

ABSTRACT
Human cytomegalovirus (HCMV) establishes latency in myeloid cells. Using the Kasumi-3 latency model, we previously showed that lytic gene expression is activated prior to the establishment of latency in these cells. The early events in infection may have a critical role in shaping the establishment of latency. Here, we have used an integrative multi-omics approach to investigate dynamic changes in host and HCMV gene expression and epigenomes at early times postinfection. Our results show dynamic changes in viral gene expression and viral chromatin. Analyses of polymerase II (Pol II), histone 3 lysine 27 acetylation (H3K27Ac), and histone 3 lysine 27 trimethylation (H3K27me3) occupancy of the viral genome showed that (i) Pol II occupancy was highest at the major IE promoter (MIEP) at 4 h postinfection. However, it was observed throughout the genome. (ii) At 24 h, H3K27Ac was localized to the major immediate early promoter/enhancer and to a possible second enhancer in the origin of replication oriLyt; (iii) viral chromatin was broadly accessible at 24 hpi. In addition, although HCMV infection activated expression of some host genes, we observed an overall loss of de novo transcription. This was associated with loss of promoter-proximal Pol II and H3K27Ac but not with changes in chromatin accessibility or a switch in modification of H3K27.
IMPORTANCE Human cytomegalovirus (HCMV) is an important human pathogen in immunocompromised hosts and developing fetuses. Current antiviral therapies are limited by toxicity and emergence of resistant strains. Our studies highlight emerging concepts that challenge current paradigms of regulation of HCMV gene expression in myeloid cells. In addition, our studies show that HCMV has a profound effect on de novo transcription and the cellular epigenome. These results may have implications for mechanisms of viral pathogenesis.
KEYWORDS: human cytomegalovirus, epigenetics, gene expression, herpesviruses
INTRODUCTION
Human cytomegalovirus (HCMV) is a ubiquitous human herpesvirus that latently infects 50% to 90% of the population. Primary infection in immunocompetent hosts is controlled by the immune response, which eliminates cells producing virus, but the virus has the ability to establish a lifelong latent infection in hematopoietic progenitor cells in the bone marrow (1–4). Reactivation of latent virus is generally asymptomatic in immunocompetent hosts, but in immunocompromised individuals, including recipients of hematopoietic stem cells and solid organ transplants, reactivation is associated with significant infectious complications, increased morbidity, and mortality (5). In addition, HCMV-infected pregnant women can transmit the virus to developing fetuses. HCMV infection in utero can result in death of the fetus or significant permanent impairment of survivors, including hearing and vision loss (6).
Infection of cells with HCMV initiates a complex interplay between the virus and the host, in which the virus attempts to commandeer the cell and transform it into a replication factory while the host tries to defend itself from attack. In some cell types, such as fibroblasts, the virus successfully overrides host defenses, replicates to high levels, and ultimately kills the cells. However, in other cell types, such as monocytes and myeloid progenitors in the bone marrow, the virus initiates its replication cycle, but the host cell overcomes the attack and shuts off viral replication (7–19). The viral genome remains in the nucleus as an extrachromosomal episome, which is heterochromatinized (10, 20–25).
HCMV has a large and complex genome of 235 kb, with the capacity to encode more than 165 open reading frames, four highly expressed long noncoding RNAs, and multiple microRNAs (reviewed in references 26 and 27). In the viral particle, the DNA is complexed with polyamines in order to achieve the high degree of compaction required to package its large genome into the capsid during virion assembly (28–30). The events that occur once the virus enters the cell have been studied most thoroughly in permissive fibroblasts (reviewed in references 31 and 32). Fusion of the viral and cellular membranes, which occurs either at the cell surface or the endosome, results in release of the capsid and tegument proteins into the cytoplasm. The capsid is transported along microtubules to the nuclear pore complex, and the viral DNA is then injected through this complex into the nucleus, where it circularizes and rapidly associates with histones (33–35). Expression of viral genes occurs in a highly regulated tripartite cascade (reviewed in reference 36). In the immediate early phase, transcription is limited to 2 major immediate early genes, IE1 and IE2, which are encoded by alternatively spliced transcripts initiated from the major immediate early promoter, and minor transcripts from three other regions: TRS1/IRS1, US3, and UL36/UL37. IE2 is the major transactivator protein required to activate transcription of the early genes, which encode enzymes required for replication of the viral DNA, as well as genes involved in immune evasion, cellular tropism, and encode transcription factors required for activation of late gene expression (37–39). Expression of the early genes permits both amplification of viral DNA and the third phase of the viral life cycle, in which the late genes encoding viral structural components are expressed and infectious viral particles are assembled. Dynamic changes in modifications of histones bound to the DNA mediated in part by the immediate early proteins are thought to regulate this process (40–43).
The events that occur in CD34+ myeloid progenitor cells, which are a site of latency, are much less well characterized. The viral tegument protein pp71, which helps to activate transcription of the IE genes through degradation of Daxx, fails to localize to the nuclei in these cells (44–49), and early studies indicated that viral gene expression was restricted to a few genes (24, 50–55), which were categorized as latency genes. However, a number of investigators have observed activation of virally encoded green fluorescent protein (GFP) reporter genes in both primary and transformed cell models of latency, and recent studies have shown that there is broad activation of viral gene expression in these cells (9–18, 56). Our own studies with the Kasumi-3 cell line have shown that viral gene expression is activated prior to the establishment of latency (7). Previous studies showed that, in latently infected cells, viral genomes are heterochromatinized with hypoacetylated and lysine 9- and lysine 27-methylated histone 3 (H3K9 and H3K27, respectively) and are associated with heterochromatin protein 1 and KAP1 (10, 20–22, 53). However, there have been limited studies on the state of viral chromatin, which is crucial to the regulation of viral gene expression, using currently available high-resolution genome-wide methods (10). In part, this is due to the difficulty in obtaining the large numbers of infected primary cells required for chromatin immunoprecipitation sequencing (ChIP-seq) analyses. This problem can be circumvented through the use of the Kasumi-3 cell model, which mimics many of the aspects of experimental HCMV latency in CD34+ myeloid progenitor cells (7, 11).
We previously analyzed expression of selected genes over the course of infection of Kasumi-3 cells (7). These studies showed that there is an initial activation of lytic gene expression in the vast majority of infected cells and low-level production of infectious virus, which is subsequently repressed in some cells to allow the establishment of latency. The early events in infection may have a critical role in shaping the establishment of latency. Here, we have used an integrative approach to investigate genome-wide regulation of both viral and cellular gene expression during the early phase of infection, which precedes the establishment of latency, in Kasumi-3 cells (Fig. 1).
FIG 1.

Schematic of the samples used in the analysis of host and HCMV transcriptomes and epigenomes in Kasumi-3 cells. (A) Kasumi-3 cells were infected with HCMV TB40/Ewt-GFP at an MOI of 1. The total cell population, which includes GFP+ and GFP− infected cells as well as uninfected cells, was collected at 4 and 24 hpi. Cells were used for RNA-seq, and Pol II, H3K27Ac, and H3K27me3 ChIP-seq analyses to study the HCMV epigenome. (B) Kasumi-3 cells were mock infected or infected with HCMV TB40/Ewt-GFP at an MOI of 1. At 24 hpi, cells were sorted to obtain GFP+ live infected cells and live mock-infected cells. Those populations were then subjected to ATAC-seq, RNA-seq, nascent RNA-seq, and Pol II, H3K27Ac and H3K27me3 ChIP-seq analyses to study the effect of HCMV infection on both host and viral epigenomes.
RESULTS
Dynamic changes in RNA expression in infected Kasumi-3 cells at early times postinfection.
To investigate the kinetics of viral gene expression in Kasumi-3 cells, we analyzed genome-wide RNA expression at 4 and 24 h postinfection (hpi) by transcriptome sequencing (RNA-seq) (Fig. 1A). In the Kasumi-3 model, 20% to 50% of the cells become infected, and the majority of the cells are therefore uninfected. To obtain a pure population of infected cells, which would allow us to investigate the effect of the virus on both the host transcriptome and epigenome, additional studies were performed on cells that were sorted for GFP expression at 24 hpi (Fig. 1B). Our results show that in unsorted cells at 4 hpi, expression of the major immediate early genes UL122 and UL123 vastly exceeded that of other genes, as expected (Fig. 2). However, even at 4 hpi, low-level expression of nearly all viral open reading frames (ORFs) was detectable (see Table S1 in the supplemental material). By 24 hpi, the pattern of viral RNA expression shifted significantly (Fig. 2 and 3), with a striking downregulation of the two major immediate early genes. Interestingly, several other genes downstream of the major IE promoter (MIEP), such as the 5-kb intron RNA (57), UL119, UL120, UL121, and UL124 (Fig. 3; Tables S1 and S2) were downregulated, and there was a correlation between distance from the MIEP and the fold change in expression. Very little expression of UL127, which is located upstream of the MIEP, was observed, consistent with the presence of the previously described insulator sequence between the MIEP and this ORF (58–60). In addition to the IE genes, UL16, UL23, UL37, UL91, UL100, UL116, UL134, UL137, US7, US33, and US34 were downregulated (see Table S2). Many early genes were expressed at relatively higher levels at 24 hpi (Table S2). Two early noncoding RNAs (ncRNAs), RNA2.7 and 1.2 (61), were the most prominent transcripts upregulated at this time (Fig. 2). However, expression of the viral late gene transactivators UL79, UL87, UL91, UL92, and UL95 (62–66) and of the late genes, such as UL32, UL75, UL99, and UL115 (62, 64, 67), was relatively low at this time. An exception was the late gene UL132 (68), which was expressed at relatively high levels at 24 hpi in Kasumi-3 cells. Changes in the relative expression of selected viral genes from the immediate early (UL122 and UL123), early (UL54), and late (UL32) phases of viral replication were confirmed by reverse transcriptase quantitative PCR (RT-qPCR) (Fig. 2C). Our results show that transcription of the viral genome was much broader than anticipated at very early times postinfection. There were changes in the expression of the transcripts between 4 and 24 hpi, including reduced expression of the IE genes and increased expression of the early genes.
FIG 2.

Expression of HCMV RNAs in infected Kasumi-3 cells at 4 and 24 hpi. (A) IGV (Integrative Genomics Viewer) of RNA-seq coverage of the entire HCMV genome at 4 and 24 hpi in infected Kasumi-3 cells. (B) Reduced-scale view of the RNA-seq coverage map to allow the visualization of all the expressed viral transcripts. Selected immediate early, early, early/late, and late genes are shown in yellow, green, pink, and blue, respectively. Kinetic classes are based on previous analyses of HCMV-infected fibroblasts (57, 61–64, 66, 67, 137–139). Three independent experiments are shown for each time. HCMV sequences were aligned to TB40/E clone TB40-BAC4 (EF999921.1), to which the GFP gene sequence was added. (C) RT-qPCR validation of changes in the relative abundance of selected viral genes between 4 and 24 hpi. Statistical significance was determined by one-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
FIG 3.

Temporal changes in the relative abundance of HCMV RNAs. (A) Volcano plot of changes in viral gene expression between 4 and 24 hpi. Significant regulated genes are shown in red (P value <0.05 and log2 fold change [FC] higher than 1 and less than −1). NS, not significant. (B) Heat map of differentially expressed genes. Values shown are Z scores of normalized reads, with a cutoff of −1 < log2 FC > 1 and adjusted P value of <0.05.
The previous analysis detected changes in the relative abundance of steady-state RNAs, which is the net result of synthesis and degradation of the RNA, and does not reveal temporal changes in activation of viral gene expression. In order to investigate these differences, we analyzed newly synthesized RNAs expressed between 24 and 28 hpi. This was accomplished by sorting GFP+ cells at 24 hpi, labeling the cells for 4 h with 5-ethynyl uridine, biotinylating the RNA, selecting the nascent transcripts with streptavidin beads, and performing RNA-seq analysis. A portion of the RNA was saved prior to bead selection for comparison of the total RNA population with the nascent RNA. Figure 4 shows tracks and heat maps of the nascent and total RNAs. As expected, many early genes involved in core replication functions, which are located in the central region of the genome, were enriched in the nascent RNA relative to total RNA at 24 hpi. These included genes involved in DNA replication (UL44, UL54, UL56, and UL57) as well as the late gene transactivators, UL79, UL87, UL91, UL92, and UL95. Interestingly, transcription of the 2.7- and 1.2-kb noncoding RNAs was reduced at this time relative to that of total RNA. We conclude from these results that, although many genes are expressed as early as 4 hpi (Table S1), there is a transition toward higher levels of transcription of early genes by 24 h after of infection in Kasumi-3 cells.
FIG 4.

HCMV genes are differentially expressed in the early stages of infection. (A) IGV tracks of nascent and total RNAs expressed in infected Kasumi-3 cells at 24 to 28 hpi. (B) Reduced-scale view of the RNA-seq coverage map to allow the visualization of all the expressed viral transcripts. (C). Heat map of the Z scores of the normalized counts of nascent and total RNAs expressed between 24 and 28 hpi with a cutoff of −1 < log2 FC > 1 and adjusted P value of <0.05.
Dynamic changes in the landscape of the viral epigenome occur at early times postinfection.
To investigate the regulation of HCMV gene expression, we analyzed occupancy of acetylated H3K27 (H3K27Ac) and trimethylated H3K27 (H3K27me3) on the viral genome at 4 and 24 hpi. These modifications were chosen for analysis because previous studies showed that (i) H3K27 can be modified with either activating acetylation or repressive methylation (69, 70), (ii) H3K27Ac is specifically localized to promoter and enhancer regions (71–73) and therefore can be used to study activation of gene expression, (iii) methylation of H3K27 has been shown to have a role in HCMV latency and reactivation (22, 25), (iv) repressive histone modifications can be detected on viral genomes at very early times postinfection (40, 41), and (v) there may be a switch from heterochromatic to euchromatic modifications as different classes of genes are activated (40, 41). In addition, we analyzed occupancy of polymerase II (Pol II) on the viral genome using an antibody that recognizes both phosphorylated and nonphosphorylated forms of the major Pol II subunit, Rpb1. These studies were performed in two to three independent experiments on both unsorted cells and GFP+ cells (Fig. 1A and B).
Pearson correlations showed a high degree of reproducibility among the replicates (Fig. 5A and B). Analysis of the tracks of representative cellular genes that are expressed (GAPDH), or not expressed (CD8A) in Kasumi-3 cells showed that, as expected, H3K27Ac, but not H3K27me3, was bound to genes that are expressed, while the opposite pattern was observed in genes that are not expressed (Fig. 5C). These results confirm the reproducibility and the validity of our analyses. At 4 hpi, we observed a major peak of Pol II bound to the MIEP (Fig. 6 and Fig. S1). However, consistent with our RNA-seq data, binding of Pol II was also apparent throughout the genome. At 24 hpi, we did not observe the peak at the MIEP, and Pol II binding was distributed uniformly throughout the viral genome.
FIG 5.
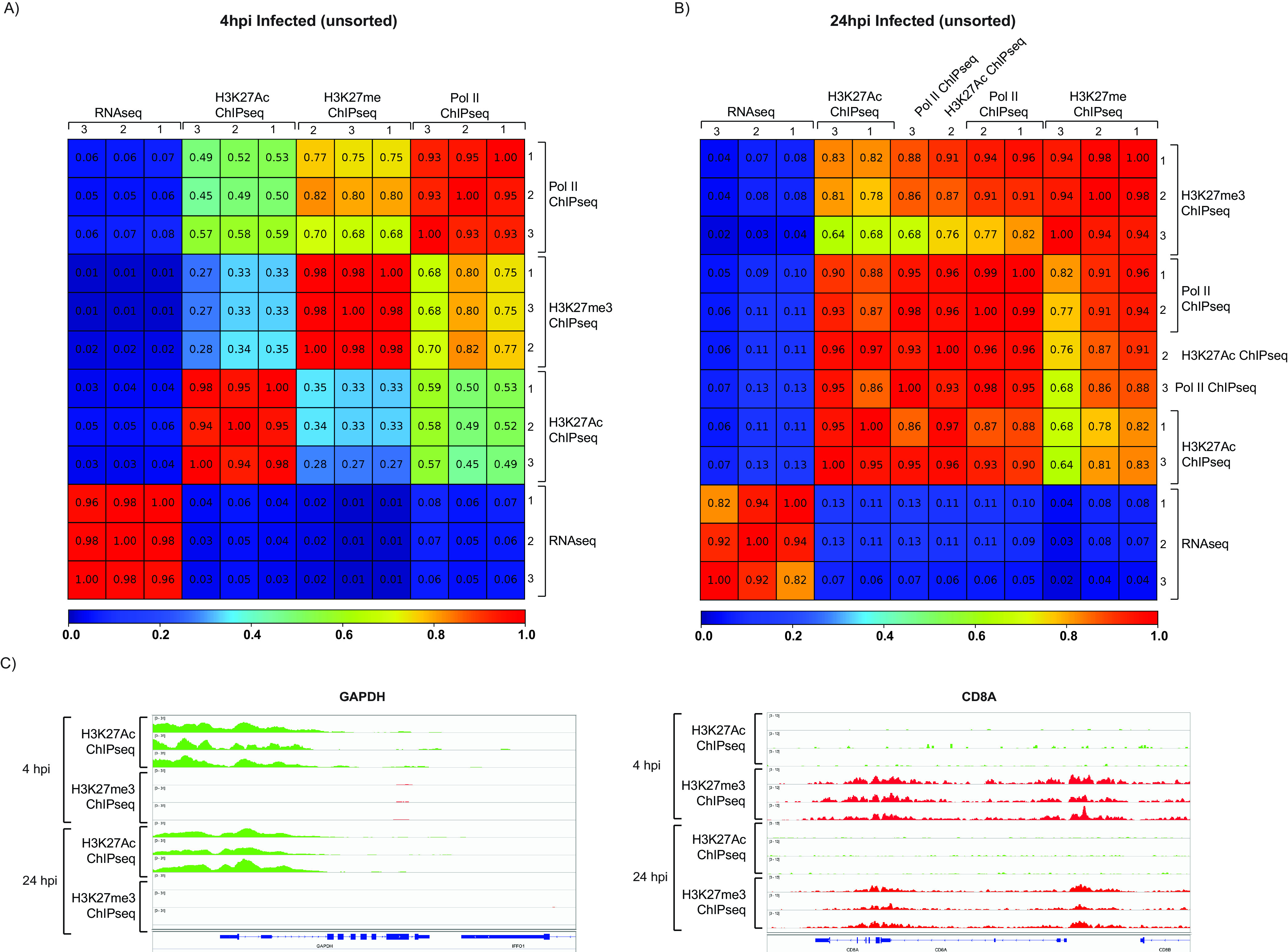
Validation of ChIP-seq analyses. Pearson correlation analyses between replicates of RNA-seq, Pol II ChIP-seq, H3K27Ac ChIP-seq, and H3K27me3 infected samples at 4 (A) and 24 (B) hpi. (C) H3K27Ac ChIP-seq (green) and H3K27me ChIP-seq (red) coverage maps for CD8A, which is not expressed, and GAPDH, which is transcriptionally active in HCMV-infected Kasumi-3 cells at 4 and 24 hpi. Tracks shown are from three independent experiments.
FIG 6.

Epigenetic landscape of the HCMV genome at 4 and 24 hpi. RNA-seq (purple), Pol II ChIP-seq (pink), H3K27Ac ChIP-seq (green), and H3K27me ChIP-seq (red) coverage maps for the entire HCMV genome (A) and zoom-in image for the MIEP (B) and oriLyt (C) regions in Kasumi-3 infected cells at 4 and 24 hpi. A representative example of three independent experiments is shown. (D) PCR-ChIP analyses of the MIEP and oriLyt regions at 4 and 24 hpi. Three biological replicates were analyzed.
Transcription of the major immediate early genes is initiated at multiple sites near the MIEP (74–76). Regulation of the upstream enhancer region through modification of histones is thought to be a major control point in determining lytic versus latent infection. At 4 hpi, we observed a major peak of H3K27Ac in the region of the MIEP and additional low-level binding throughout the genome (Fig. 6A and B and Fig. S1C). Thus, acetylation of H3K27 largely paralleled binding of Pol II to the MIEP at 4 hpi. At 24 hpi, the MIEP was still highly acetylated, despite a substantial reduction in Pol II occupancy and RNA expression. Interestingly, a new peak of H3K27Ac was observed in the oriLyt region at 24 hpi (Fig. 6A and C). Similar results were observed in cells that were sorted for GFP expression at 24 hpi (see Fig. S2). These results were not due to clustering of nucleosomes at specific regions, since H3 was dispersed throughout the genome (see Fig. S3).
H3K27me3 ChIP-seq analysis was performed to identify potential regions of transcriptional repression. A low level of H3K27me3 occupancy was observed throughout the viral genome at 4 hpi and 24 hpi. Increased methylation of H3K27 of the MIEP was observed in 2 of 3 replicates at 24 hpi (Fig. 6A and B and Fig. S1D). This coincided with decreased occupancy of Pol II and reduced expression of UL122 and UL123 transcripts. With the exception of the MIEP, H3K27me3-modified histones were not clustered to particular classes of promoters, and the pattern did not correlate with the level of RNA expression at either 4 or 24 hpi. Detection of H3K27me3 on the transcriptionally silent CD8A gene and lack of detection at the transcriptionally active GAPDH gene (Fig. 5C) served as controls to ensure the validity of the of H3K27me3 analysis. However, we cannot exclude the possibility that the detected H3K27me3 is due to background.
We then performed ChIP-PCR analyses with assays localized to the MIEP and oriLyt regions (Fig. 6D). These analyses confirmed that (i) the MIEP was H3K27-acetylated at 4 and 24 hpi, (ii) at 4 hpi, H3K27Ac occupancy was higher at MIEP than at oriLyt, (iii) at 24 hpi, H3K27Ac of MIEP and oriLyt were comparable in 2 of 3 experiments, and (iv) levels of H3K27me3 at the MIEP and oriLyt were similar at 4 hpi, but there was an increase in H3K27me3 at the MIEP in 2 of 3 replicates.
Collectively, our results suggest that acetylation of H3K27 at the MIEP and oriLyt is likely important in driving expression from these regions, while changes in modifications of H3K27 at other promoters do not play a major role in regulating viral gene expression.
Viral chromatin is open and accessible to cleavage with Tn5 transposase at 24 hpi.
Like cellular DNA, viral DNA is wrapped around nucleosomes composed of histones H2A, H2B, H3, and H4, which are compacted into higher-order structures through binding of the linker histone H1 and associated factors (33, 34). Highly compacted nucleosomes block access of RNA Pol II to the promoter and downstream regions and thus present a barrier to transcription (77). Previous studies showed that nucleosomes associated with viral genomes undergo dynamic changes over the course of infection (33, 34). We used an assay for transposase-accessible chromatin using sequencing (ATAC-seq) to identify open regions of the viral chromatin at 24 hpi. This assay uses Tn5 transposase to simultaneously cut accessible regions and tag them with sequencing adaptors. These regions can then be identified as peaks following amplification and genome-wide high-throughput sequencing (78). Because HCMV infection kills some of the cells, and the results of this assay are confounded by the presence of dead cells, we sorted for live GFP+ cells (Fig. 1B). There were some differences in the relative abundance of viral RNAs between the cells that were sorted for GFP expression versus unsorted cells (see Fig. S2A). However, these differences were not apparent in the occupancy of Pol II or H3K27Ac on the viral genome. Visualization of ATAC-seq tracks for cellular genes showed the expected sharp peaks of accessibility, which largely corresponded to the promoter regions of the genes. The GAPDH gene is shown as an example in Fig. 7B (additional genes are shown in Fig. 11). However, analysis of the viral chromatin showed a markedly different result, with a broad pattern of susceptibility to Tn5 (Fig. 7A). These results demonstrate that, in contrast to the cellular genome, the entire viral genome was open and accessible to the transposase at 24 hpi, when transcription of many viral genes was observed.
FIG 7.

HCMV chromatin is open and accessible to cleavage with Tn5 transposase in infected Kasumi-3 cells. IGV ATAC-seq coverage maps of the HCMV genome (A) and the cellular GAPDH gene (B) in GFP+ HCMV-infected cells at 1 dpi. GAPDH analysis serves as internal control for the quality of the ATAC-seq. Three independent biological replicates are shown.
Effect of HCMV infection on cellular gene expression.
We then analyzed the effect of HCMV infection on the host transcriptome by RNA-seq analysis. Because most cells remain uninfected in this system, we sorted the cells for GFP expression at 24 hpi to obtain a pure population of infected cells. Mock-infected cells were prepared in parallel and sorted for live cells (Fig. 1B). Duplicate samples showed a high degree of correlation (R2 = 0.88), validating the reproducibility of our results (Fig. 8A). We found that 1,551 genes were differentially expressed (adjusted P value <0.05, −1 < log2 fold change [FC] > 1), with 1,061 genes significantly upregulated and 490 genes downregulated (Fig. 8B).
FIG 8.

Changes in cellular gene expression induced by HCMV infection at 24 hpi. (A) Pearson correlation analyses between replicates of RNA-seq, Pol II ChIP-seq, H3K27Ac ChIP-seq, and H3K27me3 ChIP-seq from mock-infected and TB40/Ewt-GFP-infected sorted cells at 24 hpi. (B) Volcano plot showing differential gene expression between mock-infected and GFP+-infected Kasumi-3 cells at 1 dpi. Vertical lines indicate a log2 fold change (FC) of 1 and −1. (C) DAVID functional GO analysis of biological processes (BP) enrichment for downregulated and upregulated genes in HCMV-infected cells compared to mock-infected cells (adjusted P value < 0.05, −1 < log2 FC > 1). (D) RT-qPCR validation of changes in cellular gene expression induced by HCMV infection. RNAs from HCMV-infected (+) or mock-infected Kasumi-3 cells (−) were analyzed for the indicated cellular genes. Statistical significance was determined by one-way ANOVA with Tukey’s multiple-comparison test. *, P < 0.05; **, P < 0.01.
Next, we performed a pathway analysis to determine functional pathways that were significantly changed by HCMV infection. As expected, many differentially expressed genes belonged to host immune response pathways (Fig. 8C). Indeed, genes involved in antiviral defense response, including type I interferon-responsive genes, were highly upregulated. RT-qPCR was used to validate the upregulation of two interferon-responsive genes, IFIT1 and IFIT3 (Fig. 8D). HCMV infection differentially targeted genes involved in antigen presentation by major histocompatibility complex (MHC) class I and class II genes. Genes encoding MHC class I HLA-A, -B, -C, -F, and -H, which are expressed on the surfaces of all cell types and are recognized by cytotoxic CD8+ T cells, were upregulated, while genes involved in MHC class II antigen presentation, which are expressed primarily on antigen-presenting cells and are recognized by CD4+ helper T cells, were downregulated. Among these downregulated genes were the class II MHC genes for HLA-DQB1, HLA-DRB1, HLA-DMB, HLA-DMA, HLA-DRB5, HLA-DPB1, HLA-DPA1, HLA-DRA, and the MHC class II chaperone CD74 as well as for the CIITA transactivator, which regulates transcription of the MHC genes. Interestingly, the interferon gamma signaling pathway, which leads to induction of MHC gene expression, was also differentially affected, with upregulation of some genes and downregulation of others. Previous studies have shown that HCMV downregulates both class I and class II MHC antigen presentation pathways through targeted degradation of MHC proteins, blockade of peptide processing, and interference with the transport of peptide-loaded complexes to the cell surface (79, 80). Our studies confirm previous findings reporting that HCMV also blocks class II antigen presentation in myeloid cells at the level of RNA expression (19).
In addition to its effect on the immune response, HCMV infection modulated apoptotic processes both by inducing expression of genes involved in the positive regulation of apoptosis and by downregulating genes involved in the negative regulation of apoptosis. We also found that the infection upregulated genes involved in actin reorganization. Among the genes downregulated by the infection are many genes involved in the regulation of transcription, hemopoiesis, leukocyte migration, signal transduction, mitogen-activated protein kinase (MAPK) signaling, protein phosphorylation, response to cytokines, and cellular proliferation. RT-qPCR was used to validate downregulation of two of these genes, CISH (cytokine-inducible SH2 containing protein) and IL17RB, a receptor for the cytokine IL-17 (Fig. 8D).
These analyses were performed using steady-state RNA from infected and mock-infected cells, where the abundance of the RNA is the net result of de novo transcription and RNA degradation. In order to identify changes in gene expression that were due to changes in transcription alone, we analyzed the expression of newly synthesized RNA, as described above. Mock-infected cells were analyzed in parallel by sorting for live cells and then labeling the RNA. For both populations, a portion of the RNA was saved for analysis of total RNA. Thus, there were four populations: (i) nascent RNA from mock-infected cells, (ii) total RNA from mock-infected cells, (iii) nascent RNA from infected cells in which 100% of the cells were infected, and (iv) total RNA from this population of infected cells. These analyses were performed on two independent replicates. By comparing infected cells and mock-infected cells, we identified differentially expressed genes in both the nascent and steady-state RNA populations. In the total, steady-state RNA, ∼82% of the differentially expressed genes were upregulated in response to infection and ∼17% were downregulated. In contrast, nearly 50% of differentially expressed genes in the nascent RNA were downregulated (Fig. 9A and B). We conclude from these results that there is a significant loss of new transcription in infected cells.
FIG 9.

Effect of HCMV infection on de novo transcription of cellular genes. (A) Percentage of genes that were up- or downregulated at 24 hpi in total (steady state) and nascent RNAs from HCMV-infected versus mock-infected Kasumi-3 cells at 24 hpi, as determined by differential gene expression analysis (P < 0.05). (B) Heat maps of differentially expressed genes in total or nascent RNAs from mock-infected or infected cells. Values were derived from the Z scores of the normalized reads. (C) DAVID functional GO analysis of BP enrichment for upregulated and downregulated genes in nascent RNA from HCMV-infected cells compared to that in mock-infected cells.
We then performed pathway analysis on the differentially expressed genes in the nascent RNA (Fig. 9C). Similar to our previous analysis of steady-state RNA, the genes that were newly transcribed in response to infection were dominated by interferon signaling and a defense response to viral infection. Differences between the nascent and steady-state RNAs were more apparent in the genes that were downregulated in response to infection. Nascent RNAs involved in DNA replication and repair, G1/S cell cycle control, ribosomal processes, including translation, and rRNA processing were downregulated as well as genes involved in hemopoiesis, leukocyte migration, regulation of transcription, and apoptosis, which were also observed in the steady-state RNA analysis (Fig. 9C).
Viral infection dysregulates promoter-proximal binding of RNA polymerase II and H3K27 acetylation of cellular promoters and enhancers without significantly altering chromatin accessibility.
Transcription is regulated at multiple levels, including chromatin accessibility, histone modification, binding of the RNA polymerase II preinitiation complex, pausing of the polymerase, and elongation (69, 70, 77, 81). To identify changes in the cellular epigenome that were associated with changes in de novo transcription, we analyzed occupancy of Pol II, H3K27Ac, and H3K27me3 to cellular genes and chromatin accessibility in GFP+-sorted infected and in mock-infected Kasumi-3 cells sorted for live cells at 24 hpi (Fig. 1B). These analyses were conducted in duplicates or triplicates, with replicates showing a high degree of correlation (Fig. 8A). We performed metagene and heat map analyses to detect changes in occupancy of Pol II, H3K27Ac, and H3K27me3 at gene promoters (Fig. 10). These analyses revealed genome-wide loss of Pol II and H3K27Ac and significant changes in the pattern of binding of these factors to the cellular genome at 24 hpi. These differences were not due to differences in the number of reads, which were actually higher in the samples from infected cells (see Table S3). In contrast to H3K27Ac, there was a slight increase in binding of H3K27me3 to cellular promoters in HCMV-infected cells (Fig. 10A).
FIG 10.

Effect of HCMV infection on occupancy of Pol II and H3K27 at cellular promoters. (A) Metagene analyses of ChIP-seq data of Pol II, H3K27me3, and H3K27Ac promoter occupancy and H3K27Ac enhancer occupancy in HCMV-infected or mock-infected Kasumi-3 cells at 1 dpi. (B) Heat maps of Pol II, H3K27Ac, and H3K4me3 occupancy at promoter regions. TSS, transcription start site.
We then analyzed epigenetic changes in individual genes that were up- or downregulated in response to infection using Integrative Genomics Viewer (IGV) to visualize BigWig files (Fig. 11). These genes were selected from DAVID analysis of differentially expressed genes in the nascent RNAs from infected and mock-infected cells (Fig. 9). PCNA, RPL13, CD34, and MYC and NFATC2 were selected as representative genes that were downregulated in pathways involved in DNA replication, translation, hemopoiesis, and transcription, respectively (Fig. 11A and C). For all of these genes, loss of transcription was associated not only with loss of Pol II but also with loss of H3K27Ac. In contrast, there was very little change in trimethylation of H3K27 or in chromatin accessibility.
FIG 11.

Epigenetic profiles of selected cellular genes with changes in de novo transcription in response to HCMV infection. Reads per kilobase per million (RPKM) of selected genes whose de novo expression is significantly decreased (A) or increased (B) in infected versus mock-infected Kasumi-3 cells (P < 0.05). Bars show mean values plus standard deviations. Coverage maps of selected genes whose expression is decreased (C) or increased (D) in infected versus mock-infected Kasumi-3 cells. Pol II ChIP-seq (pink), H3K27Ac ChIP-seq (green), H3K27me3 ChIP-seq (red), and ATAC-seq (blue). Tracks shown are representative of two to three independent experiments.
To analyze changes in genes that were upregulated in response to infection, we examined virus defense response genes, including DHX58, RNASEL, and MX2, and the antigen presentation genes HLA-B and TAP1 (Fig. 11 B, D). Promoter-proximal occupancy of Pol II was also reduced by infection in these genes. In contrast to genes that were downregulated, the promoters of these genes retained H3K27Ac in infected cells. There were no significant changes in H3K27me3 or chromatin accessibility in response to infection. Although further confirmation is needed, our results suggest that increased expression of these genes was likely due to release of Pol II from pausing.
We performed differential peak analysis to further investigate epigenetic changes in cellular genes induced by HCMV infection. These changes were characterized according to their gene annotation, gene type, functional pathways, and correlations (Fig. 12 and 13). ATAC-seq analysis revealed that there were very few changes in chromatin accessibility. Of the >100,000 peaks observed, only 88 were significantly different between infected and mock-infected cells. Most of these localized to intergenic regions and introns (Fig. 12A), and changes in chromatin accessibility did not correlate with changes in gene expression.
FIG 12.

Localization of changes in the cellular epigenome induced by HCMV infection. (A) Gene annotation of changes in chromatin accessibility, Pol II, and H3K27Ac and H3K27me3 occupancy derived from differential peak analyses performed on genes in mock-infected versus HCMV-infected Kasumi-3 cells at 24 hpi (adjusted P value < 0.05 and −0.5 < log2 FC > 0.5). (B) Identification of gene type changes in Pol II, H3K27Ac, and H3K27me3 occupancy on entire genes and promoter-proximal regions in mock-infected and HCMV-infected Kasumi-3 cells at 24 hpi (adjusted P value < 0.05 and −0.5 < log2 FC > 0.5). (C). Venn diagrams showing correlations between changes in H3K27 versus Pol II and H3K27Ac versus H3K27me3.
FIG 13.

Pathway analysis of the effect of HCMV infection on occupancy of Pol II and H3K27 at cellular promoters. DAVID functional GO analysis of BP enrichment conducted on genes with increased and decreased Pol II (A), H3K27Ac (B), and H3K27me3 occupancy at the promoter regions of HCMV-infected cells compared to that of mock-infected cells (adjusted P value < 0.05, −0.5 < log2 FC > 0.5).
Most of the changes in Pol II occupancy occurred at promoter regions of protein-coding genes, while ∼24% of the changes occurred in introns and ∼14% were in the intergenic regions (Fig. 12B). There was a high correlation between changes in Pol II and H3K27Ac (Fig. 8A). Loss of promoter-proximal Pol II and H3K27Ac was observed at 734 and 1,984 peaks, respectively (adjusted P value <0.05 and −0.5 < log2 FC > 0.5) (Fig. 12C, top right). Pathways associated with loss of Pol II included those involved in translation, RNA processing, transcription, rRNA processing, and biogenesis (Fig. 13). Loss of promoter-proximal H3K27Ac was associated with genes involved in transcription and its regulation, DNA repair, and cellular response to DNA damage, among others (Fig. 13). We found 283 genes with reduced occupancy of both Pol II and H3K27Ac (Fig. 12C, top right), of which 49 were involved in transcription.
Increases in promoter-proximal Pol II were observed at 1,597 genes (Fig. 12C), and these were involved in regulation of transcription, cell cycle, apoptosis, and cell-cell adhesion (Fig. 13). Increased occupancy of Pol II coincided with increased promoter-proximal H3K27Ac for some genes and loss of H3K27Ac at others (Fig. 12C, top left and middle). Similarly, increases in Pol II were associated with increased transcription in some cases (e.g., RHOB and KLF2), and decreased transcription in others (e.g., RUNX1, SH2B3, TGFB1, and FHL3) (Fig. 14). An increase in Pol II associated with downregulation of transcription may be due to increased pausing of the polymerase.
FIG 14.

Increased occupancy of Pol II at promoter-proximal regions is associated with both upregulation and downregulation of gene expression. RNA-seq (purple) and Pol II ChIP-seq (pink) coverage maps for RHOB (A), KLF2 (B), RUNX1 (C), SH2B3 (D), TGFB1 (E), and FHL3 (F). Tracks shown are representative of two to three independent experiments.
Increases in H3K27Ac were observed at 426 genes (Fig. 12C), and, like the nascent RNAs that were upregulated in response to infection, many of these were associated with pathways involved in interferon signaling and the immune defense response (Fig. 13B). Most changes in H3K27Ac occurred in introns and intergenic regions rather than at the promoter regions (Fig. 12A). Since H3K27Ac is specifically localized to enhancers and promoters, the other changes in H3K27Ac we observed were most likely localized to enhancer regions, which also showed loss of H3K27Ac (Fig. 10A).
We observed increased trimethylation of H3K27 at 773 peaks and loss of H3K27me3 at 214 peaks (Fig. 12C, bottom), and there was little correlation with changes in H3K27Ac (Fig. 8A). Very few changes in H3K27me3 occurred at promoter regions (Fig. 12A), and there were only 29 promoters in which upregulation of acetylation occurred concurrently with a decrease in methylation and 22 genes where a decrease in acetylation corresponded to an increase in methylation (Fig. 12C, bottom). Changes in H3K27me3 occurred at a higher proportion of intergenic regions (Fig. 12A) and noncoding RNAs than H3K27Ac (Fig. 12B).
The observations that (i) methylation and acetylation of H3K27 occur at different regions and do not correlate well with each other (Fig. 8, 10, and 12), (ii) there was little overlap in the pathways affected by changes in H3K27Ac and H3K27me3 (Fig. 13), and (iii) very few genes showed concomitant changes in modification of H3K27 (Fig. 12C, bottom) suggest that a switch in modifications of promoter-proximal H3K27 is unlikely to play a major role in regulating the cellular response to HCMV infection.
Expression and phosphorylation of Pol II are not affected by HCMV infection.
RNA Pol II activity is regulated in part by posttranslational modifications. To investigate whether HCMV-induced dysregulation of promoter-proximal Pol II could be due to changes in Pol II protein expression or its phosphorylation status, we performed Western blot analysis of the major Pol II subunit, Rpb1, as well as the Ser5 and Ser2 phosphorylated forms of Rpb1, which are present on paused and elongating Pol II, respectively (Fig. 15). We found that neither total Pol II nor its phosphorylated forms were reduced in HCMV-infected Kasumi-3 cells at 24 hpi.
FIG 15.

Effect of HCMV infection on Pol II protein isoforms. Kasumi-3 cells were infected with TB40/Ewt-GFP, and at 1 dpi, GFP-expressing cells were purified by flow cytometry. (A) Lysates were used for Western blot (WB) analysis against Rpb1, phospho (p)-Ser2 Rpb1, and p-Ser5 Rpb1. Cyclophilin B was used as a loading control (B). Quantification of protein expression levels from three independent experiments. Error bars represent standard errors of the means (SEMs). P value <0.05 by one-tailed Student’s pairwise t test.
In summary, our analyses of the cellular epigenome show that HCMV infection induces a loss of de novo transcription. This was associated primarily with reduced occupancy of Pol II and H3K27Ac at the promoter regions of genes, rather than changes in phosphorylation of Pol II, accessibility of the chromatin, or a switch between acetylation and methylation of H3K27.
However, HCMV infection also induced expression of some genes, particularly those involved in the host defense response. Increased transcription was sometimes associated with loss of promoter-proximal Pol II, but for these genes, Pol II was released from pausing to produce full-length transcripts, and acetylation of promoter-proximal H3K27Ac was increased or maintained.
DISCUSSION
Previous studies have shown that HCMV genomes are heterochromatinized in latency models and that histones with repressive modifications are recruited onto the viral genome in the early stages of infection in permissive fibroblasts (10, 20–22, 53). This has led to the suggestion that HCMV latency is established in myeloid cells at the outset of infection through transcriptional silencing of most viral genes involved in lytic replication, due to recognition of incoming viral genomes as pathogen-associated molecular patterns (PAMPs) by the host intrinsic immune response (reviewed in references 82 and 83). While permissive cells are able to overcome this repression to activate the lytic cycle, this was thought not to occur in nonpermissive cells, such as myeloid progenitors, until they received signals from differentiation or inflammatory mediators that trigger epigenetic reprogramming of histones bound to the MIEP (reviewed in references 8 and 83).
However, more recent studies using sensitive new technologies that interrogate the entire transcriptome have shown widespread activation of viral gene expression in various models of latency, including primary CD34+ cells and monocytes (17, 18, 56, 84). These observations are not consistent with a persistence of heterochromatic marks on viral genomes following initial infection. We previously demonstrated that, although latency can be established by ∼14 days postinfection (dpi) in Kasumi-3 cells, activation of lytic gene expression precedes the establishment of latency (7).
Here, we have used a multi-omics approach to investigate temporal changes in the viral transcriptome and epigenomes over the course of infection in Kasumi-3 cells. Our initial goal was to characterize these events from the initial infection to the establishment of latency. However, there is a significant loss of viral genomes over the course of infection, both in primary myeloid cells and in Kasumi-3 cells (7, 12, 85). The resulting very low copy number precluded ChIP-seq analysis of the latent HCMV epigenome, and we therefore focused on the early events during infection of Kasumi-3 cells. We show that activation of the lytic cycle in infected Kasumi-3 cells follows a pattern of temporal activation of gene expression similar to that observed in permissive cells. Consistent with previous studies in fibroblasts, we found that at 4 hpi, RNA expression, Pol II binding, and H3K27Ac were all highest in the IE region. These results are consistent with previous studies showing that high levels of histone acetylation have an important role in activating viral gene expression (20, 21, 23, 40–42, 86, 87). However, we were surprised to find binding of Pol II and acetylation of H3K27 throughout the genome and expression of many viral RNAs, even at this early point in infection. Our results are in accordance with those of a recent study which identified transcripts initiated at start regions distributed throughout the viral genome at 4 hpi in infected fibroblasts (88).
Similar to previous analyses of infected fibroblasts which showed binding of repressive H3K9me2 to viral genes at very early times postinfection (40, 41), we observed binding of H3K27me3 to the viral genome in Kasumi-3 cells at 4 hpi, and we observed increased occupancy of H3K27me3 at the MIEP at 24 hpi, when IE gene expression was repressed. Acetylation and methylation of H3K27 are mutually exclusive. Therefore, detection of both H3K27Ac and H3K27me3 could be due to the presence of mixed populations of transcriptionally active or repressed genomes. Genomes with activating modifications may be destined to go on to lytic infection, while those with repressive modifications could be a reservoir for latency. Alternatively, there may be bivalent genomes with repressive and activating modifications on different residues in the same genome. High levels of acetylation at the enhancer regions may override repression mediated by H3K27me3 distributed throughout the genome. Finally, we cannot exclude the possibility that the H3K27me3 signal we observed on the viral genome is simply due to background. Because we observed similar levels of H3K27me3 occupancy in both cells sorted for GFP expression and those that were unsorted (see Fig. S2D in the supplemental material), we favor one of the latter explanations. Further studies, including single-cell analyses, will be required to distinguish these possibilities and to resolve the question of whether latency can be established at the outset of infection.
At 24 hpi, the patterns of RNA expression, Pol II binding, and H3K27Ac occupancy had also shifted. While the MIEP was still highly acetylated, binding of Pol II to MIEP and the abundance of the IE transcripts were reduced, as previously shown in infected fibroblasts. Previous studies have shown that MIEP activity is downregulated through acquisition of histones with repressive histone modifications (23, 40, 41). Consistent with this, we observed an increase in H3K27me3 in 2 of 3 replicates at 24 hpi. Variability in detection of H3K27me3 at the MIEP may reflect variation in the timing of transition from activation to repression of the MIEP.
Surprisingly, a number of other genes in the vicinity of the MIEP were also lower in abundance at 24 hpi. Although further studies are needed to investigate the regulation of these genes, our results suggest that the MIEP may control expression of a larger array of genes than previously recognized. The observation that the 5-kb noncoding IE RNA was downregulated in Kasumi-3 cells but not in fibroblasts (57) adds to the mystery surrounding this transcript.
Many viral genes, which fell primarily in the category of the early genes, were upregulated at 24 hpi relative to that at 4 hpi. Although most viral genes are expressed as early as 4 hpi, analysis of nascent RNAs expressed 24 to 28 hpi showed that there is also enhanced transcription of many genes during this phase of infection in Kasumi-3 cells. These included early genes involved in viral DNA replication as well as the late gene transcriptional activators.
In addition, our ATAC-seq analysis demonstrated that the entire viral genome was open and accessible to cleavage by Tn5 transposase in Kasumi-3 cells 24 hpi, and, consistent with this observation, binding of Pol II was observed throughout the genome. Previous studies have shown that viral genomes have low nucleosome occupancy during lytic infection of fibroblasts (33, 34, 37).
oriLyt may encode a second HCMV enhancer that is activated in the early phase of infection.
We observed a major switch in modification of H3K27 in the oriLyt region from hypoacetylation to hyperacetylation between 4 and 24 hpi, which correlated with enhanced transcription of early genes in the neighboring central region of the genome. oriLyt is a complex region with two major functional domains required for CMV replication, essential region I (ER1) and essential region II (ERII). ER1 contains a bidirectional promoter with binding sites for viral proteins pUL84 and IE2 and the cellular transcription factors CEBPα/β, ATF-CREB, and Sp1 (89). The oriLyt promoter drives expression of a highly abundant 4.9-kb noncoding RNA, which is expressed in both fibroblasts and CD14+ monocytes and regulates viral DNA replication (17, 90, 91). Thus, oriLyt shares many of the features of an enhancer. This putative enhancer is activated in the early phase of infection, and we speculate that it may have a role in activating early gene expression. Despite high-level acetylation of H3K27 at the oriLyt, expression of the 4.9-kb ncRNA encoded by this region was relatively low in Kasumi-3 cells at this time.
The mechanisms regulating temporal changes in expression of viral genes requires further study. Transcription of cellular genes occurs through a complex series of steps, including binding of RNA Pol II to nucleosome-depleted promoter regions, initiation of short transcripts, pausing of the polymerase, and release from pausing into the body of the gene (81). Previous studies of HCMV infection of fibroblasts have shown that Pol II pausing and elongin B-mediated release from pausing play an important role in regulating HCMV gene expression (88, 92). Additional studies have implicated the FACT complex, which is involved in nucleosome remodeling, in regulation of IE gene expression (93). Our studies suggest that (i) changes in the acetylation state of H3K27 at enhancer regions, rather than changes in accessibility of viral chromatin or a switch in promoter-proximal modification of H3K27 from methylation to acetylation, and (ii) factors that govern transcriptional elongation, rather than Pol II binding, are primarily responsible for the transition from immediate early to early gene expression in Kasumi-3 cells.
In summary, our studies show that (i) transcription of the viral genome is broadly activated early in infection, (ii) the genome is decorated with acetylated H3K27, first at the MIEP and then at oriLyt, (iii) H3K27 methylation occurs throughout the genome at 4 and 24 hpi and was generally increased at the MIEP at 24 hpi, and (iv) there is low nucleosome occupancy of the viral genome at early times postinfection. Collectively, these results suggest that chromatin changes on the HCMV genome in the early phase of latent infection resemble those that occur during lytic infection.
Previous studies have shown that Kaposi’s sarcoma-associated herpesvirus (KSHV) latency is established following a prelatency phase characterized by expression of some viral genes and genome-wide deposition of activating histone marks H3K4me3 and H3K27Ac (94). These marks are gradually replaced by heterochromatic modifications between 24 and 72 hpi in parallel with loss of viral gene expression. Further studies will be required to determine whether the HCMV epigenome undergoes a similar transition during the establishment of latency and to identify the factors that mediate heterochromatinization of viral genomes.
HCMV induces loss of de novo transcription and dysregulation of promoter-proximal Pol II and acetylation of H3K27.
Like other herpesviruses, HCMV relies on host proteins for many aspects of its gene expression, including transcription, splicing, nuclear export, and translation of viral RNAs by the ribosomes. However, herpesviruses manipulate these processes in myriad ways to favor expression of viral genes over host genes. Herpes simplex virus (HSV), a member of the alphaherpesvirus family, induces a rapid shutoff of host gene expression to specifically favor synthesis of viral RNAs by reducing promoter-proximal occupancy of Pol II at cellular genes (95, 96) and disruption of transcript termination (97, 98).
Gammaherpesviruses, including Kaposi’s sarcoma-associated herpesvirus (KSHV) and murine herpesvirus 68 (MHV-68), also specifically disrupt transcription of host cell genes by hijacking Pol II onto viral genes (99, 100). Viral endonuclease-induced degradation of host cell transcripts contributes to this process, in part through loss of Pol II subunits and TFIIB in the nucleus, while transcription of viral genes is spared through sequestration of the polymerase into viral replication compartments (100).
HCMV, a betaherpesvirus, also manipulates the host cell environment to facilitate viral gene expression. Previous studies have elucidated the ways in which HCMV manipulates cellular RNA expression, transport of mRNA out of the nucleus, and translation of mRNAs on the ribosomes (15, 19, 101–110). Although HCMV does not encode an RNA endonuclease for rapid shutoff of host gene expression, previous studies have shown downregulation of some cellular genes in response to infection (107, 111). Here, we show that HCMV profoundly dysregulates cellular transcription, recruitment of Pol II, and acetylation of H3K27 at cellular promoters. This results in loss of transcription of many genes, especially those involved in DNA replication, cell cycle control, DNA damage and repair, hemopoiesis, transcription, rRNA processing, and mRNA translation. Previous studies have shown the HCMV dysregulates the cell cycle, and that IE2 is required to block cell cycle progression at the G1/S transition (112, 113). Our studies suggest that the effect of HCMV on expression of genes that regulate the cell cycle is broader than previously appreciated.
Loss of de novo transcription was often associated with loss of H3K27Ac as well as loss of Pol II. These effects were not due to changes in accessibility of the chromatin or to an increase in methylation of H3K27. Rather, loss of Pol II and H3K27Ac bound to cellular genes occurred concomitantly with gain of Pol II/H3K27Ac on the viral genome at 24 hpi. These observations suggest that the virus encodes a factor(s) that sequesters the polymerase and/or histone modifying enzymes onto viral genomes in order to facilitate transcription of viral genes at the expense of the host. In contrast to MHV-68, this did not occur because of loss of Pol II protein expression at this early time postinfection (100). Interestingly, immune response genes, which were upregulated in response to HCMV infection, were spared from this loss of Pol II and H3K27Ac. We speculate that the host has developed a means to counteract hijacking of Pol II as part of the defense response to HCMV infection.
How does the virus commandeer host Pol II onto viral genes? The Pol II complex binds to transcription start sites in host genes through binding of TATA-binding protein (TBP) to TATA sequences in the promoter region. Gammaherpesviruses encode TBP-like proteins that activate late gene expression from noncanonical TATT promoter sequences in the late phase of infection, including EBV BcRF1 and MHV-68 ORF24 (114). HCMV late genes are also regulated by noncanonical TATT promoters, and based on its structural similarity to other herpesvirus proteins, HCMV pUL87 is thought to be a TBP-like protein that recruits Pol II to late promoters (88, 115). We speculate that a viral DNA-binding protein may also associate with the Pol II transcription apparatus at early times postinfection to redirect it to early gene promoters. It has long been known that IE2 binds to early gene promoters and interacts with a number of cellular transcriptional regulators, including AP1, CREB1, EGR1, SP1, p53, Rb, TAF4, TBP, TFIIB, and TFIID, chromatin assembly factor 1 (CAF1), and the histone acetyltransferase P/CAF (37, 116–126). Recent studies show that IE2 also drives expression of a subset of early/late and late genes that lack the TATT sequence (127). Although this study showed that IE2 did not affect cellular gene expression at late times, it is tempting to speculate that IE2 may have a role in sequestering Pol II onto viral genes at early times postinfection. Further studies are needed to investigate this question.
MATERIALS AND METHODS
Cells and virus stocks.
Kasumi-3 cells were obtained from ATCC and maintained in RPMI 1640 medium (ATCC 30-2001) supplemented with 20% fetal bovine serum (Corning) and 1,000 U/ml penicillin-streptomycin (Gibco). TB40/Ewt-GFP (128) viral stocks were produced and the titers were determined as previously described (7).
Antibodies and other reagents.
Rpb1 N-terminal domain (NTD) (D8L4Y) rabbit monoclonal antibody (MAb) 14958, phospho-Rpb1 C-terminal domain (CTD) (Ser 2) (E1Z3G) rabbit MAb 13499, phospho-Rpb1 CTD (Ser 5) (D9N5I) rabbit MAb 13523, trimethyl-histone H3 (Lys27) (C36B11) rabbit MAb 9733, and acetyl-histone H3 (Lys27) (D5E4) XP rabbit MAb 8173 were obtained by Cell Signaling Technologies. Cyclophilin B polyclonal antibody (PA1-027A) was obtained from Invitrogen, and the protein A/G PLUS-agarose (sc-2003) was purchased from Santa Cruz Biotechnology.
Western blotting.
Cells were washed with phosphate-buffered saline (PBS) and lysed with 50 mM Tris, pH 8, 150 mM NaCl, 5 mM EDTA, 0.1% SDS, 1% IGEPAL (NP-40), 0.1% Triton, 0.5% sodium deoxycholate, 1 mM dithiothreitol (DTT), cOmplete EDTA-free protease inhibitors (Roche), and PhosSTOP phosphatase inhibitor cocktail (Roche) for 30 min on ice. Lysates were then cleared at 20,000 × g for 30 min at 4°C, and protein concentrations were determined by a bicinchoninic acid (BCA) protein assay kit (Pierce). Ten micrograms of extracts per sample was run on a 4 to 20% Mini-Protean TGX stain-free gel (Bio-Rad), and the proteins were transferred to a polyvinylidene difluoride (PVDF) transfer membrane (Thermo Scientific). Membranes were blocked with Tris-buffered saline (TBS), 0.1% Tween, and 5% bovine serum albumin (BSA), and probed with the primary antibodies overnight at 4°C and with the IRDye 800CW goat anti-rabbit secondary antibody (Li-Cor Biosciences) for 1 h at room temperature. Images were acquired and quantified with the Odyssey Fc imaging system (Li-Cor). Quantification was performed on three independent biological replicates.
Infection of Kasumi-3 cells.
Kasumi-3 cells were infected with TB40/Ewt-GFP at a multiplicity of infection (MOI) of 1 as previously reported (7). Extracellular virus was inactivated at the time of harvest (4 or 24 hpi) (129), and when required, GFP-expressing cells were sorted on a FACSAria-6 laser cell sorter (BD Biosciences). Mock-infected cells were prepared in parallel with infected cells, without addition of virus. For experiments with sorted cells, mock-infected cells were sorted in parallel for live cells.
RNA extraction and RNA-seq.
At the defined times, mock-infected and infected Kasumi-3 cells (sorted or not) were collected and lysed in TRIzol. Total RNA was prepared using the Direct-zol RNA miniprep kit (Zymo Research) with an on-column DNase digestion step according to the manufacturer’s instructions. RNA was rRNA depleted with Ribo-zero (Illumina), and RNA-seq libraries were prepared with TrueSeq stranded mRNA (Illumina). RNA-seq libraries were sequenced on the Illumina NextSeq 500 sequencing system (50-bp single-end reads, 50 million reads/sample). RNA-seq analysis was conducted on three independent biological replicates.
RT-qPCR analysis.
Total RNA was reverse transcribed with the ABI high-capacity cDNA reverse transcription kit (Thermo Fisher), and relative mRNA expression was analyzed by the comparative threshold cycle (2−ΔΔCT) method. For HCMV UL122, UL123, UL54, and UL32, we used the TaqMan assays previously validated (7). For CISH and IL17R, we used TaqMan gene expression assays obtained from Life Technologies (hs 00367082 and hs 00218889, respectively) and a SYBR green-based real-time PCR assay for IFIT1 (forward, 5′-GGAATACACAACCTACTAGCC-3′, and reverse, 5′-CCAGGTCACCAGACTCCTCA-3′) and IFIT3 (forward, 5′-AAAAGCCCAACAACCCAGAAT-3′). GAPDH was used as normalization control (Life Technologies hs99999905 for TaqMan assays, and 5-ACCCACTCCTCCACCTTTGAC-3′ [forward] and 5′-CTGTTGCTGTAGCCAAATTCGT-3′ [reverse] for SYBR green).
RNA-seq analysis.
The quality of DNA reads, in FASTQ format, was evaluated using FastQC. Reads were trimmed to remove Illumina adapters from the 3′ ends using a cutadapt wrapper, Trimgalore (130) (www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Trimmed reads were aligned to the human genome (hg19) or HCMV TB40/E clone TB40-BAC4 (EF999921.1), to which the GFP gene sequence was added, using STAR (131). Read counts for each gene were calculated using the -quantMode gene counts option in STAR. Normalization and differential expression were calculated using DESeq2 (132). The cutoff for determining significantly differentially expressed genes was a false-discovery rate (FDR)-adjusted P value of less than 0.05. Pathway analyses were performed using DAVID to identify significant pathways among the differentially expressed genes.
Nascent RNA-seq analysis.
Twenty-four hours postinfection, sorted mock-infected and infected Kasumi-3 cells were resuspended at 500,000 cells/ml in the culture medium and treated with 200 μM 5-ethynyl uridine (EU) for 4 h at 37°C to label the newly synthesized RNAs. Cells were collected and resuspended in TRIzol. RNAs were prepared with the Direct-zol RNA miniprep kit (Zymo Research) accordingly to the manufacturer’s instructions that included a DNase digestion step. Nascent RNAs were purified by using the Click-iT nascent RNA capture kit (Life technologies), as indicated by the manufacturer. Briefly, for each sample, 5 μg of EU-RNA was biotinylated with 0.5 mM biotin azide for 30 min at room temperature. The reaction mixture consisted of 25 μl of Click-iT EU buffer 2×, 4 μl of 25 mM CuSO4, 2.5 μl of biotin azide (10 mM), 1.25 μl of 400 mM Click-iT reaction buffer additive 1, 1.5 μl of 400 mM Click-iT reaction buffer additive 2, 5 μg of RNA, and water to 50 μl. The biotinylated RNA was purified through precipitation in the presence of 1 μl UltraPure glycogen, 75 μl of 5 M ammonium acetate, and 700 μl of chilled 100% ethanol. RNA was then resuspended in water, and its concentration measured with a NanoDrop.
One microgram was put aside as total RNA, and 1 μg was used for the purification of the nascent RNA. Fifty microliters of Dynabeads MyOne streptavidin T1 magnetic beads were used per sample. Beads were washed three times with 500 μl of Click-iT reaction wash buffer 2. To separate the beads from the supernatants, we used the Invitrogen DynaMag-2 magnet.
After the last wash, the beads were resuspended in 50 μl of wash buffer 2. To allow the binding of the RNA to the beads, RNA was added to a mix with 125 μl Click-iT RNA binding buffer 2×, 2 μl of RNaseOUT recombinant RNase inhibitor (Thermo Fisher Scientific), and water to a final volume of 250 μl. The RNA binding reaction mixture was incubated at 68°C for 4 min and added to the bead suspension. The RNA-bead suspension was incubated at room temperature for 30 min and subjected to gently vortexing. The beads bound to the biotinylated nascent RNA were then immobilized on the DynaMag-2 magnet. Beads were washed five times with 500 μl of Click-iT reaction wash buffer 1 and five times with 500 μl of Click-iT reaction wash buffer 2. The beads were resuspended in 50 μl of Click-iT reaction wash buffer 2, and RNA captured on the beads was used as the template for cDNA synthesis. For the reverse transcriptase reaction, we used the high-capacity cDNA reverse transcription kit (Applied Biosystems). Ten microliters of 10× RT buffer, 10 μl of 10× RT random primers, 4 μl 25× deoxynucleoside triphosphate (dNTP) mix (10 mM), and 21 μl of water were added to 50 μl of the bead suspension preheated at 68°C for 5 min. The reaction mixture was brought to room temperature, we added 5 μl of MultiScribe reverse transcriptase (50 U/μl), and the reaction mixture was incubated at 37°C for 2 h while gently mixing. The reaction was stopped by incubation at 85°C for 5 min, and the cDNA released from the beads. For the reverse transcription of total RNA, we conducted two reactions with 0.5 μg of RNA each. The reaction mixture included 2 μl of 10× RT buffer, 2 μl of 10× RT random primers, 4 μl 25× dNTP mix (10 mM), 21 μl of water, and 10 μl of RNA. The reaction mixture was incubated at 25°C for 10 min, 37°C for 2 h, and 85°C for 5 min. Finally, both nascent and total RNAs were purified with the with Qiagen MinElute PCR purification kit. The cDNA was quantitated by qubit and processed into next-generation sequencing (NGS) libraries using the Swift BioSciences ACCEL-NGS 1S Plus DNA library kit in combination with the Swift BioSciences 1S unique dual indexing kit. Library quality and quantity (QC) was determined by a Bioanalyzer, and the pool of libraries was sequenced at the University of Chicago Genomics Facility on the Illumina NextSeq using Illumina reagents and protocols. Two independent biological replicates were analyzed.
ATAC-seq.
One hundred thousand mock-infected or acutely infected Kasumi-3 cells were spun down for 10 min at 2,200 rpm at 4°C and resuspended in 50 μl of a transposase mix that included 25 μl of 2× Nextera Tagment DNA buffer (Illumina), 2.5 μl Nextera Tn5 transposase (Illumina), 0.5 μl 5% digitonin (Invitrogen), and 22 μl of H2O. The reaction was carried out at 37°C for 30 min. DNA was purified with Qiagen MinElute PCR purification kit and eluted in 10 μl of buffer EB. DNA libraries were amplified by using NEBNext high-fidelity (New England BioLabs) and barcoded primers as previously described (133). The numbers of amplification cycles were calculated by qPCR and kept under 11 to prevent saturation. Libraries were purified with a Qiagen MinElute PCR purification kit and size selected with AMPure XB beads (Beckman Coulter) to have fragments of less than 1,000 bp. Library quality control was assessed by a Bioanalyzer high-sensitivity DNA analysis kit (Agilent), and the libraries were sequenced with Illumina NextSeq technology (75-bp single-end reads, 100 million reads/sample). Three independent biological replicates were analyzed.
ChIP-seq.
Chromatin immunoprecipitation (ChIP) was conducted as previously described (134) with a few variations. For each immunoprecipitation, 5 × 106 cells were washed twice with PBS, cross-linked with 1% formaldehyde in PBS for 10 min, and quenched with 0.125 M glycine for 5 min at room temperature. Cells were washed twice with PBS, resuspended in 10 ml of buffer 1 (50 mM HEPES at pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% IGEPAL, and 0.25% Triton X-100) and incubated for 10 min at 4°C on a rotator. Cells were spun down for 7 min at 2,200 rpm at 4°C and resuspended in 10 ml of buffer 2 (10 mM Tris-HCl at pH 8.0, 200 mM NaCl, 1 mM EDTA, and 0.5 mM EGTA). After an incubation of 10 min on a rotator at room temperature, the cells were centrifuged as previously described and resuspended in 1 ml of buffer 3 (10 mM Tris HCl at pH 8.0, 1 mM EDTA, and 0.1% SDS). Sonication was conducted with Covaris Focused ultrasonicator at 10% duty cycle, 140 peak, 200 cycles per burst, and 360 s to obtain fragments between 200 and 500 bp. An aliquot of the sheared chromatin (50 μl) was de-cross-linked in elution buffer (50 mM Tris-HCl at pH 8.0, 10 mM EDTA, and 1.0% SDS) for 2 h at 65°C, and the DNA was purified with a QIAquick PCR purification kit (Qiagen). The size of DNA fragments was checked on a 1% agarose gel, and the range was between 200 and 500 bp. The rest of the sonicated chromatin was cleared at maximum speed for 15 min at 4°C in a Microfuge. We added 100 μl of 10× ChIP dilution buffer (10% Triton X-100, 1 M NaCl, 1% Na-deoxycholate, and 5% N-lauroylsarcosine) to the cleared chromatin, and 100 μl was collected as input. The sample was then incubated with the designated antibody overnight at 4°C on a rotator at the concentration suggested by the manufacturer. Thirty microliters of protein A/G agarose beads was added to the sample and incubated for 4 h at 4°C on a rotator. Beads were then pelleted, washed 4 times with RIPA buffer (50 mM HEPES at pH 7.5, 500 mM LiCl, 1 mM EDTA, 1.0% IGEPAL, and 0.7% Na-deoxycholate) and once with Tris-EDTA (TE) 1× with 50 mM NaCl. Two hundred microliters of elution buffer (50 mM Tris-HCl at pH 8.0, 10 mM EDTA, and 1.0% SDS) was added to the beads and incubated at 65°C for 30 min on a shaking incubator. Beads were spun down, and the supernatant was de-cross-linked at 65°C overnight. Elution buffer was added to the input and the de-cross-linking conducted as described. Finally, the DNA was purified using the Qiagen PCR purification kit in a volume of 60 μl. Libraries were prepared with the KAPA HTP library preparation kit multiplexed with NEXTflex DNA barcodes (Bioo Scientific) and size selected with AMPure XB beads (Beckman Coulter) to have fragments 250 to 450 bp in length. Library quality control was assessed using a Bioanalyzer high-sensitivity DNA analysis kit (Agilent), and sequencing was performed with Illumina NextSeq technology (50-bp single-end reads).
ChIP-qPCR.
ChIP was performed according to the same protocol described for the ChIP-seq analysis with the only difference that the DNA was purified in a volume of 50 μl. DNA levels were measured by RT-qPCR. To amplify the MIEP and oriLyt promoters, we used custom assays designed by Thermo Fisher. The DNA amount was calculated relative to the input.
ChIP-seq analysis.
The quality of reads, in FASTQ format, was evaluated using FastQC. Reads were trimmed to remove Illumina adapters from the 3′ ends using cutadapt (135). Trimmed reads were aligned using Bowtie 2 (version 2.2.9) with default parameters to the human genome (hg19) and the HCMV TB40/E clone TB40-BAC4 sequence (EF999921.1), to which the GFP gene sequence was added. Reads mapping uniquely to the genomes were further analyzed with Hypergeometric Optimization of Motif EnRichment (HOMER, version 4.11) (136) to call Pol II, H3K27Ac, and H3K27me3 binding regions, identify regions of differential binding, and generate IGV read density tracks. Pathway analyses were performed using DAVID.
Data availability.
A track hub with the HCMV TB40/E+GFP reference genome has been made available on GitHub (https://github.com/Mary-Hummel/HCMV-TB40-E-GFP-reference-genome.git). ChIP-seq, ATAC-seq, and RNA-seq raw data sets and BigWig files can be accessed at GEO under accession number GSE164861.
ACKNOWLEDGMENTS
This work was supported by NIH P01AI112522 and by the Northwestern University Flow Cytometry Core Facility supported by a Cancer Center Support Grant (NCI CA060553). Flow cytometry cell sorting was performed on a BD FACSAria SORP system purchased through the support of NIH 1S10OD011996-01. A.P. was supported by the transition to independence grant K99CA234434-01.
We thank Scott Terhune and Katie Cataldo for help with virus preparations, Stacy Marshall for technical support, Suchitra Swaminathan for help with cell sorting, Christopher Futter and Gregory Crawford for advice with the ATAC-seq, and Pieter Faber at the University of Chicago Genomics Facility for RNA-seq and ChIP-seq library construction, sequencing, and advice.
Footnotes
Supplemental material is available online only.
Contributor Information
Eleonora Forte, Email: e-forte@northwestern.edu.
Jae U. Jung, Lerner Research Institute, Cleveland Clinic
REFERENCES
- 1.Mendelson M, Monard S, Sissons P, Sinclair J. 1996. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J Gen Virol 77:3099–3102. 10.1099/0022-1317-77-12-3099. [DOI] [PubMed] [Google Scholar]
- 2.Sindre H, Tjoonnfjord GE, Rollag H, Ranneberg-Nilsen T, Veiby OP, Beck S, Degre M, Hestdal K. 1996. Human cytomegalovirus suppression of and latency in early hematopoietic progenitor cells. Blood 88:4526–4533. 10.1182/blood.V88.12.4526.bloodjournal88124526. [DOI] [PubMed] [Google Scholar]
- 3.Minton EJ, Tysoe C, Sinclair JH, Sissons JG. 1994. Human cytomegalovirus infection of the monocyte/macrophage lineage in bone marrow. J Virol 68:4017–4021. 10.1128/JVI.68.6.4017-4021.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kondo K, Mocarski ES. 1995. Cytomegalovirus latency and latency-specific transcription in hematopoietic progenitors. Scand J Infect Dis Suppl 99:63–67. [PubMed] [Google Scholar]
- 5.Fishman JA. 2017. Infection in organ transplantation. Am J Transplant 17:856–879. 10.1111/ajt.14208. [DOI] [PubMed] [Google Scholar]
- 6.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev 26:86–102. 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forte E, Swaminathan S, Schroeder MW, Kim JY, Terhune SS, Hummel M. 2018. Tumor necrosis factor alpha induces reactivation of human cytomegalovirus independently of myeloid cell differentiation following posttranscriptional establishment of latency. mBio 9:e01560-18. 10.1128/mBio.01560-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forte E, Zhang Z, Thorp EB, Hummel M. 2020. Cytomegalovirus latency and reactivation: an intricate interplay with the host immune response. Front Cell Infect Microbiol 10:130. 10.3389/fcimb.2020.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodrum FD, Jordan CT, High K, Shenk T. 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc Natl Acad Sci U S A 99:16255–16260. 10.1073/pnas.252630899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rauwel B, Jang SM, Cassano M, Kapopoulou A, Barde I, Trono D. 2015. Release of human cytomegalovirus from latency by a KAP1/TRIM28 phosphorylation switch. Elife 4:e06068. 10.7554/eLife.06068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Connor CM, Murphy EA. 2012. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J Virol 86:9854–9865. 10.1128/JVI.01278-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodrum F, Jordan CT, Terhune SS, High K, Shenk T. 2004. Differential outcomes of human cytomegalovirus infection in primitive hematopoietic cell subpopulations. Blood 104:687–695. 10.1182/blood-2003-12-4344. [DOI] [PubMed] [Google Scholar]
- 13.Cheung AK, Abendroth A, Cunningham AL, Slobedman B. 2006. Viral gene expression during the establishment of human cytomegalovirus latent infection in myeloid progenitor cells. Blood 108:3691–3699. 10.1182/blood-2005-12-026682. [DOI] [PubMed] [Google Scholar]
- 14.Zhu D, Pan C, Sheng J, Liang H, Bian Z, Liu Y, Trang P, Wu J, Liu F, Zhang CY, Zen K. 2018. Human cytomegalovirus reprogrammes haematopoietic progenitor cells into immunosuppressive monocytes to achieve latency. Nat Microbiol 3:503–513. 10.1038/s41564-018-0131-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galinato M, Shimoda K, Aguiar A, Hennig F, Boffelli D, McVoy MA, Hertel L. 2019. Single-cell transcriptome analysis of CD34+ stem cell-derived myeloid cells infected with human cytomegalovirus. Front Microbiol 10:577. 10.3389/fmicb.2019.00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hargett D, Shenk TE. 2010. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc Natl Acad Sci U S A 107:20039–20044. 10.1073/pnas.1014509107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rossetto CC, Tarrant-Elorza M, Pari GS. 2013. cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14+ monocytes and CD34+ cells. PLoS Pathog 9:e1003366. 10.1371/journal.ppat.1003366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shnayder M, Nachshon A, Krishna B, Poole E, Boshkov A, Binyamin A, Maza I, Sinclair J, Schwartz M, Stern-Ginossar N. 2018. Defining the transcriptional landscape during cytomegalovirus latency with single-cell RNA sequencing. mBio 9:e00013-18. 10.1128/mBio.00013-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shnayder M, Nachshon A, Rozman B, Bernshtein B, Lavi M, Fein N, Poole E, Avdic S, Blyth E, Gottlieb D, Abendroth A, Slobedman B, Sinclair J, Stern-Ginossar N, Schwartz M. 2020. Single cell analysis reveals human cytomegalovirus drives latently infected cells towards an anergic-like monocyte state. Elife 9:e52168. 10.7554/eLife.52168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reeves MB, Lehner PJ, Sissons JG, Sinclair JH. 2005. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J Gen Virol 86:2949–2954. 10.1099/vir.0.81161-0. [DOI] [PubMed] [Google Scholar]
- 21.Reeves MB, MacAry PA, Lehner PJ, Sissons JG, Sinclair JH. 2005. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc Natl Acad Sci U S A 102:4140–4145. 10.1073/pnas.0408994102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abraham CG, Kulesza CA. 2013. Polycomb repressive complex 2 silences human cytomegalovirus transcription in quiescent infection models. J Virol 87:13193–13205. 10.1128/JVI.02420-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ioudinkova E, Arcangeletti MC, Rynditch A, De Conto F, Motta F, Covan S, Pinardi F, Razin SV, Chezzi C. 2006. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 384:120–128. 10.1016/j.gene.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 24.Lee SH, Albright ER, Lee JH, Jacobs D, Kalejta RF. 2015. Cellular defense against latent colonization foiled by human cytomegalovirus UL138 protein. Sci Adv 1:e1501164. 10.1126/sciadv.1501164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gan X, Wang H, Yu Y, Yi W, Zhu S, Li E, Liang Y. 2017. Epigenetically repressing human cytomegalovirus lytic infection and reactivation from latency in THP-1 model by targeting H3K9 and H3K27 histone demethylases. PLoS One 12:e0175390. 10.1371/journal.pone.0175390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Damme E, Van Loock M. 2014. Functional annotation of human cytomegalovirus gene products: an update. Front Microbiol 5:218. 10.3389/fmicb.2014.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hancock MH, Landais I, Hook LM, Grey F, Tirabassi R, Nelson JA. 2013. Cytomegalovirus-encoded miRNAs, p 68–85. In Reddehase MJ, Lemmermann N (ed), Cytomegaloviruses: from molecular pathogenesis to intervention, vol 1. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 28.Mounce BC, Olsen ME, Vignuzzi M, Connor JH. 2017. Polyamines and their role in virus infection. Microbiol Mol Biol Rev 81:e00029-17. 10.1128/MMBR.00029-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bhella D, Rixon FJ, Dargan DJ. 2000. Cryomicroscopy of human cytomegalovirus virions reveals more densely packed genomic DNA than in herpes simplex virus type 1. J Mol Biol 295:155–161. 10.1006/jmbi.1999.3344. [DOI] [PubMed] [Google Scholar]
- 30.Yu X, Jih J, Jiang J, Zhou ZH. 2017. Atomic structure of the human cytomegalovirus capsid with its securing tegument layer of pp150. Science 356:eaam6892. 10.1126/science.aam6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strang BL. 2015. Viral and cellular subnuclear structures in human cytomegalovirus-infected cells. J Gen Virol 96:239–253. 10.1099/vir.0.071084-0. [DOI] [PubMed] [Google Scholar]
- 32.Thomas M, Reuter N, Stamminger T. 2013. Multifaceted regulation of human cytomegalovirus gene expression, p 174–195. In Reddehase MJ (ed), Cytomegaloviruses: from molecular pathogenesis to intervention, vol 1. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 33.Nitzsche A, Paulus C, Nevels M. 2008. Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J Virol 82:11167–11180. 10.1128/JVI.01218-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zalckvar E, Paulus C, Tillo D, Asbach-Nitzsche A, Lubling Y, Winterling C, Strieder N, Mucke K, Goodrum F, Segal E, Nevels M. 2013. Nucleosome maps of the human cytomegalovirus genome reveal a temporal switch in chromatin organization linked to a major IE protein. Proc Natl Acad Sci U S A 110:13126–13131. 10.1073/pnas.1305548110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Albright ER, Kalejta RF. 2016. Canonical and variant forms of histone H3 are deposited onto the human cytomegalovirus genome during lytic and latent infections. J Virol 90:10309–10320. 10.1128/JVI.01220-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mocarski E. 2007. Betaherpes viral genes and their functions. In Arvin A, Campadelli-Fiume G, Mocarski E (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. https://www.ncbi.nlm.nih.gov/books/NBK47435/. [PubMed] [Google Scholar]
- 37.Adamson CS, Nevels MM. 2020. Bright and early: inhibiting human cytomegalovirus by targeting major immediate-early gene expression or protein function. Viruses 12:110. 10.3390/v12010110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meier JL, Stinski MF. 2013. Major immediate-early enhancer and its gene products, p 152–173. In Reddehase MJ (ed), Cytomegaloviruses: from molecular pathogenesis to intervention, vol 1. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 39.White EA, Spector DH. 2007. Early viral gene expression and function. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. https://www.ncbi.nlm.nih.gov/pubmed/21348120. [PubMed] [Google Scholar]
- 40.Cuevas-Bennett C, Shenk T. 2008. Dynamic histone H3 acetylation and methylation at human cytomegalovirus promoters during replication in fibroblasts. J Virol 82:9525–9536. 10.1128/JVI.00946-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Groves IJ, Reeves MB, Sinclair JH. 2009. Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic 'pre-immediate-early' repression of viral gene expression mediated by histone post-translational modification. J Gen Virol 90:2364–2374. 10.1099/vir.0.012526-0. [DOI] [PubMed] [Google Scholar]
- 42.Nevels M, Paulus C, Shenk T. 2004. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc Natl Acad Sci U S A 101:17234–17239. 10.1073/pnas.0407933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nitzsche A, Steinhausser C, Mucke K, Paulus C, Nevels M. 2012. Histone H3 lysine 4 methylation marks postreplicative human cytomegalovirus chromatin. J Virol 86:9817–9827. 10.1128/JVI.00581-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Albright ER, Kalejta RF. 2013. Myeloblastic cell lines mimic some but not all aspects of human cytomegalovirus experimental latency defined in primary CD34+ cell populations. J Virol 87:9802–9812. 10.1128/JVI.01436-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saffert RT, Penkert RR, Kalejta RF. 2010. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J Virol 84:5594–5604. 10.1128/JVI.00348-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalejta RF, Albright ER. 2020. Expanding the known functional repertoire of the human cytomegalovirus pp71 protein. Front Cell Infect Microbiol 10:95. 10.3389/fcimb.2020.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saffert RT, Kalejta RF. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J Virol 81:9109–9120. 10.1128/JVI.00827-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee JH, Kalejta RF. 2019. Human cytomegalovirus enters the primary CD34+ hematopoietic progenitor cells where it establishes latency by macropinocytosis. J Virol 93:e00452-19. 10.1128/JVI.00452-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee JH, Pasquarella JR, Kalejta RF. 2019. Cell line models for human cytomegalovirus latency faithfully mimic viral entry by macropinocytosis and endocytosis. J Virol 93:e01021-19. 10.1128/JVI.01021-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goodrum F, Reeves M, Sinclair J, High K, Shenk T. 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110:937–945. 10.1182/blood-2007-01-070078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poole E, Walther A, Raven K, Benedict CA, Mason GM, Sinclair J. 2013. The myeloid transcription factor GATA-2 regulates the viral UL144 gene during human cytomegalovirus latency in an isolate-specific manner. J Virol 87:4261–4271. 10.1128/JVI.03497-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reeves MB, Compton T. 2011. Inhibition of inflammatory interleukin-6 activity via extracellular signal-regulated kinase-mitogen-activated protein kinase signaling antagonizes human cytomegalovirus reactivation from dendritic cells. J Virol 85:12750–12758. 10.1128/JVI.05878-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reeves MB, Sinclair JH. 2010. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J Gen Virol 91:599–604. 10.1099/vir.0.015602-0. [DOI] [PubMed] [Google Scholar]
- 54.Bego M, Maciejewski J, Khaiboullina S, Pari G, St Jeor S. 2005. Characterization of an antisense transcript spanning the UL81-82 locus of human cytomegalovirus. J Virol 79:11022–11034. 10.1128/JVI.79.17.11022-11034.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Poole E, Sinclair J. 2015. Sleepless latency of human cytomegalovirus. Med Microbiol Immunol 204:421–429. 10.1007/s00430-015-0401-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng S, Caviness K, Buehler J, Smithey M, Nikolich-Zugich J, Goodrum F. 2017. Transcriptome-wide characterization of human cytomegalovirus in natural infection and experimental latency. Proc Natl Acad Sci U S A 114:E10586–E10595. 10.1073/pnas.1710522114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kulesza CA, Shenk T. 2004. Human cytomegalovirus 5-kilobase immediate-early RNA is a stable intron. J Virol 78:13182–13189. 10.1128/JVI.78.23.13182-13189.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Angulo A, Kerry D, Huang H, Borst EM, Razinsky A, Wu J, Hobom U, Messerle M, Ghazal P. 2000. Identification of a boundary domain adjacent to the potent human cytomegalovirus enhancer that represses transcription of the divergent UL127 promoter. J Virol 74:2826–2839. 10.1128/jvi.74.6.2826-2839.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lashmit PE, Lundquist CA, Meier JL, Stinski MF. 2004. Cellular repressor inhibits human cytomegalovirus transcription from the UL127 promoter. J Virol 78:5113–5123. 10.1128/jvi.78.10.5113-5123.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lundquist CA, Meier JL, Stinski MF. 1999. A strong negative transcriptional regulatory region between the human cytomegalovirus UL127 gene and the major immediate-early enhancer. J Virol 73:9039–9052. 10.1128/JVI.73.11.9039-9052.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McDonough SH, Staprans SI, Spector DH. 1985. Analysis of the major transcripts encoded by the long repeat of human cytomegalovirus strain AD169. J Virol 53:711–718. 10.1128/JVI.53.3.711-718.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Isomura H, Stinski MF, Murata T, Yamashita Y, Kanda T, Toyokuni S, Tsurumi T. 2011. The human cytomegalovirus gene products essential for late viral gene expression assemble into prereplication complexes before viral DNA replication. J Virol 85:6629–6644. 10.1128/JVI.00384-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Omoto S, Mocarski ES. 2013. Cytomegalovirus UL91 is essential for transcription of viral true late (gamma2) genes. J Virol 87:8651–8664. 10.1128/JVI.01052-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Omoto S, Mocarski ES. 2014. Transcription of true late (gamma2) cytomegalovirus genes requires UL92 function that is conserved among beta- and gammaherpesviruses. J Virol 88:120–130. 10.1128/JVI.02983-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Perng YC, Campbell JA, Lenschow DJ, Yu D. 2014. Human cytomegalovirus pUL79 is an elongation factor of RNA polymerase II for viral gene transcription. PLoS Pathog 10:e1004350. 10.1371/journal.ppat.1004350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Perng YC, Qian Z, Fehr AR, Xuan B, Yu D. 2011. The human cytomegalovirus gene UL79 is required for the accumulation of late viral transcripts. J Virol 85:4841–4852. 10.1128/JVI.02344-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chambers J, Angulo A, Amaratunga D, Guo H, Jiang Y, Wan JS, Bittner A, Frueh K, Jackson MR, Peterson PA, Erlander MG, Ghazal P. 1999. DNA microarrays of the complex human cytomegalovirus genome: profiling kinetic class with drug sensitivity of viral gene expression. J Virol 73:5757–5766. 10.1128/JVI.73.7.5757-5766.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Spaderna S, Kropff B, Kodel Y, Shen S, Coley S, Lu S, Britt W, Mach M. 2005. Deletion of gpUL132, a structural component of human cytomegalovirus, results in impaired virus replication in fibroblasts. J Virol 79:11837–11847. 10.1128/JVI.79.18.11837-11847.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gates LA, Foulds CE, O'Malley BW. 2017. Histone marks in the 'driver's seat': functional roles in steering the transcription cycle. Trends Biochem Sci 42:977–989. 10.1016/j.tibs.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kouzarides T. 2007. Chromatin modifications and their function. Cell 128:693–705. 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 71.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, Ku M, Durham T, Kellis M, Bernstein BE. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473:43–49. 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 107:21931–21936. 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470:279–283. 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Collins-McMillen D, Rak M, Buehler JC, Igarashi-Hayes S, Kamil JP, Moorman NJ, Goodrum F. 2019. Alternative promoters drive human cytomegalovirus reactivation from latency. Proc Natl Acad Sci U S A 116:17492–17497. 10.1073/pnas.1900783116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hale AE, Collins-McMillen D, Lenarcic EM, Igarashi S, Kamil JP, Goodrum F, Moorman NJ. 2020. FOXO transcription factors activate alternative major immediate early promoters to induce human cytomegalovirus reactivation. Proc Natl Acad Sci U S A 117:18764–18770. 10.1073/pnas.2002651117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Arend KC, Ziehr B, Vincent HA, Moorman NJ. 2016. Multiple transcripts encode full-length human cytomegalovirus IE1 and IE2 proteins during lytic infection. J Virol 90:8855–8865. 10.1128/JVI.00741-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Klemm SL, Shipony Z, Greenleaf WJ. 2019. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet 20:207–220. 10.1038/s41576-018-0089-8. [DOI] [PubMed] [Google Scholar]
- 78.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10:1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Johnson DC, Hegde NR. 2002. Inhibition of the MHC class II antigen presentation pathway by human cytomegalovirus. Curr Top Microbiol Immunol 269:101–115. 10.1007/978-3-642-59421-2_7. [DOI] [PubMed] [Google Scholar]
- 80.Wiertz EJ, Devlin R, Collins HL, Ressing ME. 2007. Herpesvirus interference with major histocompatibility complex class II-restricted T-cell activation. J Virol 81:4389–4396. 10.1128/JVI.01525-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen FX, Smith ER, Shilatifard A. 2018. Born to run: control of transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol 19:464–478. 10.1038/s41580-018-0010-5. [DOI] [PubMed] [Google Scholar]
- 82.Kalejta RF. 2013. Pre-immediate early tegument protein functions, p 141–151. In Reddehase MJ (ed), Cytomegaloviruses: from molecular pathogenesis to intervention, vol 1. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 83.Sinclair J. 2010. Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim Biophys Acta 1799:286–295. 10.1016/j.bbagrm.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 84.Schwartz M, Stern-Ginossar N. 2019. The transcriptome of latent human cytomegalovirus. J Virol 93:e00047-19. 10.1128/JVI.00047-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Coronel R, Takayama S, Juwono T, Hertel L. 2015. Dynamics of human cytomegalovirus infection in CD34+ hematopoietic cells and derived Langerhans-type dendritic cells. J Virol 89:5615–5632. 10.1128/JVI.00305-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Murphy JC, Fischle W, Verdin E, Sinclair JH. 2002. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J 21:1112–1120. 10.1093/emboj/21.5.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Krishna BA, Lau B, Jackson SE, Wills MR, Sinclair JH, Poole E. 2016. Transient activation of human cytomegalovirus lytic gene expression during latency allows cytotoxic T cell killing of latently infected cells. Sci Rep 6:24674. 10.1038/srep24674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Parida M, Nilson KA, Li M, Ball CB, Fuchs HA, Lawson CK, Luse DS, Meier JL, Price DH. 2019. Nucleotide resolution comparison of transcription of human cytomegalovirus and host genomes reveals universal use of RNA polymerase II elongation control driven by dissimilar core promoter elements. mBio 10:e02047-18. 10.1128/mBio.02047-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pari G. 2008. Nuts and bolts of human cytomegalovirus lytic DNA replication, p 153–166. In Shenk TE, Stinski MF (ed), Human cytomegalovirus, vol 325. Springer-Verlag, Berlin, Germany. [DOI] [PubMed] [Google Scholar]
- 90.Gatherer D, Seirafian S, Cunningham C, Holton M, Dargan DJ, Baluchova K, Hector RD, Galbraith J, Herzyk P, Wilkinson GW, Davison AJ. 2011. High-resolution human cytomegalovirus transcriptome. Proc Natl Acad Sci U S A 108:19755–19760. 10.1073/pnas.1115861108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tai-Schmiedel J, Karniely S, Lau B, Ezra A, Eliyahu E, Nachshon A, Kerr K, Suarez N, Schwartz M, Davison AJ, Stern-Ginossar N. 2020. Human cytomegalovirus long noncoding RNA4.9 regulates viral DNA replication. PLoS Pathog 16:e1008390. 10.1371/journal.ppat.1008390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hwang J, Saffert RT, Kalejta RF. 2011. Elongin B-mediated epigenetic alteration of viral chromatin correlates with efficient human cytomegalovirus gene expression and replication. mBio 2:e00023-11. 10.1128/mBio.00023-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.O'Connor CM, Nukui M, Gurova KV, Murphy EA. 2016. Inhibition of the FACT complex reduces transcription from the human cytomegalovirus major immediate early promoter in models of lytic and latent replication. J Virol 90:4249–4253. 10.1128/JVI.02501-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Toth Z, Brulois K, Jung JU. 2013. The chromatin landscape of Kaposi's sarcoma-associated herpesvirus. Viruses 5:1346–1373. 10.3390/v5051346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Birkenheuer CH, Danko CG, Baines JD. 2018. Herpes simplex virus 1 dramatically alters loading and positioning of RNA polymerase II on host genes early in infection. J Virol 92:e02184-17. 10.1128/JVI.02184-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Abrisch RG, Eidem TM, Yakovchuk P, Kugel JF, Goodrich JA. 2015. Infection by herpes simplex virus 1 causes near-complete loss of RNA polymerase II occupancy on the host cell genome. J Virol 90:2503–2513. 10.1128/JVI.02665-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rutkowski AJ, Erhard F, L'Hernault A, Bonfert T, Schilhabel M, Crump C, Rosenstiel P, Efstathiou S, Zimmer R, Friedel CC, Dolken L. 2015. Widespread disruption of host transcription termination in HSV-1 infection. Nat Commun 6:7126. 10.1038/ncomms8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang X, Hennig T, Whisnant AW, Erhard F, Prusty BK, Friedel CC, Forouzmand E, Hu W, Erber L, Chen Y, Sandri-Goldin RM, Dolken L, Shi Y. 2020. Herpes simplex virus blocks host transcription termination via the bimodal activities of ICP27. Nat Commun 11:293. 10.1038/s41467-019-14109-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen CP, Lyu Y, Chuang F, Nakano K, Izumiya C, Jin D, Campbell M, Izumiya Y. 2017. Kaposi's sarcoma-associated herpesvirus hijacks RNA polymerase II to create a viral transcriptional factory. J Virol 91:e02491-16. 10.1128/JVI.02491-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hartenian E, Gilbertson S, Federspiel JD, Cristea IM, Glaunsinger BA. 2020. RNA decay during gammaherpesvirus infection reduces RNA polymerase II occupancy of host promoters but spares viral promoters. PLoS Pathog 16:e1008269. 10.1371/journal.ppat.1008269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McKinney C, Zavadil J, Bianco C, Shiflett L, Brown S, Mohr I. 2014. Global reprogramming of the cellular translational landscape facilitates cytomegalovirus replication. Cell Rep 6:1175. 10.1016/j.celrep.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 102.Stamminger T. 2008. Interactions of human cytomegalovirus proteins with the nuclear transport machinery. Curr Top Microbiol Immunol 325:167–185. 10.1007/978-3-540-77349-8_10. [DOI] [PubMed] [Google Scholar]
- 103.Tirosh O, Cohen Y, Shitrit A, Shani O, Le-Trilling VT, Trilling M, Friedlander G, Tanenbaum M, Stern-Ginossar N. 2015. The transcription and translation landscapes during human cytomegalovirus infection reveal novel host-pathogen interactions. PLoS Pathog 11:e1005288. 10.1371/journal.ppat.1005288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhu H, Cong JP, Mamtora G, Gingeras T, Shenk T. 1998. Cellular gene expression altered by human cytomegalovirus: global monitoring with oligonucleotide arrays. Proc Natl Acad Sci U S A 95:14470–14475. 10.1073/pnas.95.24.14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhu H, Cong JP, Shenk T. 1997. Use of differential display analysis to assess the effect of human cytomegalovirus infection on the accumulation of cellular RNAs: induction of interferon-responsive RNAs. Proc Natl Acad Sci U S A 94:13985–13990. 10.1073/pnas.94.25.13985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kenzelmann M, Muhlemann K. 2000. Transcriptome analysis of fibroblast cells immediate-early after human cytomegalovirus infection. J Mol Biol 304:741–751. 10.1006/jmbi.2000.4271. [DOI] [PubMed] [Google Scholar]
- 107.Browne EP, Wing B, Coleman D, Shenk T. 2001. Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: viral block to the accumulation of antiviral mRNAs. J Virol 75:12319–12330. 10.1128/JVI.75.24.12319-12330.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Challacombe JF, Rechtsteiner A, Gottardo R, Rocha LM, Browne EP, Shenk T, Altherr MR, Brettin TS. 2004. Evaluation of the host transcriptional response to human cytomegalovirus infection. Physiol Genomics 18:51–62. 10.1152/physiolgenomics.00155.2003. [DOI] [PubMed] [Google Scholar]
- 109.Hertel L, Mocarski ES. 2004. Global analysis of host cell gene expression late during cytomegalovirus infection reveals extensive dysregulation of cell cycle gene expression and induction of pseudomitosis independent of US28 function. J Virol 78:11988–12011. 10.1128/JVI.78.21.11988-12011.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Simmen KA, Singh J, Luukkonen BG, Lopper M, Bittner A, Miller NE, Jackson MR, Compton T, Fruh K. 2001. Global modulation of cellular transcription by human cytomegalovirus is initiated by viral glycoprotein B. Proc Natl Acad Sci U S A 98:7140–7145. 10.1073/pnas.121177598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Harwardt T, Lukas S, Zenger M, Reitberger T, Danzer D, Ubner T, Munday DC, Nevels M, Paulus C. 2016. Human cytomegalovirus immediate-early 1 protein rewires upstream STAT3 to downstream STAT1 signaling switching an IL6-type to an IFNgamma-like response. PLoS Pathog 12:e1005748. 10.1371/journal.ppat.1005748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sanchez V, Spector DH. 2008. Subversion of cell cycle regulatory pathways. Curr Top Microbiol Immunol 325:243–262. 10.1007/978-3-540-77349-8_14. [DOI] [PubMed] [Google Scholar]
- 113.Petrik DT, Schmitt KP, Stinski MF. 2006. Inhibition of cellular DNA synthesis by the human cytomegalovirus IE86 protein is necessary for efficient virus replication. J Virol 80:3872–3883. 10.1128/JVI.80.8.3872-3883.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gruffat H, Marchione R, Manet E. 2016. Herpesvirus late gene expression: a viral-specific pre-initiation complex is key. Front Microbiol 7:869. 10.3389/fmicb.2016.00869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wyrwicz LS, Rychlewski L. 2007. Identification of herpes TATT-binding protein. Antiviral Res 75:167–172. 10.1016/j.antiviral.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 116.Lee SB, Lee CF, Ou DS, Dulal K, Chang LH, Ma CH, Huang CF, Zhu H, Lin YS, Juan LJ. 2011. Host-viral effects of chromatin assembly factor 1 interaction with HCMV IE2. Cell Res 21:1230–1247. 10.1038/cr.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bryant LA, Mixon P, Davidson M, Bannister AJ, Kouzarides T, Sinclair JH. 2000. The human cytomegalovirus 86-kilodalton major immediate-early protein interacts physically and functionally with histone acetyltransferase P/CAF. J Virol 74:7230–7237. 10.1128/jvi.74.16.7230-7237.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lang D, Gebert S, Arlt H, Stamminger T. 1995. Functional interaction between the human cytomegalovirus 86-kilodalton IE2 protein and the cellular transcription factor CREB. J Virol 69:6030–6037. 10.1128/JVI.69.10.6030-6037.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lukac DM, Harel NY, Tanese N, Alwine JC. 1997. TAF-like functions of human cytomegalovirus immediate-early proteins. J Virol 71:7227–7239. 10.1128/JVI.71.10.7227-7239.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lukac DM, Manuppello JR, Alwine JC. 1994. Transcriptional activation by the human cytomegalovirus immediate-early proteins: requirements for simple promoter structures and interactions with multiple components of the transcription complex. J Virol 68:5184–5193. 10.1128/JVI.68.8.5184-5193.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Caswell R, Hagemeier C, Chiou CJ, Hayward G, Kouzarides T, Sinclair J. 1993. The human cytomegalovirus 86K immediate early (IE) 2 protein requires the basic region of the TATA-box binding protein (TBP) for binding, and interacts with TBP and transcription factor TFIIB via regions of IE2 required for transcriptional regulation. J Gen Virol 74:2691–2698. 10.1099/0022-1317-74-12-2691. [DOI] [PubMed] [Google Scholar]
- 122.Yoo YD, Chiou CJ, Choi KS, Yi Y, Michelson S, Kim S, Hayward GS, Kim SJ. 1996. The IE2 regulatory protein of human cytomegalovirus induces expression of the human transforming growth factor beta1 gene through an Egr-1 binding site. J Virol 70:7062–7070. 10.1128/JVI.70.10.7062-7070.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hagemeier C, Walker S, Caswell R, Kouzarides T, Sinclair J. 1992. The human cytomegalovirus 80-kilodalton but not the 72-kilodalton immediate-early protein transactivates heterologous promoters in a TATA box-dependent mechanism and interacts directly with TFIID. J Virol 66:4452–4456. 10.1128/JVI.66.7.4452-4456.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yurochko AD, Mayo MW, Poma EE, Baldwin AS, Jr, Huang ES. 1997. Induction of the transcription factor Sp1 during human cytomegalovirus infection mediates upregulation of the p65 and p105/p50 NF-kappaB promoters. J Virol 71:4638–4648. 10.1128/JVI.71.6.4638-4648.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fortunato EA, Sommer MH, Yoder K, Spector DH. 1997. Identification of domains within the human cytomegalovirus major immediate-early 86-kilodalton protein and the retinoblastoma protein required for physical and functional interaction with each other. J Virol 71:8176–8185. 10.1128/JVI.71.11.8176-8185.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bonin LR, McDougall JK. 1997. Human cytomegalovirus IE2 86-kilodalton protein binds p53 but does not abrogate G1 checkpoint function. J Virol 71:5861–5870. 10.1128/JVI.71.8.5861-5870.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Li M, Ball CB, Collins G, Hu Q, Luse DS, Price DH, Meier JL. 2020. Human cytomegalovirus IE2 drives transcription initiation from a select subset of late infection viral promoters by host RNA polymerase II. PLoS Pathog 16:e1008402. 10.1371/journal.ppat.1008402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Umashankar M, Petrucelli A, Cicchini L, Caposio P, Kreklywich CN, Rak M, Bughio F, Goldman DC, Hamlin KL, Nelson JA, Fleming WH, Streblow DN, Goodrum F. 2011. A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection. PLoS Pathog 7:e1002444. 10.1371/journal.ppat.1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Umashankar M, Goodrum F. 2014. Hematopoietic long-term culture (hLTC) for human cytomegalovirus latency and reactivation. Methods Mol Biol 1119:99–112. 10.1007/978-1-62703-788-4_7. [DOI] [PubMed] [Google Scholar]
- 130.Martin T, Cardarelli PM, Parry GC, Felts KA, Cobb RR. 1997. Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-kappa B and AP-1. Eur J Immunol 27:1091–1097. 10.1002/eji.1830270508. [DOI] [PubMed] [Google Scholar]
- 131.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. 2015. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol 109:21.29.1–21.29.9. 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, Rendleman EJ, Ma Q, Takahashi YH, Woodfin AR, Misharin AV, Abshiru NA, Lulla RR, Saratsis AM, Kelleher NL, James CD, Shilatifard A. 2017. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med 23:493–500. 10.1038/nm.4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 136.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38:576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Qin Q, Lee SH, Liang R, Kalejta RF. 2014. Insertion of myeloid-active elements into the human cytomegalovirus major immediate early promoter is not sufficient to drive its activation upon infection of undifferentiated myeloid cells. Virology 448:125–132. 10.1016/j.virol.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Umashankar M, Rak M, Bughio F, Zagallo P, Caviness K, Goodrum FD. 2014. Antagonistic determinants controlling replicative and latent states of human cytomegalovirus infection. J Virol 88:5987–6002. 10.1128/JVI.03506-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tenney DJ, Colberg-Poley AM. 1991. Human cytomegalovirus UL36-38 and US3 immediate-early genes: temporally regulated expression of nuclear, cytoplasmic, and polysome-associated transcripts during infection. J Virol 65:6724–6734. 10.1128/JVI.65.12.6724-6734.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
A track hub with the HCMV TB40/E+GFP reference genome has been made available on GitHub (https://github.com/Mary-Hummel/HCMV-TB40-E-GFP-reference-genome.git). ChIP-seq, ATAC-seq, and RNA-seq raw data sets and BigWig files can be accessed at GEO under accession number GSE164861.