

Abstract
Age-related cognitive decline, a common component of the brain aging process, is associated with significant impairment in daily functioning and quality of life among geriatric adults. While the complexity of mechanisms underlying cognitive aging are still being elucidated, microbial exposure and the multifactorial inflammatory cascades associated with systemic infections are emerging as potential drivers of neurological senescence. The negative cognitive and neurobiological consequences of a single pathogen-associated inflammatory experience, such as that modeled through treatment with lipopolysaccharide (LPS), are well documented. Yet, the brain aging impacts of repeated, intermittent inflammatory challenges are less well studied. To extend the emerging literature assessing the impact of infection burden on cognitive function among normally aging mice, here, we repeatedly exposed adult mice to intermittent LPS challenges during the aging period. Male 10-month-old C57BL6 mice were systemically administered escalating doses of LPS once every two weeks for 2.5 months. We evaluated cognitive consequences using the non-spatial step-through inhibitory avoidance task, and both spatial working and reference memory versions of the Morris water maze. We also probed several potential mechanisms, including cortical and hippocampal cytokine/chemokine gene expression, as well as hippocampal neuronal function via extracellular field potential recordings. Though there was limited evidence for an ongoing inflammatory state in cortex and hippocampus, we observed impaired learning and memory and a disruption of hippocampal long-term potentiation. These data suggest that a history of intermittent exposure to LPS-induced inflammation is associated with subtle but significantly impaired cognition among normally aging mice. The broader impact of these findings may have important implications for standard of care involving infections in aging individuals or populations at-risk for dementia.
Keywords: Brain aging, Cognitive aging, Infection burden, Lipopolysaccharide, Systemic inflammation, Neuroinflammation, Learning and memory, Hippocampus, Synaptic plasticity, Long-term potentiation
1. Introduction
Age-related cognitive decline, namely in domains of attention, memory, executive function, and visuospatial abilities, is a common component of the brain aging process (Murman, 2015). However, some individuals experience pathological brain aging, the cognitive consequences of which can be devastating. Indeed, mild cognitive impairment (MCI), associated with a spectrum of symptoms such as forgetfulness and impaired decision making, negatively affects between 6.7 and 25.2 % of geriatric adults (Petersen et al., 2018). Further, MCI can represent a prodromal stage of dementia, a severe form of age-related cognitive decline associated with neurodegeneration (Zhang et al., 2021; Mitchell and Shiri-Feshki, 2009; Michaud et al., 2017; Anderson, 2019). The economic burden of treatment and care for older individuals afflicted with MCI and dementia is exorbitant as annual nationwide costs are estimated to be up to $300 billion (Oba et al., 2021; Ton et al., 2017; Dharmarajan and Gunturu, 2009; Sloane et al., 2002; Wong, 2020). Importantly, the magnitude of this critical issue is growing. By the year 2050, ~85 million Americans are projected to be 65+ years old, constituting >20 % of the US total population (Ortman et al., 2014). This means that the human, medical and financial costs of cognitive aging will only increase as the population ‘grays’. Better understanding of the causes of MCI are urgently needed.
Though the mechanisms driving brain aging and MCI are numerous, complex, and still being elucidated (Anderson, 2019); microbial exposure, and the multifactorial inflammatory cascades associated with systemic infections, are emerging as potential drivers of neurological senescence among elderly people (Harris and Harris, 2015; Batista et al., 2019). Indeed, infection is one of the most common causes of delirium, a short-lived state of cognitive dysfunction that is particularly impactful among aged individuals (van Gool et al., 2010). Further, evidence among older adults suggests that a higher lifetime infection burden, indicated by higher seropositivity for pathogens such as Chlamydia pnuemoniae, Mycoplasma pnuemoniae, Helicobacter pylori, cytomegalovirus, or herpes simplex virus, is associated with worse cognitive scores and more cognitive decline during aging (Katan et al., 2013; Strandberg et al., 2004).
The association between infection, cognitive change, and neurobiological hallmarks of dementia has also been reported using preclinical models. One commonly leveraged approach to study neurological consequences of infection is lipopolysaccharide (LPS)-induced inflammation (Catorce and Gevorkian, 2016). Indeed, LPS signals principally through interactions with several proteins including toll-like receptor (TLR)-4 (Lu et al., 2008), a receptor whose expression on a number of nervous system cell types has been documented in multiple species (Vaure and Liu, 2014). Studies conducted by numerous groups using a variety of exposure paradigms (i.e., single or continuous regimens, systemic or central nervous system administration) have demonstrated that LPS increases inflammatory cytokine levels in a variety of brain regions, activates microglia and astrocytes, and impairs cognition (Biesmans et al., 2013; Dunn and Swiergiel, 2005; Herber et al., 2006; Quan et al., 1994; Zhao et al., 2019; Hauss-Wegrzyniak et al., 1998). Age appears to potentiate the detrimental cognitive impacts of LPS (Chen et al., 2008; Tarr et al., 2011). Finally, some of the neurobiological consequences of even a single LPS exposure may persist weeks or even months after resolution of the initial inflammatory cascade (Bossù et al., 2012), suggesting the potential for long-lasting effects.
The neurological impacts of higher infection burden as modeled by repeated, intermittent LPS exposure are less well studied, though emerging findings suggest cognitive impairment and detrimental effects on brain, especially among transgenic mice expressing dementia-associated risk factors (Marottoli et al., 2017; McAlpine et al., 2009; Sheng et al., 2003; Sy et al., 2011), though not all investigators have observed deficits (Thygesen et al., 2018). To extend the emerging literature assessing the impact of infection burden on cognitive function among normally aging mice, here, we repeatedly exposed adult mice to intermittent LPS challenges during aging. We were intentional in employing a regimen of increasing LPS doses spaced sufficiently apart to enable us to consistently induce a modest sickness response from which mice recovered between exposures. Following the final challenge, we evaluated cognitive consequences using a battery of tests designed to capture several domains of learning and memory known to be impacted during aging and to be altered in preclinical models of dementia (Webster et al., 2014). We also probed several potential mechanisms, including brain cytokine levels and hippocampal neuronal function.
2. Materials and methods
2.1. Subjects
The present study was conducted at West Virginia University, an institution accredited by AAALAC International (Association for Assessment and Accreditation of Laboratory Animal Care). All procedures were evaluated and approved by the West Virginia University Institutional Animal Care and Use Committee. C57BL/6JNia male mice, aged 10-months, were acquired from the National Institutes on Aging Aged Rodent Colony. Female mice were not included in this study due to the known sex differences in immune system function (Engler-Chiurazzi et al., 2022; Klein and Flanagan, 2016), especially across transitions in reproductive capacity, and response to LPS (Cai et al., 2016; Kuo, 2016), in which adult male mice typically display a more robust response. The mice were maintained under laboratory conditions typical of AAALAC institutions and utilized according to ARRIVE guidelines: 12 hr light/dark cycle (light hours: 06:00 to 18:00 hrs), with an ambient room temperature of 20–26 °C and a relative humidity of 30–70 %. Animals were group-housed (3–5 per cage), in standard filter-topped, transparent cages, and provided with environmental enrichment in the form of crinkled paper strips (Uline Shipping Supplies, Allentown, PA, USA) and corncob bedding (Envigo, Indianapolis, IN, USA), as well as access to chow pellets and water ad libitum throughout the entire study.
2.2. Experimental design and treatments
The experimental design and timeline are depicted in Fig. 1. At the start of the study, there were 103 mice used for experimentation. Animals were randomly assigned to one of two treatment conditions: Vehicle (N = 52) or Intermittent LPS (N = 51). LPS (Escherichia coli 055: B5, Sigma, St. Louis, MO) was reconstituted in sterile, injectable saline (B. Braun Medical Inc, Irvine, CA); injectable saline without LPS was used as the Vehicle control. To reliably induce a moderate sickness response from which subjects made a full recovery, mice received one injection (i.p.) every 15 days whereby the dosage of LPS was progressively increased with each subsequent injection such that: Injection 1 = 0.4 mg/kg, Injection 2 = 0.8 mg/kg, Injection 3 = 1.6 mg/kg, Injection 4 = 3.2 mg/kg, Injection 5 = 6.4 mg/kg.
Fig. 1. Experimental Design and Timeline.
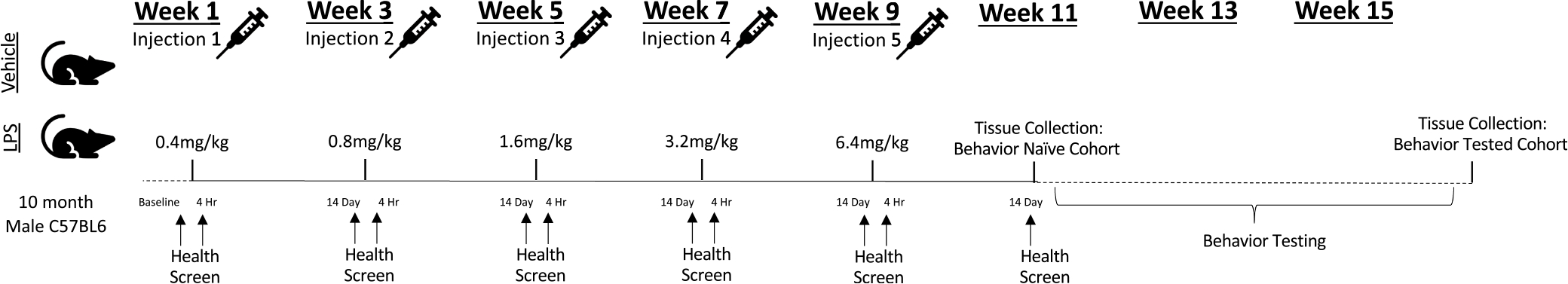
10-month-old C57BL/6JNia male mice were randomly assigned to one of two treatment conditions: Vehicle (Saline) or Intermittent LPS. To reliably induce a moderate sickness response from which subjects made a full recovery, mice received one injection every 15 days whereby the dosage of LPS was progressively increased with each subsequent injection such that: Injection 1 = 0.4 mg/kg, Injection 2 = 0.8 mg/kg, Injection 3 = 1.6 mg/kg, Injection 4 = 3.2 mg/kg, Injection 5 = 6.4 mg/kg. Health and sickness behavior was measured at baseline (one day prior to the first injection) to verify all mice displayed equivalent health without signs of sickness, at 4 h post-injection (Day 1) to verify induction of moderate sickness among LPS-treated mice, and at 14 days post-injection (one day prior to the subsequent injection) to verify a lack of sickness. Following the final injection (Injection 5), mice were randomly assigned to one of two cohorts (Behavior Naïve or Behavior Tested). Tissue was collected from the Behavior Naïve cohort approximately 2–3 weeks following the final injection, while tissue collection for the Behavior Tested cohort took place approximately 3–4 weeks later (approximately 5–6 weeks following the final injection).
2.3. Health and sickness screen
Overall health and sickness behavior within the home cage environment was assessed under a ventilated laminar flow hood in the housing room using an objective 20 point screen developed in our laboratory, as we have done previously (Doll et al., 2015). The screen was designed to provide insight into the global physical health of an animal, to be rapid and easy to administer in the home cage environment, to be minimally invasive, and to ensure consistency in scoring across the post-injection recovery period. For the screen, a subject was observed in its home cage and evaluated for 1) general appearance, 2) posture, 3) respiration, and 4) spontaneous locomotion/social interaction. Body condition (emaciation and hydration; 5) was assessed by the pinch test. Body temperature (rectal; 6) and body weight (7) changes from baseline were also measured. The screen was administered at baseline, and at 4 hr and again 14 days following each injection. Scores, the dependent variable, were determined by totaling points assigned to mice in each of the seven unique categories such that lower scores (e.g. <3) were indicative of ‘normal’ health and higher scores indicated increasing burden of sickness behavior and physiological state.
2.4. Behavioral test battery
All behavioral testing was performed beginning 15 days following Injection 5. All animals underwent all behavioral tests included in the test battery in the order in which they are described below. Behavior tests were conducted over consecutive days and administered by experimenters who were blinded to treatments the animals received. All testing occurred under ambient lighting between 06:00 to 18:00 hrs. Within a given test day, due to equipment constraints, animals were tested in batches of 8–12 mice/cohort. Animals from each treatment condition were counter-balanced across batches such that mice from each treatment group were equivalently represented in each batch. Prior to behavioral testing, each batch of animals were acclimated to the testing room environment for at least 15 min. Between each animal, the apparatus was cleaned of debris and olfactory cues using an anti-bacterial disinfectant (Virkon, Pharmacal, Naugatuck, CT) unless noted.
2.4.1. Open field
To determine spontaneous locomotor activity under full room illumination, each animal was placed into individual 16 × 16 × 15 inch3 chambers (Photobeam Activity System; San Diego Instruments, San Diego, CA, USA) and allowed to explore the arena for a 10 min trial. Horizontal and vertical movements were measured by determining the number of photo-beams disrupted by the animals, the dependent variable.
2.4.2. Step-through inhibitory avoidance
To assess single-trial aversive learning and retention (Detrait et al., 2009; Jarvik and Kopp, 1967), mice were evaluated on the step-through inhibitory (passive) avoidance task. The apparatus (Med Associates, Fairfax, VT, USA) consisted of a two-compartment device (one side under full room illumination, one side darkened with a non-transparent black plastic covering) where each compartment contained a metal rung floor that was connected to an electric shock delivery device and was separated by a door that could be raised to permit free movement of the subject between compartments. For training, each mouse was placed in the illuminated side and allowed to freely explore for 5 sec. Then, the door was raised allowing the mouse free access to the darkened chamber. When the mouse entered the darkened compartment, as defined by all four paws contained in the darkened chamber, the door was lowered and within 2 sec, the mouse was administered a 3 sec duration electric shock (0.3 mA). Any mouse that did not enter the darkened compartment within 120 sec was gently encouraged to enter the darkened compartment, whereby it was administered a foot shock. Following training of all animals, for retention testing of learned information, this procedure was repeated immediately (10 mins) to verify learning ability (Immediate Retention Trial), and 24 h later to assess memory retention (24 Hr Retention Trial). Retention trials had a maximal duration of 300 sec and no foot shocks were administered. Latency to enter the darkened compartment was recorded for all animals on each trial.
2.4.3. Hot plate
To verify nociceptive capacity, mice were exposed to the hot plate test (Bannon and Malmberg, 2007). For this test, the hot plate apparatus (Model 39; IITC, Woodland Hills, CA) was set to 55.0 °C. A subject was placed onto the hot plate surface and confined using a square plastic box (23.5 cm × 23.5 cm). The total duration of hot plate exposure was limited to 30 s (a cutoff time typically employed (Bannon and Malmberg, 2007) to provide additional dependent variable readouts, described below, as well as to dissociate apparatus removal from a given behavioral response. The dependent variable, type of and latency (s) to first nociceptive behavior, defined as flicking/shaking of a hindlimb or jumping (all four paws cease contact with the heated surface), was recorded. As well, the number of nociceptive behaviors of each subtype (hindlimb flicks/shaking or jumps) and the total number of observed nociceptive behaviors (hindlimb flicks/shaking and jumps combined) were documented.
2.4.4. Morris water maze (Reference and working memory Versions)
The Morris water maze (Morris, 1984) is a commonly-utilized, well-validated rodent test known to elucidate robust age- or AD-related changes in learning and memory (Webster et al., 2014; Baxter and Gallagher, 1996; Bizon et al., 2012). Testing was carried out according to previously published methods (Engler-Chiurazzi et al., 2011; Engler-Chiurazzi et al., 2012), with some modifications. The apparatus was a white circular tub (San Diego Instruments, San Diego, CA) filled with cool room temperature (20 °C) water made opaque with gray non-toxic tempera paint unless noted. Animal performance was documented with videos acquired from an overhead camera. Data were analyzed via a commercially available tracking system (ANY-maze, Stoelting, Chicago, IL); path length to platform, latency to escape, and swim speed were the primary outcome measures for all phases. A cued (visible platform) learning session to rule out group differences in locomotion, vision, and motivation (Stage 1) was followed by a win-stay spatial reference memory version of the hippocampal-dependent Morris maze (Stage 2) as well as a spatial delayed match-to-place adaptation to assess prefrontal cortex-dependent working memory and striatal-dependent perseveration (Stage 3). For all phases, testing was carried out by a well-trained experimenter who was blinded to treatment group status. Mice had 60 sec to locate the platform in each trial. Once they located the escape platform, they remained on it for 15 sec and were removed from the maze to a standard mouse cage warmed using heat lamps. If an animal failed to locate the escape platform, it was gently led to it after time expired. Given that aging is associated with reductions in swim speed, we analyzed performance using distance moved as the dependent variable for all phases, unless noted.
For Phase 1, water was undyed, the visible contrasting-colored escape platform was positioned ~0.5 cm above the water, and curtains obscured spatial cues. Animals were given 6 trials in one day.
For spatial testing (Phase 2–3), non-toxic water dye obscured an escape platform 1 cm submerged. Spatial cues (posters, shelving, curtains, etc.) were fixed throughout the room to aid in navigation. Animals received 6 trials/day for 7 days for both phases. For Phase 2, the platform location remained the same for all trials across all days of testing; start locations varied semi-randomly across days and trials to prevent the use of motoric, non-spatial strategies to gain water escape. Memory retention was assessed by analyzing change in outcome measures from the final trial of each day to the first trial of the following day (overnight forgetting). To verify the use of a spatial strategy and the extent of platform location localization on Phase 2, on the final testing day, an additional probe trial (platform removed) was given and additional outcome measures, number of platform location crossings and distance moved in distinct maze quadrants, was assessed.
For Phase 3, the platform location remained fixed within a day but varies across days, thus animals need to update the association between the escape platform location and the spatial cues daily to efficiently gain water escape. Group differences in outcome measures between the first trial (in which the animal learns the day’s platform location) and the second trial (Working Memory Trial; when the animal must return to the just-rewarded spatial location) reveal working memory deficits. Subsequent trials in a day allow for the determination of platform location learning/consolidation (Recent Memory Trials; Trials 3–6). On the final day of testing, a two-hour delay was imposed between trial 1 and 2 to assess working memory under increased memory demand.
2.5. Stratification strategy for tissue processing
Following the final injection (Injection 5), mice were randomly assigned to two cohorts (Behavior Naïve or Behavior Tested). Tissue was collected from a subset of the Behavior Naïve cohort following the final health/sickness behavior screen (approximately 2–3 weeks later), while tissue collection for a subset of the Behavior Tested cohort took place approximately 5–6 weeks following the final injection (approximately 4 weeks after the Behavior Naïve cohort). Subsets of tissues were collected and stored until processing, according to assay-specific methods detailed below. So as to capture the full spectrum of cognitive ability displayed within the group for each endpoint, animals within each treatment group were rank ordered based on passive avoidance performance and allocated for further assessment, as described below.
2.6. Brain cytokine expression
Under terminal isoflurane inhalation-induced anesthesia, animals were euthanized via cardiocentesis followed by rapid decapitation. As we have done previously (Carrera Arias et al., 2021; Michalovicz et al., 2019), total RNA was isolated from discrete brain regions (frontal cortex and hippocampus) and cytokine expression measured by qRT-PCR analysis. Briefly, total RNA was isolated from the brain tissue using Trizol Reagent (Thermo Fisher Scientific, Waltham, MA, USA), Phase-lock heavy gel (Eppendorf AG, Hamburg, Germany), and RNeasy mini spin columns (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. PCR analysis of the housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and of the proinflammatory mediators, C–C Motif Chemokine Ligand (CCL)2, glial fibrillary acidic protein (GFAP), interleukin (IL)-6, IL-1β, leukemia inhibitor factor (LIF), oncostatin M (OSM), and tumor necrosis factor (TNF)-α, was performed in an ABI7500 Real-Time PCR System (Thermo Fisher Scientific) in combination with TaqMan® chemistry. These targets were selected to capture a broad range of neuroinflammatory pathways known to be engaged in neurodegenerative disease states, including glycoprotein 130-cytokines, markers of astrogliosis-associated STAT3 activation and cytokine/chemokine secretion, and cytokines secreted in response to inflammasome activation (Cekanaviciute and Buckwalter, 2016; Choi et al., 2014; Damiani and O’Callaghan, 2007; Kwon and Koh, 2020; Sriram et al., 2004; Linnerbauer et al., 2020). Relative quantification of gene expression was performed using the comparative threshold (ΔΔCT) method to normalize expression changes against the GAPDH control, as well as to normalize the expression changes of Intermittent LPS-treated mice to the corresponding saline-treated controls (Behavior Naïve, Behavior Tested, or Naïve + Behavior Tested Combined). Cycle threshold values for our single housekeeping GAPDH gene were not different across experimental groups, and ranged from 18.54 to 18.90 for cortex and from 18.70 to 19.31 for hippocampus, indicating stable expression across tissues assayed.
2.7. Hippocampal slice preparation
Procedures identical to those previously published (Hunsberger et al., 2016; Sen et al., 2016; Wang and Zheng, 2015) were used for brain slices, electrophysiological recording, and data analysis. Briefly, animals were euthanized with carbon dioxide, the hippocampi were isolated, and 300-μm thick transverse slices were prepared using a Leica VT1200S Vibratome (Leica Microsystems, Wetzlar, Germany). Slices were incubated at room temperature in artificial cerebrospinal fluid (ACSF; 124 mM NaCl, 3 mM KCl, 1.2 mM MgSO4, 2.1 mM CaCl2, 1.4 mM Na2 PO4, 26 mM NaHCO2, 20 mM dextrose) saturated with 95 % O2/5% CO2 to maintain pH 7.4. After one-hour equilibration, slices were transferred into a recording chamber for electrophysiological measurements. Field potential recordings were made on 2–3 slices per mouse, and the total number of slices per group was 15, 14, 14, and 12 for Saline Naïve, LPS Naïve, Saline + Behavior, and LPS + Behavior, respectively.
2.8. Extracellular field potential recording
The slices were viewed with an Olympus BX50WI microscope equipped with a high-resolution, high-sensitivity CCD camera (Dage-MTI, Michigan City, IN). A bipolar stimulating electrode (100-μm separation, FHC, Bowdoinham, ME) was placed in the Schaffer collateral pathway. A patch pipette drawn with the P97 Brown-Flaming Puller, (Sutter Instruments, Novato, CA) and filled with ACSF (2–5 MΩ, 1.5 mm OD, 0.86 mm ID) was placed in the stratum radiatum of CA1 to record excitatory postsynaptic potentials (EPSPs). Field EPSPs (fEPSPs) were recorded in the presence of 20 μM bicuculline methochloride (Tocris, Bristol, UK) to eliminate GABAergic inhibitory inputs. All parameters including pulse duration, width, and frequency were computer controlled. Constant-current pulse intensities were controlled by a stimulus isolation unit A360 (WPI, Sarasota, FL). The data were recorded online using Clampex 10.0 software (Molecular Devices, Sunnyvale, CA). For fEPSPs recording, signals were amplified (gain 200–500), filtered (6 kHz), acquired at a sampling rate of 10 kHz with 20 s intervals using MultiClamp 700B amplifier with pClamp 10.0 software (Molecular Devices, Sunnyvale, CA). Standard off-line analyses of the data were conducted using Clampfit 10.0 software (Molecular Devices, Sunnyvale, CA).
Basal synaptic transmission represented by input–output responses and the slopes of fEPSP were plotted as a function of stimulus intensity. Paired pulse facilitation (PPF) was used to assess short-term synaptic plasticity attributed mainly to a presynaptic effect. Pairs of stimuli separated by varying intervals were delivered to the stratum radiatum at 0.05 Hz. Paired responses were averaged, and ratios of fEPSP slopes from the second stimulus (fESPS2) to fEPSP slopes from the first stimulus (fESPS1) were calculated and plotted as a function of interstimulus intervals. To assess potential changes in synaptic plasticity, long term potentiation (LTP) was evaluated after 5–10 min of stable baseline recording. LTP was induced by high frequency stimulation (HFS), which consisted of a total of 400 stimuli that were delivered as 4 discrete bursts with inter-burst intervals of 20 s. The stimuli within bursts were delivered at 10 ms intervals (100 Hz). The strength of fEPSPs was assessed by measuring the slope (initial 20–80 %) of the fEPSPs rising phase. LTP was quantified by comparing the mean of fEPSPs slope 60 min post-HFS period with the mean fEPSPs during the baseline and expressed as the percentage change from the baseline.
2.9. Statistical analyses
Data were analyzed using StatView 5.0, SPSS (version 28, SPSS Inc., Chicago, IL) and GraphPad Prism 8.0 (GraphPad, La Jolla, CA) with ANOVA where Treatment (Vehicle or Intermittent LPS) was the between factor with or without repeated measures (Injection (#1, 2, 3, 4, or 5) and Timepoint (4 hr or 14 day post-injection)), or Student’s t-test, as appropriate for a given dependent measure. Where relevant, Behavior Experience (Naïve or Tested) was included. Fisher’s LSD post-hoc two-group analyses were used in the presence of significant higher order interactions. Violations to fundamental assumptions of ANOVA (i.e., homogeneity of variance) were checked prior to additional analyses. Given the large number of animals, to support the logistical execution of the experiments and embed internal replication of findings, studies were conducted in two waves, with members of each treatment condition equally represented in each wave; Wave was added as a between factor in the analyses for each dependent variable. There were no significant interactions of Treatment with Wave for any test or variable of interest, thus all analyses presented here were conducted without the Wave variable included. Unless otherwise noted, two-tailed tests were used, p < 0.05 was considered significant and data are represented as mean ± SEM. A summary of Ns for each of the assays is described in Supplemental Fig. 1.
3. Results:
3.1. Experimental exclusions prior to behavior
To confirm a reliably moderate sickness response in the immediate hours following each LPS-induced inflammatory/infection mimic experience, as well as to verify inflammatory resolution, we evaluated the presence and magnitude of sickness behaviors at 4 h and 14 days post-injection using a 20 point scale we have previously developed (Doll et al., 2015). Only animals who presented visually healthy (i.e., low sickness score, no signs of fighting-related injuries) at baseline prior to first treatment administration were included in analyses (N = 6 did not meet this criteria). The criterion for sickness was set at a score of 3, as this was the lowest score displayed by an LPS-treated mouse after the first injection. Over the course of the 10-week injection period, five animals were found dead or were humanely euthanized due to likely injection complications (e.g. puncturing a vital organ), fighting-related injuries, or idiopathic paralysis (Vehicle = 2 and LPS-treated = 3); these mice were excluded from all analyses. As well, as our research question critically required LPS-exposed mice to mount a reliable sickness response and to make a full recovery prior to the next inflammatory challenge, two LPS-treated mice who failed to mount a sickness response immediately following two or more injections (score < 3) were excluded from all analyses. One mouse (LPS-treated) failed to make a full recovery (score > 3) from at least two injections by 14 days and was excluded.
3.2. Health and sickness screen
At baseline, one day prior to the first LPS injection, all animals displayed equivalent and low health/sickness behavior screen scores (Treatment (Vehicle or LPS) t-test; p > 0.74). Following baseline, health and sickness data were analyzed in a factorial ANOVA with mixed measures where Treatment (Vehicle or LPS) was the between factor and Injection (1, 2, 3, 4, or 5) and Timepoint (4hr or 14 day post-injection) were the repeated factors. There was a significant higher order Treatment × Injection × Timepoint interaction [F(1,348) = 10.11, p < 0.0001] (Fig. 2); when we assessed the Treatment × Timepoint interaction at each level of Injection, all interactions were significant [Injection 1: F(1,87) = 486.43, p < 0.0001; Injection 2: F(1,87) = 239.86, p < 0.0001; Injection 3: F(1,8) = 279.18, p < 0.0001; Injection 4: F(1,87) = 266.38, p < 0.0001; Injection 5: F(1,87) = 354.71, p < 0.0001]. We probed this interaction by first assessing the effect of Treatment at the 4hr-post injection timepoint. Following each injection, there was a significant Treatment effect [Injection 1: F(1,87) = 485.10, p < 0.0001; Injection 2: F(1,87) = 293.07, p < 0.0001; Injection 3: F(1,87) = 463.54, p < 0.0001; Injection 4: F(1,87) = 353.22, p < 0.0001; Injection 5: F(1,87) = 470.28, p < 0.0001], such that LPS exposure consistently moderately elevated sickness scores (means = 4.61 to 6.43) relative to Vehicle-treated mice (means = 0.37 to 0.88). We next evaluated the extent to which animals’ scores within a given group changed across time within a given injection cycle. Within Vehicle-treated mice, there were no significant effects of Timepoint, with the exception of Injection 3 [F(1,5) = 6.355, p < 0.05], in which health/sickness scores were slightly increased at the 14D timepoint (mean ± SD at 4hr = 0.66 ± 0.71; mean ± SD at 14d = 1.0 ± 0.67). Within the LPS-treated groups, there were significant effects of Timepoint for all Injection cycles [Injection 1: F(1,42) = 491.83, p < 0.0001; Injection 2: F(1,42) = 304.24, p < 0.0001; Injection 3: F(1,42) = 361.38, p < 0.0001; Injection 4: F(1,42) = 304.63, p < 0.0001; Injection 5: F(1,42) = 507.85, p < 0.0001], verifying that all LPS animals included in behavioral analyses had substantially reduced health scores by 14 days; immediately prior to the following injection.
Fig. 2. Intermittent LPS Regimen Reliably Induces Moderate Sickness Behavior.
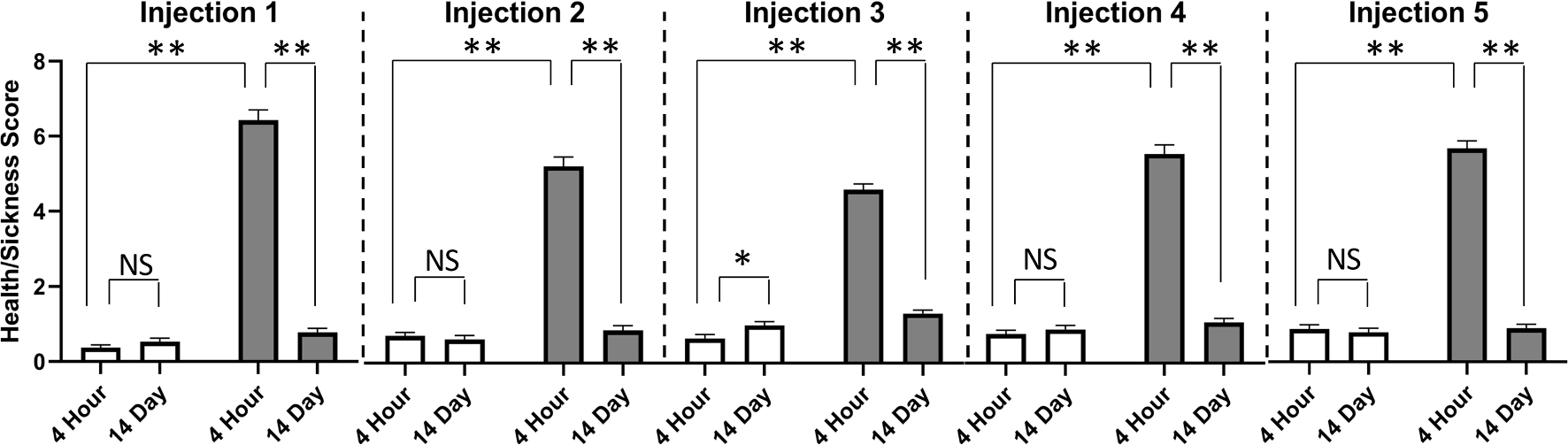
At 4 h following each injection, LPS exposure induced significantly elevated sickness behavior relative to Vehicle-treated mice. Within group comparisons across time in a given injection cycle revealed that LPS-treated mice made complete recoveries by 14 days following all five exposures, as evidenced by significantly lower sickness behavior scores at Day 14 than at 4 h post-injection. With the exception of the Injection 3 cycle, Vehicle-treated mice showed no changes in health/sickness scores across time within a given injection cycle. Mean ± SEM; * = p < 0.05; ** = p < 0.0001. Vehicle (N = 46) =; Intermittent LPS (N = 43) =.
3.3. Open field spontaneous locomotion
Open field data were determined with t-tests with Treatment (Vehicle or LPS) as the between factor for each dependent variable (movement zone). Repeated LPS exposure did not impact spontaneous locomotor or anxiety-like behavior in the open field (ps > 0.5). There were no significant group differences in beam breaks (a proxy for distance moved) in the vertical axis (rearing; Fig. 3Ai) nor in the arena (Fig. 3Aii).
Fig. 3. Intermittent LPS Exposure During Aging Subtly Impairs Cognitive Function.
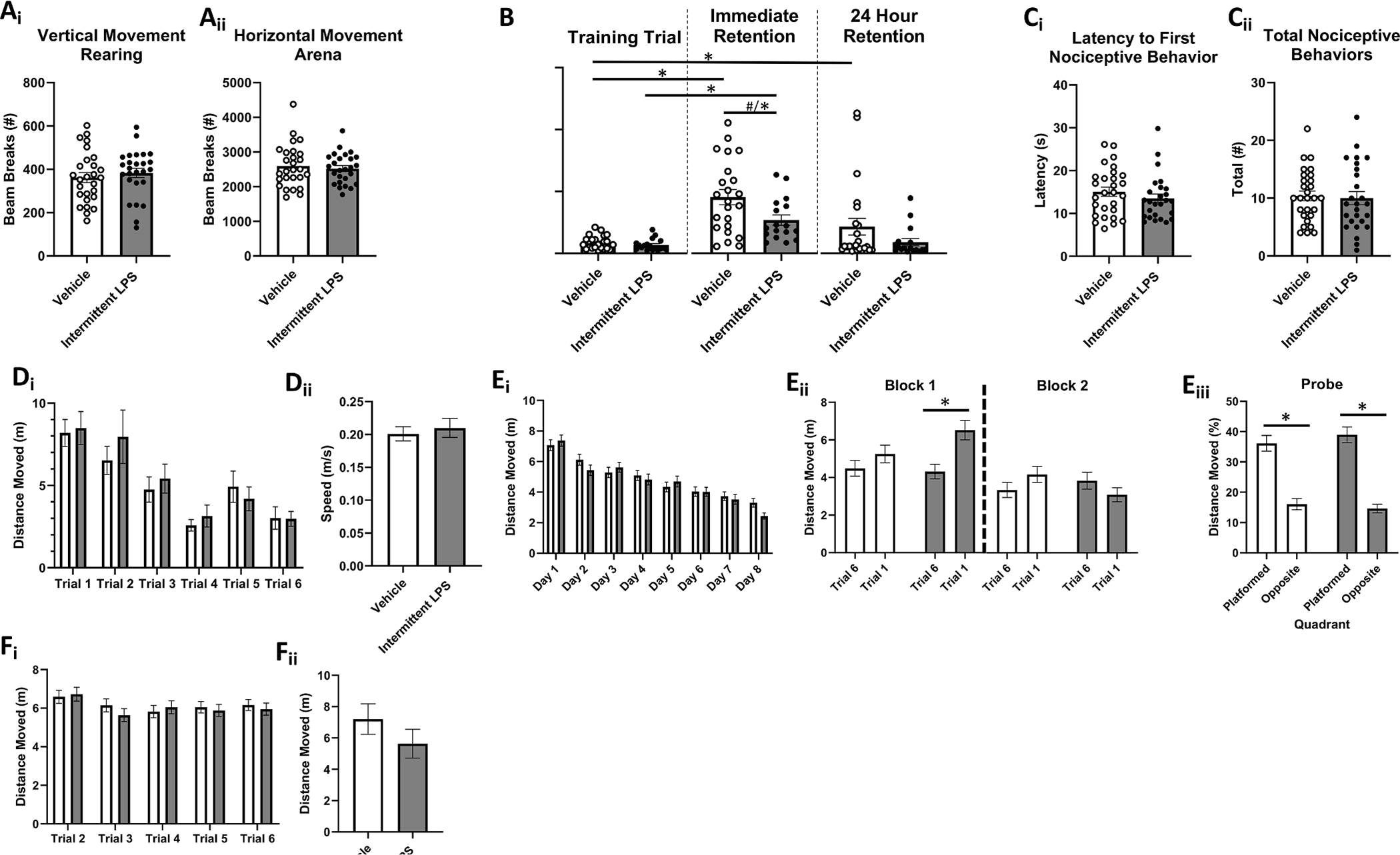
A. Open Field Locomotor Activity. There were no group differences in (Ai) vertical movement (rearing behavior) nor (Aii) horizontal movement in the arena. B. Inhibitory (Passive) Avoidance Learning and Retention. Though neither group differed in their latency to enter the darkened compartment on the learning trial prior to shock pairing (p > 0.20), LPS-treated mice showed a trend towards impaired learning on the immediate retention trial (~20 min delay) relative to Vehicle-treated mice; removal of a significant outlier revealed a significant learning impairment of LPS. Vehicle-treated, but not LPS-treated, mice displayed longer term retention of the shock-location pairing. C. Hot Plate Nociception. Groups did not differ on either (Ci) latency to display nociceptive behaviors nor (Cii) total nociceptive behaviors displayed on the hot plate test. D. Visible Platform Performance. There were no group differences in (Di) latency to platform, nor (Dii) swim speed. E. Morris Water Maze Spatial Reference Learning and Memory. Overall learning curves did not differ between the groups (Ei), and there was a lack of group differences in the extent to which mice explored the previously platformed quadrant on the Probe trial (Eiii), indicating that all mice similarly acquired the spatial location of, and utilized a spatial strategy to find, the hidden platform by the end of testing. However, LPS-treated mice displayed worse overnight forgetting of the platform location (indicated by longer swim distance on trial 1 of each day than on trial 6 of the previous day) during the early phase of training (Eii). F. Morris Water Maze Spatial Working Learning and Memory. There were no group differences in swim distance to water escape across days and trials (Fi) nor following the two-hour delay challenge (Fii). Mean ± SEM; * = p < 0.05; # = p < 0.10. Vehicle (Ns = 23–27) =; Intermittent LPS (Ns = 18–26) =.
3.4. Passive avoidance aversive learning and retention
Passive avoidance data were analyzed with t-tests for each unique trial (Learning, Immediate Retention, and 24 hr Retention) with Treatment as the Between factor. Three animals (Vehicle = 2; LPS = 1) displayed an unrepresentatively long latency to enter the darkened compartment on the learning trial (prior to shock-paired association), possibly indicative of basal differences in light-aversion/exploration motivation. To avoid attributing long latencies to enter the darkened compartment on retention trials as evidence of shock-association learning among subjects who differ at training in there basal locomotor, affective or visual capacity, these mice were excluded from all the passive avoidance analyses; their data did not impact results. Possible equipment failure of shock delivery and/or procedural errors occurred for 8 mice (Vehicle = 3, LPS = 5) and their data were excluded. As well, three animals (Vehicle = 0, LPS = 3) displayed deficits in nociception (no behavior displayed until final 3 sec of trial and/or 3 > total behaviors displayed) therefore data from these mice were excluded from analyses.
Though neither group differed in their latency to enter the darkened compartment on the learning trial prior to shock pairing (p > 0.20), LPS-treated mice showed a trend towards significantly shorter latencies to enter the shock-paired darkened compartment on the immediate retention trial (~20 min delay) relative to Vehicle-treated mice [F(1,39) = 2.87, p < 0.1] (Fig. 2B). However, this included a statistically significant outlier in the LPS group (>4 SD from group mean). Removal of this outlier revealed a statistically significant group difference [F(1,38) = 5.32, p < 0.05]. The Treatment main effect on the 24 hr retention trial was not statistically significant. We then compared performance on the learning trial vs the 24 hr retention trial within Treatment groups (t-test within each level of Treatment (Vehicle or LPS) separately with Trial as the within factor). Vehicle-treated, but not LPS-treated, mice displayed a significant Trial effect [t(22) = 4.72, p < 0.05] such that their latency to enter the darkened compartment on the 24 hr retention test was significantly longer than that of their learning trial, indicative of long-term retention of the shock-location pairing in this treatment group.
3.5. Hot plate nociception
T-tests measuring the Treatment effect (Vehicle or LPS) for the latency to nociception and distinct nociceptive behaviors were conducted. Animals that failed to display sufficient nociceptive behavior as described above were excluded from analyses. There were no treatment group differences for either latency to first nociceptive behavior (p > 0.56) nor for total nociceptive behaviors displayed (p > 0.13) during the trial (Fig. 2C).
3.6. Morris water maze spatial working and reference memory
Water maze data were analyzed for each testing phase separately (Visible platform, reference memory, and working memory), using factorial ANOVA with Treatment as the Between factor and Days and/or Trials as the within factors. One animal (Vehicle) was found dead following a day of water maze behavior testing and was excluded from all subsequent behavioral analyses. Also, one (Vehicle) mouse presented with visual deficits and was excluded.
During visible platform (Phase 1) testing, there were no Treatment × Trial interactions nor Treatment main effects for any relevant dependent variable (Latency to Platform, Distance Moved, or Speed), indicating that all mice had similar capacity to see and swim regardless of Treatment (Fig. 2D).
During the reference memory Phase 2 portion, all animals, regardless of treatment condition, showed evidence of learning, with significant main effects of Day [F(7,315) = 24.60, p < 0.0001] and Trial [F(5,225) = 3.87, p < 0.005] for distance moved (Fig. 2Ei). To assess overnight forgetting, we considered treatment group differences in performance on the final trial of each day vs the first trial of each subsequent day (Fig. 2Eii). Though the analysis with all overnight intervals included revealed no significant Trial × Treatment interactions, visual inspection of the graph revealed potential LPS retention deficits early in the training period. Thus, excluding the first overnight interval to ensure evenly sized blocks, we grouped data into early and late overnight blocks, (D2/3, D3/4, and D4/5) and (D5/6, D6/7, and D7/8), revealing a significant Block × Trial × Treatment interaction [F(1,45) = 6.21, p < 0.05]. When we probed this effect at each level of Treatment, the Block × Trial interaction was only significant for LPS-treated mice [F(1,21) = 20.13, p < 0.0005] and the Trials main effect was only significant within the first block of overnight intervals [F(1,21) = 22.47, p < 0.0001], such that average performance during the early learning phase of testing among LPS-treated mice was worse on the first trial of each day relative to their average performance on the final trial of previous days. For the probe trial, there were no group differences on any measure (percent distance moved in platformed vs opposite quadrant, platform zone entries, latency to first platform zone entry), indicating that by the end of testing, all mice regardless of treatment condition, had localized the platform location to a similar extent and employed similar search strategies to navigate to it (Fig. 2Eiii).
During the working memory Phase 3 portion, there were no higher order interactions between Days, Trials (T2-6) and Treatment (Fig. 2Fi). No significant treatment effects emerged even when we conducted a priori planned comparisons evaluating performance on the working memory trial (T2) or recent memory trials (T3-6) separately. On the 2-hour delay challenge test day (Fig. 2Fii), there were no differences between Vehicle or LPS-treated mice on the post-delay trial (T2).
3.7. Brain cytokine gene expression
To determine the extent to which intermittent LPS induced a persistent brain inflammatory response, we evaluated gene expression of a panel of cytokines, chemokines, or immune signaling markers in brains (frontal cortex and hippocampus) of both behaviorally naïve (N = 3/group) and behaviorally tested (N = 3–4/group) mice. Among behaviorally naïve mice, subjects exposed to intermittent LPS displayed significantly reduced levels of hippocampal IL1β relative to Vehicle-treated mice [t(4) = 2.88, p < 0.05](Table 1); there were no group differences in hippocampal expression of any other cytokine. Similarly, inflammatory cytokine gene expression was similar for all targets within cortex. Among behaviorally tested mice, there were no group differences in hippocampal nor cortical gene expression for any inflammatory target. Because a lack of significant differences could be explained by the analyses being underpowered due to low N (Kepple and Wickens, 2004), we calculated Cohen’s d effect sizes and used these values to conduct post-hoc power analyses of the gene expression data using the G*Power software (Supplemental Table 1A). Approximately half of the comparisons yielded at least moderate sized effects (Cohen’s d > 0.5) with low power (power 1 − β < 0.8). Combining gene expression values from each exposure group across both timepoints partially improved the issues with underpowered analyses (Supplemental Table 1B). When we combined both Naïve and Tested groups within each treatment condition and determined target expression fold changes of all LPS mice normalized to all Vehicle mice, we found that hippocampal IL-6 levels were significantly higher among mice with a history of LPS exposure [t(11) = 2.28, p < 0.05].
Table 1.
Cytokine gene expression in cognitive brain regions (Mean ± SEM; relative fold change).
| Behaviorally Naïve Mice | TNF-α |
OSM |
LIF |
CCL2 |
IL-1β |
GFAP |
IL-6 |
|||||||
| Veh | LPS | Veh | LPS | Veh | LPS | Veh | LPS | Veh | LPS | Veh | LPS | Veh | LPS | |
|
| ||||||||||||||
| Cortex | 1.08 ± 0.30 | 1.04 ± 0.08 | 1.01 ± 0.09 | 0.90 ± 0.09 | 1.09 ± 0.32 | 0.83 ± 0.23 | 1.07 ± 0.25 | 0.90 ± 0.08 | 1.02 ± 0.12 | 0.89 ± 0.18 | 1.02 ± 0.14 | 1.03 ± 0.24 | 1.05 ± 0.22 | 1.14 ± 0.17 |
| Hippocampus | 1.09 ± 0.34 | 1.09 ± 0.09 | 1.02 ± 0.12 | 0.93 ± 0.14 | 1.14 ± 0.36 | 0.95 ± 0.22 | 1.04 ± 0.21 | 0.55 ± 0.04 | 1.02 ± 0.16 | 0.54 ± 0.04* | 1.01 ± 0.08 | 1.21 ± 0.09 | 1.02 ± 0.13 | 1.25 ± 0.14 |
|
| ||||||||||||||
| Behaviorally Tested Mice | TNF-α | OSM | LIF | CCL2 | IL-1β | GFAP | IL-6 | |||||||
| Veh | LPS | Veh | LPS | Veh | LPS | Veh | LPS | Veh | LPS | Veh | LPS | Veh | LPS | |
|
| ||||||||||||||
| Cortex | 1.04 ± 0.15 | 1.15 ± 0.21 | 1.02 ± 0.10 | 1.20 ± 0.06 | 1.03 ± 0.14 | 0.72 ± 0.20 | 1.04 ± 0.17 | 0.95 ± 0.10 | 1.05 ± 0.17 | 1.18 ± 0.17 | 1.01 ± 0.09 | 1.10 ± 0.13 | 1.02 ± 0.10 | 0.89 ± 0.07 |
| Hippocampus | 1.05 ± 0.18 | 1.55 ± 0.41 | 1.00 ± 0.06 | 0.96 ± 0.09 | 1.12 ± 0.33 | 1.49 ± 0.32 | 1.03 ± 0.16 | 0.97 ± 0.02 | 1.04 ± 0.15 | 1.70 ± 0.40 | 1.00 ± 0.04 | 0.96 ± 0.03 | 1.02 ± 0.11 | 1.26 ± 0.08 |
| All Mice (Naïve & Tested) | TNF-α | OSM | LIF | CCL2 | IL-1β | GFAP | IL-6 | |||||||
| Veh | LPS | Veh | LPS | Veh | LPS | Veh | LPS | Veh | LPS | Veh | LPS | Veh | LPS | |
| Cortex | 1.07 ± 0.16 | 1.10 ± 0.11 | 1.05 ± 0.13 | 1.07 ± 0.08 | 1.07 ± 0.17 | 0.79 ± 0.16 | 1.07 ± 0.17 | 0.94 ± 0.08 | 1.04 ± 0.11 | 1.06 ± 0.13 | 1.03 ± 0.11 | 1.08 ± 0.14 | 1.03 ± 0.10 | 1.00 ± 0.10 |
| Hippocampus | 0.99 ± 0.16 | 1.22 ± 0.17 | 1.00 ± 0.06 | 0.95 ± 0.07 | 1.14 ± 0.23 | 1.22 ± 0.18 | 1.09 ± 0.19 | 1.56 ± 0.28 | 1.07 ± 0.15 | 1.04 ± 0.21 | 1.00 ± 0.04 | 1.08 ± 0.06 | 1.02 ± 0.08 | 1.26 ± 0.07* |
= p < 0.05.
3.8. Hippocampal Long-term potentiation
In order to investigate the potential mechanisms underlying behavioral changes induced by repeated LPS injections, field potential recordings were made in hippocampal slices from animals that either did not undergo behavioral testing (Behavior Naïve) or that received behavioral assessment (Behavior Tested) after repeated injections of either LPS or saline.
An input–output curve was constructed with stimulus intensity versus the slope of fEPSP elicited in response to increasing intensities of stimulation. As shown in Fig. 4A–B, the mean slope of fEPSP increased with stronger intensity of stimulus, which was confirmed by an analysis of variance that revealed a main effect of stimulus intensities [F(10,490) = 50.78, p < 0.001]. However, there was no main effect of group [F(3,49) = 0.49, p = 0.689], Treatment [F(1,49) = 0.005, p = 0.945] or behavior [F(1,49) = 1.394, p = 0.243], and no Treatment × Behavior Experience interaction [F(1,49) = 0.025, p = 0.875]. This suggests repeated, intermittent LPS injections do not affect basal synaptic transmission in the Schaffer collateral commissural pathway of hippocampus.
Fig. 4. Intermittent LPS Exposure Impacts Hippocampal Neuronal Function.
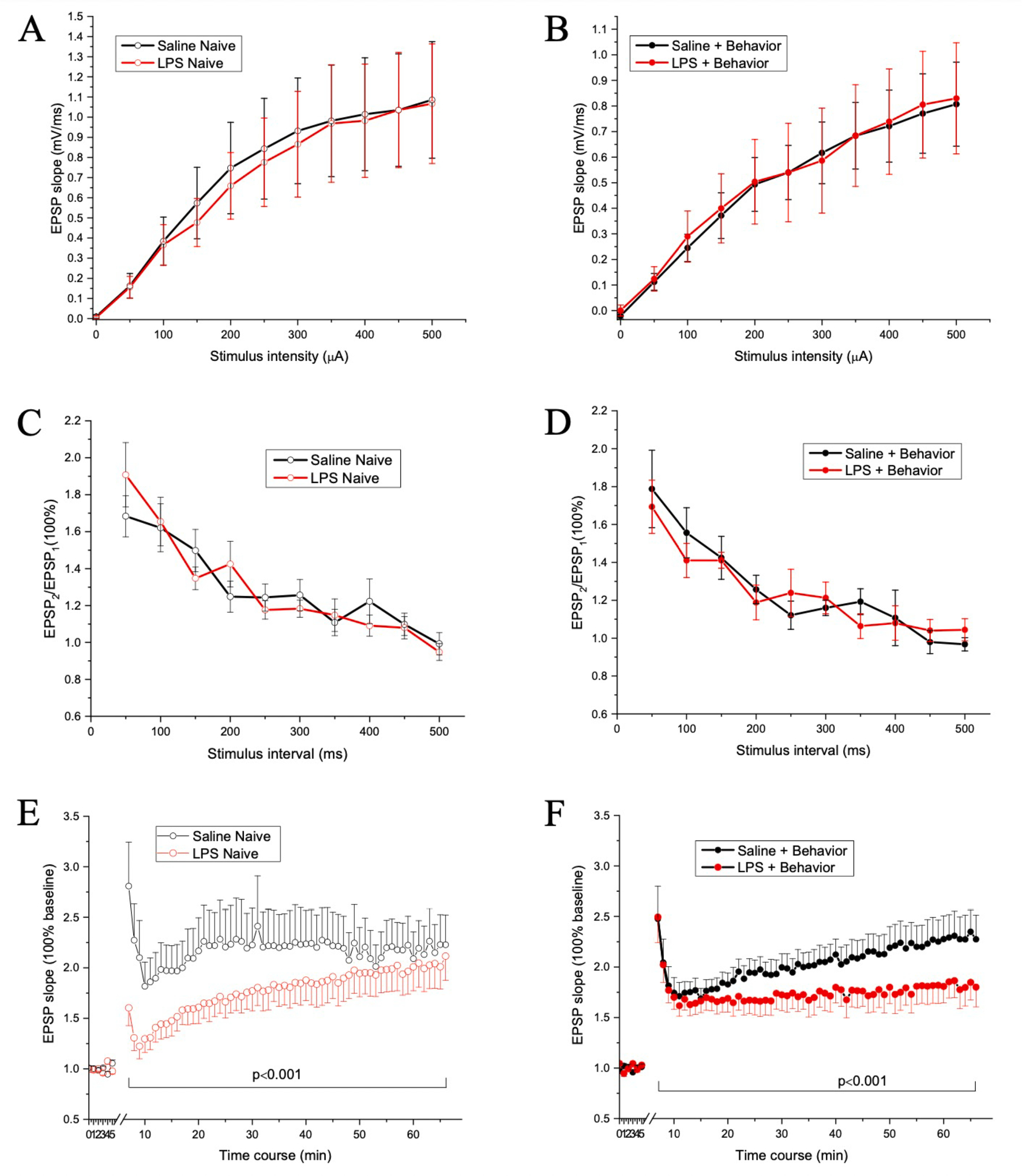
Input–output curves for Behavior Naïve (A) and Behavior Tested (B) mice. There were no group difference in input–output curves, suggesting repeated intermittent LPS injection (red lines) may not affect the basal synaptic transmission relative to Vehicle treated mice (black lines). Evoked PPF responses for Behavior Naïve (C) and Behavior Tested (D) mice. Data shown are the slope ratio (slope2/slope1) of fEPSP. Note that there was no statistical significance between the groups, indicating repeated LPS injection may not influence pre-synaptic function. Evoked LTP responses for Behavior Naïve (E) and Behavior Tested (F) mice. Data shown are the slope ratio (slope2/slope1) of fEPSP. Note that there were significant differences between the groups, specifically, between Saline Naive and LPS Naive, between Vehicle Naive and Vehicle + Behavior, but not between Intermittent LPS Naïve and Intermittent LPS Behavior. This suggests the altered LTP expression may mediate the changes in behavioral performance in mice given repeated LPS injections. N = 5 mice/group. N = number for slices is 15, 14, 14, and 12 for Vehicle Naïve, Intermittent LPS Naive, Vehicle + Behavior, and Intermittent LPS + Behavior groups, respectively.
PPF was used to assess pre-synaptic short-term synaptic plasticity. As shown in Fig. 4C–D, the slope ratio of fEPSP2/fEPSP1 in response to the interstimulus intervals was maximal at an interval of 50 ms and recovered after an interval of 350 ms. This was confirmed by an analysis of variance that revealed a main effect of interstimulus interval [F(11,550) = 50.996, p < 0.001]. However, there was no main effect of group [F(3,50) = 0.328, p = 0.805], LPS [F(1,50) = 0.007, p = 0.932] or behavior [F(1,50) = 0.047, p = 0.828], and no Treatment × Behavior Experience interaction [F(1,50) = 0.925, p = 0.341]. This indicates intermittent LPS injections do not significantly influence presynaptic function in the hippocampus and thus pre-synaptic function may not be the major contributor to the altered behavioral performance induced by repeated LPS injection.
LTP was used to assess the long-term synaptic plasticity mediated by post-synaptic function. Field potential recordings were made in the stratum radiatum of CA1 hippocampus in response to stimulation of the Schaffer collateral-commissural pathway. As shown in Fig. 4E–F, regardless of whether the mice received behavioral assessment or not, mice given repeated LPS injections exhibited smaller LTP expression compared to mice given repeated saline injections. The changes were only prominent at the duration for post-tetanic potentiation and first phase of LTP in LPS Naïve group, while the changes were long lasting in LPS + Behavior group. This was confirmed by an analysis of variance that revealed a group main effect [F(3,243) = 88.554, p < 0.001]. Post-Hoc comparisons revealed the differences were between Vehicle Naive and LPS Naive [p < 0.001], between Vehicle Naive and Vehicle + Behavior [p < 0.001], between Vehicle Naive and LPS + Behavior [p < 0.001], and between Vehicle + Behavior and LPS + Behavior [p < 0.001], however, there was no significant difference between LPS Naive and LPS + Behavior (p = 0.317). There were main effects of Treatment [F(1,243) = 232.538, p < 0.001] and Behavior Experience [F(1,243) = 6.415, p < 0.05]. Therefore, our data suggest both repeated LPS injection and behavioral experience can modulate LTP expression in hippocampal CA1 neurons, suggesting the altered LTP expression may mediate the changes in behavioral performance in mice given repeated LPS injection.
4. Discussion
Here, we evaluated the impacts of intermittent systemic inflammatory activation via LPS on cognitive performance and hippocampal neuronal function in an effort to model the neurobiological and behavioral impacts of higher lifetime infection burden. We were able to repeatedly induce moderate sickness behavior from which mice made a full recovery by systemically administering escalating doses of LPS every-two weeks. We found that experience with intermittent LPS-induced inflammation resulted in subtle but significant cognitive deficits in learning and overnight retention. We also identified that intermittent LPS reduced hippocampal long-term potentiation without impacting basal synaptic transmission. This occurred generally in the absence of an ongoing, robust brain inflammatory state (but see below discussion regarding statistical power). As detailed below, these findings support and extend what is currently known regarding the cognitive and neurobiological consequences of systemic inflammation.
LPS is a commonly deployed model to rigorously and robustly induce a systemic inflammatory response mimicking bacterial infection. We were intentional with our approach using escalating doses of LPS given every-two weeks as overcoming LPS tolerance appears to be critical to repeatedly achieving a moderate sickness response within the same organism. In a separate cohort of 15-month-old, male C57BL/6JNia mice, we exposed animals to six i.p. injections of either LPS (0.4 mg/kg) or Vehicle saline, with each injection spaced 14 days apart (Supplemental Fig. 2). We observed a significantly increased moderate sickness response of at least 3.5 points among LPS-treated mice only after the first three injections; health/sickness scores at each subsequent LPS injection were significantly lower after the first exposure. We postulate that this approach using repeated exposure to 0.4 mg/kg LPS induces endotoxin tolerance, defined in previous literature as a reduction in response to gram negative bacterial LPS after the initial stimulus (Seeley and Ghosh, 2017). Macrophage and monocyte immune cells have been reported to release less TNFα and IL-6 but release more IL-10 and transforming growth factor-β as a result of endotoxin tolerance (Biswas and Lopez-Collazo, 2009; Pena et al., 2011). This resultant switch from a pro-inflammatory to anti-inflammatory phenotype is long lived but can be reversed. Similarly, systemic LPS preconditioning with low doses (0.05 mg/kg to 0.9 mg/kg), which leads to endotoxin tolerance, is known to be neuroprotective, reduces some brain inflammatory responses, transforms microglial cells, and promotes maintained learning and memory ability during a subsequent neurological or immunological challenge (DiCarlo et al., 2001; Mizobuchi and Soma, 2021; Tasaki et al., 1997; Turner et al., 2017; Zhang et al., 2018). This portends the use of an escalating LPS dose paradigm as an effective approach to overcoming endotoxin tolerance and/or preconditioning, modeling intermittent moderate inflammatory experience, and discerning the influence a higher lifetime infection-induced inflammatory burden has on cognitive and neuronal function, as we did here. However, while our intention was to repeatedly engage a moderate inflammatory response with intermittent escalating doses of LPS, our approach may also mimic low grade inflammation postulated to contribute to age-associated pathologies and drive endotoxin tolerance (Franceschi et al., 2018). This may explain the reduced or not changed brain pro-inflammatory cytokine levels observed in our escalating dose model (discussed below).
The cognition-impairing effects of LPS are well-known. For example, a single dose of LPS can induce deficits in several domains of learning and memory, including spatial memory, object recognition, and alternation performance (Tarr et al., 2011; Kranjac et al., 2012; Sparkman et al., 2005). Similar effects have also been reported in female mice as well as in rats, indicating that these results are reproducible in several species and both sexes (Sparkman et al., 2005; Shaw et al., 2001; Zarifkar et al., 2010). For instance, a single injection of 1 mg/kg LPS given to adult male Wistar rats impaired Morris water maze performance, as indicated by increased escape latency and greater total distance travelled relative to vehicle treated animals (Shaw et al., 2001). Similar memory deficits are seen when LPS is given continuously over multiple days (Zarifkar et al., 2010). Importantly, LPS-induced cognitive deficits may persist for long durations after initial exposure, as male C57BL/6 mice continue to perform poorly on the novel object recognition test up to 28 days post-i.p. injection of 5 mg/kg LPS. Few investigators have assessed the cumulative effects of a greater history of infection-like inflammatory experiences on cognitive states of animals. Our study among wild type male mice transitioning between late adulthood to early middle age (10 months – 14 months) revealed modest cognitive impairments (poorer learning and overnight retention) with our repeated intermittent inflammatory regimen, findings which expand upon earlier observations (Marottoli et al., 2017; Sparkman et al., 2005; Lee et al., 2008) and support an important role for inflammation in impairing cognition. This observation may be especially relevant for organisms carrying increased dementia risk, including genetically at-risk individuals (Marottoli et al., 2017) as well as those of advanced age (Tarr et al., 2011) but see (Sparkman et al., 2005). It is important to note that our intermittent LPS exposure paradigm was implemented in relatively healthy mice, relatively brief in duration (~2.5 months) relative to the mouse lifespan, and represented just five peripheral inflammatory insults that induced moderate sickness. To permit sensitivity to observing potential LPS-induced cognitive impairment, our intermittent LPS paradigm was intentionally initiated at a time in life when cognitive aging deficits are not yet readily apparent in male C57BL/6J mice. Further, given the known sex differences in immune system function (Engler-Chiurazzi et al., 2022; Klein and Flanagan, 2016), especially across transitions in reproductive capacity and response to LPS (Cai et al., 2016; Kuo, 2016), and due to the intense logistical demands of testing the large numbers of mice required for our study, we only included 10–14 month old aging male mice here. We did not assess aging female mice nor did we include comparison groups of young adult mice given either treatment regimen to permit assessments of age-related cognitive change. The inclusion only of 10–14 month old male mice limits generalizability of the findings and future work will address potential sex- and/or age-specific effects of repeated, intermittent LPS exposure on cognitive performance. Finally, determining whether LPS-induced deficits would be potentiated by factors such as age at time of first exposure, number of inflammatory insults, genetic sex or sex hormones, dose of the starting LPS challenge, or duration of the repeated, intermittent paradigm is an important next step. Indeed, severity of infections likely plays an important role in the realization of LPS-induced cognitive and neurobiological consequences. Delirium is a common side-effect of severe acute infection in the elderly (van Gool et al., 2010). Similarly, endotoxin-based rodent models of sepsis, in which cognitive impairment is well established, generally employ 2–10 mg/kg doses of LPS to naïve mice. These observations are important considering that immunosenescence cascades typically render aging organisms susceptible to more frequent and more severe infections, and potentially more profound cognitive impairment (Franceschi et al., 2018; Aiello et al., 2019).
The changes in behavioral performance may be attributable to the inhibited expression of hippocampal LTP, which is widely recognized as the cellular mechanism that underlies learning and memory (Bliss and Collingridge, 1993; Brown et al., 1988). There is strong evidence that a single (Shaw et al., 2001; Anaeigoudari et al., 2016; Arai et al., 2001; Beyer et al., 2020; Commins et al., 2001) or chronic/continuous (Min et al., 2009; Wu et al., 2021; Zhang et al., 2017) LPS exposure can disrupt hippocampal synaptic plasticity and LTP, processes that support the formation and storage of new memories. For example, 24 h following a single exposure to 1 mg/kg LPS, young adult BALB/c mice had reduced synaptopodin-mRNA expression, and impaired LTP induction in CA1 hippocampal neurons (Strehl et al., 2014). In a continuous LPS administration model, male Wistar rats showed reduced monosynaptic field excitatory postsynaptic potentials in CA1 neurons three days post-LPS injection battery (0.25 mg/kg/day for six consecutive days) (Hosseini et al., 2021). To our knowledge, we are the first to examine synaptic communication consequences of repeated, intermittent exposure to LPS. Results here suggest LTP deficits can be induced with our intermittent LPS model and that they persist long after (~6 weeks) the final LPS challenge. Interestingly, a previous study (Commins et al., 2001) showed a decrease in paired-pulse facilitation, a form of short-term plasticity that is widely regarded as presynaptic in origin, 4 h following a single i.p. LPS exposure. However, we did not see this change in mice given our intermittent LPS administration regimen and several factors, including LPS source (Beyer et al., 2020), injection times (Shaw et al., 2001), and administration approach (Min et al., 2009; Costello et al., 2011), may account for these differences.
In addition to cognitive consequences, impacts of LPS-induced inflammatory challenge have been described in other models of neurological injury and disease. Indeed, cognitive impairments and exacerbation of AD-associated neuropathological features have been demonstrated in transgenic mice by several teams (Batista et al., 2019), though the extent to which LPS administration regimens used in those studies induced a cyclical pattern of sickness and recovery as our regimen did is not clear. We have previously shown that a mild LPS dose (0.1 mg/kg) given 30 mins prior to induction of a mild experimental stroke (30 min transient middle cerebral artery occlusion) exacerbated infarct size and potentiated functional deficits (Doll et al., 2015; Doll et al., 2015). We went on to show that animals that were not actively LPS-treated at the time of stroke but who had been exposed to repeat intermittent modest LPS challenge in the months prior to ischemic challenge also exhibited larger cortical infarct sizes (Russell et al., 2021). Together with the results of the current study, these collective findings suggest that more experience with systemic inflammatory challenges is associated with worsened neurological outcomes among mice as well as those exposed to age-related brain injuries. Whether behavioral and LTP-related consequences of LPS exposure are the result of direct or indirect effects of LPS is not yet clear. Though peripherally administered LPS appears to be non-brain penetrant (Banks and Robinson, 2010), the lipid A LPS fragment as well as core LPS may accumulate in circumventricular organs as well as periventricular sites and the ventral hippocampal commissure (Vargas-Caraveo et al., 2017). That CNS cells, including brain endothelial cells and astrocytes, express major LPS binding structures (i.e., TLR4 and Cluster of Differentiation (CD) 14) could indicate potential direct actions of LPS on brain substrates underlying cognitive function (Vargas-Caraveo et al., 2017). However, the possibility for indirect effects cannot be ruled out as LPS dosage seems to relate to the extent of blood–brain barrier damage (3.0 mg/kg but not lower doses increased permeability in young adult mice at 24 hr post-exposure) and increased barrier permeability is known to permit entry of a host of peripheral residing cells and factors, any one or combination of which can impact brain function (Banks et al., 2015).
Mechanisms underlying LPS-induced cognitive impairments, such as inflammatory cascade induction, are likely manifold and are actively being explored. Indeed, LPS exposure is known to cause generalized inflammation in brain areas critically involved in learning and memory (Jeong et al., 2010; Liu et al., 2018). Increased protein and gene expression levels of inflammatory cytokines, mainly IL-1β and IL-6, TLR2 and 4 mRNA, and also GFAP and Iba-1 (activated microglia) have been consistently reported in cognitive brain regions (Kranjac et al., 2012; Liu et al., 2018; Noh et al., 2014; Zhao et al., 2019) While some studies note persistent inflammation when LPS is administered repeatedly over a short timespan, such as elevated levels of IL-1β and TNF-α two weeks post-LPS withdrawal in repeatedly exposed mice (2.5 mg/kg/day for 7 days) (Salmani et al., 2021), inflammatory consequences in an intermittent inflammation model, like the one we used here, are not yet well studied. Using our repeated paradigm, initial findings revealed that hippocampal IL-1β gene expression was lower among intermittent LPS-treated mice at two weeks following the final injection (Table 1). When we combined naïve and tested groups based on exposure condition to improve statistical power, hippocampal IL-6 was higher among mice with a history of intermittent LPS exposure, suggesting a possible cytokine mechanism to account for our observations. However, there were no differences in gene expression of any other cytokine in any brain region at any timepoint (Table 1); protein levels of cytokines were not evaluated nor did we assess neural or immune cell populations in peripheral lymphoid, blood, or central nervous system compartments to discern the cellular source of these cytokines. Finally, our analyses may have been underpowered to detect significant differences should they exist in the population. Collectively, these preliminary data could indicate that display of intermittent LPS-induced cognitive changes may not require an ongoing inflammatory cascade and may define the window of therapeutic intervention. Anti-inflammatory intervention administered near LPS exposure appears to restrain detrimental consequences of this proinflammatory cascade (Liu et al., 2018; Zarifkar et al., 2010). Whether a similar intervention could prevent cognitive and neurobiological consequences of repeated, intermittent infection exposures and which specific inflammatory pathway is involved remains to be determined. Follow-up investigations in larger cohorts of mice assaying the time course of changes in brain inflammatory cytokines in this model is warranted.
Previous studies have confirmed the dual effects of cytokines on CNS (Beattie et al., 2002; Bruunsgaard et al., 1999; Ross et al., 2003). Cytokines at physiologically low levels are required for the preservation of synaptic strength at glutamatergic excitatory synapses (Beattie et al., 2002) and LTP (Ross et al., 2003). However, TNF-α over-expression impaired LTP across species such as mice (Maggio and Vlachos, 2018; Singh et al., 2019), rats (Zhou and Bickler, 2017; Rizzo et al., 2018; Prieto et al., 2019; Wall et al., 2015; Anaeigoudari et al., 2016), and rabbits (Wang, 2019), as did IL-1 over-expression (Ross et al., 2003; Bellinger et al., 1993; Hoshino et al., 2017; Katsuki et al., 1990; Lynch et al., 2004; O’Connor and Coogan, 1999). These observations are recapitulated in pathologically aging organisms, as proinflammatory cytokines, activated microglia, and other inflammatory markers are found at increased levels in brains (Akiyama et al., 2000; Shen et al., 2019) and periphery of AD patients (Ng et al., 2018). LPS-induced inflammation may contribute to LTP impairment by targeting either the function or structure of postsynaptic neurons and their dendrites. Indeed, LPS and/or the cytokines produced as a result of its administration may inhibit LTP by their actions on N-methyl D-aspartate receptors (NMDAR) or AMPA on postsynaptic neurons (Francija et al., 2019; Lin et al., 2019; Postnikova et al., 2020) and the related signaling pathways (Lonergan et al., 2004; Zubareva et al., 2020). LPS can also activate microglia and trigger a series of cascading events, which include a decrease in the occupancy of NMDAR complex in the hippocampal CA1 region and reduced glutamatergic transmission (Tanaka et al., 2006), down regulation of hippocampal synaptic proteins (Chamniansawat and Chongthammakun, 2015), reduced activity among microglia-synapse contacts and reduced synaptic plasticity (Akiyoshi et al., 2018), and alterations in dendrite morphology of hippocampal CA1 pyramidal neurons in aged mice (Richwine et al., 2008) and total spine density (Kondo et al., 2011), possibly leading to neuronal cell death (Munch et al., 2003). We did not conduct measures of spine morphology, glutamatergic receptor expression nor microglia activation in this study, thus whether intermittent LPS impacts these substrates is not yet known. Despite these limitations, our preliminary cytokine gene expression findings could suggest that inflammation resultant from intermittent LPS exposures induces structural changes in brain architecture underlying cognitive function that do not repair in the timeframe of our study. Further interrogation of the neuro-architectural consequences of intermittent LPS exposure will likely provide clarity on this potential mechanism and potentially reveal therapeutic targets.
5. Conclusions
In conclusion, we observed that repeated, intermittent exposure to systemic LPS induced modest cognitive impairments and neuronal dysfunction in early middle-aged mice, changes which took place in the absence of an ongoing brain inflammatory or sickness state. Given that conventional medical wisdom generally recommends bedrest and fluids mild-moderate spontaneous infection experiences, the broader impact of these findings may have important implications for standard of care involving infections in aging individuals. Indeed, more aggressive infection resolution interventions or the use of preventative anti-pathogen treatments may be warranted among those at high risk for dementia. Taking the collective knowledge of LPS-induced inflammation on cognitive outcomes together may warrant a re-evaluation of the current medical approaches to managing intermittent moderate infection/inflammation-causing experiences, especially among those with elevated risk for developing normal or pathological age-related cognitive decline.
Supplementary Material
Acknowledgements
This work was supported by: K01MH117343, U54GM104942, P20GM109098, P20GM103629, T32AG052375. We acknowledge the support of the West Virginia University Rodent Behavior Core.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbi.2022.12.013.
Data availability
Data will be made available on request.
References
- Aiello A, Farzaneh F, Candore G, et al. , 2019. Immunosenescence and its hallmarks: how to oppose aging strategically? A review of potential options for therapeutic intervention. Front. Immunol. 10, 2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, et al. , 2000. Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyoshi R, Wake H, Kato D, et al. , 2018. Microglia enhance synapse activity to promote local network synchronization. eNeuro 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anaeigoudari A, Soukhtanloo M, Reisi P, Beheshti F, Hosseini M, 2016. Inducible nitric oxide inhibitor aminoguanidine, ameliorates deleterious effects of lipopolysaccharide on memory and long term potentiation in rat. Life Sci. 158, 22–30. [DOI] [PubMed] [Google Scholar]
- Anderson ND, 2019. State of the science on mild cognitive impairment (MCI). CNS Spectrums 24, 78–87. [DOI] [PubMed] [Google Scholar]
- Arai K, Matsuki N, Ikegaya Y, Nishiyama N, 2001. Deterioration of spatial learning performances in lipopolysaccharide-treated mice. Japanese J. Pharmacol. 87, 195–201. [DOI] [PubMed] [Google Scholar]
- Banks WA, Gray AM, Erickson MA, et al. , 2015. Lipopolysaccharide-induced blood-brain barrier disruption: roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J. Neuroinflammation 12, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Robinson SM, 2010. Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain Behav. Immunity 24, 102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon AW, Malmberg AB 2007. Models of nociception: hot-plate, tail-flick, and formalin tests in rodents. Curr. Protoc. Neurosci., Chapter 8:Unit 8 9. [DOI] [PubMed] [Google Scholar]
- Batista CRA, Gomes GF, Candelario-Jalil E, Fiebich BL, de Oliveira ACP, 2019. Lipopolysaccharide-induced neuroinflammation as a bridge to understand neurodegeneration. Int. J. Mol. Sci. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter MG, Gallagher M, 1996. Intact spatial learning in both young and aged rats following selective removal of hippocampal cholinergic input. Behav. Neurosci. 110, 460–467. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, et al. , 2002. Control of synaptic strength by glial TNFalpha. Science 295, 2282–2285. [DOI] [PubMed] [Google Scholar]
- Bellinger FP, Madamba S, Siggins GR, 1993. Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 628, 227–234. [DOI] [PubMed] [Google Scholar]
- Beyer MMS, Lonnemann N, Remus A, Latz E, Heneka MT, Korte M, 2020. Enduring changes in neuronal function upon systemic inflammation are NLRP3 inflammasome dependent. J. Neurosci. 40, 5480–5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesmans S, Meert TF, Bouwknecht JA, et al. , 2013. Systemic immune activation leads to neuroinflammation and sickness behavior in mice. Mediat. Inflammation 2013, 271359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SK, Lopez-Collazo E, 2009. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 30, 475–487. [DOI] [PubMed] [Google Scholar]
- Bizon J, Foster T, Alexander G, Glisky E, 2012. Characterizing cognitive aging of working memory and executive function in animal models. Front. Aging Neurosci. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL, 1993. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. [DOI] [PubMed] [Google Scholar]
- Bossù P, Cutuli D, Palladino I, et al. , 2012. A single intraperitoneal injection of endotoxin in rats induces long-lasting modifications in behavior and brain protein levels of TNF-α and IL-18. J. Neuroinflammation 9, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TH, Chapman PF, Kairiss EW, Keenan CL, 1988. Long-term synaptic potentiation. Science 242, 724–728. [DOI] [PubMed] [Google Scholar]
- Bruunsgaard H, Andersen-Ranberg K, Jeune B, Pedersen AN, Skinhoj P, Pedersen BK, 1999. A high plasma concentration of TNF-alpha is associated with dementia in centenarians. J. Gerontol. A Biol. Sci. Med. Sci. 54, M357–M364. [DOI] [PubMed] [Google Scholar]
- Cai KC, van Mil S, Murray E, Mallet JF, Matar C, Ismail N, 2016. Age and sex differences in immune response following LPS treatment in mice. Brain Behav. Immunity 58, 327–337. [DOI] [PubMed] [Google Scholar]
- Carrera Arias FJ, Aenlle K, Abreu M, et al. , 2021. Modeling neuroimmune interactions in human subjects and animal models to predict subtype-specific multidrug treatments for gulf war illness. Int. J. Mol. Sci. 22, 8546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catorce MN, Gevorkian G, 2016. LPS-induced murine neuroinflammation model: main features and suitability for pre-clinical assessment of nutraceuticals. Curr. Neuropharmacol. 14, 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cekanaviciute E, Buckwalter MS, 2016. Astrocytes: integrative regulators of neuroinflammation in stroke and other neurological diseases. Neurotherapeutics 13, 685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamniansawat S, Chongthammakun S, 2015. Inhibition of hippocampal estrogen synthesis by reactive microglia leads to down-regulation of synaptic protein expression. Neurotoxicology 46, 25–34. [DOI] [PubMed] [Google Scholar]
- Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW, 2008. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav. Immunity 22, 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SS, Lee HJ, Lim I, Satoh J. i., Kim SU, 2014. Human astrocytes: secretome profiles of cytokines and chemokines. PLoS One 9, e92325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commins S, O’Neill LA, O’Mara SM, 2001. The effects of the bacterial endotoxin lipopolysaccharide on synaptic transmission and plasticity in the CA1-subiculum pathway in vivo. Neuroscience 102, 273–280. [DOI] [PubMed] [Google Scholar]
- Costello DA, Lyons A, Denieffe S, Browne TC, Cox FF, Lynch MA, 2011. Long term potentiation is impaired in membrane glycoprotein CD200-deficient mice: a role for Toll-like receptor activation. J. Biol. Chem. 286, 34722–34732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiani CL, O’Callaghan JP, 2007. Recapitulation of cell signaling events associated with astrogliosis using the brain slice preparation. J. Neurochem. 100, 720–726. [DOI] [PubMed] [Google Scholar]
- Detrait ER, Hanon E, Dardenne B, Lamberty Y, 2009. The inhibitory avoidance test optimized for discovery of cognitive enhancers. Behav. Res. Methods 41, 805–811. [DOI] [PubMed] [Google Scholar]
- Dharmarajan TS, Gunturu SG, 2009. Alzheimer’s disease: a healthcare burden of epidemic proportion. Am. Health Drug Benefits 2, 39–47. [PMC free article] [PubMed] [Google Scholar]
- DiCarlo G, Wilcock D, Henderson D, Gordon M, Morgan D, 2001. Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol. Aging 22, 1007–1012. [DOI] [PubMed] [Google Scholar]
- Doll DN, Engler-Chiurazzi EB, Lewis SE, et al. , 2015. Lipopolysaccharide exacerbates infarct size and results in worsened post-stroke behavioral outcomes. Behav. Brain Funct. 11, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll DN, Hu H, Sun J, Lewis SE, Simpkins JW, Ren X, 2015. Mitochondrial crisis in cerebrovascular endothelial cells opens the blood-brain barrier. Stroke 46, 1681–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn AJ, Swiergiel AH, 2005. Effects of interleukin-1 and endotoxin in the forced swim and tail suspension tests in mice. Pharmacol. Biochem. Behav. 81, 688–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler-Chiurazzi EB, Talboom JS, Braden BB, et al. , 2012. Continuous estrone treatment impairs spatial memory and does not impact number of basal forebrain cholinergic neurons in the surgically menopausal middle-aged rat. Horm. Behav. 62, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler-Chiurazzi EB, Chastain WH, Citron KK, Lambert LE, Kikkeri DN, Shrestha SS, 2022. Estrogen, the peripheral immune system and major depression - A reproductive lifespan perspective. Front. Behav. Neurosci. 16, 850623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler-Chiurazzi E, Tsang C, Nonnenmacher S, et al. , 2011. Tonic Premarin dose-dependently enhances memory, affects neurotrophin protein levels and alters gene expression in middle-aged rats. Neurobiol. Aging 32, 680–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A, 2018. Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 14, 576–590. [DOI] [PubMed] [Google Scholar]
- Francija E, Petrovic Z, Brkic Z, Mitic M, Radulovic J, Adzic M, 2019. Disruption of the NMDA receptor GluN2A subunit abolishes inflammation-induced depression. Behav. Brain Res. 359, 550–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SA, Harris EA, 2015. Herpes simplex virus type 1 and other pathogens are key causative factors in sporadic Alzheimer’s disease. J Alzheimers Dis. 48, 319–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauss-Wegrzyniak B, Dobrzanski P, Stoehr JD, Wenk GL, 1998. Chronic neuroinflammation in rats reproduces components of the neurobiology of Alzheimer’s disease. Brain Res. 780, 294–303. [DOI] [PubMed] [Google Scholar]
- Herber DL, Maloney JL, Roth LM, Freeman MJ, Morgan D, Gordon MN, 2006. Diverse microglial responses after intrahippocampal administration of lipopolysaccharide. Glia 53, 382–391. [DOI] [PubMed] [Google Scholar]
- Hoshino K, Hasegawa K, Kamiya H, Morimoto Y, 2017. Synapse-specific effects of IL-1beta on long-term potentiation in the mouse hippocampus. Biomed. Res. 38, 183–188. [DOI] [PubMed] [Google Scholar]
- Hosseini M, Salmani H, Baghcheghi Y, 2021. Losartan improved hippocampal long-term potentiation impairment induced by repeated LPS injection in rats. Physiol. Rep. 9, e14874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsberger HC, Wang D, Petrisko TJ, et al. , 2016. Peripherally restricted viral challenge elevates extracellular glutamate and enhances synaptic transmission in the hippocampus. J. Neurochem. 138, 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvik ME, Kopp R, 1967. An improved one-trial passive avoidance learning situation. Psychol. Rep. 21, 221–224. [DOI] [PubMed] [Google Scholar]
- Jeong H-K, Jou I, Joe E. h., 2010. Systemic LPS administration induces brain inflammation but not dopaminergic neuronal death in the substantia nigra. Exp. Mol. Med. 42, 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katan M, Moon YP, Paik MC, Sacco RL, Wright CB, Elkind MS, 2013. Infectious burden and cognitive function: the Northern Manhattan Study. Neurology 80, 1209–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuki H, Nakai S, Hirai Y, Akaji K, Kiso Y, Satoh M, 1990. Interleukin-1 beta inhibits long-term potentiation in the CA3 region of mouse hippocampal slices. Eur. J. Pharmacol. 181, 323–326. [DOI] [PubMed] [Google Scholar]
- Kepple G, Wickens TD, 2004. Design and Analysis: a Researrcher’s Handbook, 4th ed. Pearson: Prentice Hall, Upper Saddle River, New Jersey. [Google Scholar]
- Klein SL, Flanagan KL, 2016. Sex differences in immune responses. Nat. Rev. Immunol. 16, 626–638. [DOI] [PubMed] [Google Scholar]
- Kondo S, Kohsaka S, Okabe S, 2011. Long-term changes of spine dynamics and microglia after transient peripheral immune response triggered by LPS in vivo. Mol. Brain 4, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranjac D, McLinden KA, Deodati LE, Papini MR, Chumley MJ, Boehm GW, 2012. Peripheral bacterial endotoxin administration triggers both memory consolidation and reconsolidation deficits in mice. Brain Behav. Immunity 26, 109–121. [DOI] [PubMed] [Google Scholar]
- Kuo SM, 2016. Gender difference in bacteria endotoxin-induced inflammatory and anorexic responses. PLoS One 11, e0162971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HS, Koh S-H, 2020. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl. Neurodegener. 9, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Lee YK, Yuk DY, et al. , 2008. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflammation 5, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Chen X, Zhou Q, et al. , 2019. Synaptic structure and alterations in the hippocampus in neonatal rats exposed to lipopolysaccharide. Neurosci. Lett. 709, 134364. [DOI] [PubMed] [Google Scholar]
- Linnerbauer M, Wheeler MA, Quintana FJ, 2020. Astrocyte crosstalk in CNS inflammation. Neuron 108, 608–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang Y, Zheng X, et al. , 2018. Galantamine improves cognition, hippocampal inflammation, and synaptic plasticity impairments induced by lipopolysaccharide in mice. J. Neuroinflammation 15, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonergan PE, Martin DS, Horrobin DF, Lynch MA, 2004. Neuroprotective actions of eicosapentaenoic acid on lipopolysaccharide-induced dysfunction in rat hippocampus. J. Neurochem. 91, 20–29. [DOI] [PubMed] [Google Scholar]
- Lu YC, Yeh WC, Ohashi PS, 2008. LPS/TLR4 signal transduction pathway. Cytokine 42, 145–151. [DOI] [PubMed] [Google Scholar]
- Lynch AM, Walsh C, Delaney A, Nolan Y, Campbell VA, Lynch MA, 2004. Lipopolysaccharide-induced increase in signalling in hippocampus is abrogated by IL-10–a role for IL-1 beta? J. Neurochem. 88, 635–646. [DOI] [PubMed] [Google Scholar]
- Maggio N, Vlachos A, 2018. Tumor necrosis factor (TNF) modulates synaptic plasticity in a concentration-dependent manner through intracellular calcium stores. J. Mol. Med. (Berl) 96, 1039–1047. [DOI] [PubMed] [Google Scholar]
- Marottoli FM, Katsumata Y, Koster KP, Thomas R, Fardo DW, Tai LM, 2017. Peripheral inflammation, apolipoprotein E4, and amyloid-β interact to induce cognitive and cerebrovascular dysfunction. ASN Neuro 9, 1759091417719201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlpine FE, Lee J-K, Harms AS, et al. , 2009. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol. Dis. 34, 163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalovicz LT, Locker AR, Kelly KA, et al. , 2019. Corticosterone and pyridostigmine/DEET exposure attenuate peripheral cytokine expression: supporting a dominant role for neuroinflammation in a mouse model of Gulf War Illness. Neurotoxicology 70, 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud TL, Su D, Siahpush M, Murman DL, 2017. The risk of incident mild cognitive impairment and progression to dementia considering mild cognitive impairment subtypes. Dement. Geriatr. Cogn. Dis. Extra 7, 15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min SS, Quan HY, Ma J, Han JS, Jeon BH, Seol GH, 2009. Chronic brain inflammation impairs two forms of long-term potentiation in the rat hippocampal CA1 area. Neurosci. Lett. 456, 20–24. [DOI] [PubMed] [Google Scholar]
- Mitchell AJ, Shiri-Feshki M, 2009. Rate of progression of mild cognitive impairment to dementia–meta-analysis of 41 robust inception cohort studies. Acta Psychiatr. Scand. 119, 252–265. [DOI] [PubMed] [Google Scholar]
- Mizobuchi H, Soma GI, 2021. Low-dose lipopolysaccharide as an immune regulator for homeostasis maintenance in the central nervous system through transformation to neuroprotective microglia. Neural Regen. Res. 16, 1928–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R, 1984. Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 11, 47–60. [DOI] [PubMed] [Google Scholar]
- Munch G, Gasic-Milenkovic J, Dukic-Stefanovic S, et al. , 2003. Microglial activation induces cell death, inhibits neurite outgrowth and causes neurite retraction of differentiated neuroblastoma cells. Exp. Brain Res. 150, 1–8. [DOI] [PubMed] [Google Scholar]
- Murman DL, 2015. The impact of age on cognition. Semin. Hear. 36, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng A, Tam WW, Zhang MW, et al. , 2018. IL-1β, IL-6, TNF- α and CRP in elderly patients with depression or Alzheimer’s disease: systematic review and meta-analysis. Sci. Rep. 8, 12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh H, Jeon J, Seo H, 2014. Systemic injection of LPS induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem. Int. 69, 35–40. [DOI] [PubMed] [Google Scholar]
- Oba H, Kadoya Y, Okamoto H, et al. , 2021. The economic burden of dementia: evidence from a survey of households of people with dementia and their caregivers. Int. J. Environ. Res. Public Health 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor JJ, Coogan AN, 1999. Actions of the pro-inflammatory cytokine IL-1 beta on central synaptic transmission. Exp. Physiol. 84, 601–614. [PubMed] [Google Scholar]
- Ortman J, Velkoof VA, Hogan H 2014. An aging nation: the older population of the United States. US Census Bureau. [Google Scholar]
- Pena OM, Pistolic J, Raj D, Fjell CD, Hancock RE, 2011. Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J. Immunol. (Baltimore, Md: 1950) 186, 7243–7254. [DOI] [PubMed] [Google Scholar]
- Petersen RC, Lopez O, Armstrong MJ, et al. , 2018. Practice guideline update summary: mild cognitive impairment: report of the guideline development, dissemination, and implementation subcommittee of the american academy of neurology. Neurology 90, 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postnikova TY, Griflyuk AV, Ergina JL, Zubareva OE, Zaitsev AV, 2020. Administration of bacterial lipopolysaccharide during early postnatal ontogenesis induces transient impairment of long-term synaptic plasticity associated with behavioral abnormalities in young rats. Pharmaceuticals (Basel) 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto GA, Tong L, Smith ED, Cotman CW, 2019. TNFalpha and IL-1beta but not IL-18 suppresses hippocampal long-term potentiation directly at the synapse. Neurochem. Res. 44, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan N, Sundar SK, Weiss JM, 1994. Induction of interleukin-1 in various brain regions after peripheral and central injections of lipopolysaccharide. J. Neuroimmunol. 49, 125–134. [DOI] [PubMed] [Google Scholar]
- Richwine AF, Parkin AO, Buchanan JB, et al. , 2008. Architectural changes to CA1 pyramidal neurons in adult and aged mice after peripheral immune stimulation. Psychoneuroendocrinology 33, 1369–1377. [DOI] [PubMed] [Google Scholar]
- Rizzo FR, Musella A, De Vito F, et al. , 2018. Tumor necrosis factor and interleukin-1beta modulate synaptic plasticity during neuroinflammation. Neural Plast. 2018, 8430123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross FM, Allan SM, Rothwell NJ, Verkhratsky A, 2003. A dual role for interleukin-1 in LTP in mouse hippocampal slices. J. Neuroimmunol. 144, 61–67. [DOI] [PubMed] [Google Scholar]
- Russell AE, Cavendish JZ, Rai A, et al. , 2021. Intermittent lipopolysaccharide exposure significantly increases cortical infarct size and impairs autophagy. ASN Neuro 13, 1759091421991769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmani H, Hosseini M, Baghcheghi Y, Samadi-Noshahr Z, 2021. The brain consequences of systemic inflammation were not fully alleviated by ibuprofen treatment in mice. Pharmacol. Rep.: PR 73, 130–142. [DOI] [PubMed] [Google Scholar]
- Seeley JJ, Ghosh S, 2017. Molecular mechanisms of innate memory and tolerance to LPS. J Leukocyte Biol. 101, 107–119. [DOI] [PubMed] [Google Scholar]
- Sen A, Hongpaisan J, Wang D, Nelson TJ, Alkon DL, 2016. Protein Kinase C (PKC) promotes synaptogenesis through membrane accumulation of the postsynaptic density protein PSD-95. J. Biol. Chem. 291, 16462–16476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw KN, Commins S, O’Mara SM, 2001. Lipopolysaccharide causes deficits in spatial learning in the watermaze but not in BDNF expression in the rat dentate gyrus. Behav. Brain Res. 124, 47–54. [DOI] [PubMed] [Google Scholar]
- Shen XN, Niu LD, Wang YJ, et al. , 2019. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: a meta-analysis and systematic review of 170 studies. J. Neurol. Neurosurg. Psychiatry 90, 590–598. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE, 2003. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol. Dis. 14, 133–145. [DOI] [PubMed] [Google Scholar]
- Singh A, Jones OD, Mockett BG, Ohline SM, Abraham WC, 2019. Tumor necrosis factor-alpha-mediated metaplastic inhibition of LTP is constitutively engaged in an Alzheimer’s disease model. J. Neurosci. 39, 9083–9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloane PD, Zimmerman S, Suchindran C, et al. , 2002. The public health impact of Alzheimer’s disease, 2000–2050: potential implication of treatment advances. Annu. Rev. Public Health 23, 213–231. [DOI] [PubMed] [Google Scholar]
- Sparkman NL, Martin LA, Calvert WS, Boehm GW, 2005. Effects of intraperitoneal lipopolysaccharide on Morris maze performance in year-old and 2-month-old female C57BL/6J mice. Behav. Brain Res. 159, 145–151. [DOI] [PubMed] [Google Scholar]
- Sriram K, Benkovic SA, Hebert MA, Miller DB, O’Callaghan JP, 2004. Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration: key signaling pathway for astrogliosis in vivo? J. Biol. Chem. 279, 19936–19947. [DOI] [PubMed] [Google Scholar]
- Strandberg TE, Pitkala KH, Linnavuori K, Tilvis RS, 2004. Cognitive impairment and infectious burden in the elderly. Arch Gerontol. Geriatr. Suppl. 419–423. [DOI] [PubMed] [Google Scholar]
- Strehl A, Lenz M, Itsekson-Hayosh Z, et al. , 2014. Systemic inflammation is associated with a reduction in Synaptopodin expression in the mouse hippocampus. Exp. Neurol. 261, 230–235. [DOI] [PubMed] [Google Scholar]
- Sy M, Kitazawa M, Medeiros R, et al. , 2011. Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am. J. Pathol. 178, 2811–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Ide M, Shibutani T, et al. , 2006. Lipopolysaccharide-induced microglial activation induces learning and memory deficits without neuronal cell death in rats. J. Neurosci. Res. 83, 557–566. [DOI] [PubMed] [Google Scholar]
- Tarr AJ, McLinden KA, Kranjac D, Kohman RA, Amaral W, Boehm GW, 2011. The effects of age on lipopolysaccharide-induced cognitive deficits and interleukin-1β expression. Behav. Brain Res. 217, 481–485. [DOI] [PubMed] [Google Scholar]
- Tasaki K, Ruetzler CA, Ohtsuki T, Martin D, Nawashiro H, Hallenbeck JM, 1997. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 748, 267–270. [DOI] [PubMed] [Google Scholar]
- Thygesen C, Ilkjær L, Kempf SJ, et al. , 2018. Diverse protein profiles in CNS myeloid cells and CNS tissue from lipopolysaccharide- and vehicle-injected APP(SWE)/PS1 (ΔE9) transgenic mice implicate Cathepsin Z in Alzheimer’s disease. Front. Cell. Neurosci. 12, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ton TGN, DeLeire T, May SG, et al. , 2017. The financial burden and health care utilization patterns associated with amnestic mild cognitive impairment. Alzheimers Dement. 13, 217–224. [DOI] [PubMed] [Google Scholar]
- Turner RC, Naser ZJ, Lucke-Wold BP, et al. , 2017. Single low-dose lipopolysaccharide preconditioning: neuroprotective against axonal injury and modulates glial cells. Neuroimmunol. Neuroinflamm. 4, 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gool WA, van de Beek D, Eikelenboom P, 2010. Systemic infection and delirium: when cytokines and acetylcholine collide. Lancet 375, 773–775. [DOI] [PubMed] [Google Scholar]
- Vargas-Caraveo A, Sayd A, Maus SR, et al. , 2017. Lipopolysaccharide enters the rat brain by a lipoprotein-mediated transport mechanism in physiological conditions. Sci. Rep. 7, 13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaure C, Liu Y, 2014. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front. Immunol. 5, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall AM, Mukandala G, Greig NH, O’Connor JJ, 2015. Tumor necrosis factor-alpha potentiates long-term potentiation in the rat dentate gyrus after acute hypoxia. J. Neurosci. Res. 93, 815–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, 2019. Tumor necrosis factor-alpha alters electrophysiological properties of rabbit hippocampal neurons. J. Alzheimers Dis. 68, 1257–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Zheng W, 2015. Dietary cholesterol concentration affects synaptic plasticity and dendrite spine morphology of rabbit hippocampal neurons. Brain Res. 1622, 350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SJ, Bachstetter AD, Nelson PT, Schmitt FA, Van Eldik LJ, 2014. Using mice to model Alzheimer’s dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 5, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W, 2020. Economic burden of Alzheimer disease and managed care considerations. Am. J. Manag. Care 26, S177–S183. [DOI] [PubMed] [Google Scholar]
- Wu K-C, Lee C-Y, Chern Y, Lin C-J, 2021. Amelioration of lipopolysaccharide-induced memory impairment in equilibrative nucleoside transporter-2 knockout mice is accompanied by the changes in glutamatergic pathways. Brain Behav. Immunity 96, 187–199. [DOI] [PubMed] [Google Scholar]
- Zarifkar A, Choopani S, Ghasemi R, et al. , 2010. Agmatine prevents LPS-induced spatial memory impairment and hippocampal apoptosis. Eur. J. Pharmacol. 634, 84–88. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Natale G, Clouston S, 2021. Incidence of mild cognitive impairment, conversion to probable dementia, and mortality. Am. J. Alzheimers Dis. Other Demen. 36, 15333175211012235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Ji M, Liao Y, Yang J, Gao J, 2018. Endotoxin tolerance induced by lipopolysaccharide preconditioning protects against surgeryinduced cognitive impairment in aging mice. Mol. Med. Rep. 17, 3845–3852. [DOI] [PubMed] [Google Scholar]
- Zhang J, Li A, Song Z, 2017. Systemic LPS resulted in a transient hippocampus malfunction but a prolonged corpus callosum injury. BMC Anesthesiol. 17, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Bi W, Xiao S, et al. , 2019. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 9, 5790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Bickler P, 2017. Interaction of isoflurane, tumor necrosis factor-alpha and beta-amyloid on long-term potentiation in rat hippocampal slices. Anesth. Analg. 124, 582–587. [DOI] [PubMed] [Google Scholar]
- Zubareva OE, Postnikova TY, Grifluk AV, et al. , 2020. Exposure to bacterial lipopolysaccharidein early life affects the expression of ionotropic glutamate receptor genes and is accompanied by disturbances in long-term potentiation and cognitive functions in young rats. Brain Behav. Immun. 90, 3–15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.