

Abstract
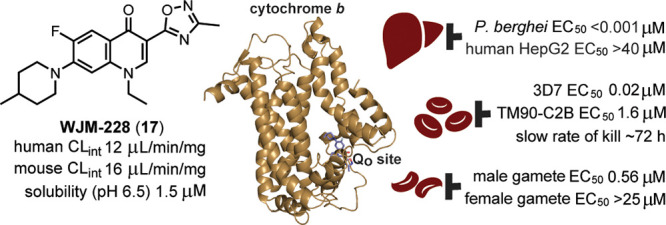
The development of new antimalarials is required because of the threat of resistance to current antimalarial therapies. To discover new antimalarial chemotypes, we screened the Janssen Jumpstarter library against the P. falciparum asexual parasite and identified the 7-N-substituted-3-oxadiazole quinolone hit class. We established the structure–activity relationship and optimized the antimalarial potency. The optimized analog WJM228 (17) showed robust metabolic stability in vitro, although the aqueous solubility was limited. Forward genetic resistance studies uncovered that WJM228 targets the Qo site of cytochrome b (cyt b), an important component of the mitochondrial electron transport chain (ETC) that is essential for pyrimidine biosynthesis and an established antimalarial target. Profiling against drug-resistant parasites confirmed that WJM228 confers resistance to the Qo site but not Qi site mutations, and in a biosensor assay, it was shown to impact the ETC via inhibition of cyt b. Consistent with other cyt b targeted antimalarials, WJM228 prevented pre-erythrocytic parasite and male gamete development and reduced asexual parasitemia in a P. berghei mouse model of malaria. Correcting the limited aqueous solubility and the high susceptibility to cyt b Qo site resistant parasites found in the clinic will be major obstacles in the future development of the 3-oxadiazole quinolone antimalarial class.
Keywords: malaria, Plasmodium, antimalarial, mitochondria, cytochrome bc1
Malaria in humans causes significant morbidity and mortality. In 2021, approximately 619,000 deaths globally resulted from malaria,1 significantly more cases than in proceeding years due to supply constraints on resources and medicines because of the COVID-19 pandemic.1 Malaria is caused by five Plasmodium species. P. falciparum is the most prevalent in sub-Saharan Africa and the deadliest, accounting for more than 90% of all deaths. P. vivax is mostly found in South East Asia and the Americas and is responsible for the relapse of malaria weeks or even months after drug treatment due to a dormant liver stage form known as the hypnozoite. P. knowlesi, P. malariae, and P. ovale are endemic to South East Asia and cause varying degrees of disease morbidity, but to date, only a few known deaths have occurred.
Malaria is transmitted from one human host to another by a blood meal taken by the female Anopheles mosquito. In the human host, the parasite has a multistage life cycle comprising a liver or pre-erythrocytic stage, a symptomatic asexual blood stage, and a transmission stage where sexual forms of the parasite known as gametocytes are transferred to the mosquito via a blood meal of an infected human. Once inside the mosquito, male and female gametes are immediately formed, which initiate the life cycle inside the mosquito host.
Malaria is combated by therapeutics that interfere with different stages of the malaria parasite life cycle. Curative therapies, which largely target the asexual blood stage, include quinoline-based antimalarials, anthracene-like antimalarials (lumefantrine and halofantrine), and artemisinin combination therapies (ACTs). Because of the reliance on a narrow set of antimalarial chemotypes, resistance to quinoline therapies (e.g., chloroquine, mefloquine, piperaquine) is widespread, while resistance has recently emerged against ACT therapies in South East Asia2 and in Africa,3 highlighting the need for new antimalarials. Toward this goal, in the past 20 years, several new antimalarial chemotypes have been under clinical development,4 but concerningly, genetic alterations conferring resistance to some of these antimalarials have been detected. Therefore, the development of new therapies with a high barrier to resistance and no cross-resistance to existing antimalarials is paramount.
The development of therapies that target multiple stages of the malaria parasite life cycle is also preferred. Compounds that target both the asexual and transmission stages of the parasite life cycle have use not only in a curative therapy but also in a therapy that controls the transmission of malaria in endemic areas. Prophylaxis therapies aimed at preventing the onset of the symptomatic asexual stage commonly target both the pre-erythrocytic and asexual stages and include drugs that target the parasite mitochondrion that have been or are under clinical development such as atovaquone (ATQ) (3) (in combination with proguanil, Malarone),5,6 the prodrug of ELQ-300 (5) (Figure 1), ELQ-331,7 and DSM265.8,9
Figure 1.

Structures of ubiquinone (2); known quinone, pyridone, and quinolone related antimalarials; and the hit compound class that is the focus of this research (1).
The mitochondrial electron transport chain (ETC) is essential for the survival of asexual blood and liver stage parasites.10 The ETC is also thought to be important for gamete development and oocyst formation in the transmission/mosquito stage.11,12 The ETC consists of several components. Unlike many other eukaryotes in Plasmodium, complex I is replaced by NADH dehydrogenase (NDH) that oxidizes ubiquinone (2) (Figure 1). In addition to NDH, glycerol-3-phosphate dehydrogenase, the malate quinone oxidoreductase, dihydroorotate dehydrogenase (DHODH), and succinate dehydrogenase (complex II) are involved in the ETC and contribute to the regeneration of ubiquinol for complex III, the so-called cytochrome bc1 complex.13 The movement of electrons through the cytochrome bc1 complex contributes to a proton gradient across the inner mitochondrial membrane (ΔΨm). Electrons are then passed via cytochrome c to complex IV, responsible for the reduction of oxygen to water. The mitochondrial ETC critically acts as an electron sink for DHODH in the biosynthesis of pyrimidine precursors, the sole essential function of the mitochondria in asexual parasites.14 DHODH is a validated antimalarial drug target and the target of the clinical candidate DSM265.8 Cytochrome bc1 (cyt bc1) (complex III) is a well-characterized and validated antimalarial target. In Plasmodium, cyt bc1 is composed of the catalytic subunits cyt b, Rieske, and cyt c1. Cyt b facilitates the oxidation of ubiquinol at the so-called Qo site and the reduction of ubiquinone (2) at the Qi site. Cyt b also facilitates the transfer of electrons from the Qo site via the Rieske and Cyt c1 proteins to cytochrome c.
Cyt bc1 is highly conserved across Plasmodium species but has significant structural divergence from the human form and is the reason cyt b is selectively targeted by antimalarials. Typically, cyt b is targeted by compounds with quinone, quinolone, or pyridone scaffolds that mimic or have a structural resemblance to the natural substrate ubiquinone (2) (Figure 1).13,15 These include antimalarials with an endochin-like scaffold (4)16,17 such as ELQ-300 (5)9 or GSK932121 (6)18 that bind to the Qi site of cyt b,19 whereas antimalarials with a decoquinate-like scaffold (7)20−23 such as ICI56,780 (8)24 and 8j from the Manetsch group (9)25 preferentially bind the Qo site of cyt b. ATQ (3) also targets the Qo site of cyt b.26 In addition to quinolone-like antimalarials, several recent phenotypic screens have uncovered that cyt b was also the target of a diverse array of structural chemotypes.27−29 A significant obstacle in the future design of cyt b targeted antimalarials is overcoming clinical resistance that arises from single point mutations in the mitochondrially encoded cyt b gene. However, it was recently discovered that ATQ (3) resistant parasites with a cyt b mutation may not be able to develop in the mosquito, and therefore, resistance may not spread in the field.30
To discover new antimalarial chemotypes, we performed a high-throughput screen of the Janssen Jumpstarter library composed of 80,000 compounds with diverse and drug-like structures against the P. falciparum asexual-stage parasite (72 h incubation). The primary screen of the library was conducted at 1 μM using a lactate dehydrogenase (LDH) assay31 with a Z’ of 0.86 and a 0.19% primary hit rate (>60% inhibition) resulting in 156 primary hits. A P. falciparum asexual-stage parasite LDH assay in a dose–response format (in duplicate) was then undertaken to confirm the activity of hit compounds (EC50 < 1 μM), and concomitantly, a counterscreen in a dose–response format was performed to ensure that hit compounds did not interfere with the LDH assay technology. The counterscreen consisted of evaluating compounds (up to a concentration of 10 μM) against recombinant bovine LDH. Subsequently, a human HepG2 cell growth assay (48 h incubation) using Cell TitreGlo31 in a dose–response format was undertaken to ensure that hit compounds were not broadly cytotoxic (EC50 cutoff was >10 μM). This process resulted in 86 confirmed hits with a hit confirmation rate of 54%. Confirmed hit compounds from this screen include the triazolopyrimidine class32 and the 7-N-substituted-3-oxadiazole quinolone (1) (Figure 1) that is the focus of the research herein.
The quinolone compound class has a core scaffold that is common to many cyt bc1 inhibitors (Figure 1). This class has distinct differences with known quinolone antimalarials, with 7-piperidine substitution, 1-N-ethyl substitution, and a 2-pyridine substituted oxadiazole in the 3-position of the quinolone. We questioned whether this class targeted the mitochondrial cyt bc1 because of the structural similarities between known cyt bc1 inhibitors and the hit compound 1. To distinguish whether the hit quinolone 1 had the same phenotype as the cyt bc1 inhibitor ATQ (3), we initially showed that the hit quinolone class had a faster rate of parasite kill compared to ATQ (3). This suggested that the hit quinolone class could act via a different mechanism than known quinolone antimalarials that target cyt bc1. Herein, we defined the structure–activity relationship of the hit quinolone 1 to optimize the antimalarial activity and to differentiate whether there is a connection between known quinolone antimalarials and the quinolone hit compound class. We also determined the mechanism of action of the quinolone class by using forward genetic resistance studies, characterizing the cross-resistance against P. falciparum cyt bc1 resistant mutants, and profiling the impact on the parasite mitochondrial electron transport chain. Finally, we characterized the multistage activity and efficacy in a P. berghei mouse model.
Results and Discussion
We aimed to determine the structure–activity relationship (SAR) of quinolone hit 1 to optimize its antimalarial activity and also to establish whether the active pharmacophore is connected to that of existing quinolone antimalarials (Figure 1). To investigate the SAR, we initially set out to determine the importance of the pyridyl group on the oxadiazole and whether the 3-oxadiazole moiety was acting as a bioisostere of the ester functionality in the 3-position of the decoquinate-like quinolones (7–9) (Figure 1) and therefore provide insight as to whether the hit and decoquinate quinolone classes have the same cyt b Qo site binding mode. Second, we explored iterations to improve aqueous solubility and the importance of the substitution on the 7-amino group to improve antimalarial activity.
Chemistry
The synthesis of 3-(1,2,4-oxadiazole)-quinolone analogs first involved the installation of the desired piperidine derivative at the 7-position of the commercially available quinolone 10 (Scheme 1). This was achieved through either direct nucleophilic aromatic substitution or esterification of the carboxylic acid (11) followed by Buchwald–Hartwig cross-coupling (12). The carboxylic acid, either directly or via ester hydrolysis, was converted to an anhydride by reaction with methyl chloroformate (13) and subsequently cyclized with N′-hydroxyacetamidine to form the desired 3-methyl-1,2,4-oxadiazole (14). Other analogs with variations at the quinolone 7-position were also synthesized following this synthetic pathway.
Scheme 1. Synthetic Route to Generate 3-(1,2,4-Oxadiazole)-quinolone Derivatives.

Reagents and conditions: (a) EtI, DMF, 70 °C, 24 h; (b) amine, DMSO, 120 °C, 3 h; (c) Pd(OAc)2, BINAP, Cs2CO3, DMF, 115 °C, 2 h; (d) LiOH, THF/H2O (1:1), rt, 2 h; (e) Et3N, methyl chloroformate, DCM, rt; and (f) N′-hydroxyacetamidine, NaOH, DMSO, 60 °C.
Structure–Activity Relationship
We initially investigated the importance of the 2-pyridyl group on the oxadiazole of the hit compound 1. The 3-pyridyl ring in the 3-position of the oxadiazole (15) showed reduced activity (EC50 0.123 μM) (Table 1) compared to 1 (EC50 0.263 μM) (Figure 1). Changing the 1,2,4-oxadiazole (1) configuration to 1,3,4-oxadiazole (16) ablated the parasite activity (EC50 > 10 μM). The significant activity disparity between the two oxadiazole systems may be explained by the subtle differences in electronic and dipole moment33 affecting hydrogen bond donor or acceptor interactions with the target protein. Replacing the pyridyl group with a methyl substituent in the 3-position of 1,2,4-oxadiazole (17, WJM-228) led to a marked increase in parasite activity (EC50 0.019 μM), showing that a large steric group in the 3-position was detrimental to activity. Accordingly, the pyridyl group in the 3-position was not further applied in this study. We next determined if other heterocyclic systems were a suitable replacement for 1,2,4-oxadiazole. It was found that the 2-pyridyl, 3-pyridyl, and 5-pyrazole groups (18, 19, and 20) were all detrimental to activity (EC50 > 10 μM), highlighting the requirement of 1,2,4-oxadiazole for parasite activity.
Table 1. Activities of 3-Substituted Derivatives.

EC50 data represent means and SDs for three or more experiments measuring LDH activity of P. falciparum 3D7 parasites following exposure to compounds for 72 h.
EC50 data represent means for two or more experiments measuring HepG2 viability over 48 h using CellTiter-Glo.
Decoquinate (7) and related quinolones (8, 9) all possess a 3-carboxylate functionality in the 3-position (Figure 1). To determine if the 1,2,4-oxadiazole in the 3-position of 17 was acting as a carboxylate or carboxamide bioisostere, we replaced 1,2,4-oxadiazole with a selection of carboxyl groups. The activity results show that the ethyl carboxylate (21) has a slight reduction in activity (EC50 0.028 μM) (Table 1) compared to 17, suggesting that 1,2,4-oxadiazole is acting as a carboxylate ester bioisostere. The analogs with a carboxamide, N-ethyl carboxamide, and N,N-diethyl carboxamide (22, 23, and 24) in the 3-position of the quinolone were all approximately 20-fold less active (EC50 0.390, 0.477, and 0.363 μM) than 17, whereas the carboxylic acid derivative (25) was inactive. N-Morpholine and N-piperazine substituted carboxamides (26 and 27) were also synthesized to improve aqueous solubility; however, these analogs were inactive. Overall, the activity data on these analogs imply that 1,2,4-oxadiazole is acting as a carboxylate ester bioisostere.
We reasoned that the ethyl ester would be susceptible to metabolism by esterases in serum and liver hepatocytes. To confirm this, in vitro metabolism using liver microsomes was collected and showed that the ethyl carboxylate derivative 21 has poor metabolic stability (human CLint 205 μL/min/mg), whereas 17 shows an enhanced metabolic stability (mouse and human CLint 16.2 and 12.4 μL/min/mg), implying that 1,2,4-oxadiazole masks the metabolism of the ethyl ester (Table 2). The quinolone scaffold is known to be readily crystalline and has inherently modest aqueous solubility. Determination of kinetic aqueous solubility established that the oxadiazole analog (17) possessed limited solubility (<1.2 μM at pH 7.4), whereas the solubility of the ethyl carboxylate analog (21) was slightly enhanced (12 μM at pH 7.4).
Table 2. Evaluation of Aqueous Solubility, In Vitro Metabolism, and LogD for Selected Compounds.
| aqueous
solubility |
liver microsomes |
hepatocytes | |||||
|---|---|---|---|---|---|---|---|
| cmpd | pH 7.4 (μM)a | pH 6.5 (μM)b | pH 1.2 (μM)c | mouse CLint (μL/min/mg) | human CLint (μL/min/mg) | rat CLint (μL/min/106 cells) | logDd |
| 17 | <1.2 | 1.5 | 4.6 | 16.2 | 12.4 | 53.8 | 2.6 |
| 21 | 22.9 | 205.2 | >92.4 | 3.2 | |||
| 41 | <1.2 | 44.1 | 101.9 | 24.7 | 4.2 | ||
| 43 | 10.4 | 193.2 | >92.4 | 4.6 | |||
Kinetic in PBS.
Kinetic FaSSIF.
Kinetic FaSSGF.
Shake-flask method.
We envisaged that the parasite potency and aqueous solubility of the quinolone scaffold could be improved with the appropriate substituted amino group installed in the 7-position of the quinolone while maintaining 1,2,4-oxadiazole in the 3-position for metabolic stability. The size of the ring system was first investigated in the 7-position, and parasite activity showed that the five-membered pyrrolidinone (28) was not tolerated (EC50 0.500 μM) whereas the seven-membered azepine (29) was tolerated (EC50 0.035 μM) although slightly less potent (EC50 0.026 μM) than the six-membered piperidine (30) (Table 3). To improve aqueous solubility, polar six-membered heterocycles including morpholine and piperazine (31, 32, and 33) were trialed in place of piperidine, but these modifications also led to a decrease in potency against the parasite (EC50 0.297, 4.57, and >10 μM), signifying that polar groups were not tolerated in this position.
Table 3. Activities of 7-Substituted 3-Oxadiazole-derivatives.

EC50 data represent means and SDs for three or more experiments measuring LDH activity of P. falciparum 3D7 parasites following exposure to compounds for 72 h.
EC50 data represent means for two or more experiments measuring HepG2 viability over 48 h using CellTiter-Glo.
Substitution on the piperidine was next explored. A methyl substituent in the 3-position of the piperidine ring (34) resulted in a 5-fold decrease in potency (EC50 0.114 μM) compared to the 4-methyl substitution (17) (Table 3), and therefore, further exploration focused on the 4-position of the piperidine ring. 4,4-Dimethyl and 4,4-cyclopropyl substitution of the piperidine ring (35 and 36) markedly reduced parasite potency (EC50 1.48 and 0.967 μM), whereas difluoro substitution (37) was tolerated (EC50 0.130 μM), and therefore, disubstitution at the 4-position was not further considered. Replacing the methyl in the 4-position of piperidine with an n-propyl group (38) resulted in a 2-fold improvement in activity (EC50 0.008 μM). Analogs with a 4-isopropyl or 4-phenyl group (39 and 40) were 2-fold less potent (EC50 0.041 and 0.030 μM) than the 4-methyl analog (17). The derivative 41 with a 4-benzyl group in the 4-position was 2-fold more potent (EC50 0.009 μM) than 17. Mimicking the steric bulk of the benzyl, a CH2-N-morpholine group (42) was 30-fold less active (EC50 0.290 μM) than the benzyl analog 41, again showing that polarity in this region was detrimental to parasite activity, impeding efforts to improve aqueous solubility.
It was found that additional polarity could not be installed on the quinolone scaffold to improve aqueous solubility. However, a 4-benzyl piperidine substituent on either the 3-oxadiazole or ethyl carboxylate scaffold (41 and 43) disturbed the crystallinity, improving the aqueous solubility (∼10 μM at pH 7.4) in comparison to the 4-methyl piperidine variants (17 and 21) (Table 2). In vitro metabolic stability was lower (human CLint 101.9 μL/min/mg; mouse CLint 44.1 μL/min/mg) for 41 with a 4-benzyl piperidine substituent compared to 17 with a 4-methyl piperidine substituent in the 7-position of the quinolone scaffold (Table 2), implying that added lipophilicity and the presence of a benzylic group on 41 were contributing to the lower metabolic stability.
To confirm that the SAR between the 3-ethyl carboxylate and the 3-oxadiazole scaffolds was interconnected, 3-ethyl carboxylate analogs with 4-benzyl-piperidine and 4-n-propyl-piperidine groups (43 and 44) in the 7-position were generated, and both derivatives showed equipotent parasite activity (EC50 0.043 and 0.030 μM) to 4-methyl piperidine counterpart 21 (Table 3). Overall, it was found that the 3-oxadiazole derivatives were generally 3- to 4-fold more potent than the 3-ethyl carboxylate orthologs and confirmed that the 3-oxadiazole moiety was a bioisostere 3-ethyl carboxylate. Although the SAR with this quinolone series is distinct, it shares similarities with the decoquinate-like scaffolds (7–9) (Figure 1).21−25 We also synthesized an analog of 17 without the N-1 ethyl group, but it was insoluble in DMSO and aqueous media and therefore could not be characterized or biologically tested. This highlights the inherent physical characteristics of the quinolone scaffold (Figure 1) and the reason a prodrug approach has been applied by other groups to improve aqueous solubility and in vivo exposure.34−36
Parasite Reduction Ratio and Asexual Stage of Arrest
The quinolone antimalarial scaffolds and cyt bc1 inhibitors such as ATQ (3) (Figure 1) are characterized by a slow-acting asexual phenotype. To determine whether the quinoline 3-oxadiazole scaffold was aligned with this phenotype, we characterized the asexual rate of kill and the stage of arrest. To do this, highly synchronized ring stage parasites were treated with vehicle control and ATQ (3), 17, and 41 at a concentration 10 times the EC50. Giemsa-stained parasites were then prepared and visualized by microscopy every 12 h of the 48 h asexual stage cycle. The results show that 17 and 41 treated parasites begin to be affected at the trophozoite stage and do not develop beyond this stage by 48 h. This stalling of trophozoite-stage parasites seemed to occur more quickly than ATQ (3) treated parasites (Figure 2BC and Figure S2).
Figure 2.
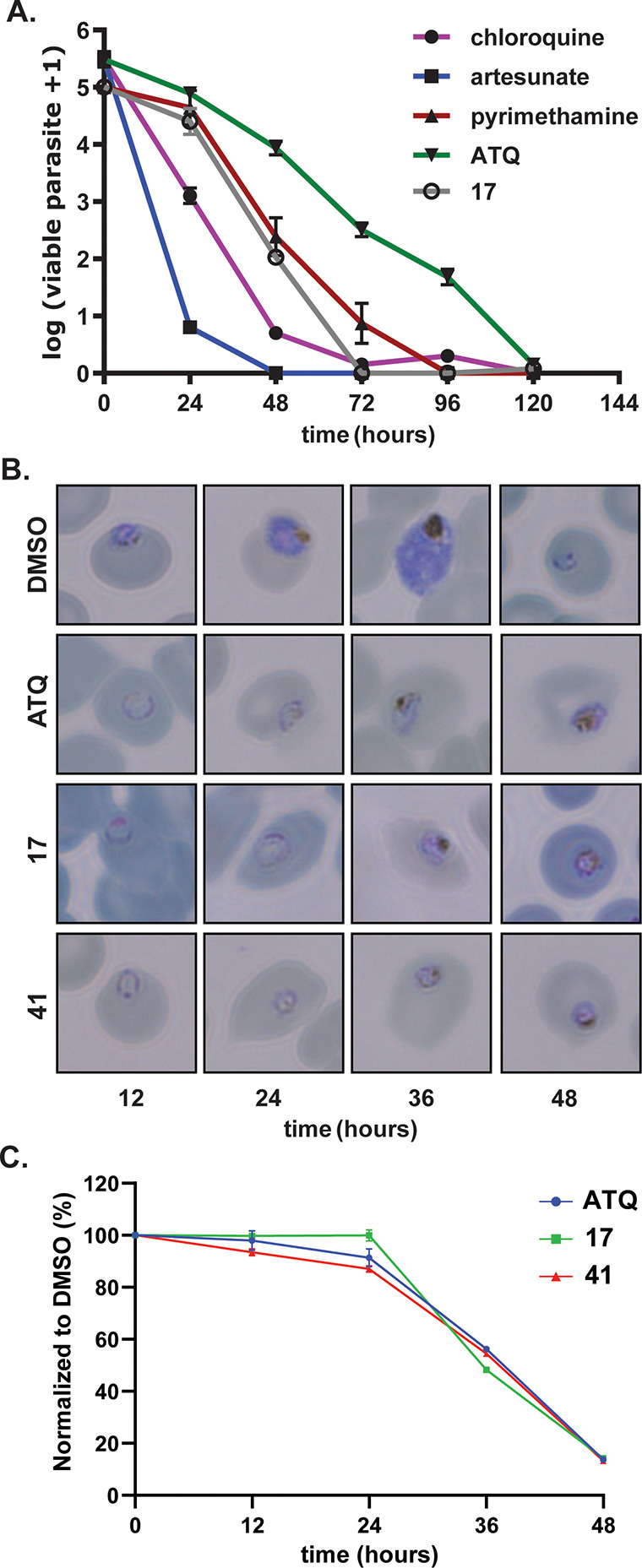
(A) Activity of 17 in a parasite reduction ratio assay in comparison to antimalarial drugs. Data represent the means and SDs of three replicate experiments using Pf 3D7 parasites in a 3H-hypoxanthine assay. (B) Representative Giemsa-stained microscopy images showing the asexual stage of arrest treated with ATQ (3), 17, and 41. Other representative images can be found in Figure S2. (C) Flow cytometry of SYBR green-stained infected RBCs. Data points represent the mean of three technical replicates analyzed via flow cytometry. Compounds in these experiments were used at a concentration 10 times the asexual EC50 value.
To characterize the rate of asexual stage arrest, we performed a parasite reduction ratio (PRR) assay following a protocol previously described.37 Briefly, ring stage parasites are treated with compound at a concentration 10 times the EC50, and then each 24 h for 120 h, the compound is washed out and drug free culture is maintained before parasitemia is determined to calculate the rate of kill. The results indicate that 17 reduced parasitemia at a moderate rate comparable with the rate of pyrimethamine (Figure 2A). Notably, 17 does not reduce parasitemia at the slow rate observed with the ATQ (3), implying that 17 is unlikely to have the same mechanism of action as ATQ (3).
Forward Genetic Resistance Study
To determine the mechanism of action of the 3-oxadiazole quinolone class, we applied drug pressure using incremental concentrations of 41 to three independent 3D7 P. falciparum parasite cultures to generate recrudescent parasites. Compound 41 was selected for this study because it has superior aqueous solubility compared to other analogs. Briefly, three parasite populations were treated with 41 at 2 times the EC50, and then the parasites were allowed to recover. This process was repeated incrementally increasing the compound concentration until recrudescent parasites were observed at 25-fold the initial EC50 value using an LDH assay. Compound 41 has an EC50 value of approximately 800 nM against all three populations (Figure 3), suggesting that each population has the same genetic aberration.
Figure 3.

Activity of 17, 41, ATQ, and ELQ300 against 41 resistant populations (with a V259L cyt b Qo site mutation). EC50 values represent an average of three experiments using the LDH assay. Error bars are SD. EC50 error values are shown in Table S2.
The genomic DNA was extracted from each parasite population resistant to 41 and then whole genome sequenced. The genome sequencing of each population revealed 17 variants in 4 or more out of the 6 samples from the 3 resistant lines compared to the wild type. The 17 variants included 16 single nucleotide variants (SNVs) and 1 indel (Table S1). Most of the variants were in low-coverage, repetitive regions or very prone to false calls. These apply to the single base deletion and 12 SNVs in the PF3D7_1036400 gene. The only dominant variant detected in six out of six samples was a single nucleotide polymorphism (SNP) in the PF3D7_MIT02300 gene encoding a V259L mutation in cyt b from cyt bc1 (complex III) (Figure S4 and Table S1). The V259L mutation was mapped to the Qo site of P. falciparum cyt b using a homology model created from an X-ray structure of Gallus gallus cyt bc1 (PDB: 3H1I) (Figure 4 and Figure S3).38 Notably, the V259L mutation was also previously found in ATQ (3) and DDD01061024 resistant parasite strains.27,39
Figure 4.
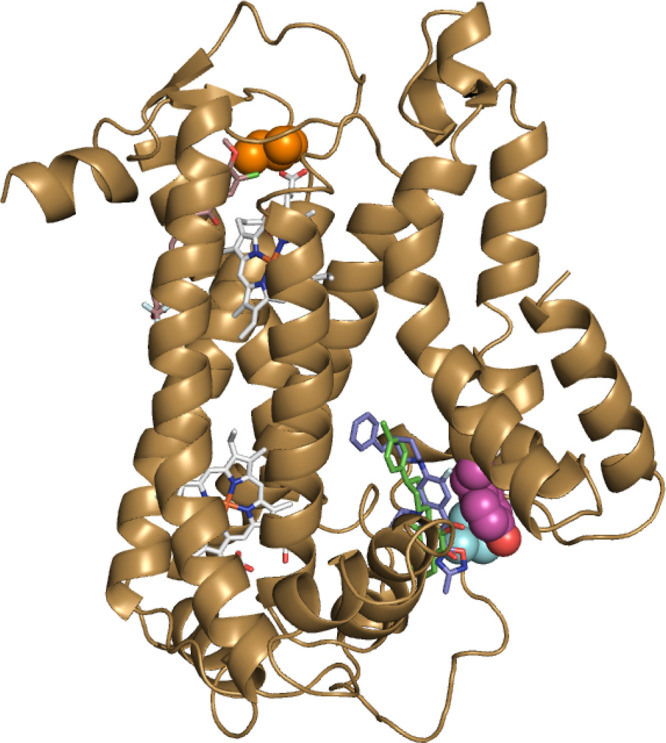
A homology model of P. falciparum cyt b showing mutations found in 41, ELQ300 (5), and ATQ (3) P. falciparum-resistant strains are shown in Figure 3 and Table 4, respectively. The homology model of P. falciparum cyt b was created from Gallus gallus cyt bc1 (PDB: 3H1I).38 TM90-C2B strain cyt b Y268S Qo site mutation is shown in magenta, Dd2 strain cyt b I22L Qi site mutation is shown in orange, and 3D7 41 resistant strain cyt b V259L Qo site mutation is shown in cyan. Heme molecules are shown in gray. The relative position of 41 (blue) is predicted by docking to the Qo site, whereas ATQ (green) and ELQ300 (brown) are overlaid using previous structural data.19,26
We next determined whether 17, ATQ (3), and ELQ-300 (5) were cross-resistant to the 41 resistant populations using an LDH assay. These assay data established that 17 was approximately 2- to 3-fold less active against 41 resistant parasites compared to the wild-type 3D7 parasites (Figure 3), demonstrating that modification in the 7-position of the quinolone did not significantly affect the level of sensitivity to the V259L mutation. ATQ (3) was 3- to 4-fold less active against 41 resistant parasites, whereas no difference in sensitivity was observed with ELQ-300 (5) against the 41 resistant parasites. These data are consistent with ATQ (3) exclusively targeting the Qo site and ELQ-300 (5) targeting the Qi site of cyt b.
Evaluation against Cyt b-Resistant Parasites
To further explore whether the oxadiazole quinolone series specifically targeted the Qo site of cyt b, we evaluated representative analogs against the ATQ (3) clinically resistant TM90-C2B strain with a Y268S mutation in the Qo site of cyt b(40,41) or the ELQ-300 (5) resistant Dd2 strain with an I22L mutation in the Qi site of cyt b(16) (mutations mapped to a model of cyt b in Figure 4). The 3-oxadiazole analogs 17 and 41 showed an approximately 150- and 50-fold respective decrease in activity against the TM90-C2B strain (Table 4) compared to the Dd2 parental strain. The 3-carboxylate derivatives 22 and 43 exhibited a 3- and 5- fold decrease in activity against the TM90-C2B strain, a significantly lower fold difference than the 3-oxadiazole analogs. All of the analogs 17, 22, 41, and 43 showed no difference in sensitivity between the ELQ-300 (5) resistant Dd2 strain and the parent Dd2 parental strain. These data agree with the genome sequencing of 41 resistant clones indicating that the quinolone analogs selectively target the Qo site of cyt b.
Table 4. Evaluation of Selected Compounds against P. falciparum Asexual Parasites Resistant to Mitochondria Targeted Drugs or Expressing ScDHODH.
| Pf cytBC1
mutant strain EC50 μM |
|||||
|---|---|---|---|---|---|
| cmpd | Pf Dd2 EC50 μMa | TM90-C2Ba | Dd2cyt b(I22L)a | SB1-A6 EC50 μMb | Dd2 ScDHODH EC50 μMb |
| 17 | 0.010 | 1.6 | 0.007 | 1.8 | 1.7 |
| 21 | 0.055 | 0.145 | 0.039 | ||
| 41 | 0.004 | 0.19 | 0.002 | 0.48 | 0.86 |
| 43 | 0.015 | 0.053 | 0.011 | ||
| ATQ | 0.001 | 5.4 | 0.005 | >10 | >10 |
| ELQ300 | 0.021 | 0.176 | |||
| DSM265 | 0.024 | 0.008 | >10 | >10 | |
Activity values against the Pf Dd2 parental line, Pf TM90-C2B strain with a Y268S mutation in the Qo site of cyt b, or Pf Dd2 strain with an I22L mutation in the Qi site cyt b using a 3H-hypoxanthine 72 h assay. EC50 values are an average of two experiments. Data for each experiment are shown in Table S3.
EC50 values represent an average of three independent experiments against the Pf SB1-A6 strain with a CNV (∼2-fold) and a C276F mutation in DHODH or Pf Dd2 expressing ScDHODH over 72 h measuring SYBR green by FACS. Dose–response curves with error values are shown in Figure S5.
Evaluation against DHODH-Resistant and ScDHODH-Expressing Parasites
DHODH and cyt bc1 are both required for the function of the mitochondrial ETC. To further demonstrate the impact of quinolone derivatives on the ETC, we evaluated the activity of the quinolone analogs 17 and 41 against the ATQ (3) and DSM265-resistant strain SB1-A6 and a Dd2 strain expressing Saccharomyces cerevisiae DHODH (ScDHODH). The SB1-A6 strain has a 2-fold amplification and a C276F SNP in the parasite DHODH gene and results in mitochondrial targeted inhibitors with reduced sensitivity.42 ScDHODH shares a low sequence identity with PfDHODH and is ubiquinone-independent, and therefore, compounds targeting the electron transport chain or DHODH confer reduced antiparasitic activity.14 Compounds 17 and 41 both showed a >200-fold decrease in activity against both the SB1-A6 strain and the Dd2 ScDHODH parasite line compared to the parental Dd2 strain (Table 4 and Figure S5). These data are consistent with the profile of the cyt bc1 inhibitor ATQ (3) and the DHODH inhibitor DSM265 against these lines, confirming that the quinolone series impact the mitochondrial ETC.
Impact on the Electron Transport Chain
To further demonstrate that the quinolone series specifically impacts cyt bc1 in the mitochondrial electron transport chain, we performed an oxygen consumption rate (OCR) assay using a Seahorse XFe96 extracellular flux analyzer.27 In this assay, the OCR at basal levels of P. falciparum 3D7 asexual parasites was determined using malate as the energy source. Compounds at a concentration of 5 μM were then added to measure the effect on the OCR. Both 17 and 41 were found to significantly reduce the OCR at levels comparable to ATQ (3), whereas DSM265 had no effect because DHODH is not involved in malate-dependent electron transport chain activity (Figure 5). The quinolone analog 33, which has no activity against P. falciparum parasites, does not affect the OCR and serves as a robust negative control. To differentiate between cyt bc1 (complex III) and complex IV activity, TMPD is added, which is an electron donator for cytochrome c and therefore bypasses the requirement of complex III in the ETC. Upon addition of TMPD, OCR levels of 17 and 41 are rescued, signifying that they act upstream of cytochrome c, consistent with them being cyt bc1 inhibitors. Finally, to confirm the effect of TMPD, NaN3, a complex IV inhibitor, is added, which results in a decrease in OCR, validating that TMPD-dependent OCR relies on complex IV. Collectively, these data indicate that the quinolone compounds 17 and 41 reduce oxygen consumption through inhibition of the mitochondrial ETC via inhibition of cyt bc1.
Figure 5.
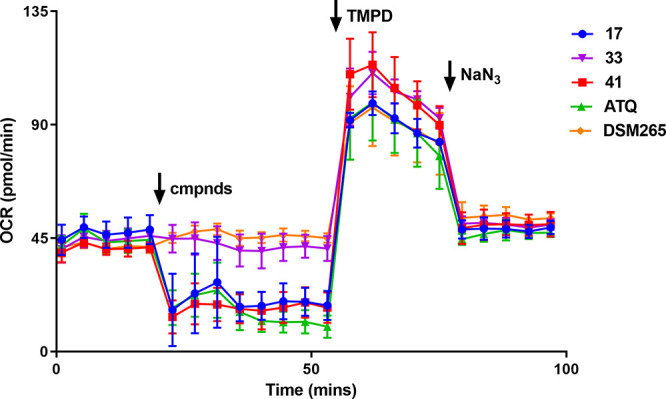
P. falciparum oxygen consumption rate (OCR) assay. Data represent the means and SDs of three technical replicates and are representative of three independent experiments. Time points and reagent injections were as follows: (1) the first five time points measured the basal level of malate-dependent OCR; (2) compounds (5 μM) were injected independently (denoted by arrow), and OCR was measured for eight time points; (3) TMPD (a cytochrome c electron donor) was injected, and OCR was measured for five time points; and (4) NaN3 (a complex IV inhibitor) was then added, and OCR was measured for five time points.
Evaluation against Drug-Resistant Parasites
To further demonstrate cyt b selectivity, a representative cohort of quinolone analogs were profiled against a selection of multidrug-resistant parasites, including parasites that are resistant to the clinical antimalarials KAF156 (CARL), DSM265 (DHODH), DDD107498 (EF2), and MMV048 (PI4K). It was found that quinolone derivatives 17, 21, 41, and 43 were equipotent to each drug-resistant strain compared to the Dd2 and NF54 wild-type parasites (Table 5). These data further support the on-target selectivity toward cyt bc1 and establish that the quinolone series is not cross-resistant to antimalarials under clinical development, including the mitochondrial pyrimidine biosynthesis inhibitor DSM265.
Table 5. Evaluation of Selected Compounds against Drug P. falciparum Strains.
| Pf multidrug-resistant strain EC50 μMa |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| cmpd | Dd2 EC50 μMa | Dd2 CARL (I1139K) | Dd2 DHODH (C276F) | Dd2 eEF2 (Y86N) | Dd2 PI4K (S743T) | K1 | NF54 | RF12 | 7G8 |
| 17 | 0.010 | 0.009 | 0.015 | 0.008 | 0.011 | 0.009 | 0.012 | 0.006 | 0.009 |
| 21 | 0.055 | 0.020 | 0.018 | 0.027 | 0.048 | 0.065 | 0.020 | 0.038 | |
| 41 | 0.004 | 0.004 | 0.005 | 0.003 | 0.005 | 0.005 | 0.005 | 0.002 | 0.003 |
| 43 | 0.015 | 0.007 | 0.006 | 0.009 | 0.014 | 0.017 | 0.005 | 0.012 | |
| KAF156 | 0.008 | 1.53 | |||||||
| DSM265 | 0.024 | 0.47 | |||||||
| DDD107498 | 0.0004 | 0.85 | |||||||
| MMV048 | 0.016 | 0.12 | |||||||
EC50 data represent means for two 3H-hypoxanthine experiments using a selection of drug-resistant P. falciparum strains following exposure to compounds in 10-point dilution series for 72 h. Data for each experiment are shown in Table S4.
Evaluation against Sexual-Stage P. falciparum Gametes and Liver-Stage Schizonts
Antimalarials that block transmission of the parasite to the mosquito are desirable as they have the potential to control the spread of malaria. Transmission blocking agents generally impact the development of sexual blood stage gametocyte or gamete, or oocyst formation in the mosquito. Mitochondrial metabolism, including reactions catalyzed by dehydrogenases that contribute electrons to the ETC, is important for the sexual stage development of Plasmodium parasites.43 This is consistent with an increased role in the ETC in oxidative phosphorylation in these stages. ATQ (3) has been reported to inhibit male gamete exflagellation but does not impact female gamete viability.12 ATQ (3) is also known to inhibit ookinete formation and prevent oocyst formation in the mosquito midgut.11
To determine if the quinolone series has transmission blocking capacity, we evaluated selected quinolone analogs 17 and 21 against male and female gametes. A dual gamete formation assay (DGFA) was used to assess the impact of compounds on the development of both male and female gametes on treatment with 17 and 21. The DGFA uses a microscopy algorithm to quantitate male exflagellation and an antibody to the female specific Pfs25 surface marker to assess female gamete viability.12 The results from the DGFA show that both 17 and 21 potently inhibit male gamete exflagellation (EC50 0.56 and 0.16 μM) but not female gamete viability (Table 6 and Figure S6). The activity observed against male gametes is less potent than that observed against asexual stage parasites. Notably, 21 is more potent than 17 against male gametes, which is in contrast with their asexual activities (Table 2).
Table 6. P. falciparum Liver Stage and Gamete Activity of Selected Compounds.
| liver stage |
transmission stage |
|||
|---|---|---|---|---|
| cmpd | Pb liver stage EEFs EC50 (SD) μMa | HepG2 EC50 μMb | male gamete EC50 μMc | female gamete EC50 μMc |
| 17 | <0.001 | >50 | 0.56 | >25 |
| 21 | 0.024 (0.003) | >50 | 0.15 | >25 |
EC50 data represent means and SDs for four technical replicates following exposure to compounds in 10-point dilution series over 48 h.
Activity was determined using a 48 h CellTiter-Glo assay.
Data represent means from four replicate experiments using NF54 parasites in a DGFA. Dose–response curves with error values are shown in Figure S6.
Compounds that disrupt the ETC including ATQ (3) and ELQ-300 (5) exhibit potent activity against exoerythrocytic forms (EEFs) of the malaria parasite and have potential as a chemopreventative therapy.9,44 To establish whether the quinolone series has activity against EEFs, we evaluated 17 and 21 against P. berghei EEFs.45 In this assay, human HepG2 hepatocytes expressing a tetraspanin CD81 receptor-GFP construct are used to enable P. berghei infection. The transformed HepG2 cells are pretreated with the compound for 18 h, and then P. berghei sporozoites expressing luciferase dissected from mosquito salivary glands are added. The infected hepatocytes are then incubated for 48 h, and EEF viability is measured by luciferase activity.44 This assay established that both 17 and 22 have potent EEF activity (EC50 < 0.001 and 0.024 μM) without affecting human host cell viability (EC50 > 50 μM) (Table 6). Collectively, the sexual-stage and pre-erythrocytic activity of the quinolone series corroborates the activity profile of cyt b Qo site targeted compounds, such as ATQ (3).
Evaluation in a P. berghei 4 Day Mouse Model
The quinolone analog 17 (WJM-228) has in vitro metabolic stability that is suitable for evaluation in a Peter’s 4 day mouse model of malaria. Compound 17 was then administered via oral gavage to mice at 20 mg/kg doses at 2, 26, 50, and 74 h post infection with P. bergheiGFP/CON asexual parasites.46 Blood samples were taken from each mouse 48, 72, and 96 h after administration of each dose, and parasitemia was measured by flow cytometry. Blood samples were also taken at 2, 6, and 22 h after the first dose to quantify compound concentration.
Bioanalysis of blood samples showed that compound 17 administered at 20 mg/kg was characterized by moderate absorption and a total plasma concentration that exceeds the in vitro EC99 of 0.030 μM of 17 for approximately 12 h (Figure S7B). Analysis of blood samples from the mouse model on day 4 showed that mice treated with compound 17 at 20 mg/kg had an average 56% decrease in erythrocytic blood-stage parasitemia (Table 7 and Figure S7A). The data imply that further optimization of 17 is required to improve aqueous solubility, absorption, and plasma half-life to achieve greater efficacy in the mouse model.
Table 7. Evaluation of 17 in a Peter’s 4 Day P. berghei Mouse Modela.
| compound | 17 | CQ | vehicle |
|---|---|---|---|
| dose (mg/kg) | 20 | 10 | |
| % parasitemiab | 8.1 | 0.02 | 18.5 |
| % reduction in parasitemiac | 56.2 | 99.8 |
P. berghei ANKA parasites expressing GFP were injected into the tail vein to infect mice on day 0. Compound 17 was administered q.d. at 20 mg/kg by p.o. 2 h after infection (day 0) and then on days 1, 2, and 3. Parasitemia was measured by flow cytometry. Figure S7 shows data for individual mice. CQ = chloroquine.
Average % parasitemia for four mice on day 4.
Average % reduction in parasitemia versus the vehicle control for four mice on day 4.
Conclusions
We identified a hit compound with a quinolone scaffold from a high-throughput screen against asexual P. falciparum parasites that has structural similarities to known quinolone antimalarials but has a unique substitution pattern not previously described. Investigation of the SAR led to the removal of the pyridyl group from the oxadiazole, which significantly improved parasite activity and engendered the frontrunner analog WJM-228 (17), whereas the incorporation of nitrogen-linked hydrophobic groups in the 7-position maintained or slightly improved parasite activity. The SAR investigation of the quinolone hit scaffold revealed that the 3-oxadiazole motif was acting as an ester bioisostere. The 3-oxadiazole was seen as an adventitious replacement of the 3-carboxylate moiety seen in other decoquinate-like scaffolds (Figure 1) because it was responsible for significantly increasing metabolic stability, although the 3-oxadiazole contributed to its limited aqueous solubility. The introduction of polar functionality in the 7-position into the scaffold led to a decrease in activity, preventing improvement in aqueous solubility. Although the metabolism of the 3-oxadiazole scaffold was robust, improvement in solubility is required to further develop the scaffold. Aqueous solubility is an inherent issue with quinolone scaffolds, and other groups have applied prodrug strategies to correct this trait to improve in vivo efficacy, which is a future consideration with this quinolone scaffold. Collectively, the SAR and structural features uncovered in this SAR study were largely aligned with those of the decoquinate-like quinolone series, and this information could be useful in further optimization efforts to improve the overall physicochemical properties of antimalarial quinolone scaffolds.
The parasite reduction ratio assay showed that the quinolone hit class exhibited a modest rate of action that significantly differed from the rate observed with ATQ. We are unsure why the quinolone class displayed a phenotype that was different from other mitochondrial targeted antimalarials. One assumption is that the high compound concentration (10 times the parasite EC50) used in the PRR assay may have resulted in the inhibition of another non-mitochondrial target providing a PRR that is not usually associated with a mitochondrial targeted antimalarial. Forward genetic resistance studies with the quinolone derivative 41 uncovered a single SNP in the Qo site of cyt b. The Qo site of cyt b was confirmed as the molecular target by showing that our quinolone series was cross-resistant to ATQ resistant parasites with a Qo site mutation but not ELQ-300 (5) resistant parasites with a Qi site mutation. Conversely, we showed that ATQ was cross-resistant to 41 resistant parasites but not ELQ-300 (5), providing further evidence that the Qo site of cyt b is the target of our quinolone series. Cyt bc1 is essential for the function of the mitochondrial ETC and pyrimidine biosynthesis in asexual-stage parasites. We further confirmed that the quinolone class impacted the mitochondrial ETC in P. falciparum parasites by inhibition of cyt bc1 (complex III) in an OCR biosensor assay. Collectively, the data provided robust evidence that the quinolone series kills the P. falciparum parasite via inhibition of cyt b.
The quinolone class has pre-erythrocytic activity corroborating the activity of other mitochondrial targeted compounds, such as ATQ (3) and ELQ-300 (5). Analogs of the quinolone class also showed modest activity against male gametes consistent with the activity of ATQ (3) and ELQ-300 (5). It is uncertain whether slow-acting mitochondrial targeted compounds would be suitable as a transmission blocking treatment to control the spread of the disease in malaria endemic regions, but mitochondrial targeted antimalarials are used as a partner drug in combination prophylaxis and curative therapies. A seemingly inherent issue with mitochondrial targeted antimalarials is cross-resistance to drug-resistant strains. The quinolone class was not cross-resistant to numerous drug-resistant parasite strains but was cross-resistant to the ATQ (3) resistant clinical TM90-C2B strain. Decoquinate-like antimalarials including our quinolone series are readily susceptible to the TM90-C2B strain, limiting their development and the impetus for the development of the Qi site selective inhibitor ELQ-300 (5). A combination of substituents described here and elsewhere,9,15,16,22−25,38 appropriately positioned on the decoquinate-like quinolone framework, may provide an avenue to enhance solubility and overcome susceptibility to cyt b resistance. Correcting these attributes will be key for the continued development of the quinolone antimalarial chemotype.
Methods
Chemistry Methods
General Chemistry Methods
NMR spectra were recorded on either a Bruker Avance DRX 300 or Bruker Ascend 300. Chemical shifts are reported in ppm on the δ scale and referenced to the appropriate solvent peak. MeOD, DMSO-d6, D2O, and CDCl3 contain H2O. Chromatography was performed with silica gel 60 (particle size 0.040–0.063 μm) using an automated CombiFlash Rf Purification System. LCMS was recorded on an Agilent LCMS system composed of an Agilent G6120B Mass Detector, 1260 Infinity G1312B Binary pump, 1260 Infinity G1367E HiPALS autosampler, and 1260 Infinity G4212B Diode Array Detector (method B). Conditions for LCMS method A were as follows: column: Luna Omega 3 μm PS C18 100 Å, LC Column 50 × 2.1 mm at 20 °C, injection volume: 2 μL, gradient: 5–100% B over 3 min (solvent A: H2O 0.1% formic acid; solvent B: ACN 0.1% formic acid), flow rate: 1.5 mL/min, detection: 100–600 nm, and acquisition time: 4.3 min. Conditions for LCMS method B were as follows: column: Poroshell 120 EC-C18, 2.1 × 30 mm, 2.7 μm at 30 °C; injection volume: 2 μL; gradient: 5–100% B over 3 min (solvent A: H2O 0.1% formic acid; solvent B: ACN 0.1% formic acid); flow rate: 0.8 mL/min; detection: 254 nm; and acquisition time: 4.1 min. Unless otherwise noted, all compounds were found to be >95% pure by this method. HRMS was acquired through the Bio21 Mass Spectrometry and Proteomics Facility using a Thermo Scientific nano-LC Q Exactive Plus mass spectrometer. Compounds 1 and 15 were procured from commercial vendors and used without further purification.
Chemistry Procedures
1-Ethyl-6-fluoro-7-(4-methyl-1-piperidyl)-3-[5-(3-pyridyl)-1,3,4-oxadiazol-2-yl]quinolin-4-one (16)
25 (50 mg, 0.15 mmol) and pyridine-3-carbohydrazide (21 mg, 0.15 mmol) were heated at reflux in phosphorus oxychloride (1 mL) for 6 h. The reaction was then basified with saturated NaHCO3 and filtered. The crude precipitate was then purified by column chromatography eluting with 100% DCM to 5% MeOH/DCM to afford 16 as a solid (4.6 mg, 7.1%). 1H NMR (300 MHz, CDCl3): δ 9.42 (s, 1H), 8.76 (dd, J 4.9, 1.6 Hz, 1H), 8.59 (s, 1H), 8.46 (dt, J 7.9, 2.0 Hz, 1H), 8.11 (d, J 13.3 Hz, 1H), 7.45 (dd, J 8.0, 4.9 Hz, 1H), 6.80 (d, J 6.9 Hz, 1H), 4.28 (q, J 7.2 Hz, 2H), 3.67 (app d, J 11.9 Hz, 2H), 2.88–2.78 (m, 2H), 1.81 (app d, J 12.6 Hz, 2H), 1.70–1.50 (m, 4H), 1.46 (td, J 11.8, 8.1 Hz, 3H), 1.03 (d, J 6.3 Hz, 3H). LCMS m/z 434.2 [M + 1]. HRMS acquired: (M + H) 434.1989; C24H24FN5O2 requires (M + H), 434.1987.
General Method C
1-Ethyl-6-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)-7-(4-methyl-1-piperidyl)quinolin-4-one (17)
Anhydrous DMSO (5 mL) was added to a mixture of N′-hydroxyacetamidine (48 mg, 0.65 mmol) and 45 (250 mg, 0.63 mmol). After 1 h, NaOH (0.6 g, 15 mmol, pellet form) was added, and the mixture was heated to 60 °C. After a further 3 h, the reaction mixture was cooled to room temperature and then diluted in H2O (10 mL). The precipitate was collected by filtration and then washed with ice-cold EtOH (1 mL) and then Et2O (2 × 1 mL) to obtain the crude product. The product was further purified by recrystallization from pyridine (1 mL) to afford 17 as a solid (38 mg, 16%). 1H NMR (300 MHz, DMSO-d6) δ 8.90 (s, 1H), 7.83 (d, J 13.5 Hz, 1H), 7.07 (d, J 7.3 Hz, 1H), 4.48 (q, J 6.9 Hz, 2H), 3.62 (app d, J 12.0 Hz, 2H), 2.85 (t, J 11.8 Hz, 2H), 2.36 (s, 3H), 1.76 (app d, J 12.8 Hz, 2H), 1.66–1.49 (m, 1H), 1.45–1.25 (m, 5H), 0.97 (d, J 6.4 Hz, 3H). LCMS m/z 371.0 [M + 1]. HRMS acquired: (M + H) 371.1878; C20H23FN4O2 requires (M + H), 371.1878.
1-Ethyl-6-fluoro-7-(4-methyl-1-piperidyl)-3-(2-pyridyl)quinolin-4-one (18)
Pd(PPh3)4 (3.9 mg, 0.0034 mmol), 47 (25 mg, 0.068 mmol), and THF (3 mL) were added to a 5 mL pressure flask with a magnetic stir bar. The mixture was stirred for 0.5 h at 20 °C, resulting in a yellow-orange slurry. 2-Pyridylzinc bromide (0.5 M solution, 0.204 mL, 0.10 mmol) was then added by a syringe. The reaction mixture was then heated at reflux for 4 h. Another portion of 2-pyridylzinc bromide (0.5 M solution, 2.04 mL, 1.00 mmol) was then added, and the reaction was stirred at reflux for 2 days. The reaction was then cooled to 20 °C, and aqueous HCl (3.0 M; 5 mL) was added to the solution. The reaction was then made to reach pH 8 by the addition of aqueous 2 M NaOH. The crude product was then extracted with Et2O (20 mL), dried with anhydrous Na2SO4, filtered, and concentrated. The crude material was then purified by column chromatography eluting with 100% CyHex to 80% EtOAc/CyHex to afford the crude product. The crude material was additionally purified by reverse phase preparatory HPLC using a gradient of 95% H2O/ACN to 100% ACN to afford 18 as a solid (3.1 mg, 12%). 1H NMR (300 MHz, CDCl3): δ 8.80 (dt, J 8.2, 1.1 Hz, 1H), 8.73 (s, 1H), 8.56 (ddd, J 4.8, 1.9, 0.9 Hz, 1H), 8.15 (d, J 13.6 Hz, 1H), 7.73 (ddd, J 8.1, 7.4, 1.9 Hz, 1H), 7.16 (ddd, J 7.5, 4.8, 1.2 Hz, 1H), 6.78 (d, J 7.0 Hz, 1H), 4.28 (q, J 7.2 Hz, 2H), 3.64 (app d, J 12.1 Hz, 2H), 2.87–2.72 (m, 2H), 1.86–1.72 (m, 2H), 1.59–1.40 (m, 6H), 1.03 (d, J 6.1 Hz, 3H). LCMS m/z 366.4 [M + 1]. HRMS acquired: (M + H) 366.1978; C22H24FN3O requires (M + H), 366.1976.
1-Ethyl-6-fluoro-7-(4-methyl-1-piperidyl)-3-(3-pyridyl)quinolin-4-one (19)
47 (25 mg, 0.068 mmol), 3-pyridylboronic acid (17 mg, 0.14 mmol), and K2CO3 (28 mg, 0.20 mmol) were dissolved in a mixture of 1,4-dioxane (3 mL) and H2O (0.5 mL) and purged with nitrogen for 10 min. Pd(PPh3)4 (7.9 mg, 0.0068 mmol) was then added, and the reaction was stirred at reflux under nitrogen for 2 h. The reaction was then cooled to 24 °C and filtered through diatomaceous earth, which was washed with EtOAc (10 mL). The crude was then concentrated and then dissolved in EtOAc (15 mL), washed with saturated NaHCO3 (10 mL) and brine (10 mL), dried with anhydrous Na2SO4, filtered, and concentrated. The crude was then purified by column chromatography eluting with 100% DCM to 7% EtOAc/DCM to obtain 19 as a solid (23 mg, 93%). 1H NMR (300 MHz, CDCl3): δ 8.73 (d, J 2.2 Hz, 1H), 8.51 (d, J 4.8 Hz, 1H), 8.18 (d, J 8.0 Hz, 1H), 8.07 (d, J 13.5 Hz, 1H), 7.67 (s, 1H), 7.32 (dd, J 8.0, 4.8 Hz, 1H), 6.74 (d, J 7.0 Hz, 1H), 4.19 (t, J 7.2 Hz, 2H), 3.63 (app d, J 11.8 Hz, 2H), 2.78 (t, J 11.3 Hz, 2H), 1.84–1.73 (m, 2H), 1.58–1.40 (m, 6H), 1.02 (d, J 6.1 Hz, 3H). LCMS m/z 366 [M + 1]. HRMS acquired: (M + H) 366.1978; C22H24FN3O requires (M + H), 366.1976.
1-Ethyl-6-fluoro-7-(4-methyl-1-piperidyl)-3-(2-methylpyrazol-3-yl)quinolin-4-one (20)
47 (25 mg, 0.068 mmol), 1-methylpyrazole-5-boronic acid pinacol ester (21 mg, 0.10 mmol), and K2CO3 (28 mg, 0.20 mmol) were dissolved in a mixture of 1,4-dioxane (3 mL) and H2O (0.5 mL) and purged with N2 gas for 10 min. Pd(PPh3)4 (7.9 mg, 0.0068 mmol) was then added, and the reaction was stirred at reflux for 2 h. The reaction was then cooled to 24 °C and filtered through diatomaceous earth, which was then washed with EtOAc (10 mL). The crude was then concentrated and then dissolved in EtOAc (15 mL), washed with saturated NaHCO3 (10 mL) and brine (10 mL), dried with anhydrous Na2SO4, filtered, and concentrated. The crude was then purified by column chromatography eluting with 100% DCM to 10% EtOAc/DCM to obtain 20 as a solid (6.4 mg, 24%). 1H NMR (300 MHz, CDCl3): δ 8.07 (d, J 13.4 Hz, 1H), 7.60 (s, 1H), 7.48 (d, J 1.9 Hz, 1H), 6.77 (d, J 7.0 Hz, 1H), 6.19 (d, J 1.9 Hz, 1H), 4.18 (q, J 7.2 Hz, 2H), 3.84 (s, 3H), 3.65 (app d, J 11.7 Hz, 2H), 2.86–2.73 (m, 2H), 1.80 (app d, J 12.6 Hz, 2H), 1.59–1.39 (m, 6H), 1.03 (d, J 6.2 Hz, 3H). LCMS m/z 369.2 [M + 1]. HRMS acquired: (M + H) 369.2088; C21H25FN4O requires (M + H), 369.2085.
Ethyl 1-Ethyl-6-fluoro-7-(4-methyl-1-piperidyl)-4-oxo-quinoline-3-carboxylate (21)
H2SO4 (98%, 2 mL) was added to a suspension of 25 (166 mg, 0.50 mmol) in EtOH (15 mL), and the mixture was heated under reflux. After 18 h, the reaction mixture was cooled to room temperature, diluted in H2O (100 mL), and extracted with DCM (3 × 30 mL). The extracts were dried over MgSO4, filtered, and concentrated. The residue was purified by column chromatography eluting with 0–10% MeOH/DCM, and the resulting residue was then triturated with Et2O (2 mL) to afford 21 as a solid (72 mg, 40%). 1H NMR (300 MHz, DMSO-d6) δ 8.60 (s, 1H), 7.76 (d, J 13.6 Hz, 1H), 7.01 (d, J 7.3 Hz, 1H), 4.38 (q, J 7.2 Hz, 2H), 4.21 (q, J 7.1 Hz, 2H), 3.64–3.53 (m, 2H), 2.92–2.71 (m, 2H), 1.81–1.69 (m, 2H), 1.65–1.45 (m, 1H), 1.42–1.16 (m, 8H), 0.97 (d, J 6.4 Hz, 3H). LCMS m/z 361.0 [M + 1]. HRMS acquired: (M + H) 361.1921; C20H25FN2O3 requires (M + H), 361.1922.
General Method D
1-Ethyl-6-fluoro-7-(4-methyl-1-piperidyl)-4-oxo-quinoline-3-carboxamide (22)
Ammonia (25% aq, 0.10 mL, 1.3 mmol) was added to a solution of 25 (35 mg, 0.090 mmol) in DCM (5 mL) with vigorous stirring. After 30 min, the reaction mixture was concentrated, and the residue was triturated with H2O (2 × 2 mL) to afford 22 as a solid (22 mg, 63%) at 85% purity. 1H NMR (300 MHz, DMSO-d6) δ 9.30 (d, J 4.8 Hz, 1H), 8.77 (s, 1H), 7.86 (d, J 13.6 Hz, 1H), 7.44 (d, J 4.8 Hz, 1H), 7.08 (d, J 7.3 Hz, 1H), 4.48 (q, J 7.1 Hz, 2H), 3.69–3.53 (m, 2H), 2.94–2.76 (m, 2H), 1.83–1.70 (m, 2H), 1.66–1.50 (m, 1H), 1.44–1.24 (m, 5H), 0.98 (d, J 6.4 Hz, 3H). LCMS m/z 332.0 [M + 1]. HRMS acquired: (M + H) 332.1769; C18H22FN3O2 requires (M + H), 332.1769.
N,1-Diethyl-6-fluoro-7-(4-methyl-1-piperidyl)-4-oxo-quinoline-3-carboxamide (24)
General method D was followed using ethylamine (70% aq, 0.10 mL, 1.8 mmol) and 25 (33 mg, 0.085 mmol) to afford 24 as a solid (21 mg, 60%) at 87% purity. 1H NMR (300 MHz, DMSO-d6) δ 9.9 (t, 1H), 8.8 (s, 1H), 7.8 (d, J 13.6 Hz, 1H), 7.1 (d, J 7.3 Hz, 1H), 4.5 (q, J 7.2 Hz, 2H), 3.7–3.5 (m, 2H), 3.4–3.3 (m, 7H), 2.9–2.8 (m, 2H), 1.8 (d, J 12.7 Hz, 2H), 1.7–1.5 (m, 1H), 1.4–1.3 (m, 5H), 1.1 (t, J 7.2 Hz, 3H), 1.0 (d, J 6.4 Hz, 3H). LCMS m/z 360.0 [M + 1]. HRMS acquired: (M + H) 360.2082; C20H26FN3O2 requires (M + H), 360.2082.
N,N,1-Triethyl-6-fluoro-7-(4-methyl-1-piperidyl)-4-oxo-quinoline-3-carboxamide (24)
General method D was followed using diethylamine (0.10 mL, 0.09 mmol and 25 (35 mg, 0.090 mmol) to afford 24 as a solid (21 mg, 51%) at 85% purity. 1H NMR (300 MHz, DMSO-d6) δ 8.1 (s, 1H), 7.7 (d, J 13.5 Hz, 1H), 7.0 (d, J 7.5 Hz, 1H), 4.5–4.3 (m, 2H), 3.7–3.5 (m, 2H), 3.5–3.1 (m, 4H), 2.9–2.7 (m, 2H), 1.8 (d, J 12.8 Hz, 2H), 1.6 (s, 1H), 1.5–1.2 (m, 5H), 1.2–0.9 (m, 9H). LCMS m/z 388.0 [M + 1]. HRMS acquired: (M + H) 388.2394; C22H30FN3O2 requires (M + H), 388.2395.
General Method A
1-Ethyl-6-fluoro-7-(4-methyl-1-piperidyl)-4-oxo-quinoline-3-carboxylic Acid (25)
7-Chloro-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic acid (2.70 g, 10.0 mmol) was added to a suspension of 4-methylpiperidine (6.00 mL, 50.7 mmol) in DMSO (20 mL) in a pressure tube, and the mixture was heated to 120 °C. After 3 h, the reaction mixture was cooled to room temperature. The reaction mixture was then diluted in H2O (50 mL) and then cooled on ice, and the precipitate was collected by filtration. The filter cake was washed successively with H2O (3 × 10 mL), cold EtOH (10 mL), and Et2O (2 × 10 mL) to obtain 25 as a white solid (2.85 g, 86%). 1H NMR (300 MHz, DMSO-d6) δ 15.40 (s, 1H), 8.95 (s, 1H), 7.91 (d, J 13.5 Hz, 1H), 7.17 (d, J 7.4 Hz, 1H), 4.59 (d, J 7.2 Hz, 2H), 3.70 (d, J 12.3 Hz, 2H), 2.92 (s, 2H), 1.78 (d, J 13.0 Hz, 2H), 1.69–1.52 (m, 1H), 1.51–1.21 (m, 5H), 0.98 (d, J 6.5 Hz, 3H). LCMS m/z 333.2 [M + 1]. HRMS acquired: (M + H) 333.1609; C18H21FN2O3 requires (M + H), 333.1609.
1-Ethyl-6-fluoro-7-(4-methyl-1-piperidyl)-3-(morpholine-4-carbonyl)quinolin-4-one Formate (26)
25 (20 mg, 0.060 mmol), HATU (34 mg, 0.090 mmol), and morpholine (0.031 mL, 0.36 mmol) were stirred in DMF (2 mL) for 4 h. The reaction was concentrated in vacuo and dissolved in EtOAc (10 mL). The organic layers were successively washed with saturated NaHCO3 (10 mL) and brine (10 mL), dried with anhydrous Na2SO4, filtered, and concentrated. The crude material was then purified by reverse phase preparatory HPLC using a gradient of 95% H2O/ACN to 100% ACN to afford 26 as a solid (21.9 mg, 91%). 1H NMR (300 MHz, CDCl3): δ 8.02 (t, J 6.7 2H), 6.96 (br s, 1H), 4.20 (q, J 7.2 Hz, 2H), 3.90–3.35 (m, 10H), 2.88 (t, J 11.6 Hz, 2H), 1.86–1.74 (m, 2H), 1.67–1.40 (m, 6H), 1.02 (d, J 5.9 Hz, 3H). LCMS m/z 402.2 [M + 1]. HRMS acquired: (M + H) 402.2187; C22H28FN3O3 requires (M + H), 402.2187.
1-Ethyl-6-fluoro-3-(4-methylpiperazine-1-carbonyl)-7-(4-methyl-1-piperidyl)quinolin-4-one (27)
The procedure used for 26 was followed using 25 (69 mg, 0.18 mmol) and 1-methylpiperazine (0.080 mL, 0.72 mmol) to afford 27 as a solid (3.8 mg, 8%). 1H NMR (300 MHz, MeOD): δ 8.28 (s, 1H), 7.92 (d, J 13.6 Hz, 1H), 7.12 (d, J 7.2 Hz, 1H), 4.44 (q, J 7.2 Hz, 2H), 3.80–3.69 (m, 2H), 3.63–3.51 (m, 2H), 3.41–3.35 (m, 2H), 3.31–3.15 (m, 3H), 3.02–2.88 (m, 5H), 1.90–1.81 (m, 2H), 1.72–1.59 (m, 1H), 1.58–1.35 (m, 5H), 1.06 (d, J 6.4 Hz, 3H). LCMS m/z 415.2 [M + 1]. HRMS acquired: (M + H) 415.2504; C23H31FN4O2 requires (M + H), 415.2503.
1-Ethyl-6-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)-7-pyrrolidin-1-yl-quinolin-4-one (28)
General method C was followed using 49 (125 mg, 0.35 mmol) and N′-hydroxyacetamidine (51 mg, 0.69 mmol) to afford 28 as a solid (24 mg, 20%). 1H NMR (300 MHz, DMSO-d6): δ 8.84 (s, 1H), 7.78 (d, J 14.6 Hz, 1H), 6.63 (d, J 7.5 Hz, 1H), 4.44 (q, J 7.0 Hz, 2H), 3.61–3.53 (m, 4H), 2.37 (s, 3H), 2.0–1.94 (m, 4H), 1.40 (t, J 7.0 Hz, 3H). LCMS m/z 343.2 [M + 1]. HRMS acquired: (M + H) 343.1567; C18H19FN4O2 requires (M + H), 343.1565.
7-(Azepan-1-yl)-1-ethyl-6-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-4-one (29)
General method C was followed using 51 (47 mg, 0.12 mmol) and N′-hydroxyacetamidine (18 mg, 0.25 mmol) to afford 29 as a solid (28 mg, 62%). 1H NMR (300 MHz, CDCl3): δ 8.51 (s, 1H), 8.10 (d, J 14.9 Hz, 1H), 6.66 (s, 1H), 4.25 (q, J 7.2 Hz, 2H), 3.58 (td, J 6.2, 1.6 Hz, 4H), 2.47 (s, 3H), 1.98–1.88 (m, 4H), 1.75–1.65 (m, 4H), 1.59 (t, J 7.2 Hz, 3H). LCMS m/z 371.2 [M + 1]. HRMS acquired: (M + H) 371.1879; C20H23FN4O2 requires (M + H), 371.1878.
1-Ethyl-6-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)-7-(1-piperidyl)quinolin-4-one (30)
General method C was followed using 53 (190 mg, 0.50 mmol) and N′-hydroxyacetamidine (37 mg, 0.50 mmol) to afford 30 as a solid (84 mg, 47%). 1H NMR (300 MHz, DMSO-d6) δ 8.88 (s, 1H), 7.82 (d, J 13.3 Hz, 1H), 7.06 (d, J 7.3 Hz, 1H), 4.47 (q, J 7.1 Hz, 2H), 3.25–3.14 (m, 4H), 2.36 (s, 3H), 1.81–1.52 (m, 6H), 1.40 (t, J 7.0 Hz, 3H). LCMS m/z 357.0 [M + 1]. HRMS acquired: (M + H) 357.1721; C19H21FN4O2 requires (M + H), 357.1721.
1-Ethyl-6-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)-7-morpholino-quinolin-4-one (31)
General method C was followed using 55 (95 mg, 0.25 mmol) and N′-hydroxyacetamidine (19 mg, 0.26 mmol) to afford 31 as a solid (15 mg, 17%). 1H NMR (300 MHz, DMSO-d6) δ 8.93 (s, 1H), 7.87 (d, J 13.5 Hz, 1H), 7.11 (d, J 7.2 Hz, 1H), 4.50 (d, J 6.8 Hz, 2H), 3.88–3.70 (m, 4H), 3.29–3.16 (m, 4H), 2.37 (s, 3H), 1.41 (t, J 6.9 Hz, 3H). LCMS m/z 358.8 [M + 1]. HRMS acquired: (M + H) 359.1514; C18H19FN4O3 requires (M + H), 359.1514.
1-Ethyl-6-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)-7-(4-methylpiperazin-1-yl)quinolin-4-one (32)
General method C was followed using 57 (100 mg, 0.255 mmol) and N′-hydroxyacetamidine (19 mg, 0.26 mmol) to afford 32 as a solid (45 mg, 47%). 1H NMR (300 MHz, DMSO-d6) δ 8.90 (s, 1H), 7.84 (d, J 13.6 Hz, 1H), 7.08 (d, J 7.2 Hz, 1H), 4.49 (q, J 7.1 Hz, 2H), 3.29–3.20 (m, 4H), 2.57–2.48 (m, 4H), 2.36 (s, 3H), 2.25 (s, 3H), 1.40 (t, J 7.0 Hz, 3H). LCMS m/z 372.0 [M + 1]. HRMS acquired: (M + H) 372.1830; C19H22FN5O2 requires (M + H), 372.1830.
7-(4-Acetylpiperazin-1-yl)-1-ethyl-6-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-4-one (33)
General method C was followed using 59 (65 mg, 0.16 mmol) and N′-hydroxyacetamidine (12 mg, 0.16 mmol) to afford 33 as a solid (38 mg, 61%). 1H NMR (300 MHz, CDCl3): δ 8.54 (s, 1H), 8.17 (d, J 13.0 Hz, 1H), 6.78 (d, J 6.8 Hz, 1H), 4.26 (q, J 7.3 Hz, 2H), 3.86 (t, J 5.1 Hz, 2H), 3.70 (t, J 5.1 Hz, 2H), 3.25 (dt, J 20.6, 5.1 Hz, 4H), 2.46 (s, 3H), 2.17 (s, 3H), 1.62–1.54 (m, 3H). LCMS m/z 400.2 [M + 1]. HRMS acquired: (M + H) 400.1783; C20H22FN5O3 requires (M + H), 400.1779.
1-Ethyl-6-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)-7-(3-methyl-1-piperidyl)quinolin-4-one (34)
General method C was followed using 63 (32 mg, 0.082 mmol) and N′-hydroxyacetamidine (12 mg, 0.16 mmol) to afford 34 as a solid (1.4 mg, 5%). 1H NMR (300 MHz, MeOD): δ 8.95 (s, 1H), 7.99 (d, J 13.5 Hz, 1H), 7.13 (d, J 7.1 Hz, 1H), 4.53 (q, J 7.3 Hz, 2H), 3.72–3.63 (m, 2H), 3.01–3.04 (m, 2H), 2.65–2.55 (m, 1H), 2.44 (s, 3H), 1.97–1.79 (m, 4H), 1.58 (t, J 7.2 Hz, 2H), 1.29–1.13 (m, 2H), 1.03 (d, J 6.5 Hz, 3H). LCMS m/z 371.2 [M + 1]. HRMS acquired: (M + H) 371.1880; C20H23FN4O2 requires (M + H), 371.1878.
7-(4,4-Dimethyl-1-piperidyl)-1-ethyl-6-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-4-one (35)
General method C was followed using 66 (50 mg, 0.12 mmol) and N′-hydroxyacetamidine (18 mg, 0.25 mmol) to afford 35 as a solid (13 mg, 27%). 1H NMR (300 MHz, CDCl3): δ 8.53 (s, 1H), 8.13 (d, J 13.4 Hz, 1H), 6.79 (d, J 6.9 Hz, 1H), 4.26 (q, J 7.2 Hz, 2H), 3.27–3.20 (m, 4H), 2.46 (s, 3H), 1.63–1.57 (m, 7H), 1.04 (s, 6H). LCMS m/z 385.4 [M + 1]. HRMS acquired: (M + H) 385.2033; C21H25FN4O2 requires (M + H), 385.2034.
7-(6-Azaspiro[2.5]octan-6-yl)-1-ethyl-6-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-4-one (36)
General method C was followed using 69 (52 mg, 0.13 mmol) and N′-hydroxyacetamidine (14 mg, 0.19 mmol) to afford 36 as a solid (18 mg, 36%). 1H NMR (300 MHz, CDCl3): δ 8.48 (s, 1H), 8.07 (d, J 13.3 Hz, 1H), 6.78 (d, J 6.9 Hz, 1H), 4.25 (q, J 7.3 Hz, 2H), 3.28 (t, J 5.4 Hz, 4H), 2.45 (s, 3H), 1.65–1.52 (m, Hz, 7H), 0.41 (s, 4H). LCMS m/z 383.2 [M + 1]. HRMS acquired: (M + H) 383.1879; C21H23FN4O2 requires (M + H), 383.1878.
7-(4,4-Difluoro-1-piperidyl)-1-ethyl-6-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-4-one (37)
General method C was followed using 72 (42 mg, 0.10 mmol) and N′-hydroxyacetamidine (15 mg, 0.20 mmol) to afford 37 as a solid (2.3 mg, 6%). 1H NMR (300 MHz, MeOD): δ 8.92 (s, 1H), 8.03 (d, J 13.3 Hz, 1H), 7.21 (d, J 7.2 Hz, 1H), 4.52 (q, J 7.2 Hz, 2H), 3.49 (t, J 5.8 Hz, 4H), 2.45 (s, 3H), 2.24 (m, 4H), 1.58 (t, J 7.1 Hz, 3H). LCMS m/z 393.4 [M + 1]. HRMS acquired: (M + H) 393.1535; C19H19F3N4O2 requires (M + H), 393.1533.
1-Ethyl-6-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)-7-(4-propyl-1-piperidyl)quinolin-4-one (38)
General method C was followed using 74 (130 mg, 0.31 mmol) and N′-hydroxyacetamidine (46 mg, 0.62 mmol) to afford 38 as a solid (52 mg, 42%). 1H NMR (300 MHz, DMSO-d6) δ 8.92 (s, 1H), 7.84 (d, J 13.5 Hz, 1H), 7.08 (d, J 7.3 Hz, 1H), 4.49 (q, J 7.2 Hz, 2H), 3.70–3.61 (m, 2H), 2.85 (t, J 11.8 Hz, 2H), 2.37 (s, 3H), 1.85–1.77 (m, 2H), 1.45–1.25 (m, 10H), 0.90 (t, J 7.0 Hz, 3H). LCMS m/z 399.4 [M + 1]. HRMS acquired: (M + H) 399.2192; C22H27FN4O2 requires (M + H), 399.2191.
1-Ethyl-6-fluoro-7-(4-isopropyl-1-piperidyl)-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-4-one (39)
General method C was followed using 76 (125 mg, 0.30 mmol) and N′-hydroxyacetamidine (44 mg, 0.60 mmol) to afford 39 as a solid (44 mg, 33%). 1H NMR (300 MHz, DMSO-d6) δ 8.92 (s, 1H), 7.85 (d, J 13.5 Hz, 1H), 7.09 (d, J 7.3 Hz, 1H), 4.49 (d, J 7.2 Hz, 2H), 3.70 (app d, J 11.9 Hz, 2H), 2.82 (t, J 11.7 Hz, 2H), 2.38 (s, 3H), 1.84–1.75 (m, 2H), 1.53–1.31 (m, 7H), 0.91 (d, J 6.7 Hz, 6H). LCMS m/z 399.2 [M + 1]. HRMS acquired: (M + H) 399.2192; C22H27FN4O2 requires (M + H), 399.2191.
1-Ethyl-6-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)-7-(4-phenyl-1-piperidyl)quinolin-4-one (40)
General method C was followed using 78 (62 mg, 0.14 mmol) and N′-hydroxyacetamidine (10 mg, 0.14 mmol) to afford 40 as a solid (36 mg, 60%). 1H NMR (300 MHz, CDCl3): δ 8.57 (s, 1H), 8.19 (d, J 13.2 Hz, 1H), 7.43–7.32 (m, 3H), 7.29–7.21 (m, 2H), 6.86 (d, J 6.9 Hz, 1H), 4.30 (q, J 7.2 Hz, 2H), 3.84 (app d, J 11.7 Hz, 2H), 2.99 (t, J 13.5 Hz, 2H), 2.81–2.71 (m, 1H), 2.49 (s, 3H), 2.13–2.02 (m, 4H), 1.64 (d, J 7.2 Hz, 3H). LCMS m/z 433.2 [M + 1]. HRMS acquired: (M + H) 433.2039; C25H25FN4O2 requires (M + H), 433.2034.
7-(4-Benzyl-1-piperidyl)-1-ethyl-6-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-4-one (41)
General method C was followed using 80 (117 mg, 0.25 mmol) and N′-hydroxyacetamidine (37 mg, 0.50 mmol) to afford 41 as a solid (54 mg, 48%). 1H NMR (300 MHz, DMSO-d6) δ 8.90 (s, 1H), 7.82 (d, J 13.5 Hz, 1H), 7.64–7.54 (m, 1H), 7.33–7.24 (m, 2H), 7.21 (d, J 7.2 Hz, 2H), 7.06 (d, J 7.2 Hz, 1H), 4.46 (q, J 7.0 Hz, 2H), 3.63 (app d, J 12.2 Hz, 2H), 2.81 (t, J 12.0 Hz, 2H), 2.59 (app d, J 6.7 Hz, 2H), 2.36 (s, 3H), 1.71 (d, J 12.6 Hz, 3H), 1.48–1.31 (t, J 7.0 Hz, 5H). LCMS m/z 447.2 [M + 1]. HRMS acquired: (M + H) 447.2194; C26H27FN4O2 requires (M + H), 447.2191.
1-Ethyl-6-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)-7-[4-(morpholinomethyl)-1-piperidyl]quinolin-4-one (42)
General method C was followed using 83 (37 mg, 0.078 mmol) and N′-hydroxyacetamidine (12 mg, 0.16 mmol) to afford 42 as a solid (6.9 mg, 19%). 1H NMR (300 MHz, DMSO-d6) δ 8.92 (s, 1H), 7.84 (d, J 13.5 Hz, 1H), 7.08 (d, J 7.3 Hz, 1H), 4.49 (q, J 7.0 Hz, 2H), 3.74–3.54 (m, 6H), 2.95–2.82 (m, 2H), 2.44–2.29 (m, 7H), 2.20 (d, J 7.0 Hz, 2H), 1.91–1.70 (m, 3H), 1.41 (t, J 7.0 Hz, 3H), 1.31 (d, J 12.4 Hz, 2H). LCMS m/z 456.4 [M + 1]. HRMS acquired: (M + H) 456.2407; C24H30FN5O3 requires (M + H), 456.2405.
General Method E
Ethyl 7-(4-Benzyl-1-piperidyl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylate (43)
A mixture of 60 (110 mg, 0.37 mmol), 3-methylpiperidine 4-benzylpiperidine (0.13 mL, 0.74 mmol), Cs2CO3 (361 mg, 1.10 mmol), rac-BINAP (23 mg, 0.037), and Pd(OAc)2 (4.1 mg, 0.019 mmol) in DMF (1 mL) was combined in a pressure tube and purged with nitrogen for 5 min. The reaction mixture was heated to 115 °C for 2 h under a nitrogen atmosphere. The reaction was then concentrated and dry loaded onto silica. The crude material was then purified by column chromatography eluting with 100% CyHex to 35% EtOAc to afford 43 as a solid (138 mg, 85%). 1H NMR (300 MHz, CDCl3): δ 8.47 (s, 1H), 8.10 (d, J 13.4 Hz, 1H), 7.36–7.30 (m, 2H), 7.27–7.16 (m, 4H), 4.42 (q, J 7.1 Hz, 2H), 4.24–4.20 (m, 2H), 3.76–3.58 (m, 2H), 2.81 (t, J 11.8 Hz, 2H), 2.65 (d, J 6.5 Hz, 2H), 1.90–1.68 (m, 4H), 1.60–1.52 (m, 4H), 1.43 (t, J 7.1 Hz, 3H). LCMS m/z 437.0 [M + 1]. HRMS acquired: (M + H) 437.2238; C26H29FN2O3 requires (M + H), 437.2235.
Ethyl 1-Ethyl-6-fluoro-4-oxo-7-(4-propyl-1-piperidyl)quinoline-3-carboxylate (44)
General method D was followed using 74 (8.0 mg, 0.19 mmol) and EtOH (0.5 mL, 0.009 mol) to afford 44 as a solid (2.2 mg, 30%). 1H NMR (300 MHz, CDCl3): δ 8.42 (s, 1H), 8.08 (d, J 13.4 Hz, 1H), 6.74 (d, J 6.9 Hz, 1H), 4.39 (q, J 7.1 Hz, 2H), 4.19 (q, J 7.2 Hz, 2H), 3.64 (app d, J 11.7 Hz, 2H), 2.78 (t, J 11.0 Hz, 2H), 1.83 (app d, J 10.1 Hz, 2H), 1.55–1.26 (m, 13H), 0.92 (t, J 6.9 Hz, 3H). LCMS m/z 389.2 [M + 1]. HRMS acquired: (M + H) 389.2237; C22H29FN2O3 requires (M + H), 389.2235.
General Method B
Methoxycarbonyl 1-Ethyl-6-fluoro-7-(4-methyl-1-piperidyl)-4-oxo-quinoline-3-carboxylate (45)
Triethylamine (0.943 mL, 6.77 mmol) was added to a suspension of 25 (1.50 g, 4.51 mmol) in DCM (50 mL) followed by methyl chloroformate (0.453 mL, 5.87 mmol). After 1 h, the reaction mixture was washed with KHSO4 (5%, 50 mL) and then NaHCO3 (5%, 50 mL), dried over MgSO4, filtered, and concentrated to afford 17 as a solid (230 mg, 98%). 1H NMR (300 MHz, CDCl3): δ 8.46 (s, 1H), 7.99 (d, J 13.4 Hz, 1H), 6.74 (d, J 6.9 Hz, 1H), 4.18–4.28 (m, 2H), 3.95 (s, 3H), 3.64 (d, J 12.3 Hz, 2H), 2.75–2.88 (m, 2H), 1.74–1.84 (m, 2H), 1.56 (t, J 7.2 Hz, 3H), 1.39–1.52 (m, 3H), δ 1.02 (d, J 6.3 Hz, 3H). LCMS m/z 391.2 [M + 1].
1-Ethyl-6-fluoro-7-(4-methyl-1-piperidyl)quinolin-4-one (46)
25 (300 mg, 0.90 mmol) in concentrated HCl (5 mL) was stirred for 7 days. The reaction was then concentrated in vacuo, dissolved in EtOAc (20 mL), and washed with saturated NaHCO3 (20 mL) and H2O (20 mL). The organics were then dried with anhydrous Na2SO4, filtered, and concentrated to obtain 46 as a white solid (240 mg, 92%). 1H NMR (300 MHz, CDCl3): δ 8.02 (d, J 13.4 Hz, 1H), 7.44 (d, J 7.7 Hz, 1H), 6.73 (d, J 7.0 Hz, 1H), 6.19 (d, J 7.7 Hz, 1H), 4.12 (q, J 7.2 Hz, 2H), 3.62 (d, J 11.8 Hz, 2H), 2.77 (t, J 12.0 Hz, 2H), 1.79 (d, J 12.3 Hz, 2H), 1.56–1.37 (m, 6H), 1.02 (d, J 6.2 Hz, 3H). LCMS m/z 289.2 [M + 1].
3-Bromo-1-ethyl-6-fluoro-7-(4-methyl-1-piperidyl)quinolin-4-one (47)
46 (240 mg, 0.81 mmol) in acetic acid (3 mL) was cooled to 10 °C. To this was added molecular bromine (0.045 mL, 0.87 mmol) in portions (0.015 mL, 3 times over 20 min), and the reaction was warmed to 20 °C and then stirred for 16 h. The reaction mixture was then concentrated and dissolved in EtOAc (20 mL), which was then washed with saturated NaHCO3 (20 mL) and brine (20 mL), dried with anhydrous Na2SO4, filtered, and concentrated. The crude was then purified by column chromatography eluting with 100% DCM to 20% EtOAc/DCM to obtain 47 as a solid (206 mg, 69%). 1H NMR (300 MHz, CDCl3): δ 8.05 (d, J 13.4 Hz, 1H), 7.88 (s, 1H), 6.70 (d, J 7.0 Hz, 1H), 4.15 (q, J 7.2 Hz, 2H), 3.64 (d, J 12.3 Hz, 2H), 2.85–2.70 (m, 2 H), 1.86–1.74 (m, 2 H), 1.57–1.36 (m, 6 H), 1.03 (d, J 6.2 Hz, 3 H). LCMS m/z 367 [M + 1].
1-Ethyl-6-fluoro-4-oxo-7-pyrrolidin-1-yl-quinoline-3-carboxylic Acid (48)
General method A was followed using 7-chloro-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic acid (200 mg, 0.742 mmol) and pyrrolidine (0.248 mL, 2.97 mmol) to afford 48 as a solid (113 mg, 50%). 1H NMR (300 MHz, DMSO-d6): δ 8.86 (s, 1H), 7.83 (d, J 14.5 Hz, 1 H), 6.69 (d, J 7.1 Hz, 1H), 4.62–4.36 (m, 2H), 3.68–3.59 (m, 4H), 2.04–1.88 (m, 4 H), 1.41 (t, J 6.8 Hz, 3H). LCMS m/z 305.2 [M + 1].
Methoxycarbonyl 1-Ethyl-6-fluoro-4-oxo-7-pyrrolidin-1-yl-quinoline-3-carboxylate (49)
General method B was followed using 48 (113 mg, 0.371 mmol) and methyl chloroformate (0.037 mL, 0.48 mmol) to afford 49 as a solid (125 mg, 93%). 1H NMR (300 MHz, CDCl3): δ 8.54–8.46 (m, 1H), 8.00 (dd, J 14.3, 4.3 Hz, 1H), 6.38 (dd, J 7.3, 3.7 Hz, 1H), 4.24 (dd, J 14.1, 7.1 Hz, 2H), 3.96 (s, 3H), 3.68–3.56 (m, 4H), 2.10–2.01 (m, 4H), 1.57 (td, J 7.3, 2.3 Hz, 3H). LCMS m/z 363.2 [M + 1].
7-(Azepan-1-yl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic Acid (50)
General method A was followed using 7-chloro-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic acid (200 mg, 0.74 mmol) and azepane (0.334 mL, 3.0 mmol) to afford 50 as a solid (41 mg, 17%). 1H NMR (300 MHz, CDCl3): δ 8.57 (s, 1H), 7.91 (d, J 14.8 Hz, 1H), 6.62 (d, J 7.3 Hz, 1H), 4.26 (q, J 7.3 Hz, 2H), 3.54–3.64 (m, 4H), 1.96–1.86 (m, 4H), 1.61–1.73 (m, 4H), 1.56 (t, J 7.3 Hz, 3H). LCMS m/z 333.2 [M + 1].
Methoxycarbonyl 7-(Azepan-1-yl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylate (51)
General method B was followed using 50 (40 mg, 0.12 mmol) and methyl chloroformate (0.012 mL, 0.16 mmol) to afford 51 as a solid (47 mg, 100%). 1H NMR (300 MHz, CDCl3): δ 8.35 (s, 1H), 7.83 (d, J 15.1 Hz, 1H), 6.47 (d, J 7.4 Hz, 1H), 4.15 (q, J 7.1 Hz, 2H), 3.92 (s, 3H), 3.60–3.42 (m, 4H), 1.96–1.79 (m, 4H), 1.61 (ap, 4H), 1.50 (t, J 7.1 Hz, 3H). LCMS m/z 391.2 [M + 1].
1-Ethyl-6-fluoro-4-oxo-7-(1-piperidyl)quinoline-3-carboxylic Acid (52)
General method A was followed using 7-chloro-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic acid (2.70 g, 10.0 mmol) and piperidine (5.00 mL, 50.6 mmol) to afford 52 as a solid (1.88 g, 59%). 1H NMR (300 MHz, DMSO-d6) δ 8.92 (s, 1H), 7.87 (d, J 13.4 Hz, 1H), 7.14 (d, J 7.3 Hz, 1H), 4.57 (q, J 7.1 Hz, 2H), 3.31–3.17 (m, 4H), 1.81–1.55 (m, 6H), 1.41 (t, J 7.0 Hz, 4H). LCMS m/z 319.0 [M + 1].
Methoxycarbonyl 1-Ethyl-6-fluoro-4-oxo-7-(1-piperidyl)quinoline-3-carboxylate (53)
General method B was followed using 52 (1.59 g, 5.00 mmol) and methyl chloroformate (0.50 mL, 6.5 mmol) to afford 53 as a solid (1.43 g, 76%). 1H NMR (300 MHz, CDCl3) δ 8.42 (s, 1H), 7.92 (d, J 13.3 Hz, 1H), 6.74 (d, J 6.9 Hz, 1H), 4.21 (q, J 7.2 Hz, 2H), 3.93 (s, 3H), 3.20 (t, J 5.3 Hz, 4H), 1.85–1.70 (m, 4H), 1.69–1.59 (m, 2H), 1.53 (t, J 7.2 Hz, 3H). LCMS m/z 376.8 [M + 1].
1-Ethyl-6-fluoro-7-morpholino-4-oxo-quinoline-3-carboxylic Acid (54)
General method A was followed using 7-chloro-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic acid (2.70 g, 10.0 mmol) and morpholine (4.50 mL, 51.4 mmol) to 54 as a solid (2.41 g, 75%). 1H NMR (300 MHz, DMSO-d6) δ 8.95 (s, 1H), 7.91 (d, J 13.5 Hz, 1H), 7.18 (d, J 7.3 Hz, 1H), 4.59 (q, J 7.1 Hz, 2H), 3.88–3.71 (m, 4H), 3.33–3.26 (m, 4H), 1.42 (t, J 7.1 Hz, 3H). LCMS m/z 320.8 [M + 1].
Methoxycarbonyl 1-Ethyl-6-fluoro-7-morpholino-4-oxo-quinoline-3-carboxylate (55)
General method B was followed using 54 (1.60 mg, 5.00 mmol) and methyl chloroformate (0.50 mL, 6.5 mmol) to afford 55 as a solid (1.41 g, 75%). 1H NMR (300 MHz, CDCl3) δ 8.48 (s, 1H), 8.03 (d, J 13.3 Hz, 1H), 6.75 (d, J 6.82 Hz, 1H), 4.25 (q, J 7.3 Hz, 2H), 3.85–3.97 (m, 7H), 3.23–3.29 (m, 4H), 1.58 (t, J 7.2 Hz, 3H). LCMS m/z 379.2 [M + 1].
1-Ethyl-6-fluoro-7-(4-methylpiperazin-1-yl)-4-oxo-quinoline-3-carboxylic Acid (55)
General method A was followed using 7-chloro-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic acid (2.70 mg, 10.0 mmol) and 1-methylpiperazine (5.50 mL, 49.6 mmol) to give 55 as a solid (2.88 g, 87%). 1H NMR (300 MHz, DMSO-d6 with 20% D2O) δ 8.80 (s, 1H), 7.89 (d, J 13.4 Hz, 1H), 7.11 (d, J 7.3 Hz, 1H), 4.47 (d, J 7.3 Hz, 2H) 3.29 (br s, 4H), 2.23 (s, 3 H), 1.41 (t, J 7.1 Hz, 3 H) (four piperazine protons obscured by DMSO peak at 2.50 ppm). LCMS m/z 334.0 [M + 1].
Methoxycarbonyl 1-Ethyl-6-fluoro-7-(4-methylpiperazin-1-yl)-4-oxo-quinoline-3-carboxylate (56)
General method B was followed using 55 (1.67 g, 5.00 mmol) and methyl chloroformate (0.50 mL, 6.5 mmol) to afford 56 as a solid (1.79 g, 91%). 1H NMR (300 MHz, CDCl3) δ 8.45 (s, 1H), 7.96 (d, J 13.3 Hz, 1H), 6.73 (d, J 6.9 Hz, 1H), 4.23 (q, J 7.2 Hz, 2H), 3.94 (s, 3H), 3.39–3.24 (m, 4H), 2.79–2.63 (m, 4H), 2.43 (s, 3H), 1.56 (t, J 7.2 Hz, 3H). LCMS molecular ion not observed.
7-(4-Acetylpiperazin-1-yl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic Acid (58)
General method A was followed using 7-chloro-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic acid (150 mg, 0.56 mmol) and 1-acetylpiperazine (285 mg, 2.23 mmol) to afford 58 as a solid (129 mg, 64%). 1H NMR (300 MHz, DMSO-d6) δ 8.97 (s, 1H), 7.96 (d, J 13.2 Hz, 1H), 7.21 (d, J 7.3 Hz, 1 H), 4.60 (d, J 7.2 Hz, 2H), 3.62–3.71 (m, 4H), 3.29–3.23 (m, 4H), 2.07 (s, 3H), 1.42 (t, J 7.1 Hz, 3H). LCMS m/z 362.2 [M + 1].
Methoxycarbonyl 7-(4-Acetylpiperazin-1-yl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylate (59)
General method B was followed using 58 (60 mg, 0.17 mmol) and methyl chloroformate (0.017 mL, 0.13 mmol) to afford 59 as a solid (32 mg, 46%). 1H NMR (300 MHz, CDCl3): δ 8.46 (s, 1 H), 8.02 (d, J 13.1 Hz, 1H), 6.76 (d, J 6.8 Hz, 1H), 4.24 (q, J 7.2 Hz, 2H), 3.95 (s, 3H), 3.88–3.82 (m, 2H), 3.75–3.67 (m, 2H), 3.20–3.32 (m, 4H), 2.17 (s, 3H) 1.60–1.53 (m, 3H). LCMS m/z 420.2 [M + 1].
Ethyl 7-Chloro-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylate (60)
7-Chloro-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic acid (1.3 g, 4.8 mmol) and iodoethane (0.78 mL, 9.6 mmol) were dissolved in DMF (10 mL) and stirred at 70 °C in a sealed tube for 24 h. The reaction was then filtered through diatomaceous earth and concentrated. The crude material was then purified by column chromatography eluting with 100% CyHex to 80% EtOAc/CyHex to afford 60 as a solid (1.2 g, 84%). 1H NMR (300 MHz, CDCl3): δ 8.59 (br s, 1H), 8.29 (d, J 9.0 Hz, 1H), 7.57 (d, J 5.6 Hz, 1H), 4.43 (q, J 7.1 Hz, 2H), 4.28 (q, J 7.2 Hz, 2H), 1.58 (t, J 7.3 Hz, 3H), 1.43 (t, J 7.1 Hz, 3H). LCMS m/z 298.2 [M + 1].
Ethyl 1-Ethyl-6-fluoro-7-(3-methyl-1-piperidyl)-4-oxo-quinoline-3-carboxylate (61)
General method E was followed using 60 (125 mg, 0.42 mmol) and 3-methylpiperidine (0.074 mL, 0.62 mmol) to afford 61 as a solid (32 mg, 21%). 1H NMR (300 MHz, CDCl3): δ 8.44 (s, 1H), 8.10 (d, J 13.4 Hz, 1H), 6.75 (d, J 6.9 Hz, 1H), 4.47–4.37, (m, 2H), 4.22 (q, J 7.2 Hz, 2H), 3.66–3.48 (m, 2H), 2.85–2.70 (m, 1H), 2.51–2.42 (m, 1H), 2.01–1.07 (m, 4H), 1.60–1.50 (m, 3H), 1.48–1.35 (m, 3H), 1.20–1.08 (m, 1H), 1.00 (d, J 6.4 Hz, 3H). LCMS m/z 361.2 [M + 1].
General Method F
1-Ethyl-6-fluoro-7-(3-methyl-1-piperidyl)-4-oxo-quinoline-3-carboxylic Acid (62)
61 (32 mg, 0.089 mmol) and LiOH (15 mg, 0.36 mmol) were stirred in a mixture of THF/H2O (1:1, 2 mL) for 16 h at 20 °C. The reaction was concentrated to remove THF and then acidified to pH 5 using a 5% citric acid solution. The aqueous layer was washed with EtOAc (3 × 7 mL). The organics were combined, washed with brine (15 mL), dried with anhydrous Na2SO4, filtered, and concentrated to obtain 62 as a solid (29 mg, 98%). 1H NMR (300 MHz, CDCl3): δ 8.67 (s, 1H) 8.05 (d, J 13.2 Hz, 1 H) 6.90–6.73 (m, 1H), 4.32 (q, J 7.27 Hz, 2H), 3.69–3.56 (m, 1H), 2.89–2.79 (m, 1H), 2.60–2.46 (m, 1H), 1.97–1.76 (m, 4H), 1.63–1.55 (m, 4H), 1.07–0.86 (m, 4H). LCMS m/z 333.4 [M + 1].
Methoxycarbonyl 1-Ethyl-6-fluoro-7-(3-methyl-1-piperidyl)-4-oxo-quinoline-3-carboxylate (63)
General method B was followed using 62 (29 mg, 0.088 mmol) and methyl chloroformate (0.013 mL, 0.17 mmol) to afford 63 as an oil (32 mg, 94%). 1H NMR (300 MHz, CDCl3): δ 8.41 (s, 1H), 7.97 (d, J 13.28 Hz, 1H), 6.72 (d, J 3.7 Hz, 1 H), 4.21–4.11 (m, 2H), 3.88 (s, 1H), 3.46 (d, J 11.9 Hz, 2H), 2.48–2.35 (m, 1H), 1.84–1.71 (m, 4H), 1.51–1.39 (m, 4H), 1.14–0.99 (m, 3H), 0.91 (d, J 6.4 Hz, 3H). LCMS m/z 391.4 [M + 1].
Ethyl 7-(4,4-Dimethyl-1-piperidyl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylate (64)
General method E was followed using 60 (100 mg, 0.34 mmol) and 4,4-dimethylpiperidine HCl (75 mg, 0.50 mmol) to afford 64 as an oil (56 mg, 45%). 1H NMR (300 MHz, CDCl3): δ 8.52 (s, 1H), 8.11 (d, J 13.5 Hz, 1H), 6.80 (d, J 3.5 Hz, 1H), 4.41 (q, J 7.1 Hz, 2H), 4.16–4.34 (m, 2H), 3.24–3.33 (m, 4H), 1.71–1.63 (m, 4H), 1.57 (t, J 7.3 Hz, 3H), 1.43 (t, J 7.1 Hz, 3H), 1.06 (s, 6H). LCMS m/z 375.2 [M + 1].
7-(4,4-Dimethyl-1-piperidyl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic Acid (65)
General method F was followed using 64 (56 mg, 0.15 mmol) to afford 65 as a solid (43 mg, 84%). 1H NMR (300 MHz, CDCl3): δ 8.65–8.55 (m, 1 H), 8.04–7.90 (m, 1H), 7.40–7.21 (m, 1H), 4.26 (q, J 7.3 Hz, 2H), 3.35–3.15 (m, 4H), 1.62–1.42 (m, 8H), 0.99 (s, 6H). LCMS m/z 347.2 [M + 1].
Methoxycarbonyl 7-(4,4-Dimethyl-1-piperidyl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylate (66)
General method B was followed using 65 (43 mg, 0.12 mmol) and methyl chloroformate (0.019 mL, 0.25 mmol) to afford 66 as an oil (50 mg, 99%). 1H NMR (300 MHz, CDCl3): δ 8.48 (s, 1H), 8.03 (d, J 13.4 Hz, 1H), 6.77 (d, J 6.9 Hz, 1H), 4.24 (q, J 7.2 Hz, 2H), 3.96 (s, 3H), 3.37–3.03 (m, 4H), 1.62–1.57 (m, 7H), 1.04 (s, 6H). LCMS m/z 405.4 [M + 1].
Ethyl 7-(6-Azaspiro[2.5]octan-6-yl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylate (67)
General method E was followed using 60 (75 mg, 0.25 mmol) and 6-azaspiro[2.5]octane HCl (56 mg, 0.38 mmol) to afford 67 as an oil (45 mg, 48%). 1H NMR (300 MHz, CDCl3): δ 8.42 (s, 1 H), 8.09 (d, J 13.4 Hz, 1 H), 6.78 (d, J 7.0 Hz, 1H), 4.40 (q, J 7.1 Hz, 2H), 4.20 (q, J 7.2 Hz, 2H), 3.36–3.17 (m, 4H), 1.67–1.48 (m, 7H), 1.42 (t, J 7.1 Hz, 3H), 0.40 (s, 4H). LCMS m/z 373.4 [M + 1].
7-(6-Azaspiro[2.5]octan-6-yl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic Acid (68)
General method F was followed using 67 (45 mg, 0.12 mmol) to afford 68 as a solid (42 mg, 100%). 1H NMR (300 MHz, CDCl3): δ 15.19 (br s, 1H), 8.66 (s, 1H), 8.01 (d, J 13.1 Hz, 1H), 6.94 (d, J 6.8 Hz, 1H), 4.33 (q, J 7.3 Hz, 2H), 3.30–3.41 (m, 4H), 1.59–1.67 (m, 7H), 0.42 (s, 4H). LCMS m/z 345.4 [M + 1].
Methoxycarbonyl 7-(6-Azaspiro[2.5]octan-6-yl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylate (69)
General method B was followed using 68 (45 mg, 0.13 mmol) and methyl chloroformate (0.020 mL, 0.26 mmol) to afford 69 as an oil (52 mg, 99%). 1H NMR (300 MHz, CDCl3): δ 8.45 (s, 1H) 7.96 (d, J 13.3 Hz, 1H) 6.77 (d, J 6.9 Hz, 1H) 4.23 (q, J 7.2 Hz, 2H) 3.94 (s, 3H) 3.35–3.20 (m, 4H) 1.62–1.53 (m, 7H) 0.40 (s, 4H). LCMS m/z 403.2 [M + 1].
Ethyl 7-(4,4-Difluoro-1-piperidyl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylate (70)
General method E was followed using 60 (125 mg, 0.42 mmol) and 4,4-difluoropiperidine HCl (86 mg, 0.55 mmol) to afford 70 as a solid (75 mg, 46%). 1H NMR (300 MHz, CDCl3): δ 8.72 (app br s, 1H), 8.11 (d, J 13.0 Hz, 1H), 6.83–6.76 (m, 1H), 4.51–4.34 (m, 4H), 3.45 (t, J 5.6 Hz, 4H), 2.31–2.10 (m, 4H), 1.58 (t, J 7.2 Hz, 3H), 1.45 (t, J 7.1 Hz, 3H). LCMS m/z 383.2 [M + 1].
7-(4,4-Difluoro-1-piperidyl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic Acid (71)
General method F was followed using 70 (75 mg, 0.20 mmol) to afford 71 as a solid (39 mg, 56%). 1H NMR (300 MHz, CDCl3): δ 14.98 (s, 1H) 8.70 (s, 1H), 8.12 (d, J 12.8 Hz, 1 H), 6.89 (d, J 6.9 Hz, 1H), 4.37–4.28 (m, 2H), 3.48–3.41 (m, 4H), 2.34–2.11 (m, 4H), 1.61 (t, J 7.3 Hz, 3H). LCMS m/z 355.2 [M + 1].
Methoxycarbonyl 7-(4,4-Difluoro-1-piperidyl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylate (72)
General method B was followed using 71 (39 mg, 0.11 mmol) and methyl chloroformate (0.017 mL, 0.22 mmol) to afford 72 as an oil (42 mg, 92%). 1H NMR (300 MHz, CDCl3): δ 8.48 (s, 1H), 8.03 (d, J 12.9 Hz, 1H), 6.80 (d, J 6.73 Hz, 1H), 4.25 (q, J 7.2 Hz, 2H), 3.95 (s, 3H), 3.42–3.35 (m, 4H), 2.32–2.12 (m, 4H), 1.59–1.54 (m, 3H). LCMS m/z 413.2 [M + 1].
1-Ethyl-6-fluoro-4-oxo-7-(4-propyl-1-piperidyl)quinoline-3-carboxylic Acid (73)
General method A was followed using 7-chloro-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic acid (250 mg, 0.93 mmol) and 4-propylpiperidine (236 mg, 1.85 mmol) to afford 73 as a solid (140 mg, 42%). 1H NMR (300 MHz, DMSO-d6) δ 15.41 (br s, 1H), 8.94 (s, 1H), 7.90 (d, J 13.4 Hz, 1H), 7.16 (d, J 7.5 Hz, 1H), 4.58 (q, J 6.7 Hz, 2H), 3.71 (app d, J 12.4 Hz, 2H), 2.90 (t, J 11.4 Hz, 2H), 1.81 (d, J 10.4 Hz, 2H), 1.46–1.24 (m, 10H), 0.90 (t, J 7.0 Hz, 3H). LCMS m/z 361.3 [M + 1].
Methoxycarbonyl 1-Ethyl-6-fluoro-4-oxo-7-(4-propyl-1-piperidyl)quinoline-3-carboxylate (74)
General method B was followed using 73 (120 mg, 0.33 mmol) and methyl chloroformate (0.033 mL, 0.43 mmol) to afford 74 as a solid (139 mg, 100%). 1H NMR (300 MHz, CDCl3): δ 8.46 (s, 1H), 7.99 (d, J 13.3 Hz, 1H), 6.77 (d, J 6.1 Hz, 1H), 4.23 (q, J 7.2 Hz, 2H), 3.95 (s, 3H), 3.72–3.59 (m, 2H), 2.82 (t, J 11.3 Hz, 2H), 1.84 (d, J 9.7 Hz, 2H), 1.56 (t, J 7.2 Hz, 3H), 1.50–1.28 (m, 7H), 0.93 (t, J 7.0 Hz, 3H). LCMS m/z 419.4 [M + 1].
1-Ethyl-6-fluoro-7-(4-isopropyl-1-piperidyl)-4-oxo-quinoline-3-carboxylic Acid (75)
General method A was followed using 7-chloro-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic acid (150 mg, 0.56 mmol) and 4-isopropylpiperidine (275 mL, 1.85 mmol) to afford 75 as a solid (180 mg, 67%). 1H NMR (300 MHz, DMSO-d6) δ 15.44 (s, 1H), 8.94 (s, 1H), 7.90 (d, J 13.5 Hz, 1H), 7.15 (d, J 7.5 Hz, 1H), 4.58 (q, J 6.9 Hz, 2H), 3.75 (d, J 13.1 Hz, 2H), 2.86 (t, J 12.3 Hz, 2H), 1.78 (d, J 12.0 Hz, 2H), 1.52–1.20 (m, 7H), 0.90 (d, J 6.7 Hz, 6H). LCMS m/z 361.3 [M + 1].
Methoxycarbonyl 1-Ethyl-6-fluoro-7-(4-isopropyl-1-piperidyl)-4-oxo-quinoline-3-carboxylate (76)
General method B was followed using 75 (120 mg, 0.33 mmol) and methyl chloroformate (0.033 mL, 0.43 mmol) to afford 76 as a solid (139 mg, 100%). 1H NMR (300 MHz, CDCl3): δ 8.87 (s, 1H), 8.57–8.46 (m, 1H), 8.10–8.04 (m, 1H), 4.33–4.20 (m, 2H), 3.97 (s, 3H), 3.79–3.68 (m, 2H), 2.95–2.77 (m, 2 H), 1.86 (app d, J 12.3 Hz, 2H), 1.65–1.50 (m, 7H), 0.96 (d, J 6.7 Hz, 6H). LCMS m/z 419.2 [M + 1].
1-Ethyl-6-fluoro-4-oxo-7-(4-phenyl-1-piperidyl)quinoline-3-carboxylic Acid (77)
General method A was followed using 7-chloro-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic acid (150 mg, 0.56 mmol) and 4-phenylpiperidine (359 mL, 2.22 mmol) to afford 77 as a solid (120 mg, 55%). 1H NMR (300 MHz, DMSO-d6) δ 8.95 (s, 1H) 7.93 (d, J 13.5 Hz, 1H), 7.37–7.27 (m, 4H), 7.27–7.15 (m, 2H), 4.60 (q, J 7.1 Hz, 2H), 3.92–3.76 (m, 2H), 3.12–2.99 (m, 2H), 2.90–2.69 (m, 1H), 2.01–1.80 (m, 4H), 1.42 (t, J 7.1 Hz, 3H). LCMS m/z 395.2 [M + 1].
Methoxycarbonyl 1-Ethyl-6-fluoro-4-oxo-7-(4-phenyl-1-piperidyl)quinoline-3-carboxylate (78)
General method B was followed using 77 (60 mg, 0.15 mmol) and methyl chloroformate (0.015 mL, 0.20 mmol) to afford 78 as a solid (125 mg, 93%). 1H NMR (300 MHz, CDCl3): δ 8.53 (s, 1H), 8.12 (d, J 13.2 Hz, 1H), 7.41–7.30 (m, 5H), 7.27–7.21 (m, 1H), 4.29 (q, J 7.2 Hz, 2H), 3.98 (s, 3H), 3.90–3.76 (m, 2H), 3.12–3.00 (m, 2H), 2.83–2.71 (m, 1H), 2.12–2.00 (m, 4H), 1.62 (t, J 7.2 Hz, 3H). LCMS m/z 453.2 [M + 1].
7-(4-Benzyl-1-piperidyl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylic Acid (79)
General method F was followed using 43 (115 mg, 0.26 mmol) to afford 79 as a solid (103 mg, 96%). 1H NMR (300 MHz, CDCl3): δ 15.19 (br s, 1H) 8.65 (s, 1H), 8.02 (d, J 13.1 Hz, 1H) 7.37–7.28 (m, 2H) 7.23–7.17 (m, 2H), 6.84 (d, J 6.4 Hz, 1H), 4.30 (q, J 6.8 Hz, 2H), 3.73 (app d, J 11.7 Hz, 2H), 2.85 (app t, J 11.8 Hz, 2H), 2.64 (d, J 6.6 Hz, 2H), 1.91–1.72 (m, 3H), 1.63–1.44 (m, 5H). LCMS m/z 409.3 [M + 1].
Methoxycarbonyl 7-(4-Benzyl-1-piperidyl)-1-ethyl-6-fluoro-4-oxo-quinoline-3-carboxylate (80)
General method B was followed using 79 (103 mg, 0.25 mmol) and methyl chloroformate (0.025 mL, 0.33 mmol) to afford 80 as a solid (117 mg, 99%). 1H NMR (300 MHz, CDCl3): δ 8.48 (s, 1H), 8.03 (d, J 13.3 Hz, 1H), 7.37–7.29 (m, 2H), 7.24–7.15 (m, 3H), 6.73 (d, J 6.9 Hz, 1H), 4.22 (q, J 7.3 Hz, 2H), 3.95 (s, 3 H), 3.74–3.60 (m, 2H), 2.77 (t, J 11.6 Hz, 2H), 2.63 (d, J 6.6 Hz, 2H), 1.88–1.70 (m, 3H), 1.56 (t, J 7.2 Hz, 4H), 1.49–1.44 (m, 2H). LCMS m/z 467.3 [M + 1].
Ethyl 1-Ethyl-6-fluoro-7-[4-(morpholinomethyl)-1-piperidyl]-4-oxo-quinoline-3-carboxylate (81)
General method E was followed using 60 (100 mg, 0.34 mmol) and 4-(4-piperidylmethyl)morpholine (95 mg, 0.50 mmol) to afford 81 as a solid (84 mg, 56%). 1H NMR (300 MHz, CDCl3): δ 8.43 (s, 1H), 8.10 (d, J 13.3 Hz, 1H), 6.75 (d, J 6.9 Hz, 1H), 4.40 (q, J 7.1 Hz, 2H), 4.20 (q, J 7.2 Hz, 2H), 3.99–3.61 (m, 6H), 3.07–2.74 (m, 4H), 2.56–2.16 (m, 5H), 2.14–1.87 (m, 2H), 1.57–1.52 (m, 3H), 1.42 (t, J 7.1 Hz, 3H). LCMS m/z 446.4 [M + 1].
1-Ethyl-6-fluoro-7-[4-(morpholinomethyl)-1-piperidyl]-4-oxo-quinoline-3-carboxylic Acid (82)
General method F was followed using 81 (40 mg, 0.090 mmol) to afford 82 as a solid (34 mg, 91%). 1H NMR (300 MHz, CDCl3): δ 15.17 (br s, 1H), 8.65 (s, 1H), 8.01 (d, J 13.1 Hz, 1H), 6.83 (d, J 6.9 Hz, 1H), 4.32 (q, J 7.2 Hz, 2H), 3.84–3.64 (m, 6H), 2.89 (t, J 11.4 Hz, 2H), 2.53–2.41 (m, 3H), 2.40–2.24 (m, 2H), 2.04–1.73 (m, 4H), 1.59 (t, J 7.2 Hz, 3H), 1.54–1.38 (m, 2H). LCMS m/z 418.3 [M + 1].
Methoxycarbonyl 1-Ethyl-6-fluoro-7-[4-(morpholinomethyl)-1-piperidyl]-4-oxo-quinoline-3-carboxylate (83)
General method B was followed using 82 (33 mg, 0.079 mmol) and methyl chloroformate (0.08 mL, 0.52 mmol) to afford 83 as a solid (37 mg, 98%). 1H NMR (300 MHz, CDCl3): δ 8.48 (s, 1H), 8.03 (d, J 13.3 Hz, 1H), 6.76 (d, J 6.9 Hz, 1H), 4.24 (q, J 7.2 Hz, 2H), 3.96 (s, 3H), 3.60–3.84 (m, 6H), 2.83 (t, J 11.3 Hz, 2H), 2.39–2.53 (m, 3H), 2.18–2.36 (m, 2H), 2.13–1.74 (m, 4H), 1.57 (t, J 7.2 Hz, 3H), 1.52–1.34 (2H). LCMS m/z 476.4 [M + 1].
Biology Methods
Primary Screen Assay
The P. falciparum 3D7 LDH growth assay was conducted with minor modifications to the previous method.47 Briefly, an inoculum of parasitized red blood cells (PRBCs) at 0.7% parasitemia and 2% hematocrit in RPMI-1640, 5% AlbuMAX, 2% d-sucrose, 0.3% glutamine, and 150 μM hypoxanthine was used for the assay. Assay plates (Greiner #781098, 384 well, white, tissue culture treated) were prepared by dispensing 40 nL of the compound (from 1 mM master stock plates) into assay plates using an Echo555 (Labcyte) such that the final concentration was 1 μM. The positive growth control was 0.1% DMSO, and the negative growth control was 100 nM chloroquine. The parasite inoculum (40 μL) was dispensed into plates containing compounds using a Multidrop Combi dispenser (Thermo Scientific). Plates were incubated at 37 °C for 72 h in an atmosphere of 5% CO2, 5% O2, and 95% N2. Following 72 h growth, plates were sealed with parafilm and frozen at −80 °C overnight. Plates were thawed at room temperature for at least 4 h prior to LDH activity being measured. Forty-five microliters of fresh LDH reaction mix (174 mM sodium l-lactate, 214 μM 3-acetyl pyridine adenine dinucleotide (APAD), 270 μM nitro blue tetrazolium chloride (NBT), 4.35 U mL–1 diaphorase, 0.7% Tween 20, and 100 mM Tris–HCl (pH 7.5)) was dispensed into each well using a Multidrop Combi dispenser. Plates were shaken to ensure mixing, and absorbance at 650 nm was measured in an EnVision (PerkinElmer) plate reader after 30 min of incubation at room temperature. Data were normalized to percent growth inhibition using positive and negative controls and analyzed using the Dotmatics 5.3 and Spotfire 7.11.1 (Tibco) software.
Confirmation Assay
To calculate the IC50 of hit compounds, a 10-point dilution series of the compounds were prepared in 384-well assay plates using an Echo555 (Labcyte). Appropriate volumes of 10 mM compound stocks were transferred into the assay plates such that the starting concentration was 1 μM, with a 1:3-fold dilution series. All wells were backfilled with DMSO to 40 nL such that this remained constant (0.1% DMSO). The positive growth control was 0.1% DMSO, and the negative growth control was 100 nM chloroquine. Parasite inoculum (40 μL) was dispensed into plates containing compounds as described above. All other culture and LDH reaction methods are as described above for the primary screen assay. IC50 values were calculated by the Dotmatics 5.3 and Spotfire 7.11.1 software using a nonlinear regression four-parameter fit analysis. The equation used is sigmoidal dose–response (variable slope), Y = bottom + (top – bottom)/(1 + 10((logEC50 – X) × hill slope)).
Bovine Recombinant LDH Assay
To eliminate the selection of false-positive hits inhibiting the biochemical readout, the primary hits were assayed against bovine recombinant LDH activity (12.5 U mL–1, Sigma L3916) under identical LDH assay reaction conditions described above for the primary screen assay.
P. falciparum 3D7 Asexual-Stage Parasite LDH Viability Assay
Compounds were predispensed in 384-well assay plates, and RPMI/AlbuMAX growth media inoculated with synchronized ring-stage P. falciparum were added. Plates were incubated for 72 h and then frozen at −80 °C overnight. LDH activity was quantified with 3-acetylpyridine adenine dinucleotide (APAD) by measuring absorbance of nitro blue tetrazolium (NBT) at 650 nm using a modified LDH assay as described above in the primary screen assay. Screening data were analyzed using the Dotmatics 5.3 and Spotfire 7.11.1 (Tibco) software. Control compound activity against 3D7 parasites was as follows: chloroquine, EC50 35 (19) nM; artesunate, EC50 8 (1) nM.
HepG2 Viability Assay
This assay was performed following the previously described protocol.31 HepG2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) in a humidified incubator at 37 °C and 5% CO2. Assay plates (Greiner #781098, 384 wells, white, tissue culture treated) were prepared by transferring appropriate volumes of 10 mM compound stocks using an Echo555 (Labcyte) such that the starting concentration was 50 μM, with a 1:2-fold dilution series. All wells were backfilled with DMSO to 100 nL such that this remained constant (0.5% DMSO). The positive growth control was 0.5% DMSO, and the negative growth control was 10 μM bortezomib. HepG2 cells were seeded with 1000 cells in 50 μL of DMEM with 10% FCS into each well of the assay plates using a Multidrop Combi dispenser (Thermo Scientific). Plates were incubated at 37 °C and 5% CO2 for 48 h. Cytotoxicity was determined using CellTiter-Glo (Promega) and calculated as a percentage using DMSO as the positive growth control and 10 μM bortezomib as the negative growth control. EC50 values were calculated by the Dotmatics 5.3 and Spotfire 7.11.1 software using a nonlinear regression four-parameter fit analysis. The equation used is sigmoidal dose–response (variable slope), Y = bottom + (top – bottom)/(1 + 10((logEC50 – X) × hill slope)).
Liver Microsome Stability
A solution of compounds in a phosphate-buffered solution (1 μM) was incubated in pooled human or mouse liver microsomes (0.5 mg/mL) for 0, 5, 20, 30, 45, and 60 min at 37 °C in the presence and absence of the NADPH regeneration system. The reaction was terminated with the addition of ice-cold acetonitrile containing system suitability standard at designated time points. The sample was centrifuged (4200 rpm) for 20 min at 20 °C, and the supernatant was half diluted in water and then analyzed by means of LC–MS/MS. The % parent compound remaining, half-life (T1/2), and clearance (CLint) were calculated using standard methodology. The experiment was performed in duplicate.
Hepatocyte Stability
A solution of the test compound in a Krebs–Henseleit buffer solution (1 μM) was incubated in pooled rat hepatocytes (1 × 106 cells/mL) for 0, 15, 30, 45, 60, 75, and 90 min at 37 °C (5% CO2, 95% relative humidity). The reaction was terminated with the addition of ice-cold acetonitrile containing system suitability standard at designated time points. The sample was centrifuged (4200 rpm) for 20 min at 20 °C, and the supernatant was half diluted in water and then analyzed using LC–MS/MS. The % parent compound remaining, half-life (T1/2), and clearance (CLint) were calculated using standard methodology. The experiment was carried out in duplicate.
Solubility
The solubility assay was performed using a miniaturized shake flask method. A solution of phosphate-buffered saline (PBS pH 7.4), standard FaSSIF (pH 6.5), or FaSSGF (pH 1.2) conditions was incubated with the compound (200 μM) at 25 °C with constant shaking (600 rpm) for 2 h. The samples were filtered using a multiscreen solubility filter plate. The filtrate was half diluted in acetonitrile. A five-point linearity curve was prepared in PBS/acetonitrile (1:1, v/v) at 200, 150, 75, 25, and 2.5 μM. Blank, linearity, and test samples (n = 2) were transferred to a UV readable plate, and the plate was scanned for absorbance. Best fit calibration curves were constructed using the calibration standards and used to determine the test sample solubility. The experiment was carried out in duplicate.
LogD
LogD at pH 7.4 was determined using a miniaturized shake flask method. A solution of a presaturated mixture of 1-octanol and phosphate-buffered saline (PBS) (1:1, v/v) and the test compound (7.5 μM) was incubated at 25 °C with constant shaking (850 rpm) for 2 h. After incubation, the organic and aqueous phases were separated, and samples of each phase were transferred to a plate for dilution. The organic phase was diluted 1000-fold, and the aqueous phase was diluted 20-fold. The samples were quantitated using LC–MS/MS. The experiment was carried out in duplicate.
P. falciparum PRR Assay
This assay was performed as previously described.37 Briefly, P. falciparum parasites were cultured in RPMI-1640 (10.3 g/L) supplemented with l-glutamine (2 mM), hypoxanthine (150 μM), HEPES (25 mM), d-glucose (12 mM), sodium bicarbonate (25.70 mM), and Albumax II (0.5% or 5 g/L). In the parasite reduction ratio (PRR) assay, an inoculum of parasitized red blood cells with 0.5% parasitemia (>80% ring stage) and 2% hematocrit was treated with 10-fold concentration of IC50 of a particular test compound such that the final concentration of the vehicle (DMSO) was 0.2%. Drug was renewed every 24 h over the entire 120 h of treatment period. Samples of parasite were taken from the treated culture every 24 h (24, 48, 72, 96, and 120 h time points). Drug was washed out twice with 5 mL of complete media each, and drug-free parasites were cultured in 96-well plates by adding fresh erythrocytes and new culture media. To quantify the number of viable parasites after treatment, threefold serial dilution was used with the above-mentioned samples after removing the drug. For each time point, 12 serial dilutions of four replicates were performed to correct experimental variations. The plates containing the limiting serial dilution cultures were maintained for 28 days, and the presence of parasite growth was detected using the P. falciparum LDH assay (described above). The number of viable parasites initially present in an aliquot was back-calculated based on the most diluted well able to render detectable growth according to the formula Xn – 1, where n is the number of wells able to render growth and X is the dilution factor (when n = 0, the number of viable parasites is estimated as zero).
Asexual Stage of Arrest Assay
This assay was performed as previously described.32,48 Briefly, a 5% d-sorbitol synchronization was performed in the cycle before assay setup. For the assay setup, ring-stage 3D7 parasites were counted and adjusted to 1% parasitemia and 2% hematocrit, and compounds were added at 10 × EC50 (ATQ 10 nM, 17 100 nM, and 41 150 nM), with plates receiving replenishment of compounds every 24 h. Plates were incubated at 37 °C, and every 12 for 48 h, 50 μL aliquots in triplicate and blood smears were taken. Aliquots were subsequently fixed in 0.25% glutaraldehyde at room temperature for 30 min before being washed and stored in PBS at 4 °C. At the conclusion of the experiment, blood pellets were stained with 50 μL of 2.5× SYBR Green (Invitrogen) for 30 min at 37 °C. Pellets were then washed in PBS before being diluted with a further 180 μL of PBS before cells (100,000) were counted on an Attune Flow Cytometer (ThermoFisher Scientific). Blood smears were fixed in methanol and stained with 10% Giemsa, and the morphology was examined using a microscope (Zeiss Axio Observer).
P. falciparum Resistant Selection
A previously described method was followed for this assay.49 Briefly, three populations of 3D7 clonal parasites were exposed to 2 × EC50 of 41 (30 nM) until parasites were assessed to be dying by Giemsa-stained thin blood smears. After recovery, parasites were then treated with the next EC50 increment. This was repeated until reaching 10 × EC50, and resistance was evaluated in an LDH growth assay as previously described.31 Resistant parasites were harvested with the addition of 0.15% saponin, and genomic DNA (gDNA) was extracted using the Qiagen Blood and Tissue kit.
Whole Genome Sequencing
Parasite gDNA was prepared and sequenced as previously described49 with the following changes: indexed libraries were pooled and diluted to 750 pM for paired-end sequencing (2 × 166 cycles) on a NextSeq 2000 instrument using the P2 300 cycle kit (Illumina) as per the manufacturer’s instruction. Sequencing read quality in fastq files was assessed using fastQC and was very good, with no need for adapter trimming. Reads were aligned to reference PlasmoDB-52_Pfalciparum3D7 with bwa-mem v0.7.17 with default parameters and filtered using Picard MarkDuplicates. Calling of single nucleotide variants (SNVs) and indels was performed with bcftools v1.13 with parameters --max-depth 800, --ploidy 1, -m. SNVs and Indels were filtered using the following criteria: QUAL >50; not an indel with length > 5 of A and T only; not present in WT parent; and in core genome. Core genome was defined to exclude telomere, centromere, and hypervariable regions50 and a region for chromosome 10 centromere but include the mitochondrial genome. When calls were filtered to those in a coding sequence, genes described as PfEMP1, rifin, stevor, or pseudogene were excluded. Only variants called in a majority of compound 41 samples, and not in the comparison samples, were considered. Copy number analysis was performed using the R package QDNaseq v1.28.051 with 1 kbp bins. Copy number bins were filtered to exclude centromere and telomere regions and by mappability of 30mers generated by GenMap.52 Structural variant calling was performed using GRIDSS v2.12.1.53,54 Calls were filtered to high-confidence events not present in the uncloned parent sample using gridss-somatic_filter.R. Regions of interest were inspected with the Integrated Genome Viewer. The data for this study have been deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB58099 (https://www.ebi.ac.uk/ena/browser/view/PRJEB58099). For more details, please see https://ena-docs.readthedocs.io/en/latest/faq/release.html.
Model of P. falciparum Cyt b
A homology model of P. falciparum cyt b was derived from the X-ray structure of Gallus gallus cyt bc1 (PDB: 3H1I)38 using the SWISS-MODEL resource.55 Qo and Qi site mutations found in drug-resistant parasites were mapped to the homology structure of P. falciparum cyt b. ATQ (3), ELQ-300 (5), ubiquinol, and ubiquinone (2) were positioned in P. falciparum cyt b according to published X-ray structures.19,26,38 The CLC Drug Discovery Workbench software (version 2.5.1) was used to model compound 41 to the homology model of P. falciparum cyt b. The input ligand 41 was built using a ligand designer, and the ligand binding site was centralized to the Qo binding site of cyt b. The ligand 41 was docked using minimalization of flexible ligand conformations while maintaining a rigid protein structure during the docking process. The ligand binding interactions of the resulting minimizations were observed using the CLC Drug Discovery visualization tool. The structural data were then exported and visualized using the PyMol Molecular Graphics System (version 2.0) software.
Drug-Resistant Asexual-Stage Assays
The rationale and description of drug-resistant parasite strains used are described by Ding et al.56 This assay format was used to evaluate compounds against P. falciparum cyt b mutant strains in Table 4 and mutant strains in Table 5. An adaptation of the previously described 3H-hypoxanthine scintillation proximity assay was used to assess compounds against drug-resistant strains.57 Parasites were cultured using a medium containing RPMI 1640 (10.44 g/L) (no hypoxanthine) supplemented with HEPES (5.94 g/L), NaHCO3 (2.1 g/L), neomycin (100 μg/mL), and Albumax II (5 g/L). An uninfected red blood cell solution (1 mL) was prepared (2.5% hematocrit) containing 50 μL of washed human erythrocytes (50% hematocrit) and mixed with 950 μL of screening medium. The screening medium (100 μL) was then added to each well of the Falcon 96-well microtiter plate. Serial drug dilutions were then prepared across the plate using an additional 100 μL of the screening medium. Infected blood (100 μL) (parasitemia of 0.3%, 2.5% hematocrit) was added to all wells in the plate with the exception of control wells. The plates were incubated at 37 °C in an atmosphere of 93% N2, 4% CO2, and 3% O2. After 48 h, 50 μL of [3H]-hypoxanthine (0.5 μCi) solution was added to each well of the plate, and plates were incubated for another 24 h. The plates were then harvested with a Betaplate cell harvester (Wallac, Zurich, Switzerland) and, after collection by filtration dilution with scintillation fluid, were counted in a Betaplate liquid scintillation counter (Wallac, Zurich, Switzerland). The results were recorded as counts per minute (cpm) per well at each drug concentration. Data were transferred into the MS Excel software and expressed as a percentage of the untreated controls. The 50% inhibitory concentration (IC50) value was evaluated by logit regression analysis.
SB1-A6 and ScDHODH Asexual-Stage Assays
Assays were performed using the Pf SB1-A6 strain with a CNV (∼2-fold) and a C276F mutation in DHODH or Pf Dd2 expressing ScDHODH. A 5% d-sorbitol synchronization was performed in the cycle before assay setup. For growth assay setup, ring-stage parasites were counted and adjusted to a final 0.5 and 2% parasitemia and hematocrit, respectively, and plated into a twofold dilution of the compounds. Plates were incubated at 37 °C for 72 h. Blood pellets were stained and analyzed by SYBR Green staining as outlined in the section above describing the asexual stage of arrest assay.
ETC Biosensor Assay
This assay was performed as previously described.27 Briefly, 3 × 108 trophozoite infected RBCs were purified using the SuperMACS II separator (Miltenyi Biotec, Germany).58 The trophozoite-stage parasites were released from erythrocytes by treating them with 0.05% (w/v) prewarmed saponin in phosphate-buffered saline (PBS) at 37 °C for 3 min. The cells were washed with prewarmed PBS three times. The parasite count was adjusted to 5 × 107 parasites/mL in a mitochondrial assay solution (MAS) buffer (mannitol (220 mM), sucrose (70 mM), KH2PO4 (10 mM), MgCl2 (5 mM), HEPES (2 mM), EGTA (1 mM), and BSA 0.2% (w/v)) supplemented with 25 mM malate and 0.002% (w/v) digitonin. Parasites were seeded at a density of 5 × 106 cells per well in a Cell-Tak-coated XFe96 cell culture plate and centrifuged (800g, 10 min, RT) with low braking to adhere the parasites to the bottom of the wells. The MAS buffer (75 μL) was added to the wells without detaching the parasite. The plate was visualized under the microscope to ensure a cell monolayer. The inhibitors or control compounds were prepared in the MAS buffer (final concentration after injection given in brackets) and loaded into ports A–C of the XFe96 sensor cartridge: port A, inhibitors/atovaquone (5 μM); port B, N,N,N′,N′-tetramethyl-p-phenylenediamine dihydrochloride (TMPD; 0.2 mM) and ascorbic acid (2 mM); and port C, sodium azide (NaN3; 10 mM). The cells were measured for oxygen consumption rate (OCR) using a Seahorse XFe96 flux analyzer (Agilent). The OCR was assessed for five cycles of 20 s mixing, 1 min waiting, and 2.5 min measuring to establish baseline OCR before any substrate injections; for five cycles of 20 s mixing and 1 min waiting followed by 2.5 min measuring after inhibitor injection from port A; and for five cycles of 20 s mixing, 1 min waiting, and 2.5 min measuring after TMPD and NaN3 injections.
Dual Gamete Formation Assay (DGFA)
P. falciparum NF54 (BEI Resources MRA-1000) asexual and sexual parasite cultures were maintained as previously described.59 Gametocyte cultures were initiated using asynchronous asexual parasites at 3% parasitemia and 4% hematocrit and in a final volume of 40 mL. Gametocyte induction was facilitated by daily medium changes with gametocyte culture medium (RPMI-1640 supplemented with 25 mM HEPES, 50 μg/mL hypoxanthine, 2 g/L NaHCO3, 5% Albumax, and 5% pooled human type A+ serum) and no addition of fresh erythrocytes. Care was taken to ensure that all gametocyte cultures were kept at 37 °C by prewarming all culture media and work surfaces. Human serum type A+ was obtained from Interstate Blood Bank (U.S.A.) from donors who had taken no aspirin for at least 2 h before drawing and no antimalarials for at least 2 weeks before drawing. Gametocyte growth was monitored by Giemsa-stained thin smears on days 2, 7, and 14. On day 2, cultures were at a high percentage asexual parasitemia (±10–15%), and by day 7, young gametocytes (stages I, II, and III) were visible. Two weeks after gametocyte induction, on days 14–16, mature stage V gametocytes were assessed by counting exflagellation centers using a Neubauer chamber. Gametocyte cultures were considered acceptable for use in the DGFA if more than 0.2% exflagellation (relative to the total erythrocyte cell number) was observed. Compounds were dispensed into 384-well plates (781091, Greiner) using an HP-D300 digital dispenser and T8 and D4 cassettes, with 0.25% DMSO as the negative control and 12.5 μM gentian violet as the positive control. Compounds were stamped at 1 μM with five technical repeats. All wells were normalized with DMSO to 0.25% of the final assay volume of 50 μL. Four copies of each plate were stamped. The DGFA was performed as described previously.59,60 Mature (stage V) gametocyte cultures with more than 0.2% exflagellation (of total erythrocytes) were diluted to 12.5 million cells per milliliter using the assay culture medium (gametocyte culture medium, except with 10% human serum and no Albumax). Working swiftly to prevent cooling of gametocyte cultures (which will result in the formation of gametes prematurely), 50 μL of the diluted gametocyte culture (625,000 cells) was dispensed into each well of a prewarmed 384-well plate already containing the compounds. Four separate gametocyte culture flasks were used for each of the four plates in the assay. The plates containing compounds and gametocytes were maintained at 37 °C for 48 or 72 h in a humidified chamber with a 92% N2/5% CO2/3% O2 gas mix. After 48 h, gamete formation was initiated by the addition of 10 μL per well ookinete medium (RPMI-1640 supplemented with 25 mM HEPES, 50 μg/mL hypoxanthine, 2 g/L NaHCO3, and 100 μM xanthurenic acid) containing 0.5 μg/mL anti-Pfs25 clone 4B7 (BEI Resources MRA-315) conjugated to a Cy3 fluorophore (GE Healthcare). Immediately after adding the ookinete medium, the plates were cooled at 4 °C for 4 min followed by 28 °C for a further 5 min. Male gamete formation was assessed by recording exflagellation immediately after the 9 min cooling step using a Nikon Ti-E widefield microscope with a 4× objective at 1.5× zoom to record a 10-frame video time-lapse over 2 s for each well. After imaging, the plates were incubated in the dark at 28 °C for a further 24 h, which ensures maximal expression of Pfs25 on the surface of female gametes. Female gamete formation was assessed by capturing a single fluorescence image for each well (using the Nikon Ti-E widefield microscope and the same magnification as before). Exflagellation and female gamete formation were quantified with the use of algorithms specifically designed with the open-source ICY Image Analysis software. Percentage inhibition for each compound was calculated by normalization to negative (Ctr1) and positive controls (Ctr2) with the following equation: % inhibition = 100 – ((test compound – Ctr2)/(Ctr1 – Ctr2) × 100). Assay performance statistics and IC50 analysis were obtained using GraphPad Prism (v8.2.1) nonlinear regression.
Exoerythrocytic Stage Assay
This assay was performed as previously described.44P. berghei is able to infect human hepatocarcinoma HepG2 cells expressing the tetraspanin CD81 receptor.61 HepG2-A16-CD81EGFP cells stably transformed to express a GFP-CD81 fusion protein were cultured at 37 °C in 5% CO2 in DMEM (Invitrogen, Carlsbad, USA) supplemented with 10% FBS, 0.29 mg/mL glutamine, 100 U penicillin, and 100 μg/mL streptomycin. HepG2-A16-CD81EGFP cells (3 × 103) in 5 μL of the medium (2 × 105 cells/mL, 5% FBS, 5 × Pen/Strep/Glu) were seeded in 1536-well plates (Greiner BioOne white solid bottom custom GNF mold) 20–26 h prior to the actual infection. Eighteen hours prior to infection, 50 nL of the compound in DMSO (0.5% final DMSO concentration per well) was transferred using the Acoustic Transfer System (ATS) (Biosero) into the assay plates (10 μM final concentration). In cases when acoustic compatible source plates were not available, the 50 nL of compounds to test was transferred with PinTool (GNF Systems). Atovaquone (5 μM) and 0.5% DMSO were used as positive and negative controls, respectively. P. berghei ANKA-GFP-Luc-SMCON sporozoites20 were freshly dissected from infected A. stephensi mosquito salivary glands and filtered twice through a through a 20 μm nylon net filter (Steriflip, Millipore). The sporozoites were resuspended in media and counted in a hemocytometer, and their final concentration was adjusted to 200 sporozoites per microliter. Also, penicillin and streptomycin were added at 5-fold increased concentration for a final 5-fold increased concentration in the well. The HepG2-A16-CD81EGFP cells were then infected with 1 × 103 sporozoites per well (5 μL) with a single tip Bottle Valve liquid handler (WDII, GNF), and the plates were spun down at 37 °C for 3 min in an Eppendorf 5810 R centrifuge with a centrifugal force of 330× on normal acceleration and brake setting. After incubation at 37 °C for 48 h, the EEF growth was quantified by a bioluminescence measurement. The increased antibiotic concentration did not interfere with the parasite or HepG2-A16-CD81EGFP growth. Atovaquone and naive wells were used as controls on each plate. The compounds were screened in a 12-point serial dilution to determine their IC50 values. Bioluminescence quantification of exo-erythrocytic forms was performed as follows: Media were removed by spinning the inverted plates at 150g for 30 s. BrightGlo (2 μL, Promega) was dispensed with the MicroFlo (BioTek) liquid handler. Immediately after the addition of the luminescence reagent, the plates were read by the Envision Multilabel Reader (PerkinElmer). IC50 values were obtained using measured bioluminescence intensity and a nonlinear variable slope four-parameter regression curve fitting model in the GraphPad Prism software (version 6).
Animal Ethics and Maintenance
The use of animals in the 4 day P. berghei mouse model (below) was approved by the Walter and Eliza Hall Institute of Medical Research Animal Ethics Committee under approval number 2020.036 and governance of the Australian code for the care and use of animals for scientific purposes. A total of 34 female ASMU:Swiss outbred mice used the in the 4 day P. berghei mouse model (below) were obtained from the Monash Animal Research Platform, Monash University, Australia. ASMU:Swiss outbred (pathogen-free) mice (female, 4 weeks old, 10–17 g) were kept in individually ventilated cages (IVCs) with corncob bedding but otherwise under standard conditions with 22 °C and 40–70% relative humidity and with Barastock Rat & Mouse pellet (WEHI Mouse Breeder Cubes, Irradiated (#102119) and Mouse custom Mash Irradiated (#102121), Ridley Agriproducts Pty. Ltd.) and water ad libitum. There was a maximum of six adults per cage with more than one mouse. The water provided to mice was filtered and acidified to pH 3 and supplied in a Hydropac pouch or water bottle. Animal enrichment (e.g., tissues, domes) was provided each week when cages were serviced. Animal technicians were responsible for the daily husbandry of the mice and serviced each cage at least twice a week. Cages were cleaned on a rotating schedule (depending on the stocking density) or as often as needed. Typically, cages of four to six mice required fortnightly cleaning, and cages housing one to three mice required monthly cleaning.
P. berghei Mouse Model
A previous protocol62 was followed for the 4 day mouse model using blood-stage P. berghei ANKA GFPcon 259 cl2 parasites.46 Briefly, P. berghei parasites were expanded in Swiss mice by infection intraperitoneally with 1 × 106P. berghei ANKA-parasitized red blood cells from frozen stocks of infected blood or parasites withdrawn from a previously infected donor mouse. The “donor” female Swiss mice were infected intraperitoneally with blood-stage P. berghei parasites constitutively expressing GFP (P. berghei ANKA GFPcon 259 cl2). Three days later, groups of four “acceptor” Swiss mice were infected intravenously (IV) with 1 × 107 parasitized erythrocytes from the “donor” mice. Two hours post infection, experimental mice (four per cohort) were left untreated (control mice) or treated orally for 4 consecutive days with test drugs formulated in a vehicle consisting of 20% DMSO/60% propylene glycol/20% water (v/v/v) or chloroquine dissolved in water. Mice were treated with compound 17 for 4 days by a q.d. dosing regimen (once a day) at 20 mg/kg/day, with the first dose given 2 h after infection. Peripheral blood samples were taken 2, 6, and 22 h after the first treatment, and parasitemia was measured by flow cytometry (proportion of GFP-positive cells in 100,000 recorded events using FACSCalibur, BD) and microscopic analysis of Giemsa-stained blood smears. Parasitemia values were averages for four mice per group and are expressed as percent parasitemia. Data were analyzed in the GraphPad Prism software (version 9.5.0).
Plasma Bioanalysis
Plasma samples collected from P. berghei infected mice (collected in an external study) were analyzed by LCMS to quantify the concentrations of compound. Samples were provided as 50 μL aliquots and stored at −80 °C. Blank plasma from noninfected mice was used for preparation of calibration standards. The extraction of the test compound from plasma samples was conducted using protein precipitation with acetonitrile. A 12-point standard curve was prepared from a stock solution of the test compound (1 mg/mL in DMSO), with subsequent dilutions from the stock in 50% acetonitrile in water (v/v). The plasma calibration standards were then prepared by spiking blank mouse plasma (50 μL) with the solution standards (10 μL) and the internal standard (IS) diazepam (10 μL of 5 μg/mL in 50% acetonitrile/water). Protein precipitation for plasma standards was carried out by the addition of acetonitrile (120 μL). Plasma samples were similarly prepared except that an additional 10 μL of acetonitrile for the protein precipitation (total volume of 130 μL) was added. Standards and samples were vortexed and then centrifuged (10,000 rpm) for 3 min. The supernatant was subsequently separated, and 3 μL was injected directly onto the column for LCMS analysis using the following instrument and conditions: instrument: Waters Xevo TQS Micro coupled to a Waters Acquity UPLC; column: Supelco Ascentis Express RP Amide column (50 × 2.1 mm, 2.7 μm); gradient cycle time: 4 min; injection vol: 3 μL; flow rate: 0.4 mL/min; and mobile phase: 0.05% formic acid in acetonitrile gradient with 0.05% formic acid in water. Data were analyzed and plotted using the GraphPad Prism software (version 9.5.0).
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia (Development Grant GNT1135421 to B.E.S. and A.F.C.; Ideas Grant GNT1182369 to G.G.v.D. and A.G.M.), Wellcome (100993/Z/13/Z to J.B.), Medicines for Malaria Venture (RD-08-2800 to J.B.; RD-18-0003 to B.E.S. and A.F.C.), the Australian Cancer Research Foundation, the Victorian State Government Operational Infrastructure Support, and the Australian Government NHMRC IRIISS. We thank the University of Melbourne for provision of a Research Scholarship to B.B. We thank and acknowledge the Australian Red Cross Lifeblood and UK NHS Blood and Transplant for the provision of fresh red blood cells, without which this research could not have been performed. We thank Assoc. Prof. Paul Gilson from the Burnet Institute and Assoc. Prof. Darren Creek from Monash University for the provision of the Dd2 ScDHODH parasite strain and the SB1-A6 parasite strain, respectively. We thank Dr. Keith Watson from the Walter and Eliza Hall Institute for the helpful advice. A.F.C. is a Howard Hughes International Scholar and an Australia Fellow of the NHMRC. B.E.S. is a Corin Centenary Fellow.
Glossary
Abbreviations
- ACT
artemisinin combination therapy
- ATQ
atovaquone
- CARL
cyclic amine resistance locus
- CQ
chloroquine
- cyt b
cytochrome b
- cyt bc1
cytochrome b complex 1
- DGFA
dual gamete formation assay
- DHODH
dihydroorotate dehydrogenase
- EEF
exoerythrocytic form
- EF2
elongation factor 2
- ETC
electron transport chain
- HTS
high-throughput screening
- LDH
lactate dehydrogenase
- NDH
NADH dehydrogenase
- OCR
oxygen consumption rate
- Pb
Plasmodium berghei
- Pf
Plasmodium falciparum
- PI4K
phosphatidyl inosine 4-kinase
- PRR
parasite reduction ratio
- Sc
Saccharomyces cerevisiae
- SNP
single-nucleotide polymorphism
- SNV
single-nucleotide variant
- TCA
tricarboxylic acid
- TMPD
N,N,N′,N′-tetramethyl-p-phenylenediamine dihydrochloride
- WJM
WEHI Janssen MMV
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.2c00607.
Activity data on compounds against P. falciparum 3D7 and drug-resistant parasites, NF54 gamete data, modeling with cyt b, mouse model and bioanalysis data, parasite microscopy repeat data, whole genome sequencing data, and compound spectra (PDF)
Author Contributions
◊ W.N. and M.G.D. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- World Malaria Report; World Health Organisation: Geneva, 2022. [Google Scholar]
- Ashley E. A.; Dhorda M.; Fairhurst R. M.; Amaratunga C.; Lim P.; Suon S.; Sreng S.; Anderson J. M.; Mao S.; Sam B.; Sopha C.; Chuor C. M.; Nguon C.; Sovannaroth S.; Pukrittayakamee S.; Jittamala P.; Chotivanich K.; Chutasmit K.; Suchatsoonthorn C.; Runcharoen R.; Hien T. T.; Thuy-Nhien N. T.; Thanh N. V.; Phu N. H.; Htut Y.; Han K. T.; Aye K. H.; Mokuolu O. A.; Olaosebikan R. R.; Folaranmi O. O.; Mayxay M.; Khanthavong M.; Hongvanthong B.; Newton P. N.; Onyamboko M. A.; Fanello C. I.; Tshefu A. K.; Mishra N.; Valecha N.; Phyo A. P.; Nosten F.; Yi P.; Tripura R.; Borrmann S.; Bashraheil M.; Peshu J.; Faiz M. A.; Ghose A.; Hossain M. A.; Samad R.; Rahman M. R.; Hasan M. M.; Islam A.; Miotto O.; Amato R.; MacInnis B.; Stalker J.; Kwiatkowski D. P.; Bozdech Z.; Jeeyapant A.; Cheah P. Y.; Sakulthaew T.; Chalk J.; Intharabut B.; Silamut K.; Lee S. J.; Vihokhern B.; Kunasol C.; Imwong M.; Tarning J.; Taylor W. J.; Yeung S.; Woodrow C. J.; Flegg J. A.; Das D.; Smith J.; Venkatesan M.; Plowe C. V.; Stepniewska K.; Guerin P. J.; Dondorp A. M.; Day N. P.; White N. J. Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2014, 371, 411–423. 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uwimana A.; Legrand E.; Stokes B. H.; Ndikumana J.-L. M.; Warsame M.; Umulisa N.; Ngamije D.; Munyaneza T.; Mazarati J.-B.; Munguti K.; Campagne P.; Criscuolo A.; Ariey F.; Murindahabi M.; Ringwald P.; Fidock D. A.; Mbituyumuremyi A.; Menard D. Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat. Med. 2020, 26, 1602–1608. 10.1038/s41591-020-1005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton T. D.; Devine S. M.; Möhrle J. J.; Laleu B.; Burrows J. N.; Charman S. A.; Creek D. J.; Sleebs B. E. The development process for discovery and clinical advancement of modern antimalarials. J. Med. Chem. 2019, 62, 10526–10562. 10.1021/acs.jmedchem.9b00761. [DOI] [PubMed] [Google Scholar]
- McKeage K.; Scott L. Atovaquone/proguanil: a review of its use for the prophylaxis of Plasmodium falciparum malaria. Drugs 2003, 63, 597–623. 10.2165/00003495-200363060-00006. [DOI] [PubMed] [Google Scholar]
- Looareesuwan S.; Chulay J. D.; Canfield C. J.; Hutchinson D. B. Malarone (atovaquone and proguanil hydrochloride): a review of its clinical development for treatment of malaria. Malarone Clinical Trials Study Group. Am. J. Trop. Med. Hyg. 1999, 60, 533–541. 10.4269/ajtmh.1999.60.533. [DOI] [PubMed] [Google Scholar]
- Smilkstein M. J.; Pou S.; Krollenbrock A.; Bleyle L. A.; Dodean R. A.; Frueh L.; Hinrichs D. J.; Li Y.; Martinson T.; Munar M. Y.; Winter R. W.; Bruzual I.; Whiteside S.; Nilsen A.; Koop D. R.; Kelly J. X.; Kappe S. H. I.; Wilder B. K.; Riscoe M. K. ELQ-331 as a prototype for extremely durable chemoprotection against malaria. Malaria J. 2019, 18, 291. 10.1186/s12936-019-2921-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips M. A.; Lotharius J.; Marsh K.; White J.; Dayan A.; White K. L.; Njoroge J. W.; El Mazouni F.; Lao Y.; Kokkonda S.; Tomchick D. R.; Deng X.; Laird T.; Bhatia S. N.; March S.; Ng C. L.; Fidock D. A.; Wittlin S.; Lafuente-Monasterio M.; Benito F. J.; Alonso L. M.; Martinez M. S.; Jimenez-Diaz M. B.; Bazaga S. F.; Angulo-Barturen I.; Haselden J. N.; Louttit J.; Cui Y.; Sridhar A.; Zeeman A. M.; Kocken C.; Sauerwein R.; Dechering K.; Avery V. M.; Duffy S.; Delves M.; Sinden R.; Ruecker A.; Wickham K. S.; Rochford R.; Gahagen J.; Iyer L.; Riccio E.; Mirsalis J.; Bathhurst I.; Rueckle T.; Ding X.; Campo B.; Leroy D.; Rogers M. J.; Rathod P. K.; Burrows J. N.; Charman S. A. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci. Transl. Med. 2015, 7, 296ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen A.; LaCrue A. N.; White K. L.; Forquer I. P.; Cross R. M.; Marfurt J.; Mather M. W.; Delves M. J.; Shackleford D. M.; Saenz F. E.; Morrisey J. M.; Steuten J.; Mutka T.; Li Y.; Wirjanata G.; Ryan E.; Duffy S.; Kelly J. X.; Sebayang B. F.; Zeeman A. M.; Noviyanti R.; Sinden R. E.; Kocken C. H. M.; Price R. N.; Avery V. M.; Angulo-Barturen I.; Jiménez-Díaz M. B.; Ferrer S.; Herreros E.; Sanz L. M.; Gamo F. J.; Bathurst I.; Burrows J. N.; Siegl P.; Guy R. K.; Winter R. W.; Vaidya A. B.; Charman S. A.; Kyle D. E.; Manetsch R.; Riscoe M. K. Quinolone-3-diarylethers: a new class of antimalarial drug. Sci. Transl. Med. 2013, 5, 177ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barata L.; Houzé P.; Boutbibe K.; Zanghi G.; Franetich J.-F.; Mazier D.; Clain J. In vitro analysis of the interaction between atovaquone and proguanil against liver stage malaria parasites. Antimicrob. Agents Chemother. 2016, 60, 4333–4335. 10.1128/AAC.01685-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler R. E.; Sinden R. E.; Pudney M. Inhibitory activity of the anti-malarial atovaquone (566C80) against ookinetes, oocysts, and sporozoites of Plasmodium berghei. J. Parasitol. 1995, 81, 452–458. 10.2307/3283831. [DOI] [PubMed] [Google Scholar]
- Ruecker A.; Mathias D. K.; Straschil U.; Churcher T. S.; Dinglasan R. R.; Leroy D.; Sinden R. E.; Delves M. J. A male and female gametocyte functional viability assay to identify biologically relevant malaria transmission-blocking drugs. Antimicrob. Agents Chemother. 2014, 58, 7292–7302. 10.1128/AAC.03666-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon G. L.; Pidathala C.; Shone A. E.; Antoine T.; Fisher N.; O’Neill P. M.; Ward S. A.; Biagini G. A. Targeting the mitochondrial electron transport chain of Plasmodium falciparum: new strategies towards the development of improved antimalarials for the elimination era. Future Med. Chem. 2013, 5, 1573–1591. 10.4155/fmc.13.121. [DOI] [PubMed] [Google Scholar]
- Painter H. J.; Morrisey J. M.; Mather M. W.; Vaidya A. B. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 2007, 446, 88–91. 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- Monastyrskyi A.; Kyle D. E.; Manetsch R. 4(1H)-pyridone and 4(1H)-quinolone derivatives as antimalarials with erythrocytic, exoerythrocytic, and transmission blocking activities. Curr. Top. Med. Chem. 2014, 14, 1693–1705. 10.2174/1568026614666140808124638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickles A. M.; de Almeida M. J.; Morrisey J. M.; Sheridan K. A.; Forquer I. P.; Nilsen A.; Winter R. W.; Burrows J. N.; Fidock D. A.; Vaidya A. B.; Riscoe M. K. Subtle changes in endochin-like quinolone structure alter the site of inhibition within the cytochrome bc1 complex of Plasmodium falciparum. Antimicrob. Agents Chemother. 2015, 59, 1977–1982. 10.1128/AAC.04149-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter R.; Kelly J. X.; Smilkstein M. J.; Hinrichs D.; Koop D. R.; Riscoe M. K. Optimization of endochin-like quinolones for antimalarial activity. Exp. Parasitol. 2011, 127, 545–551. 10.1016/j.exppara.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno J. M.; Herreros E.; Angulo-Barturen I.; Ferrer S.; Fiandor J. M.; Gamo F. J.; Gargallo-Viola D.; Derimanov G. Exploration of 4(1H)-pyridones as a novel family of potent antimalarial inhibitors of the plasmodial cytochrome bc1. Future Med. Chem. 2012, 4, 2311–2323. 10.4155/fmc.12.177. [DOI] [PubMed] [Google Scholar]
- Capper M. J.; O’Neill P. M.; Fisher N.; Strange R. W.; Moss D.; Ward S. A.; Berry N. G.; Lawrenson A. S.; Hasnain S. S.; Biagini G. A.; Antonyuk S. V. Antimalarial 4(1H)-pyridones bind to the Qi site of cytochrome bc1. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 755–760. 10.1073/pnas.1416611112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cruz F. P.; Martin C.; Buchholz K.; Lafuente-Monasterio M. J.; Rodrigues T.; Sönnichsen B.; Moreira R.; Gamo F. J.; Marti M.; Mota M. M.; Hannus M.; Prudêncio M. Drug screen targeted at Plasmodium liver stages identifies a potent multistage antimalarial drug. J. Infect. Dis. 2012, 205, 1278–1286. 10.1093/infdis/jis184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam T. G.; McNamara C. W.; Bopp S.; Dharia N. V.; Meister S.; Bonamy G. M.; Plouffe D. M.; Kato N.; McCormack S.; Bursulaya B.; Ke H.; Vaidya A. B.; Schultz P. G.; Winzeler E. A. A chemical genomic analysis of decoquinate, a Plasmodium falciparum cytochrome b inhibitor. ACS Chem. Biol. 2011, 6, 1214–1222. 10.1021/cb200105d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley R.; Leung S.; Fisher N.; Al-Helal M.; Berry N. G.; Lawrenson A. S.; Sharma R.; Shone A. E.; Ward S. A.; Biagini G. A.; O′Neill P. M. The development of quinolone esters as novel antimalarial agents targeting the Plasmodium falciparum bc1 protein complex. MedChemComm 2012, 3, 39–44. [Google Scholar]
- Zhang Y.; Clark J. A.; Connelly M. C.; Zhu F.; Min J.; Guiguemde W. A.; Pradhan A.; Iyer L.; Furimsky A.; Gow J.; Parman T.; El Mazouni F.; Phillips M. A.; Kyle D. E.; Mirsalis J.; Guy R. K. Lead Optimization of 3-carboxyl-4(1H)-quinolones to deliver orally bioavailable antimalarials. J. Med. Chem. 2012, 55, 4205–4219. 10.1021/jm201642z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maignan J. R.; Lichorowic C. L.; Giarrusso J.; Blake L. D.; Casandra D.; Mutka T. S.; LaCrue A. N.; Burrows J. N.; Willis P. A.; Kyle D. E.; Manetsch R. ICI 56,780 optimization: structure-activity relationship studies of 7-(2-phenoxyethoxy)-4(1H)-quinolones with antimalarial activity. J. Med. Chem. 2016, 59, 6943–6960. 10.1021/acs.jmedchem.6b00759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelarapu R.; Maignan J. R.; Lichorowic C. L.; Monastyrskyi A.; Mutka T. S.; LaCrue A. N.; Blake L. D.; Casandra D.; Mashkouri S.; Burrows J. N.; Willis P. A.; Kyle D. E.; Manetsch R. Design and synthesis of orally bioavailable piperazine substituted 4(1H)-quinolones with potent antimalarial activity: structure-activity and structure-property relationship studies. J. Med. Chem. 2018, 61, 1450–1473. 10.1021/acs.jmedchem.7b00738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birth D.; Kao W.-C.; Hunte C. Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat. Commun. 2014, 5, 4029. 10.1038/ncomms5029. [DOI] [PubMed] [Google Scholar]
- Hayward J. A.; Makota F. V.; Cihalova D.; Rajendran E.; Zwahlen S. M.; Shuttleworth L.; Wiedemann U.; Spry C.; Saliba K. J.; Maier A. G.; van Dooren G. G., A screen of drug-like molecules identifies chemically diverse electron transport chain inhibitors in apicomplexan parasites. bioRxiv 2022, 2022.02.13.480284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorjsuren D.; Eastman R. T.; Wicht K. J.; Jansen D.; Talley D. C.; Sigmon B. A.; Zakharov A. V.; Roncal N.; Girvin A. T.; Antonova-Koch Y.; Will P. M.; Shah P.; Sun H.; Klumpp-Thomas C.; Mok S.; Yeo T.; Meister S.; Marugan J. J.; Ross L. S.; Xu X.; Maloney D. J.; Jadhav A.; Mott B. T.; Sciotti R. J.; Winzeler E. A.; Waters N. C.; Campbell R. F.; Huang W.; Simeonov A.; Fidock D. A. Chemoprotective antimalarials identified through quantitative high-throughput screening of Plasmodium blood and liver stage parasites. Sci. Rep. 2021, 11, 2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonova-Koch Y.; Meister S.; Abraham M.; Luth M. R.; Ottilie S.; Lukens A. K.; Sakata-Kato T.; Vanaerschot M.; Owen E.; Jado J. C.; Maher S. P.; Calla J.; Plouffe D.; Zhong Y.; Chen K.; Chaumeau V.; Conway A. J.; McNamara C. W.; Ibanez M.; Gagaring K.; Serrano F. N.; Eribez K.; Taggard C. M.; Cheung A. L.; Lincoln C.; Ambachew B.; Rouillier M.; Siegel D.; Nosten F.; Kyle D. E.; Gamo F.-J.; Zhou Y.; Llinás M.; Fidock D. A.; Wirth D. F.; Burrows J.; Campo B.; Winzeler E. A. Open-source discovery of chemical leads for next-generation chemoprotective antimalarials. Science 2018, 362, eaat9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman C. D.; Siregar J. E.; Mollard V.; Vega-Rodríguez J.; Syafruddin D.; Matsuoka H.; Matsuzaki M.; Toyama T.; Sturm A.; Cozijnsen A.; Jacobs-Lorena M.; Kita K.; Marzuki S.; McFadden G. I. Parasites resistant to the antimalarial atovaquone fail to transmit by mosquitoes. Science 2016, 352, 349–353. 10.1126/science.aad9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson P. R.; Tan C.; Jarman K. E.; Lowes K. N.; Curtis J. M.; Nguyen W.; Di Rago A. E.; Bullen H. E.; Prinz B.; Duffy S.; Baell J. B.; Hutton C. A.; Jousset Subroux H.; Crabb B. S.; Avery V. M.; Cowman A. F.; Sleebs B. E. Optimization of 2-anilino 4-amino substituted quinazolines into potent antimalarial agents with oral in vivo activity. J. Med. Chem. 2017, 60, 1171–1188. 10.1021/acs.jmedchem.6b01673. [DOI] [PubMed] [Google Scholar]
- Bailey B. L.; Nguyen W.; Ngo A.; Goodman C. D.; Gancheva M. R.; Favuzza P.; Sanz L. M.; Gamo F.-J.; Lowes K. N.; McFadden G. I.; Wilson D. W.; Laleu B.; Brand S.; Jackson P. F.; Cowman A. F.; Sleebs B. E. Optimisation of 2-(N-phenyl carboxamide) triazolopyrimidine antimalarials with moderate to slow acting erythrocytic stage activity. Bioorg. Chem. 2021, 115, 105244. 10.1016/j.bioorg.2021.105244. [DOI] [PubMed] [Google Scholar]
- Boström J.; Hogner A.; Llinàs A.; Wellner E.; Plowright A. T. Oxadiazoles in medicinal chemistry. J. Med. Chem. 2012, 55, 1817–1830. 10.1021/jm2013248. [DOI] [PubMed] [Google Scholar]
- Monastyrskyi A.; Brockmeyer F.; LaCrue A. N.; Zhao Y.; Maher S. P.; Maignan J. R.; Padin-Irizarry V.; Sakhno Y. I.; Parvatkar P. T.; Asakawa A. H.; Huang L.; Casandra D.; Mashkouri S.; Kyle D. E.; Manetsch R. Aminoalkoxycarbonyloxymethyl ether prodrugs with a pH-triggered release mechanism: a case study improving the solubility, bioavailability, and efficacy of antimalarial 4(1H)-quinolones with single dose cures. J. Med. Chem. 2021, 64, 6581–6595. 10.1021/acs.jmedchem.0c01104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P. C.; Piplani M.; Mittal M.; Pahwa R. Insight into prodrugs of quinolones and fluoroquinolones. Infect. Disord. Drug Targets 2016, 16, 140–161. 10.2174/1871526516666160824153226. [DOI] [PubMed] [Google Scholar]
- Frueh L.; Li Y.; Mather M. W.; Li Q.; Pou S.; Nilsen A.; Winter R. W.; Forquer I. P.; Pershing A. M.; Xie L. H.; Smilkstein M. J.; Caridha D.; Koop D. R.; Campbell R. F.; Sciotti R. J.; Kreishman-Deitrick M.; Kelly J. X.; Vesely B.; Vaidya A. B.; Riscoe M. K. Alkoxycarbonate ester prodrugs of preclinical drug candidate ELQ-300 for prophylaxis and treatment of malaria. ACS Infect. Dis. 2017, 3, 728–735. 10.1021/acsinfecdis.7b00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L. M.; Crespo B.; De-Cózar C.; Ding X. C.; Llergo J. L.; Burrows J. N.; García-Bustos J. F.; Gamo F. J. P. falciparum in vitro killing rates allow to discriminate between different antimalarial mode-of-action. PLoS One 2012, 7, e30949 10.1371/journal.pone.0030949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Huang L.; Shulmeister V. M.; Chi Y. I.; Kim K. K.; Hung L. W.; Crofts A. R.; Berry E. A.; Kim S. H. Electron transfer by domain movement in cytochrome bc1. Nature 1998, 392, 677–684. 10.1038/33612. [DOI] [PubMed] [Google Scholar]
- Abraham M.; Gagaring K.; Martino M. L.; Vanaerschot M.; Plouffe D. M.; Calla J.; Godinez-Macias K. P.; Du A. Y.; Wree M.; Antonova-Koch Y.; Eribez K.; Luth M. R.; Ottilie S.; Fidock D. A.; McNamara C. W.; Winzeler E. A. Probing the open global health chemical diversity library for multistage-active starting points for next-generation antimalarials. ACS Infect. Dis. 2020, 6, 613–628. 10.1021/acsinfecdis.9b00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel S.; Rivero A.; Kyle D. Molecular basis of extreme resistance in Plasmodium falciparum to atovaquone and other mitochondrial inhibitors. Malar. J. 2014, 13, P81 10.1186/1475-2875-13-S1-P81. [DOI] [Google Scholar]
- Musset L.; Pradines B.; Parzy D.; Durand R.; Bigot P.; Le Bras J. Apparent absence of atovaquone/proguanil resistance in 477 Plasmodium falciparum isolates from untreated French travellers. J. Antimicrob. Chemother. 2006, 57, 110–115. 10.1093/jac/dki420. [DOI] [PubMed] [Google Scholar]
- Painter H. J.; Morrisey J. M.; Mather M. W.; Orchard L. M.; Luck C.; Smilkstein M. J.; Riscoe M. K.; Vaidya A. B.; Llinás M. Atypical molecular basis for drug resistance to mitochondrial function inhibitors in Plasmodium falciparum. Antimicrob. Agents Chemother. 2021, 65, e02143-20 10.1128/AAC.02143-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke H.; Lewis I. A.; Morrisey J. M.; McLean K. J.; Ganesan S. M.; Painter H. J.; Mather M. W.; Jacobs-Lorena M.; Llinás M.; Vaidya A. B. Genetic investigation of tricarboxylic acid metabolism during the Plasmodium falciparum life cycle. Cell Rep. 2015, 11, 164–174. 10.1016/j.celrep.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann J.; Corey V.; Scherer C. A.; Kato N.; Comer E.; Maetani M.; Antonova-Koch Y.; Reimer C.; Gagaring K.; Ibanez M.; Plouffe D.; Zeeman A.-M.; Kocken C. H. M.; McNamara C. W.; Schreiber S. L.; Campo B.; Winzeler E. A.; Meister S. High-throughput luciferase-based assay for the discovery of therapeutics that prevent malaria. ACS Infect. Dis. 2016, 2, 281–293. 10.1021/acsinfecdis.5b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke-Fayard B.; Janse C. J.; Cunha-Rodrigues M.; Ramesar J.; Büscher P.; Que I.; Löwik C.; Voshol P. J.; den Boer M. A. M.; van Duinen S. G.; Febbraio M.; Mota M. M.; Waters A. P. Murine malaria parasite sequestration: CD36 is the major receptor, but cerebral pathology is unlinked to sequestration. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 11468–11473. 10.1073/pnas.0503386102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke-Fayard B.; Trueman H.; Ramesar J.; Mendoza J.; van der Keur M.; van der Linden R.; Sinden R. E.; Waters A. P.; Janse C. J. A Plasmodium berghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol. Biochem. Parasitol. 2004, 137, 23–33. 10.1016/j.molbiopara.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Nguyen W.; Hodder A. N.; de Lezongard R. B.; Czabotar P. E.; Jarman K. E.; O’Neill M. T.; Thompson J. K.; Jousset Sabroux H.; Cowman A. F.; Boddey J. A.; Sleebs B. E. Enhanced antimalarial activity of plasmepsin V inhibitors by modification of the P(2) position of PEXEL peptidomimetics. Eur. J. Med. Chem. 2018, 154, 182–198. 10.1016/j.ejmech.2018.05.022. [DOI] [PubMed] [Google Scholar]
- Richardson L.; Ashton T. D.; Dans M. G.; Nguyen N.; Favuzza P.; Triglia T.; Hodder A.; Ngo A.; Jarman K. E.; Cowman A. F.; Sleebs B. E. Substrate peptidomimetic inhibitors of P. falciparum plasmepsin X with potent antimalarial activity. ChemMedChem 2022, 17, e202200306 10.1002/cmdc.202200306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favuzza P.; de Lera Ruiz M.; Thompson J. K.; Triglia T.; Ngo A.; Steel R. W. J.; Vavrek M.; Christensen J.; Healer J.; Boyce C.; Guo Z.; Hu M.; Khan T.; Murgolo N.; Zhao L.; Penington J. S.; Reaksudsan K.; Jarman K.; Dietrich M. H.; Richardson L.; Guo K. Y.; Lopaticki S.; Tham W. H.; Rottmann M.; Papenfuss T.; Robbins J. A.; Boddey J. A.; Sleebs B. E.; Sabroux H. J.; McCauley J. A.; Olsen D. B.; Cowman A. F. Dual plasmepsin-targeting antimalarial agents disrupt multiple stages of the malaria parasite life cycle. Cell Host Microbe 2020, 27, 642–658.e12. 10.1016/j.chom.2020.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles A.; Iqbal Z.; Vauterin P.; Pearson R.; Campino S.; Theron M.; Gould K.; Mead D.; Drury E.; O’Brien J.; Ruano Rubio V.; MacInnis B.; Mwangi J.; Samarakoon U.; Ranford-Cartwright L.; Ferdig M.; Hayton K.; Su X. Z.; Wellems T.; Rayner J.; McVean G.; Kwiatkowski D. Indels, structural variation, and recombination drive genomic diversity in Plasmodium falciparum. Genome Res. 2016, 26, 1288–1299. 10.1101/gr.203711.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinin I.; Sie D.; Bengtsson H.; van de Wiel M. A.; Olshen A. B.; van Thuijl H. F.; van Essen H. F.; Eijk P. P.; Rustenburg F.; Meijer G. A.; Reijneveld J. C.; Wesseling P.; Pinkel D.; Albertson D. G.; Ylstra B. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Res. 2014, 24, 2022–2032. 10.1101/gr.175141.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pockrandt C.; Alzamel M.; Iliopoulos C. S.; Reinert K. GenMap: ultra-fast computation of genome mappability. Bioinformatics 2020, 36, 3687–3692. 10.1093/bioinformatics/btaa222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron D. L.; Schröder J.; Penington J. S.; Do H.; Molania R.; Dobrovic A.; Speed T. P.; Papenfuss A. T. GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res. 2017, 27, 2050–2060. 10.1101/gr.222109.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron D. L.; Baber J.; Shale C.; Valle-Inclan J. E.; Besselink N.; van Hoeck A.; Janssen R.; Cuppen E.; Priestley P.; Papenfuss A. T. GRIDSS2: comprehensive characterisation of somatic structural variation using single breakend variants and structural variant phasing. Genome Biol. 2021, 22, 202. 10.1186/s13059-021-02423-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A.; Bertoni M.; Bienert S.; Studer G.; Tauriello G.; Gumienny R.; Heer F. T.; de Beer T. A. P.; Rempfer C.; Bordoli L.; Lepore R.; Schwede T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X. C.; Ubben D.; Wells T. N. C. A framework for assessing the risk of resistance for anti-malarials in development. Malar. J. 2012, 11, 292. 10.1186/1475-2875-11-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder C.; Chollet J.; Santo-Tomas J.; Scheurer C.; Wittlin S. In vitro and in vivo interaction of synthetic peroxide RBx11160 (OZ277) with piperaquine in Plasmodium models. Exp. Parasitol. 2007, 115, 296–300. 10.1016/j.exppara.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Ridgway M. C.; Cihalova D.; Maier A. G. Sex-specific separation of Plasmodium falciparum gametocyte populations. Bio-Protoc. 2021, 11, e4045 10.21769/BioProtoc.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delves M. J.; Straschil U.; Ruecker A.; Miguel-Blanco C.; Marques S.; Dufour A. C.; Baum J.; Sinden R. E. Routine in vitro culture of P. falciparum gametocytes to evaluate novel transmission-blocking interventions. Nat. Protoc. 2016, 11, 1668–1680. 10.1038/nprot.2016.096. [DOI] [PubMed] [Google Scholar]
- Delves M. J.; Miguel-Blanco C.; Matthews H.; Molina I.; Ruecker A.; Yahiya S.; Straschil U.; Abraham M.; León M. L.; Fischer O. J.; Rueda-Zubiaurre A.; Brandt J. R.; Cortés Á.; Barnard A.; Fuchter M. J.; Calderón F.; Winzeler E. A.; Sinden R. E.; Herreros E.; Gamo F. J.; Baum J. A high throughput screen for next-generation leads targeting malaria parasite transmission. Nat. Commun. 2018, 9, 3805. 10.1038/s41467-018-05777-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalaoui S.; Zougbédé S.; Charrin S.; Silvie O.; Arduise C.; Farhati K.; Boucheix C.; Mazier D.; Rubinstein E.; Froissard P. Hepatocyte permissiveness to Plasmodium infection is conveyed by a short and structurally conserved region of the CD81 large extracellular domain. PLoS Pathog. 2008, 4, e1000010 10.1371/journal.ppat.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton T. D.; Ngo A.; Favuzza P.; Bullen H. E.; Gancheva M. R.; Romeo O.; Parkyn Schneider M.; Nguyen N.; Steel R. W. J.; Duffy S.; Lowes K. N.; Sabroux H. J.; Avery V. M.; Boddey J. A.; Wilson D. W.; Cowman A. F.; Gilson P. R.; Sleebs B. E. Property activity refinement of 2-anilino 4-amino substituted quinazolines as antimalarials with fast acting asexual parasite activity. Bioorg. Chem. 2021, 117, 105359. 10.1016/j.bioorg.2021.105359. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.