

ABSTRACT
Resistance-nodulation-division (RND) superfamily efflux pumps promote antibiotic resistance in Gram-negative pathogens, but their role in Gram-positive pathogens, including methicillin-resistant Staphylococcus aureus (MRSA) is undocumented. However, recent in vitro selections for resistance of S. aureus to an antimicrobial fatty acid, linoleic acid, and an antibiotic, rhodomyrtone, identified H121Y and C116R substitution variants, respectively, in a TetR family regulator, FarR, promoting increased expression of the RND pump FarE. Hypothesizing that in vivo selection pressures have also promoted the emergence of FarR variants, we searched available genome data and found that strains with FarRH121Y from human and bovine hosts have emerged sporadically in clonal complexes (CCs) CC1, CC30, CC8, CC22, and CC97, whereas multiple FarR variants have occurred within CC5 hospital-associated (HA)-MRSA. Of these, FarRE160G and FarRE93EE were exclusive to CC5, while FarRC116Y, FarRP165L, and FarRG166D also occurred in nonrelated CCs, primarily from bovine hosts. Within CC5, FarRC116Y and FarRG166D strains were polyphyletic, each exhibiting two emergence events. FarRC116Y and FarRE160G were individually sufficient to confer increased expression of FarE and enhanced resistance to linoleic acid (LA). Isolates with FarRE93EE were most closely related to S. aureus N315 MRSA and exhibited increased resistance independently of FarRE93EE. Accumulation of pseudogenes and additional polymorphisms in FarRE93EE strains contributed to a multiresistance phenotype which included fosfomycin and fusidic acid resistance in addition to increased linoleic acid resistance. These findings underscore the remarkable adaptive capacity of CC5 MRSA, which includes the polyphyletic USA100 lineage of HA-MRSA that is endemic in the Western hemisphere and known for the acquisition of multiple resistance phenotypes.
KEYWORDS: MRSA, Staphylococcus aureus, TetR family regulator, efflux pumps, mechanisms of resistance
INTRODUCTION
Efflux pumps of the resistance-nodulation-division (RND) superfamily are well known for conferring vital resistance mechanisms toward antibiotics and chemotherapeutic agents (1). This efflux pump superfamily was discovered in Gram-negative enteric bacteria through mutations which conferred sensitivity to xenobiotic compounds such as acriflavine and acridine (2, 3), and it is now apparent that one of its major physiological functions is to facilitate colonization of the gut through efflux of bile salts or related detergents and microbial metabolites (4–8). Consequently, regulation can be complex and derives input from stress-related signals and multiple transcriptional activators (9). However, a common theme is their control by TetR family regulators (TFRs) which repress their expression in the absence of an inducing stimulus (10–12). Moreover, because RND efflux pumps can accommodate a range of structurally unrelated antimicrobial compounds, a paradigm of emergence of resistance during antimicrobial therapy of Gram-negative pathogens is attributed to mutations in the TFR repressor or its cognate DNA binding motif, leading to de-repression of the efflux pump (13–16).
This resistance paradigm is well-established in Gram-negative pathogens, but the functions of RND pumps and their contribution to the emergence of resistance is less well-defined in Gram-positive bacteria. Of particular interest, Staphylococcus aureus colonizes the nose and skin in approximately 30% of the human population but is also a leading cause of infectious morbidity and mortality and a significant threat to public health due to the emergence of resistance to multiple antimicrobial agents (17, 18). An important innate defense mechanism that deters colonization of S. aureus and other pathogens is antimicrobial unsaturated free fatty acids (uFFA), which bacteria are exposed to in secretions of the upper respiratory tract and sebaceous secretions of the skin (19, 20). Consistent with the paradigm for emergence of resistance through altered expression of RND pumps, we conducted in vitro selection for increased resistance to linoleic acid (LA) in the USA300 strain of community-acquired methicillin-resistant S. aureus (CA-MRSA), which led to the recovery of a H121Y variant in a previously uncharacterized TFR which we designated FarR; this led to enhanced resistance through increased expression of the divergently transcribed RND efflux pump FarE (21). Similarly, others have identified a FarRC116R variant that confers resistance to the plant-derived antimicrobial rhodomyrtone (22).
In related work, S. aureus MRSA strain COL was subjected to in vitro selection for increased resistance to an oxadiazole antibiotic, representing a new class of non-β-lactam antibiotics which also target penicillin-binding proteins in Gram-positive bacteria (23, 24). This promoted a T172I substitution in SACOL2566 encoding efflux pump MmpL (23), which is identical to the efflux pump FarE (SAUSA300_2489) that we have described in S. aureus USA300. Consequently, in vitro selections for enhanced resistance of S. aureus to various antimicrobial agents have identified amino acid substitutions in FarR or in the FarE/MmpL efflux pump (21–23). In view of these considerations, and the notoriety of S. aureus in acquiring resistance to antibiotics, we hypothesized that in vivo exposure to either antimicrobial therapy or host-derived antimicrobial fatty acids could be a driving force in the emergence of strains with altered RND efflux pump expression. However, unlike other TFRs which primarily repress expression of the target efflux pump, FarE was not expressed in the absence of FarR (21), making it unlikely that enhanced resistance could be achieved through mutations that inactivate farR function or expression. Here, we report on our search for amino acid substitution variants in FarR across the spectrum of S. aureus sequenced genomes. We found that variant FarR proteins have repeatedly emerged in the CC5 lineage of health care-associated MRSA (HA-MRSA), of which FarRC116Y and FarRE160G are individually sufficient to promote increased resistance to LA.
RESULTS
Assessment of FarR variation in the context of S. aureus phylogenetic diversity.
We first assessed variation in FarR across all S. aureus genomes that were designated complete (as of 2020) in the Pathosystems Resource Information Center (PATRIC) database (25) (Table S1 in the supplemental material), which currently has data on 21,099 genomes. These 574 completed genomes yielded 563 full-length FarR proteins, for which multiple alignment revealed 28 variations that could be grouped into 6 primary clusters (Table 1 and Fig. S1). The FarR clusters exhibited a high degree of conservation, with the most diverse cluster, 6, exhibiting 88% identity and 95% similarity relative to cluster 1. The relatedness of the FarR clusters and proportion of strains associated with each are shown as a Grapes plot in Fig. 1A, and these clusters are mapped on a S. aureus phylogenetic tree constructed from conceptually translated proteomes of the 574 strains using PhyloPhlAn3 (Fig. 1B). Each variant was also used to match identical proteins in the NCBI database of S. aureus subsp. aureus proteins. The number of identical protein accessions for each variant and their clonal complex associations are reported in Table 1; for contextual purposes, the total number of genomes from the PATRIC database that correspond to specific multilocus sequence type (MLST) designations are provided as a footnote in Table 1. Eleven strains did not have full-length FarR proteins due to single-nucleotide deletions, transposon or IS element insertions, and, in the case of one strain, a deletion that spanned farR and most of the farE efflux pump (Fig. 1B, Table S1).
TABLE 1.
Clonal complex association of six primary FarR clusters and associated variantsa
| FarR cluster | Primary sequence and variantsb | CC(s)c | Protein accessions (n)d |
|---|---|---|---|
| 1 | MKETDLRVIKTKKALSSSLLQLLEQQLFQTITVNQICDNALVHRTTFYKHFYDKYDLLEYLFNQLTKDYFARDISDRLNHPFQTMSDTINNKEDLREIAEFQEEDAEFNKVLKNVCIKIMHNDIKNNRDRIDIDSDIPDNLIFYIYDSLIEGFIHWIKDEKIDWPGEDIDNIFHKVINIKIK | 30, 239 | 2,649 |
| 1a | D134Y | 30 | 40 |
| 2 | MKETDLRVIKTKKALSSSLLQLLEQQLFQTITVNQICDNALVHRTTFYKHFYDKYDLLEYLFNQLTKDYFARDISDRLNHPFQTMSDTINNKEDLREIAEFQEEDAEFNKVLKNVCIKIMHNDIKNNRDRIDIDSDIPDNLIFYIYDSLIEGFIHWIKDEKIDWPGEDIDNIFHRLINIKIK | 5, 6, 7, 8, 9, 15, 25, 72, 80, 88, 97, 101, 772, 1,156 | 12,894 |
| 2a | E160G | 5 | 82 |
| 2b | G166D | 5, 8, 97 | 9 |
| 2c | E93EE | 5 | 61 |
| 2d | V115I | 5 | 3 |
| 2e | R96K | 5 | 4 |
| 2f | M85I | 395 | 11 |
| 2g | A14P | 5 (ST228) | 32 |
| 2h | S16L | 5 (ST228) | 2 |
| 2i | R77H | 97 | 11 |
| 2j | R130H | 8 | 6 |
| 2k | K110V | 8 | 2 |
| 2l | S75G | 45, 198 | 716 |
| 2m | L78I,N79T | 97 | 1 |
| 2n | S17T | 101 | 36 |
| 3 | MKETDLRVIKTKKALSSSLLQLLEQQLFQTITVNQICDNALVHRTTFYKHFYDKYDLLEYLFNQLTKDYFARDISDRLNHPFQTISDTINNKEDLREIAEFQEEDIEFNKVLKNVCIKIMHDDIKNNRDRIDIDSDVPDNLIFYIYDSLIEGFuHWIKDEKIDWPGEDIDNIFHRLINIKIK | 93 | 266 |
| 3a | I106S | Outgroup | 125 |
| 4 | MKETDLRVIKTKKALSSSLLQLLEQQLFQTITVNQICDNALVHRTTFYKHFYDKYDLLEYLFNQLTKDYFARDISDRLNHPFQTISDTINNKEDLRDIAEFQEEDAEFNKVLKNVCIKIMHNDIKNNRDRIDIDSDIPDNLIFYIYDSLIEGFMHWIKDEKIDWPGEDIDNIFHRLINIKIK | 12, 20 | 167 |
| 5 | MKETDLRVIKTKKALSSSLLQLLEQQLFQTITVNQICDNALVHRTTFYKHFYDKYDLLEYLFNQLTKDYFARDISDRLNHPFQTISDTINNKEDLRDIAEFQEEDAEFNKVLKNVCIKIMHNDIKNNRDRIDIDSDIPDNLIFYIYDSLIEGFMHWIKDEKIDWPGEEIDKIFHKVINIKIK | 1, 188, 1,148 | 1,128 |
| 5a | Q35L | 398, 2,272 | 1,530 |
| 5b | Q35L, V115F | 398 | 1 |
| 5c | D136H | 22 | 2,440 |
| 5d | D136H, I142T | 22 | 2 |
| 6 | MKETDLRVIKTKKALSSSLLQLLEQHLFQTITVNQICHNALVHRTTFYKHFYDKYDLLEYLFNQLTKAYFATDISDRLNHPFQTINDTINNKEDLQKVADFQQEDAEFNKVLKNVCIKIMNDDIKNNSDRIDVDGDIPNNLLFYIYDSLIEGFLHWIKDEKIDWPSEEIDKIFHKVINIKIK | 425 | 2 |
| 6a | D147G | 50, 130, 133, 121, 152, 350 | 841 |
| 6b | D147G, H80L | 133 | 2 |
| 6c | D147G, H155Y | 705 | 48 |
| 6d | D147G, S128C | 705, 707 | 89 |
| 6e | D147G, I154M | 59 | 481 |
CC, clonal complex.
The primary sequence of each cluster is shown. For clusters 2 to 6, amino acids which differ from those in cluster 1 are identified by bold underline. Variants are listed alphabetically and are defined by amino acid substitutions that differ from the primary cluster sequence.
Bold font designates clonal complexes that are phylogenetically separated from other strains in the same FarR cluster. For contextual purposes, the total number of genomes in the PATRIC database is 21,424, of which the numbers (n) corresponding to specific multilocus sequence types (MLSTs) are ST30 (562), ST239 (537), ST5 (3,484), ST8 (3135), ST97 (236), ST395 (3), ST228 (64), ST45 (636), ST101 (21), ST93 (56), ST12 (65), ST20 (54), ST1 (630), ST188 (201), ST1148 (1), ST398 (1,314), ST22 (2431), ST425 (20), ST50 (10), ST130 (28), ST133 (65), ST121 (250), ST152 (89), ST350 (2), ST705 (0), ST707 (8), and ST59 (481).
Number of protein accessions identical to the primary clade or intraclade variant sequences, as determined by BLASTP of each sequence versus S. aureus subsp. aureus nonredundant proteins.
FIG 1.

Grapes plot of six major FarR clusters and associated variants (A) and mapping of FarR clusters on a proteome based Staphylococcus aureus phylogenetic map (B). For the Grapes plot (A), the minimum spanning tree was constructed by alignment of 564 full-length FarR proteins using GrapeTree, which identified 6 primary clusters and associated variants. Points represent groups of identical elements, with point size correlated with number of elements on a log scale. Phylogenetic distance is scaled to two single-amino acid polymorphisms (2SAAP). The asterisk on cluster 2 (*) marks FarR variant 2c where duplication of an amino acid codon at E93 alters the alignment. For the phylogenetic map (B), conceptually translated nucleotide sequences available from PATRIC are shown for 574 S. aureus and 1 S. argenteus strains using PhyloPhlAn3. Initial phylogenetic analysis used FastTree, which was refined with RaxML. The six primary FarR clusters are mapped on ring 1, while clonal complex associations are mapped on ring 2. The colored legend for clonal complex designations is presented in the same order of appearance as on ring 2, beginning with CC5 and progressing in descending order in each column from left to right, ending with the S. argenteus outgroup.
The proteome level phylogenetic tree is broadly congruent with previous DNA-based analyses and provides insight into the evolution of FarR in concert with phylogenetic diversity. Clusters 1 and 2 were both FarRM85, while clusters 3 to 6 were FarRI85 (Fig. S1, Table 1). Cluster 3 appears to reflect the evolution of S. aureus as a species because the primary cluster 3 FarR matched 266 protein accessions, some of which occurred in ST93 S. aureus (Table 1); however, identical proteins also occurred in S. argenteus, S. schweitzeri, S. roterodami, and S. singaporensis (data not shown). Moreover, cluster 3a occurred exclusively in the S. argenteus outgroup at the bottom of the phylogenetic tree (Fig. 1B). The FarR clusters were variable in the extent to which their associated clonal complexes conform to phylogenetic divisions, with different extremes noted in clusters 5 and 6. Cluster 6 and its variants all populate a single phylogenetic branch, whereas cluster 5 strains are dispersed across the phylogenetic spectrum (Fig. 1B), which likely represents historical recombination events involving farR.
The primary cluster 2 FarR protein matched 12,894 protein accessions across 14 clonal complexes, comprising a large section of the phylogenetic spectrum (Table 1, Fig. 1B). Cluster 2 was exceptional in exhibiting 14 variants, most of which occurred in the same clonal complexes represented among primary cluster 2 strains. Two exceptions were 2f (CC395) and 2l (CC45 and CC198), adjacent to the S. argenteus outgroup. At the nucleotide level, cluster 2f from CC395 shared some polymorphisms with ST93 cluster 3 farR (Fig. S2); and, as with cluster 3 FarR, cluster 2f was also M85I (Table 1, Fig. S1). Among the other cluster 2 variants, we noted non-conserved amino acid substitutions within CC5 strains, as evidenced by clusters 2a FarRE160G and 2b FarRG166D and an amino acid duplication in cluster 2c FarRE93EE (Table 1). These represent the emergence of FarR variants within CC5, independent of phylogenetic diversity.
Multiple FarR variants occur in CC5 S. aureus.
Because our analysis did not reveal FarRH121Y or FarRC116R variants that were previously discovered through in vitro selection procedures (21, 22) we conducted homology searches with each primary FarR cluster protein to search for additional variants in the S. aureus protein database. Although no strains had FarRC116R, several had FarRC116Y, and FarRP165L was also identified (Table 2). Metadata for CC5 FarR variant strains are provided in Table S2, while non-CC5 variants are described in Table S3. FarRE93EE and FarRE160G were exclusive to CC5, while FarRC116Y, FarRP165L, and FarRG166D occurred in CC5 and one or more nonrelated CCs (Table 2, Tables S2 and S3). Among the non-CC5 variants, strains with FarRC116Y were CC1 or CC97 from bovine hosts, while FarRG166D strains were ST8 and CC97 from human and bovine sources, respectively (Table 2 and Table S3). Although FarRH121Y did not occur in CC5, it appeared sporadically in CC1, CC22, CC97, CC8 and CC30 from human and bovine hosts (Table 2, Table S3). Therefore, FarRH121Y, which we discovered through in vitro selection for increased resistance to LA, has emerged sporadically across different clonal complexes but not in CC5, while all other variants occurred in CC5. We considered that the occurrence of FarRH121Y, FarRC116Y, and FarRG166D in different clonal complexes could represent farR exchange through recombination. However, nucleotide sequence alignments revealed that each farR variant has polymorphisms that are specific to the clonal complex from which it was recovered (Fig. S3), consistent with each variant having emerged independently through point mutation in different clonal complexes.
TABLE 2.
FarR variants that occur independently of clonal complex diversitya
| Variant | No. of genomes | MLST/CC | Source |
|---|---|---|---|
| H121Y | 8 | CC1 (ST2922), CC97, CC8, ST30, CC22; (EMRSA-15) | Human (CC1, ST30, ST8, CC22) and bovine (CC97) |
| C116Y | 19 | CC1 (ST3117), CC97 (ST97, ST71, ST3221, ST3109, ST3173), CC8 | Bovine mastitis infection and milk; UK, Switzerland, Ireland, Turkey, Chile |
| 16 | ST5/CC5 | HA-MRSA; US, Canada | |
| E93EE | 61 | CC5 (ST5, ST764) | HA-MRSA; Japan, China, Thailand, Europe, US |
| E160G | 82 | ST5/CC5 | USA100 HA-MRSA; Israel, US, UK, Australia, Europe |
| P165L | 34 | ST5/CC5 | HA-MRSA; US, Canada, Egypt, UK, Europe |
| 2 | ST15 | Scotland; MSSA | |
| G166D | 5 | ST8 and CC97 | Human ST8 (US and Norway) and bovine CC97 (UK) |
| 5 | ST5 | HA-MRSA; US and Netherlands |
MLST, multilocus sequence type; CC, clonal complex; ST, sequence type; HA-MRSA, hospital-acquired MRSA; MSSA, methicillin-susceptible S. aureus.
CC5 and CC97 exhibit a disproportionate frequency of FarR variants compared to CC8.
Our data indicated that CC5 S. aureus was exceptional in exhibiting numerous FarR variants, represented by FarRE93EE, FarRC116Y, FarRE160G, FarRP165L, and FarRG166D, which cumulatively accounted for 198 protein accessions. For an approximation of the frequency of FarR variants, this would represent 5.7% of the 3,477 ST5 S. aureus genomes annotated in the PATRIC database (Table 3). Moreover, while there are a comparable number of ST8 S. aureus genomes in the PATRIC database (n = 3,125), the FarRH121Y, FarRC116Y, and FarRG166D variants which occurred in CC8 represented just 0.1% of ST8 genomes (Table 3). Strikingly, this same repertoire of FarRH121Y, FarRC116Y, and FarRG166D variants accounted for 8.1% of ST97 genomes (Table 3). Although this metadata analysis is subject to caveats, including a much smaller denominator for ST97 genomes, our analysis suggests that FarR variants are overrepresented in CC5 and CC97 compared to CC8.
TABLE 3.
Frequency of FarR variants in ST5 compared to other MLSTs
| MLSTa | No. of genomesb | FarR variants (n) | %Variantsc |
|---|---|---|---|
| 5 | 3,477 | E93EE (61), C116Y (16), E160G (82), P165L (34), G166D (5) | 5.7 |
| 97 | 234 | C116Y (16), H121Y (3), G166D (3) | 8.1 |
| 8 | 3,125 | C116Y (1), H121Y (1), G166D (2) | 0.1 |
| 1 | 631 | C116Y (2), H121Y (1) | 0.3 |
| 22 | 2,440 | H121Y (2) | 0.08 |
MLST, multilocus sequence type.
Number of genomes in S. aureus PATRIC database corresponding to each MLST.
Numerator is the sum of all FarR variants, denominator is the number of genomes.
farR variants are distributed across the S. aureus CC5 phylogeny.
Our data have identified multiple FarR variants within CC5 S. aureus. Importantly, CC5 MRSA encompass multiple clones associated with health care-associated infections in the Western hemisphere (26), including an early branching CC5-basal clade and CC5-I and CC5-II clades which emerged in the early 1970s and early 1960s, respectively, followed by expansion in the Western hemisphere (26). We therefore conducted a phylogenetic comparison of 119 CC5 strains with variant FarR proteins and 26 comparator CC5 reference genomes to assess the distribution of FarR variants within the CC5 phylogeny. Metadata for these strains are provided in Table S2, and a list of polymorphisms that distinguish each strain from the S. aureus strain JH1 reference genome is provided in Table S4. Although our analysis of FarR variation in association with genetic diversity noted that a specific FarR cluster can occur in phylogenetically distinct clonal complexes, which is suggestive of historic recombination events, no evidence of recombination involving farR was detected with ClonalFrameML (CFML); neither in the current analysis of 145 CC5 strains nor in a prior study of 598 CC5 strains (26). Thus, FarR variants within CC5 are most likely the result of independent point mutations.
CC5 strains with variant FarR proteins were predominantly MRSA, since mecA was present in 25 of 34 strains with FarRP165L, 26 of 28 strains with FarRE93EE, and all strains with FarRG166D, FarRC116Y, or FarRE160G (Fig. 2, Table S2). Strains with FarRP165L, FarRG166D, and FarRE93EE emerged in the CC5-basal clade, while FarRE160G and FarRC116Y emerged in CC5-IIA and CC5-IIB, respectively (Fig. 2), which also contains the polyphyletic USA100 lineage of HA-MRSA that is endemic in North America (26, 27). Within the CC5-basal clade, strains with FarRE93EE were most closely related to ST5 MRSA strain N315 from Japan (28), and most of these were also recovered from Japan, China, or Thailand (Fig. 2, Table S2). Conversely, FarRP165L strains in CC5-basal are mainly from North America, but also occur in the United Kingdom, Europe, and Egypt. These strains are predominantly spa type t688 irrespective of their geographic location. FarRG166D strains were polyphyletic, with one isolate from Europe being well separated from four FarRG166D strains in the United States. These latter strains were most closely related to CC5 MRSA strains ISU979 and ISU936 recovered from swine (29). FarRE160G strains emerged in CC5-IIA and were recovered primarily from the US and Israel, but also appeared in the United Kingdom, Europe, and Australia, and are predominantly spa t002, which is characteristic of the USA100 lineage of HA-MRSA. These strains were most closely related to a high-level vancomycin-resistant US strain VRS10 and strain UP109 from Peru, described as a multidrug-resistant strain belonging to the NY/Japan clone in the USA100 lineage (30, 31). FarRC116Y strains within CC5-IIB are restricted to North America but, as with FarRG166D, are polyphyletic, with spa t002 strains from the United States being distinct from spa t5258 and spa t12967 strains from Canada (27). These two distinct emergences of FarRC116Y strains are also most closely related to MRSA progenitors. Therefore, strains with variant farR genes have a global distribution and have emerged multiple times across the phylogenetic spectrum of CC5.
FIG 2.

Distribution of farR variants within the representative worldwide S. aureus CC5 population. The phylogenetic distribution of strains with variant farR genes within the CC5 phylogeny was determined through a two-phase bioinformatics analysis. The first phase consisted of comparing polymorphisms in 119 CC5 strains with single amino acid substitutions in FarR to 598 CC5 reference strains (26), from which 26 reference genomes were selected that (i) subtended the nodes to which the new strains with FarR variants attached, (ii) provided examples of sister nodes of strains with FarR variants, and (iii) provided examples of the various CC5 clades previously defined. In the second phase, these 119 FarR variant strains and 26 reference genomes were analyzed with GATK to call SNPs and indels, as well as invariant core nucleotides relative to S. aureus JH1. The resulting bi-allelic SNPs and invariant core nucleotides were analyzed by PhyML and ClonalFrameML to generate a phylogeny and correct branch length for recombination. Here, strains with variant FarR proteins are placed in the context of CC5 phylogeny. The major CC5 clades, consisting of Basal, CC5-I, and CC5-II (-IIA and -IIB), as defined previously (26), are labeled on the root axis of the dendrogram. Biosample numbers from the NCBI genome sequence entries for each strain are shown adjacent to the branch structures. These strains are listed in the same order in Table S4, which also provides their common names (where available), SCCmec genotypes, and a list of polymorphisms for each strain relative to the S. aureus JH1 reference genome (83). Columns adjacent to the Biosample numbers provide information on country of origin (A), multilocus sequence type (MLST) designation (B), FarR variant (C), and presence or absence of mecA (D).
Some FarR variant strains exhibit increased resistance to linoleic acid.
Although previous in vitro selection procedures for enhanced resistance to LA and rhodomyrtone led to the recovery of FarRH121Y and FarRC116R (21, 22) these did not occur in CC5. We therefore assessed the sensitivity of these strains to LA using the control strains USA300, the USA300-FarRH121Y strain FAR7, and the ST5 MRSA reference strain N315 (28). Because FAR7 has an MIC of >1,200 μM LA (21), we chose FAR7 as a benchmark to assess resistance. USA300 and N315 exhibited negligible growth in 1,200 μM LA, while FAR7 exhibited good growth at a 24-h endpoint. All FarRC116Y strains were resistant to 1,200 μM LA (Fig. 3A and B), as were four FarRE93EE strains we obtained (Fig. 3C) and three out of four FarRE160G variants (Fig. 3D). However, of six FarRP165L strains from the United States and Canada, only the US strain DAR948 exhibited increased resistance (Fig. 3E and F).
FIG 3.
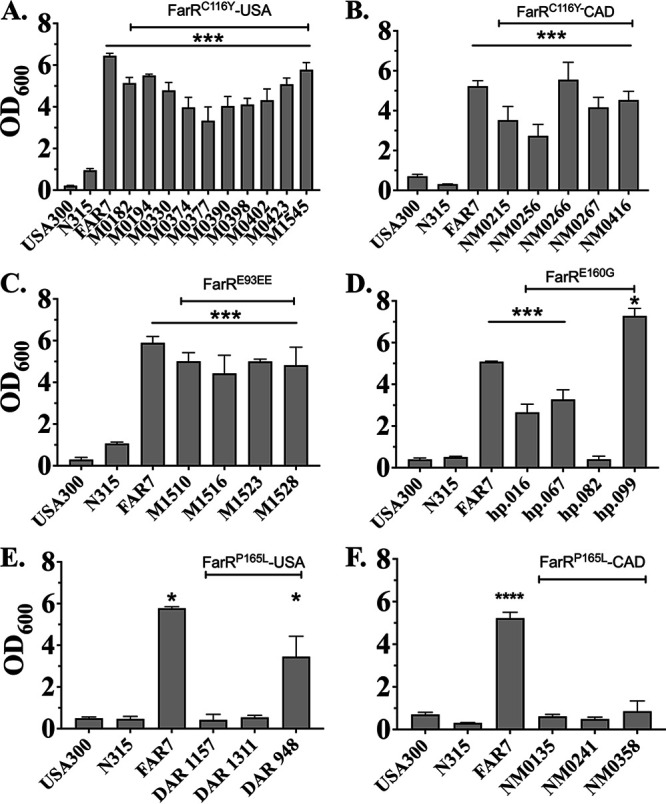
Graphs showing 24-h endpoint growth of USA100 FarR variants in tryptic soy broth (TSB) + 1,200 μM linoleic acid (LA). Cultures of USA300, N315, FAR7, or CC5 MRSA harboring either FarRC116Y from the United States (A) or Canada (B), FarRE93EE (C), FarRE160G (D), or FarRP165L from the United States (E) or Canada (F) were inoculated into triplicate tubes containing 3 mL of TSB + 1,200 μM LA + 0.1% dimethyl sulfoxide (DMSO) at an optical density at 600 nm (OD600) of 0.01, followed by incubation at 37°C with orbital shaking. Growth (OD600) was determined after 24 h. Each data point represents the mean ± standard deviation (SD) from triplicate cultures. Statistically significant differences (****, P < 0.0001; ***, P < 0.001; **, P < 0.01; *, P < 0.05) compared to N315 were determined by Tukey’s multiple-comparison test.
Representative strains were also selected to assess growth in microtiter plates containing tryptic soy broth (TSB) + 200 μM LA, which is sub-MIC for USA300. Under these conditions, the FarRH121Y strain FAR7 reached stationary phase in 7 h, at which time the ST5 HA-MRSA reference strain N315 was just emerging from an extended lag phase (Fig. 4A). The FarRE160G strain hp20814.99, which was resistant to 1,200 μM LA, matched the growth of FAR7, while the single FarRE160G strain hp20814.82 that was not resistant to 1,200 μM LA exhibited similar growth to that of N315. Strains representing the two occurrences of FarRC116Y also exhibited enhanced growth compared to N315 (Fig. 4A), as did the FarRE93EE strain M1516 (Fig. 4B). The FarRP165L strains were variable, with DAR948 and NMRSA0315 showing delayed growth relative to N315, while the growth of DAR1157 was similar to that of N315 (Fig. 4B). These data mirror the outcomes from 24-h MIC endpoints, where FarRC116Y, FarRE160G, and FarRE93EE strains which exhibited MICs of >1,200 μM LA also exhibited enhanced growth in TSB + 200 μM LA, while the FarRP165L strains had no strong association with increased resistance.
FIG 4.

Growth of representative FarR variant strains in TSB + 200 μM LA. Cultures were inoculated to OD600 = 0.01 into 96-well microtiter plates containing 200 μL of TSB supplemented with a subinhibitory concentration of 200 μM LA + 0.1% DMSO. Plates were incubated at 37°C with orbital shaking, and growth (OD600) was monitored hourly. (A) FarRC116Y (M1545, NM0256, NM0256) and FarRE160G (hp.082 and hp.099) strains. (B) FarRE93EE (M1516) and FarRP165L (DAR1157, DAR948, NM0135) strains. Growth was compared to the ST-5 HA-MRSA (health care-associated MRSA) reference strain N315 and FAR7 (FarRH121Y). Each data point represents the mean ± standard error of the mean (SEM) from 6× 200-μL wells in 96-well microtiter plates.
FarRC116Y and FarRE160G are sufficient to promote increased resistance to linoleic acid.
We previously demonstrated that complementation with plasmid pLIfarRH121Y was sufficient to promote enhanced LA resistance when transformed into strain farRΦNE, in which farR is disrupted by a transposon insertion (21). We therefore constructed pLIfarRC116Y, pLIfarRE160G, pLIfarRP165L, and pLIfarRE93EE to determine whether these variant genes confer enhanced resistance in farRΦNE. As expected, pLIfarRH121Y conferred an MIC of >1,200 μM LA, as did pLIfarRC116Y, but the other variants did not (Fig. 5A). However, when LA was reduced to 200 μM, pLIfarRE160G was able to confer growth at a 24-h endpoint (Fig. 5B). We therefore tested the ability of each variant to promote growth at a sub-MIC of 200 μM LA in 96-well microtiter plates. farRΦNE + pLI50 vehicle exhibited a lag phase of more than 20 h, while pLIfarR reduced the lag phase to ~14 h, which was further reduced to 4 to 5 h with pLIfarRH121Y or pLIfarRC116Y (Fig. 5C). The pLIfarRE160G construct also conferred a shorter lag phase relative to pLIfarR (Fig. 5C), whereas pLIfarRP165L was indistinguishable from wild-type pLIfarR, and pLIfarRE93EE conferred a longer lag phase (Fig. 5D). Therefore, we conclude that in addition to FarRH121Y, FarRC116Y and, to a lesser extent, FarRE160G are sufficient to confer increased LA resistance, whereas FarRP165L and FarRE93EE are not.
FIG 5.

farRC116Y and farRE160G are sufficient to confer either increased resistance to, or growth advantage on exposure to, LA. Growth (OD600) of farRΦNE harboring either pLI50 vehicle, pLIfarR, or variants farRH121Y, farRC116Y, farRE160G, farRP165L, or farRE93EE was assessed in TSB containing 1,200 μM (A) or 200 μM (B to D) LA + 0.1% DMSO. For panels A and B, endpoint growth (OD600) in culture tubes was assessed after 24 h, while panels C and D represent growth in microtiter plates with automated monitoring over 24 h. Each value represents the mean ± SD of triplicate 3-mL culture tubes (A and B) or the mean ± SEM of 6× 200-μL wells in 96-well microtiter plates (C and D). Statistically significant differences (***, P < 0.001; **, P < 0.01) compared to farRΦNE + pLIfarR wild type were determined by Tukey’s multiple-comparison test.
Variant farR genes promote increased expression of farE.
To assess how variant farR alleles influence expression of farE, DNA segments containing farR and the adjacent PfarE promoter were cloned in pGYlux such that luciferase expression is driven by PfarE under the control of each farR variant. Cultures of S. aureus farRΦNE containing these constructs were grown in TSB for 3h, followed by the addition of 40 μM LA and monitoring of luminescence at 30-min intervals. Cultures with farRE::lux1 (FarRH121Y) and farRE::lux2 (FarRC116Y) exhibited vastly elevated luminescence relative to wild-type farRE::lux, achieving a sharp peak after 1 h of exposure to LA and then rapidly receding (Fig. 6A and B). For farRE::lux3 (FarRE160G), luminescence was increased relative to farRE::lux after 30 and 60 min, although this was not statistically significant. We therefore conclude that, as with FarRH121Y, FarRC116Y and, to a lesser extent, FarRE160G are also sufficient to confer increased LA resistance to S. aureus clinical isolates due to increased expression of the farE efflux pump.
FIG 6.

farR variants promote increased expression of farE in a farRE::lux luciferase reporter construct. farRΦNE was transformed with either pGYfarRE::lux or variant lux1 (farRH121Y), lux2 (farRC116Y), or lux3 (farRE160G) constructs where luciferase activity is driven from the PfarE promoter under the control of wild-type or variant farR genes. Cultures were grown in 125-mL flasks containing 25 mL TSB for 3 h followed by supplementation with 40 μM LA + 0.1% DMSO. (A) Growth (OD600) and luciferase activity (relative light units [RLU]/OD600) was quantified at 30-min intervals. (B) Comparison of luciferase activity (RLU/OD600) at 1 h after addition of LA. Each data point represents the mean OD600 ± SD from triplicate flasks. Statistically significant differences (****, P < 0.0001) compared to wild-type farER were determined by Tukey’s multiple-comparison test.
Phenotypic traits associated with pseudogenes and other polymorphisms in FarRE93EE strains.
Because strains with FarRE93EE were more resistant to LA through a mechanism which was not due to the variant farR, we conducted an analysis of how they differred from CC5 reference genomes and other variant FarR strains. Strains with FarRE93EE were divided into branches A and B (Table 4, Fig. S4), with the larger B branch being subdivided such that B3 constitutes a new MLST designation, ST764. An accumulation of pseudogenes and other polymorphisms is also evident in the progression from A1 through to B3 (Table 4, Table S5).
TABLE 4.
Pseudogenes and polymorphisms in FarRE93EE variantsa
| Branch | Strains | Gene | Function | Defect |
|---|---|---|---|---|
| A and B | All except MC1031 | JH1_0212 | uhpT hexose phosphate uptake | fs@F301 (A1, A2); fs@I9 (B2, B3) |
| All | JH1_2400 | lysR family regulator | fs@I150 | |
| All except SA-1B | JH1_2130 | ilvC branched-chain amino acid synthesis | fs @E333 | |
| All | JH1_0942 | lipA lipoic acid synthase | Q302* | |
| A1 and A2 | KG-03, KG-18, KG-22, SA-1B, M1K003 | JH1_0395 | glpT glycerol-3-phosphate transporter | fs@S75; fs@S50 |
| A1 | SA-1B | JH1_0994 | lysR family regulator | Y6* |
| A2 | KG-03, KG-18, KG-22 | JH1_2112 | agrA response regulator | fs@F220 |
| JH1_2399 | hutU histidine utilization | fs@K8 | ||
| B2 | All | JH1_0092 | araC family regulator | fs@H82 |
| JH1_1680 | comE competence protein | fs@K104 | ||
| M1523, M1528, M1510, M1516 | JH1_0467 | hsdS endonuclease | fs@K374 | |
| JH1_0584 | fusA elongation factor | V90A, L461S | ||
| JH1_0586 | M20 metallopeptidase | fs@A179 | ||
| JH1_0649 | HAD family hydrolase | fs@K26 | ||
| JH1_0998 | Phospholipid binding protein | S87* | ||
| JH1_1275 | ylmH RNA binding protein | fs@E213 | ||
| JH1_1800 | Alanine dehydrogenase | fs@S340 | ||
| KUH180129 | JH1_0995 | leuA-like isopropylmalate synthase | fs@I24, K294 | |
| JH1_0997 | Membrane protein | fs@G214 | ||
| B3 | All | JH1_0842 | LysE lysine/arginine export | fs@P95 |
| JH1_1456 | Metallopeptidase | fs@G89 | ||
| JH1_2527 | Carboxylesterase/lipase | W436* | ||
| JH1_2610 | eamA efflux pump | S204* | ||
| JH1_2770 | Sulfurtransferase | fs@H284 | ||
| SH-4, SH-3, SH-2 | JH1_0584 | fusA elongation factor | H457Q | |
| SH-4, SH-3, SH-2, M19, M153-2, M392, M212, M209 | JH1_0995 | leuA-like isopropylmalate synthase | fs@I78 | |
| M19, M153-2, M392, M212, M209 | JH1_0521 | Methyltransferase | R66* | |
| JH1_1590 | Short-chain dehydrogenase | fs@A143 | ||
| JH1_2133 | leuC leucine synthesis | fs@A89 | ||
| JH1_2443 | hssR heme response regulator | fs@A128 | ||
| JH1_2484 | fmhA glycyltransferase | fs@K183 | ||
| M392, M212, M209 | JH1_0393 | mepA antimicrobial efflux | fs@S322 | |
| JH1_2112 | agrA response regulator | fs@E141 |
Bold font indicates a series of tandem genes in which inactivating mutations have accrued.
All FarRE93EE strains have a frameshift in SaurJH1_2490 encoding a LysR family regulator, which is adjacent to the hutU (JH1_2399) and hutG (JH1_2402) genes for histidine utilization. Branches A and B are distinguished by frameshifts at two sites in SaurJH1_0213 uhpT encoding a hexose phosphate transporter (Table 4 and Table S5), while Branch A strains also have a frameshift in glpT encoding a glycerol-3-phosphate transporter, and defects in these genes confer resistance to fosfomycin (32). Some strains in B2 and B3 have L461S or H457Q substitutions in FusA (Table 4), which confer resistance to fusidic acid (33, 34). Different strains in B2 and B3 also have frameshifts in co-associated genes SaurJH1_0995, JH1_0997, and JH1_0998, while strain SA-1B in branch A1 has a premature stop codon in a LysR-family regulator encoded by SaurJH1_0994. These genes, as depicted in Fig. S5, comprise an apparent operon that is divergently transcribed from the LysR regulator SaurJH1_0994. Strikingly, different strains in B2 and B3 have frameshifts within three poly(A) tracts in SaurJH1_0995, encoding the putative isopropylmalate synthase-like gene leuA (Fig. S5). Moreover, as with some of the FarRE93EE strains, the most divergent branch of the FarRP165L strains also exhibited a defect in the leuA ortholog encoded by JH1_0995 (Table S4; Fig. S5B), constituting a shared trait among some strains with FarRP165L and FarRE93EE.
In view of these observations, we queried whether loss of gene function could influence resistance to LA using the Nebraska transposon mutant library in S. aureus USA300 (35). USA300ΦΝΕ178 with a transposon insertion in the leuA-like isopropylmalate synthase exhibited increased resistance to LA (Fig. 7A). For inactivation of the lysR regulator of histidine metabolism in ΦΝΕ1511, enhanced growth was conferred in 100 and 200 μM LA, but the MIC was not altered (Fig. 7B), whereas inactivation of uhpT in ΦΝΕ1154 did not significantly alter growth or the MIC (Fig. 7C). Because the function of lysR as a regulator of histidine metabolism has not previously been addressed, we also assessed the growth of USA300 and ΦΝΕ1511 in a chemically defined medium (CDM) with either glucose (CDM-G) or histidine (CDM-H) as a carbon source. Both strains grew well in CDM with glucose, but ΦΝΕ1511 growth was significantly impaired in CDM-H, where glucose was replaced with additional histidine (Fig. 7D).
FIG 7.

Resistance and growth phenotypes of USA300 harboring transposon insertions in genes that exhibit frameshift mutations in FarRE93EE strains. Cultures of USA300 or isogenic variants ΦΝΕ178 (A), ΦΝΕ1511 (B), or ΦΝΕ1154 (C), with transposon insertions in leuA, lysR, or uhpT, respectively, were grown in TSB containing the indicated concentrations of LA + 0.1% DMSO. Each datapoint represents the mean OD600 ± SD from triplicate 3-mL tube cultures after 24 h growth. (D) Growth of USA300 and isogenic ΦΝΕ1511 (lysR::tn) in chemically defined medium containing 0.4% glucose (CDM-G) or 0.25% histidine (CDM-H) as a carbon source. All data points represent the mean ± SD from triplicate 3-mL tube cultures after 24 h growth. Statistically significant differences (****, P < 0.0001; ***, P < 0.001; *, P < 0.05) compared to S. aureus USA300 were determined by Tukey’s multiple-comparison test.
With a frameshift in uhpT, FarRE93EE strains should be deficient in using hexose-6-phosphate as a carbon source and resistant to fosfomycin. Accordingly, the growth of FarRE93EE strain M1516 was significantly impaired in CDM-G6P (CDM supplemented with glucose-6-phosphate) compared to that in CDM, whereas the ST5 HA-MRSA reference strain N315 and a FarRC116Y strain M182, both with a functional uhpT, did not exhibit impaired growth in CDM-G6P (Fig. S6A). Strain M1516 was also resistant to fosfomycin, as predicted (Fig. S6B).
DISCUSSION
We discovered FarR and the divergently transcribed efflux pump FarE through in vitro selection for increased resistance to LA (21, 36), an antimicrobial uFFA that would be encountered at sites of colonization and infection. This selected for FarRH121Y in S. aureus USA300, promoting increased expression of FarE and enhanced resistance to LA. We have now established that this same variant has emerged in S. aureus strains of diverse genetic backgrounds from both human and bovine hosts, but it did not appear to expand within any of these genetic backgrounds and it was not detected among CC5. Conversely, all other FarR variants occurred within CC5 and exhibited a broad geographic distribution. Of these, FarRC116Y and FarRE160G were individually sufficient to promote increased resistance to LA. Others have noted that in vitro selection for resistance to the lipopeptide antibiotic rhodomyrtone led to recovery of FarRC116R in a S. aureus lab strain HG001 (22). We did not detect this variant in any S. aureus strains, whereas FarRC116Y has emerged twice within CC5 MRSA, and in CC1 and CC97 strains from bovine sources. In addressing the mechanism of enhanced resistance to rhodomyrtone, it was found that highly elevated FarE expression promoted the release of membrane phospholipid (37). Consequently, mutations that promote extremely high FarE expression could exert a deleterious phenotype, which may account for the failure of strains with FarRH121Y to expand within any genetic background, while strains with FarRC116R were not detected in any S. aureus clinical isolates.
Over the past 2 decades, ST5 MRSA strains have been among the most common clones causing hospital-acquired infections in the Western hemisphere (26). This is also the principal genetic background for the emergence of high-level vancomycin resistance through the acquisition of novel d-ala ligases, as well as a common background for an intermediate resistance phenotype through the accumulation of point mutations (30, 38). However, CC5 is rivaled by CC8 for prominence of MRSA in the Western hemisphere, and with the emergence of the ST8 USA300 strain of CA-MRSA, evolutionary trends and phylogenetics of ST8 MRSA have also been extensively studied (39, 40) such that if FarR variants have become established within ST8 MRSA, they should have been found in our analysis. This underscores the remarkable adaptive capacity of ST5 MRSA, which, in addition to multiple antimicrobial resistance genes, now includes the emergence of variant FarR proteins.
Here, we have established that FarRC116Y and FarRE160G alone are sufficient to confer increased resistance to LA. It is not known whether the emergence of these variants can be attributed to exposure to antibiotics or elevated concentrations of antimicrobial fatty acids in certain niche environments. However, different in vitro selection procedures using either LA or the lipopeptide rhodomyrtone both led to the recovery of FarR variants with increased resistance (21, 22), while selection for increased resistance to an oxadiazole antibiotic promoted the recovery of a variant FarE protein (23). Exposure to antibiotics in animal-based agricultural practices promotes the emergence of resistance (41): the non-CC5 FarRC116Y and FarRH121Y variants were recovered from bovine isolates on dairy farms, while FarRG166D variants from the United States are most closely related to strains from porcine sources (29). Moreover, strains with FarRC116Y, FarRE160G, FarRE93EE, and FarRG166D all appear to have emerged within CC5 MRSA, such that repeated exposure of MRSA to antibiotics and host-derived antimicrobial fatty acids in the context of nosocomial infections could be a driving force in the emergence of FarR variants in CC5 MRSA.
It remains to be determined whether variant FarR proteins confer a competitive advantage within a particular host niche. However, the occurrence of FarRH12IY, FarRC116Y, and FarRG166D strains within CC1 and CC97 bovine hosts is suggestive of selection for the emergence of such variants in other settings besides human infections, where successful strains would be repeatedly exposed to antibiotics and host-derived antimicrobial fatty acids. Notably, bovine mastitis is often chronic, and FarR variant proteins could confer an advantage in chronic infections because LA is the most abundant antimicrobial fatty acid in tissue abscess homogenates (42). In the context of chronic infections, FarRE160G variants were recovered from patients with infectious endocarditis and cystic fibrosis (43, 44). This includes a study that assessed the evolution of antibiotic resistance in MRSA bacteremia that persisted for at least 2 weeks despite antimicrobial therapy (44). Only 2 of 48 patients conformed to these criteria, and both had infective endocarditis. From our analysis, it is evident that in both cases, the index MRSA at the onset of bacteremia were FarRE160G strains. However, we also note that FarRP165L strains were recovered from patients with cystic fibrosis at different locations in the United States (45, 46), and this variant is not sufficient to confer increased resistance to LA. As such, it is feasible that some FarR variants may promote phenotypes that have yet to be elucidated.
Although four strains that we obtained with FarRE93EE exhibited increased resistance to LA, we could not attribute this to the variant FarR. Other FarE93EE strains we identified included KG-03, which progressed to a vancomycin-intermediate resistance phenotype over the course of prolonged bacteremia therapy (47). Our data also support a trend toward a multiple-resistance phenotype in FarRE93EE strains because all of these have a defect in uhpT, while some are also defective in glpT, and these mutations promote resistance to fosfomycin (32). Fosfomycin has been evaluated as a combination therapy with daptomycin for treatment of MRSA bacteremia with endocarditis (32, 48, 49); this practice could contribute to greater future prevalence of these strains. Some FarRE93EE strains also have amino acid substitutions in FusA that confer resistance to the topical antimicrobial agent fusidic acid, including three FusAH457Q strains with high-level resistance (33), and four with FusAL461S that we obtained for this study.
Although we were not able to precisely define the mechanism by which FarRE93EE strains have increased resistance toward LA, our analysis documents an accumulation of pseudogenes, a trait associated with niche adaptation (50–53). The effects of the pseudogenes include restricted access to carbon sources due to defects in LysR, a regulator of histidine metabolism, as well as uhpT and glpT, which promote uptake of glucose-6-phosphate and glycerol-3-phosphate, respectively. A four-gene operon beginning with SaurJH1_0995, encoding a leuA-like isopropylmalate synthase, was also a target of inactivating mutations in different FarRE93EE strains. Although the FarRE93EE strains we obtained did not have defects in this gene, USA300 ΦΝΕ178, in which this gene (SAUSA300_0879) is disrupted by a transposon insertion, exhibited a significant increase in the MIC for LA, which supports our contention that increased LA resistance can occur through adaptive mechanisms.
As with FarRE93EE, FarRP165L was insufficient to confer increased resistance to LA. Nevertheless, these strains exhibited a broad geographic distribution, being recovered from the United States, Egypt, Denmark, Canada, and the United Kingdom. Most of these were spa t688, including MRSA from patients with cystic fibrosis at two locations in the United States (45, 46), and one MRSA recovered from cheese in Egypt (54). There are also several reports of ST5 spa t688 MRSA recovered from livestock and food sources, in addition to hospital settings (55–60), primarily in Egypt, Kuwait, Algeria, and Italy. Therefore, a future assessment of the co-association of FarRP165L with spa t688 MRSA is warranted.
In summary, we have defined the emergence and spread of S. aureus strains with variant FarR proteins in CC5 MRSA, of which FarRC116Y and FarRE160G confer increased resistance to LA. FarRC116Y variants emerged twice within the CC5 phylogeny but were also frequently recovered in CC97 strains from bovine hosts. In contrast, FarRE160G was unique to CC5, and some of these strains were recovered from patients with infective endocarditis and cystic fibrosis (44, 61). Future questions to address include whether such variants are more common in these patient populations and how these amino acid substitutions contribute to increased expression of FarE. TetR family regulators normally repress expression of RND superfamily efflux pumps such that enhanced efflux-mediated resistance is achieved through mutations which inactivate the TetR repressor. However, in S. aureus, FarE cannot be expressed in the absence of FarR (21, 36), and because a common trait of TetR family regulators is that their affinity for DNA is modulated by binding small molecule ligands (62), it is possible that variant amino acids modify the ligand-binding function to promote enhanced expression of FarE. Future work will address these considerations.
MATERIALS AND METHODS
Cultures, plasmids, and growth conditions.
S. aureus and Escherichia coli strains and plasmids that were constructed for this study and variant FarR strains obtained for this study are listed in Table 5. The S. aureus clinical isolates with variant FarR proteins (FarRE93EE, FarRC116Y, FarRE160G, and FarRP165L) were obtained from collections maintained by the Brigham and Women’s Hospital (Massachusetts Microbiome Center), Anthony Fisher at the University of Iowa Medical Center (43), George Golding with the Canadian Nosocomial Infection Surveillance Program and the Public Health Agency of Canada (27), and Ashley Robinson at University of Mississippi Medical Center (26, 40). A list of S. aureus genomes with variant farR genes and ST5 reference genomes used for phylogenetic construction is provided in Table S1. S. aureus and E. coli were cultured in TSB and LB, respectively, containing 15 g/L agar when required for growth on solid medium. Cultures were maintained at −80°C in 20% glycerol; when required for experimental purposes, cultures from frozen stocks were streaked on agar medium and single colonies were inoculated into polypropylene tubes containing 3 mL of the appropriate broth and grown overnight at 37°C on an orbital shaker at 220 rpm. When necessary for plasmid maintenance, TSB was supplemented with 5 μg/mL chloramphenicol or 3 μg/mL erythromycin, and LB was supplemented with 100 μg/mL ampicillin. When inoculum was needed for experimental purposes, single colonies were inoculated into 3 mL of the appropriate broth in polypropylene snap-cap tubes and were grown overnight at 37°C, rotating at 220 rpm on an orbital shaker.
TABLE 5.
S. aureus and E. coli strains and plasmidsa
| Strain or plasmid | Description | Reference or Biosample ID |
|---|---|---|
| S. aureus | ||
| RN4220 | Restriction endonuclease deficient strain capable of accepting foreign DNA (rk– mk+) | 64 |
| USA300 | CA-MRSA, wild-type strain cured of resistance plasmids | 86, 87 |
| N315 | ST5 HA-MRSA | 88 |
| FAR7 | in vitro-selected FarRH121Y variant of USA300; increased resistance to linoleic acid | 21 |
| farRΦNE | Transposon insertion in farR (SAUSA300_2490); Nebraska library mutant ΝΕ1393 was transduced into plasmid-cured USA300; ErmR | 21, 35 |
| farRΦNE pLI50 | farRΦNE with pLI50 complementation vector; Cmr, Ermr | 21 |
| farRΦNE pGYlux | farRΦNE with pGYlux luciferase reporter vector; Cmr, Ermr | 21 |
| farRΦNE pLIfarRxy | farRΦNE complemented with farRxy cloned in pLI50; farRxy segment derived from USA300, FAR7 (farRH121Y), and ST5 MRSA with farRC116Y, farRE93EE, farRE160G, or farRP165L variants; Cmr, Ermr | 21 |
| farRΦNE pGYfarRE::luxx | farRΦNE with pGYlux vector containing farRxy gene and PfarE promoter segment driving luciferase reporter expression; farRxyE segment derived from USA300 (farRE::lux), FAR7 (farRH121Y; farRE::lux1), and ST5 MRSA with farRC116Y (farRE::lux2) or farRE160G (farRE::lux3) Cmr, Ermr | This study |
| ΝΕ1154 | USA Nebraska transposon mutant library strain, containing a transposon insertion in uhpT (SAUSA300_0216). Transposon insertion was confirmed by PCR with primers NE1154_F and NE1154_R (Table 5); Ermr | 35 |
| ΝΕ1511 | USA300 Nebraska transposon mutant library strain containing a transposon insertion in putative lysR regulator of histidine metabolism (SAUSA300_2279). Transposon insertion was confirmed by PCR with primers NE1511_F and NE1511_R (Table 5); Ermr | 35 |
| ΝΕ178 | USA300 Nebraska transposon mutant library strain containing a transposon insertion in a leuA-like isopropylmalate synthase (SAUSA300_0879). Transposon insertion was confirmed by PCR with primers NE178_F and NE178_R (Table 5); Ermr | 35 |
| M0182 | FarRC116Y ST5 HA-MRSA | SAMN02325243 |
| M0194 | FarRC116Y ST5 HA-MRSA | SAMN02325252 |
| M0330 | FarRC116Y ST5 HA-MRSA | SAMN00792149 |
| M0374 | FarRC116Y ST5 HA-MRSA | SAMN00809180 |
| M0377 | FarRC116Y ST5 HA-MRSA | SAMN02325374 |
| M0390 | FarRC116Y ST5 HA-MRSA | SAMN02325384 |
| M0398 | FarRC116YST5 HA-MRSA | SAMN02325387 |
| M0402 | FarRC116Y ST5 HA-MRSA | SAMN02325390 |
| M0423 | FarRC116Y ST5 HA-MRSA | SAMN02325405 |
| M1545 | FarRC116Y ST5 HA-MRSA | SAMN02364057 |
| NMRSA0256 | FarRC116Y ST5 HA-MRSA | 27 |
| NMRSA0215 | FarRC116Y ST5 HA-MRSA | 27 |
| NMRSA0416 | FarRC116Y ST5 HA-MRSA | 27 |
| NMRSA0267 | FarRC116Y ST5 HA-MRSA | 27 |
| NMRSA0266 | FarRC116Y ST5 HA-MRSA | 27 |
| M1510 | FarRE93EE ST5 HA-MRSA | SAMN00839762 |
| M1516 | FarRE93EE ST5 HA-MRSA | SAMN02364037 |
| M1523 | FarRE93EE ST5 HA-MRSA | SAMN02364041 |
| M1528 | FarRE93EE ST5 HA-MRSA | SAMN02364045 |
| hp20814.016 | FarRE160G ST5 HA-MRSA | 61 |
| hp20814.067 | FarRE160G ST5 HA-MRSA | 61 |
| hp20814.082 | FarRE160G ST5 HA-MRSA | 61 |
| hp20814.099 | FarRE160G ST5 HA-MRSA | 61 |
| DAR 1157 | FarRP165L ST5 HA-MRSA | 26 |
| DAR 1311 | FarRP165L ST5 HA-MRSA | 26 |
| DAR 948 | FarRP165L ST5 HA-MRSA | 26 |
| NMRSA0241 | FarRP165L ST5 HA-MRSA | 27 |
| NMRSA0358 | FarRP165L ST5 HA-MRSA | 27 |
| NMRSA0135 | FarRP165L ST5 HA-MRSA | 27 |
| E. coli | ||
| DH5α | Transformation competent strain. λ−ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 recA1 endA1 hsdR17(rK− mK−) supE44 thi-1 gyrA relA1 | Invitrogen |
| Plasmid | ||
| pLI50 | E. coli–S. aureus shuttle vector; Ampr, Cmr | 89 |
| pLIfarR | pLI50 with native farR gene; Ampr, Cmr | 21 |
| pLIfarRH121Y | pLI50 with farRH121Y from S. aureus FAR7; Ampr, Cmr | 21 |
| pLIfarRC116Y | pLI50 with farRC116Y from S. aureus M1545; Ampr, Cmr | This study |
| pLIfarRE93EE | pLI50 with farRE93EE from S. aureus M1516; Ampr, Cmr | This study |
| pLIfarRE160G | pLI50 with farRE160G from S. aureus hp20814.099; Ampr, Cmr | This study |
| pLIfarRP165L | pLI50 with farRP165L from S. aureus DAR 948; Ampr, Cmr | This study |
| pGYlux | E. coli–S. aureus shuttle vector harboring promoterless luxABCDE operon; Ampr, Cmr | 66 |
| pGYfarRE::lux | pGYlux with native farR and PfarE promoter segment from USA300 cloned in pGYlux; Ampr, Cmr | This study |
| pGYfarRE::lux1 | pGYlux with farR and PfarE promoter segment from S. aureus FAR7 (H121Y) cloned in pGYlux; Ampr, Cmr | This study |
| pGYfarRE::lux2 | pGYlux with farR and PfarE promoter segment from S. aureus M1545 (C116Y) cloned in pGYlux; Ampr, Cmr | This study |
| pGYfarRE::lux3 | pGYlux with farR and PfarE promoter segment from S. aureus hp20814.99 (E160G) cloned in pGYlux; Ampr, Cmr | This study |
CA-MRSA, community-acquired MRSA; HA-MRSA, hospital-acquired MRSA.
For growth assays in chemically defined medium containing 0.4% glucose as the carbon source, CDM-G was prepared as described previously (63) from 10× stock solutions of defined l-amino acids (alanine, arginine, aspartate, cysteine, glutamate, glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine, and valine), vitamins (l-thiamine, nicotinic and pantothenic acid, biotin), salts (K2HPO4 and KH2PO4), and divalent cations (MgSO4, [NH4]2SO4, MgCl2, and CaCl2). Where indicated, CDM-G was modified by the omission of glucose and the addition of either 0.25% glucose-6-phosphate (CDM-G6P) or 0.25% l-histidine (CDM-His) as a carbon source.
Strain and plasmid construction.
Genetic manipulation of S. aureus was conducted following established guidelines (64) and as described in our previous work (21, 36, 65). Restriction enzymes and T4 DNA ligase were purchased from New England BioLabs, Taq polymerase from GenScript, kits for PCR cleanup and plasmid preparation from Geneaid, and nucleotide primers (Table 6) from Integrated DNA Technologies. All plasmids were constructed as shuttle vectors in E. coli DH5α and their integrity was confirmed by nucleotide sequencing of the cloned DNA fragments, using the primers pLI50_F and pLI50_R for genes cloned in pLI50, and pGYlux_F and pGYlux_R for products cloned in pGYlux. All shuttle vectors were then transformed by electroporation into S. aureus USA300 or isogenic derivatives using S. aureus RN4220 as an intermediate host. The PCR primers farR_F1 and farR_R1 were used to PCR-amplify variant farR genes from S. aureus clinical isolates, which were then digested with KpnI and SacI for cloning in pLI50 to create complementation plasmids in which farR is expressed from its native promoter as previously described for wild-type farR and farRH121Y (21). For luciferase reporter constructs, primers farRE::lux_F and farRE::lux_R were used to amplify a 905-bp product comprising the farR gene and adjacent PfarE promoter segment, which was then digested with BamHI and SalI for ligation into the pGYlux reporter vector (66).
TABLE 6.
PCR primer sequences
| Primer | Sequencea |
|---|---|
| pLI50_F | 5'–ATTTCCCCGAAAAGTGCC-3' |
| pLI50_R | 5'–TTTCTCGGCATAAATGCG-3' |
| pGYlux_F | 5'–CTGTTGTTTGTCGGTGAACGCT-3' |
| pGYlux_R | 5'–ATTGGGGAGGTTGGTATGTAAGC-3' |
| farR_F1 | 5'–cccggtaccTGCAGCTACAATCACTATCCATGC-3' |
| farR_R1 | 5'–cccgagctcACGGACGCTAAAACAGGTAGTCC-3' |
| farRE::lux_F | 5'–CGATAGTAGTACACGgATcCATTAACGTGTACACTATCG-3' |
| farRE::lux_R | 5'–CATTGTCAAATgTCGacGCATTTGTAGCAAGTGG-3' |
| NE178_F | 5'–GGCATGTGTACAACTATCGAGG-3' |
| NE178_R | 5'–AACATCCTACAGTGTCCGCA-3' |
| NE1511_F | 5'–TCGAAAGCACCATTCCGACT-3' |
| NE1511_R | 5'–AGTGTTTGCGCACTTGAGAAT-3' |
| NE1154_F | 5'–TTAAGCGCTTACACCGACGT-3' |
| NE1154_R | 5'–AAGTATCGGCCACGTTTCGT-3' |
Underlined lowercase nucleotides represent 5- additions for incorporation of KpnI or SacI restriction sites (farR_F1 and farR_R1, respectively) or primer-directed changes to the template sequence for incorporation of BamHI or SalI restriction sites (farRE::lux_F and farRE::lux_R, respectively).
Analysis of FarR variation in the context of S. aureus phylogenetic diversity.
We considered all available complete S. aureus genomes, and an S. argenteus genome to serve as an outgroup, from the Pathosystems Resource Integration Center (PATRIC) database as of 2020 (25). Genomes that were of low quality based on various considerations or high divergence based on mash distance were removed from consideration. Phylogenetic analysis of the S. aureus proteome, represented by conceptually translated nucleotide sequences available from PATRIC, was performed for 574 S. aureus and 1 S. argenteus strains using PhyloPhlAn3 (67). Proteins were mapped to an S. aureus reference database using Usearch (68), aligned with Muscle (69), and gappy regions in the alignment were trimmed with Trimal (70), resulting in a curated, concatenated alignment of 21,922 variable sites. Initial phylogenetic analysis used FastTree, which was refined with RaxML under the PROTCATLG model (71). 563 full-length FarR proteins from these strains were then extracted from the PATRIC database, and a minimum spanning tree was constructed by alignment of the FarR amino-acid sequences using GrapeTree (72). This identified six primary FarR sequence clusters that were mapped onto the strain phylogeny.
Identification of FarR variant proteins.
Different in vitro selection procedures promoted recovery of FarRH121Y and FarRC116R variants. As such, variants that are candidates for altering FarR function were likely to be identified through amino acid substitutions that distinguished FarR in a specific strain, from other S. aureus strains within the same clonal complex, as noted within some CC5 strains which were identified through analysis of variation across 574 genomes in the PATRIC database. For a complete analysis, each of the six primary FarR cluster sequences was used in a BLASTP search (73) to assess variation in FarR across all S. aureus genome assemblies, as well as the additional assemblies described below, to identify amino acid substitution variants that occur within specific genetic backgrounds. Additional FarR variants were identified from Sequence Read Archive (SRA) data, including MRSA from hospitals in Ontario, Canada (27) and additional strains from Europe. For this purpose, sequence data from all isolates was used to generate de novo assemblies with Spades v.3.12.0 (74). The adapters and low-quality reads were removed with Cutadapt v1.16 (75) and Sickle v1.33 (76) and the contamination screen was completed using FastQ Screen (77). Optimal k-mers were identified based on average read lengths for each genome. All assemblies were evaluated using QUAST v.5.0.1 (78). The reads were mapped back to de novo assemblies to investigate polymorphism (which is indicative of mixed cultures) using Bowtie2 v1.2.2 (79). Low-quality genome assemblies were removed from further analysis (i.e., N50 < 10,000, total assembly length outside the median sequence length of ±1 standard deviation [SD], contigs smaller than 1 kb contributing to >10% of the total assembly length).
Genotyping and phylogenetic analyses.
Multilocus sequence typing was done by scanning genome assemblies against the S. aureus MLST database (https://pubmlst.org/saureus/) using the mlst v2.10 program with default settings (https://github.com/tseemann/mlst). Detection of mecA and typing of SCCmec was done by mapping the strains’ pseudoreads to a custom-clustered database of ccr and mec gene complexes, plus SCCmec IV subtype-specific sequences (80), using SRST2 v0.2.0 (81) with the min_coverage 60 option. Staphylococcal protein A gene (spa) typing was conducted using spaTyper (https://spatyper.fortinbras.us/).
The placement of FarR variant strains within the CC5 phylogeny was assessed in a two-phase approach. First, genome assemblies from 119 strains with single amino acid substitutions in FarR (FarRE93EE, FarRC116Y, FarRE160G, FarRP165L, FarRG166D) were confirmed as CC5 using multilocus sequence typing. A total of 250,000 PE pseudoreads (100 bp each, 200-bp fragment size) were produced from these assemblies using samtools wgsim, mapped to the S. aureus JH1 reference genome with bwa, and finally realigned around indels with GATK as performed previously (26). Mapped nucleotide sites were exported as a VCF file with the ‘emit all sites’ option in GATK. Using this file, a lookup was done on the positions of 11,961 biallelic single-nucleotide polymorphisms (SNPs) identified from the 598 CC5 reference strains in the study of Challagundla et al. (26) to assign the 119 strains’ alleles at those SNPs. The SNP data from these 119 new strains were combined with those from the 598 reference strains, giving a total of 717 strains. A BioNJ tree was generated using the total number of SNP differences as a distance, and the tree was rooted with a previously identified outgroup strain, DAR3176 (26). This tree was used to select 30 reference strains which (i) subtended the nodes to which the new strains with FarR variants attached, (ii) provided examples of sister nodes of strains with FarR variants, and (iii) provided examples of the various CC5 clades previously defined.
For the second phase, a final analysis of the 117 FarR variant strains and the 30 selected reference strains was done with GATK Unified Genotyper (82) to call SNPs and indels, as well as invariant core nucleotide sites relative to the reference genome S. aureus JH1 (83). Sites present in all samples with 3 or more reads per sample were retained. This resulted in an alignment of 9,020 bi-allelic SNPs and 2,292,396 invariant core nucleotide sites that was analyzed with PhyML (HKY+G+I) (84) and then with ClonalFrameML (85) to generate a phylogeny and correct its’ branch lengths for recombination. This tree places the strains with FarR variants in the context of the CC5 phylogeny and is shown in Fig. 2.
Growth and MIC assays.
To prepare the inoculum for growth assays, single colonies of S. aureus clinical isolates were inoculated into 3 mL TSB in 12 × 75-mm polypropylene snap-cap tubes and grown overnight at 37°C on a rotary shaker. The optical density at 600 nm (OD600) of these overnight cultures was determined on a Beckman-Coulter DU 530 Spectrophotometer. For MIC determination with LA, cells were subcultured to OD600 = 0.01 in triplicate 20 × 150-mm glass tubes with plastic caps containing 3 mL TSB + 0.1% DMSO (dimethyl sulfoxide) and the indicated concentrations of LA. Tubes were placed on a rack at a 30° angle in a 37°C incubator with shaking at 220 rpm, and OD600 was measured after 24 h. For growth assays, cultures were inoculated into 96-well microtiter plates containing 200 μL TSB + 0.1% DMSO and the indicated concentrations of LA. The growth of each culture was assessed in 6 wells. Plates were incubated at 37°C on a rotary shaker (220 rpm) and OD600 was monitored at hourly intervals using a Synergy H4 Hybrid Reader (BioTek, Winooski, VT); alternately, where indicated, the plates were incubated at 37°C in the Synergy H4 Reader with automated shaking and optical density measurements taken every 15 min for 24 h. For growth in CDM-G, CDM-G6P, and CDH-His, 3-mL inoculum cultures grown overnight in the respective CDM media were subcultured to an OD600 of 0.01 into 3 mL of the appropriate medium in 20 × 150-mm glass tubes and grown as described for MIC determination, with endpoint growth (OD600) measured at 24 h.
Sensi-disc antibiotic sensitivity assay.
To test for sensitivity or resistance to fosfomycin, S. aureus cultures were grown overnight in TSB, then diluted to OD600 = 0.01 in fresh sterile TSB. This was used to saturate a sterile Q-tip swab, which was then used to swab the surface of a tryptic soy agar plate, followed by addition of a BBL Sensi-disc (Becton, Dickinson and Co.) containing fosfomycin (200 μg). Plates were incubated at 37°C for 18 h and photographed to document zones of inhibition.
Assays of reporter gene expression.
Overnight inoculum cultures containing the appropriate reporter constructs were subcultured to OD600 = 0.01 into triplicate 125-mL flasks containing 25 mL of TSB + 0.1% DMSO, and then incubated at 37°C with orbital shaking (220 rpm) for 3 h, at which point the cultures were supplemented by addition of 40 μM LA. Samples (4× 200-μL aliquots) were withdrawn at this time point (t = 0), and again at 30-min intervals, into 96-well white opaque flat-bottomed plates (Greiner Bio-One). Wells were supplemented with 20 μL of 0.1% (vol/vol) decanal in 40% ethanol and luminescence measurements were immediately taken on a Synergy H4 hybrid reader (BioTek), with 1 s of integration and a gain of 200. Synchronously, OD600 was measured in triplicate with a spectrophotometer. Data values were recorded as relative light units (RLU), corrected for background by subtracting values recorded from cultures harboring empty pGYlux. Data points were standardized for differences in growth by dividing RLU values by the recorded OD600 values of the cultures when samples were withdrawn.
ACKNOWLEDGMENTS
This work was supported through a Discovery Grant to M.J.M. from the Natural Sciences and Engineering Research Council (NSERC) of Canada. C.M.B. was the recipient of a Canada Graduate Scholarship Award from NSERC.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Nikaido H, Pagès J-M. 2012. Broad-specificity efflux pumps and their role in multidrug resistance of Gram-negative bacteria. FEMS Microbiol Rev 36:340–363. 10.1111/j.1574-6976.2011.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. 1995. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol Microbiol 16:45–55. 10.1111/j.1365-2958.1995.tb02390.x. [DOI] [PubMed] [Google Scholar]
- 3.Nakamura H. 1968. Genetic determination of resistance to acriflavine, phenethyl alcohol, and sodium dodecyl sulfate in Escherichia coli. J Bacteriol 96:987–996. 10.1128/jb.96.4.987-996.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piddock LJ. 2006. Multidrug-resistance efflux pumps: not just for resistance. Nat Rev Microbiol 4:629–636. 10.1038/nrmicro1464. [DOI] [PubMed] [Google Scholar]
- 5.Bina XR, Provenzano D, Nguyen N, Bina JE. 2008. Vibrio cholerae RND family efflux systems are required for antimicrobial resistance, optimal virulence factor production, and colonization of the infant mouse small intestine. Infect Immun 76:3595–3605. 10.1128/IAI.01620-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin J, Sahin O, Michel LO, Zhang Q. 2003. Critical role of multidrug efflux pump CmeABC in bile resistance and in vivo colonization of Campylobacter jejuni. Infect Immun 71:4250–4259. 10.1128/IAI.71.8.4250-4259.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Webber MA, Bailey AM, Blair JM, Morgan E, Stevens MP, Hinton JC, Ivens A, Wain J, Piddock LJ. 2009. The global consequence of disruption of the AcrAB-TolC efflux pump in Salmonella enterica includes reduced expression of SPI-1 and other attributes required to infect the host. J Bacteriol 191:4276–4285. 10.1128/JB.00363-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buckner MMC, Blair JMA, la Ragione RM, Newcombe J, Dwyer DJ, Ivens A, Piddock LJ. 2016. Beyond antimicrobial resistance: evidence for a distinct role of the AcrD efflux Pump in Salmonella biology. mBio 7:e01916-16. 10.1128/mBio.01916-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Routh MD, Zalucki Y, Su C-C, Zhang Q, Shafer WM, Yu EW, Yu EW. 2011. Efflux pumps of the resistance-nodulation-division family: a perspective of their structure, function, and regulation in Gram-negative bacteria. Adv Enzymol Relat Areas Mol Biol 77:109–146. 10.1002/9780470920541.ch3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans K, Adewoye L, Poole K. 2001. MexR repressor of the mexAB-oprM multidrug efflux operon of Pseudomonas aeruginosa: identification of MexR binding sites in the mexA-mexR intergenic region. J Bacteriol 183:807–812. 10.1128/JB.183.3.807-812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma D, Alberti M, Lynch C, Nikaido H, Hearst JE. 1996. The local repressor AcrR plays a modulating role in the regulation of acrAB genes of Escherichia coli by global stress signals. Mol Microbiol 19:101–112. 10.1046/j.1365-2958.1996.357881.x. [DOI] [PubMed] [Google Scholar]
- 12.Hagman KE, Shafer WM. 1995. Transcriptional control of the mtr efflux system of Neisseria gonorrhoeae. J Bacteriol 177:4162–4165. 10.1128/jb.177.14.4162-4165.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, Dzink-Fox JL, Chen M, Levy SB. 2001. Genetic characterization of highly fluoroquinolone-resistant clinical Escherichia coli strains from China: role of acrR mutations. Antimicrob Agents Chemother 45:1515–1521. 10.1128/AAC.45.5.1515-1521.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saito K, Yoneyama H, Nakae T. 1999. nalB-type mutations causing the overexpression of the MexAB-OprM efflux pump are located in the mexR gene of the Pseudomonas aeruginosa chromosome. FEMS Microbiol Lett 179:67–72. 10.1111/j.1574-6968.1999.tb08709.x. [DOI] [PubMed] [Google Scholar]
- 15.Thomas JC, Seby S, Abrams AJ, Cartee J, Lucking S, Vidyaprakash E, Schmerer M, Pham CD, Hong J, Torrone E, Cyr SS, Shafer WM, Bernstein K, Kersh EN, Gernert KM, Weinstock H, Dominguez C, Hun S, Kneupper K, Antimicrobial-Resistant Neisseria gonorrhoeae Working Group . 2019. Evidence of recent genomic evolution in gonococcal strains with decreased susceptibility to cephalosporins or azithromycin in the United States, 2014–2016. J Infect Dis 220:294–305. 10.1093/infdis/jiz079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bador J, Neuwirth C, Grangier N, Muniz M, Germé L, Bonnet J, Pillay VG, Llanes C, de Curraize C, Amoureux L. 2017. Role of AxyZ transcriptional regulator in overproduction of AxyXY-OprZ multidrug efflux system in Achromobacter species mutants selected by tobramycin. Antimicrob Agents Chemother 61:e00290-17. 10.1128/AAC.00290-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, Nouwen JL. 2005. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis 5:751–762. 10.1016/S1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- 18.Garner MJ, Carson C, Lingohr EJ, Fazil A, Edge VL, Trumble Waddell J. 2015. An assessment of antimicrobial resistant disease threats in Canada. PLoS One 10:e0125155. 10.1371/journal.pone.0125155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Do TQ, Moshkani S, Castillo P, Anunta S, Pogosyan A, Cheung A, Marbois B, Faull KF, Ernst W, Chiang SM, Fujii G, Clarke CF, Foster K, Porter E. 2008. Lipids including cholesteryl linoleate and cholesteryl arachidonate contribute to the inherent antibacterial activity of human nasal fluid. J Immunol 181:4177–4187. 10.4049/jimmunol.181.6.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takigawa H, Nakagawa H, Kuzukawa M, Mori H, Imokawa G. 2005. Deficient production of hexadecenoic acid in the skin is associated in part with the vulnerability of atopic dermatitis patients to colonization by Staphylococcus aureus. Dermatology 211:240–248. 10.1159/000087018. [DOI] [PubMed] [Google Scholar]
- 21.Alnaseri H, Arsic B, Schneider JET, Kaiser JC, Scinocca ZC, Heinrichs DE, McGavin MJ. 2015. Inducible expression of a resistance-nodulation-division-type efflux pump in Staphylococcus aureus provides resistance to linoleic and arachidonic acids. J Bacteriol 197:1893–1905. 10.1128/JB.02607-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen MT, Saising J, Tribelli PM, Nega M, Diene SM, François P, Schrenzel J, Spröer C, Bunk B, Ebner P, Hertlein T, Kumari N, Härtner T, Wistuba D, Voravuthikunchai SP, Mäder U, Ohlsen K, Götz F. 2019. Inactivation of farR causes high rhodomyrtone resistance and increased pathogenicity in Staphylococcus aureus. Front Microbiol 10:1157. 10.3389/fmicb.2019.01157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao Q, Vakulenko S, Chang M, Mobashery S. 2014. Mutations in mmpL and in cell-wall stress stimulon contribute to resistance to oxadiazole antibiotics in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 58:5841–5847. 10.1128/AAC.03501-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Daniel PI, Peng Z, Pi H, Testero SA, Ding D, Spink E, Leemans E, Boudreau MA, Yamaguchi T, Schroeder VA, Wolter WR, Llarrull LI, Song W, Lastochkin E, Kumarasiri M, Antunes NT, Espahbodi M, Lichtenwalter K, Suckow MA, Vakulenko S, Mobashery S, Chang M. 2014. Discovery of a new class of non-β-lactam inhibitors of penicillin-binding proteins with Gram-positive antibacterial activity. J Am Chem Soc 136:3664–3672. 10.1021/ja500053x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL, Gillespie JJ, Gough R, Hix D, Kenyon R, MacHi D, Mao C, Nordberg EK, Olson R, Overbeek R, Pusch GD, Shukla M, Schulman J, Stevens RL, Sullivan DE, Vonstein V, Warren A, Will R, Wilson MJC, Yoo HS, Zhang C, Zhang Y, Sobral BW. 2014. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res 42:D581–D591. 10.1093/nar/gkt1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Challagundla L, Reyes J, Rafiqullah I, Sordelli DO, Echaniz-Aviles G, Velazquez-Meza ME, Castillo-Ramírez S, Fittipaldi N, Feldgarden M, Chapman SB, Calderwood MS, Carvajal LP, Rincon S, Hanson B, Planet PJ, Arias CA, Diaz L, Robinson DA. 2018. Phylogenomic classification and the evolution of clonal complex 5 methicillin-resistant Staphylococcus aureus in the Western hemisphere. Front Microbiol 9:1901. 10.3389/fmicb.2018.01901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guthrie JL, Teatero S, Hirai S, Fortuna A, Rosen D, Mallo G.v, Campbell J, Pelude L, Golding G, Simor AE, Patel SN, McGeer A, Fittipaldi N, Delport J, Evans G, Hota S, Katz K, Lemieux C, Mertz D, Science M, Thampi N, Ontario CNISP Hospital Investigators . 2020. Genomic epidemiology of invasive methicillin-resistant Staphylococcus aureus infections among hospitalized individuals in Ontario, Canada. J Infect Dis 222:2071–2081. 10.1093/infdis/jiaa147. [DOI] [PubMed] [Google Scholar]
- 28.Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui L, Oguchi A, Aoki K, Nagai Y, Lian J, Ito T, Kanamori M, Matsumaru H, Maruyama A, Murakami H, Hosoyama A, Mizutani-Ui Y, Takahashi NK, Sawano T, Inoue R, Kaito C, Sekimizu K, Hirakawa H, Kuhara S, Goto S, Yabuzaki J, Kanehisa M, Yamashita A, Oshima K, Furuya K, Yoshino C, Shiba T, Hattori M, Ogasawara N, Hayashi H, Hiramatsu K. 2001. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357:1225–1240. 10.1016/s0140-6736(00)04403-2. [DOI] [PubMed] [Google Scholar]
- 29.Hau SJ, Bayles DO, Alt DP, Frana TS, Nicholson TL. 2017. Draft genome sequences of 63 swine-associated methicillin-resistant Staphylococcus aureus sequence type 5 isolates from the United States. Genome Announc 5:e01081-17. 10.1128/genomeA.01081-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kos VN, Desjardins CA, Griggs A, Cerqueira G, van Tonder A, Holden MTG, Godfrey P, Palmer KL, Bodi K, Mongodin EF, Wortman J, Feldgarden M, Lawley T, Gill SR, Haas BJ, Birren B, Gilmore MS. 2012. Comparative genomics of vancomycin-resistant Staphylococcus aureus strains and their positions within the clade most commonly associated with methicillin-resistant S. aureus hospital-acquired infection in the United States. mBio 3:e00112-12. 10.1128/mBio.00112-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arias CA, Reyes J, Carvajal LP, Rincon S, Diaz L, Panesso D, Ibarra G, Rios R, Munita JM, Salles MJ, Alvarez-Moreno C, Labarca J, Garcia C, Luna CM, Mejia-Villatoro C, Zurita J, Guzman-Blanco M, Rodriguez-Noriega E, Narechania A, Rojas LJ, Planet PJ, Weinstock GM, Gotuzzo E, Seas C. 2017. A prospective cohort multicenter study of molecular epidemiology and phylogenomics of Staphylococcus aureus bacteremia in nine Latin American countries. Antimicrob Agents Chemother 61:e00816-17. 10.1128/AAC.00816-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee YC, Chen PY, Wang JT, Chang SC. 2020. Prevalence of fosfomycin resistance and gene mutations in clinical isolates of methicillin-resistant Staphylococcus aureus. Antimicrob Resist Infect Control 9:135. 10.1186/s13756-020-00790-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao H, Wang X, Wang B, Xu Y, Rao L, Wan B, Guo Y, Wu X, Yu J, Chen L, Li M, Yu F. 2021. The prevalence and determinants of fusidic acid resistance among methicillin-resistant Staphylococcus aureus clinical isolates in China. Front Med (Lausanne) 8:761894. 10.3389/fmed.2021.761894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Neill AJ, Larsen AR, Henriksen AS, Chopra I. 2004. A fusidic acid-resistant epidemic strain of Staphylococcus aureus carries the fusB determinant, whereas fusA mutations are prevalent in other resistant isolates. Antimicrob Agents Chemother 48:3594–3597. 10.1128/AAC.48.9.3594-3597.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537-12. 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alnaseri H, Kuiack RC, Ferguson KA, Schneider JET, Heinrichs DE, McGavin MJ. 2019. DNA binding and sensor specificity of FarR, a novel TetR family regulator required for induction of the fatty acid efflux pump FarE in Staphylococcus aureus. J Bacteriol 201:e00602-18. 10.1128/JB.00602-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang L, Matsuo M, Calderón C, Fan SH, Ammanath AV, Fu X, Li N, Luqman A, Ullrich M, Herrmann F, Maier M, Cheng A, Zhang F, Oesterhelt F, Lämmerhofer M, Götz F. 2022. Molecular basis of rhodomyrtone resistance in Staphylococcus aureus. mBio 13:e03833-21. 10.1128/mbio.03833-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuroda M, Kuwahara-Arai K, Hiramatsu K. 2000. Identification of the up- and down-regulated genes in vancomycin-resistant Staphylococcus aureus strains Mu3 and Mu50 by cDNA differential hybridization method. Biochem Biophys Res Commun 269:485–490. 10.1006/bbrc.2000.2277. [DOI] [PubMed] [Google Scholar]
- 39.Parkhill J, Peacock SJ, Harris SR, Mohamed N, Jamrozy D, Anderson AS, Tan CY, Holden MTG. 2016. Pan-genomic perspective on the evolution of the Staphylococcus aureus USA300 epidemic. Microb Genom 2:e000058. 10.1099/mgen.0.000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Challagundla L, Luo X, Tickler IA, Didelot X, Coleman DC, Shore AC, Coombs GW, Sordelli DO, Brown EL, Skov R, Larsen AR, Reyes J, Robledo IE, Vazquez GJ, Rivera R, Fey PD, Stevenson K, Wang S-H, Kreiswirth BN, Mediavilla JR, Arias CA, Planet PJ, Nolan RL, Tenover FC, Goering RV, Robinson DA. 2018. Range expansion and the origin of USA300 North American epidemic methicillin-resistant Staphylococcus aureus. mBio 9:e02016-17. 10.1128/mBio.02016-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma C, Rokana N, Chandra M, Singh BP, Gulhane RD, Gill JPS, Ray P, Puniya AK, Panwar H. 2018. Antimicrobial resistance: its surveillance, impact, and alternative management strategies in dairy animals. Front Vet Sci 4:237. 10.3389/fvets.2017.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shryock TR, Dye ES, Kapral FA. 1992. The accumulation of bactericidal lipids in staphylococcal abscesses. J Med Microbiol 36:332–336. 10.1099/00222615-36-5-332. [DOI] [PubMed] [Google Scholar]
- 43.Fischer AJ, Singh SB, LaMarche MM, Maakestad LJ, Kienenberger ZE, Peña TA, Stoltz DA, Limoli DH. 2021. Sustained coinfections with Staphylococcus aureus and Pseudomonas aeruginosa in cystic fibrosis. Am J Respir Crit Care Med 203:328–338. 10.1164/rccm.202004-1322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu J, Gefen O, Ronin I, Bar-Meir M, Balaban NQ. 2020. Effect of tolerance on the evolution of antibiotic resistance under drug combinations. Science 367:200–204. 10.1126/science.aay3041. [DOI] [PubMed] [Google Scholar]
- 45.Ankrum A, Hall BG. 2017. Population dynamics of Staphylococcus aureus in cystic fibrosis patients to determine transmission events by use of whole-genome sequencing. J Clin Microbiol 55:2143–2152. 10.1128/JCM.00164-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernardy EE, Petit RA, Raghuram V, Alexander AM, Read TD, Goldberg JB. 2020. Genotypic and phenotypic diversity of Staphylococcus aureus isolates from cystic fibrosis patient lung infections and their interactions with Pseudomonas aeruginosa. mBio 11:e00735-20. 10.1128/mBio.00735-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuroda M, Sekizuka T, Matsui H, Ohsuga J, Ohshima T, Hanaki H. 2019. IS256-mediated overexpression of the walKR two-component system regulon contributes to reduced vancomycin susceptibility in a Staphylococcus aureus clinical isolate. Front Microbiol 10:1882. 10.3389/fmicb.2019.01882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pujol M, Miró J-M, Shaw E, Aguado J-M, San-Juan R, Puig-Asensio M, Pigrau C, Calbo E, Montejo M, Rodriguez-Álvarez R, Garcia-Pais M-J, Pintado V, Escudero-Sánchez R, Lopez-Contreras J, Morata L, Montero M, Andrés M, Pasquau J, Arenas M-M, Padilla B, Murillas J, Jover-Sáenz A, López-Cortes L-E, García-Pardo G, Gasch O, Videla S, Hereu P, Tebé C, Pallarès N, Sanllorente M, Domínguez M-Á, Càmara J, Ferrer A, Padullés A, Cuervo G, Carratalà J, MRSA Bacteremia (BACSARM) Trial Investigators . 2021. Daptomycin plus fosfomycin versus daptomycin alone for methicillin-resistant Staphylococcus aureus bacteremia and endocarditis: a randomized clinical trial. Clin Infect Dis 72:1517–1525. 10.1093/cid/ciaa1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mishra N, Lew C, Abdelhady W, Lapitan C, Proctor R, Rose W, Bayer A. 2022. Synergy mechanisms of daptomycin-fosfomycin combinations in daptomycin-susceptible and -resistant methicillin-resistant Staphylococcus aureus: in vitro, ex vivo, and in vivo metrics. Antimicrob Agents Chemother 66:e0164921. 10.1128/AAC.01649-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harris SR, Robinson C, Steward KF, Webb KS, Paillot R, Parkhill J, Holden MTG, Waller AS. 2015. Genome specialization and decay of the strangles pathogen, Streptococcus equi, is driven by persistent infection. Genome Res 25:1360–1371. 10.1101/gr.189803.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Broadbent JR, Neeno-Eckwall EC, Stahl B, Tandee K, Cai H, Morovic W, Horvath P, Heidenreich J, Perna NT, Barrangou R, Steele JL. 2012. Analysis of the Lactobacillus casei supragenome and its influence in species evolution and lifestyle adaptation. BMC Genomics 13:533. 10.1186/1471-2164-13-533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morley L, McNally A, Paszkiewicz K, Corander J, Méric G, Sheppard SK, Blom J, Manning G. 2015. Gene loss and lineage-specific restriction-modification systems associated with niche differentiation in the Campylobacter jejuni sequence type 403 clonal complex. Appl Environ Microbiol 81:3641–3647. 10.1128/AEM.00546-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guinane CM, Ben Zakour NL, Tormo-Mas MA, Weinert LA, Lowder BV, Cartwright RA, Smyth DS, Smyth CJ, Lindsay JA, Gould KA, Witney A, Hinds J, Bollback JP, Rambaut A, Penades JR, Fitzgerald JR. 2010. Evolutionary genomics of Staphylococcus aureus reveals insights into the origin and molecular basis of ruminant host adaptation. Genome Biol Evol 2:454–466. 10.1093/gbe/evq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quijada NM, Hernández M, Oniciuc EA, Eiros JM, Fernández-Natal I, Wagner M, Rodríguez-Lázaro D. 2019. Oxacillin-susceptible mecA-positive Staphylococcus aureus associated with processed food in Europe. Food Microbiol 82:107–110. 10.1016/j.fm.2019.01.021. [DOI] [PubMed] [Google Scholar]
- 55.Titouche Y, Houali K, Ruiz-Ripa L, Vingadassalon N, Nia Y, Fatihi A, Cauquil A, Bouchez P, Bouhier L, Torres C, Hennekinne JA. 2020. Enterotoxin genes and antimicrobial resistance in Staphylococcus aureus isolated from food products in Algeria. J Appl Microbiol 129:1043–1052. 10.1111/jam.14665. [DOI] [PubMed] [Google Scholar]
- 56.Boswihi S, Udo E, AlFouzan W. 2020. Antibiotic resistance and typing of the methicillin-resistant Staphylococcus aureus clones in Kuwait hospitals, 2016–2017. BMC Microbiol 20:314. 10.1186/s12866-020-02009-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.El-Ashker M, Gwida M, Monecke S, El-Gohary F, Ehricht R, Elsayed M, Akinduti P, El-Fateh M, Maurischat S. 2020. Antimicrobial resistance pattern and virulence profile of S. aureus isolated from household cattle and buffalo with mastitis in Egypt. Vet Microbiol 240:108535. 10.1016/j.vetmic.2019.108535. [DOI] [PubMed] [Google Scholar]
- 58.Vali L, Dashti AA, Mathew F, Udo EE. 2017. Characterization of heterogeneous MRSA and MSSA with reduced susceptibility to chlorhexidine in Kuwaiti hospitals. Front Microbiol 8:1359. 10.3389/fmicb.2017.01359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Basanisi M, La Bella G, Nobili G, Franconieri I, La Salandra G. 2017. Genotyping of methicillin-resistant Staphylococcus aureus (MRSA) isolated from milk and dairy products in South Italy. Food Microbiol 62:141–146. 10.1016/j.fm.2016.10.020. [DOI] [PubMed] [Google Scholar]
- 60.Parisi A, Caruso M, Normanno G, Latorre L, Sottili R, Miccolupo A, Fraccalvieri R, Santagada G. 2016. Prevalence, antimicrobial susceptibility and molecular typing of methicillin-resistant Staphylococcus aureus (MRSA) in bulk tank milk from southern Italy. Food Microbiol 58:36–42. 10.1016/j.fm.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 61.Porterfield HS, Maakestad LJ, LaMarche MM, Thurman AL, Kienenberger ZE, Pitcher NJ, Hansen AR, Zirbes CF, Boyken L, Muyskens BL, Pezzulo AA, Singh SB, Twait E, Ford B, Diekema DJ, Reeb V, Fischer AJ. 2021. MRSA strains with distinct accessory genes predominate at different ages in cystic fibrosis. Pediatr Pulmonol 56:2868–2878. 10.1002/ppul.25559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cuthbertson L, Nodwell JR. 2013. The TetR family of regulators. Microbiol Mol Biol Rev 77:440–475. 10.1128/MMBR.00018-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sheldon JR, Marolda CL, Heinrichs DE. 2014. TCA cycle activity in Staphylococcus aureus is essential for iron-regulated synthesis of staphyloferrin A, but not staphyloferrin B: the benefit of a second citrate synthase. Mol Microbiol 92:824–839. 10.1111/mmi.12593. [DOI] [PubMed] [Google Scholar]
- 64.Novick RP. 1991. Genetic systems in staphylococci. Methods Enzymol 204:587–636. 10.1016/0076-6879(91)04029-n. [DOI] [PubMed] [Google Scholar]
- 65.Kuiack RC, Veldhuizen RAW, McGavin MJ. 2020. Novel functions and signaling requirements for the GraS sensor kinase of Staphylococcus aureus in response to acidic pH. J Bacteriol 202:e00219-20. 10.1128/JB.00219-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mesak LR, Yim G, Davies J. 2009. Improved lux reporters for use in Staphylococcus aureus. Plasmid 61:182–187. 10.1016/j.plasmid.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 67.Asnicar F, Thomas AM, Beghini F, Mengoni C, Manara S, Manghi P, Zhu Q, Bolzan M, Cumbo F, May U, Sanders JG, Zolfo M, Kopylova E, Pasolli E, Knight R, Mirarab S, Huttenhower C, Segata N. 2020. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat Commun 11:2500. 10.1038/s41467-020-16366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Edgar RC, Bateman A. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 69.Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. TrimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu K, Linder CR, Warnow T. 2011. RAxML and FastTree: comparing two methods for large-scale maximum likelihood phylogeny estimation. PLoS One 6:e27731. 10.1371/journal.pone.0027731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou Z, Alikhan NF, Sergeant MJ, Luhmann N, Vaz C, Francisco AP, Carriço JA, Achtman M. 2018. GrapeTree: visualization of core genomic relationships among 100,000 bacterial pathogens. Genome Res 28:1395–1404. 10.1101/gr.232397.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sayers EW, Beck J, Bolton EE, Bourexis D, Brister JR, Canese K, Comeau DC, Funk K, Kim S, Klimke W, Marchler-Bauer A, Landrum M, Lathrop S, Lu Z, Madden TL, O'Leary N, Phan L, Rangwala SH, Schneider VA, Skripchenko Y, Wang J, Ye J, Trawick BW, Pruitt KD, Sherry ST. 2021. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 49:D10–D17. 10.1093/nar/gkaa892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin A.v, Sirotkin A.v, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 76.Joshi N, Fass J. 2011. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files (version 1.33). [Software]. Available from https://github.com/najoshi/sickle.
- 77.Wingett SW, Andrews S. 2018. FastQ Screen: a tool for multi-genome mapping and quality control. F1000Res 7:1338. 10.12688/f1000research.15931.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075. 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kaya H, Hasman H, Larsen J, Stegger M, Johannesen TB, Allesøe RL, Lemvigh CK, Aarestrup FM, Lund O, Larsen AR. 2018. SCCmec Finder, a web-based tool for typing of Staphylococcal Cassette Chromosome mec in Staphylococcus aureus using whole-genome sequence data. mSphere 3:e00612-17. 10.1128/mSphere.00612-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Inouye M, Dashnow H, Raven LA, Schultz MB, Pope BJ, Tomita T, Zobel J, Holt KE. 2014. SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med 6:90. 10.1186/s13073-014-0090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Van der Auwera G, O’Connor B. 2020. Using Docker, GATK, and WDL in Terra. Genomics in the cloud. O’Reilly Media, Inc, Sebastopol, CA. [Google Scholar]
- 83.Mwangi MM, Shang WW, Zhou Y, Sieradzki K, de Lencastre H, Richardson P, Bruce D, Rubin E, Myers E, Siggia ED, Tomasz A. 2007. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc Natl Acad Sci USA 104:9451–9456. 10.1073/pnas.0609839104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lefort V, Longueville JE, Gascuel O. 2017. SMS: Smart Model Selection in PhyML. Mol Biol Evol 34:2422–2424. 10.1093/molbev/msx149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Didelot X, Wilson DJ. 2015. ClonalFrameML: efficient inference of recombination in whole bacterial genomes. PLoS Comput Biol 11:e1004041. 10.1371/journal.pcbi.1004041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, Lin F, Lin J, Carleton HA, Mongodin EF, Sensabaugh GF, Perdreau-Remington F. 2006. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367:731–739. 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 87.Arsic B, Zhu Y, Heinrichs DE, McGavin MJ. 2012. Induction of the Staphylococcal proteolytic cascade by antimicrobial fatty acids in community acquired methicillin resistant Staphylococcus aureus. PLoS One 7:e45952. 10.1371/journal.pone.0045952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui L, Oguchi A, Aoki K-I, Nagai Y, Lian J-Q, Ito T, Kanamori M, Matsumaru H, Maruyama A, Murakami H, Hosoyama A, Mizutani-Ui Y, Takahashi NK, Sawano T, Inoue R-I, Kaito C, Sekimizu K, Hirakawa H, Kuhara S, Goto S, Yabuzaki J, Kanehisa M, Yamashita A, Oshima K, Furuya K, Yoshino C, Shiba T, Hattori M, Ogasawara N, Hayashi H, Hiramatsu K. 2001. Whole genome sequencing of methicillin-resistant Staphylococcus aureus. Lancet 357:1225–1240. 10.1016/S0140-6736(00)04403-2. [DOI] [PubMed] [Google Scholar]
- 89.Lee CY, Iandolo JJ. 1986. Lysogenic conversion of staphylococcal lipase is caused by insertion of the bacteriophage L54a genome into the lipase structural gene. J Bacteriol 166:385–391. 10.1128/jb.166.2.385-391.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Download aac.00749-22-s0001.xlsx, XLSX file, 0.03 MB (30.9KB, xlsx)
Tables S4 and S5. Download aac.00749-22-s0002.xlsx, XLSX file, 5.7 MB (5.8MB, xlsx)
Fig. S1 to S6. Download aac.00749-22-s0003.pdf, PDF file, 5.4 MB (5.6MB, pdf)
Tables S2 and S3. Download aac.00749-22-s0004.xlsx, XLSX file, 0.02 MB (26.1KB, xlsx)