

Abstract
Background
Immune-mediated necrotizing myopathy (IMNM) is an autoimmune myopathy characterised by proximal muscle weakness, high creatine kinase (CK) values, and autoantibodies recognizing 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) or the signal recognition particle (SRP). There are currently no approved therapies for IMNM and many patients experience active disease despite off-label treatment with intravenous immunoglobulin, glucocorticoids, and immunosuppressants. Detection of complement-activating anti-HMGCR and anti-SRP autoantibodies and the presence of complement deposition on the sarcolemma of non-necrotic myofibers led to the hypothesis that complement activation may be pathogenic in IMNM, therefore zilucoplan, a complement component 5 (C5) inhibitor, could be a potential therapy.
Methods
IMNM01, a phase 2, multicenter, randomised, double-blind, placebo-controlled study (NCT04025632) at 15 sites (four countries) evaluated efficacy, safety, and tolerability of zilucoplan in adult participants with anti-HMGCR or anti-SRP autoantibody-positive IMNM. Participants were randomised 1:1 to receive daily subcutaneous zilucoplan (0·3mg/kg) or placebo for eight weeks; with optional enrolment in the study open-label extension. Primary efficacy endpoint was percent change from baseline to Week 8 in CK levels. Secondary endpoints included safety.
Findings
Between 07 November 2019 and 07 January 2021, 27 participants (13 female and 14 male) received zilucoplan (n=12) or placebo (n=15) and completed the 8-week main study. At Week 8 there were no clinically relevant or statistically significant differences, despite target engagement based on mode of action, between treatment arms in mean percent change (standard deviation) of CK levels versus baseline (−9·86% [26·06] versus −20·72% [31·22] in zilucoplan [n=10] and placebo arms [n=14], p=0·46, respectively) and no clinically relevant improvement over time within the treatment arm. There were no unexpected adverse safety or tolerability findings. Treatment emergent adverse events (TEAEs) and serious TEAEs were reported in n=9 (75·0%) vs n=13 (86·7%) and n=0 (0%) and n=3 (20·0%) participants, respectively. The most frequent TEAEs were headache (n=4 in both groups [33·3% and 26·7%, respectively]) and nausea (n=3 in both groups [25·0% and 20·0%, respectively]).
Interpretation
C5 inhibition does not appear to be an effective treatment modality for IMNM. Rather than driving myofiber necrosis, complement activation may be secondary to muscle injury.
Funding
Study funded by Ra Pharmaceuticals (now part of UCB Pharma).
Introduction
Immune-mediated necrotizing myopathy (IMNM) is a clinical subtype of inflammatory myopathy with distinct clinicopathological characteristics including symmetric proximal muscle weakness, elevated muscle enzyme levels, myofiber necrosis with rare inflammatory infiltrate on muscle biopsy, and autoantibodies recognizing 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) or the signal recognition particle (SRP).(1, 2) Unfortunately, proximal muscle weakness may progress and become disabling despite treatment with intravenous immunoglobulin (IVIg), corticosteroids, and immunosuppressants.(2)
Given that not all patients with IMNM respond adequately to first-line therapies,(2) there is a need for additional treatment modalities. Several observations suggested that autoantibodies may cause muscle damage by activating complement. First, anti-HMGCR and anti-SRP autoantibody levels are associated with disease activity.(3, 4) Second, autoantibody isotypes are complement activating (5) and complement deposits are observed on the sarcolemma of non-necrotic myofibers on muscle biopsy specimens.(6) Furthermore, a passive transfer mouse model showed that anti-HMGCR and anti-SRP autoantibodies cause weakness and myofiber necrosis in the presence of complement. However, C3-deficient animals and those treated prophylactically with complement C5 inhibitors showed an attenuated disease course.(7, 8) Given these findings, we hypothesised that inhibiting the terminal complement pathway could be an effective treatment in IMNM.
Zilucoplan is a 15 amino acid peptide inhibitor of complement component C5. Zilucoplan inhibits the cleavage of C5 into its split products C5a, which is a potent anaphylatoxin, and C5b which together with complement components C6, C7, C8, and C9 forms the membrane attack complex (MAC, also called terminal complement complex or C5b-9). As zilucoplan binds the C5 protein in the region where its split product C5b interacts with C6, it also sterically hinders the formation of MAC, in addition to interfering with the cleavage of C5. Zilucoplan administration achieves complete complement inhibition within 3–6 hours of dose administration,(9) and has shown significant improvement on clinical endpoints in Phase 2 and Phase 3 trials in generalised myasthenia gravis.(10–13)
Patients with IMNM exhibit the greatest elevation of serum creatine kinase (CK) levels seen among all forms of myositis, and serum CK levels correlate well with disease activity.(14) Unlike in myopathies with less prominent tissue destruction, serum CK levels in IMNM are thought to directly reflect the degree of skeletal myocyte necrosis.(15) Therefore, CK is frequently used for routine clinical follow-up and to evaluate response to medication in patients with IMNM, in addition to clinical measures such as standardised muscle strength testing. Specifically, CK levels may increase prior to manifestation or deterioration of clinical weakness, and a decline in CK levels is often the first sign of response after treatment initiation while muscle regeneration and recovery of muscle strength may follow weeks to months later.(3)
To better understand the effect of C5 inhibition on IMNM, we conducted a randomised, double-blind, placebo-controlled, multicentre Phase 2 clinical trial to evaluate the safety, tolerability, and efficacy of subcutaneous (SC) zilucoplan 0·3 mg/kg daily in adult participants with anti-HMGCR or anti-SRP autoantibody-positive IMNM. To our knowledge, this is the first phase 2 clinical trial in participants with IMNM. The dose of zilucoplan 0·3mg/kg was selected for this study based on previously published efficacy, almost complete inhibition of the terminal complement pathway, and favourable safety in the Phase 2 study in participants with generalised myasthenia gravis (gMG).(10) This resulted in rapid, sustained and complete (97%) inhibition of the terminal complement pathway in all gMG participants receiving the 0·3mg/kg dose.
Given the reliable relationship between CK levels, disease activity, treatment response in IMNM, and the faster response and higher sensitivity of CK to effective treatment interventions compared with clinical measures,(16, 17) the main objective of this study was to assess the change in CK level after 8 weeks of zilucoplan therapy.
Methods
Study design
This study (IMNM01, NCT04025632, www.clinicaltrials.gov) was a multicenter, randomised, double-blind, placebo-controlled study to evaluate the efficacy, tolerability and safety of zilucoplan in participants with IMNM who were positive for anti-HMGCR or anti-SRP autoantibodies.
Study participants were randomised in a 1:1 ratio to receive daily SC 0·3 mg/kg zilucoplan or matching placebo (appendix p3). Randomization was stratified based on autoantibody status (anti-HMGCR+ versus anti-SRP+). The main study included a screening period of up to four weeks and an 8-week treatment period. Participants were evaluated at Baseline, and at Weeks 1, 2, 4, and 8. Participants were required to continue taking their existing standard of care medication for IMNM at the same dose levels throughout the study, including glucocorticoids, immunosuppressants, and IVIg. At the end of the main study, eligible participants had the opportunity to enter the open-label extension of the study.
Updates to the global protocol were made to include provisions for the COVID-19 pandemic, to update the statistical methods used to analyse the study objectives and endpoints and to update the inclusion and exclusion criteria. Specifically, the contraception information inclusion criteria were updated and hypersensitivity to study treatment was added as an exclusion criterion.
The study was conducted in accordance with the International Conference on Harmonisation Guideline for Good Clinical Practice and the Declaration of Helsinki. Independent ethics committees or institutional review boards provided written approval for the study protocol and all amendments.
Further details on the study, including the protocol amendments can be found in the study protocol in the Appendix p17–89.
Study participants
Participants aged ≥18 to <75 years with a clinically confirmed diagnosis of IMNM, positive serology for anti-HMGCR or anti-SRP autoantibodies, clinical evidence of weakness (≤ Grade 4 out of 5) on the Medical Research Council (MRC) Scale with manual muscle testing (MMT) in at least one proximal limb muscle group (out of either trapezius, deltoid, biceps brachii, iliopsoas, gluteus medius, gluteus maximus, quadriceps), serum total CK of >1000U/L at screening, and no change in glucocorticoids or other immunosuppressive therapies for at least 30 days prior to baseline, or anticipated to occur during the first eight weeks of the study were eligible for inclusion. Participants who had received rituximab within 90 days prior to baseline, had recently initiated IVIg treatment (first cycle <90 days prior to baseline), or had received plasma exchange within four weeks prior to baseline, were excluded from the study. Other medications were permitted while in the study, pursuant to the exclusion criteria. Participants were expected to remain on stable doses of the permitted standard of care therapy for IMNM throughout the main portion of the study and through the Week 8 visit of the open-label extension; this included glucocorticoids, immunosuppressive drugs, and IVIg. Additional details of the inclusion and exclusion criteria, including eligibility criteria for inclusion in the open-label extension, can be found in the appendix p1. Sex was participant reported.
All study participants were required to receive meningococcal vaccination at least two weeks before starting study treatment due to the potential risk of Neisseria meningitidis infection, an established risk with complement C5 inhibition or genetic C5 deficiencies.(18, 19). Participants who initiated treatment less than two weeks after receiving a meningococcal vaccine received appropriate antibiotic prophylaxis.
Randomization and blinding
Participants were randomised in a 1:1 ratio to receive daily SC doses of 0·3 mg/kg zilucoplan or a matching placebo using a computerised randomisation algorithm; randomisation was stratified based on autoantibody status (anti-HMGCR+ versus anti-SRP+).
Participants and study staff remained blinded to treatment assignments until after the data from Week 8 of the main study were reviewed, locked, and unblinded. Participants and investigators were blinded to laboratory study results including CK, alanine aminotransferase (ALT), and aspartate aminotransferase (AST) in order to prevent study unblinding.
Procedures
Following in-clinic education and training, all participants self-injected daily SC doses of zilucoplan or placebo, according to randomised treatment allocation, throughout the 8-week study period. Zilucoplan was provided in single-use prefilled syringes for self-injection using weight bracketed dosing, i.e. each of 3 fixed amounts of the drug covered a range of study participants weights (43 to 150kg).
Participants were evaluated at baseline and at Weeks 1, 2, 4, and 8. At the conclusion of the 8-week main study, all participants had the option to receive zilucoplan in the open-label extension provided they met the selection criteria for this part of the study. All participants entering the open-label extension received open-label once-daily SC zilucoplan 0·3mg/kg. Visits during the first eight weeks of the open-label extension were identical to the main study for all participants to ensure appropriate monitoring of those transitioning from placebo to active treatment. The study remained double-blinded until after the data from the main treatment period had been reviewed, locked, and unblinded.
For participants who permanently discontinued treatment with the study drug, and for those who completed the 8-week study but did not enter the open-label extension, a safety follow-up visit was performed at 40 days after the last study dose.
Outcomes
The primary endpoint was the percent change from baseline to Week 8 in CK levels. Pre-specified secondary outcomes included safety and clinical efficacy endpoints. Safety assessments included evaluations of treatment-emergent adverse events (TEAEs), clinical laboratory tests, electrocardiograms (ECGs), vital signs, and physical examinations. Efficacy assessments included minimal response based on the American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) Response Criteria Scale at Week 8, change from baseline to Week 8 in Triple Timed Up and Go (3TUG) Test (in ambulatory participants only), proximal MMT (trapezius, deltoid, biceps brachii, iliopsoas, gluteus medius, gluteus maximus, and quadriceps bilaterally), Physician Global Activity visual analogue scale (VAS), Patient Global Activity VAS, Health Assessment Questionnaire (HAQ), Myositis Disease Activity Assessment Tool (MDAAT) Extramuscular Disease Activity VAS Score, and Functional Assessment of Chronic Illness Therapy (FACIT)-Fatigue Scale. Subgroup analyses of the primary and continuous secondary efficacy variables were summarised for the ITT population for the main study based on sex (female, male), age (<55 years, ≥55 years), and stratification factor (anti-HMGCR/anti-SRP groups).
Plasma samples were analysed to confirm inhibition of the terminal complement pathway using an ex vivo antibody-sensitised sheep red blood cell (sRBC) lysis assay to assess the classical pathway of complement activation.(20)
Exploratory pharmacokinetic/pharmacodynamic outcomes included evaluation of classical complement pathway activation utilising the sRBC lysis assay.
Following the initiation of the study, the objectives and endpoints were updated to encompass evaluation of long-term efficacy, safety, and tolerability during the open label extension part of the study.
Statistical analyses
The planned enrolment was for approximately 24 participants. A sample size of 12 study participants per group yielded approximately 95% power to detect a difference in the percent reduction from baseline CK between the active and placebo groups using a Wilcoxon rank-sum test at the 2-sided 0·05 type 1 error rate. These power calculations assumed that the percent reduction in creatine kinase in the active dose group was approximately normally distributed with a mean of 80% and a standard deviation of 8%; that four of the placebo participants had a percent reduction similar to the active dose group; and the remaining eight placebo participants had a percent reduction normally distributed with a mean of 10% and a standard deviation of 8%.
Study populations
The following study populations were defined: the intention-to-treat (ITT) population included all participants randomised; the per-protocol (PP) population included all participants in the ITT population who had completed the main 8-week study period and had no major protocol deviations; the safety population included all participants who received at least 1 dose of study drug, with participants to be analysed based on the actual study treatment received.
Efficacy analysis
A two-sided stratified Wilcoxon rank sum test (Van Elteren test) was utilised in the final analysis to assess potential differences in the percentage change from baseline between treatments. The magnitude of association was expressed by Wilcoxon-Mann-Whitney odds (WMWodds) and the corresponding 95% confidence interval (CI).
ACR/EULAR minimal response at Week 8 was assessed by logistic regression model with treatment and strata (anti-HMGCR+/anti-SRP+) as fixed factors. Treatment group differences for each of the secondary efficacy change from baseline endpoints at Week 8, were assessed using an analysis of covariance model with treatment, randomization strata (anti-HMGCR+/anti-SRP+), and baseline endpoint as covariates. The least squares means (LSMs) of each treatment group and the least squares mean differences between zilucoplan and placebo were reported for the Week 8 change from baseline along with the two-sided 95% CIs and p-values.
Treatment group differences for each of the secondary efficacy change from baseline endpoints at Week 8 were assessed using a linear mixed effect model with repeated measures (MMRM) analysis of covariance (ANCOVA) with treatment and strata (anti-HMGCR+/anti-SRP+) as fixed factors and, visit, baseline score as a covariate, treatment×visit (interaction term), and baseline score×visit (interaction term) as fixed effects and participant as a random effect.
Safety analysis
Data on duration of exposure was summarised as number and percentage of study participants with cumulative study treatment duration (e.g., any duration, ≥1 week, ≥2 weeks, ≥3 weeks, etc.), duration of exposure in years (or participant exposure years [PEY])=[(min(date of last dose+40 days, last visit)-date of first dose+1)]/365 ·25. Exposure was adjusted for the 5 half-lives of active treatment, which was 40 days.
AEs were captured for the duration of the study from informed consent (SAEs only) or time of first administration, through until administration of the last study dose plus 40 days (or last visit, depending on which occurred first). TEAEs were defined as AEs starting on or after the time of first administration of study treatment. AEs were classified according to the Common Terminology Criteria for Adverse Events (CTCAE) Version 5·0. TEAE summaries were reported separately within the main and open-label extension parts of the study.
Role of the funding source
This study was funded by Ra Pharmaceuticals Inc, now part of UCB Pharma. The funding source contributed to the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, and approval of the manuscript; and the decision to submit the manuscript for publication.
Results
The study was conducted across 15 sites in the USA, UK, France, and the Netherlands, and participants were enrolled between 07 November 2019 and 07 January 2021. Twenty-seven participants were enrolled in the study and randomised (figure 1), and all received zilucoplan or placebo; all 27 participants completed the 8-week blinded study period and 25 continued to the open-label extension.
Figure 1. Study disposition.
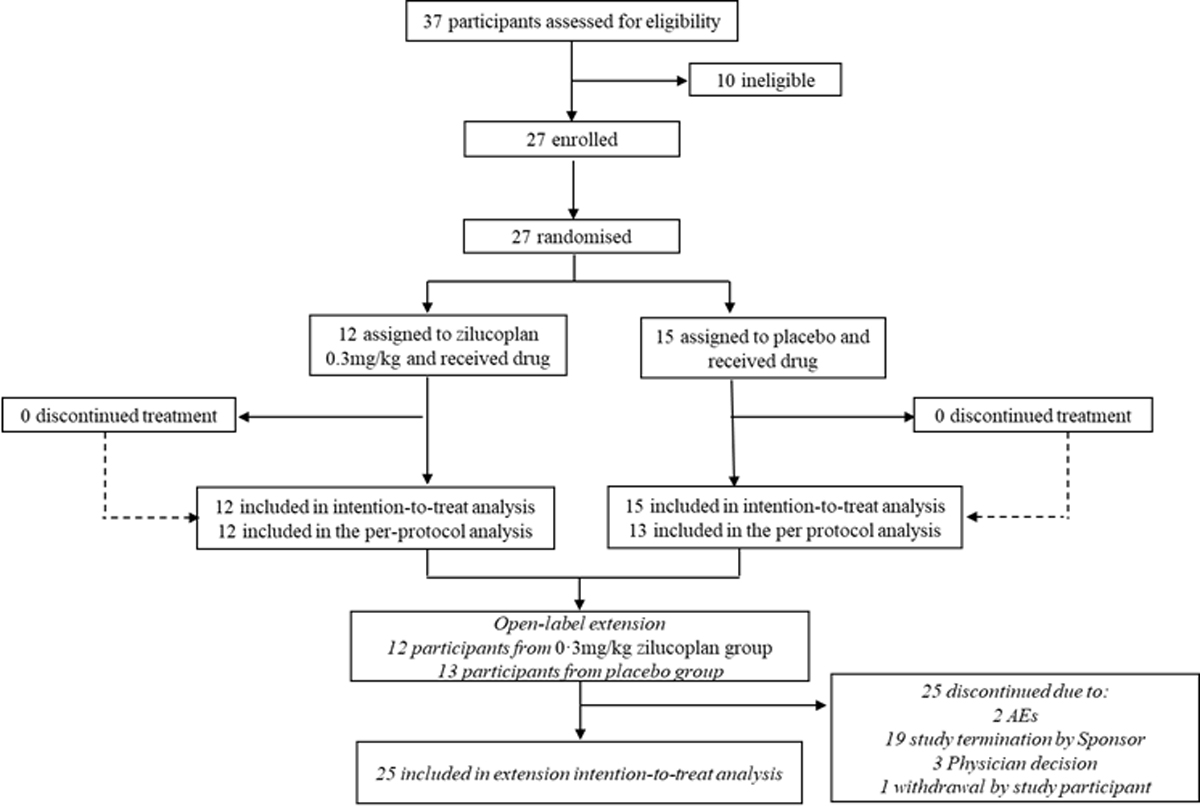
AEs, adverse events
Baseline demographic and clinical characteristics for each treatment group are shown in table 1. Overall, baseline characteristics were similar between treatment groups, with the exception of weight: compared with the placebo group, the zilucoplan group had a lower mean weight (81·1kg vs 91·5kg). The mean (SD) time since diagnosis was 35·6 (35·2) months and 21·5 (24·1) months for the zilucoplan and placebo groups, respectively. Overall, 26 study participants had previously had muscle biopsies taken supporting their diagnosis of IMNM; 25 study participants (92·6%) received prior or concomitant IMNM-related medications.
Table 1.
Demographics and Baseline Characteristics (ITT population)
| Zilucoplan 0·3mg/kg N=12 |
Placebo N=15 |
All Participants N=27 |
||
|---|---|---|---|---|
| Age (years) | ||||
| Mean (SD) | 56·9 (9·0) | 52·8 (13·6) | 54·6 (11·8) | |
| 95% CI | 51·2, 62·7 | 45·2, 60·4 | ||
| Sex | ||||
| F | n (%) | 6 (50·0) | 7 (46·7) | 13 (48·1) |
| M | n (%) | 6 (50·0) | 8 (53·3) | 14 (51·9) |
| Weight (kg) | ||||
| Mean (SD) | 81·1 (17·5) | 91·5 (27·1) | 86·9 (23·5) | |
| 95% CI | 70·0, 92·3 | 76·5, 106·5 | ||
| Countries | ||||
| France | n (%) | 2 (16·7) | 2 (13·3) | 4(14·8) |
| United Kingdom | n (%) | 2 (16·7) | 2 (13·3) | 4 (14·8) |
| Netherlands | n (%) | 0 | 1(6·7) | 1 (3·7) |
| United States | n (%) | 8 (66·7) | 10 (66·7) | 18 (66·7) |
| Race | ||||
| Black or African American | n (%) | 1 (6·7) | 3 (25·0) | 4 (14·8) |
| White | n (%) | 10 (66·7) | 7 (58·3) | 17 (63·0) |
| Unknown | n (%) | 1 (6·7) | 0 | 1 (3·7) |
| Missing | n (%) | 3 (20·0) | 2 (16·7) | 5 (18·5) |
| Months since Initial Diagnosis * | n | 12 | 14 | 26 |
| Mean (SD) | 35·6 (35·2) | 21·5 (24·1) | 28·0 (30·0) | |
| 95% CI | 13·3, 58·0 | 7·6, 35·5 | ||
| Age at Initial IMNM Diagnosis (years) † | n | 12 | 14 | 26 |
| Mean (SD) | 54·2 (9·8) | 52·2 (13·9) | 53·1 (12·0) | |
| HMGCR/SRP Antibodies | ||||
| Positive/Negative | n (%) | 10 (83·3) | 11 (73·3) | 21 (77·8) |
| Negative/Positive | n (%) | 2 (16·7) | 4 (26·7) | 6 (22·2) |
| Muscle Biopsy performed | ||||
| Y | n (%) | 11 (91·7) | 15 (100) | 26 (96.3) |
| Complement C5b-9 or C9 staining performed | n (%) | 4 (33·3) | 5 (33·3) | 9 (33·.3) |
| Positive | n (%) | 3 (25.0) | 5 (33.3) | 8 (29·6) |
| Negative | n (%) | 1 (8.3) | 0 | 1 (3·7) |
| IMNM Treatment history | ||||
| Any medication received for IMNM | n (%) | 11 (91·7) | 14 (93·3) | 25 (92·6) |
| Prednisone | n (%) | 11 (91·7) | 11 (73·3) | 22 (81·5) |
| Methotrexate | n (%) | 8 (66·7) | 7 (46.7) | 15 (55.6) |
| Azathioprine | n (%) | 3 (25·0) | 3 (20·0) | 6 (22·2) |
| Mycophenolate mofetil | n (%) | 2 (16·7) | 3 (20·0) | 5 (18·5) |
| Cyclophosphamide | n (%) | 1 (8·3) | 1 (6·7) | 2 (7·4) |
| Cyclosporine | n (%) | 0 | 0 | 0 |
| Tacrolimus | n (%) | 0 | 0 | 0 |
| Rituximab | n (%) | 3 (25·0) | 6 (40·0) | 9 (33·3) |
| Plasma Exchange | n (%) | 2 (16·7) | 3 (20·0) | 5 (18·5) |
| Intravenous immunoglobulin | n (%) | 9 (75·0) | 10 (66·7) | 19 (70·4) |
| Other | n (%) | 1 (8·3) | 3 (20·0) | 4 (14·8) |
CI=confidence interval, HMGCR=3-hydroxy-3-methyl-glutaryl-coenzyme A reductase, IMNM=Immune mediated necrotizing myopathy, SD= standard deviation, SRP=signal recognition particle.
Months since initial diagnosis was calculated as: (Date of Randomization – Date of Initial IMNM Diagnosis + 1)/30.5.
Age at initial diagnosis was calculated as: Year of Initial IMNM Diagnosis - Year of Birth.
Primary outcome
The primary efficacy endpoint in this study of change from baseline to Week 8 in CK levels was not met. There was no statistically significant difference between treatment arms Wilcoxon-Mann-Whitney odds: 0·55 95% CI: [0·19–1·57] (p=0·46) and no clinically relevant reduction in CK levels over time within treatment arms (table 2, figure 2).
Table 2.
Changes in Creatine Kinase Levels from Baseline to Week 8 (ITT population)
| Zilucoplan 0·3mg/kg N=12 |
Placebo N=15 |
|
|---|---|---|
| n=10 | n=14 | |
| Mean percent change from baseline (SD) * | −9·9 (26·1) | −20·7 (31·2) |
| Median (Min, Max) | −15·1 (−37·3, 44·5) | −16.3 (−80·0, 18·2) |
| Stratified† | ||
| p-value‡ | 0·46 | |
| Wilcoxon-Mann-Whitney odds | 0·55 | |
| 95% CI | 0·19, 1·57 |
CI=confidence interval, Max=maximum, Min=minimum, SD=standard deviation.
Week 8 CK values were not available for 3 participants (two in the zilucoplan arm and one in the placebo arm)
Primary efficacy analysis.
Based on a 2-sided Van Elteren test.
Figure 2. Percent changes in Creatine Kinase Levels from Baseline to Week 8 (ITT population).
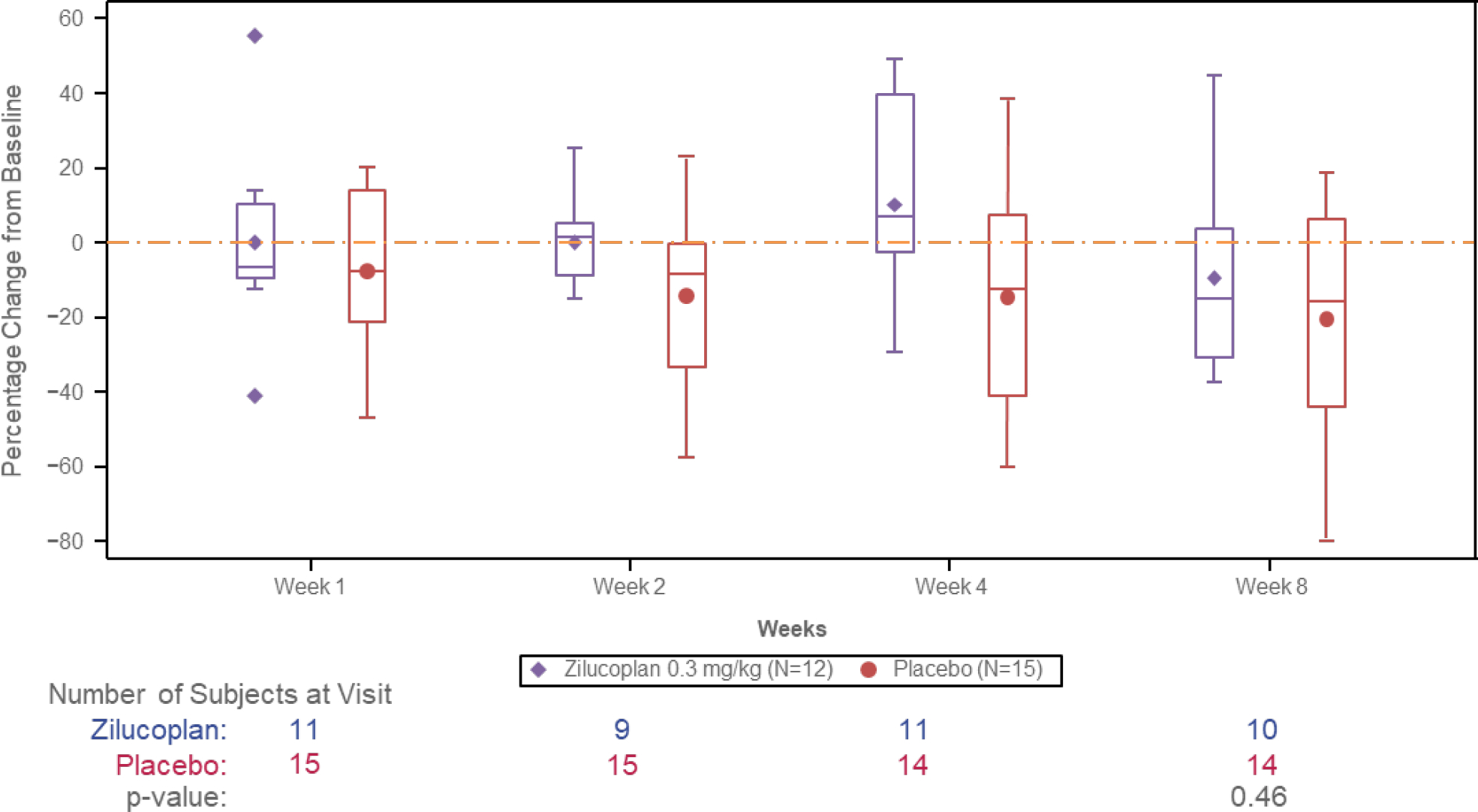
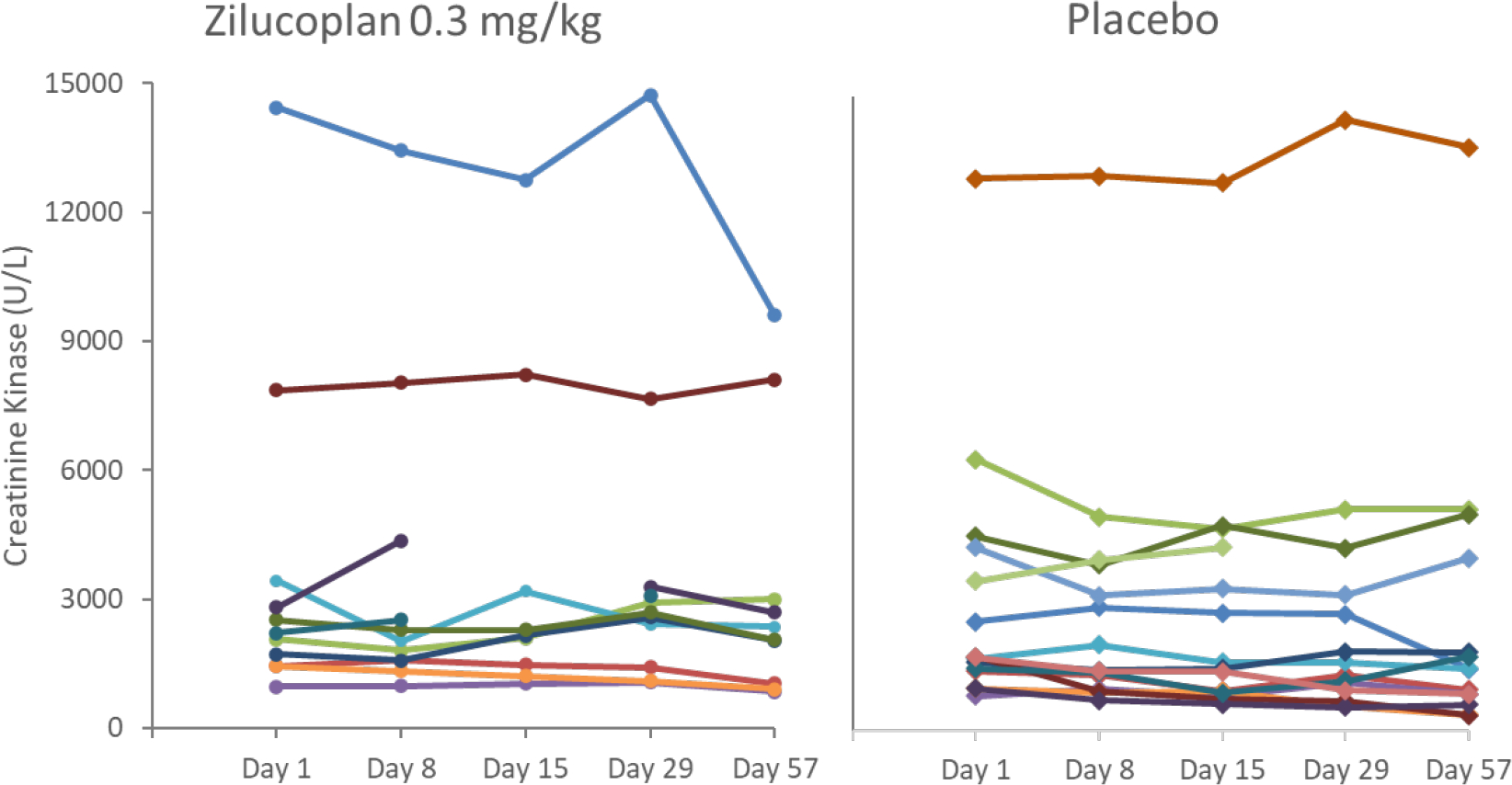
As previously described the sample size calculation was based on the assumption of 80% CK reduction; none of the participants in either group reached this level of response. Individual CK levels over the main study period are shown in figure 3. The outlier in the zilucoplan group began a course of steroids (prednisone 20 mg QD) approximately 10 days prior to the end of the treatment period, and the reduction in CK did not meet the 80% threshold.
Figure 3. Individual CK Levels (U/L) Over the Main Study Period (ITT population).
Secondary outcomes
Similarly, there were no clinically relevant differences in secondary (clinical) endpoints such as the ACR/EULAR TIS at least minimal response at Week 8 or the 3TUG test (appendix p7) between the treatment arms.
Subgroup analysis
There were no clinically relevant differences in subgroup analysis endpoints such as the change from baseline in CK to Week 4 and Week 8 by sex, age or stratification factor (appendix p4, 5 and 9–11).
Open label extension efficacy outcomes
There were also no clinically relevant changes in efficacy endpoints in the extension portion of the study.
Exploratory outcomes
Zilucoplan administration led to a sustained and complete inhibition of the terminal complement pathway measured by the sRBc lysis assay from participants on active treatment in the main study. In the zilucoplan group, baseline mean (SD) was 84·65 (34·74) compared with 3·67 (3·37) at Week 8, whereas in the placebo group, the baseline mean (SD) was 91·54 (28·13 vs 100·00 (0·00) at Week 8) (appendix p6). Two participants during the main study, one in the zilucoplan group and one in the placebo group, had low complement activity at baseline.
Safety
During the main study, for participants who received zilucoplan, the mean (SD) duration of exposure was 56·3 (3·0) days, for participants who received placebo the mean (SD) duration of exposure was 55·2 (10·1) days and for participants who received at least 1 dose of zilucoplan during the main/extension study, the mean duration of exposure was 149·2 (92·9) days.
No new or unexpected safety findings or relevant differences between the zilucoplan and placebo arms were reported overall (table 3) or by sex (appendix p12). The rate of TEAEs and serious TEAEs in the zilucoplan group was numerically lower than in the placebo group (n=9, 75·0% vs n=13, 86·7%) and n=0, 0% and n=3, 20·0%) participants, respectively). The most frequently reported TEAEs (headache and nausea) had a similar rate across both treatment groups (headache: n=4 in both groups [33·3% and 26·7%, respectively] and nausea: n=3 in both groups [25·0% and 20·0%, respectively]). No treatment-related serious TEAEs and no deaths were reported in the double-blind portion of the study. The incidence of treatment-related TEAEs was similar between treatment groups, and included headache, nausea, and vertigo. No Neisseria infections were reported in this study.
Table 3.
Summary of treatment-emergent adverse events (safety analysis population)
| Zilucoplan 0·3mg/kg N=12 n (%)* |
Placebo N=15 n (%)* |
|
|---|---|---|
| Any TEAE | 9 (75·0) | 13 (86·7) |
| Most Frequent TEAE† | ||
| Headache | 4 (33·3) | 4 (26·7) |
| Nausea | 3 (25·0) | 3 (20·0) |
| Serious TEAE | 0 | 3 (20·0) |
| TEAE Resulting in Permanent Withdrawal from Study Medication | 0 | 0 |
| Treatment-related TEAE | 4 (33·3) | 5 (33·3) |
| Headache | 2 (16·7) | 2 (13·3) |
| Nausea | 2 (16·7) | 1 (6·7) |
| Vertigo | 0 | 2 (13·3) |
| Treatment Related Serious TEAE | 0 | 0 |
| Deaths (TEAEs leading to death) | 0 | 0 |
TEAE=treatment-emergent adverse event.
n=number of participants reporting at least one TEAE in that category
TEAEs reported in >2 participants in either treatment group.
TEAEs of interest included infections and injection site reactions. Three participants [25·0%] who received zilucoplan and 2 participants [13·3%] who received placebo reported infection and infestation TEAEs during the main study (none were treatment related), and eight participants (32·0%) in the open-label extension study. One participant receiving zilucoplan during the open-label extension experienced an event of herpes zoster related to treatment. Two other study participants experienced events of acute bacterial sinusitis and sinusitis that were also considered treatment related. Mild and moderate injection site reactions were reported in 5 participants (3 in the zilucoplan group and 2 in the placebo group) in the main study and 4 participants during the open-label extension. Of these, most were treatment related (2/3 in the zilucoplan and 1/2 in the placebo group [1 participant experienced two related injection site events] in the main study and 3/4 in the open-label extension).
Open label extension safety
In the open-label extension part of the study no unexpected safety findings were reported or observed.
Discussion
In this study, C5 inhibition as a potential treatment in participants with IMNM was tested based on the hypothesis that classical complement pathway activation has a primary pathogenic role in the disease. Sustained, complete inhibition of the terminal complement pathway, was confirmed in all participants on active treatment in the double-blind period, confirming that the intended pharmacologic effect of zilucoplan administration was achieved. Daily SC self-injection of zilucoplan was well tolerated in study participants, in line with prior data in research trials in generalised myasthenia gravis.(10)
Unexpectedly, terminal complement pathway inhibition did not show an effect on either CK levels or clinical symptoms in this study. The lack of a demonstrable effect on laboratory and clinical markers of disease within a timeframe when other therapeutics such as glucocorticoids or IVIg are known to have an effect (14) suggests that complement activation may not be the primary pathomechanistic driver for disease activity in this participant population. In a recent humanized mouse model, blocking complement activation through C5 inhibition by zilucoplan protected mice from IMNM onset whereas therapeutic administration of zilucoplan following disease onset failed to significantly restore muscle strength.(8) This model also demonstrated reduced C5b9 deposits on myofibers. In combination, these preclinical and clinical findings contradict the current hypothesis that MAC deposition via the classical complement pathway activation through anti-HMGCR or anti-SRP antibodies drives the histopathological hallmark of the disease, (7, 21) and are more consistent with in vitro data showing that anti-SRP or HMGCR autoantibodies induce muscle fibre atrophy and impair myoblast fusion in complement independent mechanisms.(22)
Thus, the results of our study provide insight into the pathophysiology of IMNM in that, based on the inability of complement inhibition to reduce disease activity, the prominent presence of complement components in muscle tissue appears to be reactive rather than to cause the necrotic process. Whether or not anti-HMGCR and anti-SRP autoantibodies may be pathogenic via a non-complement mediated mechanism or are just a hallmark of the disease with no relevance to its pathobiology remains to be explored further.
In the absence of prior clinical studies in IMNM, we developed an efficient study design for evaluating a potential treatment effect in these participants. Our study allowed us to obtain results in a small number of participants over a short period of time in a placebo-controlled setting as is essential in a severe, rare disease such as IMNM. This was possible with the selection of CK as the primary endpoint, using a high threshold of 80% reduction of CK levels over the 8–week study period, in line with expectations for a treatment effect above the currently available treatment options for these participants.(14) Moreover, we identified the ACR/EULAR scale (23) as a suitable option for clinical assessment in this population, though as the study was designed to be double–blinded, CK readouts along with ALT and AST could not be provided to sites. This study design not only helped us answer the important question of whether complement inhibition is a potential treatment for IMNM participants but should also serve as a starting point for future studies on this disease.
There are a number of potential limitations of our study hindered some statistical comparisons including baseline characteristics. MAC deposition was noted on the sarcolemma of non-necrotic fibres in 8 of 9 muscle biopsy that were immunostained for MAC, however further comparisons between the zilucoplan and placebo groups may have been possible if MAC staining had been carried out in all patients. None of the 8 participants with MAC staining improved with zilucoplan during the study. The majority of study participants had previously received other treatments for IMNM, including glucocorticoids and IVIg, therefore the results may not be generalizable to treatment-naïve patients with IMNM. The study duration may not have been sufficient to allow for an effect of complement inhibition on the chosen endpoints. The chosen endpoints, notably CK, may not be sensitive to the effect of complement inhibition, though they typically respond well to glucocorticoids and IVIg treatment within the time frame of the trial. The participants included may have been a group of participants who do not respond to complement inhibition or may have been too far advanced in the course of the disease to respond to C5 inhibition.
Despite these caveats, and although the results are disappointing from the clinical perspective, our study provides valuable insight into the pathophysiology of IMNM, may support evidence-based treatment decisions in the future, and is paving a way for future clinical trials in IMNM using an efficient study design.
Supplementary Material
Research in context.
Evidence before this study
Most patients with immune-mediated necrotizing myopathy (IMNM) have autoantibodies against 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) or the signal recognition particle (SRP) that can be complement activating, and the titres of these antibodies appear to correlate with the clinical course. In addition, complement levels in IMNM muscle are higher than in other inflammatory myopathies. Passive transfer animal models using patients’ sera suggested the pathogenic potential of anti-HMGCR and anti-SRP antibodies, with disease attenuation in the context of complement deficiency and increased disease activity with complement adjunction. No randomised double-blind placebo-controlled multicentre trial had been conducted in IMNM, and no validated outcome measures for such trials had been established.
Added value of this study
Prior evidence suggested that classical pathway activation of complement could have an important role in the pathogenesis of IMNM and therefore created the possibility of terminal complement pathway inhibition as a potential therapeutic target. Our study is the first clinical trial conducted in IMNM, to our knowledge, and paves the way for the efficient design of future trials in this disease. In addition, our study provides important insights into the relevance of C5 activation in IMNM.
Implications of the available evidence
The clear results, while disappointingly negative, provide important novel data on the pathobiology of IMNM, suggesting that complement activation may not be causative and complement deposition on myofibers may be reactive rather than pathogenic.
Acknowledgements
The authors thank the participants and their families who contributed to this trial; the IMNM01 trial investigators (see List of Investigators, appendix p13–14) and study co-ordinators (see List of Study-Coordinators, appendix p15–16) and Margarita Lens, MSci, CMPP, of UCB Pharma and Fiona Woodward, PhD, of EVR consulting, contracted to UCB Pharma for publication and editorial support. In addition, the authors would like to thank Rene Bouw of UCB Pharma for his contributions to the study.
HC is supported by the National Institute for Health Research (NIHR) Biomedical Research Centre Funding Scheme. JBL held a NIHR Clinical Lectureship in Neurology (NWN/006/025/A). The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health.
Role of the funding source
This study was funded by Ra Pharmaceuticals Inc, now part of UCB Pharma.
The funding source contributed to the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, and approval of the manuscript; and the decision to submit the manuscript for publication.
Collaborators
| First and middle names | Surnames |
|---|---|
| Anthony A. | Amato* |
| Olivier | Benveniste* |
| Suur | Biliciler* |
| Hector | Chinoy* |
| Mazen M. | Dimachkie* |
| Christyn | Edmundson* |
| Miriam | Freimer* |
| Anthony | Geraci* |
| Yessar | Hussain* |
| Pedro | Machado* |
| Andrew L. | Mammen* |
| Tahseen | Mozaffar* |
| Payam | Soltanzadeh* |
| Niraja | Suresh* |
| Anneke | van der Kooi* |
| Yves | Allenbach |
| Matthew | Appleby |
| Richard J | Barohn |
| Nicolas | Champtiaux |
| Christopher | Doughty |
| Jerrica | Farias |
| Constantine | Farmakidis |
| Ali A. | Habib |
| Chafic | Karam |
| James | Lilleker |
| Samantha | Lorusso |
| Giorgia | Querin |
| Mamatha | Pasnoor |
| Iago | Pinal-Fernandez |
| Joost | Raaphorst |
| George | Ransley |
| Sami | Saba |
| Kazim | Sheikh |
| Andrew | Snedden |
| Jeffrey | Statland |
| Tuan | Vu |
Principal Investigator
Footnotes
Declaration of interests
ALM is a patent author for the anti-HMGCR test.
AAA has participated in medical advisory boards/acted as a consultant for Argenx, Ra Pharmaceuticals, Alexion, EMD Serono, OnoPharma, Horizon Therapeutics, Takeda, Johnson & Johnson (COVID-19 vaccination program)
MD has served or recently served as a consultant for Amazentis, ArgenX, Catalyst, Cello, Covance/Labcorp, CSL-Behring, EcoR1, Janssen, Kezar, Medlink, Momenta, NuFactor, Octapharma, RaPharma/UCB, Roivant Sciences Inc, RMS Medical, Sanofi Genzyme, Shire Takeda, Scholar Rock, Spark Therapeutics, Abata/Third Rock, UCB Biopharma, and UpToDate. Dr. Dimachkie received research grants or contracts or educational grants from Alexion, Alnylam Pharmaceuticals, Amicus, Biomarin, Bristol-Myers Squibb, Catalyst, Corbus, CSL-Behring, FDA/OOPD, GlaxoSmithKline, Genentech, Grifols, Kezar, Mitsubishi Tanabe Pharma, MDA, NIH, Novartis, Octapharma, Orphazyme, Ra Pharma/UCB, Sanofi Genzyme, Sarepta Therapeutics, Shire Takeda, Spark Therapeutics, UCB Biopharma / RaPharma, Viromed/Healixmith & TMA.
HC has received personal compensation for activities with Novartis, UCB, Lilly, Biogen, Orphazyme, Astra Zeneca as a speaker, advisory board member or consultancy, grants via The University of Manchester from Novartis, UCB and MedImmune, and has received travel support from Abbvie and Janssen.
JBL has received speakers fees, travel support, and/or consultancy fees from Sanofi Genzyme, Roche, and Biogen.
Yves Allenbach: has received personal compensation for activities with Lilly, and CSL-Berhing as a speaker or consultancy.
Mark Vanderkelen: Employee and stockholder of UCB Pharma
Eumorphia Delicha: Contractor of UCB Pharma
Ramin Farzaneh-Far: was an employee of Ra Pharma at the time of the study
BB, HK, PWD, CS* and OB are employees and stockholders of UCB Pharma
IPF and YH have no competing interests to declare
*Employee at the time to the study
Data sharing
Underlying data from this manuscript may be requested by qualified researchers six months after product approval in the US and/or Europe, or global development is discontinued, and 18 months after trial completion. Investigators may request access to anonymised individual patient-level data and redacted trial documents which may include: analysis-ready datasets, study protocol, annotated case report form, statistical analysis plan, dataset specifications, and clinical study report. Prior to use of the data, proposals need to be approved by an independent review panel at www.Vivli.org and a signed data sharing agreement will need to be executed. All documents are available in English only, for a pre-specified time, typically 12 months, on a password-protected portal.
References
- 1.Allenbach Y, Mammen AL, Benveniste O, Stenzel W, Immune-Mediated Necrotizing Myopathies Working G. 224th ENMC International Workshop:: Clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, 14–16 October 2016. Neuromuscul Disord. 2018;28(1):87–99. [DOI] [PubMed] [Google Scholar]
- 2.Allenbach Y, Benveniste O. Peculiar clinicopathological features of immune-mediated necrotizing myopathies. Curr Opin Rheumatol. 2018;30(6):655–63. [DOI] [PubMed] [Google Scholar]
- 3.Benveniste O, Drouot L, Jouen F, Charuel JL, Bloch-Queyrat C, Behin A, et al. Correlation of anti-signal recognition particle autoantibody levels with creatine kinase activity in patients with necrotizing myopathy. Arthritis Rheum. 2011;63(7):1961–71. [DOI] [PubMed] [Google Scholar]
- 4.Werner JL, Christopher-Stine L, Ghazarian SR, Pak KS, Kus JE, Daya NR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum. 2012;64(12):4087–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anquetil C, Boyer O, Wesner N, Benveniste O, Allenbach Y. Myositis-specific autoantibodies, a cornerstone in immune-mediated necrotizing myopathy. Autoimmun Rev. 2019;18(3):223–30. [DOI] [PubMed] [Google Scholar]
- 6.Allenbach Y, Arouche-Delaperche L, Preusse C, Radbruch H, Butler-Browne G, Champtiaux N, et al. Necrosis in anti-SRP(+) and anti-HMGCR(+)myopathies: Role of autoantibodies and complement. Neurology. 2018;90(6):e507–e17. [DOI] [PubMed] [Google Scholar]
- 7.Bergua C, Chiavelli H, Allenbach Y, Arouche-Delaperche L, Arnoult C, Bourdenet G, et al. In vivo pathogenicity of IgG from patients with anti-SRP or anti-HMGCR autoantibodies in immune-mediated necrotising myopathy. Ann Rheum Dis. 2019;78(1):131–9. [DOI] [PubMed] [Google Scholar]
- 8.Julien S, Vadysirisack D, Sayegh C, Ragunathan S, Tang Y, Briand E, et al. Prevention of Anti-HMGCR Immune-Mediated Necrotising Myopathy by C5 Complement Inhibition in a Humanised Mouse Model. Biomedicines. 2022;10(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnston Jeffrey, Ricardo Alonso, Arata Michelle, Lickliter Jason, Steven DeMarco Richard Fahrner, Hammer Robert, Newstat Beth, Roychowdhury Debasish, Sylvia Tobé Debra Winslow, Treco Douglas. A PHASE 1 MULTIPLE-DOSE CLINICAL STUDY OF RA101495, A SUBCUTANEOUSLY ADMINISTERED SYNTHETIC MACROCYCLIC PEPTIDE INHIBITOR OF COMPLEMENT C5 FOR TREATMENT OF PAROXYSMAL NOCTURNAL HEMOGLOBINURIA. 21st Congress of the European Hematology Association; Copenhagen, Denmark: 2016. [Google Scholar]
- 10.Howard JF Jr., Nowak RJ, Wolfe GI, Freimer ML, Vu TH, Hinton JL, et al. Clinical Effects of the Self-administered Subcutaneous Complement Inhibitor Zilucoplan in Patients With Moderate to Severe Generalized Myasthenia Gravis: Results of a Phase 2 Randomized, Double-Blind, Placebo-Controlled, Multicenter Clinical Trial. JAMA Neurol. 2020;77(5):582–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howard JF Jr., Vissing, Gilhus NE, Leite MI, Utsugisawa K, Duda PW, et al. Zilucoplan: An Investigational Complement C5 Inhibitor for the Treatment of Acetylcholine Receptor Autoantibody-Positive Generalized Myasthenia Gravis. Expert Opin Investig Drugs. 2021;30(5):483–93. [DOI] [PubMed] [Google Scholar]
- 12.Weiss MDGA, Hussain Y, Kaminski HJ, Leite I et al. , editor Quality of Life Outcomes in RAISE: A Double-Blind Ramdomized, Placebo-Controlled Study of Zilucoplan in GMG. American Association of Neuromuscular and Electrodiagnostic Medicine; 2022; Nashville, Tennessee: Muscle & Nerve. [Google Scholar]
- 13.Vu TGA, Hussain Y, Kaminski H, Leite MI et al. , editor Efficacy and Safety of Zilucoplan in Myasthenia Gravis: Responder Analysis From the Randomized Phase 3 RAISE Trial. American Association of Neuromuscular and Electrodiagnostic Medicine; 2022; Nashville, Tennessee: Muscle & Nerve. [Google Scholar]
- 14.Pinal-Fernandez I, Casal-Dominguez M, Mammen AL. Immune-Mediated Necrotizing Myopathy. Curr Rheumatol Rep. 2018;20(4):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allenbach Y, Drouot L, Rigolet A, Charuel JL, Jouen F, Romero NB, et al. Anti-HMGCR autoantibodies in European patients with autoimmune necrotizing myopathies: inconstant exposure to statin. Medicine (Baltimore). 2014;93(3):150–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kocoloski A, Martinez S, Moghadam-Kia S, Lacomis D, Oddis CV, Ascherman DP, et al. Role of Intravenous Immunoglobulin in Necrotizing Autoimmune Myopathy. J Clin Rheumatol. 2022;28(2):e517–e20. [DOI] [PubMed] [Google Scholar]
- 17.Wang JX, Wilkinson M, Oldmeadow C, Limaye V, Major G. Outcome predictors of immune-mediated necrotizing myopathy-a retrospective, multicentre study. Rheumatology (Oxford). 2022;61(9):3824–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El Sissy C, Rosain J, Vieira-Martins P, Bordereau P, Gruber A, Devriese M, et al. Clinical and Genetic Spectrum of a Large Cohort With Total and Sub-total Complement Deficiencies. Front Immunol. 2019;10:1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Socie G, Caby-Tosi MP, Marantz JL, Cole A, Bedrosian CL, Gasteyger C, et al. Eculizumab in paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome: 10-year pharmacovigilance analysis. Br J Haematol. 2019;185(2):297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Costabile M Measuring the 50% haemolytic complement (CH50) activity of serum. J Vis Exp. 2010(37). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abstracts of the 6(th) Congress of the European Academy of Neurology, Virtual 2020. Eur J Neurol. 2020;27 Suppl 1:1–1228. [DOI] [PubMed] [Google Scholar]
- 22.Arouche-Delaperche L, Allenbach Y, Amelin D, Preusse C, Mouly V, Mauhin W, et al. Pathogenic role of anti-signal recognition protein and anti-3-Hydroxy-3-methylglutaryl-CoA reductase antibodies in necrotizing myopathies: Myofiber atrophy and impairment of muscle regeneration in necrotizing autoimmune myopathies. Ann Neurol. 2017;81(4):538–48. [DOI] [PubMed] [Google Scholar]
- 23.Aggarwal R, Rider LG, Ruperto N, Bayat N, Erman B, Feldman BM, et al. 2016 American College of Rheumatology/European League Against Rheumatism Criteria for Minimal, Moderate, and Major Clinical Response in Adult Dermatomyositis and Polymyositis: An International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. 2017;69(5):898–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Underlying data from this manuscript may be requested by qualified researchers six months after product approval in the US and/or Europe, or global development is discontinued, and 18 months after trial completion. Investigators may request access to anonymised individual patient-level data and redacted trial documents which may include: analysis-ready datasets, study protocol, annotated case report form, statistical analysis plan, dataset specifications, and clinical study report. Prior to use of the data, proposals need to be approved by an independent review panel at www.Vivli.org and a signed data sharing agreement will need to be executed. All documents are available in English only, for a pre-specified time, typically 12 months, on a password-protected portal.