in the actual ictogenic transition, despite a substantial literature dating back 40 years regarding its importance generally in epilepsy. Here, we argue that an increase in dendritic excitability, coordinated across the population of pyramidal cells, is a key stage in ictogenesis.
Keywords: pyramidal cell, dendritic plateau potential, inhibition, chloride, potassium, ictogenesis
Understanding how seizures start is an inherently difficult scientific challenge, for multiple reasons. The activity patterns are highly complex and can rapidly spread to involve large areas of the brain, presenting significant difficulties even to identify where a seizure starts. Focusing down too far, recording from individual neurons, runs the risk of failing to see the woods for the trees. While the opposite problem (failing to see the trees at all) is perhaps a better metaphor for most clinical recordings, which are very abstracted and undersampled representations of what we would really like to see, if we aspire to take a scientific approach to managing a particular patient’s problems. All this is made more difficult by the evident variability of epileptic phenotypes.
Recently, considerable insights have been garnered from animal models, aided by the introduction of optogenetic technology. The main debate has focused upon the involvement of GABAergic interneurons 1 –5 and chloride homeostasis 6 –12 in ictogenesis. To understand this debate, it is helpful if we start by describing some notable differences between the models, and then a key point of convergence.
Differences and Commonalities Between Acute Seizure Models
Early studies of disinhibited cortical networks, induced by blocking GABAA receptors (e.g., using various penicillin derivatives, bicuculline, picrotoxin, or gabazine) provided several key insights, including an enduring model of the paroxysmal depolarizing shift, which recognized the importance of the slow kinetics of voltage-dependent Ca2+ channels and NMDA receptors. 13 –16 Another notable result was the demonstration that, in the complete absence of any restraining inhibition, network-wide pathological discharges can be entrained by a single pyramidal cell. 17 Also noteworthy is that in disinhibition models, the discharges are relatively short-lasting (mean duration ∼1 s), they propagate quickly, and one does not see extended tonic–clonic seizure-like events (SLEs), 18 which leads us to a critical point that disinhibition models tell us nothing of how GABAergic activity shapes epileptic discharges.
Instead, our current understanding of epileptic GABA involvement stems largely from 3 other experimental paradigms: (1) applying the K+ channel blocker, 4-aminopyridine (4AP), which has a disproportionate effect on certain populations of interneurons, inducing intense bursting of these cells (Figure 1); (2) removing Mg2+ ions, which enhances synaptic excitation, with only secondary effects on synaptic inhibition (Figure 2); and most recently (3) optogenetics methodology for selective stimulation, or suppression, of particular subclasses of neurons. 19 In vitro, there are very striking differences in the evolving pattern of pathological network activity in 4AP and 0 Mg2+ (see Table 1 20,21 ; note that another model, induced by raising extracellular potassium, [K+]extra, shares features with 4AP).
Figure 1.

Epileptiform activity in 4-aminopyridine. The earliest pathological activity recorded in a horizontal brain slice prepared from a young adult, wild-type mouse, following application of 100 µM 4-aminopyridine (4AP). Note the prominent early discharges in the hippocampal territories, well in advance of the first seizure-like events (SLEs), that are far more prominent in the neocortex and entorhinal cortex (not shown). Seizure-like events are small in the hippocampus and appear to be secondary generalized from the entorhinal and neocortical areas. See Codadu et al for further details. 20,21
Figure 2.

Epileptiform activity in zero Mg2+ bathing solution. The earliest pathological activity recorded in a horizontal brain slice prepared from a young adult, wild-type mouse, following wash-out of Mg2+ ions. Note the virtual absence of any hippocampal activity, until very late, when it starts to entrain the neocortical activity into what we have termed the late-recurrent discharge pattern. See Codadu et al 20,21 for further details.
Table 1.
Key Differences Between 4AP and 0 Mg2+ Models.a
| 4-Aminopyridine | 0 Magnesium | ||
|---|---|---|---|
| Primary pharmacology | Blocks voltage-gated K+ channels | Relieves voltage-dependent NMDA s blockade: enhances Glut neurotransmission | |
| Secondary pharmacology | Effects more apparent in interneurons | Reduces divalent cation shielding—shift in voltage-dependence, enhancing excitability | |
| Primary effect on cell activity | IN burst firing enhanced | Enhances synaptic summation in dendrites | |
| Early interictal activity patterns | Primary IN activity | Glutamatergic, with secondary IN activity | |
| Brain area patterns | Early | Prominent Hipp activity; also NC & EC | NC/EC IIDs; Minimal Hipp involvement |
| Intermediate | SLEs in NC, referred to Hipp | SLEs in NC/EC; Minimal Hipp involvement | |
| Late | Late recurrent discharges; Hipp led, spreading to NC/EC | Late recurrent discharges; Hipp led, spreading to NC/EC | |
| Seizure induction by optogenetic IN activation | Yes | Only in late stage | |
| Electrographic SLE pattern | Low-voltage, fast activity | Hypersynchronous-onset | |
Abbreviations: 4AP, 4-aminopyridine; EC, entorhinal cortex; Hipp, Hippocampus; IIDs, interictal discharges; IN, interneurons; NC, neocortex; SLEs, seizure-like events.
a See Codadu et al 20,21 for details; also Levesque et al 22 and de Curtis and Avoli 23 about the electrographic SLE patterns at seizure onset, and Chang et al 24 regarding SLEs being triggered in 0 Mg2+ by optogenetic interneuronal activation only once seizure-like activity is well established, and not early in that model.
Much has been made of the fact that, in 4AP, intense optogenetic activation of interneurons generally (channelrhodopsin expressed under the GAD promoter), or of the parvalbumin subpopulation alone, can induce SLEs. 3,19,24 -27 Central to the transition are 2 important ionic redistributions: a rise in intracellular chloride [Cl−]intra, which in turn can produce large secondary surges also in [K+]extra, mediated by the potassium-chloride cotransporter, KCC2. 3,28 The positive shift in the GABAergic reversal potential, EGABA, is large enough that GABAergic synaptic barrages can trigger firing—this happens during the clonic stage of seizures. 29
From these informative optogenetic experiments has arisen the idea that seizures are routinely triggered by interneuronal activation, supported by experimental and human recordings, which show intense interneuronal activity ahead of pyramidal recruitment to seizures, 30 -32 but this would be misleading.
The critical point to understand is that there is a spectrum of GABAergic dysfunction associated with seizures. The 4AP model lies at one end, where a pure GABAergic burst can trigger seizures on the background of raised [Cl−]intra and [K+]extra; at the other end are disinhibition models, when activation of the entire network can arise from stimulation of a single pyramidal cell. 17 Lying somewhere in between are the 0 Mg2+ in vitro model, in vivo models with synaptic inhibition intact, in which seizures are induced by sustained optogenetic stimulation of pyramidal cells, 5 and more pertinently, sensory-triggered seizures in humans. An important insight from the 0 Mg2+ model is that when GABAergic function is preserved (early after Mg2+ is removed; late activity patterns have compromised inhibition), even the most intense transient network stimuli do not trigger seizures (although sustained stimulation might), because the cortical microstructure favors inhibition, thereby providing a very effective restraint on activity. This protective inhibitory restraint, however, is rapidly compromised by use, and so the precise ictogenic mechanism depends both upon the level of prior interneuronal activation, and also the size of the glutamatergic drive. Given the various differences between 0 Mg2+ and 4AP, of particular note is our finding that a simple optogenetic assay of excitability (testing the postsynaptic response to a focal pyramidal cell activation, triggered using Channelrhodopsin) preempts the onset of seizure like activity in both models. 33 We describe this next.
Synergistic Positive-Feedback Forces Underlying Ictogenesis
Cortical function is often discussed in terms of the balance between inhibition and excitation, but a far better framework is the concept of “attractor states,” which derives from the field of state physics. 34 An attractor is simply a state to which the system converges from various starting points. Where multiple attractors exist, transitions from one to another are dictated by the relative “attractiveness” of each (i.e., the forces drawing the system into each attractor, and how those scale with distance), as well as the landscape of the intervening regions. One may visualize this as a map of energy valleys (the attractors) separated by ridges. The stability of a given attractor is set by negative feedback forces directed back toward its center, correcting any drift away. Forces operating in the opposite direction, by contrast, may be considered positive feedback, propelling the system to a different state. There are good mathematical models of such systems, but these are often rather high-level and abstract, and the difficulty has been to relate these to variables at the molecular or cellular level. A good case in point is the description of seizure initiation in terms of saddle-nodes, 35 and network resilience 36 or fragility 37 (essentially, these 2 latter terms are the reciprocal of each other), which mimic the tipping-point network behavior at a large scale, without specifying the molecular or cellular parameters; we here suggest a model which explains the cellular basis of this critical tipping point, prompted by our optogenetics assay. 33
We return briefly to Cl− and K+ distributions. Chloride is the main permeant anion of GABAA receptors which provide the major synaptic inhibitory drive. Importantly, these receptors are also permeable to bicarbonate ions, and since pH is buffered strongly either side of the membrane, and EBicarb is positive relative to ECl (approximately −10 mV and −60 mV respectively), there is a continuous exchange of the 2 ions—inward Cl− and outward HCO3 − movement—whenever the GABAergic conductance is high, as happens during the repeated bursts of interneuronal activity induced by 4AP. When there is also concurrent excitatory drive, the membrane potential is pushed away from ECl, increasing the driving force for inward Cl− movement, and Cl− influx is greatly accelerated. In distal dendrites, the effect is likely to be exacerbated by the small compartment size and poor diffusion. 38 The nub is that any significant level of GABAergic activation leads to progressive chloride loading of neurons, which compromises inhibitory function, making further neuronal activation more likely.
Chloride transmembrane distribution is critically coupled to potassium’s, through the action of the cation-chloride cotransporter, KCC2. The K+ gradient is fractionally steeper than that of Cl−, meaning it is dominant, leading to outward movement of both ions. Chloride-loading reduces the Cl− gradient, further favoring outward K-Cl cotransport. Consequently, after intense GABAergic activation, there occurs a secondary surge in [K+]extra. The same mechanism underlies the surge in [K+]extra that has been recorded ahead of ictal recruitment, or an ictal wavefront, at a time when there is very large GABAergic drive, but relatively little local recruitment of neurons 30 (meaning that Hodgkin-Huxley K+ extrusion is probably quite small). Subsequently, neuronal recruitment exacerbates the rise in [K+]extra, which may easily then exceed 10 mM during a seizure 39,40 (Figure 1). A key element of KCC2 function that has not been considered in ictal recruitment is that it operates quite close to its equilibrium, meaning that it may also carry the ions in the opposite direction, with relatively small changes in the concentration gradients of either ion. An interesting consequence of this is that if a subpopulation of neurons experiences an episode of chloride-loading, this leads to a local surge in [K+]extra which then causes chloride loading in other neurons; in other words, the effect is coordinated across the network. Glial buffering of [K+]extra limits this effect, and so any deficits in glial function, therefore, will make the system more susceptible to these ionic shifts.
As with raised [Cl−]intra, raising [K+]extra also constitutes positive feedback; both are caused by neuronal activity, and increase the likelihood of further activity. Another positive feedback mechanism that is likely to be critical is the presence of 2 “active” conductances in the dendrites, namely, NMDA receptors and voltage-gated Ca2+ channels (VGCCs). Like Na+ channels, these are both depolarizing conductances that are opened by depolarization, a positive feedback mechanism that gives rise to action potentials; unlike Na+ channels, NMDA receptors and VGCCs open for tens to hundreds of milliseconds, and so give rise to plateau potentials in the dendrites. 13 This transforms the output of the neuron at the threshold for the dendritic spike. If the level of glutamatergic drive is slightly below this threshold, the neuron may not fire any somatic (Hodgkin-Huxley type) action potentials, or perhaps just one or two. If on the other hand, a dendritic spike is achieved, this generally generates a high frequency burst of firing. 41 There is thus an all-or-nothing change in the output of neurons, for a fractional increase in the drive. Figure 3 provides a schematic of how these various positive and negative feedback mechanisms interact.
Figure 3.
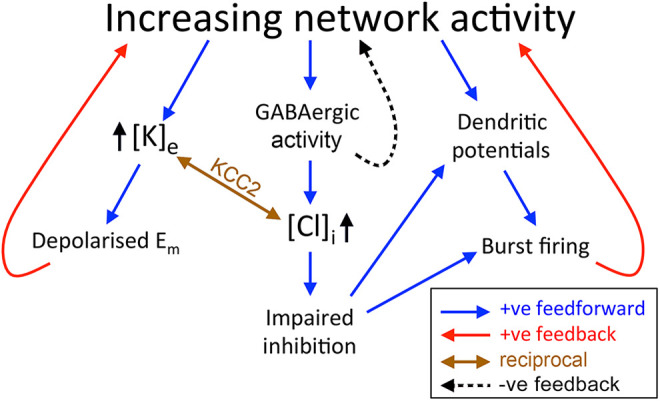
Synergistic positive feedback mechanisms. A schematic of the positive feedback mechanisms, manifest at the cellular level, which contribute to the steep-sided tipping point between physiological and ictal attractor states. An extended discussion of various negative feedback features within cortical networks, that stabilize the normal physiological state, can be found in review by Major et al. 42
We recently discovered that by monitoring dendritic excitability, using a simple optogenetic stimulation assay, the onset of seizure-like activity in all acute ictogenic models tested is presaged by a sudden step change in the response. 33 Interestingly, the centrality of dendritic spikes aligns well with computational simulations by Roger Traub and colleagues, from the 1980s, of the paroxysmal depolarizing shift, 14,16,43,44 incorporating VGCCs, although this work did not focus upon the actual transition into seizures. It is also consistent with a substantial literature associating chronic epileptic phenotypes with changes in expression of various ion channels that influence dendritic excitability, including voltage-gated Ca2+ channels, 45 INap, 46 IA, 15 Ih, 47,48 and SK-type K+ channels 49 (see also reviews 50,51 ). It squares nicely, too, with the observation that multiple antiepileptic drugs appear to act by reducing burst firing in neurons, and that the inhibitory restraint ahead of an ictal wavefront involves intense activation of both parvalbumin and somatostatin interneurons, targeting the soma and the distal dendrites, respectively. 1 Finally, various studies of neuronal bistability arising from active conductances, such as VGCCs and NMDA receptors, have been proposed to underlie a variety of other network transitions. 42,52 -55 As we have argued elsewhere, 56 the occurrence of epileptic activity exists only a short step away from normal cortical function.
This focus upon bistable states, arising from all-or-nothing dendritic spikes, has parallels with the 2 levels of activation seen in the thalamus. 57 When thalamic relay neurons (TRNs) are relatively depolarized, the low-threshold VGCCs are inactivated, and information transfer is relatively precise. In contrast, relatively hyperpolarized TRNs have VGCCs that are activatable, and so are prone to fire intense bursts of action potentials. Feedback loops involving the reticular nucleus can set up powerful oscillations, seen in both sleep spindles and absence seizures. 58 Despite there being a long-standing consensus regarding the importance of VGCCs and plateau potentials in this thalamic form of seizure, the field has rather lost sight of their importance also in cortical seizures.
In summary, this model of seizure initiation describes how several positive feedback mechanisms—raised [Cl−]intra, raised [K+]extra, and the occurrence of dendritic action potentials, which further causes a step increase in the somatic firing output with yet more glutamate being released into the local network—feed into each other. The synergistic nature of these mechanisms creates an especially steep saddle node and explains why the tipping point between normal cortical function and seizure activation can appear to occur with such speed. Importantly, the proximity to the tipping point may be assayed by recording the response to brief stimuli, to provide advance warning of imminent seizures. 33
Acknowledgments
The work was supported by grants from BBSRC (BB/P019854/1), MRC (MR/R005427/1), and Epilepsy Research UK (Celine Newman Bursary).
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work was supported by grants from BBSRC (BB/P019854/1), MRC (MR/R005427/1), and Epilepsy Research UK (Celine Newman Bursary).
ORCID iD: Andrew J. Trevelyan  https://orcid.org/0000-0001-9307-4241
https://orcid.org/0000-0001-9307-4241
References
- 1. Parrish RR, Codadu NK, Mackenzie-Gray Scott C, Trevelyan AJ. Feedforward inhibition ahead of ictal wavefronts is provided by both parvalbumin- and somatostatin-expressing interneurons. J Physiol. 2019;597(8):2297–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Trevelyan AJ, Schevon CA. How inhibition influences seizure propagation. Neuropharmacology. 2013;69:45–54. [DOI] [PubMed] [Google Scholar]
- 3. Chang M, Dian JA, Dufour S, et al. Brief activation of GABAergic interneurons initiates the transition to ictal events through post-inhibitory rebound excitation. Neurobiol Dis. 2018;109(pt A):102–116. [DOI] [PubMed] [Google Scholar]
- 4. Avoli M, De Curtis M, Gnatkovsky V, et al. Specific imbalance of excitatory/inhibitory signaling establishes seizure onset pattern in temporal lobe epilepsy. J Neurophysiol. 2016;115(6):3229–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Khoshkhoo S, Vogt D, Sohal VS. Dynamic, cell-type-specific roles for GABAergic interneurons in a mouse model of optogenetically inducible seizures. Neuron. 2017;93(2):291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pallud J, Le Van Quyen M, Bielle F, et al. Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci Transl Med. 2014;6(244):244ra289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alfonsa H, Lakey JH, Lightowlers RN, Trevelyan AJ. Cl-out is a novel cooperative optogenetic tool for extruding chloride from neurons. Nat Commun. 2016;7:13495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alfonsa H, Merricks EM, Codadu NK, et al. The contribution of raised intraneuronal chloride to epileptic network activity. J Neurosci. 2015;35(20):7715–7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dzhala VI, Kuchibhotla KV, Glykys JC, et al. Progressive NKCC1-dependent neuronal chloride accumulation during neonatal seizures. J Neurosci. 2010;30(35):11745–11761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pavlov I, Kaila K, Kullmann DM, Miles R. Cortical inhibition, pH and cell excitability in epilepsy: what are optimal targets for antiepileptic interventions? J Physiol. 2013;591(4):765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burman RJ, Selfe JS, Lee JH, et al. Excitatory GABAergic signalling is associated with benzodiazepine resistance in status epilepticus. Brain. 2019;142(11):3482–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cossart R, Bernard C, Ben-Ari Y. Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci. 2005;28(2):108–115. [DOI] [PubMed] [Google Scholar]
- 13. Schiller J, Major G, Koester HJ, Schiller Y. NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature. 2000;404(6775);285–289. [DOI] [PubMed] [Google Scholar]
- 14. Traub RD, Miles R. Neuronal Networks of the Hippocampus. Cambridge University Press; 1991. [Google Scholar]
- 15. Bernard C, Anderson A, Becker A, Poolos NP, Beck H, Johnston D. Acquired dendritic channelopathy in temporal lobe epilepsy. Science. 2004;305(5683):532–535. [DOI] [PubMed] [Google Scholar]
- 16. Traub RD, Miles R, Wong RK. Model of the origin of rhythmic population oscillations in the hippocampal slice. Science. 1989;243(4896):1319–1325. [DOI] [PubMed] [Google Scholar]
- 17. Miles R, Wong RK. Single neurones can initiate synchronized population discharge in the hippocampus. Nature. 1983;306(5941):371–373. [DOI] [PubMed] [Google Scholar]
- 18. Trevelyan AJ, Sussillo D, Yuste R. Feedforward inhibition contributes to the control of epileptiform propagation speed. J Neurosci. 2007;27(13):3383–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Magloire V, Mercier MS, Kullmann DM, Pavlov I. GABAergic interneurons in seizures: investigating causality with optogenetics. Neuroscientist. 2018;25(4):344–358:1073858418805002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Codadu NK, Parrish RR, Trevelyan AJ. Region-specific differences and areal interactions underlying transitions in epileptiform activity. J Physiol. 2019;597(7):2079–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Codadu NK, Graham RT, Burman RJ, et al. Divergent paths to seizure-like events. Physiol Rep. 2019;7(19):e14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Levesque M, Salami P, Gotman J, Avoli M. Two seizure-onset types reveal specific patterns of high-frequency oscillations in a model of temporal lobe epilepsy. J Neurosci. 2012;32(38):13264–13272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. de Curtis M, Avoli M. GABAergic networks jump-start focal seizures. Epilepsia. 2016;57(5):679–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chang M, Dufour S, Carlen PL, Valiante TA. Generation and on-demand initiation of acute ictal activity in rodent and human tissue. J Vis Exp. 2019;19(143):e57952. [DOI] [PubMed] [Google Scholar]
- 25. Levesque M, Herrington R, Hamidi S, Avoli M. Interneurons spark seizure-like activity in the entorhinal cortex. Neurobiol Dis. 2016;87:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yekhlef L, Breschi GL, Lagostena L, Russo G, Taverna S. Selective activation of parvalbumin- or somatostatin-expressing interneurons triggers epileptic seizurelike activity in mouse medial entorhinal cortex. J Neurophysiol. 2015;113(5):1616–1630. [DOI] [PubMed] [Google Scholar]
- 27. Shiri Z, Manseau F, Levesque M, Williams S, Avoli M. Interneuron activity leads to initiation of low-voltage fast-onset seizures. Ann Neurol. 2015;77(3):541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Viitanen T, Ruusuvuori E, Kaila K, Voipio J. The K+-Cl cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J Physiol. 2010;588(pt 9):1527–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ellender TJ, Raimondo JV, Irkle A, Lamsa KP, Akerman CJ. Excitatory effects of parvalbumin-expressing interneurons maintain hippocampal epileptiform activity via synchronous after discharges. J Neurosci. 2014;34(46):15208–15222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trevelyan AJ, Sussillo D, Watson BO, Yuste R. Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J Neurosci. 2006;26:12447–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schevon CA, Weiss SA, McKhann G, et al. Evidence of an inhibitory restraint of seizure activity in humans. Nat Commun. 2012;3:1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Truccolo W, Donoghue JA, Hochberg LR, et al. Single-neuron dynamics in human focal epilepsy. Nat Neurosci. 2011;14(5):635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Graham RT, Parrish RR, Alberio L, Johnson E, Trevelyan AJ. Synergistic Positive Feedback Underlying Seizure Initiation; 2021. doi:10.1101/2021.02.28.433224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hopfield JJ. Neural networks and physical systems with emergent collective computational abilities. Proc Natl Acad Sci U S A. 1982;79(8):2554–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jirsa VK, Stacey WC, Quilichini PP, Ivanov AI, Bernard C. On the nature of seizure dynamics. Brain. 2014;137(pt 8):2210–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chang WC, Kudlacek J, Hlinka J, et al. Loss of neuronal network resilience precedes seizures and determines the ictogenic nature of interictal synaptic perturbations. Nat Neurosci. 2018;21(12):1742–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li A, Huynh C, Fitzgerald Z, et al. Neural fragility as an EEG marker of the seizure onset zone. Nat Neurosc. 2021;24(10):1465–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Currin CB, Trevelyan AJ, Akerman CJ, Raimondo JV. Chloride dynamics alter the input-output properties of neurons. PLoS Comput Biol. 2020;16(5):e1007932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Somjen GG, Giacchino JL. Potassium and calcium concentrations in interstitial fluid of hippocampal formation during paroxysmal responses. J Neurophysiol. 1985;53(4):1098–1108. [DOI] [PubMed] [Google Scholar]
- 40. Somjen GG. Ions in the Brain: Normal Function, Seizures and Stroke. Oxford University Press; 2004. [Google Scholar]
- 41. Larkum ME, Zhu JJ, Sakmann B. A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature. 1999;398(6725):338–341. [DOI] [PubMed] [Google Scholar]
- 42. Major G, Larkum ME, Schiller J. Active properties of neocortical pyramidal neuron dendrites. Annu Rev Neurosci. 2013;36:1–24. [DOI] [PubMed] [Google Scholar]
- 43. Johnston D, Brown TH. Giant synaptic potential hypothesis for epileptiform activity. Science. 1982;211(4479):294–297. [DOI] [PubMed] [Google Scholar]
- 44. Wong RK, Prince DA. Dendritic mechanisms underlying penicillin-induced epileptiform activity. Science. 1979;204(4398):1228–1231. [DOI] [PubMed] [Google Scholar]
- 45. Su H, Sochivko D, Becker A, et al. Upregulation of a T-type Ca2+ channel causes a long-lasting modification of neuronal firing mode after status epilepticus. J Neurosci. 2002;22(9):3645–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kearney JA, Plummer NW, Smith MR, et al. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience. 2001;102(2):307–317. [DOI] [PubMed] [Google Scholar]
- 47. McClelland S, Flynn C, Dubé C, et al. Neuron-restrictive silencer factor-mediated hyperpolarization-activated cyclic nucleotide gated channelopathy in experimental temporal lobe epilepsy. Ann Neurol. 2011;70(3);454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Santoro B, Shah MM. Hyperpolarization-activated cyclic nucleotide-gated channels as drug targets for neurological disorders. Annu Rev Pharmacol Toxicol. 2020;60:109–131. [DOI] [PubMed] [Google Scholar]
- 49. Cai X, Wei DS, Gallagher SE, et al. Hyperexcitability of distal dendrites in hippocampal pyramidal cells after chronic partial deafferentation. J Neurosci. 2007;27(1):59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Poolos NP, Johnston D. Dendritic ion channelopathy in acquired epilepsy. Epilepsia. 2012;53(suppl 9):32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tang C-M, Thompson SM. Perturbations of dendritic excitability in epilepsy. In: Noebels J, Avoli M, Rogawski MA, Olsen R, Delgado-Escueta A, eds. Jasper’s Basic Mechanisms of the Epilepsies. National Center for Biotechnology Information; 2012:733–749. [PubMed] [Google Scholar]
- 52. Goldman MS, Levine JH, Major G, Tank DW, Seung HS. Robust persistent neural activity in a model integrator with multiple hysteretic dendrites per neuron. Cereb Cortex. 2003;13(11):1185–1195. [DOI] [PubMed] [Google Scholar]
- 53. Loewenstein Y, Mahon S, Chadderton P, et al. Bistability of cerebellar Purkinje cells modulated by sensory stimulation. Nat Neurosci. 2005;8(2):202–211. [DOI] [PubMed] [Google Scholar]
- 54. Takahashi N, Oertner TG, Hegemann P, Larkum ME. Active cortical dendrites modulate perception. Science. 2016;354(6319):1587–1590. [DOI] [PubMed] [Google Scholar]
- 55. Marder E, Abbott LF, Turrigiano GG, Liu Z, Golowasch J. Memory from the dynamics of intrinsic membrane currents. Proc Natl Acad Sci U S A. 2016;93(24):13481–13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Trevelyan AJ. Do cortical circuits need protecting from themselves? Trends Neurosci. 2016;39(8):502–511. [DOI] [PubMed] [Google Scholar]
- 57. Sherman SM, Guillery RW. Exploring the Thalamus and Its Role in Cortical Function. 2nd ed . Academic Press; 2006. [Google Scholar]
- 58. Cope DW, Di Giovanni G, Fyson SJ, et al. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. 2009;15(12):1392–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]