

Abstract
Photodynamic therapy (PDT) is an effective and low-invasive tumour therapy. However, it can induce tumour angiogenesis, which is a main factor leading to tumour recurrence and metastasis. Activin receptor-like kinase-1 (ALK1) is a key factor regulating angiogenesis. However, it remains unclear whether ALK1 plays an unusual role in low-dose PDT-induced tumour angiogenesis. In the present study, human umbilical vein endothelial cells (HUVECs) co-cultured with breast cancer MDA-MB-231 cells (termed HU-231 cells) were used to construct an experimental model of tumour angiogenesis induced by low-dose PDT. The viability, and the proliferative, invasive, migratory, as well as the tube-forming ability of the HU-231 cells were evaluated following low-dose PDT. In particular, ALK1 inhibitor and and an adenovirus against ALK1 were used to further verify the role of ALK1 in low-dose PDT-induced tumour angiogenesis. Moreover, the expression of ALK1, inhibitor of DNA binding 1 (ID1), Smad 1, p-Smad1/5, AKT and PI3K were detected in order to verify the underlying mechanisms. The findings indicated that low-dose PDT enhanced the proliferative ability of the HU-231 cells and reinforced their migratory, invasive and tube formation capacity. However, these effects were reversed with the addition of an ALK1 inhibitor or by the knockdown of ALK1 using adenovirus. These results indicated that ALK1 was involved and played a critical role in tumour angiogenesis induced by low-dose PDT. Furthermore, ALK1 was found to participate in PDT-induced tumour angiogenesis by activating the Smad1/5-ID1 pathway, as opposed to the PI3K/AKT pathway. On the whole, the present study, for the first time, to the best of our knowledge, demonstrates that ALK1 is involved in PDT-induced tumour angiogenesis. The inhibition of ALK1 can suppress PDT-induced tumour angiogenesis, which can enhance the effects of PDT and may thus provide a novel treatment strategy for PDT.
Keywords: activin receptor-like kinase-1, angiogenesis, co-culture, photodynamic therapy, tumour
Introduction
As a low-invasive tumour treatment method, photodynamic therapy (PDT) can selectively accumulate photosensitizers at the tumour site and generate a large number of reactive oxygen species under the irradiation of a specific wavelength of visible light. Subsequently, it can directly destroy tumour cells, block the microvessels of tumour tissue and activate the inflammatory and immune responses of the tumour site, which aids in tumour treatment. PDT has various advantages, such as specific targeting and high repeatability, and is a key supplement to surgical resection, chemotherapy and radiotherapy (1). It has also been shown to lead to a good response in certain tumour types, including oesophageal (2), lung (3), breast (4) and skin (5) cancer, and its use has been approved in numerous countries (1,6). However, the treatment response to certain tumour types is poor, and tumour regrowth occurs following PDT (7). In the clinical process of PDT for tumours, the low concentration of photosensitizers is used due to the particularity of tumour blood vessels. Furthermore, due to the occurrence of the physical characteristics of light, its light intensity decreases with the depth of the tumour; thus, the efficacy of PDT decreases with the increasing depth of the tumour and does not reach the lethal dose (8), that is, low-dose PDT.
As a critical part of the tumour microenvironment, tumour blood vessels are not only the channels for tumour nutrition delivery and tumour cell escape, but also play a crucial role in tumour cell invasion and metastasis, thus also greatly affecting the tolerance to treatment (1,9). In PDT for tumours, there are factors that can induce angiogenesis and reduce the therapeutic effect (1). To reduce angiogenesis induced by PDT for tumours and enhance the therapeutic effects of PDT, Ferrario et al (10) developed a model of PDT combined with anti-angiogenic drugs to treat tumours. They used PDT combined with anti-VEGF drugs for treatment, which effectively reduced the blood vessel density of breast cancer in mice and improved the cure rate of breast cancer from 39% with PDT alone to 80-90% with combination treatment (10). Furthermore, Peng et al (11) reported that the combination of PDT and anti-VEGF drugs effectively inhibited tumour angiogenesis and growth.
Previous studies have indicated that the combination of PDT with anti-angiogenic drugs provides a novel anticancer treatment strategy (12,13). Moreover, it has been hypothesized that this combination has great prospects for the development of anti-angiogenic drugs that target key molecules in the tumour angiogenesis signalling pathway. However, other studies have found that PDT combined with anti-VEGF drugs cannot effectively inhibit tumour angiogenesis for a long period of time (6,14). Drug resistance has been reported to occur at ~6 months following the use of anti-VEGF drugs, and the long-term survival rate of patients has not found to be significantly prolonged following combination treatment (6,14). Therefore, it is of utmost importance to identify key molecules in other anti-angiogenesis signalling pathways that can serve as targets in PDT combined with other therapies.
Activin receptor-like-kinase 1 (ALK1) is a type I receptor of the TGF-β family, specifically a serine/threonine kinase receptor. It is a homodimeric transmembrane glycoprotein integrated into the cell membrane, which is mainly expressed in vascular endothelial cells and other vascular-rich tissues. As a signalling pathway that differs from the VEGF signalling pathway (15), ALK1 regulates endothelial cell proliferation, migration and lumen formation, and plays a decisive role in regulating vascular development and pathological angiogenesis (16,17).
Notably, ALK1 has been widely reported in tumour blood vessels, and the expression of ALK1 is positively associated with tumour vascular density (16,18). Cunha et al (19) found that reducing the expression level of ALK1 via genetic or pharmacological approaches inhibited tumour growth and angiogenesis. Hawinkels et al (20) confirmed through in vivo and in vitro experiments on three types of tumour cells that the use of drugs targeting ALK1 effectively inhibited tumour angiogenesis and enhanced the chemotherapeutic effects. Clinical studies have found that the use of drugs targeting ALK1 can reduce the density of capillaries in tumours, inhibit various tumour growths and inhibit the metastasis of breast cancer (21,22).
There is evidence to indicate that the abnormal activation of the ALK1 signalling pathway plays a key role in the occurrence and development of tumour angiogenesis; thus, the inhibition of ALK1 may be a key strategy with which to prevent tumour angiogenesis. In order to clarify the role of ALK1 in PDT-induced tumour angiogenesis, the present study used human umbilical vein endothelial cells (HUVECs) co-cultured with breast cancer cell MDA-MB-231 (termed HU-231 cells) as the research object to better simulate the tumour intravascular environment in vivo. The present study aimed to reveal the role of the anti-angiogenesis target, ALK1, in PDT-induced tumour angiogenesis, thereby providing a theoretical basis for PDT combined with anti-ALK1 in the treatment of tumours.
Materials and methods
Cells and cell culture
The MDA-MB-231 human breast cancer cells (cat. no. SCSP-5043) were purchased from The Cell Bank of Type Culture Collection of the Chinese Academy of Sciences and HUVECs (cat. no. C0035C) were purchased from Thermo Fisher Scientific, Inc. (the cells were frozen and stored at the Chongqing Key Laboratory of Translational Medicine in Major Metabolic Diseases, Chongqing, China). The cells were routinely and individually cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% foetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and 1% penicillin and streptomycin (Gibco; Thermo Fisher Scientific, Inc.) at 37°C in 5% CO2. The HUVECs were co-cultured with the MDA-MB-231 breast cancer cells and termed HU-231 cells, which were established as experimental subjects in the present study to simulate vascular endothelial cells in the intra-tumour environment. All cell cultures were mycoplasma species-negative. The experimental groups were as follows: The ALK1 inhibitor groups (control, PDT, K02288 and PDT + K02288 group; K02288 was obtained from Sigma-Aldrich, Merck KGaA), adenovirus transfection groups (control, negative control, PDT, Ad-ALK1 and PDT + Ad-ALK1 group) and PI3K inhibitor groups (control, PDT, LY294002 and PDT + LY294002 group; LY294002 was obtained from Sigma-Aldrich, Merck KGaA). The concentrations of K02288 and LY294002 used were 5 and 10 µmol/l, respectively. Both inhibitors were used to treat the cells for 24 h.
PDT
The HU-231 cells were treated with the photosensitizer, pyropheophorbide-a methyl ester (MPPa; cat. no. P3787; Sigma-Aldrich, Merck KGaA) at the concentration of 10 µmol/l for 4 h and then washed twice with PBS. Following the addition of 2 ml DMEM (Gibco; Thermo Fisher Scientific, Inc.), the cells were irradiated for 5 sec with a wavelength of 630 nm and an energy intensity of 10 mW/cm2 using a LED laser (Chongqing Jinyu Laser Technology Co., Ltd.).
Adenovirus transfection
The HUVECs were inoculated into six-well plates at a density of 6×104 (care was taken to avoid clustering) and after 12 h, adenovirus AD-ALK1-RNAi (cat. no. 112177-1; Shanghai Genechem Co., Ltd.; https://www.genechem.com.cn/; AD-ALK1-RNAi (112177-1)-a, CCG GUC CAG AGA AGC CUA AAG UGA UCU CGA GAU CAC UUU AGG CUU CUC UGG AUU UUU G; AD-ALK1-RNAi (112177-1)-b, AAU UCA AAA AUCCAGAGA AGCCUA AAGUGAUCUCGA GAU CAC UUU AGG CUU CUC UGG A) and negative control virus CON098 (Shanghai Genechem Co., Ltd.) were added at a multiplicity of infection (MOI) of 1,000. After 12 h, the solution was changed and included MDA-MB-231 cell chambers for co-culture and subsequent experiments. The transfection efficiency was determined by measuring the fluorescence intensity and RT-qPCR.
Cell Counting Kit-8 (CCK-8) assay
The MDA-MB-231 and HUVECs were co-cultured at 5×104 cells/well in small chambers (BD Biosciences) and 24-well plates to investigate the appropriate drug concentrations and light intensities, as well as the cellular activity of the four groups of inhibitors. After 24 h, the medium of each well was discarded and 700 µl CCK-8 solution (cat. no. C0037; Beyotime Institute of Biotechnology) mixed with serum-free DMEM was added to each well and the cells were further incubated for 1 h at 37°C in 5% CO2 for a further 1 h. The optical densities (ODs) were measured using a microplate reader (Thermo Fisher Scientific, Inc.). Cell survival was calculated as follows: Cell survival (%)=(average OD of experimental group-average OD of blank group)/(average OD of control group-average OD of blank group) ×100%. The wells in the blank group contained DMEM in CCK-8 solution only.
Colony formation assay
The MDA-MB-231 and HUVECs were co-cultured in small chambers and six-well plates and treated with ALK1 inhibitor (K02288) and transfected with adenovirus, respectively. After 24 h, the HU-231 cells were counted at 800 cells/well and inoculated into 12-well plates. The culture was terminated when the appearance of colonies visible to the naked eye was observed. The colonies were fixed with 4% paraformaldehyde (cat. no. P0099; Beyotime Institute of Biotechnology) for 30 min at room temperature and stained with crystal violet solution (cat. no. C0121; Beyotime Institute of Biotechnology) for 30 min at room temperature. The colonies were photographed using an Android smartphone (Vivo X7 plus; Vivo Mobile Communication Co., Ltd.) and counted using ImageJ software (ImageJ 1.53k; National Institutes of Health).
Wound healing assay
The MDA-MB-231 cells and HUVECs were co-cultured in small chambers and six-well plates with serum-free DMEM and treated with ALK1 inhibitor (K02288; 3×105 cells/well) and transfected with adenovirus, respectively. Immediately following PDT, the cells in all groups were scored with a gun tip (10 µl). DiI cell membrane fluorescent probe orange-red (cat. no. MB4240; Meilunbio Co., Ltd.) was then added for 10 min and the cells were washed in PBS. Images were obtained immediately at 0, 24 and 48 h later using a fluorescence microscope (Carl Zeiss AG). The scratch areas were analysed using ImageJ software (ImageJ 1.53k; National Institutes of Health).
Migration and invasion assays
The HUVECs and MDA-MB-231 cells were co-cultured at 5×104 cells/well in small chambers and 24-well plates and 60 µl Matrigel gel (Corning, Inc.) diluted at 1:8 was added to the small chambers prior to cell inoculation (for the invasion assay). The cells were treated with ALK1 inhibitor (K02288) and PDT. Following 24 h of co-culture (MDA-MB-231 and HUVECs) in the adenovirus transfection group, the cells were removed from each well and inoculated into 24-well Transwell chambers at 5×104 cells/well for incubation at 37°C. After 24 h, the cells that had not invaded the surface of the Transwell chambers in each group were cleared using cotton swabs, and the invading cells in the chambers were fixed with 4% paraformaldehyde for 30 min at room temperature, washed with PBS and stained with crystal violet for 30 min at room temperature. The migration and invasion of the cells in the Transwells were observed using a Zeiss light microscope (Carl Zeiss AG). Cell counts were analysed using ImageJ software (ImageJ 1.53k; National Institutes of Health).
Tube formation assay
The MDA-MB-231 cells and HUVECs were co-cultured in small chambers and six-well plates and treated with ALK1 inhibitor (K02288) and transfected with adenovirus were incubated at 37°C and 5% CO2 for 24 h. Following incubation, cells were inoculated on confocal dishes (BD Biosciences) with plates coated with 200 µl Matrigel at a concentration of 5×105 cells per well. Tubes were incubated at 37°C and 5% CO2 for 9 h and then fixed with 4% paraformaldehyde for 30 min at room temperature, lysed with Triton-X (cat. no. P0096; Beyotime Institute of Biotechnology) for 15 min at room temperature, stained with phallacidin (cat. no. C2205S; Beyotime Institute of Biotechnology) for 30 min at room temperature, and then stained with DAPI (cat. no. C1002; Beyotime Institute of Biotechnology) for 20 min at room temperature. Tubes were photographed in five random microscope fields using a fluorescence microscope (Leica Microsystems GmbH). ImageJ Angiogenesis Analyzer software (ImageJ 1.53k; National Institutes of Health) was then used to analyse the raw images. In this process, a map of each imaging tube was generated and the number of nodes, junctions, branches and branch lengths were calculated using ImageJ software (ImageJ 1.53k; National Institutes of Health).
5-Ethynyl-2'-deoxyuridine (EdU) assay
The MDA-MB-231 cells and HUVECs were co-cultured in small chambers and six-well plates and treated with ALK1 inhibitor (K02288) and transfected with adenovirus. After 24 h, the HU-231 cells were counted as 2×104 cells/well respectively and inoculated into confocal dishes. EdU assay was then performed using an EdU kit (cat. no. C0078S; Beyotime Institute of Biotechnology) according to the manufacturer's protocol. The nuclei were stained with Hoechst 33342 for 10 min at room temperature. The cells were counted under a fluorescence microscope (Carl Zeiss AG).
Immunofluorescence staining
The HUVECs co-cultured with the MDA-MB-231 cells were treated with ALK1 inhibitor (K02288) and transfected with adenovirus. After 24 h, the HU-231 cells were counted as 2×104 cells/well respectively and inoculated into confocal dishes. After 24 h, the cells were fixed in 4% paraformaldehyde at room temperature for 15 min. The samples were then blocked with Fast Blocking Solution (cat. no. p30500; New Cell & Molecular Biotech Co., Ltd.) for 30 min at room temperature. The cell were then incubated with primary antibody ALK1 (1:150 dilution; cat. no. ab68703; Abcam) overnight at 4°C, followed by incubation with fluorescent secondary antibody goat anti-rabbit (1:500 dilution; cat. no. SA00003; Proteintech Group, lnc.) for 1 h at room temperature. Subsequently, the cells were incubated with DAPI for 10 min at room temperature. Finally, fluorescence was detected using a fluorescence microscope (Leica Microsystems GmbH).
Reverse transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted using the SteadyPure Universal RNA Extraction kit according to the manufacturer's protocol [cat. no. AG21017; Accurate Biotechnology (Hunan) Co., Ltd.] and quantified spectrophotometrically using a NanoDrop spectrophotometer (NanoDrop; Thermo Fisher Scientific, Inc.). The Evo M-MLV Mix kit with gDNA Clean for qPCR (cat. no. AG11728; Accurate Biotechnology (Hunan) Co., Ltd.) was used for reverse transcription(37°C for 15 min and 85°C for 5 sec). The primers used were the following: GAPDH forward, GCA CCG TCA AGG CTG AGA AC and reverse, TGG TGA AGA CGC CAG TGG A; ALK1 forward, CAG CCC ACG AAT CAT CTC CC and reverse, GTC CCC TGT CAC TCC ACT TC; inhibitor of DNA binding 1 (ID1) forward, ATC GCA TCT TGT GTC GCT GAA and reverse, ATT CCT CTT GCC CCC TGG AT; AKT forward, CTG TTC TTC CAC CTG TCC CG and reverse, TAA TGT GCC CGT CCT TGT CC. The SYBR-Green Premix Pro Taq HS qPCR kit according to the manufacturer's instructions (cat. no. AG11701; Accurate Biotechnology (Hunan) Co., Ltd.) and the CFX96™ real-time PCR detection system (Bio-Rad Laboratories, Inc.) were employed to perform qPCR under the following conditions: Initial hold at 95°C for 30 sec, followed by 40 cycles of denaturation at 95°C for 5 sec and annealing/extension at 60°C for 30 sec. The 2−ΔΔCq method (23) was used to analyse the relative expression levels.
Western blot analysis
Cellular proteins were collected following 24 h of treatment in each group and all cells were lysed using RIPA lysis buffer (cat. no. P0013B; Beyotime Institute of Biotechnology) containing protease inhibitor and phosphatase inhibitor for 15 min on ice. The protein content was determined using a BCA protein assay kit (cat. no. P0012; Beyotime Institute of Biotechnology). Equal amounts of protein (20 µg) were subjected to SDS-PAGE (12.5% gel) and then transferred onto a nitrocellulose membrane (Invitrogen; Thermo Fisher Scientific, Inc.) using the wet electroblotting system (Bio-Rad Laboratories, Inc.). Subsequently, the membrane was blocked with 5% non-fat milk (cat. no. P0216; Beyotime Institute of Biotechnology) for 2 h at room temperature, followed by overnight incubation at 4°C with the respective primary antibodies: Rabbit polyclonal antibody (pAb) to ALK1 (1:1,000 dilution; cat. no. ab68703; Abcam), rabbit monoclonal antibody (mAb) to phosphorylated (p-)Smad1/5 (1:1,000 dilution; cat. no. 9516T; Cell Signaling Technology, Inc.), rabbit mAb to Smad1 (1:1,000 dilution; cat. no. 6944T; Cell Signaling Technology, Inc.), rabbit pAb to ID1 (1:1,000 dilution; cat. no. ab230679; Abcam), rabbit mAb to PI3K (1:1,000 dilution; cat. no. 3011T; Cell Signaling Technology, Inc.), rabbit pAb to p-PI3K (1:1,000 dilution; cat. no. bs-6417R; Bioss), rabbit mAb to AKT (1:1,000 dilution; cat. no. 4685S; Cell Signaling Technology, Inc.), rabbit mAb to p-AKT (1:1,000 dilution; cat. no. 4060S; Cell Signaling Technology, Inc.) and rabbit mAb to β-actin (1:2,000 dilution; cat. no. AC028; ABclonal, Inc.), followed by incubation with goat anti-rabbit IgG secondary antibody (1:10,000 dilution; cat. no. ZB-2301; Zsbio) for 1 h at room temperature. The immunoreactive bands were then visualised using the Fusion FX.EDGE (Vilber Bio Imaging). Visualisation was carried out using the new Super ECL assay (Thermo Fisher Scientific, Inc.), which was then exposed to film and analysed using ImageJ software (ImageJ 1.53k; National Institutes of Health).
Statistical analysis
Data are represented as the mean ± SD. Statistical analyses were performed using GraphPad Prism9 software (GraphPad Software Inc.). The differences between two groups were analysed using a Student's t-test. Multiplegroup statistical comparisons were performed using oneway ANOVA with Tukey's multiplecomparisons test. The quantification of cell numbers, colony numbers, wound gap closure and western blot band integrated density were performed using ImageJ software (ImageJ 1.53k; National Institutes of Health). All experiments were performed in triplicate. P<0.05 was considered to indicate a statistically significant difference.
Results
Low-dose PDT induces tumour angiogenesis and ALK1 inhibitor reverses this effect
To establish a model of tumour angiogenesis induced by low-dose PDT, CCK-8 assay was used to detect the appropriate MPPa drug concentration and light intensity in the co-culture system. As illustrated in Fig. 1A, the 10 µmol/l MPPa concentration had no significant effect on cell viability, while 15 and 20 µmol/l MPPa significantly inhibited cell viability; therefore, the 10 µmol/l MPPa concentration was used in subsequent PDT experiments. Moreover, the cell viability was related to the light intensity, which was 10 MPPa µmol/l. Furthermore, when the light energy density was 0.04 J/cm2, the proliferative activity of the HU-231 cells was the strongest (Fig. 1B); thus, this light intensity was used in subsequent experiments. Additionally, when the concentration was 5 µmol/l, K02288 (ALK1 inhibitor) had no significant effect on cell viability. However, at 10, 20 and 40 µmol/l, K02288 decreased cell viability (Fig. 1C) in a concentration-dependent manner. Therefore, in the subsequent experiments, 5 µmol/l K02288 was used. The results of CCK-8 assay for the four groups of cells (the control, PDT, K02288 and PDT + K02288 group) revealed that PDT combined with K02288 significantly inhibited HU-231 cell viability compared with the control and PDT groups (Fig. 1D).
Figure 1.

Effect of PDT on the viability and proliferation of HU-231 cells. (A) Effects of various photosensitizer (MPPa) concentrations (5, 10, 15 and 20 µmol/l) on the viability of HU-231 cells. (B) Effects of different activin receptor-like kinase-1 inhibitor (K02288) concentrations (2.5, 5, 10, 20 and 40 µmol/l) on HU-231 cell viability. (C) Cell viability under different light intensities (1.6, 3.2, 4, 8, 16 and 24 ×10−2 J/cm2). (D) Cell viability of the control group, PDT group, K02288 group and PDT + K02288 group. (E and F) Cell proliferation of the control, PDT, K02288 and PDT + K02288 groups (scale bars, 200 µm). (G and H): Proliferative ability of the cells in the four groups. The red colour represents the proliferating cells stained with EdU, and the blue colour represents all cells stained with Hoechst 33342 (scale bars, 20 µm). *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. PDT, photodynamic therapy; MPPa, pyropheophorbide-a methyl ester.
Furthermore, colony forming and EdU assays were used to detect the proliferative ability of the HU-231 cells, which revealed that the colony number and proliferation rate of the low-dose PDT group were significantly higher than those of the other groups. These findings indicated that low-dose PDT enhanced the proliferative ability of the HU-231 cells. There was no significant difference between the K02288 and the control group. However, the colony number and proliferation rate of the PDT + K02288 group were significantly reduced compared with the control group and PDT group, indicating that PDT combined with K02288 weakened the proliferative ability of the cells and reversed tumour angiogenesis induced by low-dose PDT (Fig. 1E-H).
The horizontal and vertical migratory abilities of the HU-231 cells were determined using wound healing and Transwell migration assays, respectively. The results of the wound healing assay (Fig. 2A and B) revealed that the wound healing rate of the low-dose PDT group at 24 and 48 h was significantly higher than that of the control group, indicating that low-dose PDT enhanced the horizontal migratory ability of the HU-231 cells. However, the migration area was significantly reduced following combination treatment with K02288, which weakened the horizontal migratory ability of the cells. The Transwell migration assay (Fig. 2C and D) revealed that the number of migrating cells in the low-dose PDT group was significantly higher than that in the other groups, indicating that low-dose PDT enhanced the vertical migratory ability of the HUVECs co-cultured with MDA-MB-231 cells. However, following combination treatment with K02288, the number of migrating cells was significantly reduced, which weakened the vertical migratory ability of the cells.
Figure 2.

Effects of low-dose PDT on the migration, invasion and tubular formation of HU-231 cells. (A and B) Horizontal migratory ability of the cells in the control, PDT, K02288 and PDT + K02288 groups at 0, 24 and 48 h (scale bars, 200 µm). (C and D) Vertical migratory ability of the cells in the control, PDT, K02288 and PDT + K02288 groups (scale bars, 200 µm). (E and F) Invasive ability of the cells in the control, PDT, K02288 and PDT + K02288 groups (scale bars, 200 µm). (G-J) Ability of the cells to form tubes in the control, PDT, K02288 and PDT + K02288 groups (scale bars, 200 µm). *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. PDT, photodynamic therapy.
Transwell invasion assay (Fig. 2E and F) also revealed that the number of invasive cells in the low-dose PDT group was significantly higher than that in the other groups, indicating that low-dose PDT enhanced the invasive ability of the HU-231 cells. However, following combination treatment with K02288, the number of invasive cells was significantly reduced, which weakened the invasive ability of cells and reversed the promoting effects of low-dose PDT on tumour vascular invasion.
Furthermore, the tube formation assay (Fig. 2G-J) revealed that the tube-forming length, tube-forming node and tube-forming ring number of the low-dose PDT group were significantly higher than those of the other groups, indicating that low-dose PDT enhanced the tube-forming ability of the HU-231 cells. However, following combination treatment with K02288, the tube-forming length, tube-forming node and tube-forming ring number were significantly reduced, which weakened the tube-forming ability of the cells. It also reversed the effects induced by low-dose PDT, wherein the tube-forming ability of the tumour blood vessels was enhanced.
Low-dose PDT induces tumour angiogenesis and ALK1 knockdown reverses this effect
Colony formation and EdU assays were used to detect the proliferative ability of the HU-231 cells. The colony number and proliferation rate of the low-dose PDT group were significantly higher than those of the other groups. Moreover, a significant difference between the Ad-ALK1 group and the control group was observed, indicating that the knockdown of the ALK1 gene inhibited cell proliferation. After PDT was combined with Ad-ALK1, the colony number and proliferation rate significantly differed from those of the control group and PDT group, which severely weakened the cell proliferative ability and reversed the promoting effects on tumour angiogenesis induced by low-dose PDT (Fig. 3).
Figure 3.

Effects of low-dose PDT on the viability and proliferation of HU-231 cells. (A and B) Cell proliferative ability of the cells in the control group, NC group, PDT group, Ad-ALK1 group and PDT + Ad-ALK1 group. (C and D) Proliferative ability of the cells in the five groups. The red colour represents the proliferating cells stained with EdU, and the blue colour represents all cells stained with Hoechst 33342 (scale bars, 30 µm. *P<0.05, **P<0.01 and ***P<0.001. NC, negative control; Ad, adenovirus; PDT, photodynamic therapy; ALK1, activin receptor-like kinase-1.
The horizontal and vertical migratory abilities of the HU-231 cells were analysed using wound healing and Transwell migration assays, respectively. The wound healing assay (Fig. 4A and B) revealed that the wound healing rate of the low-dose PDT group at 24 and 48 h was significantly higher than that of the control group. There was no significant difference between the Ad-ALK1 group and the control group. However, the migration area of the PDT + Ad-ALK1 group was significantly reduced compared with that of the control and PDT groups, indicating the severely weakened horizontal migratory ability of the cells. Moreover, the effects induced by low-dose PDT were reversed, wherein the horizontal migratory ability of new tumour blood vessels was enhanced. The Transwell migration assay (Fig. 4C and D) also revealed that the number of migrating cells in the low-dose PDT group was significantly higher than that in the other groups. However, compared with the and PDT groups, the number of migrating cells in the PDT + Ad-ALK1 group was significantly reduced, which weakened the vertical migratory ability of the cells. It further reversed the effects of low-dose PDT, wherein the vertical migratory ability of new tumour vessels was enhanced.
Figure 4.

Effects of low-dose PDT on the migration, invasion and tubular formation of HU-231 cells. (A and B) Horizontal migratory ability of the cells in the control group, NC group, PDT group, Ad-ALK1 group and PDT + Ad-ALK1 group at 0, 24 and 48 h (scale bars, 200 µm). (C and D) Vertical migration of the cells in the control, NC, PDT, Ad-ALK1 and PDT + Ad-ALK1 groups (scale bars, 200 µm). (E and F): Vertical invasion of the cells in the control, NC, PDT, Ad-ALK1 and PDT + Ad-ALK1 groups (scale bars, 200 µm). (G-J) Ability of the cells to form tubes in the control, NC, PDT, Ad-ALK1 and PDT + Ad-ALK1 groups (scale bars, 200 µm). *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. NC, negative control; Ad, adenovirus; PDT, photodynamic therapy; ALK1, activin receptor-like kinase-1.
Transwell invasion assay (Fig. 4E and F) revealed that the number of invasive cells in the low-dose PDT group was significantly higher than that in other groups. However, compared with the control and PDT groups, the number of invasive cells in the PDT + Ad-ALK1 group was significantly reduced, which weakened the invasive ability of cells and reversed the effect induced by low-dose PDT, wherein the invasive ability of new tumour blood vessels was enhanced.
The tube formation assay (Fig. 4G-J) demonstrated that the tube length, tube node and tube ring number of the low-dose PDT group were significantly higher than those of the other groups, indicating that low-dose PDT enhanced the tube-forming ability of the HU-231 cells. However, the tube length, tube node and tube ring number of the PDT + Ad-ALK1 group were significantly reduced compared with the control and PDT groups, indicating the suppressed tube-forming ability of the cells. It also reversed the effect induced by low-dose PDT, wherein tumour angiogenesis was enhanced.
Involvement of the ALK1-Smad1/5-ID1 pathway in low-dose PDT-induced tumour angiogenesis
The aforementioned experiments verified that ALK1 was involved in the enhancement of HU-231 cell activity, proliferation, migration, invasion and tube formation abilities of the tumours induced by low-dose PDT. Furthermore, to elucidate the role of ALK1 in the enhancement of HU-231 cell activity and the ability of proliferation, migration, invasion and tube formation induced by low-dose PDT, the gene level changes (Fig. 5A and C) of ALK1 and ID1 in the control, low-dose PDT, K02288 and PDT + K02288 groups at 0, 2, 3, 6, 10 and 24 h following treatment were detected using RT-qPCR. In the low-dose PDT group, ALK1 expression was significantly increased (Fig. 5A) compared to that in the control group at 2 h following treatment. No significant differences were observed at the other time points. Furthermore, ID1 expression significantly decreased in the K02288 group and PDT + K02288 group, apart from the time point of 0 h (Fig. 5C). In addition, the gene level changes of ALK1 and ID1 were also detected at 2 h following adenovirus transfection and ALK1 knockdown. The results revealed that both ALK1 and ID1 expression levels were significantly elevated in the PDT group compared with the control group, while the decrease in ALK1 and ID1 expression levels in the Ad-ALK1 and PDT + Ad-ALK1 groups were significant compared to the control group (Fig. 5B and D).
Figure 5.

Expression of ALK1 and ID1 at different time points in each group. (A) Expression of ALK1 at different time points (0, 2, 3, 6, 10 and 24 h) in the control, PDT, K02288 and PDT + K02288 groups. (B) Expression of ALK1 at 2 h in the control, NC, PDT, Ad-ALK1 and PDT + Ad-ALK1 groups. (C) Expression of ID1 at different time points (0, 2, 3, 6, 10 and 24 h) in the control, PDT, K02288 and PDT + K02288 groups. (D) Expression of ID1 at 2 h in the control, NC, PDT, Ad-ALK1 and Ad-ALK1 + PDT groups. *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. NC, negative control; Ad, adenovirus; PDT, photodynamic therapy; ALK1, activin receptor-like kinase-1; ID1, inhibitor of DNA binding 1.
These findings indicated that during PDT-induced tumour angiogenesis, the expression of ALK1 was significantly increased at 2 h, and that of its downstream molecule, ID1, was significantly decreased following treatment with ALK1 inhibitor (at 2, 3, 6, 10 and 24 h) or ALK1 knockdown (2 h).
Subsequently, using immunofluorescence to detect the content of ALK1 in HU-231 cells, the fluorescence intensity of the PDT group was observed to be significantly stronger than that of the control group, while the fluorescence intensity significantly decreased following combination treatment with PDT with K02288 (Fig. 6A and B).
Figure 6.

The association between low-dose PDT-induced tumour angiogenesis and the ALK1-Smad1/5-ID1 pathway. (A and B) Expression of ALK1 in the control, PDT, K02288 and PDT + K02288 groups (scale bars, 50 µm). (C-F) Expression of ALK1, Smad 1, p-Smad1/5 and ID1 proteins in the control, PDT, K02288 and PDT + K02288 groups. (G-J) Expression of ALK1, Smad 1, p-Smad1/5 and ID1 proteins in the control, NC, PDT, Ad-ALK1 and PDT + Ad-ALK1 groups. *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. NC, negative control; Ad, adenovirus; p-, phosphorylated; PDT, photodynamic therapy; ALK1, activin receptor-like kinase-1; ID1, inhibitor of DNA binding 1.
Western blot analyses also revealed that the expression of ALK1, p-Smad1/5 and ID1 was upregulated in the low-dose PDT group, whereas it was inhibited following combination treatment with PDT with K02288. Following the adenovirus knockdown of ALK1, the levels of ALK1, p-Smad1/5 and ID1 were also significantly decreased compared with the control, negative control and PDT groups (Fig. 5C-J).
Furthermore, the tumour angiogenesis induced by low-dose PDT may have been attributed to the significant increase in the expression of ALK1, p-Smad1/5 and ID1. After PDT was combined with inhibitor drugs or adenovirus, the expression of the aforementioned molecules was significantly decreased, confirming that tumour angiogenesis induced by low-dose PDT may be related to this pathway.
The PI3K/AKT pathway is not involved in low-dose PDT-induced tumour angiogenesis
To determine whether tumour angiogenesis induced by low-dose PDT is related to the PI3K/AKT pathway in addition to the classic Smad pathway, the gene level changes of AKT after the combined application of ALK1 inhibitor (K02288) or the adenovirus knockdown of ALK1 was detected using RT-qPCR. AKT expression exhibited no significant change in each group at the different time points (Fig. 7A and B).
Figure 7.

The association between tumour angiogenesis induced by low-dose PDT and the PI3K/AKT pathway. (A) The expression of AKT in the control, PDT, K02288 and PDT + K02288 groups at different time points (0, 2, 3, 6, 10 and 24 h). (B) The expression of AKT at 2 h in the control, NC, PDT, Ad-ALK1 and PDT + Ad-ALK1 groups. (C and D) The protein expression of AKT, p-AKT, PI3K and p-PI3K in the control, PDT, K02288 and PDT + K02288 groups after 24 h of treatment. (E-G) The protein expression of AKT, p-AKT, PI3K and p-PI3K in the control, PDT, Ad-ALK1 and PDT + Ad-ALK1 groups after 24 h of treatment. (H) Effects of various PI3K inhibitor (LY294002) concentrations (5, 10, 20 and 40 µmol/l) on HU-231 cell viability. (I-K) Protein expression of AKT, p-AKT, PI3K and p-PI3K in the control, PDT, LY294002 and PDT + LY294002 groups after 24 h of treatment *P<0.05, **P<0.01 and ***P<0.001. ns, not significant (P>0.05); NC, negative control; Ad, adenovirus; p-, phosphorylated; PDT, photodynamic therapy; ALK1, activin receptor-like kinase-1.
Similarly, western blot analyses further revealed no significant changes in the PI3K/AKT total protein and phosphorylation levels (Fig. 7C and D) when PDT was used at a low dose alone or in combination with the ALK1 inhibitor, K02288. Moreover, following adenovirus knockdown, AKT phosphorylation was enhanced, while the PI3K and p-PI3K levels were not significantly altered, which was consistent with the observations of the inhibitor group (Fig. 7E-G). Subsequently, the PI3K inhibitor, LY294002, was used to verify whether low-dose PDT-induced tumour vascular proliferation was associated with PI3K/AKT signalling pathway. The appropriate drug concentration was first screened using CCK-8 assay and no significant effect was observed on cell viability at the concentration of 10 µmol/l (Fig. 7H); thus, this concentration was subsequently used for the subsequent western blotting experiments. In the western blot analysis, the protein expression of AKT, p-AKT, PI3K and p-PI3K was measured in four groups (control, PDT, LY294002 and PDT + LY294002 group) (Fig. 7I-K). The expression of p-PI3K and p-AKT in the LY294002 and PDT + LY294002 groups was significantly decreased compared with the control group and there was no significant difference between the two groups; similarly, there was no significant difference in the expression of p-PI3K and p-AKT between the PDT group and the control group. This suggests that the PI3K/AKT pathway may not be involved in tumour angiogenesis induced by low-dose PDT.
Discussion
PDT has unique advantages, such as minimal invasiveness, low systemic toxicity and no drug resistance (20). The main principle of PDT in cancer treatment includes the systemic or local application of a non-toxic photosensitive dye, a photosensitizer. Following the selective aggregation of the photosensitizer in tumours, the photosensitizer is stimulated by visible light at an appropriate wavelength. The presence of oxygen in cells and tissues leads to the production of cytotoxic substances and the activation of signalling pathways, thereby causing cell death and tumour tissue destruction (24). However, due to the specificity of tumour blood vessels, the concentration of photosensitizer accumulation in the tumour is low. Moreover, the light intensity decreases with the depth of the tumour. In addition, the tumour resides in a hypoxic condition, which causes PDT to affect the deep part of the tumour in a low-dose state (8). Subsequently, the induced oxidative stress and cell damage activate the tumour cell survival-promoting signalling pathway (25). This may be a potential reason for the poor response of PDT to some tumours and tumour regrowth following PDT treatment (26). Previous research has reported that low-dose PDT not only affects cell proliferation and differentiation, but also has a tumour-protecting effect (27,28), and improves the tumorigenic potential of cells (29). In vivo studies have also found that low-dose PDT increases the proliferation of cerebral vascular endothelial cells in nude mice (30). Therefore, the present study focused on the effects of low-dose PDT on the tumour vascular endothelium.
Herein, to better simulate the tumour intravascular environment in vivo, HUVECs co-cultured with breast cancer cell line MB-231 (HU-231 cells) were used as the research object. The photosensitizer concentration (10 µmol/l), combined with the light intensity (0.04 J/cm2) that can significantly increase the cell activity, was further used to simulate the tumour angiogenesis induced by low-dose PDT. Consistently, in the present study, low-dose PDT did indeed induce the proliferation of tumour vascular endothelial cells and enhanced their migration, invasion and tube formation.
It has been demonstrated that ALK1 plays a key role in the regulation of physiological and pathological angiogenesis (31). In humans, ALK1 mutation can cause type II hereditary haemorrhagic telangiectasia, which is characterized by telangiectasia, arteriovenous malformations and periodic bleeding (32). In a genetic study on mice, ALK1 gene mutation was observed to induce the death of mice at the embryonic stage, owing to vascular malformations, such as excessive fusion of capillary plexus and excessive expansion of large blood vessels (33). It has been demonstrated that the deletion of ALK1 not only inhibits the proliferation and migration of endothelial cells, but also the differentiation and maturation of vascular smooth muscle cells and the recruitment to the original vascular network formed by endothelial cells (34). Therefore, the role of ALK1 in tumour angiogenesis induced by low-dose PDT was explored herein.
The present study observed that in the co-culture of MDA-MB-231 breast cancer cell and HUVECs, after low-dose PDT was combined with ALK1 inhibitor (K02288) or the adenovirus knockdown of ALK1, the significantly enhanced proliferation, migration, invasion and tubulogenesis induced by low-dose PDT were suppressed. However, the ALK1 mRNA and protein expression levels increased following low-dose PDT treatment, particularly at 2 h after low-dose PDT treatment. These findings thus indicated that tumour blood vessels were stimulated by stress after PDT treatment and the expression of ALK1 in endothelial cells was upregulated, thereby promoting the proliferation and migration of vascular endothelial cells and participating in PDT-induced tumour angiogenesis. Therefore, the inhibition of ALK1 could enhance the efficacy of PDT in treating tumours. Based on pharmacological and genetic approaches, PDT was combined with K02288 or ALK1 knockdown, which revealed that the protein expression of ALK1 was significantly downregulated compared with the control and low-dose PDT group. This may be attributed to the fact that K02288 is a competitive inhibitor. Furthermore, combining K02288 or ALK1 knockdown significantly inhibited cell viability, proliferation, migration, invasion and tube formation. Therefore, ALK1 can promote the proliferation, migration and luminal formation of tumour vascular endothelial cells induced by low-dose PDT; however, the inhibition of related pathways can reduce the effects of low-dose PDT.
As a member of the TGF-β family, the classical signalling pathway of ALK1 is the Smad pathway. The binding of ALK1 with the BMP9/10 receptor results in the sequential activation of type II receptor BMPRII and type I receptor ALK1. This is then followed by the phosphorylation of Smad1 and Smad5 on their conservative C-terminal SXS motif through ALK1. Once phosphorylated, Smad1, Smad5 and Smad4 form heterotrimeric complexes and translocate to the nucleus (35), where they activate the transcription of downstream target genes, such as ID1, ID3, transmembrane protein 100 and other target genes. Subsequently, downstream angiogenic factors are activated, thereby regulating endothelial cell proliferation, migration and lumen formation, and regulating angiogenesis (17,36,37).
The present study observed that following low-dose PDT, the expression of ALK1, p-Smad1/5 and ID1 was upregulated. After combining PDT with K02288 and the adenovirus knockdown of ALK1, the expression of the aforementioned three genes was significantly reduced. This finding confirms that ALK1-Smad1/5-ID1, a classic SMAD pathway, may be involved in the low-dose PDT-induced vascular proliferation.
In addition to regulating the Smad pathway, ALK1 can also affect the function of the vascular endothelium through the non-Smad pathway, such as the PI3K and AKT pathway. Moreover, in a previous study, the knockout of BMP-9 receptor ALK1 in non-small cell lung cancer was reported to inhibit the growth of A549 cells in vitro and in vivo, which was related to the PI3K/Akt and Smad1/5 signalling pathways (38). In another study, in hereditary haemorrhagic telangiectasia type 2, PI3K/AKT signal transduction increased when ALK1 was absent (39). Additionally, after reducing the binding of BMP-9 and ALK1 in osteosarcoma, the PI3K/AKT pathway was activated (40). In the present study, it was observed that the phosphorylation of PI3K and AKT in the low-dose PDT group was not significantly altered. After using the PI3K inhibitor, LY294002, the results revealed that the expression of p-PI3K and p-AKT in the LY294002 group and PDT + LY294002 group was significantly decreased compared with the control group, and there was no significant difference between the two groups; similarly, there was no significant difference in the expression of p-PI3K and p-AKT between the PDT group and the control group. Furthermore, the results of RT-qPCR and western blot analysis revealed that there was no significant effect on the PI3K/AKT pathway, indicating that PDT induced changes in ALK1 to promote vascular proliferation, which was not through the PI3K/AKT pathway. Notably, it was also found that AKT phosphorylation increased after the adenovirus knockdown of ALK1, which may be due to the blocking of BMP9 signal transduction after the ALK1 knockdown. It was also observed to regulate the location and activity of PTEN, a lipid phosphatase upstream of PI3K-AKT, thus activating the PI3K-AKT pathway (39). The PI3K/Akt signalling pathway promotes tumour angiogenesis by affecting various angiogenic factors and plays a critical role in tumour development (41). Based on the findings of the present study, ALK1 gene therapy is not recommended in the future strategy of tumour PDT combined with anti-ALK1, owing to its activation of the PI3K/AKT pathway that could lead to vascular proliferation and tumour recurrence. However, a limitation of the present study is that no experiments were performed using inhibitors of Smad1/5, ID1 and AKT to fully validate the pathway.
In conclusion, the present study observed that low-dose PDT enhanced the activity of tumour vascular endothelial cells and their ability to proliferate, migrate, invade and form tubes. However. when PDT was combined with ALK1 inhibitor or Ad-ALK1 treatment, the aforementioned changes were reversed. Additionally, ALK1 participates in PDT-induced tumour angiogenesis by activating the Smad1/5-ID1 pathway, as opposed to the PI3K/AKT pathway (illustrated in Fig. 8). To the best of our knowledge, the present study is the first to demonstrate that ALK1 is involved in PDT-induced tumour angiogenesis and provides a theoretical basis for the combination of PDT with anti-ALK1 treatment and has strong clinical significance. However, another limitation of the present study is the lack of in vivo experiments. The authors aim to conduct further research on other tumour cells and also perform in vivo studies to verify these results in the future.
Figure 8.
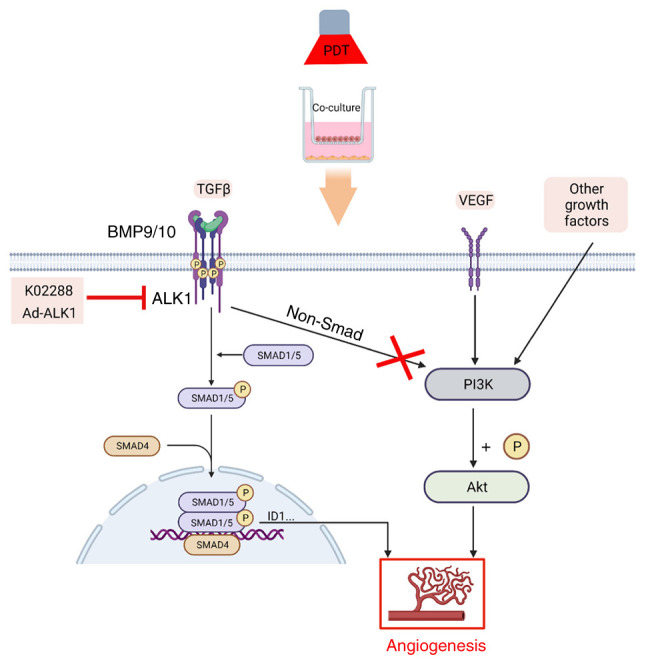
Pathway model of vascular hyperplasia induced by low-dose PDT. As shown in the figure, the treatment of human umbilical vein endothelial cells co-cultured with MDA-MB-231 breast cancer cells (termed HU-231 cells) with low-dose PDT led to endothelial cell proliferation in the co-culture system, which may be related to the ALK1-Smad1/5-ID1 signalling pathway, but not to the PI3K/AKT pathway. PDT, photodynamic therapy; ALK1, activin receptor-like kinase-1.
Acknowledgments
Not applicable.
Funding Statement
The present study was funded by the National Natural Science Foundation of China (grant nos. 81871853 and 82003306), the Natural Science Foundation of Chongqing (grant nos. cstc2019jcyj-msxmX0397 and cstc2020jcyj-msxmX0484) and the Cultivating Fund in the First Affiliated Hospital of Chongqing Medical University (grant nos. PYJJ2018-24 and PYJJ2019-214).
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
XG, DB and KL conceptualized the study. XG, KL, WH, YN and XH were involved in the study methodology. YZ and QC were involved in data validation. YZ and ST were involved in the formal analysis. QC was involved in the investigative aspects of the study. XG was involved in data curation, and in the writing and preparation of the original draft of the manuscript. KL was involved in the writing, reviewing and editing of the manuscript. DB and KL supervised the study. KL and DB were involved in funding acquisition. XG and KL confirm the authenticity of all the raw data. All authors have read and agreed to the published version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, et al. Photodynamic therapy of cancer: An update. CA Cancer J Clin. 2011;61:250–281. doi: 10.3322/caac.20114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li L, Song D, Qi L, Jiang M, Wu Y, Gan J, Cao K, Li Y, Bai Y, Zheng T. Photodynamic therapy induces human esophageal carcinoma cell pyroptosis by targeting the PKM2/caspase-8/caspase-3/GSDME axis. Cancer Lett. 2021;520:143–159. doi: 10.1016/j.canlet.2021.07.014. [DOI] [PubMed] [Google Scholar]
- 3.Simone CB, II, Cengel KA. Photodynamic therapy for lung cancer and malignant pleural mesothelioma. Semin Oncol. 2014;41:820–830. doi: 10.1053/j.seminoncol.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gamelas SR, Moura NM, Habraken Y, Piette J, Neves M, Faustino MA. Tetracationic porphyrin derivatives against human breast cancer. J Photochem Photobiol B. 2021;222:112258. doi: 10.1016/j.jphotobiol.2021.112258. [DOI] [PubMed] [Google Scholar]
- 5.Leon D, Buchegger K, Silva R, Riquelme I, Viscarra T, Mora-Lagos B, Zanella L, Schafer F, Kurachi C, Roa JC, et al. Epigallocatechin gallate enhances MAL-PDT cytotoxic effect on PDT-resistant skin cancer squamous cells. Int J Mol Sci. 2020;21:3327. doi: 10.3390/ijms21093327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broekgaarden M, Weijer R, van Gulik TM, Hamblin MR, Heger M. Tumor cell survival pathways activated by photodynamic therapy: A molecular basis for pharmacological inhibition strategies. Cancer Metastasis Rev. 2015;34:643–690. doi: 10.1007/s10555-015-9588-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wan Y, Fu LH, Li C, Lin J, Huang P. Conquering the hypoxia limitation for photodynamic therapy. Adv Mater. 2021;33:e2103978. doi: 10.1002/adma.202103978. [DOI] [PubMed] [Google Scholar]
- 8.Yue D, Cai X, Fan M, Zhu J, Tian J, Wu L, Jiang Q, Gu Z. An alternating irradiation strategy-driven combination therapy of PDT and RNAi for highly efficient inhibition of tumor growth and metastasis. Adv Healthc Mater. 2021;10:e2001850. doi: 10.1002/adhm.202001850. [DOI] [PubMed] [Google Scholar]
- 9.De Sanctis F, Ugel S, Facciponte J, Facciabene A. The dark side of tumor-associated endothelial cells. Semin Immunol. 2018;35:35–47. doi: 10.1016/j.smim.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Ferrario A, von Tiehl KF, Rucker N, Schwarz MA, Gill PS, Gomer CJ. Antiangiogenic treatment enhances photodynamic therapy responsiveness in a mouse mammary carcinoma. Cancer Res. 2000;60:4066–4069. [PubMed] [Google Scholar]
- 11.Peng CL, Lin HC, Chiang WL, Shih YH, Chiang PF, Luo TY, Cheng CC, Shieh MJ. Anti-angiogenic treatment (Bevacizumab) improves the responsiveness of photodynamic therapy in colorectal cancer. Photodiagnosis Photodyn Ther. 2018;23:111–118. doi: 10.1016/j.pdpdt.2018.06.008. [DOI] [PubMed] [Google Scholar]
- 12.Olivo M, Bhuvaneswari R, Lucky SS, Dendukuri N, Soo-Ping Thong P. Targeted therapy of cancer using photodynamic therapy in combination with multi-faceted anti-tumor modalities. Pharmaceuticals (Basel) 2010;3:1507–1529. doi: 10.3390/ph3051507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimizu K, Asai T, Oku N. Antineovascular therapy, a novel antiangiogenic approach. Expert Opin Ther Targets. 2005;9:63–76. doi: 10.1517/14728222.9.1.63. [DOI] [PubMed] [Google Scholar]
- 14.Weiss A, den Bergh Hv, Griffioen AW, Nowak-Sliwinska P. Angiogenesis inhibition for the improvement of photodynamic therapy: The revival of a promising idea. Biochim Biophys Acta. 2012;1826:53–70. doi: 10.1016/j.bbcan.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Hu-Lowe DD, Chen E, Zhang L, Watson KD, Mancuso P, Lappin P, Wickman G, Chen JH, Wang J, Jiang X, et al. Targeting activin receptor-like kinase 1 inhibits angiogenesis and tumorigenesis through a mechanism of action complementary to anti-VEGF therapies. Cancer Res. 2011;71:1362–1373. doi: 10.1158/0008-5472.CAN-10-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Vinuesa AG, Bocci M, Pietras K, Ten Dijke P. Targeting tumour vasculature by inhibiting activin receptor-like kinase (ALK)1 function. Biochem Soc Trans. 2016;44:1142–1149. doi: 10.1042/BST20160093. [DOI] [PubMed] [Google Scholar]
- 17.Roman BL, Hinck AP. ALK1 signaling in development and disease: New paradigms. Cell Mol Life Sci. 2017;74:4539–4560. doi: 10.1007/s00018-017-2636-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhatt RS, Atkins MB. Molecular pathways: Can activin-like kinase pathway inhibition enhance the limited efficacy of VEGF inhibitors? Clin Cancer Res. 2014;20:2838–2845. doi: 10.1158/1078-0432.CCR-13-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cunha SI, Pardali E, Thorikay M, Anderberg C, Hawinkels L, Goumans MJ, Seehra J, Heldin CH, ten Dijke P, Pietras K. Genetic and pharmacological targeting of activin receptor-like kinase 1 impairs tumor growth and angiogenesis. J Exp Med. 2010;207:85–100. doi: 10.1084/jem.20091309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hawinkels LJ, de Vinuesa AG, Paauwe M, Kruithof-de Julio M, Wiercinska E, Pardali E, Mezzanotte L, Keereweer S, Braumuller TM, Heijkants RC, et al. Activin receptor-like kinase 1 ligand trap reduces microvascular density and improves chemotherapy efficiency to various solid tumors. Clin Cancer Res. 2016;22:96–106. doi: 10.1158/1078-0432.CCR-15-0743. [DOI] [PubMed] [Google Scholar]
- 21.Cunha SI, Bocci M, Lövrot J, Eleftheriou N, Roswall P, Cordero E, Lindström L, Bartoschek M, Haller BK, Pearsall RS, et al. Endothelial ALK1 is a therapeutic target to block metastatic dissemination of breast cancer. Cancer Res. 2015;75:2445–2456. doi: 10.1158/0008-5472.CAN-14-3706. [DOI] [PubMed] [Google Scholar]
- 22.Goff LW, Cohen RB, Berlin JD, de Braud FG, Lyshchik A, Noberasco C, Bertolini F, Carpentieri M, Stampino CG, Abbattista A, et al. A Phase I study of the Anti-Activin Receptor-Like Kinase 1 (ALK-1) monoclonal antibody PF-03446962 in patients with advanced solid tumors. Clin Cancer Res. 2016;22:2146–2154. doi: 10.1158/1078-0432.CCR-15-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Alzeibak R, Mishchenko TA, Shilyagina NY, Balalaeva IV, Vedunova MV, Krysko DV. Targeting immunogenic cancer cell death by photodynamic therapy: Past, present and future. J Immunother Cancer. 2021;9:e001926. doi: 10.1136/jitc-2020-001926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weijer R, Clavier S, Zaal EA, Pijls MM, van Kooten RT, Vermaas K, Leen R, Jongejan A, Moerland PD, van Kampen AH, et al. Multi-OMIC profiling of survival and metabolic signaling networks in cells subjected to photodynamic therapy. Cell Mol Life Sci. 2017;74:1133–1151. doi: 10.1007/s00018-016-2401-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, Moan J, Peng Q. Photodynamic therapy. J Natl Cancer Inst. 1998;90:889–905. doi: 10.1093/jnci/90.12.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blazquez-Castro A, Carrasco E, Calvo MI, Jaén P, Stockert JC, Juarranz A, Sánz-Rodríguez F, Espada J. Protoporphyrin IX-dependent photodynamic production of endogenous ROS stimulates cell proliferation. Eur J Cell Biol. 2012;91:216–223. doi: 10.1016/j.ejcb.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Espada J, Galaz S, Sanz-Rodríguez F, Blázquez-Castro A, Stockert JC, Bagazgoitia L, Jaén P, González S, Cano A, Juarranz A. Oncogenic H-Ras and PI3K signaling can inhibit E-cadherin-dependent apoptosis and promote cell survival after photodynamic therapy in mouse keratinocytes. J Cell Physiol. 2009;219:84–93. doi: 10.1002/jcp.21652. [DOI] [PubMed] [Google Scholar]
- 29.Udartseva OO, Zhidkova OV, Ezdakova MI, Ogneva IV, Andreeva ER, Buravkova LB, Gollnick SO. Low-dose photodynamic therapy promotes angiogenic potential and increases immunogenicity of human mesenchymal stromal cells. J Photochem Photobiol B. 2019;199:111596. doi: 10.1016/j.jphotobiol.2019.111596. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Jiang F, Zhang ZG, Kalkanis SN, Hong X, deCarvalho AC, Chen J, Yang H, Robin AM, Chopp M. Low-dose photodynamic therapy increases endothelial cell proliferation and VEGF expression in nude mice brain. Lasers Med Sci. 2005;20:74–79. doi: 10.1007/s10103-005-0348-8. [DOI] [PubMed] [Google Scholar]
- 31.Muñoz-Félix JM, González-Núñez M, López-Novoa JM. ALK1-Smad1/5 signaling pathway in fibrosis development: Friend or foe? Cytokine Growth Factor Rev. 2013;24:523–537. doi: 10.1016/j.cytogfr.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 32.Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- 33.Urness LD, Sorensen LK, Li DY. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet. 2000;26:328–331. doi: 10.1038/81634. [DOI] [PubMed] [Google Scholar]
- 34.Scharpfenecker M, van Dinther M, Liu Z, van Bezooijen RL, Zhao Q, Pukac L, Löwik CW, ten Dijke P. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J Cell Sci. 2007;120:964–972. doi: 10.1242/jcs.002949. [DOI] [PubMed] [Google Scholar]
- 35.Fu Y, Wang H, Dai H, Zhu Q, Cui CP, Sun X, Li Y, Deng Z, Zhou X, Ge Y, et al. OTULIN allies with LUBAC to govern angiogenesis by editing ALK1 linear polyubiquitin. Mol Cell. 2021;81:3187–3204.e7. doi: 10.1016/j.molcel.2021.05.031. [DOI] [PubMed] [Google Scholar]
- 36.Somekawa S, Imagawa K, Hayashi H, Sakabe M, Ioka T, Sato GE, Inada K, Iwamoto T, Mori T, Uemura S, et al. Tmem100, an ALK1 receptor signaling-dependent gene essential for arterial endothelium differentiation and vascular morphogenesis. Proc Natl Acad Sci USA. 2012;109:12064–12069. doi: 10.1073/pnas.1207210109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li W, Salmon RM, Jiang H, Morrell NW. Regulation of the ALK1 ligands, BMP9 and BMP10. Biochem Soc Trans. 2016;44:1135–1141. doi: 10.1042/BST20160083. [DOI] [PubMed] [Google Scholar]
- 38.Hou X, Peng Y, Liu J, Zhong Q, Yu Z, Zhang L. Bone morphogenetic protein-9 promotes the proliferation of non-small cell lung cancer cells by activating PI3K/Akt and Smad1/5 pathways. RSC Adv. 2020;10:7214–7220. doi: 10.1039/D0RA00737D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ola R, Dubrac A, Han J, Zhang F, Fang JS, Larrivée B, Lee M, Urarte AA, Kraehling JR, Genet G, et al. PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nat Commun. 2016;7:13650. doi: 10.1038/ncomms13650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen H, Pan R, Li H, Zhang W, Ren C, Lu Q, Chen H, Zhang X, Nie Y. CHRDL2 promotes osteosarcoma cell proliferation and metastasis through the BMP-9/PI3K/AKT pathway. Cell Biol Int. 2021;45:623–632. doi: 10.1002/cbin.11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gong T, Cui L, Wang H, Wang H, Han N. Knockdown of KLF5 suppresses hypoxia-induced resistance to cisplatin in NSCLC cells by regulating HIF-1α-dependent glycolysis through inactivation of the PI3K/Akt/mTOR pathway. J Transl Med. 2018;16:164. doi: 10.1186/s12967-018-1543-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.