

Abstract
Disturbed inhibitory synaptic transmission has functional impacts on neurodevelopmental and psychiatric disorders. An essential mechanism for modulating inhibitory synaptic transmission is alteration of the postsynaptic abundance of GABAARs, which are stabilized by postsynaptic scaffold proteins and recruited by presynaptic signals. However, how GABAergic neurons trigger signals to transsynaptically recruit GABAARs remains elusive. Here, we show that UNC-43/CaMKII functions at GABAergic neurons to recruit GABAARs and modulate inhibitory synaptic transmission at C. elegans neuromuscular junctions. We demonstrate that UNC-43 promotes presynaptic MADD-4B/Punctin secretion and NRX-1α/Neurexin surface delivery. Together, MADD-4B and NRX-1α recruit postsynaptic NLG-1/Neuroligin and stabilize GABAARs. Further, the excitation of GABAergic neurons potentiates the recruitment of NLG-1-stabilized-GABAARs, which depends on UNC-43, MADD-4B, and NRX-1. These data all support that UNC-43 triggers MADD-4B and NRX-1α, which act as anterograde signals to recruit postsynaptic GABAARs. Thus, our findings elucidate a mechanism for pre- and postsynaptic communication and inhibitory synaptic transmission and plasticity.
Subject terms: Molecular neuroscience, Synaptic transmission
The pre-and postsynaptic communication is critical for faithful synaptic transmission and induction of synaptic plasticity. Here, the authors found that CaMKII functions at GABAergic neurons to recruit GABAARs by triggering anterograde signals.
Introduction
Fast synaptic inhibition is mediated by neurotransmitter GABA and GABA-activated chloride channels (GABAARs). Disturbed inhibitory synaptic transmission has functional impacts on the pathology of neurodevelopmental and psychiatric disorders (including autism spectrum disorders and depression). An essential mechanism for modulating inhibitory synaptic transmission and plasticity is altering the postsynaptic abundance of GABAARs1. Long-term potentiation and inhibition of GABAergic transmission have been associated with increased or decreased GABAAR synaptic abundance, respectively2,3.
Like other postsynaptic receptors, GABAARs at the cell surface undergo lateral diffusion and require postsynaptic scaffolds to stabilize their synaptic enrichment2–5. In mammals, GABAAR clustering and anchoring are mediated by complex inhibitory postsynaptic scaffolds major consist of gephrin, collybistin, Neuroligin-2, and LHFPL4/GARLH45–14. Previous research has shown that the GABAARs are stabilized by distinct scaffolds at C. elegans neuromuscular junctions (NMJs), including the synaptic adhesion molecule NLG-1/Neuroligin, and the FERM domain-containing protein FRM-315–18. The postsynaptic GABAARs and inhibitory postsynaptic currents are eliminated in double mutants lacking both NLG-1 and FRM-317.
Besides the postsynaptic scaffolds, the presynaptic neurons also play essential roles in positioning and clustering postsynaptic receptors. During synaptogenesis, the innervation by presynaptic neurons releases anterograde signals to induce receptor aggregation and clustering in the post-junctional membranes. For example, at mammalian cholinergic synapses, cholinergic motor neurons release extracellular proteoglycan agrin to recruit acetylcholine receptors to postsynaptic membranes19–21. Other reports have shown that GABAergic motor neurons secrete the ADAMTS-like extracellular protein MADD-4B/Punctin, which promotes the localization of GABAARs at inhibitory synapses at NMJs in C. elegans15,16. In addition, during the induction of activity-dependent synaptic plasticity, the excitation of the presynaptic neurons could trigger the recruitment of postsynaptic receptors and require anterograde signals released by presynaptic neurons. The transsynaptic recruitment of receptors has been extensively studied at excitatory synapses22–27. However, how the GABAergic neurons trigger anterograde signals to recruit GABAARs remains unclear.
Calcium/calmodulin-dependent protein kinase II (CaMKII) is a serine/threonine-specific protein kinase activated by the Ca2+/Calmodulin and functions as a ubiquitous mediator of cellular Ca2+ signals28. CaMKII is well known as an essential component of postsynaptic density (PSD) proteins at excitatory synapses29, and necessary for NMDA receptor-dependent long-term potentiation (LTP)30–36. At GABAergic synapses, postsynaptic CaMKII has been reported to phosphorylate GABAARs within the TM3-4 domain to regulate receptor insertion at the cell surface37–41. CaMKII is required for the induction of rebound potentiation in Purkinje neurons42–44 and is also required for the moderate N-methyl-Daspartate receptor (NMDAR)-activating stimuli-induced long-term potentiation of inhibition (iLTP) in hippocampal neurons2,45. Besides that, it has been known for a long time that CaMKII is expressed and functions on the presynaptic side35,46–51. However, whether the presynaptic CaMKII could transsynaptically recruit postsynaptic receptors and be involved in inhibitory synaptic transmission and plasticity remain elusive.
Here, we utilize the C. elegans NMJs as a model to study how presynaptic neurons trigger anterograde signals to recruit GABAARs at inhibitory synapses. We found that UNC-43, the C. elegans ortholog of CaMKII, is required for GABAARs recruitment and modulates inhibitory synaptic transmission. Experiments using multiple reporter fusion constructs showed that UNC-43 functions at GABAergic motor neurons to recruit GABAARs in the same pathway with NLG-1, but not FRM-3. Next, we demonstrated that UNC-43 promotes presynaptic MADD-4B secretion and cell adhesion molecule NRX-1α/Neurexin GABAergic motor neuron surface delivery. Together, MADD-4B and NRX-1α recruit postsynaptic NLG-1 and stabilize GABAARs. Further, we confirmed that the activity-dependent plasticity in the inhibitory synapses requires the presynaptic UNC-43, MADD-4B, and NRX-1α, and is mediated by the NLG-1-stabilized GABAARs. Collectively, our work elucidates how presynaptic neurons transsynaptically recruit postsynaptic receptors during synaptogenesis and activity-dependent plasticity.
Results
Presynaptic UNC-43/CaMKII regulates postsynaptic GABAARs abundance at GABAergic synapses
To study the function of CaMKII on inhibitory synaptic transmission, we took advantage of the neuromuscular junction (NMJ) as a synaptic model. In C. elegans, the majority of GABAergic neurons in the nervous system are inhibitory motor neurons that innervate body-wall muscles52–55. At the nerve cord, muscle arms elongate and form synapses with GABAergic axon terminals. As a result, the C. elegans body-wall muscles receive direct synaptic inputs from both cholinergic and GABAergic motor neurons56. To visualize endogenous GABAARs, we used a similar strategy as the previous report by inserting a tagRFP coding sequence after the signal peptide sequence of the unc-49 gene that encodes GABAAR in C. elegans using the CRISPR-cas9 genome editing system18. The tagRFP can label all three GABAAR subunits: UNC-49A, UNC-49B, and UNC-49C (Fig. 1a). To test whether the tagRFP fused GABAARs fold and function well, we performed aldicarb assays to study whether the inhibitory synaptic transmission at NMJs is altered in this tagRFP-tagged GABAARs imaging strain; very briefly, the cholinesterase inhibitor aldicarb prevents acetylcholine breakdown, and the attendant acetylcholine accumulation at synapses leads to muscle over-excitation and worm paralysis. Fundamentally, this assay can monitor the alteration of excitatory and inhibitory synaptic transmission57. During the 120 minutes of exposure to aldicarb, we found that the paralysis rate of tagRFP knock-in worms did not differ from the wild type (Supplementary Fig. 1). In contrast, the unc-49 null mutants showed accelerated paralysis compared to the wild-type worms, thus implying impairment of inhibitory synaptic transmission at NMJs (Supplementary Fig. 1) and supporting that our tagRFP-tagged GABAARs are functional.
Fig. 1. Presynaptic UNC-43/CaMKII regulates postsynaptic GABAAR abundance at GABAergic synapses.
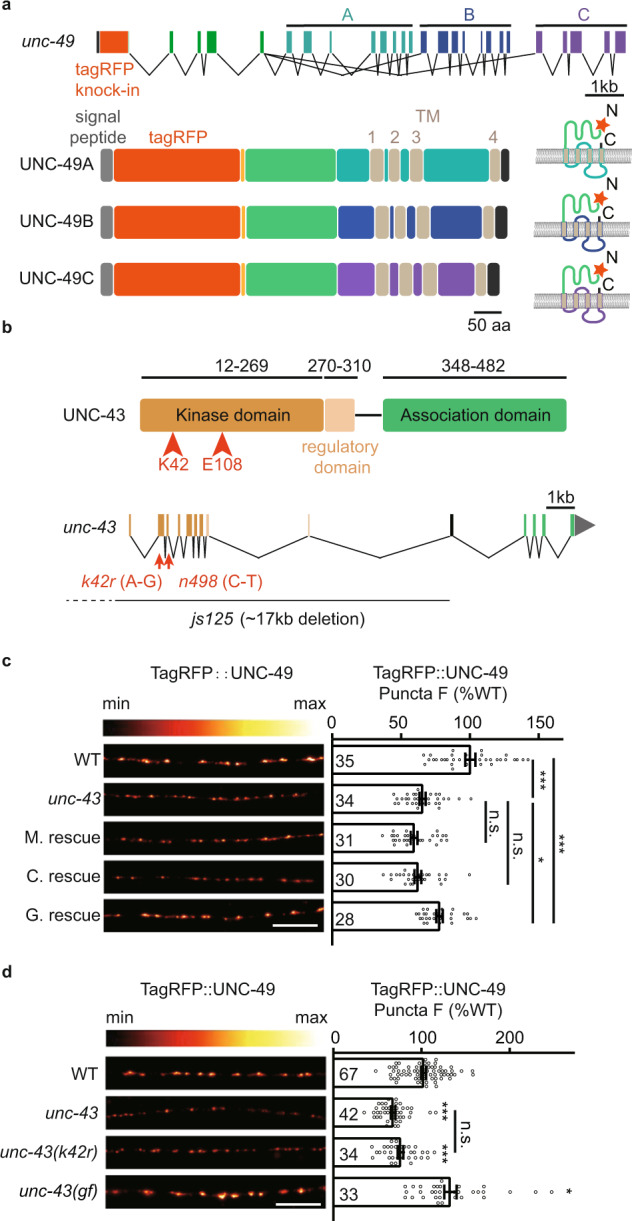
a Schematic representation of the xj1024 locus. A tagRFP coding sequence was inserted before the first exon shared by UNC-49A, UNC-49B, and UNC-49C. TagRFP consequently labels the UNC-49A, UNC-49B, and UNC-49C proteins. b The protein structure of UNC-43 is shown on top. The js125 mutation has a 17 kb deletion (upstream of the promoter to the 10th exon) that deletes most of the coding sequences. The kinase-dead mutant unc-43 (k42r) has a point mutation and causes the K42R coding variant. The gain-of-function unc-43 (gf) (n498, gain of function) mutant has a point mutation that causes single amino acid substitution E108K. c The puncta fluorescence intensity marked by TagRFP-UNC-49 (under unc-49 own promoter) in dorsal nerve cord axons was decreased in the unc-43 mutants. This defect was rescued by transgenic expression of UNC-43 in GABAergic motor neurons (G. rescue), but not by UNC-43 expression in cholinergic neurons (C. rescue) or in body-wall muscles (M. rescue). Representative images (left, scale bar 10 μm, Pseudo-color: Red Hot) and mean puncta intensities +/− SEM (right, Ns represent the number of animals tested) are shown. One-way ANOVA with post-hoc Bonferroni’s multiple comparison test. *p < 0.05, ***p < 0.001, n.s. not significant. d GABAAR fluorescence intensity is decreased in the unc-43 (k42r) kinase-dead mutants, and increased in the unc-43(gf) (n498, gain of function) mutants. Representative images (left, scale bar 10 μm, Pseudo-color: Red Hot) and mean puncta intensities + /- SEM (right, Ns represent the number of animals tested) are shown. Kruskal–Wallis test with post-hoc Dunn’s test. *p < 0.05, ***p < 0.001, n.s. not significant. For c, d, source data are provided as a Source Data file.
To determine whether UNC-43/CaMKII participates in regulating GABAAR abundance, we used the unc-43(js125) null allele50,58, which deletes 17 kb of sequence positioned downstream of the promoter and the 10th exon and lacks most of the sequence encoding UNC-43 (Fig. 1b). Note that in C. elegans, UNC-43 encodes the sole CaMKII. Quantitative imaging analysis indicated that the GABAAR puncta fluorescence intensity (puncta peak fluorescence to cord fluorescence ratio, peak-to-cord) in the unc-43 mutants was decreased by around 35% compared with wild type (Fig. 1c), suggesting that UNC-43 does affect GABAAR synaptic abundance. We also examined whether UNC-43 exerts a similar function for cholinergic receptors by measuring the synaptic abundance of two acetylcholine receptors L-AchR (UNC-29) and N-AchR (ACR-16) in the unc-43 mutant animals. However, neither of them presented decreased fluorescent intensities compared to the wild type (Supplementary Fig. 2). Together, these findings support that UNC-43 is specifically involved in maintaining GABAAR abundance at inhibitory synapses.
UNC-43 is expressed in both pre- and postsynapses at excitatory and inhibitory synapses35,46–51. At C. elegans NMJs, the excitatory motor neurons innervate and contract muscles, and also synapses onto the inhibitory motor neurons to relax the contralateral muscles to generate the sinusoidal movement53. To examine where UNC-43 functions to stabilize the postsynaptic GABAARs, we restored UNC-43 expression in GABAergic motor neurons (under the unc-25 promoter), in cholinergic motor neurons (under the unc-17 promoter), or in body-wall muscles (under the myo-3 promoter) (all in the unc-43 mutant background). We found that the decreased synaptic abundance of GABAARs in the unc-43 mutant was partially rescued by either UNC-43D or UNC-43G isoform59–61 in GABAergic motor neurons; no rescue was observed with the cholinergic neuron or body-wall muscle groups (Fig. 1c and Supplementary Fig. 3). All of the data supports that UNC-43 functions in the GABAergic motor neurons to stabilize postsynaptic GABAARs.
Since we observed a partial rescue of GABAARs recruitment defects in unc-43 mutants by expressing UNC-43 in the GABAergic motor neurons, therefore, we restored UNC-43 (UNC-43D isoform) expression in pan-neurons (under rab-3 or unc-43 promoter), in both GABAergic and cholinergic motor neurons (under unc-25 promoter+unc-17 promoter), and in both GABAergic motor neurons and muscle cells (under unc-25 promoter+myo-3 promoter) in the unc-43 mutants, and we found that the GABAARs puncta fluorescence defects in the unc-43 mutants can be fully rescued by expressing UNC-43 in pan-neurons or in both GABAergic and cholinergic motor neurons, but not with both GABAergic motor neurons and muscle cells groups (Supplementary Fig. 3). These data suggest that the cholinergic UNC-43 is also involved in GABAARs recruitment with unknown mechanisms, and it depends on the GABAergic UNC-43.
As a calcium/calmodulin-dependent protein kinase, we next studied whether its kinase activity is required for UNC-43-mediated recruitment of GABAARs. We generated a kinase-dead mutant unc-43(k42r) using CRISPR-cas9 and analyzed GABAAR abundance. The GABAAR fluorescence intensity was reduced by about 25% in unc-43(k42r) (Fig. 1d), indicating that UNC-43’s kinase activity is involved in the recruitment of GABAARs. Further, we also analyzed the GABAAR synaptic abundance in the unc-43(n498) gain-of-function allele that bears a constitutively activating mutation E108K in the active site core62. The GABAAR fluorescence intensity was significantly increased in the unc-43 mutant (Fig. 1d), supporting the conclusion that activation of UNC-43 promotes GABAARs recruitment.
UNC-43/CaMKII is required for the GABAergic synaptic localization of GABAARs
To further investigate whether the remaining GABAARs in the unc-43 mutant are correctly localized at GABAergic synapses, we labeled the GABAergic synapses by fusing the endogenous UNC-57/Endophilin in GABAergic motor neurons with GFP in the tagRFP-tagged GABAARs strain using a split GFP complementation system63,64 (Fig. 2a), and calculated the colocalization coefficient between UNC-57 and GABAARs (Fig. 2b). We found that the colocalization coefficient in the unc-43 mutant was significantly decreased compared with the wild type (0.7770 ± 0.0143 for wild type vs. 0.4848 ± 0.0316 for unc-43) (Fig. 2c, d). Further, GABAAR mislocalization could be rescued by restoring UNC-43 expression in the GABAergic motor neurons (Fig. 2c, d), indicating that presynaptic UNC-43 is required for the GABAergic synaptic localization of GABAARs.
Fig. 2. Presynaptic UNC-43/CaMKII is required for the GABAergic synaptic localization of GABAARs.

a, b Schematic illustration of labeling UNC-57 at the GABAergic motor neurons by split GFP complementary system. Seven copies of the split GFP11 were inserted into the C-terminal of unc-57 genomic loci by CRISPR-Cas9 system. The split GFP1-10 was expressed in GABAergic motor neurons by unc-25 promoters. c, d The colocalization coefficient between GABAARs and GABAergic synaptic boutons was decreased in the unc-43 mutants; this was rescued by transgenic expression of UNC-43 in GABAergic motor neurons (G. rescue). Pearson’s correlation coefficients between the intensities of GABAergic bouton marker UNC-57 (green, labeled by UNC-57-split GFP under unc-25 promoter) and postsynaptic GABAARs (magenta) were used to assess the localization of GABAARs at inhibitory synapses. Representative images (c-top, scale bar, 10 μm), corresponding line scan curve (c-bottom), and mean Pearson’s correlation coefficients +/− SEM (d, Ns represent the number of animals tested) are shown. Kruskal–Wallis test with post-hoc Dunn’s test. ***p < 0.001. Source data are provided as a Source Data file.
Presynaptic UNC-43 modulates inhibitory synaptic transmission at NMJs
To determine whether the observed GABAARs synaptic recruitment defect in the unc-43 mutants caused impairment of GABAergic synaptic transmission, we patch-clamped body-wall muscles and recorded spontaneous miniature inhibitory postsynaptic currents (mIPSCs). Consistent with the previous report50, we observed a dramatic decrease in mIPSC frequency in the unc-43 mutants (Fig. 3a, b). Besides the mIPSC frequency, the mISPC amplitude was also significantly decreased in the unc-43 mutants (Fig. 3a, c), implying a defect in postsynaptic GABAARs. Further, the decreased mIPSC frequency and amplitude could also be partially recovered by expressing UNC-43 in GABAergic neurons but not by expressing UNC-43 in muscles or cholinergic neurons (Fig. 3a–c).
Fig. 3. Presynaptic UNC-43/CaMKII modulates inhibitory synaptic transmission.

a–c Endogenous inhibitory synaptic transmission was assessed by recording mIPSCs from body-wall muscles. The defects of mIPSC in the unc-43 mutants were rescued by transgenic expression of UNC-43 in GABAergic motor neurons (G. rescue), but not by UNC-43 expression in cholinergic neurons (C. rescue) or in body-wall muscles (M. rescue). Representative mIPSC traces (a), mIPSC rates (b, Data are presented as mean values +/− SEM, Ns represent the number of animals tested.), and the cumulative fraction of mIPSC amplitude (c) are shown. Kruskal–Wallis test with post-hoc Dunn’s multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.001, n.s. not significant. d Schematic for construction of the unc-43 conditional deletion lines. Two LoxP sites were inserted into the genomic loci (at the promoter and the first intron of UNC-43 K11E8.1a to K11E8.1l) by CRISPR/Cas9, the minigene encoding Cre recombinase under unc-25 and myo-3 promoters were used to knock out unc-43 gene in GABAergic motor neurons (G.cKO) and muscle cells (M.cKO), respectively. e–g Endogenous inhibitory synaptic transmission was assessed by recording mIPSCs from body-wall muscles of unc-43 conditional knockout animals. Representative mIPSC traces (e), mIPSC rates (f, Data are presented as mean values +/− SEM, Ns represent the number of animals tested.), and the cumulative fraction of mIPSC amplitude (g) are shown. Kruskal–Wallis test with post-hoc Dunn’s multiple comparisons. *p < 0.05, ***p < 0.001, n.s. not significant. h, i The GABA-evoked currents in the unc-43 mutants were comparable to that in wild-type animals (WT). Representative responses (h) and current amplitude (i, Data are presented as mean values +/− SEM, Ns represent the number of animals tested.) are shown. Two-tailed and unpaired Student’s t-test. n.s. not significant. For b, c, f, g, i, source data are provided as a Source Data file.
To further confirm UNC-43 functions at GABAergic motor neurons to modulate inhibitory synaptic transmission, we generated worms with a conditional unc-43 deletion in GABAergic motor neurons, or in muscle cells. Briefly, we inserted two LoxP sites at the unc-43 promoter and the first intron, respectively, and drove CRE recombinase expression in the GABAergic motor neurons (under unc-25 promoter) and body-wall muscles (under myo-3 promoter) (Fig. 3d). We found that unc-43 deletion at the GABAergic motor neurons mimics the phenotype of the unc-43 knockout mutants, showing a significant decrease in mIPSC frequency and amplitude (Fig. 3e–g). Note that there were no obvious mIPSC frequency or amplitude defects in the strains with unc-43 deletion in the body-wall muscles (Fig. 3e–g). These results confirm that UNC-43 functions at GABAergic motor neurons to support inhibitory synaptic transmission.
To investigate whether the expression and/or surface delivery of GABAARs are compromised in the unc-43 mutants, we recorded the 0.2 mM and 0.5 mM GABA-evoked currents, which were unaltered in unc-43 mutants (Fig. 3h–i and Supplementary Fig. 4). Thus, the mIPSC amplitude defect in the unc-43 mutants is unlikely to be caused by decreased bulk expression and surface delivery of GABAARs. A previous study reported a minor decrease in response to pressure-applied GABA (0.2 mM) in the unc-43 mutant50. This discrepancy likely arises from the different protocols of recording. These results verified that UNC-43 is required for both GABAAR synaptic recruitment and inhibitory synaptic transmission.
To rule out the possibility that the decreased postsynaptic GABAARs in the unc-43 mutant is the secondary result of synaptic structural defects, we measured the puncta fluorescence and intensity of endogenous UNC-57/Endophilin in GABAergic motor neurons in the unc-43 mutant. We observed no significant decrease in puncta fluorescence intensities and densities between wild-type and unc-43 mutants (Supplementary Fig. 5a–c). Further, we also labeled the endogenous UNC-2/CaV2 in the GABAergic motor neurons with GFP by split GFP complementation system65. Both the fluorescence intensity and density of UNC-2-GFP fusion protein were not altered in the unc-43 mutants compared to the wild type (Supplementary Fig. 5d–f). These results suggested that the GABAergic synapse structure was unaltered.
We also ruled out the possibility that the decreased postsynaptic GABAARs in the unc-43 mutants result from diminished synaptic GABA transmission or neuropeptide release, as both a previous report and our data have demonstrated that the synaptic abundance of GABAAR was not decreased in the unc-25 mutants (which lack GABA biogenesis), snb-1 mutants, unc-13 mutants (which lack synaptic vesicle release), unc-31 mutants (which lack dense-core vesicle release), and egl-3 (which lack neuropeptide maturation) (Supplementary Fig. 6a–c)66. Further, deletion of unc-43 causes a reduction of GABAAR puncta fluorescence in both snb-1 and unc-25 mutants (Supplementary Fig. 6a–b), indicating UNC-43 recruits GABAARs independent of GABAergic neurotransmission. The increase of GABAARs puncta fluorescence in the unc-25 mutants may be caused by synaptic homeostasis. Further, GABAARs recruitment is not modulated by the BK channel SLO-150 (Supplementary Fig. 6b).
UNC-43 recruits postsynaptic NLG-1 to stabilize GABAARs
Our previous work reported that the postsynaptic GABAARs are recruited by two distinct scaffolds FRM-3 and NLG-1/neuroligin15–18. To investigate whether UNC-43 functions in a single pathway with FRM-3 or with NLG-1 to stabilize GABAARs, we generated unc-43; frm-3 and unc-43; nlg-1 double mutants and then analyzed GABAAR fluorescence intensity at synapses. We observed a further reduction of GABAAR puncta fluorescence intensity in the unc-43; frm-3 double mutants compared to the frm-3 and unc-43 single mutants (Fig. 4a). However, there was no difference in GABAAR fluorescence intensity between the unc-43; nlg-1 double mutants and the nlg-1 single mutant worms (Fig. 4a). Besides, Super-resolution microscopy studies by Sora mode showed that the GABAARs clustering was significantly decreased in both the unc-43 mutants and the nlg-1 mutants, but not in the frm-3 mutants (Supplementary Fig. 7). Further, the increase of GABAAR fluorescence intensity by unc-43(n498) gain-of-function mutation was eliminated in the nlg-1 mutant, but not in the frm-3 mutant (Fig. 4b), lending more support to the conclusion that UNC-43 functions in a single pathway with NLG-1 to recruit GABAARs.
Fig. 4. UNC-43/CaMKII acts through a single pathway with NLG-1/neuroligin to recruit GABAARs.
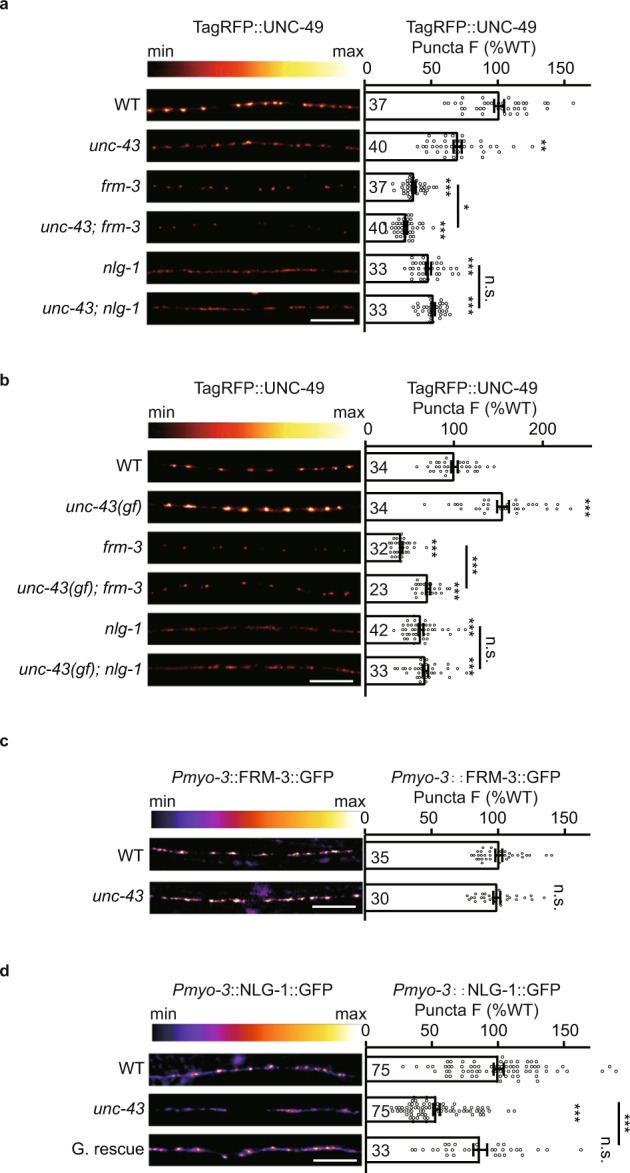
a The tagRFP-UNC-49 puncta fluorescence intensities in the unc-43; nlg-1 double mutants were comparable to the nlg-1 single mutants. Representative images (left, scale bar 10 μm, Pseudo-color: Red Hot) and mean puncta intensities +/− SEM (right, Ns represent the number of animals tested) are shown. Kruskal–Wallis test with two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli correction for multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.001, n.s. not significant. b The tagRFP-UNC-49 puncta fluorescence intensities in the unc-43 (gf); nlg-1 double mutants were comparable to the nlg-1 single mutants. Representative images (left, scale bar 10 μm, Pseudo-color: Red Hot) and mean puncta intensities +/− SEM (right, Ns represent the number of animals tested) are shown. One-way ANOVA with two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli correction for multiple comparisons. ***p < 0.001, n.s. not significant. c UNC-43 does not regulate postsynaptic FRM-3 localization. FRM-3-GFP fluorescence intensities in the body-wall muscles in wild-type and unc-43 mutants were shown. Representative images (left, scale bar 10 μm, Pseudo-color: Fire) and mean puncta intensities +/− SEM (right, Ns represent the number of animals tested) are shown. Two-tailed and unpaired Student’s t-test. n.s. not significant. d UNC-43 functions in the GABAergic neurons to stabilize postsynaptic NLG-1 localization. NLG-1-GFP fluorescence intensities in the body-wall muscles in wild-type and unc-43 mutants were shown. Representative images (left, scale bar 10 μm, Pseudo-color: Fire) and mean puncta intensities +/− SEM (right, Ns represent the number of animals tested) are shown. Kruskal–Wallis test with post-hoc Dunn’s test. ***p < 0.001, n.s. not significant. For a–d, source data are provided as a Source Data file.
Next, we tested whether UNC-43 may somehow regulate the postsynaptic localization of NLG-1. We expressed the C-terminal GFP-tagged NLG-1 (under the myo-3 promoter) in the muscle cells of wild-type and unc-43 mutant worms. The GABAARs recruitment defects of nlg-1 mutants were rescued by the transgene expression of the C-terminal GFP-tagged NLG-1 in the muscle cells (Supplementary Fig. 8). We detected a dramatic decrease in signal intensity for the NLG-1 puncta in the unc-43 mutants (Fig. 4d). Further, we found that this NLG-1 synaptic abundance defect was rescued by restoring UNC-43 expression in GABAergic motor neurons (Fig. 4d), indicating presynaptic UNC-43 is required for NLG-1 postsynaptic abundance. It is worth noting that the postsynaptic localization of FRM-3 was unaltered in the unc-43 mutants (Fig. 4c, Supplementary Fig. 8). These results suggested the possibility that UNC-43/CaMKII stabilizes synaptic GABAARs by recruiting postsynaptic NLG-1.
UNC-43 recruits postsynaptic NLG-1 and stabilizes GABAARs requiring both NRX-1α and MADD-4B
Previous studies have shown that the ADAMTS-like secretion protein MADD-4B/Punctin can be secreted by GABAergic motor neurons, and showed that MADD-4B/Punctin acts partially redundantly with presynaptic NRX-1α/Neurexin to recruit postsynaptic NLG-1 at GABAergic synapses15,16. Consistent with previous reports, we found that madd-4b and nrx-1 mutations cause an additive defect of NLG-1 synaptic localization (Fig. 5a); specifically, we detected a further decrease in NLG-1 puncta fluorescence intensity in the madd-4b; nrx-1 double mutants compared to either of the single mutants (Fig. 5a). In this context, we test whether UNC-43 may function to recruit NLG-1 in a single pathway with MADD-4B and/or NRX-1α. We analyzed the NLG-1 GABAergic synaptic abundance in the madd-4b; nrx-1; unc-43 triple mutants: the unc-43 deletion did not cause any further reduction in NLG-1 puncta fluorescence intensity as compared to the madd-4b; nrx-1 double mutants (Fig. 5a). Together, these results support that a single genetic pathway that includes UNC-43, MADD-4B, and NRX-1α is responsible for the recruitment of postsynaptic NLG-1.
Fig. 5. UNC-43/CaMKII’s recruitment of GABAARs requires both MADD-4B and NRX-1α.
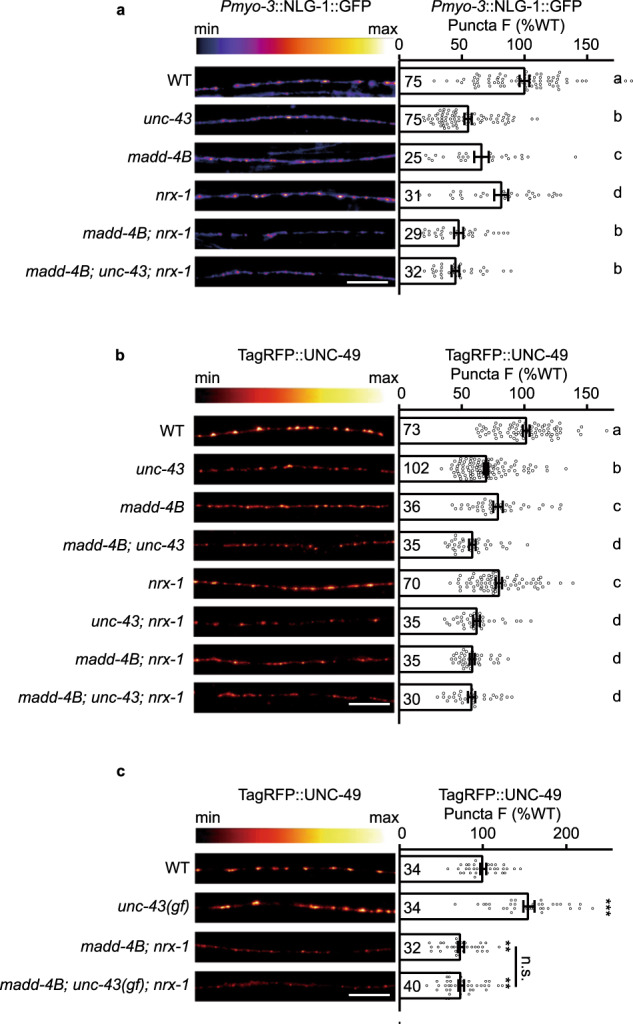
a UNC-43 functions in the same pathway with MADD-4B and NRX-1α to stabilize postsynaptic NLG-1 localization. NLG-1-GFP fluorescence intensities in the body-wall muscles in wild type and mutants were shown. Representative images (left, scale bar 10 μm, Pseudo-color: Fire) and mean puncta intensities +/− SEM (right, Ns represent the number of animals tested) are shown. The data for WT and unc-43 are the same as in Fig. 4c. Kruskal–Wallis test with two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli correction for multiple comparisons. The scatter plotted data labeled with different letters are significantly different (p < 0.05). b, c Both MADD-4B and NRX-1α are required for UNC-43’s recruitment of GABAARs. TagRFP-UNC-49 fluorescence in dorsal nerve cords in wild type and mutants is shown. Representative images (left, scale bar 10 μm, Pseudo-color: Red Hot) and mean puncta intensities +/− SEM (right, Ns represent the number of animals tested) are shown. In b, Kruskal–Wallis test with two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli correction for multiple comparisons. The scatter plotted data labeled with different letters are significantly different (p < 0.05). In c, Kruskal–Wallis test with post-hoc Dunn’s test. **p < 0.01, ***p < 0.001, n.s. not significant. For a–c, source data are provided as a Source Data file.
We then investigated whether UNC-43’s recruitment of GABAARs requires MADD-4B and NRX-1α. Consistent with previous reports15, we found that MADD-4B and NRX-1α function redundantly to recruit GABAARs: there was a further decrease in the fluorescence intensity of GABAARs fusion protein puncta in the madd-4b; nrx-1 double mutants as compared to both the madd-4b and nrx-1 single mutants (Fig. 5b). Here we observed a less severe defect of GABAARs recruitment in the madd-4b; and nrx-1 mutants than in previous reports15,16, it may be caused by different strategies to label GABAARs. Note that the deletion of unc-43 further decreases the GABAAR puncta fluorescence intensity in the madd-4b and nrx-1 single mutants (Fig. 5b), but not in the madd-4b; nrx-1 double mutants (Fig. 5b). Further, unc-43(n498) gain-of-function mutation is not able to increase the GABAAR puncta fluorescence intensity in the madd-4b; nrx-1 double mutants (Fig. 5c), indicating that UNC-43 recruits postsynaptic GABAARs requiring both MADD-4B and NRX-1α.
UNC-43 promotes MADD-4B/Punctin secretion and NRX-1α/Neurexin GABAergic motor neuron surface delivery
Since UNC-43 requires both MADD-4B and NRX-1α to recruit postsynaptic NLG-1, it is possible that UNC-43 regulates MADD-4 and NRX-1α localization. To study MADD-4B secretion, we expressed a C-terminal GFP-tagged MADD-4B fusion in GABAergic neurons (under the unc-25 promoter), and the MADD-4B-GFP fusion protein is able to rescue the GABAARs recruitment defects in the madd-4b mutants (Supplementary Fig. 8). A MADD-4B-GFP fluorescent signal was evident in both the nerve cords and coelomocytes. Coelomocytes are scavenger cells that endocytose proteins secreted into the body cavity67. Accordingly, the GFP signal in the coelomocytes indicates the secreted MADD-4B, while the fluorescent signal from the dorsal cord indicates the MADD-4B that is retained at the motor neuron axon terminals. Compared to the wild type, there was a significant decrease of MADD-4B-GFP fluorescence intensity in coelomocytes of both the unc-43 knockout and unc-43(k42r) kinase-dead mutants (Fig. 6a, b), and the MADD-4B signal in both unc-43 mutants dorsal nerve cord was significantly increased (Fig. 6c). As a result, the decrease of MADD-4B-GFP fluorescence intensity in the coelomocytes and the increase of MADD-4B-GFP fluorescence intensity in the dorsal nerve cord as shown in the unc-43 mutants indicate MADD-4B secretion by GABAergic motor neurons is partially blocked in the unc-43 mutants, thus supporting that UNC-43 promotes MADD-4B secretion and it requires UNC-43’s kinase activity.
Fig. 6. UNC-43/CaMKII promotes MADD-4B/Punctin secretion.

a Schematic representation of C-terminal GFP-tagged MADD-4B fusion under unc-25 promoter (top) and MADD-4B-GFP expressed in D-type motor neurons, meanwhile the secreted MADD-4B-GFP in body cavity was endocytosed by scavenger cell coelomocytes (bottom). b MADD-4B secretion by GABAergic motor neurons was decreased in the unc-43 knockout and unc-43 (k42r) mutants. Secretion of MADD-4B was measured by analyzing GFP fluorescence intensities in the endolysosomal compartment coelomocytes. Representative images (top, scale bar 5 μm) and mean fluorescence intensities +/− SEM (bottom, Ns represent the number of animals tested) are shown. one-way ANOVA with post-hoc Bonferroni’s multiple comparison test. **p < 0.01, n.s. not significant. c Puncta fluorescence of MADD-4B-GFP in the dorsal nerve cords was measured in the unc-43 mutants. Representative images (top, scale bar 10 μm, Pseudo-color: Fire) and mean puncta intensities +/− SEM (bottom, Ns represent the number of animals tested) are shown. One-way ANOVA with post-hoc Bonferroni’s multiple comparison test. *p < 0.05, **p < 0.01, n.s. not significant. For b, c, source data are provided as a Source Data file.
Here we ruled out the possibility that the decreased MADD-4B-GFP fluorescence intensity in coelomocytes in unc-43 mutants is caused by abnormal coelomocyte functions or general defect in presynaptic protein secretion, as the endocytosis of a constitutive GFP secreted from muscle cells or from the GABAergic motor neurons by coelomocytes was not affected by unc-43 mutation (Supplementary Fig. 9).
To study whether UNC-43 regulates the surface delivery of NRX-1α in GABAergic motor neurons, we expressed a dual-tagged NRX-1α fusion protein by inserting pHluorin at the N-terminus and wrmScarlet at the C-terminus of NRX-1α under the unc-25 promoter (Fig. 7a). pHluorin is a pH-sensitive version of GFP, and receptors with extracellular domain tagged pHluorin are fluorescent upon insertion in the plasma membrane to expose pHluorin in the extracellular environment (PH > 6). Acidification of the extracellular environment to pH 5.5 quenched all the fluorescence in both wild-type and unc-43 mutants (Supplementary Fig. 10), suggesting that all of the fluorescent signals are from the pHluorin-NRX-1 α in the plasma membrane. We observed a dramatic decrease of the pHluorin fluorescent signal in both unc-43 knockout and unc-43 (k42r) mutants (Fig. 7b, c, e), while the wrmScarlet fluorescence intensities were unaltered (Fig. 7b, d), indicating that the GABAergic motor neuron surface delivery of NRX-1α, but not its expression or axonal trafficking, was promoted by UNC-43.
Fig. 7. UNC-43/CaMKII promotes NRX-1α/neurexin GABAergic motor neuron surface localization.

a Schematic representation of N-terminal pH-sensitive GFP pHluorin-tagged and C-terminal wrmScarlet-tagged NRX-1α fusion under unc-25 promoter. pHluorin-NRX-1α are fluorescent upon insertion in the plasma membrane to expose pHluorin in the extracellular environment (pH>6). b–e The surface localization of NRX-1α is decreased in the unc-43 mutants. pHluorin and wrmScarlet dual-labeled NRX-1α was expressed in GABAergic neurons. pHluorin-NRX-1α puncta (green) fluorescence in the dorsal nerve cords was decreased in the unc-43 mutants (c). NRX-1α-wrmScarlet puncta (magenta) fluorescence in the dorsal nerve cords was unaltered in the unc-43 mutants (d). The averaged fluorescence intensity of pHluorin normalized to wrmScarlet fluorescence intensity in the unc-43 mutants (e). Representative images (top, scale bar 10 μm) and mean puncta intensities +/− SEM (bottom, Ns represent the number of animals tested) are shown. One-way ANOVA with post-hoc Bonferroni’s multiple comparison test for c, d, Kruskal–Wallis test with post-hoc Dunn’s test for e. ***p < 0.001, n.s. not significant. For c–e, source data are provided as a Source Data file.
UNC-43/CaMKII-triggered anterograde signals are required for activity-dependent plasticity at GABAergic synapses
Previous research shows that UNC-43 is required for activity-dependent plasticity at PLM-AVA synapses by regulating synaptic AMPARs trafficking after presynaptic neuron excitation or silencing30,33,68,69. Thus, we tested whether UNC-43 is also required for activity-dependent plasticity at GABAergic synapses of NMJs. To study the activity-dependent plasticity of GABAergic synapses, we optogenetically activated the GABAergic motor neurons by expressing a channelrhodopsin variant (ChIEF) in the GABAergic motor neurons (under unc-25 promoter) and delivering pulsed blue-light excitation (Fig. 8a). After 30 min of excitation, we analyzed the GABAAR synaptic abundance and found a significant increase of GABAAR puncta fluorescence intensity compared with controls without all-trans retinal (ATR), and it lasted within 2 h (Fig. 8b, c). Besides, the colocalization coefficient between the presynaptic marker UNC-57 and GABAARs was slightly but significantly increased (Fig. 8d, e). To study whether the increased GABAARs recruitment upon GABAergic motor neuron excitation potentiates inhibitory synaptic transmission, we patch-clamped body-wall muscles and recorded mIPSCs. Both the mIPSCs frequency and amplitude were significantly increased in those animals with GABAergic motor neuron excited compared to controls without all-trans retinal (ATR) (Fig. 8f–h), supporting that the inhibitory synaptic transmission was also increased. This result indicates the existence of activity-dependent synaptic plasticity at GABAergic synapses.
Fig. 8. Induction of activity-dependent plasticity at GABAergic synapses.

a Schematic illustration of the blue-light stimulation pattern. b, c The activity-dependent plasticity at GABAergic synapses lasts within two hours. Representative images (b, scale bar 10 μm, Pseudo-color: Red Hot) and the normalized tagRFP fluorescence intensity before and after GABAergic motor neuron excitation (c, Data are presented as mean values +/− SEM) are shown. n = 11(ATR(−); −30 min); n = 11(ATR(+); −30 min); n = 28 (ATR(−); 30 min); n = 25 (ATR(+); 30 min); n = 22 (ATR(−); 60 min); n = 24 (ATR(+); 60 min); n = 23 (ATR(−); 120 min); n = 23 (ATR(+); 120 min) animals. Two-tailed and unpaired Student’s t-test. ***p < 0.001, n.s. not significant. d, e The colocalization coefficient between GABAARs and GABAergic synaptic boutons was increased after excitation of GABAergic motor neurons; Pearson’s correlation coefficients between the intensities of GABAergic bouton marker UNC-57 (green, labeled by UNC-57-split GFP under unc-25 promoter) and postsynaptic GABAARs (magenta) were used to assess the localization of GABAARs at inhibitory synapses. Representative images (d-top, scale bar, 10 μm), corresponding line scan curve (d-bottom), and mean Pearson’s correlation coefficients +/− SEM (e, Ns represent the number of animals tested) are shown. Two-tailed and unpaired Student’s t-test. ***p < 0.001. f–h Endogenous inhibitory synaptic transmission was assessed by recording mIPSCs from body-wall muscles in transgenic animals expressing a channelrhodopsin variant (ChIEF) in the GABAergic motor neurons (under unc-25 promoter) after blue-light stimulation with or without all-trans retinal (ATR). Representative mIPSC traces (f), mIPSC rates (g, Data are presented as mean values +/− SEM, Ns represent the number of animals tested.), and the cumulative fraction of mIPSC amplitude (h) are shown. Two-tailed and unpaired Student’s t-test for g and Kolmogorov–Smirnov test for h. **p < 0.01, ***p < 0.001. For c, e, g, h, source data are provided as a Source Data file.
Next, we studied whether GABAergic activity-dependent plasticity requires UNC-43. We detected no increase of GABAAR puncta fluorescence at synapses afterward in the unc-43 mutant animals, and restoring UNC-43 expression in GABAergic motor neurons rescued the activity-dependent increase of GABAAR recruitment (Fig. 9a), indicating that the presynaptic UNC-43 is required for the activity-dependent plasticity at GABAergic synapses. Besides, GABAergic activity-dependent plasticity was not eliminated in the unc-43 gain-of-function mutants (Fig. 9a).
Fig. 9. UNC-43/CaMKII is required for activity-dependent plasticity at GABAergic synapses.
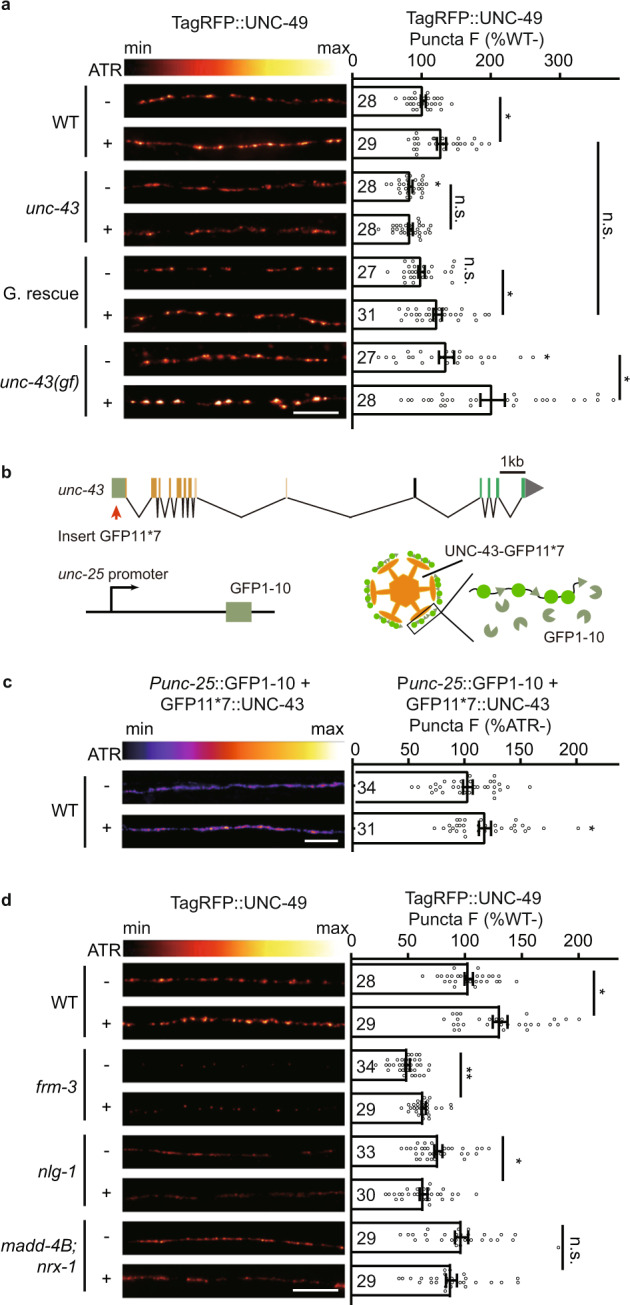
a Presynaptic UNC-43 is required for activity-dependent GABAergic plasticity. TagRFP-UNC-49 puncta fluorescence intensity was increased after GABAergic motor neuron excitation by ChIEF activation in wild type and in the unc-43 gain-of-function mutants, but not in the unc-43 loss of function mutants, and this was rescued by transgenic expression of UNC-43 in GABAergic motor neurons (G. rescue). Representative images (left, scale bar 10 μm, Pseudo-color: Red Hot) and mean puncta intensities +/− SEM (right, Ns represent the number of animals tested) are shown. Kruskal–Wallis test with two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli correction for multiple comparisons. *p < 0.05, n.s. not significant. b Schematic illustration of split GFP experimental design to label endogenous UNC-43 at GABAergic motor neurons. Seven copies of the split GFP11 were inserted into the N-terminal of unc-43 genomic loci by the CRISPR-Cas9 system. The split GFP1-10 was expressed in GABAergic motor neurons by unc-25 promoters. c The GABAergic presynaptic localization of UNC-43 was increased after GABAergic motor neuron excitation by ChIEF activation. split GFP labels endogenous UNC-43 in GABAergic neurons. Representative images (left, scale bar 10 μm, Pseudo-color: Fire) and mean puncta intensities +/− SEM (right, Ns represent the number of animals tested) are shown. Two-tailed Mann–Whitney test. *p < 0.05. d MADD-4B, NRX-1α, and NLG-1, but not FRM-3, are required for activity-dependent plasticity at GABAergic synapses. Representative images (left, scale bar 10 μm, Pseudo-color: Red Hot) and mean puncta intensities +/− SEM (right, Ns represent the number of animals tested) are shown. The data for WT is the same as in a. Kruskal–Wallis test with two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli correction for multiple comparisons. *p < 0.05, **p < 0.01, n.s. not significant.For a, c, d, source data are provided as a Source Data file.
Further, we asked whether the presynaptic localization of UNC-43 is altered upon GABAergic motor neuron excitation. To visualize endogenous UNC-43 at GABAergic motor neurons, we utilized the split GFP complementary system63,64. We used CRISPR/Cas9 system to insert a sequence coding for seven GFP11 fragments at the N-terminus of UNC-43. In parallel, the GFP1-10 fragment was constitutively expressed in the GABAergic motor neurons under the control of the unc-25 promoter (Fig. 9b) to label the endogenous localization of UNC-43 in GABAergic motor neurons. We observed a significant increase in the GFP fluorescent signal upon blue-light-stimulated GABAergic neuron excitation (Fig. 9c), supporting the conclusion that more UNC-43 are localized at the GABAergic motor neuron axon terminals upon GABAergic motor neuron stimulation.
Since UNC-43 is required for the increase of the synaptic abundance of GABAARs after excitation of GABAergic motor neurons, linking with our finding that UNC-43 acts through a single pathway with NLG-1 to recruit GABAARs, it is likely that the NLG-1-stabilized diffusing GABAARs, rather than the FRM-3-stabilized immobilized GABAARs, mediate the inhibitory activity-dependent plasticity. To test this hypothesis, we turned to the nlg-1 and frm-3 mutants. We observed an increase of GABAARs recruitment after GABAergic motor neuron excitation in the frm-3 mutants, but not in the nlg-1 mutants (Fig. 9d), which supports our hypothesis that the diffusing GABAARs stabilized by NLG-1 mediate activity-dependent plasticity at inhibitory synapses.
To study whether the UNC-43-participated activity-dependent plasticity requires MADD-4B and NRX-1α, we studied the activity-dependent plasticity in the madd-4b; nrx-1 double mutants. Similar to in the unc-43 mutants, the increase of GABAARs postsynaptic abundance post GABAergic motor neuron excitation was blocked in the madd-4b; nrx-1 double mutants (Fig. 9d). Further, the secretion of MADD-4B and the surface delivery of NRX-1α were both increased upon GABAergic neuron excitation (Supplementary Fig. 11), supporting the conclusion that MADD-4 and NRX-1α also participate in the activity-dependent plasticity at GABAergic synapses, and they are very likely to work in the single pathway with UNC-43.
Discussion
In this study, we revealed how UNC-43/CaMKII participates in transsynaptically recruiting GABAARs at NMJs. We showed that the presynaptic UNC-43/CaMKII promotes MADD-4B secretion and delivery of the cell adhesion molecule NRX-1α to the surface of GABAergic motor neurons. MADD-4B and NRX-1α act as anterograde signals to recruit postsynaptic NLG-1 and stabilize the GABAARs at inhibitory synapses. We further demonstrated that the NLG-1-stabilized GABAARs, but not the FRM-3-stabilized GABAARs, mediate activity-dependent plasticity at GABAergic synapses, and experimentally confirmed that this mediation requires UNC-43, MADD-4B, and NRX-1α. These mechanistic insights about how presynaptic neurons transsynaptically recruit postsynaptic GABAARs deepen our understanding about pre- and postsynaptic communications and inhibitory synaptic plasticity.
The presynaptic function of UNC-43/CaMKII
Most previous studies of CaMKII have focused on its facilitation of AMPAR recruitment to enhance synaptic strength at postsynapses30,33. At inhibitory synapses, CaMKII is reported to phosphorylate GABAARs and regulates their surface delivery37–41. In hippocampal neurons, the moderate N-methyl-Daspartate receptor (NMDAR)-activating stimuli cause CaMKII selectively translocating to inhibitory synapses to phosphorylate GABAAR β3S383 and recruit scaffold protein gephyrin, which promotes GABAARs accumulation and immobilization at synapses2,45,70. Besides, an acute increase in neuronal activity of cultured hippocampal neurons also promotes the phosphorylation of β3S383 by CaMKII39. Further, in cerebellar somatodendritic basket cell synapses, CaMKII is required for the rebound potentiation (RP) by phosphorylation of GABAARs receptor44.
However, many studies have indicated that CaMKII has functional impacts at presynapses35,46–51. CaMKII is enriched at presynaptic sites, and accounts for ~2% of the total synaptic vesicle protein amount71. Presynaptic injection of the membrane-impermeable CaMKII inhibitor peptide 281-309 was shown to block excitatory synaptic plasticity in cultured hippocampal neurons47 and presynaptic CaMKII was implicated in synaptogenesis and synaptic transmission at NMJs of both Drosophila and C. elegans46,50. In the present study, we identified a transsynaptic GABAAR recruitment function of presynaptic CaMKII, thus demonstrating CaMKII’s involvement in inhibitory synaptic transmission. We also show that this transsynaptic recruitment activity is supported by MADD-4B secretion and NRX-1α surface delivery and propose the hypothesis that UNC-43-triggered anterograde signals are required for the activity-dependent plasticity at inhibitory synapses. Thus, the function of CaMKII in recruiting postsynaptic GABAARs is conserved in species, and both presynaptic and postsynaptic CaMKII can recruit postsynaptic GABAARs.
Recall our finding that the extent of the synaptic GABAAR abundance defects, MADD-4B secretion defect, and NRX-1α motor neuron surface delivery were similar in the unc-43 null mutant as in worms expressing a kinase-dead mutant unc-43 (k42r) (Fig. 1d, Figs. 6 and 7). This suggests that UNC-43’s kinase activity is required to promote MADD-4B secretion and NRX-1α surface delivery to support transsynaptic recruitment of GABAARs.
NLG-1- and FRM-3-stabilized GABAARs
Previous studies have demonstrated that GABAARs are stabilized by distinct synaptic scaffolds at C. elegans NMJs: there are NLG-1-stabilized GABAARs and FRM-3-stabilized GABAARs15–18. It has been shown that the NLG-1-stabilized GABAARs and FRM-3-stabilized GABAARs together mediate inhibitory synaptic transmission15–18. However, it remains unclear which type of receptors mediate inhibitory synaptic plasticity. Here, by using the nlg-1 and frm-3 mutants, we demonstrate that NLG-1-stabilized GABAARs mediate activity-dependent plasticity at inhibitory synapses. Further, our study indicates a plausible explanation for how NLG-1-stabilized GABAARs are recruited to inhibitory postsynaptic elements upon excitation of presynaptic GABAergic motor neurons: it is likely that Ca2+ influx after GABAergic motor neuron excitation activates UNC-43, which can trigger more MADD-4B secretion and NRX-1α surface delivery. These molecules can subsequently act as anterograde signals to recruit additional NLG-1 to postsynaptic elements to somehow help stabilize the GABAARs. Given that no physical interactions have yet been reported between NLG-1 and GABAARs, the nature of this stabilization remains unclear. A recent study proposed that FRM-3 and CASK ortholog LIN-2 mediate the interaction between NLG-1 and GABAARs18. However, an additive defect in GABAARs recruitment and inhibitory synaptic transmission was observed in the frm-3; nlg-1 double mutant17. Further, UNC-43 functions in a single pathway with NLG-1 to recruit GABAARs, but not with FRM-3. In summary, the current data does not support the conclusion that NLG-1 recruits GABAARs through FRM-3.
Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and fulfilled by the Lead Contact Xia-Jing Tong (tongxj@shanghaitech.edu.cn).
Animals
All C. elegans strains were derived from the wild-type Bristol N2 (Caenorhabditis Genetics Center) strain. Worms were cultivated at 20 °C on nematode growth medium (NGM) plates seeded with Escherichia coli (E. coli) under standard conditions. The OP50 strain of E. coli was used as a food source for all experiments, except where the HB101 strain was utilized for the electrophysiology study. The well-fed young-adult hermaphrodites were used in all experiments except for coelomocyte imaging experiment in which the adult day-5 animals were used. Transgenic animals were prepared by microinjection, and integrated transgenes were isolated following UV irradiation or by single-copy insertion mediated by miniMos or CRISPR-cas9. A complete list of the strains used in this study can be found in Supplementary data 1.
Molecular information on unc-43 mutants. The js125 mutation is a deletion from 10 kb upstream of the transcription initiation site to exon 1050,58. The kinase-dead mutant unc-43(k42r) was generated by CRISPR-cas9, which caused the K42R coding variant. The gain-of-function unc-43 (n498) mutation is a point mutation that causes single amino acid substitution (E108K) and makes the kinase partially active even in the absence of Ca2+.
Molecular information on madd-4B mutants. The xj0739 mutation is a C insertion in the first exon of madd-4B, introducing an early stop codon and generating a 21 amino acids (aa) product.
Plasmids
The constructs used and created in this study are detailed in Supplementary data 2.
Primers
The primers used in this study are detailed in Supplementary data 3.
Reagents, software, and algorithms
The reagents, software, and algorithms used in this study are detailed in Supplementary data 4.
Genome editing by CRISPR-cas9
The tagRFP-T-unc-49(xj1024) knock-in was generated based on the CRISPR-cas9 technique previously described72. The tagRFP-T with 3x GS linker was inserted between Gln23 and Asp24, and an extra Gln was added before Asp24 to guarantee the UNC-49 signal peptide coding sequence integrity. The sgRNAs were designed on CRISPR (http://crispor.tefor.net/)73. The single-stranded repair template was generated by overlap extension PCR (purified and heated at 95 °C for 5 min). To remove off-target mutations, the transgenic worms were outcrossed with N2 and used for further studies.
Similarly, the unc-43(k42r) allele was also generated by CRISPR-cas9. A 99 bp single-stranded repair template was synthesized and purified before injection.
The unc-43(xj764[LoxP::unc-43p::unc-43::LoxP]) allele was generated for unc-43 conditional knockout. The first LoxP with EcoRI restriction enzyme cutting site was inserted upstream of unc-43 promoter (3 kb before exon 1 of UNC-43 transcripts K11E8.1a to K11E8.1 l), and the second LoxP with EcoRI was inserted into the first intron. Under the action of Cre recombinase driven by the tissue-specific promoter, about 3800 bp genomic DNA containing the promoter and 59 bp coding sequence of UNC-43 transcripts (K11E8.1a to K11E8.1l) including start codon was tissue-specifically removed. For the transcript K11E8.1r, the deletion of the 59 bp coding sequence causes a frameshift and an early stop codon. myo-3 and unc-25 promoters were cloned into pFX_NLS::Cre vector74. The conditional knockout animals were verified by PCR and sequencing.
The unc-57::GFP11*7(xj1544) and GFP11*7-unc-43(xj1455) allele was generated for tissue-specific labeling UNC-57 and UNC-43. unc-57::GFP11*7(xj1544), seven copies of the GFP11 coding sequence were inserted at the C-terminus of the unc-57 genomic locus. Briefly, GGGS (linker)-GFP11*7 coding sequence was inserted before the stop codon of unc-57 isoforms (T04D1.3a to T04D1.3d). For GFP11*7-unc-43(xj1455), seven copies of the GFP11 coding sequence were inserted at the N-terminus of the unc-43 genomic locus. Briefly, GGGS (linker)-GFP11*7-GGGS (linker) coding sequence was inserted after the start codon of unc-43 isoforms (K11E8.1b to K11E8.1l). In parallel, the GFP 1-10 fragment was constitutively expressed in the GABAergic motor neurons. As a result, the endogenous UNC-57 and UNC-43 localized in the GABAergic neurons can be visualized.
Generation of single-copy insertion alleles by miniMos
N2 worms were injected with 10 ng/μL of PCFJ910 plasmid-of-interest containing the promoter and open reading frame (contains a Neomycin resistance gene), 50 ng/μL of pCFJ601 (Mos1 transposase), 10 ng/μL of pGH8 (Prab-3::mCherry), 2.5 ng/μL of pCFJ90 (Pmyo-2::mCherry), and 5 ng/μL of pCFJ104 (Pmyo-3::mCherry). Animals were grown under 25 °C after injection. Neomycin (G418) was added to plates 24 h after injection at a final concentration of 1.5 μg/μL. Homozygous animals with the desired insertion were verified by PCR and sequencing.
The xjSi0009 allele encodes GFP-tagged MADD-4B driven by GABAergic motor neuron-specific unc-25 promoter. The GFP sequence was inserted after Phe711 of the MADD-4B coding sequence.
The xjSi0016 allele encodes dual-tagged NRX-1α driven by the unc-25 promoter. The pHluorin sequence was inserted between Ile29 and Ile30 of NRX-1α, and the wrmScarlet was inserted after Val1540.
Microscopy
For UNC-49, UNC-29, UNC-57, UNC-43, FRM-3, and NLG-1 puncta fluorescence imaging, images were captured using a 100x objective (NA = 1.4) on an Olympus microscope (BX53). For ACR-16, MADD-4B, and NRX-1α puncta fluorescence imaging, images were captured using a Nikon 60×1.4 NA objective on a Nikon spinning-disk confocal system (Yokogawa CSU-W1). Young-adult worms were immobilized with 30 μg/μl 2,3-Butanedione monoxime (Sigma). The maximum intensity of dorsal cord projections of Z-series stacks was obtained by Metamorph software (Molecular Devices). Line scans were analyzed in Igor Pro (WaveMetrics) using a custom script75. The mean fluorescence intensities of reference FluoSphere microspheres (0.5 μm, ThermoFisher Scientific) were measured during each experiment that controlled for changes in illumination intensities. Automatic image analysis was performed as in previous reports76. Briefly, four image parameters were defined: (1) Peak (Fpeak): Absolute peak fluorescence, is the averaged ratio of peak fluorescence to the bead standard of the cord analyzed; (2) Cord (Fcord): absolute axon fluorescence, which is the ratio of baseline fluorescence to the bead standard; (3) Puncta F: peak-to-cord magnitude, 100X (Fpeak – Fcord)/Fcord; (4) Density: is the number of peaks found per 10 μm of cord analyzed. All fluorescence values are normalized to wild-type controls to facilitate comparison. To assess the synaptic accumulation of fluorescent proteins, we used the Puncta F: peak-to-cord magnitude.
For colocalization assay, images were captured using a 100x objective (NA = 1.4) on an Olympus microscope (BX53). The maximum intensity of dorsal cord projections of Z-series stacks was obtained by ImageJ. The background was removed using the Subtract Background plugin of Image J (rolling ball radius 50 pixels). The fluorescence intensity along the cord was evaluated with the Plot Profile plugin (line width 20 pixels). For each channel, the values were normalized to the value of maximal intensity. To assess the correlations between GABAergic boutons and GABAARs, the mean of Pearson’s coefficients between the distribution of fluorescence intensity in each channel were compared in Figs. 2d and 8e.
For coelomocytes imaging, images were captured using a Nikon 60 × 1.4 NA objective on a Nikon spinning-disk confocal system (Yokogawa CSU-W1). 5-day adult worms were immobilized with 30 μg/μl 2,3-Butanedione monoxime. All fluorescence values are normalized to wild-type controls to facilitate comparison. To assess fluorescence intensities of coelomocytes, region of interests (ROIs) were traced for the coelomocytes and a background area outside of the animal in ImageJ. Maximum intensities of these ROIs were exported, and fluorescence intensity was corrected for background intensity.
Super-resolution microscopy
Super-resolution images were captured with a Nikon 60 × 1.4 NA Objective on a Nikon spinning-disk confocal system (Yokogawa CSU-W1 SoRa) based on SoRa mode. Live young adult animals were anesthetized with 30 μg/μl 2,3-Butanedione monoxime (Sigma) and the regions of dorsal cords were excited by a 561 nm laser (50% power, 400 ms exposure time). The maximum intensity of dorsal cord projections of Z-series stacks was obtained by ImageJ. The number of animals with diffusing or punctate GABAARs was assessed and analyzed by Chi-square test.
Aldicarb assay
The aldicarb assay was performed as previously described77. Briefly, 1 mM aldicarb was added to the NGM plate. More than 20 animals at the young-adult stage were picked on each plate. The paralyzed animals were counted every 10 min. At least three double-blind replicates were performed for each genotype.
Electrophysiology
Electrophysiology was conducted on dissected C. elegans as previously described17. Worms were superfused in the extracellular solution (127 mM NaCl, 5 mM KCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 20 mM glucose, 1 mM CaCl2, and 4 mM MgCl2, bubbled with 5% CO2, 95% O2) at 22 °C. Whole-cell recordings were carried out with the internal solution (105 mM CH3O3SCs, 10 mM CsCl, 15 mM CsF, 4 mM MgCl2, 5 mM EGTA, 0.25 mM CaCl2, 10 mM HEPES, and 4 mM Na2ATP). The solution was adjusted to pH 7.2 using CsOH) at 0 mV for mIPSCs.
For GABA-activated current recordings, 0.2 or 0.5 mM GABA was pressure ejected for 1.0 s onto body muscles of adult worms.
Activity-dependent plasticity assay
To prepare the standard agar plates for optogenetic activation of GABAergic motor neurons, 1.6 mM of all-trans retinal (ATR, 100 mM dissolved in ethanol) or ethanol (control) was mixed with OP50 E. coli culture and spotted on 3.5 mm NGM plates. Plates were allowed to dry for 24 h before use. L4 transgenic worms with channelrhodopsin variant CHIEF expressed in the GABAergic motor neurons were transferred to ATR plates and grown overnight. Young adult worms were received pulsed blue-light excitation (460 nm wavelength, 2.4 mW/mm2 power) at 20 Hz for 30 min, and subjected to GABAARs puncta fluorescence analysis or electrophysiology.
Statistical analysis
All data were presented as mean ± SEM (standard error of the mean) with a scatter plot. Data were analyzed with Prism 8.0 (v8.0.2, GraphPad Software, Inc.). The Kolmogorov–Smirnov test was performed to determine whether the data were normally distributed. For comparison of the two groups, a two-tailed unpaired Student’s t-test (for the normally distributed data, Figs. 3i, 4c, 8c, e, g and Supplementary Figs. 2b, d, 4, 5c, e, f,) or a two-tailed Mann–Whitney test (for the non-normal data, Fig. 9c and Supplementary Fig. 5b, 9a–b, 11a–b) was performed. For the comparison of more than two and less than six groups, one-way ANOVA with post-hoc Bonferroni’s multiple comparison test (for the normally distributed data, Figs. 1c, 6b, c, 7c, d and Supplementary Fig. 6c) or Kruskal–Wallis test with post-hoc Dunn’s test (for the non-normal data, Figs. 1d, 2d, 3b, f, 4d, 5c, 7e and Supplementary Fig. 6a) or was performed. For the comparison of more than six groups, one-way ANOVA with two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli correction for multiple comparisons (for the normally distributed data, Fig. 4b and Supplementary Figs. 3, 6b, 8) or Kruskal–Wallis test with two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli correction for multiple comparisons (for the non-normal data, Figs. 4a, 5a, b, 9a, d) was performed. For the comparison of cumulative fractions, a Kolmogorov–Smirnov test (two group, Fig. 8h) or a Kruskal–Wallis tests with post-hoc Dunn’s test (more than two group, Figs. 3c, g) were performed. For Supplementary Fig. 7b, Chi-square tests were performed. For the comparison of curves, two-way ANOVA was performed (Supplementary Fig. 1). P-value in all figures is donated as n.s. >0.05, * <0.05, ** <0.005, *** <0.001 (expect Fig. 5a, b and Supplementary Fig. 3, which marked with letters and data corresponding to scatter plots labeled with different letters are significantly different).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary files
Acknowledgements
We thank the C. elegans Genetics Stock Center, National BioResource Project (NBRP), Dr. Joshua Kaplan, Dr.Jean-Louis Bessereau, Dr. Yan Zou, and Dr. Yingchuan B. Qi for sharing strains and reagents. We also thank the Molecular Imaging Core Facility (MICF) at the School of Life Science and Technology, ShanghaiTech University for help in imaging. This work was supported by the STI2030-Major Projects (2021ZD0202500 to X.-J.T.), the National Natural Science Foundation of China (32170963 to X.-J.T.), the Science and Technology Commission of Shanghai Municipality (21ZR1481000 to X.-J.T., 19JC1414100 to X.-J.T.), and the Major International (Regional) Joint Research Project (32020103007 to S.G.).
Source data
Author contributions
Y.H., H.L., X.-T.Z, Y.W., W.-X.Z., K.-Y.Q., L.L., and M.-X.C. designed, performed, and analyzed the experiments. Y.H., X.-T.Z., W.-X.Z., K.-Y.Q., and M.-X.C. performed the aldicarb experiments, fluorescent imaging, and strain construction. H.L., Y.W., and L.L. performed electrophysiological recordings. S.G., Z.H., and X.-J.T. supervised the experimental design and data interpretation. Y.H. and X.-J.T. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Peer review
Peer review information
Nature Communications thanks Zhao-Wen Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
The full raw data that support the results of this study are available upon request. The source data underlying the quantification of Figs. 1c, d, 2d, 3b, c, f, g, 3i, 4a–d, 5a–c, 6b, c, 7c–e, 8c, e, g, h, 9a, c, d and Supplementary Figs. 1, 2b, d, 3, 4, 5b, c, e, f, 6a–c, 7b, 8, 9a, b, 11a, b are provided as a Source data file. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Yue Hao, Haowen Liu.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-023-37137-0.
References
- 1.Nusser Z, Cull-Candy S, Farrant M. Differences in synaptic GABA(A) receptor number underlie variation in GABA mini amplitude. Neuron. 1997;19:697–709. doi: 10.1016/S0896-6273(00)80382-7. [DOI] [PubMed] [Google Scholar]
- 2.Petrini EM, et al. Synaptic recruitment of gephyrin regulates surface GABAA receptor dynamics for the expression of inhibitory LTP. Nat. Commun. 2014;5:3921. doi: 10.1038/ncomms4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bannai H, et al. Activity-dependent tuning of inhibitory neurotransmission based on GABAAR diffusion dynamics. Neuron. 2009;62:670–682. doi: 10.1016/j.neuron.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 4.Dean C, Dresbach T. Neuroligins and neurexins: linking cell adhesion, synapse formation and cognitive function. Trends Neurosci. 2006;29:21–29. doi: 10.1016/j.tins.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Jacob TC, et al. Gephyrin regulates the cell surface dynamics of synaptic GABAA receptors. J. Neurosci. 2005;25:10469–10478. doi: 10.1523/JNEUROSCI.2267-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Craig AM, Banker G, Chang W, McGrath ME, Serpinskaya AS. Clustering of gephyrin at GABAergic but not glutamatergic synapses in cultured rat hippocampal neurons. J. Neurosci. 1996;16:3166–3177. doi: 10.1523/JNEUROSCI.16-10-03166.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Essrich C, Lorez M, Benson JA, Fritschy JM, Luscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat. Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- 8.Kneussel M, et al. Loss of postsynaptic GABA(A) receptor clustering in gephyrin-deficient mice. J. Neurosci. 1999;19:9289–9297. doi: 10.1523/JNEUROSCI.19-21-09289.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saiepour L, et al. Complex role of collybistin and gephyrin in GABAA receptor clustering. J. Biol. Chem. 2010;285:29623–29631. doi: 10.1074/jbc.M110.121368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davenport EC, et al. An essential role for the tetraspanin LHFPL4 in the cell-type-specific targeting and clustering of synaptic GABA(A) receptors. Cell Rep. 2017;21:70–83. doi: 10.1016/j.celrep.2017.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu M, et al. Impairment of inhibitory synapse formation and motor behavior in mice lacking the NL2 binding partner LHFPL4/GARLH4. Cell Rep. 2018;23:1691–1705. doi: 10.1016/j.celrep.2018.04.015. [DOI] [PubMed] [Google Scholar]
- 12.Yamasaki T, Hoyos-Ramirez E, Martenson JS, Morimoto-Tomita M, Tomita S. GARLH family proteins stabilize GABA(A) receptors at synapses. Neuron. 2017;93:1138–1152.e1136. doi: 10.1016/j.neuron.2017.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flores CE, et al. Activity-dependent inhibitory synapse remodeling through gephyrin phosphorylation. Proc. Natl Acad. Sci. USA. 2015;112:E65–E72. doi: 10.1073/pnas.1411170112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poulopoulos A, et al. Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron. 2009;63:628–642. doi: 10.1016/j.neuron.2009.08.023. [DOI] [PubMed] [Google Scholar]
- 15.Maro GS, et al. MADD-4/punctin and neurexin organize C. elegans GABAergic postsynapses through neuroligin. Neuron. 2015;86:1420–1432. doi: 10.1016/j.neuron.2015.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tu H, Pinan-Lucarre B, Ji T, Jospin M, Bessereau JL. C. elegans punctin clusters GABA(A) receptors via neuroligin binding and UNC-40/DCC recruitment. Neuron. 2015;86:1407–1419. doi: 10.1016/j.neuron.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 17.Tong XJ, Hu Z, Liu Y, Anderson D, Kaplan JM. A network of autism linked genes stabilizes two pools of synaptic GABA(A) receptors. Elife. 2015;4:e09648. doi: 10.7554/eLife.09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou X, et al. The netrin receptor UNC-40/DCC assembles a postsynaptic scaffold and sets the synaptic content of GABAA receptors. Nat. Commun. 2020;11:2674. doi: 10.1038/s41467-020-16473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kummer TT, Misgeld T, Sanes JR. Assembly of the postsynaptic membrane at the neuromuscular junction: paradigm lost. Curr. Opin. Neurobiol. 2006;16:74–82. doi: 10.1016/j.conb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 20.Madhavan R, Peng HB. Molecular regulation of postsynaptic differentiation at the neuromuscular junction. IUBMB Life. 2005;57:719–730. doi: 10.1080/15216540500338739. [DOI] [PubMed] [Google Scholar]
- 21.McMahan UJ. The agrin hypothesis. Cold Spring Harb. Symp. Quant. Biol. 1990;55:407–418. doi: 10.1101/SQB.1990.055.01.041. [DOI] [PubMed] [Google Scholar]
- 22.Clark BA, Cull-Candy SG. Activity-dependent recruitment of extrasynaptic NMDA receptor activation at an AMPA receptor-only synapse. J. Neurosci. 2002;22:4428–4436. doi: 10.1523/JNEUROSCI.22-11-04428.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee SJ, et al. Presynaptic neuronal pentraxin receptor organizes excitatory and inhibitory synapses. J. Neurosci. 2017;37:1062–1080. doi: 10.1523/JNEUROSCI.2768-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nam CI, Chen L. Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc. Natl Acad. Sci. USA. 2005;102:6137–6142. doi: 10.1073/pnas.0502038102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silverman JB, et al. Synaptic anchorage of AMPA receptors by cadherins through neural plakophilin-related arm protein AMPA receptor-binding protein complexes. J. Neurosci. 2007;27:8505–8516. doi: 10.1523/JNEUROSCI.1395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thalhammer A, Cingolani LA. Cell adhesion and homeostatic synaptic plasticity. Neuropharmacology. 2014;78:23–30. doi: 10.1016/j.neuropharm.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 27.Topolnik L, Congar P, Lacaille JC. Differential regulation of metabotropic glutamate receptor- and AMPA receptor-mediated dendritic Ca2+ signals by presynaptic and postsynaptic activity in hippocampal interneurons. J. Neurosci. 2005;25:990–1001. doi: 10.1523/JNEUROSCI.4388-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- 29.Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev. Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- 30.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 31.Ghosh S, Reuveni I, Lamprecht R, Barkai E. Persistent CaMKII activation mediates learning-induced long-lasting enhancement of synaptic inhibition. J. Neurosci. 2015;35:128–139. doi: 10.1523/JNEUROSCI.2123-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- 33.Hayashi Y, et al. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 34.Malenka RC, et al. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- 35.Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 36.Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- 37.Houston CM, Hosie AM, Smart TG. Distinct regulation of beta2 and beta3 subunit-containing cerebellar synaptic GABAA receptors by calcium/calmodulin-dependent protein kinase II. J. Neurosci. 2008;28:7574–7584. doi: 10.1523/JNEUROSCI.5531-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Houston CM, Lee HH, Hosie AM, Moss SJ, Smart TG. Identification of the sites for CaMK-II-dependent phosphorylation of GABA(A) receptors. J. Biol. Chem. 2007;282:17855–17865. doi: 10.1074/jbc.M611533200. [DOI] [PubMed] [Google Scholar]
- 39.Saliba RS, Kretschmannova K, Moss SJ. Activity-dependent phosphorylation of GABAA receptors regulates receptor insertion and tonic current. EMBO J. 2012;31:2937–2951. doi: 10.1038/emboj.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McDonald BJ, Moss SJ. Differential phosphorylation of intracellular domains of gamma-aminobutyric acid type A receptor subunits by calcium/calmodulin type 2-dependent protein kinase and cGMP-dependent protein kinase. J. Biol. Chem. 1994;269:18111–18117. doi: 10.1016/S0021-9258(17)32424-9. [DOI] [PubMed] [Google Scholar]
- 41.McDonald BJ, Moss SJ. Conserved phosphorylation of the intracellular domains of GABA(A) receptor beta2 and beta3 subunits by cAMP-dependent protein kinase, cGMP-dependent protein kinase protein kinase C and Ca2+/calmodulin type II-dependent protein kinase. Neuropharmacology. 1997;36:1377–1385. doi: 10.1016/S0028-3908(97)00111-1. [DOI] [PubMed] [Google Scholar]
- 42.Kano M, Kano M, Fukunaga K, Konnerth A. Ca(2+)-induced rebound potentiation of gamma-aminobutyric acid-mediated currents requires activation of Ca2+/calmodulin-dependent kinase II. Proc. Natl Acad. Sci. USA. 1996;93:13351–13356. doi: 10.1073/pnas.93.23.13351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawaguchi SY, Hirano T. Signaling cascade regulating long-term potentiation of GABA(A) receptor responsiveness in cerebellar Purkinje neurons. J. Neurosci. 2002;22:3969–3976. doi: 10.1523/JNEUROSCI.22-10-03969.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He Q, et al. Interneuron- and GABA(A) receptor-specific inhibitory synaptic plasticity in cerebellar Purkinje cells. Nat. Commun. 2015;6:7364. doi: 10.1038/ncomms8364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marsden KC, Shemesh A, Bayer KU, Carroll RC. Selective translocation of Ca2+/calmodulin protein kinase IIalpha (CaMKIIalpha) to inhibitory synapses. Proc. Natl Acad. Sci. USA. 2010;107:20559–20564. doi: 10.1073/pnas.1010346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carrillo RA, Olsen DP, Yoon KS, Keshishian H. Presynaptic activity and CaMKII modulate retrograde semaphorin signaling and synaptic refinement. Neuron. 2010;68:32–44. doi: 10.1016/j.neuron.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ninan I, Arancio O. Presynaptic CaMKII is necessary for synaptic plasticity in cultured hippocampal neurons. Neuron. 2004;42:129–141. doi: 10.1016/S0896-6273(04)00143-6. [DOI] [PubMed] [Google Scholar]
- 48.Antonov I, Kandel ER, Hawkins RD. Presynaptic and postsynaptic mechanisms of synaptic plasticity and metaplasticity during intermediate-term memory formation in Aplysia. J. Neurosci. 2010;30:5781–5791. doi: 10.1523/JNEUROSCI.4947-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoover CM, et al. A novel CaM kinase II pathway controls the location of neuropeptide release from Caenorhabditis elegans motor neurons. Genetics. 2014;196:745–765. doi: 10.1534/genetics.113.158568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Q, Chen B, Ge Q, Wang ZW. Presynaptic Ca2+/calmodulin-dependent protein kinase II modulates neurotransmitter release by activating BK channels at Caenorhabditis elegans neuromuscular junction. J. Neurosci. 2007;27:10404–10413. doi: 10.1523/JNEUROSCI.5634-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moro A, van Woerden GM, Toonen RF, Verhage M. CaMKII controls neuromodulation via neuropeptide gene expression and axonal targeting of neuropeptide vesicles. PLoS Biol. 2020;18:e3000826. doi: 10.1371/journal.pbio.3000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McIntire SL, Jorgensen E, Horvitz HR. Genes required for GABA function in Caenorhabditis elegans. Nature. 1993;364:334–337. doi: 10.1038/364334a0. [DOI] [PubMed] [Google Scholar]
- 53.McIntire SL, Jorgensen E, Kaplan J, Horvitz HR. The GABAergic nervous system of Caenorhabditis elegans. Nature. 1993;364:337–341. doi: 10.1038/364337a0. [DOI] [PubMed] [Google Scholar]
- 54.Schuske K, Beg AA, Jorgensen EM. The GABA nervous system in C. elegans. Trends Neurosci. 2004;27:407–414. doi: 10.1016/j.tins.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 55.Gendrel M, Atlas EG, Hobert O. A cellular and regulatory map of the GABAergic nervous system of C. elegans. Elife. 2016;5:e17686. doi: 10.7554/eLife.17686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Richmond JE, Jorgensen EM. One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat. Neurosci. 1999;2:791–797. doi: 10.1038/12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mahoney TR, Luo S, Nonet ML. Analysis of synaptic transmission in Caenorhabditis elegans using an aldicarb-sensitivity assay. Nat. Protoc. 2006;1:1772–1777. doi: 10.1038/nprot.2006.281. [DOI] [PubMed] [Google Scholar]
- 58.Hawasli AH, Saifee O, Liu C, Nonet ML, Crowder CM. Resistance to volatile anesthetics by mutations enhancing excitatory neurotransmitter release in Caenorhabditis elegans. Genetics. 2004;168:831–843. doi: 10.1534/genetics.104.030502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rongo C, Kaplan JM. CaMKII regulates the density of central glutamatergic synapses in vivo. Nature. 1999;402:195–199. doi: 10.1038/46065. [DOI] [PubMed] [Google Scholar]
- 60.Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell. 2005;123:849–860. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 61.Caylor RC, Jin Y, Ackley BD. The Caenorhabditis elegans voltage-gated calcium channel subunits UNC-2 and UNC-36 and the calcium-dependent kinase UNC-43/CaMKII regulate neuromuscular junction morphology. Neural Dev. 2013;8:10. doi: 10.1186/1749-8104-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park EC, Horvitz HR. Mutations with dominant effects on the behavior and morphology of the nematode Caenorhabditis elegans. Genetics. 1986;113:821–852. doi: 10.1093/genetics/113.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cabantous S, Terwilliger TC, Waldo GS. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005;23:102–107. doi: 10.1038/nbt1044. [DOI] [PubMed] [Google Scholar]
- 64.Kamiyama D, et al. Versatile protein tagging in cells with split fluorescent protein. Nat. Commun. 2016;7:11046. doi: 10.1038/ncomms11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Qian, K. Y. et al. Male pheromones modulate synaptic transmission at the C. elegans neuromuscular junction in a sexually dimorphic manner. Elife10, e67170 (2021). [DOI] [PMC free article] [PubMed]
- 66.Gally C, Bessereau JL. GABA is dispensable for the formation of junctional GABA receptor clusters in Caenorhabditis elegans. J. Neurosci. 2003;23:2591–2599. doi: 10.1523/JNEUROSCI.23-07-02591.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fares H, Greenwald I. Genetic analysis of endocytosis in Caenorhabditis elegans: coelomocyte uptake defective mutants. Genetics. 2001;159:133–145. doi: 10.1093/genetics/159.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoerndli FJ, et al. Neuronal activity and CaMKII regulate kinesin-mediated transport of synaptic AMPARs. Neuron. 2015;86:457–474. doi: 10.1016/j.neuron.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tan SE, Wenthold RJ, Soderling TR. Phosphorylation of AMPA-type glutamate receptors by calcium/calmodulin-dependent protein kinase II and protein kinase C in cultured hippocampal neurons. J. Neurosci. 1994;14:1123–1129. doi: 10.1523/JNEUROSCI.14-03-01123.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marsden KC, Beattie JB, Friedenthal J, Carroll RC. NMDA receptor activation potentiates inhibitory transmission through GABA receptor-associated protein-dependent exocytosis of GABA(A) receptors. J. Neurosci. 2007;27:14326–14337. doi: 10.1523/JNEUROSCI.4433-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Benfenati F, et al. Synaptic vesicle-associated Ca2+/calmodulin-dependent protein kinase II is a binding protein for synapsin I. Nature. 1992;359:417–420. doi: 10.1038/359417a0. [DOI] [PubMed] [Google Scholar]
- 72.Dickinson DJ, Ward JD, Reiner DJ, Goldstein B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat. Methods. 2013;10:1028–1034. doi: 10.1038/nmeth.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Concordet JP, Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018;46:W242–W245. doi: 10.1093/nar/gky354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kage-Nakadai E, et al. A conditional knockout toolkit for Caenorhabditis elegans based on the Cre/loxP recombination. PLoS ONE. 2014;9:e114680. doi: 10.1371/journal.pone.0114680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dittman JS, Kaplan JM. Factors regulating the abundance and localization of synaptobrevin in the plasma membrane. Proc. Natl Acad. Sci. USA. 2006;103:11399–11404. doi: 10.1073/pnas.0600784103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dittman JS, Kaplan JM. Behavioral impact of neurotransmitter-activated G-protein-coupled receptors: muscarinic and GABAB receptors regulate Caenorhabditis elegans locomotion. J. Neurosci. 2008;28:7104–7112. doi: 10.1523/JNEUROSCI.0378-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vashlishan AB, et al. An RNAi screen identifies genes that regulate GABA synapses. Neuron. 2008;58:346–361. doi: 10.1016/j.neuron.2008.02.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary files
Data Availability Statement
The full raw data that support the results of this study are available upon request. The source data underlying the quantification of Figs. 1c, d, 2d, 3b, c, f, g, 3i, 4a–d, 5a–c, 6b, c, 7c–e, 8c, e, g, h, 9a, c, d and Supplementary Figs. 1, 2b, d, 3, 4, 5b, c, e, f, 6a–c, 7b, 8, 9a, b, 11a, b are provided as a Source data file. Source data are provided with this paper.