

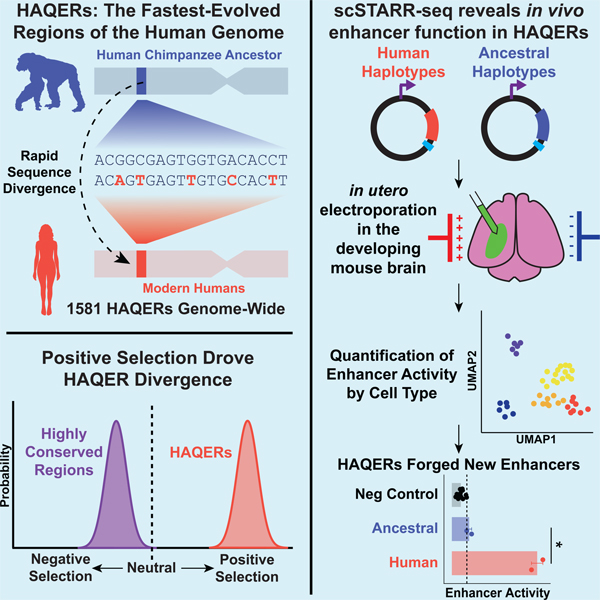
SUMMARY
Searches for the genetic underpinnings of uniquely human traits have focused on human-specific divergence in conserved genomic regions, which reflects adaptive modifications of existing functional elements. However, the study of conserved regions excludes functional elements that descended from previously neutral regions. Here we demonstrate that the fastest-evolved regions of the human genome, which we term Human Ancestor Quickly Evolved Regions (HAQERs), rapidly diverged in an episodic burst of directional positive selection prior to the human-Neanderthal split before transitioning to constraint within hominins. HAQERs are enriched for bivalent chromatin states, particularly in gastrointestinal and neurodevelopmental tissues, and genetic variants linked to neurodevelopmental disease. We developed a multiplex single-cell in vivo enhancer assay to discover that rapid sequence divergence in HAQERs generated hominin-unique enhancers in the developing cerebral cortex. We propose that a lack of pleiotropic constraints and elevated mutation rates poised HAQERs for rapid adaptation and subsequent susceptibility to disease.
Graphical Abstract
IN BRIEF
Most comparative genomics studies focus on conserved regions. However, in this study, Mangan et al. identify the fastest-evolved regions across the entire human genome and provide insights into which genomic regions underlie human-specific disease risks and adaptations.
INTRODUCTION
Humans can be distinguished from our recent great ape ancestors by many unique phenotypes, including bipedal locomotion1, craniofacial morphology2, and our remarkable cognitive capabilities3,4. Intertwined with adaptations are human-specific disease susceptibilities, including knee osteoarthritis5 and schizophrenia6. While both researchers and the public have a long-standing interest in understanding the genetic basis of human uniqueness, we have struggled to partition the millions of mutations that separate humans from our great ape ancestors into those that are neutrally evolving, and those that have significantly contributed to human-specific traits.
Initial systematic searches for the genetic basis of human traits focused on protein-coding regions to enrich for genetic changes with phenotypic effects7,8. More recent studies have identified several human-specific gene duplications that have been implicated in the expansion of the human neocortex9–11. However, humans and chimpanzees harbor few differences in amino acid sequences and it has long been hypothesized that the mutations responsible for human-specific phenotypes lie primarily in non-protein coding regulatory regions12–14.
A second generation of screens began with the insight that cross-species conservation could be utilized to enrich for functionally significant mutations in the non-protein coding genome. This allowed screens to expand from the 1% of the genome that is protein-coding to the 5% of the genome that includes highly-conserved regulatory elements. Genomic regions from these screens are termed Human Accelerated Regions (HARs)16. These screens identified HARs based on acceleration in the rate of nucleotide substitutions, positing that an increase in the rate of molecular evolution from prior constraint reflects a change in the mode of selection. Over the past 15 years, additional studies have expanded the set of HARs with the addition of more genome assemblies, specific tissues of interest, and alternative statistical methods17–20.
Many HARs act as developmental enhancers, demonstrating the feasibility of expanding beyond protein-coding regions to identify modifications to regulatory elements21,22. One example is a distal enhancer of the neurodevelopmental gene FZD8, where human-specific sequence changes increased enhancer activity in mouse embryonic brain, which was sufficient to accelerate neural precursor cell cycle dynamics and increase brain size23. As would be expected for genomic regions with important roles in neurodevelopment, mutations in HARs have been associated with schizophrenia and autism spectrum disorder6,24.
Preconditioning studies of human-specific traits on highly-conserved regions restricts analyses to 5% of the genome. However, a growing body of evidence suggests that much more of the genome is functional25,26. We propose that the combination of consortium efforts to catalog human genetic variation27 and recent advances in high-throughput functional genomic technologies28–31 provide an avenue to identify functionally significant regulatory innovations across the entire genome through the integration of comparative, population, and functional genomics.
The remaining 95% of the genome is likely to include two types of evolutionarily significant genomic regions not targeted in past studies: functional elements recurrently modified on independent lineages and functional elements unique to humans. Many distinctive characteristics of human anatomy, such as brain size, limb proportions, and craniofacial morphology are not static in non-human species, but rather are dynamic across the panoply of vertebrate life. As such, we expect many genetic determinants of these dynamic traits to be fast-evolving in both humans and non-human species and thus to exhibit function without the stringent condition of past constraint. Furthermore, regions with cross-species conservation, which have evolved under purifying selection, will not contain recently-evolved functional elements that are held under constraint only in humans. Both of these classes of regulatory innovations will be discovered in the underexplored nonconserved genome.
In this work, we integrate comparative genomics with genetic variation data from human populations to demonstrate that the fastest-evolved regions of the human genome, which we term Human Ancestor Quickly Evolved Regions (HAQERs), diverged rapidly through the combination of elevated mutation rates and positive selection. While HAQERs diverged rapidly from the human-chimpanzee ancestor, they are highly similar among extant and archaic hominins. HAQERs are enriched in bivalent domains, which are associated with spatiotemporally restricted developmental or environmentally-responsive regulatory elements. We developed in vivo scSTARR-seq as a multiplex, single-cell enhancer assay in the developing mouse cerebral cortex to demonstrate that rapid HAQER divergence forged functional elements that are exclusive to hominins. HAQERs are also enriched for disease-linked variation, suggesting an active role in shaping human-specific susceptibilities to disease.
RESULTS
Acceleration and velocity are associated with signatures of positive selection
Historically, it has been thought that higher rates of divergence in genomic regions are primarily associated with variation in the local mutation rate32, as opposed to selection. This is based on the notion that the vast majority of genetic differences between humans and great apes are selectively neutral and that positive selection is rare33.
Acceleration has been employed as a metric to mitigate the influence of local mutation rates, as a change in the rate of divergence is proposed to reflect a change in selective pressure. While this approach has been fruitfully applied to identify HARs as exciting candidates for further study in the highly-conserved genome, this strategy limits the scope of investigation to regions with an initial velocity near zero, excluding regions that have accelerated from neutrality to rapid divergence. Therefore, we sought to generalize acceleration to identify targets of positive selection in the remaining 95% of the genome.
We define the acceleration, a, of a genomic region as the difference between the current velocity of divergence, v, and the initial velocity of divergence, vo: a α Δv = v − v0. We define initial velocity, v0, as the divergence rate on the branch from the human-gorilla ancestor to the human-chimpanzee ancestor, and velocity, v, as the divergence rate on the branch from the human-chimpanzee ancestor to extant humans (Figure 1A). Both v0 and v are measured in units of genetic distance per base pair per million years, where distance is counted as the sum of substitutions, insertions, and deletions in a 500 bp window. Unlike previous work, we do not place a threshold filter on initial velocity, v0, allowing us to calculate acceleration, a, genome-wide from a syntenic alignment of the human, chimpanzee, gorilla, and orangutan reference genomes (Methods).
Figure 1: HAQERs, the fastest-evolved regions of the human genome.
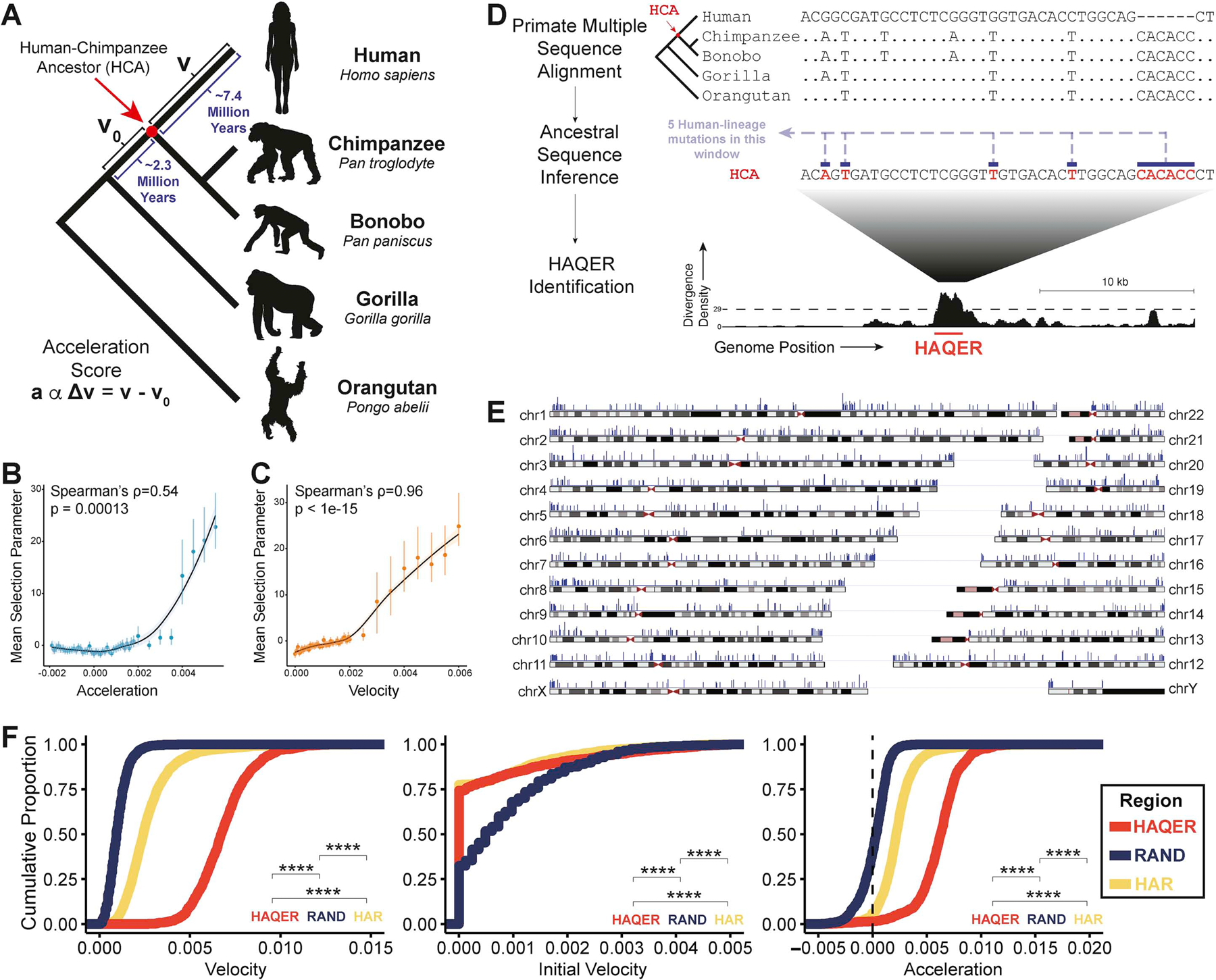
(A) We display the values of velocity (v), initial velocity (v0), and acceleration (a) in the phylogenetic context of recent human evolution. (B, C) Mean selection parameter estimates for 500bp genomic regions binned by either acceleration (B) or velocity (C). Error bars display the 95% highest density credible interval. Both acceleration and velocity correlate with signatures of selection in human populations. (D) HAQERs (Human Ancestor Quickly Evolved Regions) are identified as regions containing at least 29 mutations in a 500bp window (p < 10−6) that separate the inferred human-chimpanzee ancestor sequence from the human genome. We count insertions and deletions as one mutation regardless of their length. (E) Locations in the human genome of the 1581 HAQERs (blue markers). Marker amplitude reflects the maximum divergence density observed in each region. HAQERs are distributed across all human chromosomes and enriched near chromosome ends. (F) Cumulative distribution of velocity, initial velocity, and acceleration observed across HAQERs, Human Accelerated Regions (HARs), and random neutral proxy regions (RAND). Regions are filtered to a minimum element size of 50bp. (Bonferroni-adjusted Wilcoxon, ****; p < 0.0001). See also Figures S1–2.
To understand if acceleration is predictive of selective pressure, we analyzed the frequencies of derived alleles in African populations27 and inferred the direction and magnitude of selection acting on variants in genomic regions binned by acceleration values (Methods). Under negative selection, a derived allele is more deleterious than the ancestral allele, and thus unlikely to spread to high frequencies. Under positive selection, a derived allele is beneficial and more likely to be found at higher frequencies (Figure S1A). We implemented a statistical model to infer the mean selection parameter from derived allele frequency spectra (dAFS) using Markov chain Monte Carlo (MCMC)34 and corrected for ascertainment bias present when regions are identified based on divergence35 (Figure S1, Methods). We report that highly positive acceleration is associated with positive selection coefficients (Figure 1B) and may be an informative identifier of adaptive innovation genome-wide.
However, the most dramatically accelerated regions will still preferentially include regions with modified function and past constraint (low v0, high v) at the expense of recurrently modified functional elements (high v0, high v) and recently-functional elements from neutrally-evolving sequence (moderate v0, high v). This motivated us to examine the relationship between the current velocity of a genomic region and selection using the dAFS of variants in genomic regions binned by velocity. We observe a stronger relationship between velocity and selection than between acceleration and selection (Figure 1B, C).
We also observe a robust relationship between a and v (Figure S2A) but not between v0 and v (Figure S2B). This observation suggests that unlike at the megabase scale36, regional differences in the divergence rate at small scales are unlikely to reflect intrinsic variation in mutation rates that is stable across phylogenetic branches. Since we found that velocity and acceleration covary across the genome, we sought to disentangle the individual relationship between each of the two metrics and selection. When controlling for the other metric, we saw a strong relationship between velocity and selection, but not acceleration and selection (Figure S2C). These results indicate that rapid acceleration is associated with selection primarily in that it correlates with rapid velocity, a strong indicator of selection.
The fastest-evolved regions of the human genome
Encouraged by these findings, we implemented a computational screen to identify the most rapidly-evolved regions in the human lineage (Figure 1A). Using a syntenic genome-wide multiple alignment of great apes (human, chimpanzee, bonobo, gorilla, and orangutan), we inferred the probability of each nucleotide state in the human-chimpanzee ancestor at each alignment position (Methods). In order to more conservatively estimate genetic differences, we only considered a site divergent between the human-chimpanzee ancestor and humans when the ancestral inference estimated a base change with a probability of 80% or more (Methods). We use this conservative method to define Divergence Density as the genetic distance between the human-chimpanzee ancestor and the human genome for every 500 base pair window. If mutations were uniformly distributed across the genome at the rate observed in the fastest evolving 10 Mb genomic region, it would be unlikely to observe 29 or more mutations in a 500 bp window (p < 10−6, Bonferroni-corrected binomial, Methods). Thus, we define Human Ancestor Quickly Evolved Regions (HAQERs) as genomic regions with a Divergence Density of at least 29 evolutionary operations separating the human-chimpanzee ancestor and the human genome (Figure 1D). We identified 1581 HAQERs with an average length of 892 bp, which collectively includes ∼1.41 Mb of the human genome (Figure 1E).
As we ascertained HAQERs based on their rapid divergence, it follows that they exhibit higher velocities than either HARs or randomly selected neutral proxy regions (RAND) (Figure 1F, Figure S2D, Methods). HAQERs exhibit significantly lower initial velocity than RAND even though HAQERs were not directly ascertained based on conservation. HAQERs are also significantly more accelerated than HARs or RAND, reflecting the combination of their slightly lower initial velocity and their dramatic velocity.
HAQERs and HARs are largely independent genomic regions, with only six out of 2,733 expanded HARs24 overlapping HAQERs. One notable overlap is HAQER0035, which corresponds to HAR1, part of a well-studied RNA gene expressed in neurodevelopment15 (Figure S6C). HAQERs are also largely distinct from the fastest-evolved regions in chimpanzees and gorillas (Figure S2E). Thus, we have expanded beyond the highly conserved genome to identify over one thousand previously uncharacterized regions that represent the most rapidly-evolved regions in the human genome.
Sequence evolution in HAQERs was driven by both elevated mutation rates and directional positive selection prior to the Neanderthal split
As rapid sequence divergence in a genomic region can be generated by either variation in the local mutation rate or positive selection, we sought to determine the relative influence of these forces in HAQER evolution using recently available high-coverage human population sequencing data27. We first partitioned variants from 501 unrelated African individuals (Methods) to subsets overlapping HAQERs, HARs, RAND, ultraconserved elements (UCEs), ENCODE candidate cis-regulatory elements (cCREs)26, or missense variants (MISSENSE).
We calculated the density of polymorphic sites and divergent sites between modern humans and the inferred human-chimpanzee ancestor in these regions (Figure S3A, Methods). UCEs, which are regions that have undergone minimal sequence divergence during the last 100 million years37, exhibited very limited divergence and polymorphism density compared to RAND, whereas HAQERs exhibit significantly elevated densities of both polymorphic sites and divergent sites.
We observe the co-occurrence of HAQERs and genomic features associated with higher mutation rates, suggesting an underlying mechanism for the increased density of polymorphic sites in HAQERs. HAQERs are enriched for meiotic recombination double-stranded break hotspots (106 overlaps, 1.4x enrichment, p < 10−3), and towards the ends of chromosomes (Figure 1E, Figure S2F), both of which have been associated with elevated local mutation rates38–40. We also find that HAQERs are enriched for early replication timing41 (Figure S3C), consistent with the enrichment for meiotic recombination double-stranded break hotspots. Meiotic double-stranded breaks and subtelomeric regions are also associated with higher recombination rates42, and we observe a slight, yet significant, elevation of recombination rates in HAQERs43 (Figure S3B). GC-biased gene conversion has previously been explored as a possible contributor to the divergence observed in HARs16. We find that HAQERs demonstrate a slight enrichment for weak-to-strong divergent sites. However, this enrichment is significantly weaker than we observe in HARs (Figure S3D, E). These observations are consistent with the hypothesis that rapid HAQER divergence is driven by elevated mutation rates.
While many HAQERs appear to have elevated mutation rates, this does not rule out that these same elements harbor function and were positively selected. Indeed, we observed a significantly elevated proportion of fixed alleles relative to polymorphic alleles at sites that are divergent between the human-chimpanzee ancestor and the human genome, a statistic associated with positive selection (Figure S3F)44. To further explore positive selection as a contributing force to HAQER evolution, we constructed dAFS for each set of genomic regions. Again, HAQERs show signatures of positive selection driven by an enrichment of high-frequency derived alleles and a depletion of intermediate frequency alleles relative to RAND and the other sets of genomic regions (Figure 2A, D).
Figure 2: HAQER sequence divergence was driven by positive selection prior to the human-Neanderthal split.
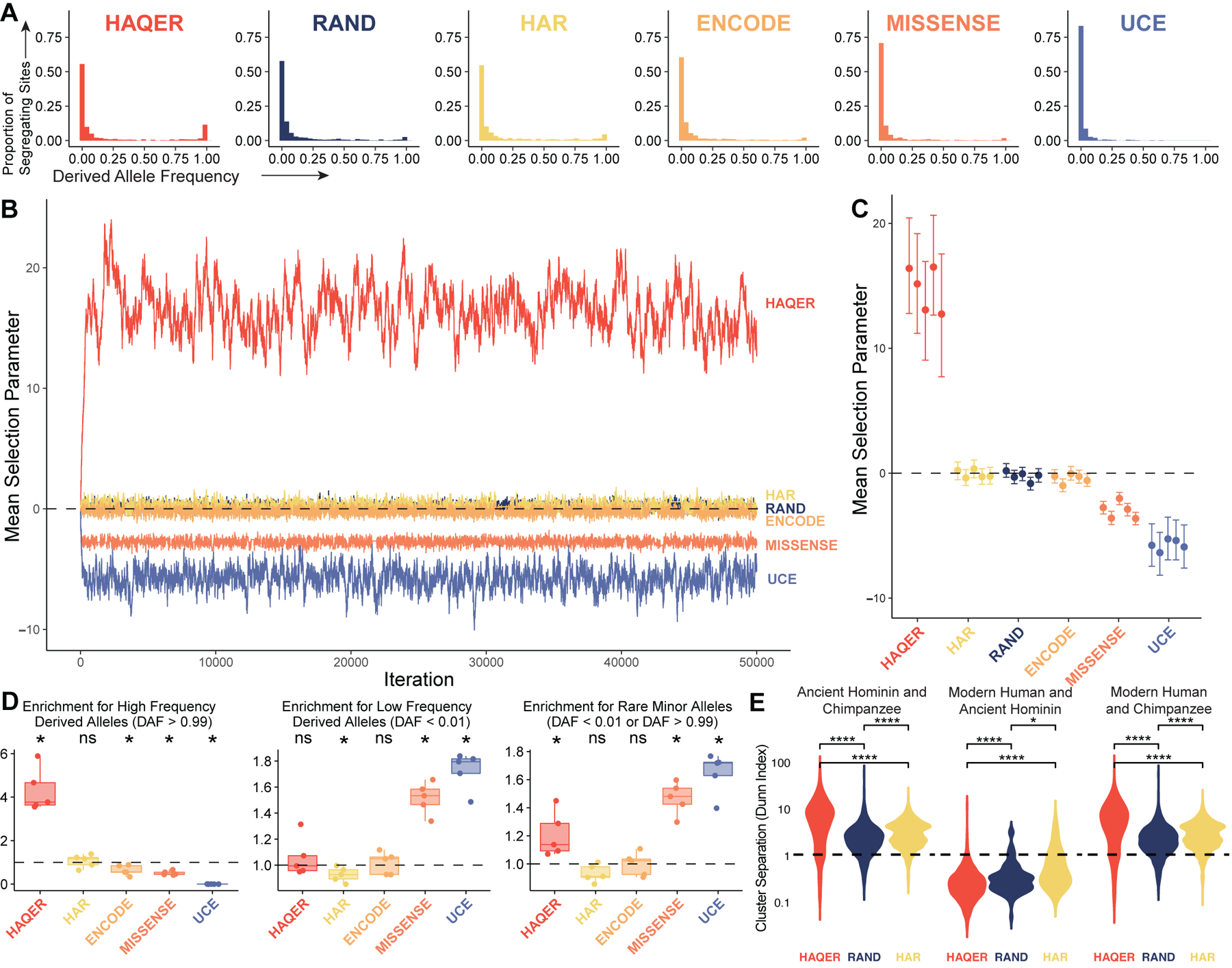
(A) Derived allele frequency spectra representing 501 individuals from African populations (1002 alleles) for segregating sites within HAQERs, RAND, HARs, ENCODE candidate cis-regulatory elements (cCREs), missense variants (MISSENSE), or ultraconserved elements (UCEs). (B) Representative MCMC trace for the mean selection parameter acting on segregating sites within each set of regions. (C) Posterior mean and 95% highest density credible intervals describing the mean selection parameters for each set of regions inferred from segregating sites from five independent populations of unrelated African individuals. (D) Enrichment for high derived allele frequency (DAF > 0.99, left), low frequency (DAF < 0.01, center), and rare minor allele (DAF < 0.01 or DAF > 0.99, right) segregating sites relative to RAND (*: p < 0.05, Bonferroni-adjusted Mann-Whitney U). Each point represents the enrichment for one population of individuals partitioned from the set of all African individuals. (E) Distribution of the cluster separation (measured as the Dunn Index) between Ancient Hominins and Chimpanzees (left), Modern Humans and Ancient Hominins (center), or Modern Humans and Chimpanzees (right). Comparisons are presented between HAQERs, RAND, and HARs (Bonferroni-adjusted Mann-Whitney U, *: p < 0.05, **: p < 0.01, ****: p < 0.0001). See also Figures S3.
To infer the magnitude of selective pressure across populations, we partitioned each dAFS into five component dAFS containing segregating variants from individuals in each of five populations (Gambian in Western Division – Mandinka, Mende in Sierra Leone, Esan in Nigeria, Yoruba in Nigeria, and Luhye in Webuye, Kenya). We evaluated the mean selection parameters acting on each population using MCMC (Figure 2B, Figure 2C; Methods). For HAQERs, the 95% credible intervals for the mean selection parameter acting on segregating sites is within the range of 12.7 to 16.5 and did not overlap intervals from any other variant set (Figure 2C). Roughly estimating the effective population size in humans at 104 individuals45, we estimate a mean selection coefficient for bases in HAQERs ranging from s = 0.000635 to 0.000825.
If HAQERs evolved under directional selection, we would expect variation between humans and chimpanzees to be much larger than the variation within humans for these regions. Alternatively, under diversifying selection, HAQER divergence between the human and chimpanzee reference genomes is instead the result of an increase in human variation without directionality. To investigate these alternatives, we analyzed the distribution of the Dunn Index, a conservative metric of cluster separation46, between clusters of modern human, ancient hominin, and chimpanzee sequences for HAQERs, HARs, and RAND (Figure 2E, Figure S3H–J). Dunn Index values of less than one suggest overlapping clusters while values greater than one suggest distinct, well-defined clusters.
HAQERs demonstrate greater cluster separation relative to RAND when comparing either humans or ancient hominin sequences to chimpanzees (Figure 2E). Significantly, most HAQERs have a Dunn Index of less than one between humans and ancient hominins, suggesting that ancient hominin HAQER sequences largely fall within the range of human variability (Figure 2E). These results are consistent with rapid directional selection in humans after the split with chimpanzees followed by a transition to constraint prior to the human-Neanderthal split.
While the dAFS model assumes infinite sites, we observe that sites in HAQERs with high derived allele frequencies exhibit an elevated proportion of transitions, which is characteristic of sites with back mutations to the ancestral state (Figure S3G, Methods). If a derived allele is advantageous for many sites in HAQERs, back mutations to the ancestral state would be deleterious by comparison, and these sites would be unlikely to drift from high to intermediate derived allele frequency. Thus, the enrichment for high frequency derived alleles observed in HAQERs may be magnified by the overabundance of fixed differences among divergent sites (itself a signifier of positive selection), an elevated mutation rate back to the ancestral state, and the maintenance of the derived state by purifying selection in modern humans.
While mutation rate variation impacts allele frequency spectra47, our results do not suggest that elevated mutation rates in neutral regions are the exclusive cause of rapid divergence in HAQERs. First, the relative depletion of intermediate frequency alleles (presented in Figure 2D as an enrichment for rare alleles) and the overabundance of fixed divergent sites compared to polymorphic sites in HAQERs is not expected in selectively neutral regions (Figure S3F). Furthermore, greater HAQER sequence cluster separation relative to RAND between modern humans and chimpanzees suggests directional evolution rather than the expansion of intraspecies variability as the cause of elevated divergence.
HAQERs are enriched in bivalent chromatin states
The conclusion that HAQERs evolved through directional, positive selection implies adaptive function in these regions. To test this hypothesis, we analyzed genome-wide patterns of enrichment and depletion in chromatin states across 127 reference epigenomes48 (Figure 3A, Figure S4A). Both HAQERs and HARs are significantly depleted in transcriptionally active chromatin states, consistent with past reports that most rapid evolution occurs outside of protein-coding regions16 and the predicted significance of noncoding regulatory regions to evolution13.
Figure 3: HAQERs are enriched in bivalent chromatin states.
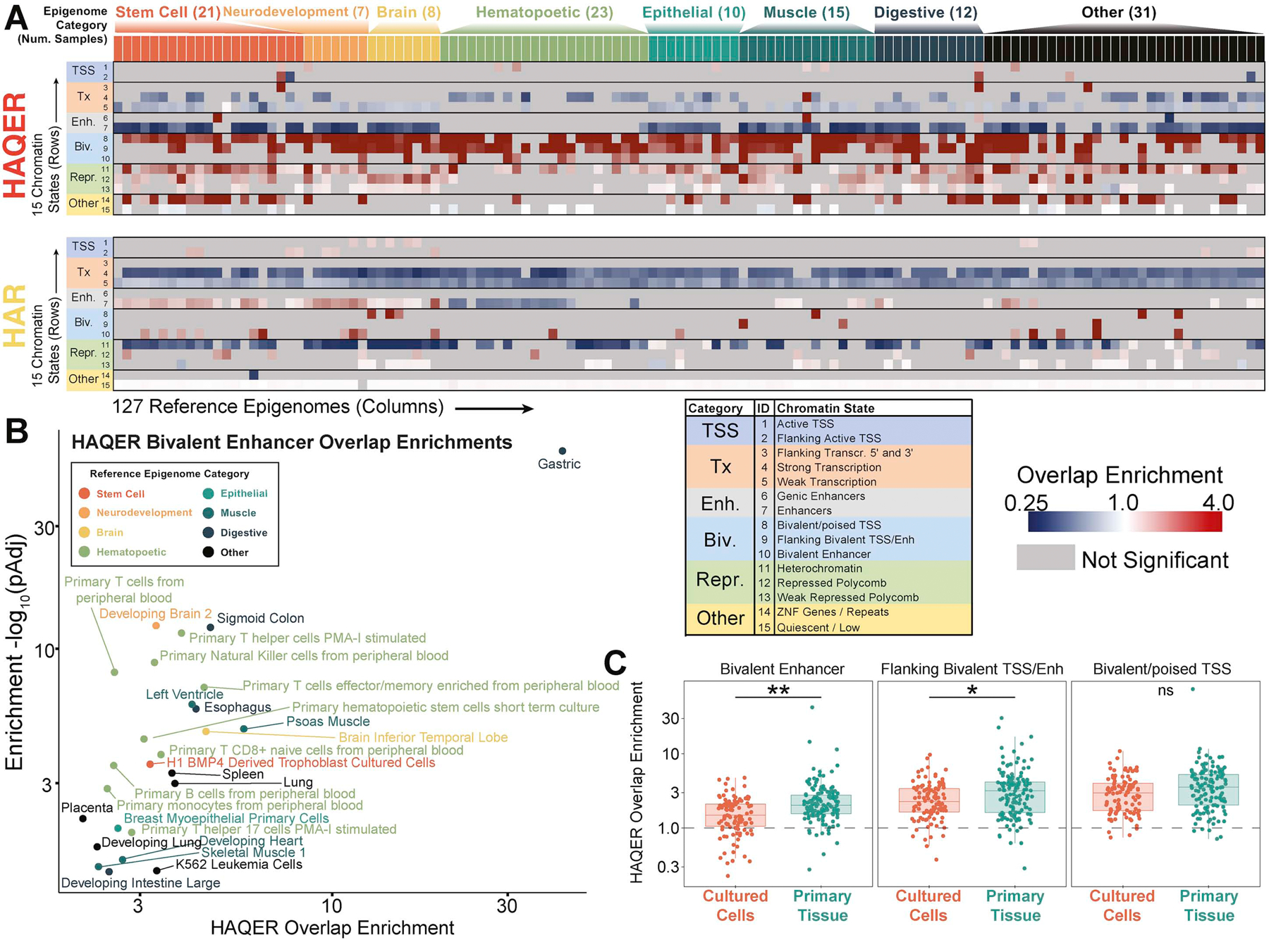
(A) Overlap enrichment/depletion matrix between HAQERs (top) or HARs (bottom) for 15 chromatin states (rows) from 127 reference epigenomes (columns). HAQERs are enriched for bivalent chromatin states but not for active enhancer and promoter states. An expanded matrix with individual sample annotations is presented in Figure S5A. (B) Volcano plot displaying significant overlap enrichments for HAQERs and the Bivalent Enhancer chromatin state in various tissues. (C) HAQER overlap enrichment for bivalent chromatin states compared between reference epigenomes derived from cultured cells and those derived from primary tissue (t-test, *: p < 0.05, **: p < 0.01). See also Figure S4.
Surprisingly, while HAQERs are not enriched for active enhancer or promoter states, they are strongly enriched for bivalent chromatin states (Figure 3A). Bivalent chromatin, which harbors both the polycomb repression mark H3K27me3 and the active promoter mark H3K4me3 and/or the active enhancer mark H3K4me1, is proposed to maintain expression of developmental and environmentally-responsive genes at low levels through active, yet rapidly reversible, silencing that allows precise activation49,50.
HAQERs are significantly enriched for bivalent chromatin states in both developing and adult primary tissues (Figure S4C). Evolutionary changes to developmental gene regulatory programs can alter adult morphology including allometric relationships. One example is gut reduction and brain expansion on the human lineage, which have been linked by the expensive tissue hypothesis51. Consistent with these dramatic changes, we observed the most significant enrichments for the bivalent enhancer chromatin state in gastrointestinal and neurodevelopmental reference epigenomes (Figure 3B, Figure S4E). As a glimpse of environmental response in adult tissues, we observe that two HAQERs transition from bivalent to active enhancer states in adult epithelial cells following exposure to dexamethasone, an anti-inflammatory glucocorticoid30 (enrichment p < 0.01, Methods).
While many of the observed bivalent states may represent domains in which individual histones bear both active and repressive modifications simultaneously (true bivalency), the observation of bivalent states in bulk ChIP-seq data may be a consequence of differential states of activation and repression in distinct cell types within heterogenous tissues (mixed cell bivalency). We observe stronger HAQER overlap enrichments for bivalent chromatin states in reference epigenomes derived from primary tissues than reference epigenomes derived from cultured cells, which represent a single cell type (Figure 3C); however, even reference epigenomes derived from cultured cells exhibit significant enrichments for bivalent states, suggesting both mixed cell and true bivalency in HAQERs.
In either scenario, genomic regions in bivalent states are likely to demonstrate more restricted spatial and temporal patterns of activity than regions with uniform active regulatory states in heterogenous tissues. In contrast to HAQERs, HARs are associated with active enhancer states (Figure S4B), which are thought to be associated with more broadly expressed genes49. Thus, enrichments for bivalency suggests that HAQERs encode gene regulatory elements with a high degree of specificity in development and environmental response.
HAQERs are enriched for recently-evolved neurodevelopmental gene regulatory elements
If the adaptive divergence observed in HAQERs underlies the innovation of developmental gene regulatory functions, we would expect differences in the epigenomic profiles between humans and closely related species. While cross-species epigenomic profiles of developing tissue are not broadly available, the developing cerebral cortex, due to its association with human cognition3, has been profiled across humans, rhesus macaques, and mice to identify putative enhancers and promoters in the human genome that were gained after the split between humans and rhesus macaques52. While HAQERs are not significantly associated with active enhancer or promoter states in the developing brain overall (Figure 3A, Figure S4A), HAQERs exhibit an enrichment for overlapping the subset of active enhancer or promoter chromatin states that were gained after the rhesus split (Figure S4D). While we observe enrichments between HAQERs and putatively gained gene regulatory activity identified across developmental stages and brain regions, HAQERs demonstrate the greatest enrichment for gained elements in the frontal lobe in late embryonic neurodevelopment (Figure S4D).
A multiplex, single-cell in vivo enhancer assay reveals hominin-specific neurodevelopmental enhancer activity in HAQERs
We identified the developing brain as a tissue of interest for in vivo analysis of HAQER function because HAQERs are enriched near genes associated with olfaction and cell recognition (Figure S2H), make 3D chromatin contacts with nervous system genes (Figure S2G), are enriched for neurodevelopmental regulatory elements gained after the human-rhesus split (Figure S4D), and are highly enriched for bivalent enhancers in the developing brain (Figure 3B). Notably, the brain has changed dramatically on the human lineage3 and is associated with many human-specific disease susceptibilities24.
Self-transcribing active regulatory region sequencing (STARR-seq) is a high-throughput sequencing-based assay in which the abundance of RNA transcripts containing a particular test sequence provides a quantitative measure of enhancer activity28. While STARR-seq has been effectively employed in cultured cell lines28,30,53, our results suggest that HAQERs function in spatiotemporally restricted contexts in highly heterogenous tissues such as late embryonic neurodevelopment54. As such, we developed in vivo single-cell STARR-seq (scSTARR-seq) to measure the enhancer activity of multiple test sequences simultaneously in developing brain tissue.
In this assay, we clone DNA sequences into a STARR-seq vector28 to form input libraries. We inject input libraries, along with a constitutive GFP transfection reporter plasmid, into embryonic mouse cerebral cortices via in utero electroporation (Figure 4A, Methods). Following dissection 16–18 hours later, we use fluorescence activated cell sorting to enrich for GFP+ cells for subsequent single-cell RNA sequencing. This approach allows us to interrogate enhancer activity in electroporated cells as well as their immediate progeny.
Figure 4: Rapid sequence divergence in HAQERs generated hominin-specific neurodevelopmental enhancers.
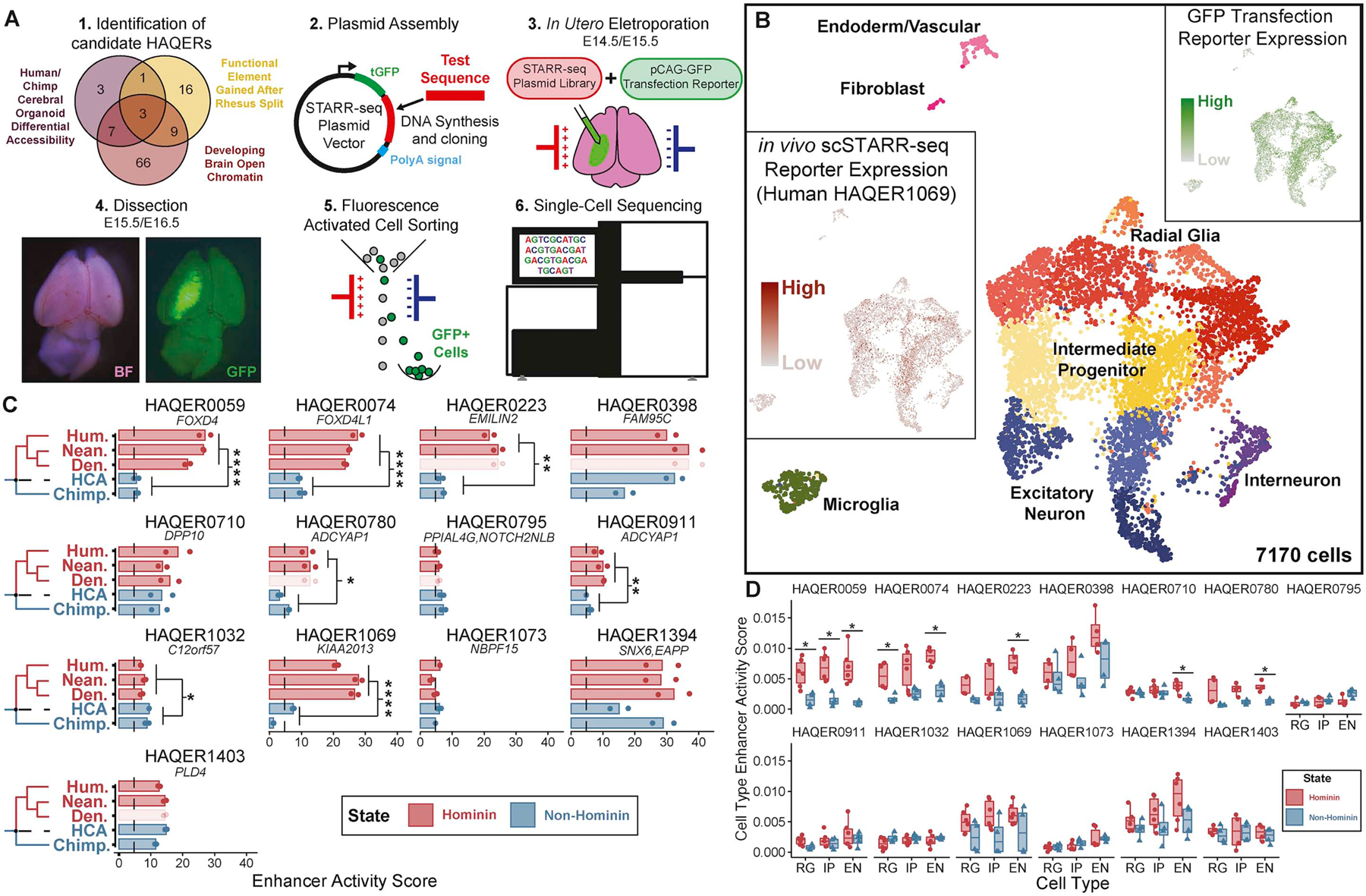
(A) Experimental design. Candidate HAQERs were prioritized based on overlaps with epigenomic datasets, cloned into a STARR-seq vector, and electroporated into developing mouse brains along with a pCAG-GFP transfection reporter. Single-cell sequencing followed dissection and FACS enrichment of GFP+ cells. (B) UMAP projection of 7,170 single cells from two scSTARR-seq experiments, labeled with metacluster identities. Inserts display cells colored by expression of the GFP transfection reporter and Human HAQER0169. (C) Enhancer activity score, defined as the input-normalized UMI count pooled across all cells per 1000 reporter UMIs, for 13 HAQERs. Nearest gene name is displayed below each HAQER ID. We display significant differences in enhancer activity between Hominin (Human, Neanderthal, Denisova) and non-Hominin (Chimpanzee, human-chimpanzee ancestor (HCA)) sequences (Bonferroni-adjusted t-test, p < 0.05). Faded bars represent sequences where Neanderthal and Denisovan had the same sequence in the 500bp genomic region and these duplicate sequences are not included in the statistical analysis. (D) Cell type enhancer activity score, or the input-normalized reporter UMI count normalized to the pCAG-GFP UMI count for each cell averaged across all cells in each metacluster (RG=Radial Glia, IP=Intermediate Progenitor, EN=Excitatory Neuron) (FDR- corrected t-test, p < 0.05). See also Figure S5–6.
To identify candidates for human-evolved neurodevelopmental regulatory elements, we identified 105 HAQERs that overlap one of three datasets: functional elements gained after the rhesus split52, open chromatin in the developing human brain48, or regions with differential chromatin accessibility between human and chimpanzee cerebral organoids, which recapitulate many features of early neurodevelopment55,56 (Figure 4A). We were able to commercially synthesize 40 of these sequences, a requirement for the analysis of extinct and ancestral alleles. We conducted a pilot assay with only the human alleles and selected the 13 with strongest signal for a full comparative analysis between the hominin (human, Neanderthal, and Denisovan) and non-hominin (chimpanzee and inferred human-chimpanzee ancestor) alleles (Supplementary Dataset 1).
We performed two independent in vivo scSTARR-seq experiments with this injection library and recovered STARR-seq reporter reads, endogenous RNA reads, and transfection reporter reads simultaneously from 7,170 single cells (Figure 4B). As these two experiments were performed at temporally close developmental timepoints (injections at E14.5 and E15.5), we observed a strong correlation between enhancer activity scores in both experiments (Figure S5E, F).
As most rapid sequence divergence in HAQERs occurred prior to the human-Neanderthal split, we expected similar patterns of enhancer activity among hominin sequences and compared enhancer activity between hominin and non-hominin sequences. Critically, 6 of the 13 HAQERs demonstrated significantly greater enhancer activity in the hominin ortholog test sequences than in the non-hominin sequences (Figure 4C, Figure S6A, B, D). Additionally, HAQER1032 showed a small but statistically significant decrease in enhancer activity in hominin orthologous sequences (Figure 4C). Many of the non-hominin sequences exhibit similar enhancer activity to random sequence negative controls. The lack of observed functionality of non-hominin alleles suggests that these HAQERs represent hominin-specific functional elements forged from previously neutrally evolving sequence, a class of elements excluded from previous comparative genomic screens reliant on functional constraint outside of humans.
We next sought to leverage the single-cell resolution of in vivo scSTARR-seq to determine the cell type specificity of enhancer activity in developing tissue for HAQERs. We annotated cell types utilizing developing brain cell atlases54,57 to calculate an enhancer activity score specific to each cell type (Figure 4B, D, Figure S5A, B, Methods). In utero electroporation preferentially targets ventricular progenitors. Thus, we observe the most GFP signal in radial glia and radial glial progeny, including intermediate progenitors and newborn excitatory neurons (Figure 4B, Figure S5C). While we resolved clusters with inhibitory neuron, microglia, and fibroblast cell type identities, these clusters exhibited limited GFP expression as they were not targeted by electroporation (Figure S5C). Therefore, to control for differences in transfection efficiency when calculating cell-type specific enhancer activity, enhancer activity scores were normalized to the amount of GFP observed in each cluster (Methods). We observed that five of our thirteen HAQER sequences demonstrated a significant increase in enhancer activity in hominin sequences in at least one cell type. While HAQER0911 and HAQER1032 exhibited significant hominin/non-hominin differences in bulk tissue, we did not observe a similar result at the metacluster level where we had less statistical power. Notably, HAQER0710 demonstrated hominin-specific enhancer activity in excitatory neurons, a result that was not visible in bulk tissue (Figure 4B,D). This result highlights the potential of single-cell technologies to uncover cell-type specific gene regulatory function in complex tissues.
As an orthogonal confirmation to our discovery of human-specific brain enhancers, we introduced the human and the human-chimpanzee ancestor sequence of HAQER0059 into an additional plasmid to test for enhancer-driven EGFP expression (Figure 5A, Methods). After in utero electroporation in the developing mouse brain, we observed robust expression of enhancer-driven EGFP for the human construct, but not the ancestral ortholog of HAQER0059 (Figure 5B, C), validating our multiplex sequencing assay with an independent fluorescence-based methodology.
Figure 5: Rapid divergence of hominin-specific neurodevelopmental enhancers near FOXD4 family genes followed multiple segmental duplications.
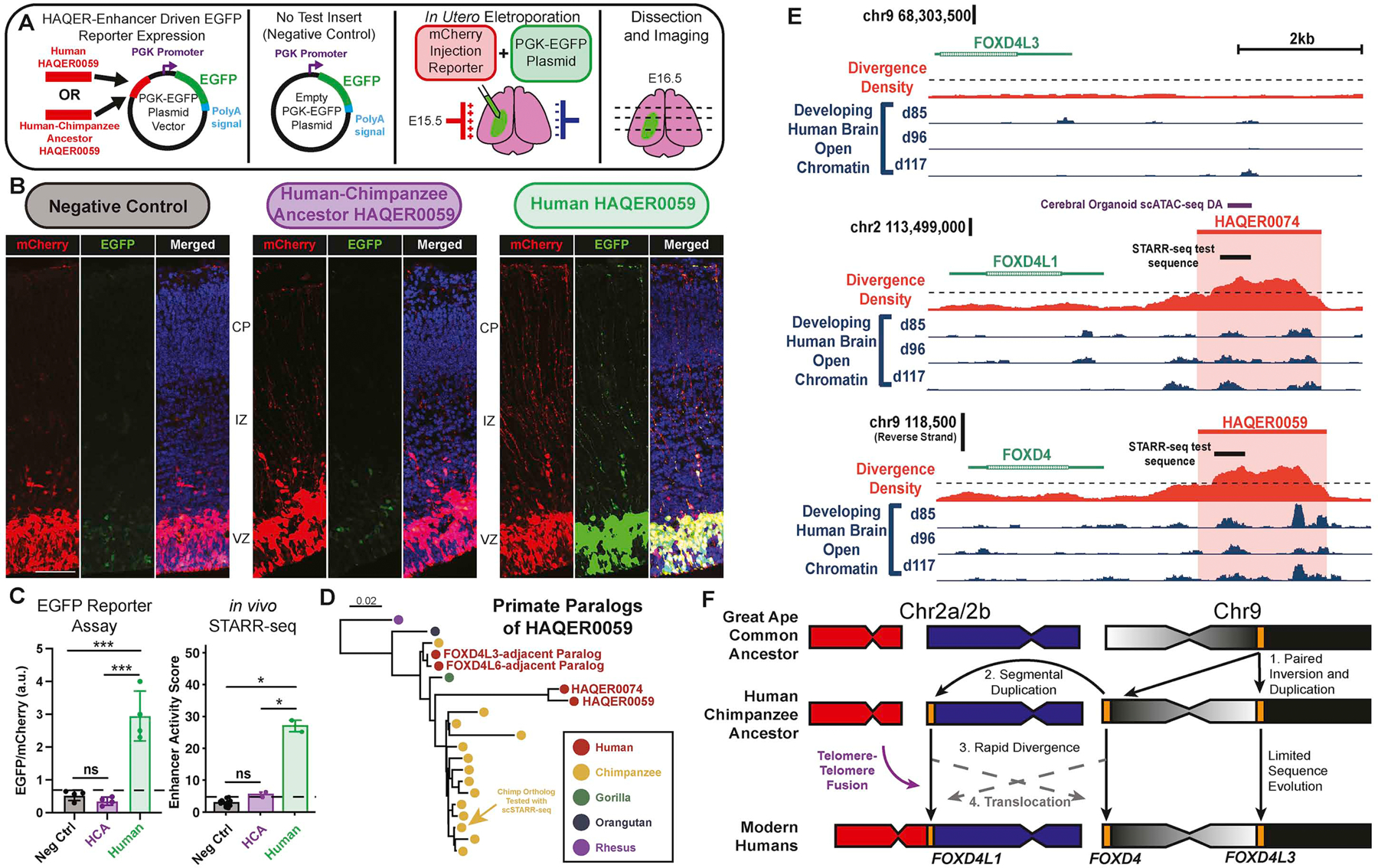
(A) Experimental design. We cloned the human or inferred human-chimpanzee ancestral sequence of HAQER0059 into an PGK-EGFP reporter plasmid and delivered plasmids to the developing cerebral cortex via in utero electroporation at E15.5 alongside an mCherry injection reporter. We performed dissection, sectioning, and imaging 24 hours later. (B) Representative images of Hoechst-stained coronal sections imaged for the mCherry injection reporter and EGFP enhancer reporter. Scale bar, 100 μm. (C) Left: Quantification of PGK-EGFP reporter signal normalized to the mCherry injection reporter for HAQER0059. Right: Corresponding in vivo STARR-seq results. (*: p<0.05, ***: p<0.001, FDR t-test. Dotted line: Negative Control Mean + 3SD). (D) Phylogeny of HAQER0059 homologs in humans and other great apes. (E) Genomic context for the paralogous regions near the genes FOXD4L3, FOXD4L1, and FOXD4. We present genomic context for the region near FOXD4, which contains HAQER0059, on the reverse strand. The region near FOXD4L3 does not have a nearby HAQER and shares synteny with the great ape ortholog. (F) A model of recent FOXD4 evolution. The great ape ortholog of the human gene FOXD4L3 generated the paralog FOXD4 in the chromosome 9 subtelomere through paired inversion and duplication. Subsequent duplication produced the paralog FOXD4L1 at the fusion site between the ancestral chromosomes 2a/2b, which formed the modern human chromosome 2. See also Figure S6.
Segmental duplication of human-specific paralogs follows rapid divergence in HAQERs
We observed that many HAQERs are contained within recent segmental duplications. This is consistent with the prevalence of differential expression between paralogous genes created by human-specific segmental duplications55,58. Two hominin-specific enhancers that we identified, HAQER0059 and HAQER0074, are located near the paralogs FOXD4 and FOXD4L1, respectively. FOXD4 encodes a forkhead-family transcription factor that is necessary for neuronal differentiation59,60 and implicated in psychiatric disorders61. The genome assemblies of mouse, gorilla, and orangutan contain one FOXD4 paralog, corresponding to the location of the human gene FOXD4L3 on chromosome 9. This suggests that one FOXD4 paralog was present in the great ape common ancestor. The short arm of the modern human chromosome 9 is inverted relative to gorilla and orangutan. In humans, FOXD4L3 is found near the inversion breakpoint and an additional paralog, FOXD4, is found at the other end of the inversion in the chromosome 9 subtelomere, suggesting a paired inversion and duplication event following the split with gorilla (Figure 5F). Chimpanzees exhibit an additional paralog in the subtelomere of chromosome 2b, suggesting an additional segmental duplication. In humans, this paralog corresponds to FOXD4L1 and is located at the site of the end-to-end fusion62 of the ancestral chromosomes 2a and 2b that formed the modern human chromosome 2 (Figure 5F). While HAQER0059 and HAQER0074 are both highly divergent from the human-chimpanzee ancestor, they exhibit 97.6% identity in the 500bp regions used as STARR-seq inserts63 (Figure 5D, E). However, the orthologous region near FOXD4L3 is not highly divergent from the ancestral sequence. While the similarity between HAQER0059 and HAQER0074 could be explained by convergent evolution, this would require over 100 parallel mutations. Thus, we propose that one of two paralogs rapidly diverged and a subsequent event translocated the highly diverged paralog to the paralogous location on the other chromosome, resulting in the same highly diverged sequence on the ends of both chromosomes 2b and 9.
Additionally, we observed 26 HAQERs in the 1q21.1–2 region containing the NBPF gene cluster (Figure S6E), which contains several human-specific segmental duplications10,64,65. NBPF genes contain Olduvai domains, which have undergone the most dramatic copy number increase of any protein-coding region in the human lineage65,66. Copy number of Olduvai domains is implicated in a dose-dependent manner with brain size, and deletions and duplications in this region are associated with microcephaly and macrocephaly, respectively67. These results are consistent with the hypothesis that adaptive increases of expression in FOXD4 and NBPF were achieved through the paired action of cis-regulatory innovation and segmental duplication. We propose that the cooperation between independent molecular mechanisms may be a common method of rapid evolution.
HAQER evolution shapes human disease susceptibility
To investigate the relationship between HAQERs and disease, we calculated if segregating variants in HAQERs are linked to SNPs that have been associated with human diseases and disorders through genome-wide association studies (GWAS). For each variant in the GWAS catalog, we used population resequencing data to identify the additional segregating sites that are in linkage disequilibrium with the reported SNP. We performed this calculation for all GWAS SNPs associated with an annotated trait to get the set of all observed linked variation for a particular GWAS trait (Figure 6, Methods). HAQERs are highly enriched for variation linked to GWAS traits including hypertension, neuroblastoma, and unipolar depression/schizophrenia/bipolar disorder (Figure 6B, Methods).
Figure 6: HAQERs are enriched near genetic variants linked to human disease.
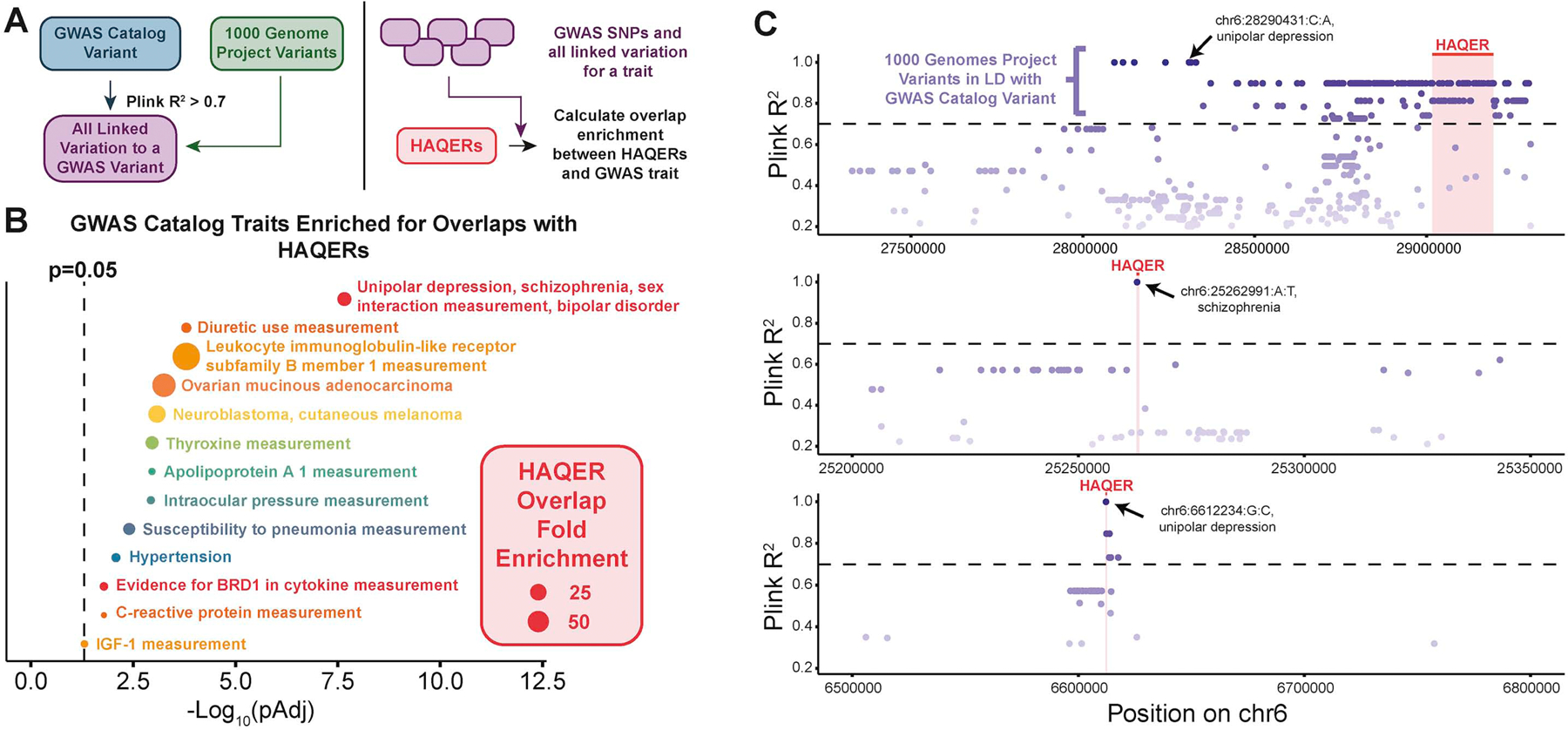
(A) Experimental design. For each GWAS catalog variant, we identified all linked variants (R2 > 0.7). We calculated overlap enrichments between HAQERs and the set of all linked variation for all SNPs associated with a GWAS trait. (B) FDR-corrected p-values for overlap enrichments between HAQERs and GWAS Catalog traits. The dotted line marks pAdj = 0.05. (C) Representative overlaps between HAQERs (red) and 1000 Genomes variants (purple) linked to GWAS catalog variants (black labels). See also Figure S6.
Variants in HAQERs could be associated with disease risk due to pleiotropic effects, where selection for an advantageous mutation in the HAQER element is accompanied by a deleterious side effect in an independent trait. In single-locus pleiotropy, the DNA segment of the HAQER element has multiple functions and the same variant that is selected for an advantageous change has an accompanying deleterious change, which is realized as a susceptibility to a disease. HAQERs do not show signs of locus-specific pleiotropy, as their single locus pleiotropy scores are much lower than HARs and similar to RAND, suggesting that many HAQERs perform more specific functions (Figure S6H, Methods). Alternatively, if HAQERs exhibit elevated haplotype lengths due to recent positive selection, we may expect HAQER disease enrichments to be the result of linkage disequilibrium-induced pleiotropy, where causal disease variants occur in elements distinct from HAQERs, but in the same haplotype. However, we do not expect elevated haplotype lengths in HAQERs, as the divergence in HAQERs had largely subsided prior to the human-Neanderthal split (Figure 2E), and selection-associated haplotype length elevation dissipates via recombination on the time scale of tens of thousands of years68. Indeed, segregating sites in HAQERs occur on smaller haplotypes than in random regions (Figure S6F,G, Methods), a reflection of their slightly elevated recombination frequency (Figure S3B). Thus, it is unlikely that HAQER disease enrichments are the result of linkage disequilibrium-induced pleiotropy. HAQER-associated disease susceptibility is not driven primarily by pleiotropic effects, as HAQERs do not exhibit significant pleiotropy either at their genomic position, or through linkage disequilibrium.
We propose that HAQERs confer disease susceptibilities in humans as they are located in genomic regions with elevated mutation rates (Figure S3B). We expect subsequent mutations to commonly occur in HAQERs and these mutations often to be deleterious and associated with disorders. It is likely that these disease susceptibilities will be specific to humans since many HAQERs are only functional in humans and, more generally, HAQERs have largely distinct gene ontology enrichments from HAQER-like regions in other species (Figure S2H). Thus, although HAQER evolution was adaptive in the human lineage, the association with disease variants suggests rapid divergence generated human-specific disease susceptibilities as consequences.
DISCUSSION
While there has been substantial disagreement in whether highly divergent regions reflect the action of natural selection69 or variation in the local mutation rate36, researchers have speculated that the careful integration of human population genetic data into comparative genomic efforts could effectively resolve the mutually confounding signatures of selection and mutation rate variation70.
While variation in local mutation rate and positive selection are often presented as mutually exclusive explanations for the generation of rapidly-evolved regions, we find evidence for both positive selection and elevated local mutation rates in HAQERs, suggesting that the combination of these two forces shaped the most divergent regions in the human genome.
Importantly, we identify that the adaptive evolution of HAQERs produced functional consequences in humans and ancient hominins. HAQERs are strongly enriched in bivalent chromatin, particularly in the gastrointestinal tract, immune system, and developing brain. We developed a multiplex single-cell enhancer assay to demonstrate that rapid sequence divergence in HAQERs forged hominin-specific gene regulatory elements.
HAQERs transitioned from rapid evolution following the human-chimpanzee ancestor to constraint among modern humans. Neanderthal and Denisovan HAQER sequences fall in the range of human variability for both sequence and function, suggesting that the rapid divergence of HAQERs largely predates this population split. While the recent accessibility of Neanderthal and Denisovan genomes has spurred substantial investigation into the differences between humans and these extinct hominins, many of the defining phenotypic transitions of the human lineage, including bipedalism and brain expansion, are shared among us. HAQERs, at the level of both sequence and function, separate humans from our great ape ancestors through rapid divergence, yet unite us as a species through modern constraint.
HAQERs and HARs show striking similarities in the anatomical specificity of their function. Both sets show enrichments for the brain and gastrointestinal tract. These consistent genomic enrichments parallel known anatomical changes on the human lineages of brain expansion and gut reduction. These two changes are proposed to have co-evolved to maintain a relatively constant basal metabolic rate51.
While HAQERs and HARs show similarities in the tissues they impact, we propose that these sets represent distinct classes of regulatory innovation during vertebrate evolution. HAQERs include de novo functional elements generated from neutral regions while HARs represent modifications of existing functional elements. This view is consistent with differences we observe between HAQERs and HARs in selection parameters, chromatin states, and pleiotropic effects. In terms of selection, HAQERs may be a better fit for a unimodal model of selection where many bases are under positive selection as a regulatory element is forged from neutral sequence. In contrast, HARs are modifications of existing functional elements and we expect their composition to be a mixture of bases under negative selection that maintain prior function and bases influenced by positive selection. Therefore, it is unsurprising that our selection model, which evaluates a selection parameter averaged across all sites, does not observe a substantial deviation from neutrality in HARs. In terms of chromatin states, HAQERs demonstrate strong and consistent enrichments for bivalent chromatin states, which are associated with spatiotemporally-restricted regulatory contexts, while HARs are associated with active enhancer states that function more broadly. Consistent with this functional specificity, we observe limited pleiotropic variation in HAQERs while HARs are substantially pleiotropic, as may be expected from modifying highly conserved active enhancers. This difference is consistent with newer and more specific functions in HAQERs compared to older and more multifunctional regulatory elements modified in HARs. Importantly, the relative contributions of gene regulatory element gain, loss, and modification to vertebrate evolution and disease remains unknown. We propose that forging functional elements from previously nonfunctional regions is likely to play an outsized role in regulatory differences between species by circumventing pleiotropic constraints that reduce the evolvability of many highly conserved developmental enhancers71.
The observation of high mutation rates in positively selected HAQERs is explained by the nonuniformity of evolvability in vertebrate genomes. As an example, populations of marine stickleback fish have independently adapted to freshwater habitats by reducing their pelvis through the deletion of a developmental enhancer72. While more than one enhancer deletion can achieve pelvic reduction73, wild populations recurrently exhibit deletions of the same enhancer located in a region that is highly susceptible to double-stranded breaks74. Often, many possible mutations can produce the same adaptive phenotype. When similarly adaptive mutations occur at different rates, mutations with higher rates of occurrence will be used preferentially for adaptation. In fact, we observe elevated mutation rates in HAQERs, and expect this pattern of elevated mutation rates in positively selected regions to be common throughout vertebrate life.
Some hypermutable regions utilized by adaptive evolution will retain their mutability in the derived state, such as regions prone to double-stranded breaks during meiosis, while other regions, including deletions at fragile sites, will not74. We propose that positively-selected regions which maintain hypermutability in the derived state will predispose organisms to disease susceptibility through subsequent deleterious mutations. Indeed, HAQERs are enriched for human genetic variants linked to diseases ranging from hypertension to neuropsychiatric disease. Thus, we anticipate a general correspondence between mutation rate, positive selection, and species-specific disease susceptibility across vertebrate evolutionary history.
Limitations of the study
As we conservatively limited our analysis to well-assembled syntenic regions to avoid the overestimation of divergence from paralog misalignment, we believe many highly-divergent regions between great apes have yet to be found. Many genome assembly gaps are located near centromeres, telomeres, and highly paralogous regions, which are also regions enriched for HAQERs. The discovery of these regions will likely require the completion of telomere-to-telomere assemblies of great ape species to resolve syntenic relationships. Second, confident ancestral sequence reconstruction of the human-chimpanzee ancestor allele requires a minimal level of identity between great ape species. Therefore, HAQERs may be missed in alignable regions where different mutations at the same base position occurred in many independent lineages. Similarly, our current method will not detect rapid evolution in positions where humans and other great apes have all independently evolved to the same derived state. Third, we focused our in vivo functional analysis on the developing brain. While we propose that HAQERs impact many anatomical locations, future work will be required to uncover how HAQER-mediated regulatory innovation impacts target gene expression and phenotypic changes across diverse tissues and stages. Finally, several HAQERs of interest overlapped simple repeat sequences. We were unable to test these HAQERs for enhancer activity due to limitations in current methods of DNA synthesis, which is required to investigate haplotypes of extinct and ancestral species.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, Craig B. Lowe (craig.lowe@duke.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All software written for this manuscript was implemented as a part of Gonomics, an ongoing effort to develop an open-source genomics platform in the Go programming language (golang). Gonomics can be accessed at https://github.com/vertgenlab/gonomics.
Raw and analyzed datasets, including browser tracks, sequencing files, multiple alignments, and variant sets used in selection analysis, have been made freely available on our lab website at https://www.vertgenlab.org/. Raw and analyzed datasets have also been deposited at GEO and are publicly available as of the date of publication at the accession number listed in the key resource table.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Stbl3 Chemically Competent E. coli | Invitrogen | Cat# C737303 |
| Chemicals, peptides, and recombinant proteins | ||
| Agencourt AMPure Beads | Beckman | Cat# A63881 |
| DNase-I | New England Biolabs | Cat# M0303S |
| EcoRI | New England Biolabs | Cat# R3101S |
| Fast Green FCF | Sigma-Aldrich | Cat# F7252 |
| FBS | ThermoFisher | Cat# 10438026 |
| Hoechst 33342 | Invitrogen | Cat# H1399 |
| NEG-50 | Richard-Allan Scientific | Epredia 6502 |
| Phusion DNA Polymerase | New England Biolabs | Cat# M0530S |
| SphI | New England Biolabs | Cat# R3182S |
| Trypsin-EDTA | Sigma-Aldrich | Cat# 59428C |
| Vectashield | Vector Laboratories | H-1000-10 |
| ZymoPURE II Plasmid Maxiprep Kit | Zymo Research | Cat# D4203 |
| Critical commercial assays | ||
| LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit | Invitrogen | Cat# L10119 |
| NovaSeq 6000 S-Prime Reagents | Illumina | Cat# 20040719 |
| Chromium Next GEM Single Cell 3’ Reagent Kit v3.1 | 10x Genomics | https://www.10xgenomics.com/support/single-cell-gene-expression/documentation/steps/library-prep/chromium-single-cell-3-reagent-kits-user-guide-v-3-1-chemistry |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | Cat# E2621L |
| NEBNext Ultra II FS DNA Library Prep Kit | New England Biolabs | Cat# E6177 |
| Deposited data | ||
| 1000 Genomes Project genomes | Byrska-Bishop et al. (2022) | https://www.internationalgenome.org/data-portal/data-collection/30x-grch38 |
| Altai Neanderthal genome | Meyer et al. (2012) | https://www.eva.mpg.de/genetics/genome-projects/neandertal/ |
| Combined Human Accelerated Region locations | Doan et al. (2016) | Table S1 of Doan et al. (2016) |
| Denisovan genome | Meyer et al. (2012) | https://www.eva.mpg.de/denisova/index.html |
| ENCODE cCRE locations and ChromHmm Datasets | Moore et al. (2020) | https://www.encodeproject.org/ |
| GWAS Catalog Variants | GWAS Catalog | https://www.ebi.ac.uk/gwas/ |
| HiCAR H1 and GM12878 | Wei et al. (2022) | GEO: GSE162819 |
| Human ChromHmm Roadmap Epigenomics data | Kundaje et al. (2015) | http://www.roadmapepigenomics.org/ |
| Human gained enhancer locations | Reilly et al. (2015) | GEO: GSE63648 |
| Individual chimpanzee genomes | Prado-Martinez et al. (2013) | https://www.ncbi.nlm.nih.gov/sra?term=SRP018689 |
| knownGene | Navarro Gonzalez et al. (2021) | https://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/ |
| Meiotic Recombination DSB hotspots | Pratto et al. (2014) | GEO: GSE59836 |
| Raw and processed sequencing reads | This study | GEO: GSE212159 |
| Recombination frequency maps | Zhou et al. (2020) |
ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/working/2013050 7 omnirecombination rates/ |
| Reference genomes: hg38, panTro6, panPan2, gorGor5, gorGor6, ponAbe3 | UCSC Genome Browser | https://hgdownload.soe.ucsc.edu/downloads.html |
| Replication timing datasets | Ding et al. (2021) | https://www.thekorenlab.org/data |
| Ultraconserved Element locations | Bejerano et al. (2004) | https://hgwdev.gi.ucsc.edu/ |
| Vindija Cave Neanderthal genome | Prüfer et al. (2017) | https://www.eva.mpg.de/neandertal/draft-neandertal-genome/data.html |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J (B6) (WT) | The Jackson Laboratory | JAX: 000664 |
| Oligonucleotides | ||
| Synthetic STARR-seq Insert Sequences | This study | Supplemental Dataset 1 |
| Targeted Enrichment Primers | This study | Supplemental Dataset 1 |
| Recombinant DNA | ||
| hSTARR-seq ORI vector | Addgene | RRID: Addgene 99296 |
| PGK-EGFP | Addgene | RRID: Addgene 169744 |
| Software and algorithms | ||
| bcl2fastq2 Conversion Software v2.20 | Illumina | https://support.illumina.com/downloads/bcl2fastq-conversion-software-v2-20.html |
| BWA 0.7.17 | Li and Durbin (2009) | https://github.com/lh3/bwa |
| CellRanger v6.0 | 10x Genomics | https://support.10xgenomics.com/ |
| ClustalW2 | (Larkin et al., 2007) | https://www.ebi.ac.uk/Tools/phylogeny/simple_phylogeny/ |
| gonomics | Vertebrate Genetics Laboratory | https://github.com/vertgenlab/gonomics |
| GraphPad Prism | GraphPad | https://www.graphpad.com/ |
| GREAT version 4.0.4 | McLean et al. (2010) | http://great.stanford.edu/public/html/ |
| ImageJ | Schindelin et al. (2012) | https://imagej.nih.gov/fiij/ |
| kentUtils | Kent et al. (2003) | https://github.com/ENCODE-DCC/kentUtils |
| lastz | Harris (2007) | https://github.com/lastz/lastz |
| multiz | Blanchette et al. (2004) | https://bio.tools/multiz |
| muscle | Edgar (2004) | https://www.ebi.ac.uk/Tools/msa/muscle/ |
| phylotree | Shank et al. (2018) | https://phylotree.hyphy.org/ |
| Plink | Purcell et al. (2007) | https://zzz.bwh.harvard.edu/plink/ |
| R version 4.0.5 | R Foundation for Statistical Computing | https://www.r-project.org/ |
| RPHAST | Hubisz et al. (2011) | https://github.com/CshlSiepelLab/RPHAST |
| Seurat v4.0 | Hao et al. (2021) | https://satijalab.org/seurat/ |
| SNPeff | Cingolani et al. (2012) | http://pcingola.github.io/SnpEff/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Wild type B6 mouse embryos at stages E14.5 and E15.5 were used for in utero electroporations for both scSTARR-seq and GFP enhancer reporter assays as described in Method Details. Embryos were assigned to experimental or control injection plasmids sequentially by position in the uterine horn. We did not restrict our scSTARR-seq or GFP enhancer reporter assays to embryos of only one sex; our data includes both developing males and females.
All experiments were performed in agreement with the guidelines from the Division of Laboratory Animal Resources from Duke University School of Medicine and the Institutional Animal Care and Use Committee of Duke University.
METHOD DETAILS
Human genetic variation preprocessing
To analyze the role of selection in shaping the fast-evolved regions of the human genome, we accessed haplotype-phased high-coverage genotype data from 2,504 human samples gathered by the 1000 Genomes Project27 from the url: https://www.internationalgenome.org/data-portal/data-collection/30x-grch38.
This genotype data underwent a series of transformations to prepare it for use in our selection analysis. First, we used gonomics: vcfFilter to retain only autosomal biallelic substitution variants in unrelated individuals. To reduce the impact of population bottlenecks introduced by human migration events, we only considered individuals from African populations (Gambian in Western Division – Mandinka, Mende in Sierra Leone, Esan in Nigeria, Yoruba in Nigeria, or Luhye in Webuye, Kenya). We implemented gonomics: vcfAncestorAnnotation to determine the ancestral allele for each variant using a pairwise alignment between the human reference sequence and the inferred Human-Chimpanzee ancestor sequence (see Ancestral state inference below). We retained variants where the ancestral and derived states could be clearly determined because one of the two alleles present in the extant human population matched the allele present in the inferred ancestral sequence. We removed polymorphic sites where neither allele matched the inferred ancestral sequence. We retained a total of 29,739,731 bi-allelic sites with genotype calls in 501 individuals (a total of 1002 alleles per site) after filtering and annotation.
We created subsets of these variants that overlap regions of interest for our comparative analyses. These regions of interest include six sets: HAQERs, HARs15, Ultraconserved Elements37 (UCEs), missense variants, ENCODE candidate cis-regulatory elements26, a random neutral proxy (RAND), which includes regions of the genome that do not overlap exons in known genes80 or ENCODE cCREs pseudorandomly selected from all ungapped bases in the hg38 assembly (gonomics: simulateBed; kentUtils: featureBits). We generated a set of missense variants from the 501 individuals from the 1000 Genomes Project using SnpEff81. We then subsampled these variant sets to contain a maximum of 1000 segregating sites for ease of computability in subsequent analyses using gonomics:vcfFilter -subset and gonomics:sampleVcf. To limit the impact of linkage disequilibrium on the shape of the derived allele frequency spectrum, we retained variants that had a minimum of 10,000 bases from any other variant in the sample set using gonomics:proximityBlockVcf. We generated derived allele frequency spectra from variant data with gonomics: vcfAfs.
For each population, we measured the proportion of three categories of derived allele frequencies (DAF): high frequency derived alleles (DAF > 0.99), low frequency derived alleles (DAF < 0.01), and rare minor alleles (DAF < 0.01 or DAF > 0.99). We then calculated the enrichment of each category as the proportion of alleles observed in each category relative to the proportion observed in our random neutral proxy set (RAND). Enrichments for a category of allele frequencies for a set of regions were calculated by a Bonferroni-adjusted Mann-Whitney U test compared to RAND (n = 5, corresponding to the five African populations).
Bayesian model design
To infer the direction and magnitude of selective pressure acting on the HAQERs, we implemented a hierarchical Bayesian model based on a statistical framework developed to infer the selective pressure acting in highly conserved genomic regions, using allele frequency data from human populations34.
We abstracted all filtered variant calls (see Human Genetic Diversity Data for Allele Frequency Analysis) for all base positions within the HAQER, or other set of genomic regions, into a set of segregating sites. We define a segregating site as a tuple, Sk, containing the quantities nk, the number of individual alleles with a genotype call for that segregating site, and ik, the number of individuals with the derived allele at the kth segregating site.
ik/nk therefore represents the derived allele frequency, or the proportion of individual sequences with the derived allele at that segregating site. Furthermore, we define S as a set of segregating sites, referred to henceforth as a derived allele frequency spectrum.
We assume that each segregating site in an allele frequency spectrum S is associated with its own selection parameter α, which is two times the product of a selection coefficient, s, and the haploid effective population size, Ne.
Therefore, the set of selection parameters corresponding to each of n segregating sites in a derived allele frequency spectrum S is represented by the vector quantity α.
We assume that each αk in α is independently selected from a normal distribution with mean μ and standard deviation σ, where the probability that an individual value α is selected follows the function f(α|μ, σ). Therefore, μ represents the mean selection parameter of a set of variants. Regions under neutral selection should exhibit μ ≈ 0 with μ < 0 and μ > 0 indicating negative and positive selection, respectively.
We also define the quantity Θ to represent the following set of parameters.
Using Bayesʹ rule, we can represent the posterior distribution of a particular parameter set given an observed allele frequency spectrum P (Θ|S) with the following equation.
Here P(S|α) represents the likelihood function of a derived allele frequency spectrum S for a given α, f(α|μ, σ) is a normally distributed prior, while g(μ) and h(σ) are hyperpriors. h(σ) is a gamma-distributed hyperprior on σ, Gamma(2, 10), and g(μ) is a normally-distributed hyperprior on μ, Normal(0, 3). This model is therefore a hierarchical Bayesian model as f(α|μ, σ), the prior distribution for the parameter set α, is governed by the hyperparameters μ and σ.
Likelihood calculations
In the Wright-Fisher model, the stationarity distribution of derived allele frequencies p can be described as a function of αk, the selection parameter for a particular segregating site, with the following equation82:
When a finite number of alleles, nk, are sampled from a population, we do not know the true derived allele frequency, but for a particular segregating site, a density function, F, can be defined as the product of the stationarity density and the binomial density of observing a segregating site at a particular discrete allele frequency ik/nk integrated over all possible derived allele frequencies p34:
The probability of observing a particular derived allele frequency ik can then be expressed as follows:
Thus, the likelihood of observing a derived allele frequency spectrum, S, for a given set of selection parameters, α, can be represented as the product of the allele frequency probability for each segregating site.
MCMC evaluation of selection parameters
We evaluated the posterior distribution P (Θ|S) with gonomics: selectionMcmc, which implements the Metropolis-Hastings algorithm, a method of Markov Chain Monte Carlo (MCMC) sampling.
The Metropolis-Hastings algorithm begins with an initial set of parameters, Θ, and draws a new set of parameters, Θʹ, based on the current parameter set. To draw this new parameter set, a new value for σ denoted σʹ is first selected as a random value from a Normal distribution, Normal(σ, sigmaStep), where σ is the value from the previous iteration and sigmaStep is a constant that may be changed for optimal parameter space exploration (we use 0.01). This makes it possible for a proposed σʹ to be less than zero, which is outside the support for h(σ) and will be evaluated to have a zero probability of acceptance. Next, a new value of μ (μʹ) is drawn from a normal distribution, Normal(μ, muStep), where muStep is a second tuning parameter that controls parameter space exploration, which we set to 0.5. We tuned sigmaStep and muStep to arrive at an acceptance probability near 0.5. We generated a proposal for α (αʹ) by drawing each from a Normal(μʹ, σʹ).
Due to symmetry in the proposal functions, where proposing μʹ and σʹ when at the current values of μ and σ would be equal to proposing μ and σ when at current values of μʹ and σʹ, we are able to reduce the acceptance probability for the candidate parameter set Θʹ to:
If a new parameter set Θʹ is accepted, it serves as the initial parameter set in the following iteration. Over many iterations, the random walk of the Θ parameter set forms a Markov Chain whose stationarity distribution represents the posterior distribution for its parameters.
We implemented gonomics: mcmcTraceStats to calculate the mean and 95% highest density credible interval for each chain, discarding the first 5,000 iterations as burn-in for variant sets overlapping regions of evolutionary interest.
Divergence-based ascertainment corrections
HAQERs, and other sets of genomic regions in our analyses, were defined based on the level of divergence between the human reference assembly and other species. This creates a systematic bias where regions in the reference assembly with low divergence are enriched for segregating sites with a low derived allele frequency. Similarly, regions in the reference assembly with a high divergence are enriched for segregating sites with a high derived allele frequency. This is because segregating sites with low derived allele frequencies are likely to appear non-divergent when sampling a single human allele (the reference assembly) and segregating sites with high derived allele frequencies are more likely to appear divergent when sampling a single human allele. This issue has been extensively explored by Kern (Kern, 2009), who describes a mathematical framework for correcting this ascertainment bias. Utilizing this framework, we introduce a corrected version of the likelihood function that is conditioned on the divergence-based ascertainment, Asc, of a set of variants:
To calculate this corrected likelihood function, we use a special case of the Kern correction where only one human allele (the allele from the reference genome) has been used for ascertainment. We represent the probability that a segregating site Sk = {ik, nk} is identified as divergent between two genomes as:
Here, nk represents the number of individuals with a genotype call for the segregating site k, including the reference genome as an additional observation of an allele. Conversely, the probability of ascertaining a segregating site in the ancestral state of a function of its allele frequency is:
Using Bayesʹ Theorem, we can then represent the corrected allele frequency probability expression as:
In this equation, the denominator represents a constant normalization factor:
We applied this correction to each segregating site in region sets generated through divergence-based criteria (i.e., HAQERs, HARs, and UCEs) by using the options - divergenceAscertainment and -includeRef in the program gonomics: selectionMcmc.
MCMC validation with synthetic datasets
In order to validate our MCMC selection model, we evaluated the ability of our model to recover known selection parameters used to generate synthetic data. To this end, we designed and implemented gonomics: simulateVcf to generate synthetic allele frequency spectra based on a particular selection parameter, α.
To generate an allele frequency spectrum, S, we generated individual segregating sites Sk with the parameters {ik, nk}. To simulate segregating sites for a particular selection parameter, α, our program first generated Beta-distributed random variates p ∈ (0, 1) from a distribution with the parameters 5000 * Beta(α = 0.001, β = 0.5). We selected these parameters so that the resulting distribution B(p) could serve as a bounding function for the allele frequency stationarity distribution φ(p|α) when α is between −10 and 10. In symbolic terms:
With this function in hand, we could then perform bounded rejection sampling to recover random variates from the stationarity distribution φ(p|α) by accepting variates from B(p) with the following probability:
This provides us with a method for generating synthetic derived allele frequencies for a set of segregating sites in a large population that are evolving under the given value of α.
To test our method we need segregating sites to be represented as finite samples from this population in the form (ik, nk). To that end, we simulate nk draws from a binomial distribution with a success probability of pk. The number of successes becomes ik. If ik were equal to 0 or nk (representing the cases where a site that is segregating in the population is not detected as segregating in the sample), this result was discarded and the process repeated with a new pk.
We generated 10 independent synthetic datasets for five values of the selection parameter α (i.e. −4, −2, ~0, 2, 4) for a total of 50 synthetic derived allele frequency spectra. As the stationarity distribution is undefined at α = 0, we used α = 0.01 to represent near neutral selection (~0). Representative spectra are displayed in Figure S1A.
To estimate selection parameters from synthetic data, we performed MCMC sampling on each dataset for 50,000 iterations starting from near neutral initial parameters Figure S1B. The mean and 95% credible intervals from the inferred posterior distributions for the mean selection parameter are displayed in Figure S1C, calculated after discarding the first 5,000 iterations as burn-in. The true value of the selection parameter used to generate each dataset is displayed as a vertical dashed line.
We implemented the program gonomics: simulateDivergentWindowsVcf to verify our ability to correct for divergence-based ascertainment biases in synthetic derived allele frequency data sets, using our special case of the Kern correction35. For each replicate experiment, we generated 1000 sets of variants, each containing 100 simulated segregating sites generated from a stationarity distribution with a fixed selection parameter α. The number of divergent sites generated in each set was then calculated, and we returned the top 1% or bottom 1% of sets ordered by the number of divergent sites. We generated 10 replicates of upper and lower divergence variant sets for three values of the selection parameter α: strong positive selection (α = 5), strong negative selection (α = −5), and neutral expectation (α = 0.01) We then used gonomics: selectionMcmc with and without the divergence-based ascertainment bias correction to assess its impact on our estimation of the mean selection parameter.
Genome-wide multiple alignment
We generated a genome-wide alignment to identify the fastest-evolved regions in the human, chimpanzee, and gorilla genome using the following assemblies: Human (Homo sapiens, hg38), Chimpanzee (Pan troglodytes; panTro6), Bonobo (Pan paniscus; panPan2), Gorilla (Gorilla gorilla, gorGor5), and Orangutan (Pongo abelii, ponAbe3).
We downloaded each reference assembly from the UCSC Genome Browser83 and generated local pairwise alignments with LASTZ84. We used the human-chimp.v2 scoring matrix with parameters (O=600 E=150 T=2 M=254 K=4500 L=4500 Y=15000)83. We then chained the local alignments together using kentUtils: axtChain85.
We took several additional steps to prevent and remove misalignments during chaining. First, to prevent the generation of chained alignments bridging assembly gaps, we chained alignments in each gapless regions of the genome independently and only considered gapless regions greater than 1 Mb of the human genome and greater than 20 kb for each of the query genomes. This filtering allowed us to ensure a large genomic context to better separate orthologs from paralogs. We also generated a custom scoring matrix (O=20 E=5):
| A | C | G | T | |
| A | 3 | −11 | −8 | −12 |
| C | −11 | 3 | −11 | −8 |
| G | −8 | −11 | 3 | −11 |
| T | −12 | −8 | −11 | 3 |
and gap penalty function:
| tableSize | 5 | ||||
| smallSize | 11 | ||||
| Position | 1 | 2 | 3 | 11 | 111 |
| qGap | 12 | 19 | 24 | 43 | 420 |
| tGap | 12 | 19 | 24 | 43 | 420 |
| bothGap | 25 | 40 | 50 | 90 | 700 |
for the axtChain program to more conservatively chain local alignments by preventing the chaining of alignments spanning large gaps in the target or query. We filtered the chains to have a minimum score of 50,000 and used kentUtils: chainNet to generate the final pairwise alignments for each alignable position of the human genome.
We used MultiZ86 to generate the multi-species genome-wide alignment and converted the output into an aligned FASTA file (gonomics: mafToFa). Subsections of this alignment were displayed using gonomics: multFaVisualizer.
Divergence velocity and acceleration analysis
To analyze the velocity and acceleration of genomic regions on the human branch we reduced our genome-wide alignment to four species: Human (Homo sapiens, hg38), Chimpanzee (Pan troglodytes; panTro6), Gorilla (Gorilla gorilla, gorGor5), and Orangutan (Pongo abelii, ponAbe3) using gonomics: faFilter and estimated the branch lengths in 500bp windows with gonomics: multiFaAcceleration.
This method estimates the branch lengths for a phylogenetic tree as the set of branch lengths that minimizes the error term Q, which represents the squared difference between the pairwise distances between the sequence of two species, D, and the patristic distance separating these two species on the tree, d, while constraining branch lengths to be non-negative87,88. We measured pairwise distances in terms of the number of differences separating two sequences, which includes both substitutions, insertions, and deletions, where each insertion or deletion counts as one difference regardless of length. As all species needed to be present in the alignment for us to calculate the branch lengths for a given region, we implemented gonomics: mafToBed to generate a BED file of all such regions.
Two branch lengths from this tree are used in the subsequent calculations: b0, which represents the distance between the human-gorilla ancestor and the human-chimpanzee ancestor, and b1, which represents the distance between the human-chimpanzee ancestor and the extant human genome assembly. We then defined the quantity v as the velocity score, or the rate of divergence over the branch b1 measured in units of mutations per site per million years of evolution. With 500 base pair windows and 7.4 million years of evolution between the human-chimpanzee ancestor and extant humans89, v can be calculated as follows:
Similarly, we define the initial velocity score v0, or the rate of divergence over the branch b0 in units of differences per site per 1 million years of evolution, as follows:
Finally, we define the quantity a, the acceleration score, as the change in velocity between branches b0 and b1:
The genome-wide average velocity score is 9.18 · 10−4 differences per site per 1 million years of evolution. Given an estimate of 7.4 million years of evolution between humans and the HCA, or a total of 14.8 million years of independent evolution separating extant humans from extant chimpanzees, our model would estimate an average sequence divergence of 1.36% in alignable regions between humans and chimpanzees, which is consistent with past estimates36. The genome-wide average acceleration score is 3.12 · 10−5.
To calculate the relationships between initial velocity, velocity, and acceleration we first pseudorandomly sampled 2.9 million 500 bp genomic windows (gonomics: bedFilter -subset). We then partitioned these genomic windows into subsets corresponding to particular ranges of velocity and acceleration scores (gonomics: bedFilter -minNameFloat/maxNameFloat, gonomics: intervalOverlap).
For each acceleration and velocity subset, we identified the biallelic SNPs that were segregating in these windows and calculated mean selection parameters associated with the given range of velocity or acceleration, as described in MCMC Evaluation of Selection Parameters. MCMC chains were run for 10,000 iteration with the first 1,000 iterations discarded as burn-in. As these regions were identified on the basis of divergence, we applied the Kern correction for divergence-based ascertainment bias for all chains.
Along with calculating initial velocity, velocity, and acceleration for 500 bp windows, we also calculate these scores for diverse sets of genomic regions where the length of genomic segments is variable (gonomics: branchLengthsMultiFaBed). We use a length of 50bp as a minimum to prevent large fluctuations in these scores seen in very small elements and use the more general equation with the genomic length, l, is a variable:
Ancestral state inference
We implemented gonomics: primateRecon to estimate the ancestral allele states using a maximum likelihood framework90 from our alignment of the human, chimpanzee, bonobo, gorilla, and orangutan genomes. We used this program to estimate both the human-chimpanzee ancestor and the human-gorilla ancestor.
We first estimated the neutral rate of evolution based on four-fold degenerate sites in codons using the knownGenes track on the UCSC Genome Browser as our gene set. We used PHAST: msa view to extract four-fold degenerate codon sites and estimated branch lengths for a fixed-topology tree using a Jukes-Cantor model of evolution91 by maximum likelihood92 (PHAST: phyloFit).
A base was determined to be present in the ancestral node if a base is present in at least two species on two independent lineages connected to the ancestral node. For alignment columns where an ancestral base was determined to be present, we first reconstructed the probabilities of A, C, G, and T in the ancestral node using the tree inferred from four-fold degenerate sites90. We then used one of two methods to assign a single base to the ancestor from these four probabilities. These distinct methods of ancestral state inference reflect the specific experimental use cases for the resulting inferred sequences. In the first method, we bias the reconstruction towards an extant species base by mandating that the sum of probabilities for the three other bases must be greater than or equal to 0.8 for the most likely base to be assigned as the ancestral state. This method produced a conservative estimation of divergent sites between modern and ancestral species and was used in the ascertainment of HAQERs, chimp-AQERs, and gorilla-AQERs. We used our second method of ancestral state inference for annotating the ancestral allele for segregating sites among modern humans. In this method, we first implemented gonomics: vcfToFa to construct a FASTA format sequence of the human reference genome where the reference allele at each segregating site is replaced with the alternate allele from a VCF format file. We then appended this sequence to our multiple alignment and treated both the reference and alternate human sequence with equal weight. we then calculated the four base probabilities for the human-chimpanzee ancestor and accepted the most likely allele as the ancestral state if its probability was greater than or equal to 99%. For uncertain positions, we assigned an N to the ancestral state to ensure that only high confidence SNPs were retained for subsequent analysis of derived allele frequencies.
HAQER Identification
We identified Human Ancestor Quickly Evolved Regions (HAQERs), or regions of the human genome with an increased density of differences when compared to the human-biased estimate of the human-chimpanzee ancestor.
We calculated the number of evolutionary operations (including substitutions, insertions, and deletions) that would be needed to convert our reconstruction of the human-chimpanzee ancestor’s genome into the human reference genome (hg38), for every 500 bp sliding window (gonomics: faFindFast). We used our reconstruction of the human-chimpanzee ancestor that conservatively uses the identity of the human base when the statistical model is uncertain (when the most likely base has a probability of less than 0.8). This results in us having high confidence in the changes on the human lineage that we do identify, which are likely to be a lower-bound on the total number of evolutionary operations that occurred in each window.
To assign statistical significance to HAQERs, we first constructed a null model by scanning the genome with a 10 Mb window to calculate the number of mutations in the fastest evolving 10 Mb section of the human genome since the split with chimpanzee. This rate of high-confidence genomic changes is 0.0126899 evolutionary operations per site, which we use as the rate of divergence μ in our null model37,93. With this rate of divergence for our null model, we are able to calculate uncorrected p-values associated with observing N changes within a 500 bp window with the R command pbinom(N − 1, 500, μ, lower.tail = FALSE, log.p = FALSE). When N = 29, our false discovery rate is 1.52096 · 10−7. We merged all overlapping 500 bp windows containing at least 29 evolutionary operations separating the human-chimpanzee reconstruction and the human reference genome (gonomics: bedFilter, bedMerge). This resulted in our final set of 1581 HAQERs.
We performed nearly identical procedures to identify the corresponding fastest-evolved regions in the chimpanzee genome (chimp-AQERs) and gorilla genome (gorilla-AQERs) using biased ancestor estimates for each of these species. We identified 2497 chimp-AQERs and 2885 gorilla-AQERs. We report overlap enrichments between HAQERs, chimp-AQERs, and gorilla-AQERs using gonomics:overlapEnrichments.
To generate ideograms for the visualization of the genomic locations of HAQERs, we converted a BED file of HAQER coordinates into a text file compatible with the UCSC Genome Graphs visualization tool such that the amplitude of each region is proportional to its maximum divergence density (gonomics: formatIdeogram). To visualize divergence density between the reconstructed human-chimpanzee ancestor and the human reference genome, we converted the BED file listing divergences for each 500 bp window into a wiggle (WIG) format track for the UCSC Genome Browser (gonomics: faFindFast, bedScoreToWig). We then converted this WIG track into a binary wiggle (bigWig) format track for final visualization on the browser (kentUtils: wigToBigWig).
Chromosome location
We generated sets of BED files containing genomic elements that are pseudorandomly generated and uniformly distributed in the human, chimpanzee, and gorilla genomes to quantify the enrichment of HAQERs near chromosome ends (gonomics: simulateBed). As chimp-AQERs and gorilla-AQERs were identified on hg38 coordinates, we used kentUtils:liftOver to project these regions onto coordinates for panTro6 and gorGor6, respectively, to measure distance from chromosome ends in the correct syntenic context for each species. We then calculated the distance to chromosome ends for both HAQERs and pseudorandom regions and compared the mean distance from the chromosome end (t-test) and proportion of elements within 5 megabases of the chromosome end (Chi-squared).
GREAT Ontology Analysis
We used the Genomic Regions Enrichment of Annotations Tool94 (GREAT) to identify enriched Gene Ontology (GO) Biological Process gene sets nearby HAQERs, chimp-AQERs, and gorilla-AQERs lifted to the human reference genome hg38. We report significant enrichments in terms of Bonferroni-adjusted Binomial p values using the whole genome as the background region.
We also used GREAT to identify GO Biological Processes enriched near 3D chromatin contact sites of HAQERs. To this end, we accessed chromatin contact sites identified in H1 hESC and GM12878 cell lines by HiCAR95. We then identified the set of all genomic regions forming distant 3D chromatin contacts with HAQERs in each cell type gonomics:intervalContacts. We report significant enrichments in terms of Bonferroni-adjusted hypergeometric p-values using the set of all chromatin contact sites for each cell line as background regions.
Mutation rate and fixation estimation
We first generated a list of all divergent positions between hg38 and the inferred human-chimpanzee ancestor (gonomics: multiFaToVcf). We then generated a set of all divergent sites that overlap specified genomic regions, including HAQERs, HARs, RAND, ENCODE, and UCE gonomics: intervalOverlap. Next, we calculated the divergent sites per base as the number of divergent sites divided by the total length in base pairs of the input set of genomic regions. Similarly, polymorphic sites per base were calculated as the number of variants from the 501 African individual subset of the 1000 Genomes Project data (see Human Genetic Diversity Data for Allele Frequency Analysis) that overlapped each set of genomic regions divided by the length in base pairs of that set.
We also intersected the set of divergent positions with the set of all polymorphic sites identified in the 501 African individual subset of the 1000 Genomes Project data (gonomics: intervalOverlap). Positions found in both sets were labeled as polymorphic divergent sites, and divergent sites not found in the 1000 Genomes Data were labeled as fixed divergent sites. We then determined the sets of fixed and polymorphic divergent sites overlapping each set of genomic regions (gonomics: intervalOverlap). We plotted the proportion of divergent sites that are polymorphic as the number of polymorphic sites divided by the sum of polymorphic and fixed divergent sites and assigned significance via the Chi-squared test of independence against the observed ratio of polymorphic divergent sites in RAND. The 2x2 contingency table for this analysis for a set of regions, X, had the dimensions {X, RAND} and {Fixed, Polymorphic}.
Recombination and replication timing
To measure recombination frequencies for regions of interest, we intersected a genome-wide recombination map estimated from all Yoruba individuals in the 1000 Genomes Project43 with HAQERs, HARs, and RAND. We also accessed a BED format file of meiotic double stranded break hotspots39 (GEO: GSE59836) and intersected this dataset with HAQERs. Overlap enrichments were quantified with gonomics:overlapEnrichments and intersecting HAQERs were identified with gonomics:intervalOverlap. We also accessed a dataset of replication timing in 300 iPSC lines41 and generated a dataset representing the average replication timing for each genomic region across these 300 iPSC lines. This dataset was then lifted to hg38 coordinates (kentUtils: liftOver) and intersected with HAQERs, HARs, and RAND gonomics: intervalOverlap.
Mutation spectrum analysis
For each genomic region of interest, we gathered the set of all divergent positions between hg38 and the inferred human-chimpanzee ancestor and partitioned this set into six classes of mutations (A → G/T → C) (G → A/C → T ) (A → T/T → A) (G → C/C → G) (A → C/T → G) (C → A/G → T ) (gonomics: divergenceSpectrum). We then calculated the proportion of HCA divergent sites that are weak to strong mutations for each genomic element (A → G/T → C or (A → C/T → G). We constructed a matrix of six values for each genomic region, with each value relating to the proportion of overlapping HCA divergent sites in each mutation class for principal component analysis in the R programming language.
Back mutation analysis
We hypothesized that while most low frequency derived alleles (DAF<0.1) will represent nearly exclusively forward mutations (in which the ancestor allele mutates to a derived variant), high frequency derived alleles (DAF>0.9) will represent a mixture of forward mutations and back mutations (in which a since diverged derived allele mutates back to the ancestral state). Forward mutations occur with unequal probabilities of transitions and transversions. We estimated the proportion of transitions in forward mutations tf to be equal to the proportion of transitions across all segregating sites in RAND (tf = tseg = 0.685; gonomics: vcfInfo). Back mutations will also occur with unequal probabilities of transitions and transversions. For the ancestral allele to be phased at a segregating site, a back mutation must revert to the ancestral allele state. We define tdiv to be equal to the proportion of transitions across all divergent sites in RAND (tdiv = 0.668). This model allows tseg to differ from tdiv, as would be the case if transition/transversion biases change over evolutionary time. However, in our analysis of the human lineage, tf and tdiv are similar. There are two scenarios in which a back mutation can occur. First, the inverse transition of a divergent transition will occur at a rate proportional to tf · tdiv. Second, the inverse transversion of a divergent transversion will occur at a rate proportional to (1 −tf )(1 −tdiv)/2, as there are two possible reverse transversions for a divergent site.
Thus, the expected proportion of transitions in back mutations will be equal to:
Based on our estimates of tf and tdiv, we estimate tb = 0.897. In other words, segregating sites that are back mutations should exhibit a quantifiable elevation in the proportion of transitions.
We use the following mixture model to estimate the relative proportion of forward and back mutations in a set of segregating sites with a proportion of transitions x:
Here f represents the proportion of forward mutations and (1 − f) represents the proportion of back mutations. We measured that segregating sites in HAQERs with DAF > 0.9 exhibit a proportion of transitions x ≈ 0.75. This figure implies that approximately 30% of segregating sites in HAQERs at DAF > 0.9 are back mutations.
Great ape genome divergence analysis
We constructed a 30-way whole-genome multiple alignment86 to analyze patterns of divergence and constraint in HAQERs. This alignment included five reference genomes: hg38, panTro6, panPan2, ponAbe3, and gorGor5. In addition to these reference genomes, we generated reference-based haploid assemblies from individuals within a species to survey intraspecific variability. To this end, we aligned the short-read sequencing data from individuals to the corresponding reference assembly and calculated the consensus allele for each position (gonomics: samConsensus).
We generated consensus sequences for three high coverage sequencing data sets from archaic hominins: a 30x coverage Denisovan genome from Denisova Cave in the Altai Mountains96, a 52x coverage Neanderthal genome also from the Denisova Cave in the Altai Mountains97, and a 30x coverage Neanderthal genome from the Vindija Cave in Croatia98. In addition to these archaic genomes, we also included the consensus sequences from 10 diverse, unrelated human individuals accessed from the 1000 Genomes Project (HG00096, HG01112, HG03052, NA18525, NA20502, HG00419, HG01879, HG01500, HG03742, NA18939 )99. Additionally, we included sequencing data from the following 12 chimpanzee individuals100, comprised of three individuals from each chimpanzee subspecies: Pan troglodytes verus (SRX243499, SRX243488, SRX243446), Pan troglodytes schweinfurthii (SRX237583, SRX237539, SRX237526), Pan troglodytes ellioti (SRX243519, SRX243518, SRX24351), and Pan troglodytes troglodytes (SRX243489, SRX243492, SRX243496).
We used the Dunn Index46 to quantify interspecies divergence in the context of intraspecies variability. We calculated the Dunn Index for each region in a set of regions as the ratio of the minimum intercluster sequence distance to the maximum intracluster distance (gonomics: dunnIndex). We restricted our Dunn Index analysis to regions with at least 5 segregating sites and where every individual had aligned sequence to the region.
Chromatin state enrichment analysis
To analyze chromatin state enrichments and depletions we used the ChromHMM classification of 127 epigenomes, which was produced as part of the Roadmap Epigenomics consortium48. We calculated the overlap enrichment and depletion between two sets of genomic elements (set 1 and set 2) in using our previously described statistical framework101 (gonomics: overlapEnrichments). We define the search space for this method as the area in the genome in which elements of set 1 and set 2 can be found, which includes all ungapped genomic regions greater than 1mb in length. If an individual genomic element from set 2 of length L were randomly distributed in the search space, the probability that it overlaps an element in set 1 can be expressed as the number of positions an element of size L can be placed in the search space that overlap an element of set 1 divided by the total number of positions in the search space in which an element of length L can be placed. The probability of observing k overlaps out of n trials, where n is equal to the number of elements in set 2, thus follows the Poisson binomial distribution. When the number of trials is large, the Poisson binomial distribution can be approximated with a normal distribution with the following mean, μ, and variance, σ2:
We report the enrichment between two sets of elements as the ratio between the observed number of overlaps and μ, the expected value of overlaps. We calculate Bonferroni-adjusted p values for enrichment and depletion with the following formulas:
We used 2C, where C is the number of comparisons, as the Bonferroni adjustment, as we tested for both enrichment and depletion for each pair of genomic elements. Significance was assigned for enrichment and depletion at p < 0.05.
To investigate the relationship between HAQER bivalent chromatin enrichments and environmental response, we accessed 17 ChromHMM datasets from untreated human A549 cells or A549 cells at various timepoints following dexamethasone (dex) treatment. These datasets were accessed from the ENCODE consortium website at the following accession numbers: ENCFF107YWL, ENCFF662GGJ, ENCFF161LGJ, ENCFF524GBP, ENCFF877NZN, ENCFF246IPY, ENCFF146UIL, ENCFF113TCU, ENCFF324PWA, ENCFF255QUQ, ENCFF052NXZ, ENCFF646AJN, ENCFF108TED, ENCFF910RII, ENCFF845TIM, ENCFF513UFQ, ENCFF418WHV. From here, we classified 410 dex-responsive bivalent enhancers as genomic regions that were in the EnhBiv state in untreated cells and in the EnhA1 or EnhA2 state in any post-treatment dataset. 2 HAQERs, HAQER0547 and HAQER0919, overlapped a dex-responsive bivalent enhancer (expected overlap: 0.32. p < 0.01, gonomics:overlapEnrichments ).
Functional annotation of HAQERs
To identify enrichments between HAQERs and gene regulatory elements gained after the rhesus split, we accessed 15-state chromHmm data from the Roadmap Epigenomics consortium48 for the active enhancer and active promoter states (7 Enh and 1 TssA) from the developing brain reference epigenomes E081 and E082. Next, we concatenated and merged BED format files with gonomics: bedMerge to produce BED files representing all regions identified as either active enhancers or active promoters in either fetal brain reference epigenome. We then used gonomics: intervalOverlap to identify promoter and enhancer regions that overlapped gene regulatory elements gained after the rhesus split52. We used gonomics: overlapEnrichments to calculate the enrichment between HAQERs and these recently-evolved regulatory elements.
To identify overlap between HAQERs and open chromatin, human fetal brain DHS-seq data was obtained from the Roadmap Epigenomics Consortium data48 from the following three individuals: GSM595920, GSM595922, and GSM595926. BAM format alignment files aligned to hg19 were disassembled to FASTQ format sequencing files using samtools:bam2fq102 and aligned to hg38 with BWA MEM103. To visualize DNase hypersensitivity sequencing (DHS-seq) data on the UCSC genome browser, we developed gonomics: samToWig to convert SAM/BAM format alignment files into WIG graphing track format. WIG files were then converted to binary bigWig files with kentUtils: wigToBigWig. We developed gonomics: bedValueWig to generate a score for each region in an input BED format file corresponding to the highest value of an input WIG file in the coordinate range of the queried region. Regions with at least 10 reads overlapping a single position were considered for further analysis as possible regions of open chromatin. Overlaps between HAQERs and other genomic regions, including functional elements gained after the rhesus split52 and differential Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) peaks from human and chimpanzee cerebral organoids56 were determined using gonomics: intervalOverlap.
Gene synthesis and plasmid preparation
Test sequences for single-cell Self-transcribing active regulatory region sequencing (scSTARR-seq) were synthesized and cloned into the STARR-seq screening vector104 using a commercial service (Twist Bioscience). We added an 8 base pair unique barcode to the 3ʹ end of each insert to differentiate between closely related orthologous sequences with short read 3ʹ RNA sequencing. Plasmids were transformed into One Shot Stbl3 chemically competent E. coli (Thermo Fisher), selected for ampicillin resistance, and amplified in Luria broth (Invitrogen) with 100 μg/mL ampicillin. Endotoxin-free plasmids were then purified with the ZymoPURE II Plasmid Maxiprep kit per manufacturer’s instructions (Zymo Research). As necessary, purified plasmids were then precipitated with 3M Na-Acetate pH 5.2, and 100% ethanol for 2 hours to achieve desirable concentrations. For in vivo STARR-seq, an equimolar solution containing each STARR-seq plasmid was prepared at a total plasmid concentration of 3 μg/mL. This pooled STARR-seq solution was then mixed with a pCAG-GFP injection reporter plasmid, which represented 1/6 of the total plasmid content of the final injection solution. Our input STARR-seq library included plasmids with a total of 77 distinct inserts: 60 corresponding to the orthologs of 13 HAQER sequences, 7 sequences used only in the analysis in Figure S5E–F, and 10 pseudorandom sequences which served as negative controls (Methods). For the PGK-EGFP enhancer reporter assay, we amplified HAQER inserts from the STARR-seq plasmid vector via polymerase chain reaction and introduced these inserts to a PGK-EGFP plasmid vector (Addgene #169744) via Gibson assembly cloning105, which we confirmed with Sanger sequencing.
In utero electroporation
In utero electroporation was performed as previously reported106. Briefly, E14.5 or E15.5 wild type B6 pregnant females were anesthetized with isoflurane. Uterine horns were exposed by making an incision in the abdomen. Each embryo was injected with 1–1.5 μl of plasmid solution (containing 0.01% fast green and 1–2 μg/μl of plasmids) and electroporated using the following parameters: five 50 ms-pulses at 50V (E14.5) or 60V (E15.5) with 950 ms pulse-interval, using platinum-plated BTX Tweezertrodes. Uterine horns were then repositioned into the abdominal cavity and the muscle and skin incisions were sutured. Dams were then placed on a heating pad for recovery and monitored.
Immunofluorescence staining and image acquisition
Brains were fixed overnight in 4% PFA-PBS at 4°C, rinsed in PBS, and submerged in 30% sucrose-PBS until sinking (24 hours). Brains were frozen in NEG-50 medium (Richard-Allan Scientific) and cryostat sections (20 μm) were prepared and stored at −80°C until use. Sections were washed 3 times 10 minutes with PBS and incubated 1 μg/ml Hoechst 33342 (Invitrogen) for 30 minutes at room temperature. Sections were then mounted using Vectashield (Vector Laboratories) as mounting media. Images were acquired with a Zeiss Axio Observer Z.1 microscope coupled with an apotome2. Image measurements and quantifications were blindly performed using Fiji107. Statistical significance was assigned by 2-way ANOVA in GraphPad Prism. We analyzed anatomically comparable regions from sections from 2 embryos from 2 IUEs (n=4) per injection construct.
Fluorescence activated cell sorting
Electroporated brains were harvested after approximately 18 hours and dissected in ice-cold sterile PBS. Meninges were removed and GFP+ portions of the cortices were incubated at 37°C for 10 minutes in 0.25% trypsin-EDTA supplemented with 0.1% DNAse I (New England Biolabs cat# M0303S). Following incubation, the trypsin solution was removed and replaced with ice-cold 10% FBS/HBSS/Propidium iodide (Invitrogen) supplemented with 0.01% DNAse I. A single cell suspension was then generated by trituration with a fire-polished glass pipette and filtered with a 30 μm cell strainer. Cells were then stained with the LIVE/DEAD Near-IR Dead Cell Stain per manufacturer’s instructions (Thermo Fisher). Following staining, viable GFP+ cells were bulk sorted using a FACS Aria II cytometer (BD Biosciences).
scSTARR-seq reporter read targeted enrichment
In order to enrich reporter read sequences from cDNA generated from endogenous mouse mRNA and STARR-seq reporter RNA, we performed a three-step PCR reaction based on a 10x targeted enrichment protocol developed by Gasperini et. al108. We began with approximately 10–13 ng of unfragmented scRNA-seq cDNA and performed qPCR-monitored 50 μl Phusion PCR (annealing temp 62°C, 1.5μl DMSO) with the following primers:
F1: tGFPOuter 5- ATGGCTAGCAAAGGAGAAGAACTCT -3
R1: R1-PCR1 5- ACACTCTTTCCCTACACGACG -3
Following 1x Agencourt AMPure XP bead cleanup (Beckman Coulter), 2 μl of cleaned product was amplified in a subsequent 50μl Phusion reaction (12 cycles) with the following primers:
F2: tGFPInner 5-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCTTGTTGAATTAGATTGATCT -3
R2: RP5 5-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACG -3
Following 1x AMPure cleanup, 2μl of cleaned product was used in a third 50 μl Phusion reaction (12 cycles), with the following primers:
F3: 5- CAAGCAGAAGACGGCATACGAGATIIIIIIIIIIGTCTCGTGGGCTCGG -3 (standard NEXTERA P7 indexing primer)
R3: Same as R2.
Following this reaction, final libraries were cleaned once more with 1X AMPure and quantified using the Bioanalyzer.
scSTARR-seq sequencing and preprocessing
Up to 10,000 GFP+ cells were captured per lane of a 10X Chromium device and single cell libraries were prepared using protocols from the Chromium Next GEM Single Cell 3ʹ Reagent Kits v3.1 (Rev D) User Guide (10X Genomics, Inc.). Final libraries were quantified using the Bioanalyzer (Agilent) according to manufacturer’s protocols. Prior to enzymatic fragmentation, an aliquot of cDNA was separated for targeted enrichment. Final 10X libraries were sequenced using the NovaSeq 6000 S-Prime reagents (Illumina; R1 28, I1 10, I2 10, R2 90). Reporter-targeted enrichment libraries were sequenced independently on the Illumina NextSeq platform. Fastq files from both libraries were recovered with bcl2fastq (v2.20.0.422, Illumina). For targeted enrichment libraries, we used the following bases mask: Y28n*,n*,I8n*,Y75n*. Output fastq files from both the unenriched and reporter-targeted enrichment libraries across multiple lanes were concatenated together for downstream analysis.
For input normalization, we used the NEBNext Ultra II FS DNA Library Prep Kit (New England Biolabs) to sequence our STARR-seq injection plasmid library and sequenced the resulting library on the Illumina iSeq 100 platform. Fastq reads from the input library were aligned using the Burrows-Wheeler Aligner (BWA) to a custom STARR-seq reference genome including the sequence of the mouse reference mm10 with additional FASTA records containing the sequences of each STARR-seq reporter construct and the pCAG-GFP injection reporter sequence. For an input STARR-seq library with n constructs, the input normalization factor Cs for an individual STARR-seq construct, s, was then calculated as the ratio of the expected number of reads from s if all n constructs were present at equimolar concentration in the input library to the observed number of reads from s: Cs = Es/Os where:
Constructs with an input normalization factor greater than 5 (indicating a greater than 5-fold depletion that equimolar expectation in the input library) were excluded from all subsequent analysis.
GFP+ cells were pooled from all embryos in each experiment to control for batch effects associated with anatomical differences in electroporation and dissection. We sequenced 3494 single cells from the first library, which was comprised of GFP+ cells pooled from 6 embryos from a singlemouse injected at E14.5. The second library, which was composed of GFP+ cells from 9 embryos injected at E15.5, yielded 3676 single-cell transcriptomes.
Enhancer activity quantification
To score enhancer activity from scSTARR-seq data, we implemented gonomics: fastqFilter -collapseUmi to remove unique molecular identifier (UMI) duplicates from our 10x libraries. We then used gonomics: fastqFormat -singleCell to parse the cell barcode and UMI from R1 into the read name for the R2 fastq. We then used the BWA to align reads to our custom STARR-seq reference genome described above. The enhancer activity score for each construct was then calculated as the input-normalized UMI count per 1000 total reporter UMI counts. To determine the basal level of transcription from the STARR-seq plasmid vector, we synthesized 10 plasmids with inserts of 500bp of pseudorandom DNA sequences generated using gonomics: randSeq. To guard against spurious enhancer activity present in pseudorandom sequences, we used six of the ten negative control constructs with the lowest enhancer activity scores to determine a limit of detection for enhancer activity, which we defined as the three standard deviations above the average enhancer activity score from these six pseudorandom sequences. We attempted to generate STARR-seq orthologous (human, Neanderthal, Denisova, chimpanzee, HCA) test sequences for each region of interest. However, some ortholog pairs exhibited the same sequence for the 500bp region of interest. Duplicate constructs were not included in statistical analysis, but are still displayed as faded bars in Figure 4C.
Single-cell cluster identification and cell-type specific enhancer activity quantification
Count matrices were produced using CellRanger v6.0 (10x Genomics) with the custom reference genome described above. Subsequent analysis for cluster identification was performed in Seurat v4.0109. For each library, cells were removed which contained 200 or fewer genes or more than 5,000 genes. Each library was independently normalized and 2,000 highly variable features were identified for each library. Cells across independent libraries were integrated for joint analysis via canonical correlation analysis110. Variation in gene expression based on cell-cycle related genes was regressed from cluster analysis in dataset scaling using an annotated set of G2M and S phase related genes provided in Seurat. k-nearest neighbors (k=20) were calculated in the space of significant principal components (in this case, 30 principal components) and clustering was performed with the Louvain-Jaccard method. Visualizations were generated in uniform manifold approximation and projection (UMAP) space111. We identified the top 10 positive markers for each cluster and manually assigned cluster identities based on marker gene expression in two previously published neurodevelopmental single-cell atlases in mouse57 and human54. Multiple clusters corresponding to the same cell type (ex. Excitatory Neuron I-IV) were pooled as metaclusters for subsequent analysis.
To perform cell-type specific enhancer activity quantification, reads from each library aligned to the custom STARR-seq reference genome described above were sorted by cell barcode using gonomics: mergeSort -singleCellBx and input-normalized count matrices were generated with gonomics: scCount. Input-normalized count matrices were then partitioned by metacluster using cluster identities determined for each cell barcode by Seurat. Cells with fewer than 4 pCAG-GFP UMIs were discarded. The input-normalized reporter UMI counts for each cell were then further normalized to the pCAG-GFP UMI count for that cell. The cell type enhancer activity score was then calculated as the average transfection-normalized, input-normalized UMI count per cell in each cluster.
Enhancer paralog phylogenetic analysis
We began with the hg38 human reference sequence for HAQER0059 and gathered the sequences for all paralogs in the human (hg38), chimpanzee (panTro6), gorilla (gorGor5), orangutan (ponAbe3), and rhesus (rheMac10) assemblies as identified with BLAT63. From here, we used gonomics: faFormat -revComp for all reverse strand sequences before aligning all forward-strand paralogous sequences with muscle112. We then constructed a Newick-format phylogenetic tree from this alignment with ClustalW2113. Finally, we visualized phylogenies with phylotree114.
GWAS catalog trait enrichment analysis
We first sampled the set of all GWAS Catalog variants that report an association in European populations to obtain a record for each SNP115. We retained only those variants that also appeared as segregating among individuals in the GBR subpopulation in the 1000 Genomes Project variant set27. To generate a comprehensive list of possible causal variants, we used plink −−r2116 to identify all other 1000 Genomes Project variants in linkage disequilibrium (Plink R2 > 0.7) with each GWAS Catalog variant.
We then merged the set of all GWAS variants and linked variation for each mapped trait from the Experimental Factor Ontology from the GWAS Catalog association table and calculated overlap enrichment between this merged set of variants and HAQERs gonomics: overlapEnrichments. We report significant enrichments for mapped traits with an FDR-adjusted p < 0.05.
We also calculated the distributions of the number of all possible causal variants (including a GWAS variant and all linked variation (Plink R2 > 0.7)) and median distance of each linked variant to its corresponding GWAS variant for all possible causal variants overlapping HAQERs, HARs, or RAND.
The observed disease enrichments are unlikely to be influenced solely by haplotype structure or density of linked variation around GWAS variants, as these features were similar between RAND and HAQERs (Figure S6F,G).
Horizontal pleiotropy score quantification
We accessed a dataset of 1,183,386 human genetic variants annotated with LD-corrected horizontal number of traits pleiotropy scores ( generated by Jordan et al.117). We intersected these variant sets with HAQERs, RAND, and HARs and compared the distribution of PLD scores in variants overlapping each set of genomic regions.
Briefly, Jordan et al. leveraged PheWAS relationships between genetic variants and human traits to calculate as the expected value of the number of statistically independent traits for which a given variant is associated in a set of 100 traits. This approach starts with a matrix Zraw of Z-scores associating each genetic variant to a human trait. Many clinical traits exhibit covariance as a result of either partially redundant or ambiguous terminology (i.e. Alzheimer’s Disease and Dementia) or vertical pleiotropy (i.e. a causal relationship between traits, such as between hypertension and heart disease). Thus, corrects for covariance between traits by applying the following Mahalanobis whitening transformation to Zraw:
where Σ is the covariance matrix of Zraw. The result of this transformation is that the covariance matrix of the resulting matrix Z will be equal to the identity matrix, indicating no covariance between traits. Pn for a variant n is then calculated as the scaled number of whitened traits significantly associated with the variant n:
Where H(zi-2) is the Heaviside step function, which is equal to 1 when |zi| > 2 and 0 otherwise. The term 100/l scales the number of significantly associated whitened traits by the number of traits l and the constant 100 so that the resulting term represents the expected value of the number of significantly associated whitened traits in a dataset of 100 traits. Finally, this value is corrected for linkage disequilibrium with the following transformation:
where x is the LD score of the variant position and βn is the regression coefficient for LD on Pn.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters were reported either in individual figures or corresponding figure legends. Statistical details of experiments can be found in Method details. All statistical analyses were performed in R or Go.
Supplementary Material
(A) Synthetic allele frequency spectra ordered by value of the selection parameter used to generate each spectrum. (B) Representative Markov chain Monte Carlo (MCMC) traces for the mean selection parameter acting on variants from synthetic derived allele frequency spectra. Traces are colored by the value of the input selection parameter used to simulate each spectra. (C) Mean and 95% highest density credible intervals for the posterior distribution of the mean selection parameter for 50 synthetic variant sets, 10 for each input selection parameter. Posterior estimates are displayed from 50,000 iterations with the first 5,000 iterations discarded as burn-in. 47/50 (94%) of traces contained the true input selection parameter in the 95% credible interval. Traces where the input selection parameter was not contained in the credible interval are marked with an asterisk. Distribution of MCMC trace means for the mean selection parameter with and without the Kern correction for divergence-based ascertainment bias are displayed for the least divergent 1% of variant sets (D) or most divergent 1% (E). Mean selection parameters are estimated as the posterior mean of 10,000 iterations with the first 1,000 iterations discarded as burn-in.
(A, B) Pearson correlations between velocity and acceleration (A) or initial velocity (B) for 2,902,532 500bp genomic regions. (C) Heatmap of mean selection parameters for sets of variants overlapping all genomic regions binned by velocity and acceleration score. Mean selection parameters are estimated as the posterior mean of 10,000 iterations with the first 1,000 iterations discarded as burn-in. (D) Cumulative proportions of velocity, initial velocity, and acceleration for variants overlapping regions of interest. Here HARs are split into the original HARs (oHAR15) and the expanded HARs (eHAR24). (Bonferroni-adjusted Wilcoxon. ***: p < 0.001; ****: p < 0.0001) (E) Phylogenetic context of HAQERs, chimp-AQERs, and gorilla-AQERs. Venn diagram displays overlaps between rapidly-evolved primate regions (****: p < 0.001 for overlap enrichment between these sets of genomic regions). (F) Observed over expected proportion of elements within 5mb of chromosome ends. Expected proportion was estimated from randomly distributed regions in each genome. (G) GREAT ontology enrichments for HAQER chromatin contact sites in H1 hESCs (red) and GM12878 (yellow). One ontology term, innate immune response, was significant in both cell types. (H) GREAT ontology enrichments for HAQERs, chimp-AQERs, and gorilla-AQERs.
(A) Density of sites divergent between the modern human sequence and the inferred Human-Chimpanzee Ancestor (HCA) sequence against the density of polymorphic sites observed in 501 unrelated African individuals for Human Ancestor Quickly Evolved Regions (HAQERs), pseudorandomly selected neutral proxy regions (RAND), human accelerated regions (HARs), ENCODE candidate cis-regulatory elements (ENCODE), and ultraconserved elements (UCEs). (B) Distribution of recombination frequency in HAQER, RAND, and HARs (Bonferroni-adjusted Wilcoxon). (C) Distribution of replication timing in HAQERs, HARs, and RAND (Bonferroni-adjusted Wilcoxon) (D) Proportion of HCA divergent sites that are weak to strong mutations (A to G/T to C or A to C/T to G) in HAQERs, HARs, and RAND. (E) Principal component analysis of the mutation spectrum for all HAQERs. HAQERs do not demonstrate distinct mutation spectrum subtypes. (F) Proportion of HCA divergent sites that are polymorphic (observed as segregating in the 501 unrelated African individuals from A) as opposed to fixed (not observed as segregating). The dotted line represents the proportion of polymorphic variants in RAND (1,069 polymorphic sites, 8,487 fixed sites) (Chi-squared test). (G) Proportion of segregating sites that are transitions in segregating sites from five African populations binned by low derived allele frequency (DAF < 0.1) and high derived allele frequency (DAF > 0.9) (Wilcoxon). (H) The samConsensus program generates consensus sequences from individuals calling substitutions (represented as red positions) from sequencing reads aligned to a reference genome and editing the corresponding positions of the reference assembly. (I) Phylogenetic history and geographic distribution of 30 great ape genomes used to construct the multiple alignment. (J) A representative 100bp block of sequence sampled from the 30-way whole genome alignment. (**: p < 0.01; ***: p < 0.001; ****: p < 0.0001).
(A) Overlap enrichment/depletion matrix between HAQERs (left) or HARs (right) and chromatin states from 127 reference epigenomes with sample level annotation. Both sets are depleted from transcribed regions, this reflects the ascertainment bias that some HAR studies specifically excluded coding regions (Prabhakar et al., 2006). (B) Comparisons between HAQER and HAR overlap enrichment scores for each chromatin state across 127 reference epigenomes quantified by Bonferroni-adjusted t-test (****: p < 0.0001). (C) Comparison of HAQER overlap enrichment between adult and developing tissue-derived reference epigenomes. (D) HAQERs are enriched for overlaps with active promoters and active enhancers gained after rhesus split, defined as increased H3K27ac or H3K4me2 ChIP-seq signal in the developing human brain relative to mouse and rhesus macaque52 (Bonferroni-adjusted overlap enrichment; ****: p < 0.0001, *: p < 0.05). (E) Volcano plot of significant overlap enrichments for HAQERs (left) or HARs (right) for 7 gene regulatory chromatin states.
(A) UMAP representation of 7,170 cells from scSTARR-seq in the developing mouse brain. Insert UMAP labels cells from two independent STARR-seq experiments, performed at E14.5 and E15.5. (B) Gene expression for canonical markers of each cell identity. (C) pCAG-GFP transfection reporter expression in each cell cluster. As radial glia and their progeny were targeted, limited GFP expression is observed in the inhibitory neuron, microglia, fibroblast, and vascular clusters. (D) Dotplot representation of the top five marker genes by cluster. The pCAG-GFP transfection reporter, which was electroporated preferentially into radial glia, was identified as a radial glial marker. (E) Pearson correlation between enhancer activity score measurements for each test sequence at the E14.5 and E15.5 injection timepoints. (F) Pearson correlation between the input normalization factor and enhancer activity score estimates for all test sequences with input normalization factors below 5.
Each panel displays the GENCODE gene track, open chromatin in the developing human brain48, and divergence density in the genomic context of HAQERs with neurodevelopmental function. The dashed line corresponds to a divergence density of 29 mutations per 500 bases, the statistical threshold for HAQER identification. Also labeled are the positions of STARR-seq test sequences used in this study, Human Accelerated Regions24, functional elements gained after the rhesus split52, and differentially accessible (DA) scATAC-seq sites in cerebral organoids56. The genomic context is shown for: (A) HAQER0710, a hominin-specific enhancer in the locus of DPP10, an autism spectrum disorder-related gene75; (B) HAQER0223, a hominin-specific enhancer in the locus of EMILIN276; and (C) HAQER0035, which corresponds to HAR1, a previously described rapidly-evolving region15. (D) The genomic context for the gene ADCYAP1, which harbors three HAQERs. HAQER0780 and HAQER0911 were both observed as hominin-specific neurodevelopmental enhancers. ADCYAP1 is associated with neurodevelopment77, human evolution78, and psychiatric disease79. (E) Genome browser snapshot of a 10 megabase region of hg38 chr1. Members of the NBPF gene family are highlighted in yellow. (F) Distributions of the number of variants in significant linkage disequilibrium (Plink R2 > 0.7) for each disease-linked variant overlapping HAQERs, RAND, and HARs (Bonferroni-adjusted Wilcoxon; ****: p < 0.0001, **: p < 0.01). (G) Distributions of the median distance of significantly linked variants (Plink R2 > 0.7) to corresponding GWAS variants for disease-linked variants overlapping HAQERs, RAND, and HARs (Bonferroni-adjusted Wilcoxon; ****: p < 0.0001). (H) Proportion of variants overlapping HAQERs, RAND, and HARs with significant (p < 0.05) HOPS horizontal pleiotropy scores (Chi-squared; ****: p < 0.0001).
HIGHLIGHTS.
HAQERs are human genomic regions highly divergent from the human-chimpanzee ancestor.
HAQERs evolved under elevated mutation rates and positive selection.
HAQERs are enriched for bivalent chromatin and disease-linked variation.
HAQER divergence forged hominin-unique enhancers in the developing cerebral cortex.
ACKNOWLEDGEMENTS
We thank the Duke Human Vaccine Institute Research Flow Cytometry and Viral Genome Analysis core facilities. Shataakshi Dube and Scott Soderling generously provided the pCAG-GFP plasmid. We thank Douglas A. Marchuk, Christiana Fauci, Chelsea R. Shoben, Seth Weaver, Shae Simpson, and Yanting Luo for critical feedback. This research was supported by the Duke Whitehead Scholarship, National Human Genome Research Institute (R35HG011332), the Sigma Xi Grants in Aid of Research Program, North Carolina Biotechnology Center (2016-IDG-1013; 2020-IIG-2109), and the Triangle Center for Evolutionary Medicine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
ADDITIONAL RESOURCES
The raw data and analyzed results are available at our website: https://vertgenlab.org/.
References
- 1.Sockol MD, Raichlen DA, and Pontzer H (2007). Chimpanzee locomotor energetics and the origin of human bipedalism. Proc. Natl. Acad. Sci 104, 12265–12269. 10.1073/pnas.0703267104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vick S-J, Waller BM, Parr LA, Smith Pasqualini MC, and Bard KA (2007). A Cross-species Comparison of Facial Morphology and Movement in Humans and Chimpanzees Using the Facial Action Coding System (FACS). J. Nonverbal Behav 31, 1–20. 10.1007/s10919-006-0017-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geschwind DH, and Rakic P (2013). Cortical Evolution: Judge the Brain by Its Cover. Neuron 80, 633–647. 10.1016/j.neuron.2013.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silver DL (2016). Genomic divergence and brain evolution: How regulatory DNA influences development of the cerebral cortex. BioEssays News Rev. Mol. Cell. Dev. Biol 38, 162–171. 10.1002/bies.201500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richard D, Liu Z, Cao J, Kiapour AM, Willen J, Yarlagadda S, Jagoda E, Kolachalama VB, Sieker JT, Chang GH, et al. (2020). Evolutionary Selection and Constraint on Human Knee Chondrocyte Regulation Impacts Osteoarthritis Risk. Cell 181, 362–381.e28. 10.1016/j.cell.2020.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu K, Schadt EE, Pollard KS, Roussos P, and Dudley JT (2015). Genomic and Network Patterns of Schizophrenia Genetic Variation in Human Evolutionary Accelerated Regions. Mol. Biol. Evol 32, 1148–1160. 10.1093/molbev/msv031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark AG, Glanowski S, Nielsen R, Thomas PD, Kejariwal A, Todd MA, Tanenbaum DM, Civello D, Lu F, Murphy B, et al. (2003). Inferring Nonneutral Evolution from Human-Chimp-Mouse Orthologous Gene Trios. Science 10.1126/science.1088821. [DOI] [PubMed] [Google Scholar]
- 8.Nielsen R, Bustamante C, Clark AG, Glanowski S, Sackton TB, Hubisz MJ, Fledel-Alon A, Tanenbaum DM, Civello D, White TJ, et al. (2005). A Scan for Positively Selected Genes in the Genomes of Humans and Chimpanzees. PLOS Biol 3, e170. 10.1371/journal.pbio.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Florio M, Heide M, Pinson A, Brandl H, Albert M, Winkler S, Wimberger P, Huttner WB, and Hiller M (2018). Evolution and cell-type specificity of human-specific genes preferentially expressed in progenitors of fetal neocortex. eLife 7, e32332. 10.7554/eLife.32332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fiddes IT, Lodewijk GA, Mooring M, Bosworth CM, Ewing AD, Mantalas GL, Novak AM, Bout A. van den, Bishara A, Rosenkrantz JL, et al. (2018). Human-Specific NOTCH2NL Genes Affect Notch Signaling and Cortical Neurogenesis. Cell 173, 1356–1369.e22. 10.1016/j.cell.2018.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heide M, Haffner C, Murayama A, Kurotaki Y, Shinohara H, Okano H, Sasaki E, and Huttner WB (2020). Human-specific ARHGAP11B increases size and folding of primate neocortex in the fetal marmoset. Science 369, 546–550. 10.1126/science.abb2401. [DOI] [PubMed] [Google Scholar]
- 12.King MC, and Wilson AC (1975). Evolution at two levels in humans and chimpanzees. Science 188, 107–116. 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- 13.Wray GA (2007). The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet 8, 206–216. 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
- 14.Carroll SB (2008). Evo-Devo and an Expanding Evolutionary Synthesis: A Genetic Theory of Morphological Evolution. Cell 134, 25–36. 10.1016/j.cell.2008.06.030. [DOI] [PubMed] [Google Scholar]
- 15.Pollard KS, Salama SR, Lambert N, Lambot M-A, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, et al. (2006). An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443, 167. 10.1038/nature05113. [DOI] [PubMed] [Google Scholar]
- 16.Pollard KS, Salama SR, King B, Kern AD, Dreszer T, Katzman S, Siepel A, Pedersen JS, Bejerano G, Baertsch R, et al. (2006). Forces Shaping the Fastest Evolving Regions in the Human Genome. PLOS Genet 2, e168. 10.1371/journal.pgen.0020168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bird CP, Stranger BE, Liu M, Thomas DJ, Ingle CE, Beazley C, Miller W, Hurles ME, and Dermitzakis ET (2007). Fast-evolving noncoding sequences in the human genome. Genome Biol 8, R118. 10.1186/gb-2007-8-6-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, et al. (2011). A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482. 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prabhakar S, Noonan JP, Pääbo S, and Rubin EM (2006). Accelerated evolution of conserved noncoding sequences in humans. Science 314, 786. 10.1126/science.1130738. [DOI] [PubMed] [Google Scholar]
- 20.Gittelman RM, Hun E, Ay F, Madeoy J, Pennacchio L, Noble WS, Hawkins RD, and Akey JM (2015). Comprehensive identification and analysis of human accelerated regulatory DNA. Genome Res 25, 1245–1255. 10.1101/gr.192591.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Capra JA, Erwin GD, McKinsey G, Rubenstein JLR, and Pollard KS (2013). Many human accelerated regions are developmental enhancers. Philos. Trans. R. Soc. Lond. B. Biol. Sci 368, 20130025. 10.1098/rstb.2013.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Girskis KM, Stergachis AB, DeGennaro EM, Doan RN, Qian X, Johnson MB, Wang PP, Sejourne GM, Nagy MA, Pollina EA, et al. (2021). Rewiring of human neurodevelopmental gene regulatory programs by human accelerated regions. Neuron 10.1016/j.neuron.2021.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyd JL, Skove SL, Rouanet JP, Pilaz L-J, Bepler T, Gordân R, Wray GA, and Silver DL (2015). Human-chimpanzee differences in a FZD8 enhancer alter cell-cycle dynamics in the developing neocortex. Curr. Biol. CB 25, 772–779. 10.1016/j.cub.2015.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doan RN, Bae B-I, Cubelos B, Chang C, Hossain AA, Al-Saad S, Mukaddes NM, Oner O, Al-Saffar M, Balkhy S, et al. (2016). Mutations in Human Accelerated Regions Disrupt Cognition and Social Behavior. Cell 167, 341–354.e12. 10.1016/j.cell.2016.08.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, Kaul R, et al. (2020). Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710. 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Byrska-Bishop M, Evani US, Zhao X, Basile AO, Abel HJ, Regier AA, Corvelo A, Clarke WE, Musunuri R, Nagulapalli K, et al. (2022). High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell 185, 3426–3440.e19. 10.1016/j.cell.2022.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arnold CD, Gerlach D, Stelzer C, Boryń ŁM, Rath M, and Stark A (2013). Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339, 1074–1077. 10.1126/science.1232542. [DOI] [PubMed] [Google Scholar]
- 29.Shen SQ, Myers CA, Hughes AEO, Byrne LC, Flannery JG, and Corbo JC (2016). Massively parallel cis-regulatory analysis in the mammalian central nervous system. Genome Res 26, 238–255. 10.1101/gr.193789.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson GD, Barrera A, McDowell IC, D’Ippolito AM, Majoros WH, Vockley CM, Wang X, Allen AS, and Reddy TE (2018). Human genome-wide measurement of drug-responsive regulatory activity. Nat. Commun 9, 1–9. 10.1038/s41467-018-07607-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein JC, Agarwal V, Inoue F, Keith A, Martin B, Kircher M, Ahituv N, and Shendure J (2020). A systematic evaluation of the design and context dependencies of massively parallel reporter assays. Nat. Methods 17, 1083–1091. 10.1038/s41592-020-0965-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ellegren H, Smith NG, and Webster MT (2003). Mutation rate variation in the mammalian genome. Curr. Opin. Genet. Dev 13, 562–568. 10.1016/j.gde.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 33.Kimura M (1983). The Neutral Theory of Molecular Evolution (Cambridge University Press; ). [Google Scholar]
- 34.Katzman S, Kern AD, Bejerano G, Fewell G, Fulton L, Wilson RK, Salama SR, and Haussler D (2007). Human Genome Ultraconserved Elements Are Ultraselected. Science 317, 915–915. 10.1126/science.1142430. [DOI] [PubMed] [Google Scholar]
- 35.Kern AD (2009). Correcting the Site Frequency Spectrum for Divergence-Based Ascertainment. PLOS ONE 4, e5152. 10.1371/journal.pone.0005152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Initial sequence of the chimpanzee genome and comparison with the human genome (2005). Nature 437, 69–87. 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 37.Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, and Haussler D (2004). Ultraconserved Elements in the Human Genome. Science 304, 1321–1325. 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- 38.Brown CA, Murray AW, and Verstrepen KJ (2010). Rapid Expansion and Functional Divergence of Subtelomeric Gene Families in Yeasts. Curr. Biol 20, 895–903. 10.1016/j.cub.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pratto F, Brick K, Khil P, Smagulova F, Petukhova GV, and Camerini-Otero RD (2014). Recombination initiation maps of individual human genomes. Science 346, 1256442. 10.1126/science.1256442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arbeithuber B, Betancourt AJ, Ebner T, and Tiemann-Boege I (2015). Crossovers are associated with mutation and biased gene conversion at recombination hotspots. Proc. Natl. Acad. Sci 112, 2109–2114. 10.1073/pnas.1416622112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding Q, Edwards MM, Wang N, Zhu X, Bracci AN, Hulke ML, Hu Y, Tong Y, Hsiao J, Charvet CJ, et al. (2021). The genetic architecture of DNA replication timing in human pluripotent stem cells. Nat. Commun 12, 6746. 10.1038/s41467-021-27115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jensen-Seaman MI, Furey TS, Payseur BA, Lu Y, Roskin KM, Chen C-F, Thomas MA, Haussler D, and Jacob HJ (2004). Comparative Recombination Rates in the Rat, Mouse, and Human Genomes. Genome Res 14, 528–538. 10.1101/gr.1970304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou Y, Browning BL, and Browning SR (2020). Population-Specific Recombination Maps from Segments of Identity by Descent. Am. J. Hum. Genet 107, 137–148. 10.1016/j.ajhg.2020.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andolfatto P (2005). Adaptive evolution of non-coding DNA in Drosophila. Nature 437, 1149–1152. 10.1038/nature04107. [DOI] [PubMed] [Google Scholar]
- 45.Tenesa A, Navarro P, Hayes BJ, Duffy DL, Clarke GM, Goddard ME, and Visscher PM (2007). Recent human effective population size estimated from linkage disequilibrium. Genome Res 17, 520–526. 10.1101/gr.6023607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dunn JC (1973). A Fuzzy Relative of the ISODATA Process and Its Use in Detecting Compact Well-Separated Clusters. J. Cybern 3, 32–57. 10.1080/01969727308546046. [DOI] [Google Scholar]
- 47.Harpak A, Bhaskar A, and Pritchard JK (2016). Mutation Rate Variation is a Primary Determinant of the Distribution of Allele Frequencies in Humans. PLOS Genet 12, e1006489. 10.1371/journal.pgen.1006489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. (2006). A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 125, 315–326. 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 50.Voigt P, Tee W-W, and Reinberg D (2013). A double take on bivalent promoters. Genes Dev 27, 1318–1338. 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aiello LC, and Wheeler P (1995). The Expensive-Tissue Hypothesis: The Brain and the Digestive System in Human and Primate Evolution. Curr. Anthropol 36, 199–221. [Google Scholar]
- 52.Reilly SK, Yin J, Ayoub AE, Emera D, Leng J, Cotney J, Sarro R, Rakic P, and Noonan JP (2015). Evolutionary changes in promoter and enhancer activity during human corticogenesis. Science 347, 1155–1159. 10.1126/science.1260943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klein JC, Keith A, Agarwal V, Durham T, and Shendure J (2018). Functional characterization of enhancer evolution in the primate lineage. Genome Biol 19. 10.1186/s13059-018-1473-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, Haeussler M, Sandoval-Espinosa C, Liu SJ, Velmeshev D, et al. (2017). Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323. 10.1126/science.aap8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pollen AA, Bhaduri A, Andrews MG, Nowakowski TJ, Meyerson OS, Mostajo-Radji MA, Di Lullo E, Alvarado B, Bedolli M, Dougherty ML, et al. (2019). Establishing Cerebral Organoids as Models of Human-Specific Brain Evolution. Cell 176, 743–756.e17. 10.1016/j.cell.2019.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kanton S, Boyle MJ, He Z, Santel M, Weigert A, Sanchís-Calleja F, Guijarro P, Sidow L, Fleck JS, Han D, et al. (2019). Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574, 418–422. 10.1038/s41586-019-1654-9. [DOI] [PubMed] [Google Scholar]
- 57.La Manno G, Siletti K, Furlan A, Gyllborg D, Vinsland E, Mossi Albiach A, Mattsson Langseth C, Khven I, Lederer AR, Dratva LM, et al. (2021). Molecular architecture of the developing mouse brain. Nature 596, 92–96. 10.1038/s41586-021-03775-x. [DOI] [PubMed] [Google Scholar]
- 58.Shew CJ, Carmona-Mora P, Soto DC, Mastoras M, Roberts E, Rosas J, Jagannathan D, Kaya G, O’Geen H, and Dennis MY (2021). Diverse Molecular Mechanisms Contribute to Differential Expression of Human Duplicated Genes. Mol. Biol. Evol 38, 3060–3077. 10.1093/molbev/msab131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neilson KM, Klein SL, Mhaske P, Mood K, Daar IO, and Moody SA (2012). Specific domains of FoxD4/5 activate and repress neural transcription factor genes to control the progression of immature neural ectoderm to differentiating neural plate. Dev. Biol 365, 363–375. 10.1016/j.ydbio.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sherman JH, Karpinski BA, Fralish MS, Cappuzzo JM, Dhindsa DS, Thal AG, Moody SA, LaMantia AS, and Maynard TM (2017). Foxd4 is essential for establishing neural cell fate and for neuronal differentiation. genesis 55, e23031. 10.1002/dvg.23031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Minoretti P, Arra M, Emanuele E, Olivieri V, Aldeghi A, Politi P, Martinelli V, Pesenti S, and Falcone C (2007). A W148R mutation in the human FOXD4 gene segregating with dilated cardiomyopathy, obsessive-compulsive disorder, and suicidality. Int. J. Mol. Med 19, 369–372. 10.3892/ijmm.19.3.369. [DOI] [PubMed] [Google Scholar]
- 62.Fan Y, Newman T, Linardopoulou E, and Trask BJ (2002). Gene content and function of the ancestral chromosome fusion site in human chromosome 2q13–2q14.1 and paralogous regions. Genome Res 12, 1663–1672. 10.1101/gr.338402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kent WJ (2002). BLAT—The BLAST-Like Alignment Tool. Genome Res 12, 656–664. 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Suzuki IK, Gacquer D, Van Heurck R, Kumar D, Wojno M, Bilheu A, Herpoel A, Lambert N, Cheron J, Polleux F, et al. (2018). Human-Specific NOTCH2NL Genes Expand Cortical Neurogenesis through Delta/Notch Regulation. Cell 173, 1370–1384.e16. 10.1016/j.cell.2018.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fiddes IT, Pollen AA, Davis JM, and Sikela JM (2019). Paired involvement of human-specific Olduvai domains and NOTCH2NL genes in human brain evolution. Hum. Genet 138, 715–721. 10.1007/s00439-019-02018-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.O’Bleness MS, Dickens CM, Dumas LJ, Kehrer-Sawatzki H, Wyckoff GJ, and Sikela JM (2012). Evolutionary History and Genome Organization of DUF1220 Protein Domains. G3 GenesGenomesGenetics 2, 977–986. 10.1534/g3.112.003061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dumas LJ, O’Bleness MS, Davis JM, Dickens CM, Anderson N, Keeney JG, Jackson J, Sikela M, Raznahan A, Giedd J, et al. (2012). DUF1220-domain copy number implicated in human brain-size pathology and evolution. Am. J. Hum. Genet 91, 444–454. 10.1016/j.ajhg.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, Shamovsky O, Palma A, Mikkelsen TS, Altshuler D, and Lander ES (2006). Positive Natural Selection in the Human Lineage. Science 312, 1614–1620. 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- 69.Haygood R, Fedrigo O, Hanson B, Yokoyama K-D, and Wray GA (2007). Promoter regions of many neural- and nutrition-related genes have experienced positive selection during human evolution. Nat. Genet 39, 1140–1144. 10.1038/ng2104. [DOI] [PubMed] [Google Scholar]
- 70.Taylor MS, Massingham T, Hayashizaki Y, Carninci P, Goldman N, and Semple CAM (2008). Rapidly evolving human promoter regions. Nat. Genet 40, 1262–1263. 10.1038/ng1108-1262. [DOI] [PubMed] [Google Scholar]
- 71.Fuqua T, Jordan J, van Breugel ME, Halavatyi A, Tischer C, Polidoro P, Abe N, Tsai A, Mann RS, Stern DL, et al. (2020). Dense and pleiotropic regulatory information in a developmental enhancer. Nature 587, 235–239. 10.1038/s41586-020-2816-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chan YF, Marks ME, Jones FC, Villarreal G, Shapiro MD, Brady SD, Southwick AM, Absher DM, Grimwood J, Schmutz J, et al. (2010). Adaptive Evolution of Pelvic Reduction in Sticklebacks by Recurrent Deletion of a Pitx1 Enhancer. Science 327, 302–305. 10.1126/science.1182213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thompson AC, Capellini TD, Guenther CA, Chan YF, Infante CR, Menke DB, and Kingsley DM (2018). A novel enhancer near the Pitx1 gene influences development and evolution of pelvic appendages in vertebrates. eLife 7, e38555. 10.7554/eLife.38555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xie KT, Wang G, Thompson AC, Wucherpfennig JI, Reimchen TE, MacColl ADC, Schluter D, Bell MA, Vasquez KM, and Kingsley DM (2019). DNA fragility in the parallel evolution of pelvic reduction in stickleback fish. Science 363, 81–84. 10.1126/science.aan1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Girirajan S, Dennis MY, Baker C, Malig M, Coe BP, Campbell CD, Mark K, Vu TH, Alkan C, Cheng Z, et al. (2013). Refinement and Discovery of New Hotspots of Copy-Number Variation Associated with Autism Spectrum Disorder. Am. J. Hum. Genet 92, 221–237. 10.1016/j.ajhg.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Paulitti A, Andreuzzi E, Bizzotto D, Pellicani R, Tarticchio G, Marastoni S, Pastrello C, Jurisica I, Ligresti G, Bucciotti F, et al. (2018). The ablation of the matricellular protein EMILIN2 causes defective vascularization due to impaired EGFR-dependent IL-8 production affecting tumor growth. Oncogene 37, 3399–3414. 10.1038/s41388-017-0107-x. [DOI] [PubMed] [Google Scholar]
- 77.Watanabe J, Nakamachi T, Matsuno R, Hayashi D, Nakamura M, Kikuyama S, Nakajo S, and Shioda S (2007). Localization, characterization and function of pituitary adenylate cyclase-activating polypeptide during brain development. Peptides 28, 1713–1719. 10.1016/j.peptides.2007.06.029. [DOI] [PubMed] [Google Scholar]
- 78.Wang Y, Qian Y, Yang S, Shi H, Liao C, Zheng H-K, Wang J, Lin AA, Cavalli-Sforza LL, Underhill PA, et al. (2005). Accelerated Evolution of the Pituitary Adenylate Cyclase-Activating Polypeptide Precursor Gene During Human Origin. Genetics 170, 801–806. 10.1534/genetics.105.040527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K, Norrholm SD, Kilaru V, Smith AK, Myers AJ, et al. (2011). Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature 470, 492–497. 10.1038/nature09856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Navarro Gonzalez J, Zweig As, Speir Ml, Schmelter D, Rosenbloom Kr, Raney Bj, Powell Cc, Nassar Lr, Maulding Nd, Lee Cm, et al. (2021). The UCSC Genome Browser database: 2021 update. Nucleic Acids Res 49. 10.1093/nar/gkaa1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, and Ruden DM (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92. 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wright S (1984). Evolution and the Genetics of Populations, Volume 2: Theory of Gene Frequencies (University of Chicago Press; ). [Google Scholar]
- 83.Karolchik D, Hinrichs AS, and Kent WJ (2012). The UCSC Genome Browser. Curr. Protoc. Bioinforma Chapter 1, Unit1.4. 10.1002/0471250953.bi0104s40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harris RS (2007). Improved Pairwise Alignmnet of Genomic DNA
- 85.Kent WJ, Baertsch R, Hinrichs A, Miller W, and Haussler D (2003). Evolution’s cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. U. S. A 100, 11484–11489. 10.1073/pnas.1932072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AFA, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. (2004). Aligning Multiple Genomic Sequences With the Threaded Blockset Aligner. Genome Res 14, 708–715. 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Felsenstein J (1997). An Alternating Least Squares Approach to Inferring Phylogenies from Pairwise Distances. Syst. Biol 46, 101–111. 10.1093/sysbio/46.1.101. [DOI] [PubMed] [Google Scholar]
- 88.Fitch WM, and Margoliash E (1967). Construction of Phylogenetic Trees. Science 155, 279–284. 10.1126/science.155.3760.279. [DOI] [PubMed] [Google Scholar]
- 89.Schrago CG, and Voloch CM (2013). The precision of the hominid timescale estimated by relaxed clock methods. J. Evol. Biol 26, 746–755. 10.1111/jeb.12076. [DOI] [PubMed] [Google Scholar]
- 90.Yang Z, Kumar S, and Nei M (1995). A new method of inference of ancestral nucleotide and amino acid sequences. Genetics 141, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jukes TH, and Cantor CR (1969). CHAPTER 24 - Evolution of Protein Molecules. In Mammalian Protein Metabolism, Munro HN, ed. (Academic Press; ), pp. 21–132. 10.1016/B978-1-4832-3211-9.50009-7. [DOI] [Google Scholar]
- 92.Hubisz MJ, Pollard KS, and Siepel A (2011). PHAST and RPHAST: phylogenetic analysis with space/time models. Brief. Bioinform 12, 41–51. 10.1093/bib/bbq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kimura M (1968). Evolutionary Rate at the Molecular Level. Nature 217, 624–626. 10.1038/217624a0. [DOI] [PubMed] [Google Scholar]
- 94.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501. 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wei X, Xiang Y, Peters DT, Marius C, Sun T, Shan R, Ou J, Lin X, Yue F, Li W, et al. (2022). HiCAR is a robust and sensitive method to analyze open-chromatin-associated genome organization. Mol. Cell 82, 1225–1238.e6. 10.1016/j.molcel.2022.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Meyer M, Kircher M, Gansauge M-T, Li H, Racimo F, Mallick S, Schraiber JG, Jay F, Prüfer K, Filippo C. de, et al. (2012). A High-Coverage Genome Sequence from an Archaic Denisovan Individual. Science 338, 222–226. 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Prüfer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, de Filippo C, et al. (2014). The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49. 10.1038/nature12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Prüfer K, de Filippo C, Grote S, Mafessoni F, Korlević P, Hajdinjak M, Vernot B, Skov L, Hsieh P, Peyrégne S, et al. (2017). A high-coverage Neandertal genome from Vindija Cave in Croatia. Science 358, 655–658. 10.1126/science.aao1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Prado-Martinez J, Sudmant PH, Kidd JM, Li H, Kelley JL, Lorente-Galdos B, Veeramah KR, Woerner AE, O’Connor TD, Santpere G, et al. (2013). Great ape genetic diversity and population history. Nature 499, 471–475. 10.1038/nature12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lowe CB, Kellis M, Siepel A, Raney BJ, Clamp M, Salama SR, Kingsley DM, Lindblad-Toh K, and Haussler D (2011). Three periods of regulatory innovation during vertebrate evolution. Science 333, 1019–1024. 10.1126/science.1202702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vockley CM, D’Ippolito AM, McDowell IC, Majoros WH, Safi A, Song L, Crawford GE, and Reddy TE (2016). Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome. Cell 166, 1269–1281.e19. 10.1016/j.cell.2016.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 106.Saito T, and Nakatsuji N (2001). Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev. Biol 240, 237–246. 10.1006/dbio.2001.0439. [DOI] [PubMed] [Google Scholar]
- 107.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gasperini M, Hill AJ, McFaline-Figueroa JL, Martin B, Kim S, Zhang MD, Jackson D, Leith A, Schreiber J, Noble WS, et al. (2019). A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 176, 377–390.e19. 10.1016/j.cell.2018.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2019). Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–44. 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 112.Edgar RC (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113. 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. (2007). Clustal W and Clustal X version 2.0. Bioinforma. Oxf. Engl 23, 2947–2948. 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 114.Shank SD, Weaver S, and Kosakovsky Pond SL (2018). phylotree.js - a JavaScript library for application development and interactive data visualization in phylogenetics. BMC Bioinformatics 19, 276. 10.1186/s12859-018-2283-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. (2019). The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47, D1005–D1012. 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. (2007). PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jordan DM, Verbanck M, and Do R (2019). HOPS: a quantitative score reveals pervasive horizontal pleiotropy in human genetic variation is driven by extreme polygenicity of human traits and diseases. Genome Biol 20, 222. 10.1186/s13059-019-1844-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Synthetic allele frequency spectra ordered by value of the selection parameter used to generate each spectrum. (B) Representative Markov chain Monte Carlo (MCMC) traces for the mean selection parameter acting on variants from synthetic derived allele frequency spectra. Traces are colored by the value of the input selection parameter used to simulate each spectra. (C) Mean and 95% highest density credible intervals for the posterior distribution of the mean selection parameter for 50 synthetic variant sets, 10 for each input selection parameter. Posterior estimates are displayed from 50,000 iterations with the first 5,000 iterations discarded as burn-in. 47/50 (94%) of traces contained the true input selection parameter in the 95% credible interval. Traces where the input selection parameter was not contained in the credible interval are marked with an asterisk. Distribution of MCMC trace means for the mean selection parameter with and without the Kern correction for divergence-based ascertainment bias are displayed for the least divergent 1% of variant sets (D) or most divergent 1% (E). Mean selection parameters are estimated as the posterior mean of 10,000 iterations with the first 1,000 iterations discarded as burn-in.
(A, B) Pearson correlations between velocity and acceleration (A) or initial velocity (B) for 2,902,532 500bp genomic regions. (C) Heatmap of mean selection parameters for sets of variants overlapping all genomic regions binned by velocity and acceleration score. Mean selection parameters are estimated as the posterior mean of 10,000 iterations with the first 1,000 iterations discarded as burn-in. (D) Cumulative proportions of velocity, initial velocity, and acceleration for variants overlapping regions of interest. Here HARs are split into the original HARs (oHAR15) and the expanded HARs (eHAR24). (Bonferroni-adjusted Wilcoxon. ***: p < 0.001; ****: p < 0.0001) (E) Phylogenetic context of HAQERs, chimp-AQERs, and gorilla-AQERs. Venn diagram displays overlaps between rapidly-evolved primate regions (****: p < 0.001 for overlap enrichment between these sets of genomic regions). (F) Observed over expected proportion of elements within 5mb of chromosome ends. Expected proportion was estimated from randomly distributed regions in each genome. (G) GREAT ontology enrichments for HAQER chromatin contact sites in H1 hESCs (red) and GM12878 (yellow). One ontology term, innate immune response, was significant in both cell types. (H) GREAT ontology enrichments for HAQERs, chimp-AQERs, and gorilla-AQERs.
(A) Density of sites divergent between the modern human sequence and the inferred Human-Chimpanzee Ancestor (HCA) sequence against the density of polymorphic sites observed in 501 unrelated African individuals for Human Ancestor Quickly Evolved Regions (HAQERs), pseudorandomly selected neutral proxy regions (RAND), human accelerated regions (HARs), ENCODE candidate cis-regulatory elements (ENCODE), and ultraconserved elements (UCEs). (B) Distribution of recombination frequency in HAQER, RAND, and HARs (Bonferroni-adjusted Wilcoxon). (C) Distribution of replication timing in HAQERs, HARs, and RAND (Bonferroni-adjusted Wilcoxon) (D) Proportion of HCA divergent sites that are weak to strong mutations (A to G/T to C or A to C/T to G) in HAQERs, HARs, and RAND. (E) Principal component analysis of the mutation spectrum for all HAQERs. HAQERs do not demonstrate distinct mutation spectrum subtypes. (F) Proportion of HCA divergent sites that are polymorphic (observed as segregating in the 501 unrelated African individuals from A) as opposed to fixed (not observed as segregating). The dotted line represents the proportion of polymorphic variants in RAND (1,069 polymorphic sites, 8,487 fixed sites) (Chi-squared test). (G) Proportion of segregating sites that are transitions in segregating sites from five African populations binned by low derived allele frequency (DAF < 0.1) and high derived allele frequency (DAF > 0.9) (Wilcoxon). (H) The samConsensus program generates consensus sequences from individuals calling substitutions (represented as red positions) from sequencing reads aligned to a reference genome and editing the corresponding positions of the reference assembly. (I) Phylogenetic history and geographic distribution of 30 great ape genomes used to construct the multiple alignment. (J) A representative 100bp block of sequence sampled from the 30-way whole genome alignment. (**: p < 0.01; ***: p < 0.001; ****: p < 0.0001).
(A) Overlap enrichment/depletion matrix between HAQERs (left) or HARs (right) and chromatin states from 127 reference epigenomes with sample level annotation. Both sets are depleted from transcribed regions, this reflects the ascertainment bias that some HAR studies specifically excluded coding regions (Prabhakar et al., 2006). (B) Comparisons between HAQER and HAR overlap enrichment scores for each chromatin state across 127 reference epigenomes quantified by Bonferroni-adjusted t-test (****: p < 0.0001). (C) Comparison of HAQER overlap enrichment between adult and developing tissue-derived reference epigenomes. (D) HAQERs are enriched for overlaps with active promoters and active enhancers gained after rhesus split, defined as increased H3K27ac or H3K4me2 ChIP-seq signal in the developing human brain relative to mouse and rhesus macaque52 (Bonferroni-adjusted overlap enrichment; ****: p < 0.0001, *: p < 0.05). (E) Volcano plot of significant overlap enrichments for HAQERs (left) or HARs (right) for 7 gene regulatory chromatin states.
(A) UMAP representation of 7,170 cells from scSTARR-seq in the developing mouse brain. Insert UMAP labels cells from two independent STARR-seq experiments, performed at E14.5 and E15.5. (B) Gene expression for canonical markers of each cell identity. (C) pCAG-GFP transfection reporter expression in each cell cluster. As radial glia and their progeny were targeted, limited GFP expression is observed in the inhibitory neuron, microglia, fibroblast, and vascular clusters. (D) Dotplot representation of the top five marker genes by cluster. The pCAG-GFP transfection reporter, which was electroporated preferentially into radial glia, was identified as a radial glial marker. (E) Pearson correlation between enhancer activity score measurements for each test sequence at the E14.5 and E15.5 injection timepoints. (F) Pearson correlation between the input normalization factor and enhancer activity score estimates for all test sequences with input normalization factors below 5.
Each panel displays the GENCODE gene track, open chromatin in the developing human brain48, and divergence density in the genomic context of HAQERs with neurodevelopmental function. The dashed line corresponds to a divergence density of 29 mutations per 500 bases, the statistical threshold for HAQER identification. Also labeled are the positions of STARR-seq test sequences used in this study, Human Accelerated Regions24, functional elements gained after the rhesus split52, and differentially accessible (DA) scATAC-seq sites in cerebral organoids56. The genomic context is shown for: (A) HAQER0710, a hominin-specific enhancer in the locus of DPP10, an autism spectrum disorder-related gene75; (B) HAQER0223, a hominin-specific enhancer in the locus of EMILIN276; and (C) HAQER0035, which corresponds to HAR1, a previously described rapidly-evolving region15. (D) The genomic context for the gene ADCYAP1, which harbors three HAQERs. HAQER0780 and HAQER0911 were both observed as hominin-specific neurodevelopmental enhancers. ADCYAP1 is associated with neurodevelopment77, human evolution78, and psychiatric disease79. (E) Genome browser snapshot of a 10 megabase region of hg38 chr1. Members of the NBPF gene family are highlighted in yellow. (F) Distributions of the number of variants in significant linkage disequilibrium (Plink R2 > 0.7) for each disease-linked variant overlapping HAQERs, RAND, and HARs (Bonferroni-adjusted Wilcoxon; ****: p < 0.0001, **: p < 0.01). (G) Distributions of the median distance of significantly linked variants (Plink R2 > 0.7) to corresponding GWAS variants for disease-linked variants overlapping HAQERs, RAND, and HARs (Bonferroni-adjusted Wilcoxon; ****: p < 0.0001). (H) Proportion of variants overlapping HAQERs, RAND, and HARs with significant (p < 0.05) HOPS horizontal pleiotropy scores (Chi-squared; ****: p < 0.0001).
Data Availability Statement
All software written for this manuscript was implemented as a part of Gonomics, an ongoing effort to develop an open-source genomics platform in the Go programming language (golang). Gonomics can be accessed at https://github.com/vertgenlab/gonomics.
Raw and analyzed datasets, including browser tracks, sequencing files, multiple alignments, and variant sets used in selection analysis, have been made freely available on our lab website at https://www.vertgenlab.org/. Raw and analyzed datasets have also been deposited at GEO and are publicly available as of the date of publication at the accession number listed in the key resource table.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Stbl3 Chemically Competent E. coli | Invitrogen | Cat# C737303 |
| Chemicals, peptides, and recombinant proteins | ||
| Agencourt AMPure Beads | Beckman | Cat# A63881 |
| DNase-I | New England Biolabs | Cat# M0303S |
| EcoRI | New England Biolabs | Cat# R3101S |
| Fast Green FCF | Sigma-Aldrich | Cat# F7252 |
| FBS | ThermoFisher | Cat# 10438026 |
| Hoechst 33342 | Invitrogen | Cat# H1399 |
| NEG-50 | Richard-Allan Scientific | Epredia 6502 |
| Phusion DNA Polymerase | New England Biolabs | Cat# M0530S |
| SphI | New England Biolabs | Cat# R3182S |
| Trypsin-EDTA | Sigma-Aldrich | Cat# 59428C |
| Vectashield | Vector Laboratories | H-1000-10 |
| ZymoPURE II Plasmid Maxiprep Kit | Zymo Research | Cat# D4203 |
| Critical commercial assays | ||
| LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit | Invitrogen | Cat# L10119 |
| NovaSeq 6000 S-Prime Reagents | Illumina | Cat# 20040719 |
| Chromium Next GEM Single Cell 3’ Reagent Kit v3.1 | 10x Genomics | https://www.10xgenomics.com/support/single-cell-gene-expression/documentation/steps/library-prep/chromium-single-cell-3-reagent-kits-user-guide-v-3-1-chemistry |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | Cat# E2621L |
| NEBNext Ultra II FS DNA Library Prep Kit | New England Biolabs | Cat# E6177 |
| Deposited data | ||
| 1000 Genomes Project genomes | Byrska-Bishop et al. (2022) | https://www.internationalgenome.org/data-portal/data-collection/30x-grch38 |
| Altai Neanderthal genome | Meyer et al. (2012) | https://www.eva.mpg.de/genetics/genome-projects/neandertal/ |
| Combined Human Accelerated Region locations | Doan et al. (2016) | Table S1 of Doan et al. (2016) |
| Denisovan genome | Meyer et al. (2012) | https://www.eva.mpg.de/denisova/index.html |
| ENCODE cCRE locations and ChromHmm Datasets | Moore et al. (2020) | https://www.encodeproject.org/ |
| GWAS Catalog Variants | GWAS Catalog | https://www.ebi.ac.uk/gwas/ |
| HiCAR H1 and GM12878 | Wei et al. (2022) | GEO: GSE162819 |
| Human ChromHmm Roadmap Epigenomics data | Kundaje et al. (2015) | http://www.roadmapepigenomics.org/ |
| Human gained enhancer locations | Reilly et al. (2015) | GEO: GSE63648 |
| Individual chimpanzee genomes | Prado-Martinez et al. (2013) | https://www.ncbi.nlm.nih.gov/sra?term=SRP018689 |
| knownGene | Navarro Gonzalez et al. (2021) | https://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/ |
| Meiotic Recombination DSB hotspots | Pratto et al. (2014) | GEO: GSE59836 |
| Raw and processed sequencing reads | This study | GEO: GSE212159 |
| Recombination frequency maps | Zhou et al. (2020) |
ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/working/2013050 7 omnirecombination rates/ |
| Reference genomes: hg38, panTro6, panPan2, gorGor5, gorGor6, ponAbe3 | UCSC Genome Browser | https://hgdownload.soe.ucsc.edu/downloads.html |
| Replication timing datasets | Ding et al. (2021) | https://www.thekorenlab.org/data |
| Ultraconserved Element locations | Bejerano et al. (2004) | https://hgwdev.gi.ucsc.edu/ |
| Vindija Cave Neanderthal genome | Prüfer et al. (2017) | https://www.eva.mpg.de/neandertal/draft-neandertal-genome/data.html |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J (B6) (WT) | The Jackson Laboratory | JAX: 000664 |
| Oligonucleotides | ||
| Synthetic STARR-seq Insert Sequences | This study | Supplemental Dataset 1 |
| Targeted Enrichment Primers | This study | Supplemental Dataset 1 |
| Recombinant DNA | ||
| hSTARR-seq ORI vector | Addgene | RRID: Addgene 99296 |
| PGK-EGFP | Addgene | RRID: Addgene 169744 |
| Software and algorithms | ||
| bcl2fastq2 Conversion Software v2.20 | Illumina | https://support.illumina.com/downloads/bcl2fastq-conversion-software-v2-20.html |
| BWA 0.7.17 | Li and Durbin (2009) | https://github.com/lh3/bwa |
| CellRanger v6.0 | 10x Genomics | https://support.10xgenomics.com/ |
| ClustalW2 | (Larkin et al., 2007) | https://www.ebi.ac.uk/Tools/phylogeny/simple_phylogeny/ |
| gonomics | Vertebrate Genetics Laboratory | https://github.com/vertgenlab/gonomics |
| GraphPad Prism | GraphPad | https://www.graphpad.com/ |
| GREAT version 4.0.4 | McLean et al. (2010) | http://great.stanford.edu/public/html/ |
| ImageJ | Schindelin et al. (2012) | https://imagej.nih.gov/fiij/ |
| kentUtils | Kent et al. (2003) | https://github.com/ENCODE-DCC/kentUtils |
| lastz | Harris (2007) | https://github.com/lastz/lastz |
| multiz | Blanchette et al. (2004) | https://bio.tools/multiz |
| muscle | Edgar (2004) | https://www.ebi.ac.uk/Tools/msa/muscle/ |
| phylotree | Shank et al. (2018) | https://phylotree.hyphy.org/ |
| Plink | Purcell et al. (2007) | https://zzz.bwh.harvard.edu/plink/ |
| R version 4.0.5 | R Foundation for Statistical Computing | https://www.r-project.org/ |
| RPHAST | Hubisz et al. (2011) | https://github.com/CshlSiepelLab/RPHAST |
| Seurat v4.0 | Hao et al. (2021) | https://satijalab.org/seurat/ |
| SNPeff | Cingolani et al. (2012) | http://pcingola.github.io/SnpEff/ |