

Abstract
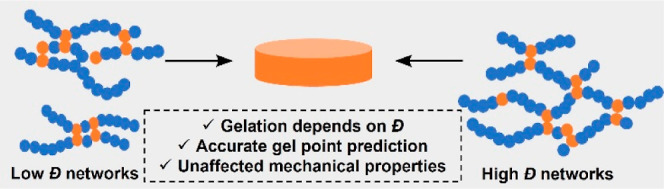
We investigated gelation in photoinduced atom transfer radical polymerization (ATRP) as a function of Cu catalyst loading and thus primary chain dispersity. Using parallel polymerizations of methyl acrylate with and without the addition of a divinyl crosslinker (1,6-hexanediol diacrylate), the approximate values of molecular weights and dispersities of the primary chains at incipient gelation were obtained. In accordance with the Flory–Stockmayer theory, experimental gelation occurred at gradually lower conversions when the dispersity of the primary chains increased while maintaining a constant monomer/initiator/crosslinker ratio. Theoretical gel points were then calculated using the measured experimental values of dispersity and initiation efficiency. An empirical modification to the Flory–Stockmayer equation for ATRP was implemented, resulting in more accurate predictions of the gel point. Increasing the dispersity of the primary chains was found not to affect the distance between the theoretical and experimental gel points and hence the extent of intramolecular cyclization. Furthermore, the mechanical properties of the networks, such as equilibrium swelling ratio and shear storage modulus showed little variation with catalyst loading and depended primarily on the crosslinking density.
Introduction
Crosslinked polymer networks, such as thermosets, elastomers, or (hydro)gels are materials of crucial industrial importance. One of the main ways to synthesize polymer networks is free radical polymerization (FRP) of vinyl monomers with small amounts of divinyl crosslinkers; such a reaction leads to highly branched chains which eventually form an infinite network at the so-called “gel point”.1−3
Crosslinking in FRP can be described by the classical Flory–Stockmayer (FS) mean-field theory3−5 which predicts that at the critical moment of gelation,
| 1 |
where vc is the weight-average number of crosslinks per primary chain (equal to 1 at incipient gelation), ρ is the fraction of the double bonds residing on the divinyl crosslinker, pc is the conversion of the double bonds, and DPw is the weight-average degree of polymerization of the primary chains in the absence of crosslinks. In FRP, polymers with high molecular weights (MWs) form immediately in the reaction; hence, gelation typically occurs at low conversions. However, the experimental gel points are still 1–2 orders of magnitude higher than those predicted by the FS theory; this is mainly caused by the intramolecular crosslinking and cyclization reactions, not accounted for by the theory,2,3,5 which result in the formation of local microgels and spatially inhomogeneous networks.6,7
Among countless polymer architectures enabled by the development of reversible deactivation radical polymerization (RDRP), these techniques have also introduced a fundamentally different mechanism of gelation.3,8,9 In RDRP, fast initiation and linear growth of uniform chains result in a greatly delayed (or even avoided) gelation and slow formation of branched polymer chains, leading to more homogeneous networks than in FRP. Indeed, experimental gel points in RDRP are observed at higher monomer conversions as well as closer to the theoretical values than in FRP due to less intramolecular cyclization and no microgelation.7−9 This effect is universal regardless of the activation/deactivation mechanism and was observed in nitroxide-mediated polymerization,10,11 atom transfer radical polymerization (ATRP),12−16 and reversible addition-fragmentation transfer (RAFT) polymerization.17−19
By applying the FS theory to RDRP systems, Gao and Matyjaszewski derived an expression to calculate theoretical gel points (see the Supporting Information for derivation)8,16
| 2 |
where pc is the conversion of vinyl bonds at incipient gelation, [PC]t is the instantaneous concentration of primary chains at the gel point, [X]0 is the initial concentration of the crosslinker, and D̵ is the dispersity of the primary chains in the absence of crosslinks. Furthermore, [PC]t can be approximated as [I]0 × IEt, where [I]0 is the initial concentration of the alkyl halide initiator in ATRP (or the chain transfer agent in RAFT), and IEt is the initiation efficiency (i.e., Mn,theo/Mn,exp) at the gel point.
From eq 2, gelation in ATRP depends on the initial ratio of the initiator and crosslinker and on D̵ of the primary chains at the gel point. Interestingly, D̵ in ATRP can be tuned by changing the catalyst loading in activator regeneration ATRP methods such as activator regenerated by electron transfer (ARGET)20−23 or photoinduced ATRP.24,25 Indeed, Li et al. have previously reported that decreasing the catalyst loading in ARGET ATRP of methyl acrylate (MA) with a crosslinker led to earlier gelation due to the higher D̵ of the primary chains, however without comparing theoretical and experimental gel points.26
Notably, the difference between the theoretical and experimental gel points can be an important and useful parameter. This is because the intramolecular cyclization events, predominantly responsible for this discrepancy, result in topological defects (loops) in the network structure, which have a detrimental effect on mechanical properties such as swelling and rubber-like elasticity.27 While some strategies to determine and even limit loop formation in step-growth polymerization and vulcanized networks have been developed,28−31 similar approaches to quantify cyclization in chain-growth polymerization remain elusive. So far, the only method was proposed by Rosselgong and Armes who used 1H and 13C NMR to quantify the extent of intramolecular cyclization in branched PMMA copolymerized with a disulfide dimethacrylate crosslinker by RAFT.32,33 However, this method can be only employed for soluble branched polymers (i.e., pregelation) containing disulfide groups.
Gel point analysis can thus serve as an indirect way to estimate the loop content in gels/networks, but it requires careful calculation and comparison of the experimental and theoretical values. Notably, the effect of primary chain dispersity must be considered as higher D̵ leads to earlier gelation (Scheme 1). Unfortunately, theoretical gel points calculated by eq 2 are usually underestimated due to the long-debated inapplicability of the FS theory to predict gelation in RDRP. More accurate gel points were obtained from computer simulation methods such as kinetic modeling or Monte Carlo and found to be significantly higher and closer to experiments than those calculated by the FS theory.12,15,34−37
Scheme 1. Illustration of the Effect of (a) Primary Chain Dispersity and (b) Intramolecular Cyclization on Gelation and Network Formation in ATRP.
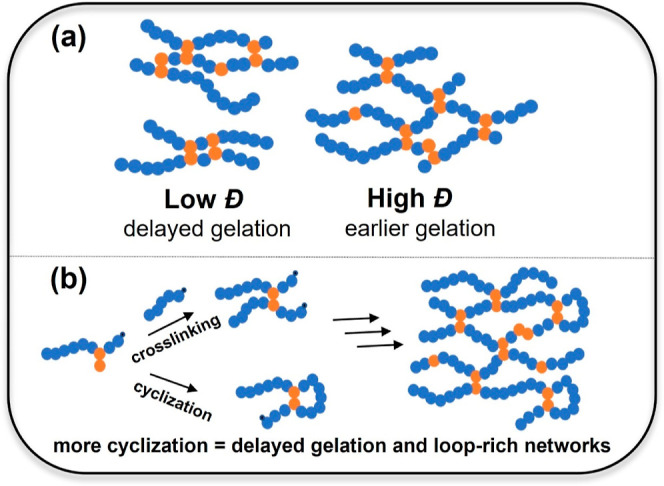
Additionally, reports on the structural characterization of networks prepared by RDRP techniques have only recently started to emerge. For example, Appel et al. proposed a gel point normalization method in order to account for crosslinks lost due to intramolecular cyclization and rationalize the synthesis of branched/network architectures by RAFT.38 Konkolewicz and Matyjaszewski employed degradable crosslinkers to compare the structure and mechanical properties of networks prepared by ATRP and RAFT.39,40 However, an interdependence between gelation kinetics, primary chain dispersity, and physical properties of networks from RDRP has not yet been studied.
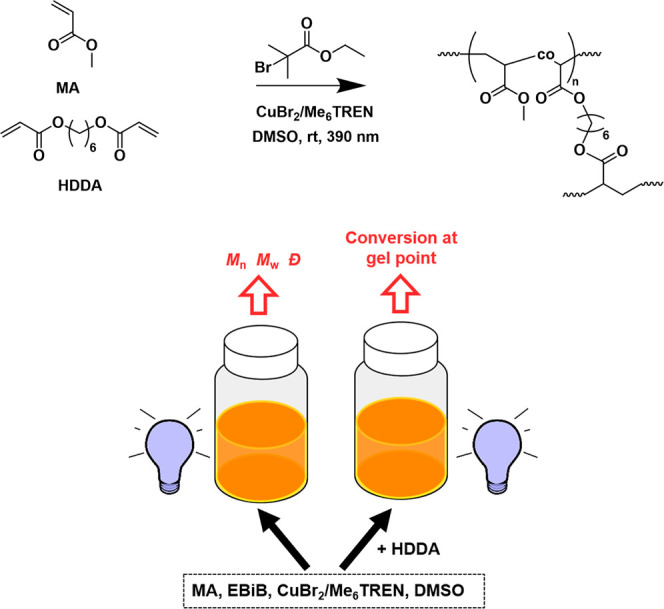
In this work, we investigate the effect of catalyst loading, and thus primary chain dispersity, on both experimental and theoretical gel points as well as on the mechanical properties of poly(methyl acrylate) (PMA) networks prepared by ATRP. A parallel reaction setup was used where two identical polymerizations are conducted simultaneously, with the only difference being the addition of a crosslinker (Scheme 2). In this way, the gel point conversion as well as MW and D̵ of the primary chains could be measured at once (assuming that the primary chains in the networks are comparable with linear polymers obtained under identical conditions). Photoinduced ATRP of MA in dimethyl sulfoxide (DMSO) with 1,6-hexanediol diacrylate (HDDA) as a crosslinker was chosen as a model reaction due to the possibility of quickly turning both reactions off immediately after gelation. Moreover, photoinduced ATRP allows facile tuning of dispersity by changing the Cu catalyst loading.24,25
Scheme 2. Overall Reaction Scheme and Illustration of the Experimental Setup for Parallel Photoinduced ATRP of MA with and without a Crosslinker.
Experimental Part
Materials
MA (99%, Alfa Aesar), ethyl α-bromoisobutyrate (EBiB, 98%, Alfa Aesar), tris 2-(dimethylamino)ethyl amine (Me6TREN, 98%, Alfa Aesar), copper(II) bromide (CuBr2, 99%, Alfa Aesar), HDDA (99%, Alfa Aesar), DMSO (Fischer), and dimethylformamide (DMF, Fischer) were used as received. MA and HDDA were passed through a column of basic alumina to remove the inhibitor before polymerization.
Gel Point Determination in Photoinduced ATRP of MA
A 20 mL sample vial was charged with 10 mL of MA (110 mmol, 100 equiv), 10 mL of DMSO, 0.163 mL (1.11 mmol, 1 equiv) of EBiB, and an appropriate amount of CuBr2/Me6TREN stock solution in DMSO to obtain the desired catalyst loading. A few drops of DMF were added as an internal NMR standard. The contents of the sample vial were then divided by half into two separate vials, and an appropriate amount of HDDA was added to one of the vials, namely, either 0.373 mL (1.67 mmol, 3 equiv) or 0.187 mL (0.832 mmol, 1.5 equiv). Both vials were fitted with a rubber seal and degassed with nitrogen for 15 min. After the initial sample was taken, each vial was irradiated by a Kessil PR106L-390 LED lamp (52 W, λmax = 390 nm, set to 100% intensity). Samples were taken at time intervals for 1H NMR and gel permeation chromatography (GPC) measurements to determine monomer conversion in both reactions and the MW and dispersity of the primary chains (non-crosslinking reaction). The reactions were conducted until a gel was observed in the polymerization with a crosslinker, defined as the moment when the reaction mixture lost its mobility upon vial inversion. Both reactions were then turned off, and a final sample was taken from the nongelled reaction.
Preparation of Fully Developed PMA Networks
A 20 mL sample vial was charged with 7 mL of MA (77.7 mmol, 100 equiv), 7 mL of DMSO, and 0.114 mL of EBiB (0.78 mmol, 1 equiv.) and stirred for 10 min 3 mL portions of this mixture were added to three vials with no screw top to make triplicate gel samples for each composition. An appropriate amount of HDDA was added to each vial, either 0.056 mL (0.25 mmol, 1.5 equiv) or 0.112 mL (0.5 mmol, 3 equiv). A few drops of DMF were added to the remaining 5 mL of MA solution to allow the reaction to be monitored for conversion by NMR. The appropriate amount of CuBr2/Me6TREN stock solution was added to the vials to reach the desired catalyst loading. The vials were fitted with a rubber seal and degassed with nitrogen for 30 min. The samples were irradiated by a Kessil PR106L-390 LED lamp (52 W, λmax = 390 nm, set to 50% intensity) for 10 h. The gels were removed from the vials and immediately washed and dried.
Swelling Analysis
The whole gel disks were washed in 4 × 15 mL of acetone, leaving each washing cycle overnight to ensure that all sol fraction was removed. After the final washing cycle, the mass of the swollen gel disk was recorded as mswollen. The disk was then dried in air overnight and then in a vacuum oven for 24 h. The dry gel disk mass was recorded as mdry. The equilibrium swelling ratio (ESR) was calculated as ESR = mswollen/mdry. The measurements were performed in triplicate.
Instrumentation
1H NMR (Bruker AVANCE 400 MHz) was used to determine monomer conversions in CDCl3 using DMF as an internal reference. GPC measurements were performed on Agilent 1260 Infinity fitted with an autosampler, dual-angle light scattering system, viscometer, and refractometer, with 2 × PL gel 5 μM Mixed D columns and a guard column, with THF as the mobile phase (kinetic samples) or with 2 × PolarGel-M 8 μM columns and DMF as the mobile phase (fully developed networks pregelation). MWs were calculated using linear poly(methyl methacrylate) standards. Oscillatory rheology measurements were carried out using a TA Instruments Discovery HR-3 rheometer fitted with a 20 mm crosshatched parallel-plate geometry and a crosshatched base plate. Dry disk-shaped network samples with a thickness of 2.5 mm were assessed under a constant axial force of 1.5 N. Frequency sweeps were carried out at 25 °C over a range of 0.01–10 rad–1 at a constant strain of 1%.
Results and Discussion
Experimental Gel Points
Photoinduced ATRP of MA in DMSO with the Me6TREN catalyst was conducted at two different [EBiB]/[HDDA] ratios, namely, 1:3 and 1:1.5, and five different catalyst loadings, namely, 200, 100, 50, 20, and 10 ppm, versus the monomer (Table 1). A single DPtarget = 100 was used as it should not significantly influence the gel point as suggested by eq 2 and observed experimentally.37 Importantly, to minimize the effect of dilution on the occurrence of intramolecular cyclization,12,16,35 the MA/DMSO ratio was kept constant at 1:1 (v/v), resulting in a relatively high initial monomer concentration [MA]0 = 5.55 M. Each crosslinking polymerization was accompanied by an analogous reaction conducted in the absence of HDDA. Both polymerizations were allowed to run until gel was observed in the vial containing the crosslinker.
Table 1. Overview of the Parallel Photoinduced ATRP Reactions with Various Catalyst Loadings Conducted in This Work.
| [MA]/[EBiB]/[HDDA] | CuBr2/Me6TREN (ppm) | with crosslinker |
no crosslinker |
|||||
|---|---|---|---|---|---|---|---|---|
| entry | apc,exp | gelation time (min) | bMn,theo | bMn,GPC | cIE | bD̵ | ||
| 1 | 100:1:3 | 200 | 0.62 | 25 | 5890 | 6970 | 0.85 | 1.19 |
| 2 | 100:1:3 | 100 | 0.55 | 30 | 4640 | 5270 | 0.88 | 1.29 |
| 3 | 100:1:3 | 50 | 0.50 | 50 | 4970 | 6120 | 0.81 | 1.30 |
| 4 | 100:1:3 | 20 | 0.50 | 70 | 4520 | 5040 | 0.90 | 1.69 |
| 5 | 100:1:3 | 10 | 0.48 | 105 | 4360 | 4470 | 0.98 | 2.16 |
| 6 | 100:1:1.5 | 200 | 0.77 | 35 | 6830 | 7540 | 0.91 | 1.17 |
| 7 | 100:1:1.5 | 100 | 0.72 | 31 | 6530 | 7110 | 0.92 | 1.20 |
| 8 | 100:1:1.5 | 50 | 0.70 | 35 | 6170 | 7530 | 0.82 | 1.25 |
| 9 | 100:1:1.5 | 20 | 0.63 | 70 | 5320 | 6180 | 0.86 | 1.62 |
| 10 | 100:1:1.5 | 10 | 0.63 | 110 | 5840 | 6100 | 0.96 | 1.86 |
Estimated conversion at the gel point, based on data from before gelation and from a corresponding linear polymerization.
Measured by GPC for a sample taken at a conversion closest to the gel point in a corresponding non-crosslinked reaction. All GPC traces are shown in Figures S1–S10.
IE = Mn,theo/Mn,GPC.
Figure 1a,c shows the conversion versus reaction time plots for all studied conditions. In all cases, the reaction with HDDA was a little slower than its linear counterpart, most likely due to the lower kp of HDDA and a slightly higher DPtarget caused by the addition of a difunctional crosslinker. Decreasing the catalyst loading resulted in slower reactions, which was expected as, in activator regeneration ATRP methods, the overall rate of polymerization (Rp) is controlled by the rate of reduction of CuII to CuI which, in turn, depends on the initial concentration of CuII.41 GPC traces of all linear polymers show monomodal distributions with MWs close to the theoretical values (Table 1 and Figures S1–S10). The initiation efficiencies (IEt = Mn,theo/Mn,GPC) varied between 0.81 and 0.98, and dispersities between 1.17 and 2.16. Notably, the IEt values remain constant or are slightly higher for reactions with less catalyst, indicating good control over the polymerization at low catalyst loadings. Indeed, while decreasing the catalyst loading in ATRP with activator regeneration leads to higher D̵ (due to slower deactivation), it does not result in excessive termination as the radical concentration also decreases with less catalyst (as reflected by slower polymerization).41 This is in line with previous works on dispersity tuning in ATRP which showed no loss of control at Cu catalyst loadings down to 10 ppm versus the monomer.22
Figure 1.

(a,c) Conversion of the vinyl bonds in photoinduced ATRP of MA with (open symbols) and without (closed symbols) the presence of the HDDA crosslinker performed at various CuBr2/Me6TREN catalyst loadings. The final open star symbols in polymerizations with HDDA denote the estimated gel point; (b,d) dependence of dispersity and experimental gel points on the catalyst loading. Dispersities were recorded by GPC of the polymerization without a crosslinker at a conversion closest to the experimental gel point. The corresponding GPC traces are shown in Figures S1–S10.
Gelation was monitored by taking samples at time intervals from both reactions and measuring the conversion of the double bonds (MA and HDDA) by 1H NMR. Once the system gelled, it was no longer possible to take samples, so the gel point was estimated (open star symbols in Figure 1a,c) based on the previous measurements and the data from the parallel linear polymerization. Due to the abovementioned differences in polymerization rates, gelation took longer with less catalyst; however, it occurred at gradually lower conversions. Specifically, at [EBiB]:[HDDA] ratio = 1:3, conversion at the gel point (pc,exp, Table 1) was 62% when the polymerization was run with 200 ppm of the catalyst (resulting in D̵ = 1.19) and decreased to 48% at 10 ppm catalyst (D̵ = 2.16). When the [EBiB]/[HDDA] ratio was lowered to 1:1.5, gel points were naturally higher, but displayed the same trend, that is, gelation occurred at 77% conversion with 200 ppm of the catalyst (D̵ = 1.17), decreasing to 63% with 10 ppm (D̵ = 1.86). This trend is in agreement with the previous report of Li et al.26 as well as with the FS theory.4,5 When less catalyst is used, the rate of deactivation decreases and more monomer/crosslinker units are added to a growing radical during each activation/deactivation cycle, leading to the formation of more high-MW components and thus broadening the MW distribution. These high MW chains will proportionally contain more crosslinks, resulting in earlier gelation than in a homogeneous (i.e., monodisperse) system (see Scheme 1).5
Theoretical Gel Points
As mentioned in the Introduction, the discrepancy between theoretical and experimental gel points is caused primarily by the intramolecular cyclization events occurring in any crosslinking polymerization. In RDRP, experimental gel points are typically observed closer to the theoretical values than in FRP; however, the correct determination of the theoretical gel points is not straightforward. FS theory has long been considered insufficient for the prediction of gel points in RDRP, and more accurate values are usually obtained by various computer simulation methods.12,15,34−37
Assuming monodisperse chains, eq 2 predicts that the gel point will depend solely on the [I]0/[X]0 ratio and reach values of 0.41 and 0.58 for [EBiB]/[HDDA] = 1:3 and 1:1.5, respectively, as indicated by the dashed lines in Figure 2. In order to more precisely calculate gel points for the investigated conditions, we corrected these values using the IEt and D̵ from linear polymerizations (Table 1). This allows us to calculate Flory-Stockmayer theoretical gel points (pc,theo,FS) for each catalyst loading/primary chain dispersity. We note that initiation efficiencies do not vary by more than 0.17, so the effect of D̵ (ranging from 1.17 to 2.16) is more pronounced.
Figure 2.

Comparison of experimental (pc,exp) and theoretical FS (pc,theo,FS) gel points calculated from eq 2 using D̵ and IEt values from linear polymerizations. (a) [MA]/[EBiB]/[HDDA] = 100:1:3; (b) [MA]/[EBiB]/[HDDA] = 100:1:1.5. Dashed lines correspond to the ideal case when D̵ and IEt = 1.
However, such calculated theoretical gel points differ quite significantly from the experimental values (Figure 2). Previously, Gao et al. used kinetic simulations (Predici) to determine theoretical gel points in ATRP of MA with a crosslinker (assuming ideal living polymerization, i.e., no termination and dispersity given by Poisson distribution D̵ = 1 + 1/DPn) and obtained the following empirical expression12
| 3 |
where X = [X]0/[I]0. Applying eq 3 to our system gives pc,theo,emp of 0.58 and 0.82 for [EBiB]/[HDDA] ratios of 1:3 and 1.5, respectively. Both are higher not only than our experimental values but also than the “ideal” cases calculated for D̵ and IEt = 1 from eq 2, highlighting the different scaling of pc,theo with [X]0 in RDRP than that predicted by the FS theory.
Interestingly, for typical
values of X, eq 3 can be approximated with
an excellent agreement by a simple expression  (see Figure S11), suggesting a more general relationship. Indeed, looking at a logarithmic
form of eq 2
(see Figure S11), suggesting a more general relationship. Indeed, looking at a logarithmic
form of eq 2
| 4 |
where f is the crosslinker
functionality. The second term on the right-hand side of eq 4, namely, 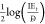 , equals 0 when both IEt and D̵ = 1. Indeed, the maximum gel
point should occur at D̵ = 1 (i.e., monodisperse
primary chains) and decrease with higher D̵ (and/or lower IEt), as predicted by
the FS theory and shown experimentally in this work and previously.26
, equals 0 when both IEt and D̵ = 1. Indeed, the maximum gel
point should occur at D̵ = 1 (i.e., monodisperse
primary chains) and decrease with higher D̵ (and/or lower IEt), as predicted by
the FS theory and shown experimentally in this work and previously.26
The first term, namely, 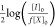 , should therefore give values of pc,theo identical to eq 3 for a given [I]0/[X]0 ratio, corresponding to a maximum
possible gel point in the system, that is, for IEt = 1 and D̵ = 1, similar to those obtained
in simulations.12,15,34−36 However, this is not the case as all simulated gel
points are consistently higher by a factor of
, should therefore give values of pc,theo identical to eq 3 for a given [I]0/[X]0 ratio, corresponding to a maximum
possible gel point in the system, that is, for IEt = 1 and D̵ = 1, similar to those obtained
in simulations.12,15,34−36 However, this is not the case as all simulated gel
points are consistently higher by a factor of 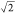 than those calculated from the FS theory
(eq 2).
than those calculated from the FS theory
(eq 2).
Therefore, we propose a modification of eq 4 to correct for this discrepancy, namely
| 5 |
or
| 6 |
When f = 2, this further
reduces to 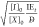 , reflecting the relationship suggested
before (Figure S11). Equation 5 or 6 gives the same
values as the empirical eq 3 when IEt and D̵ are both equal to unity but allows us to calculate pc,theo,emp for any IEt and D̵ value.
, reflecting the relationship suggested
before (Figure S11). Equation 5 or 6 gives the same
values as the empirical eq 3 when IEt and D̵ are both equal to unity but allows us to calculate pc,theo,emp for any IEt and D̵ value.
Inserting our experimental data of IEt and D̵ in eq 6 gives pc,theo,emp values much closer to the experimental gel points than those calculated previously from eq 2 (Figure 3 and Table 2). However, it should be stressed that while eqs 5 and 6 allow very accurate gel point predictions, in line with previously reported computer simulations, they should be treated as empirical and approximate. Their full derivation would probably require a modification of the FS theory to account for “living” polymerization conditions.37 This is beyond the scope of the current study but will be investigated in the future.
Figure 3.

Comparison of experimental (pc,exp) and theoretical (pc,theo,emp) gel points calculated from eq 6 using D̵ and IEt values from linear polymerizations. (a) [MA]/[EBiB]/[HDDA] = 100:1:3; (b) [MA]/[EBiB]/[HDDA] = 100:1:1.5. Dashed lines correspond to the ideal case when D̵ and IEt = 1.
Table 2. Comparison of Experimental and Theoretical Gel Points Calculated from eqs 2 and 6.
| entry | [EBiB]0/[HDDA]0 | [cat] (ppm) | IEt | D̵ | apc,exp | bpc,theo,FS | cpc,theo,emp |
|---|---|---|---|---|---|---|---|
| 1 | 1:3 | 200 | 0.85 | 1.19 | 0.62 | 0.34 | 0.49 |
| 2 | 1:3 | 100 | 0.88 | 1.29 | 0.55 | 0.34 | 0.47 |
| 3 | 1:3 | 50 | 0.81 | 1.30 | 0.50 | 0.32 | 0.45 |
| 4 | 1:3 | 20 | 0.90 | 1.69 | 0.50 | 0.30 | 0.42 |
| 5 | 1:3 | 10 | 0.98 | 2.16 | 0.48 | 0.27 | 0.39 |
| 6 | 1:1.5 | 200 | 0.91 | 1.17 | 0.77 | 0.51 | 0.72 |
| 7 | 1:1.5 | 100 | 0.92 | 1.20 | 0.72 | 0.51 | 0.71 |
| 8 | 1:1.5 | 50 | 0.82 | 1.25 | 0.70 | 0.47 | 0.66 |
| 9 | 1:1.5 | 20 | 0.86 | 1.62 | 0.63 | 0.42 | 0.59 |
| 10 | 1:1.5 | 10 | 0.96 | 1.86 | 0.63 | 0.41 | 0.59 |
Nevertheless, such calculated theoretical gel points still follow the assumptions of the FS theory, namely, equal reactivity of all vinyl bonds and no intramolecular cyclization. As such, they stay below the experimental values, with the average ratio between the theoretical and experimental values pc,theo,emp/pc,exp = 0.850 ± 0.056 and 0.948 ± 0.02 for [HDDA]0 = 3 and 1.5, respectively, reflecting more intramolecular cyclization at higher crosslinker concentrations. Importantly, this ratio is not influenced by dispersity: even though increasing D̵ of the primary chains leads to earlier gelation, it should not affect the relative probability of inter- and intramolecular reactions and hence the number of effective crosslinks.
Mechanical Properties of PMA Networks
Finally, the impact of changing the catalyst loading/dispersity on the properties of PMA networks was investigated. A series of gels were prepared by continuing the polymerization past the gel point for 10 h to maximize the monomer conversion in all reactions (see Table 3 and Figure S12). Networks were synthesized with 20, 50, and 100 ppm of the catalyst for each crosslinker content (namely, [EBiB]0/[HDDA]0 = 1:1.5 and 1:3). The obtained network samples were thoroughly washed in acetone and then left in acetone for 24 h to fully swell. The samples were then dried to measure their ESR (Figure 4a). An expected difference in ESR between networks with [HDDA]0 = 1.5 and 3 was clearly visible. However, no difference in swelling was observed for networks with [HDDA]0 = 3 prepared at various catalyst concentrations as all samples showed ESR ≈ 3.5. Networks with [HDDA]0 = 1.5 showed slightly more variation, with ESR ≈ 10, 11, and 15 for 20, 50, and 100 ppm of the catalyst, respectively. This is similar to the recent work by Wanasinghe et al. who reported higher ESR in PMA networks synthesized by PET–RAFT, which displayed lower D̵ of the primary chains than analogous networks prepared under traditional RAFT conditions and attributed this difference to their more homogeneous structure.40 However, given the larger measurement error in networks with [HDDA]0 = 1.5, this discrepancy is not substantial.
Table 3. ESR and Storage Shear Modulus of PMA Networks Prepared at Different Crosslinker and Catalyst Concentrations.
| entry | [EBiB]0/[HDDA]0 | [cat] (ppm) | aconversion (%) | bESR | cG′ at 0.1 rad s–1 (MPa) |
|---|---|---|---|---|---|
| 1 | 1:3 | 100 | >95 | 3.5 ± 0.04 | 0.03 |
| 2 | 1:3 | 50 | 90 | 3.7 ± 0.8 | 0.03 |
| 3 | 1:3 | 20 | 89 | 3.6 ± 0.5 | 0.04 |
| 4 | 1:1.5 | 100 | >95 | 15.4 ± 1.8 | 0.005 |
| 5 | 1:1.5 | 50 | >95 | 11.3 ± 1.0 | 0.007 |
| 6 | 1:1.5 | 20 | 83 | 10.0 ± 1.4 | 0.012 |
Measured by 1H NMR for reactions without HDDA after 10 h.
Average value from three measurements, determined gravimetrically by swelling in excess acetone for 4 × 24 h.
Determined by oscillatory rheology for dry networks.
Figure 4.

(a) ESRs in acetone and (b) shear storage moduli of dry PMA networks synthesized by photoinduced ATRP with different catalyst loadings. All samples were prepared in triplicate, and shown data are average from at least three separate measurements.
These results were further supported by a rheological analysis of the shear storage moduli (G′) of dried networks (Figure 4b). At low angular frequency (0.1 rad/s), corresponding to the relaxed state, virtually no difference in G′ of networks with [HDDA]0 = 3 and varied dispersities can be seen, corroborating similar crosslinking density suggested by the swelling analysis. Again, more variation was observed in the [HDDA]0 = 1.5 series of networks. G′ increased with decreasing catalyst loading, from 0.005 MPa for 100 ppm to 0.012 MPa for 20 ppm of the catalyst. However, this difference is relatively small, especially compared with the modulus difference between the samples with different crosslinker contents. Therefore, the effect of changing the dispersity of primary chains on the elastic response of the network is not significant and should only be considered when the material has a low overall crosslinking density. At a higher [X]0, gelation occurs earlier in the reaction, and the distribution of the crosslinks is more uniform throughout the gel, leading to robust and reproducible networks. Similarly, reducing the catalyst loading also leads to gelation occurring earlier and increases the G′ value of the resulting gel due to the more even crosslink distribution. This effect is apparent in the series of gels with [X]0 = 1.5. At a higher [X]0, however, the influence of D̵ becomes less important as there are enough crosslinks to maintain uniform crosslinking density and produce better-defined networks.
Overall, both swelling analysis and rheology confirm that the dispersity of the primary chains does not greatly affect the properties of polymer networks made by ATRP, which are mainly controlled by the initial [I]0/[X]0 ratio. Only at low [X]0, when gelation occurs at high conversions, increased dispersity of the primary chains can influence the properties by allowing more crosslinks to develop earlier. Furthermore, the occurrence of inevitable intramolecular cyclization, which affects the effective crosslinking density, is not influenced by the primary chain dispersity and is constant for a given [I]0/[X]0 ratio, as evidenced by gel point analysis.
Conclusions
In summary, we investigated how experimental and theoretical gel points are affected by the dispersity of the primary chains in photoinduced ATRP of MA with a diacrylate crosslinker. Decreasing the catalyst loading has two effects on the kinetics of network formation: it results in slower polymerization, that is, longer gelation time; however, it happens at a lower conversion due to the presence of more high MW components and earlier incorporation of the critical number of crosslinks in the primary chains with higher dispersity.
Careful analysis of the theoretical gel points and previously reported kinetic models of gelation in ATRP allowed us to propose a modified FS equation to accurately predict gelation in RDRP techniques. A comparison of theoretical and experimental gel points, combined with the analysis of mechanical properties of the synthesized networks showed that crosslinking density and network structure are not significantly influenced by the MW distribution of the strands and depend primarily on the crosslinker content.
These results can help to better design networks/gels synthesized by ATRP and other RDRP techniques, as well as guide efforts to control or even eliminate the occurrence of intramolecular cyclization reactions in chain-growth polymerization, leading to defect-free polymer networks. From a more practical perspective, Cu catalyst loading in ATRP can be diminished in network synthesis without a significant impact on their mechanical properties, which can be useful, for example, in the preparation of hydrogels for biomedical applications.
Acknowledgments
This work was supported by the RSC Research Enablement grant no. E21-2719779284 and EPSRC New Investigator Award grant no. EP/W034778/1. H.J. acknowledges funding within the RSC Undergraduate Summer Bursary no. U21-2424523550. V.R. thanks Infineum Ltd. for funding an undergraduate summer bursary. M.K. thanks Dr. Wojciech Kopeć (Max Planck Institute for Multidisciplinary Sciences) for helpful discussions. Analytical facilities were provided through the Material and Chemical Characterization Facility (MC2) at the University of Bath.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.macromol.2c02159.
The authors declare no competing financial interest.
Supplementary Material
References
- Gu Y.; Zhao J.; Johnson J. A. Polymer Networks: From Plastics and Gels to Porous Frameworks. Angew. Chem., Int. Ed. 2020, 59, 5022–5049. 10.1002/anie.201902900. [DOI] [PubMed] [Google Scholar]
- Matsumoto A.Free-radical crosslinking polymerization and copolymerization of multivinyl compounds. In Synthesis and Photosynthesis; Springer Berlin Heidelberg: Berlin, Heidelberg, 1995; pp 41–80. [Google Scholar]
- Gao Y.; Zhou D.; Lyu J.; Xu Q.; Newland B.; Matyjaszewski K.; Tai H.; Wang W. Complex polymer architectures through free-radical polymerization of multivinyl monomers. Nat. Rev. Chem 2020, 4, 194–212. 10.1038/s41570-020-0170-7. [DOI] [PubMed] [Google Scholar]
- Stockmayer W. H. Theory of Molecular Size Distribution and Gel Formation in Branched Polymers II. General Cross Linking. J. Chem. Phys. 1944, 12, 125–131. 10.1063/1.1723922. [DOI] [Google Scholar]
- Flory P. J.Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, 1953. [Google Scholar]
- Seiffert S. Origin of nanostructural inhomogeneity in polymer-network gels. Polym. Chem. 2017, 8, 4472–4487. 10.1039/c7py01035d. [DOI] [Google Scholar]
- Yu Q.; Xu S.; Zhang H.; Ding Y.; Zhu S. Comparison of reaction kinetics and gelation behaviors in atom transfer, reversible addition–fragmentation chain transfer and conventional free radical copolymerization of oligo(ethylene glycol) methyl ether methacrylate and oligo(ethylene glycol) dimethacrylate. Polymer 2009, 50, 3488–3494. 10.1016/j.polymer.2009.05.032. [DOI] [Google Scholar]
- Gao H.; Matyjaszewski K. Synthesis of functional polymers with controlled architecture by CRP of monomers in the presence of cross-linkers: From stars to gels. Prog. Polym. Sci. 2009, 34, 317–350. 10.1016/j.progpolymsci.2009.01.001. [DOI] [Google Scholar]
- Bagheri A.; Fellows C. M.; Boyer C. Reversible Deactivation Radical Polymerization: From Polymer Network Synthesis to 3D Printing. Adv. Sci. 2021, 8, 2003701. 10.1002/advs.202003701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide N.; Fukuda T. Nitroxide-Controlled Free-Radical Copolymerization of Vinyl and Divinyl Monomers. Evaluation of Pendant-Vinyl Reactivity. Macromolecules 1997, 30, 4268–4271. 10.1021/ma9700946. [DOI] [Google Scholar]
- Ide N.; Fukuda T. Nitroxide-Controlled Free-Radical Copolymerization of Vinyl and Divinyl Monomers. 2. Gelation. Macromolecules 1999, 32, 95–99. 10.1021/ma9805349. [DOI] [Google Scholar]
- Gao H.; Min K.; Matyjaszewski K. Determination of Gel Point during Atom Transfer Radical Copolymerization with Cross-Linker. Macromolecules 2007, 40, 7763–7770. 10.1021/ma071324b. [DOI] [Google Scholar]
- Gao H.; Miasnikova A.; Matyjaszewski K. Effect of Cross-Linker Reactivity on Experimental Gel Points during ATRcP of Monomer and Cross-Linker. Macromolecules 2008, 41, 7843–7849. 10.1021/ma801724z. [DOI] [Google Scholar]
- Gao H.; Li W.; Matyjaszewski K. Synthesis of Polyacrylate Networks by ATRP: Parameters Influencing Experimental Gel Points. Macromolecules 2008, 41, 2335–2340. 10.1021/ma702823b. [DOI] [Google Scholar]
- Gao H.; Polanowski P.; Matyjaszewski K. Gelation in Living Copolymerization of Monomer and Divinyl Cross-Linker: Comparison of ATRP Experiments with Monte Carlo Simulations. Macromolecules 2009, 42, 5925–5932. 10.1021/ma901005d. [DOI] [Google Scholar]
- Gao H.; Li W.; Min K.; Matyjaszewski K.. Gelation in Atom Transfer Radical Copolymerization with a Divinyl Cross-linker. In Controlled/Living Radical Polymerization: Progress in ATRP; American Chemical Society, 2009; Vol. 1023, pp 203–213. [Google Scholar]
- Moad G. RAFT (Reversible addition-fragmentation chain transfer) crosslinking (co)polymerization of multi-olefinic monomers to form polymer networks. Polym. Int. 2015, 64, 15–24. 10.1002/pi.4767. [DOI] [Google Scholar]
- Lin F.-Y.; Yan M.; Cochran E. W. Gelation Suppression in RAFT Polymerization. Macromolecules 2019, 52, 7005–7015. 10.1021/acs.macromol.9b00707. [DOI] [Google Scholar]
- Wang R.; Luo Y.; Li B.-G.; Zhu S. Modeling of Branching and Gelation in RAFT Copolymerization of Vinyl/Divinyl Systems. Macromolecules 2009, 42, 85–94. 10.1021/ma802006c. [DOI] [Google Scholar]
- Listak J.; Jakubowski W.; Mueller L.; Plichta A.; Matyjaszewski K.; Bockstaller M. R. Effect of Symmetry of Molecular Weight Distribution in Block Copolymers on Formation of “Metastable” Morphologies. Macromolecules 2008, 41, 5919–5927. 10.1021/ma800816j. [DOI] [Google Scholar]
- Plichta A.; Zhong M.; Li W.; Elsen A. M.; Matyjaszewski K. Tuning Dispersity in Diblock Copolymers Using ARGET ATRP. Macromol. Chem. Phys. 2012, 213, 2659–2668. 10.1002/macp.201200461. [DOI] [Google Scholar]
- Wang Z.; Yan J.; Liu T.; Wei Q.; Li S.; Olszewski M.; Wu J.; Sobieski J.; Fantin M.; Bockstaller M. R.; Matyjaszewski K. Control of Dispersity and Grafting Density of Particle Brushes by Variation of ATRP Catalyst Concentration. ACS Macro Lett. 2019, 8, 859–864. 10.1021/acsmacrolett.9b00405. [DOI] [PubMed] [Google Scholar]
- Yin R.; Wang Z.; Bockstaller M. R.; Matyjaszewski K. Tuning dispersity of linear polymers and polymeric brushes grown from nanoparticles by atom transfer radical polymerization. Polym. Chem. 2021, 12, 6071–6082. 10.1039/d1py01178b. [DOI] [Google Scholar]
- Whitfield R.; Parkatzidis K.; Rolland M.; Truong N. P.; Anastasaki A. Tuning Dispersity by Photoinduced Atom Transfer Radical Polymerisation: Monomodal Distributions with ppm Copper Concentration. Angew. Chem., Int. Ed. 2019, 58, 13323–13328. 10.1002/anie.201906471. [DOI] [PubMed] [Google Scholar]
- Rolland M.; Lohmann V.; Whitfield R.; Truong N. P.; Anastasaki A. Understanding dispersity control in photo-atom transfer radical polymerization: Effect of degree of polymerization and kinetic evaluation. J. Polym. Sci. 2021, 59, 2502–2509. 10.1002/pol.20210319. [DOI] [Google Scholar]
- Li W.; Gao H.; Matyjaszewski K. Influence of Initiation Efficiency and Polydispersity of Primary Chains on Gelation during Atom Transfer Radical Copolymerization of Monomer and Cross-Linker. Macromolecules 2009, 42, 927–932. 10.1021/ma8025833. [DOI] [Google Scholar]
- Gu Y.; Zhao J.; Johnson J. A. A (Macro)Molecular-Level Understanding of Polymer Network Topology. Trends Chem. 2019, 1, 318–334. 10.1016/j.trechm.2019.02.017. [DOI] [Google Scholar]
- Zhou H.; Woo J.; Cok A. M.; Wang M.; Olsen B. D.; Johnson J. A. Counting primary loops in polymer gels. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 19119–19124. 10.1073/pnas.1213169109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M.; Wang R.; Kawamoto K.; Olsen B. D.; Johnson J. A. Quantifying the impact of molecular defects on polymer network elasticity. Science 2016, 353, 1264. 10.1126/science.aag0184. [DOI] [PubMed] [Google Scholar]
- Gu Y.; Kawamoto K.; Zhong M.; Chen M.; Hore M. J. A.; Jordan A. M.; Korley L. T. J.; Olsen B. D.; Johnson J. A. Semibatch monomer addition as a general method to tune and enhance the mechanics of polymer networks via loop-defect control. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 4875–4880. 10.1073/pnas.1620985114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wang R.; Gu Y.; Sourakov A.; Olsen B. D.; Johnson J. A. Counting loops in sidechain-crosslinked polymers from elastic solids to single-chain nanoparticles. Chem. Sci. 2019, 10, 5332–5337. 10.1039/c9sc01297d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosselgong J.; Armes S. P. Quantification of Intramolecular Cyclization in Branched Copolymers by 1H NMR Spectroscopy. Macromolecules 2012, 45, 2731–2737. 10.1021/ma3002609. [DOI] [Google Scholar]
- Rosselgong J.; Armes S. P. Extent of intramolecular cyclization in RAFT-synthesized methacrylic branched copolymers using 13C NMR spectroscopy. Polym. Chem. 2015, 6, 1143–1149. 10.1039/c4py01441c. [DOI] [Google Scholar]
- Polanowski P.; Jeszka J. K.; Li W.; Matyjaszewski K. Effect of dilution on branching and gelation in living copolymerization of monomer and divinyl cross-linker: Modeling using dynamic lattice liquid model (DLL) and Flory–Stockmayer (FS) model. Polymer 2011, 52, 5092–5101. 10.1016/j.polymer.2011.08.055. [DOI] [Google Scholar]
- Polanowski P.; Jeszka J. K.; Krysiak K.; Matyjaszewski K. Influence of intramolecular crosslinking on gelation in living copolymerization of monomer and divinyl cross-linker. Monte Carlo simulation studies. Polymer 2015, 79, 171–178. 10.1016/j.polymer.2015.10.018. [DOI] [Google Scholar]
- Lyu J.; Gao Y.; Zhang Z.; Greiser U.; Polanowski P.; Jeszka J. K.; Matyjaszewski K.; Tai H.; Wang W. Monte Carlo Simulations of Atom Transfer Radical (Homo)polymerization of Divinyl Monomers: Applicability of Flory–Stockmayer Theory. Macromolecules 2018, 51, 6673–6681. 10.1021/acs.macromol.8b01630. [DOI] [Google Scholar]
- Lyu J.; Li Y.; Li Z.; Polanowski P.; Jeszka J. K.; Matyjaszewski K.; Wang W. Modelling Development in Radical (Co)Polymerization of Multivinyl Monomers. Angew. Chem., Int. Ed. 2023, 62, e202212235 10.1002/anie.202212235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann J. L.; Rossi R. L.; Smith A. A. A.; Appel E. A. Universal Scaling Behavior during Network Formation in Controlled Radical Polymerizations. Macromolecules 2019, 52, 9456–9465. 10.1021/acs.macromol.9b02109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert J.; Wanasinghe S. V.; Matyjaszewski K.; Konkolewicz D. Are RAFT and ATRP Universally Interchangeable Polymerization Methods in Network Formation?. Macromolecules 2021, 54, 8331–8340. 10.1021/acs.macromol.1c01587. [DOI] [Google Scholar]
- Wanasinghe S. V.; Sun M.; Yehl K.; Cuthbert J.; Matyjaszewski K.; Konkolewicz D. PET-RAFT Increases Uniformity in Polymer Networks. ACS Macro Lett. 2022, 11, 1156–1161. 10.1021/acsmacrolett.2c00448. [DOI] [PubMed] [Google Scholar]
- Matyjaszewski K.; Jakubowski W.; Min K.; Tang W.; Huang J.; Braunecker W. A.; Tsarevsky N. V. Diminishing Catalyst Concentration in Atom Transfer Radical Polymerization with Reducing Agents. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 15309–15314. 10.1073/pnas.0602675103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.