

Summary
The conserved regulon of Heat Shock Factor 1 in budding yeast contains chaperones for general protein folding as well as Zinc Finger Protein Zpr1, whose essential role in archaea and eukaryotes remains unknown. Here, we show that Zpr1 depletion causes acute proteotoxicity driven by biosynthesis of misfolded eukaryotic translation elongation factor 1A (eEF1A). Prolonged Zpr1 depletion leads to eEF1A insufficiency thereby inducing the integrated stress response and inhibiting protein synthesis. Strikingly, we show using two distinct biochemical reconstitution approaches that Zpr1 enables eEF1A to achieve a conformational state resistant to protease digestion. Lastly, we use a ColabFold model of the Zpr1-eEF1A complex to reveal a folding mechanism mediated by Zpr1’s zinc finger and alpha helical hairpin structures. Our work uncovers the long sought-after function of Zpr1 as a bespoke chaperone tailored to the biogenesis of one of the most abundant proteins in the cell.
Keywords: Chaperone, heat shock response, misfolding, translation elongation, integrated stress response
Graphical Abstract
Introduction
Chaperones facilitate folding of structurally complex proteins into their native states by minimizing the potential of folding intermediates to aggregate in the crowded cellular environment1,2. Basal levels of chaperones enable their housekeeping role but conditions that induce protein misfolding, such as heat shock, lead to elevated synthesis of certain chaperones, many of which are eponymously called Heat shock proteins (Hsps)3. In eukaryotes, the pioneering example of this type of mechanism for maintaining protein homeostasis (proteostasis) was the transcriptional regulation of chaperone genes by Heat shock factor 1 (Hsf1). More recently, we used genomics to define a conserved Hsf1 regulon in the budding yeast Saccharomyces cerevisiae and human cells that primarily consists of genes for cytosolic Hsp70 and Hsp90 family chaperones, as well as their co-chaperones and nucleotide exchange factors4. A complementary study identified the Zinc Finger Protein 1 (ZPR1) gene as an Hsf1 target in yeast by showing that its heat-shock inducibility was only out-ranked by two cytosolic Hsp70s5. Despite this strong gene regulatory association with chaperones, the Zpr1 protein has not to our knowledge been previously linked to proteostasis.
ZPR1 genes are found in organisms spanning the eukaryotic and archaeal domains of life where they perform an essential but poorly-defined role. A recent next-generation sequencing screen of essential genes in the archaea Sulfolobus islandicus noted that its particular homolog remains the only essential gene common to archaea and eukarya whose exact function is still unclear6. Zpr1 was initially identified by its interaction with the cytosolic region of the Epidermal Growth Factor Receptor (EGFR)7. This interaction was evidently disrupted by stimulation with EGF and suggested a role for Zpr1 in EGFR signaling. Subsequent work has shown that Zpr1 pull-downs from human cell extracts contain the Survival Motor Neurons (SMN) protein8, which plays an important role in pre-mRNA splicing9 and whose loss is responsible for the onset of spinal muscular atrophy (SMA)10, a severe neuromuscular disease. An intriguing positive correlation between Zpr1 and SMN levels has been observed using mouse models and cells derived from SMA patients11. More strikingly, engineered Zpr1 overexpression can restore SMN levels and alleviate disease phenotypes12. However, due to the absence of receptor tyrosine kinases in fungi13 and the lack of a detectable SMN homolog in S. cerevisiae14, mammalian studies of Zpr1 have failed to provide a satisfying mechanistic explanation for its core essential function.
We considered the possibility that Zpr1’s presence among the chaperone cadre of Hsf1’s regulon provides a clue to its essential function in the eukaryotic and archaeal domains. Most intriguing in this regard were reports of yeast and mammalian Zpr1 binding to eukaryotic translation elongation factor 1A (eEF1A)15,16. eEF1A is a highly abundant, multi-domain G protein that in its GTP-bound state delivers aminoacylated tRNAs to the ribosome A site17. Following conversion to the GDP-bound state during the elongation cycle, eEF1A is recycled by eEF1B. Historically, active eEF1A has never been successfully purified as a recombinant protein from bacteria, while eEF1A purified from its native source is known to be unstable in the absence of high glycerol (>25%)18,19. The current picture of Zpr1’s functional relationship to eEF1A is arguably confusing but not inconsistent with our hypothesis. Namely, Zpr1 has been shown to bind purified eEF1A and even outcompete eEF1B binding to eEF1A, thus implying that Zpr1 inhibits eEF1A’s activity as a translation factor16. At apparent odds with this view, additional mutagenesis experiments showed that mutations disrupting Zpr1-eEF1A interaction in fact lead to reduced cell growth16, which implies that Zpr1 somehow promotes eEF1A’s essential function in the cell.
Here, we used unbiased genomics, proteomic interactor analysis, cell microscopy, structure-guided mutagenesis, and in vitro reconstitution approaches to show that Zpr1’s essential function in the budding yeast Saccharomyces cerevisiae comes from its biochemical activity as a chaperone dedicated to eEF1A biogenesis.
Results
Zpr1 is a bona fide component of the Hsf1 proteostasis network
Loss of cytosolic chaperones or GET pathway components in the budding yeast S. cerevisiae induces severe folding stress20, which is manifested by chronic Hsf1 activation associated with protein aggregation. We began by asking if auxin inducible degradation (AID) of Zpr1 leads to similar phenotypes; thus, we tagged Zpr1 with a C-terminal mini-AID (mAID) degron in cells capable of expressing the engineered E3 ubiquitin ligase variant OsTir1(F74G)21, yielding a combination of genetic elements referred to as ZPR1-AID. By treating ZPR1-AID cells with the E3-specific auxin derivative, 5-Ph-IAA, we were able to rapidly induce Zpr1 depletion (Figure 1A), leading to cell growth arrest (Figure 1B). To monitor the effect of Zpr1 depletion on proteostasis, we first introduced a GFP transcriptional reporter of Hsf1 activity into ZPR1-AID cells and measured its activity by flow cytometry. This analysis revealed that Zpr1 depletion causes rapid and persistent Hsf1 activation that is comparable in magnitude to the strongest Hsf1 stressor alleles defined by genome-wide screening (e.g. ssa2Δ)20 (Figure 1C, S1A). Next, we introduced an mCherry-tagged version of Hsp42, a small Hsp that sequesters misfolded proteins22, and looked by microscopy for punctae as evidence of protein aggregation in the cytosol. This approach for visualizing folding stress showed that Zpr1 depletion results in rapid, robust, and persistent accumulation of cytosolic protein aggregates (Figure 1D). In sum, these findings demonstrate Zpr1’s ability to stave off proteostasis collapse and help buttress its high standing among the other gene targets of Hsf1, which are primarily chaperones.
Figure 1. Zpr1 is a bona fide component of the Hsf1 proteostasis network. See also Figure S1.

(A) ZPR1-AID cells in late-log phase were treated with 5 μM 5-Ph-IAA or DMSO and lysates were analyzed via SDS-PAGE and immunoblotting (IB). (B) Spot growth assay on YPD plates with and without 5 μM 5-Ph-IAA. (C) Strains carrying the Hsf1 reporter were analyzed by flow cytometry post-treatment with 5 μM 5-Ph-IAA or DMSO. Bar graphs show median GFP intensity values normalized to cell size and to the average median GFP intensity of ZPR1 (E3 only) DMSO control cells at 0 min. Error bars represent standard deviation from three replicates. Representative histograms are shown. Untreated ssa2Δ cells served as a positive control. (D) ZPR1-AID cells expressing Hsp42-mCherry were imaged after treatment with 5 μM 5-Ph-IAA or DMSO. Confocal micrographs are normalized to the same intensity and cell borders are outlined. Scale bar: 2.5 μm.
Zpr1 depletion acutely disrupts eEF1A biogenesis leading to proteotoxicity
To define Zpr1’s place within the Hsf1 proteostasis network, we hypothesized that Zpr1’s known interaction with eEF1A15,16 reflects its previously overlooked role as an eEF1A chaperone. eEF1A comprises approximately 2% of the total yeast proteome, a staggering contribution to the mass of the cell that is only outweighed by a few glycolytic enzymes23. Thus, high rates of eEF1A biosynthesis could pose a significant load on the protein folding machinery, which becomes manifest in the absence of Zpr1 as proteotoxicity caused by misfolding and aggregation of newly-synthesized eEF1A.
First, we reasoned that elevated formation of misfolded eEF1A species should be accompanied by their degradation by the ubiquitin-proteasome system (UPS). We measured the fate of eEF1A synthesized shortly after acute Zpr1 depletion using a radioactive pulse-chase assay. After treatment with 5-Ph-IAA for 30 minutes, ZPR1-AID cells were briefly pulsed with 35S-methionine and then chased with excess unlabeled methionine. A significant fraction of eEF1A synthesized following Zpr1 depletion, but not mock treatment, became rapidly degraded in a proteasome-dependent manner (Figure 2A–B). Additionally, prolonged periods of Zpr1 depletion were accompanied by decreased cellular levels of endogenously expressed Tef1-GFP (eEF1A in S. cerevisiae is encoded by the paralogous genes TEF1 and TEF2) (Figure S1D), an expected product of its inefficient biogenesis barring compensatory mechanisms.
Figure 2. Zpr1 depletion leads to rapid misfolding of nascent eEF1A. See also Figure S1.

(A) Representative results of radioactive pulse-chase analysis of eEF1A in ZPR1-AID cells. eEF1A was immunoprecipitated at the indicated chase time and analyzed via SDS-PAGE and autoradiography. Quantified band intensity is displayed relative to time 0. (B) As in part A, but ZPR1-AID cells were additionally treated with either DMSO or 40 μM bortezomib and 5 μM epoxomicin before pulse-chase analysis. (C) ZPR1-AID cells expressing endogenously tagged Tef1-GFP and Hsp42-mCherry were analyzed as in Figure 1D. See Figure S1E for micrographs of mock-treated cells in the same experiment. Scale bar: 2.5 μm. (D) ZPR1-AID cells expressing endogenously tagged Tef1-GFP under the GAL1 promoter were grown in synthetic media containing galactose (SGal). Cells were subsequently transferred to fresh SGal or SD for one hour, treated (one hour; 5 μM 5-Ph-IAA or DMSO), and imaged. Scale bar: 2.5 μm. (E) Tef-Off cells carrying the Hsf1 reporter were grown in SGal before back-dilution into fresh SGal or SD for one hour and analyzed by flow cytometry post-treatment with 5 μM 5-Ph-IAA or DMSO, or post-shift to 39°C. Cells were analyzed as in Figure 1C. Bar graphs depict median GFP intensity values normalized to cell size and the average median GFP intensity of Tef-Off Galactose + DMSO cells at 0 min. Error bars represent standard deviation from three replicates. Representative histograms are shown below. (F) Tef-Off cells expressing Hsp42-mCherry were grown in SGal before being transferred to fresh SGal or SD media for one hour prior to Zpr1 depletion and microscopy analysis as in Figure 1D. Scale bar: 2.5 μm.
Second, we found complementary evidence of elevated eEF1A misfolding by analyzing the localization of endogenously expressed Tef1-GFP: within one hour of Zpr1 depletion GFP signal concentrated at sites coincident with Hsp42-positive puncta (Figure 2C, S1C, S1E). A key prediction of our model is that eEF1A found in these aggregates originates from misfolded biosynthetic intermediates rather than mature Tef1-GFP that becomes aggregation-prone following Zpr1 depletion. To distinguish between these possibilities, we devised a transcriptional shut-off assay in which we combined ZPR1-AID with conditional expression of Tef1-GFP from the GAL1 promoter at the TEF1 locus. One hour of Zpr1 depletion during growth in galactose resulted in Tef1-GFP puncta (Figure 2D), thus recapitulating the earlier observation with endogenous Tef1-GFP. By contrast, we observed no puncta formation if depletion was preceded by a one hour period of GAL1 promoter shut-off in glucose (Figure 2D). Together with our earlier pulse-chase analysis, these data suggest that Zpr1 prevents misfolding of eEF1A biosynthetic intermediates but is dispensable for conformational stability of mature eEF1A.
Third, to specifically test if Zpr1 depletion causes proteotoxicity via accumulation of misfolded eEF1A biosynthetic intermediates, we engineered GAL1 promoter-driven TEF1 and TEF2 expression from their endogenous loci in ZPR1-AID cells (a combination of genetic elements we term ‘Tef-Off’). Tef-Off cells grew robustly on media containing galactose, but adding glucose caused growth arrest (Figure S1F–G). As a positive control, we monitored the effect of Zpr1 depletion on Hsf1 reporter activity in Tef-Off cells cultured in galactose media and observed a strong increase in Hsf1 activity (Figure 2E). Consistent with our hypothesis, if Tef-Off cells were first shifted to glucose for one hour prior to Zpr1 depletion, Hsf1 activation became completely suppressed (Figure 2E). Importantly, this period of transcriptional eEF1A shutoff did not abolish Hsf1 induction by an ensuing heat shock at 39°C (Figure 2E), indicating that our reporter is not trivially compromised under these conditions. Nonetheless, eEF1A depletion on its own, by comparison to control ZPR1-AID cells (Figure S1H), did somewhat reduce basal Hsf1 activity, possibly by decreasing the translational burden on protein folding (Figure 2E). Lastly, we expressed Hsp42-mCherry in Tef-Off cells to test if bulk protein aggregation induced by Zpr1 depletion is also driven by ongoing eEF1A biosynthesis. We observed Hsp42-mCherry puncta if we induced depletion in galactose-grown cells, but this effect was abolished if cells were first shifted to growth in glucose (Figure 2F). These results demonstrate that eEF1A biogenesis is the primary driver of proteotoxic stress induced by Zpr1 depletion.
Zpr1 depletion induces the Integrated Stress Response (ISR)
Despite its magnitude, eEF1A proteotoxicity occurred much earlier following Zpr1 depletion relative to the ultimate cell growth arrest phenotype (Figure S1B, S2F), suggesting acute eEF1A proteotoxicity is not the sole cause of growth arrest. To gain insight into these temporal dynamics, we defined changes in the transcriptome specific to treatment of ZPR1-AID cells with 5-Ph-IAA but not its DMSO carrier (Figure 3A, S2A). mRNA-seq analysis of the early time points revealed elevated levels of Hsf1 gene targets, corroborating our findings using the Hsf1 reporter (Figure 3A). We further noted that this immediate-early response was dampened over time, a likely product of its negative feedback structure as in the case of Hsf1 activation by heat shock24,25.
Figure 3. Decreased eEF1A abundance following prolonged Zpr1 depletion induces the Integrated Stress Response. See also Figure S2.

(A) ZPR1-AID cells were treated with 5 μM 5-Ph-IAA and subjected to mRNA sequencing in triplicate. Each point denotes a gene in the indicated target category, where the gene abundance Z-score is obtained by gene-wise mean centering and scaling to the standard deviation across all timepoints using degPatterns. See STAR methods for further details (B) Indicated strains were grown in media with 2% glucose and 5 μM 5-Ph-IAA for the indicated times and lysates were analyzed via SDS-PAGE and immunoblotting. Plots show normalized signals as mean ± standard deviation of three replicates. (C) The indicated strains were grown in YPGal media then shifted to YPD containing 5 μM 5-Ph-IAA for the indicated times. Shown are A260 traces of cell extracts following fractionation by sucrose gradient centrifugation. (D) The indicated strains were analyzed as in part C. (E) Genome browser views of GCN4 and ACT1 ORFs showing normalized, aligned ribosome protected footprints from ribosome profiling of indicated strains.
Intriguingly, at later depletion time points of our gene expression analysis we observed increased expression of many Gcn4 targets for amino acid biosynthesis (Figure 3A, S2A). Gcn4 drives the transcriptional output of the integrated stress response (ISR), a conserved adaptation to translational stressors, including amino acid starvation and ribosome collisions26–28. The ISR in yeast is mediated by activation of the protein kinase Gcn2, which phosphorylates the translation initiation factor eIF2α and thereby globally inhibits translation initiation while de-repressing translation of the GCN4 mRNA26. We found that Zpr1 depletion using a combined transcriptional repression and AID strategy (prGAL1-ZPR1-AID or prGAL1-AID-ZPR1) induced several ISR-like hallmarks of translational stress. First, we measured the effect of Zpr1 depletion on eIF2α phosphorylation by immunoblotting and saw a marked increase in Gcn2-dependent phosphorylation of eIF2α over time (Figure 3B, S2B). Second, sucrose gradient profiling revealed a decrease in the polysome to monosome (P/M) ratio upon Zpr1 depletion, a tell-tale sign of reduced translation initiation (Figure 3C, S2C). Lastly, ribosome profiling of Zpr1-depleted cells revealed a dramatic increase in ribosome-protected footprints (RPFs) mapping to the ORF region of GCN4 mRNAs, which strongly suggests that the induction of Gcn4 targets we observed by mRNA-seq was mediated by Gcn4 translational de-repression (Figure 3E). Taken together, these data suggest that prolonged Zpr1 depletion results in terminal growth arrest by inducing an unresolved form of the ISR that leads to severe inhibition of protein synthesis.
Engineered eEF1A depletion phenocopies ISR induction
To gain insight into how Zpr1 depletion leads to ISR induction, we considered a model wherein induction stems from reduced concentrations of mature eEF1A below a threshold level required for its optimal function. To test this model, we engineered a system that decouples eEF1A-associated proteotoxicity from loss of eEF1A function. Specifically, we transcriptionally shut-off TEF1 expression using the GAL1 promoter and simultaneously induced depletion of existing Tef2-AID (prGAL1-TEF1, TEF2-AID) (Figure S2D, S2F). Importantly, as in the related Tef-Off background, this method of eEF1A depletion was not associated with Hsf1 activation (Figure S2E). By contrast, after four hours of eEF1A depletion, we observed a marked accumulation of phospho-eIF2α (Figure 3B, S2B). Similarly, sucrose gradient profiling revealed a decrease in the P/M ratio at later eEF1A depletion times (Figure 3D, S2C). Taken together, these results strongly support the model in which loss of eEF1A in itself is sufficient to cause both the ISR and growth arrest in Zpr1-depleted cells.
Newly-synthesized eEF1A fluxes through Zpr1 in vivo
Zpr1 has previously been shown to bind eEF1A15,16 but the mechanistic significance of this observation has remained obscure. Our previous data show that Zpr1 behaves de facto as a chaperone for newly-synthesized eEF1A and suggest that the interaction between the two actually represents a transient biosynthetic intermediate en route to a mature eEF1A product. To test this prediction, we combined our previous radiolabeled pulse-chase analysis with native immunoprecipitation of endogenously tagged Zpr1-3ˣFLAG. Consistent with our model, we detected rapid flux of a single radiolabeled species that co-migrated with an eEF1A standard immunoprecipitated under denaturing conditions (Figure 4A). To complement this view of dynamic Zpr1 interactors, we also affinity-purified Zpr1-3ˣFLAG complexes under steady-state, non-radioactive conditions and analyzed them by SDS-PAGE (Figure 4B, S3A) and mass spectrometry (Figure 4C). Besides the dominant presence of eEF1A, we also found strong hits among components of the cytosolic Hsp70 and Hsp90 folding systems, and various large and small ribosomal subunit proteins; the latter interactions explain the presence of large ribosome-like particles in the purified material by negative stain electron microscopy (Figure S3B–C). In sum, our interactor analysis suggests that Zpr1 transiently interacts with newly-synthesized eEF1A as part of a larger chaperone network.
Figure 4. Zpr1 transiently interacts with newly-synthesized eEF1A to enable eEF1A conversion to a protease-resistant state. See also Figures S3 and S5.

(A) Pulse-chase flux analysis of newly synthesized eEF1A through Zpr1–3ˣFLAG, as described in STAR Methods. Shown is an autoradiograph of anti-FLAG immunoprecipitations resolved by SDS-PAGE. “Std.”: Anti-eEF1A immunoprecipitation as described in Figure 2A. (B) Anti-FLAG immunoprecipitations were analyzed via SDS-PAGE and coomassie staining. Shown are two of four technical replicates that were analyzed by mass spectrometry as described in part C. (C) Mass spectrometry analysis of samples described in part B. Dashed line: false discovery rate (FDR) at the adjusted p-value of 0.05. (D) Stability of 35S-Methionine-radiolabeled eEF1A synthesized in extracts derived from ZPR1-AID cells following either one hour treatment with 5 μM 5-Ph-IAA or DMSO was determined by a trypsin digestion assay as described in STAR Methods. Shown is a representative autoradiograph (top) and immunoblot (bottom) of quenched material resolved by SDS-PAGE. (E) As in part D but in Zpr1-depleted extracts supplemented with recombinant Zpr1 at the indicated final concentrations before eEF1A translation. “Tryp.-Res. eEF1A” indicates a stable, trypsin-resistant fragment of similar size to untreated eEF1A. (F) Stability of pure eEF1A (1 μM) following incubation with wild-type Zpr1 in the presence of GTP was determined by a trypsin digestion assay as described in STAR Methods. Shown is a representative immunoblot of quenched material resolved by SDS-PAGE.
Biochemical reconstitution of eEF1A folding by Zpr1
eEF1A folding to our knowledge has not been studied in vitro but the inability to express folded eEF1A from various species as a recombinant protein in Escherichia coli has previously been noted (W. Merrick and T. Kinzy, personal communication). Instead, eEF1A of several species has been successfully purified out of native extracts and its conformational stability assessed by sensitivity to trypsin digestion18,19,29. Using these historical insights, we developed two complementary in vitro systems for studying the biochemical effects of Zpr1 on eEF1A folding. First, we used a trypsin protection assay to monitor the conformational state of radiolabeled eEF1A synthesized in yeast extracts. Full-length eEF1A made by control extracts from DMSO-treated ZPR1-AID cells was largely resistant to trypsin, decreasing only slightly in molecular weight but otherwise remaining stable for up to 60 minutes (Figure 4D). In contrast, Zpr1-depleted extracts yielded eEF1A that was ultrasensitive to trypsin digestion, collapsing into a series of low molecular weight digestion products within two minutes (Figure 4D). Critically, restoring Zpr1-depleted extracts with near-physiological concentrations of recombinant Zpr1-3ˣFLAG (Figure S5F) prior to in vitro translation rescued eEF1A protease resistance to the wild-type level (Figure 4E). We asked if Zpr1 confers protease protection by stably binding to eEF1A but found two lines of evidence arguing against it. First, the highly abundant population of unlabeled eEF1A that was synthesized in intact cells prior to lysis remained largely trypsin resistant regardless of Zpr1’s presence or absence in the extracts (Figure 4D). Second, quantitative immunodepletion of Zpr1 from extracts after eEF1A translation left behind the majority of radiolabeled eEF1A in a form that remained protease resistant (Figure S3D–E). Thus, rather than acquiring trypsin resistance by stably binding Zpr1, folding flux of newly synthesized eEF1A through Zpr1 enables its maturation to trypsin resistance in yeast extracts.
Unlike native eEF1A in cell extracts, purified eEF1A from various species is known to be sensitive to trypsin digestion at low concentrations of glycerol (below ~25%)18,19,29. Moreover, the loss of eEF1A’s activity in vitro as an elongation factor in low glycerol can be counteracted by formation of ternary complexes with GTP and charged tRNAs. We took advantage of these historical observations to test if Zpr1 can minimally function as an eEF1A stabilizing factor in vitro. Thus, we first purified eEF1A to homogeneity from wild-type yeast extracts (Figure S3F) and showed that it was trypsin sensitive at ~14% glycerol (Figure 4F) as expected. Strikingly, addition of Zpr1 in the presence of GTP but not GDP induced eEF1A protease resistance, while GTP alone was not sufficient to induce stabilization (Figure 4F, S3G). In sum, our two biochemical reconstitution approaches demonstrate Zpr1’s intrinsic biochemical ability to induce eEF1A conversion into a protease-resistant conformation.
Defining the mechanism of eEF1A folding by Zpr1
What is the structural basis of Zpr1’s function as an eEF1A-specific chaperone? Zpr1 comprises two tandem halves (N-halfZpr1 and Zpr1C-half) that are closely related to each other in both sequence and structure. X-ray crystallography of the mouse Zpr1 homolog revealed that each half is made up of a zinc finger (ZnFN and ZnFC, respectively) followed by a traditional double stranded beta-helix domain carrying an alpha-helical hairpin (aHHN and aHHC) insert unique to Zpr1 homologs16. To examine the individual contributions of the two Zpr1 halves on eEF1A biogenesis, we first confirmed an earlier finding that plasmid-based overexpression of Zpr1C-half (but not N-halfZpr1) was able to restore viability to zpr1Δ cells, albeit resulting in a severe growth defect (Figure S4A). Consistent with this, Zpr1C-half was only marginally able to suppress the induction of Hsf1 activity following depletion of endogenous Zpr1 in ZPR1-AID cells (Figure S4B). Interactome analysis of FLAG-tagged versions of N-halfZpr1 and Zpr1C-half relative to a similarly expressed full-length Zpr1 control (ie. as a 2nd copy) revealed that neither half alone could efficiently capture eEF1A or the other interactors (Figures S4C–G).
To help guide our structure-function analysis of Zpr1, we turned to ColabFold, an emerging computational tool for predicting protein complex structures that is gaining in popularity30 (Supplementary Item 4). This approach led to a high-confidence model in which the C-terminal half of Zpr1 forms extensive complementary interactions with the three individually folded domains (the nucleotide-binding DI, DII, and DIII) of eEF1A (Figure S5A). Specifically, ZnFC is intercalated between DI and DIII, while aHHC makes contacts with the interface between DI and DII (Figure 5A–B). Forced computational linkage of eEF1A with N-halfZpr1 led to a similar structural model, albeit of relatively lower confidence, consistent with the high degree of sequence and structural similarity between the two halves (Figure S4H–I).
Figure 5. Zpr1 uses a zinc finger and alpha-helical hairpin-based chaperoning mechanism. See also Figures S4, S5, and S6.

(A) Ribbon diagram of ColabFold-Fold Multimer model showing C-terminal half of Zpr1 (purple) bound to eEF1A (colored by domain) with the N-terminal half of Zpr1 removed for clarity and with further emphasis on ZnFC. The inset shows four hydrophobic residues of ZnFC in stick representation (V317, I318, I319, M320) and neighboring residues within 5Å in eEF1A domain I (blue) and domain III (green). (B) The same model as in A but rotated to emphasize aHHC. The inset shows two glutamate residues of aHHC in stick representation (E391, E399) and neighboring lysines in eEF1A domain I (blue). (C) ZPR1-AID cells expressing the Hsf1 reporter and carrying the indicated plasmid-borne Zpr1 mutants were analyzed by flow cytometry post-treatment with 5 μM 5-Ph-IAA or DMSO. Bar graphs show median GFP intensity values normalized to untreated empty vector (EV) cells. Error bars represent standard deviation from three replicates. (D) Stability of 35S-Methionine-radiolabeled eEF1A synthesized in extracts derived from ZPR1-AID cells treated with 5 μM 5-Ph-IAA for one hour and supplemented with recombinant Zpr1 ZnF mutants before eEF1A translation was determined by a trypsin digestion assay as described in the STAR Methods. Shown is a representative autoradiograph of quenched material resolved by SDS-PAGE. “Tryp.-Res. eEF1A” indicates a stable, trypsin-resistant fragment of similar size to untreated eEF1A. (E) As in part C, but with aHH mutants. (F) Stability of pure eEF1A ( 1 μM) following incubation with wild-type Zpr1 or indicated Zpr1 ZnF mutants in the presence of GTP was determined as described in STAR Methods. Shown is a representative immunoblot of quenched material resolved by SDS-PAGE. (G) As in part F, but with indicated Zpr1 aHH mutants analyzed.
The presence of a continuous stretch of four conserved and hydrophobic residues on each ZnF caught our attention; by assuming an alternating pattern along one of the ZnF beta-strands, these residues mask opposing hydrophobic patches on DI and DIII (Figure 5A). To look for supporting evidence that this eEF1A inter-domain interface is aggregation prone, we analyzed by microscopy yeast cells expressing various truncations eEF1A as GFP-fusions. Most notably, we found that constructs lacking DIII were soluble even in the absence of Zpr1, while those containing DIII were constitutively aggregated (Figure S6). The second major point of contact predicted by the ColabFold model involves the alpha helical hairpin (aHH) of Zpr1 at the DI-DII interface of eEF1A. Two highly conserved glutamate residues on each aHH stood out, which in the model interact with positively charged residues on DI (Figure 5B).
To test the importance of ZnF and aHH contacts in the Zpr1 chaperone mechanism, we used site-directed mutagenesis to ablate residues at the four hydrophobic positions either in ZnFN alone (Zpr1Zn-N), ZnFC alone (Zpr1Zn-C), or both (Zpr1Zn-NC). We similarly tested the importance of the aHH contacts by mutating the conserved glutamate residues in aHHN alone (Zpr1aH-N), aHHC alone (Zpr1aH-C), or both (Zpr1aH-NC). All mutations were first introduced into a ZPR1 low-copy plasmid expression cassette (with endogenous promoter and terminator elements) that robustly restored viability to zpr1 deletes unlike the empty vector control (Figure S5B). We found Zpr1Zn-C and Zpr1Zn-NC failed to complement but that Zpr1Zn-N resulted in partial complementation and somewhat slower growth relative to the wild-type Zpr1 control (Figure S5B). Notably, all mutants were comparably expressed to the Zpr1 wild-type control (Figure S5C–D). Furthermore, the ZnFC and ZnFNC mutations abolished the ability of plasmid-borne Zpr1 to suppress Hsf1 induction following depletion of endogenous Zpr1 in ZPR1-AID cells (Figure 5C, S5E), suggesting the C-terminal ZnF is critical for eEF1A folding. Despite its ability to partially complement, ZnFN appeared to be an equally strong Hsf1 stressor allele (Figure 5C, S5E). Phenotypic analysis of the aHH alleles led to complementary conclusions. We observed full complementation by Zpr1aH-N while Zpr1aH-C and Zpr1aH-NC failed to restore viability to zpr1 deletes (Figure S5B) consistent with their inability to suppress Hsf1 induction following Zpr1 depletion (Figure 5C, S5E).
Next, we analyzed the effects of ZnF and aHH mutations on Zpr1’s activity as an eEF1A chaperone using our two biochemical reconstitution assays. Zpr1Zn-N-3ˣFLAG could restore eEF1A trypsin resistance close to wild type in both assays, while neither Zpr1Zn-C-3ˣFLAG nor Zpr1Zn-NC-3ˣFLAG could restore eEF1A protection at the physiological Zpr1 concentration (~100 nM) (Figure 5D, 5F). At the highest concentrations tested (~2.5 μM), Zpr1Zn-C-3ˣFLAG yielded some trypsin-resistant eEF1A, while Zpr1Zn-NC-3ˣFLAG remained fully inactive (Figure 5D, 5F) showing that both ZnFs contribute to Zpr1’s function, but the contribution of the C-terminal half is more crucial. The aHH mutants supported a similar dominant role for the C-terminal half, with neither Zpr1aH-C-3ˣFLAG nor Zpr1aH-NC-3ˣFLAG restoring eEF1A trypsin resistance until present at higher than physiological concentration (Figure 5E, 5G). In sum, our structure-function analysis uncovered an eEF1A chaperoning mechanism driven primarily by C-terminal Zpr1 ZnF and aHH that is at the core of Zpr1’s essential function in the cell.
Discussion
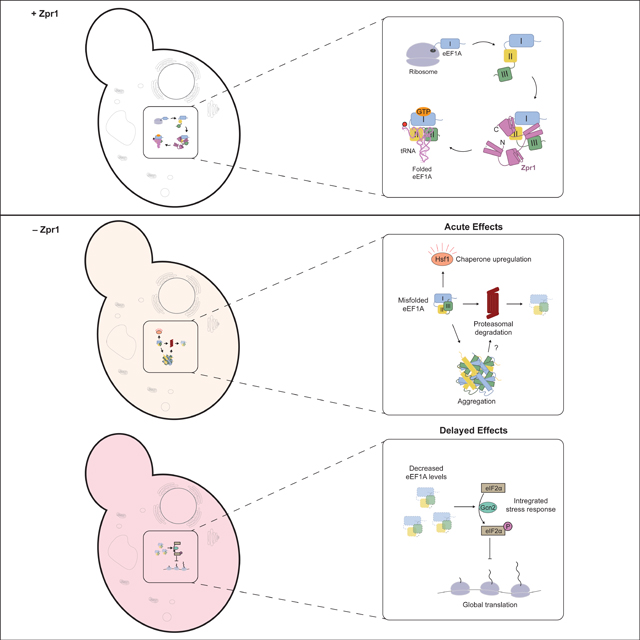
Defining the molecular basis of Zpr1’s essential function has been historically challenging. A pioneering study using fission yeast argued for Zpr1’s essential and conserved role in nucleolar function after finding a defect in rRNA biogenesis following zpr1+ gene transcriptional shut-off31. Subsequent study of budding yeast zpr1 temperature-sensitive (ts) alleles did not support this conclusion, suggesting instead that the original nuclear link was a sequelae of growth arrest following slow Zpr1 depletion (~12h)32. More critically, by analyzing zpr1ts interactions with other genes and their effects on Zpr1 cell localization, this latter study concluded that the essential role of Zpr1 is cytosolic and linked to eEF1A32. In a more recent budding yeast genomics study, the ZPR1 gene was identified as a top heat-shock induced target of Hsf15, suggesting a connection to proteostasis but adding a retrospective complexity to interpretation of zpr1ts phenotypes at restrictive temperature. To overcome these historical obstacles, we used modern functional genomic tools to profile yeast cells engineered for acute degradation of Zpr1 at physiological temperature. We found immediate Hsf1 regulon induction followed by delayed induction of the integrated stress response (ISR) (Figure 3A). Proteostasis stress following Zpr1 depletion is driven by biosynthesis of unstable eEF1A biosynthetic intermediates, which themselves are subject to proteasomal degradation and cytosolic aggregation (Figure 2). By contrast, depletion of functional eEF1A is sufficient to explain the induction of the ISR via Gcn2-dependent eIF2α phosphorylation leading to translational attenuation (Figures 3B, 3D). In sum, our cell biology analysis revealed that Zpr1 is an essential eEF1A biogenesis factor whose function cannot be bypassed by upregulating general chaperones or by decreasing their substrate load. Thus, like most other essential genes common to eukarya and archaea6, Zpr1 enables information flow along the central dogma of molecular biology.
To study the biochemical effects of Zpr1 on eEF1A biogenesis, we first used a yeast cell-free system to demonstrate Zpr1’s central role in converting newly-synthesized eEF1A to a trypsin-resistant state akin to that of native eEF1A (Figure 4D–E). Next, we obtained pure eEF1A from yeast cell extracts and showed that it is destabilized in 10% glycerol but that Zpr1 induces trypsin resistance in the presence of GTP but not GDP (Figure 4F, S3G). Lastly, we used a ColabFold model of the Zpr1-eEF1A complex to find evidence for a working model in which Zpr1 uses zinc-finger (ZnF) and alpha-helical hairpin (aHH) regions to help organize a near-native arrangement of eEF1A domains (Figure 5). Thus, we propose that Zpr1 is a bespoke chaperone tailored to eEF1A biogenesis (Figure 6). The term “molecular chaperone” was originally used to evoke the function of a specific histone-DNA assembly factor33 before becoming an umbrella term for a more eclectic group that includes chaperonins and Hsp70/90 proteins for general protein folding. Within this expanded view, Zpr1’s mechanistic role can be compared to that of Cdc123, an essential protein that facilitates eukaryotic translation initiation factor 2 (eIF2) biogenesis34. Cdc123 interacts with an eEF1A-like subunit of eIF2 via its DIII35. We have shown that the homologous domain of eEF1A is extremely aggregation-prone and confers a growth defect to cells when expressed alone or together with DII in yeast (Figure S6B, S6D). An appealing feature of our ColabFold model is that it confidently predicts a “place-holder” structural role for the Zpr1 ZnFC at a DI-DIII hydrophobic interface. More broadly, it will be interesting to see if other eEF1A-like proteins involved in translation (e.g. Sup35, Hbs1, Ski7 in yeast) depend on chaperones that share this point of mechanistic convergence between Zpr1 and Cdc123.
Figure 6. Mechanistic model of Zpr1-assisted eEF1A biogenesis.

Cartoon schematic emphasizing essential role of Zpr1 C-terminal ZnFs and aHHs in formation of correct tertiary interactions between eEF1A domains following protein synthesis. See text for further details.
Eukaryotes rely on elaborate chaperone networks to assist the folding of structurally complex proteomes. From bacteria to mammals, chaperone machineries have increased in copy number and abundance but remained fundamentally unchanged. The ability of core chaperones in eukaryotes to carry their client burden is instead enabled by co-chaperone expansion. A particularly relevant example to our study is that of Cns1 and Hgh1, two Hsp90 co-chaperones that work together to facilitate eukaryotic elongation factor 2 (eEF2) biogenesis36,37. Our Zpr1 interactor analysis also suggests that eEF1A biogenesis is a collaborative process involving general chaperones, as well as ribosomes (Figure 4C). Consistent with this notion, we observed substrate flux through Zpr1 using the yeast cell-free system but we found no evidence of catalysis using Zpr1 and purified eEF1A alone as approximately stoichiometric levels of Zpr1 were required to achieve near-quantitative protection (Figures 4F, 5F–G). This sets the stage for future bottom-up reconstitution approaches to identify additional eEF1A biogenesis factors.
A detailed view of how Zpr1 helps eEF1A substrates achieve their conformational maturity is still lacking. Our data favor the simplest possibility in which Zpr1 uses its ZnF and aHH to help organize the three eEF1A domains, which themselves were folded by earlier steps in the biogenesis process. The precise role of GTP in this process remains to be determined but we speculate that it enables eEF1A’s DI domain to assume a tertiary conformation with the other domains thereby making them as an ensemble poised for downstream interactions with amino-acid charged tRNAs and translating ribosomes. Failure to do so might result in the inappropriate exposure of a DIII hydrophobic surface and explain eEF1A aggregation and recognition by the ubiquitin proteasome system in cells depleted of Zpr1 (Figure 2). A related unresolved issue pertains to the dynamics of the two Zpr1 halves during the chaperoning mechanism. Our ColabFold structure-guided mutagenesis has demonstrated the relative supremacy of the C-terminal ZnF and aHH (Figure 5, S5B). This view is also consistent with the ability of the C-terminal half alone to weakly complement the inviability of zpr1 null cells, as others have shown previously and we have confirmed here (Figure S4A)15. Intriguingly, crystal structure of the mouse Zpr1 shows numerous structural differences between homologous regions of the two halves16, which might account for the significantly weaker confidence of ColabFold in the predicted N-halfZpr1-eEF1A model. Thus we favor the idea that the N-terminal half of Zpr1 is a functionally-divergent “co-chaperone” for the C-terminal half, in a conceptually similar way that Hsp110 is a co-chaperone for the sequence-related Hsp7038,39. Future testing of this hypothesis should be facilitated by direct structural analysis of Zpr1-eEF1A complexes and development of new in vitro eEF1A folding assays more sensitive to loss-of-function mutations in the N-terminal half.
From the more general point of interest in translational control, our work offers strains engineered for depletion of either Zpr1 or eEF1A (Tef-Off) as a resource for dissecting mechanisms of Gcn2 kinase activation by translation stress. Decades of research on this topic has led to a handful of non-mutually exclusive models. One of the earliest proposed mechanisms was that deacylated tRNAs, which increase in abundance upon amino acid starvation, are direct activators of Gcn2 kinase activity40. A drop in eEF1A levels in our depletion strains could indirectly activate Gcn2 by causing a build-up of uncharged tRNAs that are normally protected from spontaneous de-acylation by their interaction with excess eEF1A. More recently, another interesting model has emerged in which ribosome collisions enable assembly and activation of two upstream Gcn2 regulators (Gcn1 and Gcn20) on the collided disome41. A third model of Gcn2 activation invokes the latent capacity of unoccupied P-stalk proteins to directly stimulate Gcn242,43. It is attractive to speculate that a decrease in eEF1A levels leads to a lower occupancy of the P-stalk, exposing the P1/P2 stalk proteins to stimulate Gcn2 activity. Lastly, previous work has shown that eEF1A directly competes with uncharged tRNAs for binding to Gcn2 thereby blocking their ability to stimulate eIF2α phosphorylation in vitro44. Thus, a decrease in eEF1A levels could allow existing tRNAs to more effectively compete for Gcn2 binding.
Finally, our work has new implications for translational research on mammalian Zpr1. Specifically, Zpr1+/− heterozygous mice show signs of accelerated motor neuron loss with age45. Given that the mouse Zpr1 gene can complement yeast zpr1 deletes31, it is possible that defects in eEF1A biogenesis in Zpr1+/− mice put an increased burden on motor neuron proteostasis, either directly or via downstream effects on the translation apparatus. There are numerous mouse models of neurodegeneration caused by genetic defects in translation that lead to proteostasis disruption (e.g. by ribosome stalling)46. Most tellingly, tissue specific loss eEF1A has been shown to cause motor neuron degeneration in wasted (wst) mice47. Thus, future studies of Zpr1+/− mice should look for pleiotropic effects of disrupted eEF1A biogenesis as supporting evidence for our parsimonious etiology model.
Limitations of the Study
Our experiments suggest that eEF1A misfolding is the sole driver of proteotoxic stress in Zpr1-depleted cells, but they don’t rule out the possibility that Zpr1 has additional clients. The ColabFold model of the Zpr1-eEF1A complex presumptively represents the structure of eEF1A as a folding intermediate and accurately predicted the importance of the Zpr1C-half ZnF and aHH contributions to eEF1A folding, but this inherently static model is unable to speak to any structural dynamics in either Zpr1 or eEF1A that may represent crucial steps in the underlying chaperone mechanism. Specifically, the model fails to provide a structural explanation for the importance of N-halfZpr1 in eEF1A biogenesis, which is evident by the inability of Zpr1C-half to robustly complement inviability of zpr1 deletes. Future studies will need to further analyze the molecular details of N-halfZpr1 contributions to the eEF1A folding mechanism.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources or reagents should be directed to the lead contact, Vladimir Denic (vdenic@mcb.harvard.edu).
Materials Availability
The plasmids and yeast strains generated in this study may be requested from the lead contact.
Data and Code Availability
Raw RNA-sequencing data and ribosome profiling data were deposited in the GEO database and are publicly available under accession numbers GSE212387 and GSE212389. In addition, all data present in this study will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Yeast Strains
All strains used in this study are listed in the Key Resources Table. Cells were cultured on solid agar plates or in liquid culture at 3°C unless otherwise indicated.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIE R |
|---|---|---|
| Antibodies | ||
| rabbit-anti-eEF1A polyclonal | Kerafast Inc. | Cat#ED7001 |
| rabbit-anti-EIF2S1 phospho S51 [E90] recombinant (eIF2α phospho-Ser51) | Abcam | Cat#ab321 57 |
| mouse-anti-FLAG M2 monoclonal | MilliporeSigma | Cat#F3165 |
| mouse-anti-PGK1 [22C5D8] monoclonal | ThermoFisher Scientific | Cat#459250 |
| rabbit-anti-hexokinase polyclonal | US Biological Life Sciences | Cat#H2035–03 |
| goat-anti-rabbit IgG (H + L) HRP conjugate polyclonal | BioRad | Cat#17065 15 |
| goat-anti-rabbit IgG IRDye 680LT polyclonal | Cat#92668021 | |
| donkey-anti-mouse IgG IRDye 680RD polyclonal | LI-COR Biosciences | Cat#92668072 |
| donkey-anti-mouse IgG IRDye 800CW polyclonal | LI-COR Biosciences | Cat#92632212 |
| goat-anti-rabbit IgG IRDye 800CW polyclonal | LI-COR Biosciences | Cat#92632211 |
| LI-COR Biosciences | Cat#92632219 | |
| anti-GFP, N-terminal antibody produced in rabbit | MilliporeSigma | Cat#G1544 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| β-estradiol | Sigma-Aldrich | Cat#E8875; CAS: 50-28–2 |
| 5-Ph-IAA | MedChemExpress | Cat#HY134653; CAS: 168649–238 |
| 16% Formaldehyde (w/v), Methanol-free | Pierce™ | Cat#28908 |
| Cycloheximide | MilliporeSigma | Cat#01810;CAS: 6681–9 |
| Turbo™ DNAse (2 U/μL) | ThermoFisher Scientific | Cat#AM2238 |
| 3-Indoleacetic acid (auxin) | MilliporeSigma | Cat#I3750; CAS: 8751–4 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail tablets | Roche | Cat#04693 159001 |
| Micrococcal nuclease | NEB | Cat#M0247 S |
| EGTA (ethylene glycol bis(2-aminoethyl ether)-N,N,N’,N’-tetraacetic acid) 0.5 M in aqueous solution pH 8.0 | Thermo Scientific Chemicals | Cat#J60767 |
| rATP, 100 mM | Promega | Cat#E6011 |
| rGTP, 100mM | Promega | Cat#E6031 |
| Creatine phosphate | Roche | Cat#10621 714001; CAS: 71519–72-7 |
| Amino acid mixture, 1mM minus methionine | Promega | Cat#L9961 |
| Creatine kinase | Roche | Cat#10127 566001 |
| RiboGuard™ RNase Inhibitor | Lucigen (Biosearch Technologies) | Cat#RG90 925 |
| StabilizedEasyTag™ L-[35S]-Methionine | PerkinElmer | Cat#NEG7 09A |
| Harringtonine | Cat#15361; CAS: 26833–85-2 | |
| Sequencing Grade Modified Trypsin | Promega | Cat#V5113 |
| Tris(2-carboxyethyl)phosphine hydrochloride solution 0.5M pH 7.0 (TCEP) | MilliporeSigma | Cat#64654 7; CAS: 51805–45-9 |
| Dynabeads™ Protein G | Invitrogen | Cat#10004 D |
| 3X FLAG® Peptide | Sigma-Aldrich | Cat#F4799 |
| Isopropyl-β-D-thiogalactopyranoside (IPTG)- Dioxane free | US Biological Life Sciences | Cat#I8500; CAS: 36793–1 |
| Imidazole | Thermo Scientific Chemicals | Cat#A1022 1; CAS: 288–32-4 |
| Uranyl formate | Electron Microscopy Sciences | Cat#22450 |
| Zymolyase (R) 20T Arthrobacter luteus | amsbio | Cat#120491–1 |
| Ficoll® PM 400 | Sigma-Aldrich | Cat#F4375 |
| D-Sorbitol | Sigma-Aldrich | Cat#S6021; CAS: 50-70–4 |
| SUPERase-In™ | Thermo Fisher Scientific | Cat#AM2694 |
| Benzonase Nuclease | MilliporeSigma | Cat#E1014 −25KU |
| Phosphatase Inhibitor Cocktail 3 | Sigma-Aldrich | Cat#P0044 −1ML |
| Pepstatin A | Roche | Cat#11359053001; CAS:26305-03-3 |
| DEAE Cellulose resin | Biophoretics | Cat#B45059.02; CAS:901334-7 |
| CM Sepharose Fast Flow | Cytiva | Cat#17071 901 |
| Whatman filter paper | Cytiva | Cat#10011 10 |
| Critical Commercial Assays | ||
| Pierce™ BCA Protein Assay Kit | Thermo Scientific | Cat#23227 |
| mMESSAGE mMACHINE™ T7 Transcription Kit | Invitrogen | Cat#AM1344 |
| SuperSignal™ West Femto | Thermo Fisher Scientific | Cat#34095 |
| Deposited Data | ||
| Transcriptome profiling time course of ZPR1-depleted S.cerevisiae cells | GSE212387 | |
| Ribosome profiling of ZPR1-depleted S.cerevisiae cells | This paper | GSE212389 |
| Experimental Models: Organisms/Strains | ||
| W303 MATa leu2–3,112 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 | Solís et al., 2016 | W303 (VDY465) |
| BY4741 MATa his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 HO::ADH1pOsTIR1-URA3 | Schuller et al., 2017 | yCW30 |
| BY4741 MATa his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 HO::ADH1p-OsTIR1-URA3 SkHIS3-GAL1p-mAID-ZPR1 | This paper | yCW74 |
| W303 MATa leu2–3,112::pZ4EV-OsTIR1(F74G)_pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 | This paper | VDY5843 |
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 ZPR1-mAID-3ˣFLAG::kanMX | This paper | VDY5844 |
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 HIS3::prGAL1-ZPR1-mAID-3ˣFLAG::kanMX | This paper | VDY5912 |
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1::4xHSE-GFP::URA3 ade2–1 his3–11,15 ZPR1-mAID-3ˣFLAG::kanMX | This paper | VDY5892 |
| W303 MATa leu2–3,112::pZ4EV-OsTIR1(F74G)_pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1::4xHSE-GFP::URA3 ade2–1 his3–11,15 | This paper | VDY5893 |
| W303 MATa leu2–3,112 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 ZPR1–3ˣFLAG::kanMX | This paper | VDY5906 |
| W303 MATa leu2–3,112::pZ4EV-OsTIR1(F74G)_pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 HIS3::prGAL1-TEF1 TEF2-mAID-3ˣFLAG::kanMX | This paper | VDY6173 |
| W303 MATa leu2–3,112::pZ4EV-OsTIR1(F74G)_pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 gcn2Δ::TRP1 | This paper | VDY6174 |
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 | This paper | VDY5913 |
| HIS3::prGAL1-ZPR1-mAID-3ˣFLAG::kanMX gcn2Δ::TRP1 | ||
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 ZPR1-mAID-3ˣFLAG::kanMX TEF1-yeGFP::hphMX | This paper | VDY6175 |
| W303 MATa leu2–3,112::pZ4EV-OsTIR1(F74G)_pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 TEF1-yeGFP::hphMX | This paper | VDY6176 |
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 ZPR1-mAID-3ˣFLAG::kanMX HIS3::prGAL1-TEF1-yeGFP::hphMX | This paper | VDY6178 |
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 ZPR1-mAID-3ˣFLAG::kanMX TEF1-yeGFP::hphMX, HSP42-mCherry::HIS3 | This paper | VDY6179 |
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 ZPR1-mAID-3ˣFLAG::kanMX HSP42-mCherry::HIS3 | VDY6186 | |
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1::4xHSE-GFP::URA3 ade2–1 his3–11,15 ZPR1-mAID-3ˣFLAG::kanMX HIS3::prGAL1-TEF1 TRP1::prGAL1-TEF2 | This paper | VDY6188 |
| W303 MATa leu2–3,112::pZ4EV-OsTIR1(F74G)_pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1::4xHSE-GFP::URA3 ade2–1 his3–11,15 HIS3::prGAL1-TEF1 TEF2-mAID-3ˣFLAG::kanMX | This paper | VDY6189 |
| W303 MATa leu2-3,112 trp1-1 his3-11,15 ura3Δ0 zpr1Δ::kanMX + pVD2592 | This paper | VDY6194 |
| W303 MATa leu2–3,112::pZ4EV-OsTIR1(F74G)_pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1::4xHSE-GFP::URA3 ade2–1 his3–11,15 ssa2Δ::HIS3 | This paper | VDY6202 |
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1::prGAL1-FL eEF1A-GFP::URA3 ade2–1 his3–11,15 ZPR1-mAID-3ˣFLAG::kanMX | This paper | VDY6203 |
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1::prGAL1-DI-GFP::URA3 ade2–1 his3–11,15 ZPR1-mAID-3ˣFLAG::kanMX | This paper | VDY6209 |
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1::prGAL1-DI + DII-GFP::URA3 ade2–1 his3–11,15 ZPR1-mAID-3ˣFLAG::kanMX | This paper | VDY6210 |
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1::prGAL1-DII-GFP::URA3 ade2–1 his3–11,15 ZPR1-mAID-3ˣFLAG::kanMX | This paper | VDY6211 |
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1::prGAL1-DII + DIII-GFP::URA3 ade2–1 his3–11,15 ZPR1-mAID-3ˣFLAG::kanMX | This paper | VDY6212 |
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1::prGAL1-DIII-GFP::URA3 ade2–1 his3–11,15 ZPR1-mAID-3ˣFLAG::kanMX | This paper | VDY6213 |
| W303 MATa leu2–3,112::pZPR1-ZPR1–3xFLAG::LEU2 trp1–1 can1-100 ura3–1 ade2–1 his3–11,15 | This paper | VDY5907 |
| W303 MATa leu2–3,112::pZPR1-ZPR1 N-half -3xFLAG::LEU2 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 | This paper | VDY5915 |
| W303 MATa leu2–3,112::pZPR1-ZPR1 C-half -3xFLAG::LEU2 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 | This paper | VDY5916 |
| W303 MATa leu2–3, 112::pZ4EV-OsTIR(F74G) pACT1-Z4Ev-ATF_CgLEU2 trp1–1 can1–100 ura3–1::4xHSE-GFP::URA3 ade2–1 his3–11,15 ZPR1-mAID-3ˣFLAG::kanMX HIS3::prGAL1-TEF1 TRP1::prGAL1-TEF2 HSP42-mCherry::natMX | This paper | VDY6249 |
| Oligonucleotides | ||
| GGGGGTAATACGACTCACTATAGGGAGAatttcatacacaatataaacga ttgccaccATGGGTAAAGAGAAGTCTCAC | This paper | oVD13102 |
| TTTTTTTTTTTTTTTTTTTTttacatctacactgttgttatcagtcgggcTCA TTTCTTAGCAGCCTTTTGAGCAGCC | This paper | oVD13103 |
| TAAGATTTAAATATAAAAGATATGCAACTAGAAAAGTCTTATCAA TCTCC TAATACGACTCACTATAGGG | This paper | oVD12937 |
| ATGGGTAAAGAGAAGTCTCACATTAACG | oVD12932 | |
| CACAGAGATACATATTATACCTATACCGTTAAGAAATAGGATAG AAAATAagatctGTTTAGCTTGCCTC | This paper | oVD12090 |
| TATGTAAAGAGGAAGAGGAAAGGGGGAGGTGATACCGAGGCC AACGATCGTAATACGACTCACTATAGGG | This paper | oVD12091 |
| gataagcttgatatcgaattcctgcagcccCGGATTAGAAGCCGCCGAGCGG GTGACAGC | This paper | oVD13075 |
| cgcggtggcggccgctctagaactagtggatcccccACAAAATGCTTCACAACC CTGATG | This paper | oVD13074 |
| CGTTACAAGTATCCAAGCCTGAAACG | This paper | oVD13112 |
| TATAAATGTATGTATGTGTGTATAAACAGATACGATATTAATACG ACTCACTATAGGGAG | This paper | oVD13113 |
| Recombinant DNA | ||
| pET16b-10xHis-3C-ZPR1–3ˣFLAG | This paper | pVD2627 |
| pRS414:ZPR1 | This paper | pVD2592 |
| pRS414:ZPR1Zn-C-3ˣFLAG | This paper | pVD2639 |
| pRS414:ZPR1Zn-NC-3ˣFLAG | This paper | pVD2642 |
| pET16b-10xHis-3C-ZPR1Zn-C-3ˣFLAG | This paper | pVD2650 |
| pET16b-10xHis-3C-ZPR1Zn-NC-3ˣFLAG | This paper | pVD2653 |
| pRS424:ZPR1 | This paper | pVD2323 |
| pRS424:N-halfZPR1 | This paper | pVD2324 |
| pRS424:ZPR1C-half | This paper | pVD2325 |
| pRS406:prGAL1-DI-GFP | This paper | pVD2659 |
| pRS406:prGAL1-DI + DII-GFP | This paper | pVD2660 |
| pRS406:prGAL1-DII-GFP | This paper | pVD2661 |
| pRS406:prGAL1-DII + DIII-GFP | This paper | pVD2662 |
| pRS406:prGAL1-DIII-GFP | This paper | pVD2663 |
| pRS414:ZPR1WT-3ˣFLAG | This paper | pVD2680 |
| pRS414:ZPR1Zn-N-3ˣFLAG | This paper | pVD2750 |
| pRS414:ZPR1aH-N-3ˣFLAG | This paper | pVD2753 |
| pRS414:ZPR1aH-C-3ˣFLAG | This paper | pvD2751 |
| pRS414:ZPR1aH-NC-3ˣFLAG | This paper | pvD2755 |
| pET16b-10xHis-3C-ZPR1Zn-N-3ˣFLAG | This paper | pvD2758 |
| pET16b-10xHis-3C-ZPR1aH-C-3ˣFLAG | This paper | pvD2759 |
| pET16b-10xHis-3C-ZPR1aH-NC-3ˣFLAG | This paper | pvD2763 |
| Software and Algorithms | ||
| FastQC | https://github.com/s-andrews/FastQC | Version 0.11.5 |
| STAR | Dobin et al., 2013 | Version 2.7.0 |
| DESeq2 | Love et al., 2014 | Version 1.34.0 |
| seqtk | https://github.com/lh3/seqtk | Version 1.3-r106 |
| cutadapt | https://github.com/marcelm/cutadapt | Version 2.0 |
| deepTools | https://github.com/deeptools/deepTools | Version 3.3.0 |
| DEGreport | https://github.com/lpantano/DEGreport | Version 1.30.0 |
| BioConductor: flowCore | DOI: 10.18129/B9.bioc.flowCore | Bioconductor version: Release (3.15) |
| BioConductor: flowViz | DOI: 10.18129/B9.bioc.flowViz | Bioconductor version: Release (3.15) |
METHOD DETAILS
Plasmids
All plasmids used in this study are listed in the Key Resources Table. Sequences cloned into pRS plasmids were inserted into the multiple cloning site (MCS) using Gibson assembly. For 5-FOA shuffles and yeast genome plasmid integrations, plasmids were derived from the pRS400 series of vectors described in ref48.
Antibodies
All antibodies used in this study are listed in the Key Resources Table. For immunoblotting, primary antibodies were used at the indicated dilution and secondary antibodies were used 1:3,000 (HRP) or 1:10,000 (fluorescent). Primary antibodies include the following: rabbit-anti-eEF1A ([1:10,000 - IB], ED7001, Kerafast Inc., Boston, MA), rabbit-anti-EIF2S1 phospho S51 (eIF2α phospho Ser51) ([1:1000 - IB], ab32157, Abcam), mouse-anti-FLAG M2 ([1:2000–1:5000 - IB], F3165, MilliporeSigma, Burlington, MA), mouse-anti-PGK1 ([1:2000 - IB], 459250, Thermo Fisher Scientific, Waltham, MA), rabbit-anti-Hexokinase ([1:2000 - IB], U.S. Biological Life Sciences, Swampscott, MA), rabbit-anti-GFP ([1:1000 - IB], G1544–100UG, MilliporeSigma, Burlington, MA).
Immunoblotting
Samples were separated via SDS-PAGE and transferred to nitrocellulose before blocking with 5% skim milk in TBST (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% Tween 20, 0.25 mM EDTA) for 60 minutes. Membranes were probed with primary antibodies overnight at 4°C with antibody dilutions indicated in the Antibodies section. After three rinses in TBST, membranes were probed with secondary antibodies at dilutions indicated in the Antibodies section. HRP-conjugated antibodies were visualized using Super Signal West Femto (Thermo Fisher Scientific). Fluorescent secondaries were visualized on a LI-COR Odyssey scanner. Quantification of band intensity was done using ImageJ.
Autoradiography
Samples were separated via SDS-PAGE. Gels were fixed for 30 minutes in fixing solution (50% methanol and 10% acetic acid) and then rinsed for 30 minutes in drying solution (30% methanol and 5% glycerol) before drying under vacuum for 2 hours at 80°C. Phosphor screens (GE Healthcare) were exposed to dried gels overnight and then scanned on a Sapphire Biomolecular Imager (Azure Biosciences). Quantification of band intensity was done using ImageJ.
Microscopy
Hsp42-mCherry/Tef1-GFP aggregation assay
VDY6175, VDY6176, VDY6179, VDY6181, and VDY6186 cells were grown to saturation overnight in synthetic complete (SC) media containing 2% glucose. Cells were back-diluted to an OD600 of 0.1 in SC + 2% glucose and 1 μM beta-estradiol and grown for 3–4 hours to reach an OD600 ~0.5 using an Eppendorf Thermomixer R set at 30°C and mixing at 1400 RPM. Cells were subsequently back-diluted to an OD600 of 0.1 in SC + 2% glucose + 1 μM beta-estradiol and 5-Ph-IAA (or equivalent volume of DMSO, the 5-Ph-IAA solvent) was added to a final concentration of 5 μM. Cells were then grown for 1–3 hours using a thermomixer before being immediately fixed in ice-cold 4% paraformaldehyde (PFA) in PBS pH 7.4 for 15 minutes. Fixed cells were imaged with a Nikon TI inverted microscope using a 100x oil-immersion objective (1.45 NA), a Yokogawa dual spinning disk confocal unit and a Hamamatsu ImagEM EM-CCD camera. Images were acquired using MetaMorph software. A 488 nm laser was used for imaging GFP and a 594 nm laser for mCherry fluorescent protein. 0.25-micron slices with a total depth of 8 microns were used for Z stack acquisition. Maximum intensity z-projections were used to generate the final images from collected Z stacks.
Newly synthesized Tef1-GFP aggregates
VDY6178 cells were grown to saturation overnight in SC + 2% raffinose. Cells were back-diluted to an OD600 of 0.1 in SC + 2% galactose + 1 μM beta-estradiol and grown for 3 hours. 0.1 OD600 units were spun down and resuspended in either SC + 2% galactose or SC + 2% glucose (for transcriptional shutoff of the TEF1 locus) and grown for 1 hour in a thermomixer set at 30°C and mixing at 1400 RPM. Samples were then split 1:1 with respective media and 5-Ph-IAA or DMSO was added to a final concentration of 5 μM. Cells were subsequently grown for 1 hour prior to fixation with ice-cold 4% PFA for 15 minutes. Images were acquired and analyzed as stated above.
eEF1A truncation constructs
VDY5844 cells expressing GFP-tagged constructs of full-length eEF1A/Tef1 (VDY6203), DI (VDY6209), DI + DII (VDY6210), DII (VDY6211), DII + DIII (VDY6212), and DIII (VDY6213) were grown to saturation in SC + 2% raffinose. Cells were subsequently back-diluted to an OD600 0.1 in SC + 2% raffinose + 1 μM beta-estradiol and grown for 4 hours and then split 1:1 with fresh media and induced by adding 20% galactose to a final concentration of 2% galactose (w/v). Cells were grown in inducing media concurrent with the addition of DMSO or 5 fixation with ice-cold 4% PFA. Samples were imaged as described above.
Flow cytometry
For all flow cytometry experiments, we used the FACSymphony A3 analyzer (BD Biosciences) and measured ≥10,000 cells for each sample.
4xHSE-GFP reporter of Hsf1 activity
VDY5892, VDY5893, VDY6189, and VDY6202 were grown to saturation overnight in SC + 2% glucose or 2% galactose for VDY6189. Cells were next back-diluted to an OD600 of 0.1 in their respective medium containing 1 μM beta-estradiol and grown for 3–4 hours until an OD600 ~0.5 was reached. VDY5892, VDY5893, and VDY6202 cells were back-diluted to an OD600 of 0.1 and either DMSO or 5-Ph-IAA was added to VDY5892 and VDY5893 cells to a final concentration of 5 μM. 0.1 OD600 units of VDY6189 cells were spun down and resuspended in either SC + 2% galactose or SC + 2% glucose media (for transcriptional shutoff of the TEF1 locus) containing 1 μM beta-estradiol. DMSO or 5-Ph-IAA was added to a final concentration of 5 μM. 1 mL of cells of each strain were grown in a thermomixer and at the indicated time points, 200 μL of cells were aliquoted into a second tube and cycloheximide was added to a final concentration of 50 μg/mL to arrest translation. Aliquoted samples were grown for 1 hour at 30°C for fluorophore maturation and subsequently measured using a FACSymphony A3 analyzer (BD Biosciences) using the 488 nm laser (FITC). 10,000 cells were measured for each sample and medians/histograms were collected using Bioconductor packages flowCore and flowViz.
Zpr1 mutant Hsf1 activity
VDY 5844 (Zpr1-AID cells containing the 4xHSE-GFP reporter) were transformed with low-copy, TRP1-based vectors, either empty (EV) or carrying 3ˣFLAG-tagged wild-type (WT) Zpr1 or 3ˣFLAG-tagged Zpr1 mutants (Zpr1Zn-N: I76A, I77A, I78A, M79A; Zpr1Zn-C: V317A, I318A, I319A, M320A; Zpr1Zn-NC: I76A, I77A, I78A, M79A, V317A, I318A, I319A, M320A; Zpr1aH-C: E391A, E399A; Zpr1aH-NC: E148A, D156A, E391A, E399A). Cells were grown to saturation in SD-Trp media. Cells were next back-diluted into SD-Trp media containing 1 μM beta-estradiol and grown for ~2–3 hours until an OD600 ~0.3–0.4 was reached. Cells were diluted in half into SD-Trp media supplemented with 1 μM beta-estradiol and DMSO or 5 μM 5-Ph-IAA and grown for 2 hours. Cycloheximide was added to a final concentration of 50 μg/mL for 1 hour to arrest translation. Samples were measured using a FACSymphony A3 analyzer (BD Biosciences) using the 488 nm laser (FITC). 20,000 cells were measured for each sample and medians/histograms were collected using Bioconductor packages flowCore and flowViz.
Tef-Off Hsf1 activity
VDY6188 was grown overnight to saturation in SC + 2% galactose media. Cells were back-diluted to an OD600 of 0.1 and grown for 4 hours (in the presence of 1 μM beta-estradiol) until an OD600 ~0.5 was reached. 0.1 OD600 units were spun down and resuspended in either SC + 2% galactose or SC + 2% glucose (for transcriptional shutoff of the TEF1/2 loci) and grown for 1 hour in a thermomixer set at 30°C and mixing at 1400 RPM. After 1 hour, cells from each sample (in either galactose-containing media or glucose-containing media) were split into three different tubes and fresh media was added to reach a final volume of 1 mL. Each tube either received DMSO, 5-Ph-IAA (5 μM final concentration), or was grown in a thermomixer set at 39°C (to induce heat shock) with shaking (1400 RPM). Samples with DMSO or 5-Ph-IAA were grown at 30°C in a thermomixer shaking at 1400 RPM. At the indicated time points, 200 μL of cells were aliquoted into a second tube and cycloheximide was added to a final concentration of 50 μg/mL to arrest translation. Aliquoted samples were grown for 1 hour at 30°C for fluorophore maturation and subsequently measured using a FACSymphony A3 analyzer (BD Biosciences) using the 488 nm laser (FITC). 10,000 cells were measured for each sample and medians/histograms were collected using Bioconductor packages flowCore and flowViz.
Tef1-GFP levels
VDY6175 and VDY6176 cells were grown to saturation in SC + 2% glucose media. Cells were subsequently back-diluted to an OD600 0.1 in SC + 2% glucose + 1 μM beta-estradiol and grown for 3–4 hours until an OD600 0.5 was reached. Cells were back-diluted once more to an OD600 of 0.1 and 5-Ph-IAA or DMSO was added to a final concentration of 5 μM. At the indicated time points, translation was arrested with cycloheximide (50 μg/mL) and samples were immediately measured using a FACSymphony A3 analyzer (BD Biosciences) using the 488 nm laser (FITC). 10,000 cells were measured for each sample and medians/histograms were collected using Bioconductor packages flowCore and flowViz.
Growth Assays
Spot assays
Cells were grown to mid-log phase and back-diluted to an OD600 of 0.4 (undiluted, first spot) before serial dilutions (1:10) and spotting on indicated plates. Images were taken after 48 hours of growth (Figure 1B and S6D) or 72 hours of growth (S4A).
Growth curves
Cells were grown to mid-log phase before back-dilution to an OD600 of 0.01 in 200 μL of SC + 2% glucose (and 5-Ph-IAA where indicated or SC + 2% galactose (and DMSO where indicated) in 96-well plates. Cells were grown for two days shaking at 30°C with OD600 measurements taken every 10 minutes. Experiments were done in triplicate and the mean of these samples was used to produce the final plot.
Polysome Profiling
VDY5912 and VDY6173 were grown logarithmically in YPG + 1 μM beta-estradiol and shifted to YPD with 5 μM 5-Ph-IAA for the indicated times. Cells were back-diluted at the time of YPD and 5-Ph-IAA addition to reach OD600 of 0.4–0.6 at collection. Cells were collected by rapid vacuum filtration and snap freezing in liquid nitrogen. Frozen cell paste was mixed with frozen polysome lysis buffer (20 mM Tris pH 8.0, 140 mM KCl, 5 mM MgCl2, 100 μg/mL cycloheximide, 1 mM DTT, 1% Triton X-100, 0.025 U/μL Turbo DNAse [AM2238, Thermo Fisher Scientific]) at a ratio of 1 mL lysis buffer per 500 OD600 units. Frozen cells and buffer were cryogenically lysed using a Retsch MM 400 mixer mill and kept frozen at −80°C. Frozen cell lysate powder was thawed for 2 minutes at 30°C with intermittent flicking and placed on ice. Lysate was clarified by centrifugation for 10 minutes at 16,000 x g and 4°C. The RNA concentration of diluted supernatant was measured using a NanoDrop 1000 (Thermo Fisher Scientific) to normalize across samples. 200 μL of supernatant totaling 1 mg RNA was run on linear 10–50% sucrose gradients in a Beckmann Coulter SW-41 rotor for 3 hours at 30,000 RPM and 4°C. Gradients were fractionated using a BioComp Gradient Station ip and absorbance at 260 nm was measured during fractionation. The area under the A260 curve was estimated using R. The monosome region was defined by identifying the local minimum on either side of the 80S monosome peak. The polysome region was defined as everything from the local minimum to the right of the monosome peak until the bottom of the gradient. The “E3 only” control in Figure 3D is the same as in Figure 3C but with genotypes of different loci emphasized for clarity.
Preparation of Ribosome Profiling Libraries
yCW3049 and yCW74 were grown in YPGR (2% galactose and 2% raffinose) overnight at 30°C, harvested by centrifugation, washed, and then resuspended in 1L YPD containing 0.5 mM auxin (3-indoleacetic acid, MilliporeSigma). Note, the AID system in these strains is distinct from all others used in this study and relies on a constitutively-expressed E3 ligase without the F74G mutation. Cells were grown in YPD + auxin for ~8 hours, harvested at OD600 ~0.15, and flash frozen in liquid nitrogen. Isolation of ribosome-protected footprints and generation of libraries for Illumina sequencing is described in detail elsewhere50.
Zpr1–3ˣFLAG immunoprecipitation and mass spectrometry
ZPR1–3ˣFLAG growth and lysis
Both endogenous ZPR1 (VDY465), ZPR1–3ˣFLAG (VDY5906), and ectopic ZPR1–3ˣFLAG (VDY5907), N-halfZPR1–3ˣFLAG (VDY5915), ZPR1C-half-3ˣFLAG (VDY5916) introduced at the LEU2 locus were grown in large cultures to OD 1.8 in liquid YPD media. 3L of cells were spun down at 3000 x g for 15 minutes at 4°C, washed in 50 mL ddH2O twice and resuspended in 1 mL per gram of pellet of resuspension buffer (1.2% PVP-40, 20 mM HEPES pH 7.4, 1x protease inhibitor cocktail [Roche], 1% solution P [2 mg Pepstatin A, 90 mg PMSF, 5 mL 100% ethanol], 1 mM DTT). Cells were spun down twice at 3000 x g for 15 minutes to remove all buffer. Cell paste was placed into a syringe and pushed into a 50 mL conical tube filled with liquid nitrogen to snap freeze as “noodles”. Liquid nitrogen was decanted from the tube and frozen “noodles” were stored at −80°C until being cryogenically lysed using a Retsch PM100 ball mill.
ZPR1–3ˣFLAG immunoprecipitations
Approximately 250 mg of grindate was thawed in 1 mL of HIP buffer (20 mM HEPES-KOH pH 7.4, 110 mM KOAc, 2 mM MgCl2, 0.5% Triton X-100, 0.1% Tween 20, 1 mM DTT, 4 U/mL SUPERase-In [ThermoFisher], 1x protease inhibitor cocktail [Roche], 1% Solution P [2 mg Pepstatin A, 90 mg PMSF, 5 mL 100% ethanol]), vortexed briefly, and put on ice. Lysate was passed through a Whatman 25 mm GD/X Disposable filter [Cat No. 6888–2527]. Equal volume of lysates was added to 62.5 μL Protein G dynabeads pre-bound to 3.75 μL of anti-FLAG M2 antibody and agitated for 30 minutes at 4°C. Beads were washed 3x in HIP buffer. After the final wash, protein was eluted from beads with two pooled incubations of 30 μL 1 mg/mL 3ˣFLAG peptide in HIP buffer at room temperature for 15 min.
Mass Spectrometry Sample Preparation
Sample preparation for mass spectrometry was performed by the Harvard Center for Mass Spectrometry. All samples were loaded onto a 10 kDa filter (Pall, TX) and were washed 3 times with 150 μL distilled water. Next, samples were reduced and alkylated, followed by centrifugation through the filter at 1000 RPM for 2 minutes after each step to remove chemicals. Finally, remaining proteins were dissolved on the top of the filter in 100uL 50 mM TEAB (triethylammonium bicarbonate), followed by trypsin (Promega, WA) digests for 3 hours at 38°C. Digested samples were spun through the filter and peptide samples were dried under vacuum before adjusting to 20 μL and analyzing 10 μL via mass spectrometry.
Mass Spectrometry Analysis
LC-MS/MS was performed on a Lumos Tribrid Orbitrap Mass Spectrometer (ThermoFisher, San Jose, CA) equipped with Ultimate 3000 (Thermo-Fisher, CA) nano-HPLC. Peptides were separated onto a 150 μm inner diameter microcapillary trapping column packed with approximately 2cm of C18 Reprosil resin (5 μm, 100 Å, Dr. Maisch GmbH, Germany) followed by a PharmaFluidics (Gent, Belgium) 50 cm analytical column. Separation was achieved through applying a gradient from 5– 27% acetonitrile in 0.1% formic acid over 90 min at 200 nL min−1. Electrospray ionization was enabled by applying a voltage of 2 kV using a home-made electrode junction at the end of the microcapillary column and sprayed from metal tips (PepSep, Denmark). The mass spectrometry survey scan was performed in the Orbitrap in the range of 400–1,800 m/z at a resolution of 6 × 104, followed by the selection of the twenty most intense ions (TOP20) for CID-MS2 fragmentation in the Ion trap using a precursor isolation width window of 2 m/z, AGC setting of 10,000, and a maximum ion accumulation of 100 ms. Singly charged ion species were not subjected to CID fragmentation. Normalized collision energy was set to 35 V and an activation time of 10 ms. Ions in a 10 ppm m/z window around ions selected for MS2 were excluded from further selection for fragmentation for 60 seconds.
Zpr1 depletion RNA-seq time course
Cells grown to mid-log phase in 1 μM beta-estradiol were treated with 5 μM 5-Ph-IAA for the indicated times. Cells were rapidly harvested by centrifugation at 20,000 x g and snap-frozen in liquid nitrogen after removing supernatant. Frozen cell pellets were sent to Azenta Life Sciences (Chelmsford, MA, USA) for isolation of total RNA, polyA enrichment, and sample QC before preparation of Illumina sequencing libraries for pair-end sequencing using an Illumina HiSeq. RNA-seq of samples was performed in triplicate. Each point in Figure 3A denotes a gene, where the gene abundance Z-score is obtained by gene-wise mean centering and scaling to the standard deviation across all timepoints using degPatterns.
Yeast in vitro transcription and translation
Generation of capped mRNA
Capped mRNAs were in vitro transcribed from the appropriate PCR products using the mMESSAGE mMACHINE T7 kit (Invitrogen) as described previously51. PCR products encoding TEF2 were generated from pVD2609 using primers oVD13102 and oVD13103.
Preparation of cell-free translation extracts
Yeast in vitro translation (IVT) extracts were prepared as follows. Briefly, 1.5 L YPD media was inoculated with the indicated strains (VDY465 [WT], VDY5844 [ZPR1-AID], or VDY5906 [ZPR1–3ˣFLAG) at OD600 0.1 and grown to OD600 1.8–2.0. In experiments with Zpr1-depleted extracts, VDY5906 was treated with 5 μM 5-Ph-IAA for 1 hour immediately before collection. Cells were collected via centrifugation at 3,000 RPM in a JS-4.2 rotor for 15 minutes at 4°C. Cells were rinsed once in cold DEPC-treated water and then once in cold Buffer A (30 mM HEPES pH 7.4, 100 mM KoAc, 2 mM Mg(OAc)2) plus 2 mM DTT. Cells were mixed with 1 mL Buffer A with 2 mM DTT, 14 % glycerol (Buffer ADG), and 2x cOmplete protease inhibitor cocktail (PIC) tablet (Roche, Basel, Switzerland) per 6 grams of cell weight. Cell paste was then frozen in liquid nitrogen drop by drop and lysed in a Retsch PM100 ball mill and stored at −80°C. Powder was thawed at 4°C followed by addition of 2 mL Buffer ADG + PIC. Thawed lysate was then centrifuged at 13,336 x g for 10 minutes at 4°C. Supernatant was centrifuged at 49,000 RPM in a SW-55 rotor for 30 minutes at 4°C and the clear supernatant was taken while avoiding the upper lipid layer. The collected supernatant was run over five sequential Hi-Trap desalting columns (GE Healthcare, Chicago, IL, USA) on an AKTA Pure FPLC system. Fractions with Abs260 > 50 were pooled and snap frozen in liquid nitrogen. Before translation reactions, all translation extracts were treated with 2.4 μL 40 mM CaCl2 and 1 μL micrococcal nuclease (NEB) per 200 μL extract for 10 minutes at room temperature followed by treatment with 3.6 μL 100 mM EGTA for 5 minutes.
eEF1A Trypsin Protection Time Course
Reactions for yeast IVT trypsin protection assay time courses contained 7.5 μL of nuclease-treated extracts, 2 μL Buffer ADG, 2.5 μL of 6x translation buffer (132 mM HEPES-KOH, pH 7.4, 720 mM KOAc, 9 mM Mg(OAc)2, 4.5 mM ATP, 0.6 mM GTP, 150 mM creatine phosphate [Roche], 0.24 mM of each amino acid but lacking methionine [Promega, Madison, WI], 10.2 mM DTT), 0.5 μL creatine kinase [20 mg/mL in 50% glycerol, Roche], 0.5 μL RiboGuard [Lucigen, Middleton, WI, USA], 1 μL 35S-Methionine [to final 0.66 μCi/μL]; PerkinElmer, Waltham, MA, USA], and 1 μL of 200 ng/μL capped mRNA. Reactions were incubated for 45 minutes at room temperature and then 1.66 μL of 500 μM Harringtonine (Cayman Chemicals, Ann Arbor, MI, USA) in Buffer ADG was added and incubated for 15 minutes to allow run-off of all translating ribosomes. Cycloheximide was added to 100 μg/mL and an aliquot was taken for “Before Trypsin” gel samples. Trypsin (Promega) was added to 50 μg/mL and incubated for the indicated times before quenching with an equal volume of 70°C 2x SDS/Urea sample buffer (40 mM Tris-HCl pH 6.8, 8 M Urea, 5% SDS, 0.1 mM EDTA, 1% Beta-mercaptoethanol, 0.1 mg/mL bromophenol blue, 20% glycerol) with 2x EDTA-free protease inhibitor cocktail (Roche). Samples were analyzed via SDS-PAGE, autoradiography, and immunoblotting.
Reaction recipes for trypsin protection assays with recombinant Zpr1–3ˣFLAG proteins in yeast IVT extracts were the same as in time courses with the following modifications: 2 μL Buffer ADG was replaced with either 2 μL of 7.5x Zpr1–3ˣFLAG purified protein (to final concentration indicated in figures) or 2 μL Zpr1–3ˣFLAG buffer (50 mM Tris-HCl pH 8.0, 200 mM NaCl, 10% glycerol, 0.5 mM TCEP). All reactions were treated with trypsin for five minutes before quenching with an equal volume of 70°C 2x SDS/Urea sample buffer and analyzing via SDS-PAGE, autoradiography, and immunoblotting.
Zpr1–3ˣFLAG immunodepletion
Reaction recipes for Zpr1–3ˣFLAG immunodepletion from yeast IVT reactions were the same as for eEF1A trypsin protection time courses. After 45 minutes of translation at room temperature cycloheximide was added to 100 μg/mL and reactions placed on ice. Reactions were added to pre-chilled tubes containing 20 μL protein G Dynabeads pre-conjugated to 1 μL anti-FLAG (M2) antibody per time point. Samples were agitated for 20 minutes at 4°C, and the unbound material was moved to a new tube and treated with 50μg/mL trypsin. Aliquots were removed at the indicated times and quenched via equal volume of 2x SDS/Urea sample buffer pre-heated to 70°C, and then samples were then analyzed via SDS-PAGE, autoradiography, and immunoblotting.
Recombinant Protein Expression and Purification
Zpr1 Protein Expression
Zpr1–3ˣFLAG constructs were cloned into pET16b vectors with N-terminal 10xHis-3c tag and a 3ˣFLAG C-terminal tag. Rosetta (DE3) cells were transformed with these vectors and grown in Terrific Broth (TB) media until OD 0.4. The cultures were then cooled down at 4°C for 1 hour and supplemented with 1 mM IPTG (US Biological Life Sciences) to induce protein expression at 16°C for 16 hours. Cells were harvested at 3500 x g for 20min and resuspended in PBS. After a second spin, cell pellets were frozen in liquid nitrogen and stored at −80 until purification.
Zpr1 Protein Purification
Cell pellets were thawed at 4°C, resuspended in B1 Lysis Buffer (20mM sodium phosphate pH 8.0, 300mM NaCl, 20mM imidazole) supplemented with benzonase (Millipore Sigma), 5 mM Beta-mercaptoethanol, Protease inhibitor cocktail and 0.2 mM PMSF. Cells were lysed with 5 passes on an emulsiflex cell disruptor at ~12–15k psi. Lysed cells were spun down at 28,000 x g for 30 min at 4°C in a JLA 16.25 rotor. Supernatant was loaded on an equilibrated 5 mL HisTrap HP column. The column was washed with 10 column volumes of B1 buffer before eluting with a linear gradient of B2 Elution Buffer (20mM sodium phosphate pH 8.0, 300 mM NaCl, 500 mM imidazole, 5 mM Beta-mercaptoethanol) over 5 column volumes. Peak elution fractions were pooled and concentrated to 2.5 mL with a 10 kDa cutoff filter before desalting with a PD10 desalting column into B3 Low-salt Anion Exchange Buffer (50 mM Tris pH 8.0, 50 mM NaCl, 5 mM Beta-mercaptoethanol). Desalted samples were applied to an equilibrated 5 mL HiTrap Q FF column, washed with 5 column volumes of B3 buffer, and eluted with a linear gradient of B4 High-salt Anion Exchange Buffer (50 mM Tris pH 8.0, 1 M NaCl, 5 mM Beta-mercaptoethanol) over 20 column volumes. Peak fractions were pooled and concentrated to <500 μL and applied to an equilibrated Superdex 200 10/300 column for gel filtration in B5 SEC buffer (50mM Tris pH 8.0, 200mM NaCl, 10% glycerol, 0.5mM TCEP). Peak fractions were aliquoted, frozen in liquid nitrogen and stored at −80°C.
Purification of native eEF1A from yeast
Growth and lysis
VDY465 (WT) cells were grown in large cultures to OD 1.8 in liquid YPD media. 3 L of cells were spun down at 3500 x g for 20 minutes at 4°C, washed in 50 mL ddH2O twice and resuspended in 1 mL per gram of pellet of resuspension buffer (1.2% PVP-40, 20 mM HEPES pH 7.4, 1x protease inhibitor cocktail [Roche], 1% solution P [2 mg Pepstatin A, 90 mg PMSF, 5 mL 100% ethanol], 1 mM DTT). Cells were spun down twice at 3500 x g for 10 minutes to remove all buffer. Cell paste was placed into a syringe and was frozen as “noodles” in a 50 mL conical tube filled with liquid nitrogen. Liquid nitrogen was decanted from the tube and frozen “noodles” were stored at −80°C until cryogenically lysed using a Retsch PM100 ball mill.
Purification
8 g of frozen grindate was thawed in 80 mL I-100 buffer supplemented with 1x protease inhibitor cocktail [Roche] (20 mM Tris pH 7.5, 0.1 mM EDTA, 100 mM KCl, 25% glycerol, 1 mM DTT). Lysate was spun at 8,000 x g for 5 min at 4°C in a JA 25.5 rotor. Supernatant was transferred into new tubes and spun at 20,000 x g for 15 min at 4°C in JA 25.5 rotor. Supernatant was then spun at 54,400 RPM for 106 min at 4°C in TI 70 ultracentrifuge rotor. Supernatant was incubated with 10 g pre-equilibrated DEAE resin (in buffer I-100) for 1 hour while agitated on a tilt table at 4°C. Unbound fraction was removed by centrifugation. The resin was washed with an additional 20 mL I-100 buffer, spun down and the wash was added to the unbound fraction. The unbound fraction and wash was incubated with 60 mL pre-equilibrated CM-sepharose slurry (in buffer I-100) for 1 hour while agitated on a tilt table at 4°C. A buchner funnel with Whatman type 1 paper (~11 micron pore size) was used to separate the unbound fraction. The resin was washed with 40 mL I-100 before removing from the buchner funnel, resuspending in 60 mL I-100 with solid KCl supplemented to 500 mM final concentration of KCl, and incubating for 1 hour while agitated on a tilt table at 4°C. Eluate was collected using the buchner funnel, and resin was washed with an additional 20 mL buffer I-100 supplemented with solid KCl for a final concentration of 500mM KCl, which was combined with the eluate. Eluate was concentrated with a 30 kDa cutoff concentrator to ~5 mL, filtered with a 0.22 micron filter and desalted into I-50 buffer (20 mM Tris pH 7.5, 0.1 mM EDTA, 50 mM KCl, 25% glycerol, 1 mM DTT) using 4 × 5 mL sequential HiTrap desalting columns. ~8 mL of flow-through was collected and applied onto a Source 15S 4.6/100 column. The column was washed with 40 CVs of I-50 buffer before eluting with a linear gradient of I-300 buffer (20 mM Tris pH 7.5, 0.1 mM EDTA, 300 mM KCl, 25% glycerol, 1 mM DTT) over 20 CVs. Peak fractions containing eEF1A were pooled and concentrated to ~2 mL with a 30 kDa cutoff concentrator. Sample was applied onto a HiPrep 16/60 Sephacryl S-100 column. Native eEF1A eluted as the second peak. Fractions were pooled and concentrated with a 30 kDa cutoff concentrator. Aliquots were frozen in liquid nitrogen and stored at −80°C.
Native eEF1A trypsin protection assay
eEF1A was diluted to 1.33 μM into Buffer ADG (~14% glycerol) for 5 min at room temperature. Aliquots were added to tubes containing the indicated components for a final concentration of 1.11 μM eEF1A, 1.11 mM GTP/GDP (if present), and 1.11x the indicated concentration of Zpr1–3xFLAG recombinant protein (if present) and incubated for 30 min at room temperature. Trypsin was added to a final concentration of 50 μg/mL and so the final concentration of components would be 1 μM eEF1A, 1 mM GTP/GDP (if present), and 1x the indicated concentration of Zpr1–3xFLAG recombinant protein (if present) for 5 min at room temperature. Reactions were stopped by diluting in 2x SDS/Urea sample buffer and analyzed via SDS-PAGE and immunoblotting.
Zpr1 and Tef1-depletion time course
VDY 5843 (E3-only), VDY 5912 (pGAL1-ZPR1-AID), VDY 6173 (pGAL1-Tef1, Tef2-AID), and VDY 5913 (pGAL1-ZPR1-AID, Δgcn2) were grown to saturation in SGal-Met liquid media. Cells were back-diluted in SGal-Met liquid media with 1 μM beta-estradiol overnight until mid-log phase. Cells were then back-diluted into SGal-Met liquid media with 1 μM beta-estradiol, 2% glucose and 5 μM 5-Ph-IAA. Cells were kept at log phase OD by periodic dilution throughout the time course. 1 OD600 of mid-log-phase cells were harvested at each time point and resuspended in 200 μL media. 20 μCi of 35S-methionine (Perkin Elmer EasyTag) was added and cells were incubated at 30°C for 10 min. 1 μL of 50 mg/mL cycloheximide (Sigma) was added to stop translation. Proteins were precipitated by the addition of TCA to 10% and incubated overnight at 4°C. Pellets were washed twice in acetone and dried before resuspending in 100 μL urea-SDS buffer (6 M urea, 2% SDS, 50mM Tris pH 7.5, 1 mM EDTA) with phosphatase inhibitor cocktail 3 (sigma). Cells were lysed by bead beating and then heated to 65°C for 10 min before spinning at max speed in a tabletop centrifuge. Total protein was estimated with a BCA kit (23227, ThermoFisher Scientific) and samples were adjusted to an equal amount of total protein before adding SDS sample buffer (62.5 mM Tris pH 6.8, 2% glycerol, 1% w/v SDS, 2.5% v/v Beta-mercaptoethanol). Proteins were separated by SDS-PAGE and analyzed by immunoblotting with the indicated antibodies.
Negative stain electron microscopy of Zpr1–3ˣFLAG IPs
Samples were stained using a 1.5% (w/v) uranyl formate solution and imaged on a Philips CM10 electron microscope, operated at 100 kV and at 46,000x magnification. The microscope is equipped with a Gatan UltraScan 894 (2k x 2k) CCD camera. 2D processing was conducted using Simplified Application Managing Utilities of EM Labs (SAMUEL) software.
Pulse-chase analysis of eEF1A stability
Cells were grown to mid-log phase in liquid SD-Met media with 1μM beta-estradiol and resuspended at 5 OD600 in fresh media containing additional 5μM 5-Ph-IAA. Following further growth at 30°C for 30 minutes with agitation, cells were pulse labeled with 20μCi/(original)OD600 of 35S-methionine mix for an additional 10 minutes. Labeling was terminated by addition of 40 μL/mL of chase mix (125mM methionine, 25mM cysteine, 5% yeast extract). 2.5 (original)OD600 were removed at each chase timepoint and immediately precipitated with TCA (20% w/v) on ice. Denatured cell material was collected by centrifugation and washed twice with −20°C 100% acetone and briefly air dried. Lysates were prepared by resuspending dried samples in 100 μL of 6M urea, 1% SDS, 50mM Tris pH 7.5, and 1mM EDTA followed by glass bead beating. Following heat denaturation at 65°C for 10 minutes, lysates were diluted with 1 mL of 50mM Tris pH 7.5, 150mM NaCl, 0.1mM EDTA, and 0.5% Tween-20 and centrifuged. Cleared supernatants were immunoprecipitated with protein G dynabeads pre-bound to an eEF1A-specific antibody (Kerafast). Bound material was washed twice with IP buffer, followed by two more washes with IP buffer lacking detergent before being eluted with SDS-PAGE sample loading buffer. ZPR1-AID cells in Figure 2B lacked the drug-efflux pump Pdr5 (pdr5Δ).
Substrate flux through Zpr1 in vivo
ZPR1–3ˣFLAG cells were grown overnight to mid-log phase in SD-Met media. 15 OD600 of cells were harvested by centrifugation, then washed with ddH20 and incubated with 1.5 mL of pre-spheroplasting buffer (10 mM DTT, 100 mM Tris pH 9.4) for 10 minutes at RT with agitation. After harvesting by centrifugation, pre-treated cells were resuspended in 0.5 mL spheroplasting buffer (SD-Met with 1 M sorbitol, 10 mM Tris pH 7.5, 50 μg/mL Zymolyase 20T [amsbio, Cambridge, MA, USA]). Spheroplasting was carried out at 30°C with agitation and monitored by OD600 drop following 1/100 sample dilution into water until it reached >95% efficiency by 30 minutes of incubation. Spheroplasts were harvested by gently overlaying them onto 0.5 mL of RT sucrose cushion (0.8 M sucrose, 1.5% Ficoll 400, 20 mM HEPES pH 7.4) and centrifugation at 3000 x g for 3 minutes at RT. Following resuspension in 3 mL of radio-labeling buffer (SD-Met 1 M sorbitol), a 5 minute pulse phase was initiated by addition of 45 uL of label 35S-Methionine (30 μCi/OD600) and incubation at 30°C with agitation. After addition of chase mix (see pulse-chase analysis of eEF1A stability), 0.7 mL samples were removed at the indicated times and placed into individual glass beakers on ice until completion of time course. 0.5 mL of each sample were transferred to cooled Eppendorfs before harvesting the spheroplasts for 30 seconds at 4°C and 16,000 x g. After removing the supernatant, labeled spheroplasts were hypotonically lysed by resuspending them in 1 mL of cold HIP buffer with protease and RNAse inhibitors (see Zpr1–3ˣFLAG immunoprecipitation and mass spectrometry). Following centrifugation for 1 minute at 4°C and 16,000 x g, 0.9 mL of each cleared lysate was added to pre-chilled tubes containing 25 uL Dynabeads with 2 ul of previously-bound mouse anti-FLAG (M2) antibody. After IP for 30 minutes at 4°C with agitation, beads were washed 4 × 1 mL with HIP buffer and eluted with SDS-PAGE sample buffer. Eluted material was analyzed by SDS-PAGE followed by autoradiography.
ColabFold modeling of a Zpr1-eEF1A complex
ColabFold version 1.3.0 was used with the default parameters with msa_mode: “MMseqs2 (UniRef+Environmental)”, and model_type: “AlphaFold2-multimer-v2” to model the S. cerevisiae Zpr1 and eEF1A complex. Sequences were obtained from Uniprot. The residue boundaries for the S. cerevisiae N-terminal half of Zpr1 modeled with eEF1A were 1–266. The residue boundaries for the S. cerevisiae C-terminal half of Zpr1 modeled with eEF1A were 267–486. Models are provided in Supplementary Item 4.
QUANTIFICATION AND STATISTICAL ANALYSIS
Flow cytometry analysis
Samples were gated for live cells and single cells using BioConductor packages flowCore and flowViz. Each sample was normalized to cell size by dividing the raw GFP intensity (FITC.A) by side scatter (SSC.A). Data was log transformed for histograms and bar graphs.
Differential protein abundance analysis of Zpr1–3ˣFLAG IP/MS
The normalized and scaled protein-level abundances for all replicates and conditions was generated by the Harvard Center for Mass Spectrometry using Proteome Discoverer software (ThermoFisher Scientific, version 2.4) as follows. Assignments of MS/MS spectra were performed using the Sequest HT algorithm by searching the data against a UniProt protein sequence dataset of S. cerevisiae proteins, and including all entries from common contaminants such as E. coli, S. Aureus, human keratins and other common lab contaminants. Quantitative analyses between samples were performed by LFQ (label free quantitation). Sequest HT searches were performed using a 10 ppm precursor ion tolerance and requiring each peptide’s N/C-termini to adhere with trypsin specificity, while allowing up to two missed cleavages. Methionine oxidation (+15.99492 Da) was set as variable modification. A MS2 spectra assignment false discovery rate (FDR) of 1% on both the protein and peptide level was achieved by applying the target-decoy database search by use of Percolator. To detect statistically significant differences in protein abundance, the FDR was calculated on the Empirical Bayes moderated t-statistic, implemented using the R package limma (v.3.50.0)52. A protein was defined as significant if FDR < 0.05. The results of this analysis are provided in Supplementary Tables S2 and S3.
Differential gene expression analysis of RNA sequencing data
Raw reads from Illumina HiSeq were checked for read quality using FastQC (v. 0.11.5) and aligned to the reference genome S288c (v. R64–3-1) using the RNA-seq aligner STAR (v.2.7.0)53. Gene read counts obtained from STAR were passed to the Bioconductor package DESeq2 (v. 1.34.0)54 to identify differentially expressed genes after ZPR1 depletion relative to the DMSO vehicle control (Benjamini-Hochberg FDR < 0.05).
RNA sequencing time series analysis
Raw read counts obtained post-alignment using STAR at 30 minutes, 1.5 hours and 3 hours of Zpr1 depletion from ZPR1-AID cells were normalized and regularized-log (rlog) transformed using DESeq2 54. Results of DESeq2 analysis are shown in Supplementary Table S1. To identify if Hsf1 targets (28 targets obtained from ref4) or Gcn4 targets (163 positively regulated targets obtained from SGD) clustered in a particular temporal pattern, the clustering tool, degPatterns, from the DEGreport (v.1.30.0) package was run on the normalized, rlog read counts filtered to include only the genes in the Hsf1 or Gcn4 target list.
Immunoblot and autoradiography quantification
Immunoblots and autoradiographs were quantified using the BandPeakQuantification ImageJ (FIJI) plugin (dx.doi.org/10.17504/protocols.io.7vghn3w).
Ribosome profiling analysis
The four random nucleotides present in the RT primers used for library generation were trimmed from the 5’ end of reads with seqtk (https://github.com/lh3/seqtk) using options “trimfq -b 4”. Next, 3’ adapter sequences were removed using cutadapt (https://cutadapt.readthedocs.io/en/stable/index.html). Reads were then aligned to non-coding RNA sequences (rRNA, tRNA, snRNA, snoRNA, and ncRNA) derived from the Saccharomyces cerevisiae Genome Database S288C reference sequence using STAR (version 2.7.0e)53 with parameter “--outReadsUnmapped Fastx”. Reads that did not align to non-coding RNA were mapped to the R64–1-1 S288C reference genome (SacCer3) using STAR (version 2.7.0e) with parameters “--outFilterIntronMotifs RemoveNoncanonical --outFilterMismatchNmax 2 --outFilterMultimapNmax 1”. Normalized gene coverage tracks were generated from the resulting BAM files using the bamCoverage tool of the deepTools suite55 with option “--normalizeUsing CPM”. Gene coverage tracks were visualized using the Integrative Genomics Viewer (version 2.12.3)56.
Supplementary Material
Table S1. Results of DESeq2 analysis of mRNA sequencing data. Related to Figure 3.
Table S2. Results of Zpr1–3ˣFLAG IP/MS and Empirical Bayes analysis of protein abundance. Related to Figure 4.
Table S3. Results of N-halfZpr1–3ˣFLAG and Zpr1C-half-3ˣFLAG IP/MS and Empirical Bayes analysis of protein abundance. Related to Figure S4.
These supplemental data folders contain five PDB formatted structures sorted by avg. pLDDT and complexes are sorted by pTMscore. Also included are plots of the model quality, plots of the MSA coverage, a parameter log file, A3M formatted input MSA, a predicted_aligned_error_v1.json and a scores.json for each model which contains an array (list of lists) for PAE, a list with the average pLDDT and the pTMscore, and a BibTeX file with citations for all used tools and database.
Highlight 1:
Zpr1 depletion-induced proteotoxicity is due to misfolding of newly synthesized eEF1A
Highlight 2:
Prolonged Zpr1 depletion generates translational stress due to eEF1A insufficiency
Highlight 3:
Biochemical reconstitution of eEF1A biogenesis and folding requires Zpr1
Highlight 4:
Zpr1 uses zinc finger and alpha-helical hairpin structures to chaperone eEF1A
Utilizing a range of in vivo and in vitro approaches, Sabbarini et al. uncover the elusive function of the essential and highly conserved Zinc-Finger Protein 1 (ZPR1) gene. They report that Zpr1 serves as a dedicated eEF1A chaperone, using its zinc finger and alpha-helical hairpin structures to facilitate eEF1A biogenesis.
Acknowledgements
We thank David Pincus, Jack Taunton, Jonathan Weissman, Matt Waldor, William Merrick, and Satoshi Kimura for useful discussions; Satoshi Kimura for help with polysome profiling; Carmen Chan and María Angélica Bravo Núñez for useful discussions and critical reading of the manuscript; and Bogdan Budnik for help with mass spectrometry sample preparation and analysis.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Balchin D, Hayer-Hartl M, and Hartl FU (2016). In vivo aspects of protein folding and quality control. Science 353, aac4354. [DOI] [PubMed] [Google Scholar]
- 2.Hartl FU (1996). Molecular chaperones in cellular protein folding. Nature 381, 571–580. 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 3.Lindquist S, and Craig EA (1988). THE HEAT-SHOCK PROTEINS. Annu. Rev. Genet 22, 631–677. 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 4.Solís EJ, Pandey JP, Zheng X, Jin DX, Gupta PB, Airoldi EM, Pincus D, and Denic V. (2016). Defining the Essential Function of Yeast Hsf1 Reveals a Compact Transcriptional Program for Maintaining Eukaryotic Proteostasis. Mol. Cell 63, 60–71. 10.1016/j.molcel.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pincus D, Anandhakumar J, Thiru P, Guertin MJ, Erkine AM, and Gross DS (2018). Genetic and epigenetic determinants establish a continuum of Hsf1 occupancy and activity across the yeast genome. Mol. Biol. Cell 29, 3168–3182. 10.1091/mbc.E18-06-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang C, Phillips APR, Wipfler RL, Olsen GJ, and Whitaker RJ (2018). The essential genome of the crenarchaeal model Sulfolobus islandicus. Nat. Commun 9, 4908. 10.1038/s41467-018-07379-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galcheva-Gargova Z, Konstantinov KN, Wu I-H, Klier FG, Barrett T, and Davis RJ (1996). Binding of Zinc Finger Protein ZPR1 to the Epidermal Growth Factor Receptor. Science 272, 1797–1802. 10.1126/science.272.5269.1797. [DOI] [PubMed] [Google Scholar]
- 8.Gangwani L, Mikrut M, Theroux S, Sharma M, and Davis RJ (2001). Spinal muscular atrophy disrupts the interaction of ZPR1 with the SMN protein. Nat. Cell Biol 3, 376–383. 10.1038/35070059. [DOI] [PubMed] [Google Scholar]
- 9.Pellizzoni L, Kataoka N, Charroux B, and Dreyfuss G. (1998). A Novel Function for SMN, the Spinal Muscular Atrophy Disease Gene Product, in Pre-mRNA Splicing. Cell 95, 615–624. 10.1016/S0092-8674(00)81632-3. [DOI] [PubMed] [Google Scholar]
- 10.Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165. 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 11.Ahmad S, Wang Y, Shaik GM, Burghes AH, and Gangwani L. (2012). The zinc finger protein ZPR1 is a potential modifier of spinal muscular atrophy. Hum. Mol. Genet 21, 2745–2758. 10.1093/hmg/dds102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kannan A., Jiang X., He L., Ahmad S., and Gangwani L. (2020). ZPR1 prevents R-loop accumulation, upregulates SMN2 expression and rescues spinal muscular atrophy. Brain 143, 69–93. 10.1093/brain/awz373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller WT (2012). Tyrosine kinase signaling and the emergence of multicellularity. Biochim. Biophys. Acta BBA - Mol. Cell Res 1823, 1053–1057. 10.1016/j.bbamcr.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mier P, and Pérez-Pulido AJ (2012). Fungal Smn and Spf30 homologues are mainly present in filamentous fungi and genomes with many introns: Implications for spinal muscular atrophy. Gene 491, 135–141. 10.1016/j.gene.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 15.Gangwani L, Mikrut M, Galcheva-Gargova Z, and Davis RJ (1998). Interaction of ZPR1 with Translation Elongation Factor-1α in Proliferating Cells. J. Cell Biol 143, 1471–1484. 10.1083/jcb.143.6.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mishra AK, Gangwani L, Davis RJ, and Lambright DG (2007). Structural insights into the interaction of the evolutionarily conserved ZPR1 domain tandem with eukaryotic EF1A, receptors, and SMN complexes. Proc. Natl. Acad. Sci 104, 13930–13935. 10.1073/pnas.0704915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dever TE, Dinman JD, and Green R. (2018). Translation Elongation and Recoding in Eukaryotes. Cold Spring Harb. Perspect. Biol 10, a032649. 10.1101/cshperspect.a032649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwasaki K, Nagata S, Mizumoto K, and Kaziro Y. (1974). The Purification of Low Molecular Weight Form of Polypeptide Elongation Factor 1 from Pig Liver. J. Biol. Chem 249, 5008–5010. 10.1016/S0021-9258(19)42421-6. [DOI] [PubMed] [Google Scholar]
- 19.Slobin LI, Clark RV, and Olson MOJ (1981). Functional and structural studies on a tryptic fragment of eucaryotic elongation factor Tu from rabbit reticulocytes. Biochemistry 20, 5761–5767. 10.1021/bi00523a019. [DOI] [PubMed] [Google Scholar]
- 20.Brandman O, Stewart-Ornstein J, Wong D, Larson A, Williams CC, Li G-W, Zhou S, King D, Shen PS, Weibezahn J, et al. (2012). A Ribosome-Bound Quality Control Complex Triggers Degradation of Nascent Peptides and Signals Translation Stress. Cell 151, 1042–1054. 10.1016/j.cell.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yesbolatova A, Saito Y, Kitamoto N, Makino-Itou H, Ajima R, Nakano R, Nakaoka H, Fukui K, Gamo K, Tominari Y, et al. (2020). The auxin-inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nat. Commun 11, 5701. 10.1038/s41467-020-19532-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Specht S, Miller SB, Mogk A, and Bukau B. (2011). Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae. J. Cell Biol 195, 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang M, Herrmann CJ, Simonovic M, Szklarczyk D, and Mering C. (2015). Version 4.0 of PaxDb: Protein abundance data, integrated across model organisms, tissues, and cell-lines. PROTEOMICS 15, 3163–3168. 10.1002/pmic.201400441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krakowiak J, Zheng X, Patel N, Feder ZA, Anandhakumar J, Valerius K, Gross DS, Khalil AS, and Pincus D. (2018). Hsf1 and Hsp70 constitute a two-component feedback loop that regulates the yeast heat shock response. eLife 7, e31668. 10.7554/eLife.31668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng X, Krakowiak J, Patel N, Beyzavi A, Ezike J, Khalil AS, and Pincus D. (2016). Dynamic control of Hsf1 during heat shock by a chaperone switch and phosphorylation. eLife 5, e18638. 10.7554/eLife.18638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinnebusch AG (2005). TRANSLATIONAL REGULATION OF GCN4 AND THE GENERAL AMINO ACID CONTROL OF YEAST. Annu. Rev. Microbiol 59, 407–450. 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- 27.Wu CC-C, Peterson A, Zinshteyn B, Regot S, and Green R. (2020). Ribosome Collisions Trigger General Stress Responses to Regulate Cell Fate. Cell 182, 404–416.e14. 10.1016/j.cell.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan LL, and Zaher HS (2021). Ribosome quality control antagonizes the activation of the integrated stress response on colliding ribosomes. Mol. Cell 81, 614–628.e4. 10.1016/j.molcel.2020.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagata S, Iwasaki K, and Kaziro Y. (1976). Interaction of the low molecular weight form of elongation factor 1 with guanine nucleotides and aminoacyl-tRNA. Arch. Biochem. Biophys 172, 168–177. 10.1016/0003-9861(76)90063-1. [DOI] [PubMed] [Google Scholar]
- 30.Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, and Steinegger M. (2022). ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682. 10.1038/s41592-022-01488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galcheva-Gargova Z, Gangwani L, Konstantinov KN, Mikrut M, Theroux SJ, Enoch T, and Davis RJ (1998). The Cytoplasmic Zinc Finger Protein ZPR1 Accumulates in the Nucleolus of Proliferating Cells. Mol. Biol. Cell 9, 2963–2971. 10.1091/mbc.9.10.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ansari H., Greco G., and Luban J. (2002). Cyclophilin A Peptidyl-Prolyl Isomerase Activity Promotes Zpr1 Nuclear Export. Mol. Cell. Biol 22, 6993–7003. 10.1128/MCB.22.20.6993-7003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laskey RA, Honda BM, Mills AD, and Finch JT (1978). Nucleosomes are assembled by an acidic protein which binds histones and transfers them to DNA. Nature 275, 416–420. 10.1038/275416a0. [DOI] [PubMed] [Google Scholar]
- 34.Perzlmaier AF, Richter F, and Seufert W. (2013). Translation Initiation Requires Cell Division Cycle 123 (Cdc123) to Facilitate Biogenesis of the Eukaryotic Initiation Factor 2 (eIF2). J. Biol. Chem 288, 21537–21546. 10.1074/jbc.M113.472290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Panvert M, Dubiez E, Arnold L, Perez J, Mechulam Y, Seufert W, and Schmitt E. (2015). Cdc123, a Cell Cycle Regulator Needed for eIF2 Assembly, Is an ATP-Grasp Protein with Unique Features. Structure 23, 1596–1608. 10.1016/j.str.2015.06.014. [DOI] [PubMed] [Google Scholar]
- 36.Mönkemeyer L, Klaips CL, Balchin D, Körner R, Hartl FU, and Bracher A. (2019). Chaperone Function of Hgh1 in the Biogenesis of Eukaryotic Elongation Factor 2. Mol. Cell 74, 88–100.e9. 10.1016/j.molcel.2019.01.034. [DOI] [PubMed] [Google Scholar]
- 37.Schopf FH, Huber EM, Dodt C, Lopez A, Biebl MM, Rutz DA, Mühlhofer M, Richter G, Madl T, Sattler M, et al. (2019). The Co-chaperone Cns1 and the Recruiter Protein Hgh1 Link Hsp90 to Translation Elongation via Chaperoning Elongation Factor 2. Mol. Cell 74, 73–87.e8. 10.1016/j.molcel.2019.02.011. [DOI] [PubMed] [Google Scholar]
- 38.Shaner L, and Morano KA (2007). All in the family: atypical Hsp70 chaperones are conserved modulators of Hsp70 activity. Cell Stress Chaperones 12, 1. 10.1379/CSC-245R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Polier S, Dragovic Z, Hartl FU, and Bracher A. (2008). Structural Basis for the Cooperation of Hsp70 and Hsp110 Chaperones in Protein Folding. Cell 133, 1068–1079. 10.1016/j.cell.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 40.Dong J, Qiu H, Garcia-Barrio M, Anderson J, and Hinnebusch AG (2000). Uncharged tRNA Activates GCN2 by Displacing the Protein Kinase Moiety from a Bipartite tRNA-Binding Domain. Mol. Cell 6, 269–279. 10.1016/S1097-2765(00)00028-9. [DOI] [PubMed] [Google Scholar]
- 41.Pochopien AA, Beckert B, Kasvandik S, Berninghausen O, Beckmann R, Tenson T, and Wilson DN (2021). Structure of Gcn1 bound to stalled and colliding 80S ribosomes. Proc. Natl. Acad. Sci 118, e2022756118. 10.1073/pnas.2022756118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harding HP, Ordonez A, Allen F, Parts L, Inglis AJ, Williams RL, and Ron D. (2019). The ribosomal P-stalk couples amino acid starvation to GCN2 activation in mammalian cells. eLife 8, e50149. 10.7554/eLife.50149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inglis AJ, Masson GR, Shao S, Perisic O, McLaughlin SH, Hegde RS, and Williams RL (2019). Activation of GCN2 by the ribosomal P-stalk. Proc. Natl. Acad. Sci 116, 4946–4954. 10.1073/pnas.1813352116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Visweswaraiah J, Lageix S, Castilho BA, Izotova L, Kinzy TG, Hinnebusch AG, and Sattlegger E. (2011). Evidence That Eukaryotic Translation Elongation Factor 1A (eEF1A) Binds the Gcn2 Protein C Terminus and Inhibits Gcn2 Activity. J. Biol. Chem 286, 36568–36579. 10.1074/jbc.M111.248898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doran B, Gherbesi N, Hendricks G, Flavell RA, Davis RJ, and Gangwani L. (2006). Deficiency of the zinc finger protein ZPR1 causes neurodegeneration. Proc. Natl. Acad. Sci 103, 7471–7475. 10.1073/pnas.0602057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ishimura R, Nagy G, Dotu I, Zhou H, Yang X-L, Schimmel P, Senju S, Nishimura Y, Chuang JH, and Ackerman SL (2014). Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 345, 455–459. 10.1126/science.1249749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chambers DM, Peters J, and Abbott CM (1998). The lethal mutation of the mouse wasted ( wst ) is a deletion that abolishes expression of a tissue-specific isoform of translation elongation factor 1α, encoded by the Eef1a2 gene. Proc. Natl. Acad. Sci 95, 4463–4468. 10.1073/pnas.95.8.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brachmann CB., Davies A., Cost GJ., Caputo E., Li J., Hiete P., and Boeke JD. (1998). Designer deletion strains derived fromSaccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132. . [DOI] [PubMed] [Google Scholar]
- 49.Schuller AP, Wu CC-C, Dever TE, Buskirk AR, and Green R. (2017). eIF5A Functions Globally in Translation Elongation and Termination. Mol. Cell 66, 194–205.e5. 10.1016/j.molcel.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shafieinouri M, Membreno BS, and Wu CC-C (2022). High-Resolution Ribosome Profiling for Determining Ribosome Functional States During Translation Elongation. In The Integrated Stress Response Methods in Molecular Biology., Matějů D. and Chao JA, eds. (Springer US; ), pp. 173–186. 10.1007/978-1-0716-1975-9_11. [DOI] [PubMed] [Google Scholar]
- 51.Schuldiner M, Metz J, Schmid V, Denic V, Rakwalska M, Schmitt HD, Schwappach B, and Weissman JS (2008). The GET Complex Mediates Insertion of Tail-Anchored Proteins into the ER Membrane. Cell 134, 634–645. 10.1016/j.cell.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T. (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191. 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Results of DESeq2 analysis of mRNA sequencing data. Related to Figure 3.
Table S2. Results of Zpr1–3ˣFLAG IP/MS and Empirical Bayes analysis of protein abundance. Related to Figure 4.
Table S3. Results of N-halfZpr1–3ˣFLAG and Zpr1C-half-3ˣFLAG IP/MS and Empirical Bayes analysis of protein abundance. Related to Figure S4.
These supplemental data folders contain five PDB formatted structures sorted by avg. pLDDT and complexes are sorted by pTMscore. Also included are plots of the model quality, plots of the MSA coverage, a parameter log file, A3M formatted input MSA, a predicted_aligned_error_v1.json and a scores.json for each model which contains an array (list of lists) for PAE, a list with the average pLDDT and the pTMscore, and a BibTeX file with citations for all used tools and database.
Data Availability Statement
Raw RNA-sequencing data and ribosome profiling data were deposited in the GEO database and are publicly available under accession numbers GSE212387 and GSE212389. In addition, all data present in this study will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.