

Abstract
Background
Cellular immunotherapies for cancer represent a means by which a patient’s immune system can be augmented with high numbers of tumor-specific T cells. Chimeric antigen receptor (CAR) therapy involves genetic engineering to ‘redirect’ peripheral T cells to tumor targets, showing remarkable potency in blood cancers. However, due to several resistance mechanisms, CAR-T cell therapies remain ineffective in solid tumors. We and others have shown the tumor microenvironment harbors a distinct metabolic landscape that produces a barrier to immune cell function. Further, altered differentiation of T cells within tumors induces defects in mitochondrial biogenesis, resulting in severe cell-intrinsic metabolic deficiencies. While we and others have shown murine T cell receptor (TCR)-transgenic cells can be improved through enhanced mitochondrial biogenesis, we sought to determine whether human CAR-T cells could be enabled through a metabolic reprogramming approach.
Materials and methods
Anti-EGFR CAR-T cells were infused in NSG mice which bore A549 tumors. The tumor infiltrating lymphocytes were analyzed for exhaustion and metabolic deficiencies. Lentiviruses carrying PPAR-gamma coactivator 1α (PGC-1α), PGC-1αS571A and NT-PGC-1α constructs were used to co-transduce T cells with anti-EGFR CAR lentiviruses. We performed metabolic analysis via flow cytometry and Seahorse analysis in vitro as well as RNA sequencing. Finally, we treated therapeutically A549-carrying NSG mice with either PGC-1α or NT-PGC-1α anti-EGFR CAR-T cells. We also analyzed the differences in the tumor-infiltrating CAR-T cells when PGC-1α is co-expressed.
Results
Here, in this study, we show that an inhibition resistant, engineered version of PGC-1α, can metabolically reprogram human CAR-T cells. Transcriptomic profiling of PGC-1α-transduced CAR-T cells showed this approach effectively induced mitochondrial biogenesis, but also upregulated programs associated with effector functions. Treatment of immunodeficient animals bearing human solid tumors with these cells resulted in substantially improved in vivo efficacy. In contrast, a truncated version of PGC-1α, NT-PGC-1α, did not improve the in vivo outcomes.
Conclusions
Our data further support a role for metabolic reprogramming in immunomodulatory treatments and highlight the utility of genes like PGC-1α as attractive candidates to include in cargo along with chimeric receptors or TCRs for cell therapy of solid tumors.
Keywords: Receptors, Chimeric Antigen; Cell Engineering; Immunotherapy, Adoptive; Translational Medical Research
WHAT IS ALREADY KNOWN ON THIS TOPIC
The efficacy of chimeric antigen receptor (CAR)-T cell therapy in solid tumors has been limited due to various barriers, including an immunosuppressive tumor microenvironment (TME). The TME represents a metabolically dearth zone leading to tumor infiltrating T cells that are hypofunctional with severe metabolic defects. A key driver of these altered effects is the repression of PPAR gamma coactivator 1α (PGC-1α), a transcriptional coactivator that promotes mitochondrial health in tumor infiltrating T cells. We and others have previously shown overexpression of PGC-1α in T cells can show enhanced antitumor responses in murine models. However, it has not been tested whether a similar approach would successfully overcome the metabolic barriers responsible for poor immune responses in solid tumor models for human CAR-T cell therapy.
WHAT THIS STUDY ADDS
In this study, we show that it is feasible to engineer human CAR T cells to express an inhibition resistant PGC-1α gene. We also prove that this approach bolsters memory and metabolism leading to improved responses in a human tumor xenograft model.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Our study supports the use of genetic-based metabolic immune modulation in bolstering the immune response of CAR T-cell therapy against solid tumors and opens the possibility of other similar avenues being tested to achieve improved therapeutic responses.
Background
The treatment paradigm for cancer has been fundamentally altered by advances in immunotherapy. Several approaches have been successful in the treatment of various types of cancer, although most prominent among these has been the use of monoclonal antibody-mediated blockade of co-inhibitory ‘checkpoint’ molecules like cytotoxic T-lymphocytes-associated protein 4 (CTLA-4) and programmed cell death protein-1 (PD-1).1 These treatments work to ‘re-awaken’ a patient’s immune system and alleviate tumor-induced immune suppression. However, it has become clear that in order for these therapies to function effectively, patients must harbor a pre-existing immune response to their tumor that is capable of being reinvigorated.2 Thus, while the responses can be remarkable, the response rates to checkpoint blockade remain low.
Cellular therapies for cancer represent an attractive strategy in which in vitro engineering can be used to either numerically enhance tumor-specific T cells by expanding tumor-infiltrating lymphocytes (TIL) or to genetically redirect healthier, peripheral T cells to tumor targets using chimeric antigen receptors (CAR).3 In the latter case, CAR-T cells are capable of recognizing a surface tumor antigen via non-major histocompatibility complex restricted binding and dramatically reduce tumor burden. CAR-T cells are extremely effective in certain hematologic malignancies, leading to Food and Drug Administration approval for CD19-directed CAR-T cells in 2018 for B-ALL which was followed by several approvals for lymphomas and myeloma.4–8
However, despite the remarkable responses in blood cancers, CAR-T cells have shown limited efficacy in solid tumors. There are many potential reasons for these failures, including the lack of truly solid tumor-specific surface antigens, poor trafficking, and the presence of immunosuppressive factors and populations in the solid tumor microenvironment.9 We and others have demonstrated that the solid tumor microenvironment also harbors a distinct metabolic landscape, driven primarily by the energetic needs of cancer cells’ incessant proliferation. Cancer cells deprive the local milieu of essential nutrients like glucose, oxygen, and glutamine, and promote the build-up of toxic byproducts like lactic acid, reactive oxygen species, and other catabolites.10 In this environment, therapeutic T cells may be receiving all of the activating signals they need to function but lack the energetic currency needed to perform them. Further, the alternative differentiation signals infiltrating T cells received within the tumor microenvironment drive them away from long-lived T cells capable of self-renewal (memory) and instead toward antigen-dependent, poorly functional T cells with limited survival capacity (exhausted). We and others have shown that the process of exhaustion has strong metabolic underpinnings, such that terminally exhausted T cells harbor mitochondrial defects that prevent their function and self-renewal.11 A key driver of this altered metabolic fate is the repression of the PPAR gamma coactivator 1α (PGC-1α), a transcriptional coactivator that coordinates mitochondrial biogenesis, fusion, and antioxidant activities to promote mitochondrial health and capacity. PGC-1α is repressed in tumor infiltrating T cells not only at the transcriptional level but also by phosphorylation by Akt. Indeed, in mice we have previously shown that PGC-1α can be expressed in T cell receptor-transgenic (TCR-Tg) T cells to promote greater therapeutic responses.12 Dumauthioz et al showed similar findings and added that PGC-1α can increase the memory pool of adoptive cell therapy.13 Further, others have shown that stimulation of the PGC-1α pathway can promote increased responses to checkpoint blockade therapy.14
Thus, we sought to determine whether solid tumor targeting, human CAR-T cell therapy can be enabled using PGC-1α-mediated mitochondrial reprogramming. We show that solid tumor targeting CAR-T cells succumb to similar metabolic insufficiencies as endogenous T cells as we have previously observed. Further, we demonstrate that the use of an engineered PGC-1α molecule, resistant to post-translational regulation, can endow T cells with potent mitochondrial reprogramming. This mitochondrial reprogramming not only drives more effector-like programs but also promotes a more long-lived memory state, which leads to superior therapeutic efficacy.
Materials and methods
Mice
All animal work was done in accordance with the University of Pittsburgh Institutional Animal Care and Use Committee, certified by the Association for Assessment and Accreditation of Laboratory Animal Care (Ref:20077737). Procedures were performed under their guidelines. NOD.Cg-Prkdc scid ll2rg tm1Wjl /SzJ/Arc (NSG) mice were obtained from the Jackson Laboratory.
Tumor implantation
A549 lung carcinoma cell line was cultured in D10 media (Dulbecco’s Modified Eagle Medium, 10% fetal bovine serum, 2 mM L-glut, PenStrep, Non-Essential Amino Acids, 1 mM sodium pyruvate, 5 mM HEPES, β-mercaptoethanol). NSG mice were injected with 2 million A549 cells on day 0. Tumor volumes were calculated by measuring length, width and height every 3 days with digital calipers. Mice were treated with T cells when tumor volumes reached approximately 100 mm3. Number of T cells was always adjusted based on % CAR positivity.
Anti-EGFR CAR-T cell generation via lentiviral transduction
X-VIVO 15 media (Lonza) supplemented with 5% human AB serum (Valley Biomedical), 10 mM HEPES buffer and GlutaMAX (Gibco) were used for in vitro culture of CAR-T cells. Frozen peripheral blood mononuclear cells (PBMCs) were thawed and then activated with anti-CD3 and anti-CD28 antibodies (both at 50 ng/mL) in the presence of 250 IU/mL human interleukin (IL)-2 (CellGenix) for 24 hours, followed by lentiviral transduction with one or two viruses at the same time at a Multiplicity of Infection of 20. The lentiviruses were provided by bluebird bio (Patent International Publication Number WO 2021/252782 A1). Cells were initially cultured in normal flasks and subsequently transferred to G-Rex 6M Well Plate (Wilson Wolf) on day 4 post activation for rapid expansion. Cells were then harvested on day 10 for further analysis or adoptive transfer.
Anti-CD20 CAR-T cell generation via retroviral transduction
A total of 4 million 293GP cells were plated on collagen I coated 10 cm plates. Cells were transfected with plasmids carrying the gene of interest and the helper plasmid (encoding the RD114 envelope protein) using Lipofectamine 3000 (Thermo Fisher) as per the manufacturer’s protocol. Viral particles were collected 48 hours after transfection. Frozen PBMCs were thawed and T cells were isolated with the BioLegend MojoSort human CD3 isolation kit. The T cells were then activated with TransAct (Miltenyi Biotec, 1:100) in the presence of 200 IU/mL human IL-2 (CellGenix) for 48 hours, followed by retroviral transduction. Briefly, the retrovirus was preloaded through spin-transduction (2 hours, 2000 g) on a non-tissue culture retronectin-coated plate. Then T cells were added and were spun down for 10 min. The T cells were incubated for 48 hours in media as above and then were (Fluorescence-activated cell sorting)FACS sorted for CAR+cells. After sorting, the T cells were transferred to a G-Rex 6M Well Plate (Wilson Wolf) for expansion. Cells were then harvested on day 10 for further analysis.
Metabolic assays
In vitro expanded CAR-T cells were plated on Cell-Tak-coated Seahorse Bioanalyzer XFe96 culture plates at the concentration of 100,000 cells per well in the presence of assay media, which comprises minimal DMEM supplemented with 1% Bovine Serum Albumin and 2 mM glutamine, 1 mM sodium pyruvate and 25 mM glucose. In a standard Seahorse mito stress test, oligomycin (2 µM), carbonyl cyanide p-trifluoromethoxyphenylhydrazone (0.5 µM), 2-deoxy-d-glucose (10 mM), and rotenone/antimycin A (0.5 µM) were injected sequentially to measure the maximal oxygen respiration rate. For flow cytometric metabolic assays, cells were incubated with 20 nM MitoTracker Deep Red FM for 15 min at 4°C.
Respiration due to exogenous fatty acids
Fatty acid oxidation in the cells because of exogenous fatty acids were measured using the Seahorse analyzer described above. BSA 30 µL (0.17 mM) or Palm:BSA (1 mM) conjugate was added to the well in XFe96 plate Seahorse plate before Oxygen Consumption Rate (OCR) measurement. ‘Basal respiration due to exogenous fatty acid’ was calculated as a measure of the average value of basal respiration in the BSA group subtracted from the basal respiration value for each well of Palm:BSA group.
In vitro killing assay
For killing assays, the CAR-T cells were co-cultured with target A549-cells expressing luciferase at different ratios overnight. In the morning, 0.2 mg/mL of luciferin was added to each well and the luminescence was read by a plate reader. The % killing activity was normalized to the level of signal from the wells that target cells were co-cultured with mock-transduced T cells (% killing=100×(signal from mock-TD T cell well – signal from CAR-T cell well)/signal from mock-TD cell well).
Long-term CAR T-cell culture
CAR T cells were cultured for up to 7 weeks in X-VIVO media after the end of expansion. Cells were cultured in X-VIVO 15 media (Lonza) supplemented with 5% human AB serum (Valley Biomedical), 10 mM HEPES buffer and 2 mM GlutaMAX (Gibco); and further supplemented with 200 IU/mL of human IL-2. Cell numbers were counted every alternate day and plotted as a measure of fold expansion.
Tumor-infiltrating CAR-T cell analysis
Spleens and tumors were harvested from NSG mice 10–14 days after CAR-T cell transfer. To obtain single-cell suspensions, spleens were mechanically disrupted and treated with a red cell lysis buffer. Excised, whole tumors were injected using syringes with 2 mg/mL collagenase type IV, 2 U/mL hyluronidase (Dispase), and 10 U/mL DNase I in buffered X-VIVO 15 and incubated for 30 min at 37°C. Tumors were then disrupted and filtered for flow cytometric analysis. For functional readouts, cells were activated through phorbol 12-myristate 13-acetate and ionomycin for 6 hours while under the effect of GolgiPlug (BD), before intracellular staining of cytokine production. Single-cell suspensions were stained and run on either BD LSRFortessa or BD Accuri C6. Antibodies were obtained from the following companies: BioLegend anti-CD4 (clone OKT4), anti-CD8 (clone HIT8a), anti-PD-1 (clone EH12.2H7), anti-Tim3 (clone F38-2E2), anti-interferon (IFN)-g (clone 4S.B3), anti-tumor necrosis factor-a (clone Mab11), anti-CD45RA (clone HI100), anti-CCR7 (clone G043H7). MitoTracker Deep Red FM was purchased from Thermo Fisher. Anti-Lag-3 was purchased from eBioscience (clone 3DS223H). EGFR protein conjugated to Allophycocyanin was obtained by CREATIVE BIOMART (EGFR-692HA). Intracellular staining was performed using the Foxp3 Fix/Perm buffer set (eBioscience). Flow cytometry data were analyzed with FlowJo V.10 software.
Real-time quantitative PCR
Total RNA was extracted using TRIzol Reagent (Invitrogen) and complementary DNA (cDNA) was transcribed using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems), according to manufacturer’s protocol. Transcript levels were measured with SYBR Green (Thermo Fisher Scientific) and using primers specific to genes of interest. The cDNA concentration was normalized per samples relative to cyclophilin B. All experiments were performed in technical replicates.
RNA sequencing
Total RNA was isolated from in vitro expanded CAR-T cells and HEK293T cells using RNeasy Mini Kit (Qiagen). Purified RNA was sequenced from the UPMC Genome Center or the Health Sciences Sequencing Core at Children’s Hospital of Pittsburgh. RNA sequencing analysis was performed using CLC Genomics and R. Only genes that had a false discovery rate (FDR) p value<0.05 were considered significant. Volcano plot was constructed with all genes with a p value<0.05 but genes with an FDR p value<0.05 were marked with red.
Statistical analysis
The data presented in the figures are mean±SEM. For paired data either paired t-test or Wilcoxon matched-pairs signed-rank test was used, depending on the data distribution. For unpaired data t-test or Wilcoxon rank-sum test was used. Survival data are presented as Kaplan-Meier survival curves and analyzed with the non-parametric log-rank test. Tumor growth curves were analyzed with repeated measures analysis of variance. All analysis was completed with Prism V.7 software (GraphPad) and confirmed with Stata V.17.0. A value of p<0.05 was considered statistically significant. In the figures, standard designations of significance were given; *p<0.005, **p<0.01, ***p<0.001. The specific analysis used per figure in the manuscript can be found within the legends.
Results
We have previously shown that tumor-infiltrating T cells as well as adoptively transferred antigen-specific T cells succumb to metabolic insufficiency, characterized by loss of functional mitochondria.12 So, we first sought to determine if therapeutic human T cells, entering a hostile solid tumor microenvironment, would succumb to the same types of deficiencies. This was also critical given that CAR constructs generally carry co-stimulatory signaling domains that might influence metabolism.15 We employed an EGFR-targeted CAR lentiviral construct, based on a cetuximab-derived scFv and carrying a 4-1BB signaling domain (figure 1A), to generate therapeutic human CAR-T cells. This CAR has been reported before.16 Due to relatively average transduction rates, we mostly purified the CARs with FACS. The CAR positivity was assessed at the end of expansion and found to be consistent (online supplemental figure 1A). We first confirmed the in vitro functionality of these CARs using an in vitro killing assay (figure 1B). We then used these cells to treat A549 tumors xenografted into NOD-scid IL-2R gamma null (NSG) mice. Mice bearing 100 mm3 tumors were infused with 107 CAR-T cells and no therapeutic efficacy was noted (figure 1C). To investigate the reasons behind this phenomenon, we repeated the experiment and analyzed the metabolic status of the tumor-infiltrate 14 days post cell infusion. Analogous to our previous data using therapeutic OT-I T cells in B16OVA bearing mice, human EGFR-specific CAR-T cells infiltrating A549 tumors became metabolically exhausted, possessing lower MitoTracker FM staining compared with splenic T cells (figure 1D). This was concomitant with loss of immune function and upregulation of inhibitory receptors (figure 1E, F). Thus, just as observed in murine TCR-Tg T cells, human CAR-T cells responding to their antigen in the solid tumor microenvironment succumb to metabolic deficiencies as they progress to terminal exhaustion.
Figure 1.
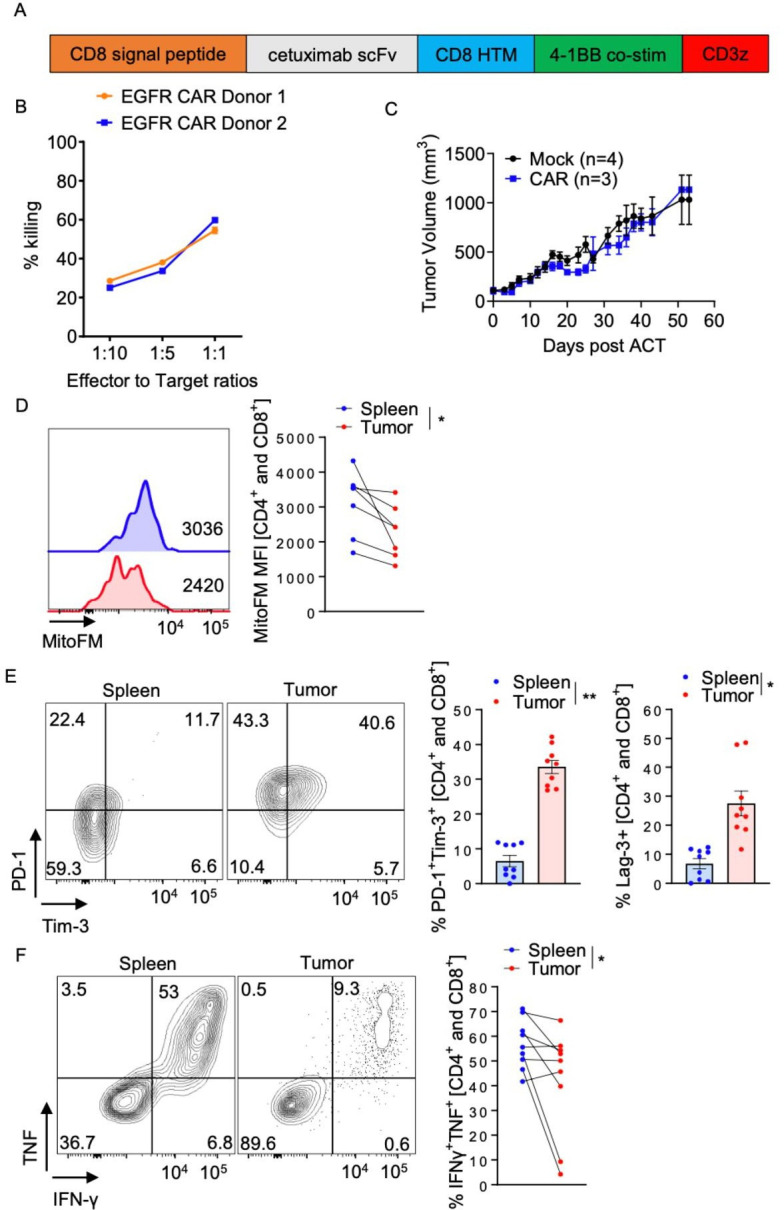
hEGFR-targeted CARs with 4-1BB co-stimulatory domains exhibit suppressed mitochondrial mass and exhaustion in the tumor microenvironment. (A) Structure of the anti-EGFR CAR that was used for the experiments, (B) in vitro killing assay of EGFR-specific CAR-T cells incubated overnight with A549 cells at the reported concentrations, (C) 107 hEGFR-targeted CARs were injected in NSG mice carrying 100 mm3 tumors, (D) MitoTracker FM staining of CAR-T cells in tumor infiltrating lymphocyte (TIL) preparations from A549 tumors and respective spleens, (E) PD-1, Tim-3, and Lag-3 staining in CAR-T cells infiltrating A549 tumors and the respective spleens. (F) Cytokine production of TIL after 6 hours of ex vivo restimulation with phorbol 12-myristate 13-acetate and ionomycin. All experiments were repeated with three different donors. Plot B is representative of three different experiments and plots C–E contain all data from all donors. Each dot represents one mouse. Statistics are Wilcoxon matched-pairs signed-rank test. *p<0.05, **p<0.01. CAR, chimeric antigen receptors; IFN, interferon; PD-1, programmed cell death protein-1; TNF, tumor necrosis factor; ACT, adoptive cell therapy
jitc-2022-006522supp002.pdf (604.3KB, pdf)
We next wanted to determine if metabolic reprogramming could be employed to prevent this loss of intratumoral function. We have previously shown that murine and human T cells infiltrating tumors repressed the expression of the PGC-1α, a transcriptional coactivator that coordinates mitochondrial biogenesis, fusion, and antioxidant activity.12 We and others have also shown that PGC-1α can be negatively regulated by the Akt signaling pathway, both indirectly (through repression of Foxo family member activity) and directly (through an inhibitory phosphorylation on S571).17 18 Thus, we engineered a variant of PGC-1α containing a point mutation at S571 (S571A), rendering it insensitive to Akt-mediated inhibition (figure 2A). Lentiviral transduction of HEK293T cells with wild-type (WT) versus S571A PPARGC1A revealed the latter had higher transcriptional activity, using a panel of PGC-1α target genes, including CPT1A/B, NFE2L2, PPARG, TFAM, and ESRRA (figure 2B). Thus, we chose to move forward with this engineered, inhibition-resistant PGC-1α to metabolically reprogram CAR-T cells for immunotherapy. At the same time, we evaluated a truncated form of PGC-1α, termed NT-PGC-1α. This isoform is much smaller than PGC-1α and thus would render the cell engineering process easier. Despite its much smaller size, previous studies suggest NT-PGC1α retains the transcriptional activation and nuclear receptor binding domains of PGC-1α.19
Figure 2.

An Akt-resistant form of PGC-1α and NT-PGC-1α improve the mitochondrial fitness of human CAR T cells in vitro. (A) Schema of different versions of PGC-1α, (B) real-time-PCR analysis of target genes from HEK293T cells transduced with different versions of PGC-1α, (C) MitoTracker staining of CAR-T cells carrying the PGC-1α construct or the NT-PGC-1α construct in vitro, (D) Seahorse analysis of CAR T cells carrying the PGC-1α or the NT-PGC-1α construct in vitro. The MitoFM experiments were repeated with five donors. The Seahorse analysis is representative of three donors each. Statistics are paired t-test (C) and Wilcoxon rank-sum test (D). *p<0.05, **p<0.01. CAR, chimeric antigen receptors; PGC-1α, PPAR gamma coactivator 1α.
Human peripheral blood T cells were activated and lentivirally transduced with the anti-EGFR CAR construct alone, with the anti-EGFR CAR construct in combination with a second lentivirus expressing PGC-1αS571A or with the anti-EGFR CAR construct linked with NT-PGC-1α with a 2A sequence (online supplemental figure 1B). Cells were FACS-sorted to increase purity. Flow cytometric analysis of metabolic capacity revealed that transduced T cells with either PGC-1αS571A or NT-PGC-1α had higher mitochondrial mass after 10 days of expansion (figure 2C). Metabolic profiling via Seahorse analysis showed that, consistent with our work and others, overexpression of PGC-1αS571A in CAR-T cells imparted these cells with high respiratory capacity (figure 2D), a metabolic state consistent with long-lived, memory T cells. NT-PGC-1α exhibited a similar effect. Similar increase in glycolytic capacity was noted for both constructs (online supplemental figure 2A, B). Additionally, since it has been previously shown that the 41BB signaling domain used in the CAR construction induces mitochondrial biogenesis,20 we tested mitochondrial capacity of PGC-1α overexpressing CAR T cells on a CD28 co-stimulation backbone. As the anti-EGFR CAR-T was proprietary and not available with a CD28 co-stimulation domain, we used an anti-CD20 CAR-T retroviral construct. Metabolic profiling via Seahorse analysis showed that, overexpression of PGC-1αS571A was sufficient to induce enhanced respiratory capacity on a CAR-T cell with a CD28 co-stimulation domain (online supplemental figure 2C). Furthermore, for the case of PGC-1αS571A overexpressing CAR-T cells we also showed that fatty acid oxidation is increased (online supplemental figure 2D). Finally, post-transduction we cultured the PGC-1αS571A overexpressing CAR-T cells for 6 days under 1.5% hypoxia and the mitochondrial mass benefit was retained (online supplemental figure 2E).
jitc-2022-006522supp003.pdf (710.3KB, pdf)
We then employed RNA sequencing to determine the transcriptional programs associated with PGC-1αS571A transgenesis. Three independent donors were used to generate CAR-T cells expressing PGC-1αS571A, NT-PGC-1α, or the CAR alone, respectively, at day 10 post activation/expansion. Principal component analysis plot analysis showed that whereas PGC-1αS571A has a substantial effect on the T-cell transcriptome, NT-PGC-1α upregulated only a handful of genes (figure 3A, E). Differential expression analysis showed that genes involved in mitochondrial biogenesis as well as IFN signaling were upregulated by PGC-1αS571A transgenesis (figure 3B). Notably, PGC-1αS571A not only coordinated mitochondrial biogenesis but also the expression of fuel choice enzymes (figure 3C), which suggests PGC-1αS571A transgenesis programs T cells to be generally more competitive for exogenous nutrients. In addition, PGC-1αS571A-transduced T cells had higher expression of genes driving effector function (figure 3D). The full list of differentially expressed genes are listed in online supplementary figure 3. Unfortunately, the NT-PGC-1α construct did not produce any of these differences with only a few genes differing between cells (figure 3F).
Figure 3.

PGC-1αS571A, but not NT-PGC1α, induces mitochondrial and immunologic changes within CAR-T cells. (A) Principal component analysis (PCA) of PGC-1αS571A CARs. (B) Differential expressed genes on a volcano plot. FDR, false discovery rate genes with an FDR p value<0.05 were marked with red. (C, D) GSEA of PGC-1αS571A CARs with heatmaps for the top differentially expressed genes in these pathways (E) PCA of NT-PGC-1α CARs (F) All genes that differed statistically in the NT-PGC-1α CARs versus WT CAR analysis. CAR, chimeric antigen receptors; PGC-1α, PPAR gamma coactivator 1α; WT, wild-type; GSEA, Gene Set Enrichment Analysis.
We next sought to determine the efficacy of PGC-1αS571A reprogrammed human CAR-T cells in vivo against their native, tumor-bound antigen. NSG mice bearing 100 mm3 A549 tumors were infused with 107 WT or PGC-1αS571A-transduced anti-EGFR CAR-T cells. While unmanipulated CAR-T cells had no effect on tumor growth and survival, PGC-1αS571A-transduced CAR-T cells had dramatically improved antitumor efficacy (figure 4A), extending survival for all mice (figure 4B). Consistent with our previous data demonstrating that NT-PGC-1α failed to effectively transcriptionally reprogram T cells compared with the full-length construct, CAR-T cells transduced with NT-PGC-1α showed no in vivo efficacy (figure 4C). Surprisingly, analysis of PGC-1αS571A tumor infiltrating CAR-T cells did not reveal any difference in MitoFM staining compared with WT CAR-T cells (figure 4D). A probable reason for this finding is that we found a diminished mCherry signal in the ex vivo analysis of the tumor CAR-T cells on day 10 (online supplemental figure 4A). However when we compared the mCherry+ cells to the mCherry– cells in the same microenvironment the higher MitoFM staining was retained within the mCherry+ fraction (online supplemental figure 4B). Without restricting to the mCherry+ fraction, CAR-T cells exhibited improved capacity to secrete cytokines, despite the fact that these cells expressed similar levels of canonical ‘exhaustion’ co-inhibitory markers like PD-1 and Tim-3 (figure 4E, F), an identical functional and surface phenotype to murine PGC-1α-transduced TCR-Tg T cells. Notably, even within the tumor microenvironment, these T cells bore hallmarks of memory, as shown by expression of CD45RA and CCR7, suggesting their reprogramming allowed them to maintain some hallmarks of stemness associated with long-lived T cells (figure 4G). Thus, through use of an engineered, highly transcriptionally active PGC-1α construct, mitochondrial reprogramming could be imparted to CAR-T cells, allowing their function within the metabolically restrictive tumor microenvironment. Lastly, to alleviate some concerns about clinical manufacturing we assessed for the evidence of possible difficulties in manufacturing or any possibility of transformation. The PGC-1αS571A cells exhibited similar short-term expansion (online supplemental figure 4C), long-term expansion (online supplemental figure 4D) and apoptosis (online supplemental figure 4E) when compared with the WT CAR-T cells.
Figure 4.

Anti-EGFR PGC-1αS571A CAR-T cells, but not NT-PGC1α CAR-T cells, exhibit significant antitumor efficacy in vivo driven by increased cytokine production and memory formation. (A, B) Tumor growth curve and survival of A549-bearing NSG mice treated with 107 anti-EGFR PGC-1αS571A CAR-T cells when tumors reached 100 mm3 (arrow). (C) Tumor growth curve as in A but using 107 anti-EGFR NT-PGC-1α CAR-T cells. (D) MitoTracker FM staining of TIL from tumors treated with unmodified or PGC-1αS571A CAR-T cells. (E) PD-1 and Tim-3 staining from TIL, (F) Cytokine from TIL after 6 hours ex vivo restimulation with phorbol 12-myristate 13-acetate and ionomycin, (G) Memory markers of TIL from day 10. The PGC-1αS571A growth curve was repeated with two donors while the TIL analysis was performed once across multiple mice. The NT-PGC-1α growth curve was repeated with three donors. Statistics are repeated-measures analysis of variance (A, C), log-rank test (B), and Wilcoxon rank-sum test (D–G). *p<0.05, **p<0.01 ***p<0.001. CAR, chimeric antigen receptors; IFN, interferon; PD-1, programmed cell death protein-1; PGC-1α, PPAR gamma coactivator 1α; TIL, tumor-infiltrating lymphocyte; TNF, tumor necrosis factor.
jitc-2022-006522supp005.pdf (491.8KB, pdf)
Discussion
T-cell therapies hold great promise for the immunotherapy of cancer, as they provide an opportunity to directly engineer immune cells to not only recognize and lyse targets after infusion, but to persist as memory cells and guard against recurrence. However, how to endow a T cell with memory potential while still allowing it to behave as an effector for the purposes of killing has been a matter of intense debate. Alterations in the cytokine milieu used to expand T cells and changes in co-stimulatory domains on chimeric receptors have been proposed as means to achieve increased longevity.15 21
Long-lived cells, of any lineage, are associated with increased mitochondrial capacity, and thus it is rather unsurprising that naïve and memory T cells, which have the potential to persist for a lifetime, are characterized by a distinct mitochondrial state.22 This mitochondrial capacity is one that can help define metabolic sufficiency in various environments, and indeed we have shown that losses of mitochondrial sufficiency underlie altered differentiation to dysfunctional, exhausted fates and are targets for the improvement of immunotherapy.11 Thus, it may be that long-lived memory character can be imparted to a cell directly by changing its metabolism, which was the subject of our study here.
Mitochondrial biogenesis is a complex physiologic process, coordinating gene transcription both in the nucleus and the mitochondria. Thus, a single transcription factor is incapable of propagating these complex signals. PGC-1α, rather, acts as a coactivator to collaborate with several transcription factors to achieve this ultimate goal. Indeed, our transcriptional profiling revealed upregulation of rate-limiting enzymes in carbohydrate, amino acid, and lipid metabolism, as well as mitochondrial transporters needed to both import and export metabolites. We also found that by overexpressing this gene involved in mitochondrial biogenesis, we also biased the differentiation of these cells, upregulating genes involved in memory. This was not a complete reprogramming of cell fate: cells still bore markers of terminal exhaustion when they entered the tumor (PD-1, Tim-3, etc), but carried increased functional capacity that was capable of mediating superior antitumor responses. While responding tumors did ultimately relapse, we theorize that this could be due to loss of the CAR antigen on constant antigen exposure.
We evaluated in parallel the NT-PGC-1α because its size would facilitate engineering of the cells compared with the full length PGC-1α. However, to our surprise, the transcriptional profiling of these cells was barely different than the WT CAR. Additionally, they did not exhibit any increase of efficacy in vivo. Although NT-PGC-1α drove mitochondrial benefits in the cells, it is likely that the missing sections of PGC-1α are the ones driving the global alterations in the transcriptional program of the cells. These data highlight that mitochondrial mass, although important, cannot drive benefit by itself, unless it is accompanied by changes in other transcriptional and memory states.
Notably, we took advantage here of the wealth of knowledge of PGC-1α regulation to program superior metabolic sufficiency in solid tumor-targeted, human CAR-T cells. We engineered this molecule to lack an inhibitory Akt phosphorylation site, a key alteration which would render it resistant to regulation by a signaling pathway known to be highly upregulated in hypermetabolic in vitro cultures as well as due to persistent stimulation in vivo. Future studies will seek to further refine our engineering approach to generate not only more active constructs, but those that may be capable of exogenous control.
jitc-2022-006522supp001.pdf (79.8KB, pdf)
jitc-2022-006522supp004.pdf (713KB, pdf)
Acknowledgments
The authors would like to thank those in the flow cytometry and animal facilities of the UPMC Hillman Cancer Center, supported in part by grant P30CA047904 (NIH).
Footnotes
Twitter: @KonLontos, @DelgoffeLab
KL and YW contributed equally.
Contributors: KL performed the majority of the NT-PGC1-α experiments, the transcriptional profiling experiments on 293T cells, the experiments for Figure 1, made the figures and helped write the manuscript. YW performed the majority of the PGC-1a CAR-T experiments and helped write the manuscript. SKJ performed CAR-T cell metabolic profiling and created figures. ATF and AK performed CAR-T therapy experiments and metabolic profiling. MJW performed the RNAseq analysis. AVM and YW assisted with ex vivo sample processing and analysis. EC and BB provided the lentiviral vectors for the anti-EGFR CAR and the PPPARGC1A mutants. GMD conceived of the study, obtained research funding, oversaw the research, and wrote the manuscript. All authors within the UPMC Hillman Cancer Center had full access to the data. All authors approved the manuscript and agreed to proceed with publication.
Funding: This work was supported by TL1 TR001858 to KL, T32CA082084 and F31AI14997-01 to MJW, a National Institutes of Health (NIH) Director’s New Innovator Award (DP2AI136598); the Alliance for Cancer Gene Therapy/Swim Across America, the Sy Holzer Endowed Immunotherapy Fund, the Mark Foundation for Cancer Research Emerging Leader Award, and a Sponsored Research Agreement from bluebird bio to GMD.
Competing interests: GMD has filed patent applications around the use of PGC-1α as a metabolic reprogramming agent in cellular therapies. BB was an employee of 2seventy bio during the period of the research and owns stock of the same company. EC was an employee of 2seventy bio during the period of the research.
Provenance and peer review: Not commissioned; internally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
Not applicable.
References
- 1.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018;359:1350–5. 10.1126/science.aar4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang AC, Postow MA, Orlowski RJ, et al. T-Cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017;545:60–5. 10.1038/nature22079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med 2018;379:64–73. 10.1056/NEJMra1706169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med 2019;380:45–56. 10.1056/NEJMoa1804980 [DOI] [PubMed] [Google Scholar]
- 5.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 2018;378:439–48. 10.1056/NEJMoa1709866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377:2531–44. 10.1056/NEJMoa1707447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munshi NC, Anderson LD, Shah N, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med 2021;384:705–16. 10.1056/NEJMoa2024850 [DOI] [PubMed] [Google Scholar]
- 8.Berdeja JG, Madduri D, Usmani SZ, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet 2021;398:314–24. 10.1016/S0140-6736(21)00933-8 [DOI] [PubMed] [Google Scholar]
- 9.Delgoffe GM, Xu C, Mackall CL, et al. The role of exhaustion in car T cell therapy. Cancer Cell 2021;39:885–8. 10.1016/j.ccell.2021.06.012 [DOI] [PubMed] [Google Scholar]
- 10.DePeaux K, Delgoffe GM. Metabolic barriers to cancer immunotherapy. Nat Rev Immunol 2021;21:785–97. 10.1038/s41577-021-00541-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scharping NE, Rivadeneira DB, Menk AV, et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol 2021;22:205–15. 10.1038/s41590-020-00834-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scharping NE, Menk AV, Moreci RS, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 2016;45:374–88. 10.1016/j.immuni.2016.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dumauthioz N, Tschumi B, Wenes M, et al. Enforced PGC-1α expression promotes CD8 T cell fitness, memory formation and antitumor immunity. Cell Mol Immunol 2021;18:1761–71. 10.1038/s41423-020-0365-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chowdhury PS, Chamoto K, Kumar A, et al. PPAR-induced fatty acid oxidation in T cells increases the number of tumor-reactive CD8+ T cells and facilitates anti-PD-1 therapy. Cancer Immunol Res 2018;6:1375–87. 10.1158/2326-6066.CIR-18-0095 [DOI] [PubMed] [Google Scholar]
- 15.Kawalekar OU, O’ Connor RS, Fraietta JA, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in car T cells. Immunity 2016;44. 10.1016/j.immuni.2016.02.023 [DOI] [PubMed] [Google Scholar]
- 16.Hooper K, Havens K, Krostag A-R, et al. Knockout of cblb greatly enhances anti-tumor activity of CAR T cells. Blood 2018;132:338. 10.1182/blood-2018-99-119247 [DOI] [Google Scholar]
- 17.Li X, Monks B, Ge Q, et al. Akt/Pkb regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature 2007;447:1012–6. 10.1038/nature05861 [DOI] [PubMed] [Google Scholar]
- 18.Southgate RJ, Bruce CR, Carey AL, et al. Pgc-1Alpha gene expression is down-regulated by Akt- mediated phosphorylation and nuclear exclusion of FOXO1 in insulin-stimulated skeletal muscle. FASEB J 2005;19:2072–4. 10.1096/fj.05-3993fje [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Huypens P, Adamson AW, et al. Alternative mRNA splicing produces a novel biologically active short isoform of PGC-1alpha. J Biol Chem 2009;284:32813–26. 10.1074/jbc.M109.037556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Menk AV, Scharping NE, Rivadeneira DB, et al. 4-1Bb costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med 2018;215:1091–100. 10.1084/jem.20171068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alizadeh D, Wong RA, Yang X, et al. Il15 enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol Res 2019;7:759–72. 10.1158/2326-6066.CIR-18-0466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buck MD, O’Sullivan D, Klein Geltink RI, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 2016;166:63–76. 10.1016/j.cell.2016.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2022-006522supp002.pdf (604.3KB, pdf)
jitc-2022-006522supp003.pdf (710.3KB, pdf)
jitc-2022-006522supp005.pdf (491.8KB, pdf)
jitc-2022-006522supp001.pdf (79.8KB, pdf)
jitc-2022-006522supp004.pdf (713KB, pdf)
Data Availability Statement
Data are available upon reasonable request.