

Summary
Our machine-learning framework, brain and organoid manifold alignment (BOMA), first performs a global alignment of developmental gene expression data between brains and organoids. It then applies manifold learning to locally refine the alignment, revealing conserved and specific developmental trajectories across brains and organoids. Using BOMA, we found that human cortical organoids better align with certain brain cortical regions than with other non-cortical regions, implying organoid-preserved developmental gene expression programs specific to brain regions. Additionally, our alignment of non-human primate and human brains reveals highly conserved gene expression around birth. Also, we integrated and analyzed developmental single-cell RNA sequencing (scRNA-seq) data of human brains and organoids, showing conserved and specific cell trajectories and clusters. Further identification of expressed genes of such clusters and enrichment analyses reveal brain- or organoid-specific developmental functions and pathways. Finally, we experimentally validated important specific expressed genes through the use of immunofluorescence. BOMA is open-source available as a web tool for community use.
Keywords: comparative analysis of brains and organoids, developmental gene expression patterns, cell-type functional genomics, manifold alignment tool and webapp
Graphical abstract
Highlights
-
•
Manifold alignment for comparing gene expression of organoids with developing brains
-
•
Global alignment by given development time and local refinement by common manifolds
-
•
Developmental similarity of brain regions and organoids in human and non-human primates
-
•
Conserved and specific cell trajectories and genes across brains and organoids
Motivation
Organoids have become valuable models for understanding cellular and molecular mechanisms in human development, including development of brains. However, whether developmental gene expression programs are preserved between human organoids and brains, especially in specific cell types, remains unclear. Importantly, there is a lack of effective computational approaches for comparative data analyses between organoids and developing human brains. To address this, we developed a machine-learning framework for comparative gene expression analysis of brains and organoids to identify conserved and specific developmental trajectories as well as developmentally expressed genes and functions, especially at cellular resolution.
He et al. develop a machine-learning framework, brain and organoid manifold alignment (BOMA), for comparative gene expression analysis of brains and organoids. Its applications reveal conserved and specific developmental trajectories across brain regions and organoids of humans and non-human primates, as well as developmentally expressed genes and gene functions.
Introduction
The development of human brains, especially during the early periods, remains poorly understood.1,2,3 Understanding how neural stem cells differentiate into the myriad cell types that form the brain, especially at the molecular level, such as gene expression and its regulatory mechanisms, will shed light on the human brain development and potentially further help understand the etiology of neurodevelopmental diseases. Several large collaborative consortia have been carried out to generate large-scale next-generation sequencing data in human brains, aiming to provide functional genomic resources for understanding molecular mechanisms of human brain and brain development. For example, BrainSpan4 collected ∼600 tissue samples from 48 postmortem human brains ranging from prenatal to adult age groups and measured the transcriptomic and epigenomic data across developmental stages and brain regions. PsychENCODE1,5 generated multi-omics data for approximately 2,000 postmortem brains, aiming to understand functional genomics and gene regulation in human adult brains and neuropsychiatric disorders. These consortia provided valuable public resources to decipher the developmental functional genomics and gene regulation in the human brain. However, the postmortem brain samples serve only as snapshots of the brain at different stages, whereas brain development is a dynamic process that requires crosstalk among various genes, cell types, brain regions, and environments.6
Because it is quite challenging to measure in vivo molecular activities such as gene expression in human brains, animals such as rodents and non-human primates (NHPs) have been used as models for studying molecular mechanisms during brain development. For example, Zhu et al.7 have thoroughly compared the bulk RNA sequencing (RNA-seq) data of brain development between humans and rhesus macaques at multiple brain regions and time points. Particularly, they performed non-negative matrix factorization to linearly factorize the gene expression matrix into five biologically meaningful “transcriptomic signatures,” which were then compared between humans and NHPs. Such comparisons demonstrated the usefulness of NHP models for studying brain development. However, their results also highlighted the divergence of molecular mechanisms across species. This is also supported by previous studies that noticed that using animals as models is insufficient because brain maturation is specific to its developmental context,4,8 and human brains have specific developmental programs that allow, for example, a dramatic size expansion when compared with other primates.9
To solve these challenges, emerging 3D brain culture technologies, such as organoids, have been developed. These cultures utilize embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs) and differentiate them into 3D human brain models.10 An intriguing discovery is that iPSCs follow intrinsic programs and extrinsic cues to form 3D forebrain organoids (3DOs) that can be maintained for at least 40 weeks and even over 2 years, with a transcriptomic signature corresponding to “birth” at ∼28 weeks of culture.11,12 Organoids as brain models, although in their early developing stages, have already found numerous medical applications. For example, Park et al. used 1,300 organoids to model the human brain and conducted drug screening for Alzheimer’s disease.13 However, to what extent the in-vitro-cultured organoids preserve the in vivo complex dynamic process remains a question,14 with contradictory conclusions that have been made by the community. For example, Gordon et al.11 cultured organoids for up to 694 days and used transition mapping (TMAP), a rank-rank hypergeometric test-based method, to map the organoids’ bulk RNA-seq datasets with BrainSpan RNA-seq datasets and demonstrated that organoid culture could reproduce several developmental milestones of in vivo brain development even at mid- to late-fetal stages. Velasco et al.15 performed single-cell RNA-seq (scRNA-seq) on 166,242 cells isolated from 21 organoids and showed that organoids can virtually indistinguishably reproduce the cell diversity of the human cerebral cortex. On the other hand, Pollen et al.9 compared human primary tissues versus human organoids using canonical correlation analysis (CCA)16 and co-clustering of the mixture cells from both origins. They found that organoids maintained the composition of cell types but varied in the cell percentages and concluded that using organoids as brain models is promising, but the organoid protocols need future improvements to better preserve the brain cell-type fractions and cell functions.9,17 Bhaduri et al.18 compared single-cell gene expression data of samples across different developmental periods and multiple cortical areas with organoids and found that cellular stress pathways have been activated in organoids, which impairs cell-type specification during organoid development. All these studies highlight the promise of using organoids as models for brain developmental research; however, until now, the fidelity of organoid models has still been under debate. One of the attributed reasons is the lack of dedicated computational approaches for integrative and comparative analysis of gene expression across developmental stages between brains and organoids, especially for single-cell datasets.
In particular, the comparative analysis of developmental data between brains and organoids can be viewed by machine learning as an alignment problem across multiple datasets. For instance, manifold alignment,19,20 a popular machine-learning technique, projects samples from multiple datasets onto a common latent space via mapping manifolds across datasets, e.g., multi-omics datasets.21 The neighboring samples on the latent space suggest that they can be aligned and thus share similar features (or distant samples for unaligned). In general, manifold alignment algorithms22 can be supervised or unsupervised depending on whether the sample correspondence is provided (supervised) or not (unsupervised). A supervised approach needs predefined correspondence between the samples across two datasets.23 For example, ManiNetCluster24 embeds samples into a latent manifold space and aligns them by minimizing the overall distances between corresponding samples. An unsupervised approach does not require correspondence; instead it learns the correspondence across multiple datasets.25 For example, MATCHER21 performs linear trajectory alignment based on latent Gaussian process; MMD-MA26 maximizes mean discrepancy on a kernel space; UnionCom27 uses matrix optimization to match the distance matrices of each dataset; and SCOT28 incorporates Gromov Wasserstein-based optimal transport to align single-cell datasets. However, unsupervised approaches, in general, automatically assume a shared underlying structure among the aligned datasets,29 which might not always be true. Besides, none of these manifold alignment methods considered prior time information across samples in development that can likely help increase performance and interpretability30 of the alignment. The developmental data for brains or organoids typically provide prior time information on developmental stages, e.g., postconceptional weeks (PCWs) of developing brains and cultured days of organoids. Such prior time information, though at low resolution, may help predict initial correspondences globally across samples from different datasets, in contrast to the fully unsupervised fashion. Building on such initial correspondence, further manifold alignment can then refine the alignment to reveal higher resolution and local timing by the manifold shapes that have been widely used to uncover pseudo-timings.31 However, to the best of our knowledge, manifold alignment has yet been applied for integrative and comparative analysis of brain and organoid data, especially for development and single cells.
In this article, we developed a manifold-learning framework, brain and organoid manifold alignment (BOMA), to align developmental gene expression data across human brains and organoids, aiming to better understand conserved and specific gene expression and functions. In particular, BOMA adopts a semi-supervised manifold alignment manner. That is, using prior timing information from datasets, we first perform a global alignment at a coarse-grained level to generate a correspondence matrix among samples. Next, using the correspondence matrix, we apply manifold alignment approaches to locally refine the global alignment of samples across datasets. The aligned samples finally reveal developmental trajectories with higher resolution pseudo-timing information. The aligned and unaligned samples aim to uncover conserved and specific developmental trajectories across human brains and organoids. We first demonstrated an application of BOMA by aligning bulk RNA-seq gene expression datasets and observed a similar developing trend as in the original respective publications.11 By aligning organoids with different human brain regions, we also found that organoids are more similar to certain brain regions at specific time points. We also aligned the scRNA-seq data of human versus chimpanzee organoids and observed a delayed development of human organoids compared with chimpanzee organoids. Finally, we compared recent time-series scRNA-seq datasets between human brains and human organoids in development. We found both common and uniquely expressed genes between the brains and organoids at resolutions of cell types across developmental stages. Moreover, we experimentally validated the expression of genes displaying differences between brains and organoids in selected cell types. BOMA is also available as an open-source web tool for community use.
Results
BOMA framework for comparative analyses of gene expression data between brains and organoids
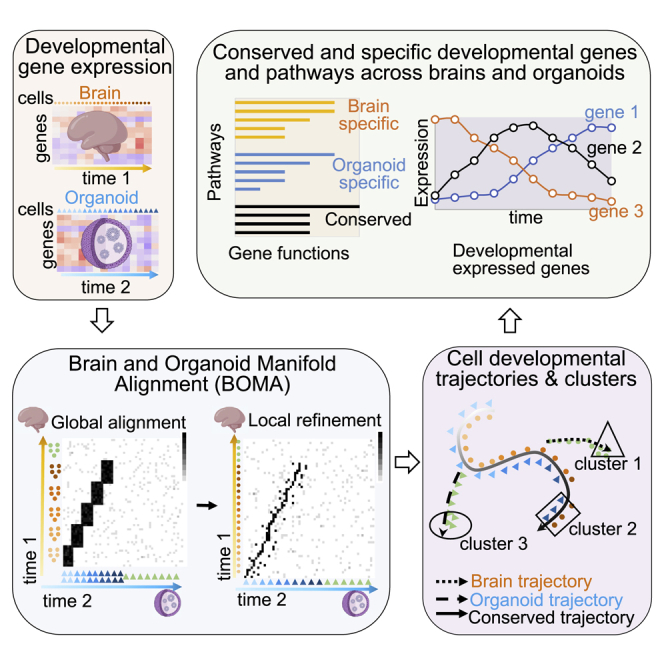
As shown in Figure 1, BOMA inputs developmental gene expression matrices of the brain and organoid samples (e.g., tissues, cells). First, it uses global alignment to align the samples and initialize a sample-wise correspondence matrix at a coarse-grain level (e.g., via manifold warping). Second, BOMA performs a manifold alignment using the correspondence matrix as the initial alignment. This step finds shared manifolds of the samples and maps them onto a common manifold space. The manifold shapes of the samples on the space are expected to uncover various developmental trajectories, which can be either conserved across brains and organoids (aligned samples) or be brain/organoid specific (unaligned samples). Finally, BOMA clusters the samples on those trajectories and finds underlying differentially expressed genes (DEGs), enriched gene functions, and associated phenotypes for each cluster, providing a deeper understanding of developmental functional genomics in brains versus organoids. The full description of the BOMA model is available in the STAR Methods.
Figure 1.

Brain and organoid manifold alignment (BOMA), a computational framework for comparative analyses of developmental gene expression data between brains and organoids
(A) BOMA inputs multiple developmental gene expression datasets (genes by samples) from brains and organoids. The samples are ordered by prior timing information in development.
(B) Step 1: global alignment to infer the correspondences of samples across the datasets at a coarse-grain level.
(C) Step 2: local alignment to refine the alignment and map samples onto a common manifold space.
(D) Clustering and functional analysis of aligned samples on the common space, e.g., brain-organoid conserved (square) or specific (circle and triangle) clusters and developmental trajectories (black curves). Downstream analyses of those clusters can discover differentially expressed genes, enriched gene functions, and associated phenotypes. GO, Gene Ontology.
To demonstrate BOMA as a framework for comparative analysis of brains and organoid development, we carried out several experiments in the following sections. We first applied BOMA on bulk RNA-seq datasets, including human brains, NHP brains, and human organoids. We further demonstrated the utility of BOMA in aligning scRNA-seq datasets, which includes single-cell data integrated from multiple independent studies. We also benchmark several state-of-the-art alignment tools using these datasets.
Spatiotemporal conservation and divergence of gene expression between organoid and brain regions
Recent landmark studies compared gene expression between the human brain and organoid development.11 However, our understanding of where and when gene expression in various brain regions is conserved or different from organoids is still unclear. To this end, we applied BOMA to align developmental gene expression data of human brains and organoids at the bulk tissue level. The brain dataset includes brain tissue samples (dataset 1, n = 460; Table S1) from 16 human brain regions (Table S2). The organoid dataset includes organoids from a recently published long-term-cultured “human cortical spheroid (hCS)” organoid bulk RNA-seq dataset11 (n = 62, dataset 6).
Our alignment shows these brain and organoid samples primarily follow a shared trajectory on the common space, indicating potential conservation during their development (Figure 2A). In particular, as shown in Figure 2B, the organoid samples from 25 to 250 days were aligned with the brain tissue samples at prenatal stages.11 At 300 days, the organoid samples started to gain postnatal signatures, indicated by their high alignment scores with postnatal brain samples (Figure 2B). Also, organoids after 350 days were not well aligned with any brain samples, which indicates that the late-stage organoids may differ from postnatal brain development. This observation was consistent with a recent comparison of brain and organoid development.11
Figure 2.

Spatiotemporal conservation and divergence of gene expression between organoid and brain regions
(A) Aligned human brain4 and organoid11 samples on the common space from BOMA. Human brain samples are from BrainSpan and are colored orange by developmental stages. The stages were described by Kang et al.32 and characterize the periods of embryo to develop into adulthood brains. Organoid samples are colored blue by cultured days. t, time; PCW, postconception weeks; M, month; Y, year.
(B) Correlation plot shows the similarity (quantified as “local alignment score”; STAR Methods) of aligned samples. Each dot is the averaged similarity across all pairs of samples at the specific developmental time points. Both the color and the size of the dots represent the local alignment score.
(C) Pairwise local alignment scores between organoids with brain samples from the OFC and mediodorsal nucleus of thalmus (MD).
(D) Averaged BOMA alignment scores between organoids versus the 16 brain regions. To calculate the averaged alignment score, for each organoid sample, its distance to the nearest sample from a certain brain region was used to calculate the local alignment score. The local alignment score was then weighted by the global alignment score of each brain region (See STAR Methods). The weighted alignment score of organoids from the same time point were averaged to show in the heatmap. Brain regions abbreviations are listed in Table S2.
Furthermore, we also interrogated which human brain regions are most similar to organoids in development. To answer this, we assessed the BOMA alignment of brain samples from each individual region with the organoid samples (Figures S1A–S1C). As expected, distinct alignment patterns were found for different brain regions, with cortical areas aligning better with hCS organoids than non-cortical brain regions during early developmental stages up to 200 days (Figures 2C and 2D). At 200 days, the alignment scores of cortical regions are significantly higher than non-cortical regions (two-sided t test, p = 0.000696). To find which genes are potentially driving the alignment, we correlated individual gene expression with organoid pseudo-time (z axis in Figure 2A) and identified 54 genes significantly correlated with the pseudo-time as summarized in Figure S1D. We also identified cortical (n = 51) and non-cortical marker genes (n = 75) by finding significantly upregulated genes across the two regions. Interestingly, many more genes upregulated during organoid development were also significantly highly expressed in cortical areas compared with non-cortical regions (n = 9 versus n = 1). Besides, we did not observe any significant overlap between the pseudo-time-correlated genes with the marker genes of each individual cortical area (Figure S1D). However, our alignment shows that within the neocortex, several cortical areas like the orbital prefrontal cortex (OFC), posterior inferior parietal cortex, primary auditory cortex (AIC), and superior temporal cortex (STC) may be better aligned with cortical organoids than other cortical areas up to 100 days (two-sided t test, p = 0.00108; Figures 2D and S1E). It is important to note that because several brain regions did not have samples from stage 2, we removed stage 2 from all the regions for the BOMA alignment. As a result, organoids at 25 days cannot align well with any brain samples. Therefore, these results suggest that cortical organoids specifically preserve brain-regional development at certain stages (i.e., spatiotemporal conservation) instead of mimicking the whole brain development. Interestingly, they also suggest that organoids are transcriptomically closer to certain neocortical areas, particularly perisylvian and orbital frontal areas, than they are to other neocortical areas.
Developmental gene expression discrepancies between human and chimpanzee organoids
We also applied BOMA to align human and NHP brains, revealing their conserved developmental gene expression across species. Specifically, we aligned rhesus macaque brain samples (n = 366)7 with human brain samples (n = 460) from BrainSpan using BOMA. We found those samples were aligned most closely around the time of birth (Figure S2A), indicating that brain gene expression across two species becomes more similar at the bulk tissue level perinatally.7
To deepen our understanding of the conservation and specificity of developmental gene expression at cellular resolution across human and NHP organoids, we applied BOMA to developmental scRNA-seq data and aligned single cells of human organoids (number of cells: n = 47,130, dataset 733) versus chimpanzee organoids (n = 26,228, six time points, dataset 7). BOMA alignment was performed on the pseudo-cells identified by a similar approach as the study generated the datasets,33 aiming to combat single-cell expression noises. Each pseudo-cell represents a group of cells with similar gene expression patterns. In total, 938 human and 483 chimpanzee organoid pseudo-cells were generated for BOMA. Our analysis shows that these pseudo-cells from the two species organoids were aligned in general to a common trajectory, which indicates cross-species developmental similarity (Figure 3A). However, some discrepancies could also be observed. First, compared with human cells, chimpanzee cells were shifted toward a later time over the maturational trajectory (toward the left in Figure 3A), suggesting that chimpanzee organoids were developing faster than human organoids (Figure 3B). The observed protracted maturation of human organoids is in line with the previous study33 and was also observed in other cross-species comparison studies on organoids34,35 as well as on 2D cultures.36,37 Second, we noticed that two sets of chimpanzee cells (Chimpanzee_1 and Chimpanzee_2; Figure 3A, right panel) could not be well aligned with any human cells. To understand the functional relevance of these two sets of cells (Data S1), we first identified each cell set by their coordinates. We then extracted upregulated genes using Presto38,39 to compare cells from each set with all other. Finally, functional enrichment analysis was performed using these genes (Figure 3C). Genes upregulated in Chimpanzee_2 were mostly enriched with brain developmental functions (false discovery rate [FDR] <10e−5). For example, the most significantly enriched term, “neuron projection morphogenesis,” is related to the maturation of neurons and circuit assembly.40 These observations again indicate faster maturation of chimpanzee organoids. On the other hand, upregulated genes in Chimpanzee_1 at early time points (0 and 4 days) were enriched in cell division processes (e.g., cell cycle, chromatin remodeling, etc.). These genes suggest that Chimpanzee_1 is likely an intermediate cell type between pluripotent stem cell and neural progenitor, as these cells express genes associated with pluripotency (e.g., POU5F1, DSG2), early embryonic development and patterning (e.g., NKX1-2, DSP, NR6A1, MGST1), neural tube development (e.g., CTHRC1, PKDCC, PRTG), and brain development/neural progenitors (e.g., TDGF1, TGFB2, PODXL, LIN28A, FOXH1, FSTL1). They also express neuron-specific genes and neuron-enriched genes (e.g., RIT2, DNMT3B, MAP1B). In support of this, Kanton et al.’s33 original analysis of these data also indicated the presence of neuroectoderm cells.
Figure 3.

Developmental gene expression alignment between human and chimpanzee organoids
(A) The samples of human and chimpanzee organoid cells33 (visualized by pseudo-cells) in the common space after BOMA alignment. Human and chimpanzee organoid cells were plotted separately for comparison. The dot colors represent the experimental time points. The dashed line in the left panel shows the direction of the developing trajectory. Two chimpanzee organoid-specific clusters (Chimpanzee_1 and Chimpanzee_2) are highlighted in the right panel.
(B) Averaged pseudo-time between human and chimpanzee organoids. Pseudo-time is defined as the x axis coordinates in (A). Each dot represents the averaged pseudo-time of samples at a certain time point (0–120 days).
(C) Functional enrichments of the chimpanzee organoid-specific cluster marker genes.
Therefore, our BOMA alignment revealed a developmental gene expression similarity between human and chimpanzee organoids. Our analysis also uncovered the cross-species discrepancy in neural development and cellular functions. These results demonstrate the capability of BOMA to compare emerging organoid single-cell data and provide insights into underlying cellular and molecular mechanisms driving neurodevelopment.
Cell-type-level conservation in development between human brains and organoids derived from ESCs
To broaden BOMA applications to single-cell datasets of human brains versus organoids, we first benchmarked BOMA on two particular single-cell datasets. The comparison of human brains and organoids, especially at the cell-type level, will greatly advance our understanding of how well in-vitro-cultured organoids model the in vivo human brain. We first aligned single-cell data from cortical regions of postmortem human brains (n = 4,261, dataset 3)41 with those of organoids differentiated from a well-established human ESC line (H9, n = 11,048, dataset 7).33 This human brain dataset covers prenatal samples across 6–32 PCWs, while the organoid dataset includes organoids from 0 days up to 4 months in vitro. Before performing the alignment, human brain pseudo-cells (n = 490) and organoid pseudo-cells (n = 497) were generated from both datasets.
Our earlier analysis of bulk RNA-seq datasets (Figure 2B) shows that organoids up to 4 months could be aligned across prenatal developmental periods, indicating the developmental time ranges of these two separate studies are comparable. Thus, we aligned these two scRNA-seq datasets and identified five cell clusters in the common space (STAR Methods). Each cluster represents a group of cells that likely have similar functions (Figure 4A). Interestingly, each individual cluster contains cells from both brains and organoids, suggesting that the in vitro organoids are likely composed of the major cell types in the human brain. To understand functions underlying those clusters, we calculated the cell types enriched in each cluster. For a given cluster, the enrichments were performed for the cluster cells from brains and organoids separately, and we observed that the enriched cell types in each cluster were generally matched (Figure 4B). For instance, cluster 1 was mainly enriched for early developing cells, such as radial glia (RGs), and oligodendrocyte progenitor cells (OPCs); cluster 2 was associated with intermediate progenitor cells (IPCs) and newborn excitatory neurons (nENs); cluster 3 was mainly mapped to excitatory neurons (ENs); cluster 4 was mainly mapped to inhibitory neurons (INs); and cluster 5 was mainly mapped to endothelial cells. The details of cell-type annotations can be found in Table S3. It is worth pointing out that we only performed coarse clustering to show the high-level correspondence of cell types between aligned brains and organoids. Further sub-clustering reveals more refined cell-type enrichments of each sub-cluster (Figure S2B).
Figure 4.

Alignment of developmental gene expression between human brains and human ESC organoids
(A) The human brain41 and organoid33 cells (visualized by pseudo-cells) on the common space after BOMA alignment. Left: human brains. Right: human organoids. Aligned cells were grouped into 5 clusters.
(B) The associated cell types of each cluster from the enrichment analysis (hypergeometric test; STAR Methods). ComplexHeatmap42 was used to plot the significance of associated cell types for each cluster. U, early time point cells of unknown cell types.41
(C) The associated developmental stages (time points) of cell clusters (hypergeometric test; STAR Methods). The stages were described by Kang et al.32 to characterize the periods of human embryo to develop into adulthood brains. t, time; PCW, postconception weeks. Dots represent associations with Benjamini-Hochberg (BH)-adjusted p values <0.05.
Moreover, it is also important to look at matching developmental timing between brains and organoids. Determining the corresponding developmental periods during which cells are generated and specified in organoids will greatly benefit the design of culturing experiments. To address this, we identified the associated developmental stages of each cluster by calculating the significance of cells overlapping between each stage versus each cluster (STAR Methods). In general, we observed that each cluster was associated with different developmental stages (Figure 4C). Together with the fact that clusters are composed by different cell types, this indicated the dynamic maturation of cell types across development. Interestingly, we observed that brain and organoid development follows a similar pattern, which again supports the developmental conservation between two datasets. However, discrepancies were observed for cluster 5, which is significantly associated with microglia and endothelial and mural cells in the brains, but with RGs in the organoids (Figure 4B), reflecting the fact that these cell types have distinct origins from neurons and glia in the brain. Also, cluster 5 is associated with later developmental stages in brains (>24 PCWs) but with earlier cultured time points in organoids (<1 month; Figure 4C). This suggests that brain cells in this cluster are more mature than organoid cells. This observation was supported by BOMA-aligned cells on the common space in Figure 4A, where brain cells stretched longer in this cluster than organoid cells.
Besides, we tested robustness of BOMA using this dataset (Figure 4). To do this, we challenged BOMA by intentionally adding mismatched regional cells/cell types (red blood cells [RBCs]) (Figure S2C) or removing certain cell types (Figure S2D). Our results show that BOMA performs reasonably well under those challenges in terms of preserving shared developmental trajectory and identifying cell-type-specific branches.
Large-scale alignment of integrated datasets in human brains and organoids derived from iPSCs
Brain organoids differentiated from iPSCs have been used extensively to model human brain development and developmental disorders.18,43,44 Here, we tested BOMA’s performance for aligning large-scale datasets of human brains versus both iPSC- and ESC-derived organoids. In particular, we integrated scRNA-seq datasets from multiple studies to align single cells of human brains and human brain organoids (STAR Methods). The integrated datasets have 57 human brain samples and 28 iPSC- or ESC-derived organoids. The brain data contain 175,334 cells across 5.85–37 PCWs, while the organoid data contain 187,179 cells across 21–105 cultured days. Similar to previous analyses, we first clustered cells into pseudo-cells (1,018 in brains, 872 in organoids) to remove stochastic noise and, afterward, evaluated the batch effects across datasets. t-distributed stochastic neighbor embedding (tSNE) plots show that minimum batch effects persist after reducing cells to pseudo-cells (Figures S3A and S3B, top panels). We then input pseudo-cells into BOMA for alignment. We found that BOMA aligns the two large-scale integrated datasets reasonably well, showing aligned cell trajectories with similar cell-type distributions between brain and organoid cells in the common space (Figures 5A, S3A, and S3B, bottom panels). For instance, OPCs were embedded in the middle, and ENs, INs, and RGs were aligned in a separate branch, while IPCs spread across both ENs and RG cell branches. Expectedly, even less batch effects were observed after BOMA alignment (Figures S3A and S3B).
Figure 5.

Large-scale alignment of integrated scRNA-seq datasets in human brains and organoids from multiple studies
Five scRNA-seq datasets of human brains18,41,45 and organoids18,44 were applied.
(A) The human brain and organoid cells (visualized by pseudo-cells) on the common space after BOMA alignment. Left: brains. Right: organoids. The dots are colored by given cell types from the datasets.
(B) Experimental time correspondence between aligned intermediate progenitor cells (IPCs) versus excitatory neurons (ENs)/radial glia (RGs) within organoids samples.
(C) Inferred developmental trajectory for ENs based on their coordinates on the common space. Top: brains. Bottom: organoids.
(D) Trajectory segments versus prior development stages (experimental timepoints). Human brain cells from segments earlier than stage 6 were grouped together due to the limited sample sizes. Mann-Kendall trend test for mean values of each segment was used to test the trending significance.
(E) The enriched functions and pathways of genes significantly upregulated in organoid ENs, genes upregulated in brain ENs, and genes expressed in both brains and organoids.
Progenitor cells, such as IPCs, can divide and differentiate into postmitotic ENs in the developing cerebral cortex. This suggests that IPCs should align with neurons on the same maturational trajectory. To test whether this is true, we compared the developmental distribution of cultured IPCs with aligned ENs and RGs within organoid samples. Interestingly, we did observe a time shift between IPCs with ENs (e.g., IPCs of 3 weeks can align with ENs of 10 weeks, IPCs of 8 or 10 weeks can align with ENs of 15 weeks, etc.) but not between IPCs and RGs (Figure 5B). The differences in alignment of IPCs with RGs versus ENs make sense given the timing of the events of neuronal cortical development. RGs divide asymmetrically to produce either two RGs or one RG daughter cell and one IPC. IPCs then undergo symmetric divisions to produce postmitotic neurons that migrate to their proper cortical layers.
Moreover, we benchmarked other state-of-the-art methods and compared them with BOMA. Although Seurat16 (Figure S3C) and Liger46 (Figure S3D) can perform alignment at the single-cell level, both failed to identify the developmental trajectories. Several other manifold-based alignment methods (UnionCom,27 SCOT,28 MMD-MA26) can map the pseudo-cells into a manifold space, but the cell types were not embedded closely (Figures S3E–S3G). MetaNeighbor,47 a correlation-based method for characterizing cell-type replicability across scRNA-seq datasets, had computational memory issues when applied on all cells within this dataset and was unable to identify cell-type replicability on a 10% sub-sampled dataset (Figure 3H). In summary, BOMA outperforms other platforms in terms of both finding aligned cell trajectories and discovering cell-type developmental conservation across large-scale human brain and organoid datasets.
Brain-organoid aligned trajectory analysis reveals conserved and distinct developmentally expressed genes in specific cell types
Aligned cell trajectories by BOMA between human brains and organoids show developmental processes across various cell types. To further understand the gene expression programs driving cell-type maturation, we identified maturation trajectories based on the coordinates of cells corresponding to each cell type in the common space, such as ENs (Figure 5C) and IPCs (Figure S4A). Then, we identified the DEGs across the cell-type trajectory between brains and organoids. The enrichment analysis of those DEGs revealed conserved and specific developmental functions of the cell type across brains and organoids (STAR Methods).
The cell-type trajectories revealed the pseudo-times of individual cells during development (i.e., cell positions over the trajectory), hypothetically providing higher timing resolution than the prior timing information. By cutting the trajectory into segments and correlating them with the developing stages, we found that the segments of such pseudo-times significantly correlate with real developmental stages (Figures 5D for ENs with adjusted p = 0.0275 in brains and p = 0.0002 in organoids, and S4A for IPC trajectory), which suggests that this trajectory (pseudo-time) captures the real developmental maturation of cell types.
We then identified the DEGs for each segment along each cell type’s trajectory (STAR Methods). We identified 549 organoid and 310 brain upregulated genes that were differentially expressed within at least one segment of the EN’s trajectory (Data S2). Functional enrichment of these DEGs showed that organoid upregulated genes were mapped to chemical stress response, which is supported by a previous study18 (Figure 5E). On the other hand, the brain upregulated genes were mapped to brain development processes, as expected.
To validate the differential expression of some of these DEGs, we performed immunofluorescence in the developing human neocortex and human organoids at different stages of differentiation (Figure S4B). We found that the expression changes of important genes across stages (percentage of expressed cells) are greatly consistent with our results. SATB2, encoding a transcription factor defining cortical neuron projection identity,48 and POU3F2, encoding a transcription factor important for primate RG expansion and differentiation,49 displayed only low levels of expression throughout the development period of the organoids by BOMA and were identified as significantly upregulated in excitatory cells of the human neocortex compared with human organoids at late stages (∼19 PCW) (Figures 6A and 6B; Benjamini-Hochberg [BH]-adjusted Wilcoxon rank-sum test p = 1e−2). Consistent with these results, immunofluorescent staining followed by quantitative analyses of human organoids across 8, 10.5, and 14 weeks of differentiation, corresponding to segments 7, 8, and 9, respectively, showed only a small proportion of SATB2+ (<1.5%; Figures 6C and S18B) or POU3F2+ (∼3%; Figures 6F and S18C) cells, whereas immunofluorescent staining of human tissue confirmed the enrichment of SATB2+ and POU3F2+ cells in the cortical plate at 19 PCWs compared with organoids at 14 weeks (Figures 6D and 6E with t test, p = 0.0038, and 6G and 6H with t test, p = 0.001). SATB2 and POU3F2 are expressed in excitatory upper layer cortical neurons in both the human brain50,51 and cortical organoids,48,49,52 which are formed after the appearance of deep-layer neurons that express markers such as TBR1 and BCL11B (also known as CTIP2). At the ages examined, our organoids have predominantly TBR1+ or BCL11B+ cells in the region surrounding the progenitor-rich (SOX2+) zone, indicating that at these stages, deep-layer, but not superficial-layer, neurons have been formed (Figure S4B, top panel). We expect that analysis of older organoid datasets (greater than 15 weeks in vitro) using BOMA would show increased numbers of these two cell populations, as has been shown by other studies using immunostaining52 or bulk RNA-seq approaches.11
Figure 6.

Experimental validation of developmental expression of predicted brain- and organoid-specific genes
(A and B) Developmental expression profiles of SATB2, POU3F2, and PSMB5 mRNAs in human organoids (A) and human neocortex (B), determined by BOMA. Time correspondence for each segment ID can be found in Figure 5D.
(C, F, and I) Immunostaining of cortical organoids (n = 3) revealed percentages of cells expressing SATB2 (C), POU3F2 (F), and PSMB5 (I) during the maturation at 8, 10.5, and 14 weeks.
(D, G, and J) Quantification of SATB2+, POU3F2+, and PSMB5+ cells showed significant enrichment for human cortical plate (PCW 19, correspond to segment 9) compared with organoids (14 weeks, corresponding to segment 9). Differences between organoid and cortical plate were tested using unpaired t test with Welch’s correction, p = 0.0038 for SATB2+ cells, p = 0.001 for POU3F2+ cells, and p = 0.0011 for PSMB5+ cells.
(E, H, and K) Representative images of organoid and human brain sections. Scale bar: 50 μm.
On the other hand, PSMB5, encoding a 20S proteasome subunit, demonstrated consistent but slightly decreasing expression across the maturation trajectories by BOMA (Figure 6A) and was validated by immunostaining of human organoids (Figures 6I and S18E; ∼10% of cells). Although PSMB5 exhibited higher expression than SATB2 or POU3F2 at earlier stages in both organoids and brains, in later stages, it exhibited similar levels of expression to those genes only in the brain (Figures 6A and 6B). Consistent with this trend, we saw similar proportions of cells that were positive for SATB2, POU3F2, or PSMB5 in 19 PCW human cortical plate by immunostaining (Figures 6D–6J). Significantly more PSMB5+ cells were found to be enriched in human neocortex (t test, p = 0.0011) but mostly in deep-layer and sub-plate neurons (Figure 6K), suggesting that human organoids may have a lower abundance of sub-plate neurons at this in vitro stage. Another possibility is that while many cells in the organoid express PSMB5 at the mRNA level, they do not express high levels of PSMB5 protein or that the organoids used to generate these datasets contained larger cortical plate regions and thus higher proportions of PSMB5+ cells.
Discussion
In this work, we present BOMA as a framework for comparative analysis of gene expression between brains and organoids, with an attempt to understand the genomic regulations during their development. Our evaluation of BOMA on both bulk tissue and single-cell datasets demonstrated its scalability. Spatiotemporal and species-wise gene expression patterns have been observed by our alignment. Genes differentially expressed across cell types and developmental stages were also identified by our scRNA-seq analysis. Although we only focused on comparing RNA-seq datasets between brains and organoids, BOMA can be easily applied to compare pairs of any samples (RNA-seq or other modalities). Hence, we provide a web tool of BOMA for general community use.
Compared with existing methodology for comparative analysis between scRNA-seq data, BOMA’s semi-supervised approach performed better than the unsupervised methods as suggested by our results. The global alignment step not only improves the model interpretability but also improves BOMA’s capabilities to find aligned developmental trajectories. scRNA-seq data are, in general, noisy and stochastic, and the pseudo-bulk methods we benchmarked demonstrated that these approaches can diminish scRNA-seq noises as well as combat batch effects across datasets. Future development of more accurate scRNA-seq technologies will potentially improve the alignment. Also, the scRNA-seq datasets were integrated from multiple published studies, so the input of BOMA can be confounded by various experimental factors, for instance sample-wise batch effects, organoid culturing periods, sample sizes, sample time, sequencing depths, etc. As showed in the results, BOMA significantly reduced these confounders and demonstrated superior performance for integrative analysis of multiple studies. In addition, as a framework, BOMA can easily incorporate other existing alignment methods (e.g., manifold warping, CCA, etc.). BOMA’s supervised manner allows the correspondences between sample pairs to be incorporated into the alignment as prior knowledge. For example, users can define any correspondence information based on their own domain knowledge. Cell correspondences generated by other alignment tools (e.g., Seurat, Liger, etc.) can also be incorporated as prior knowledge of BOMA by defining the correspondence matrices. The experiments of intentionally inserting mismatched brain regions and cell types demonstrated the robustness of BOMA. However, including RBCs does make alignment more challenging, with fewer common trajectories observed between brains and organoids (Figure S2C versus Figure 4A). One possible future solution is to run BOMA multiple times. For example, we can first run BOMA once to detect aligned/unaligned cells. Then, for aligned cells, we can run BOMA again to discover shared developmental trajectory. However, for unaligned cells, we can apply manifold learning and dimensionality reduction technics (e.g., diffusion map, etc.) to discover dataset-specific trajectories on the reduced latent space. In terms of alignment metrics, we considered both the local distances and direction of global trajectories. It is important to consider both since neither can capture the alignment quality separately. However, our current way of designing the global similarity is simply calculating the cosine similarity between vectors of aligned trajectories. More complex approaches (e.g., Procrustes analysis,53 etc.) considering the shape of the aligned trajectories might be useful to better capture the global similarity.
Our manifold alignment analysis showed gene expression similarities between organoids and brains, demonstrating the viability of using organoids to understand human brain development.15 However, differences were also observed in the comparative analysis, which suggests that future protocol optimizations are needed.9 Our data indicate that, compared with developing brain tissue, organoids contain relatively fewer superficial-layer neurons (SATB2+POU3F2+) and fewer PSMB5+ cells, which in human tissue appear to enrich among deep-layer and sub-plate neurons. Optimization of earlier organoid protocols has shown that reducing oxidative stress within organoids by cutting or slicing can improve long-term maintenance of neural progenitor populations, leading to expansion of cortical plate-/sub-plate-like regions, more distinct lamination, and increased abundance of superficial layer neurons.54,55,56 Using BOMA to analyze future scRNA-seq datasets from organoids generated using these recently optimized protocols would provide better indication of how similar organoids are to the developing human brain. Additionally, future analyses could compare datasets from organoids generated using different protocols to determine whether certain approaches better recapitulate specific features of brain development, such as formation of long-range projections or more abundant numbers of outer RGs. This information would allow researchers to choose the organoid system best suited to their research questions.
Previous studies have demonstrated the wide application of organoids as experimental models for drug screening of diseases.13,57,58 Other studies have also shown using patient-derived organoid (PDO) platforms to improve preclinical drug discovery in personalized medicine.59 Recent clinical trials are moving toward cell therapy of diseases using lab-cultured organoids.60,61,62 All these reports suggested unprecedented opportunities for organoids in both lab research and clinical treatment. Thus, we believe that BOMA, which allows a deeper understanding of the gene regulatory mechanisms underlying the cultured organoids, will benefit future clinical studies.
Limitations of the study
Our evaluation of BOMA was only based on limited samples from limited cultured periods, with limited numbers of pseudo-cells. Future studies using longer-cultured organoids and more samples are recommended for better comparative analysis between brains and organoids.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-BRN2 | Santa Cruz | Cat#SC-393324 |
| anti-PSMB5 | Novus Bio | Cat#NBP-13820 |
| anti-SATB2 | Gen Way | Cat#20-372-60065 |
| anti-TBR1 | Abcam | Cat#ab31940 |
| anti-CTIP2 | Abcam | Cat#ab18465 |
| Deposited data | ||
| Brain bulk RNA-seq (BrainSpan) | Li et al.4 | http://evolution.psychencode.org/ |
| Human Brain scRNA-seq dataset 1 | Polioudaks et al.63 | http://solo.bmap.ucla.edu/shiny/webapp/ |
| Human Brain scRNA-seq dataset 2 | Nowakowski et al.41 | https://cells.ucsc.edu/?ds=cortex-dev |
| Human Brain scRNA-seq dataset 3 | Trevino et al.45 | https://scbrainregulation.su.domains/ |
| Human Brain scRNA-seq dataset 4 | Bhaduri et al.18 | https://organoidreportcard.cells.ucsc.edu |
| Human Organoid bulk RNA-seq | Gordon et al.11 | http://solo.bmap.ucla.edu/shiny/GECO/ |
| Human Organoid scRNA-seq dataset 1 | Kanton et al.33 | https://bioinf.eva.mpg.de/shiny/sample-apps/scApeX/ |
| Human Organoid scRNA-seq dataset 2 | Birey et al.44 | GEO:GSE93811 |
| Human Organoid scRNA-seq dataset 3 | Bhaduri et al.18 | https://organoidreportcard.cells.ucsc.edu |
| Data 1 | This work | Zenodo(https://doi.org/10.5281/zenodo.7236202) |
| Data 2 | This work | Zenodo(https://doi.org/10.5281/zenodo.7236202) |
| Data 3 | This work | Zenodo(https://doi.org/10.5281/zenodo.7236202) |
| Experimental models: Cell lines | ||
| Human iPSC | WiCell Research Institute, Madison, USA | WC031i-5907-6 |
| Software and algorithms | ||
| Python (versions 3.7) | Python | https://www.python.org |
| R (version4.0) | R | https://www.r-project.org/ |
| BOMA codes | https://github.com/daifengwanglab/BOMA | Zenodo(https://doi.org/10.5281/zenodo.7556083) |
Resource availability
Lead contact
Requests for further information should be directed to the lead contact, Daifeng Wang (daifeng.wang@wisc.edu).
Materials availability
This study did not generate new materials.
Experimental model and subject details
Human WC5907 iPSC line64 was maintained on mouse embryonic fibroblast feeder layers as described65 and differentiated into organoids as carried using a published protocol.66 Briefly, iPSCs were lifted using dispase (0.4 mg mL−1) and transferred to low attachment flasks (Greiner Bio-One) in hESC media plus two SMAD inhibitors SB-431542 and LDN-193189 from days 0–5. Organoids were then switched to neural medium plus growth factors EGF (20 ng mL−1; R&D Systems) and FGF2 (20 ng mL−1; WiCell) from days 6–24. After 24 days, organoids were cultured in neural medium supplemented with growth factors BDNF (20 ng mL−1; Peprotech) and GDNF (20 ng mL−1; Peprotech) until day 43 with media changes every 2-3days. Organoids were collected at 8, 10.5, and 14 weeks of differentiation and fixed with 4% PFA overnight. They were then washed with PBS 3x for 15min, and transferred to a 30% sucrose solution for 48hrs. Organoids were embedded in OCT and 30% sucrose (1:1) and stored in −80 freezer until analysis.
Method details
Brain-organoid manifold alignment (BOMA)
Emerging organoids have been widely used as models to mimic complex brain development. We developed BOMA pipeline to use manifolds to align gene expression data between brain and organoid samples (e.g., tissues, cells) (Figure 1). Such brain-organoid expression data alignment from BOMA aims to uncover conserved (aligned) and specific (unaligned) developmental gene expression patterns across brains and organoids. Our further downstream analyses of such expression patterns allow a deeper understanding of developmental functional genomics at both tissue and cell-type levels, especially in organoids.
Suppose that we want to compare two developmental gene expression datasets (e.g., brains vs. organoids) matrices, and , where is the number of genes, and are the number of samples within each dataset, is a -dimensional vector representing the expression levels of genes in the sample in , and is also a -dimensional vector representing the expression levels of genes in the sample in . The samples of {} and {} are ordered by prior timing information if available. BOMA carries out the alignment by two major steps. In Step 1, BOMA globally aligns brain and organoid samples, based on prior timing (or any sequential) information of samples. Such prior timing information is typically at low resolution, e.g., only cultured days available for many cells in organoids. This global alignment establishes the initial correspondence across brain and organoid samples. In Step 2, from such initial correspondence, BOMA applies manifold learning to locally refine the alignment and co-embed brain and organoid samples onto a common manifold space. The manifold shapes of the samples on the space are expected to uncover various developmental trajectories, which can be either conserved across brains and organoids (aligned samples) or brain/organoid specific (unaligned samples). Furthermore, the manifold shapes from the space are expected to form developmental trajectories, revealing potential pseudo times among samples. Such pseudo times, at a refined high resolution, provide unobserved timing from prior information.
BOMA Step 1 - Global Alignment: This step aligns X and Y at a coarse-grained level and initializes the correspondence matrix () for the next step. Primarily, we introduce two popular methods for global alignment.
-
1)
Dynamic Time Warping (DTW). DTW finds the optimal set(s) of aligned samples (∗) between and by minimizing the sum of distances between all aligned sample pairs:
where is the distance between the ith and jth samples of X and Y, is the set of all possible alignments between the two datasets. Distance of the samples x and y used here is defined by , where is the Pearson Correlation.
Specifically, we used R package, dtw67 to perform the DTW alignment. In this work, we chose the constraint as ‘open begin and end’, which means that two sequential datasets can be unaligned at the beginning and end. The aligned samples from DTW can be used to initialize a corresponding matrix among samples, , where Wij = 1 if samples are aligned, and = 0 otherwise.
-
2)
Correlation based kNNgraph: This method first calculates the Pearson Correlation of each sample pair and then constructs a k-nearest neighbor graph (kNNgraph) by linking each sample with its k (a hyperparameter) most correlated neighbors in the other dataset. The adjacency matrix of the constructed kNNgraph can thus be used as the correspondence matrix .
In real application, if timing information is available, the global alignment of BOMA can be carried out by manifold warping or dynamic time warping as we demonstrated in aligning developmental bulk tissue data. However, if priori timing information is unavailable, the global alignment step of BOMA can learn the correspondences across samples that can be used for the following local alignment. To this end, the users can choose the correlation based kNNgraph. Besides the two methods mentioned above, this step can also be accomplished by other methods, e.g., Liger,46 which uses Nonlinear Matrix Factorization(NMF) for single-cell alignment; Seurat,16 which aligns single cells by identifying anchor genes.
BOMA Step 2 - Local Alignment: this step performs a manifold alignment of and using the correspondence matrix () from Step 1 as the initial alignment. Specifically, manifold alignment finds shared manifolds of samples from and and maps them onto a common space. The proximate samples on this space suggest well aligned, whereas distant samples for unaligned. To this end, it aims to find the functions and that minimize the following loss function to map the samples onto the common space:
where is the correspondence between and from Step 1. It can be weighted, or it can be binary (e.g. 0: aligned, 1: unaligned) as in this work. and are two neighborhood similarity matrices, which were generated by kNNgraph. is a scalar, which constitutes the trade-off between the alignment across datasets and preserving manifolds within datasets. By default, we set equals 0.5. Here, we use nonlinear manifold alignment (NMA) to solve the above optimization problem. NMA is non-parametric and directly estimates the coordinates of samples on the common space from optimal alignment via eigen-decomposition.24 Also, we implement NMA in this step using our previous method and tool, ManiNetCluster.24
After mapping samples onto the common manifold space by BOMA, we can simply calculate the Euclidean distances of samples on the space, i.e., for Samples and . A local alignment score between samples and can be then defined by . The high alignment scores suggest a well aligned pair of samples. However, the local alignment score can only evaluate the local similarity between a pair of aligned samples. When the time information is available, we want to ensure the two aligned trajectories evolve toward the same direction across time. To capture the global similarity between two aligned datasets (e.g., brains and organoids), we consider the direction of their aligned trajectories. This global similarity can be defined by the cosine of the angle between the two aligned trajectories , where A and B are vectors that represent the directions of two aligned trajectories after BOMA alignment. and are the L2 norm of these two vectors. A higher value of means the two trajectories have more similar directions with each other. In this work, when the time information is available (e.g., DTW), we define A and B as vectors pointing from the earliest timepoint to the latest timepoint of the aligned samples. However, when the time information is not available (e.g., correlation based kNNgraph for single-cell datasets alignment), we simply set . Finally, we use as a weight factor to adjust the loal similarity score , and define a BOMA alignment score () to capture both the local and global alignment similarity.
Gene expression datasets of brains and organoids
As summarized in Table S1, we collected recently published RNA-seq gene expression datasets for brains and organoids, covering both bulk tissues and single cells across differential developmental stages.
Briefly, Dataset 14 contains bulk-tissue RNA-seq of 826 samples from 16 regions of human brains (n = 460) and 9 regions of RM brains(n = 366) in Brainspan and PsychENCODE projects. Dataset 263 contains single-cell RNA-seq (scRNA-seq) of 40,000 cells from human brain germinal zone and developing cortex regions between 17 and 18 Postconceptional Weeks (PCWs). Dataset 341 contains scRNA-seq data of 4,261 cells in human brains between 6 and 32 PCWs. Dataset 445 contains scRNA-seq data of 57,868 cells from four human brain primary samples at different developmental stages between 16 and 24 PCWs. Dataset 518 includes scRNA-seq of 136,254 cells from human brain samples collected at 14,18,22 PCWs. Dataset 611 is from the cultured organoid samples and includes bulk RNA-seq data of 62 samples from ten time points between 50 days to ∼ two years. Dataset 733 contains scRNA-seq of 73,358 cells in organoids from human or chimpanzee between 0 days to four months. Dataset 844 contains scRNA-seq of 11,838 cells from organoids cultured for 105 days. Dataset 918 contains scRNA-seq of 189,346 organoid cells of culturing time spanning 3–10 weeks.
Identification of human brain developmental genes
We identified a set of genes related to human brain development at both tissue- and cell-type levels, as input features for BOMA alignment (Figure S5A). First, we used the bulk RNA-seq data in BrainSpan4 to predict co-expression gene modules within each brain tissue (region) by WGCNA.68 We identified 1,191 co-expression gene modules in total. Genes from the same module are co-expressed at certain tissue across the development, suggesting that they are likely co-regulated and thus involved in similar biological processes, so we term them as ‘development modules’. Second, we applied Scanpy69 on the single-cell RNA-seq dataset from Dataset 263 (Table S1) to identify developmental expressed genes at the cell-type level. Specifically, for each of 11 cell-types, we compared this cell-type with all other cell-types and found cell-type differentially expressed genes (adjust p value <0.01 and log fold change >1). After iterating through 11 cell-types, we identified 2,032 cell-type expressed genes in total. Third, we overlapped each development module from a tissue-type with each cell-type expressed gene set and performed hypergeometric tests (R function phyper()) to determine the significance of the developmental gene overlaps between the tissue-type and the cell-type (tissue-cell-type pair). We also adjusted the p values of tests using ‘Benjamini-Hochberg procedure (BH)’. We selected the overlapped genes of tissue-cell-type pairs with adjusted p < 0.01 as significant overlapped gene sets. Finally, we obtained 1,533 genes as human brain developmental genes (Data 3).
scRNA-seq data pre-processing
We used Seurat16 to preprocess all applied scRNA-seq datasets. In particular, we removed the cells expressing less than 200 genes and the genes expressed within less than 30 cells. The rest cells were filtered by mitochondrial genes to be less than 10. The preprocessed datasets were then log2 transformed.
Compared to bulk RNA-seq, scRNA-seq is noisy with random effects. To address this, recent studies70,71 used pseudo-bulk methods to aggregate single-cells across biological replicates and improved downstream differential expression gene analyses. Therefore, we also applied the pseudo-bulk methods33,70,71 to create pseudo-cells from single cells. Specifically, we first grouped single cells into cell clusters. Each cluster represents one pseudo cell, and its expression levels are the averaged gene expression of cells within the cluster. This step can also balance the sample sizes across datasets, e.g., numbers of pseudo cells.
In particular, we benchmarked two major pseudo-bulk methods, PCA-based33 and Seurat, and found the one for each application leading to a better BOMA alignment. The PCA based method calculated the principal components (PCs) of single cells, and then hierarchically clustered (R function ‘hclust’) single-cells with the top 20 PCs to generate pseudo-cells, i.e., cell clusters. We used the function FindClusters() in Seurat for clustering single-cells as Seurat-based method. We used the PCA-based method for the analyses in Figures 3 and 4, which were consistent with the paper generating the data.33 However, we benchmarked the latter method and found it works better than the PCA-based method, so we used the Seurat-based method for the analysis in Figure 5.
To determine how the alignment is affected by the number of pseudo-cells, using the dataset of Figure 5, we tested different numbers of pseudo-cells by adjusting the ‘resolution’ parameter in the Seurat FindClusters() function. In this way, we generated pseudo-cells that varied from ∼1,000 to ∼10,000 (Figure S5B, top panel). To evaluate the alignment accuracy, within the aligned common manifold space, we calculated the pairwise distances of pseudo-cells of the same cell-type. Specifically, the coordinates of pseudo-cells were standardized per pseudo-cell, then distances between pseudo-cells of the same cell-type were averaged. Interestingly, the experiment result shows BOMA is scalable to the number of pseudo-cells, with the pairwise distances not significantly affected (Figure S5B, bottom panel). Considering this characteristic, and in order to balance the number of pseudo-cells across datasets, we set a lower resolution for datasets with more cells and set a higher resolution for datasets with fewer cells for the later analysis (Table S1). In particular, for organoid data, we set the resolution values as 10 for Dataset 8 and 1 for Dataset 9; for brain data, we set the resolution values as 10 for Dataset 3, 5 for Dataset 4 and 1 for Dataset 5.
Gene set enrichment analysis
We used Metascape72 to perform the gene set enrichment analysis. The enriched categories include KEGG pathways, Gene Ontology (GO) terms, protein-protein interactions, and diseases (via DisGeNET). The false discovery rates (FDRs, q-values) were used to quantify the enrichment significance.
Clustering BOMA-aligned samples and differentially expressed genes of clusters
We applied the Spectral clustering from Python package ‘sklearn’73 to cluster aligned samples on the common space, based on their alignment scores. The number of clusters can be adjusted by tuning clustering parameters or by further sub-clustering on existing coarse clusters (Figure S2B). We also identified differentially expressed genes (DEGs) of clusters. To this end, we used Presto38,39 to perform the Wilcoxon rank-sum test and auROC analysis by comparing cells from each cluster with all others cells in the dataset.
Harmonization of cell types across datasets
Cell type names may vary across studies. For instance, cell types from Organoid Dataset 7 are broad and different from many refined types in human brain. To solve this, we reassigned the types of the cells in Dataset 7 using the human brain cell-types in Dataset 3 by the ‘TransferData’ function in R package Seurat.16 We also merged some sub-cell-types to their broader types, e.g., EN-PFC1 to EN-PFC. Besides, even different studies for brains or organoids can have different sub-cell-types. To make cell types across these studies comparable, we grouped annotated cell-types from each study into common major cell-types (Table S4) for downstream comparative analyses. Also, for Dataset 8 without cell-type information, we annotated cell types using known cell-type marker genes44 with Seurat.
Hypergeometric enrichment of cell-types and developmental time stages
For the cell clusters from BOMA applications to single cell data, we calculated their cell-type enrichments (or developmental timepoint enrichments), revealing possible cellular and developmental functions of the clusters. In particular, a hypergeometric test was performed for such enrichment analysis, with the p values being calculated as:
where is the total number of cells, is the total number of cells of a certain cell-type (or cells from a certain developmental timepoint), is the number of cells in the cluster and is the number of cluster cells of certain cell-type (time point) in the cluster. Finally, we corrected the p values using BH method and selected p < 0.05 as a significant threshold for enrichments.
Trajectory analysis for BOMA alignment
Since BOMA applies the manifolds to align single cells between brains and organoids, the manifold shapes from aligned cells are expected to reveal potential developmental trajectories. To further identify such trajectories, we used SCORPIUS74 to infer the developmental trajectory for each cell-type on the common space. Primarily, for each cell-type, we input the 3D coordinates of its cells on the common space from BOMA to the infer_trajectory() function of SCORPIUS (maximum iteration of 100) to output the trajectory. To determine a root on the trajectory, we first cut the trajectory into 10 continuous segments. Each cell was assigned to the closest segment based on the distance. Then a developmental time for each segment can be determined by averaging the prior times of all cells in that segment. We then assign the segment with minimum averaged time as the root. Besides, for each cell type, we also used FindMarkers() in Seurat16 to identify differentially expressed genes in the type’s cells of each segment, i.e., “Segment cell-type DEGs” implying development-stage-specific gene expression patterns at the cell-type level. To allow gene expression values to be comparable across datasets, the IntegrateData() function of Seurat was used to integrate datasets by identify a set of anchor genes.
Experimental validation of genes in specific cell types and developmental stages
Fixed organoids were cryosectioned (17um) and stained with antibodies against proteins and markers of interest as described.66 Organoid sections were washed with PBST (PBS containing 0.1% Triton X-100) and blocked in blocking buffer (10% normal goat serum (Sigma-Aldrich) and 0.3% Triton X-100 in PBS) for 1 h at room temperature. Primary antibodies - anti-BRN2 (mouse, 1:500, Santa Cruz, SC-393324), anti-PSMB5 (rabbit, 1:1000, Novus Bio, NBP-13820), or anti-SATB2 (mouse, 1:100, Gen Way, 20-372-60065), anti-SOX2 (Mouse, 1:500, Abgent, Am2048a), anti-TBR1 (Rabbit, 1:1000, Abcam, Ab31940), or anti-CTIP2 (Rat, 1:500, Abcam, ab18465) were diluted in blocking buffer and incubated with the organoid sections overnight at 4°C. Sections were then washed 4 × 5 min with PBST. Alexa Fluor secondary antibodies (Thermo Fisher Scientific) were diluted in blocking buffer and incubated with organoid sections for 35 min at room temperature. Organoid sections were washed 4 × 5 min with PBST and counterstained with DAPI. They were then washed 2 × 5 min with PBST. Sections were scanned and visualized using either a Nikon A1 confocal microscope (Nikon) or an AxioImager Z2 ApTome microscope (Zeiss). The numbers of marker positive cells were quantified by unbiased stereology using StereoInvestigator software (MicroBrightField, Inc) as described75 PCW 19 human neocortex was fixed in 10% neutral buffered formalin at 4°C for 72 h, cryoprotected with incubation in successive solutions of 10%, 20%, and 30% sucrose, and stored in 30% sucrose +0.1% sodium azide. For validation experiments, PCW 19 human neocortex was embedded in Optimal Cutting Temperature (OCT) compound, cryosectioned at 30um thickness, and mounted on TOMO adhesion slides (Matsunami Glass USA #TOM-11/90). Sections were washed in PBS (2 × 15 min) and incubated in blocking solution containing 5% (v/v) normal donkey serum (Jackson ImmunoResearch Laboratories) and 0.3% (v/v) Triton X-100 in PBS for 30 min at room temperature. Primary antibodies - anti-BRN2 (mouse, 1:500, Santa Cruz, SC-393324), anti-PSMB5 (rabbit, 1:1000, Novus Bio, NBP-13820) or anti-SATB2 (mouse,1:100, Gen Way, 20-372-60065) were diluted in blocking solution and incubated with tissue sections for 24 h at 4°C. Sections were washed with PBST (1X PBS +0.3% Triton X-100) prior to being incubated with the appropriate fluorophore-conjugated secondary antibodies (Jackson ImmunoResearch Labs) for 30 min at room temperature. All secondary antibodies were raised in donkey and diluted at 1:250 in blocking solution. Sections were washed with PBST (3 × 5 min), treated with Autofluorescence Eliminator Reagent (Millipore #2160) according to manufacturer instructions, and coverslipped with Vectashield Plus Antifade Mounting Medium (Vector Laboratories #H-1000). Human neocortical samples were imaged on a Nikon A1 confocal microscope. z stack images taken at 20× magnification with a step size of 2um were imaged from n = 3 sections. CellProfiler software was utilized to quantify positive cells. Difference significance between organoid and human cortical plate marker positive cell percentages was test by unpaired t-test with Welch’s correction.
Quantification and statistical analysis
Hypergeometric test was used to identify human brain developmental genes and determine associated developmental stages of cell clusters. P-values was adjusted by Benjamini-Hochberg method to keep the significance level <0.05. Wilcoxon rank-sum test and auROC analysis were used to identify DEGs. Unpaired Student’s t test with Welch’s correction was used to report the P-value when comparing differences between organoid and cortical plate. Two-side t-test was used for all the paired comparisons.
Acknowledgments
The authors would like to thank the Biomedical Computing Group in the Department of Biostatistics and Medical Informatics at the University of Wisconsin at Madison for providing computing resources. We also thank the Intellectual and Developmental Disabilities Research Center in Waisman Center for valuable comments. This work was supported by National Institutes of Health grants R01AG067025, RF1MH128695, R21NS127432, U01MH116492, R21NS128761, and R03NS123969 to D.W.; R01MH116582, R01NS105200, DOD GRANT13453162, and Jenni and Kyle Professorship to X.Z., R01MH116582-S1 to S.O.S., R01HD106197 to D.W. and A.M.M.S.; P50HD105353 to Waisman Center; NARSAD Young Investigator Grant #28721 from the Brain & Behavior Research Foundation to A.M.M.S.; National Science Foundation Career Award 2144475 to D.W.; and the start-up funding to D.W. from the Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison.
Author contributions
D.W. designed and directed the study. C.H. performed research and analyzed data. A.M.M.S. and X.Z. designed the experiment validation. N.C.K. developed the BOMA web app and provided customized scripts. S.O.S. and R.R. performed experimental validation and interpreted experimental results. C.L.S. contributed to experimental validation and results interpretation. C.Y. benchmarked alignment tools. M.S. and X.H. contributed to data analysis and web app development. Q.C., A.M.M.S., and X.Z. helped design the study. C.H., A.M.M.S., C.L.S., S.O.S., R.R., S.K., X.Z., and D.W. wrote and edited the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Published: February 15, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2023.100409.
Supplemental information
Data and code availability
-
•
This paper analyzes existing and publicly available data. All the datasets used and generated in our study were deposited in Zenodo (https://doi.org/10.5281/zenodo.7236202) and they are publicly available as of the date of publication. Datasets include supplementary Data S1. Marker genes of two chimpanzee organoids specific clusters, related to Figure 3, Data S2. Differentially expressed genes within excitatory neurons of human brains or organoids, related to Figure 5, Data S3. 1,533 human brain development-related genes, related to STAR Methods from this work.
-
•
All original codes have been deposited in Zenodo (https://doi.org/10.5281/zenodo.7556083) and are also publicly available at GitHub (https://github.com/daifengwanglab/BOMA). A web app of BOMA is available at http://daifengwanglab.org/boma-webapp/.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.PsychENCODE Consortium. Akbarian S., Liu C., Knowles J.A., Vaccarino F.M., Farnham P.J., Crawford G.E., Jaffe A.E., Pinto D., Dracheva S., et al. The PsychENCODE project. Nat. Neurosci. 2015;18:1707–1712. doi: 10.1038/nn.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhaduri A., Sandoval-Espinosa C., Otero-Garcia M., Oh I., Yin R., Eze U.C., Nowakowski T.J., Kriegstein A.R. An atlas of cortical arealization identifies dynamic molecular signatures. Nature. 2021;598:200–204. doi: 10.1038/s41586-021-03910-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tasic B., Yao Z., Graybuck L.T., Smith K.A., Nguyen T.N., Bertagnolli D., Goldy J., Garren E., Economo M.N., Viswanathan S., et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature. 2018;563:72–78. doi: 10.1038/s41586-018-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li M., Santpere G., Imamura Kawasawa Y., Evgrafov O.V., Gulden F.O., Pochareddy S., Sunkin S.M., Li Z., Shin Y., Zhu Y., et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science. 2018;362:eaat7615. doi: 10.1126/science.aat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jourdon A., Scuderi S., Capauto D., Abyzov A., Vaccarino F.M. PsychENCODE and beyond: transcriptomics and epigenomics of brain development and organoids. Neuropsychopharmacology. 2021;46:70–85. doi: 10.1038/s41386-020-0763-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marton R.M., Pașca S.P. Organoid and assembloid technologies for investigating cellular crosstalk in human brain development and disease. Trends Cell Biol. 2020;30:133–143. doi: 10.1016/j.tcb.2019.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Zhu Y., Sousa A.M.M., Gao T., Skarica M., Li M., Santpere G., Esteller-Cucala P., Juan D., Ferrández-Peral L., Gulden F.O., et al. Spatiotemporal transcriptomic divergence across human and macaque brain development. Science. 2018;362 doi: 10.1126/science.aat8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keil J.M., Qalieh A., Kwan K.Y. Brain transcriptome databases: a user’s guide. J. Neurosci. 2018;38:2399–2412. doi: 10.1523/JNEUROSCI.1930-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pollen A.A., Bhaduri A., Andrews M.G., Nowakowski T.J., Meyerson O.S., Mostajo-Radji M.A., Di Lullo E., Alvarado B., Bedolli M., Dougherty M.L., et al. Establishing cerebral organoids as models of human-specific brain evolution. Cell. 2019;176:743–756.e17. doi: 10.1016/j.cell.2019.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pașca S.P. The rise of three-dimensional human brain cultures. Nature. 2018;553:437–445. doi: 10.1038/nature25032. [DOI] [PubMed] [Google Scholar]
- 11.Gordon A., Yoon S.J., Tran S.S., Makinson C.D., Park J.Y., Andersen J., Valencia A.M., Horvath S., Xiao X., Huguenard J.R., et al. Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci. 2021;24:331–342. doi: 10.1038/s41593-021-00802-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amiri A., Coppola G., Scuderi S., Wu F., Roychowdhury T., Liu F., Pochareddy S., Shin Y., Safi A., Song L., et al. Transcriptome and epigenome landscape of human cortical development modeled in organoids. Science. 2018;362:eaat6720. doi: 10.1126/science.aat6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park J.C., Jang S.Y., Lee D., Lee J., Kang U., Chang H., Kim H.J., Han S.H., Seo J., Choi M., et al. A logical network-based drug-screening platform for Alzheimer’s disease representing pathological features of human brain organoids. Nat. Commun. 2021;12:280. doi: 10.1038/s41467-020-20440-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopez-Tobon A., Caporale N., Trattaro S., Testa G. In: Stem Cell Epigenetics Translational Epigenetics. Meshorer E., Testa G., editors. Academic Press; 2020. Three-dimensional models of human brain development; pp. 257–278. [DOI] [Google Scholar]
- 15.Velasco S., Kedaigle A.J., Simmons S.K., Nash A., Rocha M., Quadrato G., Paulsen B., Nguyen L., Adiconis X., Regev A., et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature. 2019;570:523–527. doi: 10.1038/s41586-019-1289-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salick M.R., Lubeck E., Riesselman A., Kaykas A. The future of cerebral organoids in drug discovery. Semin. Cell Dev. Biol. 2021;111:67–73. doi: 10.1016/j.semcdb.2020.05.024. [DOI] [PubMed] [Google Scholar]
- 18.Bhaduri A., Andrews M.G., Mancia Leon W., Jung D., Shin D., Allen D., Jung D., Schmunk G., Haeussler M., Salma J., et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature. 2020;578:142–148. doi: 10.1038/s41586-020-1962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hou C., Nie F., Wang H., Yi D., Zhang C. Learning high-dimensional correspondence via manifold learning and local approximation. Neural Comput. Appl. 2014;24:1555–1568. doi: 10.1007/s00521-013-1369-z. [DOI] [Google Scholar]
- 20.Ham J., Lee D., Saul L. Learning high dimensional correspondences from low dimensional manifolds. Work. Contin. from Labeled to Unlabeled Data Mach. Learn. Data Min. 2003:34–41. [Google Scholar]
- 21.Welch J.D., Hartemink A.J., Prins J.F. MATCHER: manifold alignment reveals correspondence between single cell transcriptome and epigenome dynamics. Genome Biol. 2017;18:138. doi: 10.1186/s13059-017-1269-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh R., Demetci P., Bonora G., Ramani V., Lee C., Fang H., Duan Z., Deng X., Shendure J., Disteche C., Noble W.S. Unsupervised manifold alignment for single-cell multi-omics data. Proc. 11th ACM Int. Conf. Bioinformatics, Comput. Biol. Heal. Informatics, BCB. 2020;2020:1–10. doi: 10.1145/3388440.3412410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C., Mahadevan S. A general framework for manifold alignment. AAAI Fall Symp. - Tech. Rep. FS- 2009;09–04:79–86. [Google Scholar]
- 24.Nguyen N.D., Blaby I.K., Wang D. ManiNetCluster: a novel manifold learning approach to reveal the functional links between gene networks. BMC Genom. 2019;20:1003. doi: 10.1186/s12864-019-6329-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C., Mahadevan S. IJCAI International Joint Conference on Artificial Intelligence. 2009. Manifold alignment without correspondence; pp. 1273–1278. [Google Scholar]
- 26.Liu J., Huang Y., Singh R., Vert J.P., Noble W.S. Jointly embedding multiple single-cell omics measurements. Algorithms Bioinform. 2019;143 doi: 10.4230/LIPIcs.WABI.2019.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao K., Bai X., Hong Y., Wan L. Unsupervised topological alignment for single-cell multi-omics integration. Bioinformatics. 2020;36:I48–I56. doi: 10.1093/BIOINFORMATICS/BTAA443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Demetci P., Santorella R., Sandstede B., Stafford Noble W., Singh R. Gromov-Wasserstein optimal transport to align single-cell multi-omics data. bioRxiv. 2020 doi: 10.1101/2020.04.28.066787. Preprint at. [DOI] [Google Scholar]
- 29.Cao K., Hong Y., Wan L. Manifold alignment for heterogeneous single-cell multi-omics data integration using Pamona. Bioinformatics. 2021;38:211–219. doi: 10.1093/bioinformatics/btab594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tran T.N., Bader G.D. Tempora: cell trajectory inference using time-series single-cell RNA sequencing data. PLoS Comput. Biol. 2020;16 doi: 10.1371/JOURNAL.PCBI.1008205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hruby A., Hu F.B., Frank B., Hu M.D., PhD M. The epidemiology of obesity: a big picture. Pharmacoeconomics. 2015;33:673–689. doi: 10.1007/s40273-014-0243-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang H.J., Kawasawa Y.I., Cheng F., Zhu Y., Xu X., Li M., Sousa A.M.M., Pletikos M., Meyer K.A., Sedmak G., et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanton S., Boyle M.J., He Z., Santel M., Weigert A., Sanchís-Calleja F., Guijarro P., Sidow L., Fleck J.S., Han D., et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature. 2019;574:418–422. doi: 10.1038/s41586-019-1654-9. [DOI] [PubMed] [Google Scholar]
- 34.Bakken T.E., Miller J.A., Ding S.L., Sunkin S.M., Smith K.A., Ng L., Szafer A., Dalley R.A., Royall J.J., Lemon T., et al. A comprehensive transcriptional map of primate brain development. Nature. 2016;535:367–375. doi: 10.1038/nature18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leigh S.R. Brain growth, life history, and cognition in primate and human evolution. Am. J. Primatol. 2004;62:139–164. doi: 10.1002/ajp.20012. [DOI] [PubMed] [Google Scholar]
- 36.Marchetto M.C., Hrvoj-Mihic B., Kerman B.E., Yu D.X., Vadodaria K.C., Linker S.B., Narvaiza I., Santos R., Denli A.M., Mendes A.P., et al. Species-specific maturation profiles of human, chimpanzee and bonobo neural cells. Elife. 2019;8 doi: 10.7554/eLife.37527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Otani T., Marchetto M.C., Gage F.H., Simons B.D., Livesey F.J. 2D and 3D stem cell models of primate cortical development identify species-specific differences in progenitor behavior contributing to brain size. Cell Stem Cell. 2016;18:467–480. doi: 10.1016/j.stem.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korsunsky I., Nathan A., Millard N., Raychaudhuri S. Presto scales Wilcoxon and auROC analyses to millions of observations. bioRxiv. 2019 doi: 10.1101/653253. Preprint at. [DOI] [Google Scholar]
- 39.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mérot Y., Rétaux S., Heng J.I.T. Molecular mechanisms of projection neuron production and maturation in the developing cerebral cortex. Semin. Cell Dev. Biol. 2009;20:726–734. doi: 10.1016/j.semcdb.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 41.Nowakowski T.J., Bhaduri A., Pollen A.A., Alvarado B., Mostajo-Radji M.A., Di Lullo E., Haeussler M., Sandoval-Espinosa C., Liu S.J., Velmeshev D., et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science. 2017;358:1318–1323. doi: 10.1126/science.aap8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 43.Hackett C.H., Fortier L.A. Embryonic stem cells and iPS cells: sources and characteristics. Vet. Clin. N. Am. Equine Pract. 2011;27:233–242. doi: 10.1016/j.cveq.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Birey F., Andersen J., Makinson C.D., Islam S., Wei W., Huber N., Fan H.C., Metzler K.R.C., Panagiotakos G., Thom N., et al. Assembly of functionally integrated human forebrain spheroids. Nature. 2017;545:54–59. doi: 10.1038/nature22330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trevino A.E., Müller F., Andersen J., Sundaram L., Kathiria A., Shcherbina A., Farh K., Chang H.Y., Pașca A.M., Kundaje A., et al. Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell. 2021;184:5053–5069.e23. doi: 10.1016/j.cell.2021.07.039. [DOI] [PubMed] [Google Scholar]
- 46.Welch J.D., Kozareva V., Ferreira A., Vanderburg C., Martin C., Macosko E.Z. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell. 2019;177:1873–1887.e17. doi: 10.1016/j.cell.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crow M., Paul A., Ballouz S., Huang Z.J., Gillis J. Characterizing the replicability of cell types defined by single cell RNA-sequencing data using MetaNeighbor. Nat. Commun. 2018;9:884. doi: 10.1038/s41467-018-03282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Renner M., Lancaster M.A., Bian S., Choi H., Ku T., Peer A., Chung K., Knoblich J.A. Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 2017;36:1316–1329. doi: 10.15252/embj.201694700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kadoshima T., Sakaguchi H., Nakano T., Soen M., Ando S., Eiraku M., Sasai Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. USA. 2013;110:20284–20289. doi: 10.1073/pnas.1315710110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alcamo E.A., Chirivella L., Dautzenberg M., Dobreva G., Fariñas I., Grosschedl R., McConnell S.K. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron. 2008;57:364–377. doi: 10.1016/j.neuron.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 51.Britanova O., de Juan Romero C., Cheung A., Kwan K.Y., Schwark M., Gyorgy A., Vogel T., Akopov S., Mitkovski M., Agoston D., et al. Satb2 is a postmitotic determinant for upper-layer neuron specification in the neocortex. Neuron. 2008;57:378–392. doi: 10.1016/j.neuron.2007.12.028. [DOI] [PubMed] [Google Scholar]
- 52.Paşca A.M., Sloan S.A., Clarke L.E., Tian Y., Makinson C.D., Huber N., Kim C.H., Park J.Y., O’Rourke N.A., Nguyen K.D., et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods. 2015;12:671–678. doi: 10.1038/nmeth.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kendall D.G. A survey of the statistical theory of shape. Stat. Sci. 1989;4 doi: 10.1214/ss/1177012582. [DOI] [Google Scholar]
- 54.Qian X., Su Y., Adam C.D., Deutschmann A.U., Pather S.R., Goldberg E.M., Su K., Li S., Lu L., Jacob F., et al. Sliced human cortical organoids for modeling distinct cortical layer formation. Cell Stem Cell. 2020;26:766–781.e9. doi: 10.1016/j.stem.2020.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giandomenico S.L., Mierau S.B., Gibbons G.M., Wenger L.M.D., Masullo L., Sit T., Sutcliffe M., Boulanger J., Tripodi M., Derivery E., et al. Cerebral organoids at the air–liquid interface generate diverse nerve tracts with functional output. Nat. Neurosci. 2019;22:669–679. doi: 10.1038/s41593-019-0350-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Watanabe M., Buth J.E., Vishlaghi N., de la Torre-Ubieta L., Taxidis J., Khakh B.S., Coppola G., Pearson C.A., Yamauchi K., Gong D., et al. Self-Organized cerebral organoids with human-specific features predict effective drugs to combat zika virus infection. Cell Rep. 2017;21:517–532. doi: 10.1016/j.celrep.2017.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Driehuis E., Kretzschmar K., Clevers H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat. Protoc. 2020;15:3380–3409. doi: 10.1038/s41596-020-0379-4. [DOI] [PubMed] [Google Scholar]
- 58.Calandrini C., van Hooff S.R., Paassen I., Ayyildiz D., Derakhshan S., Dolman M.E.M., Langenberg K.P.S., van de Ven M., de Heus C., Liv N., et al. Organoid-based drug screening reveals neddylation as therapeutic target for malignant rhabdoid tumors. Cell Rep. 2021;36 doi: 10.1016/j.celrep.2021.109568. [DOI] [PubMed] [Google Scholar]
- 59.Bose S., Clevers H., Shen X. Promises and challenges of organoid-guided precision medicine. Med. 2021;2:1011–1026. doi: 10.1016/j.medj.2021.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cruz-Acuña R., Quirós M., Farkas A.E., Dedhia P.H., Huang S., Siuda D., García-Hernández V., Miller A.J., Spence J.R., Nusrat A., García A.J. Synthetic hydrogels for human intestinal organoid generation and colonic wound repair. Nat. Cell Biol. 2017;19:1326–1335. doi: 10.1038/ncb3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kitano K., Schwartz D.M., Zhou H., Gilpin S.E., Wojtkiewicz G.R., Ren X., Sommer C.A., Capilla A.V., Mathisen D.J., Goldstein A.M., et al. Bioengineering of functional human induced pluripotent stem cell-derived intestinal grafts. Nat. Commun. 2017;8:765. doi: 10.1038/s41467-017-00779-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huch M., Dorrell C., Boj S.F., Van Es J.H., Li V.S.W., Van De Wetering M., Sato T., Hamer K., Sasaki N., Finegold M.J., et al. In vitro expansion of single Lgr5 + liver stem cells induced by Wnt-driven regeneration. Nature. 2013;494:247–250. doi: 10.1038/nature11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Polioudakis D., de la Torre-Ubieta L., Langerman J., Elkins A.G., Shi X., Stein J.L., Vuong C.K., Nichterwitz S., Gevorgian M., Opland C.K., et al. A single-cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron. 2019;103:785–801.e8. doi: 10.1016/j.neuron.2019.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yin Y., Petersen A.J., Soref C., Richards W.D., Ludwig T., Taapken S., Berndt E., Zhang S.C., Bhattacharyya A. Generation of seven induced pluripotent stem cell lines from neonates of different ethnic backgrounds. Stem Cell Res. 2019;34:101365. doi: 10.1016/j.scr.2018.101365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li M., Shin J., Risgaard R.D., Parries M.J., Wang J., Chasman D., Liu S., Roy S., Bhattacharyya A., Zhao X. Identification of FMR1-regulated molecular networks in human neurodevelopment. Genome Res. 2020;30:361–374. doi: 10.1101/gr.251405.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sloan S.A., Andersen J., Pașca A.M., Birey F., Pașca S.P. Generation and assembly of human brain region–specific three-dimensional cultures. Nat. Protoc. 2018;13:2062–2085. doi: 10.1038/s41596-018-0032-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giorgino T. Computing and visualizing dynamic time warping alignments in R: the dtw package. J. Stat. Software. 2009;31:1–24. doi: 10.18637/jss.v031.i07. [DOI] [Google Scholar]
- 68.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Squair J.W., Gautier M., Kathe C., Anderson M.A., James N.D., Hutson T.H., Hudelle R., Qaiser T., Matson K.J.E., Barraud Q., et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 2021;12:5692. doi: 10.1038/s41467-021-25960-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lun A.T.L., Marioni J.C. Overcoming confounding plate effects in differential expression analyses of single-cell RNA-seq data. Biostatistics. 2017;18:451–464. doi: 10.1093/biostatistics/kxw055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barupal D.K., Fiehn O. Generating the blood exposome database using a comprehensive text mining and database fusion approach. Environ. Health Perspect. 2019;127:97008–102830. doi: 10.1289/EHP4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schlitzer A., Sivakamasundari V., Chen J., Sumatoh H.R.B., Schreuder J., Lum J., Malleret B., Zhang S., Larbi A., Zolezzi F., et al. Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat. Immunol. 2015;16:718–728. doi: 10.1038/ni.3200. [DOI] [PubMed] [Google Scholar]
- 75.Li Y., Stockton M.E., Bhuiyan I., Eisinger B.E., Gao Y., Miller J.L., Bhattacharyya A., Zhao X. MDM2 inhibition rescues neurogenic and cognitive deficits in a mouse model of fragile X syndrome. Sci. Transl. Med. 2016;8:336ra61. doi: 10.1126/scitranslmed.aad9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing and publicly available data. All the datasets used and generated in our study were deposited in Zenodo (https://doi.org/10.5281/zenodo.7236202) and they are publicly available as of the date of publication. Datasets include supplementary Data S1. Marker genes of two chimpanzee organoids specific clusters, related to Figure 3, Data S2. Differentially expressed genes within excitatory neurons of human brains or organoids, related to Figure 5, Data S3. 1,533 human brain development-related genes, related to STAR Methods from this work.
-
•
All original codes have been deposited in Zenodo (https://doi.org/10.5281/zenodo.7556083) and are also publicly available at GitHub (https://github.com/daifengwanglab/BOMA). A web app of BOMA is available at http://daifengwanglab.org/boma-webapp/.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.