

Abstract
Redox processes are at the heart of synthetic methods that rely on either electrochemistry or photoredox catalysis, but how do electrochemistry and photoredox catalysis compare? Both approaches provide access to high energy intermediates (e.g. radicals) that enable bond formations not constrained by the rules of ionic or 2 electron (e) mechanisms. Instead, they enable 1e mechanisms capable of bypassing electronic or steric limitations and protecting group requirements, thus enabling synthetic chemists to disconnect molecules in new and different ways. However, while providing access to similar intermediates, electrochemistry and photoredox catalysis differ in several physical chemistry principles. Understanding those differences can be key to designing new transformations and forging new bond disconnections. This review aims to highlight these differences and similarities between electrochemistry and photoredox catalysis by comparing their underlying physical chemistry principles and describing their impact on electrochemical and photochemical methods.
Graphical Abstract
1. INTRODUCTION
Electronic perturbation of molecules for selective bond construction and cleavage is the modus operandi of chemical synthesis, with polar or 2 electron (e) bond disconnections historically dominating retrosynthetic analyses used by organic chemists to construct complex molecules from smaller, simpler building blocks (Scheme 1A).1 This synthetic tradition stems from the perception that radical intermediates are difficult to control, but the recent renaissance of one electron bond disconnections (Scheme 1B) in organic synthesis highlights the importance of radical-based logic.2,3,4,5,6,7 In fact, one electron disconnections have been shown to greatly simplify the synthesis of complex organic structures as traditional protecting and functional group interconversions or redox manipulations can be avoided.8 As a result, synthetic methods based on radical intermediates often provide a more direct access to a desired product as well as enable new strategies to assemble complex organic structures. A number of complex total syntheses have already demonstrated the use of one electron bond disconnections as a key strategy.9,10
Scheme 1.

Illustration of 2e vs 1e disconnection approaches for retrosynthesis.
Photoredox and electrochemical synthetic methods have emerged as reliable tools to control the reactivity and selectivity of radical intermediates. Their methods leverage one electron disconnections, either by relying on the innate reactivity of radicals and coupling partners, or by intercepting these radicals with a catalyst, often a transition metal-based catalyst,11,12,13 to achieve a desired bond formation. Additionally, photoredox and electrochemical methods can access radical-polar cross-over mechanisms (Scheme 1) which enable bond formations between both a radical and a polar building block.
Prior to discussing synthetic examples comparing electrochemical and photochemical methods, some nomenclature related to interpreting synthetic schemes is presented. The electrode materials are depicted under the reaction arrow in synthetic schemes and the polarity of the electrode is denoted a (+) sign for the anode, a (−) sign for the cathode; the use of one or two vertical lines (“|” or “||”) denotes an undivided or divided cell setup, respectively. For example, “Zn(+) | Pt(−)” indicates the electrolysis uses an Zn anode and a Pt cathode in an undivided cell. For electrochemical methods using a carbon-based electrode, we use the following nomenclature: GRC for graphite carbon, GLC for glassy carbon, RVC for reticulated vitreous carbon, in order to distinguish between them. Additionally, a current (i) or current density (j) is provided for electrolysis performed under constant current electrolysis (CCE); an electrode potential (Ec or Ea, for the cathode or anode potential, respectively) is provided for constant potential electrolysis (CPE), and cell potential (Ucell) for constant voltage electrolysis. A complete list of abbreviations and symbols is provided at the end of this review.
2. SCOPE OF REVIEW
With both photoredox catalysis and electrochemistry taking center stage in the field of radical chemistry, organic chemists may wonder how photoredox catalysis and electrochemistry differ and in which cases one approach might be advantageous over the other. Individually, the topics of photochemistry14,15,16,17,18 and electrochemistry19,20,21,22,23,24,25,26,27 have been extensively reviewed. In contrast, reviews of direct comparisons between photoredox catalysis and electrochemistry are limited.28,29 This review aims to provide a critical comparison between photoredox catalysis and electrochemistry by: (1) discussing and comparing the basic concepts of photoredox catalysis and electrochemistry in Section 3, and (2) by comparing photoredox and electrochemical approaches of selected synthetic transformations in Section 4. We specifically aim to highlight how the choice of either photoredox or electrochemical approach can impact the reaction outcome of a given functional group conversion or bond formation/cleavage step. Therefore, transformations for which only one approach has been reported are not discussed herein. Additionally, photochemical transformations which do not proceed via a redox mechanism (e.g. energy transfer) are out of scope, and only those that involve photoredox catalysis will be included in our comparison with electrochemical synthetic transformations. We also note that electrochemical and photochemical transformations involving enzymes/biocatalysts are out of scope of this review and the reader should consult these references for insight on this field of catalysis.30,31,32,33,34,35,36 Finally, Section 5 focuses on emerging topics along with an outlook. We believe that the community will benefit from insights into these similarities and differences, thus being able to leverage either method to their advantage, as well as realize new or unique opportunities for the development of new transformations,37 including asymmetric reactions38,39,40,41,42,43,44,45,46,47,48,49
3. BASICS OF ELECTROCHEMISTRY & PHOTOREDOX CATALYSIS
How do photoredox and electrochemistry compare? What are their similarities and differences? To begin answering these seemingly simple questions, we first need to think about how radicals are formed, as well as realize that their lifetime50 and reactivity can differ greatly (i.e. radicals may behave as nucleophiles or electrophiles).51,52,53,54 In photoredox catalysis, radicals are generated via the interaction of the photocatalyst with the substrate/intermediate. Thus, the concentration of radicals in solution is relatively low compared to direct electrolysis – in the absence of a redox mediator55 – where high concentration of radicals can be achieved at the interface of the solution and working electrode. This fundamental difference in the generation of radicals in photoredox and electrochemical reactions impacts how redox processes in either field are designed. In the following sections, we will review the basic principles of electrochemical and photoredox mechanisms to highlight the advantages and disadvantages of either approach when performing overall (a) redox neutral, (b) net reductive, or (c) net oxidative transformations.
3.1. Basics of Electrochemistry
Electrolysis experiments utilize electrical energy to apply a potential across a pair of electrodes immersed into a solution containing the components to be electrolyzed (Scheme 2A). In conventional electrolysis experiments, four key features are always present: (1) an oxidation reaction occurs at the anode, (2) a reduction reaction occurs at the cathode, and (3) conservation of charge in solution implies that the rates of interfacial electron transfer at the anode and cathode must be equal, leading to a balanced, redox-neutral net reaction, (4) the solution must have an electrical resistance low enough to enable current flow between the two electrodes — this typically requires the use of a soluble supporting electrolyte in the reaction mixture. In many organic electrochemical transformations, only one of the two electrodes (the working electrode) generates a useful product. On the counter electrode, a non-productive reaction takes place. For net oxidative reactions, hydrogen evolution (i.e. proton reduction to H2) at the counter-electrode is the most common non-productive reaction. For net reductive reactions, sacrificial oxidation of an amine or the anode itself (e.g. Zn, Mg, Al, Cu) are commonly encountered counter-electrode reactions. Later sections will discuss details of each of the above topics. While many electrolysis can be performed using an undivided cell (Scheme 2A), the separation of the two half reactions (oxidation at anode and reduction at the cathode) may be desirable if a starting material, intermediate, or product is susceptible to undesired reaction at the opposite electrode relative to where it is generated or consumed. Separation of the two half-reaction can be accomplished via the use of a divided cell (Scheme 2B). Nonetheless, in order to maintain conservation of charge in solution, a method to accommodate the movement of ions from one half cell to the other is required. This can is achieved by interfacing the two solutions from the two half cells via a salt bridge, or more commonly, by directly interfacing the solutions by using a separator, such as a sintered-glass frit, a porous ceramic, a porous polymer sheet, or a semi-permeable ion-selective membrane (e.g. Nafion™). Polymeric membranes can exhibit selective transport56 of either cations (cation exchange membranes)57 or anions (anion exchange membranes).58,59 For example, Nafion™ — a perfluorinated sulfonic acid-based membrane — is one of the most frequently used cation exchange membrane in divided cell electrolysis for organic synthesis due to its excellent thermal and mechanical stability. Other cation exchange membrane materials include sulfonated polymers based on polystyrenes, polyimides, and poly(arylene ethers). Anion exchange membranes include those based on polyketones, poly(arylene)s or polyolefins linked to alkyltrimethylammonium, cyclic ammonium, multi-substituted imidazolium, tetrakis-aminophosphonium and cobaltocenium cations, as well as complexes between crown ethers or terpyridine with a metal cation-based salt.
Scheme 2.
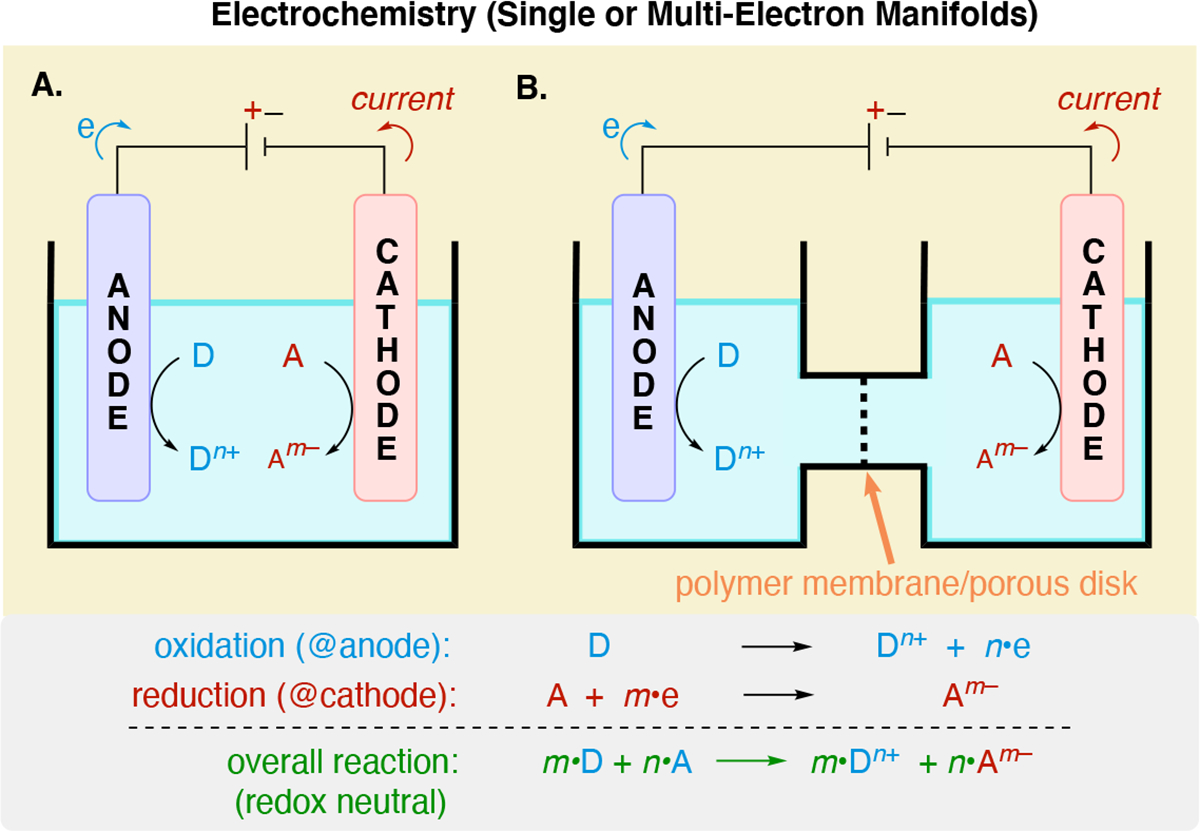
Schematic representation of an electrolysis experiment illustrating the two half-reactions. Note that the overall reaction is a linear combination of the two half-reaction, illustrating that redox neutrality must be maintained for (A) an undivided cell and (B) a divided cell.
3.2. Common Mechanisms in Electrochemistry
In the context of electrolysis, various reaction mechanisms exist including those outlined in Scheme 3. The nomenclature introduced by Testa and Reinmuth is commonly used in classifying mechanisms of electrochemical transformations by using the letters E and C to denote steps involving electron transfer and chemical steps, respectively.60,61 The presence or absence of a chemical step (C) either before or after the electron transfer step (E), as well as whether the step is reversible or irreversible are all options. Subscripts r and i are used to denote whether the E or C step is reversible or irreversible, respectively (e.g. Ci for an irreversible chemical step).
Scheme 3.
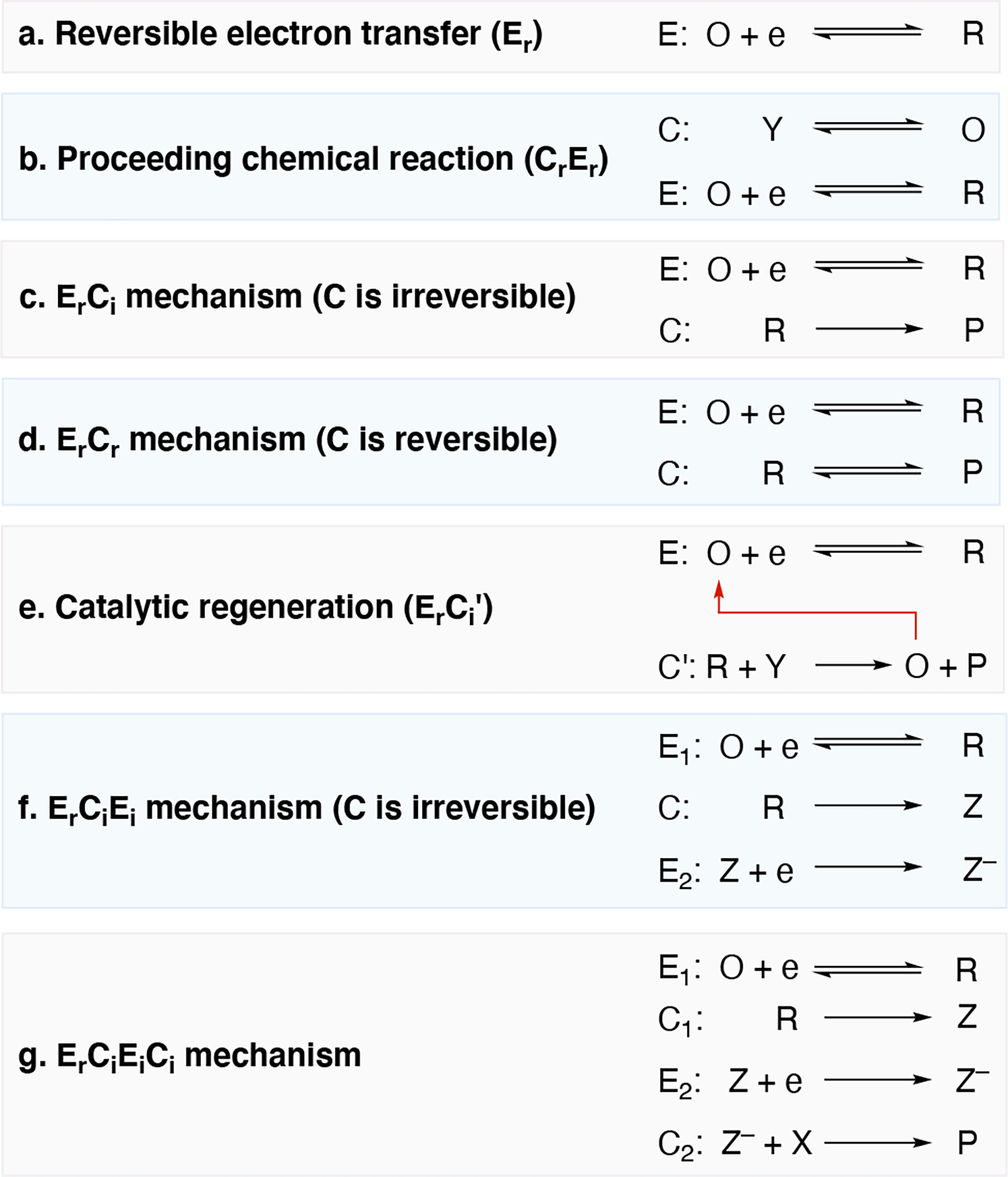
Illustration of various electrode mechanisms. O and R are the oxidized and reduced form of the substrate undergoing a reduction event, e is an electron, Z is a generic intermediate formed during a chemical step, Y and P are a starting material and a product, respectively. The subscripts r and i are used to denote whether the E or C step is reversible or irreversible, respectively. Ci’ denotes an irreversible chemical step that regenerates a catalytic mediator.
Cyclic voltammetry (Figure 1) is an excellent method to measure the potential at which a redox event takes place but also gain insight in the mechanism and reversibility of the reaction.62 Figure 1 illustrates cyclic voltammetry data for a reversible reduction event, along with several key descriptors: peak anodic current (i,pa), peak cathodic current (ip,c), anodic peak potential (Ep,a), cathodic peak potential (Ep,c). Finally, the half-wave potential (E1/2) for reversible systems is simply obtained by taking the average between the anodic and cathodic peak potential, explicitly, E1/2 = 0.5•(Epa + Epc). The Nernst equation (equation 1a) describes the partitioning between the oxidized (O) and reduced (R) form of an analyte undergoing a reversible redox process at an electrode in an unstirred solution at a given applied potential (E) based on its standard potential (E°), where R is the ideal gas constant (R = 8.31447 J•mol−1•K−1), F is Faraday’s constant (note F = 96487 C•mol−1) and n is the number of electrons involved in the redox event (A + n e → An−). E1/2 can provide an approximation for E°. We refer the reader to tutorial reviews63 and books64,65 for a more detailed treatise of the topic.
| (1a) |
Figure 1.

Simulated cyclic voltammogram data (2nd cycle) for a reversible reduction event illustrating several key parameters: anodic peak current (ip,a), cathodic peak current (ip,c), anodic peak potential (Ep,a), cathodic peak potential (Ep,c), and the half-wave potential (E1/2).
Returning to the topic of cyclic voltammetry as a tool to discern reaction mechanism,63,66 Figure 2 illustrates various simulated cyclic voltammograms corresponding to the prototypical mechanisms outline in Scheme 3.63 The ratio of ip,a to ip,c from the cyclic voltammograms measured at various scan rates (ν) as well as the peak separation between Ep,a and Ep,c can provide insight into the reversible nature of the redox process and information regarding the underpinning mechanism such as those in Scheme 3. For example, a reversible redox event (E) with no subsequent chemical step (Scheme 3, scenario a), will have ip,a/ip,c = 1, such as shown in Figure 1. Scheme 3, scenario b illustrates a mechanism in which a chemical step is followed by an electron transfer step (CE mechanism), where the faster the forward rate constant of the reversible chemical step (Cr), the more reversible the voltammogram becomes (i.e. ip,a/ip,c approaches 1), as shown the color progression from blue to dark green to lime green to orange in Figure 2, middle. Figure 2, top, illustrates the scenario where a reversible electron transfer step precedes the chemical step (ErCi mechanism, Scheme 3, scenario c), where increasing the scan rate restores reversibility. Scenarios c and d in Scheme 3 differ based on whether the electrochemical step is reversible (ErCi, Scheme 3, scenario c) or irreversible (ErCr, Scheme 3, scenario d). Multielectron transfer processes via sequential redox events can occur, such as two reversible redox events (ErEr) shown in Figure 2, bottom. More elaborate mechanisms exist, such as an ECE mechanism in which a second electron transfer event occurs after the initial EC events. Variations of the ECE mechanism exist where the 2nd electron transfer event (E2) may occur at a potential that is more or less thermodynamically facile than the first electron transfer event (E1), again which require cyclic voltammetry experiments to discern them apart. Scenario e in Scheme 3 illustrates an EC’ mechanism in which the catalytic species utilized in the first step is regenerated in the 2nd step.
Figure 2.

(Left) Simulated cyclic voltammograms using DigiElech simulation software for three common mechanisms. The currents are normalized. (Top) ErCi mechanism: increasing the scan rate (from ν = 0.1 (red) to 1 (green) to 10 V•s−1 (blue)) restores reversibility (rate constant for the Ci step k = 5 s−1). (Middle) CrEr mechanism: the faster the forward rate constant of the Cr step, the more reversible the voltammogram (Keq = 0.1, kf = 1 (blue), 10 (dark green), 100 (lime green), 1000 s−1 (red)). (Bottom) ErEr mechanism: as the separations between the two reduction potentials (ΔE1/2) decreases, the peaks merge to become a single two-electron peak. ΔE1/2 = −0.05 (dark blue), 0 (light blue), 0.05 (dark green), 0.1 (lime green), 0.15 (orange), and 0.2 V (red). Figure reproduced from ref 63. Copyright 2018 American Chemical Society.
We note that the cyclic voltammetry data shown in Figure 1 and 2 are for freely diffusing species at the electrode but scenarios where the starting material or product of the redox event is adsorbed onto the electrode are also possible. The peak separation between Ep,a and Ep,c, as well as the effect of scan rate on each of these potential provides insight into discerning between these scenarios. For a fully reversible redox event involving free diffusing species, the following three criteria should be met: (1) the peak anodic and cathodic currents (ip,a and ip,c, respectively) should be of equal magnitude (specifically, ip,a = −ip,c); (2) a plot of ip vs ν1/2 should be linear; and (3) the peak anodic and cathodic potentials (Ep,a, and Ep,c) should be independent of the scan rate. Deviations from linear behavior for plots of ip vs ν1/2 may be indicative that either (a) the electrochemical process is quasi-reversible, or (b) the species undergoing the redox event is surface-adsorbed (not freely diffusing). The peak to peak separation between Ep,a, and Ep,c will change as a function of scan rate for quasi-reversible processes, in contrast to a surface adsorbed species which will have this value constant.63
Advanced electrochemical and spectroscopic techniques can be useful to characterize these electrocatalytic systems.67,68,69,70,71,72 While this nomenclature was originally developed to describe mechanistic scenarios in the context of electrochemistry, the reader should realize that these classifications may be useful for describing photoredox reactions, as well as those that rely on stoichiometric redox reagents, thus enabling ease of comparison between these systems.
It is not uncommon that only the oxidation or only the reduction process is of interest, while the balancing redox event pertains to redox chemistry on the solvent, electrolyte, or a sacrificial species (reagent or electrode); these are termed unpaired electrolysis. Towards maximizing energy use and minimizing waste, electrochemical reactions can be designed in which both the reduction and oxidation events lead to useful chemistry (paired electrolysis).73,74,75,76 Scheme 4 illustrates common electrolysis reaction manifolds whereby the overall reaction can be broken down into a combination of oxidation and reduction reactions (half-reactions).77 Scheme 4A illustrates divergent paired electrolysis whereby the starting material can either be oxidized or reduced, leading to two different products. An example is the oxidation of glucose to gluconate accompanied by the reduction of glucose to sorbitol, a process that has been utilized on industrial scale.78,79 Parallel paired electrolysis (Scheme 4B) utilizes two different starting materials, one being oxidized and the other being reduced, in which the intermediates generated at each electrode do not interact with one another and leads to two different products (P1 and P2). An example of parallel paired electrolysis is the BASF synthesis of phthalide from dimethyl phthalate, where the cathodic reduction of dimethyl phthalate is paired with the simultaneous oxidation of various organic anodic depolarizers (e.g. 4-tert-butyltoluene).80 Convergent paired electrolysis (Scheme 4C) brings intermediates generated from both electrodes together to form one product. To enable the convergent reactivity, convergent paired electrolysis usually utilizes a 2e redox event to generate an ionic intermediate which have longer lifetimes than radicals. The Shono oxidation81,82 is one such example. An amine undergoes 2e oxidation at the anode to generate the corresponding iminium, which then reacts with an alkoxide nucleophile that was generated at the cathode through the reduction of protons (hydrogen generation) from an alcohol solvent. Sequential paired electrolysis (domino electrolysis) processes (Scheme 4D) are reactions that utilize redox events at both electrodes in a linear, sequential mechanism to generate the desired product. An example of a sequential paired electrolysis is the oxidation of oximes to nitrile oxides followed by their reduction to the corresponding nitrile.83,84 We note that the sequential paired electrolysis illustrated in Scheme 4D is shown to initiate at the anode with an oxidation process. The opposite scenario in which the initial redox event occurs at the cathode followed by a redox event at the anode falls into the same category (we chose to arbitrarily illustrate only one of the two scenarios for simplicity).
Scheme 4.
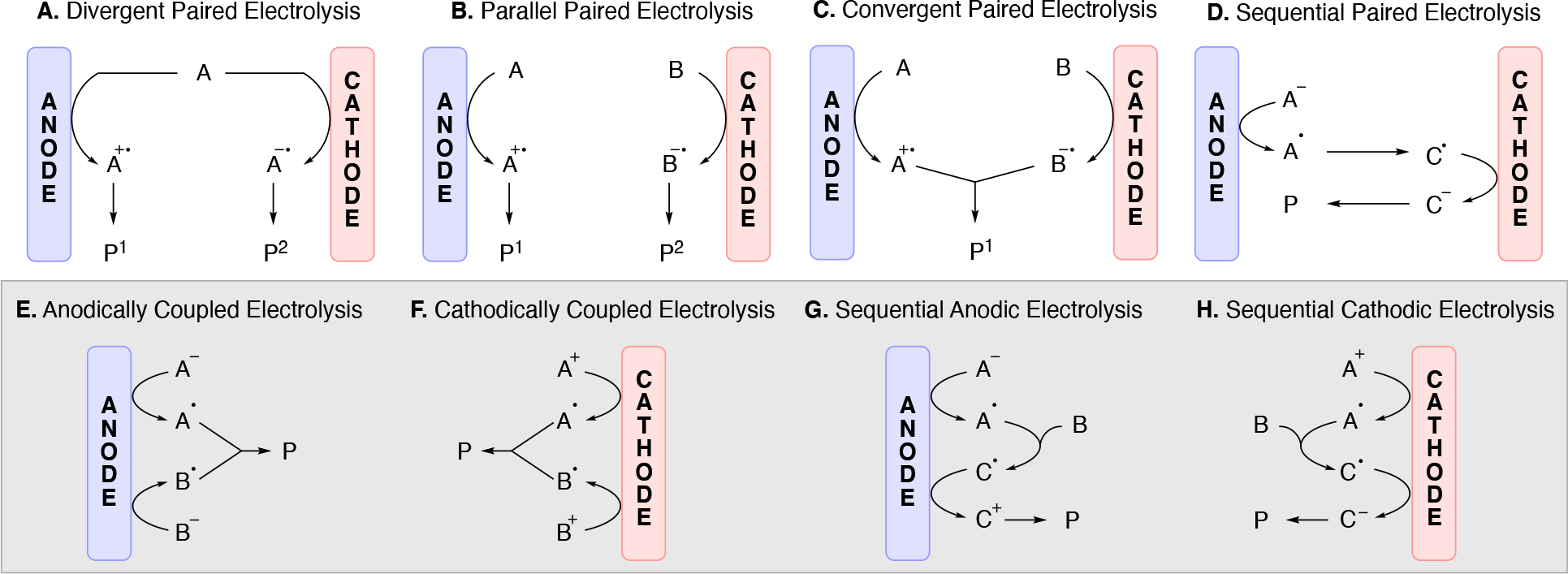
Illustration of several common electrolysis reaction manifolds: (A) divergent paired electrolysis, (B) parallel paired electrolysis, (C) convergent paired electrolysis, (D) sequential paired electrolysis (domino electrolysis), (E) anodically coupled electrolysis (F) cathodically coupled electrolysis, (G) sequential anodic electrolysis, and (H) sequential cathodic electrolysis. [All redox events are shown as single electron transfer events but redox events involving more than one electron also fall into these categories].
Anodically coupled electrolysis (Scheme 4E) illustrates a type of EEC mechanism whereby two different substrates (A and B) each individually undergo an oxidation event at the anode and their subsequent bimolecular coupling results in the desired product (P). The analogous cathodically coupled process is illustrated in Scheme 4F. The reaction mechanism illustrated in Scheme 4E relies on the individual oxidation potentials of A and B to be close to one another in order to appropriately match the rate of production of radicals A• and B•. The analogous statement is true for the reducing potentials of A and B in the context of Scheme 4F). Lin's chlorotrifluoromethylation of alkenes85 discussed in Section 4.8.1 is an example of a reaction proceeding via an anodically coupled electrolysis mechanism, while the electrochemical hindered amine synthesis86 discussed in Section 4.10 is an example of a cathodically coupled electrolysis mechanism. Sequential anodic and sequential cathodic electrolysis (Scheme 4G and Scheme 4H, respectively) highlight an example of the classic ECEC mechanism. This approach has been utilized for numerous vicinal difunctionalization of alkenes (see Section 4.8).
3.3. Basics of Photoredox Catalysis
In contrast to electrochemistry, photoredox catalysis cannot physically separate the two half reactions of a redox process since both half reactions have to occur at the same photocatalyst, and not at two different electrodes that are spatially separated. A photoredox catalyst (PC) can absorb a photon of appropriate wavelength to generate an excited state (PC*) which can then interact with a substrate via: (1) energy transfer87 (which is out of scope of this review), (2) photoinduced electron transfer (PET), or (3) atom transfer. PET is achieved by PC* participating in either an oxidative or reductive quenching process (Scheme 5). This quenching process occurs via single electron transfer (SET) with an electron acceptor (A) or donor (D), where the acceptor and donor can be any combination of a substrate, an intermediate or a terminal oxidant/reductant.
Scheme 5.
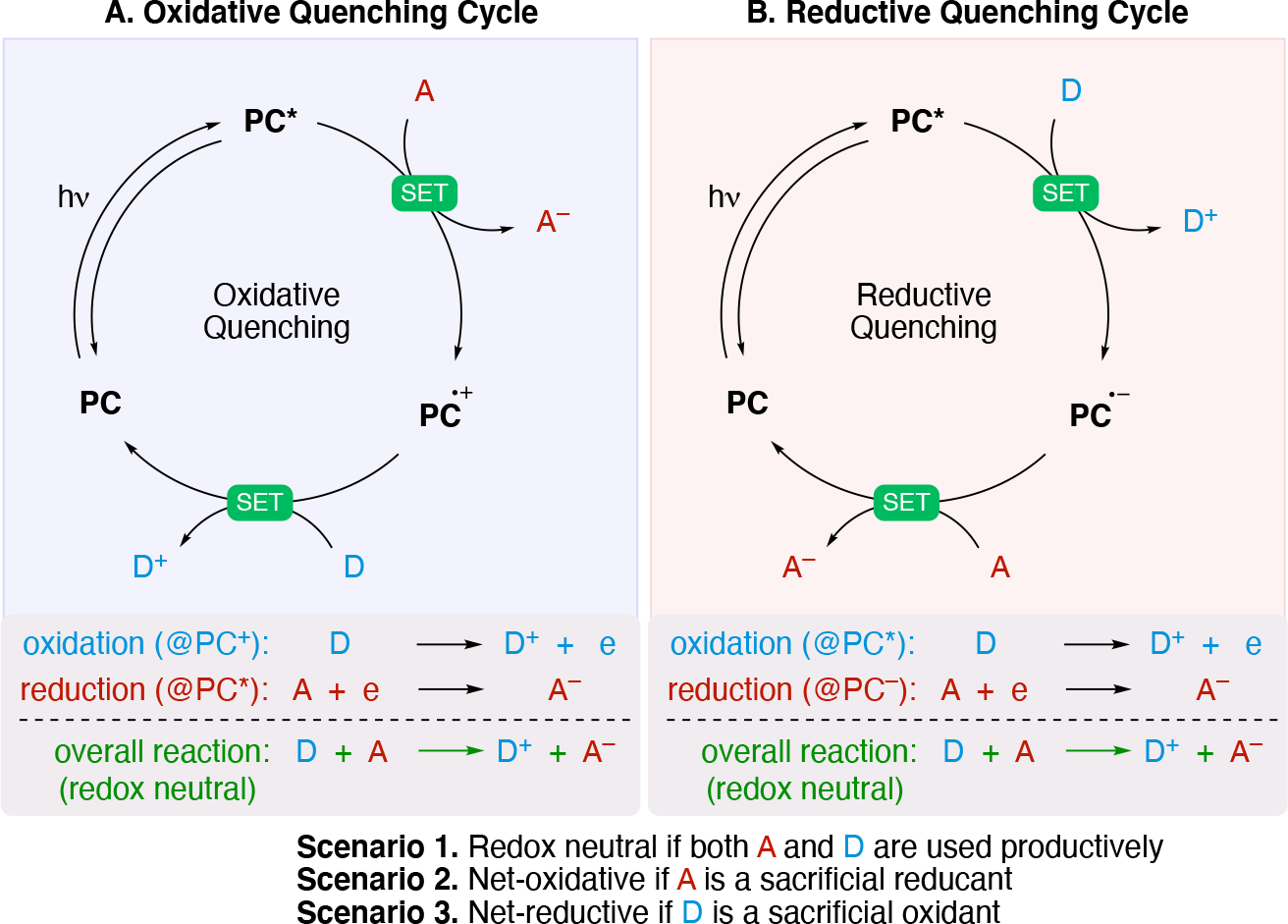
Illustration of (A) an oxidative and (B) a reductive quenching photoredox cycle.
3.4. Common Mechanisms in Photoredox Catalysis
In an oxidative quenching cycle (Scheme 5A), SET from PC* to an acceptor (A) results in ground state PC•+ and the reduced form of the acceptor (A−). A subsequent SET from a donor (D) to PC•+, results in the regeneration of the photocatalyst (PC) and the oxidized form of the donor (D+). Alternatively, in a reductive quenching cycle (Scheme 5B), SET from the donor (D) to PC* results in ground state PC•− and the oxidized form of the donor (D+). A subsequent SET from PC•−, to an acceptor (A) results in the regeneration of the photocatalyst and A−. Productive chemistry is leveraged from these (radical) intermediates A− and D+ via direct reaction or interception of these radicals with other catalysts (e.g. transition-metal catalyst for cross coupling reactions). Photoredox reactions fall into one of three categories: (scenario 1, Scheme 5) redox neutral process (where radical A+ and D− combine or originate from the same structure), or (scenario 2, Scheme 5) a net oxidative, or (scenario 3, Scheme 5) net reductive transformation where the radicals may react with a different starting material. Net oxidative transformations rely on a terminal oxidant (sacrificial acceptor of electrons) whereas net reductive process requires a terminal reductant (sacrificial source of electrons).
Scheme 6 illustrates a common reaction manifold observed in photoredox catalysis, namely radical-polar crossover mechanisms,88,89 which again is classifiable into either an oxidative or a reductive quenching variation (Scheme 6A and Scheme 6B, respectively). In this reaction manifold, the generation of the radical is followed by radical reactivity, generating a new radical intermediate which then undergoes SET with the photocatalyst to generate a cationic or an anionic intermediate primed for downstream polar reactivity. The polar reactivity can be as simple as loss of a proton/leaving group to generate an alkene, or reaction with a nucleophile/electrophile to generate a new bond. Radical-polar crossover is a common strategy for vicinal alkene difunctionalization, including ring formation reactions where a nucleophile or an electrophile is tethered to the substrate to facilitate intramolecular reactivity from a cationic or an anionic intermediate, respectively.
Scheme 6.

Illustrations of radical-polar crossover mechanisms via (A) an oxidative quenching cycle and (B) a reductive quenching cycle. RP = radical progenitor, PC = photocatalyst, SET = single electron transfer, using alkenes as the radical acceptor in the initial radical-substrate bond forming event. E and C denote electron transfer and chemical steps, respectively, in the mechanism.
3.5. Comparison Between Electrochemistry and Photoredox Mechanisms
Having introduced the various common electrochemical and photoredox mechanisms, we can now draw analogies between the two systems. Redox neutrality in the overall balanced equation is required for both photoredox and electrochemistry (Scheme 5 and Scheme 2, respectively). In electrochemistry the half-reactions (oxidation at the anode and reduction at the cathode) must add to a balanced, redox neutral process to obey the conservation of charge principle. In photoredox, redox neutrality is necessary in order to regenerate the photocatalyst (i.e. catalyst turnover). As a result, direct comparison between the various mechanisms discussed in section 3.2 and 3.4 are possible.
The redox neutral photocatalytic cycle shown in Scheme 5, scenario 1, where both the acceptor and donor in are used in productive manner and are both incorporated into the same product, is analogous to the convergent paired electrolysis (Scheme 4C). Redox neutral processes are more challenging in electrochemistry since the reduction and oxidation events occur at different electrodes and would require diffusion of the radical from one electrode to the other, usually unlikely given the short lifetime of a radical and the relatively long required for it to traverse the inter-electrode distance in a typical bulk electrolysis cell. A handful of examples on how to solve this problem have begun to emerge using microflow electrochemistry and alternating polarity electrolysis (see Section 5.1.2). To address this limitation, radical intermediates can be intercepted by a catalyst (such as a transition-metal catalyst) to generate a longer-lived intermediate for downstream chemistry. Scheme 5, scenario 2 illustrates a net oxidative process, in which the unproductive process is the reduction of an acceptor. This is analogous to an unpaired electrolysis in which a non-productive reduction occurs at the cathode, (e.g. proton reduction to generate hydrogen). Analogously, the net reductive process form scenario 3 in Scheme 5 in which the oxidation reaction (D to D+) is an unproductive reaction (e.g. oxidation of an amine) is analogous to an unpaired reduction electrolysis (e.g. using a sacrificial anode as the unproductive oxidation half reaction). The radical-polar cross over reactions from Scheme 6B correspond to the sequential paired electrolysis illustrated in Scheme 4D. The opposite polarity to Scheme 4D (i.e. the cathodic event preceding the anodic event) would be the direct analogy to the radical-polar crossover mechanism in Scheme 6A. Therefore, these radical-polar crossover mechanisms can also be classified as ECEC mechanisms.
In direct electrolysis, redox events occur at the interface of the electrode and solution (typically a heterogenous process) which can lead to a localized, high concentration of radicals. In contrast, redox chemistry in photoredox catalysis occurs in solution (homogenous) and provides a low concentration of radicals. As a result, coupling of two transient radicals is feasible in electrochemistry if they are generated at the same electrode, whereas this is more challenging in photoredox catalysis. Thus, anodically and cathodically coupled electrolysis shown in Scheme 4E & Scheme 4F, respectively, are commonly achieved using electrochemistry. In contrast, these are rarer in photoredox catalysis as it would require the use of at least one persistent radical (increased radical lifetime) to increase the likelihood of the bimolecular encounter event for radical-radical couplings.50 Finally, doubly reductive (or doubly oxidative) processes such as the sequential electrolysis mechanisms shown in Scheme 4G & Scheme 4H, are typically unfeasible in photoredox due to the low statistical chance that the radical lifetime is long enough for encountering the low concentration of the redox agent (e.g. photoexcited photocatalyst). In contrast, such events can be achieved more easily in electrochemistry, since both the rate of redox events can be much higher (high current density) and the radical is generated at the electrode-solution interface (highly localized). For noteworthy examples of doubly reductive processes via photoredox catalysis, see Section 5.2.
Radical-polar crossover mechanisms are another strategy widely used in photoredox whereby radical intermediates undergo a redox event to generate an ionic (cationic/anionic) intermediate that affords increased lifetime and the ability to react orthogonally to the radical manifold. In contrast, redox neutral processes are simpler to achieve with photoredox since the photocatalyst can act both as reductant and oxidant in the same cycle which can occur in the same spatial location (within diffusion distance attainable in one turnover of the photocatalyst). Examples of sequential redox chemistry with one redox event occurring at each electrode on the path to product generation do exist but are usually limited to an intermediate traversing between the two electrodes being a closed-shell species (i.e. not a radical) generated via a net 2e electron redox event instead of a net 1e redox event.
3.6. Terminal Oxidants & Reductants
In order to carry out net-oxidative and net-reductive transformations (Scheme 5, scenarios 2 & 3 in photoredox, and unpaired anodic oxidation & unpaired cathodic reductions), the use of terminal oxidants and terminal reductants are required. Some common terminal reductants employed in photoredox include Hantzsch ester,90 1-benzyl-1,4-dihydronicotinamide (BNAH), 1,4-dihydronicotinamide adenine dinucleotide (NADH), amines (Et3N, i-Pr2NEt, i-Pr2NH, Cy2NMe, Ar3N), i-PrOH, ascorbic acid, PPh3,91 and oxalate (via (C2O4)2− → 2 CO2 + 2e−). Common terminal oxidants92 include alkyl peroxides (e.g. DTBP), H2O2,93 O2, BrCCl3,94,95 oxoammoniums, peracids (e.g. peracetic acid), persulfates (e.g. (NH4)2S2O8, Na2S2O8), CuII salts96,97 (e.g. Cu(OAc)2 and Cu(O2CCF3)2). In the context of electrochemistry, in addition to the above redox reagents, the anode can also be used as a terminal reductant if oxidation of the anode material is sufficiently facile. Materials like Zn, Al, Mg, Cu can be used as sacrificial anodes.98 Upon donating one or more electrons from the sacrificial (metal) anode to a species at the cathode, the oxidized form of the anode metal will desorb from the anode into solution. The cation may dissolve into the solution or precipitate out as a salt (Scheme 7A), and results in the anode losing mass. In either photoredox or electrochemistry settings, the byproduct of these terminal oxidants/reductants is not always innocent. For example, oxidation of amines typically results in iminium or enamine formation (Scheme 7B) which can interfere with the desired chemistry.99 In electrochemistry, the use of a sacrificial metal anode will release metal cations that may participate (enhance) in the catalysis100,101,102,103 (e.g. acting as a Lewis-acid), or plate out on the cathode via reductive processes and ultimately decrease the current efficiency. Proton reduction to generate hydrogen is a common sacrificial reduction process at the cathode (Scheme 7C) which can result in the pH increasing in the reaction mixture as the reaction progresses. Electrochemical proton reduction generally relies on the use of a cathode with low overpotential for proton reduction (e.g. Pt) to minimize the cell potential and avoid side reactions. The use of divided cells to isolate the unproductive half-reaction, including its byproducts, can be a useful strategy to avoid it from interfering in the productive half-reaction. Hydrogen evolution has also been employed in the context of photoredox catalysis to turn over the catalyst via a non-productive reduction, for example by using a cobaloxime catalyst (Scheme 7D).104,105,106,107
Scheme 7.
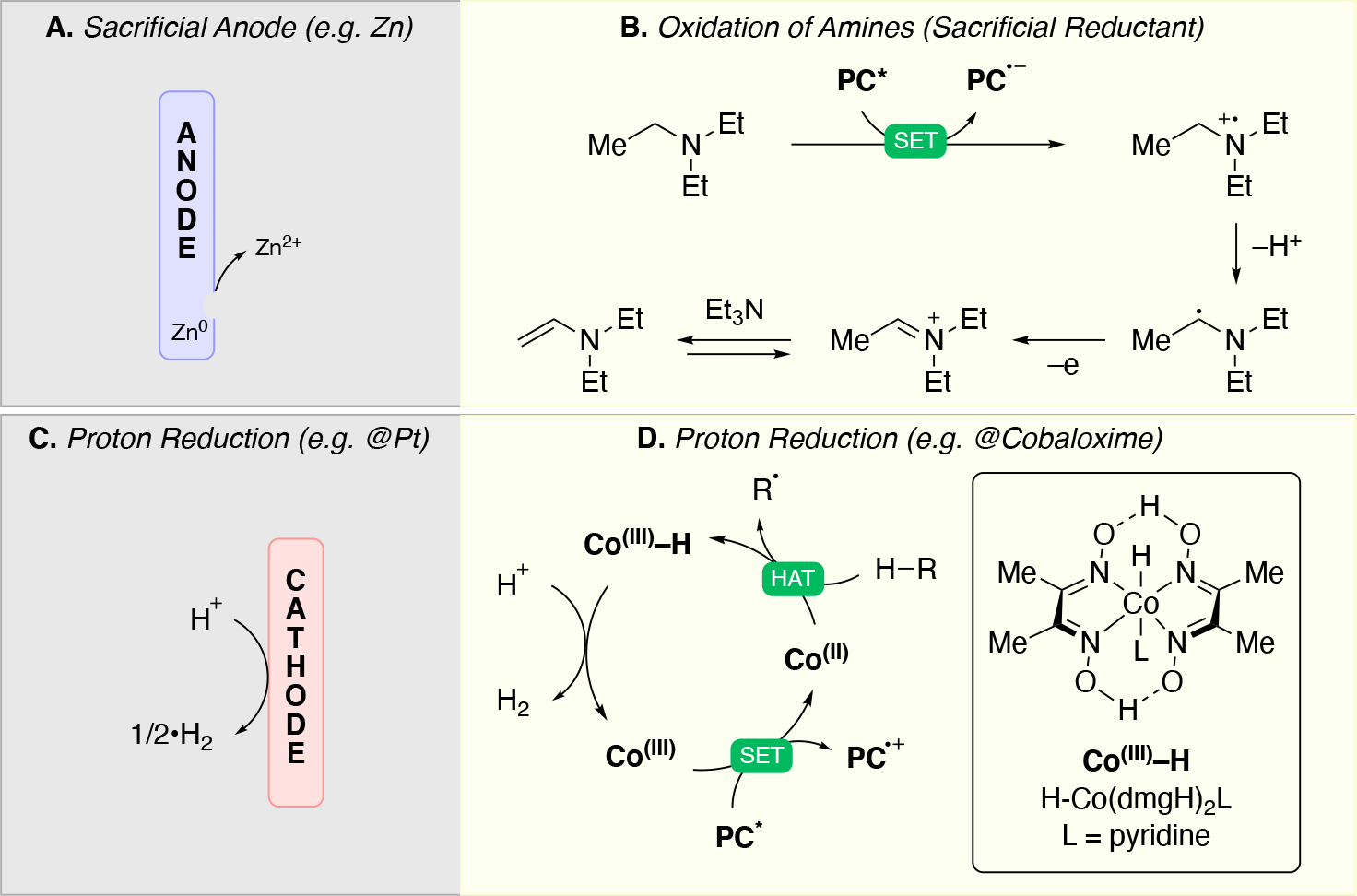
Illustration of a few common sacrificial half-reactions: (A) sacrificial anode (oxidation of metals), (B) sacrificial oxidation of amines, (C) reduction of protons to evolve hydrogen, and (D) proton reduction to evolve hydrogen via cobaloxime catalyst.
3.7. Driving Forces for Electron Transfer and Related Considerations
In section 3.1 to 3.5, we highlighted the similarities and differences between photoredox and electrochemical reaction mechanisms by emphasizing their advantages and disadvantages. This basic overview should enable the reader to select one or the other approach for their synthetic transformation of choice. However, in order to design a photoredox or an electrochemical process, a deeper understanding of the fundamentals of electron transfer and their driving force is useful, as it informs selection of photocatalyst, or electrode material and electrolyte.
Given that these two fields focus on the conversion of electron energy to chemical energy, its relevance to organic chemistry is that electrical and light energy can be converted to chemical energy (in kcal•mol−1) via interconversions of electron volt (eV) and light wavelength (λ) using the Nernst and Planck-Einstein equations respectively. Thus, we believe that developing an understanding of the relationships between eV, λ, and kcal•mol−1 (Scheme 8) will help organic chemists to appreciate and develop new ways of using electrochemistry and photoredox catalysis for chemical synthesis. Despite this similarity, the mechanisms for electron transfer in both methods are quite different, namely, that photoredox is typically limited to single electron transfer events, while electrochemistry can leverage multi-electron redox steps.
Scheme 8.
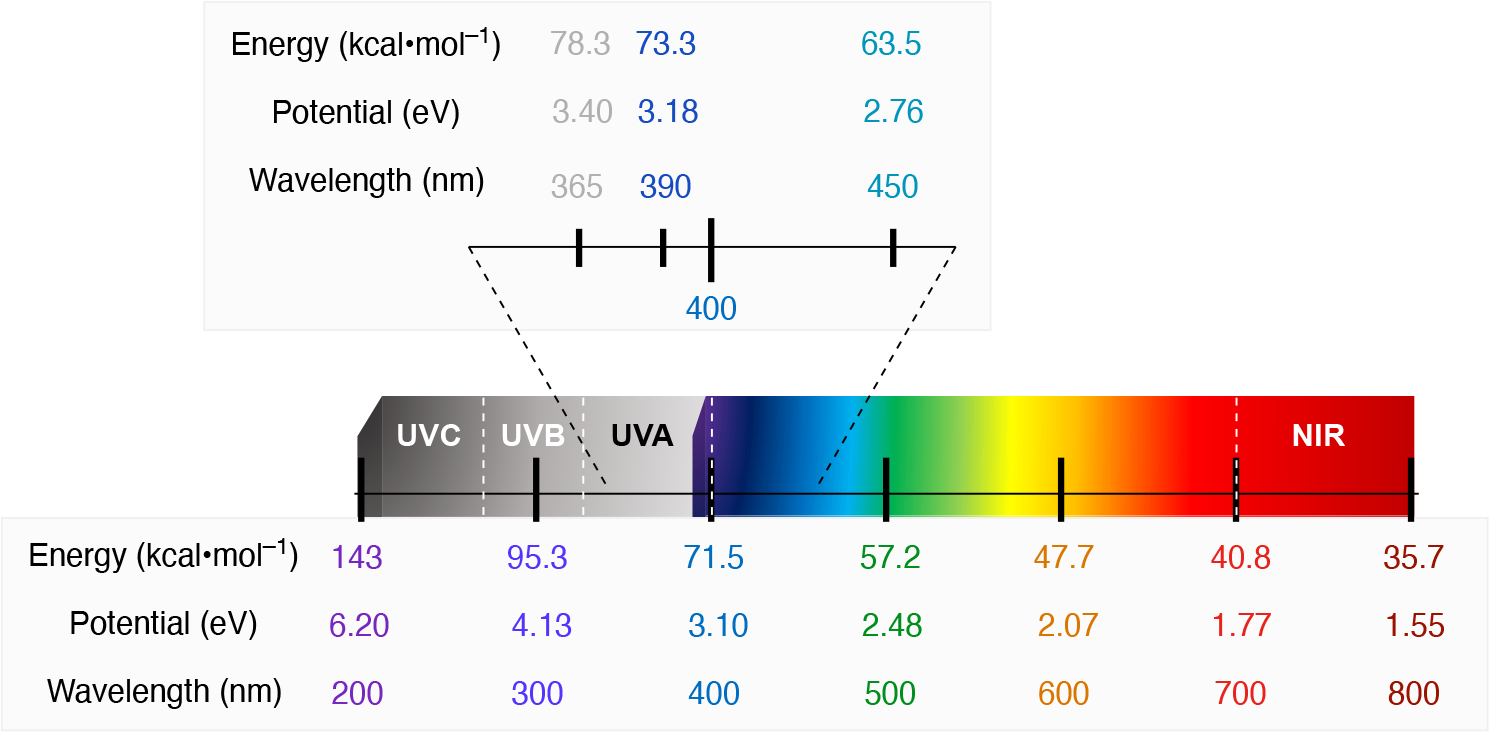
The relationship between chemical, electrical, and light energy. Insert shows a comparison of these relationships for three commonly-used wavelengths for photoredox catalysis. UV and NIR stand for ultraviolet (< 400 nm) and near-infrared (> 700 nm) respectively.
3.7.1. Photophysical and Electrochemical Considerations for Photoredox Catalysts
The photophysical processes of photoredox catalysts are arguably the most important factors in determining their photochemical reactivity (e.g. redox potentials, excited state lifetime). The behavior of the electronically excited photocatalyst dictates the feasibility and rates of electron transfer (ET), which in turn impacts substrate activation. Thus, any improvements to the efficiencies of reaction methodologies, or developments in catalyst design usually stem from a thorough analysis and understanding of photochemical mechanisms. While covering these photophysical processes in detail is beyond the scope of this review, this section will highlight the fundamentals of excited-state electron transfer and summarize the practical considerations that factor into choosing an appropriate transition-metal or organic photoredox catalyst. Readers interested in a deeper examination of the photophysics involving photoredox catalysis should consult a recent review by McCusker.108
A representation of the photophysics associated with the electron transfer processes for a general photocatalyst in its ground state (1A) is illustrated in a simplified state energy diagram (Figure 3A). 1A is electronically excited upon absorption of light (+hν), which then promotes an electron from its ground state (S0) to a higher energy singlet state. Vibrational relaxation of the promoted electron by non-radiative pathways leads to the lowest energy singlet excited state (S1) of 1A (1A*). The fate of this electron in 1A* is dependent on its radiative (light emission) and non-radiative (heat emission) pathways. Relaxation of the S1 via the fluorescence (radiative, −hν) or internal conversion (non-radiative) returns the excited electron to S0. Alternatively, the electronically excited electron in 1A* can transition to a lower energy triplet excited state (T1) by a spin-forbidden, non-radiative process called intersystem crossing (ISC) to form triplet species 3A*. Because ISC is a spin-forbidden transition, the lifetime for the triplet state 3A* is usually longer than its excited state singlet state 1A*. If no ET occurs, 3A* can relax to 1A via similar radiative (phosphorescence) and non-radiative pathways. Alternatively, energy transfer (EnT) mechanisms — Förster and Dexter — may be operative for a photocatalyzed transformation whereby an energy acceptor molecule (e.g. substrate) quenches a photoexcited chromophore. Distinguishing EnT and ET mechanisms typically requires time-resolved experiments, namely transient absorption spectroscopy.108 For the remainder of this review, we will assume ET as the major contributor to photocatalyzed transformations but also highlight examples where energy transfer mechanisms are invoked. Additionally, Figure 3B illustrates the merger between electrochemical potentials and the energies associated with the electronically excited and ground states of photoredox catalysis. The following discussions of the mathematical principles of electron transfer will use the energy state diagrams in Figures 3A and 3B as a graphical representation.
Figure 3.
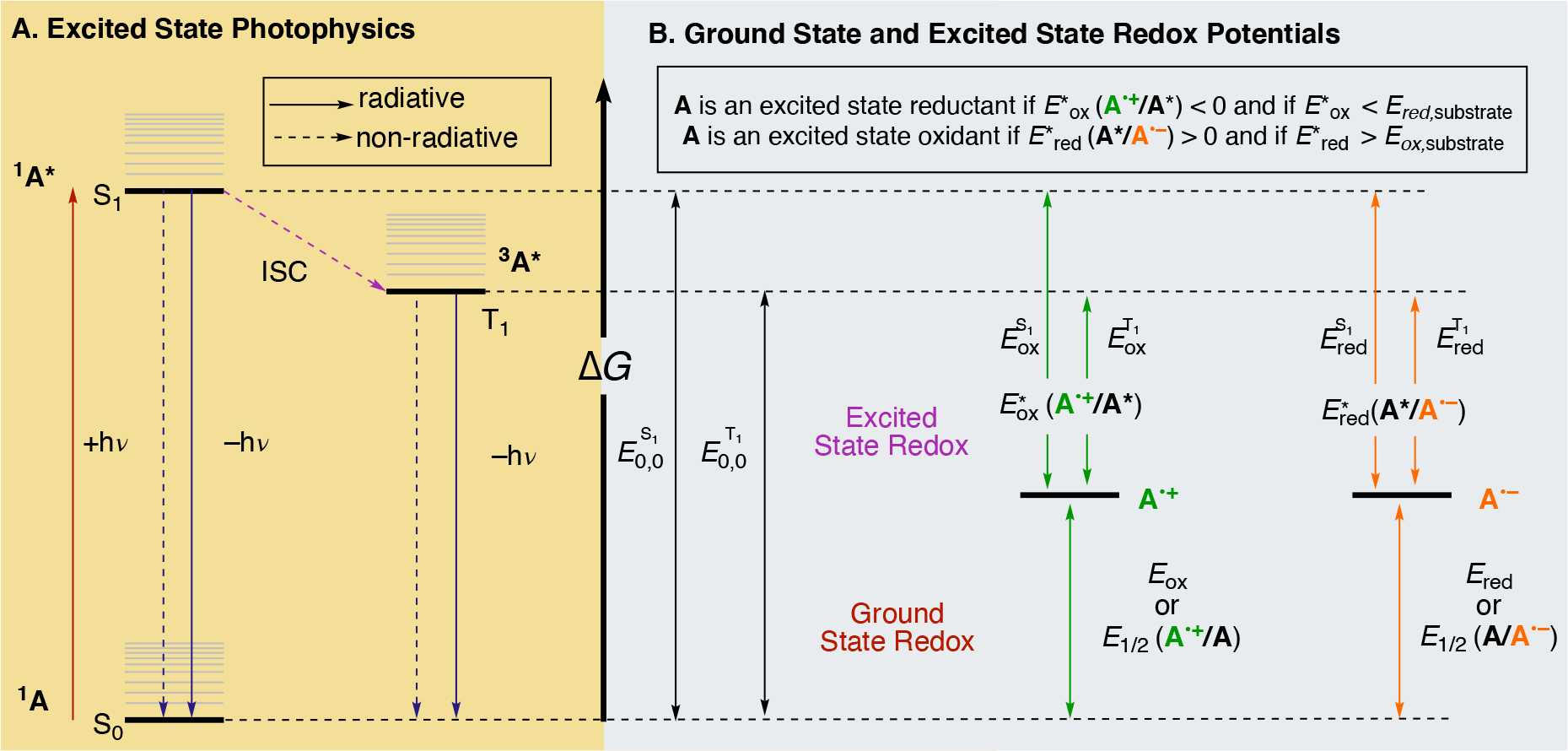
The intersection between photophysical (A) and electrochemical (B) properties of photocatalyst A. Diagram adapted from Romero and Nicewicz.18
In photoredox catalysis, obtaining appreciable substrate activation depends on effective PET. One important factor is the lifetime of the photoexcited catalyst, as the short-lived catalytic photo-oxidant or -reductant may be unable to engage in appreciable electron transfer. Thus, the empirical generalization is that the longer the lifetime of 1A* and 3A*, the greater the likelihood that photoexcited 1A* will undergo PET. For the excited state singlet 1A*, its lifetime is best approximated as its fluorescence lifetime (τf), which is measured using time-resolved emission spectroscopy. This assumption holds that photon emission occurs on faster time scales than non-radiative pathways, and in general, PET is observed when τf ≥ 1 ns because the rate of excited state decay is greater than the rate of diffusion.18 However, to accurately determine the likelihood of PET from the 1A* state, the fluorescence quantum yield (ϕf) must be measured as it reveals the likelihood of non-radiative deactivation pathways (i.e. internal conversion) or the prevalence of ISC from 1A* to 3A*. Confirmation of the latter mechanism is obtained if a high ISC quantum yield (ϕISC) is observed and would suggest that PET occurs from the 3A* state. Since decay from the triplet state via phosphorescence or internal conversion is much slower (a consequence of symmetry-forbidden transitions), the lifetime of 3A* will be several orders longer than that of 1A*.
Another important consideration is a thermodynamically favorable redox potential match between the donor and acceptor species associated with the reaction shown in equation 1b.
| (1b) |
In order to quantify the excited state and the ground state redox potentials for photoredox catalysts, both spectroscopy and electrochemistry are used. Equation 2, better known as the Gibbs energy of photoinduced electron transfer (ΔGPET), is a generalized equation typically used to determine the thermodynamic favorability of PET from A* to D (equation 1) and considers both the excited state (E0,0) and ground state (E°) energies of the species involved in photoinduced electron transfer (Figure 4). F in equation 2 is the Faraday constant (23.061 kcal•V−1•mol−1), while E1/2(A/A•−) and E1/2(D•+/D) are the reduction and oxidation potentials respectively for a single electron acceptor (A) and single electron donor (D).
| (2) |
Figure 4.
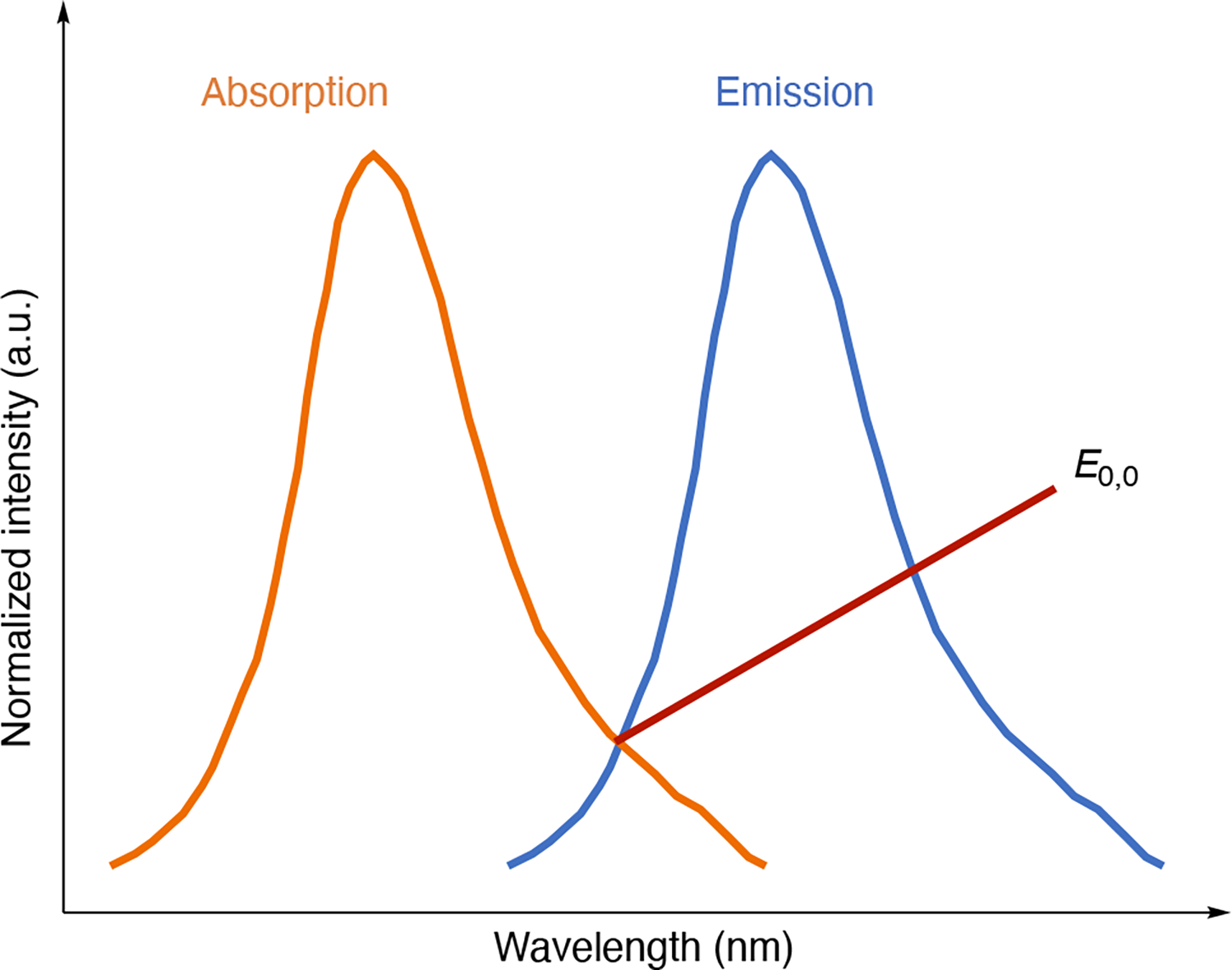
Illustration of how one estimates E0,0 of a photoredox catalyst from overlaid normalized absorption and emission spectra.
The excited state energy of the S1 state can be experimentally estimated from either 1) the intersection between the normalized absorption and emission spectra or 2) the midpoint between the absorption and emission maxima. The excited state energy of the T1 state is less trivial to determine and is often estimated using the phosphorescence maximum obtained under cryogenic temperatures.18 The work term (w) in equation 3 considers the solvent-dependent Coulombic attractions, but is typically omitted due to its relative insignificance in polar solvents. However, these Coulombic interactions have nontrivial impact on redox potentials in nonpolar solvents.109 Nevertheless, a good first approximation of ΔGPET can be obtained through this relationship between ground and excited state redox properties.
| (3) |
Electrochemistry is an invaluable tool for measuring ground-state redox potentials of organic and inorganic species. The free energy of electron transfer in the ground state is given by equation 1, where F is the Faraday constant (23.061 kcal•V−1•mol−1), and E1/2(A/A•−) and E1/2(D•+/D) are the reduction and oxidation potentials respectively for a single electron acceptor A and single electron donor D. We use the convention of writing half reactions as net single electron reductions; thus, oxidation potential of a donor refers to the half reaction D → D•+ + e, whereas the reduction potential of an acceptor molecule refers to the half reaction A + e → A•−.
In photoredox catalysis, the excited state chromophore (A*) acts as either an oxidant or as a reductant, and thus it is important to obtain its excited state reduction and oxidation potentials (Figure 3B). These values can be derived from equations 4 and 5, which describe the relationship between the energies of A and A*.
| (4) |
| (5) |
With and , we can finally determine the thermodynamic favorability of PET in photoredox catalysis. For A* and a generic substrate B, equation 2 can be further generalized for the two redox events by merging with equations 4 and 5. Equations 6 and 7 describe the free energy of PET for an excited state photooxidant and photoreductant respectively. If photoinduced oxidation of B is favorable, then . Similarly, if photoinduced reduction of B is to be thermodynamically allowed, then .
| (6) |
| (7) |
An important consideration for productive electron transfer is a hypothetical overpotential intrinsic to a photoexcited photoredox catalyst and the corresponding substrate. A moderate overpotential likely accelerates electron transfer, with Marcus theory predicting that the rate of electron transfer is at its maximum when −ΔGET = λ (i.e. the reorganization energy associated with reorganization of the nuclei from the equilibrium geometry of the reactants to the equilibrium geometry of the products, including reorganization of the solvent).110 However, drops in ET rates are observed at more negative free energies (the Marcus inverted region), with a nonadiabatic back electron transfer process being a putative relaxation mechanism (see next section for details).111,112,113,114,115 This observation also holds for proton-coupled electron transfer mechanisms (PCET).116
3.7.2. Comparison of Electron Transfer in Electrochemistry and Photoredox
The theory underlying electron transfer has been established for many decades and we draw attention to Marcus theory in particular and the reviews that discuss this topic in greater detail.117,118,119,120,121,122,123 A fundamental premise of Marcus theory is that the kinetics of single electron transfer can be derived from the potential energy wells associated with reactants and products. For a generic single electron donor and acceptor, there are three hypothetical scenarios for SET: (1) the donor being in the excited state and the acceptor being in the ground state (equation 1), (2) the acceptor being in the excited state and the donor in the ground state, and (3) both donor and acceptor are in their electronic ground states. Figure 5A illustrates the scenario for an ergoneutral SET, namely when the Gibbs free energy for electron transfer is zero, which leads to the barrier for SET (ΔGǂ) being the energy difference between the intersection of the substrate and product parabolas on the reaction coordinate and their absolute minima (ΔGǂ = λ/4). The ΔGǂ values for exergonic and endergonic SET processes vary (Figures 5B and 5C respectively) and are best determined using equation 8, which describes the barrier ΔGǂ as a function of the Gibbs free energy, ΔG, associated with the SET, and the reorganization energy (λ).
| (8) |
Figure 5.
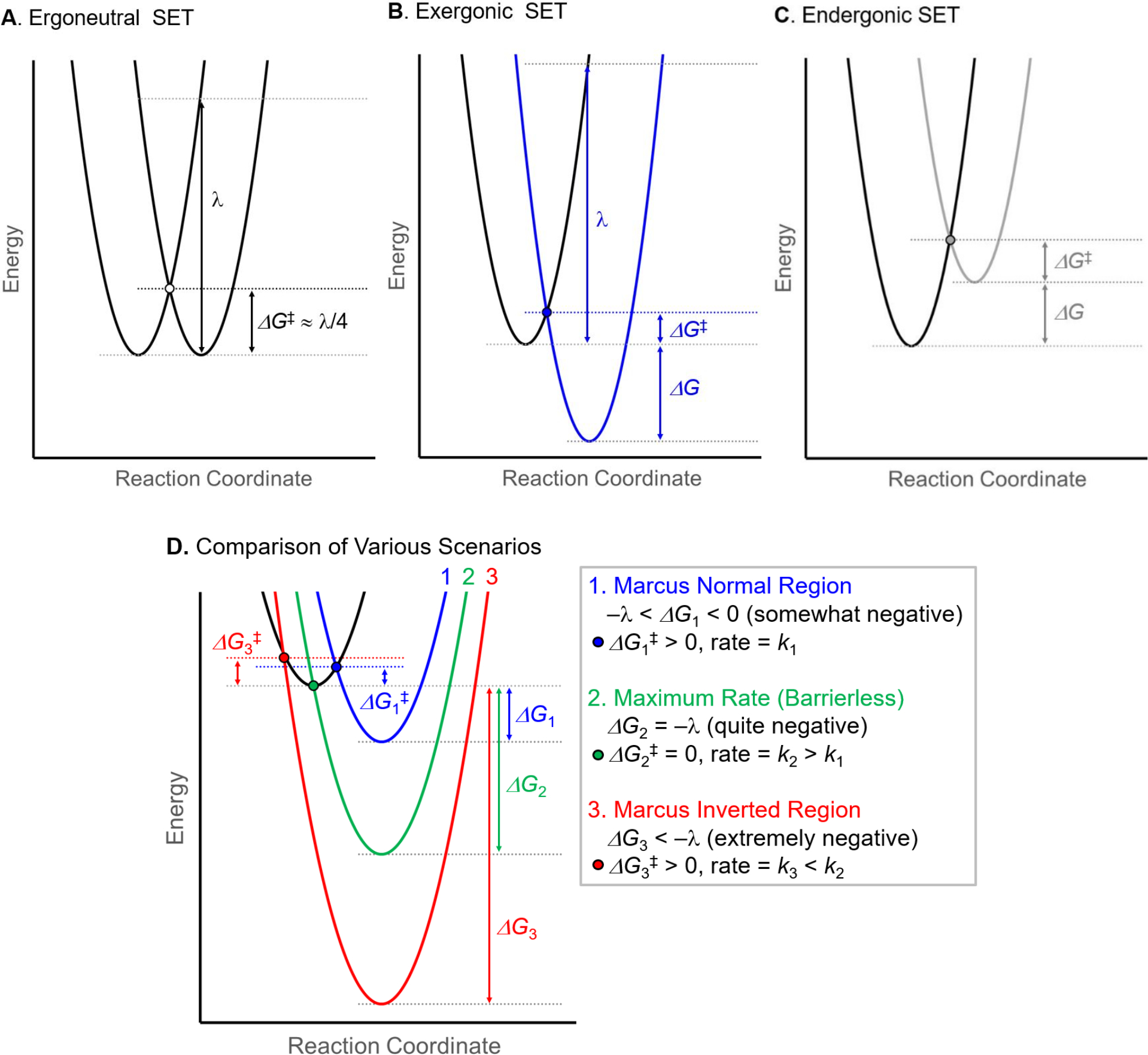
Graphical depiction of Marcus theory for (A) ergoneutral, (B) exergonic, and (C) endergonic single electron transfers. (D) A comparison of the potential energy curves for exergonic electron transfer involving the Marcus normal region (blue), a barrierless SET process (green), and slowed SET in the Marcus inverted region (red).
The rate of electron transfer predicted by Marcus theory is determined using equation 9. Note: ħ is Planck’s constant, HA,B represents the electronic coupling between the initial and final states, λ is the reorganization energy, kB is the Boltzmann constant (1.38064852 × 10−23 m2•kg•s−2•K−1), T is temperature (in Kelvin), and ΔG is the Gibbs free energy for SET. We encourage the reader to consult the aforementioned reviews for detailed analyses of SET using this equation.
| (9) |
Figure 5D illustrates the relationship between ΔGǂ and ΔG for SET through a comparison of three exergonic SET processes: (1) one with a small driving force (ΔG1 somewhat negative, as is shown in Figure 5B) which has a small barrier (ΔGǂ1) and is within in the Marcus normal region; (2) a barrierless SET event (ΔGǂ2 = 0) when the reaction driving force of ΔG2 = −λ, resulting in barrierless ET; (3) a scenario where a highly exergonic reaction (ΔG3 extremely negative, ΔG3 < −λ) will slow down SET — namely, the Marcus inverted region. This last situation is unique as it appears counterintuitive: reaction rates, including those for electron transfer, usually increase with decreasing ΔG values. However, in the Marcus inverted region for electron transfer, SET rates become slower, even if the reaction is highly exergonic, such that ΔGǂ3 > ΔGǂ1.
Scheme 9 illustrates the frontier molecular orbital of a substrate and those of a photocatalyst, and the Fermi level associated with the potential of the electrode, as well as the Gibbs free energy driving forces related to electron transfer (ΔGET). Equation 10 illustrates the relationship between the change in Gibbs free energy (ΔG) and the thermodynamically required cell potential (Ee), Faraday’s constant (F), and the number of electrons transferred (n) per molecule.124
| (10) |
Scheme 9.
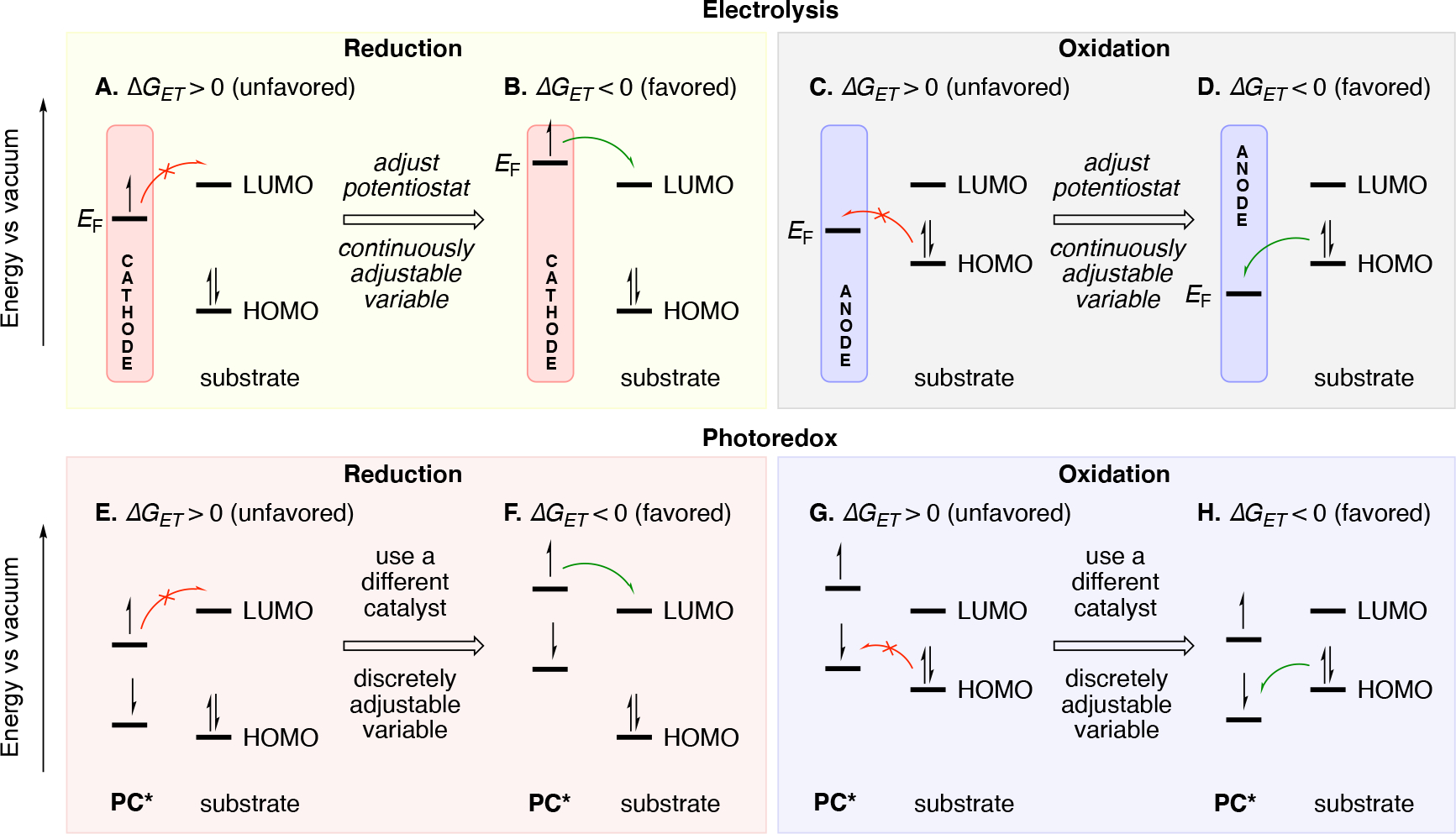
Illustration of various scenarios for associated with electron transfer events in electrochemistry and photoredox catalysis. For photoredox catalysis, only excited state electron transfer is illustrated. Additionally, unproductive back electron transfer can be operative, but is omitted from this figure for clarity.
In the context of an electrochemical reduction event, when the electron transfer from the electrode to the substrate (reduction) is favorable, the Fermi level of the cathode is at a higher energy than that of the LUMO of the substrate. Analogously the singly occupied molecular orbital (SOMO) containing the electron in the photocatalyst to be transferred to substrate should be higher than the LUMO of the substrate. For oxidation of a substrate, the Fermi level on the anode should be lower than that of the HOMO of the substrate. Analogously, the SOMO of the catalyst accepting the electron from the substrate should be lower in energy than that of the HOMO of the substrate. We note that the above criteria result in a negative change in Gibbs free energy, which at first hand the reader may think always implies it is spontaneous, but this is not always the case. Orbital overlap restrictions associated with the electron transfer can lead to cases (inverted Marcus regime) where the electron transfer becomes more challenging due to the change in Gibbs free energy being too negative.117,118 121 We also note that small positive Gibbs free energy associated with electron transfer can still lead to electron transfer if it is coupled with a subsequent irreversible chemical step (EC mechanism Scheme 3C).
Loosely speaking, electrochemistry has an advantage of being able to render any redox process thermodynamically favorable since it is possible to adjust the Fermi level of the electrode (Scheme 9) by simply adjusting the potential through external controls using a potentiostat (limitations on the electrochemical operating window do exist based on the choice of solvent, electrolyte, and electrode). In contrast, the analogous change in photoredox catalysis requires utilizing a different photocatalyst which has a set of more favorable redox properties, or varying the photocatalyst counterion (applicable only to solvents with low dielectric constants).109 An additional key difference is that the Fermi level of the electrodes can be adjusted in a continuous manner (Scheme 10A), whereas this variable cannot be adjusted in a continuous manner in photoredox catalysis, nor is it as simply adjusted (Scheme 10B). Careful choice of the photoredox catalyst is required in order to achieve the desired driving force (ΔGSET) for electron transfer from the photocatalyst to the substrate since the oxidizing or reducing potential of the photocatalyst are intrinsic properties (as discussed in Section 3.7.1). In order to achieve different redox potentials, a new catalyst must be chosen. This may require synthesis of a new catalyst or potentially there may be no catalyst structure known to have the desired redox potential. Thus, electrochemistry enables control over the electrochemical potential at each electrode, whereas photoredox catalysts do not allow for impromptu changes of its reduction and oxidation potentials since both are intrinsic properties of its different redox states. Finally, unlike photoredox catalysis which typically only enables a single electron to be transferred, electrochemistry usually enables multi-electron transfers as well as single electron transfer events (for a discussion of sequential single electron transfers, see Section 5).
Scheme 10.
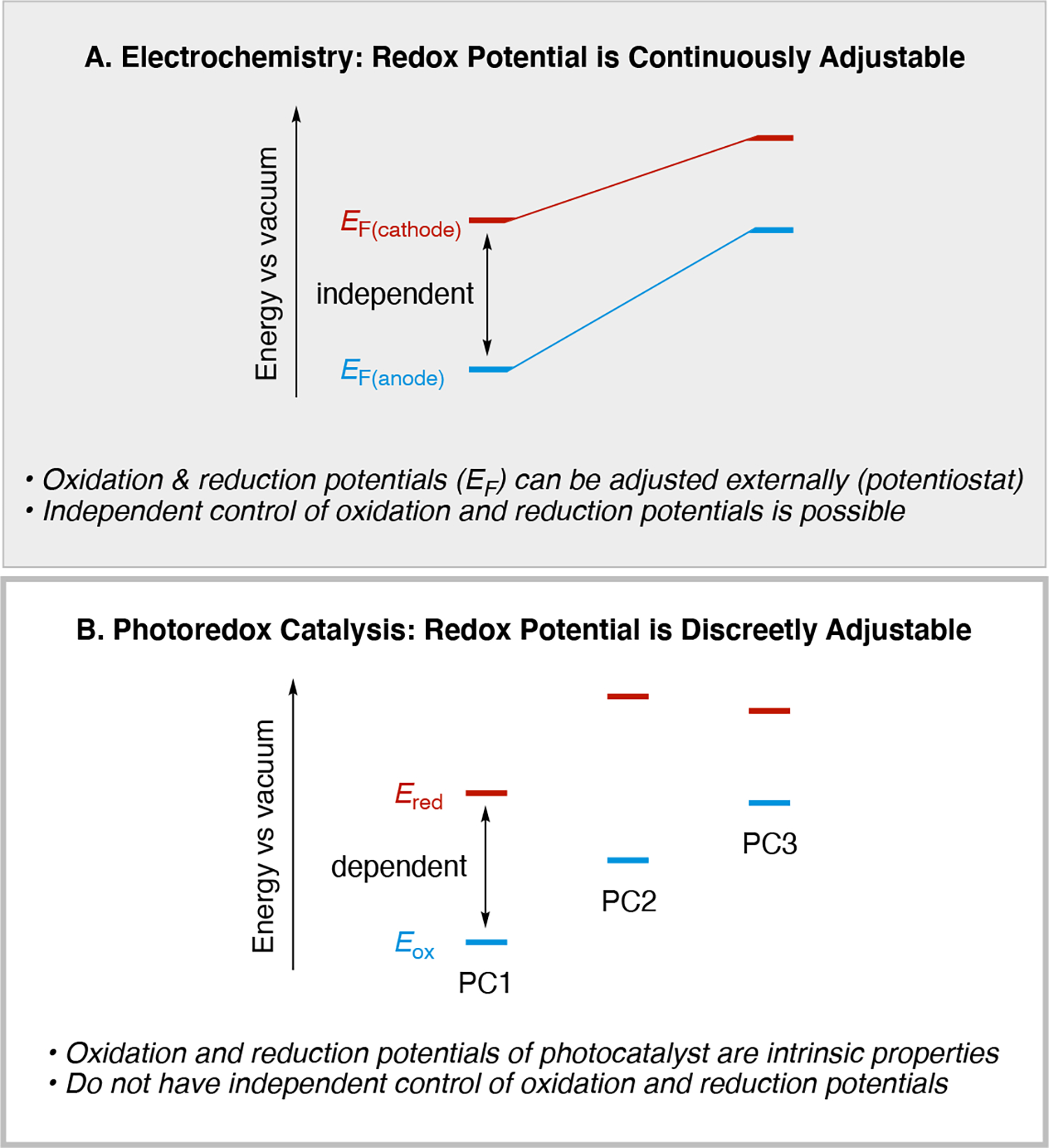
Illustration of how control over the redox potentials differs between (A) electrochemistry and (B) photochemistry. Note: EF(cathode) is the applied potential at the cathode and EF(anode) is the applied potential at the anode. Ered = E(PC*/PC•−) or E(PC•+/PC), and Eox = E(PC*/PC•+) or E(PC•−/PC), depending on the situation.
An underappreciated difference between electrochemistry and photoredox catalysis is the relationship between the energy source and its flux (rate). In photoredox catalysis, the energy inputted (wavelength) is decoupled from the photon flux (rate). In contrast, these are not decoupled in electrochemistry as evident from Ohm’s law (see section 3.7.7). For example, a large negative potential can be applied at the cathode potential but the only way to control the flux rate (current) is to either increase or decrease the resistance of the solution (solved with modifications to supporting electrolyte concentration or mass transport conditions). In contrast, photoredox catalysis enables the use of high-energy photons alongside a photocatalyst capable of being strongly reducing in its excited state, while still being able to control the rate externally via the photon flux by the selection of the light source’s power (Watts; e.g. 0.1 W vs 10 W).
3.7.3. Common Classes of Photocatalysts
Photoredox catalysts used in organic synthesis are typically transition-metal complexes or organic dyes. Ruthenium125 and iridium126 catalysts are often preferred due to their long-lived, redox-active triplet metal-to-ligand charge transfer (3MLCT) states and high reaction stabilities (Scheme 11A). However, the relative scarcity of these precious metals has invigorated the search for reliable alternatives that center on the more-abundant first-row transition metals.127,128,129 Alternatively, organic dyes18,130,131,132,133,134 provide a main-group centered solution, with customizable scaffolds enabling divergent oxidative and reductive PET (Scheme 11B). Several of the most commonly used photocatalysts and their respective redox potentials are shown in Scheme 11.135 Readers interested in alternate catalysts are highly encouraged to consult aforementioned references.
Scheme 11.
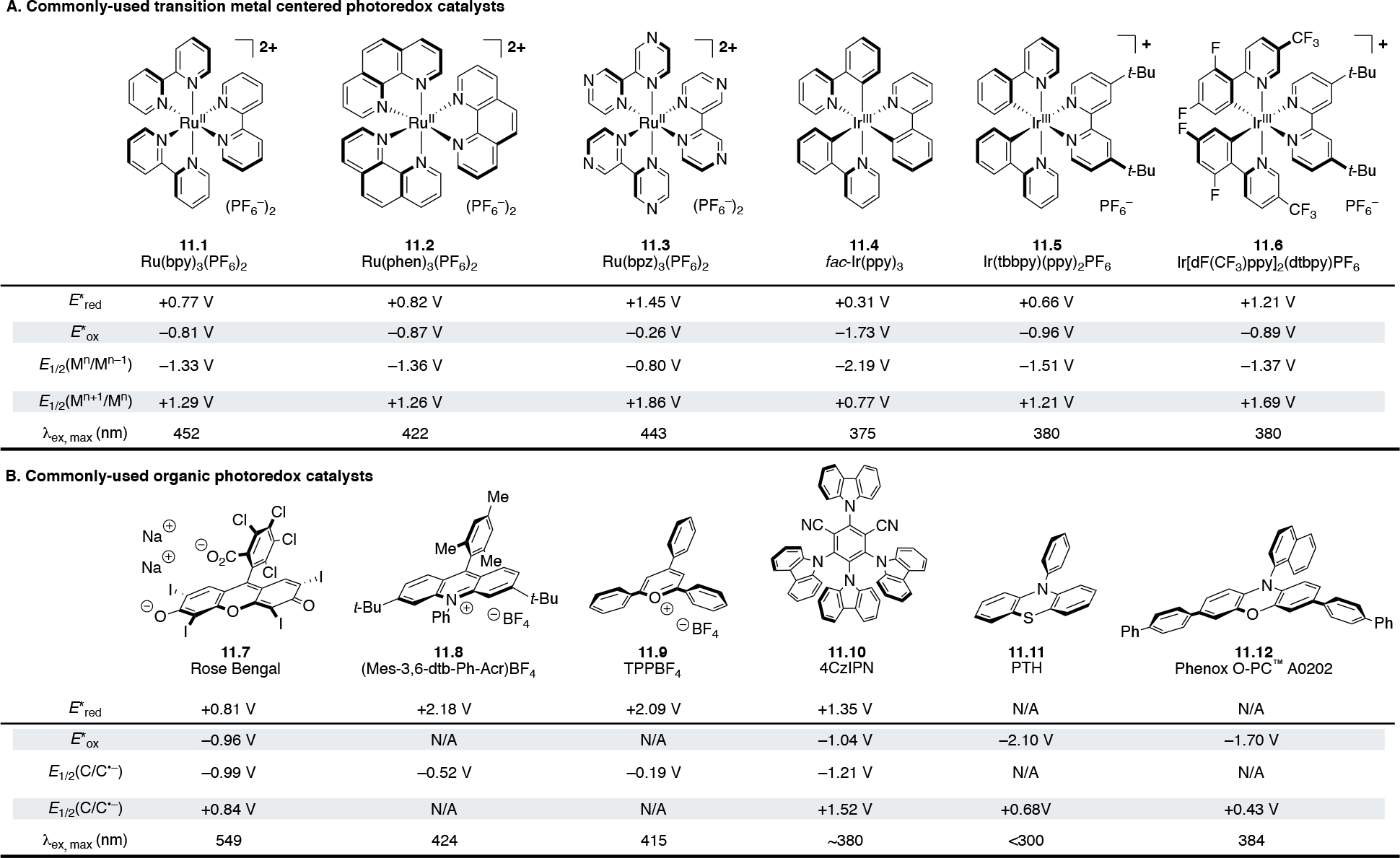
Selected photoredox catalysts along with their photochemical and electrochemical proprieties. All potentials are measured versus the saturated calomel electrode (SCE).
3.7.4. Electrode Materials
A key additional feature of electrochemistry, compared to photoredox catalysis, is the use of electrodes to facilitate redox processes at the electrode-solution interface (Scheme 2). The choice of electrode material can influence the reaction outcome in terms of yield, current efficiency, chemoselectivity, stereoselectivity, mechanism of electron transfer, and the number of electrons transferred to the substrate.136,137,138,139 Thus, the choice of electrode can be a critical parameter in optimizing electrochemical transformations. Electrodes can range from metals to carbon materials and beyond. Carbon based electrodes come in various forms, graphite (GRC), glassy carbon (GLC), reticulated vitreous carbon (RVC),140 and carbon felt are all commonly available. Graphite is one of the most commonly used anode material. RVC is a porous form of glassy carbon but suffers from being brittle, thus substitutes such as carbon felt are sometimes used, particularly on larger scale. Electrode selection may be influence in part by their robustness to the reaction condition. Electrode degradation of Pt,141 graphite,142,143 glassy carbon, 144 and boron doped diamond (BDD)145 anodes have been characterized in the literature. Cathodic corrosion can also result in degradation of cathodes and details on its process and mitigation strategies have been recently reviewed.146
Electrodes can be either passive or active. Active electrode materials have electrocatalytically active species on the surface of the electrode and akin to immobilized redox-active catalyst. These active electrodes can impart unique reactivity/selectivity beyond simple electron transfer. Active electrodes include NiOOH generated through the in situ oxidation of a Ni anode147,148,149,150 under alkaline conditions, and PbO2 anodes can be generated under acidic conditions. Waldvogel has demonstrated active anodes using Mo in hexafluoroisopropanol (HFIP),151 as well as active Ni in HFIP, for dehydrogenative arene couplings.152
The choice of electrode can play a key role in the energy required to facilitate electron transfer processes. Figure 6 illustrates the concept of overpotential for a redox event under a given set of conditions. Namely that even if the change in Gibbs free energy associated with a certain reaction is negative (thermodynamics), it is not given that the reaction will proceed at an appreciable rate (kinetics) – reaction barriers must be taken into account. Thus, additional potential, beyond the thermodynamically required potential (E1/2), is used to drive the reaction forward at an appreciable rate. This additional potential is referred to as the overpotential (η) and is given by equation 11:
| (11) |
where Eapp is the applied potential and E1/2 is the half-potential (thermodynamically required potential), each quantified relative to the same reference potential (Eref). The overpotential associated with the reaction occurring at the electrode is dependent on the electrode material (as well as reaction conditions). The overpotential associated with key sacrificial transformations such as hydrogen evolution reaction (HER, equation 12) and oxygen evolution reaction (OER, equation 13) is dependent on the electrode material (see Table 1 for a summary).
| (12) |
| (13) |
Figure 6.

Illustration of the breakdown of the cell potential as a function of the thermodynamic potentials and overpotentials for oxidation (left hand side) and reduction (right hand side) processes.
Table 1.
Conductivity and overpotential (HER and HOR) associated with various electrode materials.
| Electrode Material | Conductivity[a] /104 S•cm−1 |
η for H2 Evolution[b] in H2O /V | η for H2 Evolution in MeOH /V | η for O2 Evolution[d] in H2O /V |
|---|---|---|---|---|
| Ag | 68.17 | −0.59,172 −0.46,159 −0.444[h]154 | −0.21172 | 0.61173 |
| Al | 41.37 | −0.58159 | ||
| Au | 48.76 | −0.12,172 −0.430[h]154 | −0.20172 | 0.96173 |
| Be | 33.11 | −0.63160 | ||
| Bi | 0.93 | −0.33172 | −0.32172 | |
| Cd | 14.71 | −0.99,159 −1.225[i]154 | 0.80173 | |
| Co | 17.86 | −0.3 to −0.4174 | 0.39173 | |
| Cu | 64.81 | −0.46,155 −0.57,160 −0.60,159 −0.720[j] 154 | −0.32155, −1.1[m]137c | 0.58173 |
| Fe | 11.67 | −0.40159 | 0.41173 | |
| Ga | 7.35 | −0.63[c] 175 | ||
| Hg | 1.04 | −1.04,159 −1.475[k] 154 | ||
| In | 12.50 | −0.80160 | ||
| Ir | 21 | −0.030[j] 154 | ||
| Mg | 24.7 | |||
| Mo | 20.62 | −0.13,172 −0.30160 | −0.28172 | |
| Nb | 6.58 | −0.65,160 −0.80[k] 154 | ||
| Ni | 16.32 | −0.32,160 −0.434[h] 154 | 0.61173 | |
| Pb | 5.21 | −0.85,160 −0.91,159 −1.311[k] 154 | −1.7[m] 139 | 0.80173 |
| Pd | 10.22 | −0.01,172 −0.09,161 −0.044[h] 154 | −0.01172 | 0.89173 |
| Pt | 10.42 | −0.27,172 −0.09,159 −0.043[k] 154 | −0.19172 | 1.11173 |
| Pt Plated | NA | −0.01172 | −0.01172 | 0.46173 |
| Rh | 23.3 | −0.08,159 −0.042[l] 154 | ||
| Sn | 8.70 | −0.81176, −0.877[l] 154 | −1.3[m] | |
| Ta | 8.20 | −0.20,172 −0.41160 | −0.36172 | |
| Ti | 2.6 | −0.767[h] 154 | ||
| Tl | 6.67 | −0.61,172 −1.05160 | −0.44172 | |
| W | 20.75 | −0.11,172 −0.27159 | −0.32172 | |
| Zn | 18.3 | −1.092[k] 154 | ||
| Stainless Steel | 1.40177 | −0.42178 | 0.28178 | |
| Graphite | 0.0003, 0.4, 2.6 | −0.47159 | 0.50173 | |
| BDD | 10−6 to 0.002 | −1.5[e]179 | 1 to 2 [f] 179,180, 181 | |
| Glassy Carbon (RVC) | 0.02 to 0.10 | −1.13[g]182 | ||
| Leaded Bronze CuSn7Pb15 |
−1.5[m] 139 | |||
| PbO2 | −0.996[l] 154 |
conductivity data from reference 183 and 184, measured at 273 K (except Hg, glassy carbon, BDD which were measured at 298 K), unless stated otherwise.
HER overpotentials measured at measured in 1 M HCl or H2SO4 in solvent at 298 K and 1 mA•cm−2, unless stated otherwise
measured at 2 × 10−4 A•cm−2.
OER measured at 1 mA•cm−2 298 K, 1 M KOH in H2O, unless stated otherwise.
0.5 M H2SO4.
Value significantly depends on electrode doping and pre-treatment procedure.
pH 3.4.
measured in 1 M H2SO4 at 298 K and 200 mA•m−2.
measured in 0.25 M H2SO4 at 298 K and 200 mA•m−2.
measured in 0.5 M H2SO4 at 298 K and 200 mA•m−2.
measured in 1 M HCl at 298 K and 200 mA•m−2.
measured in 4 M H2SO4 at 298 K and 200 mA•m−2.
recorded in 2% H2SO4 in MeOH at 2 mA•cm−2.
We note that data for various electrodes based on alloy materials is available.153,154 It should be emphasized that these overpotentials are also dependent on other reaction parameters such concentration of the analyte/intermediate undergoing the redox event, solvent,155,156,157,158 supporting electrolyte,159 current density,160,161 temperature,162 as well as additional additives.137,163,164 Both the HER and the OER can be used as a non-productive reaction at the counter electrode when performing organic electrosynthesis. Experimentalist leverage electrode materials with low overpotential for these reactions if HER or OER is desired. In contrast, if the HER or OER is undesirable, choosing an electrode which has a high overpotential for these unproductive reactions may be desirable. Electrodes with high overpotential for proton reduction include Pb, leaded bronze,139 PbO2,165,166 Hg, Cd, Zn, as well as BDD.138,167,168,169 Hence, these are common choices in reductive electrolysis such as reductive dimerization of acrylonitrile to adiponitrile (Cd cathode),170 reduction of oximes (Hg, Pb or leaded bronze cathodes),171 deprotection of Ts groups on nitrogen (Hg cathode, see Section 4.18.2). For example, Lehnherr and Rovis leveraged the use of a Zn cathode, associated with a high overpotential for the HER, to avoid unproductive H+ reduction and favor productive reduction of substrates to access hindered primary amines via a reductive coupling of iminiums and cyanoheteroarenes (see Section 4.10). The authors demonstrated that other cathode materials with lower HER overpotential, such as Pt, were less efficient due to competing proton reduction.
The choice of electrode, electrolyte, and solvent can also influence the stereochemical outcome of reactions. An example of that is the electrochemical reduction of oximes (e.g. 12.1) to generate amines (Scheme 12),171 where the use of a Hg cathode favors the formation of diastereomer 12.2, whereas the use of a Pb cathode favors formation of diastereomer 12.3.
Scheme 12.

Illustration of diastereoselectivity change upon using different reaction conditions.
The product selectivity can also be influenced by the choice of electrode. The classic example is the use of graphite vs Pt in the oxidation of alkyl carboxylic acid. Pt favors the 1e oxidation process leading to Kolbe185 electrolysis, a 1e decarboxylative oxidation process leading to the generation of an alkyl radical, ultimately resulting in the dimerization of the alkyl radical. In contrast, the use of graphite results in a 2e oxidation process (Hofer-Moest reaction),186,187 forming an alkyl carbocation which can be trapped by nucleophiles such as acetate to afford an ester product (Scheme 13). While the photoredox catalyzed (1e) oxidative decarboxylation is well precedented, examples of dimerizing the resulting radical is not well precedented due to the low concentration of radicals in photoredox catalysis. A noteworthy photochemical example exists where dimerization of the radical generated from a carboxylic acid occurs, but it leverages a high-photon density light source (500 W Xenon lamp) which may provide atypically high concentration of the key radical.188
Scheme 13.
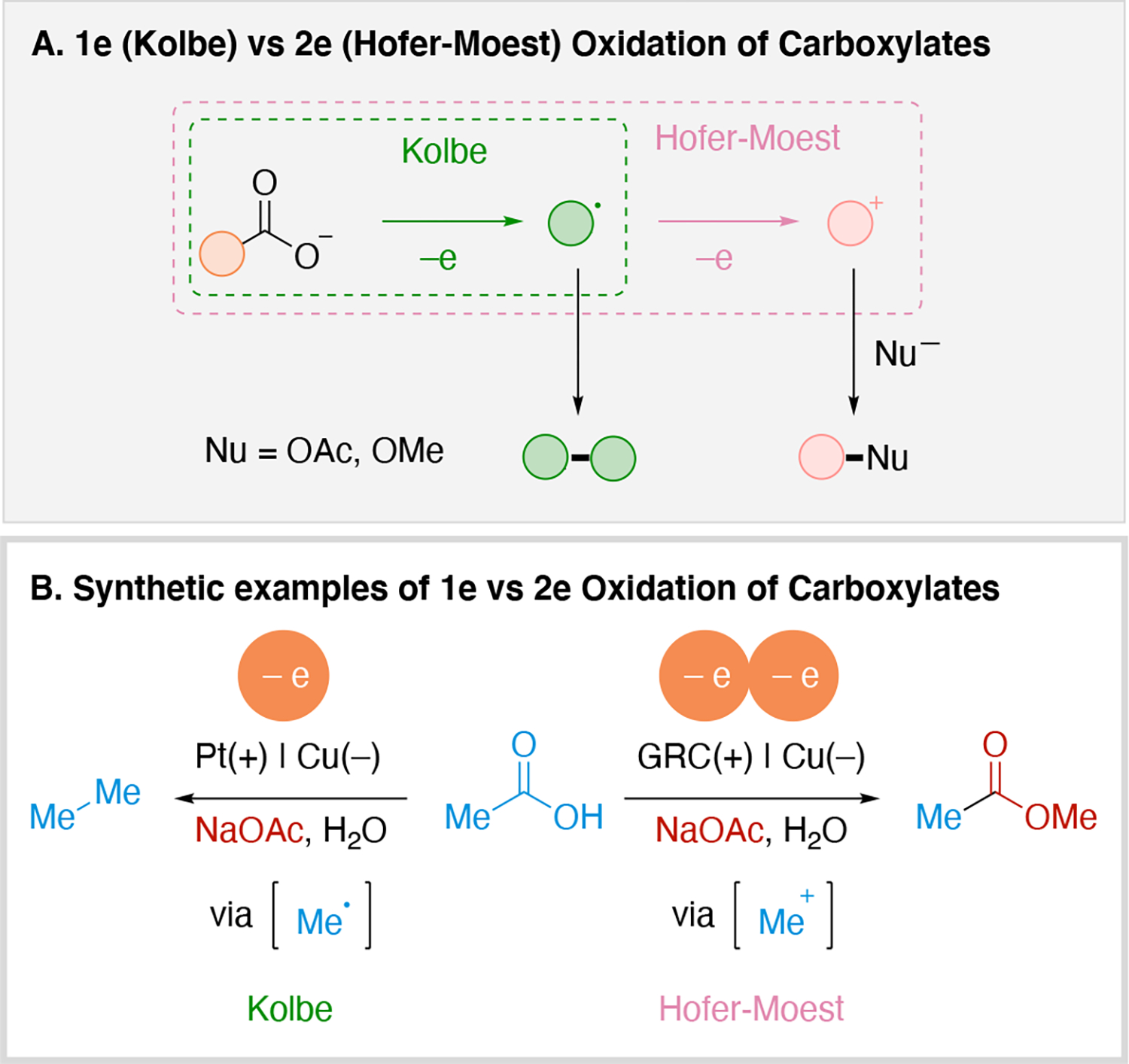
Illustration of 1e vs 2e oxidation of carboxylates.
Product selectivity in reduction chemistries can also arise based on the choice of electrode. For example, the reduction of acetone under protic conditions will result in mixtures that are dominated by isopropanol or propane depending on whether a Pb or Cu cathode is utilized. The reduction of acetone using lead cathode in acidic solution, predominantly leads to isopropanol formation, whereas the use of a zinc or copper cathode results in formation of propane.189
Finally, it is also possible to alter the regioselectivity outcome of reactions through the choice of an electrode material. One example is the reductive coupling of allyl chlorides and aldehydes shown in Scheme 14 which can be carried out in a micro-flow reactor.190 The use of Pt vs Ag cathode results in a reversal in regioselectivity, namely a 41:59 γ:α selectivity (14.1:14.2) with Pt whereas Ag cathode under identical conditions affords a reversal in the major isomer, specifically obtaining a 87:13 γ:α selectivity (Scheme 14A). Additionally, the use of laminar flow is demonstrated to be critical in achieving this selectivity reversal. It has been observed that the electrochemical allylation with aldehydes that have a higher reduction potential than the allylic halides results in predominantly the γ-adduct; in contrast, the α-adduct is obtained when the aldehyde is more easily reduced than the allylic chloride. By using laminar flow, streams of the two incoming reagents via inlet 1 and inlet 2 (Scheme 14A), can afford a choice as to which reagent is first exposed to the cathode, thus facilitating its preferential reduction to amplify this effect in flow mode A; in contrast flow mode B is unable to reverse the selectivity due to the aldehyde being reduced preferentially over the allylic chloride (Scheme 14C). For comparison to fully mixed reaction mixtures, the batch conditions using Pt cathode provide 29:71 ratio of 14.1:14.2 products in a combined 69% yield, highlighting the selectivity effect by using Laminar flow mode.
Scheme 14.

Illustration of regioselectivity change upon using different electrode materials as well as the effect on selectivity as a result of leveraging laminar flow delivering either allylic chloride or benzaldehyde reagent to the cathode.
3.7.5. Electrolytes and the Electric Double Layer
The addition of a supporting electrolyte (a salt) is typically required in order to minimize the resistance of the solution in organic electrosynthesis to enable the passage of an electrical current between the electrodes. The electrolyte should ideally be highly soluble and completely dissociate into cations and anions in the reaction mixture. Common cations in electrolytes include Li+, K+, tetraalkylammonium (R4N+) and sometimes tetraalkyl phosphonium (R4P+). Common anions in electrolytes include Cl−, Br−, PF6−, BF4−, TsO−, BzO−, AcO−, TfO−, ClO4−, (SO2CF3)2N−, B(ArF)4− and MeOSO2−. The electrolyte composition and concentration, as well as the pH of the mixture, all can influence the nature of the electrode-electrolyte interface which can influence rates of electron transfer as well as product selectivity outcomes. The interface between the electrode and the bulk solution features a unique environment. Not only is the solution experiencing a potential gradient which can induce polarization effects (e.g. alignment of dipole of the substrate) but this also leads to the formation of an electric double layer (EDL), also referred to as the Helmholtz double layer (Scheme 15A). While a number of models have been put forward (e.g. Helmholtz, Stern, Gouy-Champan),191 the details can depend on experimental conditions such as electrode material, solvent and electrolyte composition. The negative potential of the cathode provides a driving force for cations to self-assemble on the surface of the cathode, with a second layer of ions comprising of anions to balance charge, thus forming the EDL. In cases where large excess of supporting electrolyte is present relative to substrate, the electrolyte comprises of significant portion of these cations and anions in the EDL, in which the substrate may be interspersed. The choice of the electrolyte can govern whether solvent, protons, or a specific substrate can interpenetrate this EDL, thus imparting selectivity in which substrate may be closer to the electrode and thus facilitating electron transfer with those substrates. Tuning the atomic/molecular radii of the electrolyte’s cation and anion can impart effects on the redox potential of species in solution as well as the reaction outcome in part due to changing the polarity of the EDL (polar vs greasy) as well as ion-pairing strengths.192,193 Small cations have a higher charge density, which leads to stronger interaction with anions in solution. Tetraalkyl ammonium cations can be tuned in size simply by changing the alkyl chain length (e.g. NMe4, NEt4, NBu4 cations, Scheme 15B).194 Analogously smaller anions, such as chloride, interact strongly with cationic species unlike large, non-coordinating anions such as tetrakis(3,5-trifluoromethylphenyl)borate (B(ArF)4) anions (Scheme 15C). Solubility considerations in choosing an electrolyte are critical in order to ensure it can contribute to lowering the resistance of the reaction mixture.195,196
Scheme 15.
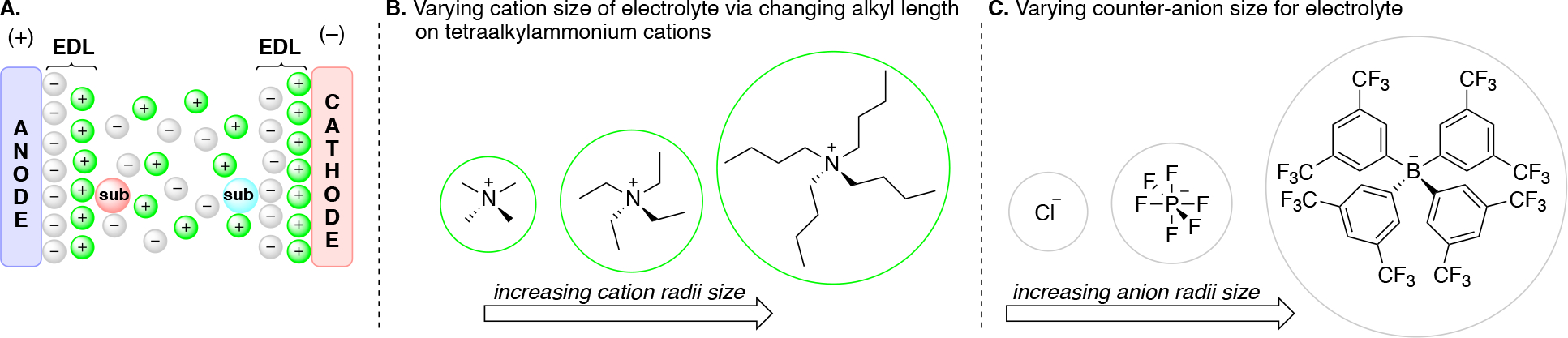
(A) A simplified representation of the electric double layer at the anode and cathode. (B) Schematic representation of cation size increasing with longer alkylchains on tetraalkylammonium derived electrolytes. (C) Schematic representation of increasing anion radii size from Cl to PF6 to B(ArF)4 anions.
Quaternary ammonium salts are often used to avoid proton reductions at the cathode, as these can help exclude protons from the EDL and effectively aid in increasing the overpotential for proton reduction associated with the electrode material used.163 These EDL related effects can loosely be thought of analogous to solvent effects imparting changes in reactivity behavior and the EDL is a unique aspect not present in photoredox catalysis. The choice of electrolyte also influences the double layer capacitance which can impact current efficiency in electrochemical transformation, and this double layer capacitance197 can play an important role in optimizing parameters for alternating current electrolysis.198,199,200 It is therefore no surprise that the choice of electrolytes can influence the reaction outcomes (yield, selectivity, current efficiencies,201 etc.),202 including in bioelectrochemistries in part related to the Hofmeister effect.203 Advances in the interfacing of analytical techniques have even allowed for in situ probing of electrode-electrolyte interface by time-of-flight secondary ion mass spectrometry (ToF-SIMS),204 as well as understanding electrode fouling in situ.205 Other recent advances in photoelectron-based spectroelectrochemical methods are enabling the study of these electrode/electrolyte interface effects.206
3.7.6. Mechanisms of Catalyst-Mediated Electrolysis
Electrocatalysis is the term traditionally used to refer to reactions in which the electrode is chemically involved in the catalytic process.207 The properties (e.g. chemical, crystallographic features, surface defects) of the electrode material may impart catalytic efficiency and selectivity, the bulk properties of the electrode may not be as important as its surface properties which directly interface with the reaction mixture. In contrast, the use of molecular catalysts refers to the use of molecule either in solution or immobilized on the electrode surface (monolayer or multilayer) to impart catalytic properties on the reaction.207 The use of a catalyst in electrochemical transformations can facilitate either an electron transfer step and/or chemical step.208 Schemes 16A–E illustrates various scenarios in which the catalysts itself may be redox-innocent (Scheme 16A–C) or redox active (Scheme 16D–E).209 Scheme 16A illustrates how a catalyst may bind to substrate to increase the HOMO energy of the substrate, analogously a catalyst can bind substrate and lower the LUMO energy thus facilitating reduction (Scheme 16B). Another example of redox innocent catalysis may involve catalyzing chemical steps after the redox event (Scheme C). In contrast, redox-active catalysts undergo redox events.210,211,212 Scheme 16E illustrates the use of a redox mediator to oxidize a substrate (facilitating only redox events), whereas (Scheme 16D) illustrates a redox-active catalyst also participating in catalyzing chemical steps (Scheme 16E). While Scheme 16A, 16C–E illustrate anodic processes, the analogous cathodic scenarios are also possible scenarios.
Scheme 16.
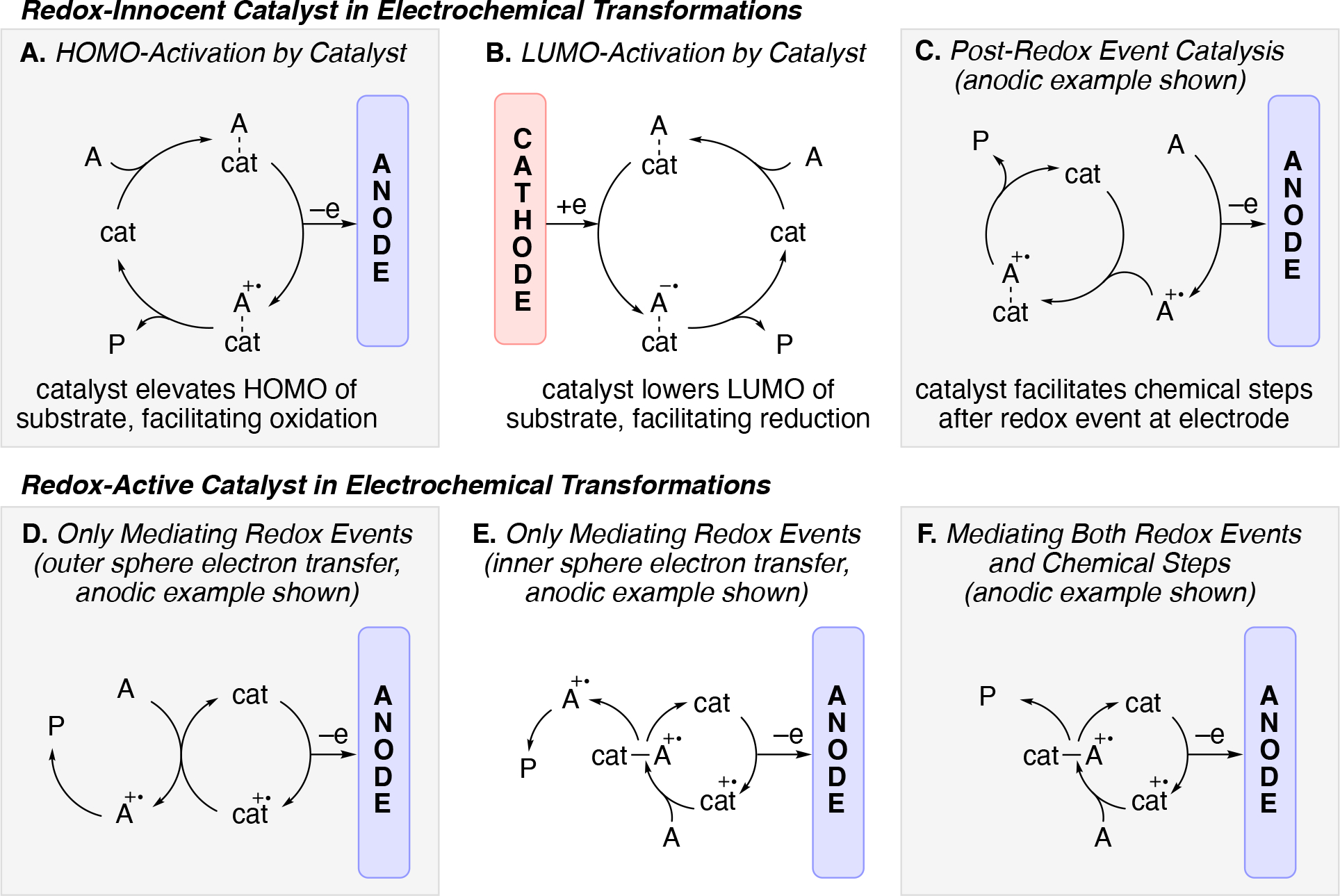
Illustration of several common catalyzed manifolds of electrochemical reactions using redox-innocent catalysts (A, B) and redox-active catalysts (C, D, E). (A) HOMO-activation of substrate by a catalyst (facilitates oxidation), (B) LUMO-activation of substrate by a catalyst (facilitates reduction), (C) catalyst only facilitates redox events (outer sphere electron transfer) (D) and (E) catalyst only facilitates redox event (either outer or inner-sphere electron transfer), (F) catalyst facilitates both redox and chemical events. [All redox events are shown as single electron events but redox events involving >one electron also fall into these categories].
The use of a redox-active catalyst in electrochemical transformations can impart a number of benefits.210,211 The passivation of electrode surfaces commonly observed in direct electrolysis can be suppressed. Passivation or electrode fouling can lead to significant increase in resistance during electrolysis and hamper scalability of such reactions. Redox reactions at electrodes are often slow electron transfer processes, particularly when the redox event imparts a bond breaking/forming event and involves more than one reactant/product and subsequent redox events, due to the presence of a large overpotential. In order to drive reactions at meaningful rate a large excess of energy must be applied to overcome this overpotential.210 This overpotential problem (and associated increased energy consumption) is exacerbated upon scaling up direct electrolysis, thus the use of a redox mediator is often sought to decrease this overpotential (and reduce energy consumption), typically resulting in improved selectivity for the functional group targeted to undergo the redox event as a result of the lower cell potential. The selectivity and reactivity between the various functional groups in the substrate is mainly controlled by the difference in the redox potentials of the various groups and that of the electrode potential. In contrast, the use of redox mediator may bind substrate to facilitate redox at a specific functional group (inner sphere electron transfer) thereby impart further improved selectivity. It should be emphasized that it is not uncommon to use a redox mediator with a standard potential that is significantly lower (up to ca. 600 mV) than that of the substrate’s standard potential if the redox process on the substrate is followed by a fast and irreversible chemical reaction, highlighting how to render the process milder compared to direct electrolysis. The number of electrons in the redox event associated with the substrate can be controlled by the choice of the redox mediator, which can be beneficial when aiming for selectively performing a 1e vs 2e redox process (e.g. ferrocene vs quinone, respectively).213 From a green chemistry perspective, the use of stoichiometric redox reagents is problematic not only in terms of the waste generated from these reagents at the end of the reaction but also the additional waste/energy required to separate these from the products. In contrast to reactions using a stoichiometric redox reagent where the concentration of that reagent varies as a function of the extent of reaction, this is not the case in electrochemical reactions using a redox mediator where its relative concentration can be maintained at an optimized concentration. The partitioning of the total concentration of the redox mediator can be estimated by using the Nernst equation64 which takes into account both the applied potential and the standard half-cell potential associated with the redox mediator. Direct electrolysis faces a related problem of having a significant concentration gradient as a function of distance from the electrode which may be problematic in some context. Given the above benefits, it is of no surprise that significant efforts have been devoted in the development of redox-active catalysts for electrochemical transformations. Redox mediators should have the following desirable characteristics: (1) both its oxidized and reduced form should be chemically stable, else catalytic activity will be lost, (2) the electron transfer with the electrode and the substrate should be fast and reversible, (3) redox events with solvent or other compounds in the reaction mixture, aside from the targeted substrate, should not occur, and (4) both the oxidized and reduced form of the mediator should be soluble in the reaction mixture (except if it is a biphasic system). A variety of methods have been developed for simulating and interpreting experimental data, including cyclic voltammetry, towards elucidating reaction mechanism of catalyzed (and uncatalyzed) electrochemical transformations.68,214,215,216,217
A few redox mediators are highlighted here in Scheme 17, but a more comprehensive list can be found in other reviews.55,210,211,212,218,219 Polycyclic aromatic hydrocarbons and viologens serve as redox mediators for electrochemical reductions, while quinones (typically used in the presence of a proton source)220,221 metallocenes (e.g. ferrocene derivatives),222,223,224,225 and triarylamines226 are used for oxidation chemistries. Aminoxyl derivatives like TEMPO and ketoABNO are prototypical oxidation catalysts via their oxidized oxoammonium form for hydride abstraction in alcohol oxidation chemistries.212,227 TEMPO and structurally related analogs have also been used in the diazidation of alkenes,228 as well as Pd-catalyzed Wacker oxidation of alkenes.229 N-hydroxyphthalimides (typically used in the presence of base) have been used for hydrogen atom transfer (HAT) catalysts for generation of benzylic and allylic radicals from their corresponding C–H precursors. Other commonly used HAT catalysts include quinuclidine (and related structures like DABCO and 3-acetoxyquinuclidine),230,231,232 thiols,233 benzoate salts (Na or Bu4N as the cation),234 thiobenzoates,235 sulfonamides,236 phosphates,237,238 halides,17 zwitterionic amidates, or alcohols,239,240 as well as others,241,242,243,244,245 all of which can be oxidized either by the anode or a photoredox catalyst to generate their oxidized form capable of doing hydrogen atom abstraction. These method of indirect HAT are in contrast to direct HAT approaches such as those using photocatalysts like decatungstate derivatives (e.g. Na4W10O32, (Bu4N)4W10O32) which can rely on triplet reactivity for HAT.246,247 Other prototypical redox mediators include halides, metallocenes (ferrocenes and cobaltacenes) and more recently the use of organic dyes more commonly associated with photocatalysis are emerging as useful classes as well (e.g. methylene blue, acridiniums, cyclopropeniums, dicyanoanthracene, perylenediimides).
Scheme 17.
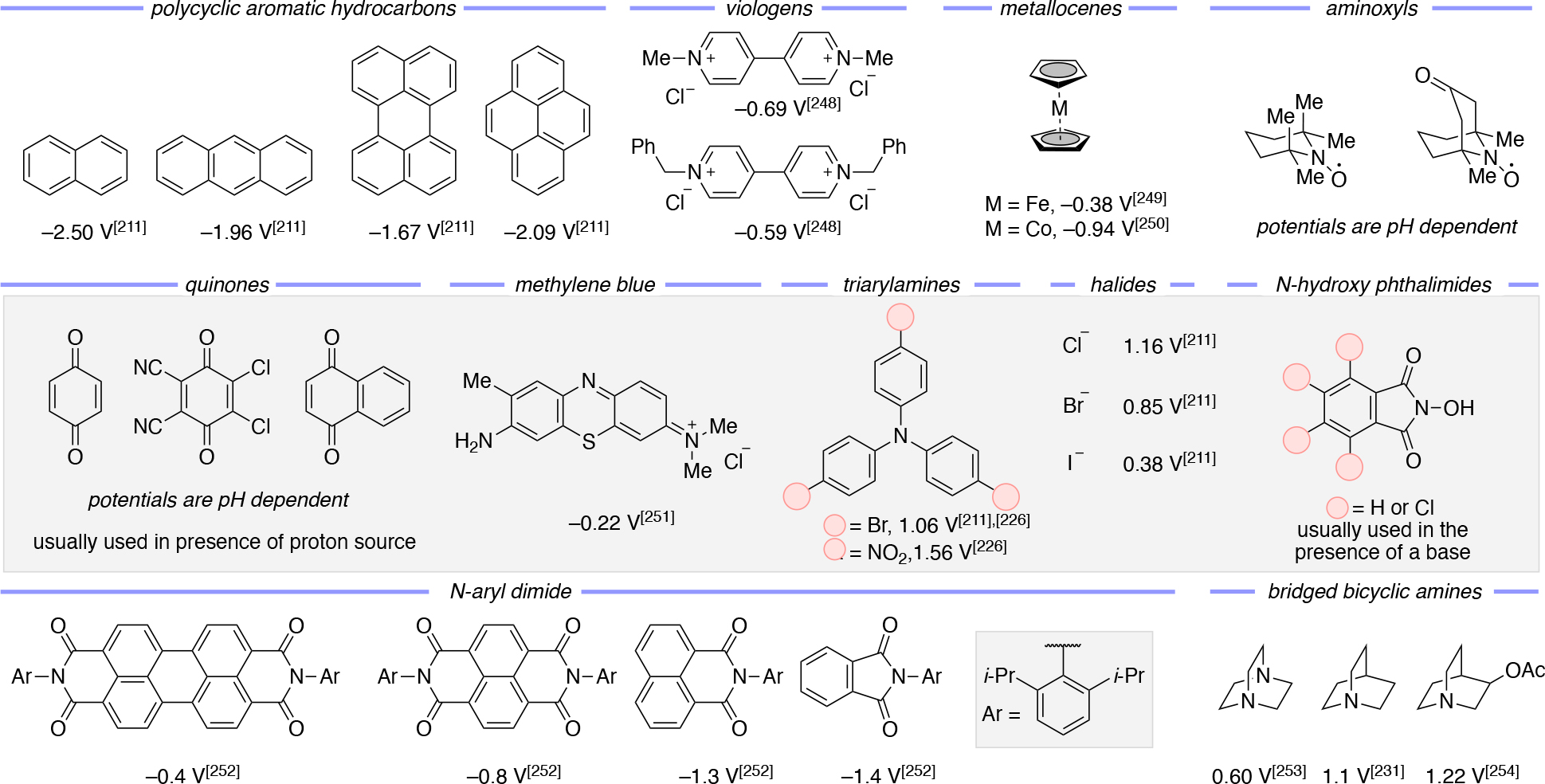
Structures of some key redox mediators along with their redox potentials (in V vs SCE).211,226,231,248,249,250,251,252,253,254,255
3.7.7. Quantifying Efficiency and Other Key Metrics
The internal energy efficiency of photo and electrochemical reactions are captured by the quantum yield256,257,258 and faradaic efficiency,64,259,260 respectively. The quantum yield of a radiation-induced process refers to the number of times a specific event occurs per photon absorbed by the system.261 For a photochemical reaction, the quantum yields can be calculated for product formation or for starting material consumption. The quantum yield for product formation (ϕproduct) is defined the number of product molecules generated divided by the number of photons absorbed by the starting material (equation 14b). Analogously, the quantum yield for starting material consumption (ϕreactant) is defined the number of reactant molecules consumed divided by the number of photons absorbed by the reactant.
| (14a) |
| (14b) |
A quantum yield less than 1 is the result of non-productive consumption of photons. This can arise from numerous types of events such as the photocatalyst excited state relaxing to ground state via either an internal conversion mechanism (e.g. conversion of energy into heat via vibrational modes of the molecule), luminescence (e.g. fluorescence from a singlet excited state or phosphorescence from a triplet excited state), back electron transfer (e.g. electron transfer from the acceptor back to the ground state oxidized form of the photocatalyst after a reductive quenching event), or other side reactions which by definition do not lead to product formation. A quantum yield greater than 1 can indicate the presence of a chain reaction mechanism (e.g. radical chain, hole catalysis).262 Finally we note that the quantum yield can depend on reaction conditions, such as concentration, temperature, as well as the extent of conversion.263
To quantify the efficiency of electrochemical reactions, we can either measure the instantaneous current efficiency (, equation 15), or the overall charge efficiency (), as known as the Faradaic efficiency (equation 16.25,264 The instantaneous current efficiency can be thought of as a ratio of the current that participates in generating desired product, the productive current (), and the total current which takes into account the current that participates in unproductive reactions, the unproductive current (). In contrast, the overall charge efficiency (), sometimes referred to as coulometric efficiency,265 is the is the charge consumed to form the desired product and is the total charge consumed. Thus, the current efficiency and Faradaic efficiency are each a number between 0 and 1. Although these metrics each can be reported as a percentage.25,264 A current efficiency of unity represents perfect current efficiency, where only the desired process is occurring for each charge transfer event at the electrode. For most electrochemical reactions, current consumption efficiency is usually < 100%, with side reactions or non-faradaic processes (e.g. double layer charging) consume current parasitically.
| (15) |
| (16) |
The relationship between current and charge is described below using Ohm’s law, where the potential of a cell (, given in volts [V]) is equal to the product of the current (i, given in amperes [A]) and its resistance (R, given in ohms [Ω]), as shown by equation 17. We note that the cell potential is the difference between the anode and cathode potential ( and , respectively).
| (17) |
From equation 17 it becomes clear that if the electrical resistance of a reaction is changing over the course of the reaction (e.g. as starting material is consumed, electrical resistance increases are common), then it is impossible to keep both the current and the potential constant for the entire duration of the reaction. Hence, organic electrochemical transformations are typically performed using one of the following techniques: (a) constant current electrolysis (CCE), also known as galvanostatic electrolysis, in which the current is constant throughout the duration of the reaction (the potential is allowed to vary); (b) constant cell voltage, in which the cell voltage () is constant throughout the duration of the reaction (the current is allowed to vary); or (c) constant potential electrolysis (CPE), also known as potentiostatic electrolysis, in which the potential of the working electrode is held constant throughout the duration of the reaction (either the potential of the anode () is kept constant, or the potential of the cathode () is kept constant) while the current is allow to vary over time.
Constant current electrolysis is operationally simpler when exploring new substrates/reactions because no prior knowledge is required with regards to the threshold potential required to affect a redox event. The potential will dynamically adjust until a redox event occurs at each of the electrode-solution interface to enable current flow through the solution. These experiments will usually exhibit an increase in the cell potential () as the reaction progresses due to a gradual decrease in concentration of the starting materials (species able to undergo the more facile redox events), as illustrated in Figure 7. As a result, the increased cell potential can lead to undesired side reactions, particularly near the end of electrolysis. In contrast, using constant potential electrolysis not only requires the user to know what potential is necessary to affect the redox event targeted but also requires the use of a reference electrode to accurately achieve this electrode potential. Constant potential electrolysis does have the advantage of being able to dial in a potential that is just enough to reduce or oxidize the functional group of interest, while keeping the magnitude of the potential small enough to minimize side reactions. Improved selectivity can be achieved with CPE but can require significantly longer reaction times to achieve the same level of starting material consumption compared to CCE.
Figure 7.

Illustration of how the electrode potential (anodic potential in this example) rises as a function of conversion in a constant current electrolysis in order to adjust to the depletion of substrate using an anodic oxidation as the example. Figure reproduced from ref 266. Copyright 2020. Wiley-VCH Verlag GmbH & Co. KGaA.
Faraday’s Law (equation 18) illustrates that the total charge, Q (given in Coulombs [C]; note 1 C = 1 A•s), passing through the electrochemical cell is proportional to the absolute amount of analyte (, given in moles), where n is the number of electrons per molecule of analyte required for the redox event, and F is the Faraday constant (given in faradays [F]; note 1 F = 96487 C•mol−1). The charge is given by equation 19 which is the integral of the current as a function of time (t, given in seconds [s]) from t = 0 (start of electrolysis) to the end of the electrolysis ().
Faraday’s Law:
| (18) |
| (19) |
For CCE:
| (20) |
Using equation 19, it is apparent that total charge is the integration area of the change in current over time (Figure 8B and 8D). As a corollary, when the current is constant for the entire duration of the electrolysis, such as in CCE, equation 19 simplifies to equation 20 (Figure 8A):
Figure 8.
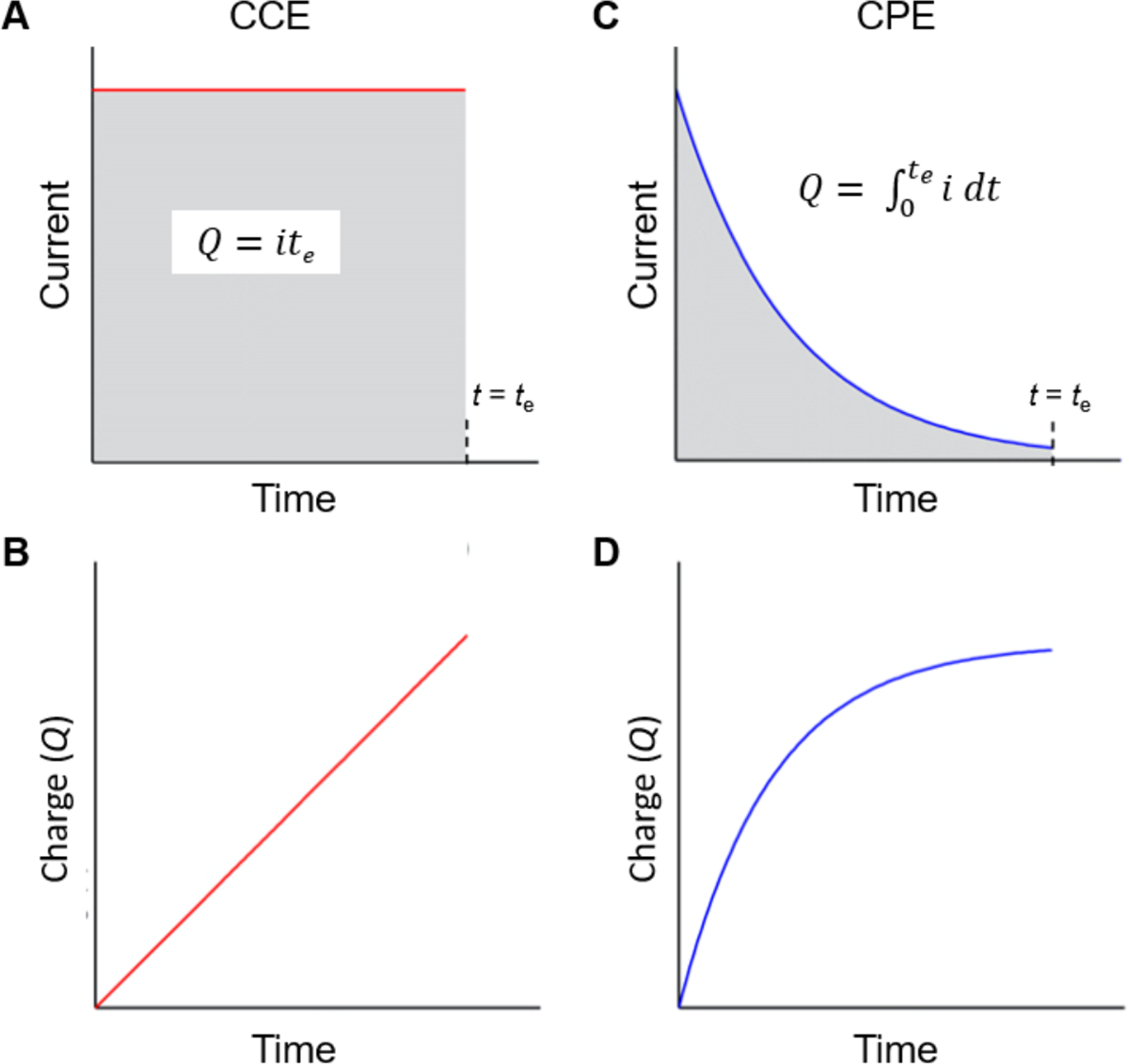
Temporal profile of the current and charge for constant current electrolysis (A and B, respectively) and constant potential electrolysis (C and D, respectively).
Since the consumption of starting materials (analyte) is directly proportional to the total charge passed through the solution (Faraday’s law), the rate of starting material consumption will differ whether operating the reaction under CCE or CPE. For a unimolecular process where the electron transfer is the rate determining step (charge transfer limiting regime), CCE will result in the rate of starting material consumption being zeroth order since current is directly proportional to the rate (Figure 8C). In contrast, the same reaction operated using CPE will exhibit 1st order kinetics due to the current exponentially decreasing as the extent of reaction increases (Figure 8D). Equation 21 describes this exponential decay, where is the limiting current at the onset of electrolysis, and k is a rate constant that is directly proportional to both the surface area of the electrode immersed in solution and mass transport parameters, and inversely proportional to the volume of the solution, and t is time.267
For CPE:
| (21) |
Analogous to how the total charge is a useful metric in electrochemistry, the concept of photon equivalents268,269,270 is a useful metric for scaling up photochemical reactions. The absorption spectrum of a reaction may change with time (e.g. darkening of reaction mixture is common), which in turn can impact the yield by changing the instantaneous photoabsorption efficiency and quantum yield. To empirically account for these deviations, the cumulative value of photon equivalents is determined with respect to product yield. This correlation establishes photon equivalents as a scaling factor to enable translation between different photoreactors of the same scale, or between different scales of the same reactor. We refer the reader to published works on this topic for further details.268
3.7.8. Mass Transfer
Both photochemistry and electrochemistry reactions are interfacial in nature, thus aspects of mass transport will influence their reaction rate. In the context of electrochemistry there are three types of mass transport124 phenomena: diffusion, convection, and migration. Diffusion relates to the movement of a species due to a concertation gradient. Transport of a species from a region of higher concentration to one of lower concentration occurs naturally to minimize concentration gradients. Convection is the movement of a species due to an external mechanical force (e.g. stirring of the solution, shaking of the reaction vessel, moving the electrodes (or an immersed light source) through the reaction mixture, sparging the reaction mixture in which the bubbles of the gas being sparged into the solution provides mixing). Migration is the movement of charged species along the electric gradient due to coulombic forces (e.g. positively charged ions are attracted to the cathode, and negatively charged ions are attracted to the anode). Migration becomes an insignificant contributor to mass transport in electrolysis when a high concentration of an inert electrolyte is present relative to the substrate concentration since the electric field imparted by the applied potential between the two electrodes is “screened” by the electrolyte (i.e. the charge will be transported mainly by the electrolyte). As for photochemical reactions, only convection and diffusion contribute to the mass transport mechanisms since there are no externally applied electrical gradients, thus migration is not a contributor to mass transport.
In the context of electrochemistry, the substrate (reactant, intermediate, or redox mediator) will have to migrate from the bulk phase of the solution to the electrode-solution interface region in order to undergo a redox event, and subsequently diffuse back into the bulk solution (Figure 9A). In some cases, redox events may require that the substance be adsorbed on the electrode, thus adsorption/desorption rates may come into play as well. Mass transfer regime or charge transfer regimes may dominate, as in the rate determining step may vary depending on the transformation, the reactor design, the mode of operation (e.g. CCE vs CPE), or reaction parameters (e.g. current density or electrode potential). In the extreme scenario, as the electrode polarization increases, the ratio of starting material to product in the electrode surface region (as governed by the Nernst equation) will be increasingly biased towards product, and in the extreme scenario, mass transport will dominate the rate. Thus, the improved mixing available in a flow reactor can be greatly beneficial compared to batch reactors for reducing material processing times (Figure 9B). In a contrasting scenario, when the electrode polarization is barely sufficient for charge transfer with the substrate, then charge transfer will dominate and govern rates of reaction (charge transfer controlled regime).
Figure 9.
A) Illustration of mass transport of the substrate into the Nernst layer, adsorption, electron transfer to the substrate generating the product, desorption of the product, and its mass transport out of the Nernst layer. (B) Illustration of two extreme rate determining step scenarios, namely charge-transfer controlled or mass-transport controlled regimes. Figure reproduced from ref 272. Copyright 2019 American Chemical Society.
Given that the redox event occurring at the electrode cannot occur faster than the analyte reaches the electrode surface (quantified by the mass transfer coefficient, km), this implies that a mass transfer limited regime governs the limiting rate of such reactions. The cell current () is for reactions that are mass transfer controlled (assuming a current efficiency of 1 for the desired reaction) is given by equation 22271:
| (22) |
where Ae is the electrode area, c is the concentration of the analyte (reactant) in the cell. The fractional conversion (for mass transfer limited reactions) as a function of time (t) can be obtained from equation 23:271
| (23) |
where is mass transfer coefficient, is the electrode area, and is the total reaction volume. From the above equation it becomes clear that efficient mass transport to the electrode as well as a high ratio of electrode surface area to reaction volume lead to faster conversion. An important parameter in optimizing electrolysis reactions is the current density (j), which is given by equation 24 (valid for cells with uniform current density), where i is the current (in amperes) passed through the electrode and is the geometric surface area of the electrode exposed to the solution (given in cm2).
| (24) |
In order to maximize productivity, high current densities are desirable, but this can lead to side reactions and thus decreased yields of the desired product, therefore it is usually necessary to optimize for this tradeoff. For example, if the redox event occurring at the electrode leads to radical formation, then a high current density equates to a high concentration of radicals formed at the electrode. This high current density may facilitate dimerization processes or multi-electron transfer events associated with the same substrate. The use of a redox mediator is a useful strategy to minimize such undesired reaction pathways by enabling redox events to occur in the bulk solution (homogeneous) instead of occurring at a heterogeneous interface (solid-liquid interface of the electrode with the solution). Redox mediators also enable control of the number of electrons transferred to substrate (e.g. this is an effective strategy to favor single electron transfer in cases where attempting to avoid multi-electron transfer events that occur in the absence of the mediator due to the high current density at the electron-solution interface).
Scaling up electrochemistry in batch naturally faces significant challenges since the ratio of the solution-exposed electrode surface area to the reactor volume is difficult to maintain as the scale increases; in contrast, flow electrochemistry can enable scaling up of the reaction via simple cell stacking in order to maintain very high electrode surface area to reactor volume ratios. Similarly, scaling up photochemistry is best suited for flow setups instead of batch in order to maximize the ratio of the surface area that is impinged by light relative to the reaction volume.
3.8. Flow Chemistry and Its Benefits for Scaling Up
Flow chemistry can provide significant advantages compared to batch mode reactions when it comes to scaling up photochemical or electrochemical reactions.272 Numerous reasons exist to use flow chemistry for conducting photoredox catalysis or electrochemistry, including: (1) improved irradiation of the reaction mixture (applicable to photochemistry), (2) reliable scale-up, (3) improved reaction selectivity and increased reproducibility, (4) fast mixing, (5) fast heat-exchange, (6) improved mass transfer for multiphase chemistry (e.g. gas-liquid), and (7) increased safety of operation.273
The high surface area-to-volume ratio of flow reactors not only provides excellent heat transfer and control over mixing, but more importantly can address mass transfer limitations encountered in batch, as mentioned in the previous section. For example, Beer’s law and its implication for light penetration269,274 into the solution for photochemical reactions can limit the reaction rate due to being in a photon-limited regime where only a fraction of the solution is exposed to light to excite the photocatalyst, as illustrated in Figures 10A and 10B. The distance, or path length (L), to which light can penetrate into the solution achieving a certain level of transmittance (T), is governed by the concentration (c) and molar absorptivity (ε) of the photocatalyst as illustrated by equation 25, for reactions when the only absorbing specie in solution at that wavelength is the photocatalyst. We note that transmittance of a solution is the transmitted intensity (I) divided by the incident intensity (Io), and this is sometimes expressed as a percentage (%T). The absorbance of a solution is simply the product of the concentration (c), molar absorptivity (ε) and the path length.
| (25) |
Figure 10.
(A) Top: Attenuation of light as a function of pathlength through a solution containing Ru(bpy)3(PF6)2 (ε = 13000 cm−1•M−1 at λ = 452 nm)275, bottom: generic effect of light attenuation with increasing concentration of catalyst or using catalysts with increasingly larger e. (B) Comparison of light transmission between batch and (C) flow photochemistry.
The use of reactors with large volume to surface area ratio (typical of batch reactors) results in only a small portion being illuminated due to the light attention as shown in Figures 10A and 10B. The result is non-homogenous illumination of the batch, with excess-photon regions (bright zones) and photon-starved regions (dark zones). In order to drive the reaction to complete, over-irradiation is necessary leading to increased reaction times, which can cause the formation of byproducts. In contrast, the use of flow reactors for photochemistry can enable lower volume to surface area ratios, providing nearly homogenous illumination of the reaction mixture with no dark zones (no photon-starved regions), thus leading to efficient reactions (Figure 10C). The use of continuous stirred tank reactors (CSTRs) interfaced with a laser and a lens for light diffusion have been explored276 as a way to address these light penetration challenges towards scaling up photochemistry on commercial scale. This approach is attractive from the point of view of being compatible with heterogenous mixtures in contrast to photo-flow which are best suited for homogenous conditions.
Below we illustrate several key benefits imparted by flow chemistry for performing electrosynthesis compared to batch operation. Based on Ohm’s law, equation 29 illustrates the ohmic potential drop () between electrodes is equal to the product of the ohmic resistance () and the current (i). The ohmic resistance of an electrochemical cell is linearly proportional to the inter-electrode distance (d) where a is the cross-sectional area between the two electrodes where current is flowing and ρ is the resistivity (given in ohms•meter). The alternate form of this equation is shown on the far right-hand side of equation 26 using solution conductivity, κ, given in siemens (S) per meter; the siemens is the reciprocal of the ohm (Ω), i.e., 1 S = 1•ohm−1).
| (26) |
Equation 26 illustrates why minimizing the inter-electrode separation distance is critical to minimizing the ohmic resistance and thus the ohmic potential drop. An increase in inter-electrode distance, will result in increased resistance which in turn increases the cell potential. Undesirable side reaction may occur as a result of loss of selectivity associated with larger cell potentials. In typical laboratory scale batch electrolysis experiments, such as those utilizing the popular Electrasyn 2.0 setup by IKA equipped with 5 mL or 10 mL reaction volume vial sizes, the inter-electrode distance is typically ca. 0.5 cm. The use of additional electrolyte may be required to offset the increased resistance imposed when increasing the inter-electrode spacing or when introducing a separating material (e.g. semi-permeable membrane, sintered-glass frit) such as in divided cell electrolysis. Flow chemistry for organic electrosynthetic chemistry can enable advantages such as smaller inter-electrode spacing (e.g. <1 mm) which in turn has been demonstrated to enable significant decrease, and even elimination, of supporting electrolytes, highlighting benefits compared to batch mode operations.271,277,278,279,280,281,282
The electrical resistance associated with the ohmic drop results in heating of the solution when current passes through it, resulting in a phenomenon known as Joule heating. The classical description of Joule heating involves the conversion of electrical current energy converted into heat as it flows through a resistor, and this concept can be adapted to electrochemistry by assuming that the solution is the bulk resistor. According to Joule’s first law, the Joule heat is directly proportional to the ohmic drop resistance (), the magnitude of the current passing through the solution, and the time duration (t) for which current is passed, as shown in equation 27.283 Flow chemistry provides the benefit of efficient heat-transfer, and given the smaller inter-electrode spacing compared to traditional batch setups, Joule heating is minimized.
| (27) |
A benefit of using electrochemistry is that electrons are used as the redox reagent instead of traditional redox reagents with significantly higher molecular weights and potentially toxic and/or hazardous properties. This green chemistry aspect must be considered in conjunction with the total energy required for running the process. The energy consumption for the cell is mainly dictated by the cell resistance and the operating current density. Equation 28 illustrates how the cell potential () is the sum of the thermodynamically required potential (), the sum of the overpotential (η) associated with the redox process at each electrode, and the potential associated with the ohmic cell resistance () and the current (i).
| (28) |
The energy consumption for an electrochemical reactor, given in units W•s•mol−1, can be obtained from the instantaneous current efficiency and the cell potential () using equation 29,284 where n is the number of electrons per molecule of analyte (stoichiometry), and F is the Faraday constant; thus it becomes clear that minimizing the cell potential and maximizing the current efficiency are desirable towards minimizing the energy consumption.
| (29) |
Electrochemical flow reactions are typically carried out at constant current using the optimal conditions for the analogous batch reaction (e.g. the choice of electrode, solvent, electrolyte, current density, etc.) as a starting point. These parameters enable fast translation of the batch reaction to a flow system. The total charge (Q) is given by equation 30,285 where i is the current in amperes, is the flow rate in mL•min−1, c is the concentration of the substrate in solution in Molar units and F is Faraday’s constant (1608.0889 min•A•mol−1). This equation can be used to determine the ratio of the flow rate to current.
| (30) |
Flow cell operation can be either be in single-pass mode (continuous processing), or in recirculating mode (semi-batch process), as illustrated in Figure 11.271,285 In single-pass mode, high conversion must be obtained through the flow cell. In order to achieve this, the flow rate and current need to be adjusted to achieve such outcome and has the drawback of potentially having difficulties in handling gas formation (e.g. H2 evolution). On the other hand, recirculating mode can easily handle gas as the flow rate can be adjusted high enough that saturation of the solution is not reached and venting of the gas is allowed in the tank that holds the reaction mixture outside of the flow cell. Recirculating mode requires that the reaction mixture be recirculated until high conversion is reached and is usually easier to develop into a process than single pass operation (which requires full conversion upon exiting the cell after a single pass).
Figure 11.

An illustrative comparison between (A) single-pass mode flow cell operation and (B) recirculation-mode flow cell operation.
In a flow setting, the relationship between the cell current (), assuming 100% current efficiency for the desired reaction, and the fractional conversion (X) can be estimated using Faraday’s law by translating equation 30 to arrive at equation 31:271
| (31) |
where is the volumetric flow rate, is the concentration of the reactant at the cell inlet, and X is the fractional conversion. The fractional conversion can be estimated using equation 32:271
| (32) |
Where and are the concentrations of the reactant at the inlet and outlet of the cell, km is the mass transfer constant, w and L are the width and length of the solution channel, respectively.
A key metric to measure productivity of a continuous-processing reactor, including those for photochemical and electrochemistry transformations, is the space-time yield (STY).124,286,287 This metric is used across all of flow chemistry. Equation 33 illustrates how to calculate the space-time yield (), which is the mass of product (mp) formed per unit of time, t, and reactor volume (Vr):288
| (33) |
A range of reactor designs271,289 can be leveraged for continuous processing of electrochemical reactions, from microreactors,290,291 parallel plate flow reactors,292 pipe cell (cylindrical electrode rod inside a tube) reactor (ideal for high pressure/high temperature reactions),293 solid polymer electrolyte (SPE) (also called MEA) reactor, 271,294 bipolar disk stack cell,271 rotating electrode reactors (various electrode geometries include rotating cylinder, rotating cone, rotating hemispherical electrodes, with cylindrical geometries being most common),295,296 and continuous stirred tank reactors (CSTR) connected in series.271,291 Similarly, continuous processing reactors for photochemistry range in design from plug flow reactors (PFR), CSTR, packed bed reactors, falling film reactors and even tube in tube reactors.297 Depending on the type of reaction, each reactor may afford advantages as well as disadvantages associated with the reaction conditions required (homo vs heterogenous reaction mixture, high pressure vs ambient, high vs low temperature, single vs multi-phasic systems, etc.). Other unique reactor design have also successfully been implemented on large scale such as in the use of stacked (graphite) disks in the anodic methoxylation of toluene derivatives by BASF (14 tons/batch),298,299 or the use of Swiss-roll300 reactors in an alcohol oxidation (diacetone-L-sorbose to diacetone-2-keto-L-gulonic acid, 2 tons per day) as part of the production of Vitamin C.
The application of electrochemistry on large scale in the context of industrial chemistry has been realized for decades and a summary of such examples has been reviewed.170,298,291,301,302,303 Additional green chemistry aspects can exist when using electrochemistry,304,305,306,307,308,309,310,311 and in some cases circumvent the use of stoichiometric, potentially hazardous/toxic redox agents by relying on electricity as the source of the most economical reduction/oxidation agent, the electron, simply by adjusting the potential at the working electrode.
3.9. Tools for Studying Reaction Mechanisms
The approaches for gaining insight into the reaction mechanism for a photoredox312 or electromediated313,314,315,316 process share similarities. Characterization of ground state species by common analytical techniques such as NMR and IR spectroscopy can provide structural and quantitative information, insight into equilibria distribution of starting materials, persistent intermediates and products. UV-Vis absorption spectroscopy can provide insight into the absorption features of the individual substrates and catalyst, but just as importantly experiments where combinations of substrate and catalysts aid in gaining insight whether (1) electron donor-acceptor (EDA)317 complexes are forming, or (2) whether a bathochromic shift occurs in the substrate absorption as a result of interacting with a catalyst,318 or (3) increased absorption occurs upon complexation to the catalysts.319 Cyclic voltammetry63,320 enables the measurement of redox potentials of starting materials as well as photocatalysts. Steady-state and time-resolved emission spectroscopy are useful for obtaining Stern-Volmer plots,108,321 which provide insight into which species quench the excited state and the mechanism by which quenching occurs (static vs dynamic quenching, or combination of both).108,312 It is important to emphasize that the observation of quenching of an excited state via Stern-Volmer luminescence quenching alone cannot conclusively discern between electron transfer and energy transfer mechanisms.108 Repeating these experiments using solvents of varying polarities can help discern between electron and energy transfer since electron transfer involved charge separation while energy transfer does not. Thus, rates of quenching are largely solvent independent for energy transfer unlike electron transfer processes. Other experiments such as the observation (or lack thereof) a correlation between the rate of quenching vs the triplet energy of the sensitizer, or measurement of redox potentials of the photocatalyst and the substrate quenching the excited state of the photocatalyst can aid in delineating between the two mechanisms.108,312 Transient absorption spectroscopy via time-resolved pump–probe experiments is another great technique for understanding electron and energy transfer processes. Transient absorption spectroscopy enables the characterization of events occurring on the picosecond timescale and has been used to understand metallaphotoredox catalysis (e.g. dual catalysis based on a transition metal, such as Ni, and a photoredox catalyst) – particularly in differentiating between photoinduced electron transfer and energy transfer mediated processes in reductive elimination events.322,323,324 Recently transient laser spectroscopy, in combination with steady state photochemical measurements as well as electrochemical methods enabled for the experimental characterization photoredox chemistry involving α-aminoarylation.325
Reaction progress kinetics (RPK)326,327,328 can be particularly useful in elucidating reaction mechanism and tools such as in situ LED-NMR329,330 spectroscopy (Figure 12) and in situ electrochemistry-NMR331,332,333,334,335,336,337,338,339,340 (Figure 13) spectroscopy not only provide kinetic but structural information while being intrinsically quantitative. Critical advances from the original design by Berliner341 were made by Gschwind342 through improvements in the illumination strategy. These methods can be especially useful for compounds that are formed only under the presence of light or an electric potential as well as those which are volatile (e.g. CO). 343 The measurement of quantum yields can also be done via in situ LED-NMR spectroscopy344 towards understanding mechanistic aspects such as whether the reaction is photocatalyzed or photoinduced343 and obtaining evidence of a chain reaction process. The collection of both NMR and UV-vis absorption spectroscopy via in situ LED-NMR has been developed by Gschwind345 providing a tool for characterizing species which may be difficult to observe via NMR spectroscopy (e.g. signal broadening due to paramagnetic species or characterization of species that are NMR silent). Analogous to in situ LED-NMR spectroscopy tools and techniques, in situ electrochemistry-NMR (EC-NMR) has been developed.331 A variety of approaches in the realization of the setup exist, some place the working electrode in the NMR detection zone (in the cross-sectional path of the radiofrequency coil) (Figure 12A), while others minimize interference by having the working electrode directly above this region (Figure 12B). The setup shown in Figure 12A has been used to measure cyclic voltammetry of benzoquinone and simultaneously record 1H NMR spectra. The setup in Figure 12B has been used to study the electrooxidiation of ascorbic acid to dehydroascorbic acid. There are drawbacks to in situ NMR-based tools, including that they cannot stir the reaction mixture and face challenges in studying heterogenous reactions.
Figure 12.
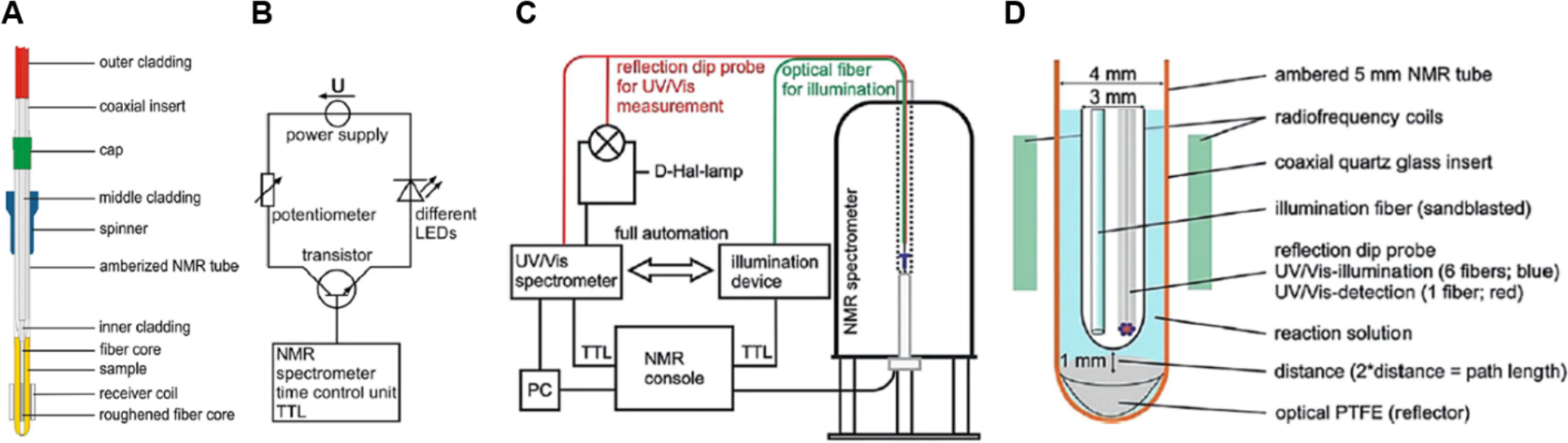
(A,B) in situ LED-NMR spectroscopy setup and (C,D) in situ UV-LED-NMR spectroscopy setup. (A) Schematic diagram illustrating fiber optic cable inserted into a coaxial insert which centers the illumination in a 5 mm NMR tube. (B) Schematic diagram illustrating the power supply, LED-source and NMR spectrometer time control unit. (C) Schematic diagram illustrating UV-NMR setup (D) Illumination fiber and reflection dip probe inside the NMR. [Figures A & B reproduced from ref 352. Copyright 2016 American Chemical Society. Figures C & D reproduced from ref 345. Copyright 2018 Wiley-VCH Verlag GmbH & Co. KGaA.]
Figure 13.
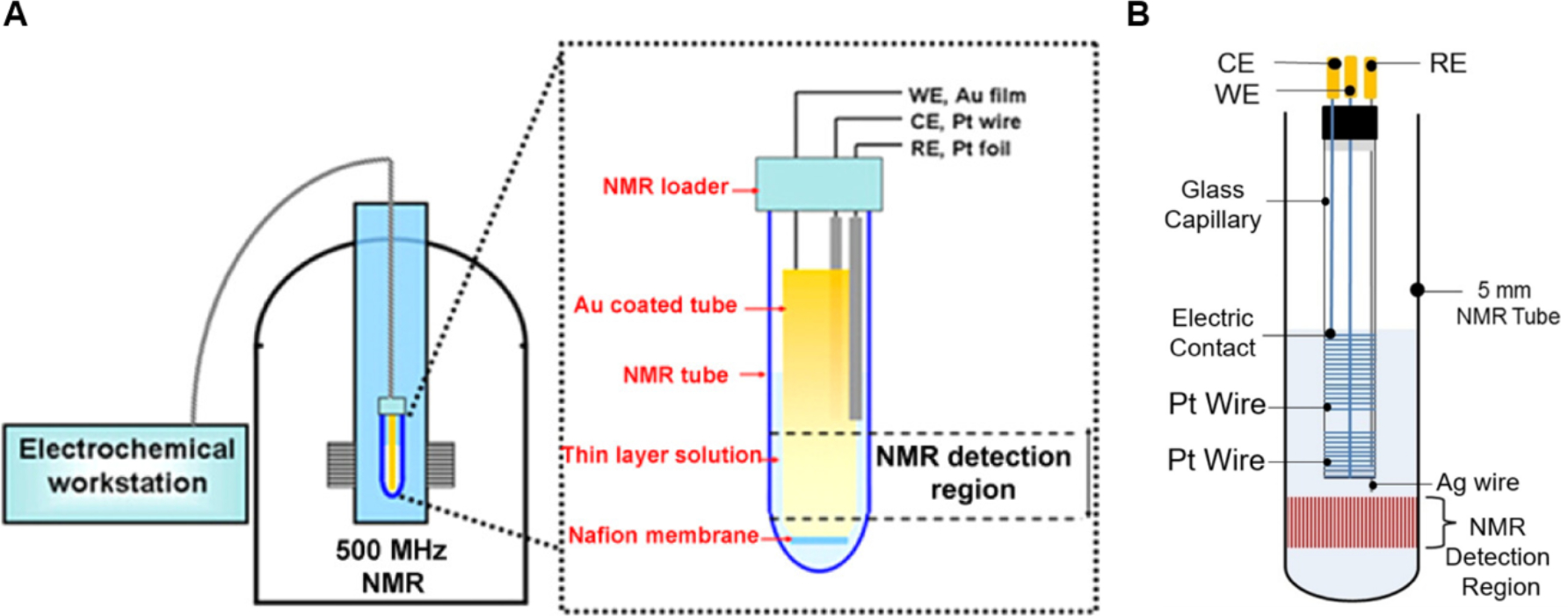
(A) Schematic representation of an in situ EC-NMR spectroscopy setup with the working electrode being in the NMR detection region (inset illustrates NMR tube with working electrode (WE), counter electrode (CE) and reference electrode (RE), along with a Nafion™ membrane providing a divided cell. (B) Schematic representation of an in situ EC-NMR spectroscopy setup with the working electrode being outside of the NMR detection region (inset illustrates NMR tube with working electrode (WE), counter electrode (CE) and reference electrode (RE). [Figure A reproduced from ref 339. Copyright 2017 American Chemical Society. Figure B reproduced from ref 340 Copyright 2019 Elsevier B.V.]
In order to profile heterogenous reaction mixtures, other methods must be employed. In situ Raman346 or in situ FT-IR spectroscopy347,348 can be used to monitor reaction mixtures which are simultaneously irradiated. This approach has also been demonstrated to capture kinetic data, although these techniques provide significantly less structural information and require calibration curves to extract quantitative information. These methods do however benefit from being able to study heterogenous reactions as forced convection (stirring) can be applied. Ex situ techniques, such as collection of reaction aliquots and characterization of those via offline HPLC or NMR spectroscopy, are also useful for studying heterogenous reactions, although species produced only under photolysis or electrolysis or unstable/volatile species may not be observed, thus may not be representative of the components of the reaction mixture. Online Flow-NMR techniques are a way to handle heterogenous mixtures, with the benefit of being able to externalize the reaction vessel relative to the NMR magnet, while continuously pumping a sample from that mixture into the spectrometer through an NMR flow tube within a standard NMR probe, before returning it to the reaction vessel. This enables mixing, irradiation, addition of reagents and temperature control. Online flow-NMR has been used in the context of studying photochemical,349,350,351,352,353,354 as well as electrochemical355 transformations.
UV-Vis spectroscopy is another useful process analytical technology tool for collecting reaction time-course data;356 modest structural information can limit the usefulness of this technique, although examples of its application to studying photochemical reactions are widely reported.357,358,359 Spectroelectrochemistry67,360,361,362,363,364 merges a spectroscopic technique (e.g. IR spectroscopy or UV-Vis absorption) with electrochemistry to spectroscopically characterize species generated by a redox event at a transparent electrode (e.g. indium-tin oxides, ITO) or a mesh electrode (e.g. Pt mesh). This enables the characterization of radical intermediates towards understanding their structure as well as their lifetime. Electrochemistry-mass spectrometry (EC-MS) is another analytical tool that has provided useful insight into products of photochemical365 and redox events.366 This method is useful for elucidating mechanistic aspect of electrosynthetic transformations, as well as mimicking oxidative metabolism367,368,369 and studying corrosion processes.
EPR spectroscopy is another great method of characterizing radicals. If the radical species of interest can be generated with high enough concentration in a steady state approach, the sensitivity of the detection method may enable direct characterization of the radical despite the radical having a short lifetime. If this is not the case, radical species can be trapped using radical trapping agents (e.g. DMPO = 5,5-dimethyl-1-pyrroline-N-oxide) and BMPO = 5-tert-butoxycarbonyl 5-methyl-1-pyrroline-N-oxide) to generate a longer-lived radical species & accumulate its concentration which can be characterized by EPR (Scheme 18).370,371,372 Advanced techniques such as photo-electron paramagnetic resonance (photo-EPR) and electrochemical-EPR techniques373 can enable the characterization of transiently formed species with unpaired electrons and has been realized as an in situ tool for probing redox chemistry. Coupled in situ NMR and EPR studies have also been used to study rates of electron transfer and electrolyte decomposition in redox flow batteries.374
Scheme 18.
Illustration of an EPR spin trapping experiment.
While the use of radical inhibitors like TEMPO, ABNO, BHT, or benzoquinone to inhibit product formation can be used as evidence that is consistent with a radical mechanism, it does not provide evidence for a radical mechanism. Positive evidence for a radical mechanism via indirect methods for product generation occurring via a radical intermediate include the use of radical probes (Scheme 19). Radical probes rely on a unimolecular bond formation/cleavage to occur if passing through a key radical intermediate and is reflected in the product outcome (e.g. opening of cyclopropyl ring when passing through a cyclopropyl carbinyl radical intermediate). In some cases, the desired radical reactivity may have comparable rates to the reactivity of the radical probe and thus lead to a mixture of products, which can then be used to benchmark the rate of the desired radical reactivity versus that of the radical reactivity built in the radical probe – these are radical clock experiments.375,376,377 An example of using TEMPO to inhibit a radical pathway is illustrated below in a set of control experiments as part of a study on the 1,2-diazidation of alkenes by the Lin group (Scheme 19A). Anodic oxidation of a mixture of 4-tert-butylstyrene (19.1) and sodium azide typically results in a distribution of dimer 19.3 as well as hydroxazide 19.4 (arising from over oxidation and subsequent attack by water) as well as diazide 19.5. In contrast to these major products, reactions run in the presence of TEMPO suppressed the above mentioned products and instead provided azidooxygenation product 19.6, demonstrating TEMPO’s ability to inhibit the typical carbon centered radical reactivity.378 Subsequent report by Lin group with optimized conditions for this reaction led to an azidooxygenation of alkene method using NaN3 and TEMPO under constant potential electrolysis, such as the conversion of 4-tert-butylstyrene (19.1) to product 19.6.379 The use of a radical probe such as vinylcyclopropyl derivative 19.7 affords product 19.8 which provides supporting evidence for the intermediacy of radical intermediate 19.8 which undergoes rapid ring opening to form 19.9 (Scheme 19B). The azidooxygenation of substrate 19.10 postulated to proceed via intermediate 19.11 provides for a competition reaction between radical annihilation with TEMPO to furnish product 19.12 and intramolecular 5-exo-trig cyclization to product 19.13. Experimentally, the dominant pathway leads to product 19.12. These results suggest that the radical coupling between the intermediate 19.12 and TEMPO• occurs at a rate between 106 and 1011 s−1. The use of radical probes is also quite prevalent in photoredox catalysis to support the intermediacy of radical intermediates.380,381,382
Scheme 19.
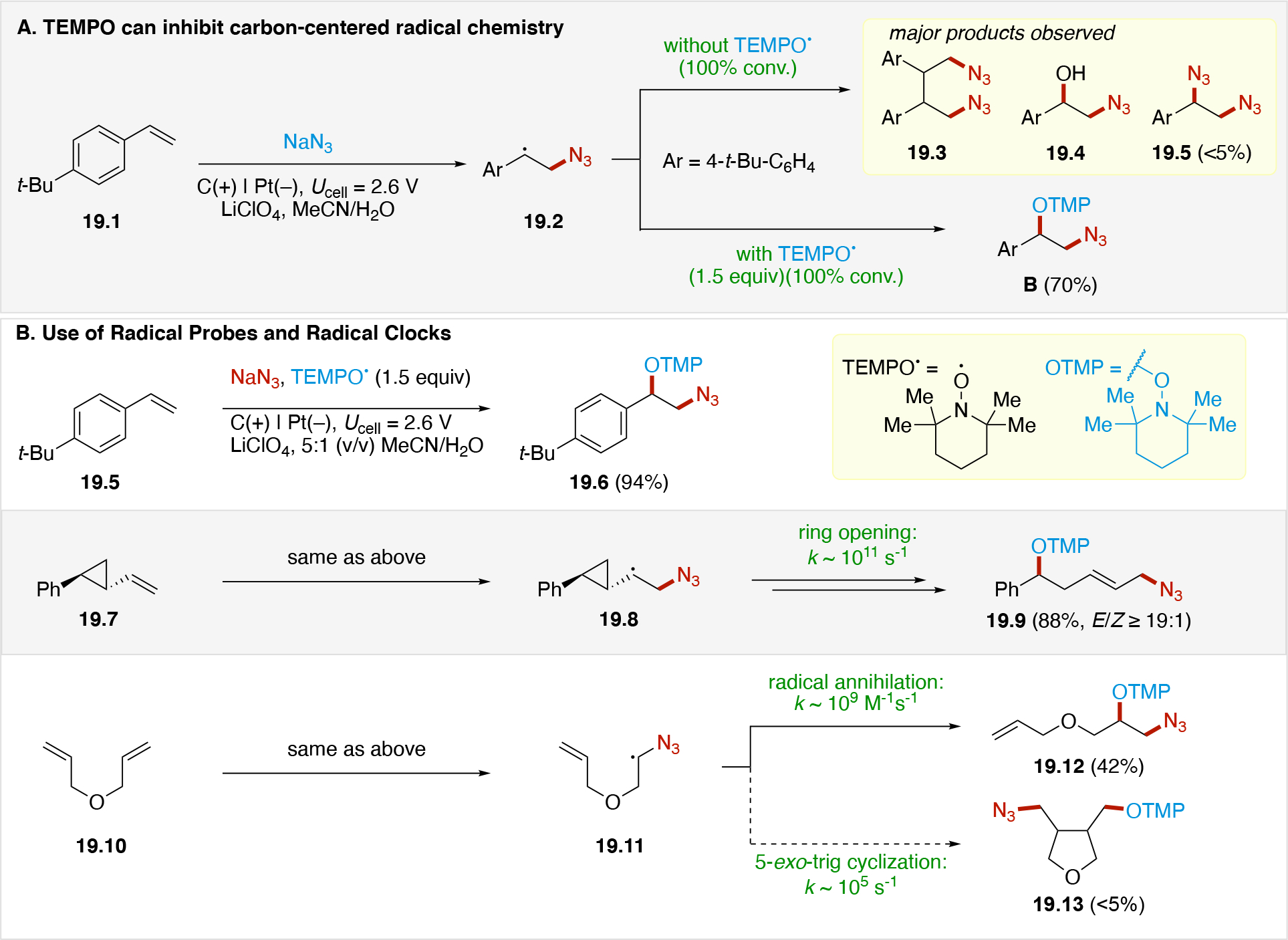
(A) Illustration of using stoichiometric TEMPO to inhibit a radical reaction. (B) Illustration of a radical probes and clocks to support a mechanism proceeding via a radical intermediate and estimate rate constants in a mechanistic study.
3.10. High Throughput Experimentation (HTE)
High throughput experimentation (HTE) is a powerful approach for both reaction discovery and optimization to do more with less material in a faster timescale.383 Both reaction discovery and optimization benefit from the exploration of combinations of reaction variables (e.g. ligands, catalysts, reagents, solvents) including relative stoichiometries (e.g. metal-ligand ratios, ratios of two coupling partners, solvent ratios, catalyst loading). While chemical biology, including protein engineering, has used HTE via arrayed plate-based setups (e.g. 96-well plate), only in the past decade or so has synthetic chemistry emerged in such miniaturized formats for reaction discovery and optimization through the development of HTE workflows. Pharmaceutical and agrochemical companies now routinely leverage arrays of 8 mm × 30 mm glass vial inserts in 24-well 4 rows × 6 column array) or 96-well (8 rows × 12 column array) metal microtiter plates in which reactions can be carried out on ~100 μL volume which translates to 10 μmol scale for 0.1 M limiting substrate concentration. These microtiter plates have the standard SBS footprint and vial spacing (well positions) established by the former Society for Biomedical Sciences (SBS), now the Society for Laboratory Automation and Screening (SLAS),384 which enables interfacing with standard multi-channel pipettors or liquid handling robots.385 The use of pre-plated libraries of solids (e.g. ligands, catalysts, reagents like acids/bases) enable incorporation of screening such parameters within the HTE workflow, whereby stock solutions of the substrate and/or additional reagents/catalysts can be rapidly dosed into these pre-plated libraries to enable efficient HTE.
While a broad range of thermally mediated synthetic transformations can easily be explored (e.g. metal catalyzed cross-couplings), slightly more elaborate equipment is necessary to enable the interfacing of these setups with gases to enable HTE to carry out hydrogenations, hydroformylations, carbonylations, etc.386,387 With the emergence of photoredox catalysis in the mid 2010’s, it became clear that having a method to introduce light into the reactions in an HTE setup would be beneficial for reaction discovery and optimization (Figure 14). DiRocco reported388 a method of interfacing such 24-well and 96-well plates with blue LED arrays and demonstrated its applicability in the developing a photoredox late-stage methylation of heterocycles (Minisci reaction) for pharmaceutically relevant targets. The light is introduced from below the reaction vial through circular openings in bottom of the metal microtiter plate using an array of LEDs (one LED per reaction well). Commercialized setups now offer such microtiter plate as well as a broad range of wavelength (365 nm and 450 nm being the most used, although longer wavelength applications are emerging) for the LED arrays, as well as the ability to control the light intensity. Recently, Kalyani, Lehnherr, and Lin reported389 equipment (HTe−chem) that enables electrochemistry to be performed within such microtiter 24-well plate (Figure 15). The HTe−chem was demonstrated to enable screening of various currents or potentials (depending if operated in constant current or constant potential mode), total charge, electrode materials, in addition to more common reaction parameters such as solvent, concentration, temperature, regent combinations (including electrolyte choice), etc. Since the HTe−chem setup utilizes the footprint and well spacing of standard 24-well microtiter plates, its incorporation into HTE workflows was seamless, including the ability to introduce light using LED arrays analogous to photoredox setup for HTE, thus enabling electrophotochemistry HTE screens. Reaction mixing for reactions in 24-well plates can accomplished using rotary magnetic stirring, larger 96-well plates typically require the use of tumble stirrers.390 Larger arrays of reactions have been achieved in 384- and 1536-well plate formats for nanomolar scale screening although to date this has been limited to thermal or photochemical transformations.391,392
Figure 14.
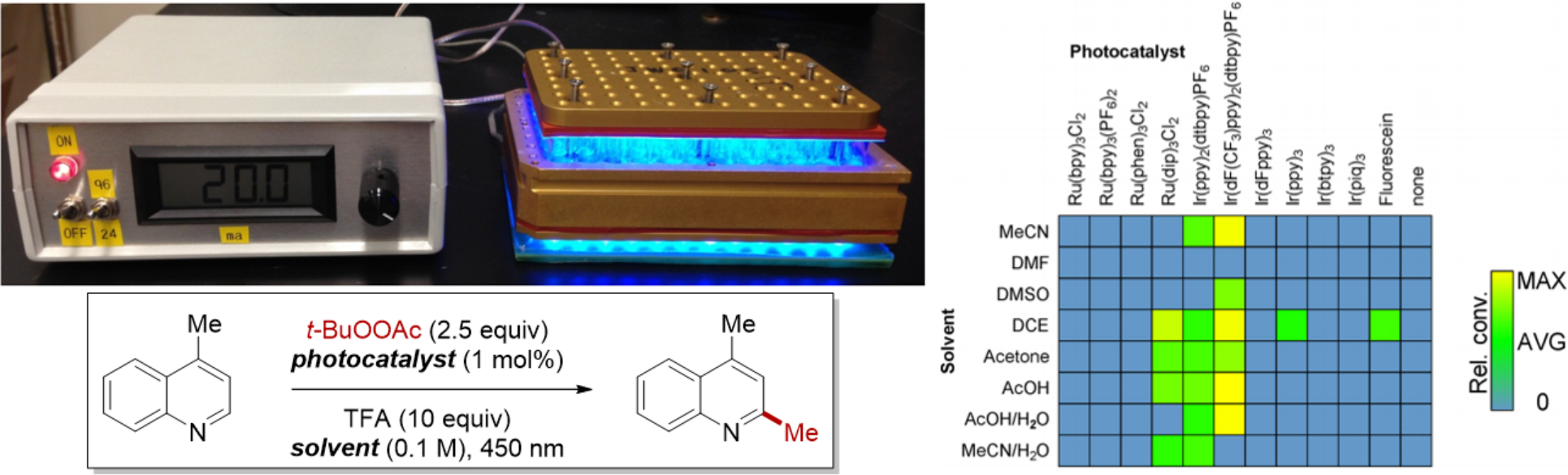
(Top left) photograph of HTE photoredox setup for 24-well (not shown) or 96-well plate screening (bottom left) photoredox methylation of lepidine (right) HTE screening results (relative conversion) for 6 different solvents and 12 different photocatalysts. [Left figure reproduced from ref 388. Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA.]. [Right figure reproduced from ref 386. Copyright 2017 American Chemical Society.].
Figure 15.
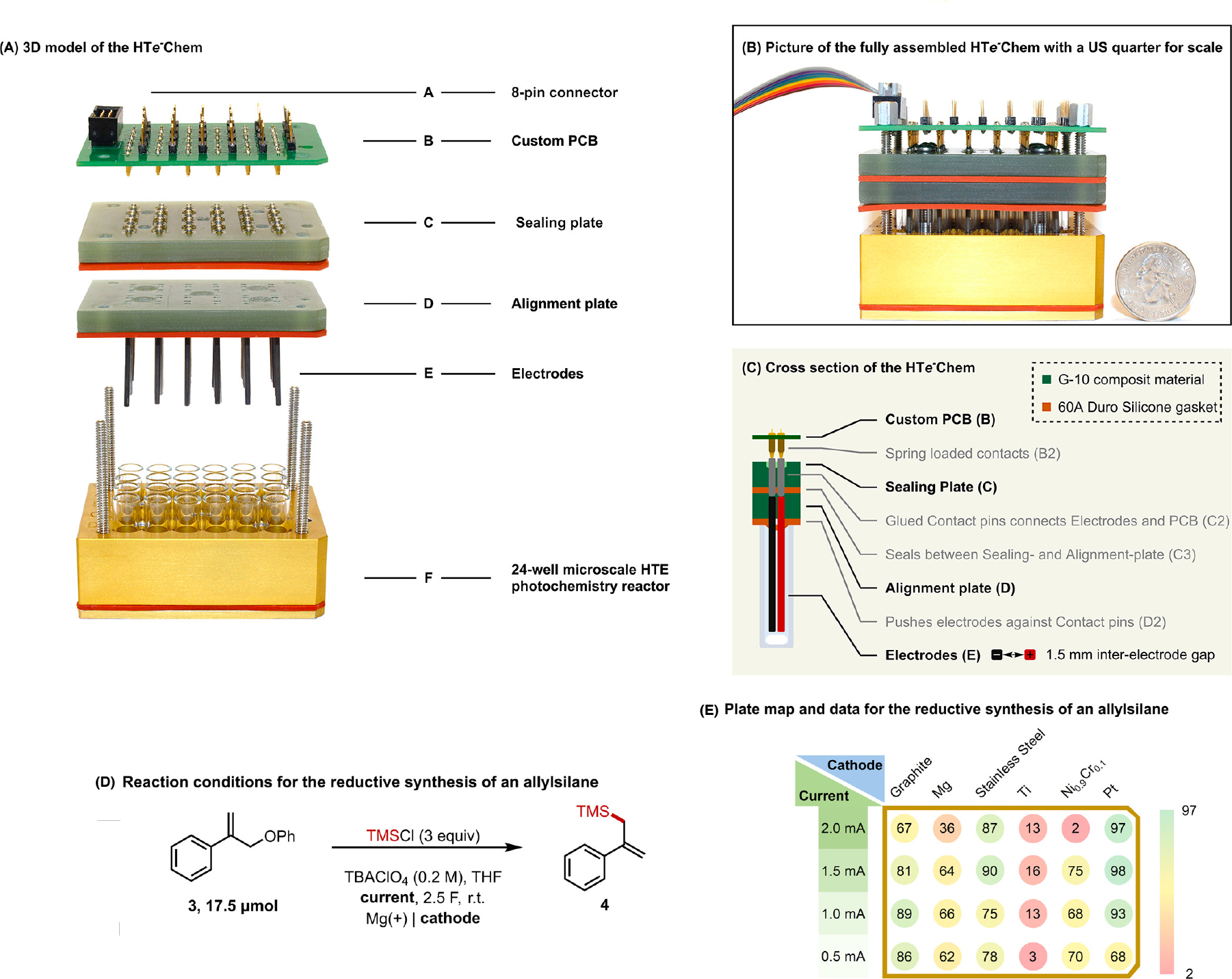
(A) Design and assembly of the HTe−chem (B) photograph of the fully assembled HTE HTe−chem setup with the constant potential top plate alongside a US quarter for scale (C) cross section of the HTe−chem (D) Reductive allyl silane synthesis and (E) assay yield data obtained from exploring 4 different current densities and 6 different cathode materials in the HTe−chem. [Figures reproduced from ref 389. Copyright 2021 American Chemical Society].
Setups for preparative lab scale commonly encountered in synthetic methodology publications (~1 mmol scale) vary greatly from lab to lab in both electrochemistry and photochemistry. For example, photochemistry is routinely run using Kessil lamp setups with or without fans for air cooling in which the vial to lamp distance can greatly influence reaction temperature and photon flux,393 while non-standardized electrochemical setups can have vastly different cell geometries, electrode dimensions, shapes, and inter-distance spacings, ultimately bring about challenges in the ease of uptake of these synthetic methods. With the advance of HTE equipment for both photo and electrochemistry, the advance of standardized reactors for larger scale (ca. 0.1 mol to 4 mol scale, 1 mL to 40 mL reaction volumes) have also emerged as useful tools for validation of HTE leads by carrying out preparative synthesis, including substrate scope. These standardized reactors have greatly facilitated the ease in reproducing results from one lab to another by controlling the critical parameters mentioned above in the equipment setup. A few notable setups include the Penn PhD m1/m2 photoreactors,394,395 EvoluChem™ (by HepatoChem),396 TAK-120 photoreactors (air or liquid cooled),397 while Kessil offers the PR160 Rig to enable reproducible positioning of lamps and air-cooling fans. In terms of electrochemistry, the most widely used setup is IKA’s ElectraSyn 2.023,313,398 which has made entry into electrochemistry facile as well as providing a standardized reaction setup for ease of reproducibility. The Electrasyn 2.0 can be interface with either a single reaction holder or a carousel for up to 6 parallel reactions, or through the use of their e-Hive accessory, up to 24 reactions can be run in parallel, although certain limitations exist, including a non-standard footprint / vial spacing that render it challenging to interface with standardized HTE workflows. We also note important contributions by Yudin in developing a 16-well parallel electrochemical setup for parallel electrosynthesis on ~1 mmol scale.399 Parallel reactor setups were also latter reported by Toshiki & Atsushi (5 reactions, ~1 mmol scale)400, and Waldvogel (8 reactions, ~1 mmol scale),401 as well as others.402 Beeler403 and Stephenson404 individually reported microfluidic based screening platforms for screening photochemical reactions.
3.11. Common Redox-activatable Radical Precursors
The generation of radicals on organic substrates is commonly accomplished via either an electron transfer event or atom transfer reaction (e.g. HAT),405,406,407,408,409 although other mechanisms such as halogen atom transfer (XAT)410,411,412,413,414,415,416,417 or ET/EnT mediated strain release mechanisms exist418 (Scheme 20).
Scheme 20.
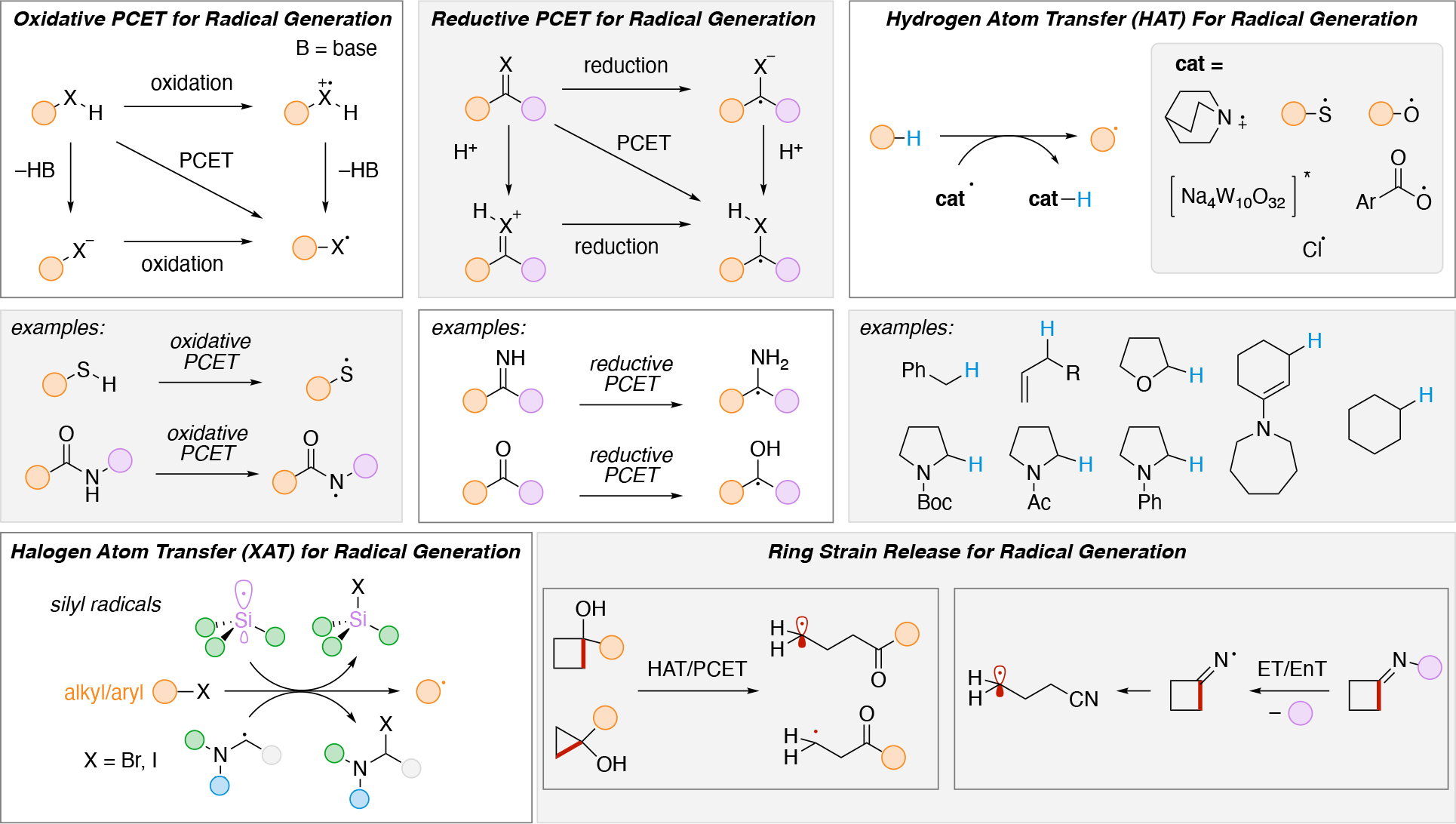
Illustration of oxidative and reductive PCET as well as HAT processes to generate radicals.
Certain functional groups provide synthetic handles to site-selectively generate radicals on organic molecules (Scheme 21). Various names have been proposed in the literature for these groups, such as radical precursor group (RPG) or electroauxilliaries. For example, carboxylic acids,419 particularly under basic conditions, can be utilized to generate radicals via a decarboxylation process via a single electron oxidation process. The related N-hydroxyphthalimide esters420,421,422,423,424,425 (redox active esters) can undergo a single electron reduction to release CO2, phthalimide and the corresponding alkyl radical. Many other groups exist, including oxalates426,427 as activating groups for alcohols for radical generation, alkyl BF3K428 salts, alkyl silicates,428,429,430,431,432 benzylsilanes, allylsilanes, alkyl halides (α-halo esters/ketones),433,434,435,436,437,438 aryl halides. Deaminative radical generation processes include the use of pyridiniums439,440, tetraalkylammonium salts, and electron-rich imines.441 The use of silanes (or trialkyltin) α to heteroatoms (e.g. N, O, and S) results in raising the HOMO energy and thus facilitate oxidative radical generation, examples include α-amino silanes, α-silyl ethers and α-silylthioethers;442,443 the oxidation potential relative to the des-silyl analog is largest in magnitude for O as the α-heteroatom, and smallest for S (see Scheme 21 below).443,444,445 The generation of radicals can also be accomplished via HAT,446,447,448,449 including those via oxidative proton coupled electron transfer or reductive PCET.450,451,452,453
Scheme 21.

Common radical precursors for organic synthesis. All oxidation426,432,438,441,444,454,455,456,457,458,459,460,461,462,463,464,465,466,467 and reduction86,420,433,434,435,436,437,438,468,469,470,471,472,473,474,475,476,477,478,479,480,481 potentials provided are in V vs SCE.255
3.12. Summary
For a summary see Table 2.
Table 2.
Comparative summary of electrosynthesis and photoredox catalysis.
| Feature | Electrosynthesis | Photoredox Catalysis |
|---|---|---|
| underlying principle | redox chemistry | redox chemistry |
| number of electrons in redox event | 1e or 2e are common (>2e possible), the use of a redox mediator can enable control over this number | typically 1e (two sequential 1e transfer events are postulated in only a limited number of examples) |
| generation of radical | Direct electrolysis (DE): redox event at electrode interface, within electrochemical double layer (localized) Mediated electrolysis (ME): redox event with redox mediator (electrocatalyst) can do redox within bulk | redox event via interaction with photocatalyst (excited or ground state), substrate-catalyst distance ≤ 20 Å,482 delocalized throughout solution but only within the photon-accessible regions of the solution (Beer-Lambert law)483 |
| concentration of radicals | DE: high, localized at electrode interface/electrochemical double layer ME: low, delocalized throughout the bulk of the solution | low, localized but only within the photon-accessible regions of the solution unless generating persistent radical or proceeding through a radical chain mechanism which enables diffusion/propagation into the bulk solution |
| productivity throughput (space-time yield) | limited by current density, flow electrochemistry is often best suited for maximizing space-time yield; electron flux (current) is coupled to potential (Ohm’s law) | limited by photon flux absorbed by photocatalyst, photo-flow best suited for maximizing space-time yield; photon flux is not coupled to photocatalyst redox potential |
| efficiency range of redox potential accessible | faradaic efficiency (current efficiency) can be adjusted in a continuous manner via external control, limited by solvent/electrolyte compatibility (electrochemical window); | quantum yield can be adjusted in discrete manner (different photocatalysts), redox potential limited by photocatalyst identity (i.e. may be unable to synthesize a catalyst with the desired redox potentials); |
| independent control over oxidation and reduction potentials | yes (oxidation and reduction occur at different electrodes) | no (oxidation and reduction occur with the same photocatalyst) |
| can oxidation and reduction events be separated | oxidation occurs at anode, reduction occurs at cathode, electrode separation distance spatially separates these events. A divided cell can provide additional physical separation of the two redox events (e.g. using a porous frit). Tailored membranes can also restrict permeability based on chemical nature of the ion (ion-selective membranes). | oxidation and reduction event both occur at the photocatalyst (one in the excited state and one in the ground state after the initial redox event from the excited state – spatially separation is likely limited by diffusion distance travelled between these two events |
| net oxidative transformation | requires terminal oxidant (e.g. reduction of H+ from protic solvent to H2 gas) for sacrificial reduction | requires terminal oxidant (e.g. air, persulfate salts, or reduction of H+ from protic solvent to H2 gas using co-catalyst such as cobaloximes) |
| net reductive transformation | requires sacrificial electrode (e.g. Zn anode) or terminal reductant (e.g. Et3N) | requires terminal reductant (e.g. Et3N) |
| redox neutral transformation | challenging (lifetime of radical intermediate typically not long enough to travel from one electrode to the other) – requires use of a cell with small inter-electrode spacing (<100 μm) or alternating potential/current electrolysis | facile, usually better suited for photoredox catalysis than for electrosynthesis |
| radical-radical coupling | achievable via DE to have a high local concentration of radicals | requires the use of a persistent radical |
4. SYNTHETIC COMPARISONS BETWEEN PHOTOREDOX CATALYSIS AND ELECTROCHEMISTRY METHODOLOGIES
4.1. Introduction
The second half of this review aims to detail several key synthetic transformations shared by both photochemistry and electrochemistry, with a particular focus on the key disconnections and intermediates associated with the respective strategies. For the photoredox examples chosen, we explicitly limited these reactions to ones that proceed via ET, and omit EnT-mediated reactions, although relevant references are included. The classification of reactions as net reductive, net oxidative, or net redox-neutral is denoted in the synthetic schemes using orange circles marked with +e+e, −e−e, and ±e, respectively. The list of transformations included in these sections are inclusive to the overlap of photoredox and electrochemistry, and we hope that these comparisons will illuminate the subtle limitations that are associated with synthetic translations between the two methods.
4.2. Decarboxylation
Synthetic methods that rely on redox triggered decarboxylation event are pillars in both photoredox484 and electrochemical chemistries.485 In the context of electrochemistry, as mentioned in an earlier section, the choice of electrode material or reaction conditions, or the use of a redox mediator can aid in selectively achieving either a 1e oxidation (Kolbe process; graphite anode, low current densities) or a 2e oxidation (Hofer-Moest process, Pt or glassy carbon anode, high current densities) to access the corresponding radical vs cationic decarboxylated intermediates, respectively. In contrast photoredox is typically limited to single electron transfer events, thus the 1e oxidation chemistry underpins most methods reported to date. The following sections highlight some of these powerful transformations to form carbon–carbon bonds as well as carbon–heteroatom bonds.
4.2.1. Decarboxylative Etherification
Alkoxy aryl ethers and dialkyl ethers are common motifs in bioactive molecules486, with the Williamson ether synthesis used to access primary alkyl ethers from activated alkyl halides and alcohols. However, accessing hindered variants of these structures can be difficult as unproductive elimination byproducts predominate from SN1 intermediates. To overcome this challenge, alternative functional handles alongside one- or two-electron redox activation pathways have been explored for decarboxylative etherification (Schemes 22 and 23).
Scheme 22.

A representative substrate scope for decarboxylative etherification methods via electrochemical and photoredox methods.
Scheme 23.
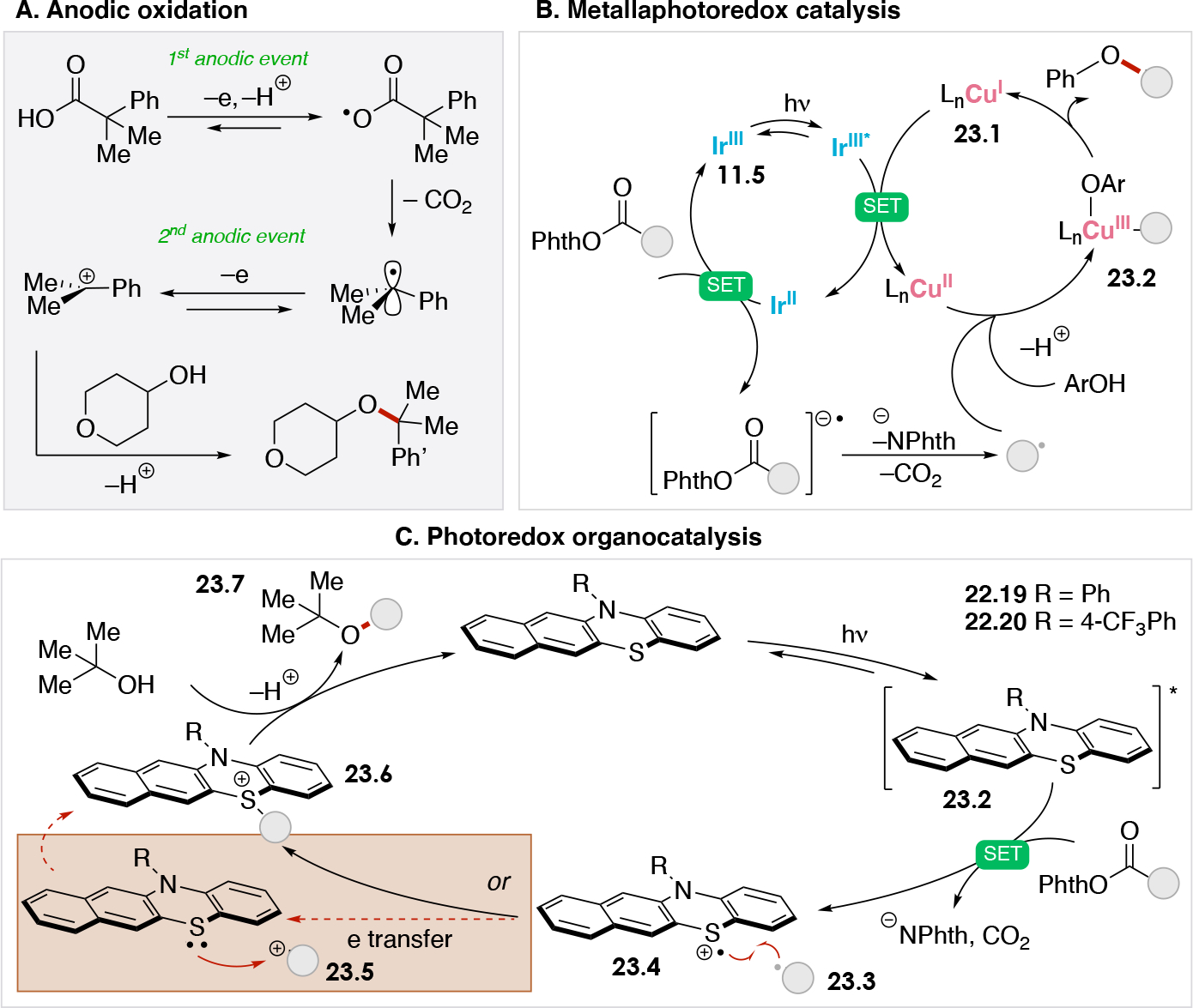
(A) Anodic oxidation produces the key alkyl carbocation for ether formation. (B) The putative metallaphotoredox mechanism for decarboxylative etherification by Hu. (C) The putative tandem photoredox and organocatalytic mechanism for decarboxylative etherification by Nagao and Ohmiya.
Baran, Blackmond and co-workers provide an electrochemical approach to this problem in their report of a modern-day Hofer–Moest reaction, in which dialkylethers are synthesized via two-electron anodic oxidation of carboxylic acids under galvanostatic electrolysis.487 The first oxidation of the carboxylate initiates decarboxylation and yields the alkyl radical, which then undergoes a second oxidation to reveal the key alkyl carbocation intermediate (Scheme 23A). Optimizing this process involved overcoming radical byproduct or elimination pathways, which were solved by using DCM as the reaction solvent. Furthermore, unproductive hydration and cathodic DCM reduction were overcome by the addition of molecular sieves and a sacrificial oxidant (AgPF6). This method is applicable to a broad range of tertiary, secondary and primary acids; partner alcohol generality and the use of water as a nucleophile is also observed. Yields range from low to excellent and are dependent on the choice of oxidant (AgClO4 vs. AgPF6). Additionally, these reactions are scalable, with respective gram-scale etherification and hydroxylation of tertiary acids obtained in good yield.
Hu and co-workers report a tandem photoredox and copper catalyzed decarboxylative C(sp3)–O cross coupling between alkyl N-hydroxyphthalimide (NHPI) esters and phenols in the presence of Et3N (Scheme 22).488 The key intermediate in this transformation is an alkyl radical formed from single electron reduction of the NHPI ester by reduced photocatalyst and subsequent radical fragmentation. A photocatalyst screen revealed that the more reducing heteroleptic iridium photocatalyst Ir[(dtbbpy)(ppy)2]PF6 (11.5) [dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine; ppy = 2-phenylpyridine] was necessary for initiating NHPI esters (E1/2red ≤ −1.26 V vs. SCE in MeCN) as the more oxidizing catalyst 11.6 was less efficient in effecting the transformation. (Scheme 23B). Furthermore, a CuI species is necessary as yields drop when starting with CuII. Stern-Volmer studies revealed that photoexcited 11.5 oxidizes a CuI-Et3N complex (23.1), which undergo ligand exchange with the phenol before trapping the alkyl radical. The resultant CuIII alkyl aryloxide (23.2) then undergoes facile reductive elimination to furnish the desired product. The phenol scope is general, with good electronic tolerance observed when coupled to cyclohexyl bromide. Secondary and primary alkyl NHPI esters were well tolerated, with moderate to excellent yields obtained. Tertiary NHPI esters were limited to non-planarizable radicals (e.g. 1-phenylcyclopropane-1-NHPI ester (22.9) as low yields (< 30%) were observed for NHPI esters derived from pivalic acid and 1-methylcyclohexanoic acid. An organophotoredox variant of this transformation was recently reported by Nagao, Ohmiya and co-workers, wherein the decarboxylative coupling of NHPI esters and aliphatic alcohols is accomplished using a N-aryl phenothiazine derivative (22.20), without the prior Cu co-catalyst requirement489. Photoexcited 22.20 (23.2) initiates the reductive decarboxylation of the NHPI ester, forming an alkyl radical (23.3) that can recombine with the cation radical of 22 (23.4). Alternatively, the alkyl radical can be further oxidized to the alkyl carbocation (23.5) — this pathway is supported by mechanistic studies involving alkyl migration that forms the more stable carbocation — which is then trapped by 22.20 to form an alkyl sulfonium intermediate 23.6. Nucleophilic displacement by an alcohol then reveals the desired unsymmetrical dialkyl ether 23.7. This method tolerates a wide range of tertiary and secondary NHPI esters, as well as numerous functionalized primary and secondary alcohols, with good chemoselectivity for the primary O-substitution over secondary O-attack. Nagao, Ohmiya and coworkers followed up this work by coupling hindered ethers and NHPI esters across an alkene, thus broadening the potential disconnections available for unsymmetrical ether synthesis.490
4.3. Minisci Reaction with Heterocycles
Photoredox and electrochemical methods have been used to form alkyl radicals employed in the Minisci reaction, with the former strategy receiving the majority of attention. Phipps and co-workers have recently reviewed both approaches491; thus, our survey of this topic will only briefly compare and contrast arene trifluoromethylation as the prototypical example. Readers interested in Minisci-type photoredox and electrochemical strategies for heteroarene alkylation492,493,494,495,496,497, hydroxymethylation498,499,500, carbamoylation501,502, and asymmetric alkylation503,504,505,506, as well as new photoelectrochemical approaches507,508,509 are encouraged to explore the listed references.
4.3.1. Arene C–H Trifluoromethylation
The trifluoromethyl group is a privileged motif in drug discovery as its incorporation into small molecules improves their metabolic stability and can promote desirable conformational changes for better target binding.510,511 (Hetero)arene trifluoromethylation typically proceeds by the formation of trifluoromethyl radical (•CF3), which can be generated by single-electron oxidation or reduction (Scheme 24). In 1993, Mallouk and co-workers reported the first example of a photochemical arene trifluoromethylation using superstoichiometric TiO2 photooxidant and silver trifluoroacetate as the CF3 source.512 The mechanism of the transformation proceeds via single electron oxidation of trifluoroacetate promotes decarboxylation and subsequent formation of •CF3, which then traps an arene and forms the C–CF3 bond. Consecutive oxidation and deprotonation then reveal the trifluoromethylated product. While a novel mechanism at the time, a small scope (8 substrates) and moderate-to-low yields highlight the limitations of this method. In 2011, MacMillan and co-workers improved on Mallouk’s initial report, in which a wide range of hetero(arenes) are efficiently trifluoromethylated in good-to-excellent yield using trifluoromethanesulfonyl chloride (TfCl).475 This transformation forms •CF3 from TfCl via single electron reduction from a photoexcited RuII catalyst (Ru(phen)3, 11.2), and releases equivalents of SO2 and chloride byproducts. Monotrifluoromethylation was observed, with bis-trifluoromethylation only becoming competitive when higher amounts of TfCl were used. Observed regioselectivities trended with the most-electron rich C–H position, as moderate-to-low regioselectivities were observed for substrates without significant electronic bias. MacMillan and co-workers highlight the generality of their method through late-stage functionalization of several biologically active molecules, most notably Lipitor, in which its three trifluoromethylated regioisomers were individually isolated using supercritical fluid chromatography. While effective source of •CF3, TfCl is prohibitively expensive to use on scale, motivating Stephenson and co-workers to use the combination of trifluoroacetic anhydride (TFAA) and pyridine N-oxides alongside Ru(bpy)3(PF6)2.513 Mixing these reagents forms a donor-acceptor adduct which then undergoes single electron reduction to reveal •CF3 and single equivalents of CO2 and pyridine byproducts. The yields for this transformation are lower (vs. MacMillan’s precedent) with bis-trifluoromethylation being problematic for several substrates, but this transformation was efficiently scaled on batch (0.12 mol) and flow (0.12 and 0.60 mol). Further mechanistic insight revealed that direct photoactivation of an EDA complex generates •CF3, thus enabling arene trifluoromethylation.514 The efficacy of the EDA complex was dependent on arene identity, as less electron-rich arenes gave minimal trifluoromethylation product in the absence of photocatalyst. The measured quantum yield of 0.87 suggests that presumptive chain propagation processes are not dominant. Finally, Stephenson and co-workers demonstrated the trifluoromethylation of N-Boc pyrrole on a kilogram scale using a flow system, obtaining the desired product 24.16 in 50% yield and 81% purity (0.95 kg) after 48 h.
Scheme 24.

A representative substrate scope for arene trifluoromethylation via electrochemical and photoredox methods.
Electrochemical approaches to arene trifluoromethylation have mainly focused on zinc trifluoromethylsulfinate (ZnTFMS) as the •CF3 source. Blackmond, Baran and co-workers first reported the use of direct electrolysis (25 mA constant current with 1.62 F•mol−1 passed; carbon cloth anode; carbon auxiliary cathode; potential drift from +0.7 to +1.3 V; Et4NClO4 electrolyte) to oxidize ZnTFMS and access requisite •CF3 for heteroarene trifluromethylation in a divided cell.515 Yields for this transformation were generally higher than their tert-butyl hydroperoxide(TBHP)-initiated method, and regioselectivity was observed for substrates with strong electronic bias. Reaction progress kinetic analysis reveals that ZnTFMS is consumed slower under the electrooxidative conditions relative to TBHP, which in turn minimizes unproductive side reactions and enable lower ZnTFMS concentrations (1.4 equiv vs. 2.0–3.0 equiv) to be used. An alternative system was recently reported by Zeng and co-workers, in which bromide oxidation to bromine is used to convert ZnTFMS to TfBr, which then is reduced to •CF3, SO2 and bromide. 516 This strategy bypasses the direct electrolysis of ZnTFMS, which in turns enables the use of an undivided cell (platinum net anode; graphite plate cathode; constant current electrolysis, j = 5 mA cm−2) without a supporting electrolyte.
Noting the potential of merging the two approaches, Ackermann and co-workers used an acridinium photocatalyst to oxidize Langlois reagent, which fragments to •CF3 and SO2, and an undivided hydrogen-evolving electrochemical cell to oxidize the resultant acridine, promote product formation, and regenerate the photocatalyst (Scheme 25).517 Control experiments reveal that the absence of either photocatalyst or electrical potential is detrimental to product formation (<10%). This strategy enables the trifluoromethylation of electron-rich arenes, heteroarenes, and the late-stage functionalization of several bioactive molecules in moderate-to-good yields. Thus, this example demonstrates the complementarity of photoredox catalysis and electrochemistry as the deficiencies present in individual systems are overcome through their merger.
Scheme 25.

A representative scope and putative mechanism for the electrophotochemical arene trifluoromethylation.
4.4. α-Cyanation of Amines
The functionalization of pyrrolidine serves as a great example of the contrasts that can be seen in the synthetic methods based on electrochemistry and photoredox chemistry. The α-functionalization of amines in the context of photoredox and electrochemistry leverage different intermediates (Scheme 26). A wealth of photoredox methods to achieve the α-functionalization of 2° & 3°-amines are known518,519,520,521 and these by in large rely on the generation of α-amino radicals, either from a sequential oxidation-deprotonation or a direct HAT process or an oxidative (1e) decarboxylation event. In contrast to the net 1e oxidation events used in photoredox, the oxidation of amines under electrochemical conditions is typically challenging to stop at the 1e oxidation intermediate (α-amino radical), and thus the subsequent oxidation (net 2e oxidation) is typically observed, resulting in the generation of iminiums. The prototypical example is the Shono oxidation522,523 which achieves an α-methyoxylation of an amine via the in situ generation of the iminium. Electrochemical iminium formation via anodic oxidation of (N-acyl) amines can be achieved via a cation pool method.522,523,524,525,526,527,528 The use of a redox mediator can enable for α-oxygenation of amine (protected as a carbamate) as demonstrated by Stahl.529 We note that reductive generation of α-amino radical from imine precursors represents an alternative approach to generated α-functionalized amines.530,531,532 Electrochemical reductive generation of α-amino radicals from iminiums has been used to form C–C bond via reaction with acrylates533,534,535,536 as well as for dimerizations.537,538
Scheme 26.
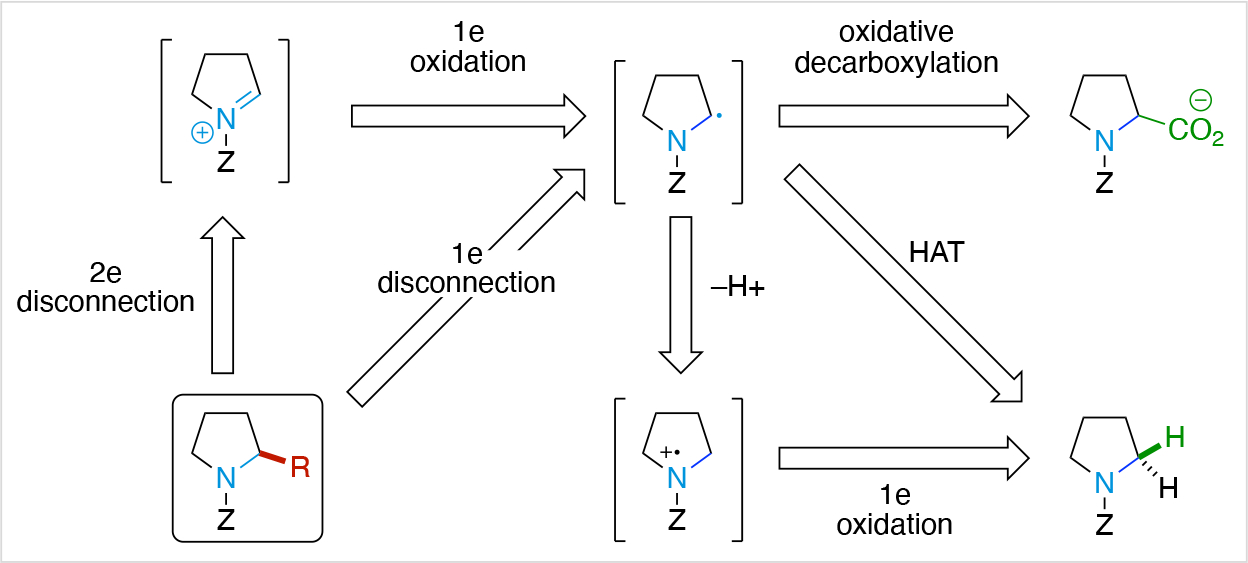
Illustration of 1e vs 2e disconnection for the α-functionalization of amines.
The α-cyanation of cyclic amines can be accomplished in numerous ways either via one electron or 2 electron redox chemistries, or via HAT processes as summarized in Scheme 26. Early electrochemical methods focus on cation pool or direct oxidation approaches.539,540,541,542 Onomura reported an undivided cell electrolysis method using TMSCN, MeSO3H, and graphite carbon (GRC) electrodes at 0 °C (method A1).543 The acid was essential for the generation of product and was proposed to control the equilibrium between HCN and CN− in addition to suppressing enamine formation from the proposed iminium intermediate. Tajima has also reported a direct oxidative cyanation of imines using constant potential electrolysis, Pt electrodes and polystyrene-supported quaternary ammonium cyanide (PS-NMe3CN, method A2).544 Stahl leveraged a redox-active catalysts to electrochemically generate an oxammonium catalyst capable of facilitating the oxidation of amines to iminiums that serve as the key intermediate undergoing nucleophilic attack by cyanide (method A3).545 The use of a redox mediator provides for milder reaction conditions compared to the direct oxidation methods and thus enables a great functional group tolerance in the substrate scope. The closest related photoredox method is one that uses rose bengal and air, either in the presence (methods B2) or absence (method B3) of graphene oxide (GO)546 to access iminiums for nucleophilic cyanation. Emmert and Oderinde reported an Ir-catalyzed photoredox method using NaCN and air as the terminal oxidant (Method B8), again proposed to proceed via an iminium intermediate.547
Strikingly different mechanistic strategies exist for the numerous photoredox methods that are proposed to access α-amino radicals. Oxidative deborylation (method B1)548 or decarboxylation (method B4 and B5) followed by cyanation of the α-amino radical using TMSCN (method B1) or hypervalent iodine reagent cyanobenziodoxolone (CBX) in the presence of CsOBz (method B4)480 or CsHCO3 (method B5).549 The generation of alkyl radicals via photoredox decarboxylation processes are widely known and was employed as α-cyanation strategy by Gonzalez-Gomez leveraging TsCN as the cyanation reagent (method B7).550 The direct C–H functionalization approach, bypassing the requirement for pre-functionalization of the amine can be accomplished using hydrogen abstraction strategies using an HAT catalyst (BDE of α-amino C–H bonds typically range from 89–94 kcal•mol−1).551 Inoue reported a method using benzophenone for HAT to generate the requisite α-amino radical from the corresponding amine with subsequent cyanation using TsCN in the absence or presence of a hindered pyridine base (methods B10 & B11, respectively).552 Another HAT approach was reported by Kanai and Oisaki, this time making use of BINOL phosphoric acid (1,1′-binaphthyl-2,2′-diyl hydrogen phosphate), TsCN and an Ir-photocatalyst [Ir(dFCF3ppy)2(5,5′-dCF3bpy)(PF6); note that dFCF3ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine, 5,5′-dCF3bpy = 5,5′-bis(trifluoromethyl)-2,2′-bipyridine] (method B9).553 Other methods include the use of Pd-porphyrin catalyzed oxidative cyanation method using O2 as the terminal oxidant (method B12),554 Rueping reported methods of using an Ir-photocatalyst (method B13; note: tbppy = 2-(4-tert-butylphenyl)-pyridine)),555 or a metal oxide photocatalyst (TiO2 or ZnO),556,557 or Rose Bengal (method B15)558 to access iminium intermediates for nucleophilic cyanation, similar to strategies reported by Stephenson using a Ru-photocatalyst (method B14)94,559 or those by Che using a gold photocatalyst.560 The α-cyanation of amines presented in the synthetic scheme (Scheme 27) highlights the breadth of mechanistic approaches leveraged to achieve these products and their proposed mechanisms are respectively shown in Scheme 28.
Scheme 27.
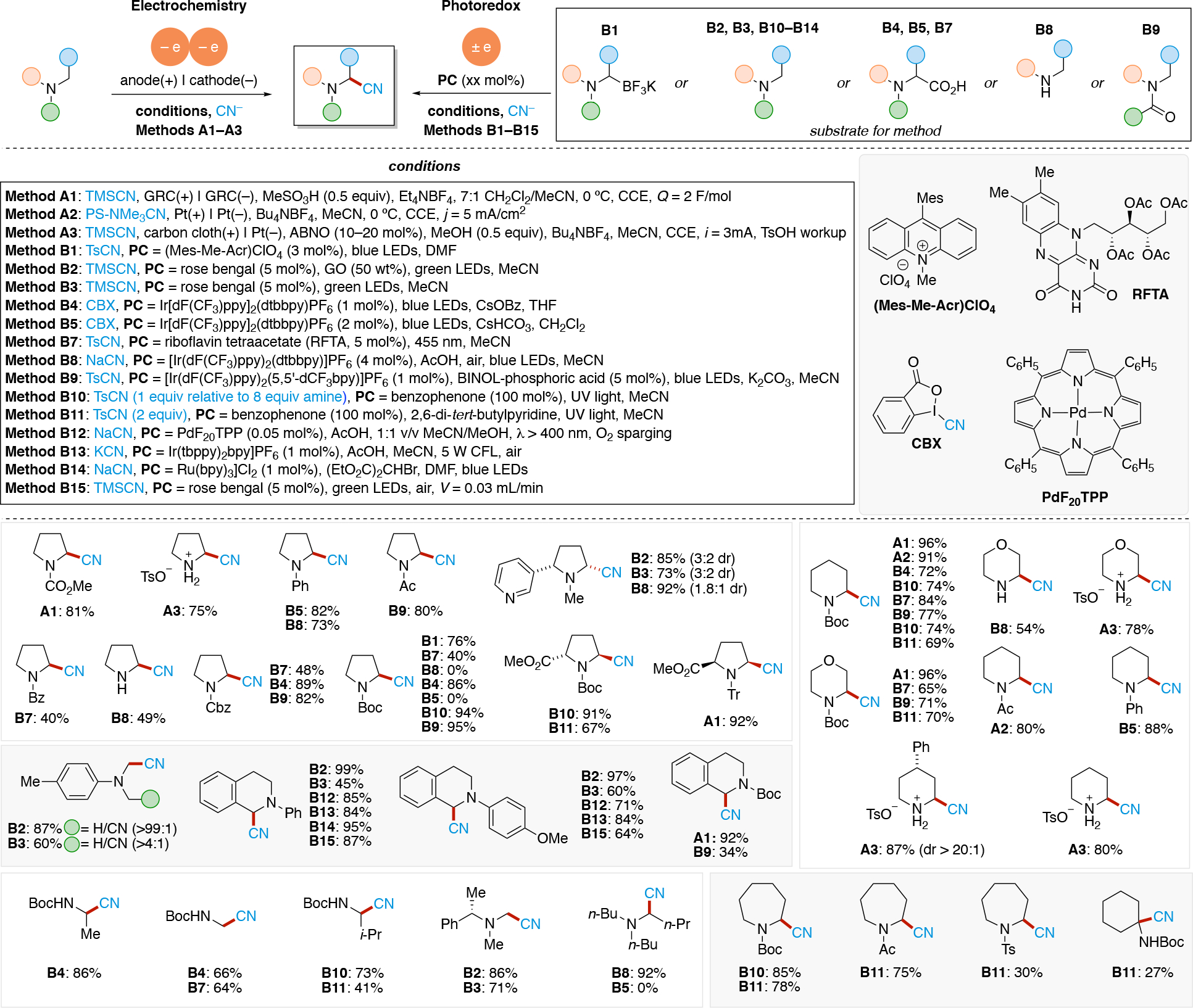
Representative examples of electrochemical (Method A) and photoredox methods (Method B) for the α-cyanation of amines.
Scheme 28.

Selected mechanisms for amine α-cyanation discussed Scheme 27: (A) electrochemical amine oxidation to iminiums; (B) indirect electrochemical oxidation using an oxoammonium; (C) photoredox oxidation of amines to iminiums; (D,E) photoredox decarboxylation via SET from a carboxylate or PCET from carboxylic acid (F,G) direct and indirect HAT using benzophenone and phosphate derived radicals, respectively.
4.5. Dehydrogenation of N-Heterocycles
The dehydrogenation of N-heterocycles such as tetrahydroquinoline can be accomplished under oxidative conditions in the presence of a redox-active TEMPO-catalyst. Comparable yields are obtained using either electrochemical561 or photochemical oxidative conditions relying on a Ni-coated TiO2 photocatalyst.562 Tetrahydroquinoline derivatives as well as related indoline (2,3-dihydroindole), imidazoline, 1,4-dihydropyridines can be oxidized. Interestingly, incomplete oxidation of tetrahydroisoquinoline was observed to result in mixtures of the isoquinoline and 3,4-dihydroquinoline. Partial oxidation of N-arylated tetrahydroisoquinoline using Ru(bpy)3(PF6)2-photoredox and CCl4 as the sacrificial oxidant to access iminium salts.563
4.6. Nickel-Catalyzed Cross-Couplings
The synthetic utility of nickel-catalyzed cross-couplings has recently grown alongside the advances in understanding the radical-based mechanisms of odd-electron Ni species. When combined with the radical-forming pathways associated with photoredox catalysis and electrochemistry, Ni catalysis enables the discovery of new reaction platforms complementary to palladium-catalyzed methods.564 Three examples — C(sp2)–N, C(sp2)–S, and reductive C(sp2)–C(sp3) cross-couplings — are presented.
4.6.1. C(sp2)–N Bond Cross-Coupling
The Buchwald-Hartwig amination is the gold standard for arene C–N cross couplings, with applications ranging from medicinal chemistry to materials research.565 However, because of the high costs associated with Pd and the specialized ligands involved, there is a demand for alternative, low-cost methods. One important solution is the development of Ni-catalyzed methods that use photoredox and electrochemical strategies to overcome redox barriers. A tandem photoredox- and nickel-catalyzed method (termed Ni metallaphotoredox hereafter) was reported by MacMillan, Buchwald and co-workers, in which an IrIII photocatalyst and NiII pre-catalyst were used to couple secondary and primary amines to aryl bromides and iodides in good to excellent yields under ambient conditions (Schemes 30 and 31).566 No specialized ligand was used in this transformation as the presumed ligands on the nickel center are the halide and amine nucleophile with the latter existing in equilibrium speciation states.567 This transformation was subjected to high-throughput screening by collaborators at Merck & Co., Inc., Kenilworth, NJ, USA. C–N cross-coupling with piperidine was observed for 78% of complex, drug-like aryl halides studied, highlighting its utility for high-throughput experimentation (HTE). The putative mechanism proceeds by oxidative addition of the aryl halide into a Ni0 species to form NiII oxidative addition complex 30.1 that undergoes ligand exchange with an amine to form a NiII aryl imido complex (30.2) (Scheme 30A). This intermediate is then oxidized to NiIII species 30.3 by photoinduced electron transfer to excited state heteroleptic IrIII photocatalyst Ir[dF(CF)3ppy]2(dtbbpy)PF6 (11.6). This NiIII species then undergoes facile reductive elimination to yield the desired aryl amination product. The two catalytic cycles converge with electron transfer from the reduced IrII photocatalyst to the NiI intermediate. Buchwald and co-workers then adapted this method to a flow system with Ru(bpy)3(PF6)2 replacing Ir[dF(CF)3ppy]2(dtbbpy)PF6 as the photocatalyst.568 Gram-scale C–N cross-couplings with (hetero)arenes were demonstrated, with 2.21 g of commercial anesthetic tetracaine synthesized in 84% yield with a 10-minute residence time. Johannes and Oderinde reported a similar transformation, albeit with dtbbpy as a supporting ligand on Ni and is limited to aniline, sulfonamide, and benzylamine nucleophiles (Scheme 30B).569 An alternative mechanism is proposed, in which reductive quenching of photoexcited Ir[dF(CF)3ppy]2(dtbbpy)PF6 by aniline generates an aniline cation radical that is deprotonated to form an anilinyl radical (30.4). Trapping this open shell species at a NiI center forms a NiII imido intermediate (30.5) that is reduced to NiI by IrII before undergoing oxidative addition with the aryl halide. Facile reductive elimination from the NiIII aryl imido complex 30.6 yields the desired arylamine. Further investigation of this mechanism by Miyake and co-workers suggest that either direct UV photoexcitation of NiIIBr2-tris(amino) species570 or Förster-type energy transfer from Ru(bpy)3Cl2567 can facilitate the same transformation. More recently, MacMillan and co-workers present a revised mechanism in which aryl C–N cross-coupling proceeds via a Ni(I/III) dark cycle and photoreduction of off-cycle NiII species initiates and sustains the catalytic Ni pathway (Scheme 30C).571 Stern-Volmer and transient absorption spectroscopy studies show that DABCO is the primary quencher of photoexcited IrIII, which generates a IrII reductant. Reduction of resting-state NiII species 30.7 by IrII initiates the NiI/Ni(/III) cycle responsible for C–N coupling, as suggested by stoichiometric reductive elimination and quantum yield studies. Because NiII reduction is the rate-determining step, photocatalyst ligand modifications to obtain greater reductive overpotentials led to 37-fold and 10-fold increases in initial rate and quantum yield of product formation respectively. Nocera and co-worker independently provide evidence for the NiI/NiIII cycle, as C–N cross-coupling is observed when a catalytic amount of Zn reductant is used in place of a photoredox catalyst, albeit for a limited substrate scope.572 Taken together, these results suggest a complex mechanistic landscape in which modifications to catalyst, reagent and irradiation wavelength result in C–N coupling operating by different mechanisms and that a dark catalytic cycle (thermally sustained cycle, free of requiring light) may be operational with light serving to drive photoredox chemistry to bring off-cycle Ni species onto the catalytic cycle.
Scheme 30.

(A) The original mechanism proposed for nickel metallaphotoredox-catalyzed C–N cross coupling. (B) An alternate C–N cross coupling mechanism for aniline substrates proposed by Oderinde. (C) Current revised mechanism for nickel metallaphotoredox-catalyzed C–N cross coupling. Note: blue SET boxes represent a specific coupled electron transfer even between IrIII* and organometallic intermediate 30.2.
Scheme 31.
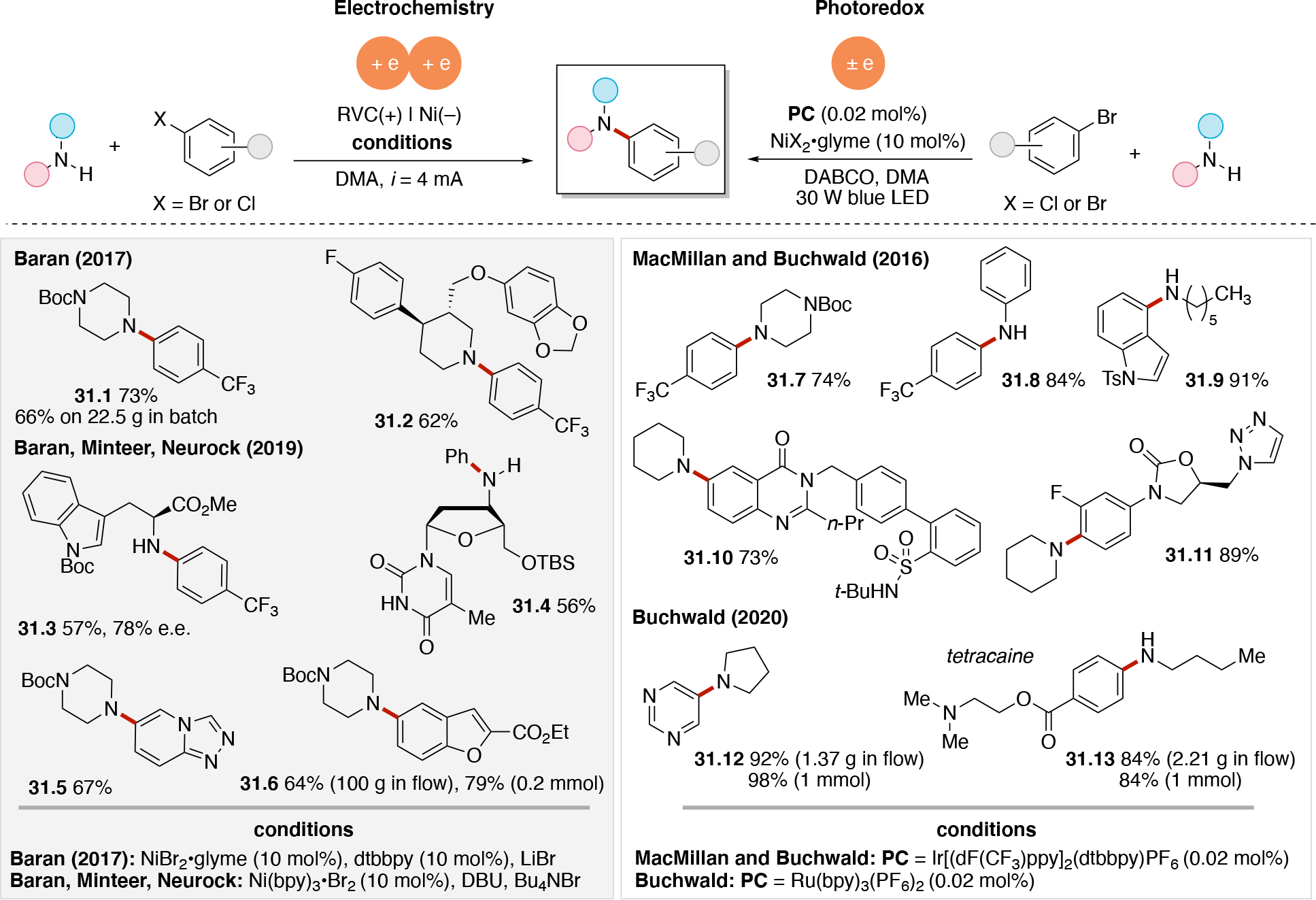
Representative examples of electrocatalysis and photoredox catalysis for nickel-catalyzed C–N cross coupling.
The electrochemical approach to Ni-catalyzed arene C–N cross coupling was developed by Baran and co-workers using catalytic NiBr2-dtbbpy in an undivided, galvanostatic cell.573 This method enables the coupling of hetero(aryl) bromides with 1°, 2°, and 3° amines in yields comparable to photoredox and palladium catalysis. This method was also scalable, with the coupled product of 4-bromobenzotrifluoride and N-Boc piperazine obtained in 66% yield. Further mechanistic investigation was carried out by Baran, Minteer, Neurock, and co-workers in which UV-Vis spectroscopy, cyclic and square wave voltammetry, and DFT computations were used to probe the elementary steps for the transformation (Scheme 31).574 A NiI/NiIII pathway was found to be operative, with a NiI-halide species 32.1 formed via cathodic reduction from NiII undergoing oxidative addition with the aryl halide to form a NiIII intermediate (32.2). This complex can substitute a halide for an amine nucleophile, setting up the NiIII aryl imido complex (32.3) for reductive elimination to yield the desired product. However, this ligand exchange step is the minor pathway in this transformation, as 32.2 forms stable NiII intermediate 32.4 via facile comproportionation with 32.1 or through cathodic reduction. Ligand exchange at 32.4 is the rate-determining step and is dependent on base loading; the resultant NiII aryl imido complex 32.5 can then be oxidized to the key reductive elimination adduct 32.3 Thus, the overarching role of electrical current is to regenerate catalytically-active NiI/NiIII intermediates. With a better understanding of mechanism, Baran, Minteer and Neurock were able to expand the nucleophile scope to amino acid esters, nucleoside analogs, and oligopeptides and the arene scope to aryl chlorides. Additionally, they demonstrate the use of flow chemistry to synthesize a precursor to the FDA-approved antidepressant vilazodone, obtaining the aminated product in 64% yield on a 100 g scale.
4.6.2. Arene C–S Cross-Coupling
The cross-coupling of aryl halides and thiols has historically been a challenge for transition metal catalysis as catalysis deactivation via the strong coordination of thiolates necessitates the use of specialized ligands, high catalyst loadings and/or high reaction temperatures for successful C–S bond formation.575 As a result, redox strategies using nickel catalysis have been developed to overcome some of these issues (Scheme 33). Oderinde, Johannes and co-workers circumvent this problem by using oxidative photoredox catalysis to generate thiyl radicals that participate in a NiI/NiIII catalytic cycle to provide aryl sulfides from aryl iodides.576 Benzylthiols, thiophenols, alkyl thiols and cysteine derivatives were competent nucleophiles, with the only low-yielding substrate being the sterically-hindered tertiary thiol. The electrochemical counterpart to this method was reported by the Wang577 and Mei578 groups independently, in which cathodic reduction generates active Ni0 and thiolate from thiol with concomitant hydrogen evolution, while diametrical anodic oxidation provides the active thiyl radicals from the thiolate. Both undivided cell systems use LiBr, amide solvents, and nickel foam as the electrolyte, solvent, and cathode respectively, however they diverge in choice of anode and electrochemical set-up. Wang uses graphite felt (anode) under potentiostatic electrolysis, whereas Mei uses a sacrificial magnesium (anode) under a constant current. A notable side reaction pathway observed in the electrochemical set-ups is the formation of disulfides, which presumably forms via thiyl dimerization. This side product is not appreciably observed under photoredox conditions because thiyl radicals are catalytically formed.
Scheme 33.
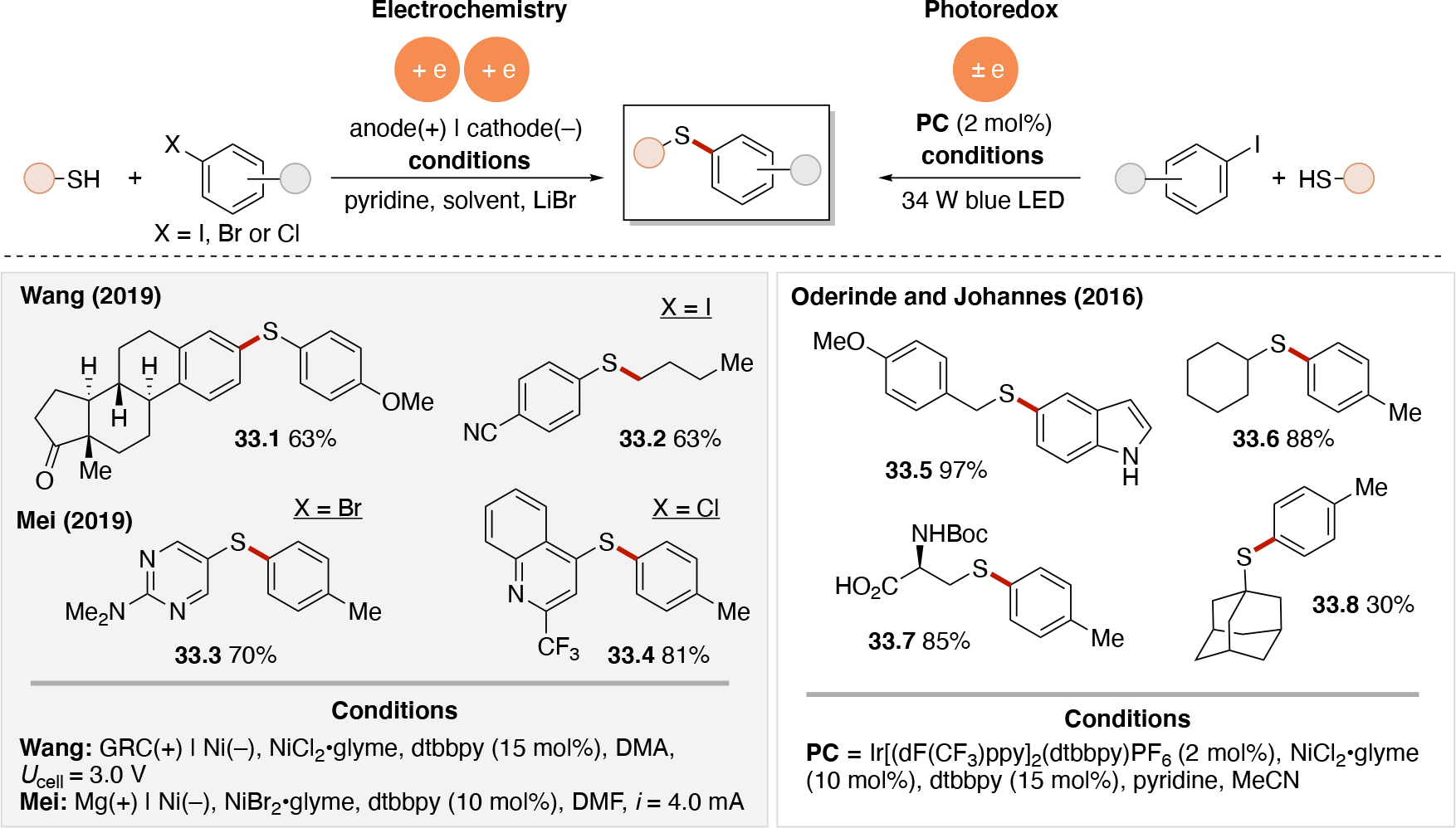
Electrochemical and photoredox approaches to activating nickel-catalyzed aryl thiolation.
4.6.3. C(sp2)–C(sp3) Cross-Coupling
The facile and general construction of alkyl-aryl bonds via transition metal catalysis is a longstanding challenge of organic synthesis and the development of redox-centered pathways have provided important contributions towards solving this problem and streamlined its use for drug discovery.579 In particular, we highlight photoredox and electrochemical strategies involving the coupling of aryl halides with either (1) alkyl trifluoroborates (Schemes 34 and 35) or (2) alkyl halides (Schemes 36 and 37).
Scheme 34.

Representative examples of Ni-metallaphotoredox catalyzed cross-coupling of alkyl trifluoroborates and aryl halides, with respective putative mechanisms depicted.
Scheme 35.

Representative examples of electrochemically-mediated nickel-catalyzed cross-coupling of alkyl trifluoroborates and aryl halides alongside its putative mechanism.
Scheme 36.
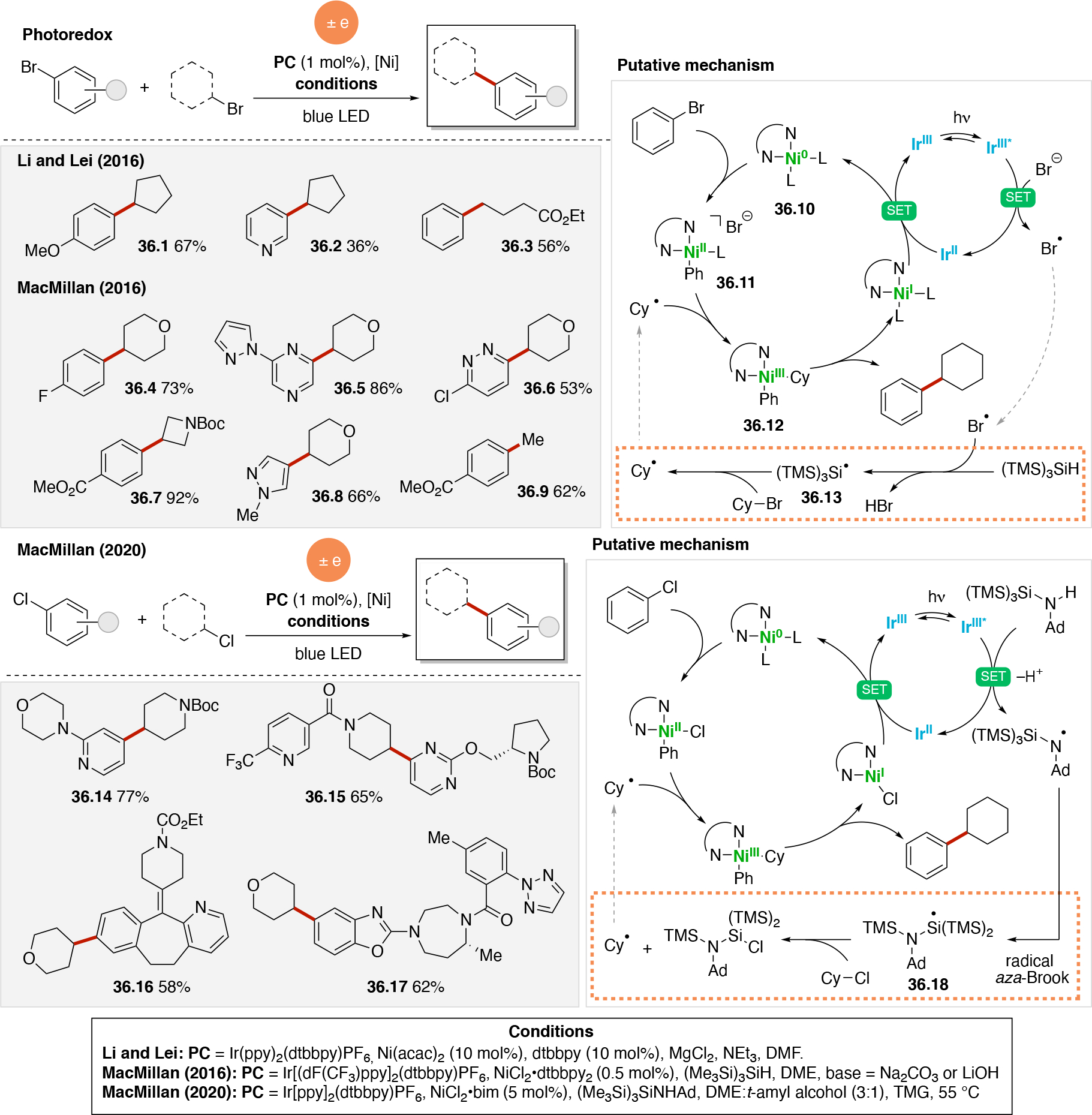
Representative examples of nickel-metallaphotoredox catalyzed cross-electrophile coupling of alkyl and aryl halides, with respective putative mechanisms depicted.
Scheme 37.
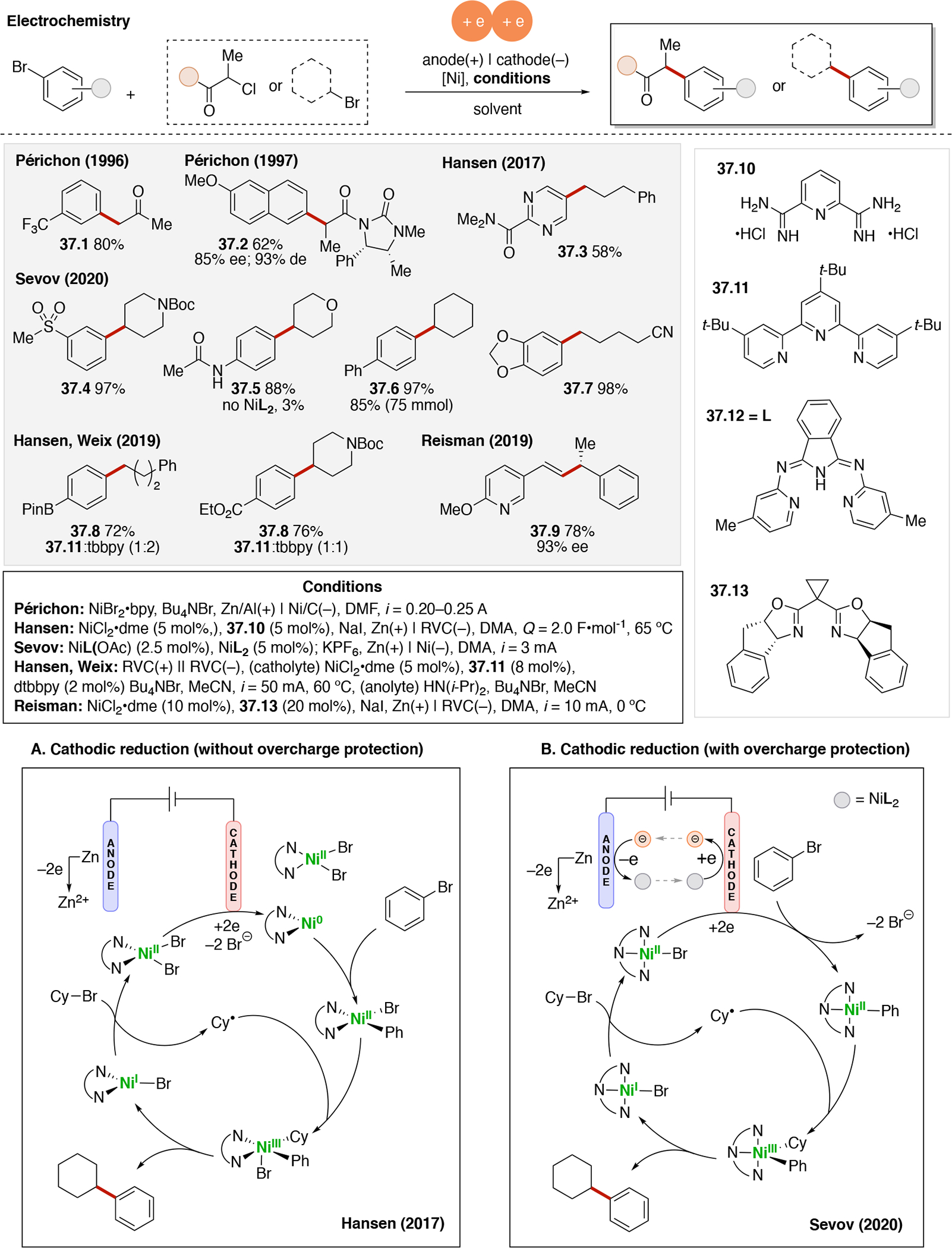
Representative examples of electrochemical methods for nickel-catalyzed cross-electrophile coupling of alkyl and aryl halides, with respective putative mechanisms depicted.
4.6.3.1. C(sp2)–C(sp3) Couplings Between Aryl Halides and Alkyl Trifluoroborates
Palladium-catalyzed cross-couplings between aryl halides and alkyl boron nucleophiles is a classical strategy for accessing C(sp2)–C(sp3) bonds580,581,582, with recent methods developed for enantiospecific couplings.583,584 Despite its successes, Pd-catalyzed reactions generally require high temperatures or additives to accelerate the slow alkyl transmetalation step, which is potentially problematic for secondary alkyl boron nucleophiles due to the potential for β-hydride elimination at the palladium center. One way to alleviate this problem is to use analogous tetravalent organotrifluoroborates, which undergo cleaner transmetalation and are less susceptible to degradative oxidation relative to trivalent boron nucleophiles.585,586 However, a more general solution is single electron transmetalation, where an alkyl radical generated via single electron oxidation of the parent alkyl trifluoroborate can add to a transition metal center to generate an odd-electron organometallic species.587,588 Molander and coworkers report a Ni-metallaphotoredox method for generating stabilized benzyl radicals from single electron oxidation of a corresponding alkyl BF3K species and coupling it with aryl bromides.589 A wide range of benzylic BF3K species and aryl bromides with differing electronic properties are tolerated in this transformation, with the products obtained in moderate to excellent yield. Follow-up work by Molander and coworkers expanded the scope to include secondary alkyl trifluoroborates590,591 — a key limitation of previously described Pd-catalyzed methods — as well as α-alkoxy-,592,593,594 α-amino,595 α-hydroxyalkyl-,596 and α-trifluoromethyltrifluoroborates.597 Notably, the use of alkyltrifluoroborates enables selective C(sp2)–C(sp3) cross-couplings even for aryl halides containing boronic acids, boronic esters or MIDA boronates.598 The putative mechanism is as follows: The photoredox-generated open shell alkyl radical 34.5 readily enters the catalytic cycle by adding to a NiII-arene oxidative addition complex 34.6 to form a NiIII intermediate 34.7 that readily undergoes reductive elimination to provide the desired C(sp2)–C(sp3) coupling product 34.8. Concomitant reduction of the resultant NiI complex 34.9 to Ni0 by an IrII species closes the two catalytic cycles. However, follow-up computational studies by Kozlowski and Molander reveal that an alternate mechanism may exists, where a Ni0-tbbpy species 34.15 intercepts an alkyl radical 34.16 to form a NiI species 34.17, which then participates in haloarene oxidative addition and subsequent reductive elimination to furnish the C(sp2)–C(sp3) coupled product.599 This revision likens the mechanism to aryl-alkyl Ni-catalyzed Negishi600,601 and Kumada602,603 couplings, where the participation of alkyl radicals is shown. This study also proposes that asymmetry at the C(sp3) center can be induced by using a sterically biased chiral bisoxazoline ligand; reversible alkyl radical fragmentation from the NiIII intermediate leads to enantioconvergent dynamic kinetic resolution and the formation of the desired chiral species. In order to activate tertiary alkyl trifluoroborates to aryl halides, Molander and Primer switched from neutral bipyridine scaffold on Ni to the anionic 2,2,6,6-tetramethyl-3,5-heptanedionate (TMHD).604 This change enables the coupling of several tertiary alkyltrifluoroborates with electron-deficient bromoarenes; however, electron-rich bromoarenes and N-heteroaryl bromides were inefficient coupling partners. The effectiveness of this ligand switch was detailed by Gutierrez, Molander and coworkers, in which computational studies suggest that in the anionic TMHD-Ni system, aryl-alkyl coupling occurs via an outer-sphere reductive elimination (34.24), whereas inner-sphere reductive elimination is operative when the neutral tbbpy-Ni system is used.605 This distinction arises from the steric congestion at the aryl-alkyl-halo- NiIII intermediate, where the cross-coupling barrier is at least 10 kcal•mol−1 higher for acyclic tertiary alkyls. Taken together, these studies add to the growing field of work incorporating alkyl radicals into transition metal catalysis.428,606
Electrochemical methods for the coupling of alkyltrifluoroborates and aryl halides have been relatively underdeveloped due to the difficulties in minimizing undesirable Ni-associated homocoupling side reactions, but a recent report by Liu and coworkers using convergent paired electrolysis to effect the coupling of benzyltrifluoroborates and aryl halides suggests that this strategy can mimic redox-neutral photoredox catalysis.607 Using galvanostatic electrolysis, C(sp2)–C(sp3) coupled products are obtained from electronically distinct aryl bromides and benzyltrifluoroborates in moderate to excellent yields, with the compatibility of aryl chlorides and vinyl bromides as coupling partners displayed. Notably, one advantage of this electrochemical method is its scalability, as the coupling of benzyltrifluoroborate and methyl 4-bromobenzoate can be scaled (up to 3.0 g) with minimal yield drop (93% to 86%). A key distinction of this transformation is that mechanistic studies of cathodic reduction suggest that only NiII/NiI reduction is operative, which rules out the involvement of a Ni0 species as proposed by the Kozlowski and Molander mechanism. Thus, the putative mechanism involves a NiI/NiIII cycle, where cathodic reduction of a NiII-tbbpy intermediate 35.7 forms a NiI species 35.8 that readily undergoes oxidative addition with an aryl halide to form an aryl-bishalo Ni intermediate 35.9. Further cathodic reduction of 35.9 and halide extrusion leads to formation of the corresponding NiII complex 35.10, which intercepts the benzyl radical — generated from anodic oxidation of the benzyl trifluoroborate — to form the aryl-alkyl-halo NiIII intermediate 35.11. Reductive elimination furnishes the desired C(sp2)–C(sp3) coupled product and completes the catalytic cycle. Despite their success with benzyl trifluoroborates, Liu and coworkers were unable to use other alkyl trifluoroborates, which is a current limitation, but future developments in this field should enable greater generality of both coupling partners.
4.6.3.2. C(sp2)–C(sp3) Cross-Electrophile Couplings Between Aryl and Alkyl Halides
Transition-metal catalyzed cross-electrophile couplings (XECs) between aryl and alkyl halides typically involves the use of stoichiometric metal reductants (e.g. Zn, Mn), with odd-electron organometallic species and radical intermediates implicated in the transformation.608 Thus, replacing these chemical reductants with photoredox- or electrochemical- generated electrons offer new XEC strategies (Schemes 36 and 37). Lei, Lu, and co-workers report a metallaphotoredox XEC between aryl and alkyl bromides using triethylamine as the terminal reductant, and obtained moderate to good yields for the desired products, albeit with limited substrate generality.609 The mechanism proposed is similar to the seminal XEC report by Weix and co-workers610 with IrIII photocatalyst 11.5 facilitating NiI/Ni0 reduction via the IrIII/IrII cycle. A more general metallaphotoredox approach was reported by MacMillan and co-workers in which IrIII photocatalyst 11.6 and NiCl2•dtbbpy are used to effect C(sp2)–C(sp3) XEC between alkyl and aryl bromides in good to excellent yields.611 The putative mechanism operates by a Ni0 complex 36.10 that undergoes oxidative addition with the aryl halide to form NiII species 36.11, which then intercepts an alkyl radical formed in situ to yield the NiIII complex 36.12. This alkyl radical is formed via halogen abstraction by a silyl radical 36.13 originating from tris(trimethylsilyl)silane (TTMSS); bromine radical generated from the bromide oxidation by photoexcited 11.6 participates in H-atom abstraction from TTMSS to form 36.13. Reductive elimination from 36.12 yields the desired C(sp2)–C(sp3) XEC product, and a NiI intermediate that closes the nickel and photoredox cycles by oxidizing IrII. Notably, five-membered heterocycles are compatible electrophiles, which have historically been difficult for palladium-catalyzed cross-coupling reactions. This strategy was also applicable to activated alkyl halides such as α-chloro carbonyls612 and bromodifluoromethane.613 More recently, MacMillan and co-workers reported the XEC of aryl chlorides and alkyl chlorides through photoredox-mediated generation of a polarity-matched aza-silyl radical 36.18 to mediate effective chlorine atom abstraction from unactivated alkyl chlorides.614 The scope of alkyl chlorides and aryl chlorides is broad, and good functional group tolerance is observed. Additionally, the late-stage functionalization of several pharmaceuticals is shown, with desired C(sp2)– C(sp3) coupling obtained in the presence of triazoles, amides, sulfones, and carbamates — functional groups common in medicinal chemistry settings.
The electrochemical approach to XEC615 was first detailed by Périchon and co-workers, which couples activated alkyl halides (e.g. α-chloro carbonyls) and aryl bromides/iodides) using superstoichiometric Ni(bpy)2Br2 under constant current electrolysis in an undivided cell.616 Two-electron reduction of NiII to Ni0 is observed and this species participates in oxidative addition with the aryl halide to an aryl-NiII intermediate that can intercept an α-carbonyl radical originating from cathodic reduction of the parent α-chloro carbonyl. This NiIII species rapidly undergoes reductive elimination to furnish the desired C(sp2)–C(sp3) coupled product. Slow addition of the alkyl halide was necessary for circumventing unproductive alkyl halide homocoupling. The racemization of chiral methyl 2-chloropropionate suggests a radical pathway for the formation of the α-carbonyl radical and using a substrate with chiral auxiliary enables the diastereo- and enantioselective C(sp2)–C(sp3) couplings.617 Hansen and co-workers extended the method to include unactivated alkyl halides, with the main modifications being the use of 4,4′-dimethoxy-2–2′-bipyridine (dmbpy) as the supporting ligand on Ni, higher constant current (10 mA; 2.0 F•mol−1), and higher temperatures (65 °C) (Scheme 37A).618 This XEC method is scalable up to 6.5 mmol, with a constant current of 2.0 F•mol−1 and 10 mol% of NiCl2•dmbpy. The reaction is sensitive to the rate of NiII reduction, as the use of high currents leads to low yield, poor selectivity, and low mass balance. Pyridine-amidine and 2,6-bis(amidine)pyridine (37.10) ligands were also used as they enabled higher yields for certain substrate classes (e.g. heteroaryl bromides) relative to dmbpy. Sevov and co-workers improved further on this system by introducing a redox shuttle to mitigate unproductive overcharge electron transfer that leads to catalyst degradation and undesirable protodehalogenated and homocoupling arene byproducts (Scheme 37B).619 The reaction was performed under constant current conditions (3 mA; 2.5 F•mol−1) in an undivided cell, with a tridentate bis-(pyridylamino)isoindoline[(BPyI), 37.12]-Ni species and a Ni(BPyI)2 redox mediator found to be the most effective co-catalyst system. The overcharge protection afforded by Ni(BPyI)2 enables XEC for redox sensitive groups such as sulfones and nitriles and challenging secondary alkyl halides at ambient temperatures and in good yields; without the redox mediator, yields are typically < 20%. Furthermore, the redox mediator enabled this transformation to be scaled in batch (75 mmol) with a high 400 mA current, providing the XEC product in 85% yield after 12 h. However, a potential downside may be a decrease in charge efficiency, as degenerate shuttling requires approximately an additional 0.5 equivalents of e (2.5 F•mol−1).
The methods discussed thus far require sacrificial anodes that produce stochiometric metal waste, leading to reproducibility issues that limit its application on larger scales. Hansen, Weix and co-workers offer a workaround by using diisopropylamine as the terminal reductant, albeit in a divided cell run at a constant current (25 mA; 2.0 F•mol−1), with substrates and catalyst segregated in the cathodic chamber and the reductant isolated in the anodic chamber.620 A dual ligand system — terpyridine (tpy) and bipyridine (bpy) — were used for Ni in this method, with varying ratios of each species influencing product yield. Qualitative analysis suggests that substrates with fast alkyl halide activation benefit from a lower tpy:bpy loading whereas high levels of aryl homocoupling can be circumvented using a higher tpy:bpy ratio.
Reisman and co-workers demonstrated C(sp2)–C(sp3) XEC can be rendered enantioselective, albeit through Ni-catalyzed reductive alkenylation between benzyl halides and vinyl bromides.621 A chiral indanyl- substituted bis(oxazoline) (37.13) catalyst enables moderate to excellent yields with good to excellent ee under constant current electrolysis (10 mA; 2.0 F•mol−1) in an undivided cell. This transformation could be scaled up on a gram scale by increasing the current to 100 mA with only minor losses in yield and enantioselectivity for a XEC between (E)-1-(2-bromovinyl)-4-methoxybenzene and (1-chloroethyl)benzene (83% yield, 91% ee). While the electrochemical and photoredox XECs may appear similar, the mechanisms are distinct because of the net electron flow involved in each respective transformation. In the electrochemical mechanism, a minimum input of 2 electrons via cathodic reduction is necessary to reduce a NiII pre-catalyst to generate a catalytically-active Ni species whereas in photoredox catalysis, a one-electron mechanism is invoked for generating Ni0 from NiI, with net neutral electron flow. More recently, Li and coworkers report a dehydroxylative method for alkyl-aryl XEC that proceeds through an anodic Appel reaction to yield an alkyl bromide intermediate that readily engages in the electrochemically-mediated Ni-catalyzed XEC with aryl bromides in accordance with the previously-discussed mechanism.622
4.6.4. Arene C–P Cross-Coupling
C(sp3)–PV and C(sp2)–PV bonds are important synthetic motifs that are found in pharmaceuticals623,624,625,626, agrochemicals, organic materials627, ligands for asymmetric synthesis,628 and as organocatalysts629,630,631,632,633. Precursors to the synthesis of these functional motifs via redox pathways involve the single electron oxidation of diaryl and dialkyl phosphine oxides, as well as organophosphites to generate phosphinoyl radicals after deprotonation. The formation of phosphinoyl radicals via photochemical pathways have been previously studied634 and their generation via photoredox or electrochemical methods opens up new reactivity pathways for these reactive intermediates. For this review, we will specifically highlight nickel-catalyzed phosphorylation of aryl halides via redox strategies (Schemes 38 and 39), but readers should consult the following reviews for convergent and divergent phosphorylation methods between photoredox catalysis635,636 and electrochemistry637.
Scheme 38.
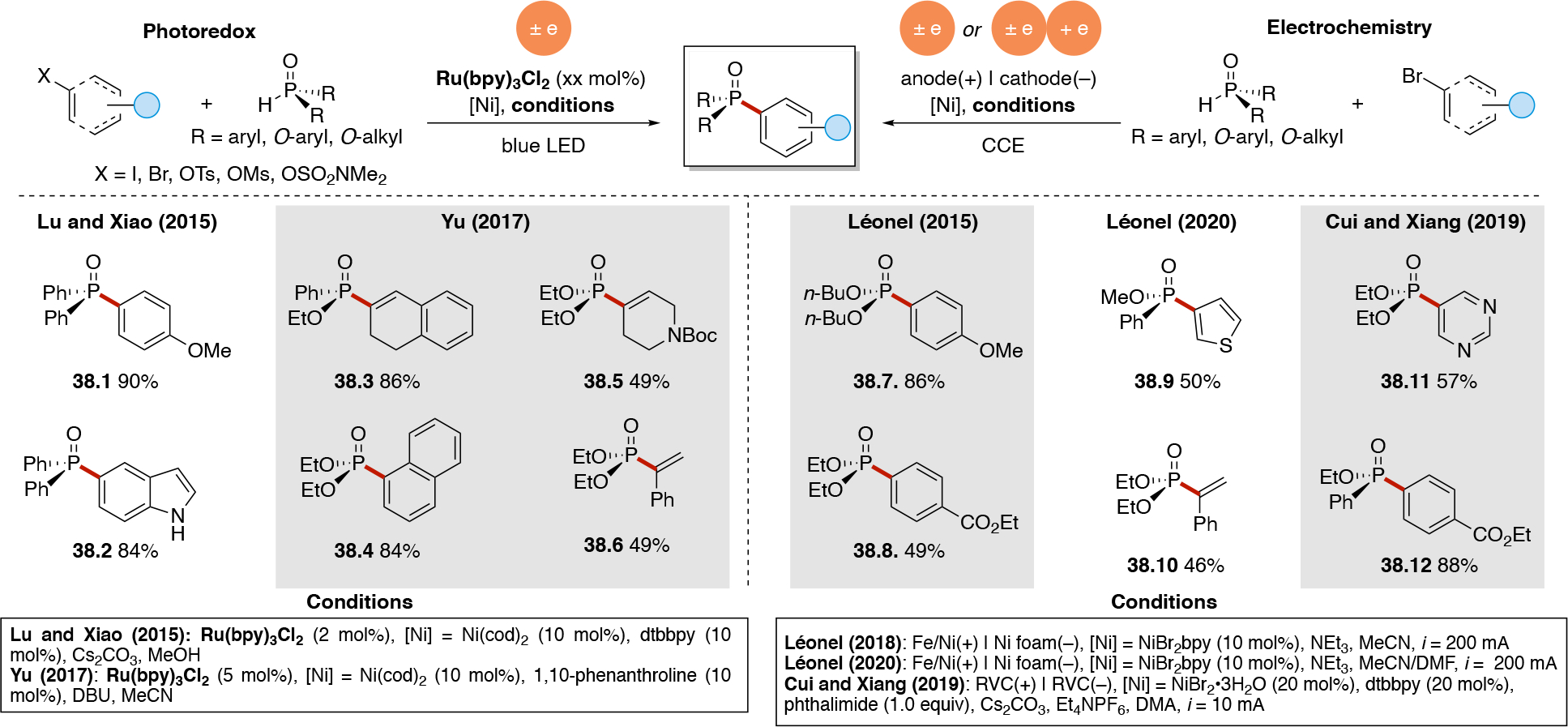
Photoredox and electrochemical arene C–P cross-coupling methods.
Scheme 39.

Proposed mechanism for photochemical and electrochemical arene C–P cross-couplings.
In 2015, Lu, Xiao and co-workers report a metallaphotoredox method using Ni(cod)2 and Ru(bpy)3Cl2 to couple aryl iodides with H-diarylphosphine oxides.638 The arene scope is tolerant of electronic changes and compatible with heterocycles such as 5-methoxyindole and 2-iodopyridine. The putative mechanism is as follows (Scheme 39A): Ru(bpy)32+ photoexcitation and subsequent oxidation of a trivalent phosphinic acid 39.1 (the preferred tautomer relative to the alternative pentavalent H-diarylphosphine oxide 39.2) forms a phosphine centered cation radical that yields a phosphinoyl radical 39.3 upon deprotonation. Concomitant oxidative addition of the aryl iodide with Ni0 leads to an nickelarene intermediate 39.4 that intercepts the phosphinoyl radical to form a NiIII complex 39.5, which then undergoes reductive elimination to yield the desired triarylphosphine oxide. Regeneration of active Ni0 from NiI via oxidation of a RuI species back to ground state RuII closes the catalytic cycles. The main limitation of this work is its reliance on H-diarylphosphine oxides, as H-dicyclohexylphosphine oxide and ethyl H-phenylphosphinate were incompatible coupling partners. This is rationalized by the preference of electron-rich secondary phosphine oxides to exist in its pentavalent form, which negates the possibility of single electron oxidation by photoexcited Ru(bpy)32+.639 Yu and co-workers improve on this precedent by extending the method to aryl and vinyl pseudohalides, as well as expanding the nucleophile scope to include H-diaryl and H-dialkyl phosphonates.640 The reaction scope is broad for alkenyl tosylates with moderate to excellent yields observed; however, the range of compatible aryl tosylates is rather limited but examples of C–P bond formation with an aryl mesylate and an aryl sulfamate are shown. The mechanism of this transformation is similar to the one described by Lu, Xiao and coworkers, however, Yu notes that a side pathway in which the phosphinoyl radical adds to Ni0 before arene oxidative addition with a NiI intermediate 39.6 cannot be discounted (Scheme 39B).
The electrochemical counterpart to this transformation was first reported by Léonel and co-workers, in which the nickel-catalyzed coupling of aryl bromides with H-dialkylphosphites is accomplished under galvanostatic conditions (i = 200 mA).641 This strategy enables the use of a NiII catalyst, which is reduced to Ni0 to facilitate oxidative addition with the aryl halide. A wide range of aryl bromides are compatible with this transformation, and good tolerance is observed for arene electronics, albeit with reduced yields observed when ortho-substitution or carbonyl functional groups are present. A smaller scope of compatible H-dialkylphosphites is presented, but high yields are observed, except when a loss in conversion is observed with bulky isopropyl groups. Léonel and co-workers follow up on this work by demonstrating the synthesis of aryl and vinyl phosphinates when using alkyl H-phenylphosphinates as the coupling partner.642 Cui, Xiang and co-workers report a similar transformation coupling aryl bromides with H-dialkyl phosphites, except that non-sacrificial RVC electrodes are used with a ten-fold reduction in current (i = 10 mA).643 This change enables cleaner reactions with previously problematic electron-rich substrates and eliminates the need of a sacrificial anode. Additionally, the synthesis of alkyl-diarylphosphinates and triarylphosphine oxides from alkyl H-arylphosphinate and H-diarylphosphine oxides is shown respectively. The putative mechanisms proposed by Xiang and Léonel diverge on active oxidation states of nickel and on the formation of the phosphinoyl radical. In Léonel’s example, a NiI species 39.7 —formed via cathodic reduction of an arylnickelII intermediate 39.8 — intercepts the trivalent H-dimethylphosphite before undergoing deprotonation to arrive at an aryl-nickel-dimethylphosphonate ate complex 39.9 (Scheme 39C). Anodic oxidation, followed by reductive elimination, yields the product and closes the catalytic cycle; thus, the possibility of a phosphinoyl radical is not considered. By contrast, Cui, Xiang and coworkers propose a mechanism identical to aforementioned photoredox conditions, wherein the active catalytic cycle is a Ni0/NiII/NiIII system, with 1 and 2 e reduction of respective NiI (39.10) and NiII (39.11) intermediates regenerating active Ni0 (Scheme 39D). Additionally, two-step anodic oxidation and deprotonation purportedly generates active phosphinoyl radical that joins the catalytic cycle through addition to either a NiII or Ni0 intermediate. More recently, Rueping and coworkers provide a route to unsymmetrical triarylphosphines via sequential paired electrolysis.644 Their method couples aryl halides and H-diphenylphosphine oxide in moderate to excellent yields, with H-dialkylphosphites also viable phosphorus partners.
4.7. Synthesis of Sulfones, Sulfonamides and Sulfuryl Fluorides
SulfurVI motifs such as sulfones and sulfonamides are important functional groups in medicinal chemistry and agrochemistry645, and have been used as bioisosteres in drug design to overcome issues related to metabolic stability and binding affinity.646 Additionally, sulfonyl fluorides are an emergent class of biocompatible electrophiles for click chemistry647 and are important functional groups for probing the activity of oxygen-, nitrogen- and sulfur-based nucleophiles of amino acid residues within proteins to enable the mapping of enzyme binding sites and protein–protein interactions.648,649,650 Photoredox catalysis and electrochemistry offer similar and complementary pathways towards these structural motifs, with divergences observed in reaction mechanism or bond disconnection. For this review, we omit a discussion of vinyl sulfone synthesis via oxidative decarboxylation of cinnamic acids, as this topic was recently covered by Verschueren and De Borggraeve.28 Additionally, readers should consult a minireview by Wu and coworkers on photocatalyzed methods involving the redox-induced trapping of SO2 with alkyl and aryl radicals.651 The use of electrochemistry can also enable for the selective oxidation of thiols to sulfinic esters (R-SO-OR1) without over-oxidizing to the corresponding sulfonate ester (R-SO2-OR1).652 Both electrochemistry661 and photoredox catalysis653,654,655,656,657,658,659 methods to oxidize thiols have been reported to access symmetrical disulfides, and recent advances have also provided methods for accessing unsymmetrical disulfides.660,662
4.7.1. Oxidation of Thioethers to Sulfones and Sulfoxides
Oxidation of thioethers has been realized using electrochemistry to reach either the corresponding sulfoxide (R-SO-R1) or further oxidized to the sulfone (R-SO2-R1). Noël used electrochemical microflow cell with Fe electrodes in MeCN (conditions A1 & A2, Scheme 40),661 while Xu reported a batch method using Pt electrodes in 1/1 (v/v) AcOH/HFIP (conditions A3 & A4, Scheme 40).662 Both methods rely on proton reduction at the cathode as the sacrificial reduction. The electrochemical generation of H2O2 from O2 and H+ to turnover chloroperoxidase for the enantioselective generation of (R)-methylphenylsulfoxide (>98.5% ee) from thioanisole has been reported by Liese as part of an electroenzymatic method using a carbon felt cathode in a divided cell.663 In the context of photoredox catalysis, the use of polyoxometalates as photocatalyst for oxidation prove to be useful once again, this time a decavanadate (TPPV10 = (Ph4P)4H2[V10O28]) enabled oxidation of thioethers to the corresponding sulfoxide or sulfone with high levels of selectivity simply by using a different reaction solvent mixture – methylethylketone (MEK) enables access to sulfones while MEK/water mixtures enables access to sulfoxides.664 The water suppresses sulfone formation presumably preventing MEK-derived peroxide formation or decomposing MEK-derived peroxides faster than they can react with sulfoxides (conditions B1 and B2, Scheme 40). Photoredox using a uranyl photocatalyst (UO2(OAc)2•2H2O) can achieve the same overall transformations (conditions B3 and B4, Scheme 40).665 The quantum yield for uranyl photocatalyzed oxidation of 4-Me-C6H4-S-n-Bu to its corresponding sulfoxide and sulfone were both determined to be >1 (ϕ = 7.6 and 8.2, respectively), suggesting a chain mechanism was operational. Pd-porphyrin (Pd-F10TPP) photocatalyzed selective oxidation of sulfides to sulfoxides was reported by Che (method B5).554 The use of less common electrodes such as CoFe layered double hydroxide electrodes has also been demonstrated to be useful for the oxidation of sulfides to sulfoxide via electrochemical means and successfully minimizing the formation of related sulfone impurities.666 X-ray spectroscopy revealed that the CoFe-layered double hydroxide electrode transformed into amorphous CoFe-oxyhydroxide under the reaction conditions as the conversion evolved, yet the catalytic properties were demonstrated sustainable for 10 cycles.
Scheme 40.
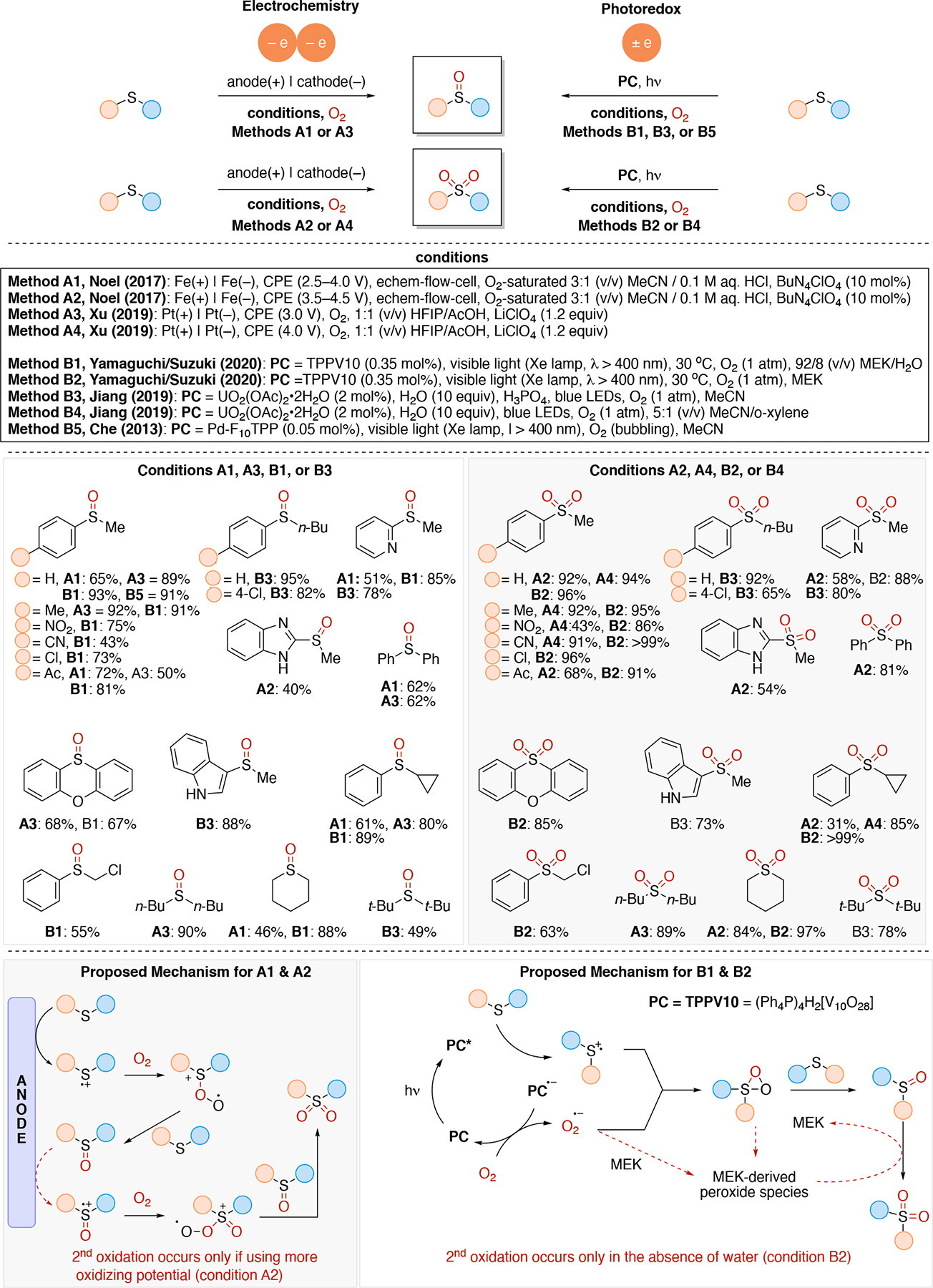
Selective oxidation of thioethers to sulfoxides or sulfones.
4.7.2. Synthesis of Vinyl Sulfones
Vinyl sulfones are common motifs in medicinal chemistry due to their synthetic versatility667,668, use as cysteine-selective inhibitors of proteases669 and phosphatases670, and emerging application as a molecular tag for the omic sciences.671,672 While classic routes towards their synthesis have been established through aldehyde condensation or transition metal-catalyzed methods, accessing vinyl sulfones via redox pathways establishes a useful C–S bond disconnection between alkenes and sulfinates or sulfonyl hydrazides (Scheme 41). Photoredox methods are centered on the single electron oxidation of SIV aryl and alkyl sulfinates, which generates a sulfonyl radical primed for addition to an alkene partner. König and co-workers use photoexcited Eosin Y to generate an aryl sulfonyl radical 42.1, which readily adds to a styrene partner at the least sterically-hindered carbon to form intermediate 42.2 before HAT to a stoichiometric radical acceptor (typically nitrobenzene) delivers the desired vinyl sulfone (Scheme 42).673 Trapping of species 42.2 by TEMPO provided evidence for this mechanistic pathway. The preliminary König report was limited to aryl sulfinates, but their follow-up manuscript expanded the method to include both linear, branched, and cyclic alkyl sulfinates.674 This observation is especially interesting as SO2 extrusion is observed in synthetic applications of alkyl sulfinates that involve chemical oxidation.675,676,677,678 However, attempts to expand the alkene scope beyond styrenyl cores provided difficult. König and co-workers also updated their understanding of the mechanism, in which transient absorption spectroscopy showed that the triplet excited state of Eosin Y undergoes oxidative quenching with nitrobenzene (as opposed to the earlier hypothesis of a reductive Eosin Y quench with sulfinate), which then forms the sulfinate-oxidizing Eosin Y cation radical.
Scheme 41.

Representative examples of vinyl sulfone synthesis using alkenes via photoredox and electrochemical methods.
Scheme 42.
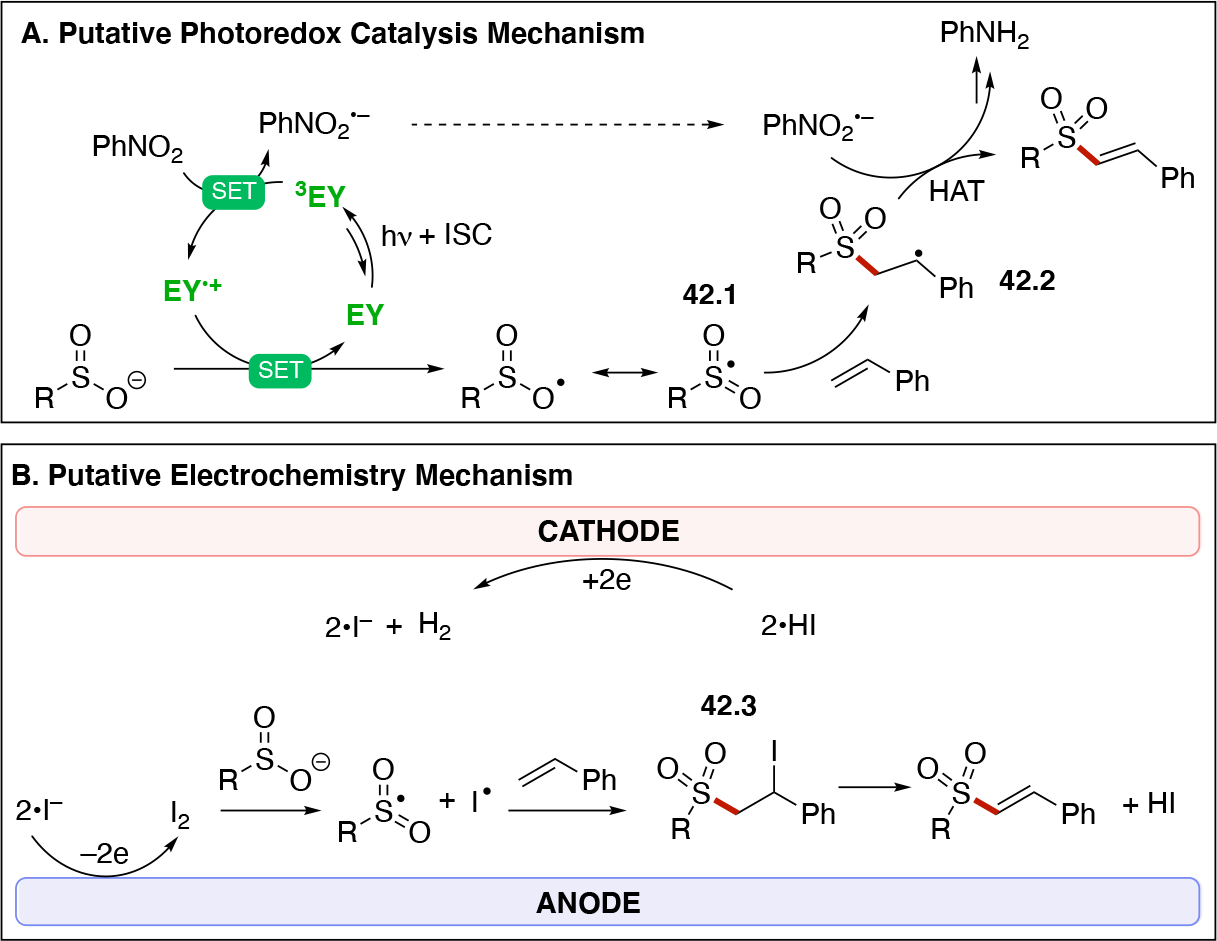
Putative mechanisms for vinyl sulfone synthesis via (A) photoredox catalysis and (B) electrochemistry.
The electrochemical counterpart of this method was reported by Yuan and co-workers, where aryl sulfinates are coupled with styrenes in an undivided cell equipped with a graphite anode-nickel cathode pair and N-tetrabutylammonium iodide as a supporting electrolyte and a redox mediator.679 The reaction mechanism is similar to photoredox conditions with a few differences: anodic oxidation of iodide to I2 reacts with the sulfinate to form a sulfonyl iodide which fragments to the reactive aryl sulfonyl radical. Intercepting this intermediate with an alkene, followed by iodine radical, forms 42.3, which undergoes deprotonation-elimination to yield the desired product and an equivalent of hydriodic acid that is reduced at the anode to both evolve H2 and regenerate iodide. An analogous electrochemical method using SVI sulfonyl hydrazides as an alternate sulfur coupling partner was reported by Terenťev and co-workers.680
An interesting divergence between photoredox and electrochemical methods arises when terminal alkynes are used as the coupling partner (Scheme 43). Lei and co-workers report an Eosin Y-photocatalyzed method for the synthesis of α-vinyl sulfones, where aryl sulfonyl radicals generated from the parent sulfinates add to terminal alkynes with Markovnikov selectivity (Scheme 43A).681 A wide range of aromatic alkynes provide desired product in good to excellent yields, but diminished yields were observed for aliphatic alkynes. The putative mechanism proceeds via reductive quenching of excited state Eosin Y by in situ-generated sulfinate 43.12, which forms the key sulfonyl radical intermediate 43.13. Eosin Y radical anion then reduces the terminal alkyne to the alkyne radical anion, which can undergo sulfonyl radical trapping and protonation sequence to arrive at the desired product. Alternatively, the same product can also be formed via alkyne trapping of the sulfonyl radical (43.14) followed by a reduction–protonation sequence. However, the electrochemical coupling of aromatic alkynes and sulfonyl hydrazides under potentiostatic conditions results in alkynyl sulfones (Scheme 43B).682 The divergence in product formation is rationalized by the redox activity of iodine, as the absence of N-tetrabutylammonium iodide is detrimental. The anodic oxidation of iodide to iodine leads to the formation of the key sulfonyl radical intermediate, which has two potential reaction pathways. The first pathway proceeds via anti-Markovnikov addition of the sulfonyl radical to the alkyne, followed by iodine radical trapping and base-promoted hydriodic acid elimination to reveal the sulfonyl alkyne. Alternatively, sulfonyl radical addition to in situ-formed alkynyl iodide followed by iodide radical extrusion also leads to the desired product. For both pathways, cathodic proton reduction closes the redox cycle. Mechanistic studies suggest that both reaction pathways are likely operative, with O2 accelerating the formation of sulfonyl alkyne.
Scheme 43.
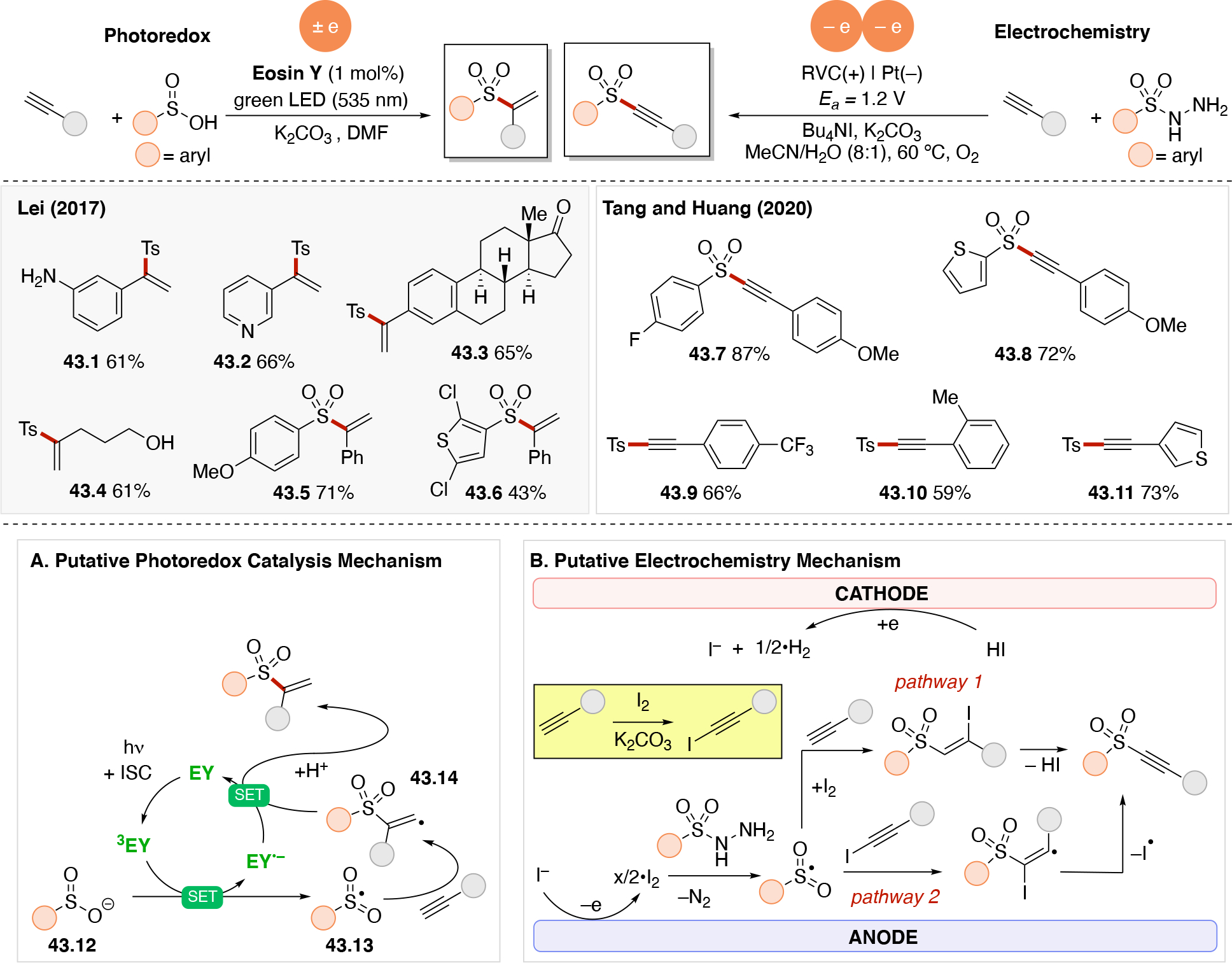
Representative examples of vinyl sulfone synthesis using alkynes via photoredox and electrochemical methods, with corresponding putative mechanisms (A) and (B).
4.7.3. Synthesis of β-Keto or Hydroxy Sulfones
The synthetic versatility of β-keto sulfones,683 especially as an auxiliary group684 for the synthesis of C–C bonds makes it a useful motif for total synthesis.685 As a result, photoredox and electrochemical strategies for β-keto sulfone synthesis with alkynes have been developed (Scheme 44). Photoredox methods for synthesizing this functional moiety have centered coupling alkenes or alkynes with catalytically-formed sulfonyl radicals under oxidative conditions. Cai and coworkers demonstrate that the reductive quenching of a stoichiometric iodide source by Ru(bpy)32+ enables the synthesis of β-keto sulfones using a wide range of sulfonyl hydrazides and aryl alkynes.686 Trapping the alkyne partner with catalytically generated iodine radical forms an unstable vinyl radical that reacts with oxygen to form a short-lived vinyl hydroperoxide that is readily reduced with excess iodide to the vinyl enolate, which then tautomerizes to the desired product. In the same year, Yang, Wang and coworkers report a similar synthesis of β-keto sulfones using styrenes and sulfinic acids as the reaction partners under oxidative conditions with Eosin Y as the photocatalyst.687 tert-Butyl hydroperoxide fragmentation by oxidative quenching of excited state Eosin Y forms tert-butoxyl radical, which undergoes HAT with sulfinic acid to form the alkene-trapping sulfonyl radical. Oxidation of this intermediate by Eosin Y cation radical generates a benzylic cation that is trapped by either by hydroxide or water to yield the benzyl alcohol. This hypothesis is supported by the observation that when an α-substituted styrene is used, the reaction stops at the tertiary alcohol. Downstream oxidation of this α-functionalized benzyl alcohol furnishes the aryl ketone. An electrochemical route for the synthesis of aryl β-keto sulfones from aryl methyl ketones or aryl alkynes was reported by Yavari and Shaabanzadeh, with a terminal oxidant necessary for competent reactivity with the latter substrate.688 When aryl methyl ketones are used, trapping of oxidatively formed sulfonyl radical by the enol tautomer yields a benzylic radical that is later oxidized to the desired ketone by catalytic iodine formed under galvanostatic conditions. If aryl alkynes are used, the oxidation of a vinyl sulfone sp2-centered carbon radical by tert-butyl hydroperoxide leads to the desired product in a manner similar to the mechanism proposed by Yang, Wang and coworkers.687
Scheme 44.
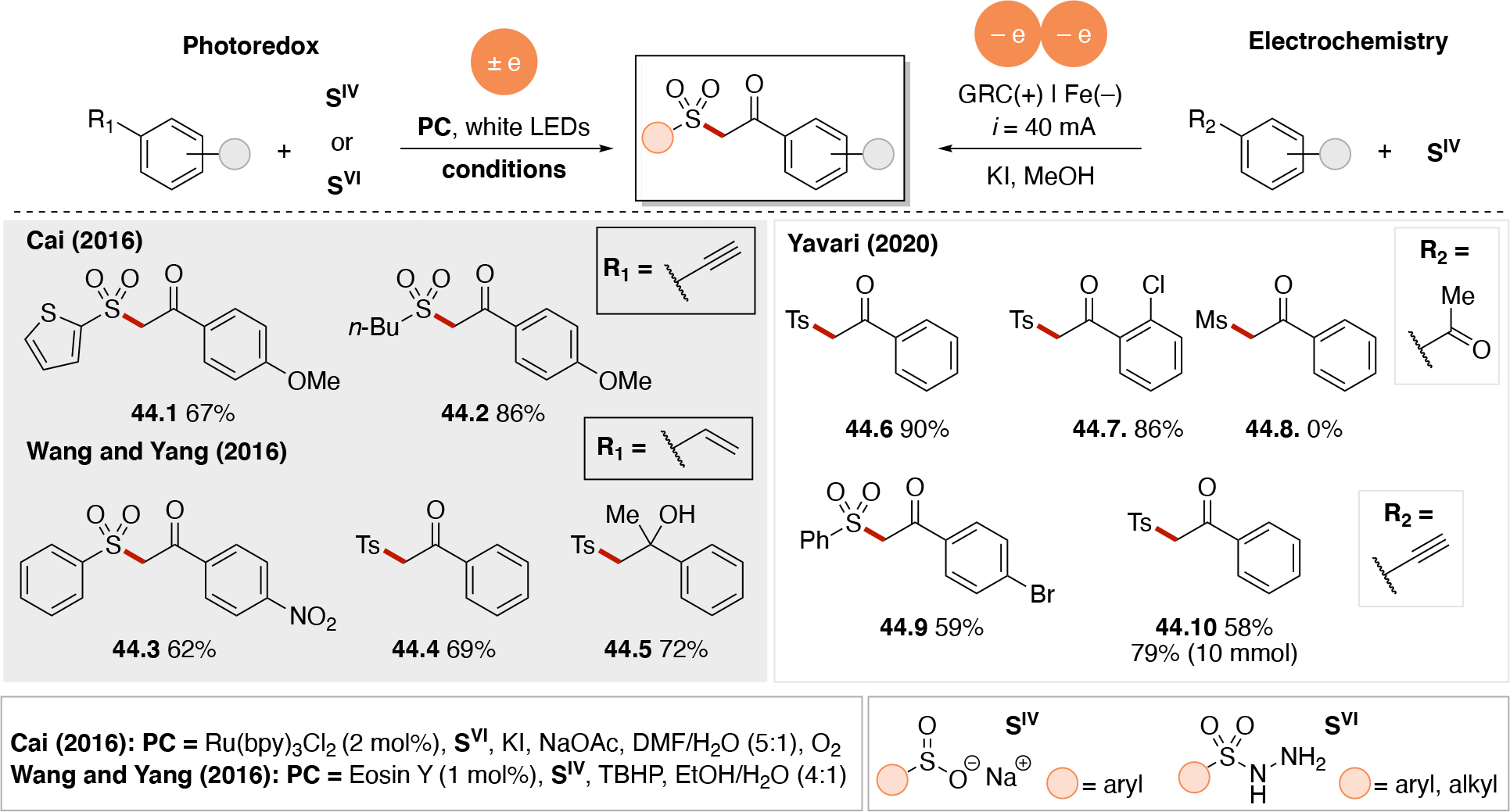
Representative examples of β-keto sulfone synthesis via photoredox and electrochemical methods.
The synthesis of β-hydroxy sulfones — the reduced variant of the parent ketone — can also be accessed via photoredox and electrochemical pathways (Scheme 45). Reiser and coworkers demonstrate the photoredox-catalyzed synthesis of β-hydroxy sulfones arising from styrene, sulfonyl chlorides and water as the reaction partners.689 Oxidative quenching of photoexcited 11.4 by sulfonyl chlorides forms a sulfonyl radical that traps a partner styrene. Oxidation of the resultant benzylic radical by an IrIV species closes the photoredox cycle and generates a benzylic carbocation that is then trapped by water — isotopic labeling with H218O supports this hypothesis. This reaction is broadly applicable to a range of styrenes and sulfonyl chloride partners, however a major limitation of this method is the lack of reactivity with aliphatic alkenes. The electrochemical counterpart to this method was reported by Chen, Chang and coworkers in which sodium sulfinates were used instead of sulfonyl chlorides.690 As is common with the earlier examples, iodide is used as the redox mediator for this transformation, which leads to broad compatibility with a wide range of styrene coupling partners. Except for this key difference, the reaction mechanism mirrors Reiser’s reported transformation.
Scheme 45.

Representative examples of hydroxy sulfone synthesis via photoredox and electrochemical methods.
4.7.4. Arene Sulfonylation
Aryl sulfones are an important component of numerous pharmaceuticals and agrochemicals691, and new redox-mediated strategies towards the C(sp2)–SO2R bond disconnection are welcome additions to a growing field of synthetic methods. Photoredox methods have largely centered on improving the Ni-catalyzed C–S coupling of (hetero)aryl halides and sulfinates, beyond the limitations of thermally-activated precedents.692 Rueping and co-workers reported the first photoredox example using iridium photocatalyst 46.16, where a broad range of (hetero)aryl halides were coupled to aryl sulfinates in moderate to excellent yield.693 Additional examples of sulfinate coupling to aryl iodides and chlorides, as well as vinyl bromides was demonstrated. Within a similar timeframe, two other similar strategies using Ru(bpy)32+ were independently reported by 1) Manolikakes and coworkers,694 and 2) Molander, Gutierrez, and coworkers.695 The former example focuses on expanding the scope of compatible aryl iodides with aryl and alkyl sulfinates whereas the latter strategy builds on the Rueping work and provides computational analysis of the reaction mechanism. The mechanism is as follows (Scheme 46A): the formation of a sulfonyl radical 46.17 by reductive quenching of photoexcited catalyst by a sulfinate salt is intercepted by a Ni0 species to form a NiI–sulfinate species (46.18) that readily undergoes oxidative addition with an aryl halide to yield a NiIII intermediate (46.19). Reductive elimination of the two coupling partners furnishes the desired product and a NiI species (46.20) that turned over by reductively quenched photocatalyst to complete both catalytic cycles. Interestingly, Molander and Gutierrez note the presence of an aryl sulfide side product, which they hypothesize forms via a pathway involving stepwise NiIII-sulfonyl bond dissociation, light-promoted reduction of the sulfonyl radical, and recombination with off-cycle NiII aryl halide intermediate. A photocatalyst-free method has also been reported, with Zhang, Yan and coworkers demonstrating that irradiating EDA complexes that form between aryl halide and sulfinate with 365 nm light can induce the formation of desired aryl sulfone.696 This strategy works well for electron-deficient aryl bromides and iodides, however significantly reduced reactivity observed when electron-rich arenes are used, which is consistent with electronic restraints observed for photochemical reactions involving EDA complexes.697 Photoredox methods are not limited to aryl halides as a collaborative effort between the Willis, Dixon, Paton, Schofield, and Smith groups resulted in the oxidative C–H sulfonylation of anilines using iridium photocatalyst 11.6 and potassium persulfate as the terminal oxidant.698 A wide range of electron-rich disubstituted anilines are sulfonylated in moderate to excellent yield, with observed site-selectivities mirroring arene electronics, although steric effects can induce greater para-selectivity. Coupled with a general alkyl and aryl sulfinate scope, this method can be used for late-stage C–H sulfonylation of multiple drug candidates — an important strategy for diversifying known biomolecules for the discovery of new therapeutic leads. The proposed mechanism suggests that two independent photocatalyst cycles are operative; oxidative quenching of 11.6 by persulfate generates an oxidizing IrIV intermediate 46.21. Reductive quenching of two equivalents of this Ir species respective oxidizes the aniline to the aniline cation radical 46.22 and the sulfinate to the sulfonyl radical. Radical coupling of these two oxidized intermediates then furnishes the desired product. Additionally, the photoredox-catalyzed, in situ formation of aryl sulfinates from (hetero)aryl halides and thiourea dioxide enables the formation of (hetero)aryl sulfones and sulfonamides in two steps.699
Scheme 46.
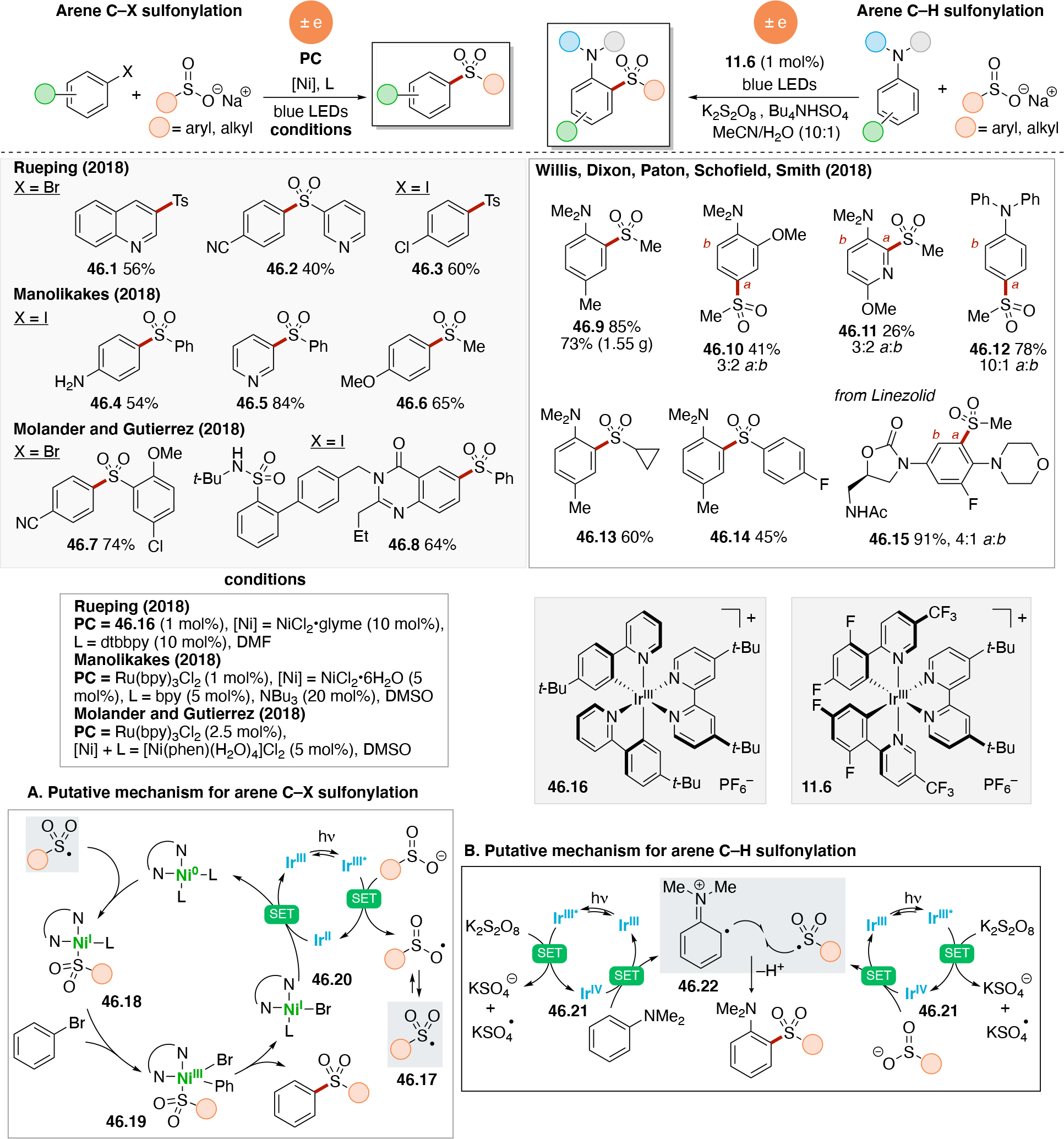
Representative examples of photoredox catalyzed methods for the synthesis of aryl sulfones.
Electrochemical methods predominantly focus on C–S bond formations involving oxidative (hetero)arene C–H sulfonylation with either SIV or SVI reagents (Scheme 47). Lei, Huang and coworkers report a galvanostatic method using aryl sulfonyl hydrazides and hetero(arenes) as coupling partners in an undivided cell.700 A broad scope of functionalized benzofurans were successfully sulfonylated, with C2 selectivity observed; other arenes and N-heterocycles are also amenable to sulfonylation using this strategy (Scheme 47A). Oxidative conversion of the sulfonyl hydrazide generates the sulfonyl radical, which adds to the (hetero)arene to form a stabilized carbon radical. Subsequent rearomatization via stepwise oxidation-deprotonation yields the (hetero)aryl sulfone. Other methods for arene C–H sulfonylation use more easily-accessible aryl and alkylate sulfinates as coupling partners. A majority of the early methods for phenol sulfonylation were developed by Nematollahi and coworkers, who used potentiostatic electrolysis to sulfonylate quinone-based substrates.701,702,703 Waldvogel and coworkers offer a galvanostatic alternative for mequinol and guaiacol derivatives, with moderate yields observed for an appreciable arene and sulfinate scope.704 A key advantage of this method is its scalability, as quadrupling the reaction scale from 5.0 mmol to 20 mmol gave the desired product with minimal change in the product yield. This work was quickly followed up with an expansion of the substrate scope to include a large scope of electron-rich arenes, especially aniline derivatives.705 A similar strategy is independently disclosed by Li, Song and co-workers, with its focus entirely on N,N-disubstituted anilines; this method is closely related to the photoredox arene sulfonylation strategy from the Willis, Dixon, Paton, Schofield, and Smith groups (Scheme 47B).706 Additionally, Oh and coworkers expand the heteroarene scope to include 2H-indazoles with highly-selective C3 sulfonylation observed.707
Scheme 47.
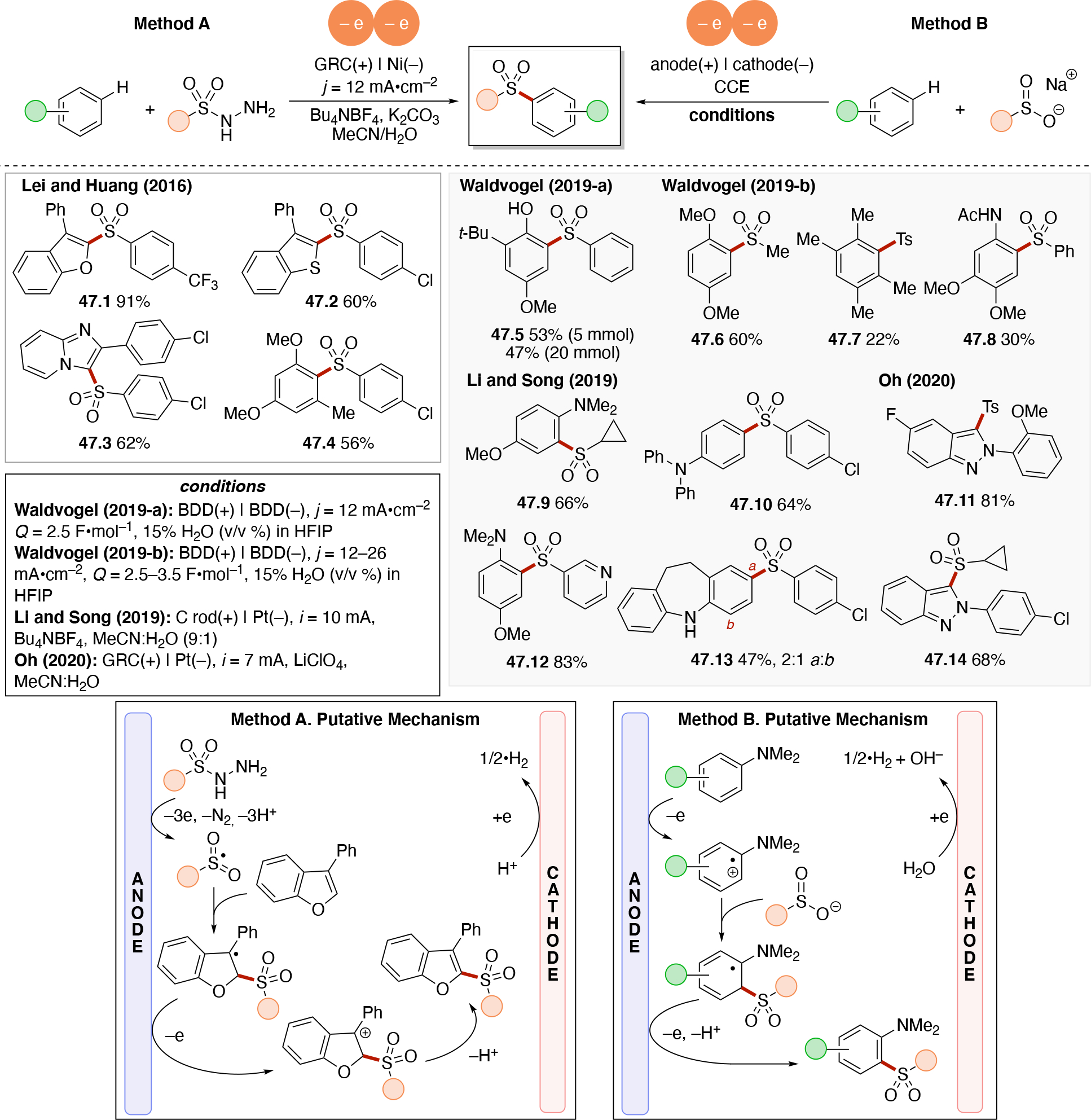
Representative examples of electrochemical methods for the synthesis of aryl sulfones.
4.7.5. Sulfonamide Synthesis
The frequency of sulfonamide use in numerous pharmaceuticals and its favorable pharmacological properties are well-documented in medicinal chemistry.708,709,710,711 Furthermore, sulfonamides are classic carboxylic acid and carboxamide bioisosteres with the added benefit of having a tunable pKa for the sulfonamide N–H through substituent design.712,713 Traditional methods of sulfonamide synthesis involve reacting sulfonyl chlorides with a partner amine714, with transition metal-catalyzed methods enabling greater flexibility over reaction partners for the construction of (hetero)arylsulfonamides.715,716 Despite these advances, photoredox and electrochemical methods have the potential to improve existing methods or to create new C–S and S–N bond disconnections for sulfonamide synthesis (Scheme 48). One example of the former strategy is shown by Zeng, Little, and co-workers, where the oxidative amination of sodium sulfinates in the presence of catalytic iodide enables the synthesis of sulfonamides in moderate to good yield.717 While chemical oxidation methods exist for the same transformation718,719,720,721,722, this galvanostatic method uses less net oxidant than the previous strategies that rely on stoichiometric peroxide or iodide. Mechanistic studies suggest that reaction pathways involving the formation of sulfonyl iodide or N-iodoamine (48.14) are viable, with the former mirroring classic sulfonamide syntheses whereas the sulfinate displacement of the iodide from the latter intermediate would furnish the desired product (Scheme 48A). A more interesting use of electrochemistry for sulfonamide synthesis is reported by Noël and co-workers, who use an electrochemical flow cell to oxidatively couple thiols with amines under potentiostatic conditions.277a A broad scope of thiol and amine partners was demonstrated with moderate to excellent yields. Notably, the lower oxidation potentials of both thiols and amines allows for the inclusion of oxidation-sensitive functionalities such as alcohols, alkenes, alkynes, and arenes in the product structure. Mechanistic studies suggest that concomitant oxidation of amine and thiol form the corresponding amine cation radical and disulfide respectively, which then readily form a sulfenamide intermediate (48.15). Treatment of an isolable sulfenamide to the reaction conditions furnishes the desired sulfonamide, thus demonstrating its role in the transformation. Radical trapping experiments yield products that hint at the formation of aminium radical intermediates, thus supporting the putative mechanism.
Scheme 48.
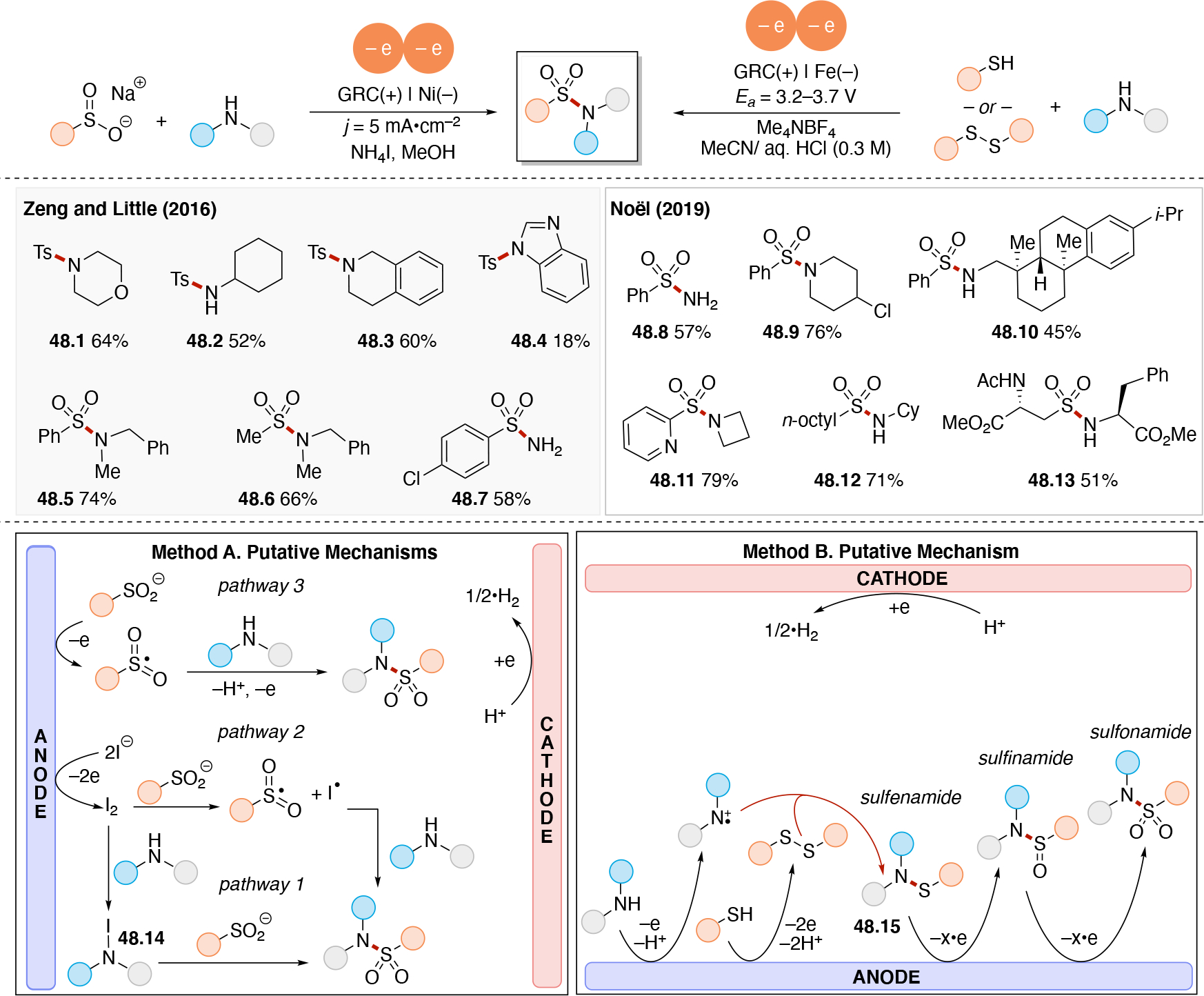
Representative examples of electrochemical methods for the two-component synthesis of sulfonamides with corresponding mechanisms depicted.
More recently, Waldvogel and coworkers report an electrochemical, three-component coupling of electron-rich alkoxyarenes, alkylamines, and SO2 to yield sulfonamides in low to good yields when using BDD electrodes under galvanostatic conditions (Scheme 49).723 Regioselectivity for arene sulfoamidation is substrate-dependent and occurs at most electron-rich C(sp2)–H site for the highly substituted arenes. Additionally, both secondary and primary amines are competent reaction partners, but reaction conversions can be low when using the latter substrate class. This transformation can be scaled up from 0.6 mmol to 8.0 mmol with a slight improvement in yield when using a divided H-cell. The putative mechanism of the reaction is as follows: anodic oxidation of the arene generates an arene cation radical, which is trapped by an amidosulfinate intermediate (49.5)—a Lewis pair adduct originating from SO2 and amine. Subsequent stepwise oxidation and deprotonation furnish the desired product.
Scheme 49.

The electrochemical, three-component synthesis of sulfonamides with the corresponding mechanism depicted.
Given that the electrochemical strategies require multiple oxidation steps — a current limitation of photoredox catalysis—other bond disconnections must be considered. One such method is the sulfonamide synthesis reported by Gouverneur and coworkers, which focuses instead on the C–S bond disconnection between sulfamoyl chlorides and alkene coupling partners (Scheme 50).724 Broad compatibility of sulfamoyl chlorides and electron-deficient alkenes with the reaction conditions is demonstrated; interestingly, using an alkyne coupling partner provides the (Z)-vinyl sulfonamide with high selectivity (9:1 Z:E). However, a major limitation of this transformation is the lack of reactivity when unactivated or electron-rich alkenes are used — an opportunity for further reaction development. A putative mechanism is as follows: tris-(trimethylsilyl)silane (TTMSS) reductively quenches photoexcited Eosin Y to form a Si-centered radical (50.5) after subsequent deprotonation. Intermediate 50.5 then forms the sulfamoyl radical 50.6 from the parent sulfamoyl chloride via halogen atom transfer; 50.6 then adds to the alkene with Giese-type regioselectivity to form the stabilized radical 50.7. While a reduction-protonation sequence involving Eosin Y radical anion or radical chain propagation with TTMSS are both viable pathways for converting 50.7 to the desired coupled product, radical trapping and deuterium labeling studies suggest that the latter mechanism dominates. It is also worthwhile to note the report by MacMillan and coworkers on a photosensitized Ni-catalyzed method for C–N coupling between aryl halides and sulfonamides.725 While general compatibility between the two general reaction partners is observed, this strategy’s reliance on energy-transfer sensitization instead of photoinduced redox changes precludes further discussion of this transformation.
Scheme 50.
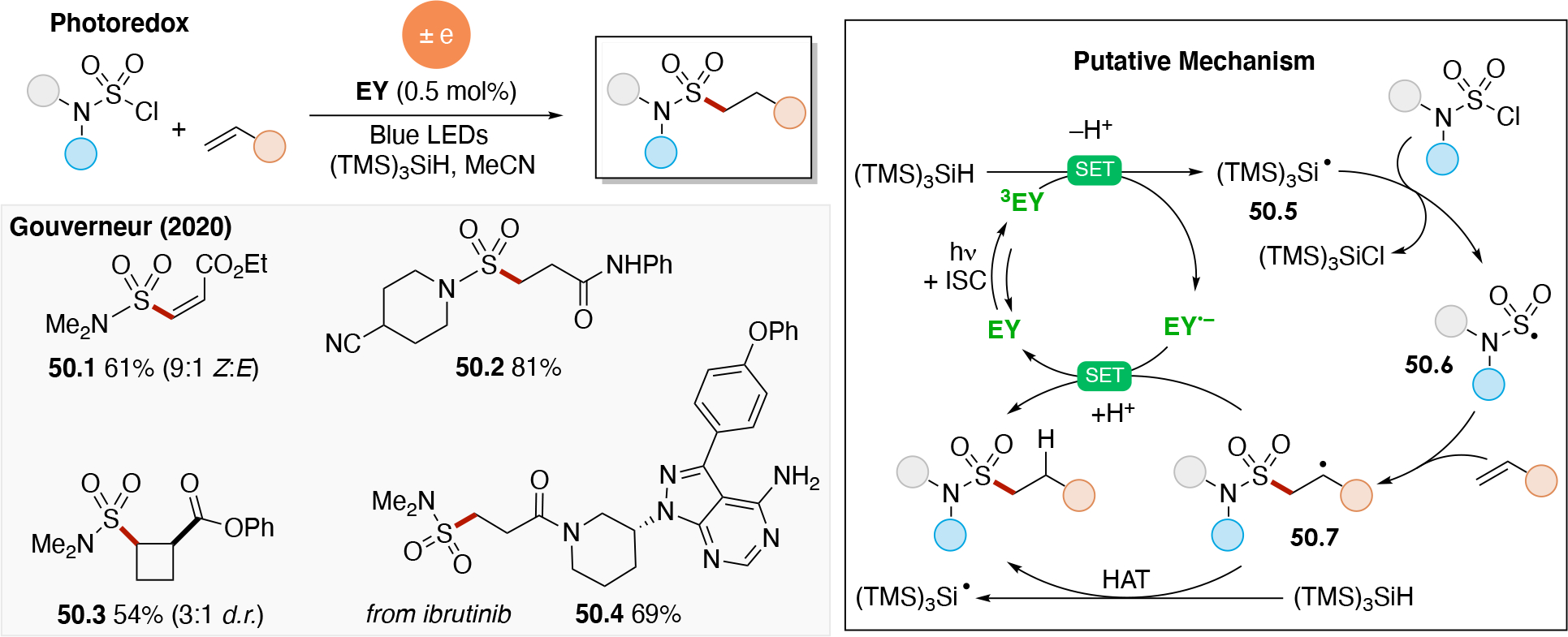
The photoredox-catalyzed synthesis of sulfonamides with the corresponding mechanism depicted.
4.7.6. Sulfonyl Fluoride Synthesis
Developing new methods for the synthesis of sulfonyl fluorides is an important goal due to their emerging importance of in organic synthesis and chemical biology. Current strategies rely primarily on chloride/fluoride exchange on reactive sulfonyl chlorides726,727,728 or transition metal-catalyzed strategies729,730 that require atom-inefficient amounts of electrophilic fluorine reagents. Therefore, applying redox strategies to this synthetic challenge can enable new disconnections for sulfonyl fluoride synthesis (Scheme 51). A key example is the electrochemically-mediated synthesis of sulfonyl fluorides reported by Noël and coworkers, where thiols and disulfides are oxidatively coupled with potassium fluorides under galvanostatic conditions (Scheme 51A).731 Whereas stoichiometric chemical methods for the synthesis of sulfonyl fluorides from disulfides require excess amounts of electrophilic fluorine (>6 equiv. of Selectfluor)732, the Noël method uses a biphasic system to form in situ HF-pyridine, which diffuses into acetonitrile to maintain equilibrium amounts of nucleophilic fluoride in the organic layer.
Scheme 51.
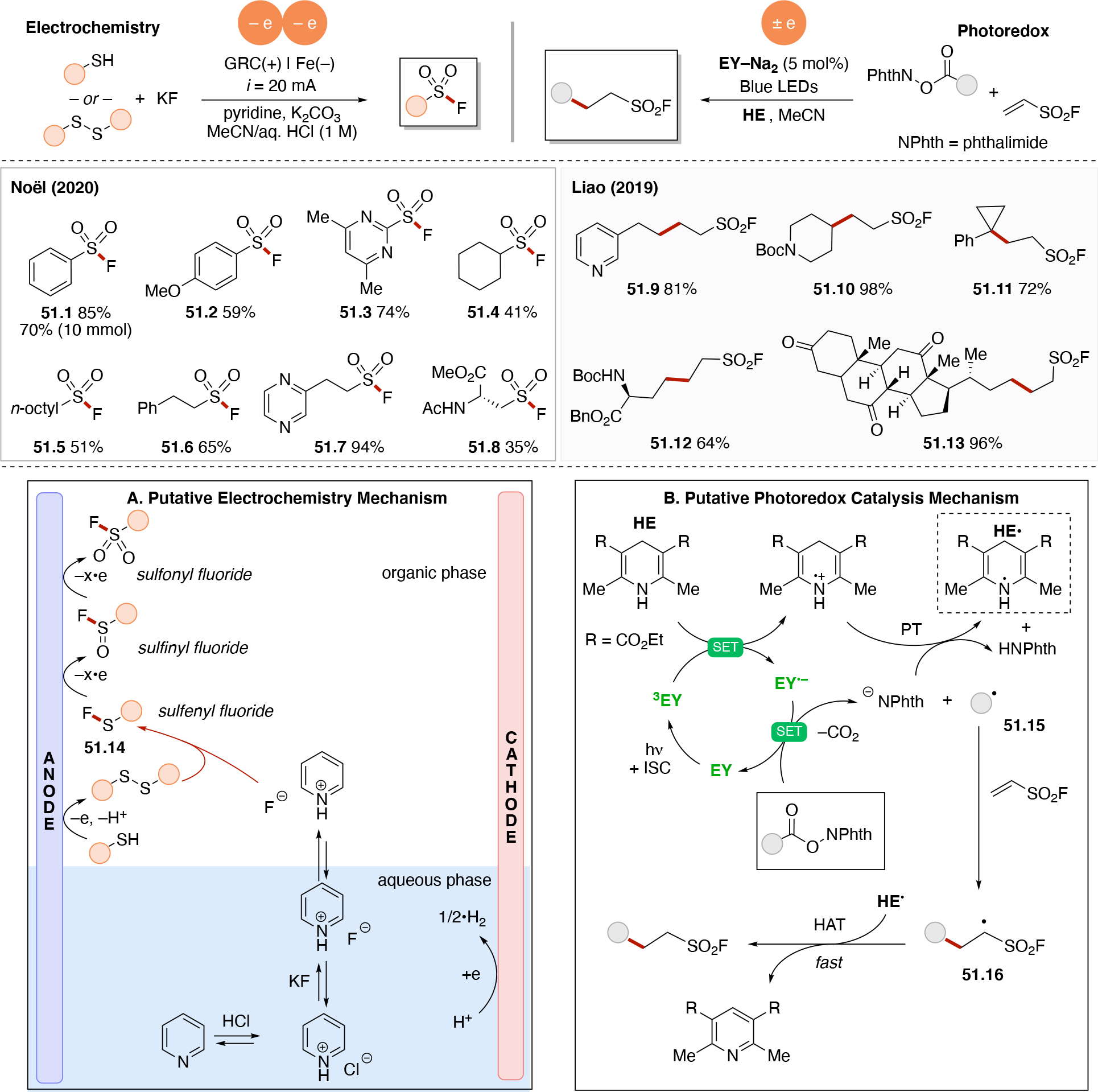
Representative examples of photoredox and electrochemical methods for the two-component synthesis of sulfonyl fluorides with corresponding mechanisms depicted.
A wide range of aryl and alkyl thiols are compatible with this transformation, with moderate to excellent yields observed. In several cases, the instability of the sulfonyl fluoride requires extraneous trapping by phenol to yield the phenyl sulfonate. Mechanistic studies suggest that the disulfide is oxidized to the sulfur cation radical, which then traps fluoride to yield a short-lived sulfenyl fluoride (51.14) that is further oxidized to the sulfonyl fluoride at the anode; no conversion to the desired product is observed when electrophilic fluorine sources are used. Kinetic experiments suggest that extended reaction times due to mass transfer limitations are prominent in batch electrochemical experiments, which can be overcome by using an electrochemical microflow reactor with small interelectrode gaps; this switch enables the synthesis of benzenesulfonyl fluoride from thiophenol in 5 minutes.
While an analogous photoredox strategy remains to be developed, a complementary route for the synthesis of aliphatic sulfonyl fluorides is attainted through the coupling of alkyl radicals and ethene sulfonyl fluoride (ESF), a commercially available precursor.733 Liao and coworkers report a method to that couples alkyl radicals — formed via photoredox-induced decarboxylation of redox-active N-hydroxyphthalimide esters — with ESF.734 An extensive range of primary, secondary and tertiary NHPI esters were successfully used, with good to excellent yields observed. In particular, carboxylic acid moieties in natural products and pharmaceuticals are functional handles for efficient conversion to the corresponding sulfonyl fluoride. Interestingly, the mild reaction conditions enable isolation of the parent sulfonyl fluorides with minimal loss of fidelity. The putative mechanism for this transformation involves reductive quenching of Eosin Y by Hantzsch ester (HE) to form Eosin Y radical anion which induces the decarboxylation of the NHPI ester (Scheme 51B). Subsequent trapping of the resultant alkyl radical 51.15 by ESF, followed by HAT to the ESF-alkyl adduct 51.16 from a HE radical or an amine base furnishes the aliphatic sulfonyl fluoride.
4.8. Alkene Mono- and Difunctionalizations
The difunctionalization of alkenes has been an area of intense research both in the context of electrolysis and photocatalysis.209,735,736 Many approaches have relied on the addition of a radical to an alkene, followed by oxidation of the newly generated radical intermediate to enable a radical-polar crossover mechanism and subsequent nucleophilic attack of the cationic species. Alternatively, the initial radical intermediate can be trapped by transition metal catalysts (e.g., Cu or Ni) followed by a more traditional cross-coupling approach to form the 2nd bond of the difunctionalization via a reductive elimination process. Homo-difunctionalizations using electrochemistry have been reported, including diazidation,378,737,738 diamination,739 dichlorination,740,741,742 difluorination,743 and dihydroxylation.744 Photoredox diaminations are also known.745,746,747 Vicinal heterofunctionalization of alkenes have also been extensively studies and subject to reviews,104,748 including those relying on dual catalytic systems involving both a photocatalyst and a transition metal (e.g., Ni or Cu). Asymmetric electrochemical cyanophosphinoylation and cyanosulfonylation of vinylarenes have been reported,749 and bear similarity to the photoredox asymmetric cyanofluoroalkylation of alkenes,750 and cyanotrifluoromethylation of alkenes,751,752 as well as the thermal enantioselective copper-catalyzed cyanotrifluoromethylation of alkenes.753 In these examples of alkene difunctionalizations these methods do not yet have a photochemical or electrochemical counterpart.
Alternatively, similar radical-polar crossover mechanisms for alkene difunctionalization can be accomplished without transition-metal co-catalysts. Lin and coworkers report a recent example of using an electroreduction approach to first reduce alkyl halides to alkyl radicals, which after coupling to alkenes in an anti-Markovnikov fashion, can be reduced again to form a benzyl carbanion which readily traps an available electrophile—DMF, MeCN and CO2 respectively, to yield carboformylation, hydroalkylation, and carbocarboxylation products.754 This approach is conceptually similar to analogous photoredox methods, in which substrate activation by a photoredox catalyst generates a reactive intermediate that can then trap alkenes to afford the benzyl radical, followed by consecutive one-electron reduction and electrophile trapping. Comparable hydroformylation755,756, hydroalkylation454n,757,758,759,760, carboalkylation761, hydrocarboxylation762, and carbocarboxylation763 products are obtained, with key divergences being the coupling partners as well as product regioselectivity.
4.8.1. Vicinal Dichlorination of Alkenes
Vicinal dichlorination of alkenes has been realized using several electrochemical and photoredox methods (Scheme 52). Lin reported740 a method relying on stoichiometric MgCl2 (3 equiv) as the chloride source in the presence of Mn(OTf)2 which was proposed to generate a MnII chloride salt which upon anodic oxidation forms the chlorinating MnIII chloride reagent. Morandi and Waldvogel reported741 a related approach in which they merged shuttle catalysis and paired electrolysis to achieve the 1,2-dichlorination of alkenes using either 1,2-dichloroethane or 1,1,1,2-tetrachloroethane as the chlorine source. These chlorinated ethane derivatives can be reduced to generate chloride and the corresponding alkene in situ, while the opposite electrode (anode) oxidizes the chloride (or a MnII chloride salt analogous to the Lin chemistry above) to generate the chlorinating reagent. Sequential paired electrolysis provides method of halogenating alkenes without the use of more toxic, hazardous Cl2 or Br2 reagents and was also demonstrated to enable 1,2-dibromination of alkenes using 1,2-dibromoethane instead of 1,2-dichloroethane. Photochemical 1,2-dichlorination of alkene using white LEDs was reported by Wan742 using stoichioimetric CuCl2 for aryl substituted alkenes, while alkyl substituted alkenes could be dichlorinated using catalytic CuCl2 in the presence of HCl and air (as the terminal oxidant). This chemistry relies on photoexcitiaton of CuCl2 (or LnCuCl3− species when exogenous chloride is present such as in HCl)128,764 to enable a ligand-to-metal charge transfer (LMCT) and subsequent generation of chlorine radical and CuCl.
Scheme 52.
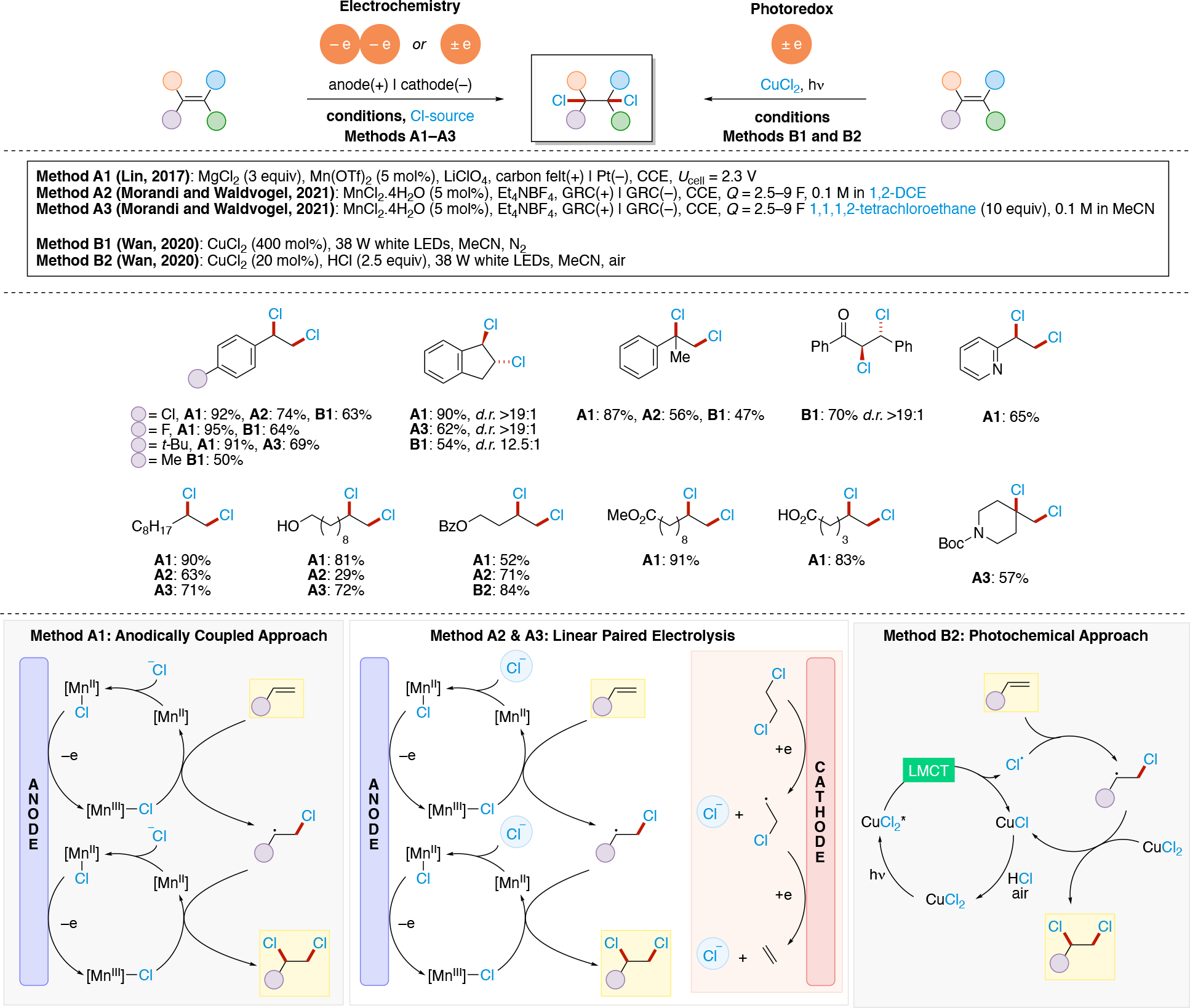
Vicinal dichlorination of alkenes.
4.8.2. Chloro-trifluoromethylation
The vicinal difunctionalization of alkenes involving trifluoromethylation765 has been extensively explored (Scheme 53).766,767 Chlorotrifluoromethylation of olefins using electrochemistry was reported by Lin85 using a manganese acetate as a pre-catalyst, stoichiometric MgCl2 as the source of chloride and sodium trifluoromethylsulfinate as the CF3 source. The reaction was proposed to operate via anodically coupled approach in which oxidation of the trifluoromethylsulfinate generates the CF3 radical followed by addition to the alkene to generate intermediate 53.1. Concurrently, MnII is oxidized to MnIII, generating a MnIII–Cl persistent radical able to chlorinate intermediate 53.1. Han reported a redox neutral approach based on Ru-photoredox and trifluoromethanesulfonyl chloride.768 Reduction of CF3SO2Cl generates the CF3 radical and subsequent reaction with the alkene affords radical intermediate 53.2 which is oxidized to the corresponding cation (53.3) and trapped by chloride. The scope of the photoredox method appears to perform better with alkenes lacking aryl substituents whereas the electrochemical approach performs well with either aliphatic or aryl functionalized olefins. Additionally, the photoredox method is limited to terminal olefins whereas internal alkenes can be functionalized using the electrochemical strategy.
Scheme 53.
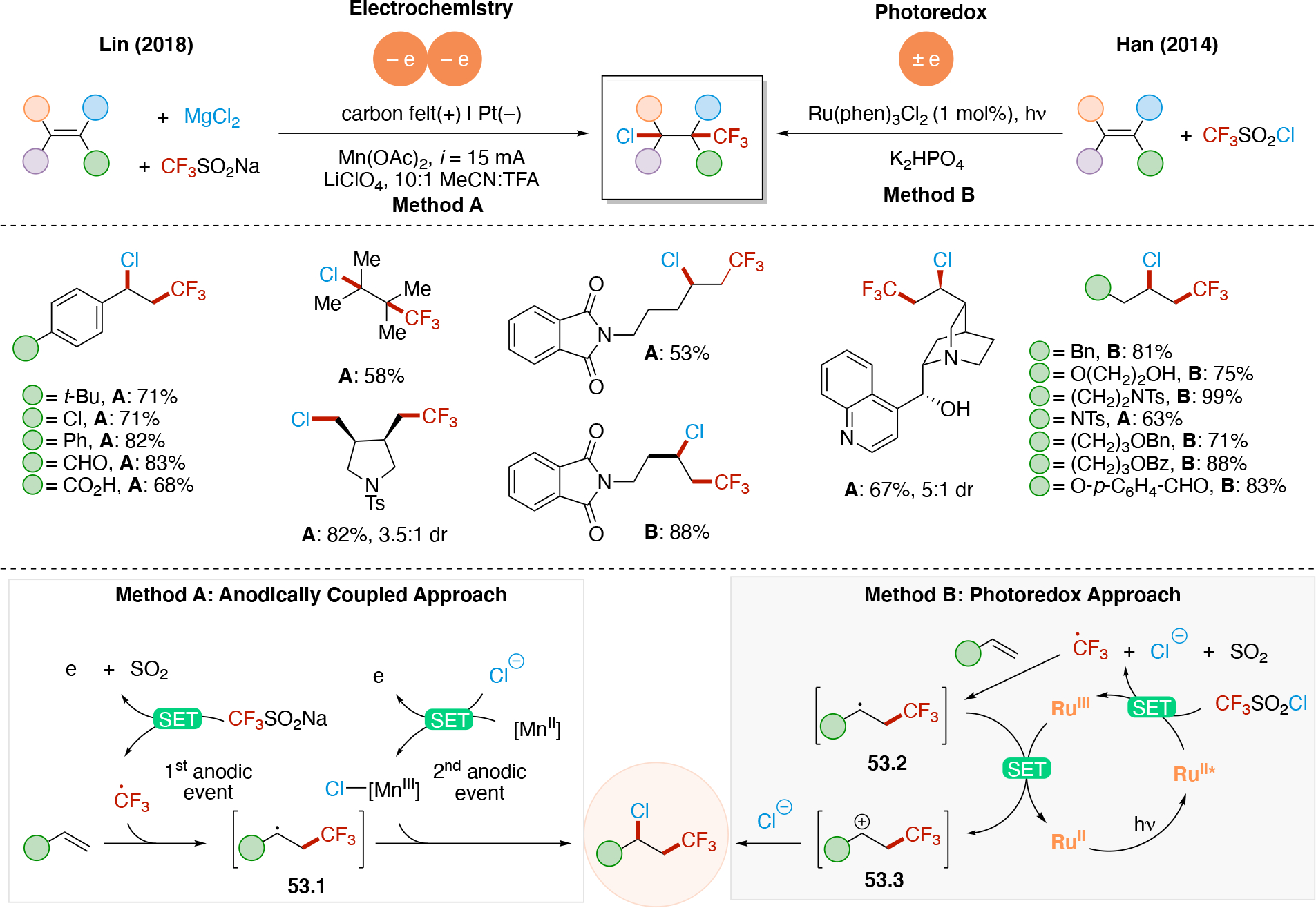
Chlorotrifluoromethylation of alkenes.
4.8.3. Aminotrifluoromethylation
Vicinal amino-trifluoromethylation of alkenes can be accomplished via anodically coupled electrolysis769 and redox-neutral photoredox catalytic methods (Scheme 54).770,771 Oxidative generation of CF3 radical from sodium triflinate, addition of the radical to the styrene and subsequent oxidation of the benzylic radical enables benzylic cation generation, which is trapped by MeCN solvent in a Ritter-type amination to generate the product. In contrast a reductive generation of the CF3 radical is utilized in the photoredox method, ultimately arriving at the analogous benzylic radical that is oxidized by the photocatalyst and analogous subsequent steps to the electrochemical method to produce the vicinal amino-trifluoromethylated product.
Scheme 54.
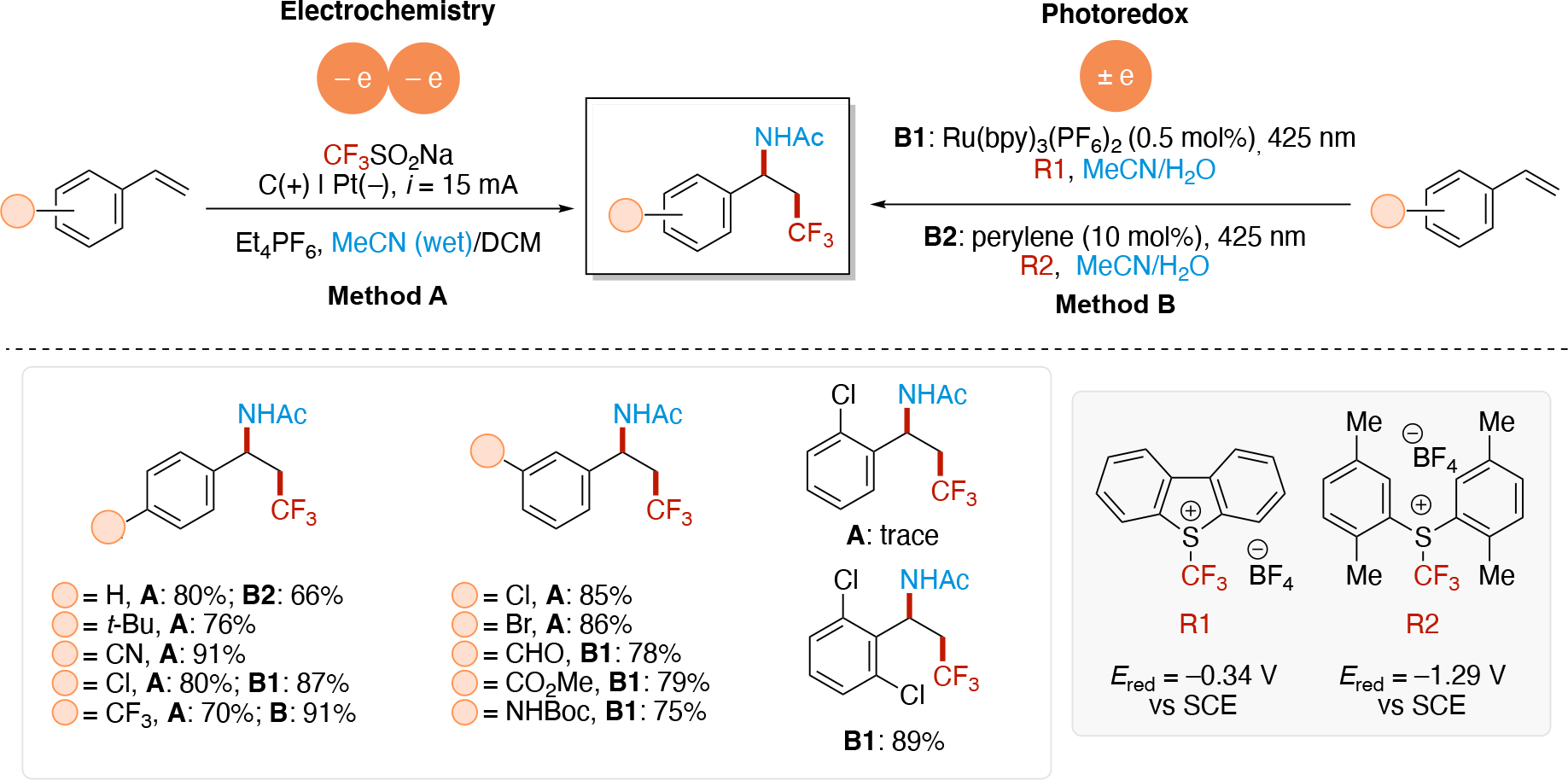
Aminotrifluoromethylation of styrenes.
4.8.4. Hydroxytrifluoromethylation
Leveraging the same mechanistic manifolds as the aminotrifluoromethylation of alkenes in Section 4.8.2, the vicinal oxytrifluoromethylation of alkenes can be accomplished via electrochemistry772 as well as photoredox (Scheme 55).773 The benzylic carbocation is directly trapped by water to afford the benzylic alcohol with a beta-CF3 group for styrene derivatives.
Scheme 55.
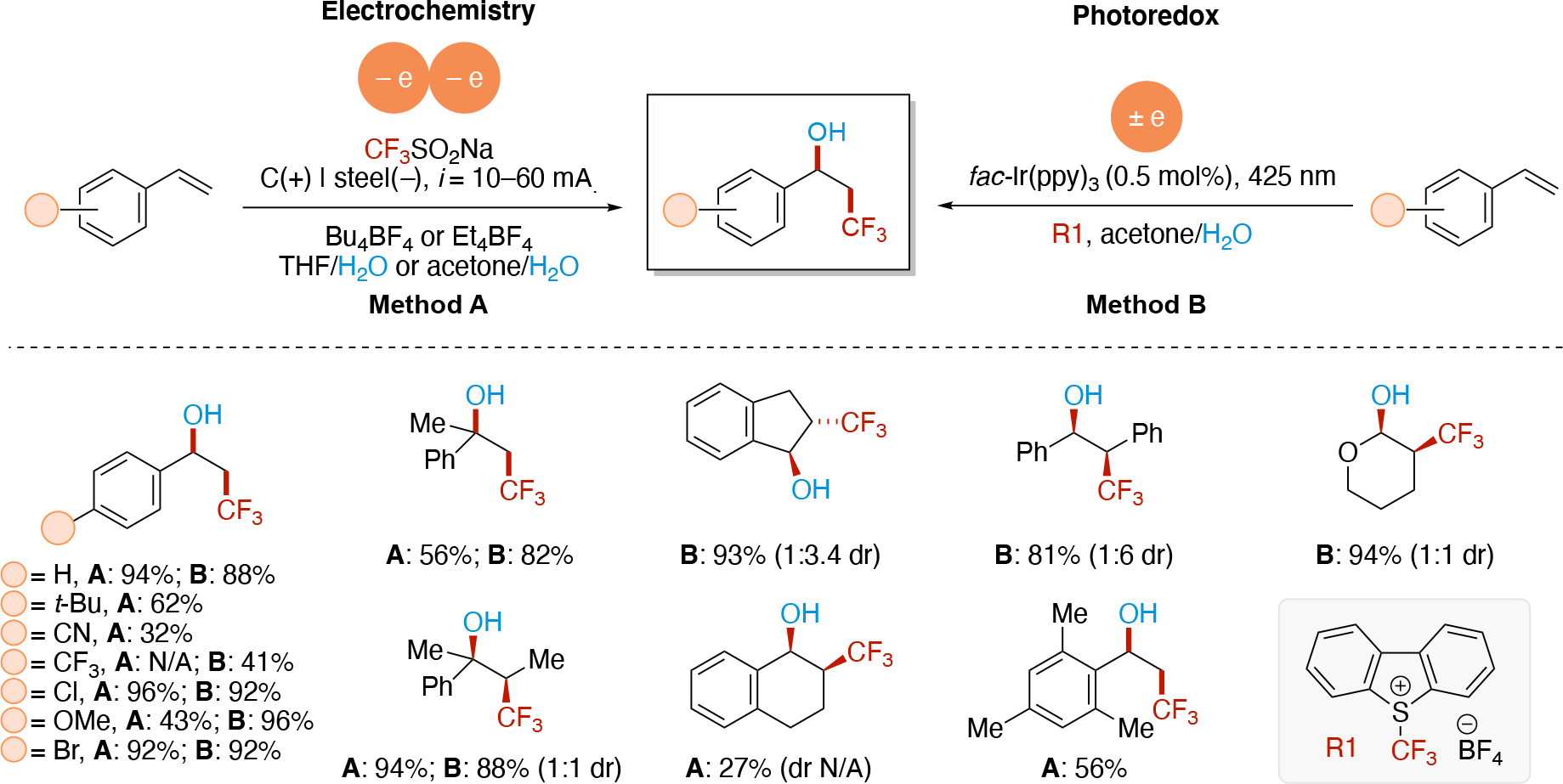
Olefin hydroxytrifluoromethylation.
4.8.5. Hydrofunctionalization of Alkenes
Hydrofunctionalization of alkenes have been realized to introduce a functional group and a hydrogen atom across the alkene.774 For example the addition of an azide to an alkene has been realized in a photoredox hydroazidation,775 while no electrochemical method has yet been reported.
The asymmetric synthesis of benzylic nitriles has been achieved electrochemically via a hydrocyanation of alkenes776 and via photoredox using NHPI esters (Scheme 56).777 In each case the catalyst leverages a chiral box ligand bound to Cu which is postulated to trap a benzylic radical, the copper catalyst also requires oxidation (either prior or after trapping the radical) to ultimately generate a R–CuIII–CN intermediate for facile reductive elimination and formation of the C–CN bond. The photoredox process is redox neutral with the reduction event being photoredox-induced NHPI-decarboxylation to generate the benzylic radical. In contrast, the electrochemical process operates via anodically coupled processes where the 2nd oxidation event oxidizes the CoII to CoIII, enabling reaction with the silane to form a CoIII-hydride capable of HAT to the alkene. A closely related photoredox mediated asymmetric carbocyanation778 of styrene has also been reported using NHPI esters to deliver a carbon centered radical to the alkene followed by a Cu–bis-oxazole catalyzed asymmetric cyanation.
Scheme 56.

Olefin hydrocyanation.
4.8.6. Olefin Hydroxyfunctionalization
Alkene hydration by electrochemical and photoredox methods proceeds via the alkene cation radical, albeit with interesting divergences in hydroxylation regioselectivity (Schemes 57 and 58). Aiwen Lei and coworkers report two methods for photoredox-catalyzed anti-Markovnikov alkene oxidation wherein co-catalyst choice dictates the final product oxidation state (Scheme 57).779,780 Single electron oxidation of the alkene generates the olefin cation radical, which is preferentially hydroxylated at the terminal position to yield 57.12. When a cobaloxime species (57.13) in used, a terminal aldehyde is obtained from styrenyl derivatives via hydrogen gas evolution that involves stepwise reduction and protonation steps — the possibility of PCET is not suggested but is plausible. When diphenyl disulfide is used, HAT from thiophenol to 57.12 results in the terminal alcohol.
Scheme 57.
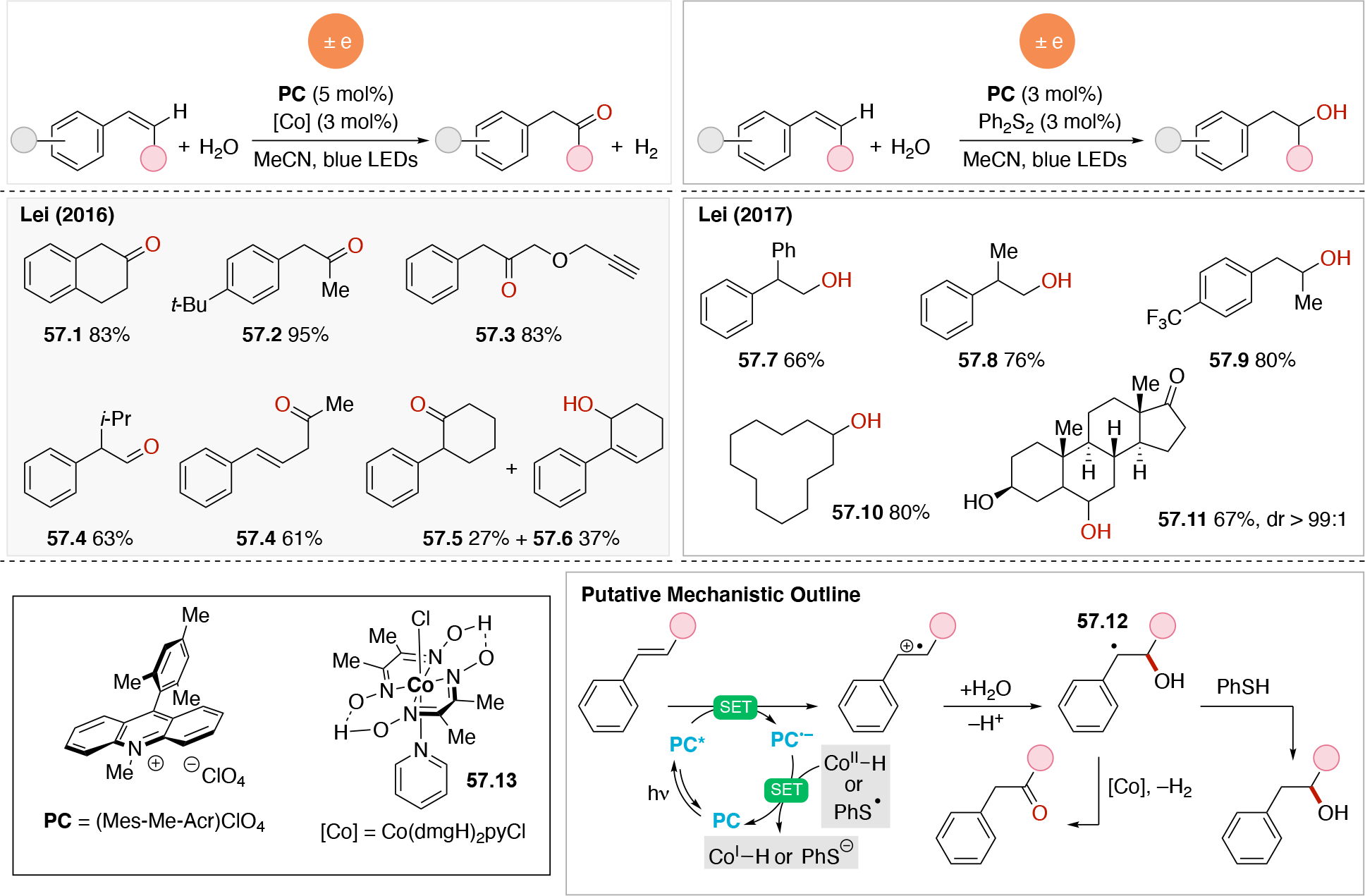
Photoredox-catalyzed olefin oxidation and hydration with corresponding mechanisms.
Scheme 58.
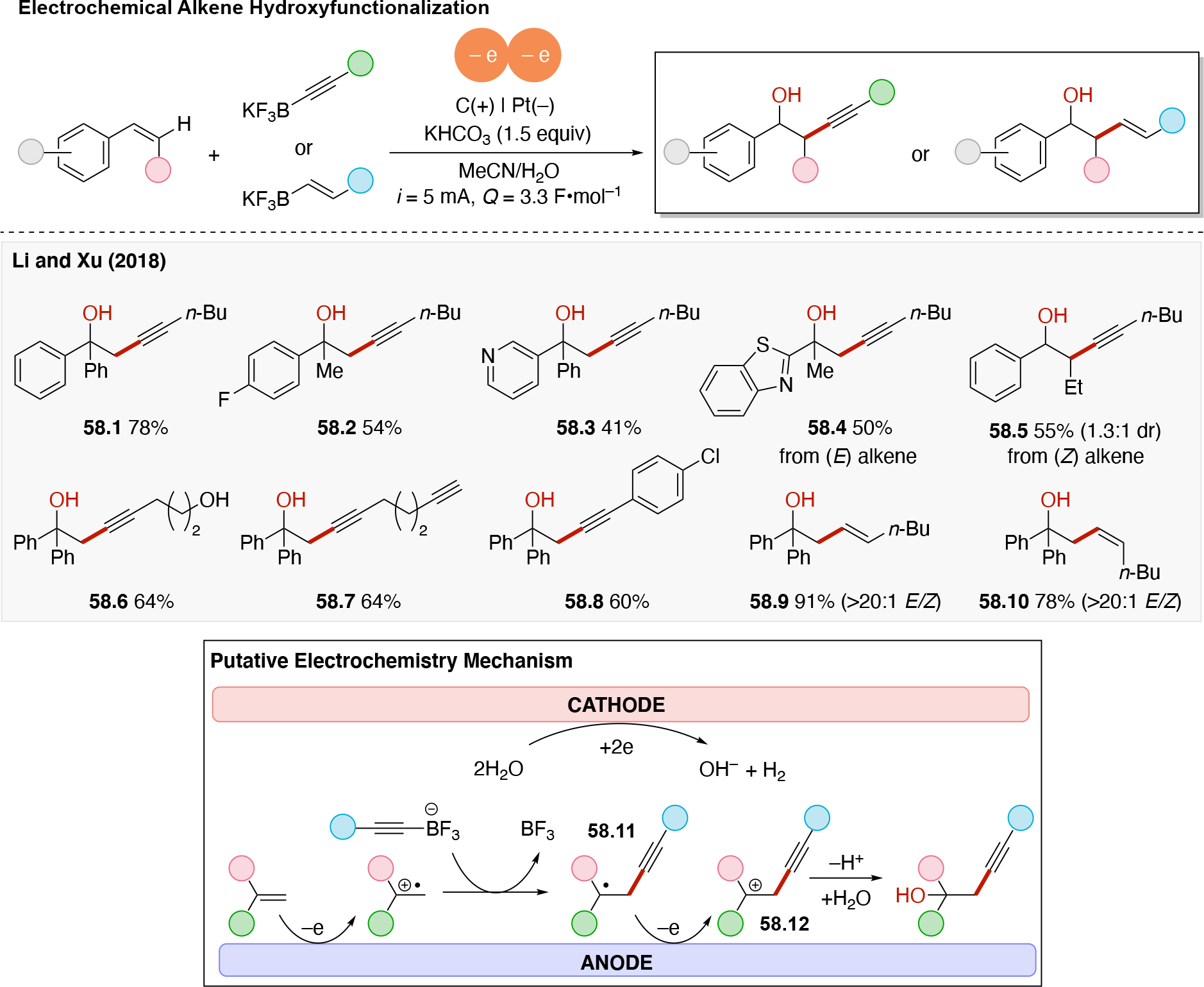
Electrochemical alkyne carbohydroxylation with corresponding mechanism.
An interesting divergence in hydration regioselectivity is observed in the electrochemically-mediated alkene carbohydroxylation reported by Li, Xu and co-workers, with terminal alkenylation and olefination by potassium organotrifluoroborate precursors observed alongside hydration at the internal carbon site (Scheme 58).781 Subjecting the alkynyltrifluoroborate to similar photoredox conditions only results in terminal olefin oxidation, thus suggesting that water outcompetes carbon nucleophile trapping. This outcome is likely due to the unique electrode surface effects; the build-up of anionic organotrifluoroborate at the positively charged anode surface, which then reacts efficiently with the olefin radical cation. Subsequent anodic oxidation of the resultant alkyl radical (58.11) results in a stabilized tertiary carbocation (58.12) that readily undergoes hydration to furnish the observed carbohydroxylation product.
4.9. Reductive Coupling of Ammonium Salts with CO2
Reductive coupling of trialkylbenzylammonium salts with CO2 can provide access to carboxylic acids (Scheme 59). Electrochemically driven direct anodic reduction of trimethylbenzylammonium bromide salts at a carbon cloth anode in DMF under CO2 atmosphere under constant cell potential of −4.5 V can provide benzylic carboxylic acids (Scheme 59).782 The byproduct of the anodic reaction releases Me3N, which acts as a terminal reductant, thus the reaction did not require the use of a sacrificial anode. The choice of counter-anion in the electrochemical method influences the yield, while the use of the Cl and BF4 salt results in ~20% decreased yield of phenylacetic acid, only 25% yield is obtained with the iodide salt (no mention of the outcome with triflate salts). The photoredox catalyzed method783 employs benzyltrialkylammonium triflate salts as starting materials in the DMF in the presence of a mixtures of cesium and sodium carbonate bases (2 and 1 equivalents, respectively) and affords comparable yields and scope, both methods also tolerating heterocycles such as thiophene and furan derivatives although devoid of nitrogen heterocycles or amines. Benzyl halides are also suitable precursors for an electrochemical reductive coupling with CO2 to access arylcarboxylic acids.784,785,786
Scheme 59.
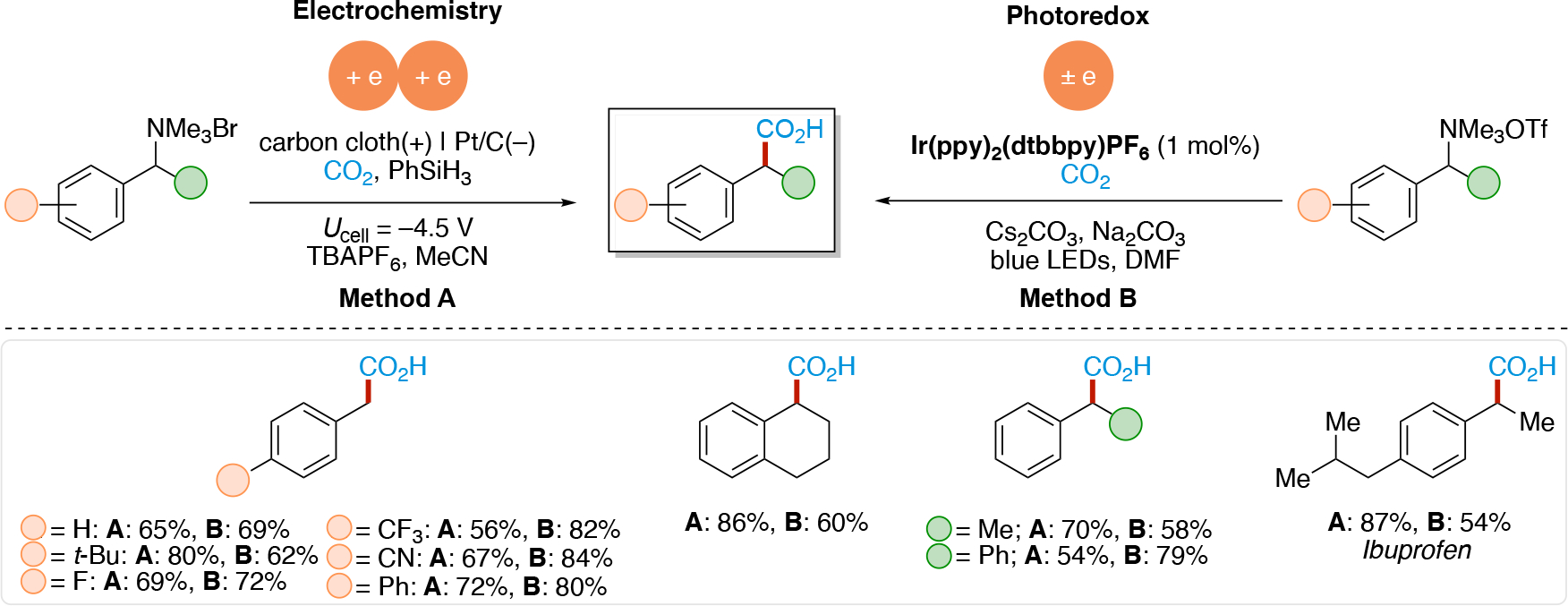
Reductive coupling of CO2 with trialkylbenzylammonium salts to access benzylcarboxylic acids.
4.10. Hindered Primary Amine Synthesis
Recently the Rovis group in collaboration with Merck & Co., Inc., Kenilworth, NJ, USA reported three synthetic methods to access hindered primary and secondary amines via a radical-radical coupling between cyanoheteroarenes and α-amino radicals derived from either iminiums or oximes (Scheme 60).86,787,788 These methods enable the synthesis of primary amines with a fully substituted α-carbon center with broad functional group tolerance, including the incorporation of heterocycles and various heteroatom containing groups.
Scheme 60.
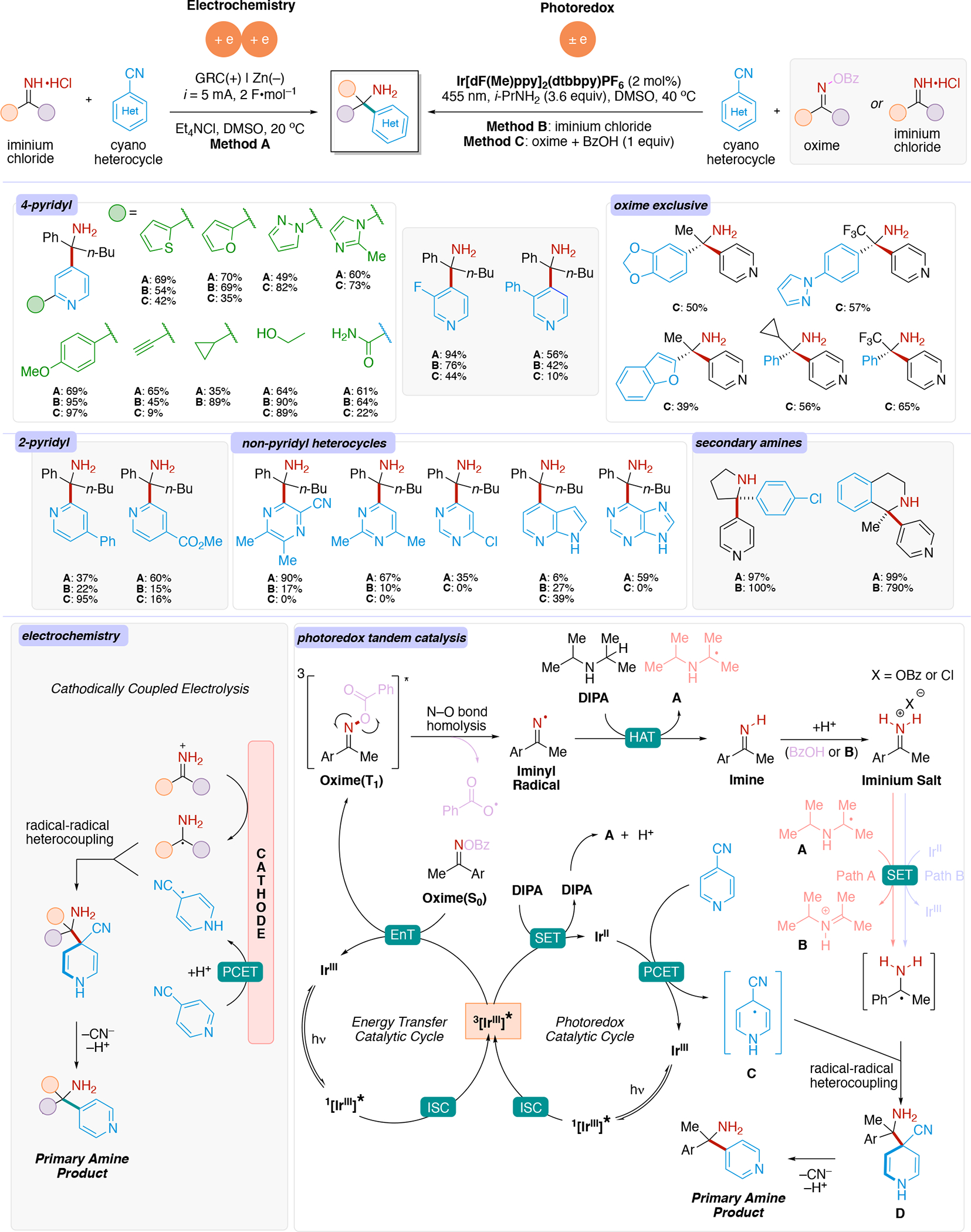
Synthesis of hindered amines from cyanoheterocycles and iminiums chlorides (or oximes).
In comparing over scope of the greater than 60 hindered amines accessed via the three synthetic methods, complementarity in terms of yield is clearly observed. Whenever one method has short comings in terms of yield, one or both of the other methods prevail to afford useful yields of the desired amine. Reflecting on the scope, the electron rich cyanoarenes perform better using the oxime photochemistry, whereas electron deficient cyanoarenes are better suited for electrochemical reductive coupling with iminiums. Beyond cyanopyridyl substrates, azaindole performed significantly better in the photochemistry from the oximes than the electrochemical coupling method. In contrast, pyrazine, pyrimidines, and purine derived products were better suited for synthesis via electrolysis.
In terms of the electronics on the α-amino-benzyl radical coupling partner, differences are also observed between methods. Analysis of the Hammett series of electronically varied oxime substrates indicates a rather flat response in terms of yield whereas electron-rich iminiums (e.g. 4-MeO- and 4-Me-substituted substrates) clearly perform better than electron-poor iminiums (e.g. 4-CF3- and 4-Cl-substituted substrates) regardless of using photochemistry or electrochemistry. The use of an ortho-methyl substituent on the phenyl ring of the iminium substrate was demonstrated to improve coupling yields in the case of iminium substrates (photo- or electrochemical methods) as a result of a conformational change that raises the energy of the LUMO. This substitution pattern on the oxime substrates had the negative consequence of dramatically increasing the T1 to S0 energy (ca. 74 kcal•mol−1) for the Z-oxime to be outside the range that could be photosensitized by the photocatalyst utilized (ca. 56 kcal•mol−1 for PC1), explaining why incomplete consumption of the oxime was observed in those transformations.789
Despite sharing similarities in the mechanistic pathway, the differences in the three methods are explained by the subtle changes in the conditions. First, the electrolytic method where the substrate undergoes the reduction event at the electrode results in a high local concentration of reduced substrate (α-amino radical) at the electrode, whereas photochemistry has significantly lower concentrations of these species. As a consequence, dimerization of the α-amino radical to generate 1,2-diamine impurities is suppressed under photochemical methods compared to the electrolysis method. While the photochemical methods rely on the photocatalyst for the driving force of the reduction process of the substrate (e.g. cyanoarene), the electrolysis does not have a fixed reduction potential – the electrode is adjusted to a more negative potential until it finds something to reduce in order to maintain constant current.790 This is not the case for the photocatalysis where thermodynamically neutral or endothermic processes may be unfavorable to the point of not being able to activate that substrate towards reduction and coupling (back electron transfer from the substrate to the catalysts may outcompete radical-radical coupling). Finally, the putative iminium generated from the oxime starting material is the iminium benzoate salt, whereas in the case of starting from the iminium substrate directly, it is the iminium chloride salt. Mechanistically, the identity of the counteranion imparts subtle differences in the downstream coupling chemistry, particularly due to the difference in the reduction potential of these two iminium salts. It is well known that the reduction potential for PCET events can be modulated via the strength of the acid (i.e. different pKa) according to Bordwell’s equation.451,791 This likely explains the differences in the cyanoarene electronic preferences when comparing oxime vs iminium photocatalytic methods presented herein keeping in mind that the acid for these PCET is the iminium salt (benzoate vs chloride).
While iminium substrates can be readily accessed from arylnitriles in a single step via addition of organometallic reagents (e.g. alkyllithiums) followed by salt formation using anhydrous HCl, these conditions are usually incompatible with functional groups prone to react with strong nucleophiles/bases (e.g. esters). This is where the oximes substrates provide unique access to the necessary requisite starting material bearing those functional groups or heterocycles (e.g. benzofuran). Additionally, the synthesis of primary amines with α-CF3 groups is again uniquely suited for the oxime route in contrast those relying on iminium substrates. Similarly, substrates where the iminium is hydrolytically unstable (strongly electron deficient bearing substrates), or those that are challenging to crystallize during the HCl salt formation (α-cyclopropyl or heterocycle bearing-derivatives), or where the requisite arylnitrile staring material is not available are better suited for the oxime chemistry by utilizing readily available requisite ketones, generating the oxime substrate under mild conditions and avoiding the above stability/crystallization issues.
These products generated from the methods in Scheme 60 are high utility building blocks for medicinal chemists and this synthetic method enables efficient and modular exploration of chemical space. The starting materials are readily available and can be stored long term, ready to be utilized to access libraries via a permutation approach to explore chemical space to provide bi-directional building blocks.792 In fact, a recent report demonstrated the use of the above reaction for parallel synthesis of a library of hindered amines using the high throughput experimentation electrochemistry equipment (HTe−chem) described in Section 3.10.389 With the advances in high throughput experimentation for parallel synthesis and direct to biology workflows793 that utilize reaction mixtures coming from solvents like DMSO directly into bioassays, these synthetic methods may accelerate the exploration of chemical space to test biological hypotheses to solve important medical and agrochemical challenges.
An alternate photoredox method for accessing hindered primary amines was reported by Gilmore, in which they describe three examples of a three-component coupling between 4-cyanopyridine, ammonia and a (hetero)arylketone (Scheme 61).788 The reaction uses Hantzsch ester (2.2. equiv) as the terminal reductant, stoichiometric TFA in 60 °C MeCN and Ru(bpy)3Cl2•6H2O as the photocatalyst. While it remains unclear how broad the scope is for this transformation, is does look promising as the (hetero)arylketones are structurally varied — specifically, 3-acetylpyridine, 2-acetylfuran, and 4-fluoroacetophenone each provide their corresponding hindered amine in good to excellent yield (72%, 80%, and 99%, respectively). The reaction is proposed to proceed via in situ formation of an iminium, with its 1e reduction is enabled by photoredox catalysis to generated the transient α-amino radical for coupling with 4-cyanopyridine.
Scheme 61.
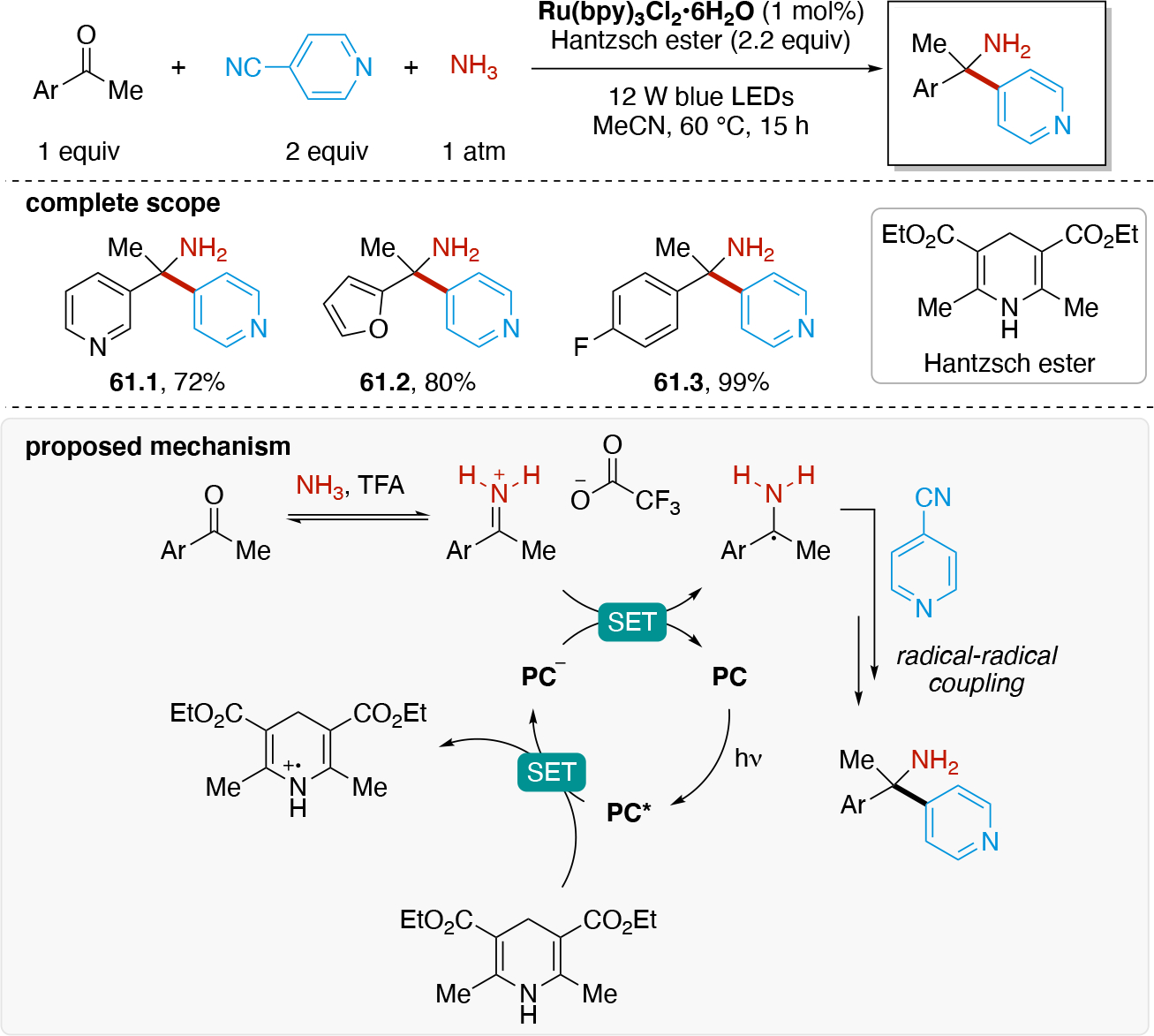
Reductive arylations of (hetero)aryl ketones to generate primary amines with α-tri-substituted centers and its proposed mechanism.
4.11. Oxidative Arene Functionalization
Oxidative arene functionalization occurs either by oxidation of a redox-active mediator or by direct oxidation of the arene or one of its electron-rich substituents. Photoredox and electrochemical methods for intramolecular dehydrogenative lactonization and lactamization of arene C–H bonds, which occur by the former mechanism, have been recently covered28 and are not discussed further. This section will instead cover examples involving arene oxidation.
4.11.1. Arene C–H Amination
Anodic oxidation of aromatics has rich history794,795 but practical applications were limited due to unproductive oxidation of either coupling partner or of the product. One such example is aryl C–H amination of electron-rich arenes with N-centered nucleophiles, in which both coupling partners are susceptible to overoxidation. Yoshida and co-workers approached this problem by demonstrating the electrochemical coupling of electron-rich arenes and pyridine under constant current electrolysis (Scheme 62A).796 The key intermediate is an oxidatively-generated arene cation radical, which traps pyridine to yield an arylpyridinium intermediate that is deprotected to the aniline after a secondary Zincke reaction. Good to excellent yields were observed (65 to 99%) for a range of electron-rich arenes, with site-selectivity ranging from excellent para-amination selectivity relative to the most electron-rich substituent to slight para:ortho selectivity (1.3:1 to 2.3:1) relative to the methoxy group. A follow-up publication showing N-aryl imidazole synthesis via arene cation radical trapping with N-methylsulfonateimidazoles followed by piperidine deprotection (Scheme 62B).797 A key limitation of this method is preferential amination of benzylic C–H bonds. Arene amination with primary amines was also discovered later, but is limited to heterocyclic, N-protected primary alkylamines bearing β or γ N/O-functionality; these reveal the arylamine when deprotected with ethylenediamine (Scheme 62C).798 In all three methods, the reactions were run in a LiClO4/MeCN or Bu4NBF4/MeCN solution (0.1 to 0.3 M) under a constant current (8.0 mA, 2.5 F•mol−1) in an H-type divided cell equipped with a carbon felt anode and a platinum plate cathode. A photoredox complement to Yoshida’s work was developed by Nicewicz and co-workers, in which electron-rich arenes were aminated with azoles and ammonium carbamate using a highly oxidizing 9-mesityl-3,6-di-tert-butyl-10-phenylacridinium tetrafluoroborate (Mes-3,6-dtb-Ph-Acr, 11.8, PC-1) photoredox catalyst and TEMPO as a co-catalyst under an oxygen atmosphere (Scheme 62D).799 N-arylazole and aniline yields were generally moderate to excellent (26 to 99%) for a greater range of electron-rich arenes and azoles, with no benzylic amination observed. Aminated products were either obtained as single regioisomers or with moderate to excellent para:ortho site-selectivity. The reaction proceeds by PET from the arene to photoexcited PC-1, generating the key arene cation radical which is then trapped by the nucleophile. Co-catalyst TEMPO or molecular oxygen act as the oxidants in the transformation, and presumably turn over photocatalyst cycle. A follow-up work by Nicewicz and co-workers expand the amine scope to primary amines using PC-2, with amino acids, linear and branched amines, hindered amines, benzylamines and complex amines tolerated (Scheme 62E).800
Scheme 62.
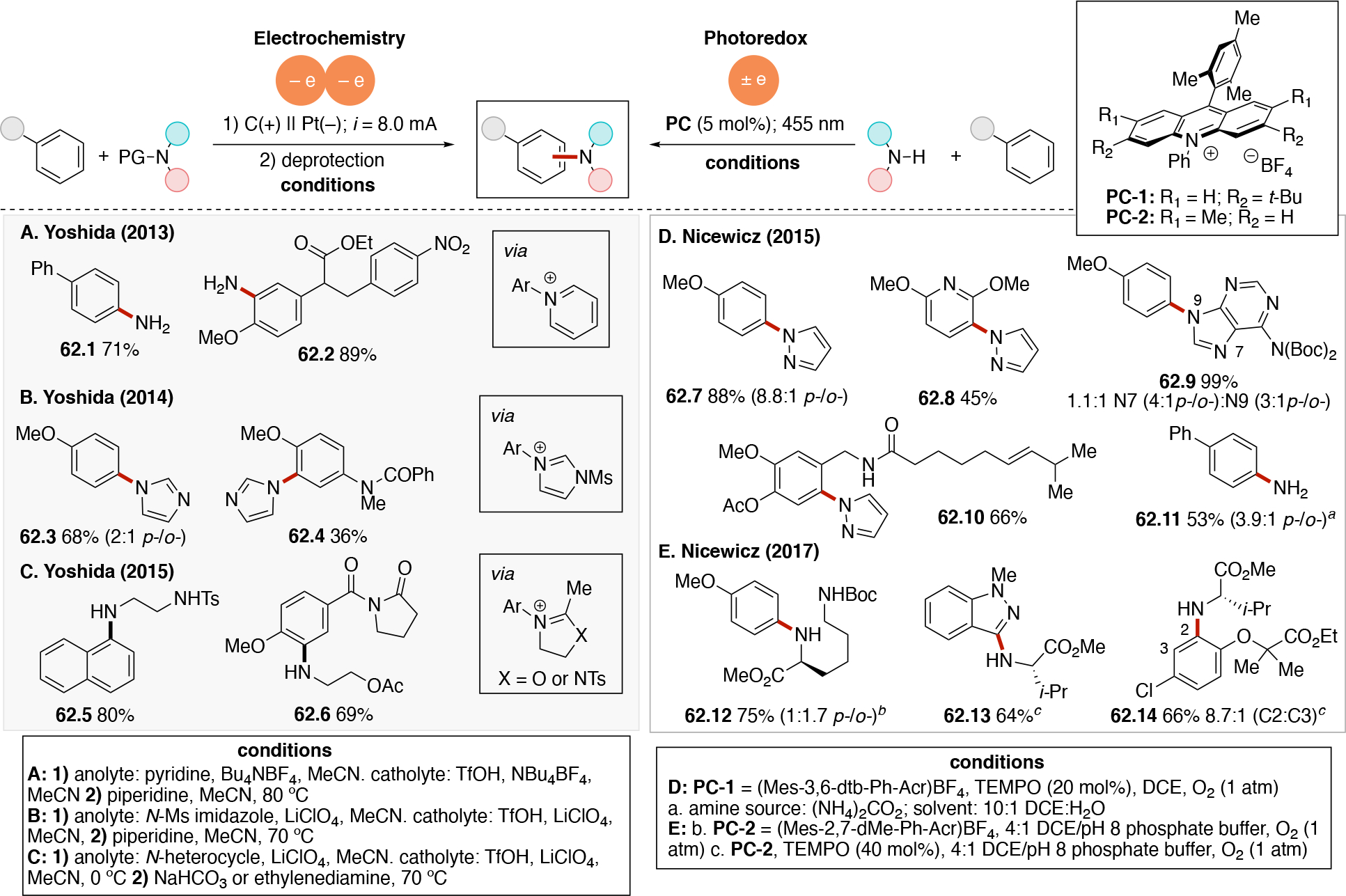
Representative examples of arene C–H amination via electrochemical and photoredox methods.
4.11.2. Arene C–H Cyanation
The direct C–H cyanation of hetero(arenes) enables facile access to the aryl cyano group, which is a versatile synthon typically used in functional group interconversion (FGI) to access aryl carbonyl, imine and heterocycle scaffolds. Several redox-centered methods for benzonitrile synthesis, have been developed (Scheme 63) with Nicewicz and co-workers reporting the cyanation of electron-rich aromatics using an acridinium photoredox catalyst (11.8).801 This reaction occurs via the formation of a key arene cation radical intermediate, with minimal oxidation of the cyanide detected under reaction conditions. Simple and complex (hetero)aromatics are functionalized with moderate to excellent para:ortho selectivity relative to the most electron-rich substituent. The key to successful cyanation is the use of trimethylsilyl cyanide, as it afforded the slow release of cyanide which would otherwise be competitively oxidized if present in excess. An electrochemical counterpart was reported by Gooßen, Tschulik and co-workers, where sodium cyanide is used to trap arene cation radicals formed via anodic oxidation.802 A relatively diverse substrate scope is reported, with lower para:ortho selectivity observed for substrates similar to the photoredox method. Platinum electrodes were used under constant current (20 mA), and two-electron anodic oxidation was coupled to cathodic hydrogen evolution. A key observation in this transformation is the necessary passivation of the platinum electrodes by cyanide, which the authors hypothesize leads to greater substrate scope diversity.
Scheme 63.

Representative examples of arene C–H cyanation via electrochemical and photoredox methods.
4.11.3. Arene C–18F Fluorination
The direct radiofluorination of aryl C–H bonds enables the development of 18F-labeled small molecules and their application towards positron emission tomography. However, a limitation is the requirement for short reaction times, as t1/2(18F) is 109.8 min. Thus, redox-centered methods for arene C–H 18F-fluorination must account for 18F lifetime limitations (Scheme 64). Sadeghi and co-workers attempted the radiofluorination of an analog of the nonsteroidal anti-inflammatory drug (NSAID) celecoxib via anodic oxidation of the heteroaromatic ring.803 The electrolysis was performed under pulsating potentiostatic conditions (working phase of 2.7 V vs Ag/Ag+ for 4 s, then recovery phase pulse of 0.3 V vs Ag/Ag+ for 1 s) with platinum electrodes and Bu4NClO4 as the electrolyte. The radiofluorination was carried out in MeCN under carrier-added conditions (i.e. 18F reagent has residual 19F) with Et4N[18F]•4H[18F], and afforded the 18F-labeled arene in 70 minutes (0.8–2.0 radiochemical yield [RCY, decay-corrected]). This radiofluorinated celecoxib analog was shown to have good metabolic stability and demonstrated promising pharmacokinetics for future PET studies. However, its low molar activity (3 Ci•mmol−1) prevents its use as an imaging agent, a problem that could be resolved through the use of no-carrier added 18F sources. A photoredox-catalyzed arene C–H radiofluorination method was reported by Nicewicz, Li and coworkers, which builds upon their arene C–H functionalization strategy with highly oxidizing acridinium photoredox catalysts.804 RCYs for this reaction ranged from 4.9 ± 0.5 to 50 ± 11% as simple and complex electron-rich (hetero)arenes are radiofluorinated with no-carrier added [18]tetrabutylammonium fluoride ([18F]TBAF) with high para-selectivity relative to the most electron-rich substituent. The molar activity obtained for radiofluorinated diphenyl ether is 1.37 Ci/μmol (yield: 38.2 ± 10% RCY), which suggests that imaging agents for positron emission tomography can be developed. The utility of this method was demonstrated with the synthesis of [18F]DOPA, and the application of radiolabeled NSAID [18F]fenoprofen and tyrosine analog 64.5 to the imaging of inflammation and cancer tumors respectively in mouse models.
Scheme 64.
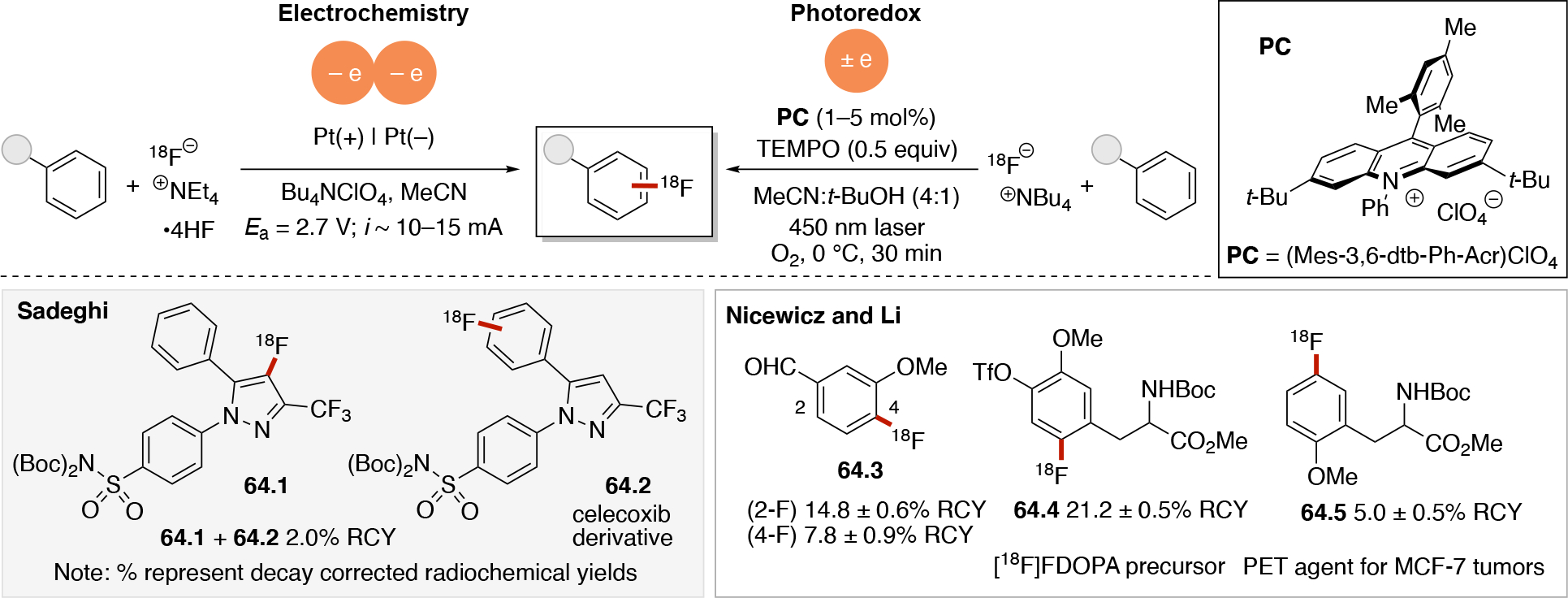
Representative examples of arene C–H radiofluorination via electrochemical and photoredox methods.
4.11.4. Thiolation/Heterocycle Formation
The synthesis of benzothiazoles from benzothioamides via redox methods proceeds by a sulfur-centered radical (thioamidyl) through oxidative mechanisms (Scheme 65). The electrochemical strategy reported by Song and Xu uses indirect electrolysis with catalytic TEMPO as the main redox agent in an undivided cell under constant current (10 mA; RVC anode, Pt plate cathode; Bu4NBF4 electrolyte; MeOH/MeCN (1:1) solvent).805 A wide range of functionalities are tolerated with this method, which directly contrasts with a previous method using harsher direct electrolysis conditions.806 Yields obtained are moderate to excellent for most substrates, with high to exquisite regioselectivity observed. An oxoammonium intermediate formed via the electrochemical oxidation of TEMPO traps the benzothiazole at sulfur to form a weak S–O bond. This species is deprotonated and undergoes rapid S–O bond homolysis (BDE ~ 12.5 kcal•mol−1) to form the thioamidyl radical, which rapidly cyclizes to the azacyclohexadienyl radical. DFT computations suggest that the regioselectivity observed arises from the kinetics of radical cyclization and the overall thermodynamic favorability of the azacyclohexadienyl radical regioisomer. Rearomatization via proton and electron loss furnishes the desired benzothiazole. A photoredox counterpart to this reaction was developed by Wu and Lei, in which a two-catalyst system — Ru(bpy)3(PF6)2 and cobaloxime 65.14 — were employed, with moderate to quantitative yields and excellent regioselectivity observed for a diverse substrate scope.807 Experimental studies of the reaction mechanism established that deprotonation of benzothioamides was necessary to generate a thioamide anion species that is readily oxidized by photoexcited Ru(bpy)32+ to the thioamidyl radical which subsequently cyclizes. Reduced Ru(bpy)3+ can transfer an electron to the CoIII-species 65.14, which forms a CoII intermediate capable of oxidizing the azacyclohexadienyl radical, which rearomatizes by deprotonation to the desired benzothioamide. The CoI species then intercepts two protons in a stepwise manner to generate H2 and regenerate 65.14.
Scheme 65.
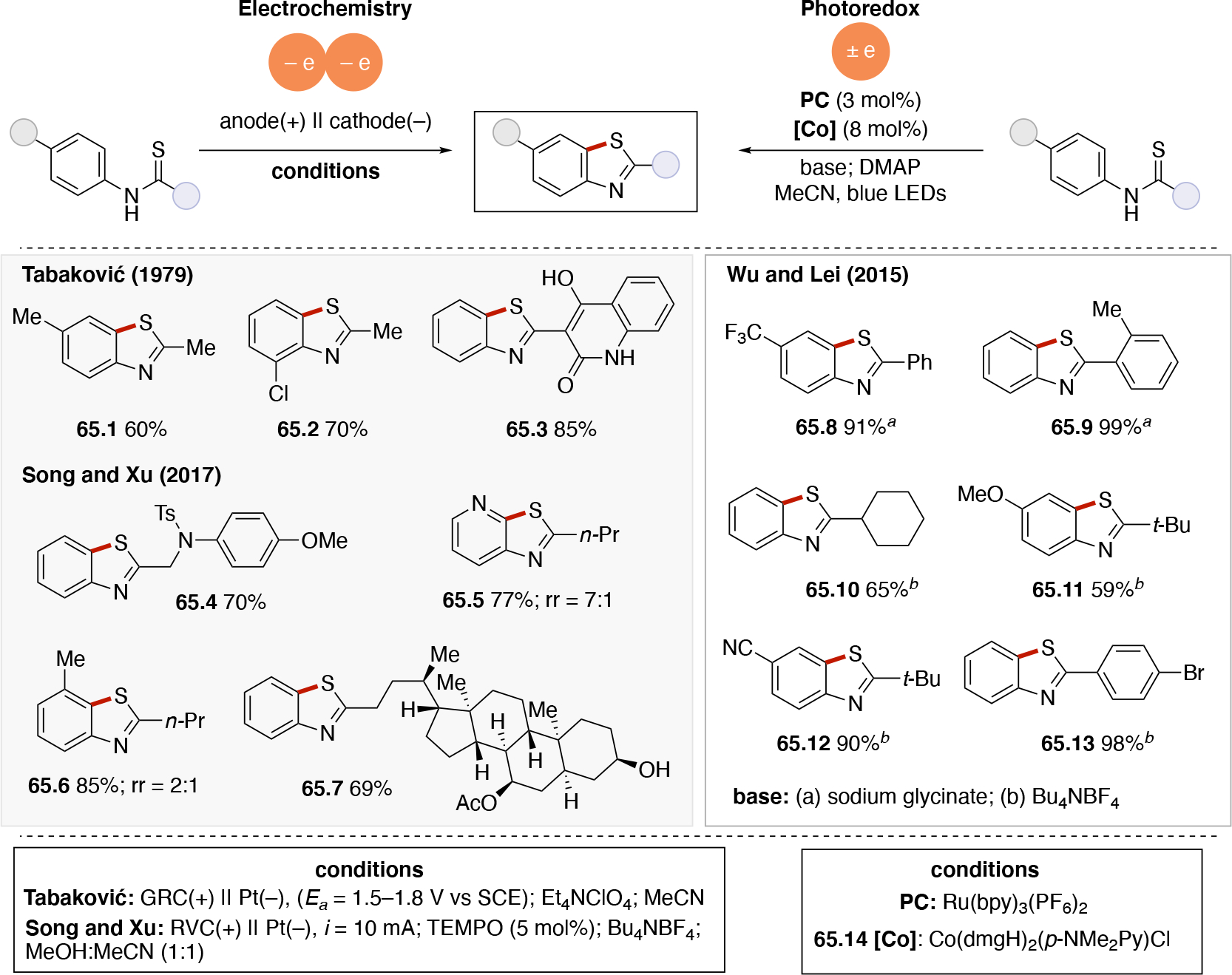
Representative examples of intramolecular arene C–H thiolation via electrochemical and photoredox methods.
4.11.5. Intramolecular Oxidative Biaryl Couplings
The synthesis of polycyclic aromatic hydrocarbons (PAHs) and graphene nanoribbons, including small molecule models of graphene and nanotubes, often leverage oxidative cyclodehydrogenation reactions (e.g. Scholl-oxidation).808,809,810,811,812 These reactions convert ortho-arylene precursors to larger aromatic compounds via a net dehydrogenative biaryl coupling using stoichiometric oxidants (e.g. DDQ).813 Non-photochemical conditions relying on the use of stoichiometric oxidants (e.g. DDQ) can render the reaction purification challenging.814,815 Thus the use of catalytic DDQ which can be regenerated electrochemically provides benefits towards addressing the above problem as was reported by Hilt in the oxidative biaryl coupling shown in Scheme 66.220 Since the reduction of DDQ to generate the corresponding dihydroquinone (DDH) is a net 2e/2H+ reduction event, a source of protons is required. Brønsted acid, such as MeSO3H or TFA, not only provide the necessary protons but also shift the redox potentials associated with the cycling between DDQ and DDH, as is typical of quinone redox chemistry.816 The Mallory reaction uses light to transform ortho-arylene compounds to their dehydrogenative biaryl coupled analog via the use of light and oxidants such as iodine or oxygen.817 The reaction is proposed to proceed via a photoinduced 6π-electrocyclization with subsequent oxidative aromatization via hydrogen abstraction.817,818 For example photochemical conditions B1 in Scheme 66 were leveraged to access various triphenylenes.819 The Katz-modified Mallory reaction leverages propylene oxide as an additive to scavenge the HI byproduct, thus suppressing acid-mediated rearrangements/side-products.820 The application of these conditions (method B2 in Scheme 66) were demonstrated in the synthesis of 2-methyltriphenylene.821
Scheme 66.

Intramolecular oxidative biaryl couplings.
Electrochemical cross-dehydrogenation C–C couplings are now widely reported. Oxidative homo- and hetero-coupling of phenols822,823 has been extensively studies using electrochemistry, particularly by Waldvogel.822,824,825 the use of a BDD anode and HFIP as the solvent enabled for homocoupling of phenols to access 2,2’-biphenols826 as well as 4,4’-biphenols (Schemes 67 and 68).827 The use of graphite electrodes and fluorinated additives such as TFA was found to also provide for useful conditions to oxidatively dimerize phenols via electrolysis (Scheme 67, method A2). A templating strategy of pre-forming a tetraborate salt containing the phenol enabled anodic oxidation of these salts to the biphenol including phenols lacking a para-substituent relative to the hydroxyl group (Scheme 67, method A4).828 Demonstration of oxidative dimerization of phenols in flow using glassy carbon anode and stainless steel cathode (j = 60 mA•cm−2, Q = 0.8F) under supporting-electrolyte-free in conditions using HFIP with 5 vol % pyridine at 0 °C achieved 59% yield of 3,3′,5,5′-tetramethyl-2,2′-biphenol with a productivity of 9.60 g/h (Scheme 67, method A3).829 Kozlowski reported a photocatalyzed method based on acridinium photocatalysts achieving both homo- and hetero-cross couplings (Schemes 67 and 68).830 Both methods achieve similar products, and are proposed to proceed via a similar reaction mechanism, specifically via a radical−neutral coupling in which a 1e oxidation of the phenol generates a phenoxyl radical intermediate followed by reaction with an unoxidized (neutral) phenol. In the context of electrochemistry, the anode facilitates the oxidation, while the photochemical method relies on an acridinium photocatalyst [(Mes-Me-Acr(BF4)], a biphenyl derivative (4,4’-di-tert-butylbiphenyl) as a redox mediator, and oxygen (from air) as the terminal oxidant, as shown in Scheme 66. The synthesis of unsymmetrical biphenols can be accessed using similar photochemical and electrochemical831 conditions, with the photochemical methods usually providing improved yields.
Scheme 67.
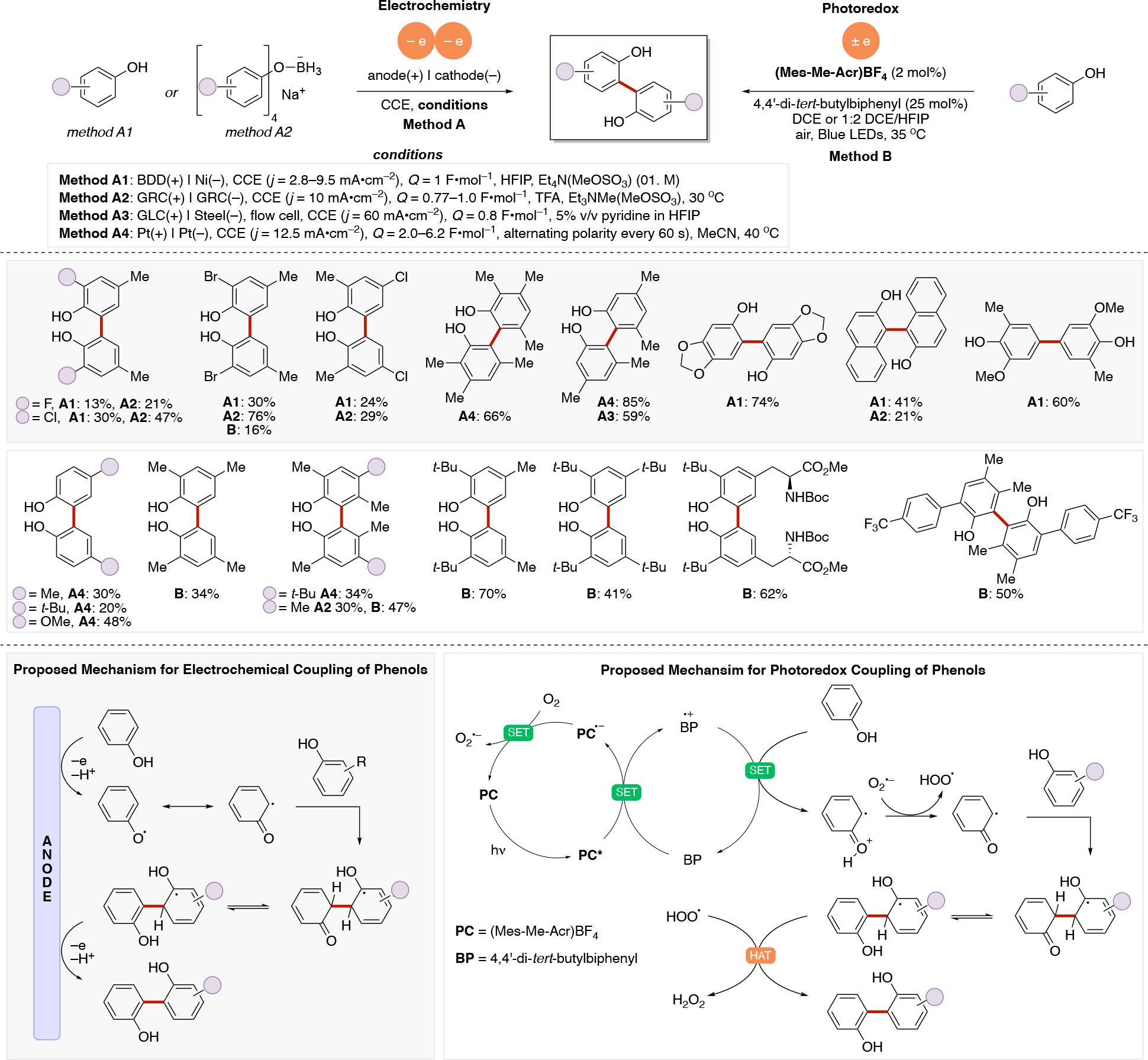
Oxidative homo-coupling of phenols.
Scheme 68.

Oxidative cross-coupling of phenols.
Müllen, Gu, and Ma recently reported an electrochemical synthesis and deposition of polycyclic aromatic hydrocarbon films on ITO electrodes for optoelectronic applications (Scheme 69).832 While thermal conditions typically rely on stoichiometric use of DDQ as the oxidant usually in solvent mixtures of chlorinated solvents and TFA, the electrochemical method could be carried out directly at the electrode or mediated by 5 mol% of DDQ in the presence of TFA in CH2Cl2 solutions. Morin has accessed related PAH structures using a photochemical cyclodehydrochlorination reaction833 with the benefit of being able to obtain preparative amounts of the products in contrasts to the electrodeposited films (Scheme 69).
Scheme 69.
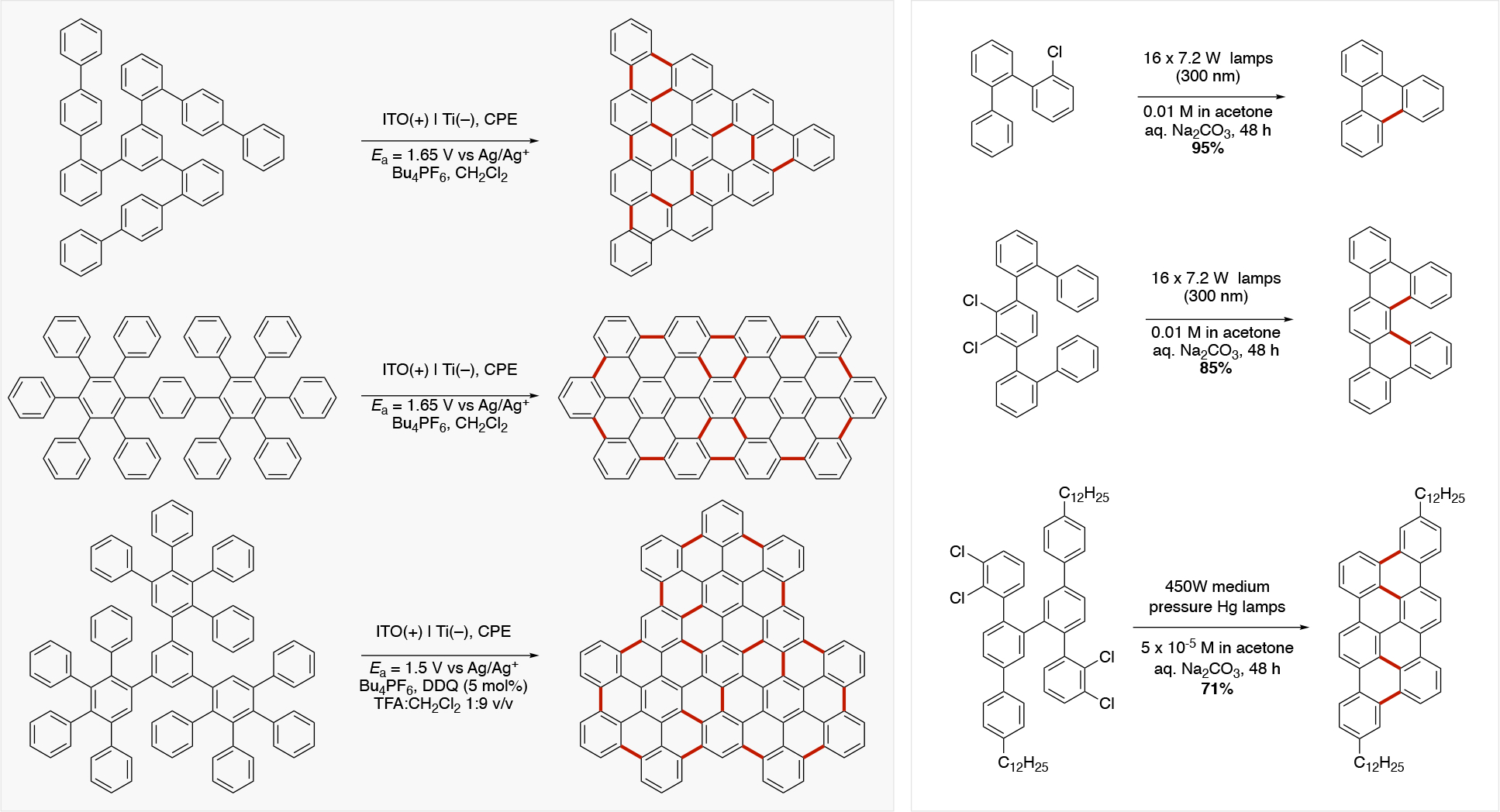
(Left) Electrochemical synthesis of polycyclic aromatic hydrocarbons as films on ITO electrodes. (Right) Photochemical cyclodehydrochlorination to PAH derivatives.
4.12. C(sp2)–F Activation
4.12.1. Arene Hydrodefluorination
The strategic inclusion of fluorine atoms within aromatic scaffolds of pharmaceuticals is a common practice in drug discovery because it potentially enhances drug potency and improves the metabolic stability of electron-rich phenyl rings.834 However, because site-selective aryl fluorination is challenging, conventional syntheses of fluorinated pharmaceuticals start with regio-defined fluorinated precursors.835,836 An alternative strategy is site-selective hydrodefluorination of perfluorinated arenes — which are readily synthesized through the well-established LaMar process involving F2 gas or through electrochemical fluorination837 — to access partially fluorinated aromatic scaffolds. Weaver and co-workers report a method for photoredox-mediated aromatic hydrodefluorination using the reducing photocatalyst Ir(ppy)3 and sacrificial amine (Scheme 70).838 This strategy proceeds by reductive quenching of photoexcited Ir(ppy)3 by a donor amine to form an IrII species that then reduces the perfluorinated (hetero)arene to a perfluoro(hetero)aryl radical anion 70.10. Fluoride extrusion, followed by HAT to the resulting aryl radical 70.11 reveals the desired product. HDF regioselectivity is largely dictated by the electronic stability of the resultant radical anion; computational studies of the arene σ* SOMO show that hydrodefluorination site-selectivity correlates with the arene sites containing the greatest electron density and the longest C–F bonds.839 Di- and tri-hydrodefluorination is observed, albeit limited to electron-deficient substrates. Since this initial report, Weaver and co-workers have expanded the scope of trapping agent for the perfluoro(hetero)aryl radical to alkenes840, (hetero)arenes841, alkynes842, nitroalkanes843, oxazolones844, and prenyl and allyl electrophiles845. In several examples840,823, stepwise carbo- and hydrodefluorination were used to quickly construct functionalized difluoroaryl scaffolds. More recently, Chu and coworkers show that fluoroarene radicals generated via photoredox catalysis can be used in palladium catalysis, with a net cross-electrophile coupling with aryl bromides and triflates.846 This transformation highlights the potential for new metallaphotoredox couplings for radical species generated via photoreduction.
Scheme 70.
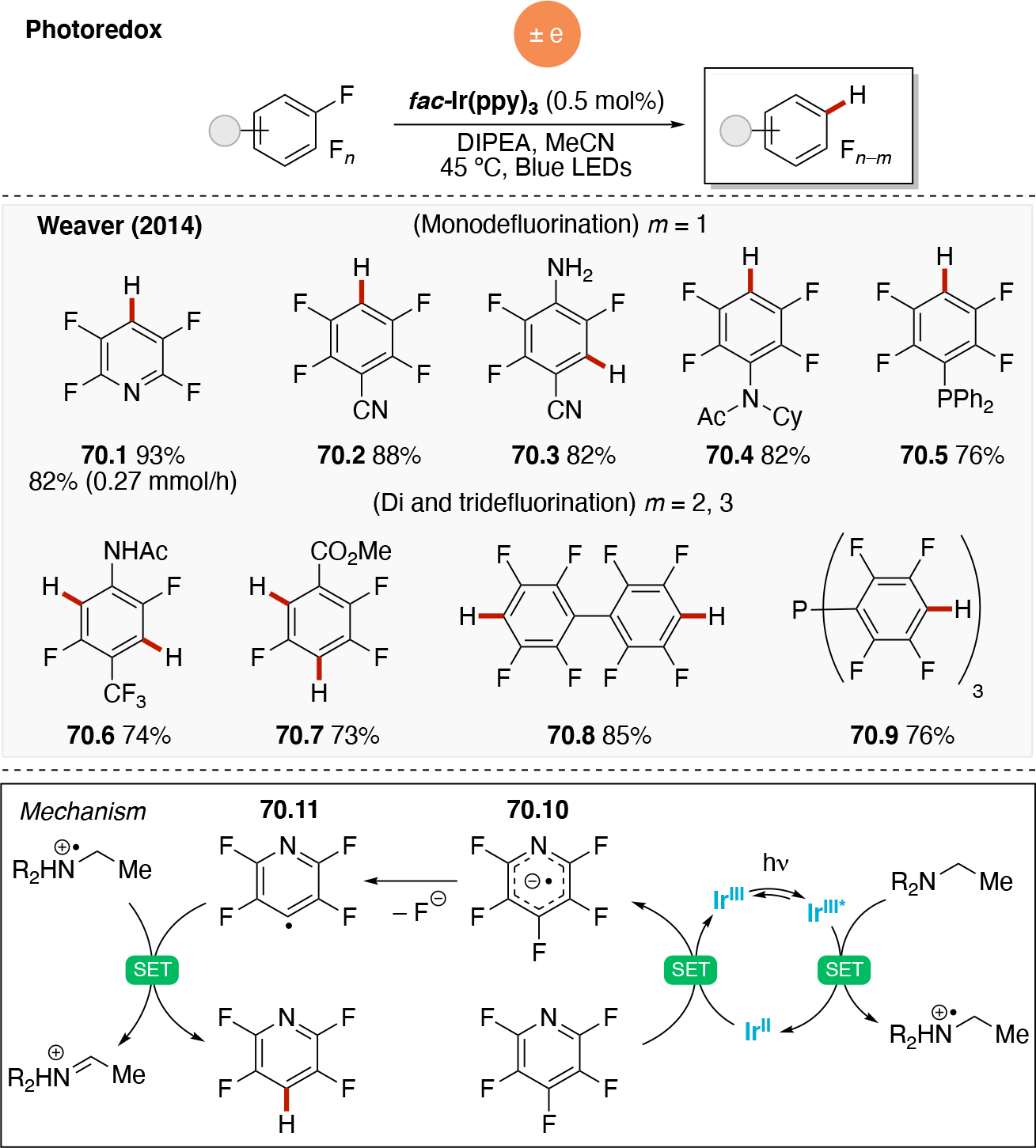
Photoredox-catalyzed hydrodefluorination of perfluorinated arenes, with the corresponding mechanism.
While strategic hydrodefluorination is widely employed in photoredox catalysis, far fewer electrochemical methods have been reported (Scheme 71). Drakesmith reported the reductive hydrodefluorination of pentafluorobenzoic acid under acidic conditions.847 Cathodic reduction with a potential of −1.20 V vs SCE resulted in excellent conversion to 2,3,5,6-tetrafluorobenzoic acid (71.1, 66%), with further reduction of product to 2,3,5,6-tetrafluorobenzyl alcohol (71.2) and unproductive reduction of substrate to pentafluorobenzyl alcohol (71.3) observed (20% and 6% respectively). Increasing the negative potential up to −1.30 V led to full substrate conversion, albeit with increased amounts of 71.2 (from 20% up to 48%) and 71.3 (from 6% up to 24%). Despite this example, reports of perfluorinated arene hydrodefluorination via electrochemical methods are sparse. On the other hand, electrochemical reductions of less fluorinated arenes are more common. Vajtner and Kariv-Miller report the potentiostatic hydrodefluorination of 1,3-difluorobenzene and fluorobenzene using tetraalkylammonium salts as the electron mediator at the Hg cathode surface.848 Using N,N-dimethylpyrrolidinium tetrafluoroborate ((DMP)BF4), the direct electrolysis of 1,3-difluorobenzene in diglyme-H2O afforded 71.4 in 85% yield; reactions at higher current densities led to competitive double dehydrofluorination of 1,3-difluorobenzene to 71.5. Treating fluorobenzene to similar electroreduction conditions (j = 1.25 mA•cm−1) afforded benzene in 76% yield. This concept was improved on by Huang and co-workers, who use sodium borohydride as a sacrificial reductant to accelerate arene hydrodefluorination under galvanostatic conditions.849 Using Pt electrodes in an undivided cell containing 0.2 M Bu4NBF4 solution (20 mA), a range of electron-rich and electron-neutral fluoroarenes were efficiently hydrodefluorinated (yields 70–99%) whereas full defluorination was observed for di- and trifluorinated arenes. Deuterium labeling studies suggest that the solvent or tetraalkylammonium electrolyte counterion is the proton donor in this reaction and that direct HAT from borohydride to the fluoroarene anion radical is a minor reaction pathway.
Scheme 71.
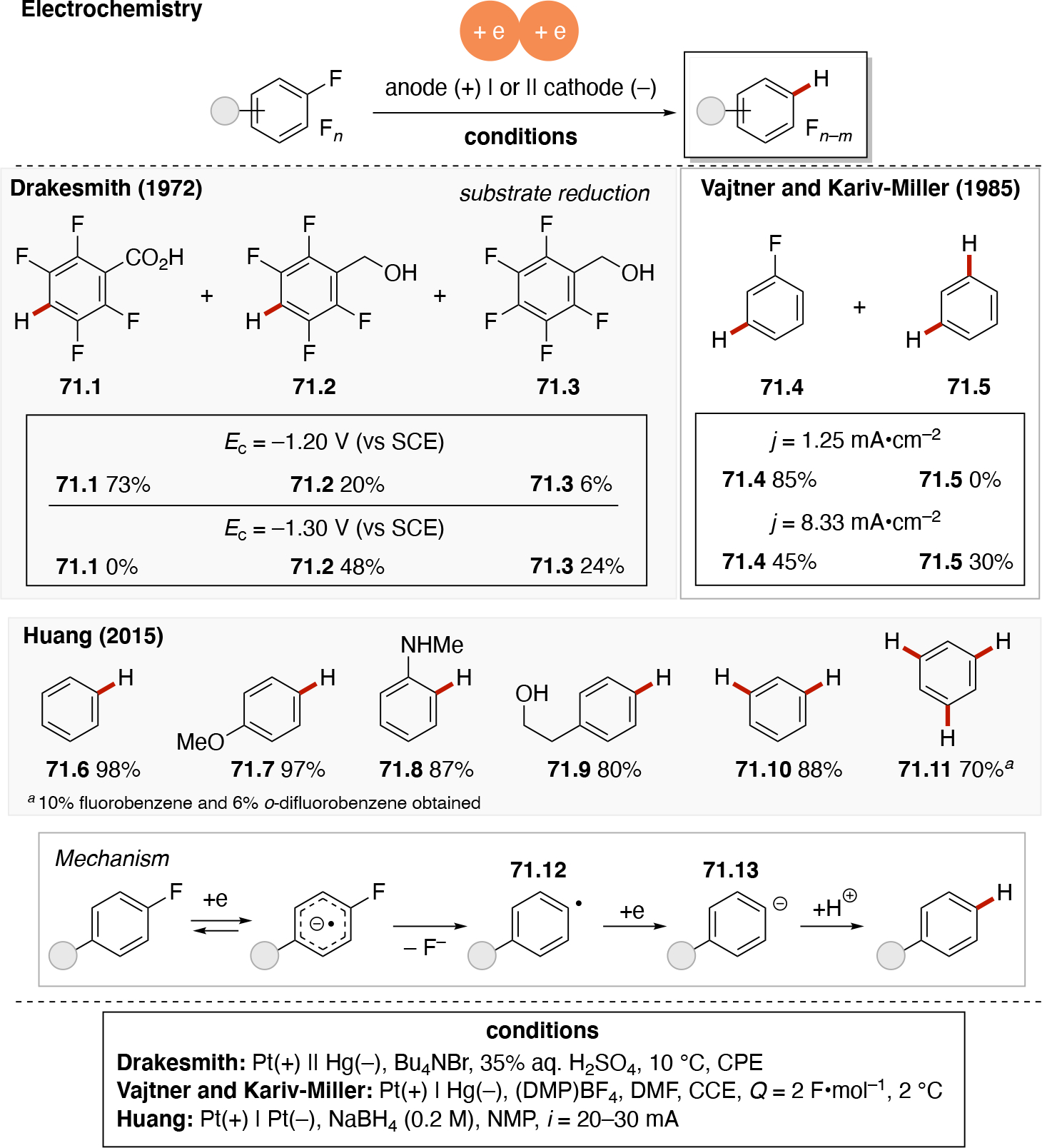
Electrochemical hydrodefluorination of arenes with the corresponding mechanism.
The putative mechanism for hydrodefluorination is an ECEC process: single electron reduction of fluoroarene forms arene radical anion, from which fluoride expulsion occurs (71.12). Facile reduction of 71.12 to 71.13 then occurs, followed by protonation to the desired product. The second hydrodefluorination event is likely slower than first ECEC process as suggested by the need for either a higher overpotential or current density for multiple defluorinations.
4.12.2. Defluorinative Carboxylation of gem-Difluoroalkenes with CO2
Redox methods for C−F bond carboxylation of gem-difluoroalkenes can be achieved with high selectivity for the Z- product (Scheme 72). Wu and Zhou reported an electrochemical method relying on a Pt cathode and sacrificial Ni anode in DMF containing Bu4NI as electrolyte in the presence of 1 atmosphere of CO2.850 The mechanism was proposed to follow an ECEC mechanism in which the gem-difluoroalkene was reduced to the radical anion, followed by reaction with CO2 and a subsequent reduction and loss of fluoride to generate the Z-carboxylated alkene product. The analogous photoredox transformation has been achieved using an Ir-photocatalyst in the presence of a Pd co-catalyst using Pd(PPh3)4 as its pre-catalyst.851 The mechanism initially mirrors that of the electrochemical pathway in that the gem-difluoroalkene is reduced to its radical anion by the reduced formed of the photocatalyst (obtained via reductive quenching by Hünig’s base) but then the mechanism is proposed to diverge. Loss of fluoride and interception of the fluorovinyl radical 72.1 by Pd0 is proposed to afford a vinyl-PdI species which bind CO2 and enables C–C bond formation via a migratory insertion elimination process, SET from the reduced form of the photocatalyst to the PdI regenerates Pd0. Feng and co-workers propose that reversible addition of radical 72.1 to Pd0 is responsible for the interconversion between E- and Z-isomers, as the more kinetically labile form of intermediate 72.2 participates in the carboxylation step. However, it is also conceivable that the E → Z isomerization of these α-fluoroacrylic acids can occur via EnT sensitization by the photocatalyst.852
Scheme 72.

Defluorinative carboxylation of gem-difluoroalkenes with CO2.
4.13. Aryltrifluoromethyl C–F Activation
The aryl difluoromethyl substituent is an important bioisostere853 as it imparts a lipophilicity increase relative to a methyl analog and also demonstrates hydrogen bonding ability similar to an O–H group.854 Furthermore, benzylic difluoroalkyl motifs are gaining interest due to improved metabolic stability855 and the difluoroethyl moiety is an effective bioisostere for aryl methoxy substituents.856 An attractive route to these pharmacophores involves the reduction of benzotrifluorides, which forms the radical anion (73.5) leading to fluoride extrusion to reveal the aryldifluoromethyl radical (73.6), which can either undergo further reduction/fluoride extrusion or participate in further functionalization. The electrochemical reduction of benzotrifluorides is stepwise, with the potentials for aryl-CF3, -CHF2, and -CH2F groups existing within a small potential window, with complete reduction to the methyl usually observed.857,858,859 Attempts at interrupting the defluorination at the RCHF2 or RCH2F moiety exist, but are limited. Périchon and co-workers demonstrated that the electroreductive coupling of benzotrifluoride and 4-fluorobenzotrifluoride with carbonyl species occurs in moderate to good yield, with only the monodefluorinated product observed under constant current electrolysis in an undivided cell (2.2 F•mol−1).860 While an important proof-of-concept, the electrophile scope is limited to solvents (acetone and DMF) or dissolved CO2, as successful coupling requires the generation of a RC6H5CF2− (R = H or F) anion (73.7). Bordeau and co-workers explored the electroreductive silylative defluorination of m-bis(trifuoromethyl)benzene (m-BTFMB) and observed the defluorinative mono- and di-silylation of one CF3 group.861 Defluorination selectivity is dependent on the amount of current passed (optimal current > 2.0 F•mol−1) and the difference in reduction potentials between the trifluoro and the difluorosilyl moieties. Small differences in Ered (< 70 mV) led to unselective product distribution, whereas co-solvent-enabled ΔEred values > 150 mV led to high yields of 73.8 and 73.9 from benzotrifluoride and m-BTFMB respectively (undivided cell; 2.4 F•mol−1). Difluorosilylated m-trifluoromethylphenol (73.11) and aniline (73.12) were also obtained in excellent yields with minimal disilylation. Thus, these methods highlight the predominance of two stepwise, one-electron reduction pathways in benzotrifluoride defluorinative functionalization (Scheme 74A)
Scheme 74.
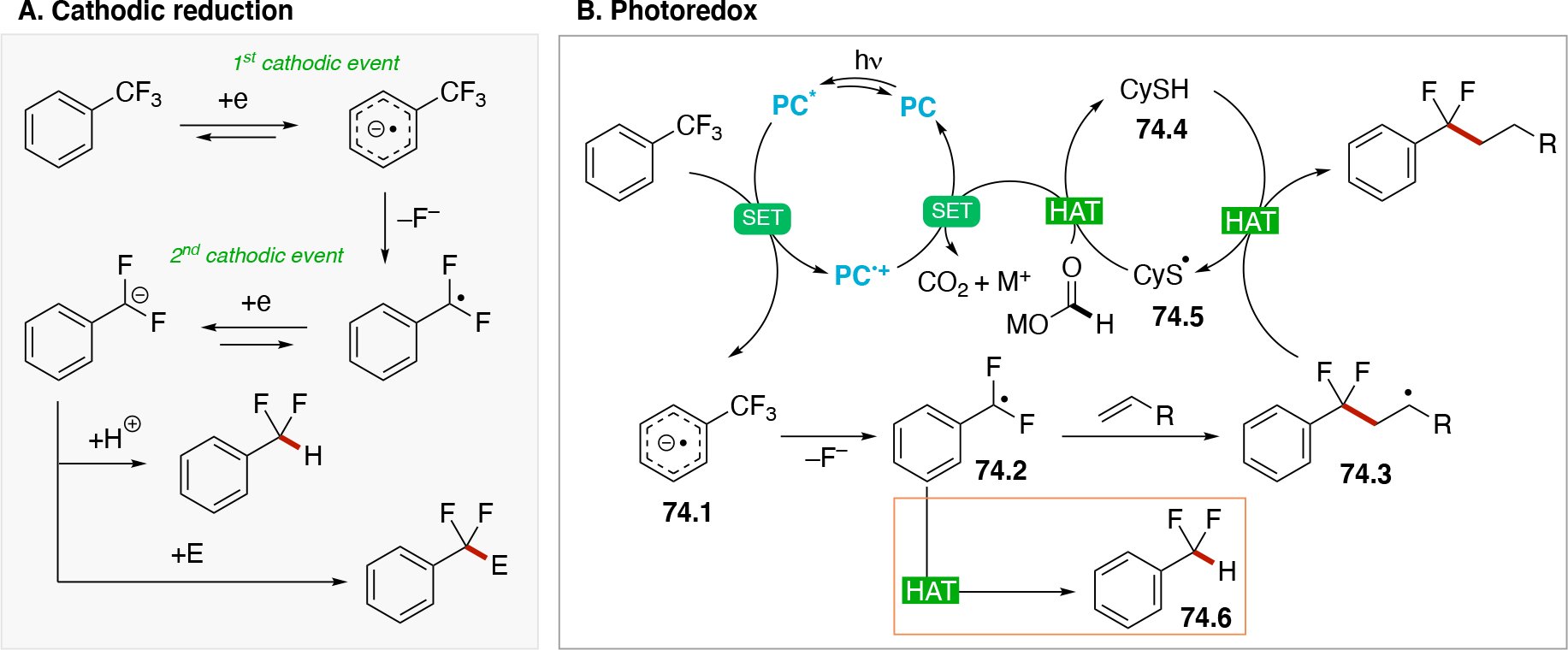
A comparison of (A) electrochemical and (B) photoredox mechanisms for the defluorinative functionalization of benzotrifluorides.
While the selective functionalization of aryl-CF3 groups has been a challenge through electrolysis, photoredox catalysis offers a complementary CF3 activation mode that enables successful monodefluorinative functionalization (Schemes 74B and 75). König and co-workers reported the first example of a photoredox-catalyzed benzotrifluoride defluoroalkylation, in which electron-deficient benzotrifluorides are coupled to N-aryl methacrylamides with assistance from an in situ generated Lewis acid to yield oxindole products in moderate to excellent yield.862 This Lewis acid is formed from protonated 2,2,6,6-tetramethylpiperidine (TMP) and pinacolborane, and is key to this transformation as its exclusion inhibits desired reactivity. Jui and co-workers build upon this work with a procedure for benzotrifluoride defluoroalkylation catalyzed by organic photoreductant N-phenylphenothiazine (11.11, PTH) and cyclohexanethiol, in which electron-deficient benzotrifluorides are coupled to unactivated alkenes in moderate to excellent yields.863 Instead of overreduction, catalytic single electron transfer from an excited state PTH forms the arene radical anion 74.1, which rapidly undergoes defluorination to form arene radical 74.2. This electrophilic species can trap an alkene to form 74.3, which reacts with a thiol cocatalyst (74.4) to furnish desired product. The photocatalyst and thiol catalytic cycles are turned over by the concomitant oxidation of net reductant sodium formate by oxidized PTH and HAT from oxidized formate to thiyl 74.5. Alternatively, radical 74.2 can undergo HAT with a thiol co-catalyst 74.3 to yield the hydrodefluorination product (74.6). Jui and co-workers follow up this initial report with an expansion of the substrate scope to unactivated benzotrifluorides, which is enabled by phenoxazine 11.12 and conducting the reaction at elevated temperatures (100 °C).864 This method was applied to the facile syntheses of benzylic difluoroalkyl motifs found in bioactive difluoroalkylaromatic molecules (yields ≥ 63%, 1–2 steps); prior literature routes usually require ≥ 3 steps with expensive fluorinating agents and suffer from low yields. In the same paper, Jui and co-workers also demonstrate the synthesis of aryl difluoromethanes in moderate to good yield through the use of cesium formate as the H-atom source; only monodefluorination is observed in this hydrodefluorination (HDF) method. Gouverneur, Noël, and co-workers expand on this HDF strategy by targeting electron-poor trifluoromethyl (hetero)arenes, which would furnish electron-poor difluoromethyl (hetero)aryl cores prevalent in pharmacophores.865 This transformation is performed at room temperature, with yields ranging from moderate to good; more electron-deficient arenes proved slightly more problematic as bis-defluorination was observed. An isophthalonitrile-based organic photoredox catalyst (4-DPA-IPN) was used alongside a 4-hydroxythiophenol (4-HTP) co-catalyst, in which thiol oxidation by photoexcited 4-DPA-IPN yields its reduced counterpart, the species implicated in the arene reduction; this mechanism was supported by Stern-Volmer quenching and radical trapping studies. This method was also applied to continuous-flow conditions, with the HDF of 4-(trifluoromethyl)benzonitrile performed on a 14.6 mmol scale in 69% yield.
Scheme 75.

Photoredox-catalyzed defluorinative functionalization of benzotrifluorides with the corresponding mechanism.
4.14. Birch Reduction
The two-electron reduction of aromatics to cyclohexadienes (Birch reduction) is traditionally carried out with sodium or lithium metal in ammonia (E°= − 3.29 V (Li), −2.95 V (Na) vs. SCE), and proceeds by a key radical anion intermediate.866 Despite its synthetic utility, the necessity of super stoichiometric alkali metal and ammonia limits the scalability of the Birch reduction for industrial applications. Thus, milder electrolysis and photoredox-centered methods offer new replacements for metal reductions (Scheme 76). Baran, Minteer, Neurock, and co-workers report an electrochemical alternative that uses a constant current (10 mA) undivided cell with a sacrificial Mg (anode) and galvanized steel wire (cathode) at room temperature or sacrificial aluminum (anode) and zinc (cathode) at −78 °C.867 The key to obtaining reproducible yields was the use of a phosphoramide additive (tris(pyrrolidino)phosphoramide, TPPA) and 1,3-dimethylurea (DMU) as the proton source. TPPA was especially crucial in minimizing electrode passivation by unproductive Li deposition and THF electrolysis. Computational and experimental studies suggest that two-electron reduction occurring at the cathode is favored over a Li0 pathway. The scalability of this transformation was demonstrated with TBS-protected p-cresol in batch and flow (>10 g) with minimal loss in reaction efficiency. Because the Birch reduction is a two-electron process, two-photon systems for photoredox catalysis have been developed to harness either a tandem EnT-ET excitation pathway or two consecutive photoredox cycles (see Section 5.2 for a discussion of two-photon photoredox catalysis). The former mechanism was invoked in the Birch-type reduction of hetero(arenes) reported by König and co-workers, in which Dexter energy transfer between a polyarene and a photoexcited IrIII chromophore generates a triplet polyarene that can be reduced by a separate IrII species that is generated via amine oxidation by photoexcited IrIII.868 The resultant arene radical anion then undergoes two-step HAT and protonation to yield the reduced arene. A wide range of poly(hetero)aromatic substrates were competent substrates for reduction, with anthracenes and phenanthrenes reduced to the respective dihydro products. The photoreduction of naphthalenes formed both the dihydro- and tetrahydro products, with their respective ratios dependent on reaction times and substitution. The reduction selectivities of azo derivatives (e.g. acridine, quinoline) mirror the patterns observed for the carbocycle variants. More recently, a photoredox-catalyzed Birch reduction method operative via two consecutive PET steps was reported by Miyake and co-workers, in which benzo[ghi]perylene monoimides (BPI) were used as photocatalysts.869 While the scope of compatible (hetero)arenes is similar to the electrochemical precedent, yields were generally lower, reaction times are much longer (4–6 days) and multiple additions of photocatalyst (3–5 additions after 24 h) were required to obtain synthetically useful yields. Superstoichiometric amounts of hydroxide are required to generate a photocatalyst-hydroxide complex, which presumably eliminates hydroxyl radical upon irradiation (405 nm) to generate BPI radical anion (BPI•−). Irradiation of the BPI intermediate leads to excited-state BPI•− (BPI•−*), which is capable of reducing (hetero)arenes (E°comp= −2.43 V to −4.28 V vs SCE). Mechanistically, this process merges the energy of two-photons to generate a highly reducing catalytic species.
Scheme 76.
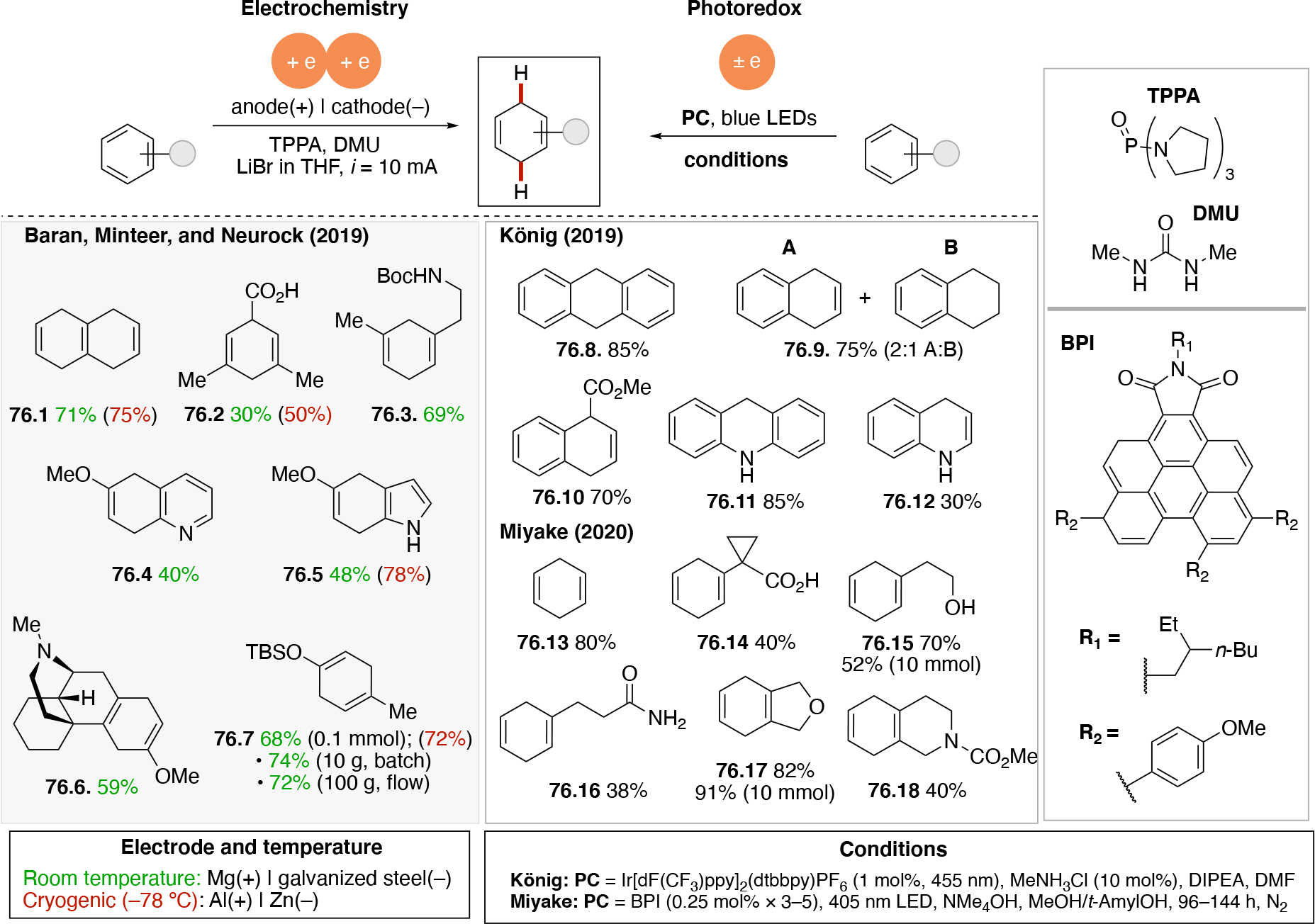
A comparison of electrochemical and photoredox methods for Birch reduction.
4.15. Aliphatic C–H Oxidation
Regioselective functionalization of alkyl C–H bonds is a fundamental goal of organic synthesis as it offers new disconnection patterns for introducing molecular complexity within aliphatic architectures. One approach is the development of redox-activatable catalysts to oxidize strong C–H bonds, with photoredox and electrochemical methods being key to generating high-energy catalytic intermediates (Scheme 77). In 1983, Masui reported870 the use of N-hydroxyphthalimide and superstoichiometric pyridine in acetonitrile for anodic oxidation of weak C–H bonds (e.g. benzylic and allylic) using NaClO4 electrolyte and glassy carbon electrodes under constant potential electrolysis. A range of substrates were demonstrated including the conversion of cyclohexene to cyclohex-2-en-1-one in 44% yield, with additional examples reported in a 1985 publication.871 In 2016, Baran and co-workers reported the electrochemical oxidation of allylic C–H bonds using a redox-active N-hydroxytetrachlorophthalimide (Cl4NHPI) under a constant current (10 mA•mol−1) in an undivided cell with pyridine, tert-butyl hydroperoxide, and acetone as the base, terminal oxidant and solvent respectively.872 This transformation is applicable to (a)cyclic alkenes, monoterpenes, diterpenes, triterpenes, sesquiterpenes and steroids, providing enones from allylic substrates. Deprotonation and subsequent electrochemical oxidation of Cl4NHPI generates a N-hydroxylphthalimide radical capable of generating a stable allylic radical via HAT. Trapping of this species by electrochemically generated tert-butyl hydroperoxyl radical leads to its collapse to the enone upon elimination of tert-butanol. Cathodic reduction of protonated pyridine liberates the base and H2 gas, effectively turning over the catalytic cycle. This transformation was also adapted to the process-scale (up to 100 g) allylic oxidation of steroids and terpenes using graphite electrodes. A follow-up manuscript by Baran and co-workers on the aerobic oxidation of unactivated C–H bonds used a redox-active quinuclidine as the HAT reagent alongside an RVC anode and a nickel foam cathode under galvanostatic conditions (25 mA•mol−1) in MeCN.232 Secondary C–H bonds are oxidized to the ketone, with high selectivities for activated 2° C–H bonds and significantly lower selectivities for electronically indistinguishable 2° C–H bonds. Tertiary C–H bonds are oxidized to the corresponding alcohols. The mechanism for this transformation is similar, with anodic oxidation of quinuclidine necessary to generate catalytically-active quinuclidine cation radical capable of HAT. More recently, Baran, Neurock and coworkers report N-ammonium ylides as a new class of tunable redox mediators for aliphatic C–H oxidation, with substrate-specific formation of the corresponding alcohol, carbonyl species, or mixture of both products. These ylides enable new or improved regioselective oxidations of several steroid, cyclohexane and decalin frameworks; for the most part the oxidations observed in this systems mirror established electrochemical methods or other chemical oxidants.873
Scheme 77.
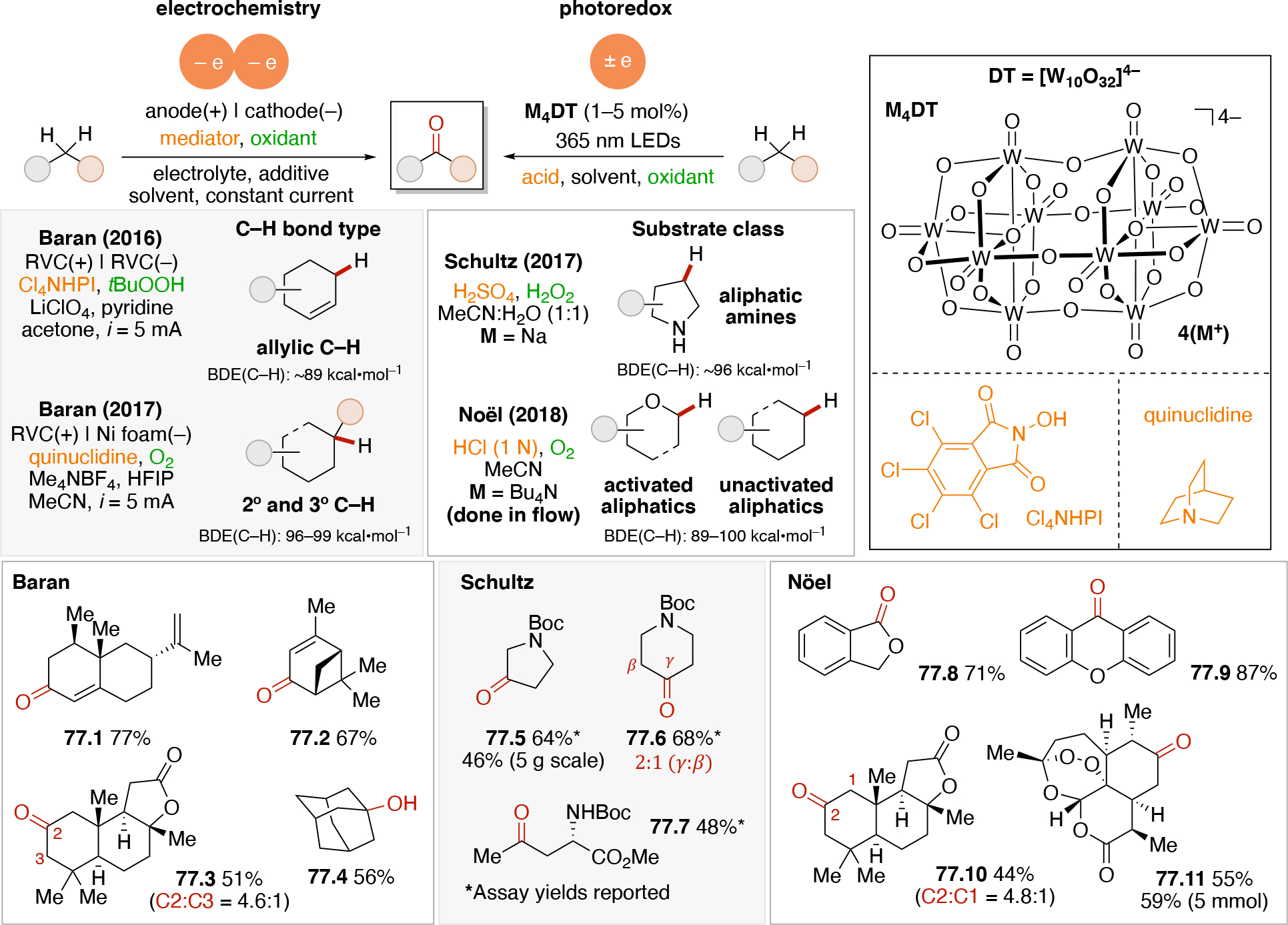
A comparison of electrochemical and photoredox methods for aliphatic C–H oxidation.
The photoredox counterpart for this transformation was independently reported by the Noël group874 and Schultz and co-workers at Merck & Co., Inc., Kenilworth, NJ, USA (Scheme 77).875 Both research groups use a decatungstate (DT) photooxidant (1 to 5 mol%, 365 nm LED irradiation) to initiate HAT at an electron-rich alkyl C–H bond with oxidative trapping of the resultant radical with either molecular oxygen or peroxide. Notably, Noël’s reactions were run under continuous flow conditions. Site-selectivity is substrate-specific for 2° C–H bonds in Noël’s work, with the oxidation occurring at the most sterically-accessible electron-rich C–H bonds for a range of substrates. Similar trends are observed in Schultz’s work with aliphatic amines; protonation of free amines deactivates its a and (in some cases) β positions such that oxidation is observed at either the β or γ sites δ. Mechanistic studies using in situ LED-NMR show the formation of 3-hydroperoxypyrrolidine from pyrrolidine, which Schultz and co-workers suggest is the intermediate that funnels to the ketone. Finally, both Schultz and Noël demonstrate the amenability of the method for scale up, with the former obtaining N-Boc pyrrolidin-3-one (77.5) in 46% yield on a 5 g scale and the latter obtaining artemisitone-9 (77.11) in 59% yield on a 5 mmol scale. The photocatalytic strategy can be adapted to a metal–organic framework wherein the integration of a copper photosensitizer and an iron-centered oxidation site enables the aerobic oxidation of benzylic C–H bonds to a ketone product.876 Efforts towards the merger of the two catalytic strategies are relatively limited, but a photo-electrochemical method from Sayama and coworkers enables the oxidation of cyclohexane to cyclohexanol and cyclohexanone (KA oil) under aerobic conditions using a WO3 photoanode and simulated solar light (10 mW•cm−2).877 The method provides with high partial oxidation selectivity (99%) and high current utilization ratio (76%) at room temperature and atmospheric pressure of air achieving photon to current efficiencies at 365 and 420 nm of 57% and 24%, respectively.
4.15.1. Benzylic Etherification
In contrast to C–H oxygenation, the etherification of benzylic C–H bonds typically proceeds via direct oxidation of the aromatic substructure, either through SET to generate a radical cation (photoredox), or two stepwise SET to form a benzyl carbocation (electrochemistry) (Scheme 78). Benzylic C–H bonds are heavily acidified upon single-electron oxidation878, thus the formation of a benzylic C-centered radical is a common intermediate between the two methods. Sharma and coworkers summarize recent progress in the photochemical and electrochemical C–H functionalization of benzylic C–H bonds, and readers are encouraged to refer to this reference for more information.879 To highlight the key divergences exist between these two systems, the outcomes of benzylic etherification are compared. Yoon and co-workers demonstrate that a photoredox-generated electron-rich benzylic radical undergoes further oxidation by a CuII species to form a benzylic carbocation that is then trapped by a partner alcohol to furnish the benzyl ether after facile deprotonation.880 The substrate scope is strictly limited to oxyarenes because the formation of a putative quinone methide intermediate is necessary to stabilize the benzylic carbocation. Despite this requirement, exquisite site-selectivity is afforded and trends with the predicted stability of the quinone methide intermediate; this enables highly selective functionalization of p-alkoxy-activated benzylic C–H bonds even when weaker benzylic C−H bonds are present. Additionally, the alcohol scope is impressive with a range of functionalities well-tolerated, notably serine, hexose and pentose derivatives, and terpene alcohols. In complex substrates with preexisting stereochemical bias, high diastereoselectivity of C–O bond formation is observed. Lei and co-workers report an electrochemical counterpart to benzylic etherification, in which direct arene electrolysis under mild basic conditions forms the requisite benzylic carbocation that is trapped by a partner alcohol881. Using a constant current (10 mA), indane is successfully coupled to a range of alcohols, albeit lacking the sensitive functionality tolerated in Yoon’s examples. This is likely due to potential overoxidation given that the highly oxidizing potentials are required. While this method can be applied to arenes with multiple alkyl substituents, it cannot tolerate oxyarenes, which presumably dearomatize and dimerize under direct anodic electrolysis.882
Scheme 78.
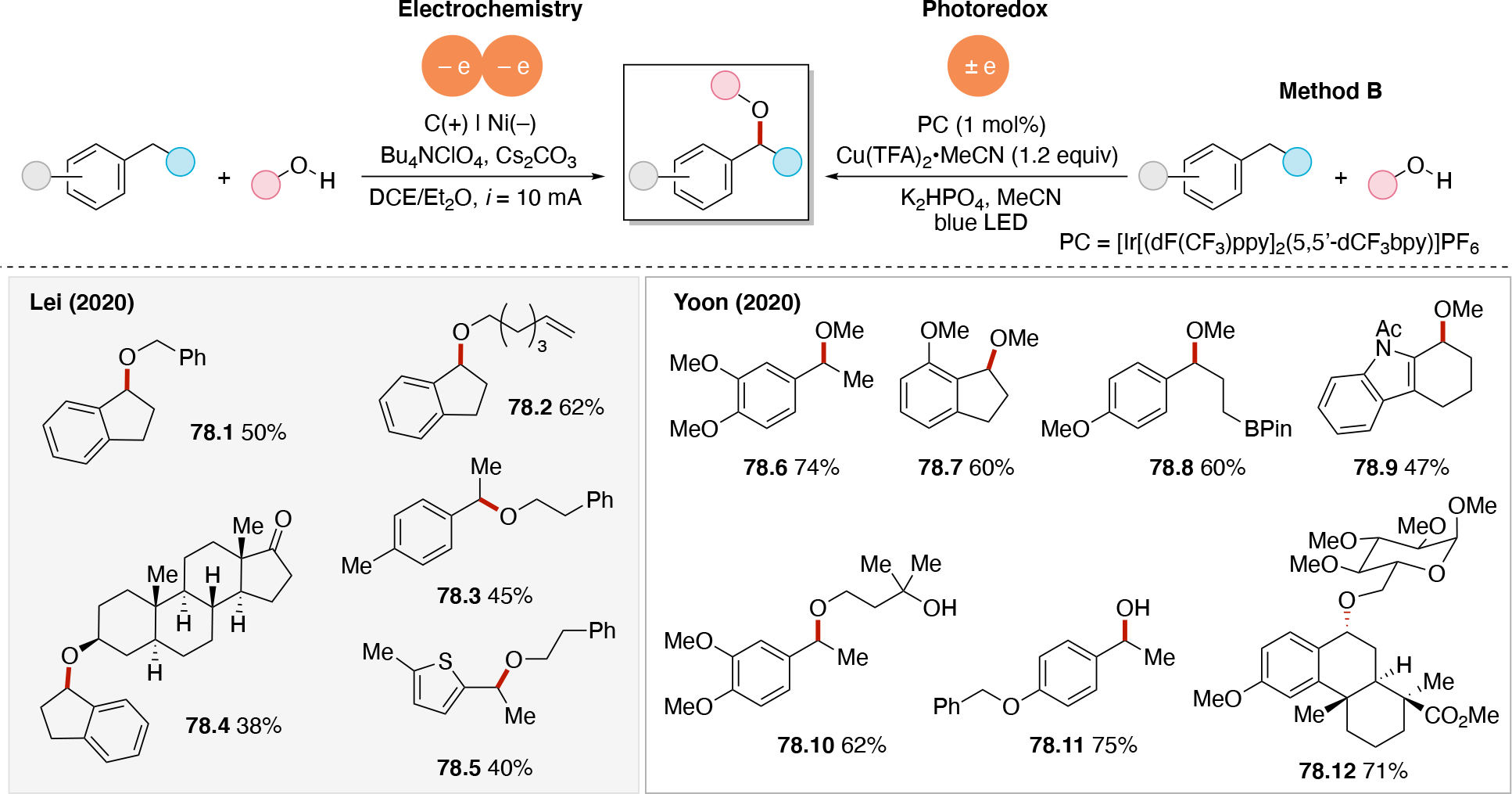
A comparison of electrochemical and photoredox methods for benzylic etherification.
4.15.2. Aliphatic C–H Fluorination
The direct fluorination of aliphatic C–H bonds is an important synthetic tool because hydrogen-to-fluorine substitution has the ability to impart desirable potency, conformation, metabolism and membrane permeability effects onto pharmaceutical targets834,856 and enable the synthesis of radiotracers using 18F for biological and disease mechanisms through PET imaging.883 Letcka and co-workers reported the use of UVB (280–315 nm) activated 1,2,4,5-tetracyanobenzene (79.10) to catalyze the fluorination of aliphatic884 and benzylic885 C–H bonds using Selectfluor as the electrophilic fluorine source. While mechanism data is only presented for benzylic systems, Letcka proposes that fluorination occurs via the formation of radical cations rather than HAT from a photoactivated intermediate. Britton and co-workers demonstrate the use of a HAT mechanism, where a UVA photoactivated DT abstracts a hydrogen atom from an electron-rich C–H bond; the carbon radical is then fluorinated through fluorine transfer from N-fluorobenzenesulfonimide (NFSI).886 Sorensen and co-workers present a visible-light alternative using a photoactivated uranyl cation operating by the same mechanism (HAT), albeit with abbreviated substrate tolerance.887 In the examples presented, simple and complex aliphatic C–H bonds are fluorinated with preference for 3° C–H bonds and sterically-accessible electron-rich C–H bonds in examples containing 2° C–H bonds. Britton and co-workers are able to leverage this selectivity using their DT system to enable the 18F-fluorination of 3° C–H bonds in branched aliphatic amino acids and peptides using [18F]NFSI as the radiofluorine source; radiolabeled amino acids were to image glioma xenografts through in vivo PET imaging.888,889 An electrochemical counterpart was reported by Baran and co-workers, in which unactivated 3° and 2° C–H bonds are selectively fluorinated using Selectfluor as the fluorine source and electrolyte, in addition to using sodium nitrate as the initial redox mediator under galvanostatic electrolysis (3 mA, 0.25–3.0 F•mol−1).890 Site-selectivities are similar to those obtained under photocatalytic conditions, and the di/tri-fluorination of unsubstituted adamantanes was observed. The transformation was amenable to large scale 3° C–H fluorination of a protected amino acid (100 g), which was isolated in 78% yield. Mechanistic studies suggest that the reaction proceeds by a radical mechanism due to the observation of > 100% current efficiency. This suggests that radical propagation is occurring, with the amine cation radical of defluorinated Selectfluor acting as an initiator.
More recently, radical-polar crossover methods in photoredox catalysis are opening new polarity-reversed disconnections for aliphatic C–H fluorination by enabling the formation and trapping of a carbocation by nucleophilic fluoride (Scheme 80). Independently-published methods from the Doyle891 and Musacchio892 groups demonstrate the mono- and difluorination of benzylic C–H bonds, with a photoredox-activated chemical oxidant generating a benzyl radical via HAT; single-electron reduction and generation of a radical precursor—tert-butyl peroxybenzoate to an O-centered radical for Musacchio and N–acyloxyphthalimide to methyl radical for Doyle. Subsequent oxidation of the radical by the photoredox catalyst furnishes the carbocation required for fluoride trapping. Substrate scope is broad for both methods, with yields ranging from low to excellent. Musacchio and coworkers demonstrate a sizeable scope for benzylic difluorination, with low to moderate yields obtained. Allylic and aliphatic C–H fluorination are more efficient using Doyle’s method, but yields currently remain low. Other heteroatom-centered nucleophiles are amenable to this transformation, as well as arenes, which follow Friedel-Crafts reactivity patterns.
Scheme 80.
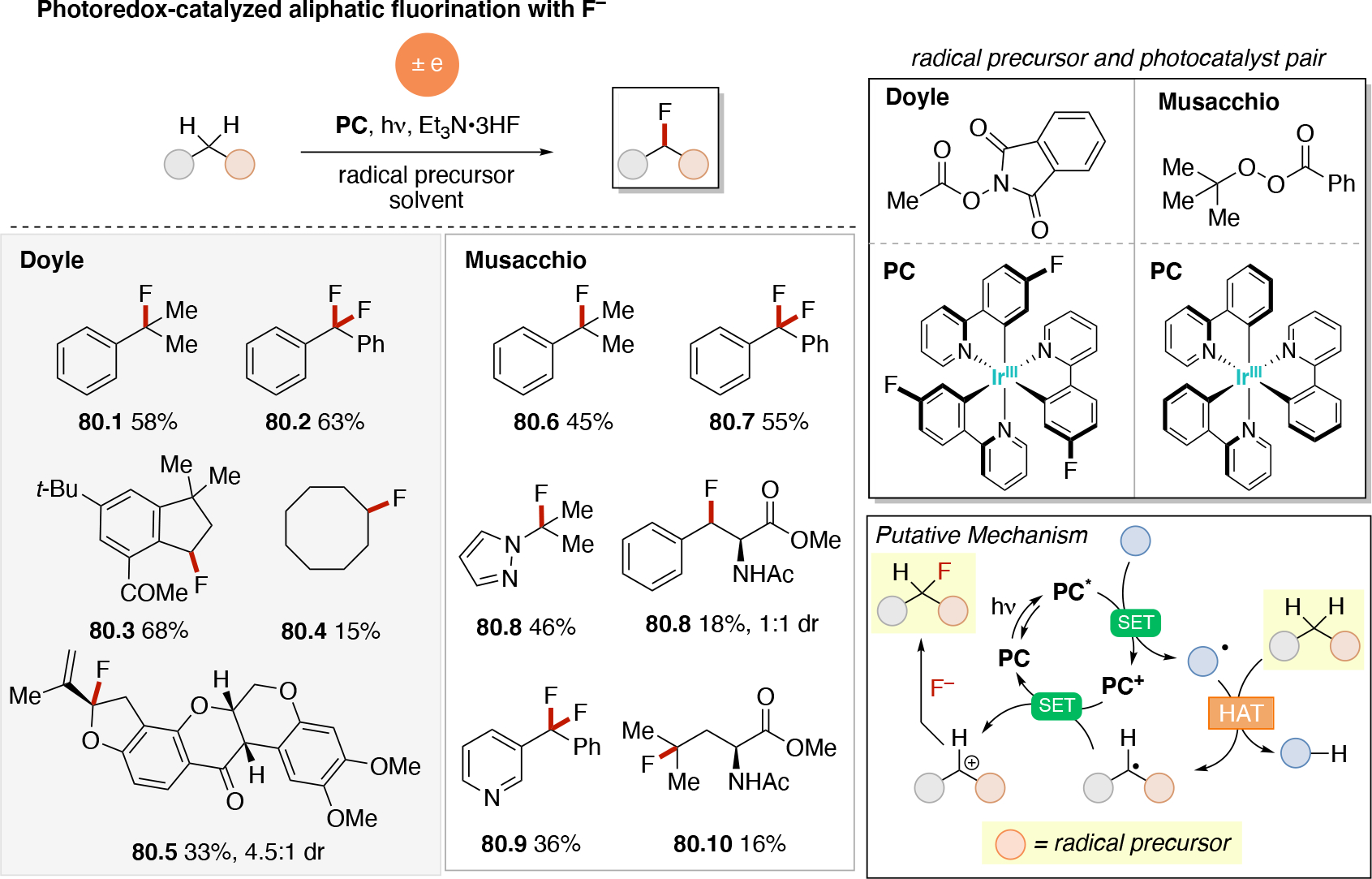
Representative examples of photoredox-catalyzed alkyl fluorination using nucleophilic fluoride.
4.15.3. Nitrogen-centered 1,5-Hydrogen Atom Transfer
Photochemical and electrochemical methods have been used to initiate the intramolecular 1,5-hydrogen atom transfer (1,5-HAT) mechanism to generate reactive C-centered radicals from alkyl C–H bonds to N-centered aminyl or amidyl radicals (Scheme 81). In 2016, Rovis893 and Knowles894 independently reported the generation of amidyl radicals via oxidation-initiated photoredox catalysis. Rovis and co-workers propose that their major reaction pathway is the direct oxidation of deprotonated alkyl trifluoroacetamide by a photoexcited iridium catalyst which reveals the trifluoroacetamidyl radical, a hypothesis supported with Stern-Volmer luminescence quenching studies. On the other hand, Knowles and co-workers propose that their system requires a concerted PCET mechanism in which oxidation of a N-aryl amide-phosphate, hydrogen-bonded complex by an excited state iridium photocatalyst furnishes the desired N-aryl amidyl radical; Stern-Volmer luminescence quenching and NMR studies support their hypothesis. The resultant C-centered radical then participates in Giese-type reactions with electron-deficient olefins, with closure of the catalytic cycle accomplished via two-step SET from reduced IrII and protonation of the resulting anion. Both the Rovis and Knowles studies describe broad substrate scopes with good functional group tolerance and show high selectivity for 1,5-HAT over 1,6-HAT. Rovis and co-workers use this regioselectivity preference to perform a stepwise, double C–H functionalization of an alkyl trifluoroacetamide, in which the 1,5-position relative to the N–H bond is alkylated before 1,6-alkylation. Knowles and co-workers show that multiple alkyl N-aryl amides and an alkyl sulfonamide are competent substrates, but observe significantly diminished yields when an alkyl tert-butyl carbamate is used. Additionally, Knowles and co-workers demonstrate intermolecular C–H activation, with alkylation of cyclohexane, tetrahydrofuran, and N-Boc pyrrolidine (at the 2-position for the latter two substrates) observed.
Scheme 81.
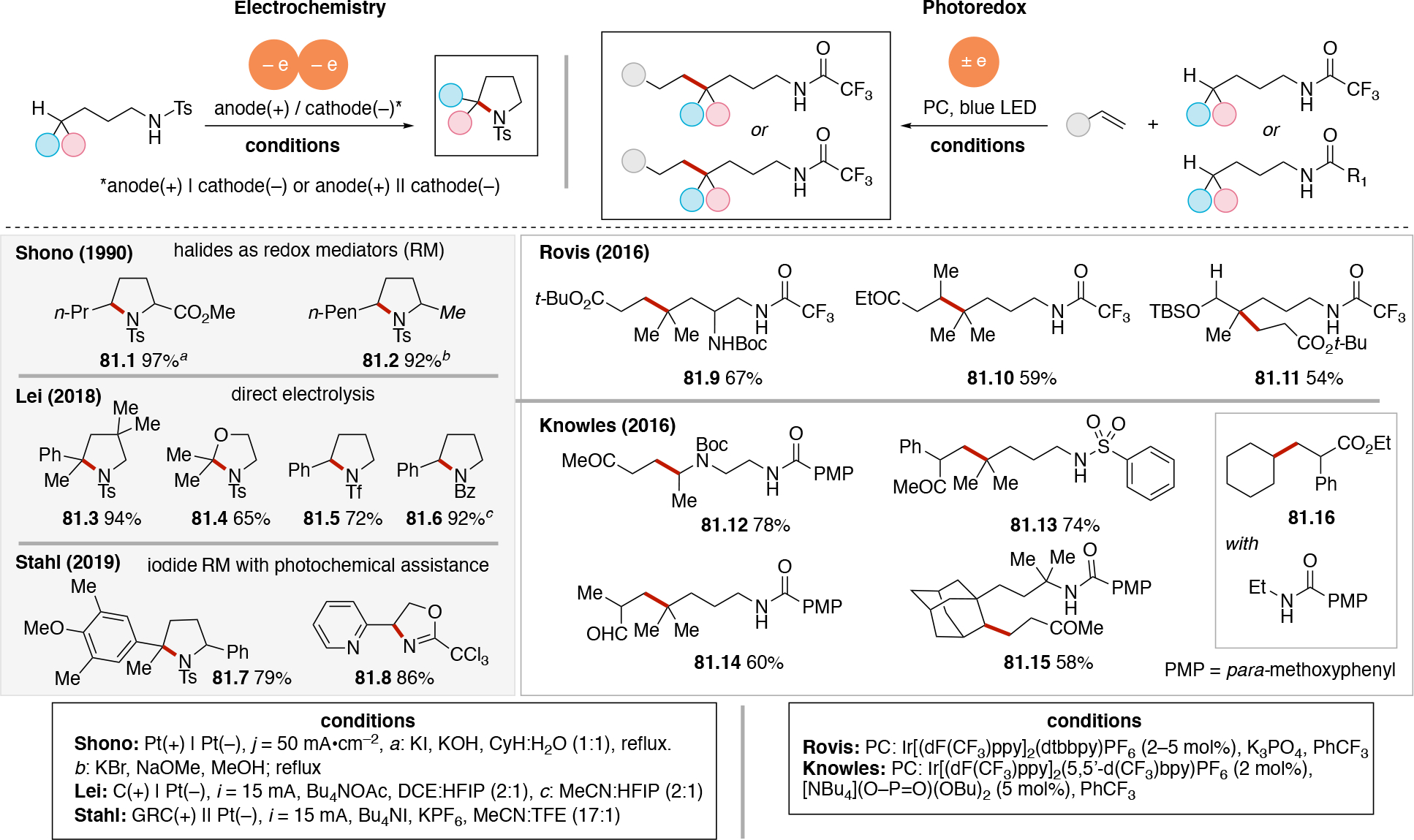
A comparison of electrochemical and photoredox methods for alkyl C–H functionalization via nitrogen-centered 1,5-hydrogen atom transfer.
The electrochemical approach to amidyl-based 1,5-HAT initiation proceeds via halogen-mediated formation of an N-halo intermediate or via a PCET mechanism initiated by base-promoted electrolysis. However, this strategy diverges from the photoredox examples as the synthetic pathways discussed are electrochemical alternatives to the Hoffmann-Loffler-Freytag (HLF) reaction. 895 Shono and co-workers report the synthesis of N-tosyl pyrrolidine that proceeds via bromide-mediated electrolysis.896 Constant current anodic oxidation of potassium bromide or iodide (j = 50 mA•cm−1) in either a refluxing water-cyclohexane mixture or in refluxing methanol provides the desired N-tosyl pyrrolidines selectively. The mechanism of this transformation proceeds via the formation of the N-halo tosylamine. Homolytic cleavage of the N–I bond reveals an N-centered radical, from which 1,5-HAT, C–I bond formation and subsequent ring closure furnish the N-tosyl pyrrolidine via a mechanism reminiscent of the HLF reaction. Alternatively, Lei and co-workers show that galvanostatic electrolysis can initiate an acetate-mediated PCET process to generate the requisite N-centered radical for the HLF reaction.897 While a stepwise deprotonation-oxidation mechanism may be operative if solvent-derived hexafluoroisopropoxide is formed, control studies suggest that the acetate-based PCET pathway is operative. Additionally, electrolysis serves an additional purpose — anodic oxidation of the resultant C-centered radical forms the requisite carbocation that leads to nucleophilic trapping of the tosylamine — due to the absence of halide. Lastly, a range of sulfonylamines and benzamides can be used in this transformation, thus highlighting its N-protecting group versatility. Stahl and co-workers show that the strategic use of photochemistry can facilitate the photochemical cleavage of weak nitrogen-halogen bonds and enable the use of iodide as a redox mediator.898 Iodide has a narrower redox window for mediator regeneration, which enables greater method compatibility for substrates containing electron-rich functional groups. Additionally, this method can also be used to generate N-centered imidate-based radicals for the synthesis of oxazolines and 1,2-amino alcohols.
4.15.4. Aminoxyl-mediated Alcohol Oxidation
The oxidation of alcohols to aldehydes, ketones and carboxylic acids is a key transformation in organic synthesis, and the convergence of photoredox catalysis and electrocatalysis on the use of organic aminoxyl species (Schemes 82–84).899 The low redox potentials required to oxidize aminoxyls to the catalytically-active oxoammonium (0.506 V. vs. SCE) leads to selective carbonyl formation at with low overpotentials900, thereby minimizing undesirable oxidative pathways and allowing a broader range of functional groups to be used concurrently alongside the alcohol of interest (Scheme 82A). There are two broad reactivity patterns for alcohol oxidation with oxoammoniums: under basic conditions, deprotonation and N–O adduct formation at the nitrogen first occurs before intramolecular hydride transfer yields the carbonyl and hydroxylamine products. Under acidic conditions, a bimolecular hydride transfer occurs instead (Scheme 82B). These two differing pathways have important implications for site-selectivity, as the oxidation selectivity between secondary and primary alcohols can be mediated with pH changes. Additionally, comproportionation-1e oxidation of the oxammonium and the hydroxylamine form of the electrocatalyst to afford two molecules of the nitroxyl via a base mediated process has also been postulated.901
Scheme 82.
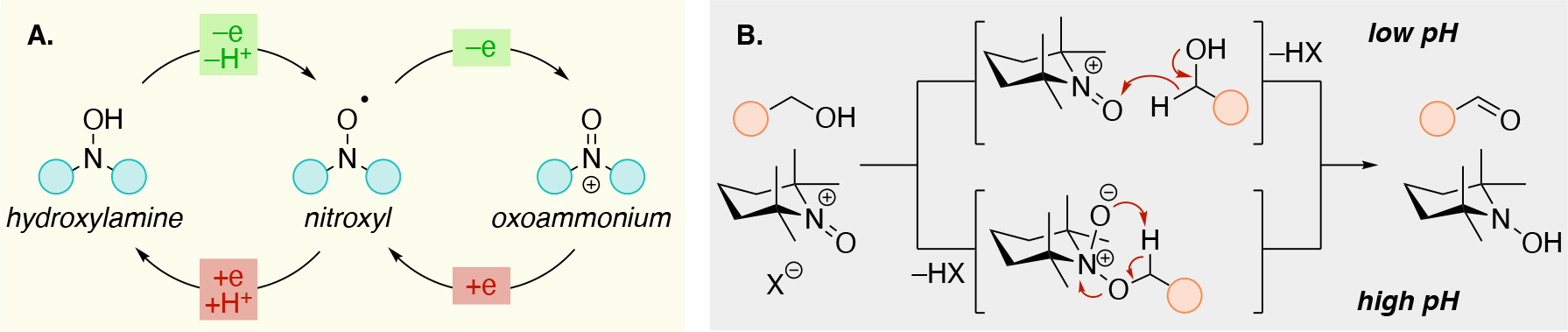
(A) The redox pathways between the hydroxylamine, nitroxyl and oxoammonium states of aminoxyl oxidants. (B) The pH-dependent divergence in mechanism for oxoammonium-based alcohol oxidation.
Scheme 84.
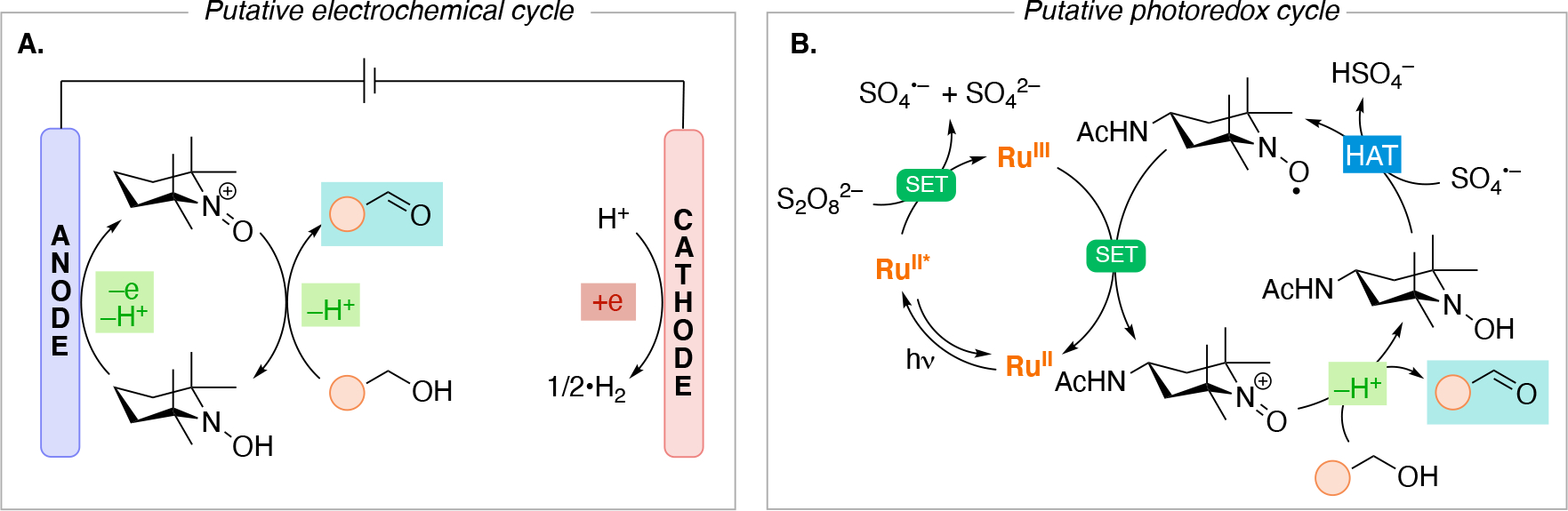
Representative putative mechanisms for the oxoammonium-based oxidation via (A) electrochemical and (B) photoredox pathways.
The first example of nitroxyl-mediated electrooxidation was reported by Semmelhack and coworkers, in which alcohols are selectively oxidized to aldehydes and ketones using catalytic amounts of TEMPO (Scheme 83).902 The direct conversion of aldehydes to nitriles can also be accomplished, with TEMPOnium-mediated oxidation of an imine intermediate leading to the desired product.903,904 Extension of this methodology to the synthesis of carboxylic acids was first achieved on biomass-type substrates under basic conditions from the parent alcohol905,906 (Anelli-type oxidation), but more recent advances by Stahl and coworkers using 4-acetamido-TEMPO (ACT), which upon electrooxidation forms the corresponding oxoammonium to facilitate Pinnick-type oxidation of aldehydes to the corresponding acid in addition to Anelli-type oxidations.907 Systematic evaluation of electrocatalytic alcohol oxidation with monocyclic and bicyclic nitroxyl radicals has also been explored, with mechanistic data demonstrating that the favorable oxidation potentials of cheap monocyclic nitroxyls (notably ACT) increase their electrocatalytic efficiency relative to more expensive bicyclic nitroxyls (Scheme 84A).908 This result highlights the difference in reactivity between chemically-mediated oxidation (e.g. bleach) in which catalytic activity for bicyclic nitroxyl appears to be superior to their monocyclic variants due to reduced steric hindrance around the active nitroxyl/oxoammonium group.909
Scheme 83.
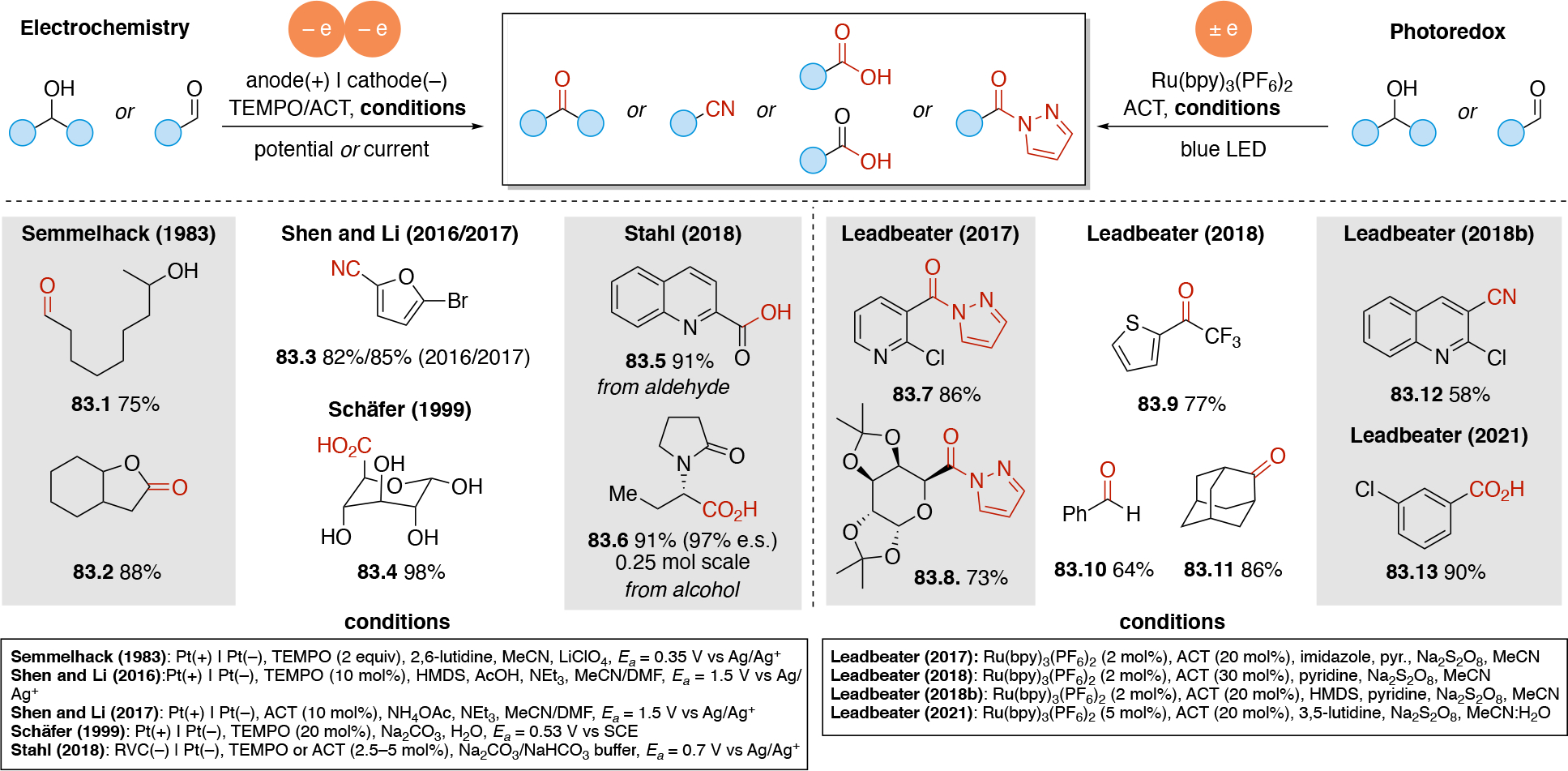
A comparison of photoredox and electrochemical methods for oxoammonium-based alcohol and aldehyde oxidation.
Photoredox catalysis can also be used to mediate the aminoxyl/oxoammonium cycle, albeit with a stoichiometric chemical oxidant (Schemes 83 and 84B). Leadbeater and coworkers report the synthesis of amides910, aldehydes and ketones911, nitriles912, and carboxylic acids913 from the parent alcohols and/or aldehydes with Ru(bpy)3(PF6)2 as the photoredox catalyst, ACT as the nitroxyl co-catalyst, and persulfate salts as the stoichiometric oxidant. All of the aforementioned synthetic methods employ a variant of this strategy and a general mechanism is as follows: the photoexcited Ru chromophore reduces persulfate to sulfate anion and sulfate radical anion, and forms a RuIII intermediate that oxidizes the parent nitroxyl to the key oxoammonium catalyst, hence closing the photoredox cycle. The resulting hydroxylamine can undergo HAT with the persulfate radical anion to regenerate ACT and close the nitroxyl redox cycle.
4.16. Hole Catalysis
4.16.1. SET-Triggered [4+2] and [2+2] Cycloadditions
Radical chain reactions can be initiated by light (photoinduced catalysis) or electrochemical means. Photoinduced catalysis refers to “photochemical reaction generates an active species that participates in thermal catalytic reactions. The quantum yield may exceed unity and the overall free energy of the catalyzed reaction must be negative.”914 Electrocatalytic reactions are chain processes where the electrochemical transformation only requires a catalytic amount of current, relative to the amount of starting material consumed. The chain mechanism in either the photoinduced catalysis or electrocatalytic reactions requires that the intermediate and/or product that is photochemically or electrochemically generated can activate the starting material, thus propagating the chain process. Several examples are shown below in SET-triggered cycloadditions.915
The Diels-Alder reaction, [4+2] cycloaddition, enables the construction of 6 membered rings from a diene and a dienophile alkene, with redox-triggered methods enabling C–C bond formation between species of differing ground state electronic compatibilities (Scheme 85). Chiba and coworkers reported916 an anodic SET-triggered cycloaddition variant of the classic Diels-Alder, providing an umpolung approach to electronically mismatched coupling partners. The olefin geometry on the anethole has a minor impact on the diastereoselective outcome of the reaction. trans-Styrene provides cleanly the trans product, whereas the cis-anethole results in 1: >10 cis/trans ratio. A photoinitiated method917 using TiO2 and 365 nm light provides comparable results and a slight broader scope under otherwise nearly identical solvent/electrolyte conditions, illustrating the similarities because the chemistry relies on “hole-catalysis” via a chain mechanism. The Ru-photocatalyzed variant disclosed by Yoon and coworkers disclosed enables the same transformation with a broader substrate scope (method B1).918,919,920
Scheme 85.
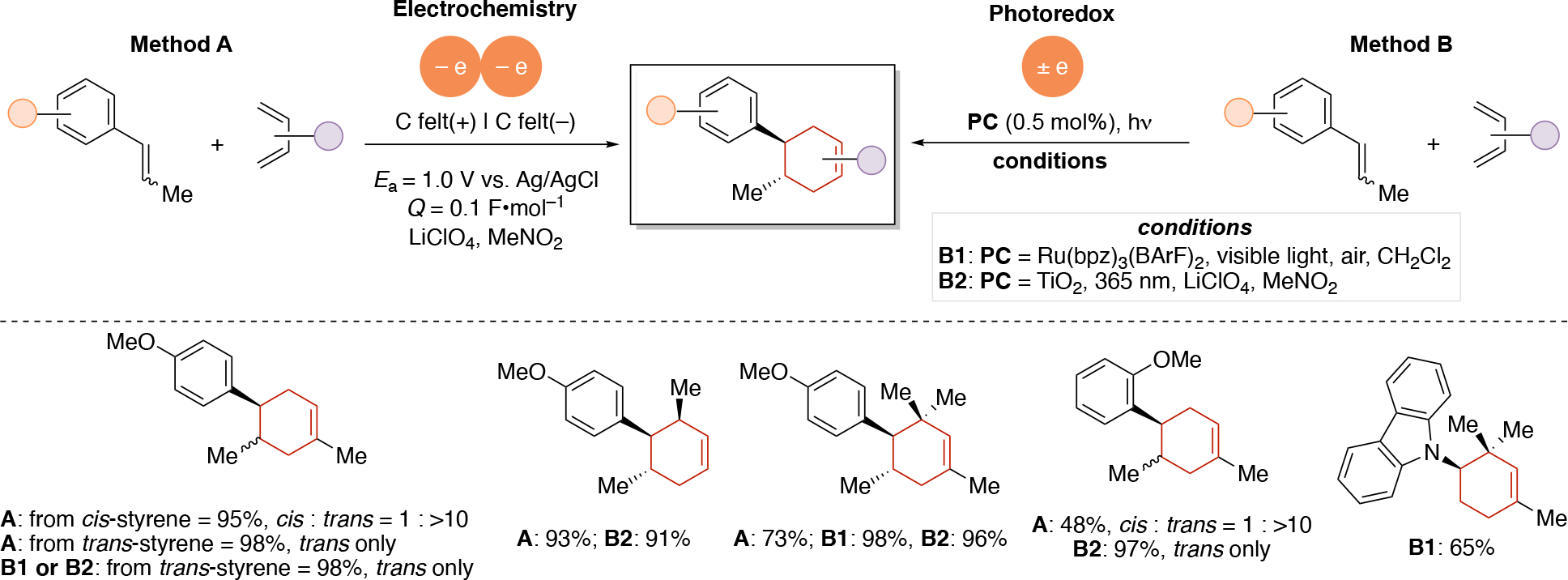
A comparison of photoredox and electrochemical methods for redox-triggered [4+2] cycloadditions.
Anion radical chain [2+2] cycloadditions of tethered enones were first reported by Krische and Bauld using chemical reductants921,922,923,924, and they later reported an electrochemical variant by Krische and Bauld in 2002 using electrolysis in a divided cell to provide mixtures of products, including observation of a [4+2]-cycloaddition side product (Scheme 86).925 This preliminary study serves as inspiration for the photoredox variant developed by Yoon and coworkers in 2008, where significantly improved [2+2] cycloaddition selectivity and diastereoselectivity are observed (relative to the electrochemical method).926
Scheme 86.

A comparison of photoredox and electrochemical methods for redox-triggered [2+2] cycloadditions.
4.16.2. Redox Triggered [3+2] Cycloadditions
Cyclopropylamines provide a synthetic handle for redox triggered intermolecular [3+2] cycloaddition with olefins by either electrochemical927 or photoredox methods.382 The oxidation of the lone pair, provides a radical exocyclic and alpha to the cyclopropyl ring, which enables for ring opening to relieve ring strain via formation of a distonic radical cation928 in which the primary carbon centered-radical can attack an olefin, and after cyclization, ultimately leads to the formation of a five membered ring. The radical cation 5-membered intermediate can either undergo back electron transfer from the reduced photocatalysts (redox neutral process) or alternatively can propagate a chain process via oxidation of the starting material cyclopropylamine. The proposed radical chain process was supported by the measurement of quantum yields (ϕ) > 1 (ϕ = 1.44 to 1.68).929 In the context of the electrochemical methods, the chain process enables for the use of catalytic amounts of total charge and indeed this was verified experimentally (0.34 equiv. of charge is required).927 Scheme 87 illustrates the ability to access functionalized cyclopentylamine derivatives using styrenes or acrylamide as olefin partners for intramolecular reaction with cyclopropylamines. The diastereoselectivities are typically low, but may still be useful for the synthetic needs of medicinal chemists. Products containing 5,5-fused rings can be obtained if the cyclopropylamine is a 2-azabicyclo[3.1.0]hexane (a 3,5-fused system). Booker-Milburn and Aggarwal recently reported930 a related diastereoselective photoredox-catalyzed [3+2] cycloaddition of N-sulfonyl cyclopropylamines with electron-deficient olefins to access trans-cyclopentanes bearing a sulfonamide.
Scheme 87.

Intermolecular [3+2] cycloaddition of cyclopropylamines with olefins.
4.16.3. Redox Triggered Newman-Kwart Rearrangement
The conversion of a phenol to a thiophenol can be accomplished in a multi-step manner via the Newman-Kwart rearrangement.931,932 Traditionally Newman-Kwart rearrangement leverages high temperature (200–300 °C) to overcome the high energy barrier required to convert an O-aryl thiocarbamate (88.1) to a S-aryl carbamate (88.2) via spirocyclic compound 88.3 (Scheme 88) which can lead to low yields due to undesired side-reactions. Recently, oxidatively triggered O- to S-rearrangement via intermediate 88.4 followed a reduction step to obtain the desired product can enable the redox neutral transformation at room temperature with high yields. An electrochemical approach using CCE, carbon anode in HFIP containing Bu4NClO4 was reported by Francke in 2018,280 who also subsequently reported a detailed mechanistic study.933 HFIP was critical to the success of the reaction, as other solvents (e.g. TFE, MeOH, CH3CN or CH2Cl2) give poor results. This can in part be understood due to HFIP’s high anodic stability and low pKa which enables it to participate in sacrificial proton reduction without interfering in oxidative processes.934 The adaptation of these batch conditions to electrolyte-free flow electrochemical conditions were successfully demonstrated with productivity of > 2 g•h−1. Noteworthy is the use of catalytic amounts of charge (Q = 0.3 F•mol−1) to obtain 95% yield of 4-methoxy-thiophenolcarbamate, supporting a radical chain process. The activation barriers, as calculated by DFT, do not sufficiently explain the observed substrate-specific reactivity outcomes for the electrochemical Newman-Kwart rearrangement, and the full understanding also requires consideration of the thermodynamically favorable equilibrium forming an off-cycle dimer (88.5). The hydrogen-bonding from HFIP to the key transition state 88.6 was also postulated to both decrease the activation barrier of the rearrangement and improve the thermodynamic driving force for the final reduction step to generate 88.2. Nicewicz reported the use of a 2,4,6-tri(p-tolyl)pyrylium tetrafluoroborate photocatalyzed method in 2015 for the Newman-Kwart rearrangement935 and a subsequent detailed mechanistic study.936 The use of chemical oxidant, namely cerium ammonium nitrate937 or persulfate938 has also been demonstrated to affect such rearrangements. A key difference between the electrochemical and photochemical Newman-Kwart rearrangement is related to the back-electron transfer to either 88.5 or 88.7, either events resulting in termination of the chain process. While turnover of the photocatalysts necessitates a reduction event from PC−, the electrochemical system does not suffer from this restriction. This is highlighted by the increased concentration resulting in lower efficiency only in the photochemical and not the electrochemical variant of the Newman-Kwart rearrangement.
Scheme 88.

Redox triggered Newman-Kwart rearrangement.
4.17. Polymer Chemistry
4.17.1. RAFT Polymerizations
Reversible addition–fragmentation chain transfer (RAFT) polymerizations are radical-based living polymerizations that operate via degenerative chain transfer processes involving xanthate, dithiocarbamate, dithioester, or trithiocarbonate-based chain transfer agents (CTA).939,940 Original developments of this strategy centered on the use of free radical initiators (e.g. azobisisobutyronitrile; AIBN) to initiate the RAFT process and while photoinitiated RAFT was initially demonstrated with thiocarbonylthio compounds941, a lack of end-group fidelity due to UV-light photolysis necessitated new developments in photocontrolled RAFT methodologies (Scheme 89). Boyer and co-workers reported a photoredox-controlled RAFT polymerization strategy, wherein PET via oxidative quenching of photoexcited fac-Ir(ppy)3 by thiocarbonylthio-capped species enables an off-cycle RAFT process. Upon the removal of light stimulus, concomitant reduction of the IrIV catalyst to IrIII and oxidation of the thiocarbonylthiolate group results in efficient end-capping of the growing polymer (a process now known as PET-RAFT).942 Stern-Volmer analysis confirmed that thiocarbonylthiolate compounds quench the excited state of fac-Ir(ppy)3, although with differing efficiencies. The dithiobenzoate 4-cyano-4(phenylcarbonothioylthio)pentanoic acid (89.1) is quenched very rapidly relative to trithiocarbonates 2-(n-butyltrithiocarbonate)-propionic acid (89.2) and benzylsulfanyl-thiocarbonylthiosulfanyl propionic acid (89.3), as well as xanthate 89.4. Initiators 89.1, 89.2, and 89.3 were used for PET-RAFT polymerization of conjugated monomers, with optimal monomer-initiator compatibility determined empirically. Most PET-RAFT polymerizations were accomplished using ≥ 5 ppm of photocatalyst (relative to monomer), and excellent control over polymer weight and dispersity (Ð < 1.21) was observed. Higher light intensities (4.8 W) led to faster polymer kinetics, but most polymerizations on smaller scale could be accomplished with a 1 W blue LED. More importantly, polymerization is solely controlled by light, with light on-off studies demonstrating that conversion only occurs during the irradiation phase. PET-RAFT also enables the living polymerization of high molecular weight polymers, as shown by the synthesis of poly(methylacrylate) with conversions (≥ 93%) with low dispersity943 (Ð = 1.08–1.09) were demonstrated for Mn ranging from 1.8 kDa to 2.18 MDa (with 1 ppm of photocatalyst).
Scheme 89.

Representative examples of PET-RAFT and putative mechanism.
While 89.1, 89.2, and 89.3 were ideal initiators and CTAs for conjugated monomers, they were less efficient in initiating the polymerization of unconjugated monomers (e.g. vinyl acetate). However, xanthate 89.4 enables PET-RAFT of unconjugated monomers, albeit with reduced monomer conversion (65–81%) and requiring slightly higher catalyst loadings (5–10 ppm). Nevertheless, good agreement between theoretical and experimental molecular weights is observed, and the polymers obtained have low dispersities (Ð < 1.4).
A feature of RAFT polymerization is the ability to synthesize block copolymers, thus chain-end fidelity was studied. Polymer chains subjected to PET-RAFT typically retained chain end fidelity for 5 cycles, with decreases starting at the 6th cycle. This observation enabled the application of PET-RAFT towards the controlled synthesis of ultrahigh molecular weight (Mn > 300 kDa) triblock copolymers using a range of conjugated polymer macroinitiators (Ð < 1.30). Another important advance of PET-RAFT is its efficacy in the presence of oxygen as oxygen-mediated deactivation of radical pathways is a problem associated with traditional radical polymerization methods. While an inhibitory period was observed (3–4 h), the polymerization of methyl acrylate (MA) and methyl methacrylate (MMA) in non-degassed reaction solution proceeded with very similar kinetics to the degassed solution. Additionally, no substantial differences between polymer dispersity was observed, thus highlighting robustness of PET-RAFT. These observations enabled the synthesis of di- and tri-block polymers in degassed solution. PET-RAFT has since grown rapidly and has been expanded to include a range of photocatalysts944,945,946,947 and applications.948,949,950
The electrochemical analog to PET-RAFT (namely, eRAFT) by comparison, is largely underdeveloped. Johannsmann and co-workers first demonstrated that using a trithiocarbonate CTA in ammonium persulfate-mediated polymerization of N-isopropylacrylamide improves the homogeneity of the resultant hydrogel, thus suggesting greater control over intermolecular crosslinking (Scheme 90).951 However, this preliminary study lacked thorough mechanistic investigations on the role of initiators in eRAFT. This inspired Matyjaszewski and co-workers to use electrochemically-mediated activated radical initiation to trigger eRAFT polymerization.952 A limitation of traditional RAFT polymerization is the irreversible nature of radical mediators, which necessitates their continuous initiation in order to sustain polymerization amid inevitable chain termination. Noting this, Matyjaszewski and co-workers reduced benzoyl peroxide (BPO) and diazonium salt 90.1 to initiate RAFT polymerization. While BPO activation enabled the potentiostatic polymerization of MMA, the process was inefficient due to the overlap in reduction potentials for BPO and CTA agent used (90.2). This led to the use of easily-reducible diazonium 90.1, which has minimal overlap with 90.2 (−0.1 V vs. SCE, ~ 1 V more positive than 90.2). Potentiostatic conditions proved problematic with 90.1 due to undesirable electrografting of aryl radicals on the electrode surface, thus a switch to galvanostatic conditions was adopted. This change enables the eRAFT with n-butyl acrylate (BA) at low applied currents (−50 to −200 μA), and provides controlled polymer growth with good conversion (58–80%) and low dispersities (Ð < 1.5). Lower applied current currents led to slower but more controlled polymer growth. Block co-polymers of acrylates were also synthesized using eRAFT, with molecular weights and dispersities similar to traditional RAFT polymerization, thus demonstrating the chain-end fidelity of polymers obtained via eRAFT. Attempts at bypassing the use of electrochemically-initiated radicals through the use of electrochemical mediator to activate CTA agents have been shown with mixed success. Matyjaszewski and co-workers identified meso-tetraphenylporphyrin as a potential mediator, with the polyacrylates obtained with low dispersities and good experimental molecular weight agreement with theoretical values. However, these polymerizations are slow and gradual decomposition of meso-tetraphenylporphyrin is observed.953 Yan and co-workers identified a coenzyme-mediated system, wherein electrochemically-switching between polymerization-active nicotinamide adenine dinucleotide (NADH) (90.3) and polymerization-inactive NAD+ (90.4) enables temporal control over eRAFT.954 Using CTA agent 90.5, the controlled growth of polyMMA (Ð = 1.07, ~1.2% divergence from Mn, theo) is obtained under potentiostatic conditions (Eapp = −0.65 V) with high monomer conversion observed after 6 hours. Intermittent electrochemical potential switching between −0.65 and −0.40 V showed polymer growth during the former whereas a stop in polymerization is observed when the lower reducing potential is applied. This eRAFT method can be applied to a range of conjugated and unconjugated monomers, with high monomer conversions, low dispersities (Ð ≤ 1.21) and good molecular weight agreement (< 23% disparity) for high molecular weight polymer (up to 42 kDa) observed. This method can also be used to synthesize amphiphilic co-block polymers, which form self-assembled bowl-shaped vesicles in water.
Scheme 90.

Representative examples of eRAFT and respective putative mechanisms.
4.17.2. Atom Transfer Radical Polymerization
Atom transfer radical polymerization (ATRP) is a synthetic strategy for the precise construction of well-defined architectures polymers through high levels of control over polymer molecular weights and molecular weight distribution (Ð < 1.5). Its success depends on maintaining a dynamic equilibrium between propagating radicals and dormant macromolecular alkyl halides through halide transfer between the organic species and a transition metal catalyst, with controlled polymer chain growth occurring when the concentration of active propagating species is kept low. In the case of copper-based catalysts, stochiometric reductants can be used to regenerate active CuIBr complexes from CuIIBr2 species via electron transfer processes, which enables external control over polymerization rates and minimizes homopolymerization of block copolymers955. Matyjaszewski and co-workers reported the first example of electrochemical ATRP (eATRP), a method that enables dynamically modulation of the equilibrium between CuIBr/Me6TREN (Me6TREN = tris[2-(dimethylamino)ethyl]amine) and CuIIBr2/Me6TREN speciation (Scheme 91).956 ATRP of methyl acrylate with an ethyl 2-bromopropionate initiator carried out with a constant potential (Ec) of −0.69 V vs. Ag+/Ag — the E1/2 for the Cu catalyst couple — in a divided cell shows approximately 80% conversion within 2 h, with linear, first-order kinetic behavior and excellent correlation between experimental and theoretical weight-average molecular weights (MW,exp and MW,theo respectively) observed. Additionally, constant decreases in Ð (Ðmin = 1.06) and a narrow, monomodal distribution of polymeric species were noted with increasing monomer conversion — features indicative of a living polymerization. The electrochemical potential is then used as a dynamic polymerization “on-off” switch via repetitive stepping of Ec between −0.69 and −0.40 V; at the former potential, CuI speciation is favored leading to polymerization whereas in the latter situation, the CuII state predominates, resulting in polymerization deactivation. Throughout the on-off cycling process, strong correlation between MW,exp and MW,theo, low Ð values, and the consistent growth of a high molecular weight polymer were observed, thus confirming the dynamic nature of eATRP. Matyjaszewski and co-workers also successfully applied this strategy to the controlled polymerization of the acidic monomer methacrylic acid (MAA) under acid, aqueous conditions, which is a historical limitation of ATRP957. The inhibitive irreversible chain-end PMAA cyclization involving a carboxylate and a tertiary alkyl bromide was initially observed, but three changes — using Cl as chain-end halogen, lowering the pH to 0.9, and using an applied potential of −0.18 V for rate acceleration — enables the controlled synthesis of PMAA with high monomer conversion and low Ð. Electrochemical cycling was also performed by cycling Eapp between −0.2 and 0.8 V vs SCE; negligible conversion was observed for the off-period whereas steady growth of a high MW polymer during the on-period was detected. Additional details regarding the growth and application of eATRP are summarized by Matyjaszewski and co-workers in a recent review.958
Scheme 91.

Representative examples of eATRP and putative mechanism.
While eATRP relies on redox modulation of CuI/CuII speciation, initial attempts at photoredox-controlled ATRP used transition-metal based photocatalysts that operate via reductive959 or oxidative960,961 quenching of the excited state chromophore (Scheme 92). These methods pioneered the use of light as a tool for controlling living radical polymerizations, and the work from Hawker and co-workers being especially notable for producing polymers with low dispersities (Ð < 1.50) and with good control over its molecular weight. However, recent methodologies have shifted towards the use of organic photocatalysts to initiate and sustain polymerization (O-ATRP). Fors, Hawker and co-workers report the metal-free, light-mediated polymerization of methyl methacrylate using the chromophore PTH (11.1) and the ATRP initiator ethyl α-bromophenylacetate (EBPA) with good agreement between MW,exp and MW,theo observed and low Ð values (Ð < 1.35) obtained.962 The O-ATRP process only occurs upon irradiation as light “on-off” studies show no conversion during the dark phases. More importantly, linear increases in MW vs. conversion are observed despite multiple light “on-off” cycles, and chain-end fidelity is maintained when the light stimulus is removed. Success of this method depends on the highly reducing nature of excited state PTH (Ered = −2.10 V vs. SCE) and less oxidizing PTH cation radical (PTH•+, Eox = 0.68 V vs. SCE) as using fac-Ir(ppy)3 — a transition-metal photocatalyst with lower reducing ability (Ered = −1.73 V vs. SCE) and a higher IrIV/IrIII oxidation potential (Eox = 0.77 V vs. SCE) — results in uncontrolled polymerization (Ð = 3.69) of dimethylaminoethyl methacrylate due to side reactions arising from competitive amine oxidation. Lastly, the resultant homopolymer from O-ATRP can be used to form multiple block copolymers with acrylates or styrene in high molecular weight (MW > 20 kDa) with good dispersity (Ð = 1.06 – 1.31). Miyake and co-workers improve on this methodology by developing diaryl dihydrophenazine (DDP) photoreductants (e.g. 92.1) that enables the use of visible light for initiating O-ATRP, with Ð improvements observed for polymerizations of similar scale, with a slight loss in polymerization control (Ð = 1.54) occurring during the synthesis of a high MW polymer (Mn = 85.5 kDa).963 Computational analysis suggests that efficient reduction from excited state DDP species requires SOMO electron localization on one aryl species rather on both rings. This prediction is confirmed by the development of 2-naphthyl and 1-naphthyl DDP species — photoreductants with spatially separated excited-state SOMOs — that enable the synthesis of PMMA with dispersities that rival metal-based ATRP catalysts (Ð = 1.03–1.08). Further catalyst development from the Miyake led to the development of tailored phenoxazine species (e.g. 11.12)964 capable of excited-state reduction for use in organic ATRP965 as well as for synthetic applications.567
Scheme 92.

Representative examples of PET-ATRP and O-ATRP, with the putative mechanism provided.
4.17.3. Living-Type Cationic Polymerizations.
Cationic polymerization is a widely-used strategy for chain growth polymerization by which an initiator generates a cationic charge on a monomer, which then transfers the charge to another monomer in a chainwise fashion to form a polymer (chain growth). While initially developed using discrete Brønsted and Lewis acids, electrochemical966,967 and photochemical968,969,970,971,972 methods have been used to initiate cationic polymerization, with recent photoinitiated strategies involving activated olefins demonstrate the characteristics of living-type polymerizations (Scheme 93).973,974 However, stimuli-mediated control over cationic polymerization was not demonstrated until a report by Fors and coworkers involving a dithiocarbamate CTA (93.1) and an oxidizing 2,4,6-p-methoxyphenylpyrylium tetrafluoroborate (93.2) photocatalyst.975 This photoredox process enables the high conversion, living-type polymerization of isobutyl vinyl ether (IBVE), with low dispersities (Ð = 1.19 to 1.37) and good agreement between the experimental and theoretical number-average molecular weights (Mn,exp and Mn,theo respectively). Temporal control was obtained using light on-off studies, with high conversions only observed in the light-on periods, and initial polymerization rates were found to be linearly dependent on light intensity. Multiple vinyl ether monomers were polymerized with good Mn,exp and Mn,theo agreement and low Ð values, and chain-end fidelity was demonstrated via the successful formation of a block copolymer of ethyl vinyl ether and IBVE with narrow dispersity. The putative mechanism is as follows: PET from the CTA to the photoexcited 93.2 generates a dithiocarbamate radical and the polymerization-initiating oxocarbenium ion (Scheme 94A). During the light-off phase, the photoredox cycle closes with electron transfer from reduced photocatalyst to dithiocarbamate radical, which forms the anion necessary to cap polymer chain growth. Further mechanistic insights into this system revealed the importance of photocatalyst and CTA to successful photocontrolled living-type cationic polymerization, as respective photoinitiation and uncontrolled polymerization are observed with either the substitution or exclusion of CTA 93.1 and pyrylium 93.2.976
Scheme 93.
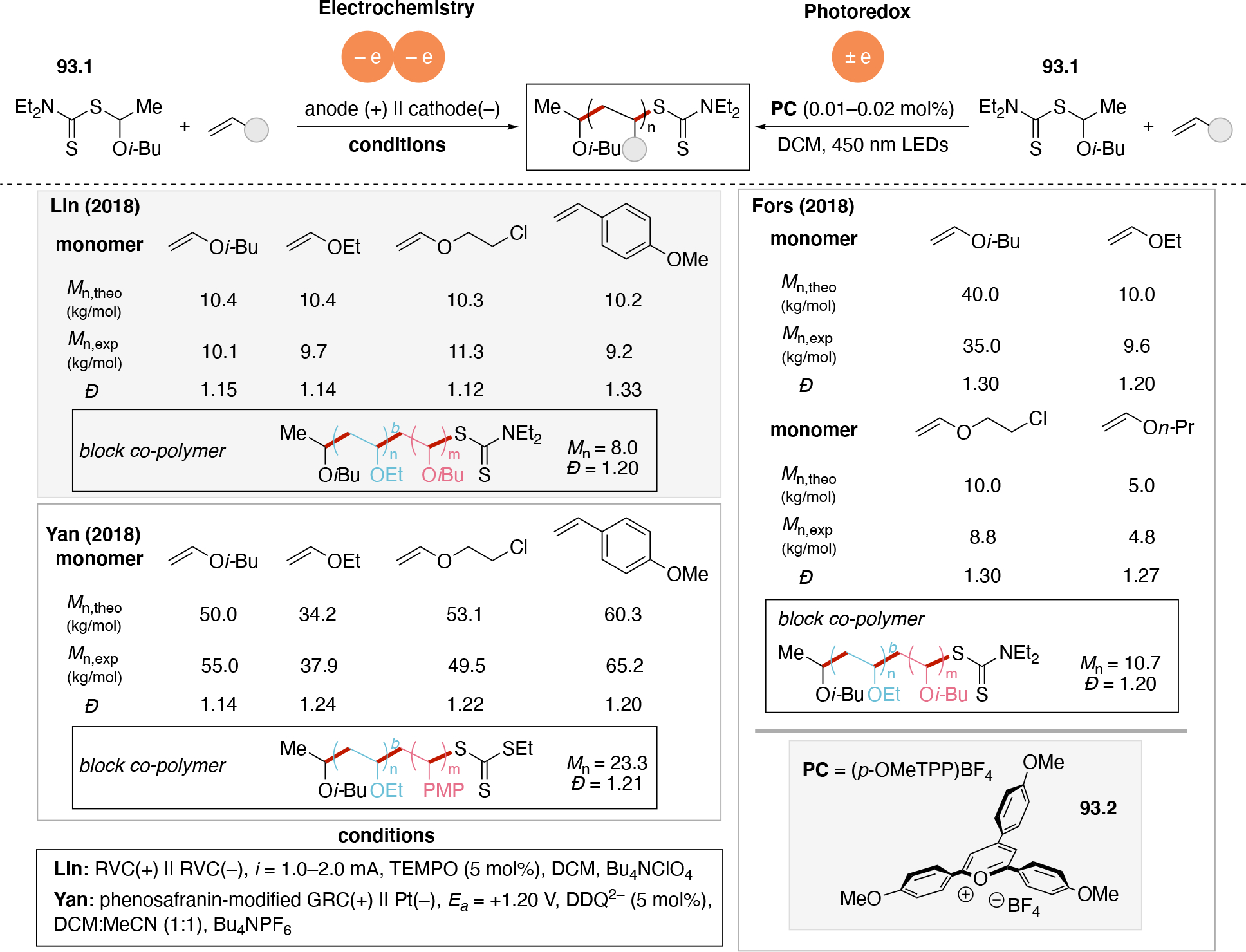
A comparison of electrochemical and photoredox methods for cationic polymerization.
Scheme 94.
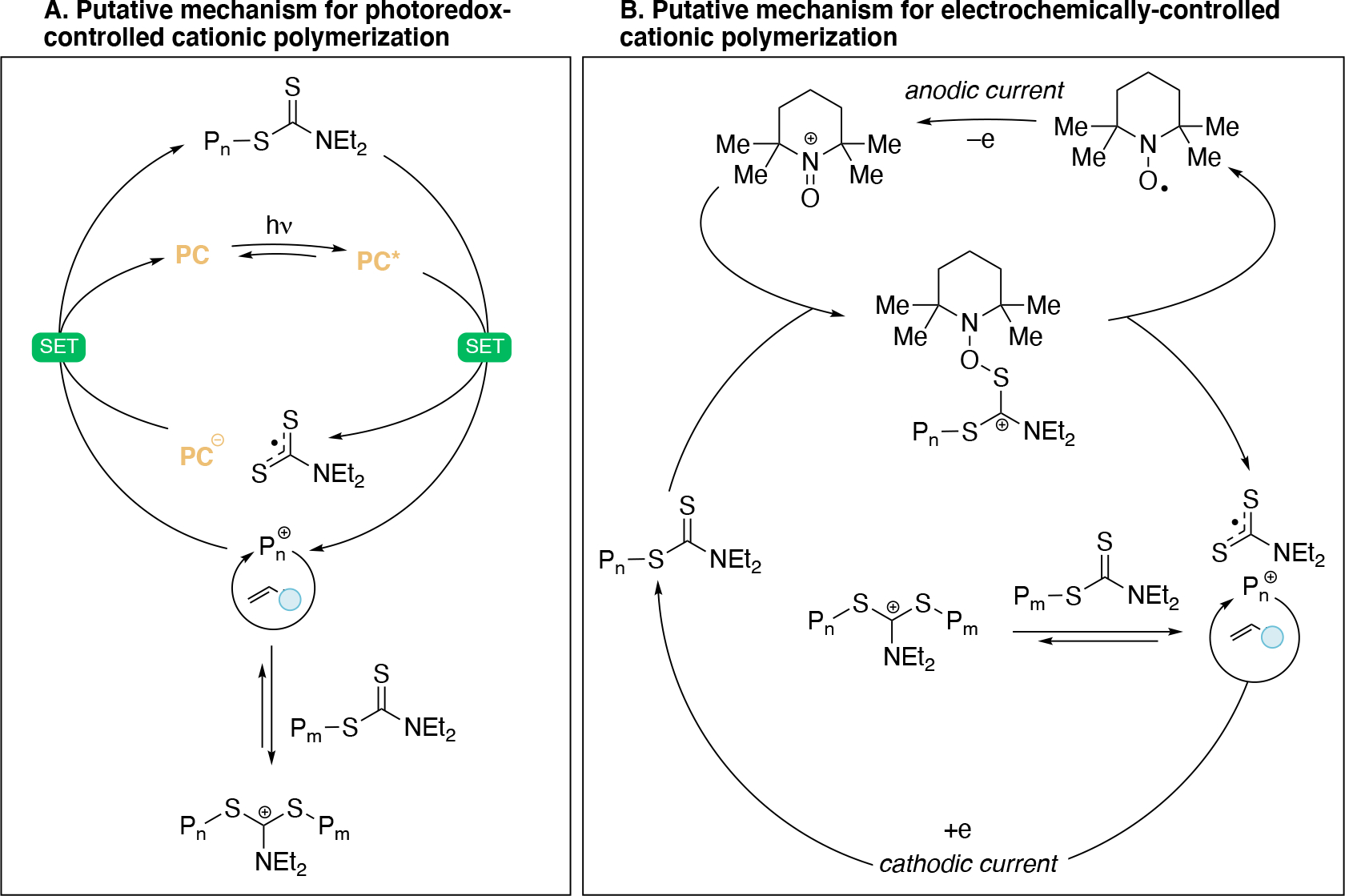
A comparison of putative mechanisms for (A) photoredox and (B) electrochemical cationic polymerizations.
The electrochemically-controlled polymerization of vinyl ethers was accomplished by Lin, Fors, and coworkers, in which electrooxidation mediates the activation of dithiocarbamate CTA responsible for initiating vinyl ether polymerization.977 Under constant potential conditions (divided cell; Ea = 325 mV vs. Fc+/Fc), the cationic polymerization of IBVE was observed, but bimodal molecular weight distributions and broad Ð values persist. This uncontrolled polymerization was attributed to unproductive polymer plating at the electrodes, thus a redox mediator TEMPO was used to control homogenous CTA electrooxidation. Using these conditions, a polymer with low Ð and matching Mn,exp and Mn,theo values was obtained; this latter observation indicates a controlled cationic polymerization is operative. Furthermore, polymers with lower Ð values and higher conversions were obtained when run under galvanostatic conditions (1 mA) relative to the potential of the TEMPO redox couple. Temporal control over polymer growth was obtained by using an alternating sequence of an oxidizing current (1 mA) followed by a reducing potential (Ec = −875 mV vs Fc/Fc+); high chain growth was only observed during the oxidizing current phase, with minimal to no background growth observed when the reducing potential was applied. Various vinyl ether and styrenyl monomers were polymerized with good Mn,exp and Mn,theo agreement and low Ð values, and two distinct vinyl ether monomers were used to form a diblock copolymer with narrow dispersity. The putative mechanism is as follows: electrochemical oxidation of TEMPO leads to an oxoammonium that traps the CTA and forms a polymerization-initiating oxocarbenium ion, a dithiocarbamate radical, and TEMPO after fragmentation (Scheme 94B). Applying a reducing potential then reduces the dithiocarbamate radical, which in turn caps the propagating polymer chain. A similar strategy was independently reported by Yan and co-workers, where instead of TEMPO the 2e cycling between 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and its dianion (DDQ2−) was used to mediate the electrochemically-controlled, living-type, cationic polymerization of vinyl ethers and styrenes.978 Cathodic oxidation of DDQ2− to DDQ under a constant potential (Ea = +1.20 V) initiates SET from the CTA-capped polymer and subsequent cationic RAFT polymerization. The resultant semiquinone DDQ radical anion can also participate in SET with the CTA-capped polymer to initiate the cationic polymerization process. High monomer conversions (> 90%) to polymers with low dispersities (Ð > 1.25) and good Mn,exp and Mn,theo agreement were observed, and the synthesis of a vinyl ether–styrene co-block polymer was demonstrated.
4.17.4. Activating Olefin Metathesis
Olefin metathesis (OM) is a fundamental organic transformation with broad application across scientific disciplines. Advancements in catalyst design and rigorous mechanistic studies have resulted in the development of efficient catalysts — Mo and W imido alkylidenes979 and Ru-NHC complexes980 — for a broad range of substrates. However, because alkenes and OM-catalysts are redox-active species, substrate SET via photoredox- or electrochemically-induced oxidation can initiate OM (Scheme 95).
Scheme 95.

A comparison of electrochemical and photoredox methods for substrate-activated olefin metathesis.
4.17.4.1. Substrate Activation
The electrochemical oxidation of electron-rich enol ether species and application towards intermolecular OM was shown by Chiba and coworkers, in which 4-methoxybut-3-enylbenzene (95.1) was anodically oxidized via potentiostatic electrolysis (1.0 F•mol−1) and coupled to 1-hexene (95.2) to yield 3-butenylbenzene (95.3) in 78% yield.981 Alkyl enol ethers were also tolerated as 1-methoxy-tridec-1-ene (95.4) was converted to tridec-1-ene (95.5) easily (76% yield). The mechanism of this transformation proceeds by the oxidation of the enol ether, followed by intermolecular C–C bond formation with a partner alkene to generate the less sterically-hindered four-membered radical cation ring, which fragments to yield the metathesis product and a new enol ether cation radical — this intermediate can initiate another metathesis event by intermolecular ET from the parent enol ether (Scheme 95A). This concept was applied to OM-polymerization by Boydston and coworkers, in which photoredox-catalyzed oxidation of various enol ethers were used to initiate the ring-opening metathesis polymerization (ROMP) of norbornene.982 Polymerizations in DCM initiated by methyl vinyl ether oxidation demonstrated dispersity (Ð) values between 1.3 and 1.7 while also showing a consistent correlation between the Mn,exp and Mn,theo values respectively, with the former being higher than expected for observed monomer/initiator ratios and % conversions. A gradual increase in Mn was observed with increasing monomer conversion, which is suggestive of a chain growth ROMP mechanism, however the lack of precise linearity suggests that this process is not a controlled living ROMP that is characteristic of fast-initiating Ru-based metathesis catalysts (Scheme 95B). This strategy was also applied to the ROMP of endo-dicyclopentadiene (DCPD) to form linear polydicyclopentadiene (polyPCPD).983 This transformation is unattainable with metal-based catalysts due to their propensity to activate the cyclopentane towards polymer cross-linking. While monomer conversion was low (endo-DCPD: 20%, Mn = 5.1 kDa; exo-DCPD: 20%, Mn = 10.8 kDa), the polymer was obtained with low dispersity (Ð = 1.5 and 1.4 respectively). and high purity. Mechanism studies involving endo- and exo-DCPD and their dihydro-analogs suggest that the low conversion observed for DCPD arises from unproductive quenching pathways of the alkene rather than steric impedance.
4.17.4.2. Catalyst Activation
The electrochemical activation of molybdenum and tungsten chlorides towards OM was explored by Petit and coworkers, in which they demonstrated that the reduction of WCl6 in the presence of DCM generates the requisite metallocarbene initiator.984,985 The electrolysis is performed in an undivided cell (Pt anode; Al cathode) under a constant potential (Ec = −0.9 V vs. SCE), with the generation of reduced WCl6− crucial for the formation of Cl4W=CH2 (96.1); supporting electrolyte was deleterious towards reactivity. Cross metathesis involving equimolar concentration of 1- and 2- pentene proceeded with 93% selectivity and a 70.5 h−1 turnover rate for the alkene monomers, providing propene and 3-heptene in highest yield (15.7% for both) for a distribution of OM products. The catalyst was also sufficiently stable to undergo two additional reloadings of the olefin, albeit with lower 2-pentene conversion. MoCl5 catalysts were also studied but demonstrated lower turnover rates and selectivity. More recently, Bielawski and co-workers report a method for potentiostatic control over Ru-based olefin metathesis with a modified Hoveyda–Grubbs catalyst containing a redox-active quinone built into NHC ligand (96.2).986 Electrochemical reduction of the quinone to its radical anion deactivates the catalyst towards olefin metathesis by stabilizing the ruthenacyclobutane resting state of the catalytic cycle and thus suppressing retro-[2+2] cycloaddition step. This strategy was applied to the ring closing metathesis (96.3) and ROMP (96.4), with approximately a seven-fold decrease in relative reaction rates upon single electron reduction
A more general method for catalyst oxidation-initiated OM was demonstrated using photoredox catalysis, wherein bis-NHC-Ru complex (97.1) and 2,4,6-triphenylpyrylium tetrafluoroborate (11.9) were used to spatiotemporally control olefin metathesis (Scheme 97).987 Coordinatively saturated 97.1 is inactive, but oxidation by photoexcited 11.9 induces the dissociation of a carbene cation radical, thus forming a catalytically-active, 14-e Ru species (97.2). Ring closing and cross metathesis reactions proceeded smoothly (46–90% yields), and ROMP was demonstrated for cyclooctadiene, norbonadiene, dicyclopentadiene, and a range of norbornene derivatives. Light on-off studies with diethyl diallylmalonate show maximal reactivity only in the presence of light, whereas the dark phase only provided minimal yields (0–3%). Additionally, the macroscopic patterning of poly(dicyclopentadiene) and microscopic patterning of poly(norbornadiene) were accomplished with photomasks, thus demonstrating that photoredox catalysis enables high spatiotemporal control over Ru-controlled OM.
Scheme 97.
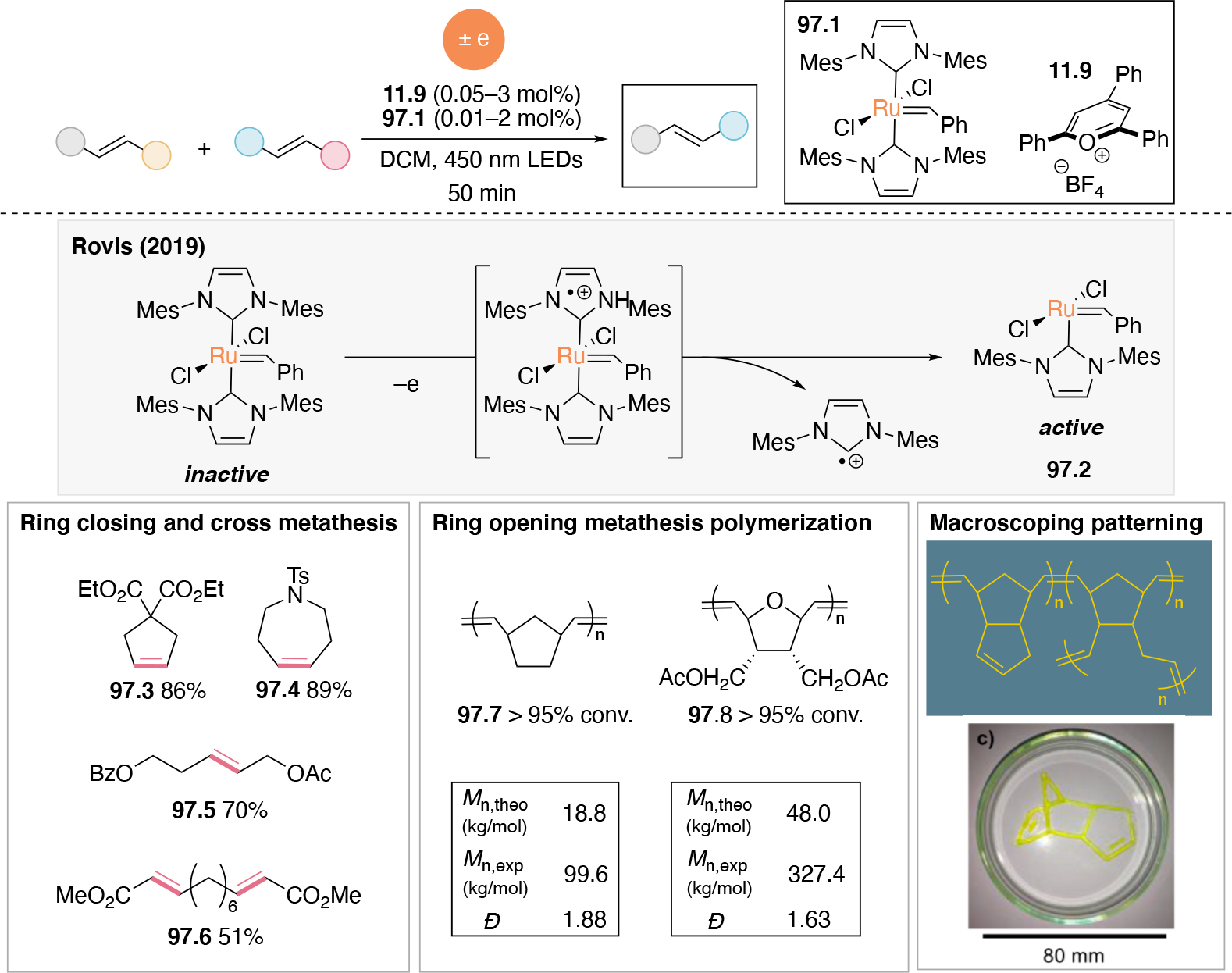
A photoredox method for catalyst-activated olefin metathesis. Macroscopic patterning picture reproduced from ref 987. Copyright 2019 American Chemical Society.
4.17.5. Lignin Degradation.
Lignin is a key component of lignocellulosic biomass (20–35% by weight; > 40% by energy content) and is the only biopolymer to have a high percentage of aromatic monomers within its structure.988 Thus, lignin depolymerization offers an opportunity to harness renewable sources of organic feedstocks. An efficient method for converting biomass to aromatic commodity chemicals involves depolymerization of the β-O-4 linkage in lignin (Scheme 98). The two major strategies in Scheme 98 show that controlling site-selectivity of alcohol oxidation within the β-O-4 linkage to the corresponding ketone or carboxylic acid is key as it enables access to differing monomeric fragmentation outcomes.
Scheme 98.
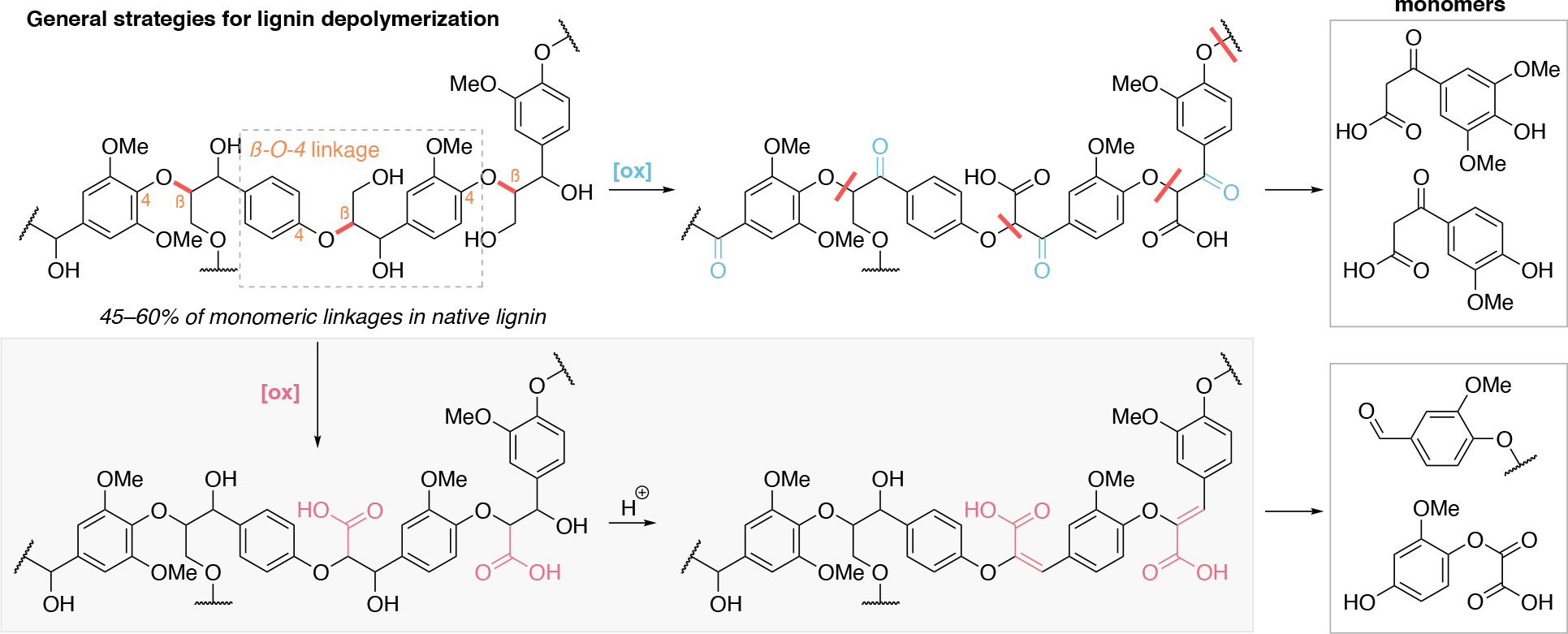
General oxidative strategies for lignin depolymerization.
Photochemical approaches towards this goal focus on exploiting the β-O-4 cleavage of oxidized lignin and its corresponding model systems (Scheme 99). This transformation proceeds first by oxidation of 1-phenylethanol groups in its backbone to the 1-phenylethanone moiety through stoichiometric aminoxyl oxidants989, catalytic palladium990,991,992, or electrochemistry.993 This carbonyl is crucial for reactivity as proton-assisted single electron reduction forms an α-hydroxyl radical intermediate which readily fragments to the corresponding ketone and phenol after H-atom transfer. Yields for β-O-4 cleavage within lignin model systems are typically good (most > 65%) and its application to native lignin yields approximately 43 wt% of oligomeric units and up to 55% of low molecular weight species.993 There are exceptions to this approach, which include inducing an O-4’ cleavage in lignin model systems through an arene cation radical intermediate994 or a PCET strategy to directly cleave β-O-4 linkages in native lignin without necessitating substrate oxidation.995
Scheme 99.

Representative photoredox strategies for oxidative lignin depolymerization.
An electrochemical strategy towards lignin depolymerization was developed by Stahl and co-workers using bulk electrolysis by first oxidizing the 1° alcohols of lignin model systems to either the aldehyde or the carboxylic acid, which can undergo a retro-aldol C–C bond cleavage of the β-O-4 linkage (Scheme 100). 996 This transformation was carried out using TEMPO and/or 4-acetamido-TEMPO (100.1) as the catalytic oxidant and a constant potential (0.80 V) undivided cell in basic solution (pH 10 to 12.5). In lignin model systems without β-O-4 cleavage, high yields were observed for the oxidation of 1° and 2° alcohols to the carboxylic acid and ketone respectively. Running the electrolysis at lower pH (9.2) results in full oxidation of 1° alcohol with minimal β-O-4 cleavage. However, the basic conditions used for electrolysis were not translatable towards lignin depolymerization as only low amounts of lignin monomers were obtained (≤ 8 wt%) relative to the mass of initial lignin, with monomer oligomerization and oxidative degradation products observed. Thus, Stahl and co-workers developed a two-step basic electrochemical oxidation/acid-promoted depolymerization sequence, where higher amounts of lignin monomers were obtained (29 wt% with H2O/formic acid) from the depolymerization of oxidized lignin. Syringyl (100.4–100.6) and guaiacyl-based (100.7–100.9) species were comprised the majority of the monomers, with the resultant aldehydes indicative of β-O-4 fragmentation whereas the observation of diketone and carboxylic acid monomers suggests alternative cleavage mechanisms. The cleavage of C(sp2)–C(sp3) σ-bonds in lignin has also recently been accomplished via a coupling a dye-sensitized photoelectrochemical cell system with a hydrogen atom transfer (HAT) mediator, namely 4-ACT.997
Scheme 100.
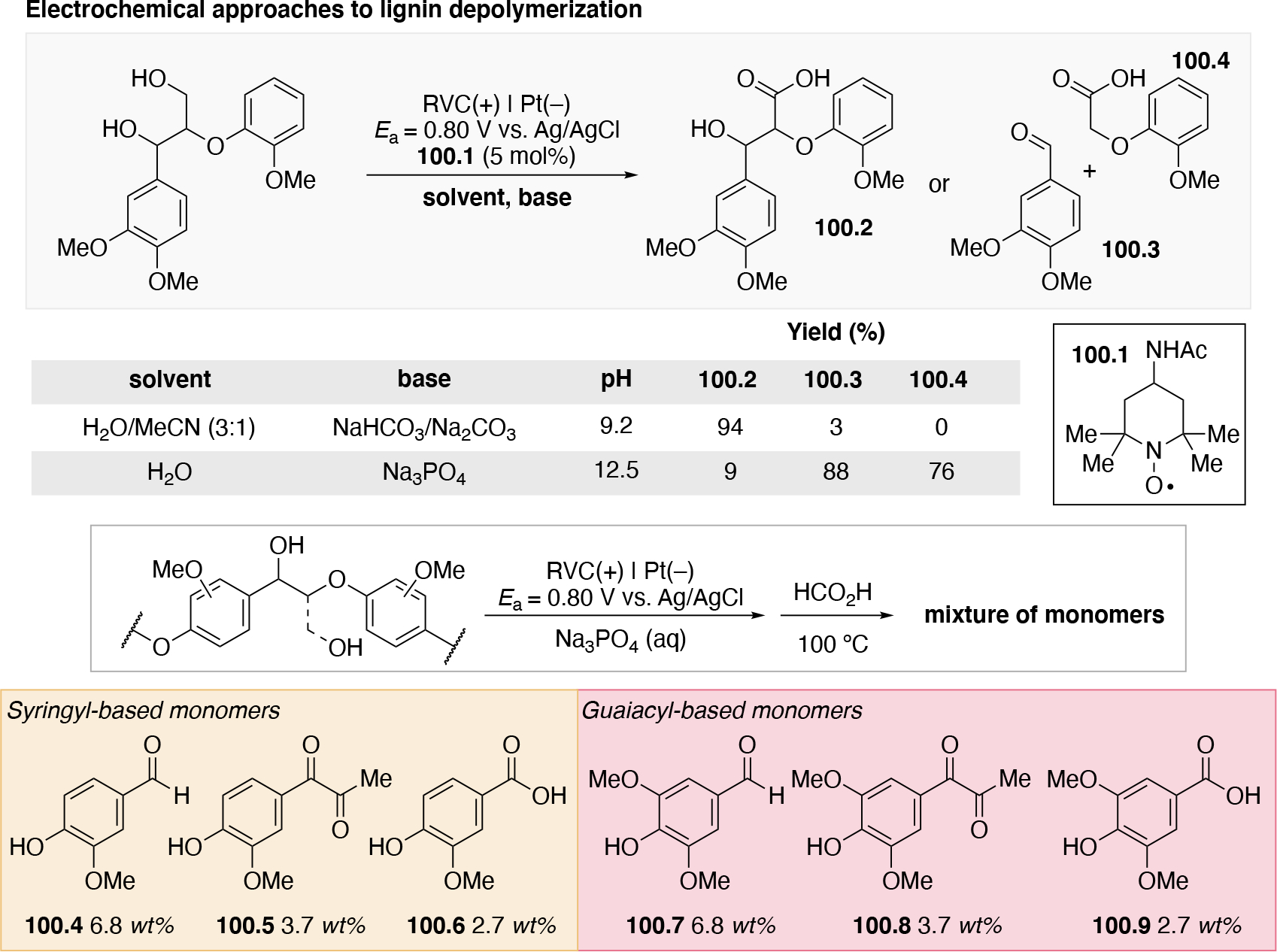
Representative photoredox strategies for oxidative lignin depolymerization.
4.18. Deprotection via Redox Chemistries
Not only can electrochemistry facilitate key bond forming steps towards building complex molecules but it can also be useful for protecting group manipulations, such as removal of protecting groups (e.g. Ts, PMB, Bz on oxygen or nitrogen),998,999 as well as their installation,1000,1001 as well as cleavage of C–C bonds.1002 Electrochemical removal of numerous other protecting groups such as benzyl groups, benzyloxycarbonyl groups, dithioketals,1003 and ester hydrolysis, are known.998,999 Oxidative dealkylation of amines has been reported both by electrochemical1004 and photochemical1005,1006 methods, as well as a related reductive dealkylation of amides,1007 and sulfonamides.1008
4.18.1. Removal of PMB Groups.
Chemical removal of PMB groups proceed primarily through harsh oxidative strategies (e.g. DDQ is used as a stoichiometric oxidant) and redox-mediated strategies improve on this process by enabling the use of milder oxidants (Schemes 101 and 102). Weinreb reported a preparative scale direct electrochemical oxidative method for PMB group deprotection (Scheme 101, method A1) using divided cell.1009 Steckhan reported the use of tris(4-bromophenyl)amine as a redox mediator for the oxidative removal of PMB groups for two substrates (Scheme 101, method A2) using a divided cell.1010 Zheng and Little reported a novel triarylimidazole as a redox mediator for the electrochemical oxidative PMB group removal but focused on obtaining the yield of the aldehyde byproduct and not on that of the alcohol.1011 Brown has reported an electrochemical deprotection in flow (Scheme 101, method A3) using an undivided cell.1012 Stephenson reported an Ir-photoredox catalyzed deprotection of PMB groups using BrCCl3 as the terminal oxidant (Scheme 101, method B1).1013 Cheng reported the use of eosin Y as a photoredox catalyst for the oxidative cleavage of PMB ethers to reveal alcohols using a mixture of H2O2 and NaHSO4 as the terminal oxidant (Scheme 101, method B2).93 The same transformation was reported by Woo using an acridinium photocatalyst and (NH4)2S2O8 and air the terminal oxidants (Scheme 101, method B3).1014 Most of these methods provide comparable yields and afford high selectivity for deprotection of PMB ethers in the presence of other protecting groups (alcohols protected with THP and TBS groups, or amines protected with a Piv, Boc, or Ts group). The electrochemical anodic oxidation methods for PMB removal rely on a net 2 electron oxidation,1011,1015 whereas the photoredox methods leverage 1e oxidation followed by a subsequent HAT event to arrive at the same benzylic cation prone to hydrolysis. The application of these anodic oxidation for the removal of para-methoxyphenyl (PMP) groups from the anomeric alcohol of mono-, di- and tri-saccharides has been reported by d'Alarcao.1016 A closely related method for the deprotection of 4-methoxythioethers is also known using electrochemistry.1017 Kappe reported a photochemical generation of 4-methoxybenzyl bromide for telescoped PMB group installation on amines, the method relies on halogenation of toluene using BrCCl3, 365 nm irradiation, and benzophenone as an HAT catalyst.1018
Scheme 101.
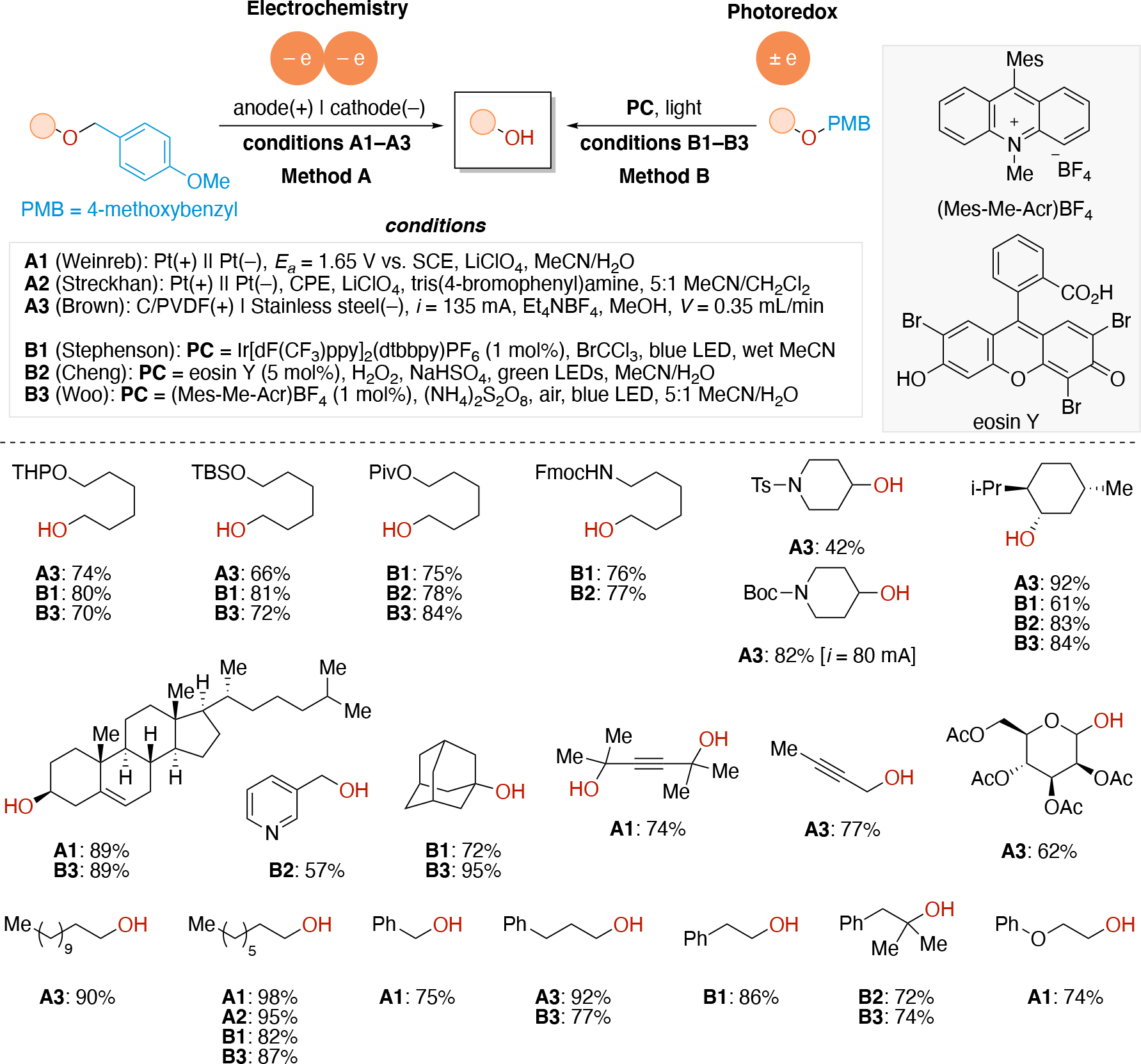
Oxidative deprotection of PMB groups.
Scheme 102.
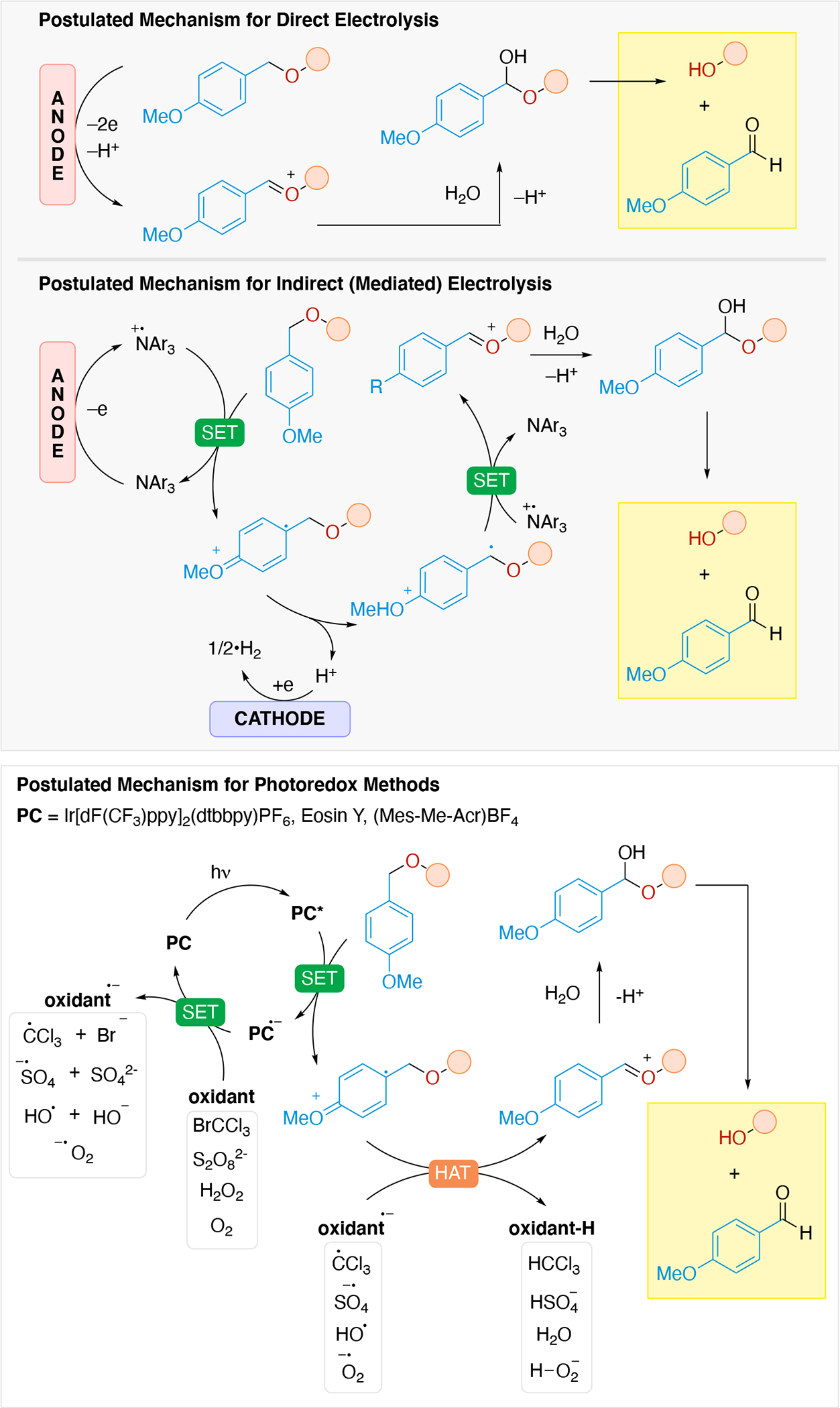
Proposed mechanisms related to oxidative deprotection of PMB groups.
4.18.2. Desulfonylation of Tosyl Protected Amines.
Reductive cleavage of a tosyl (Ts) protecting group on aliphatic amines, aromatic amines, and alcohols is known, typically in aprotic solvents such as DMF or MeCN and the presence of an alkylammonium electrolyte. The ease of reductive cleavage for S–O and S–N bond in tosyl-protected species is as follows: Ts–O–Ar > Ts–O–Alkyl, Ts–NH–Ar > Ts–NH–Alkyl > Ts–NH–CH(Alkyl)–CO2H (in descending order).1019 Electrochemical methods of desulfonylation of tosylprotected amines has been known for decades. Horner and Neuman reported electrochemical conditions in 1965 relying on a Hg pool cathode.1020,1021 Constant current electrolysis using a divided cell and a lead cathode was reported by Iwasaki and Matsumoto in 1973.1022 While the literature has numerous reports of related conditions utilizing a Hg pool cathode1023,1024,1025,1026,1027,1028,1029,1030,1031,1032, select examples using RVC exist which are more practical and greener alternatives (Scheme 103, methods A1 & A2),1033 and method A3).1034 Silvestri reported constant potential electrolysis using compact graphite cathode in a divided cell (Nafion™ 324 cation exchange membrane) in batch mode, as well as a related continuous batch recirculation mode using a filter press two-compartment microflow cell to effect detosylation of tetratosylcyclen to cyclen reaching as high as 80% yield with 55% faradaic efficiency.1035 The use of redox mediators such as pyrene and naphthalene1036 can provide conditions that operate at less reducing potentials. More recently the use of Pt as a cathode and naphthalene as a redox mediator (catalytically generating naphthalide in situ) was demonstrated to provide milder conditions (method A4).1037 Naphthalenesulfonamides are a milder alternative to tosylsulfonamide protecting groups as they can be deprotected with lower reducing potentials (typically by about 200–400 mV).1038 Monteiro realized that p-toluenesulfonyl groups attached to the nitrogen of tryptophan- and histidine side-chain residues of amino acids have reduction potentials that are less negative than those linked to the nitrogen of an aliphatic amine. This enables the development of potential-selective deprotection of p-toluenesulfonyl protected tryptophan- and histidine side-chain residues in the presence of the p-toluenesulfonyl protected α-amino group in good yields using an divided cell with Et3N•HCl as the proton source and Et4NCl electrolyte in acetonitrile.1039 Liu and Xiao reported a photochemical methods for Ts deprotection of tosylamines relying on Hantzsch ester as the terminal reductant (Scheme 103, method B1).1040 Song and Wang reported an Ir-photocatalyzed tosyl deprotection of N-heterocycles that has selectivity for the N-aryl over N-alkyl Ts-group as shown by monodeprotection of bis-N-Ts-tryptoline (Scheme 103, conditions B2).1041 As already described in the sequential 2e reduction section, Nicewicz reported the photocatalyzed method based on oxidizing an amine using an acridinium photocatalyst to generate a persistent radical anion derived from the acridinium catalyst, which upon photoexcitation produces a highly potent reductant capable of reductively cleaving the Ts–N bond of Ts-protected amines (Scheme 103, method B3).1042 The use of hydride reagents as the terminal reductant was also demonstrated in the context of photoredox catalysis for the Ts deprotection.1043,1044,1045
Scheme 103.
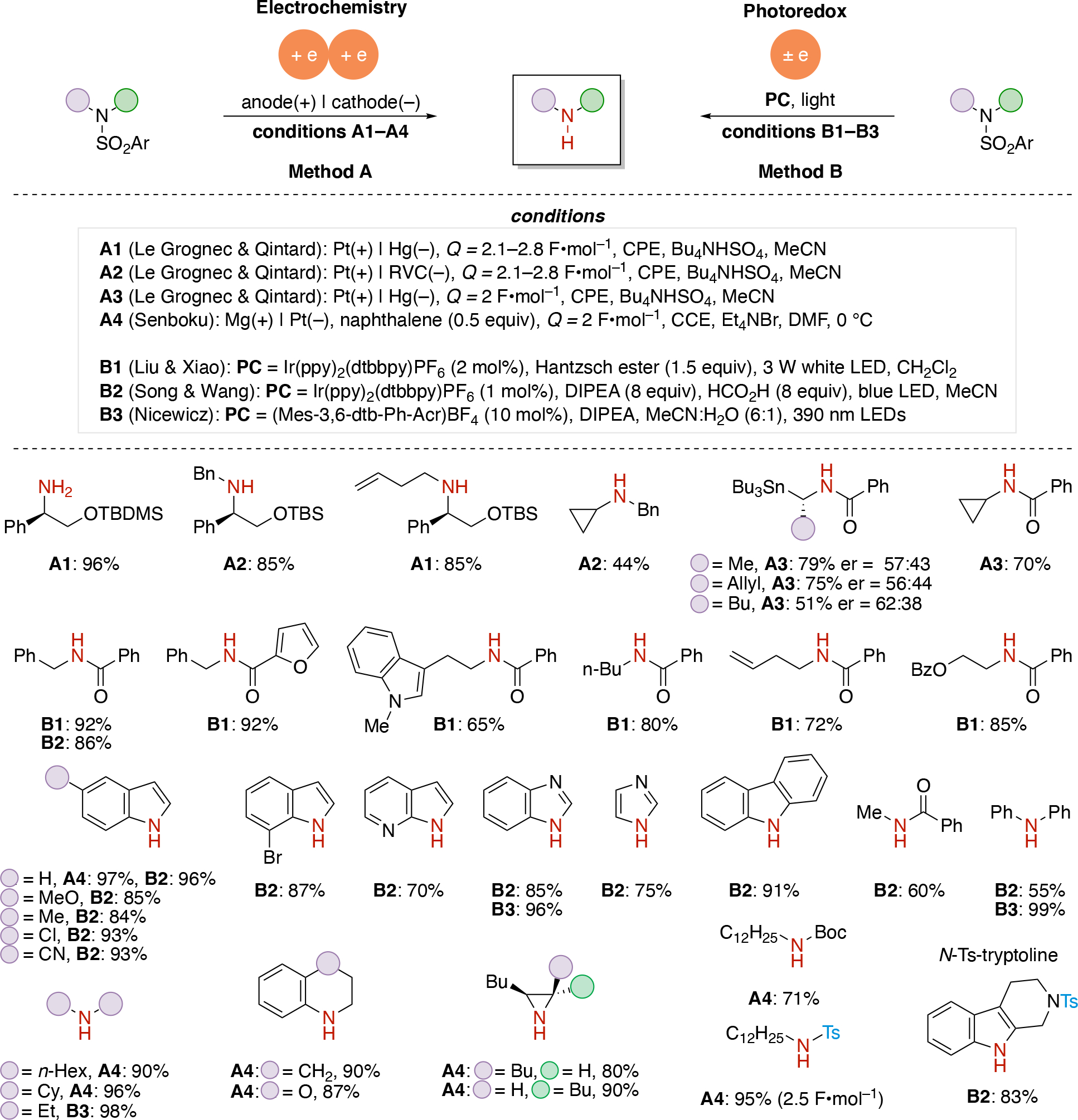
Representative methods for amine desulfonylation via electrochemical and photoredox pathways.
4.19. Formylation of Aryl Halides
The formylation of aryl halides to synthesize benzaldehyde derivatives can be done via electrolysis in the present of DMF as a formylating agent (Scheme 104, methods A1–A4).1046 The stainless steel cathode is pretreated by freshly electrodepositing Zn, Cd, or even tin which facilitate the subsequent electrolytic formylation reaction carried out on that cathode, while a sacrificial anode, such as Mg, balances the redox chemistry. A Pd catalyzed electrochemical formylation of aryl halides was reported by Carelli.1047 Mariano and Wang reported a nickel and organic dye co-catalyzed formylation of aryl halides, aryl triflates and, vinyl triflates using diethoxyacetic acid as a formyl equivalent via a decarobylxation process (Scheme 104, method B1).1048 Doyle reported a related approach using 1,3-dioxolane as a formyl equivalent leveraging HAT and Ni-catalysis (Scheme 104, method B2).1049 Fu reported a photoredox decarboxylation of glyoxylic acid in the presence of aryl halides and a Pd catalyst to achieve their formylation (Scheme 104, method B3).1050 Analogous to the formylation method by Mariano and Wang described above, the same groups reported a hydroformylation of styrenes.755a
Scheme 104.

Formylation of aryl halides to access benzaldehydes and their derivatives.
4.20. Olefin Aziridination and Cyclopropanation
4.20.1. Olefin Aziridination
In 2001 the Yudin group reported an electrochemical olefin aziridination using N-aminophthalimide under constant potential electrolysis in a divided cell (Scheme 105, method A1).1051,1052 It was proposed that the anodic oxidation of N-aminophthalimide generates the corresponding nitrene intermediate based on the observation that the aziridination of cis-2-hexen-1-ol is stereospecific, yielding exclusive formation of the cis-aziridine (no yield was provided). Interestingly, the choice of the anode material was critical – no aziridine product was observed when the optimal Pt anode was replaced by a graphite anode – perhaps hinting at analogies to electrode material impacting 1e vs 2e oxidation process as observed in electrochemical decarboxylations (i.e. Kolbe vs Hofer-Moest electrolysis). Zeng and Little reported an olefin aziridination using N-aminophthalimide and tetrabutylammonium iodide as a redox mediator (Scheme 105, method A2), although control experiments support ambient laboratory light contributing to the success of the reaction.1053 The lack of stereoretention in the aziridination, namely that both cis-stilbene and trans-stilbene afford the cis-aziridine product, suggests the reaction proceeds via a stepwise process, and rules out the involvement of a singlet nitrene. Reactions in the presence of TEMPO as a radical inhibitor, or the use of I2 in the absence of light and electrolysis afforded low yield (5–11%) of the desired azridine product, suggesting a radical pathway as well as ambient laboratory light providing a beneficial role for the reaction outcome. Cheng reported the aziridination of olefins using a partially fluorinated sulfonamide reagent under constant potential electrolysis (Scheme 105, method A3)1054 and Xu reported a photoredox approach to olefin aziridination using an Ir-photocatalyst and an N-protected-1-aminopyridinium reagent for a redox neutral approach (Scheme 105, method B1).1055 Reduction of the N-protected-1-aminopyridinium reagent was proposed to release the N-protected amino radical able to add to alkenes and the subsequent intermediate was oxidized by the photocatalyst to generate a carbocation, enabling C–N bond formation to generate the aziridine. The associated mechanisms in the methods described above are shown in Scheme 106.
Scheme 105.
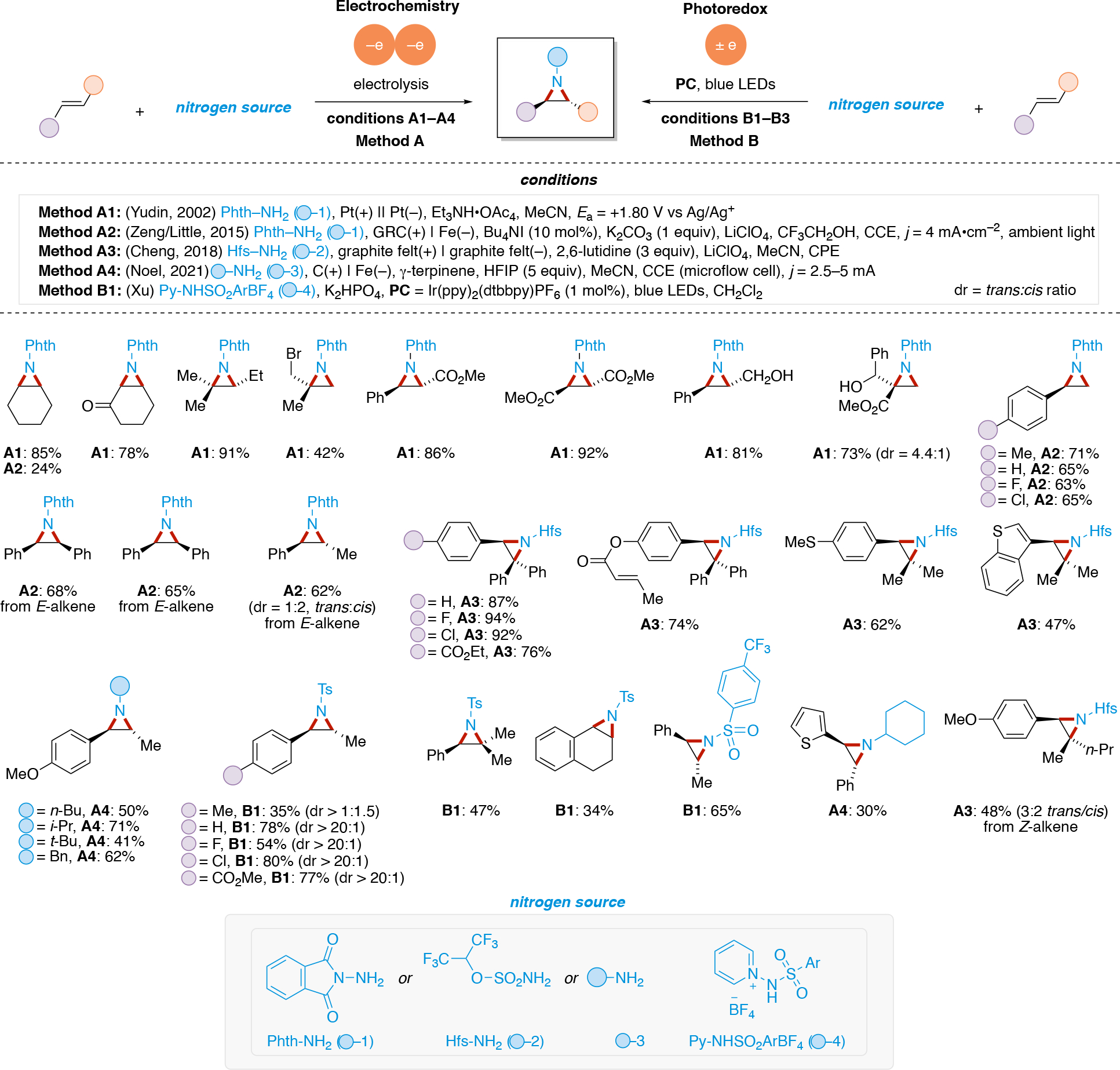
Olefin aziridination.
Scheme 106.
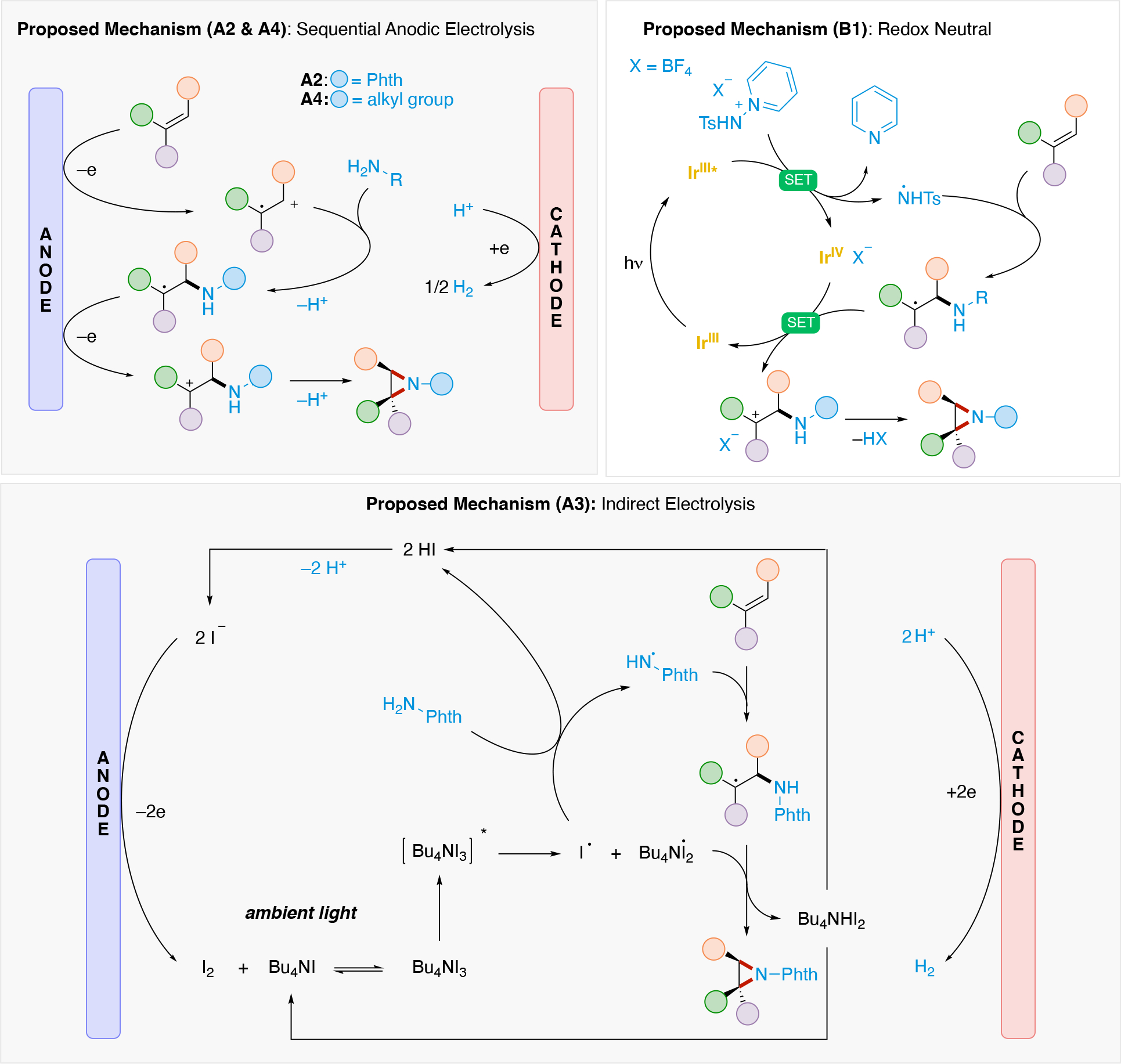
Proposed mechanisms associated with olefin aziridinations shown in Scheme 105.
4.20.2. Olefin Cyclopropanation
The cyclopropanation of alkenes via electrochemistry1056 or photoredox catalysis1057 have similarities in retrosynthetic disconnection (i.e. one and two carbon synthons for the cyclopropane), but the methods have stark differences. The two general electrochemical strategies for cyclopropane synthesis are (1) direct substrate electrolysis (Scheme 107) and (2) indirect electrolysis of redox-active species (Scheme 108). The following methods for substrate activation primarily involve cathodic reduction as oxidative methods are still underdeveloped but a few exceptions exist.1058,1059 A classic electrochemical method of synthesizing cyclopropanes is cathodic reduction of either 1,3-dibromoalkanes or 1,3-di(methanesulfonate)alkanes to yield 1,3-difunctionalized alkanes. Electrolysis has proven useful for the reduction of alkyl halides and the formation of organometallic reagents in situ.1060,1061,1062,1063 While the use of Zn dust to reduce 1,3-dibromopropane to afford cyclopropane in 56% yield was reported in 1882,1064 its electrochemical analog was reported 85 years later by Rifi in 1967.1065 The method involves potentiostatic (Ec = −2.0 ± 0.1 V vs SCE), two-electron reduction of 3-bromopropyltriethylammonium bromide, which forms a highly reactive carbanion that undergoes intramolecular substitution at the alkylammonium to eject triethylamine and form cyclopropane (yield not reported). Similar conditions were used to reduce 1,3-dibromo-1,3-dimethylcyclobutane and 1,3-dibromo-2,2-di(bromomethyl)propane to the corresponding 1,3-dimethylbicyclo[1.1.0]butane and spiro[2.2]pentane in 55% and 50% yield respectively. Reduction of 1,3-dibromopropane under similar conditions furnishes cyclopropane as a product, but attempts at quantifying the yield proved difficult. Further elaboration of this concept was reported by Shono in which galvanostatic reduction of 1,3-dimethanesulfonates using a divided cell provides a range of cyclobutanes in moderate yields (50–75%).1066 The key to obtaining these high yields was the slow addition of substrate to the cathodic chamber, which minimizes undesirable substrate decomposition (presumably to favor intramolecular cyclization over bimolecular oligomerization). Another strategy for electrolytic substrate activation is to generate stabilized carbenes (e.g. dihalocarbene);1067 this was accomplished on a preparative scale by Fritz and Kornrumpf,1068,1069,1070 as well as Baizer and Chruma.1071 A route to gem-dichlorocyclopropanes following a similar strategy have been reported by Petrosyan and coworkers using CCl4 and alkenes in chloroform containing tetraalkylammonium electrolyte in a divided cell using a lead cathode.1072 A synthetically useful electrochemical analog to the Simmons-Smith reaction was first shown by Périchon and coworkers, who report the synthesis of cyclopropanes via dihalomethane reduction in the presence of alkenes within an undivided cell.1073 The results suggest in situ formation of a zinc carbenoid (CH2ZnI2), an intermediate observed in the Simmons-Smith reaction. This leads to two reported conditions, with the difference being whether exogenous ZnBr2 was absent or present prior to electrolysis, respectively, to accelerate the formation of reactive zinc carbenoid.
Scheme 107.
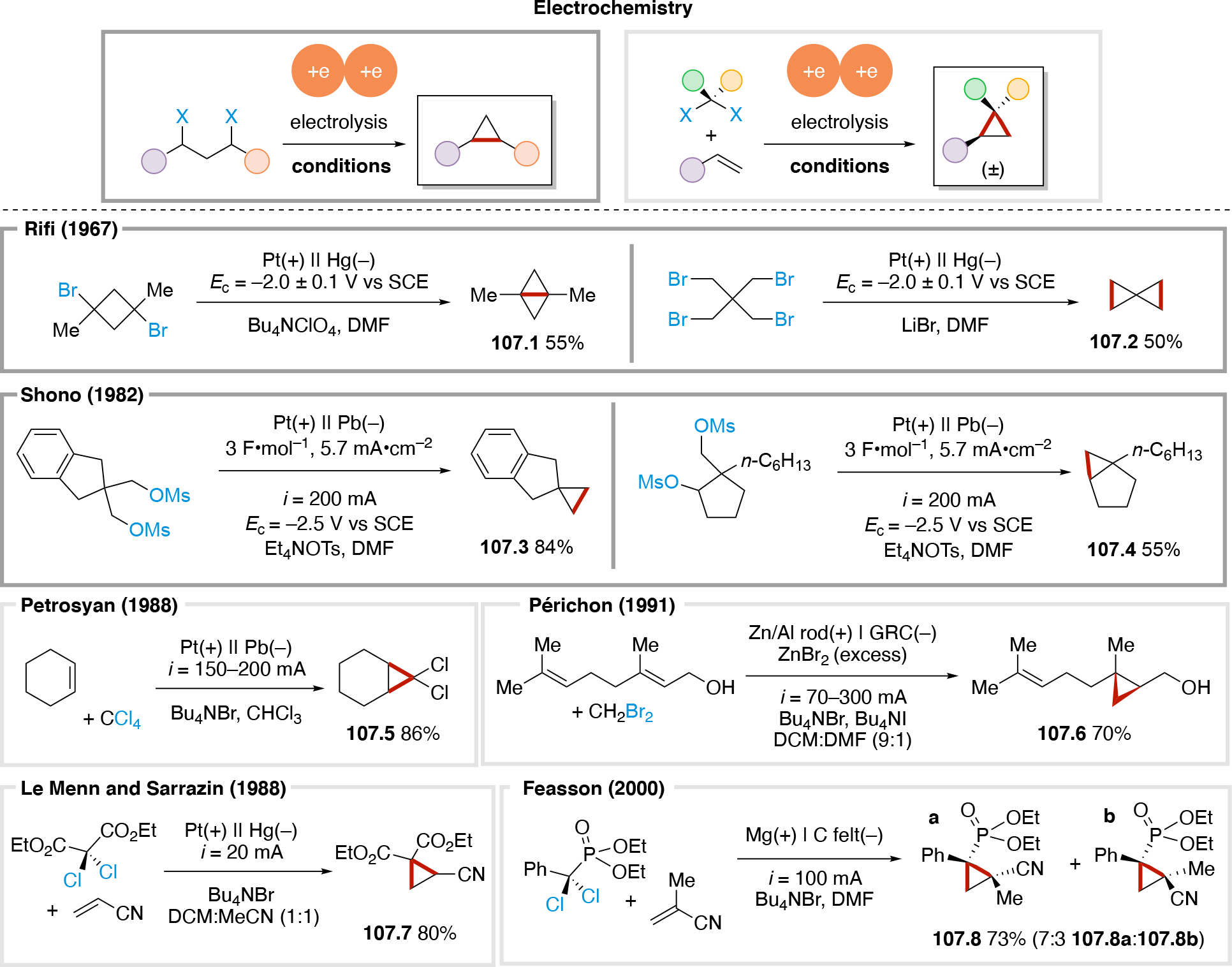
Electrochemical methods for cyclopropane synthesis via substrate reduction.
Scheme 108.
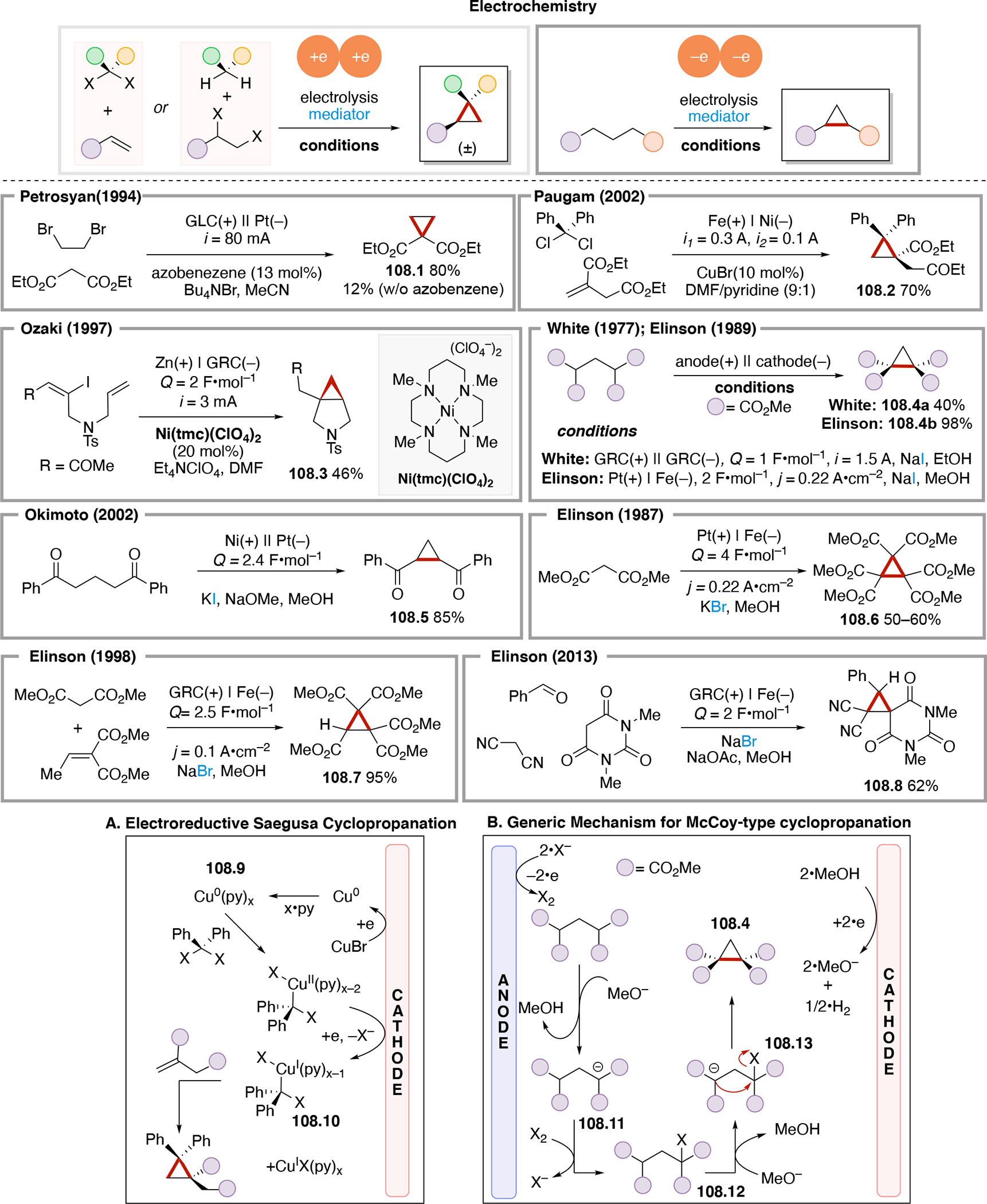
Mediator-centered electrochemical methods for cyclopropane synthesis
Electroreductive formation of stabilized carbanions is another effective strategy for olefin cyclopropanation.1074 Le Menn and Sarrazin show that galvanostatic electrolysis of dibromomalonate and dichloromalonate in the presence of Michael acceptors enables the synthesis of substituted cyclopropanes in good yield (≥ 70%); solvent choice is crucial as unproductive protonation of halocarbanions can be a problem.1075 Expansion of this strategy to phosphonate-stabilized carbanions was carried out by Feasson and coworkers, where galvanostatic reduction of tetraethyl dichloromethylbis(phosphonate) generates a stabilized carbanion that adds to (meth)acrylate, (meth)acrylonitrile, and methyl vinyl ketone in moderate to good yields.1076 This method is extended to the galvanostatic reduction of diethyl α,α-dichlorobenzylphosphonate in the presence of Michael acceptors yields α-aryl, β-substituted cyclopropylphosphonates, although low diastereoselectivity is observed.1077 A different activation strategy is the formation of carbonyl-stabilized carbanions, which Petrosyan and coworkers demonstrate in an electrochemical Perkin-type reaction between 1,3-dicarbonyl compounds and 1,2-dihaloalkanes to form substituted cyclopropanes.1078 While yields were moderate to low under direct substrate electrolysis, using an electrogenerated azobenzene base significantly improves reaction yields (e.g. from 12% to 80%).
This success of redox-active reagents is translated to other cyclopropane syntheses, and is notable in the galvanostatic cyclopropanation of Michael acceptors with stabilized 1,1-dichloroalkanes reported by Paugam and coworkers, in which an electrogenerated low-valent copper species catalyzes the reaction (Scheme 108A).1079,1080,1081 The electroreduction of CuBr (i1 = 0.3 A) forms redox-active Cu0 species (108.9) in situ, which undergoes oxidative addition into the substrate C–Cl bond and subsequent reduction (i2 = 0.1 A) to form a discrete copper carbenoid species (108.10) analogous to Cu0 cyclopropanation methods first reported by Saegusa.1082,1083,1084,1085 A nickel-catalyzed variant was also disclosed by Paugam and coworkers during the development of their copper-catalyzed method.1086 Additionally, a tandem intramolecular radical [2+1] cycloaddition facilitated by electroreductive formation of NiI was demonstrated by Ozaki and coworkers, providing bicyclic [3.1.0] derivatives in moderate yields.1087
Another distinguishing factor of cyclopropanation strategies for redox mediator-centered electrolysis is the reliance on the electrochemical oxidation of redox mediators, in contrast to the methods involving direct substrate reduction. These transformations follow McCoy-type cyclopropanation, as in situ halogenation of stabilized carbanions enables base-catalyzed condensation with Michael acceptors (Scheme 108B). White demonstrated the first example of this via the electrooxidative cyclization of tetramethyl-propane-1,1,3,3-tetracarboxylate with sodium iodide as a mediator to form tetramethyl-cyclopropane-1,1,2,2-tetracarboxylate (108.4) in 40% yield.1088 Cathodic reduction of the O–H proton of ethanol generates H2 and ethoxide enabling deprotonation of acidic protons from the substrate to form stabilized carbanion 108.11. This anion traps iodine — generated via anodic oxidation of iodide — to furnish an alkyl iodide (108.12). A second deprotonation event of intermediate 108.12 forms a carbanion (108.13) that readily cyclizes to product 108.4 by a SN2-type ring closure. The yields and scope of tetramethyl-propane-1,1,3,3-tetracarboxylates was improved on by Elinson and coworkers,1089 and Okimoto and coworkers expand this transformation to the cyclization of 1,3-diaroylpropanes.1090 This strategy can also be applied to the cyclotrimerization of dimethylmalonate1091 and ethylcyanoacetate1092 through a repeating stepwise deprotonation-halogenation-halogen elimination sequence culminating in cyclopropane synthesis. Applying halide redox mediators to the coupling of malonic acid derivatives and Michael acceptors eliminates the need for direct reduction of substrate as the transformation proceeds mechanistically similar to McCoy-type cyclopropanation. This strategy has been extensively covered by Elinson and coworkers.1093,1094,1095,1096,1097,1098,1099,1100,1101 Variations of this method have been applied to the spiro-coupling of barbituric acid derivatives with benzylidenemalononitriles and benzylidenecyanoacetates.1102 Lastly, redox-mediated electrosynthesis of functionalized cyclopropanes via three-component coupling of aromatic aldehyde, malononitrile, and dimethylmalonate has been shown by Elinson and coworkers.1103 Knoevenagel condensation of aldehyde onto malononitrile forms an alkylidenemalononitrile that then participates in the McCoy-type cyclopropanation sequence with dimethylmalonate as outlined previously. This method is also compatible with barbituric acid derivatives as the coupling partner to form spirocyclopropylbarbiturates.1104
The first disclosure of a photoredox-catalyzed alkene cyclopropanation using 1,1-dihaloreagents was reported by Guo in 2015 using dibromomalonates (R1, Scheme 109) as the one-carbon synthon and activated styrenes as the reaction partner.1105 Double single electron reduction and single halogen extrusion of the dibromomalonate by photoactivated Ru(bpy)32+ purportedly yields a stabilized carbanion that undergoes Michael addition and subsequent bromide extrusion to reveal the cyclopropane ring. Interestingly, stereoconvergence is observed in this transformation, as only the trans-product 109.1 is observed when using either Z- and E- styrene isomers. A more general photoredox method for stereoconvergent cyclopropanation was reported by Suero in 2017 using diiodomethane (R2, Scheme 109).1106 A range of internal (1,2-disubstituted and 1,2,3-trisubstituted) alkenes are compatible, but terminal alkenes (e.g. 4-methoxystyrene or 1,1-diphenylethylene) provided low yields of cyclopropane or open-chain alkyl iodides. The scope was later extended to also include Michael acceptors (e.g. α,β-unsaturated aldehydes and ketones) but ester derivatives are not tolerated.1107 The postulated mechanism (Scheme 109A) for both methods proceeds via reductive quenching of the RuII photocatalyst to RuI, followed by reduction of diiodomethane (Ered = −1.44 V vs SCE) to turn over the photocatalyst and generate iodomethyl radical (109.13), which can add to the alkene. The resulting radical intermediate (109.14) is sufficiently long-lived to allow equilibration between rotamers, thus leading to ring closure to the trans-product and concomitant release of I•. Expanding on the versatility of the di-iodomethyl reagents, Charette and coworkers report the use of 1,1-diiiodo-pinacol boronate (R3, Scheme 109) in the photoredox-catalyzed cyclopropanation of styrenes using 350 nm light and xanthone as the photocatalyst to afford borocyclopropanes.1108 The reaction performed well with unsubstituted styrenes or α-substituted styrenes, but fewer examples of α,β-disubstituted styrenes (e.g. β-methylstyrene) were reported. Overall, this transformation is useful as the boryl group provides a functionalization handle for further diversification of the cyclopropane via cross-coupling or oxidation strategies.
Scheme 109.
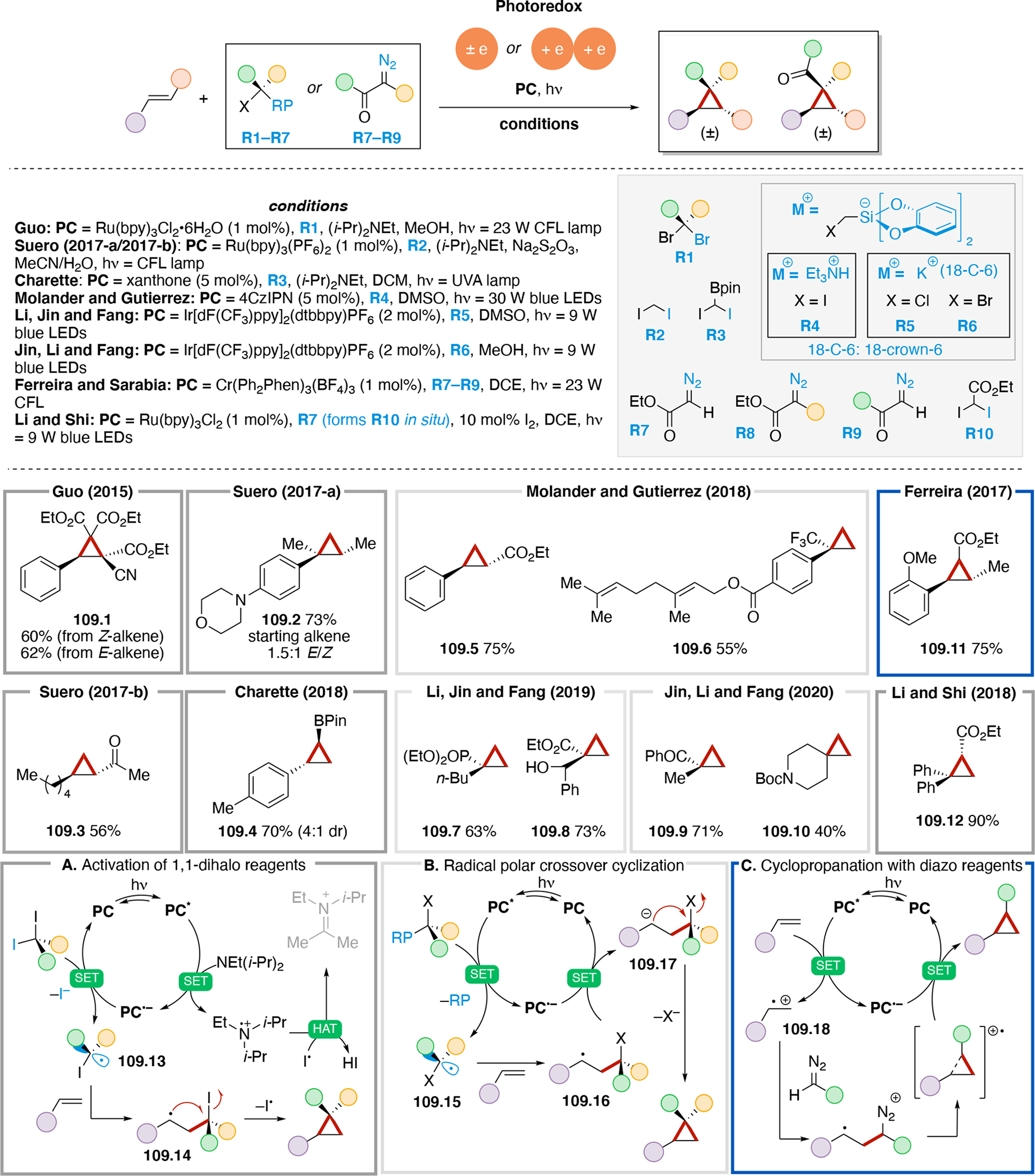
Photoredox methods for olefin cyclopropanation via 1,1-difunctionalized carbon synthons.
Complementary one-carbon synthons can be obtained by replacing one halogen in prototypical 1,1 dihalo reagents with a redox-active precursor, as shown by Molander, Gutierrez and coworkers, where a photoredox method for olefin cyclopropanation proceeds via redox-neutral radical polar crossover, (Scheme 109B).761 The method relies on benchtop stable triethylammonium bis(catecholato)iodomethylsilicate reagent (R4, Scheme 109), which is synthesized in two steps from chloromethyltrimethoxysilane. Using organophotocatalyst 4CzIPN (11.10), a wide range of substituted alkenes are tolerated with impressive stereoconvergence in cases when a starting mixture of E/Z isomers are used. Later developments of chloromethylsilicates1109 (R5, Scheme 109) and bromomethylsilicates1110 (R6, Scheme 109) variants enabling greater generality in substrate scope. The mechanism here diverges from Suero’s method, as the reductive quenching of photoexcited Ru(bpy)32+ is used to generate halomethyl radical (109.15) from the silicate, which adds to the alkene to form a stabilized radical (109.16). SET from the resultant RuI species to the alkyl radical leads to a radical-polar crossover event to turn over the photocatalyst and generate an alkyl carbanion (109.17), which displaces the iodide via SN2-type ring closure.
Diazo compounds are a widely-used one-carbon synthon in transition-metal catalyzed cyclopropanations,1111,1112 and early reports by Pérez-Prieto and Stiriba have shown that benzophenones can act as triplet sensitizers to generate triplet carbenes from diazo precursors.1113,1114 A different activation mechanism was accomplished by Ferreira and Sarabia, in which a chromium photocatalyst is used to generate styrenyl radical cations (109.18) via single electron oxidation to enable the cyclopropanation of styrenes with ethyl diazoacetate (R7, Scheme 109), α-alkyl diazo esters (R8, Scheme 109) and diazo arylketones (R9, Scheme 109).1115 An alternate mechanism for the involvement of ethyl diazoacetate in photoredox-catalyzed cyclopropanation was reported by Li in 2018 using catalytic I2 as an activating agent to form ethyl 1,1-diiodoacetate (R10, Scheme 109) in situ that participates in the photoredox reaction.1116
A mechanistically-distinct radical addition-polar cyclization cascade (Scheme 110) was reported by Aggarwal, Noble and coworkers, where 4CzIPN-catalyzed decarboxylation of carboxylic acids generates a reactive alkyl radical (110.9) that combines with an alkene to form a stabilized carbon-centered radical intermediate (110.10).1117 This reaction is distinct from Molander’s example in that the halide nucleofuge is installed in the alkene (as a homoallyl chloride) rather than in the one-carbon synthon. Despite requiring an alkenyl Bpin or ester substituent to stabilize formation of tertiary carbanion 110.11, the scope of the transformation is quite general, and the chloride nucleofuge can be substituted for a tosylate. Expansion of this strategy to incorporate other radical precursors and generalize the substrate scope was accomplished by Molander and Kelly,1118,1119 as well as Li, Jin and Fang.1120 Lastly, visible light activation of aryldiazoacetates1121 and 2,2-diiodoacetates1122 enable the cyclopropanation of styrenes, but a discussion of their mechanisms is outside the scope of this review.
Scheme 110.
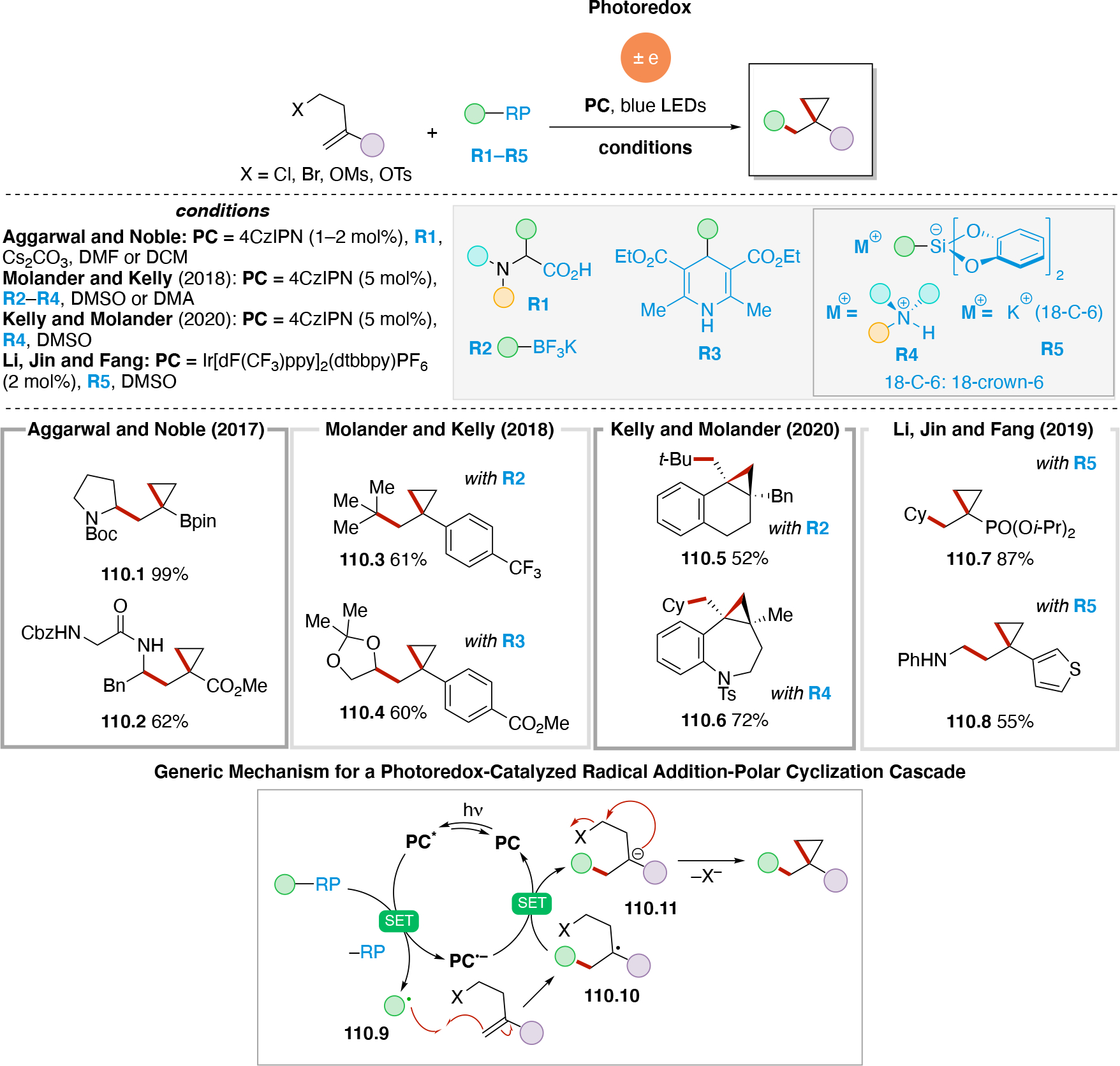
Photoredox methods for olefin cyclopropanation via the merger of radical precursors with alkenes containing a built-in electrophile.
4.21. Ring Expansions
4.21.1. Semi-pinacol Rearrangement for Ring Expansion from Vinyl Alcohols.
Simple ring expansion from vinyl alcohols has been realized via electrochemical1123,1124,1125 and photochemical1126 methods. Below we describe related method that achieve functionalization of the alkene as well as ring expansion, such as trifluoromethylation or sulfonylation, from allylic alcohols.
4.21.2. Functionalization-Ring Expansion of Allylic Alcohols via Semi-Pinacol Rearrangements
4.21.2.1. Trifluoromethylative Ring Expansion
Semi-pinacol ring expansion-trifluoromethylation has been realized using electrochemistry using allylic alcohols (Scheme 111, method A11127 and method A3)1128 or their silylated derivatives (Scheme 111, method A2).1129 Two closely related photoredox catalytic methods have also been reported starting from the alcohols which are in situ converted to the silyl ether (Scheme 111, method B1)1130 or simply used as is (Scheme 111, method B2).1131 The photoredox approaches use a redox neutral strategy to a reductively generate of the trifluoromethyl radical, followed by its addition to an alkene, subsequent oxidation of the transient radical to the corresponding cation (radical-polar crossover) and finally ring expansion via a 1,2-alkylshift to afford the desired product. This is in contrast to the electrochemical method where two sequential oxidative events are necessary, specifically anodic oxidation of Langlois’ reagent (CF3SO2Na) enables trifluoromethyl radical generation after SO2 extrusion, with the rest of the mechanism being analogous to the photoredox cycle where a subsequent oxidation of the radical intermediate is required for the radical-polar crossover and 1,2-alkylshift to achieve the ring expansion.
Scheme 111.
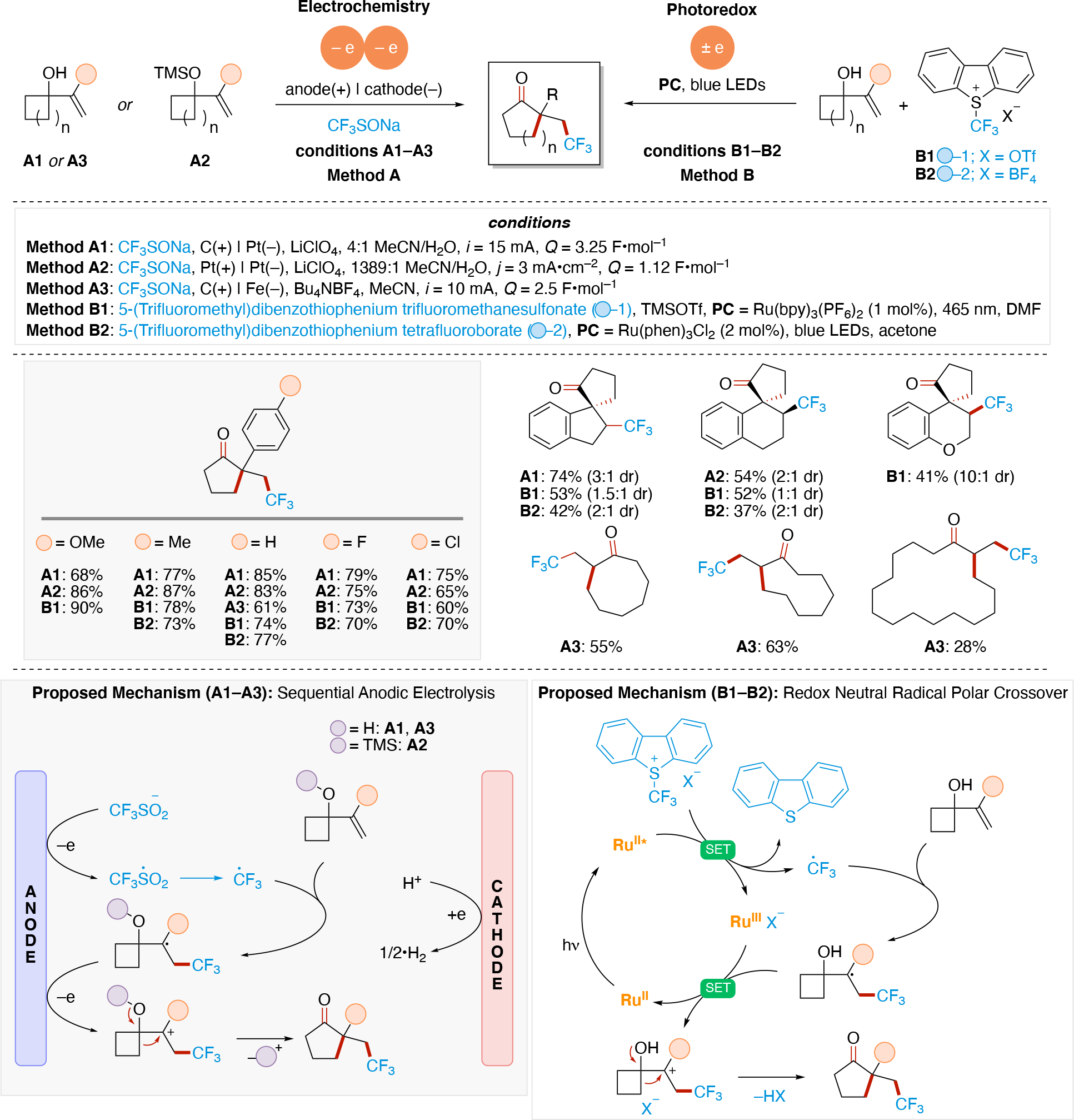
Trifluoromethylative semi-pinacol ring expansion of allylic alcohols.
4.21.2.2. Sulfonylative Ring Expansions
Analogous to the ring expansion-trifluoromethylation presented in the previous section, other functional groups besides a trifluoromethyl group can be introduced via this approach, such as sulfones. Electrochemical sulfonylative semi-pinacol ring expansion via an oxidation approach can be accomplished using sodium p-toluene sulfonate under constant current electrolysis (Scheme 112, method A1).1127 Kim demonstrated the use of p-toluenesulfonyl hydrazide in a sulfonylation/semipinacol rearrangement sequences of alkenylcyclobutanols using electrolysis relying on NaI as both the electrolyte and redox mediator (Scheme 112, Method A2).1132 A related semi-pinacol rearrangement method which installs a phenylselium instead of an arylsulfone group via electrolysis enables the cyclopentanone-selenium products to undergo a subsequent Dowd–Beckwith-type ring-expansion to access the corresponding one-carbon ring-expanded ketones.1133 In contrast to sequential anodic electrolysis approach of method A1, a photoredox neutral approach using photocatalysis was reported in which TsCl is reduced to generate the requisite sulfonyl radical (Scheme 112, method B1)1134 or three-component coupling leveraging oxidative approach (Scheme 112, method B2).1135
Scheme 112.
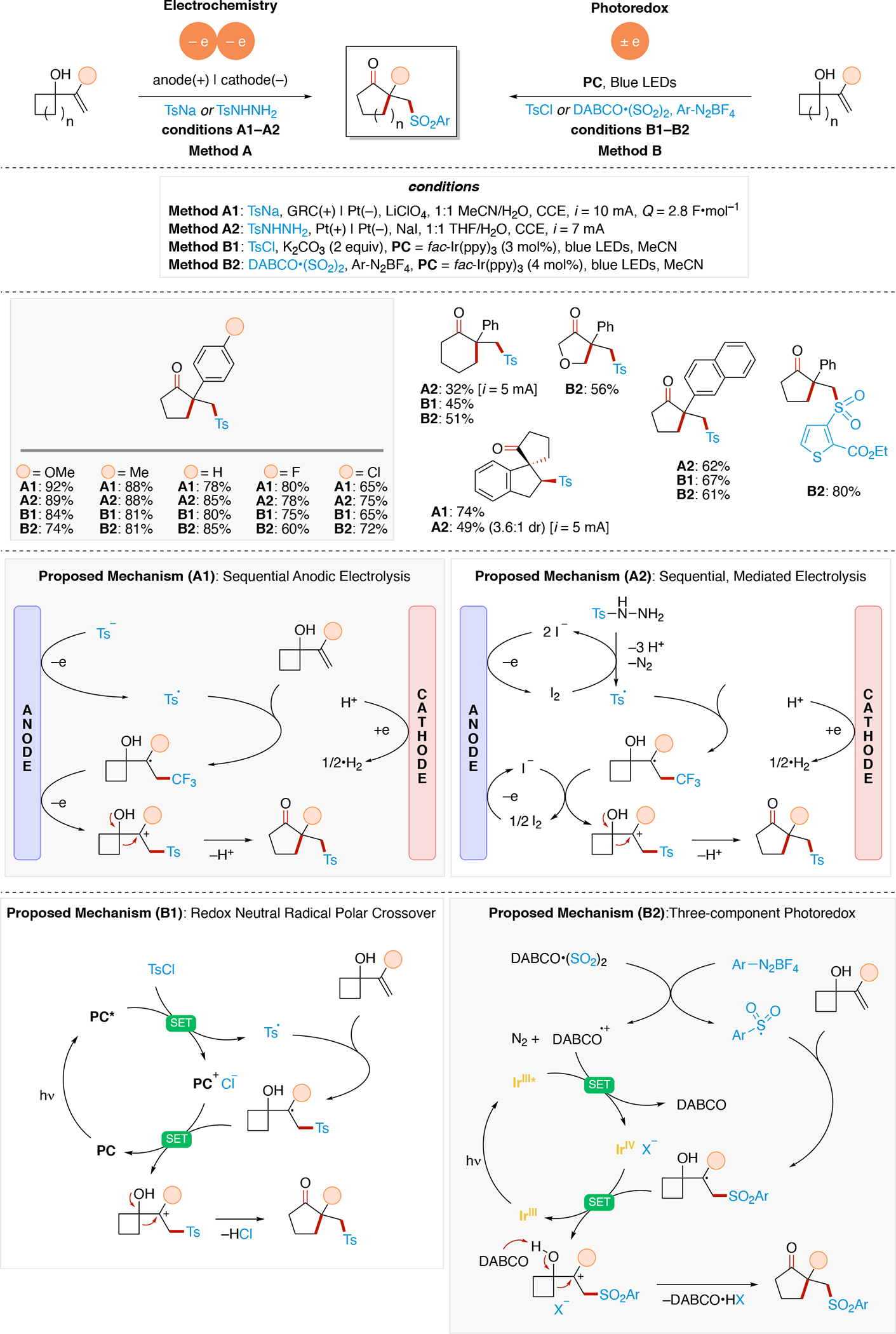
Sulfonylative semi-pinacol ring expansion of allylic alcohols.
Related arylative ring expansion processes have been reported using aryl diazoniums.1136 Related ring opening chemistries from cycloketone oxime derivatives are numerous using photoredox chemistries but underexplored using electrochemical means.1137 Electrochemical ring opening of epoxides using mixtures of chloroform and carbon tetrachloride to obtain chlorohydrins are known. Ring opening arylation of aziridines and epoxides using a Ni/photoredox cross-electrophile coupling strategy has been reported by Doyle.1138,1139 Opening of aziridines using photoredox to install an azide, halides, ethers, alkyl or dihydropyridyl groups has also been reported.1140,1141,1142 The generation of radicals derived from epoxides and aziridines for their addition to alkenes and acrylates has been reported.1143
5. EMERGING TOPICS AND OPPORTUNITIES
5.1. Redox Neutral Electrochemistry
5.1.1. Microfluidic Electrochemistry for Redox-Neutral Reactions.
While redox neutral processes are common in photoredox chemistry, for the most part, they have been elusive in the electrochemical literature in the context of organic synthesis. This is in part due to the lifetime of radicals and the location at which they are generated. In photoredox catalysis, the photocatalyst can act both as an oxidant and a reductant (one role in the excited state and the other role in the ground state). Thus, oxidation and reduction can effectively occur in the same location in the reaction mixture. In contrast, electrochemical transformations (in the absence of a redox mediator) are localized, namely oxidation events at the anode and the reduction events at the cathode. Thus, a redox neutral transformation would require intermediates generated at one electrode to live long enough for migration to the other electrode for the opposite redox event to occur. Typical batch electrolysis setups have an interelectrode separation distance that is simply too large (mm to cm distances) to enable migration of electrochemically generated species from one electrode to another on the timescale compatible with the lifetimes of organic radicals used in synthesis. Equation 34 illustrates the problem, namely that the characteristic time, t, is governed by the inter-electrode distance (d), and the molecular diffusivity (D).124,267,1144,1145
| (34) |
The problem of short radical liftetimes relative to the long duration require to migrate between electrode pairs in traditional bulk electrolysis setups is further exasperated due to mixing inefficiencies in batch settings. In contrast the use of flow chemistry can both enable efficient mixing and also provide a small electrode spacing (e.g. 25 μm) that together redox neutral electrochemical processes can be realized. Buchwald and Jensen1146 recently demonstrated the use of microfluidic electrochemistry to achieve five different redox neutral transformations previously demonstrated using photoredox (Schemes 113 and 114). A 25 μm interelectrode distance provides a short enough spacing to enable diffusion of persistent radicals derived from electron deficient cyanobenzenes from the cathode to the anode. The diffusion time is on the subsecond timescale, which is substantially shorter than the lifetime of the radical. Scheme 113 illustrates electrochemical redox neutral decarboxylative arylation (Scheme 113, top), α-amino-arylation (Scheme 113, middle), and deborylative arylation (Scheme 113, bottom), all of which proceed via convergent paired electrolysis. The reactions are complimentary to their redox neutral photoredox counterparts.457,1147,1148,1149,1150 The use of HAT catalysts or redox mediators can also be incorporated into redox neutral transformations. Scheme 114 (top) illustrates an allylic arylation using a thiol as an HAT catalyst, while the bottom of Scheme 114 illustrates a Minisci-type reaction using a redox mediator to facilitate the oxidation of the key pyridinium intermediate. Photoredox counterparts to these reactions operate similarly. Specifically, the allylic arylation utilizes the same thiol HAT catalyst,1151 while the Minisci-type reaction uses a Brønsted acid co-catalyst.491,495,1152 Redox neutral convergent electrolysis C–O couplings catalyzed by Ni/dtbbpy were also accomplished using this microfluidic electrochemical method, contrasting to MacMillan’s photochemical report which was proposed to operate via energy transfer.1153
Scheme 113.

Redox neutral transformations: (top) decarboxylative arylation, (middle) α-amino-arylation, (bottom) deborylative arylation. (Note: SET events are depicted by arrows touching an electrode).
Scheme 114.
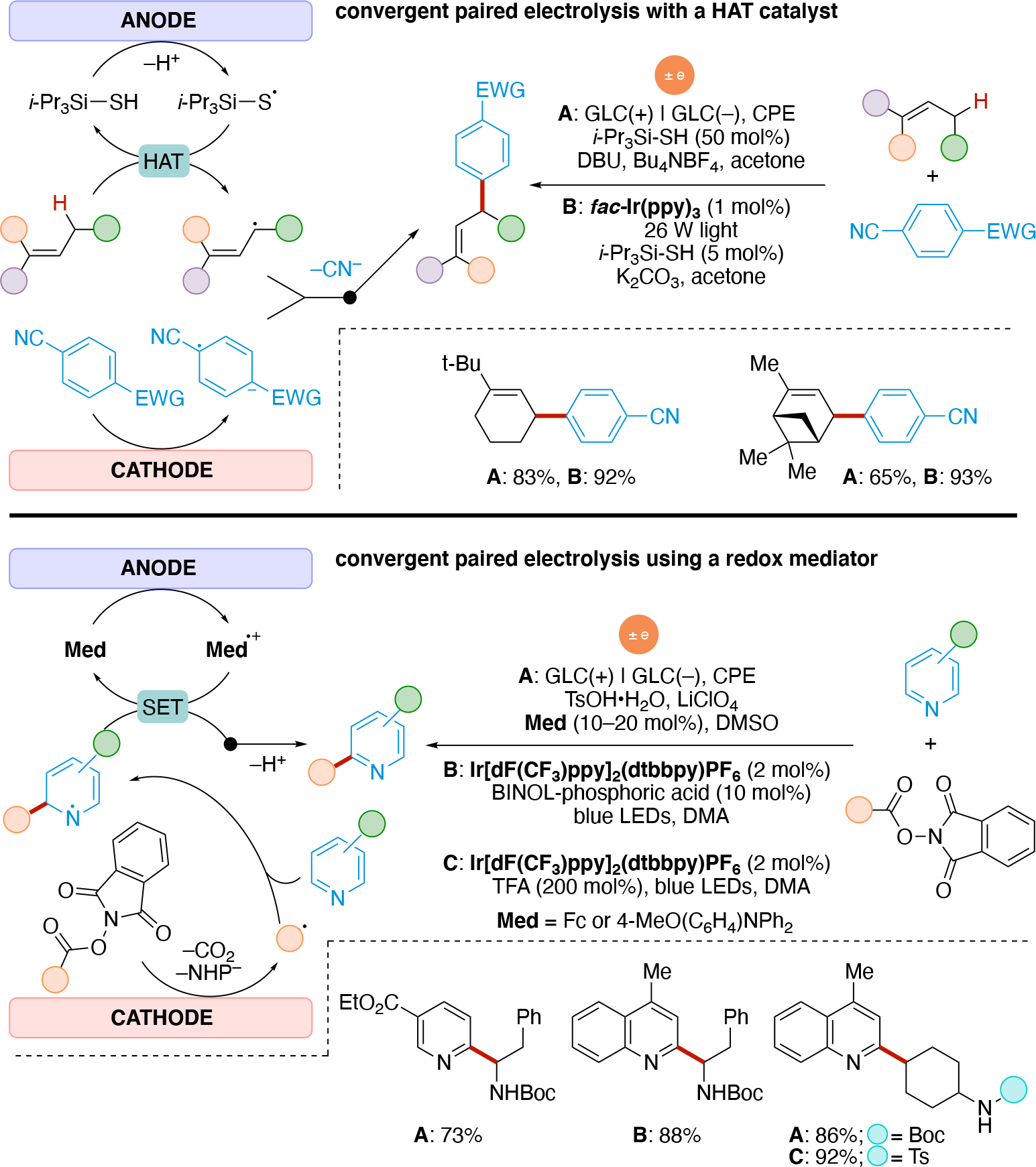
Redox neutral transformations: (top) allylic arylation using a thiol HAT catalyst, (bottom) Minisci reaction.
5.1.2. Alternating Polarity Electrolysis.
The use of alternating current electrolysis is not a new concept1154,1155,1156 however, it is an underutilized method in the context of organic synthesis for redox neutral transformations (Figure 16).1157,1158 Alternating polarity electrolysis relies on alternating the polarity of the electrode (e.g. from an oxidizing to a reducing potential) to accomplish both an oxidation and a reduction event at the same electrode. Under the term of alternating polarity electrolysis, we can sub-classify this concept into alternating current and alternating potential electrolysis (ACE or APE, respectively). A quiet time where no potential (no current) is applied to the electrode can be used in between switching the polarity of the electrode. Figure 16A illustrates the alternating current profile of a single electrode as a function of time for a “square wave” ACE (a sinusoidal function for ACE has also been used for synthesis). APE is analogous to ACE, with the only difference being that the reaction is operated at a constant potential (instead of constant current) when a potential is applied across the electrodes during the non-quiet periods (Figure 16B). In ACE, the amplitude of the wave is defined by the peak current (ip) and is alternated between +ip and −ip. Analogously, APE relies on alternating between peak voltages +Up and −Up. The duration of the applied current (or applied potential) is characterized by time t1 prior to switching polarity. In some examples, it may be beneficial to introduce a quiet period of duration t2, in which no potential (no current) is applied to the electrode in between the periods of applying opposing current (or opposing potential) (Figures 15C and 15D). This quiet period is introduced to enable the newly generated radical intermediate enough time to react with the substrate (as well as enable diffusion of key reactants, intermediates, products, or electrolyte ions away from the electrode). If no quiet period is present, the reversal of the electrode polarity may result in imparting the reverse redox event on the intermediate that was generated in the prior pulse of opposite polarity (e.g. a radical generated from a reduction event of a starting material may be reversibly re-oxidized to the initial starting material). If the quiet period is too long, the transiently generated radical intermediate may not live long enough to undergo the opposing redox process to form product. Thus, careful attention must be devoted to optimizing both t1 and the duration of the quiet period (t2) for the successful outcome of redox neutral transformations via ACE or APE. Diffusion models1159 and electron transfer1160 models for describing alternating current electrolysis have been developed.
Figure 16.
Alternating current electrolysis (A) and alternating potential electrolysis (B) without a quiet time or with quiet times (C and D respectively).
The trifluoromethylation of heterocycles (pyrroles, furans and thiophenes) and electron rich benzene derivatives has been realized using APE through a sequential paired electrolysis mechanism (Scheme 115).198 The reduction of CF3SOCl generates the CF3 radical along with SO2 and Cl−. Addition of the CF3 radical to the (hetero)arene provides a key intermediate that undergoes subsequent oxidation and loss of proton (net re-aromatization) to generate the desired trifluoromethylated (hetero)arene product. MacMillan reported the use the analogous redox neutral photocatalytic trifluoromethylation of (hetero)arenes using CF3SOCl.468o
Scheme 115.

Trifluoromethylation via alternating potential electrolysis (method A) compared to a redox neutral photocatalytic method (method B). Reaction mechanism associated with APE is illustrated on the bottom. (Note: SET events are depicted by arrows touching an electrode).
Pulsed potential electrolysis has also been recently used in the context of optimizing the electrohydrodimerization of acrylonitrile to synthesize adiponitrile, a precursor to Nylon 6,6.1161 The conversion of acrylonitrile to adiponitrile, which has been used on metric ton scale, is a net reductive process, and is traditionally carried out under continuous electrolysis conditions. The pulsed potential was found beneficial towards suppressing undesired side reactions.
Other synthetic examples of using ACE includes the synthesis of mixed disulfides via sulfur-sulfur bond metathesis from symmetrical disulfide starting materials.1162 Alternating current electrolysis can also be used in transition-metal catalysis as recently showcased with several examples of Ni-catalyzed cross-couplings,1163 including amination (C–N coupling), etherification (C–O coupling), and esterification of aromatic bromides. Selected examples using this alternating current electrolysis provided higher yields and selectivity compared to those with direct current electrolysis. The improvements were attributed to the alternating current facilitating turnover limiting steps (either oxidative addition or reductive elimination steps) while not having to rely on intermediates generated at one electrode, which may be short lived, having to migrate to the opposite electrode to achieve productive chemistry. Alternating current electrolysis has also been used for the synthesis of phenol from benzene with high selectivity and higher current efficiency than direct current (DC) electrolysis. These results were in part due to generating reactive oxygen-containing species (e.g. HO•) both during the oxidation (via oxidation of water) and reduction (via reduction of O2) segments of the AC electrolysis. Inversion of product selectivity1164 in benzylic C–H oxidation processes have also been demonstrated when comparing direct current vs alternating current electrolysis, such as in the oxidation of 4-methylanisole (Scheme 116).1165 Very recently Kawamata and Baran also demonstrated the application of alternating current electrolysis for the reduction of carbonyls such as those found in imides, aldehydes, thioesters, and α,β-unsaturated esters. Improved chemoselectivity between different carbonyls was demonstrated using alternating current electrolysis relative to direct current electrolysis.1166 The electrochemical synthesis of various metal-NHC complexes1167 based on Cu, Pd, Au, Ni, and Fe has also been realized1168,1169,1170,1171,1172. Alternating polarity electrolysis has been used to minimize the reliance on mass transport of intermediates generated at opposite electrodes towards product formation of metal-NHC complexes, such as CuI-NHC from Cu electrodes and IMes•HCl in MeCN.1173 Optimization of the switching frequency could suppress the build-up of metal dendrites that cause short-circuiting.
Scheme 116.
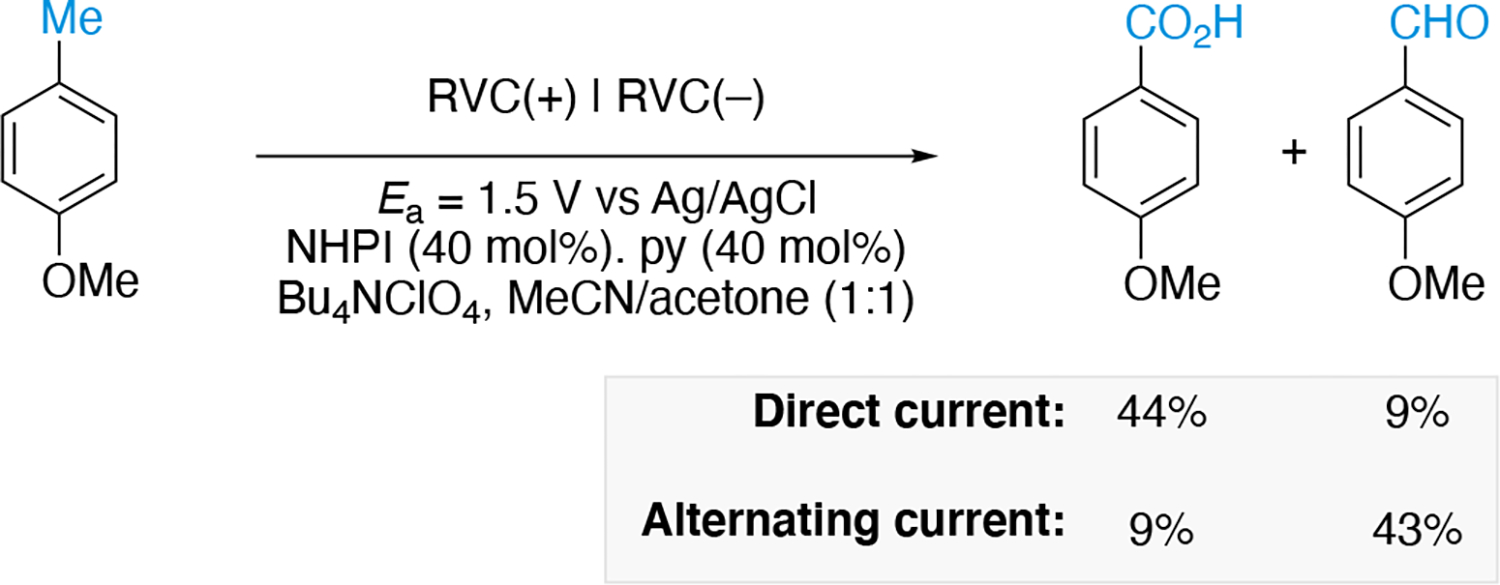
Product selectivity effect dependent on using direct vs alternating current electrolysis.
5.1.3. Bipolar Electrodes
Related to alternating current electrolysis and alternating potential electrolysis used to conduct redox neutral transformations at a single electrode, the use of bipolar electrodes (BPE) can achieve redox neutral transformations.1174,1175,1176,1177 “A bipolar electrode is an electrically conductive material that promotes electrochemical reactions at its extremities (poles) even in the absence of a direct ohmic contact.” 1178 The use of bipolar electrodes can be viewed as a wireless technique in which a potential is applied across a set of driving (feeder) electrodes interfaced with the electrolyte solution that contains the bipolar electrode, The bipolar electrode facilitates oxidation and reduction at its opposite anodic and cathodic poles. A key point is that the poles of a bipolar electrode are oriented in the opposite polarity of the driving electrodes. This is denoted in Scheme 117 below in which the driving cathode induced the closest extremity of the BPE to become the anodic pole (performing oxidation of an analyte in solution denoted by the blue arrow). The red arrow denotes the reduction process occurring at the cathodic pole of the BPE facing towards the anode driving electrode in the right-hand side of Scheme 117. The potential drop over a BPE (ΔUBPE) is given by equation 35, where ΔUtotal is the potential drop between the driving electrode, lBPE is the length of the BPE, and lchannel is the interelectrode spacing between the driving electrodes.320
| (35) |
Scheme 117.
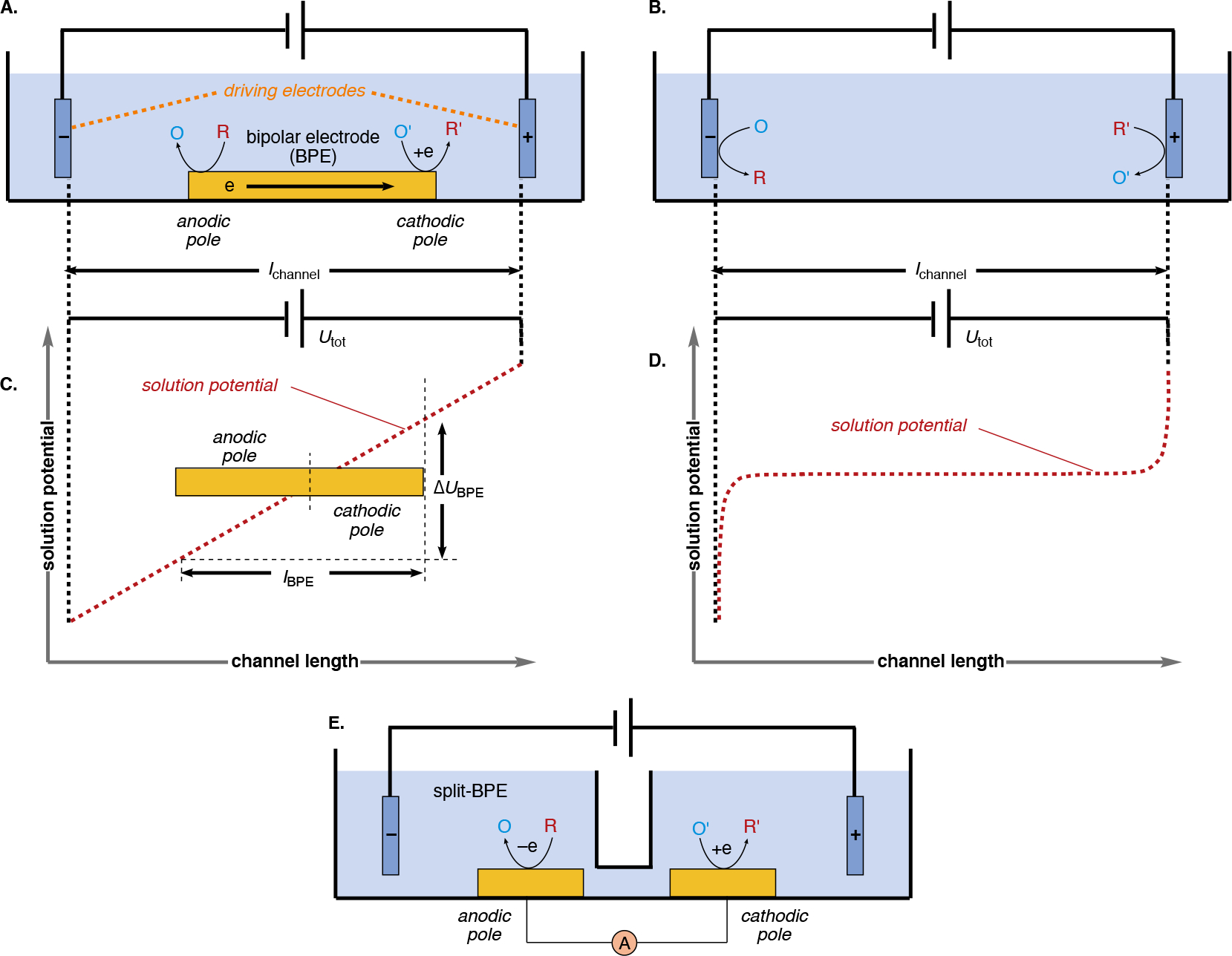
A comparison of the experimental set-ups for open bipolar electrolysis (A) and conventional electrolysis (B). An approximately linear gradient electric field is present in the bulk of the solution between bipolar electrodes due to the low concentration of supporting electrolyte (C), while in conventional electrolysis electric fields are localized at the electrode surface (~exponential decay over tens of Angstroms) as a high concentration of supporting electrolyte is used (D). (E) An example of a closed, split bipolar electrode setup. Note: O, O', R, and R' represent oxidized and reduced species occurring at either the anodic pole of the BPE (facing the cathode driving electrode), or the cathodic pole of the BPE (facing the anode driving electrode).
Several key differences are present in BPE electrochemistry (Scheme 117A) compared to a conventional electrolysis setup (Scheme 117B) that enable the redox chemistry to occur at the BPE and not the driving electrodes.1179 The supporting electrolyte concentration is lower in BPE electrochemistry compared to conventional electrochemistry which results in a high electric field in the bulk of the solution (Scheme 117C) in contrast to the conventional electrochemistry where no significant electric field is present in the bulk (Scheme 117D) as it is localized to the electrode-solution interface. Finally, the potential on the electrode is a gradient on a BPE, in contrast to being uniform in a conventional electrolysis setup. The BPE material is usually selected to have a significantly lower kinetic barrier (i.e. lower overpotential) than the driving electrodes when used in an open BPE setup. Alternatively, multi-compartment setup (i.e. a closed BPE setup) can be utilized in which an insulating wall separates the two driving electrodes. 1180
Bipolar electrochemistry can simplify micro- and nanofabrication of materials such as directional growth of Cu microwires, asymmetric modification of materials, generation of compositional gradients, as well as applications in sensing technologies.1178 To date BPE has been demonstrated to be suitable for the rapid screening of electrocatalysts for the oxygen reduction reaction and has been applied for the concomitant H2 and O2 evolution from aqueous media at the same electrode. Despite these advances, bipolar electrochemistry has not yet made significant impact on organic synthesis. Of the few impactful synthetic examples relevant to organic synthesis using BPE, the synthesis of propylene oxide from propylene gas and sodium bromide electrolyte solution has been reported in flow using a packed bed reactor.1181 Despite the low current efficiency (15 to 55%) due to competing processes such as bromate formation, relatively high space time yields of 10 to 100 kg•m−3•h−1 were achieved near ambient pressures. BPE have made impact in enabling parallel screening of electrocatalysts1182 and will likely continue to bring new impactful directions in the future. A challenge in using BPE for electroorganic synthesis is that in situ monitoring of the current is challenging.320 A noteworthy synthetic application of bipolar electrodes, particularly split bipolar electrodes (s-PBE), is the C(sp3)–H fluorination of triphenylmethane in a U-shaped cell.1183 In contrast to bipolar electrodes, a split bipolar electrode employs an insulator shielding wall in the middle of the BPE, which augments the potential drop around the split electrodes and enables monitoring of the electrical current (Scheme 117E). The reaction is postulated to proceed via a net 2e benzylic oxidation leading to the trityl cation, enabling subsequent nucleophilic fluorination from the fluoride provided by the CsF electrolyte.
5.2. Two Photon-Mediated Photoredox Catalysis
In most photoredox catalytic cycles, the one-photon excitation of a ground state chromophore is typically invoked for the formation of its excited state counterpart. In terms of common excitation wavelengths, irradiation with 365 nm, 390 nm, and 450 nm light leads to respective theoretical upper limits of 3.4, 3.2, and 2.8 eV for electron transfer, although practical values end up lower due to vibrational relaxation and nonradiative pathways.1184 One approach for overcoming this barrier is to use the energy of two photons in a single catalytic cycle, which is best illustrated in the development of challenging aryl halide reductions (Scheme 118), in which the energy of two photon is required to access a reducing excited state chromophore. It is important to note that photoredox-induced aryl reductions have been used as a benchmark,1185 and recent developments in ligand design have led to the bis-cyclometalated iridium photocatalysts with electron-rich beta-diketiminate ancillary ligands, which are potent one-photon photoreductants and accomplish the proto-dehalogenation of bromo-, chloro-, and fluoroarenes, as well as alkyl bromides.1186 Photoexcitation of polysulfide anions (S42−) have also been demonstrated to enable single electron reduction of haloarenes to generate aryl radicals for protodehalogenation, borylation and (hetero)biaryl cross-couplings.1187 However, for the purposes of this review, this section will focus mainly two-photon strategies.
Scheme 118.
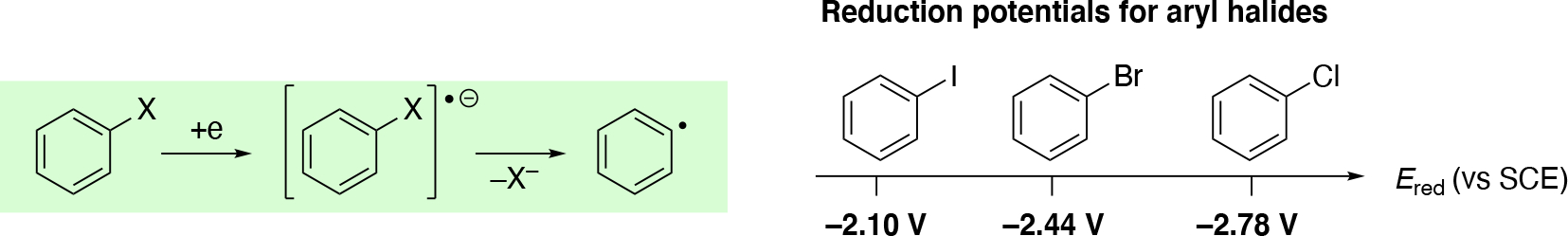
The common mechanism for aryl radical formation (left) and relative reduction potentials for aryl halides (right). Potentials are from reference 438.
One strategy is to access the excited state of a radical anion species, which would then serve as a more potent reductant than the ground state counterpart. König and co-workers demonstrate this concept with a perylene diimide derivative (PDI, 115.1) to accomplish aryl halide reduction via a consecutive PET (conPET).1188 Reductive quenching of photoexcited 119.1 by a sacrificial amine forms a PDI radical anion (119.2), which is photoexcited with blue light to reveal a potent single-electron reductant (119.3) capable of reducing electron-deficient aryl iodides, bromides, and chlorides (Scheme 119). Furthermore, König and co-workers show that the aryl radical intermediate is an effective trap for pyrroles, furnishing the C–C bond forming product at the 2-position of the pyrrole. A follow-up manuscript from the König group uses the redox-dependent absorption of rhodamine 6G (Rh-6G, 119.6) species to enable two-photon, chromoselective reduction of polyhalogenated arenes.1189 Green-light excitation of ground state 119.6 — which can also be excited with blue light — and subsequent reductive quenching yields Rh-6G radical anion (119.7), which reduces electron deficient aryl halides (Ered ≤ −1.0 V vs. SCE). However, blue light irradiation of 119.7 results in the formation of excited state 119.8, which capable of reducing more electron-rich aryl halides (Ered ≤ −2.4 V vs. SCE). The utility of this method was highlighted by chemoselective reduction of polybrominated arenes, wherein monoarylation was observed in the presence of green light whereas diarylated product is obtained when blue light is used.
Scheme 119.
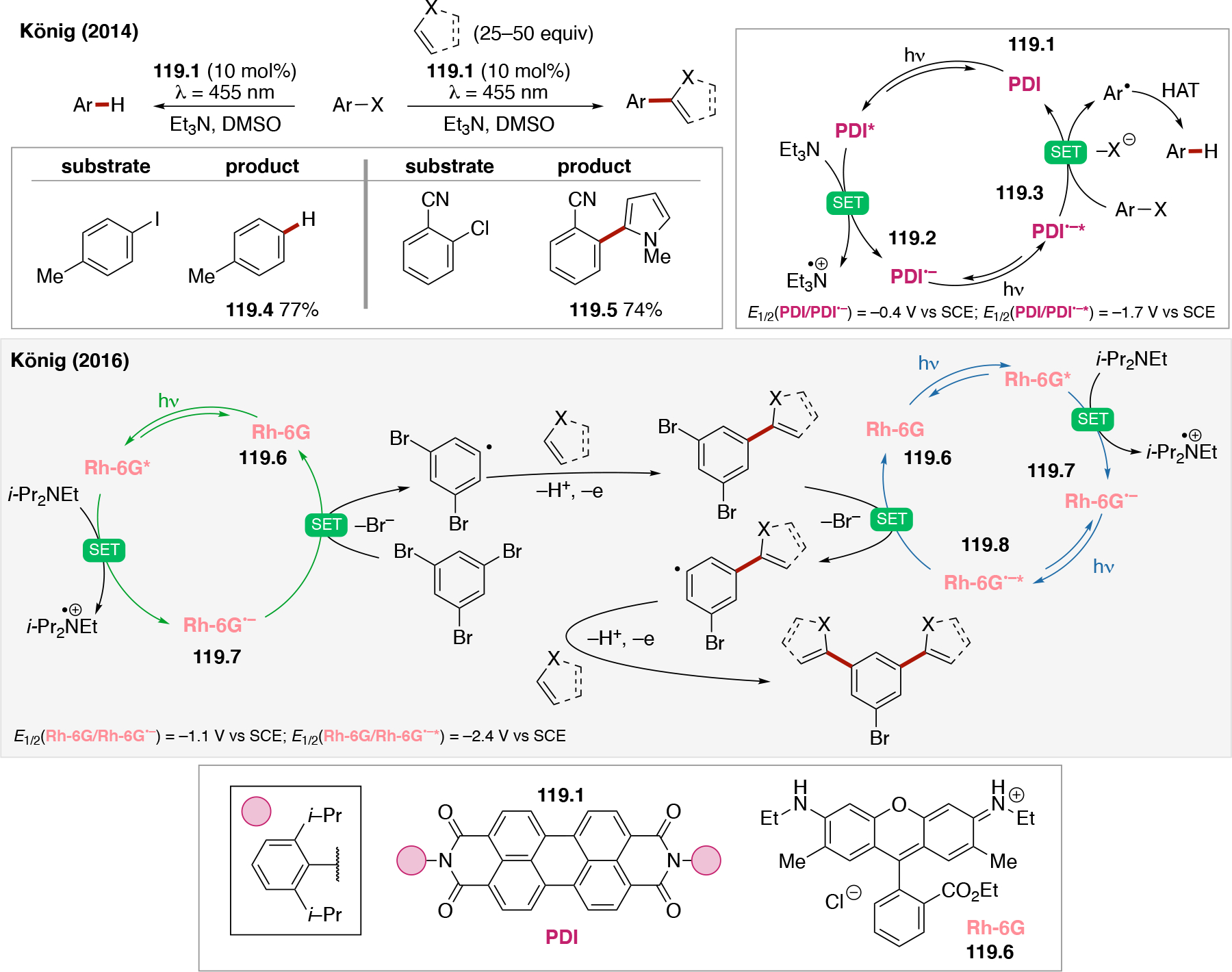
Representative examples of conPET and chromoselective reductions for aryl halides.
Two-photon chromophore excitation can also be applied to transition-metal photoredox catalysts (Scheme 120). Wenger and co-workers demonstrate the formation of a hydrated electron (120.3) in the presence of a sacrificial reductant when a water-soluble tris-sulfonylated Ir(ppy)3 derivative (120.4), is subjected to two laser pulses — first at 430 nm, then at 532 nm — to generate the long-lived excited-state triplet of 120.4 and to promote 120.3 production.1190 This method efficiently applied to the dehalogenation of chloroacetate, monodefluorination of 4-(trifluoromethyl)benzoate and debenzylation of benzyltrimethylammonium cation. Polyzos and co-workers show that the non-innocence of the tbbpy ligand on the IrIII photocatalyst (Ir-1, 11.5) leads to its dearomatization in the presence of HAT-capable reductants and results in the formation of an IrIII chromophore (Ir-2, 120.9) with a greater excited state oxidation potential (E(PC+/PC*) −1.70 to −3.0 V vs. SCE) than excited state 11.5 (−0.96 V vs. SCE).1191 This enables the reductive dehalogenation of aryl halides and the reductive formal hydrogenation of 1,1-diaryl olefins and α- or β-substituted styrenes, with the latter method also enabling the anti-Markovnikov alkene hydrofunctionalization with ketones.1192
Scheme 120.
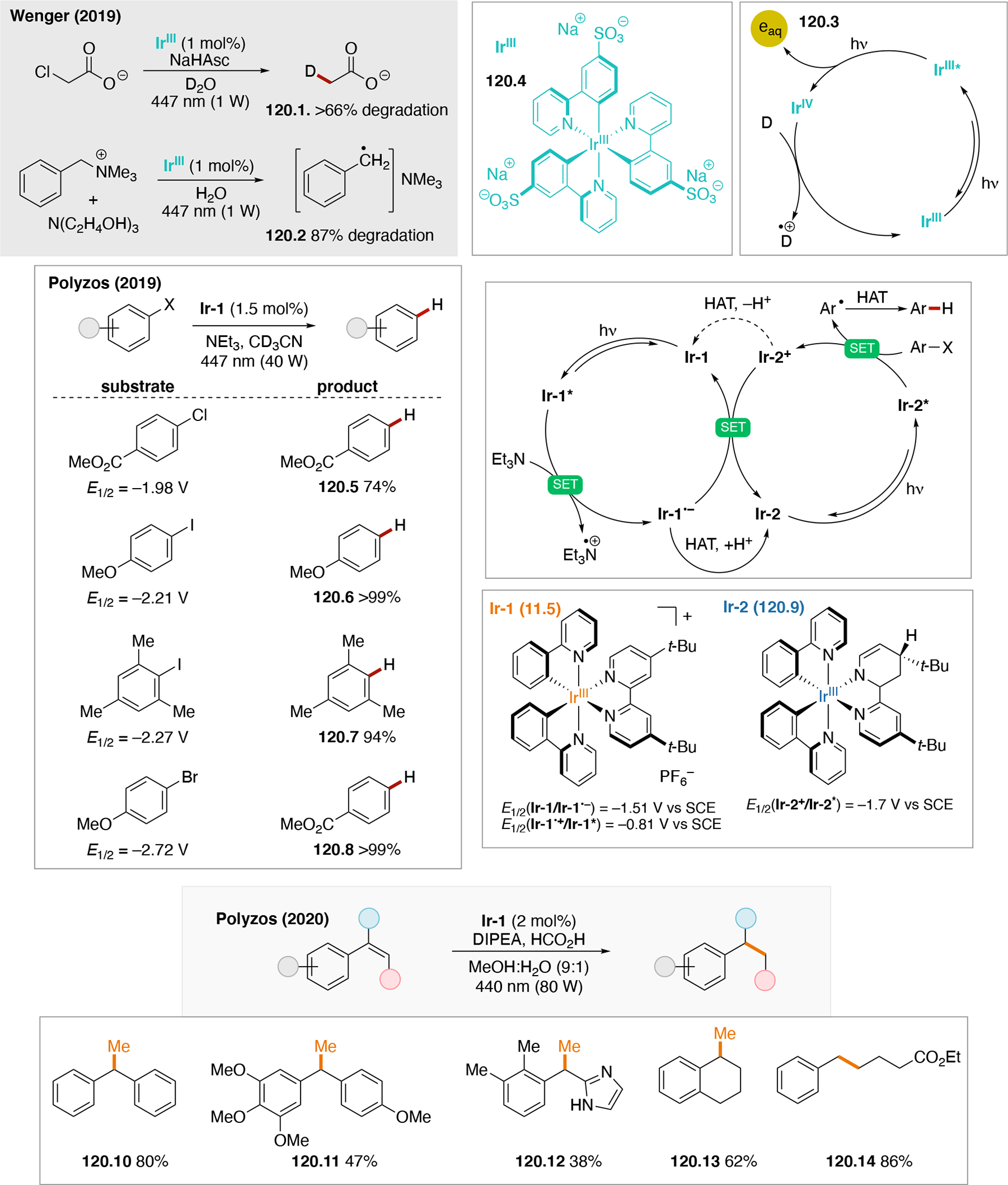
Representative examples of two-photon excitation of a chromophore to access highly reducing photoredox organometallic catalysts.
Lastly, this two-photon absorption strategy can also be applied to the excitation of neutral radical organic chromophore, as shown by Nicewicz and co-workers (Scheme 121).1042 Reductive quenching of excited state acridinium (121.9) by a sacrificial tertiary amine leads to a stable acridine radical (121.10) that can be photoexcited with 390 nm light to access two excited states — a lower energy doublet and a higher energy twisted intramolecular charge-transfer (121.11) with respective oxidation potentials of −2.91 and −3.36 V vs. SCE. This highly-reducing photoexcited acridine is capable of electron-rich aryl bromide and chloride protodehalogenation, as well as reductive detosylation of N-aryl and N-alkyl tosylamines — a notable deprotection strategy that is historically limited to strong acids, dissolving metal, and low-valent transition metal reductions (electrochemical strategies exist, see section 4.18.2). Overall, these methods demonstrate the growing interest in multiphoton excitations for accessing highly reducing species for photoredox catalysis and may yet reveal new highly oxidizing chromophores.
Scheme 121.

Representative examples of two-photon excitation of an organic chromophore to access highly reducing photoredox catalysts.
5.3. Electrophotochemistry – Combining the Best of Both Worlds
Combining aspects of photoredox and electrochemistry dates back decades – in fact electrophotochemistry has been investigated since the 1970s. The field is experiencing a resurgence of interest, especially within the context of organic synthesis, with several recent minireviews/highlights1193,1194,1195 providing an overview. It is important to clearly distinguish electrophotochemistry (EPC) from photoelectrochemistry (PEC).29,1196,1197 Photoelectrochemistry — sometimes referred to as interfacial photoelectrochemistry (iPEC)1193 — refers to light-induced sensitization of a semiconductor electrode material (e.g. photoanode) to generate a photocurrent which promotes a chemical transformation.1198 In contrast, electrophotochemistry — sometimes referred to as electrochemically mediated photoredoxcatalysis (e-PRC)1193 — refers to the use of electrochemistry to generate an intermediate via a redox event, which is then photoexcited. This photoexcited intermediate is usually a catalyst which experiences highly potent redox properties (e.g. photoexcited radical anion of 9,10-dicyanoanthracene is highly reducing, Eox* = −3.2 V vs SCE; photoexcited dicationic trisaminocyclopropeniums can be highly oxidizing, Ered* = +3.3 V vs SCE). We also note a third category exists in which the electrochemistry and photochemistry have separate, discrete roles, termed decoupled photoelectrochemistry (dPEC), but few examples of synthetic transformations via this strategy have been disclosed.898,1193,1199 Additionally, electrophotochemistry has the potential to make photochemical reactions greener by replacing sacrificial reductants or oxidants used in photoredox reactions. The redox event at an electrode can be used for photocatalyst turnover, which when coupled to desirable half-reactions such as hydrogen evolution, can minimize waste production.
A more synthetic-focused reason for using electrophotochemistry is its ability to generate highly reductive/oxidative species under mild conditions (Schemes 122A and 122B). Lambert and Nuckolls demonstrated that cyclopropenium perchlorate photocatalyst 122.1 could be oxidized to its dicationic form and upon its photoexcitation would generate a highly oxidizing catalyst (ca. +3.3 V vs SCE) enabling oxidative C–H functionalization of unactivated arenes for C–N bond formation (Scheme 122A).1200 Lambert and Huang also report a electrophotochemical cation radical accelerated SNAr994a (CRA-SNAr) of unactivated fluorobenzenes with nitrogen and oxygen nucleophiles, which is enabled by pairing a DDQ photocatalyst with CPE.1201 This method is best compared to analogous photooxidatively-mediated CRA-SNAr reported by Nicewicz1202 and Zhou1203, where highly oxidizing xanthylium and acridiniums are used to facilitate similar transformations. Wickens and coworkers also demonstrate that PTH (11.11) can be electrochemically oxidized to its corresponding cation radical, which is sufficiently oxidizing to generate arene cation radicals from benzene, toluene, chlorobenzene, xylenes, and mesitylene for arene C–H amination with pyrazoles.1204 Lambert recently demonstrated an electrophotochemical C–H heterofunctionalization approach using DDQ as the catalyst and blue LEDs in an undivided cell electrolysis setting.1205 The use of water, alcohols, or carboxylic acids as a nucleophile provided direct access to phenols, arylethers, and aryl esters, respectively, via C–O bond formation. Further extension with nitrogen nucleophiles was demonstrated with benzamide, ethyl carbamate, urea, acetamide, and sulfonamide derivatives, via C–N bond formation.
Scheme 122.
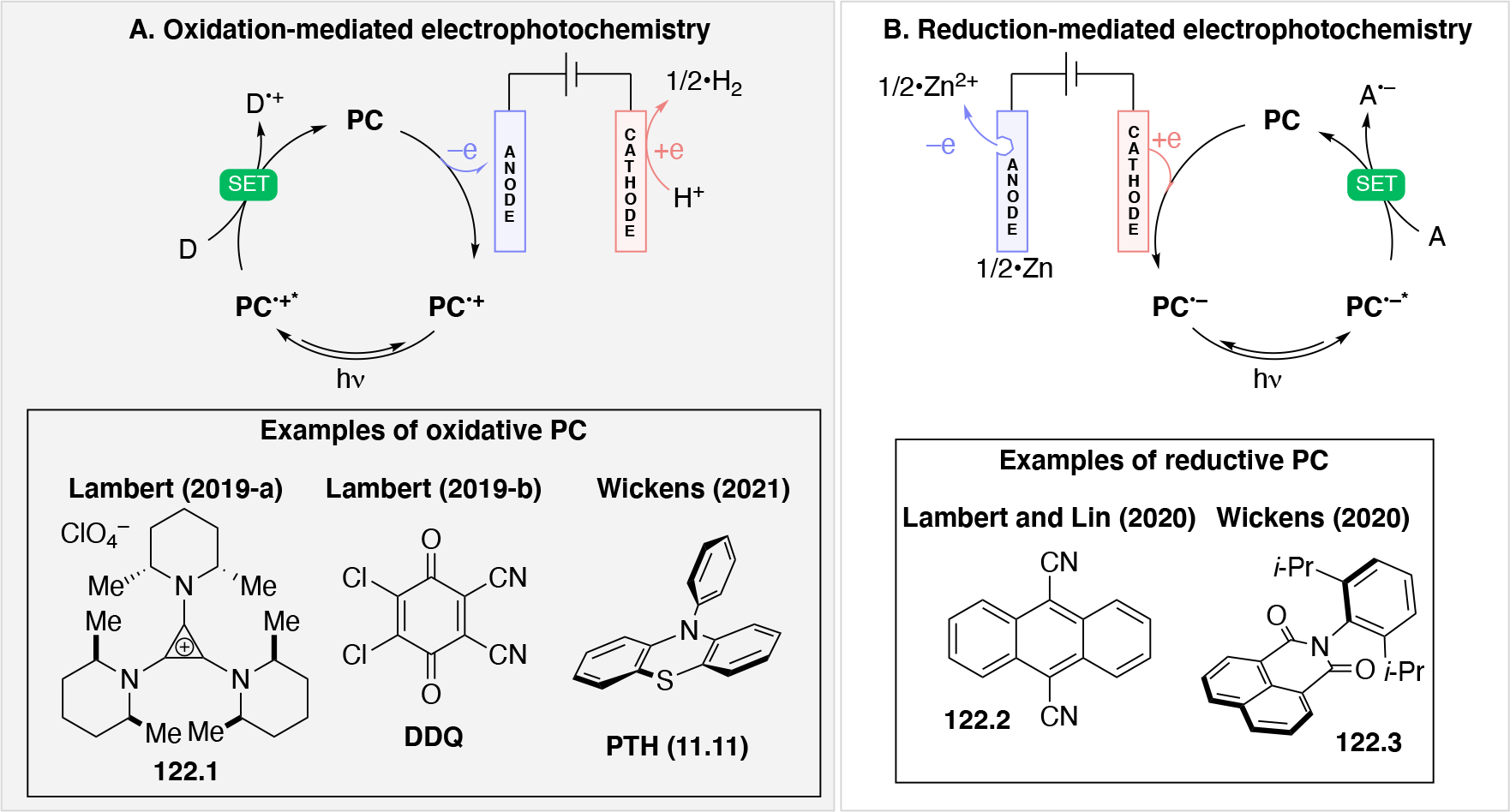
Oxidative and reductive electrophotochemistry along with selected examples of catalysts.
Electrophotochemistry can also be used to generate highly reductive catalysts (Scheme 122B). One example is from the Lambert and Lin labs, where CPE is used to reduce 9,10-dicyanoanthracene (122.2) to its radical anion, which is then photoexcited to generate a highly reducing catalyst (ca. −3.20 V vs SCE) to enable reduction of unactivated aryl chlorides and bromides, and the subsequent trapping of the resultant aryl radical with boron, tin and heteroaryl electrophiles.1206 The electrophotochemical reduction of aryl chlorides was also independently reported by Wickens and coworkers, where a highly reducing catalyst (ca. −3.3 V vs SCE) is formed via photoirradiation of an electrochemically-reduced N-aryl naphthalene imide (122.3) radical anion.248h
5.4. Emerging Alternatives to Blue-light Activated Photoredox
There is a growing interest in using low-energy light as improved reaction media penetration can potentially enable efficient photoredox reactions on scale and allow for biological applications that do not suffer from phototoxicity associated with high-energy light.1207,1208 However, the challenge with this approach is that most widely-used photoredox catalysts require near-UV (365 nm = 78.3 kcal•mol−1, 390 nm = 73.3 kcal•mol−1) and blue (450 nm = 63.5 kcal•mol−1) light for activation.1209,1210
5.4.1. Triplet Fusion Upconversion.
Triplet fusion upconversion (TFU) is a strategy that converts low-energy photons to higher energy photons through triplet donor (sensitizers) and acceptor (annihilator) molecules1211 (Scheme 123). Two long-lived triplet sensitizers (3S) formed through stepwise 1MLCT excitation and ISC, or by direct 3MLCT, transfer their energies to two discrete annihilators (1A) to form two long-lived triplet annihilators (3A). Triplet annihilation between two 3A* species generates a higher energy singlet exciton on one annihilator (1A*) and a quenched partner (1A), with the restriction that E(1A*) < 2 x E(3A*). The excited singlet 1A* can then either emit a high energy photon via fluorescence or participate in single-electron redox transfer as a photoredox catalyst. Congreve, Rovis, Campos and co-workers recently reported near-infrared (NIR) photoredox catalysis using Pd- and Pt-based sensitizers and organic annihilators (Scheme 124).1212 Using palladiumII octabutoxyphthalocyanine (124.1) as the sensitizer with furanyldiketopyrrolopyrrole (124.2) as an annihilator1213 enables upconversion of NIR photons (730 nm, 38 kcal•mol−1) to orange region (λem,max ~ 550 nm) whereas using the sensitizer platinumII tetraphenyltetranaphthoporphyrin (124.3)1214,1215 coupled with the annihilator, tetrakis(tert-butyl)perylene (124.4) enables NIR-to-blue photon upconversion (λem,max ~ 460 nm). The former system successfully enables Eosin Y-mediated hydrodehalogenation and reductive cyclization, as well as Rose Bengal-catalyzed amine oxidation1216 and Eosin Y catalyzed1217 radical cyclization. The TFU system with 124.3 and 124.4 were successfully used to excite Ru(bpy)32+ for an intramolecular [2+2] reaction926, and additional experiments demonstrate that the two-component system is capable of photoredox catalysis. More importantly, this TFU system demonstrates the enhanced material penetrance of NIR light, as the photo-ATRP of MMA1218 was demonstrated in the presence of several visible light-absorbing barriers; only NIR irradiation generated a freestanding gel of crosslinker-doped MMA (5% ethylene glycol dimethacrylate).
Scheme 123.

Representative comparisons of energy for UV, blue and NIR light (left) and a general schematic for triplet fusion upconversion (right).
Scheme 124.

Triplet fusion upconversion for photoredox catalysis.
TFU has also been successfully applied to aqueous systems (Scheme 125), as demonstrated by Wenger and co-workers in their green light (532 nm)-mediated monodehalogenation of trichloroacetate using Ru(bpy)32+ as a sensitizer and the water-soluble annihilator anthracene-9-propionate (125.2).1219 Interestingly, this reaction proceeds in air-saturated water despite the facile quenching of triplet sensitizer or annihilator by oxygen gas to form singlet oxygen. This observation is rationalized by the low O2 solubility in water (~0.27 mM), which is tenfold lower than in common organic solvents.1220 Wenger and co-workers build on this work by reporting methods for blue (420 nm)-to-near UV upconversion λem,max ~320 nm) and photoreduction using sulfonylated IrIII catalyst 125.3 as the sensitizer and the annihilators 1,5-naphthalenedisulfonate (125.4) and naproxen (125.5).1221 The latter method was applied towards the reductive debromination of 4-bromo-2-chloro-5-fluorobenzoate and debenzylation of benzyltrimethylammoniums in water, owing to the highly reducing nature of photoexcited singlet naproxen (−2.7 V vs. SCE). With these examples, the future of TFU for photoredox catalysis is bright, with sensitizer and annihilator development driving advances in this rapidly growing field.
Scheme 125.
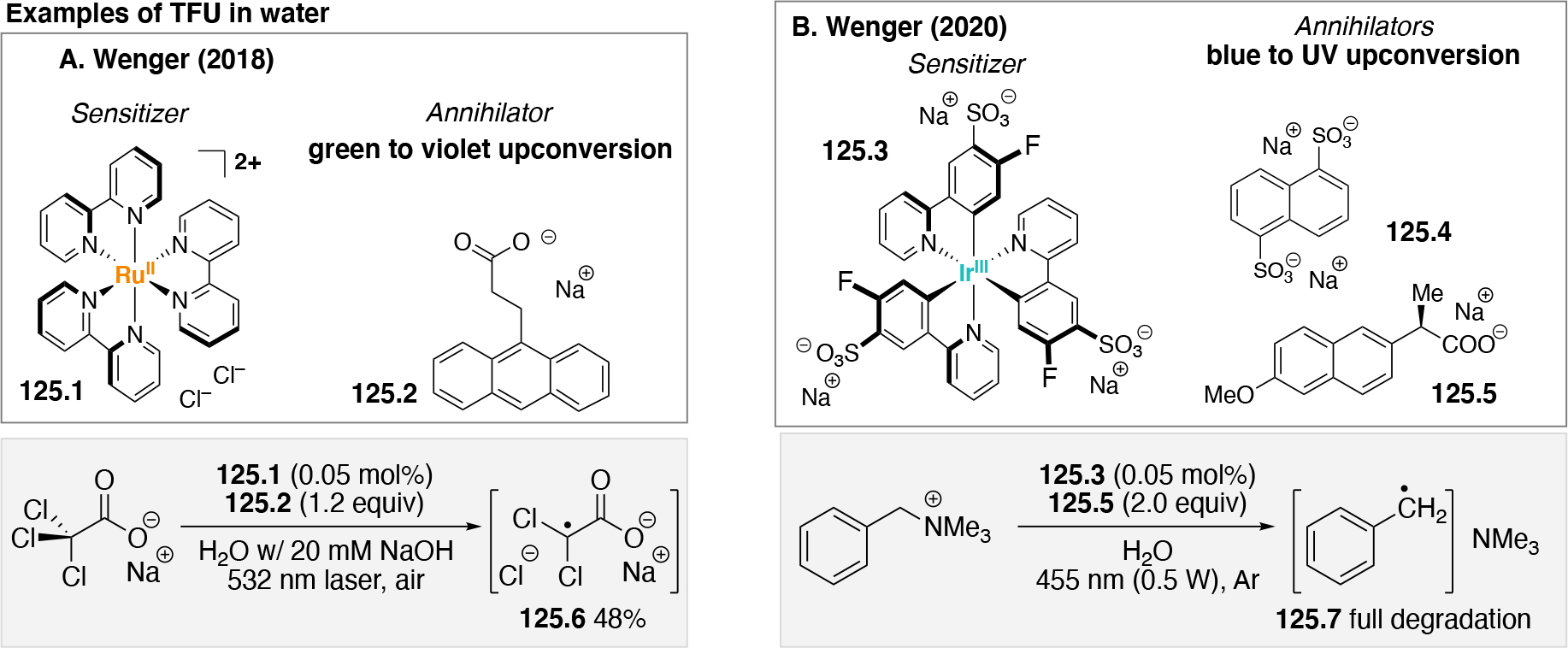
Triplet fusion upconversion in aqueous systems.
5.4.2. Photoredox Catalysis with DR/NIR-absorbing Chromophores.
An alternative strategy is to use deep red (DR; λ = 660 nm) and NIR-light absorbing chromophores to generate excited-state species capable of electron transfer (Scheme 126). Turro and co-workers report dihydrogen formation using a DR-excited dirhodiumII photosensitizer; this catalyst performs two step-wise DR-mediated oxidations to generate a doubly reduced Rh2 species (126.1) capable of converting two protons into dihydrogen.1222 More recently, Rovis, Joe, and co-workers report the development of DR and NIR-photoredox catalysis using OsII chromophores (126.2–126.4).1223 The versatility afforded by these photoredox catalysts is observed in successful redox-based photopolymerizations, redox-initiated synthesis, and metallaphotoredox transformations. Additionally, the excited and ground state redox potentials of these photocatalysts can be modulated through modifications to the terpyridine, azine, and phenanthroline ligand scaffolds, thus enabling their rational design. Lastly, NIR photoredox catalysis enables mole scale arene trifluoromethylation using a batch reactor. This observation is notable given that arene trifluoromethylation yields drop when using blue light and Ru(bpy)32+, whereas using NIR with an Os chromophore (126.4) leads to a consistent yield increase for scale-comparative transformations. A concurrent report by Gianetti and co-workers show that a helical carbenium ion (126.8) operates as a DR organic photoredox catalyst.1224 These recent reports are encouraging signs for future development of photoredox reactions using DR and NIR light to mediate electron-transfer mechanisms for organic synthesis.
Scheme 126.
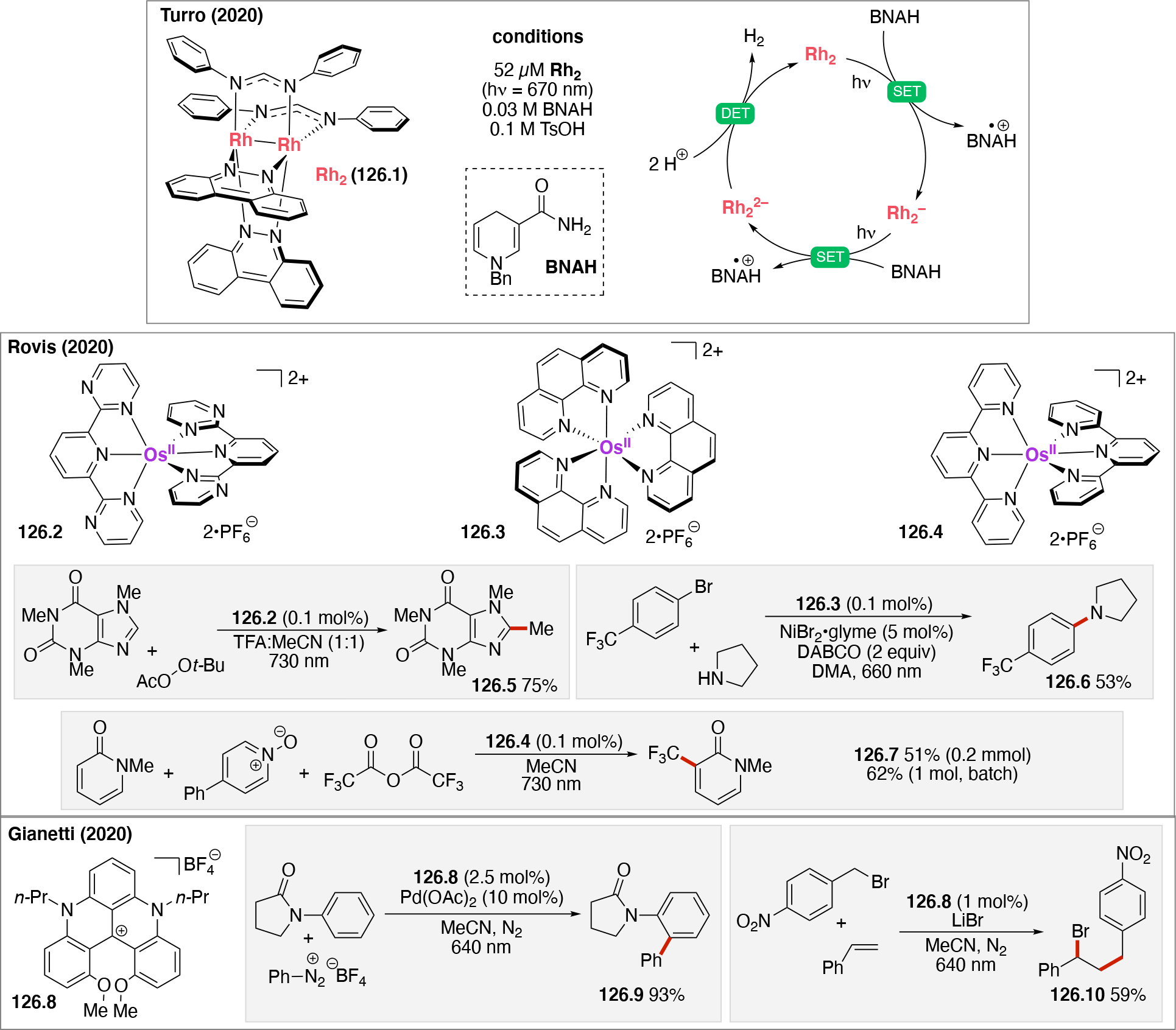
Representative examples of photoredox catalysis with DR/NIR-absorbing chromophores.
5.5. Singlet Fission.
Singlet fission is in effect the reverse process of triplet-triplet fusion upconversion. Singlet fission is the process of using one photon to generate a singlet which then spontaneously generates two triplets. The energetics require that the energy gap between the S1 and So state are greater than twice the energy gap between the T1 and So state. While singlet fission1225,1226,1227,1228,1229,1230,1231,1232 continues to be of interest within the solar cell community due to the promise of improving photon-to-current conversion efficiencies beyond the 30% Shockley–Queisser limit, applications in synthetic photocatalysis are limited. Intramolecular singlet fission (iSF) whereby two chromophores are covalently linked to enable the generation of two triplets on two chromophores in close proximity has been demonstrated, for example in pentacene dimers.1225 A key example is from Steigerwald, Sfeir, Campos, and coworkers, where a bridged pentacene dimer undergoes near perfect iSF in ~ 220 ps to yield two long-lived pentacene triplets (270 ns).1233 The mechanism for iSF is highly dependent on the linker length and identity, as iSF can lead to the formation of the triplet pair directly from the photoexcited pentacene or it may require access to charge-transfer states (Scheme 127A).1234 A discussion of the photophysical considerations is outside the scope of this review. We envision that iSF can be leveraged to generate two separate electron hole/donor pairs, and thus may enable new ways to carry out doubly reductive (or doubly oxidative) transformations using a single photocatalyst, akin to common electrochemical reactions occurring at a single electrode (Schemes 127B and C). A photocatalytic process relying on singlet fission could enable new transformation or selectivities not attainable via electrochemistry, potentially even utilizing a combination of redox and energy transfer (triplet-sensitization) processes (Schemes 127D and E).
Scheme 127.
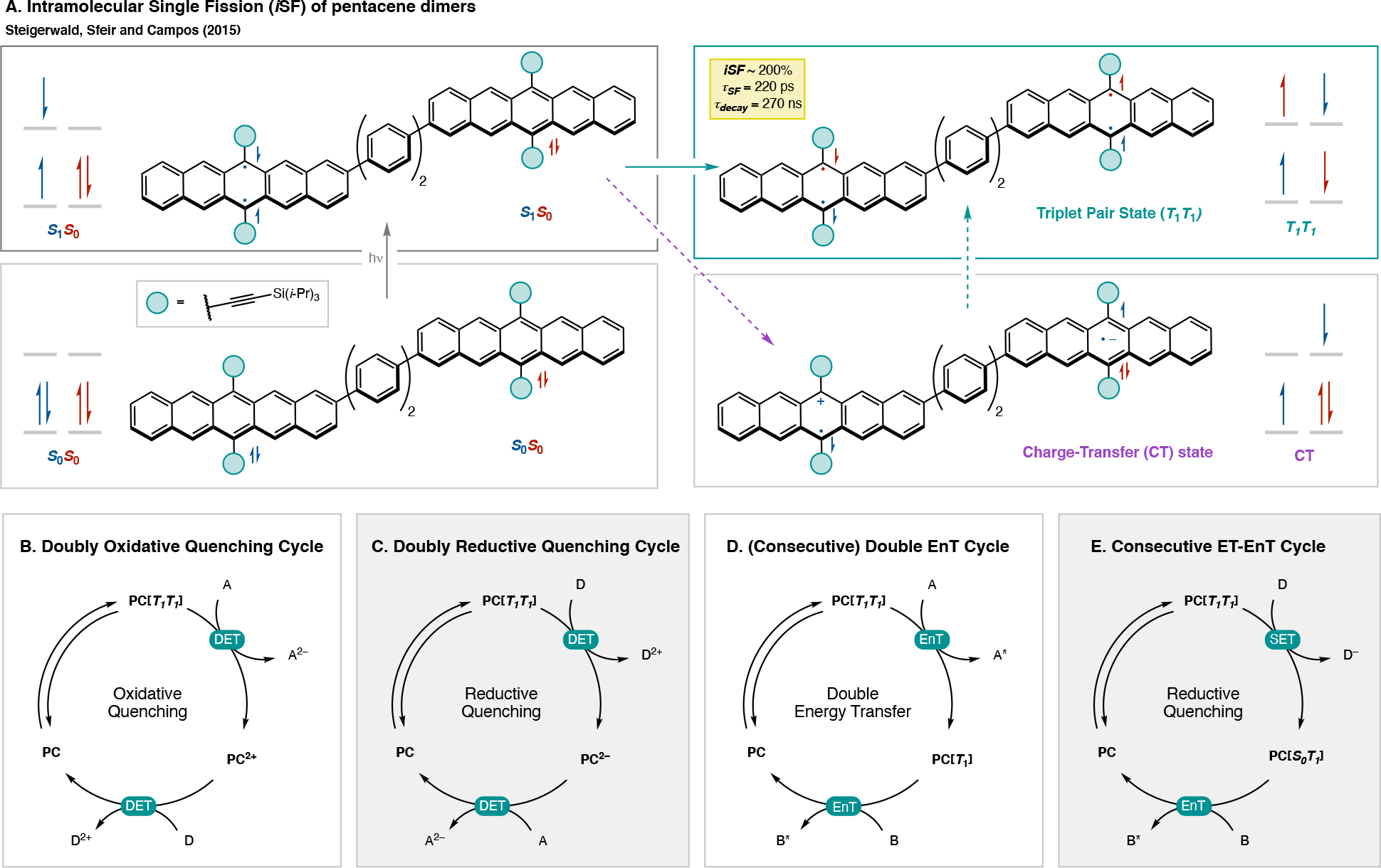
(A) The mechanism for intramolecular singlet fission for bridged pentacene dimers. (B–D): Potential outcomes when leveraging two triplets from singlet fission for (B) a doubly oxidative quenching cycle, (C) a doubly reductive quenching cycle (2e redox events), (D) double energy transfer (EnT) cycle, (E) a consecutive ET-EnT cycle (the analogous process where the EnT precedes the ET step can also be envisioned).
5.6. Machine Learning/QSAR.
The use of machine learning coupled with automation tools for the discovery and optimization of chemical reactions is an attractive direction.1235,1236 Predictive models for the classification of excited states of heteroleptic IrIII-photocatalysts based have recently been developed.1237,1238 While thermodynamic based predictors like redox potential can be used to rationalized outcomes of reactions using various photocatalysts, the kinetic components related to quenching of the excited state photocatalyst by various substrates is also a critical factor. To being to address this, predictive models recently have been developed.1239 Applications of quantitative structure activity relationship (QSAR) models using electroanalytical parameters are emerging and may impart new machine learning approaches to developing new transformations as well as understanding trends and limits of existing synthetic methods.1240,1241,1242 Design of experiments (DoE) techniques are routinely applied in industrial applications for the optimization of reactions. Examples of using DoE techniques in the context of optimizing electrochemical reactions have been reported to successfully optimize factors such as stir rate, inter-electrode spacing, concentration of reactants, total charge, and current density.1243 Machine learning algorithms have been used to build predictive models for optimization of membranes,1244 cathode materials,1245 as well as electrolyte composition.1246 Progress towards being able to predict the outcome (reaction yield) of electro-synthetic experiments using machine learning algorithms based on multiple electrochemical parameters (electro-descriptors) such as onset potential, Tafel slope, and effective voltage were demonstrated in the context of a variety of transformations ranging from phosphorylation of tetrahydroquinolines, aziridination of alkenes, TEMPO-mediated dehydrogenation of N-Heterocycles.1247 Machine learning algorithms have also been demonstrated successful in the context of optimizing of the dimerization of acrylonitrile to form adiponitrile via alternating current electrolysis.1161 In this case the duration of the pulse as well as the current density and the presence/absence of a quiet period were critical parameters and were optimized via artificial intelligence to minimize side reactions and maximize yield. Advances in predictive models related to photoredox chemistry such as excited state energies, redox properties and Stern-Volmer quenching rates have emerged with an aim to discovering and understanding reactions.1239,1248
6. Concluding Remarks and Outlook
This review provides a detailed comparison between electrochemistry and photochemistry, focusing primarily on the fundamental aspects pertinent to both methods. While we present key synthetic transformations shared by both photochemistry and electrochemistry, there is a much larger volume of chemical space in which there is a lack of corresponding overlap between either photoredox or electrochemistry. We believe that this empty space signals opportunities for the discovery and development of complementary synthetic methods — each with their own advantages and limitations — and further contributions of unexpected discoveries (e.g. side reactions/unexpected reactions, new reaction concepts, novel catalysts, improved functional group tolerance).
The wealth of synthetic methods presented in this review not only highlights the creativity and utility of redox chemistries (i.e. electrochemistry and photoredox catalysis) but also supports their kinship. Subtle limitations may arise when trying to translate a photoredox method into an electrosynthetic method (and vice versa) and detailed mechanistic studies should help improve synthetic translations between the two strategies. For example, the generation of intermediates at the electrodes after the first redox event may be prone to undergo a second redox event (i.e. net doubly oxidative process as in the Hofer-Moest decarboxylation to the carbocation instead of the alkyl radical as observed in Kolbe electrolysis). The choice of electrode material and/or the current density can aid in tuning the reaction between the 2e pathway (Hofer-Moest is favored by the use of graphite anode and high current densities) or the 1e process (Kolbe electrolysis is favored by the use of Pt anode and lower current densities in the decarboxylation example above). Additionally, redox mediators can be used to suppress 2e redox chemistry and the effect of the electrode material on the redox outcome of the substrate. Likewise, consecutive electron transfers via emerging photoredox strategies can help translate electrochemical reactions that redox-neutral photoredox methods struggle with.
The traditional limitation of electrochemical synthetic methods to net-reductive and net-oxidative transformations is due to technological limitations imposed by electrochemical cell designs and available operating modes. In particular, the large inter-electrode distance (>0.5 cm) prevents transiently generated radical intermediates from efficient migration from one electrode to the other to achieve a redox neutral transformation. Thus, recent advances in electrochemical microflow cell designs and the re-emergence of alternating potential/current electrolysis provide two useful pathways forward to achieve redox neutral reactions which have been typically better suited for photoredox catalysis. The APE/ACE approach has the benefit of not being restricted to homogenous solution required for the electrochemical microflow cell. Continuous processing via adaptation of APE/ACE into a CSTR will enable the processing of heterogenous/slurry mixtures in net redox neutral processes (avoiding clogging issues with heterogeneous reactions that plague flow cells). An exciting path forward is the integration of electrochemistry and photochemistry — namely, electrophotochemistry — in which an electrochemically-generated persistent intermediate is photoexcited to enable access to highly potent oxidants and reductants to bypass the use of direct electrolysis with extreme potentials. We expect new electrocatalysts with tuned radical lifetimes and redox potentials (pre- and post-photoexcitation) will accelerate discoveries in this area of research. We believe that major advances await in both redox-neutral electrochemistry and electrophotochemistry are areas and new developments in these fields will result in the discovery of new and improved synthetic transformations. Engineering innovations are also key to success for both fields. Improved cell design to modulate and control light input reproducibly and improvements membrane technologies are necessary. The ability to screen and monitor electrochemical and electrophotochemistry reactions in a high-throughput fashion is necessary and currently lags behind photochemical solutions.391
While at first sight one may postulate that photocatalysis and electrochemistry would differ in activation methods, given that electrochemistry involves only the ground state potential energy surface, whereas photochemistry also involves both the ground and excited state potential energy surfaces. However, this is not necessarily correct. While the photocatalyst may operate on the excited potential energy surface (PES), the redox event with substrates/reagents results in oxidized/reduced forms of the substrate/reagent still on the ground state energy surface. Only in photochemical examples where no photocatalyst is used and direct excitation of the substrate/reagent leads to the desired redox process via an excited state or excited EDA complex will the substrate be on the excited state PES. The use of energy transfer in photocatalysis enables for tandem processes in which both an EnT catalytic cycle and a photoredox cycle may operate,1249 something that is not achievable using electrochemistry alone – yet nothing precludes a user from combining photosensitization and electrochemistry. EnT can be useful not only for alkene isomerization,1250,1251 photoderacemization processes,1252 rearrangements or 4n p-cycloadditions/electrocyclizations (e.g. [2+2]-cycloadditions) but also for fragmentations reactions such as σ-bond cleavage (e.g. N–O bond cleavage of oximes),787,1137 as well as accessing spin selective transformations (triplet vs singlet reactivity such as in nitrene chemistry1253 and reductive elimination from transition metal complexes.1153,1254 Additionally, the electron flux (current) is coupled to potential (Ohm’s law) for electrochemistry, while the photon flux is not coupled to photocatalyst redox potential in photoredox catalysis. However, because the redox window accessible for a photocatalyst is limited by the input energy of an absorption wavelength, limitations will surface if one uses higher energy photons (shorter wavelength) than conventionally-used irradiation sources (e.g. hν < 365 nm). Specifically, organic substrates start to become competitive absorbers of light relative to the photocatalyst, which may lead to decreased efficiency and undesirable side reactions.
Perhaps one of the most significant differences in the comparison is that reduction (at cathode) and oxidation (at anode) are spatially separated in electrochemistry, unless performing alternating current/potential electrolysis in which both redox events occur at the same electrode. This contrasts with photochemistry where both redox events occur at the photocatalyst, thus occur spatially close, namely not significantly further than the distance the photocatalyst can diffuse during the lifetime between the first and the second (and opposite) redox event associated with the photocatalyst’s catalytic cycle. The other major difference being that multielectron redox events are more easily achieved via electrochemistry although we have highlighted several examples of consecutive SET events via photochemistry. Additionally, the use of high-powered light sources (e.g. lasers) may enable the concentration of excited states to be high enough to render ternary reaction events (two excited photocatalyst being able to simultaneously transfer an electron each to the substrate to achieve a multi-electron transfer event).
More trivial differences are related to reaction mixture composition; including the common use of electrolytes in electrochemistry to reduce the resistance of the reaction mixture to facilitate conductivity between the two electrodes; whereas no electrolyte is needed in photochemistry. Electrochemical reactions can also proceed in the absence of an electrolyte when the solution resistance between the two electrodes is low enough (e.g. very small electrode separation or the reagents used in the reaction mixture can provide the needed mixtures of low resistance). Green chemistry aspects unique to electrochemistry revolve about the amount of electrolyte used, often stoichiometric or super-stoichiometric relative to the limiting reagent. Towards addressing this challenge, solutions have either focused on recycling1255 of the electrolyte or minimizing/eliminating it altogether via leveraging flow electrolysis where the electrode separation distance is small enough that solution resistance in the absence of electrolyte is negligible.
While the focus has been on the pathway leading to the desired product and metrics such as yield and efficiency (e.g. the Faradaic efficiency in electrochemistry and the quantum yield in photochemistry), what happens in the non-productive pathways and how do those compare between electrochemistry and photochemistry? In electrochemistry, the non-productive consumption of electricity includes redox events of species not associated with productive pathway but also with non-Faradaic current consumption (charging of the double layer) and external energy loss due to electrical resistance in the setup (leading to Joule heating). In photochemistry, photons can be wasted due to the photocatalyst not absorbing the photon, or absorbing the photon to reach an excited state but the excited state of the photocatalyst returning to the ground state via (a) internal conversion (photon to thermal heat generation) or (b) emission of light, either via fluorescence or phosphorescence depending on the nature of the excited state (e.g. S1 vs T1). Undesired redox events may also consume photonic energy via the photocatalysts oxidizing or reducing a reagent/intermediate in a non-productive manner. Energy loss associated with the light source (e.g. low efficiency in electrical to light energy conversion can also result in low energy efficiency of the setup and significant heat generation at the light source.
Twenty years into the 21st century and it is undeniable that we are experiencing a renaissance in both electrochemistry and photochemistry. Many challenges lie ahead of us, not only in the synthetic realm but also global environmental challenges such as climate change, energy crisis and pollution. Research in photocatalysis and electrochemistry are starting to yield innovations as solutions to the aforementioned problems. For example, converting greenhouse gases such as CO2 into valuable feedstock chemicals1256,1257 via CO2 fixation1258 methods is a first step in shifting away from commodities based on fossil fuels, 1259 and the use of water splitting1260 is a key step towards greener fuel sources (e.g. water powered fuel cells replacing fossil fuel combustion engines). Both hydrogen evolution reaction1261,1262 and oxygen evolution reactions1263 are subjects of years of intensive research.1264 Additionally, the merger of photo- and electrochemistry in the context of photoactive electrodes (photoelectrochemistry) also plays a role in solar fuel research.1265 Furthermore, energy conversion technologies, including those related to solar and wind energy conversion to electrical energy, energy storage technologies (fuels & batteries, including redox flow batteries) continue to be a challenge of which both photo- and electrochemistry are crucial in providing solutions.1266,1267,1268,1269 Additionally, both photo- and electrochemistry have long played an important role in water treatment/remediation via the oxidation of organic pollutants in water,1270,1271,1272,1273 and more recently in the desalination of water.1274,1275 This need is most urgently felt in the remediation of per- and polyfluoroalkyl substances (PFAS) — termed “forever chemicals” due to their persistence in the environment and their inertness towards breaking down in the environment — as their presence poses significant adverse health effects (e.g. cancer, immunotoxicity, metabolic diseases, and neurodevelopmental disorders). One promising solution is the electrochemical oxidation of PFAS,1276 and this breakthrough is likely linked to the recent popularization of BDD electrodes, which have excellent resistance to fouling under high current density conditions. While non-exhaustive, we hope that these topics clearly solidify the need for continued research in both photoredox and electrochemical methods to address pressing needs of global significance.
Scheme 29.
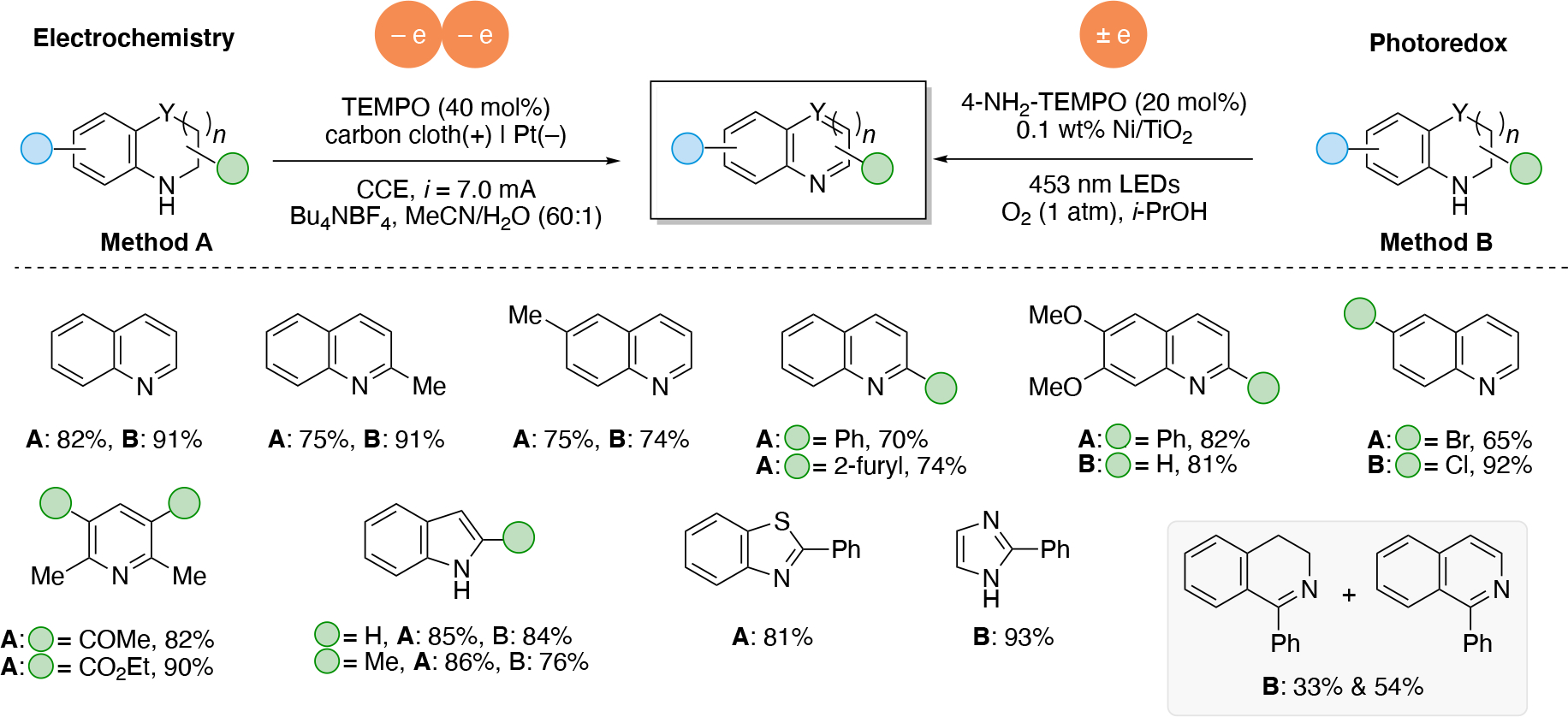
Dehydrogenation/aromatization of N-heterocycles via electrochemical or photoredox methods.
Scheme 32.

The putative mechanism for electrochemically-mediated nickel-catalyzed C–N cross coupling.
Scheme 73.
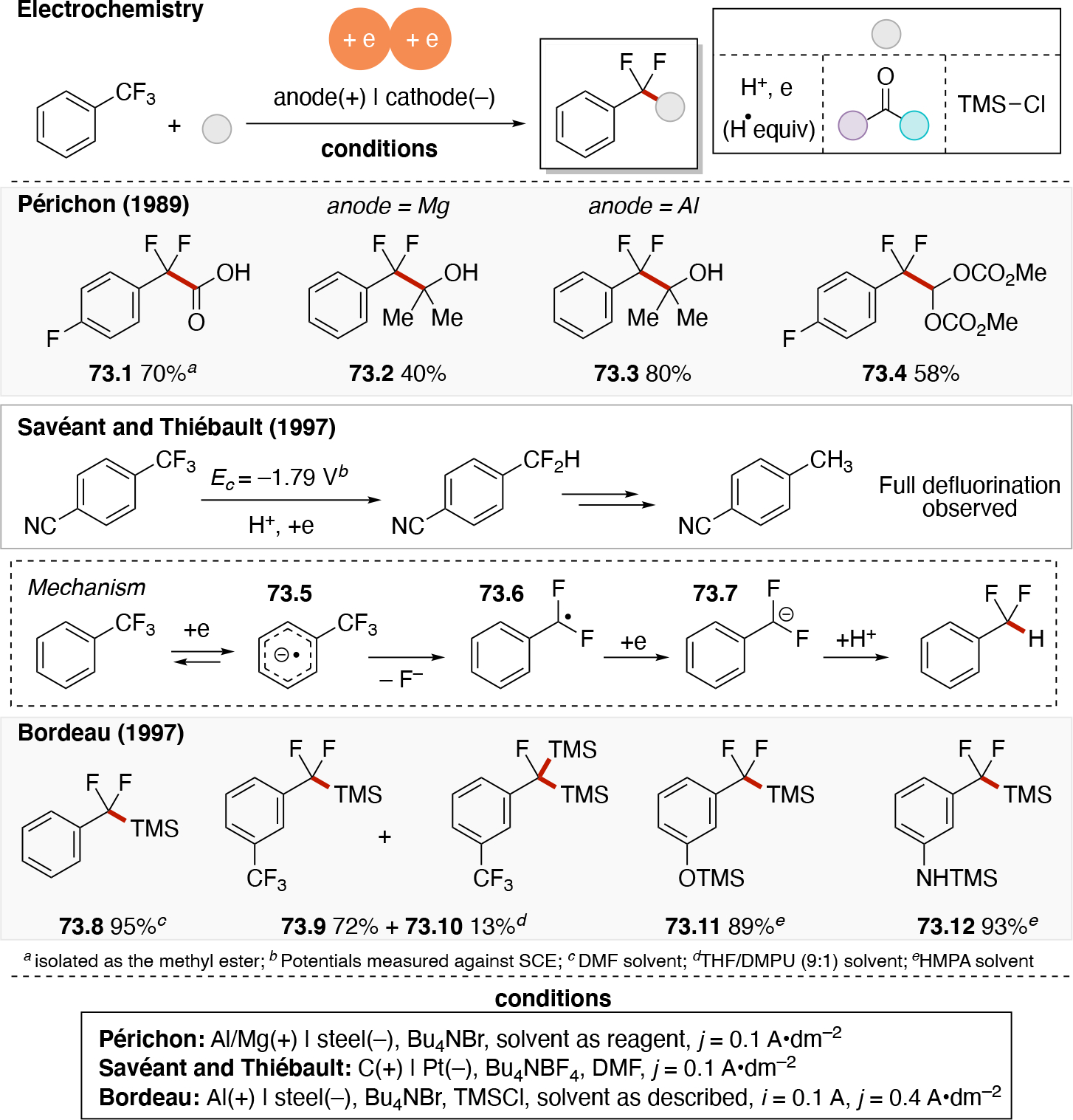
Electrochemical defluorinative functionalization of benzotrifluorides with the corresponding mechanism.
Scheme 79.

A comparison of electrochemical and photoredox methods for aliphatic C–H fluorination with electrophilic fluorine sources.
Scheme 96.
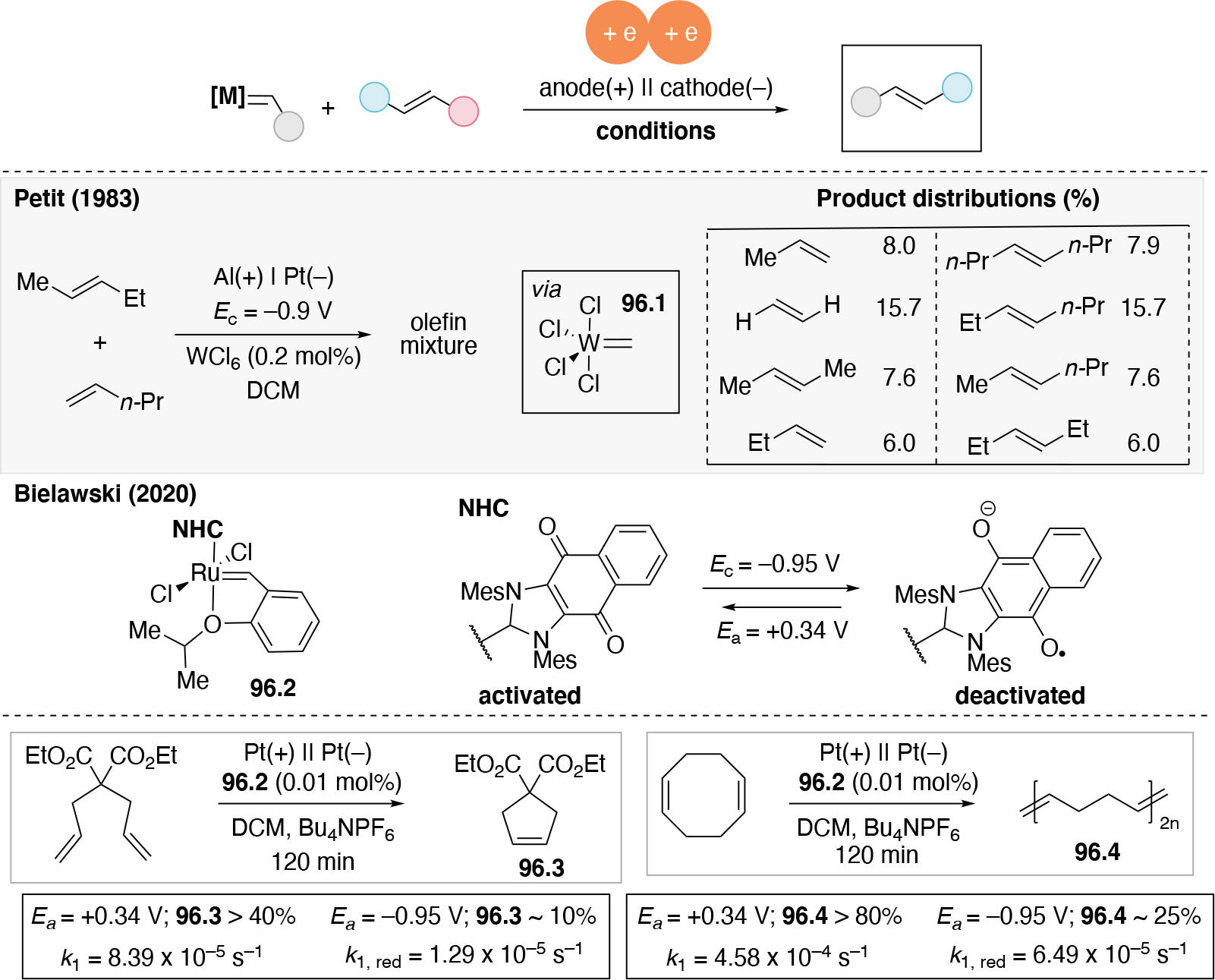
A comparison of electrochemical methods for catalyst-activated olefin metathesis.
ACKNOWLEDGEMENTS
T.R acknowledges NIGMS (GM125206) for financial support. We would like to thank Daniel A. DiRocco (Merck & Co., Inc., Kenilworth, NJ, USA) for insightful discussions and constructive feedback during the preparation of this review. D.L. thanks Byron K. Peters, Nadine Kuhl and François Lévesque (all at Merck & Co., Inc., Kenilworth, NJ, USA) for critical reading of the manuscript and constructive feedback.
ABBREVIATIONS
- 1,5-HAT
1,5-hydrogen atom transfer
- 18-C-6
18-crown-6, CAS 17455–13–9
- 4CzIPN
1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene, CAS 1416881–52–1
- 4-DPA-IPN
2,4,5,6-tetrakis(diphenylamino)isophthalonitrile, CAS 1846598–27–3
- 4-HTP
4-hydroxythiophenol
- 5,5′-dCF3bpy
5,5′-bis(trifluoromethyl)-2,2′-bipyridine
- A
acceptor molecule
- ABNO
9-azabicyclo[3.3.1]nonane N-oxyl
- AC
alternating current
- Ac
acetyl
- ACE
alternating current electrolysis
- AcOH
acetic acid
- Acr
acridinium
- ACT
4-acetamido-TEMPO
- Ad
adamantly
- AIBN
azobisisobutyronitrile
- APE
alternating potential electrolysis
- Ar
aryl
- ATRP
atom transfer radical polymerization
- BA
n-butyl acrylate
- B(ArF)4
Tetrakis(3,5-bis(trifluoromethyl)phenyl)borate
- BDE
bond dissociation energy
- BDD
boron-doped diamond
- BHT
butylated hydroxytoluene; 2,6-di-tert-butyl-4-methylphenol
- BMPO
5-tert-butoxycarbonyl 5-methyl-1-pyrroline-N-oxide
- Bn
benzyl
- BNAH
1-benzyl-1,4-dihydronicotinamide
- Boc
tert-butyloxycarbonyl
- BPE
bipolar electrode
- BPI
benzo[ghi]perylene monoimides
- bpm
2,2′-bipyrimidine
- BPO
benzoyl peroxide
- bpy
2,2′-bipyridine
- BPyI
bis(pyridylamino)isoindoline
- bpz
2,2′-bipyrazine
- BrCCl3
bromotrichloromethane
- n-Bu
n-butyl
- Bz
benzoyl
- cat
catalyst
- CBX
cyanobenziodoxolone
- cbz
carboxybenzyl
- CCE
constant current electrolysis
- CCl4
carbon tetrachloride
- CE
counter electrode
- CF3
trifluoromethyl
- CFL
compact fluorescent light
- Cl4NHPI
N-hydroxytetrachlorophthalimide
- Co(dmgH)2(p-Me2NPy)Cl
chloro[(4-dimethylamino)pyridine]bis(dimethylglyoximato)cobalt(III), CAS 483979–48-2
- Co(dmgH)2pyCl
chloro(pyridine)bis(dimethylglyoximato)cobalt(III), CAS 23295–32–1
- conPET
consecutive photoinduced electron transfer
- CPE
constant potential electrolysis
- CRA-SNAr
cation radical accelerated SNAr
- Cr(Ph2phen)3(BF4)3
chromium(III) tris(4,7-diphenyl-1,10-phenanthroline-κN1,κN10)-, (OC-6–11)-, tetrafluoroborate (1:3), CAS 1808257–86–4
- CsOBz
cesium benzoate
- CSTR
continuous stirred tank reactor
- CTA
chain transfer agent
- CV
cyclic voltammetry
- Cy
cyclohexyl
- Cy2NMe
N,N-dicyclohexylmethylamine
- CySH
cycohexylthiol
- D
donor molecule
- DBU
1,8-Diazabicyclo[5.4.0]undec-7-ene, CAS 6674–22-2
- DC
direct current
- DCE
1,2-dichloroethane
- DCM
dichloromethane
- DCPD
endo-dicyclopentadiene
- DDP
diaryl dihydrophenazine
- DDQ
2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- DDH
2,3-dichloro-5,6-dicyano-p-hydroquinone
- DE
direct electrolysis
- DET
double electron transfer
- dF(CF3)ppy
2-(2,4-difluorophenyl)-5-trifluoromethylpyridine
- DFT
density functional theory
- diglyme
diethylene glycol dimethyl ether
- DIPEA
N,N-diisopropylethylamine
- DMA
dimethylacetamide
- DME
dimethoxyethane
- DMF
dimethylformamide
- DMPO
5,5-dimethyl-1-pyrroline-N-oxide
- DMSO
dimethylsulfoxide
- dPEC
decoupled photoelectrochemistry
- DoE
design of experiments
- dmbpy
4–4′-dimethoxy-2–2′-bipyridine
- DMF
dimethylformamide
- (DMP)BF4
N,N-dimethylpyrrolidinium tetrafluoroborate
- DMU
1,3-dimethylurea
- dr
diastereomeric ratio
- DR
deep red (e.g., λ = 660 nm)
- DT
decatungstate
- DTBP
di-tert-butylperoxide
- dtbbpy
4,4′-di-tert-butyl-2,2′-bipyridine
- e
electron
- eATRP
electrochemical atom transfer radical polymerization
- EBPA
ethyl α-bromophenylacetate
- EC
electrochemistry
- EDA
electron donor-acceptor
- EDL
electric double layer
- ee
enantiomeric excess
- EnT
energy transfer
- Eosin Y
2′,4′,5′,7′-tetrabromofluorescein
- EY-Na2
Eosin Y disodium, CAS 17372–87-1
- E pa
anodic peak potential
- E pc
cathodic peak potential
- EPC
electrophotochemistry
- EPR
electron paramagnetic resonance
- er
enantiomeric ratio
- ESF
ethylene sulfonyl fluoride
- ET
electron transfer
- Et
ethyl
- Et3N
triethylamine
- EtOH
ethanol
- e-PRC
electrochemically mediated photoredox catalysis
- eV
electron volt
- fac
facial
- Fc
ferrocene
- Fc+
ferrocenium
- Fppy
2-(2,4-difluorophenyl)pyridine
- FT
Fourier transform
- ΔGPET
Gibbs free energy of photoinduced electron transfer
- GO
graphene oxide
- GLC
glassy carbon
- glyme
1,2-dimethoxyethane
- GRC
graphite carbon
- H+
hydrogen ion
- H2O2
hydrogen peroxide
- HAT
hydrogen atom transfer
- HDF
hydrodefluorination
- HE
Hantzsch ester
- HER
hydrogen evolution reaction
- HFIP
hexafluoroisopropanol
- HLF
Hoffmann-Loffler-Freytag
- HOMO
highest occupied molecular orbital
- HPLC
high-performance liquid chromatography
- HTE
high-throughput experimentation
- i-Pr
isopropyl
- iPEC
interfacial photoelectrochemistry
- IBVE
isobutyl vinyl ether
- IR
infrared
- i p,a
peak anodic current
- i p,c
peak cathodic current
- i-Pr2NEt
N,N-diisopropylethylamine
- i-Pr2NH
N,N-diisopropylamine
- i-PrOH
isopropanol
- ISC
intersystem crossing
- Ir[dF(CF3)ppy]2(dtbbpy)PF6
[4,4′-bis(1,1-dimethylethyl)-2,2′-bipyridine-N1,N1′]bis[3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl-N]phenyl-C]Iridium(III) hexafluorophosphate, CAS 870987–63-6
- [Ir(dFCF3ppy)2-(5,5′-dCF3bpy)]PF6
[5,5′-Bis(trifluoromethyl)-2,2′-bipyridine-κN,κN]bis[3,5-difluoro-2-[5- (trifluoromethyl)-2-pyridinyl-κN]phenyl]Iridium(III) hexafluorophosphate, CAS 1973375–72-2
- Ir(dF(Me)ppy)2(dtbbpy)PF6
[bis[2-(2,4-difluorophenyl)-5-methylpyridine-N,C20]-4,40-di-tert-butyl-2,20-bipyridine iridium(III)] hexafluorophosphate, CAS 1335047–34-1
- Ir[p-F(t-Bu)-ppy]3
tris-(4-(tert-butyl)-2-(5-fluoro-phenyl)-pyridine)-iridium(III)], CAS 1311386–93-2
- Ir(ppy)3
tris[2-phenylpyridinato-C2,N]iridium(III), CAS 94928–86-6
- Ir(ppy)2(dtbbpy)PF6
[4,4′-bis(1,1-dimethylethyl)-2,2′-bipyridine-N1,N1′]bis[2-(2-pyridinyl-N)phenyl-C]iridium(III) hexafluorophosphate, CAS 676525–77-2
- [Ir(tbppy)2(bpy)]PF6
[(2-(4-tert-butylphenyl)-pyridine)2(2,2'-bipyridine)]iridium(III) hexafluorophosphate, CAS 1352428–78–4
- iSF
intramolecular singlet fission
- ITO
indium tin oxide
- KA oil
ketone-alcohol oil
- LED
light emitting diode
- LMCT
ligand to metal charge transfer
- LUMO
lowest unoccupied molecular orbital
- MA
methyl acrylate
- ME
mediated electrolysis
- Me
methyl
- MeCN
acetonitrile
- Me6TREN
tris[2-(dimethylamino)ethyl]amine, CAS 33527–91-2
- MEK
methylethylketone
- MeOH
methanol
- (Mes-2,7-dMe-Ph-Acr)BF4
9-mesityl-2,7-dimethyl-10-phenylacridinium tetrafluoroborate, CAS 1621020–00-5
- (Mes-3,6-dtb-Ph-Acr)BF4
9-mesityl-3,6-di-tert-butyl-10-phenylacridinium tetrafluoroborate, CAS 1810004–87-5
- (Mes-3,6-dtb-Ph-Acr)ClO4
9-mesityl-3,6-di-tert-butyl-10-phenylacridinium perchlorate
- (Mes-Me-Acr)BF4
9-Mesityl-10-methylacridinium tetrafluoroborate, CAS 1442433–71-7
- (Mes-Me-Acr)ClO4
9-Mesityl-10-methylacridinium perchlorate, CAS 674783–97-2
- (Mes-Ph-Acr)BF4
9-Mesityl-10-phenylacridinium tetrafluoroborate, CAS 1621019–96-2
- MeSO3H
methanesulfonic acid
- MIDA
N-methyliminodiacetic acid
- MLCT
metal to ligand charge transfer
- MMA
methyl methacrylate
- M n,exp
experimental number-average molecular weights
- M n,theo
theoretical number-average molecular weights (determined by computational methods)
- MS
mass spectrometry
- MTES
methyl(triethyl)ammonium sulfate (MeNEt3)(OSO2OMe)
- M W,exp
experimental weight-average molecular weights
- M W,theo
theoretical weight-average molecular weights (determined by computational methods)
- NADH
1,4-dihydronicotinamide adenine dinucleotide
- NBu4
tetra-n-butylammonium
- NEt4
tetraethylammonium
- NFSI
N-fluorobenzenesulfonimide
- NHC
N-heterocyclic carbene
- NHPI
N-hydroxyphthalimide
- NIR
near infrared
- NMe4
tetramethylammonium
- NMR
nuclear magnetic resonance
- NSAID
nonsteroidal anti-inflammatory drug
- OAc
acetate
- O2CCF3
trifluoroacetate
- OER
oxygen evolution reaction
- OM
olefin metathesis
- P
product
- PAHs
polycyclic aromatic hydrocarbons
- PC
photocatalyst
- PC*
electronically excited photocatalyst
- PCET
proton-coupled electron transfer
- PEC
photoelectrochemistry
- PES
potential energy surface
- PET
photoinduced electron transfer
- PFAS
per- and polyfluorinated alkyl substances
- PFR
plug flow reactor
- PG
protecting group
- Ph
phenyl
- phen
1,10-phenanthroline
- Phenox O-PC™ A0202
3,7-Di(4-biphenyl) 1-naphthalene-10-phenoxazine, CAS 1987900–95-7
- Phth
phthalimide
- Piv
pivaloyl
- PMB
para-methoxybenzyl
- PMP
para-methoxyphenyl
- PMMA
poly(methyl methylacrylate)
- PNO
pyridine-N-oxide
- polyPCPD polydicyclopentadiene
(p-OMeTPP)BF4 2,4,6-Tris(4-methoxyphenyl)pyrylium tetrafluoroborate, CAS 580–34-7
- PPh3
triphenylphosphine
- ppy
2-phenylpyridine
- PS-NMe3CN
polystyrene-supported quaternary ammonium cyanide
- PTH
10-phenylphenothiazine, CAS 7152–42-3
- py
pyridine
- QSAR
quantitative structure-activity relationship
- R
rest
- RAFT
reversible addition-fragmentation chain transfer
- RCY
radiochemical yield
- RE
reference electrode
- RFTA
riboflavin tetraacetate
- RPG
radical precursor group
- RPK
reaction progress kinetics
- ROMP
ring-opening metathesis polymerization
- rr
regiosiomeric ratio
- Ru(bpy)3Cl2•6H2O
tris(2,2′-bipyridyl)dichlororuthenium(II) hexahydrate, CAS 50525–27-4
- Ru(bpy)3(PF6)2
tris(2,2′-bipyridine)ruthenium(II) hexafluorophosphate, CAS 60804–74–2
- Ru(bpz)3(BArF)2
tris(2,2′-bipyrazine)ruthenium(II)tetrakis[3,5-bis(trifluoromethyl)phenyl]borate, CAS 1350432–81–3
- Ru(bpz)3(PF6)2
tris(2,2′-bipyrazine)ruthenium(II) hexafluorophosphate, CAS 80907–56–8
- Ru(bpz)3Cl2
tris(1,10-phenanthroline)ruthenium(II) dichloride, CAS 23570–43–6
- Ru(phen)3(PF6)2
tris(1,10-phenanthroline)ruthenium(II) hexafluorophosphate, CAS 60804–75–3
- RVC
reticulated vitreous carbon
- SBS
Society for Biometrical Sciences
- SCE
saturated calomel electrode
- Selectfluor
1-Chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate, CAS 140681–55–6
- SET
single electron transfer
- SLAS
Society for Laboratory Automation and Screening
- SOMO
singly occupied molecular orbital
- SPE
solid polymer electrode
- STY
space-time yield
- TBAF
tetra-n-butylammonium fluoride
- t-Bu
tert-butyl
- TBHP
tert-butyl hydroperoxide
- tbppy
2-(4-tert-butylphenyl)-pyridine
- TBS
tert-butyldimethylsilyl
- TEMPO
2,2,6,6-tetramethylpiperidin-1-yl)oxyl
- TFA
trifluoroacetic acid
- TFAA
trifluoroacetic anhydride
- TfCl
trifluoromethanesulfonyl chloride
- TFU
triplet fusion upconversion
- THF
tetrahydrofuran
- tmc
tetramethylcyclam, 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane
- TMG
1,1,3,3-tetramethylguanidine
- TMHD
2,2,6,6-tetramethyl-3,5-heptanedionate
- TMP
2,2,6,6-tetramethylpiperidine
- TMS
trimethylsilyl
- TMPA
tris(2-pyridylmethyl)amine, CAS 16858–01-8
- ToF-SIMS
time-of-flight secondary ion mass spectrometry
- Tol
toluene
- TPPA
tris(pyrrolidino)phosphoramide
- TPPBF4
2,4,6-Triphenylpyrylium tetrafluoroborate, CAS 448–61–3
- TPPV10
tetrakis(tetraphenylphosphonium) dihydrogen decavanadate, (Ph4P)4H2[V10O28], CAS 138521–25–2
- tpy
2,2′:6′,2′′-terpyridine
- TREN
tris(2-aminoethyl)amine, CAS 4097–89–6
- Ts
toluenesulfonyl, tosyl
- TTA
triplet-triplet annihilation
- TTMSS
tris(trimethylsilyl)silane
- UV
ultraviolet
- Vis
visible
- v/v
volume per volume
- WE
working electrode
- XAT
halogen atom transfer
- XEC
cross-electrophile coupling
- ZnTFMS
zinc trifluoromethylsulfinate
Symbols
- |
undivided cell
- ‖
divided cell
- Ð
dispersity
- Eo
standard potential
- E 0,0
excited state energy of an electronically excited fluorophore in its lowest energy vibrational state
- E1/2
half-wave potential
- Ea
anode potential
- Ec
cathode potential
- Ee
thermodynamically required cell potential
- Eox
oxidation potential
- Ep/2
half-peak potential
- E p,a
peak anodic potential
- E p,c
peak cathodic potential
- E red
reduction potential
- ε
molar extinction coefficient
- η
overpotential
- F
Faraday constant (23.061 kcal•V−1•mol−1 or 96485 C•mol−1)
- ϕ
quantum yield
- i
current
- i p,a
anodic peak current
- ip,c
cathodic peak current
- j
current density
- l BPE
length of bipolar electrode
- l channel
length of channel between driving electrodes
- λ
wavelength (photochemistry)
- λ
reorganization energy (electron transfer)
- Q
total charge
- R
ideal gas constant (8.31447 J•mol−1•K−1)
- τ
lifetime
- U cell
cell potential
- ν
scan rate
volumetric flow rate
- V r
reactor volume
Biographies
Nicholas E. S. Tay was born in Johor Bahru, Malaysia in 1992 and immigrated to the United States of America in 2008. He attended Messiah College, obtaining a B.S. in Chemistry with Departmental Honors in 2014, after carrying out research on microelectrode development for zinc selenide surfaces with Professor Alison R. Noble. He also conducted synthetic organic research in the lab of Professor Steve Buchwald at the Massachusetts Institute of Technology in the summer of 2013. Nicholas began graduate studies at the University of North Carolina at Chapel Hill in 2014, where he joined the lab of Professor David A. Nicewicz and developed new methodologies for organic-photoredox-catalyzed C–H and C–O functionalizations of (hetero)arenes as a National Science Foundation Graduate Research Fellow. He completed his PhD in 2019 and then began his postdoctoral studies with Professor Tomislav Rovis at Columbia University. His research in the Rovis group has focused on the development of new chemical tools for deep red and near-infrared photoredox catalysts, with a particular focus on applications for process chemistry and chemical biology.
Dan Lehnherr received his BSc degree from the University of Victoria (Canada), where he carried out undergraduate research on photochemistry topics in the laboratory of Prof. Peter Wan. He obtained his PhD from the University of Alberta under the mentorship of Prof. Rik R. Tykwinski (Chemistry) and Prof. Frank A. Hegmann (Physics) developing conjugated organic materials for optoelectronic applications (e.g. photodetectors, thin film transistors) and dimeric pentacenes as a platform for investigating intramolecular singlet fission for solar cell technologies. He was an NSERC Postdoctoral Fellow at Harvard University with Prof. Eric N. Jacobsen focusing on organocatalysis and reaction mechanism elucidation where he developed dimeric thiourea catalysts for cooperative anion-binding catalysis. Subsequently he carried out postdoctoral research at Cornell University with Prof. William R. Dichtel developing synthetic methods to foldamers and nanographenes and studying their properties. Since 2016 he has been in the Catalysis Group within Process Research and Development at Merck & Co., Inc., Kenilworth, NJ, USA.
Tomislav Rovis was born in Zagreb in the former Yugoslavia but was largely raised in Southern Ontario, Canada. Following his undergraduate studies at the University of Toronto, he earned his Ph.D. degree at the same institution in 1998 under the direction of Professor Mark Lautens. From 1998–2000, he was an NSERC postdoctoral fellow at Harvard University with Professor David A. Evans. In 2000, he began his independent career at Colorado State University and was promoted in 2005 to Associate Professor and in 2008 to Professor and John K. Stille Chair in Chemistry. His group’s accomplishments have been recognized by a number of awards including an NSF CAREER and a Roche Excellence in Chemistry award. He has been named a GlaxoSmithKline Scholar, Amgen Young Investigator, Eli Lilly Grantee, Alfred P. Sloan Fellow, Monfort Professor at Colorado State University, Fellow of the American Association for the Advancement of Science, Katritzky Young Investigator in Heterocyclic Chemistry, and an Arthur C. Cope Scholar. In 2016, he moved to Columbia University where he is currently the Samuel Latham Mitchill Professor of Chemistry.
Footnotes
The authors declare no competing financial interest.
References
- 1.Yan M; Lo JC; Edwards JT; Baran PS Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc. 2016, 138, 12692–12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curran DP The Design and Application of Free Radical Chain Reactions in Organic Synthesis. Part 2. Synthesis 1988, 7, 489–513. [Google Scholar]
- 3.Jasperse CP; Curran DP; Fevig TL Radical Reactions in Natural Product Synthesis. Chem. Rev. 1991, 91, 1237–1286. [Google Scholar]
- 4.Stephenson CRJ; Studer A; Curran DP The Renaissance of Organic Radical Chemistry – Deja vu All over Again. Beilstein J. Org. Chem. 2013, 9, 2778–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Studer A; Curran DP Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem. Int. Ed. 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]
- 6.Smith JM; Harwood SJ; Baran PS Radical Retrosynthesis. Acc. Chem. Res. 2018, 51, 1807–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petzold D; Giedyk M; Chatterjee A; König B A Retrosynthetic Approach for Photocatalysis. Eur. J. Org. Chem. 2020, 1193–1244. [Google Scholar]
- 8.For a discussion on redox economy, see: Burns NZ; Baran PS; Hoffmann RW Redox Economy in Organic Synthesis. Angew. Chem. Int. Ed. 2009, 48, 2854–2867.
- 9.Romero KJ; Galliher MS; Pratt DA; Stephenson CRJ Radicals in natural product synthesis. Chem. Soc. Rev. 2018, 47, 7851–7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shatskiy A; Lundberg H; Kärkäs MD Organic Electrosynthesis: Applications in Complex Molecule Synthesis. ChemElectroChem 2019, 6, 4067–4092. [Google Scholar]
- 11.Duñach E; Franco D; Olivero S Carbon−Carbon Bond Formation with Electrogenerated Nickel and Palladium Complexes. Eur. J. Org. Chem. 2003, 1605–1622. [Google Scholar]
- 12.Lu J; Wang Y; McCallum T; Fu N Harnessing Radical Chemistry via Electrochemical Transition Metal Catalysis. iScience 2020, 23, 101796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Twilton J; Le CC; Zhang P; Shaw MH; Evans RW; MacMillan DWC. The merger of Transition Metal and Photocatalysis. Nat. Rev. Chem. 2017, 1. 0052. [Google Scholar]
- 14.Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5233–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang C-S; Dixneuf PH; Soulé J-F Photoredox Catalysis for Building C–C Bonds from C(sp2)–H Bonds. Chem. Rev. 2018, 118, 7532–7585. [DOI] [PubMed] [Google Scholar]
- 17.Troian-Gautier L; Turlington MD; Wehlin SAM; Maurer AB; Brady MD; Swords WB; Meyer GJ Halide Photoredox Chemistry. Chem. Rev. 2019, 119, 4628–4683. [DOI] [PubMed] [Google Scholar]
- 18.Romero NA; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- 19.Lund H A Century of Organic Electrochemistry. J. Electrochem. Soc. 2002, 149, S21. [Google Scholar]
- 20.Yan M; Kawamata Y; Baran PS Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117, 13230–13319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiebe A; Gieshoff T; Möhle S; Rodrigo E; Zirbes M; Waldvogel SR Electrifying Organic Synthesis. Angew. Chem. Int. Ed. 2018, 57, 5594–5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Möhle S; Zirbes M; Rodrigo E; Gieshoff T; Wiebe A; Waldvogel SR Modern Electrochemical Aspects for the Synthesis of Value-Added Organic Products. Angew. Chem. Int. Ed. 2018, 57, 6018–6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kingston C; Palkowitz MD; Takahira Y; Vantourout JC; Peters BK; Kawamata Y; Baran PS A Survival Guide for the “Electro-curious”. Acc. Chem. Res. 2020, 53, 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pollok D; Waldvogel SR Electro-Organic Synthesis – A 21st Century Technique. Chem. Sci. 2020, 11, 12386–12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schotten C; Nicholls TP; Bourne RA; Kapur N; Nguyen BN; Willans CE Making Electrochemistry Easily Accessible to the Synthetic Chemist. Green Chem. 2020, 22, 3358–3375. [Google Scholar]
- 26.Zhu C; Ang NWJ; Meyer TH; Qiu Y; Ackermann L Organic Electrochemistry: Molecular Syntheses with Potential. ACS Cent. Sci. 2021, 7, 415–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moeller K Synthetic Applications of Anodic Electrochemistry. Tetrahedron 2000, 56, 9527–9554. [Google Scholar]
- 28.Verschueren RH; De Borggraeve WM Electrochemistry and Photoredox Catalysis: A Comparative Evaluation in Organic Synthesis. Molecules 2019, 24, 2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J; Lu L; Wood D; Lin S New Redox Strategies in Organic Synthesis by Means of Electrochemistry and Photochemistry. ACS Cent. Sci. 2020, 6, 1317–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toogood HS; Scrutton NS Chapter Sixteen - Thermal, Electrochemical and Photochemical Reactions Involving Catalytically Versatile Ene Reductase Enzymes. The Enzymes, 2020, 47, 491–515. [DOI] [PubMed] [Google Scholar]
- 31.Hyster TK Radical Biocatalysis: Using Non-Natural Single Electron Transfer Mechanisms to Access New Enzymatic Functions Synlett 2019, 30, 248–254. [Google Scholar]
- 32.Björn LO Photoenzymes and Related Topics: An Update. Photochem. Photobiol. 2018, 94, 459–465. [DOI] [PubMed] [Google Scholar]
- 33.Sandoval BA Hyster TK Emerging Strategies for Expanding the Toolbox of Enzymes in Biocatalysis. Curr. Opin. Chem. Biol. 2020, 55, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H; Dong F; Minteer SD The Progress and Outlook of Bioelectrocatalysis for the Production of Chemicals, Fuels and Materials. Nat. Catal. 2020, 3, 225–244. [Google Scholar]
- 35.Chen H; Simoska O; Lim K; Grattieri M; Yuan M; Dong F; Lee YS; Beaver K; Weliwatte S; Gaffney EM Minteer, S. D.; Fundamentals, Applications, and Future Directions of Bioelectrocatalysis. Chem. Rev. 2020, 120, 12903–12993. [DOI] [PubMed] [Google Scholar]
- 36.Electrocatalysis and Bioelectrocatalysis – Distinction Without a Difference. NanoEnergy, 2016, 29, 466–475. [Google Scholar]
- 37.Tang S; Zeng L; Lei A Oxidative R1–H/R2–H Cross-Coupling with Hydrogen Evolution. J. Am. Chem. Soc. 2018, 140, 13128–13135. [DOI] [PubMed] [Google Scholar]
- 38.Chang X; Zhang Q; Guo C Asymmetric Electrochemical Transformations. Angew. Chem. Int. Ed. 2020, 59, 12612–12622. [DOI] [PubMed] [Google Scholar]
- 39.Ghosh M; Shinde VS; Rueping M A Review of Asymmetric Synthetic Organic Electrochemistry and Electrocatalysis: Concepts, Applications, Recent Developments and Future Directions. Beilstein J. Org. Chem. 2019, 15, 2710–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Margarita C; Lundberg H Recent Advances in Asymmetric Catalytic Electrosynthesis. Catalysts 2020, 10, 982. [Google Scholar]
- 41.Lin Q; Li L; Luo S Asymmetric Electrochemical Catalysis. Chem. Eur. J. 2019, 25, 10033–10044. [DOI] [PubMed] [Google Scholar]
- 42.Lin Q; Luo S Tailoring Radicals by Asymmetric Electrochemical Catalysis. Org. Chem. Front. 2020, 7, 2997–3000. [Google Scholar]
- 43.Huo H; Shen X; Wang C; Zhang L; Röse P; Chen L-A; Harms K; Marsch M; Hilt G; Meggers E Asymmetric Photoredox Transition-Metal Catalysis Activated by Visible Light. Nature 2014, 515, 100–103. [DOI] [PubMed] [Google Scholar]
- 44.Jiang C; Chen W; Zheng W-H; Lu H Advances in Asymmetric Visible-Light Photocatalysis, 2015–2019. Org. Biomol. Chem. 2019, 17, 8673–8689. [DOI] [PubMed] [Google Scholar]
- 45.Wang C; Lu Z Catalytic Enantioselective Organic Transformations via Visible Light Photocatalysis. Org. Chem. Front. 2015, 2, 179–190. [Google Scholar]
- 46.Brimioulle R; Lenhart D; Maturi MM; Bach T Enantioselective Catalysis of Photochemical Reactions. Angew. Chem. Int. Ed. 2015, 54, 3872–3890. [DOI] [PubMed] [Google Scholar]
- 47.Zhang H-H; Chen H; Zhu C; Yu S A Review of Enantioselective Dual Transition Metal/Photoredox Catalysis. Sci. China Chem. 2020, 63, 637–647. [Google Scholar]
- 48.Saha D; Catalytic Enantioselective Radical Transformations Enabled by Visible Light. Chem. Eur. J. 2020, 15, 2129–2152. [DOI] [PubMed] [Google Scholar]
- 49.Wang C; Qin J; Shen X; Riedel R; Harms K; Meggers E Asymmetric Radical-Radical Cross-Coupling through Visible-Light-Activated Iridium Catalysis. Angew. Chem. Int. Ed. 2016, 55, 685–688. [DOI] [PubMed] [Google Scholar]
- 50.Leifert D; Studer A The Persistent Radical Effect in Organic Synthesis. Angew. Chem. Int. Ed. 2020, 59, 74–108. [DOI] [PubMed] [Google Scholar]
- 51.Roberts BP; Polarity-Reversal Catalysis of Hydrogen-Atom Abstraction Reactions: Concepts and Applications in Organic Chemistry. Chem. Soc. Rev. 1999, 28, 25–35. [Google Scholar]
- 52.De Vleeschouwer F; Van Speybroeck V; Waroquier M; Geerlings P; De Proft F Electrophilicity and Nucleophilicity Index for Radicals. Org. Lett. 2007, 9, 2721–2724. [DOI] [PubMed] [Google Scholar]
- 53.Héberger K; Lopata A Assessment of Nucleophilicity and Electrophilicity of Radicals, and of Polar and Enthalpy Effects on Radical Addition Reactions. J. Org. Chem. 1998, 63, 8646–8653. [Google Scholar]
- 54.Fischer H; Radom L Factors Controlling the Addition of Carbon-Centered Radicals to Alkenes – An Experimental and Theoretical Perspective. Angew. Chem. Int. Ed. 2001, 40, 1340–1371. [DOI] [PubMed] [Google Scholar]
- 55.Francke R; Little RD Redox Catalysis in Organic Electrosynthesis: Basic Principles and Recent Developments. Chem. Soc. Rev. 2014, 43, 2492–2521. [DOI] [PubMed] [Google Scholar]
- 56.Luo T; Abdu S; Wessling M Selectivity of Ion Exchange Membranes: A Review. J. Membr. Sci. 2018, 555, 429–454. [Google Scholar]
- 57.Yee RSL; Rozendal RA; Zhang K; Ladewig BP Cost Effective Cation Exchange Membranes: A Review. Chem. Eng. Res. Des. 2012, 90, 950–959. [Google Scholar]
- 58.You W, Noonan KJT and Coates GW, Alkaline-Stable Anion Exchange Membranes: A Review of Synthetic Approaches. Prog. Polym. Sci. 2020, 100, 101177. [Google Scholar]
- 59.Maurya S; Shin S-H; Kim Y; Moon S-H A Review on Recent Developments of Anion Exchange Membranes for Fuel Cells and Redox Flow Batteries. RSC Adv. 2015, 5, 37206–37230. [Google Scholar]
- 60.For discussion of the E and C notation, see: Testa A; Reinmuth W Anal. Chem. 1961, 33, 1320–1324.
- 61.Mairanovsky VG On the Abbreviated Notation of Electrode Reactions. J. Electroanal. Chem. 1979, 97, 103–105. [Google Scholar]
- 62.There are two common conventions for plotting cyclic voltammetry data, namely the IUPAC and US conventions. In the IUPAC convention, potential (represented by the x-axis) increases going from left to right, whereas in the US convention, potential (represented by the x-axis) decreases from left to right. As a consequence, when comparing identical data plotted in each convention, the data will appear rotated by 180°.
- 63.Elgrishi N; Rountree KJ; McCarthy BD; Rountree ES; Eisenhart TT; Dempsey JL A Practical Beginner’s Guide to Cyclic Voltammetry. J. Chem. Ed. 2018, 95, 197–206. [Google Scholar]
- 64.Bard AJ; Faulkner LR Electrochemical Methods: Fundamentals and Applications, 2nd Edition; John Wiley & Sons: New York, 2001. [Google Scholar]
- 65.Marken F; Neudeck A; Bond AM (2005) Cyclic Voltammetry. In: Scholz F (eds) Electroanalytical Methods. Springer, Berlin, Heidelberg. 10.1007/978-3-662-04757-6_4 [DOI] [Google Scholar]
- 66.Molina A; López-Tenés M; Laborda E Unified theoretical treatment of the Eirrev, CE, EC and CEC Mechanisms Under Voltammetric Conditions. Electrochemistry Commun. 2018, 92, 48–55. [Google Scholar]
- 67.Lee KJ; Elgrishi N; Kandemir B; Dempsey JL Electrochemical and spectroscopic methods for evaluating molecular electrocatalysts. Nat. Rev. Chem. 2017, 1, 0039. [Google Scholar]
- 68.Costentin C; Savéant J-M Multielectron, Multistep Molecular Catalysis of Electrochemical Reactions: Benchmarking of Homogeneous Catalysts ChemElectroChem 2014, 1, 1226–1236. [Google Scholar]
- 69.Mann MA; Helfrick JC Jr.; Bottomley LA Diagnostic Criteria for Identifying an ECE Mechanism with Cyclic SquareWave Voltammetry. J. Electrochem. Soc. 2016, 163, H3101–H3109. [Google Scholar]
- 70.Nematollahi D, Forooghi Z, ECEC and ECE-Type Mechanisms in Electrochemical Oxidation of 4-Substituted Catechols in the Presence of 4-Hydroxy-6-methyl-2-pyrone. Electroanalysis 2003, 15, 1639–1644. [Google Scholar]
- 71.Feldberg SW; Jeftic L Nuances of the ECE Mechanism. IV. Theory of Cyclic Voltammetry and Chronoamperometry and the Electrochemical Reduction of Hexacyanochromate(III). J. Phys. Chem. 1972, 76, 2439–2446. [Google Scholar]
- 72.Okada Y Akaba R; Chiba K EC-backward-E Electrochemistry Supported by an Alkoxyphenyl Group. Tetrahedron 2009, 50, 5413–5416. [Google Scholar]
- 73.Aust N; Kirste A Paired Electrosynthesis. In Encyclopedia of Applied Electrochemistry; Kreysa G, Ota K, Savinell RF, Eds; Springer: New York, 2014. DOI: 10.1007/978-1-4419-6996-5_370 [DOI] [Google Scholar]
- 74.Ibanez JG; Frotana-Uribe BA; Vasquez-Medrano R Paired Electrochemical Processes: Overview, Systematization, Selection Criteria, Design Strategies, and Projection. J. Mex. Chem. Soc. 2016, 60, 247–260. [Google Scholar]
- 75.Paddon CA; Atobe M; Fuchigami T; He P Watts P; Haswell SJ; Pritchard GJ; Bull SD; Marken F Towards Paired and Coupled Electrode Reactions for Clean Organic Microreactor Electrosyntheses. J. Appl. Electrochem. 2006, 36, 617–634. [Google Scholar]
- 76.Vantourout JC From Bench to Plant: An Opportunity for Transition Metal Paired Electrocatalysis. Org. Process Res. Dev. 2021. DOI: 10.1021/acs.oprd.1c00046 [DOI] [Google Scholar]
- 77.Hilt G Basic Strategies and Types of Applications in Organic Electrochemistry. ChemElectroChem 2020, 7, 395–405. [Google Scholar]
- 78.Baizer MM In Organic Electrochemistry; Lund H; Baizer MM, Eds.; Marcel Dekker Inc.: New York, 1991, pp. 1265–1282. [Google Scholar]
- 79.Park K; Pintauro PN; Baizer MM; Nobe K Flow Reactor Studies of the Paired Electro-Oxidation and Electroreduction of Glucose. J. Electrochem. Soc. 1985, 132, 1850–1855. [Google Scholar]
- 80.Hermann Pütter n.; Hannebaum H Preparation of Phthalides. US Patent 6,063,256, May 16, 2000.
- 81.Shono T Electroorganic chemistry in organic synthesis. Tetrahedron 1984, 40, 811–850. [Google Scholar]
- 82.Jones AM; Banks CE The Shono-Type Electroorganic Oxidation of Unfunctionalised Amides. Carbon–Carbon Bond Formation via Electrogenerated N-Acyliminium Ions. Beilstein J. Org. Chem. 2014, 10, 3056–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.For an example, see: Hartmer MF, Waldvogel SR Electroorganic Synthesis of Nitriles via a Halogen-Free Domino Oxidation–Reduction Sequence. Chem. Commun. 2015, 51, 16346–16348.
- 84.Gütz C; Stenglein A; Waldvogel SR Highly Modular Flow Cell for Electroorganic Synthesis. Org. Process. Res. Dev. 2017, 21, 771–778. [Google Scholar]
- 85.Ye K-Y; Pombar G; Fu N; Sauer GS; Keresztes I; Lin S Anodically Coupled Electrolysis for the Heterodifunctionalization of Alkenes. J. Am. Chem. Soc. 2018, 140, 2438–2441 [DOI] [PubMed] [Google Scholar]
- 86.Lehnherr D; Lam Y.-h.; Nicastri MC; Liu J; Newman JA; Regalado EL; DiRocco DA; Rovis T Electrochemical Synthesis of Hindered Primary and Secondary Amines via Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2020, 142, 468–478. [DOI] [PubMed] [Google Scholar]
- 87.Strieth-Kalthoff F; James MJ; Teders M; Pitzer L; Glorius F Energy Transfer Catalysis Mediated by Visible Light: Principles, Applications, Directions. Chem. Soc. Rev. 2018, 47, 7190–7202. [DOI] [PubMed] [Google Scholar]
- 88.Wiles RJ; Molander GA Photoredox-Mediated Net-Neutral Radical/Polar Crossover Reactions. Isr. J. Chem. 2020, 60, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sharma S; Singh J; Sharma A Visible Light Assisted Radical-Polar/Polar-Radical Crossover Reactions in Organic Synthesis. Adv. Synth. Catal. 2021, 363, 3146–3169. [Google Scholar]
- 90.Wang P-Z; Chen J-R; Xiao W-J Hantzsch Esters: An Emerging Versatile Class of Reagents in Photoredox Catalyzed Organic Synthesis. Org. Biomol. Chem. 2019, 17, 6936–6951. [DOI] [PubMed] [Google Scholar]
- 91.Jiang M; Yang H; Lefebvre Q; Su J; Fu H Olefination of Alkyl Halides with Aldehydes by Merging Visible-Light Photoredox Catalysis and Organophosphorus Chemistry. iScience 2018, 6, 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reed NL; Yoon TP Oxidase Reactions in Photoredox Catalysis. Chem. Soc. Rev. 2021, 50, 2954–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu Z; Zhang Y; Cai Z; Sun H; Cheng X Photoredox Removal of p-Methoxybenzyl Ether Protecting Group with Hydrogen Peroxide as Terminal Oxidant. Adv. Synth. Catal. 2015, 357, 589–593. [Google Scholar]
- 94.Freeman DB; Furst L; Condie AG; Stephenson CRJ Functionally Diverse Nucleophilic Trapping of Iminium Intermediates Generated Utilizing Visible Light. Org. Lett. 2012, 14, 94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Franz JF; Kraus WB; Zeitler K No Photocatalyst Required – Versatile, Visible Light Mediated Transformations With Polyhalomethanes. Chem. Commun. 2015, 51, 8280–8284. [DOI] [PubMed] [Google Scholar]
- 96.Reed NL; Herman MI; Miltchev VP; Yoon TP Photocatalytic Oxyamination of Alkenes: Copper(II) Salts as Terminal Oxidants in Photoredox Catalysis. Org. Lett. 2018, 20, 7345–7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Reed NL; Lutovsky GA; Yoon TP Copper-Mediated Radical–Polar Crossover Enables Photocatalytic Oxidative Functionalization of Sterically Bulky Alkenes. J. Am. Chem. Soc. 2021, 143, 6065–6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.The use of tetraethylammonium fluoride as an additive (or the electrolyte) can enable sequestration of the Mg2+ ions via precipitation of MgF2, see: Genesty M; Thobie C; Gautier A; Degrand C Electrochemical Synthesis in an Undivided Cell of Aromatic Mono and Dichalcogeno Derivatives by SRN1 Substitution Reactions in MeCN. J. Appl. Electrochem. 1993, 23, 1125–1131.
- 99.Furst L; Matsuura BS; Narayanam JMR; Tucker JW; Stephenson CRJ Visible Light-Mediated Intermolecular C-H Functionalization of Electron-Rich Heterocycles with Malonates. Org. Lett. 2010, 12, 3104–3107. [DOI] [PubMed] [Google Scholar]
- 100.Manabe S; Wong CM; Sevov CS Direct and Scalable Electroreduction of Triphenylphosphine Oxide to Triphenylphosphine. J. Am. Chem. Soc. 2020, 142, 3024–3031. [DOI] [PubMed] [Google Scholar]
- 101.Lu L; Siu JC; Lai Y; Lin S An Electroreductive Approach to Silyl Radical Chemistry via Strong Si–Cl Bond Activation. J. Am. Chem. Soc. 2020, 142, 21272–21278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Palma A; Frontana-uribe BA; Cárdenas J; Saloma M Electrochemical synthesis of α,β-(E)-unsaturated esters in undivided cell (Horner–Emmons reaction). Electrochem. Commun. 2003, 5, 455–459. [Google Scholar]
- 103.Lu W; Nedelec JY; Folest JC; Perichon J Electrochemical Coupling of Activated Olefins and Alkyl Dihalides: Formation of Cyclic Compounds. J. Org. Chem. 1990, 55, 2503–2507. [Google Scholar]
- 104.Cartwright KC; Davies AM; Tunge JA Cobaloxime-Catalyzed Hydrogen Evolution in Photoredox-Facilitated Small-Molecule Functionalization. Eur. J. Org. Chem. 2019, 1245–1258. [Google Scholar]
- 105.Kojima M; Matsunaga S The Merger of Photoredox and Cobalt Catalysis. Trends in Chemistry 2020, 2, 410–426. [Google Scholar]
- 106.We draw attention to the reader that additional mechanisms for hydrogen evolution using cobaloxime catalyst have been reported in addition to the one shown in Scheme 7D, see reference 104.
- 107.Dempsey JL; Brunschwig BS; Winkler JR; Gray HB Hydrogen Evolution Catalyzed by Cobaloximes. Acc. Chem. Res. 2009, 42, 1995–2004. [DOI] [PubMed] [Google Scholar]
- 108.Arias-Rotondo DM; McCusker JK The Photophysics of Photoredox Catalysis: A Roadmap for Catalyst Design. Chem. Soc. Rev. 2016, 45, 5803–5820. [DOI] [PubMed] [Google Scholar]
- 109.Farney EP; Chapman SJ; Swords WB; Torelli MD; Hamers RJ; Yoon TP Discovery and Elucidation of Counteranion Dependence in Photoredox Catalysis. J. Am. Chem. Soc. 2019, 141, 6385–6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Marcus RA Electron Transfer Reactions in Chemistry: Theory and Experiment (Nobel Lecture). Angew. Chem. Int. Ed. 1993, 32, 1111–1121. [Google Scholar]
- 111.Miller JR; Beitz JV; Huddleston RK Effect of Free Energy on Rates of Electron Transfer between Molecules. J. Am. Chem. Soc. 1984, 106, 5057–5068. [Google Scholar]
- 112.Ohno T; Yoshimura A; Mataga N Bell-Shaped Energy Gap Dependence of Backward Electron-Transfer Rate of Geminate Radical Pairs Produced by Electron-Transfer Quenching of Ruthenium(II) Complexes by Aromatic Amines. J. Phys. Chem. 1986, 90, 3295–3297. [Google Scholar]
- 113.Gould IR; Ege D; Mattes SL; Farid S Return Electron Transfer within Geminate Radical Ion Pairs. Observation of the Marcus Inverted Region. J. Am. Chem. Soc. 1987, 109, 3794–3796 [Google Scholar]
- 114.Ohno T; Yoshimura A; Shioyama H; Mataga N Energy Gap Dependence of Spin-Inverted Electron Transfer within Geminate Radical Pairs Formed by the Quenching of Phosphorescent States in Polar Solvents. J. Phys. Chem. 1987, 91, 4365–4370. [Google Scholar]
- 115.Closs GL; Miller JR Intramolecular Long-Distance Electron Transfer in Organic Molecules. Science 1988, 240, 440–447. [DOI] [PubMed] [Google Scholar]
- 116.Parada GA; Goldsmith ZK; Kolmar S; Rimgard BP; Mercado BQ; Hammarström L; Hammes-Schiffer S; Mayer JM Concerted Proton-Electron Transfer Reactions in the Marcus Inverted Region. Science 2019, 364, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Marcus RA On the Theory of Oxidation-Reduction Reactions Involving Electron Transfer. J. Chem. Phys. 1956, 24, 966. [Google Scholar]
- 118.Marcus RA On the Theory of Electron-Transfer Reaction VI. Unified Treatment of Homogeneous and Electrode Reactions. J. Chem. Phys. 1965, 43, 679. [Google Scholar]
- 119.Siders P; Marcus RA Quantum Effects for Electron-Transfer Reactions in the “Inverted Region”. J. Am. Chem. Soc. 1981, 103, 748–752. [Google Scholar]
- 120.Marcus RA Electron Transfer Reactions in Chemistry. Theory and Experiment. Rev. Mod. Phys. 1993, 65, 599–610 [Google Scholar]
- 121.Barbara PF; Meyer TJ; Ratner MA Contemporary Issues in Electron Transfer Research. J. Phys. Chem. 1996, 100, 13148–13168. [Google Scholar]
- 122.Bard AJ; Faulkner LR Kinetics of Electrode Reactions. In Electrochemical Methods: Fundamentals and Applications; John Wiley & Sons: New York, 2001; pp 87–136. [Google Scholar]
- 123.Appleby AJ; Zagal JH Free Energy Relationships in Electrochemistry: A History That Started in 1935. J. Solid State Electrochem. 2011, 15, 1811–1832. [Google Scholar]
- 124.Browne WR Electrochemistry,; Oxford University Press: New York, 2018. [Google Scholar]
- 125.Teegardin K; Day JI; Chan J; Weaver J Advances in Photocatalysis: A Microreview of Visible Light Mediated Ruthenium and Iridium Catalyzed Organic Transformations. Org. Process Res. Dev. 2016, 20, 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Singh A; Teegardin K; Kelly M; Prasad KS; Krishnan S; Weaver JD Facile Synthesis and Complete Characterization of Homoleptic and Heteroleptic Cyclometalated Iridium(III) Complexes for Photocatalysis. J. Organomet. Chem. 2015, 776, 51–59. [Google Scholar]
- 127.Larsen CB; Wenger OS Photoredox Catalysis with Metal Complexes Made from Earth-Abundant Elements. Chem. Eur. J. 2018, 24, 2039–2058. [DOI] [PubMed] [Google Scholar]
- 128.Hossain A; Bhattacharyya A; Reiser O Copper’s Rapid Ascent in Visible-Light Photoredox Catalysis. Science 2019, 364, eaav9713. [DOI] [PubMed] [Google Scholar]
- 129.Hockin BM; Li C; Robertson N; Zysman-Colman E Photoredox Catalysts Based on Earth-Abundant Metal Complexes. Catal. Sci. Technol. 2019, 9, 889–915. [Google Scholar]
- 130.Lee Y; Kwon MS Emerging Organic Photoredox Catalysts for Organic Transformations. Eur. J. Org. Chem. 2020, 6028–6043. [Google Scholar]
- 131.Zilate B; Fischer C; Sparr C Design and Application of Aminoacridinium Organophotoredox Catalysts. Chem. Commun. 2020, 56, 1767–1775. [DOI] [PubMed] [Google Scholar]
- 132.Bryden MA; Zysman-Colman E Organic Thermally Activated Delayed Fluorescence (TADF) compounds used in photocatalysis. Chem. Soc. Rev. 2021, 50, 7587–7680. [DOI] [PubMed] [Google Scholar]
- 133.Vega-Peñaloza A; Mateos J; Companyó X; Escudero-Casao M; Dell'Amico L A Rational Approach to Organo-Photocatalysis: Novel Designs and Structure-Property Relationships. Angew. Chem. Int. Ed. 2021, 60, 1082–1097. [DOI] [PubMed] [Google Scholar]
- 134.Bell JD; Murphy JA Recent Advances in Visible Light-Activated Radical Coupling Reactions Triggered by (I) Ruthenium, (II) Iridium and (III) Organic Photoredox Agents. Chem. Soc. Rev. 2021, DOI: 10.1039/D1CS00311A. [DOI] [PubMed] [Google Scholar]
- 135.Wu Y; Kim D; Teets TS Photophysical Properties and Redox Potentials of Photosensitizers for Organic Photoredox Transformations. Synlett 2021, DOI: 10.1055/a-1390-9065. [DOI] [Google Scholar]
- 136.Heard D; Lennox A Electrode Materials in Modern Organic Electrochemistry. Angew. Chem. Int. Ed. 2020, 59, 18866–18884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Couper AM; Pletcher D; Walsh FC Electrode Materials for Electrosynthesis. Chem. Rev. 1990, 90, 837–865. [Google Scholar]
- 138.Lips S; Waldvogel SR Use of Boron-Doped Diamond Electrodes in Electro-Organic Synthesis. ChemElectroChem 2019, 6, 1649–1660. [Google Scholar]
- 139.Gütz C; Grimaudo V; Holtkamp M; Hartmer M; Werra J, Frensemeier L, Kehl A, Karst U, Broekmann P, Waldvogel SR Leaded Bronze: An Innovative Lead Substitute for Cathodic Electrosynthesis. ChemElectroChem 2018, 5, 247–252. [Google Scholar]
- 140.Friedrich JM; Ponce-de-León C; Reade GW; Walsh FC Reticulated Vitreous Carbon as an Electrode Material. J. Electroanal. Chem. 2004, 561, 203–217. [Google Scholar]
- 141.Bittles JA; Littauer EL Anodic Corrosion and Passivation of Pt in Cl− solution. Corrosion Science 1978, 10, 29–41. [Google Scholar]
- 142.Hudson MJ; Hunter-Fujita FR; Peckett JW; Smith PM Electrochemically Prepared Colloidal, Oxidised Graphite. J. Mater. Chem. 1997, 7, 301–305. [Google Scholar]
- 143.Srivastava SK; Pionteck J Recent Advances in Preparation, Structure, Properties and Applications of Graphite Oxide. J. Nanosci. Nanotechnol. 2015, 15, 1984–2000. [DOI] [PubMed] [Google Scholar]
- 144.Rapeckia T Nowicka AM; Dontena M; Scholz F Stojek Z Activity Changes of Glassy Carbon Electrodes Caused by Their Exposure to OH• Radicals. Electrochem. Commun 2010, 12, 1531–1534. [Google Scholar]
- 145.Duo I; Levy-Clement C, C.; Fujishima A; Comninellis C Electron Transfer Kinetics on Boron-Doped Diamond Part I: Influence of Anodic Treatment. J. Appl. Electrochem. 2004, 34, 935. [Google Scholar]
- 146.Wirtanen T; Prenzel T; Tessonnier J-P; Waldvogel SR Cathodic Corrosion of Metal Electrodes—How to Prevent It in Electroorganic Synthesis. Chem. Rev. 2021, DOI: 10.1021/acs.chemrev.1c00148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zirbes M; Schmitt D; Beiser N; Pitton D; Hoffmann T; Waldvogel SR Anodic Degradation of Lignin at Active Transition Metal-based Alloys and Performance-enhanced Anodes. ChemElectroChem, 2019, 6, 155–161. [Google Scholar]
- 148.Schmitt D; Regenbrecht C; Hartmer M; Stecker F; Waldvogel SR Highly Selective Generation of Vanillin by Anodic Degradation of Lignin: A Combined Approach of Electrochemistry and Product Isolation by Adsorption. Beilstein J. Org. Chem. 2015, 11, 473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zirbes M; Quadri LL; Breiner M; Stenglein A; Bomm A; Schade W; Waldvogel SR High-Temperature Electrolysis of Kraft Lignin for Selective Vanillin Formation. ACS Sustainable Chem. Eng. 2020, 8, 7300–7307. [Google Scholar]
- 150.Lyalin BV; Petrosyan VA Oxidation of Organic Compounds on NiOOH Electrode. Russ J. Electrochem. 2010, 46, 1199–1214. [Google Scholar]
- 151.Beil SB; Mgller T; Sillart SB; Franzmann P; Bomm A; Holtkamp M; Karst U; Schade W; Waldvogel SR Active Molybdenum-Based Anode for Dehydrogenative Coupling Reactions. Angew. Chem. Int. Ed. 2017, 57, 2450–2454. [DOI] [PubMed] [Google Scholar]
- 152.Beil SB; Breiner M; Schulz L; Schüll A; Müller T; Schollmeyer D; Bomm A; Holtkamp M; Karst U; Schade W; Waldvogel SR About the Selectivity and Reactivity of Active Nickel Electrodes in C–C Coupling Reactions. RSC Adv. 2020, 10, 14249–14253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Safizadeh F; Ghali E; Houlachi G Int. J. Hydrogen Energy 2015, 40, 256–274. [Google Scholar]
- 154.Miscellaneous Electrical Materials. Materials Handbook, 2nd ed.; Springer: London, 2008; p 566–567. DOI: 10.1007/978-1-84628-669-8_9 [DOI] [Google Scholar]
- 155.Bockris JOM; Ignatowicz S Studies in Electrolytic Polarisation (IV). The Effect of the Solvent on the Hydrogen Overpotential (ii). Trans. Faraday Soc. 1948, 44, 519–527. [Google Scholar]
- 156.Bockris JO Effect of the Solvent on Hydrogen Overpotential Nature, 1946, 158, 584–584. [Google Scholar]
- 157.Hickling A; Salt FW Studies in Hydrogen Overvoltage at High Current Densities. Part II. The Influence of the Solvent. Trans. Faraday Soc. 1941, 37, 224–231. [Google Scholar]
- 158.Bockris JOM Electrolytic Polarisation: III. The Effect of the Solvent on the Oxygen Overpotential. Faraday Discuss. 1947, 1, 229–235. [Google Scholar]
- 159.Hickling A; Salt FW Studies in Hydrogen Overvoltage at High Current Densities: Part I—The Influence of Electrode Material, Current Density, and Time, in Aqueous Solution. Trans. Faraday Soc. 1940, 36, 1226–1235. [Google Scholar]
- 160.Bockris JOM Electrolytic Polarisation—I. the Overpotential of Hydrogen on Some Less Common Metals at High Current Densities. Influence of Current Density and Time. Trans. Faraday Soc. 1947, 43, 417–429. [Google Scholar]
- 161.Bockris JOM; Azzam AM The Kinetics of the Hydrogen Evolution Reaction at High Current Densities. Trans. Faraday Soc. 1952, 48, 145–160. [Google Scholar]
- 162.Tang Z; Liao L; Zheng Y; Kang J; Chen Y Temperature Effect on Hydrogen Evolution Reaction at Au Electrode. Chin. J. Chem. Phys. 2012, 25, 469–474. [Google Scholar]
- 163.Edinger C, Grimaudo V, Broekmann P, Waldvogel SR, Stabilizing Lead Cathodes with Diammonium Salt Additives in the Deoxygenation of Aromatic Amides. ChemElectroChem 2014, 1, 1018–1022. [Google Scholar]
- 164.Kulisch J; Nieger M; Stecker F; Fischer A; Waldvogel SR Efficient and Stereodivergent Electrochemical Synthesis of Optically Pure Menthylamines. Angew. Chem. Int. Ed. 2011, 50, 5564–5567. [DOI] [PubMed] [Google Scholar]
- 165.Lee CH; Kanan MW Controlling H+ vs CO2 Reduction Selectivity on Pb Electrodes. ACS Catal. 2015, 5, 465–469. [Google Scholar]
- 166.Carr JP; Hampson NA Lead Dioxide Electrode. Chem Rev. 1972, 72, 679–703. [Google Scholar]
- 167.Gütz C; Herold S; Waldvogel SR “Chapter 5: Novel Electrode Materials in Electroorganic Synthesis. ” In Modern Electrosynthetic Methods in Organic Chemistry; (Eds. Marken F, F., Atobe M, M., Eds; CRC Press: New York: ), 2018, pp. 97–126, CRC Press. [Google Scholar]
- 168.Panizza M; Cerisola G Application of Diamond Electrodes to Electrochemical Processes. Electochimica Acta 2005, 51, 191–199. [Google Scholar]
- 169.Macpherson JV A Practical Guide to Using Boron Doped Diamond in Electrochemical Research. Phys. Chem. Chem. Phys. 2015, 17, 2935–2949. [DOI] [PubMed] [Google Scholar]
- 170.Botte GG Electrochemical Manufacturing in the Chemical Industry. Electrochem. Soc. Interface 2014, 23, 49. [Google Scholar]
- 171.Edinger C; Kulisch J; Waldvogel SR Stereoselective Cathodic Synthesis of 8-Substituted (1R,3R,4S)-Menthylamines. Beilstein J. Org. Chem. 2015, 11, 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bockris JOM; Parsons R Studies in Electrolytic Polarisation. Part V.–Hydrogen Overpotential in Methanolic Solution. Trans. Faraday Soc. 1948, 44, 860–872. [Google Scholar]
- 173.Hickling A; Hill S Oxygen Overvoltage. Part I. the Influence of Electrode Material, Current Density, and Time in Aqueous Solution. Faraday Discuss. 1947, 1, 236–246. [Google Scholar]
- 174.Rojas M; Fan CL; Miao HJ; Piron DL Characterization of Hydrogen Evolution on Cobalt Electrodeposits in Water Electrolysis. J. Appl. Electrochem. 1992, 22, 1135–1141. [Google Scholar]
- 175.Bowden FP; O’Connor EA The Change in the Area and Catalytic Activity of Metallic Surfaces on Passing From the Solid to the Liquid State. Proc. R. Soc. London Ser. A 1930, 128, 317–329. [Google Scholar]
- 176.Noninski CI; Ivanov IP; Vassileva-Dimova M Hydrogen Overvoltage on Tin Self-Cleaning Rotating Electrode (SRE) in Absence and Presence of Surface Active Ions. J. Electrochem. Soc. 1983, 130, 1836. [Google Scholar]
- 177.Mitchell BS An Introduction to Materials Engineering and Science, Wiley, Hoboken, 2003. [Google Scholar]
- 178.Hu X; Tian X; Lin Y-W; Wang Z RSC Adv. 2019, 9, 31563–31571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Katsuki N; Takahashi E; Toyoda M; Kurosu T; Iida M; Wakita S; Nishiki Y; Shimamune T Water Electrolysis Using Diamond Thin-Film Electrodes. J. Electrochem. Soc. 1998, 145, 2358. [Google Scholar]
- 180.Martin HB; Argoitia A; Landau U; Anderson AB; Angus JC Hydrogen and Oxygen Evolution on Boron-Doped Diamond Electrodes. J. Electrochem. Soc. 1996, 143, L133. [Google Scholar]
- 181.Santana MHP; Faria LAD; Boodts JFC Electrochemical Characterisation and Oxygen Evolution at a Heavily Boron Doped Diamond Electrode. Electrochim. Acta 2005, 50, 2017–2027. [Google Scholar]
- 182.Koca A Hydrogen Evolution Reaction on Glassy Carbon Electrode Modified With Titanyl Phthalocyanines. Int. J. Hydrogen Energy 2009, 34, 2107–2112. [Google Scholar]
- 183.Walsh FC Electrode Reactions in Metal Finishing.Trans. IMF 1991, 69, 107–110. [Google Scholar]
- 184.Lide DR, Ed., CRC Handbook of Chemistry & Physics, CRC Press, Boca Raton, FL, 2004. [Google Scholar]
- 185.Kolbe H, Untersuchungen über die Elektrolyse organischer Verbindungen, Justus Liebigs Ann. Chem. 1849, 69, 257–294. [Google Scholar]
- 186.Hofer H and Moest M, Ueber die Bildung von Alkoholen bei der Elektrolyse fettsaurer Salze, Justus Liebigs Ann. Chem. 1902, 323, 284–323. [Google Scholar]
- 187.Wang Z, in Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2010. DOI: 10.1002/9780470638859.conrr322 [DOI] [Google Scholar]
- 188.Kodama T; Kubo M; Shinji, Ohkubo K; Tobisu M Phenylene-Bridged bis(Benzimidazolium) (BBIm2+): A Dicationic Organic Photoredox Catalyst. Chem. Sci. 2020, 11, 12109–12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sekine T; Yamura A; Sugino K Mechanism of Hydrocarbon Formation in the Electrolytic Reduction of Acetone in Aqueous Sulfuric Acid. J. Electrochem. Soc. 1965, 112, 439–443. [Google Scholar]
- 190.Amemiya F; Fuse K; Fuchigami T; Atobe M Chemoselective Reaction System Using a Two Inlet Micro-Flow Reactor: Application to Carbonylallylation. Chem. Commun. 2010, 46, 2730–2732. [DOI] [PubMed] [Google Scholar]
- 191.Parsons R The Electrical Double Layer: Recent Experimental and Theoretical Developments. Chem. Rev. 1990, 90, 813–826. [Google Scholar]
- 192.Birda Matthew J., M. J.; Iyodab T; Bonuraa N; Bakalisa J; Ledbettera AJ; Millera JR Effects of Electrolytes on Redox Potentials Through Ion Pairing. J. Electroanal.Chem. 2017, 804, 107–115. [Google Scholar]
- 193.DeBlase CR; Hernández-Burgos K; Rotter JM; Fortman DJ; dos S Abreu D; Timm RA; Diógenes ICN; Kubota LT; Abruña HD; Dichtel R, W. R. Cation-Dependent Stabilization of Electrogenerated Naphthalene Diimide Dianions in Porous Polymer Thin Films and Their Application to Electrical Energy Storage. Angew. Chem. Int. Ed. 2015, 54, 13225–13229. [DOI] [PubMed] [Google Scholar]
- 194.Kurzweil P Chapter 19 - Electrochemical Double-Layer Capacitors. In Electrochemical Energy Storage for Renewable Sources and Grid Balancing; Moseley PT, Garche J, Eds.; Elsevier: Amsterdam, 2015; pp 345–407. [Google Scholar]
- 195.House HO; Feng E; Peet NP Comparison of Various Tetraalkylammonium Salts as Supporting Electrolytes in Organic Electrochemical Reactions. J. Org. Chem. 1971, 36, 2371–2375. [Google Scholar]
- 196.Westbroek P 1 - Fundamentals of Electrochemistry. In Analytical Electrochemistry in Textiles; Westbroek P, Priniotakis G, Kiekens P, Eds.; Woodhead Publishing Series in Textiles; Woodhead Publishing, 2005; pp 3–36. [Google Scholar]
- 197.Morales DM; Risch M Seven Steps to Reliable Cyclic Voltammetry Measurements for the Determination of Double Layer Capacitance. J. Phys. Energy 2021, 3, 034013. [Google Scholar]
- 198.Rodrigo S; Um C; Mixdorf JC; Gunasekera D; Nguyen HM; Luo L Alternating Current Electrolysis for Organic Electrosynthesis: Trifluoromethylation of (Hetero)arenes. Org. Lett. 2020, 22, 6719–6723. [DOI] [PubMed] [Google Scholar]
- 199.Bump DD; Remick AE Studies on Alternating Current Electrolysis: V. Double Layer Resistance and Electrode Area as Factors in Electrode Kinetics. J. Electrochem. Soc. 1964, 111, 981. [Google Scholar]
- 200.Remick AE; Mccormick HW Studies on Alternating-Current Electrolysis: III. Effects of Concentration on Polarization Capacity and Polarization Resistance. J. Electrochem. Soc. 1955, 102, 534. [Google Scholar]
- 201.Sterten Å; Solli PA; Skybakmoen E Influence of Electrolyte Impurities on Current Efficiency in Aluminium Electrolysis Cells. J. Appl. Electrochem. 1998, 28, 781–789. [Google Scholar]
- 202.de Salles Pupo MM; Kortlever R Electrolyte Effects on the Electrochemical Reduction of CO2. ChemPhysChem, 2019, 20, 2926–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Carucci C; Salis A; Magner E Electrolyte Effects on Enzyme Electrochemistry. Curr. Opin. Electrochem. 2017, 5, 158–164. [Google Scholar]
- 204.Liu B; Yu X-Y; Zhu Z; Hua X; Yang L; Wang Z In situ Chemical Probing of the Electrode–Electrolyte Interface by ToF-SIMS. Lab Chip, 2014,14, 855–859. [DOI] [PubMed] [Google Scholar]
- 205.Fangmeyer J; Behrens A; Gleede B; Waldvogel SR; Karst U Mass-Spectrometric Imaging of Electrode Surfaces—a View on Electrochemical Side Reactions. Angew. Chem. Int. Ed. 2020, 59, 20428–20433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Wong RA; Yokota Y; Wakisaka M; Kim Y Probing Consequences of Anion-Dictated Electrochemistry on the Electrode/Monolayer/Electrolyte Interfacial Properties. Nat. Commun. 2020, 11, 4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Savéant J-M Molecular Catalysis of Electrochemical Reactions. Mechanistic Aspects. Chem. Rev. 2008, 108, 2348–2378. [DOI] [PubMed] [Google Scholar]
- 208.Novaes T, L. F.; Liu J; Shen Y; Lu L; Meinhardt M, J.; Lin S Electrocatalysis as an Enabling Technology for Organic Synthesis. Chem. Soc. Rev. 2021, 50, 7941–8002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Sauer GS; Lin S An Electrocatalytic Approach to the Radical Difunctionalization of Alkenes. ACS Catal. 2018, 8, 5175–5187. [Google Scholar]
- 210.Steckhan E Organic Syntheses with Electrochemically Regenerable Redox Systems. In Electrochemistry I; Steckhan E, Ed.; Topics in Current Chemistry; Springer: Berlin, Heidelberg, 1987; pp 1–69. 10.1007/3-540-17871-6_11. [DOI] [Google Scholar]
- 211.Steckhan E Indirect Electroorganic Syntheses-A Modern Chapter of Organic Electrochemistry. Angew. Chem. Int. Ed. Engl. 1986, 25, 683–701. [Google Scholar]
- 212.Nutting JE; Rafiee M; Stahl SS Tetramethylpiperidine N-Oxyl (TEMPO), Phthalimide N-Oxyl (PINO), and Related N-Oxyl Species: Electrochemical Properties and Their Use in Electrocatalytic Reactions. Chem. Rev. 2018, 118, 4834–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wienkamp R; Steckhan E Indirect Electrochemical Regeneration of NADH by a Bipyridinerhodium(I) Complex as Electron-Transfer Agent. Angew. Chem. Int. Ed. Engl. 1982, 21, 782–783. [Google Scholar]
- 214.Britz D; Strutwolf J Digital Simulations in Electrochemistry, Springer International Publishing, Cham Switzerland, 4th ed., 2016. [Google Scholar]
- 215.Messersmith SJ Cyclic Voltammetry Simulations with DigiSim Software: An Upper-Level Undergraduate Experiment. J. Chem. Educ. 2014, 91, 1498–1500. [Google Scholar]
- 216.Brown JH Development and Use of a Cyclic Voltammetry Simulator To Introduce Undergraduate Students to Electrochemical Simulations. J. Chem. Educ. 2015, 92, 1490–1496 [Google Scholar]
- 217.Wang S; Wang J; Gao Y Development and Use of an Open-Source, User-Friendly Package To Simulate Voltammetry Experiments. J. Chem. Educ. 2017, 94, 1567–1570. [Google Scholar]
- 218.Ogibin YN; Elinson MN; Nikishin GI Mediator Oxidation Systems in Organic Electrosynthesis. Russ. Chem. Rev. 2009, 78 (2), 89. [Google Scholar]
- 219.Kyriacou D (1994) Indirect Electroorganic Reactions (Indirect Electrolysis). In: Modern Electroorganic Chemistry. Springer Lab Manual. Springer, Berlin, Heidelberg. 10.1007/978-3-642-78677-8_4 [DOI] [Google Scholar]
- 220.Röse P; Emge S; König CA; Hilt G Efficient Oxidative Coupling of Arenes via Electrochemical Regeneration of 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) under Mild Reaction Conditions. Adv. Synth. Catal. 2017, 359, 1359–1372. [Google Scholar]
- 221.Luca OR; Wang T; Konezny SJ; Batista VS; Crabtree RH; DDQ as an Electrocatalyst for Aminedehydrogenation, a Model System for Virtual Hydrogen Storage. New J. Chem. 2011, 35, 998–999. [Google Scholar]
- 222.Lennox AJJ; Nutting JE; Stahl SS Selective Electrochemical Generation of Benzylic Radicals Enabled by Ferrocene-Based Electron-Transfer Mediators. Chem. Sci. 2018, 9, 356–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Walker BR; Manabe S; Brusoe AT; Sevov CS Mediator-Enabled Electrocatalysis with Ligandless Copper for Anaerobic Chan–Lam Coupling Reactions. J. Am. Chem. Soc. 2021, 143, 6257–6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Silva NPRA, M. E.; Pombeiro AJL; Fraústo da Silva JJR; Herrmann R; Deus N; Castilho TJ; Silva MFCG Redox Potential and Substituent Effects at Ferrocene Derivatives. Estimates of Hammett σp and Taft Polar σ Substituent Constants. J. Organomet. Chem. 1991, 421, 75–90. [Google Scholar]
- 225.Silva NPRA, M. E.; Pombeiro AJL; Fraústo da Silva JJR; Herrmann R; Deus N; Bozak E, R. Redox Potential and Substituent Effects in Ferrocene Derivatives: II. J. Organomet. Chem. 1994, 480, 81–90. [Google Scholar]
- 226.Dapperheld S; Steckan E; Brinkhaus K-HG; Esch T Substituted Triarylamine Cation-Radical Redox Systems - Synthesis, Electrochemical and Spectroscopic Properties, Hammet Behavior, and Suitability as Redox Catalysts. Chem. Ber. 1991, 124, 2557–2567. [Google Scholar]
- 227.Wang F; Stahl SS Electrochemical Oxidation of Organic Molecules at Lower Overpotential: Accessing Broader Functional Group Compatibility with Electron−Proton Transfer Mediators. Acc. Chem. Res. 2020, 53, 561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Siu JC Lin S Catalyzing Electrosynthesis: A Homogeneous Electrocatalytic Approach to Reaction Discovery. Acc. Chem. Res. 2020, 53, 547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Mitsudo K; Kaide T; Nakamoto E; Yoshida K; Tanaka H Electrochemical Generation of Cationic Pd Catalysts and Application to Pd/TEMPO Double-Mediatory Electrooxidative Wacker-Type Reactions. J. Am. Chem. Soc. 2007, 129, 2246–2247. [DOI] [PubMed] [Google Scholar]
- 230.Le C; Liang Y; Evans RW; Li X; MacMillan DWC Selective sp3 C–H Akylation via Polarity-Match-Based Cross-Coupling. Nature 2017, 547, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Twilton J; Christensen M; DiRocco DA; Ruck R; Davies IW; MacMillan DWC Selective Hydrogen Atom Abstraction through Induced Bond Polarization: Direct α-Arylation of Alcohols Through Photoredox, HAT, and Nickel Catalysis. Angew. Chem. Int. Ed. 2018, 57, 5369–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kawamata Y; Yan M; Liu Z; Bao D-H; Chen J; Starr JT; Baran PS Scalable, Electrochemical Oxidation of Unactivated C–H Bonds. J. Am. Chem. Soc. 2017, 139, 7448–7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Hager D; MacMillan DWC Activation of C−H Bonds via the Merger of Photoredox and Organocatalysis: A Coupling of Benzylic Ethers with Schiff Bases. J. Am. Chem. Soc. 2014, 136, 16986–16989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Mukherjee S; Maji B; Tlahuext-Aca A; Glorius F Visible-Light-Promoted Activation of Unactivated C(sp3)–H Bonds and Their Selective Trifluoromethylthiolation. J. Am. Chem. Soc. 2018, 138, 16200–16203. [DOI] [PubMed] [Google Scholar]
- 235.Kobayashi F; Fujita M; Ide T; Ito Y; Yamashita K; Egami H; Hamashima Y Dual-Role Catalysis by Thiobenzoic Acid in Cα−H Arylation under Photoirradiation. ACS Catal. 2021, 11, 82–87. [Google Scholar]
- 236.Tanaka H; Sakai K; Kawamura A; Oisaki K; Kanai M Sulfonamides As New Hydrogen Atom Transfer (HAT) Catalysts for Photoredox Allylic and Benzylic C–H Arylations. Chem. Commun. 2018, 54, 3215–3218. [DOI] [PubMed] [Google Scholar]
- 237.Margrey KA; Czaplyski WL; Nicewicz DA; Alexanian EJ A General Strategy for Aliphatic C−H Functionalization Enabled by Organic Photoredox Catalysis. J. Am. Chem. Soc. 2018, 140, 4213–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Morton CM; Zhu Q; Ripberger H; Troian-Gautier L; Toa ZSD; Knowles RR; Alexanian EJ C−H Alkylation via Multisite-Proton-Coupled Electron Transfer of an Aliphatic C−H Bond. J. Am. Chem. Soc. 2019, 141, 13253–13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Hu A; Guo J-J; Pan H; Zuo Z Selective Functionalization of Methane, Ethane, and Higher Alkanes by Cerium Photocatalysis. Science 2018, 361, 668–672. [DOI] [PubMed] [Google Scholar]
- 240.An Q; Wang Z; Chen Y; Wang X; Zhang K; Pan H; Liu W; Zuo Z Cerium Catalyzed C−H Functionalizations of Alkanes Utilizing Alcohols as Hydrogen Atom Transfer Agents. J. Am. Chem. Soc. 2020, 142, 6216–6226. [DOI] [PubMed] [Google Scholar]
- 241.Deng H-P; Zhou Q; Wu J Microtubing-Reactor-Assisted Aliphatic C−H Functionalization with HCl as a Hydrogen-Atom Transfer Catalyst Precursor in Conjunction with an Organic Photoredox Catalyst. Angew. Chem. Int. Ed. 2018, 57, 12661–12665. [DOI] [PubMed] [Google Scholar]
- 242.Rohe S; Morris AO; McCallum T; Barriault L Hydrogen Atom Transfer Reactions via Photoredox Catalyzed Chlorine Atom Generation. Angew. Chem. Int. Ed. 2018, 57, 15664–15669. [DOI] [PubMed] [Google Scholar]
- 243.Shen Y; Gu Y; Martin R sp3 C−H Arylation and Alkylation Enabled by the Synergy of Triplet Excited Ketones and Nickel Catalysts. J. Am. Chem. Soc. 2018, 140, 12200–12209. [DOI] [PubMed] [Google Scholar]
- 244.Santos MS; Corrêa AG; Paixão MW; König B C(sp3)−C(sp3) Cross-Coupling of Alkyl Bromides and Ethers Mediated by Metal and Visible Light Photoredox Catalysis. Adv. Synth. Catal. 2020, 362, 2367–2372. [Google Scholar]
- 245.Ryder ASH; Cunningham WB; Ballantyne G; Mules T; Kinsella AG; Turner-Dore J; Alder CM; Edwards LJ; McKay BSJ; Grayson MN; et al. Photocatalytic α-Tertiary Amine Synthesis via C−H Alkylation of Unmasked Primary Amines. Angew. Chem. Int. Ed. 2020, 59, 14986–14991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Perry IB; Brewer TF; Sarver PJ; Schultz DM; DiRocco, MacMillan DWC Direct Arylation of Strong Aliphatic C–H Bonds. Nature, 2018, 560, 70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Sarver PJ; Bacauanu V; Schultz DM; DiRocco DA; Lam Y.-.h.; Sherer EC; MacMillan DWC The Merger of Decatungstate and Copper Catalysis To Enable Aliphatic C(sp3)–H Trifluoromethylation. Nat. Chem. 2020, 12, 459–457. [DOI] [PubMed] [Google Scholar]
- 248.Bird CL; Kuhn AT Electrochemistry of the Viologens. Chem. Soc. Rev. 1981, 10, 49–82. [Google Scholar]
- 249.Pavlishchuk VV; Addison AW Conversion Constants for Redox Potentials Measured versus Different Reference Electrodes in Acetonitrile Solutions at 25°C. Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar]
- 250.Geiger WE Electroreduction of Cobaltocene. Evidence for a Metallocene Anion. J. Am. Chem. Soc. 1974, 96, 2632–2634. [Google Scholar]
- 251.Sund CJ; McMasters S; Crittenden SR; Harrell LE; Sumner JJ Effect of Electron Mediators on Current Generation and Fermentation in a Microbial Fuel Cell. Appl. Microbiol. Biotechnol. 2007, 76, 561–568. [DOI] [PubMed] [Google Scholar]
- 252.Cowper NGW; Chernowsky CP; Williams OP; Wickens ZK Potent Reductants via Electron-Primed Photoredox Catalysis: Unlocking Aryl Chlorides for Radical Coupling. J. Am. Chem. Soc. 2020, 142, 2093–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Nelsen SF; Hintz PJ Electrochemical Oxidation of Tertiary Amines. Effect of Structure upon Reversibility. J. Am. Chem. Soc. 1972, 94, 7114–7117. [Google Scholar]
- 254.Shaw MH; Shurtleff VW; Terrett JA; Cuthbertson JD; MacMillan DWC Native Functionality in Triple Catalytic Cross-Coupling: sp3 C–H Bonds as Latent Nucleophiles. Science 2016, 352, 1304–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Potentials reported against Ag/Ag+ in the literature were converted to vs SCE by subtracting 45 mV.
- 256.Pitre SP; McTiernan CD; Vine W; DiPucchio R; Grenier M; Scaiano JC Visible-Light Actinometry and Intermittent Illumination as Convenient Tools to Study Ru(bpy)3Cl2 Mediated Photoredox Transformations. Sci. Rep. 2015, 5, 16397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Hatchard CG; Parker CA A New Sensitive Chemical Actinometer – II. Potassium Ferrioxalate as a Standard Chemical Actinometer. Proc. R. Soc. London, Ser. A 1956, 325, 518–536. [Google Scholar]
- 258.Kirk AD; Namasivayam C Errors in Ferrioxalate Actinometry. Anal. Chem. 1983, 55, 2428–2429. [Google Scholar]
- 259.Faraday M Experimental Researches in Electricity, Phil. Trans. Roy. Soc. London, 1833, 123, 39. [Google Scholar]
- 260.Ehl RG; Inde AJ Faraday's Electrochemical Laws and the Determination of Equivalent Weights. J. Chem. Educ. 1954, 31, 226–232. [Google Scholar]
- 261.Braslavsky SE Glossary of Terms Used In Photochemistry 3rd Edition. Pure Appl. Chem. 2007, 79, 293–465. [Google Scholar]
- 262.Cismesia MA; Yoon TP Characterizing chain processes in visible light photoredox catalysis. Chem. Sci. 2015, 6, 5426–5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Yayla HG; Peng F; Mangion IK; McLaughlin M; Campeau L-C; Davies IW; DiRocco DA; Knowles RR Discovery and Mechanistic Study of a Photocatalytic Indoline Dehydrogenation for the Synthesis of Elbasvir. Chem. Sci. 2016, 7, 2066–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Walsh FC; Perry B Fundamental Definitions and Concepts. Trans. of the IMF, 1992, 70, 87–89, DOI: 10.1080/00202967.1992.11870949. [DOI] [Google Scholar]
- 265.Bard AJ; Inzelt G; Scholz F (Eds.) in Electrochemical Dictionary (2nd Edition), Springer, New York, 2021, p 166. DOI: 10.1007/978-3-642-29551-5. [DOI] [Google Scholar]
- 266.Francke R; Gonzalez L; Little RD; Moeller KD Chapter 79: Electrons, Electrodes, and the Transformation of Organic Molecules. In Surface and Interface Science: Applications of Surface Science II, First Edition, Wandelt K, Ed.; Wiley: New York, p. 875; DOI: 10.1002/9783527822508.ch79 [DOI] [Google Scholar]
- 267.Heineman WR; Kissinger PT Large-Amplitude Controlled-Potential Techniques. In Laboratory Techniques in Electroanalytical Chemistry, 2nd edition; Kissinger PT; Heineman WR, Eds.; Marcel Dekker, Inc.: New York, 1990; pp. 51–126. [Google Scholar]
- 268.Corcoran EB; McMullen JP; Lévesque F; Wismer MK; Naber JR Photon Equivalents as a Parameter for Scaling Photoredox Reactions in Flow: Translation of Photocatalytic C−N Cross-Coupling from Lab Scale to Multikilogram Scale. Angew. Chem. Int. Ed. 2020, 59, 11964–11968. [DOI] [PubMed] [Google Scholar]
- 269.Bonfield HE; Knauber T; Lévesque F; Moschetta EG; Susanne F; Edwards LJ Photons as a 21st Century Reagent. Nat. Commun. 2020, 11, 804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Lévesque F; Di Maso MJ; Narsimhan K; Wismer MK; Naber JR Design of a Kilogram Scale, Plug Flow Photoreactor Enabled by High Power LEDs. Org. Process Res. Dev. 2020, 24, 2935–2940. [Google Scholar]
- 271.Pletcher D; Green RA; Brown RDC Flow Electrolysis Cells for the Synthetic Organic Chemistry Laboratory. Chem. Rev. 2018, 118, 4573–4591. [DOI] [PubMed] [Google Scholar]
- 272.Noël T; Cao Y; Laudadio G The Fundamentals Behind the Use of Flow Reactors in Electrochemistry. Acc. Chem. Res. 2019, 52, 2858–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Cambié D; Bottecchia C; Straathof NJW; Hessel V; Noël T Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem. Rev. 2016, 116, 10276–10341. [DOI] [PubMed] [Google Scholar]
- 274.Sambiagio C; Noël T Flow Photochemistry: Shine Some Light on Those Tubes! Trends in Chem. 2020, 2, 92–106. [Google Scholar]
- 275.Juris A; Balzani V; Belser P; von Zelewsky A Characterization of the Excited State Properties of Some New Photosensitizers of the Ruthenium (Polypyridine) Family. Chem. Helv. Acta 1981, 64, 2175–2182. [Google Scholar]
- 276.Harper KC; Moschetta EG; Bordawekar SV; Wittenberger SJ A Laser Driven Flow Chemistry Platform for Scaling Photochemical Reactions with Visible Light. ACS Cent. Sci. 2019, 5, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Laudadio G; Barmpoutsis E; Schotten C; Struik L; Govaerts S; Browne DL; Noël T Sulfonamide Synthesis through Electrochemical Oxidative Coupling of Amines and Thiols. J. Am. Chem. Soc. 2019, 141, 5664–5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Horcajada R; Okajima M; Suga S; Yoshida J -i. Microflow Electroorganic Synthesis Without Supporting Electrolyte. Chem. Commun. 2005, 1303–1305. [DOI] [PubMed] [Google Scholar]
- 279.van Melis CGW; Penny MR; Garcia AD; Petti A; Dobbs AP; Hilton ST; Lam K Supporting-Electrolyte-Free Electrochemical Methoxymethylation of Alcohols Using a 3D-Printed Electrosynthesis Continuous Flow Cell System. ChemElectroChem 2019, 6, 4144–4148. [Google Scholar]
- 280.Broese T; Roesel AF; Prudlik A; Francke R An Electrocatalytic Newman−Kwart-type Rearrangement. Org. Lett. 2018, 20, 7483–7487. [DOI] [PubMed] [Google Scholar]
- 281.van Melis CGW; Penny MR; Garcia AD; Petti A; Dobbs AP; Hilton ST; Lam K Supporting-Electrolyte-Free Electrochemical Methoxymethylation of Alcohols Using a 3DPrinted Electrosynthesis Continuous Flow Cell System. ChemElectroChem 2019, 6, 4144–4148. [Google Scholar]
- 282.He P; Watts P; Marken F; Haswell SJ Electrolyte Free Electro-Organic Synthesis: The Cathodic Dimerisation of 4-Nitrobenzylbromide in a Micro-Gap Flow Cell. Electrochem. Commun. 2005, 7, 918–924. [Google Scholar]
- 283.Thomson W LXVI. On the Mechanical Theory of Electrolysis. The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science, 1851, 2, 429–444. [Google Scholar]
- 284.Scott K Process intensification: An Electrochemical Perspective. Renewable Sustainable Energy Rev.2018, 81, 1406–1426. [Google Scholar]
- 285.Maljuric S; Jud W; Kappe CO; Cantillo D Translating Batch Electrochemistry to Single-Pass Continuous Flow Conditions: An Organic Chemist’s Guide. J. Flow Chem. 2020, 10, 181–190. [Google Scholar]
- 286.Gutmann B; Cantillo D; Kappe CO Continuous-Flow Technology—A Tool for the Safe Manufacturing of Active Pharmaceutical Ingredients. Angew. Chem. Int. Ed. 2015, 54, 6688–6729. [DOI] [PubMed] [Google Scholar]
- 287.Perry SC; de León CP; Walsh FC Review—The Design, Performance and Continuing Development of Electrochemical Reactors for Clean Electrosynthesis. J. Electrochem. Soc. 2020, 167, 155525. [Google Scholar]
- 288.Jüttner K Chapter 1 Technical Scale of Electrochemistry. In Encyclopedia of Electrochemistry: Vol. 5 Electrochemical Engineering. Edited by Bard AJ and Stratmann M Macdonald DD, Schmuki P, Eds.; Wiley: Weinheim, 2007. DOI: 10.1002/9783527610426.bard050001. [DOI] [Google Scholar]
- 289.Walsh FC; Ponce de León C Progress in Electrochemical Flow Reactors for Laboratory and Pilot Scale Processing. Electrochim. Acta 2018, 280, 121–148. [Google Scholar]
- 290.Atobe M; Tateno H; Matsumura Y Applications of Flow Microreactors in Electrosynthetic Processes. Chem. Rev. 2018, 118, 4541–4572. [DOI] [PubMed] [Google Scholar]
- 291.Cardoso DSP; Šljukić B; Santos DMF; Sequeira CAC Organic Electrosynthesis: From Laboratorial Practice to Industrial Applications. Org. Process Res. Dev. 2017, 21, 1213–1226. [Google Scholar]
- 292.Arenas LF; de León CP; Walsh FC Critical Review—The Versatile Plane Parallel Electrode Geometry: An Illustrated Review. J. Electrochem. Soc. 2020, 167, 023504. [Google Scholar]
- 293.Jud W; Kappe CO; Cantillo D A Continuous Flow Cell for High-Temperature/High-Pressure Electroorganic Synthesis. ChemElectroChem 2020, 7, 2777–2783. [Google Scholar]
- 294.Jörissen J Ion Exchange Membranes as Solid Polymer Electrolytes (SPE) in Electro-Organic Syntheses without Supporting Electrolyte. Electrochim. Acta 1996, 41, 553–562. [Google Scholar]
- 295.Gabe DR, Walsh FC The Rotating Cylinder Electrode: A Review of Development. J. Appl. Electrochem. 1983, 13, 3–21. [Google Scholar]
- 296.Gabe DR; Wilcox GD Vonzalez-Garcia J; Walsh FC The Rotating Cylinder Electrode: Its Continued Development and Application J. Appl. Electrochem. 1998, 28, 759–780. [Google Scholar]
- 297.Sambiagio C; Timothy Noël T Flow Photochemistry: Shine Some Light on Those Tubes! Trends in Chemistry, 2020, 2, 92–106. [Google Scholar]
- 298.Pütter H, Industrial Electroorganic Chemistry. In Organic Electrochemistry, 4th edition; Lund H; Hammerich O Eds.; Marcel Dekker: New York, 2001. [Google Scholar]
- 299.Steckhan E; Arns T; Heineman WR; Hilt G; Hoormann D; Jörissen J; Kröner L; Lewall B; Pütter H Chemosphere 2001, 43, 63–73. [DOI] [PubMed] [Google Scholar]
- 300.Robertson PM; Berg P; Reimann H; Schleich K; Seiler P Application of the "Swiss-Roll" Electrolysis Cell in Vitamin-C Production. J. Electrochem. Soc. 1983, 130, 591–596. [Google Scholar]
- 301.Industrial Electrochemistry, Second Edition, Pletcher D, Walsh FC, Eds.; Springer: London, 1993. DOI: 10.1007/978-94-011-2154-5. [DOI] [Google Scholar]
- 302.Leech MC; Garcia AD; Petti A; Dobbs AP; Lam K Organic electrosynthesis: from academia to industry. React. Chem. Eng. 2020, 5, 977–990. [Google Scholar]
- 303.Tanbouza N; Ollevier T; Lam K Bridging Lab and Industry with Flow Electrochemistry. iScience, 2020, 23, 101720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Connelly NG; Geiger WE Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. [DOI] [PubMed] [Google Scholar]
- 305.Ibanez JG; Fitch A; Frontana-Uribe BA; Vasquez-Medrano R Green Electrochemistry. In Encyclopedia of Applied Electrochemistry. Kreysa G, Ota K, Savinell RF, Eds; Springer: New York, 2014. DOI: 10.1007/978-1-4419-6996-5_132. [DOI] [Google Scholar]
- 306.Frontana-Uribe BA; Little RD; Ibanez JG; Palmad A; Vasquez-Medranoc R Organic Electrosynthesis: A Promising Green Methodology in Organic Chemistry. Green Chem. 2010, 12, 2099–2119. [Google Scholar]
- 307.Orella MJ; Román-Leshkov Y; Brushett FR Emerging Opportunities for Electrochemical Processingto Enable Sustainable Chemical Manufacturing . Curr. Opin. Chem. Eng. 2018, 20, 159–167. [Google Scholar]
- 308.Dallinger D; Kappe CO Why Flow Means Green – Evaluating the Merits of Continuous Processing in the Context of Sustainability. Curr. Opin. Green Sustain. 2017, 19, 6–12. [Google Scholar]
- 309.Crisenza GEM; Melchiorre P Chemistry Glows Green With Photoredox Catalysis. Nat. Commun. 2020, 11, 803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Horn EJ; Rosen BR; Baran PS Synthetic Organic Electrochemistry: An Enabling and Innately Sustainable Method. ACS Cent. Sci. 2016, 2, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Kärkäs MD Electrochemical strategies for C–H functionalization and C–N bond formation. Chem. Soc. Rev. 2018, 47, 5786–5865. [DOI] [PubMed] [Google Scholar]
- 312.For a review on guiding principles to studying reaction mechanisms of photocatalytic reactions, see: Buzzetti L; Crisenza GEM; Melchiorre P Mechanistic Studies in Photocatalysis. Angew. Chem. Int. Ed. 2019, 58, 3730–3747.
- 313.Yan M; Kawamata Y; Baran PS Synthetic Organic Electrochemistry: Calling All Engineers. Angew. Chem. Int. Ed. 2018, 57, 4149–4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Grimshaw J Electrochemical Reactions and Mechanisms in Organic Chemistry; Elsevier: New York: 2000. DOI: 10.1016/B978-0-444-72007-8.X5000-9. [DOI] [Google Scholar]
- 315.Costentin C; Savéant J-M Concepts and Tools for Mechanism and Selectivity Analysis in Synthetic Organic Electrochemistry. Proc. Natl. Acad. Sci. USA 2019, 116, 11147–11152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Yang Y; Xiong Y; Zeng R; Lu X; Krumov M; Huang X; Xu W; Wang H; DiSalvo FJ; Brock JD; Muller DA; Abruña HD Operando Methods in Electrocatalysis. ACS Catal. 2021, 11, 1136–1178. [Google Scholar]
- 317.Lima CGS; de M. Lima T; Duarte M; Jurberg ID; Paixão MW Organic Synthesis Enabled by Light-Irradiation of EDA Complexes: Theoretical Background and Synthetic Applications. ACS Catalysis 2016, 6, 1389–1407. [Google Scholar]
- 318.Brimioulle R; Bach T Enantioselective Lewis Acid Catalysis of Intramolecular Enone [2+2] Photocycloaddition Reactions. Science 2013, 342, 840–843. [DOI] [PubMed] [Google Scholar]
- 319.Huang X; Quinn TR; Harms K; Webster RD; Zhang L; Wiest O; Meggers E Direct Visible-Light-Excited Asymmetric Lewis Acid Catalysis of Intermolecular [2+2] Photocycloadditions. J. Am. Chem. Soc. 2017, 139, 9120–9123. [DOI] [PubMed] [Google Scholar]
- 320.Sandford C; Edwards MA; Klunder KJ; Hickey DP; Li M; Barman K; Sigman MS; White HS; Minteer SD A Synthetic Chemist’s Guide to Electroanalytical Tools for Studying Reaction Mechanisms. Chem. Sci. 2019, 10, 6404–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Kuijpers KPL; Bottecchia C; Cambié D; Drummen K; König NJ; Noël T A Fully Automated Continuous-Flow Platform for Fluorescence Quenching Studies and Stern–Volmer Analysis. Angew. Chem. Int. Ed. 2018, 57, 11278–11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Shields BJ; Kudisch B; Scholes GD; Doyle AG Long-Lived Charge-Transfer States of Nickel(II) Aryl Halide Complexes Facilitate Bimolecular Photoinduced Electron Transfer. J. Am. Chem. Soc. 2018, 140, 8, 3035–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Ting SI; Garakyaraghi S; Taliaferro CM; Shields BJ; Scholes GD; Castellano FN; Doyle AG 3d-d Excited States of Ni(II) Complexes Relevant to Photoredox Catalysis: Spectroscopic Identification and Mechanistic Implications. J. Am. Chem. Soc. 2020, 142, 5800–5810. [DOI] [PubMed] [Google Scholar]
- 324.Tian L; Till NA; Kudisch B; MacMillan DWC; Scholes GD Transient Absorption Spectroscopy Offers Mechanistic Insights for an Iridium/Nickel-Catalyzed C–O Coupling. J. Am. Chem. Soc. 2020, 142, 4555–4559. [DOI] [PubMed] [Google Scholar]
- 325.Stevenson BG; Spielvogel EH; Loiaconi EA; Wambua VM; Nakhamiyayev RV; Swierk JR Mechanistic Investigations of an α-Aminoarylation Photoredox Reaction. J. Am. Chem. Soc. 2021, 143, 8878–8885. [DOI] [PubMed] [Google Scholar]
- 326.Blackmond DG Kinetic Profiling of Catalytic Organic Reactions as a Mechanistic Tool. J. Am. Chem. Soc. 2015, 137, 10852–10866. [DOI] [PubMed] [Google Scholar]
- 327.Blackmond DG Reaction Progress Kinetic Analysis: A Powerful Methodology for Mechanistic Studies of Complex Catalytic Reactions. Angew. Chem. Int. Ed. 2005, 44, 4302–4320. [DOI] [PubMed] [Google Scholar]
- 328.Burés J A Simple Graphical Method to Determine the Order in Catalyst. Angew. Chem. Int. Ed. 2016, 55, 2028–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Ji Y; DiRocco DA; Kind J; Thiele CM; Gschwind RM; Reibarkh M LED-Illuminated NMR Spectroscopy: A Practical Tool for Mechanistic Studies of Photochemical Reactions. ChemPhotoChem 2019, 3, 984–992. [Google Scholar]
- 330.Nitschke P; Lokesh N; Gschwind RM Combination of Illumination and High Resolution NMR Spectroscopy: Key Features and Practical Aspects, Photochemical Applications, and New Concepts. Prog. Nucl. Magn. Reson. Spectrosc. 2019, 114–115, 86–134. [DOI] [PubMed] [Google Scholar]
- 331.Ye Y; Tong J In Situ Electrochemical Nuclear Magnetic Resonance Spectroscopy for Electrocatalysis: Challenges and Prospects.Curr. Opin. Electrochem. 2017, 4, 60–68. [Google Scholar]
- 332.Richards JA; Evans DH Flow Cell for Electrolysis Within the Probe of a Nuclear Magnetic Resonance Spectrometer. Anal. Chem. 1975, 47, 964–966. [Google Scholar]
- 333.Neudert R; Strofer E; Bremser FV On-Line NMR in Process Engineering. Magn. Reson. Chem. 1986, 24, 1089–1092. [Google Scholar]
- 334.Richter JB; Eßbach C; Senkovska I; Kaskel S; Brunner E Quantitative in situ 13C NMR Studies of the Electro-Catalytic Oxidation of Ethanol. Chem. Commun. 2019, 55, 6042–6045. [DOI] [PubMed] [Google Scholar]
- 335.Cao S-H; Liu S; Sun H-J; Huang L; Ni Z-R; Jiang W-L; Zhan M; Zhou Z-Y; Sun S-G; Chen Z Versatile, Robust, and Facile Approach for in Situ Monitoring Electrocatalytic Processes through Liquid Electrochemical NMR Spectroscopy. Anal. Chem. 2019, 91, 1686–1691 [DOI] [PubMed] [Google Scholar]
- 336.Webster RD In Situ Electrochemical-NMR Spectroscopy. Reduction of Aromatic Halides. Anal. Chem. 2004, 76, 1603–1610. [DOI] [PubMed] [Google Scholar]
- 337.Boisseau R; Bussy U; Giraudeau P; Boujtita M In Situ Ultrafast 2D NMR Spectroelectrochemistry for Real-Time Monitoring of Redox Reactions. Anal. Chem. 2015, 87, 372–375. [DOI] [PubMed] [Google Scholar]
- 338.Huang L; Sorte EG; Sun S-G; Tong YYJ A Straightforward Implementation of In Situ Solution Electrochemical 13C NMR Spectroscopy for Studying Reactions on Commercial Electrocatalysts: Ethanol Oxidation. Chem. Commun. 2015, 51, 8086–8088. [DOI] [PubMed] [Google Scholar]
- 339.Cao S-H; Ni Z-R; Huang L; Sun H-J; Tang B; Lin L-J; Huang Y-Q; Zhou Z-Y; Sun S-G; Chen Z In Situ Monitoring Potential-Dependent Electrochemical Process by Liquid NMR Spectroelectrochemical Determination: A Proof-of-Concept Study. Anal. Chem. 2017, 89, 3810–3813. [DOI] [PubMed] [Google Scholar]
- 340.daSilva PF; Gomes BF; Lobo CMS; Keng LH; Júnior Q; Danieli E; Carmo M; Blümich B; Colnago LA; Electrochemical NMR Spectroscopy: Electrode Construction and Magnetic Sample Stirring. Microchem. J. 2019, 146, 658–663. [Google Scholar]
- 341.Scheffler JE; Cottrell CE; Berliner LJ An Inexpensive, Versatile Sample Illuminator for Photo-CIDNP on Any NMR Spectrometer. J. Magn. Reson. 1985, 63, 199–201. [Google Scholar]
- 342.Feldmeier C; Bartling H; Riedle E; Gschwind RM LED Based NMR Illumination Device for Mechanistic Studies on Photochemical Reactions – Versatile and Simple, yet Surprisingly Powerful. J. Magn. Reson. 2013, 232, 39–44. [DOI] [PubMed] [Google Scholar]
- 343.Lehnherr D; Ji Y; Neel AJ; Cohen RD; Brunskill APJ; Yang J; Reibarkh M Discovery of a Photoinduced Dark Catalytic Cycle Using in Situ LED-NMR Spectroscopy. J. Am. Chem. Soc 2019, 140, 13843–13853. [DOI] [PubMed] [Google Scholar]
- 344.Ji Y; DiRocco DA; Hong CM; Wismer MK; Reibarkh M Facile Quantum Yield Determination via NMR Actinometry. Org. Lett. 2018, 20, 2156–215. [DOI] [PubMed] [Google Scholar]
- 345.Seegerer A; Nitschke P; Gschwind RM Combined In Situ Illumination-NMR-UV/Vis Spectroscopy: A New Mechanistic Tool in Photochemistry. Angew. Chem. Int. Ed. 2018, 57, 7493–7497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Rößler M; Huth PU; Liauw MA Process Analytical Technology (PAT) as a Versatile Tool for Real-Time Monitoring and Kinetic Evaluation of Photocatalytic Reactions. React. Chem. Eng. 2020, 5, 1992–2002. [Google Scholar]
- 347.Pieber B; Malik JA; Cavedon C; Gisbertz S; Savateev A; Cruz D; Heil T; Zhang G; Seeberger PH Semi-heterogeneous Dual Nickel/Photocatalysis Using Carbon Nitrides: Esterification of Carboxylic Acids with Aryl Halides. Angew. Chem. Int. Ed. 2019, 58, 9575–580. [DOI] [PubMed] [Google Scholar]
- 348.Malik JA; Madani A; Pieber B; Seeberger PH Evidence for Photocatalyst Involvement in Oxidative Additions of Nickel-Catalyzed Carboxylate O-Arylations. J. Am. Chem. Soc. 2020, 142, 11042–11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Hall AMR; Broomfield-Tagg R; Camilleri M; Carbery DR; Codina A; Whittaker DTE; Coombes S; Lowe JP; Hintermair U Online Monitoring of a Photocatalytic Reaction by Real-Time High Resolution FlowNMR Spectroscopy. Chem. Commun. 2018, 54, 30–33. [DOI] [PubMed] [Google Scholar]
- 350.Gomez MV; Juan A; Jimenez-Marquez F; de la Hoz A; Velders AH Illumination of Nanoliter-NMR Spectroscopy Chips for Real-Time Photochemical Reaction Monitoring. Anal. Chem. 2018, 90, 1542–1546. [DOI] [PubMed] [Google Scholar]
- 351.Foley DA; Dunn AL; Zell MT Reaction Monitoring Using Online vs Tube NMR Spectroscopy: Seriously Different Results. Magn. Reson. Chem. 2016, 54, 451–456. [DOI] [PubMed] [Google Scholar]
- 352.Bliumkin L; Majumdar RD; Soong R; Adamo A; Abbatt JPD; Zhao R; Reiner E; Simpson AJ Development of an in Situ NMR Photoreactor To Study Environmental Photochemistry. Environ. Sci. Technol. 2016, 50, 5506–5516. [DOI] [PubMed] [Google Scholar]
- 353.Emmanuel N; Mendoza C; Winter M; Horn CR; Vizza A; Dreesen L; Heinrichs B; Monbaliu J-CM Scalable Photocatalytic Oxidation of Methionine under Continuous-Flow Conditions. Org. Process Res. Dev. 2017, 21, 1435–1438. [Google Scholar]
- 354.Wu B; Majumdar RD; Lysak DH; Biswas RG; Tabatabaei-Anaraki M; Jenne A; You X; Soong R; Lane D; Helm PA; Codina A; Decker V; Simpson MJ; Simpson AJ Towards Real-Time Kinetic Monitoring of Wastewater Treatment: A Case Study of Sunlight and Ozone Treatment of Unconcentrated Wastewater Using Flow NMR. Chem. Eng. J. 2021, 405, 126696. [Google Scholar]
- 355.Albert K Dreher E-L; Straub H; Rieker A Monitoring electrochemical reactions by 13C NMR spectroscopy. Mag. Res. Chem. 1987, 25, 919–922. [Google Scholar]
- 356.Liauw MA; Baylor LC; O’Rourke PE Chapter 4: UV-visible Spectroscopy for On-line Analysis. In Process Analytical Technology: Spectroscopic Tools and Implementation Strategies for the Chemical and Pharmaceutical Industries, 2nd edition, Bakeev KA, Ed.; 2010, Wiley: New York. DOI: 10.1002/9780470689592.ch4 [DOI] [Google Scholar]
- 357.Stadler E; Eibel A; Fast D; Freißmuth H; Holly C; Wiech M; Moszner N; Gescheidt G A Versatile Method for the Determination of Photochemical Quantum Yields via Online UV-Vis Spectroscopy. Photochem. Photobiol. Sci. 2018, 17, 660–669. [DOI] [PubMed] [Google Scholar]
- 358.Gombár M; Józsa É; Braun M; Ősz K Construction of a Photochemical Reactor Combining a CCD Spectrophotometer and a LED Radiation Source. Photochem. Photobiol. Sci. 2012, 11, 1592–1595. [DOI] [PubMed] [Google Scholar]
- 359.Lehóczki T; Józsa É; Osz K Ferrioxalate Actinometry With Online Spectrophotometric Detection. J. Photochem. Photobiol. A 2013, 251, 63–68. [Google Scholar]
- 360.Ashley K; Pons S Infrared Spectroelectrochemistry. Chem. Rev. 1988, 88, 673–695. [Google Scholar]
- 361.Krejčik M, Daněk M, Hartl F Simple Construction of an Infrared Optically Transparent Thin-Layer Electrochemical Cell: Applications to the Redox Reactions of Ferrocene, Mn2(CO)10 and Mn(CO)3(3,5-di-t-butyl-catecholate)−. J. Electroanal. Chem. Interfacial Electrochem. 1991, 317, 179–187. [Google Scholar]
- 362.Kaim W; Fiedler J Spectroelectrochemistry: The Best of Two Worlds. Chem. Soc. Rev. 2009, 38, 3373–3382. [DOI] [PubMed] [Google Scholar]
- 363.Zhai Y; Zhu Z; Zhou S; Zhu C; Dong S Recent Advances in Spectroelectrochemistry. Nanoscale, 2018, 10, 3089–3111. [DOI] [PubMed] [Google Scholar]
- 364.Lozeman JJA; Führer P; Olthuis W; Odijk M Spectroelectrochemistry, the Future of Visualizing Electrode Processes by Hyphenating Electrochemistry with Spectroscopic Techniques. Analyst 2020, 145, 2482–2509. [DOI] [PubMed] [Google Scholar]
- 365.Han Z; Gu X; Wang S; Liu L; Wang Y; Zhao Z; Yu Z Time-Resolved In Situ Monitoring of Photocatalytic Reactions by Probe Electrospray Ionization Mass Spectrometry. Analyst 2020, 145, 3313–3319. [DOI] [PubMed] [Google Scholar]
- 366.Herl T; Matyski F-M Recent Developments in Electrochemistry-Mass Spectrometry. ChemElectroChem 2020, 7, 2498–2512. [Google Scholar]
- 367.Stalder R; Roth GP Preparative Microfluidic Electrosynthesis of Drug Metabolites. ACS Med. Chem. Lett. 2013, 4, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Bussy U; Boujtita M Advances in the Electrochemical Simulation of Oxidation Reactions Mediated by Cytochrome P450. Chem. Res. Toxicol. 2014, 27, 1652–1668. [DOI] [PubMed] [Google Scholar]
- 369.Genovino J; Sames D; Hamann LG; Touré B Accessing Drug Metabolites via Transition-Metal Catalyzed C−H Oxidation: The Liver as Synthetic Inspiration. Angew. Chem. Int. Ed. 2016, 55, 14218–14238. [DOI] [PubMed] [Google Scholar]
- 370.“Spin Trapping and Spin Labelling Studied Using EPR Spectroscopy” in Encyclopedia of Spectroscopy and Spectrometry (Third Edition), Arroyo CM Elsevier, 2017, pp. 209–217. DOI: 10.1016/B978-0-12-803224-4.00291-0. [DOI] [Google Scholar]
- 371.Electron Spin Resonance Spectroscopy | Specialized Techniques” in Encyclopedia of Analytical Science (Second Edition). Rhodes CJ; Morris H 2016, pp. 308–314. DOI: 10.1016/B0-12-369397-7/00118-7 [DOI] [Google Scholar]
- 372.Davies MJ Detection and Characterisation of Radicals Using Electron Paramagnetic Resonance (EPR) Spin Trapping and Related Methods. Methods, 2016, 109, 21–30. [DOI] [PubMed] [Google Scholar]
- 373.Toybenshlak M; Carmieli R A New and Robust Method for In-situ EPR Electrochemistry. Isr. J. Chem. 2019, 59, 1020–1026. [Google Scholar]
- 374.Zhao EW; Jónsson E; Jethwa RB; Hey D; Lyu D; Brookfield A; Klusener PAA; Collison D; Grey CP Coupled In Situ NMR and EPR Studies Reveal the Electron Transfer Rate and Electrolyte Decomposition in Redox Flow Batteries. J. Am. Chem. Soc. 2021, 143, 1885–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Griller D; Ingold KU Free-Radical Clocks. Acc. Chem. Res. 1980, 13, 317–323. [Google Scholar]
- 376.Newcomb M Competition Methods and Scales for Alkyl Radical Reaction Kinetics. Tetrahedron 1993, 49, 1151–1176. [Google Scholar]
- 377.Newcomb M (2012) Radical Kinetics and Clocks. In Encyclopedia of Radicals in Chemistry, Biology and Materials, John Wiley & Sons, Inc., Hoboken, NJ, USA, DOI: 10.1002/9781119953678. [DOI] [Google Scholar]
- 378.Fu N; Sauer GS; Saha A; Loo A; Lin S Metal-Catalyzed Electrochemical Diazidation of Alkenes. Science, 2017, 357, 575–579. [DOI] [PubMed] [Google Scholar]
- 379.Siu JC; Sauer GS; Saha A; Macey RL; Fu N; Chauvire T; Lancaster KM; Lin S Electrochemical Azidooxygenation of Alkenes Mediated by a TEMPO-N3 Charge-Transfer Complex. J. Am. Chem. Soc. 2018, 140, 12511–12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Miyazawa K; Koike T; Akita M Regiospecific Intermolecular Aminohydroxylation of Olefins by Photoredox Catalysis. Chem. Eur. J. 2015, 21, 11677–11680. [DOI] [PubMed] [Google Scholar]
- 381.Qin Q; Yu S Visible-Light-Promoted Remote C(sp3)–H Amidation and Chlorination. Org. Lett. 2015, 17, 1894–1897. [DOI] [PubMed] [Google Scholar]
- 382.Maity S; Zhu M; Shinabery RS; Zheng N Intermolecular [3+2] Cycloaddition of Cyclopropylamines with Olefins by Visible-Light Photocatalysis. Angew. Chem. Int. Ed. 2012, 51, 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Mennen SM; Alhambra C; Allen CL; Barberis M; Berritt S; Brandt TA; Campbell AD; Castañón J; Cherney AH; Christensen M; et al. The Evolution of High-Throughput Experimentation in Pharmaceutical Development and Perspectives on the Future. Org. Process Res. Dev. 2019, 23, 1213–1242. [Google Scholar]
- 384.ANSI/SLAS Microplate Standards, https://www.slas.org/education/ansi-slas-microplate-standards/ (accessed April 4, 2021).
- 385.Shevlin M Practical High-Throughput Experimentation for Chemists. ACS Med. Chem. Lett. 2017, 8, 601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Shultz CS; Krska SW Unlocking the Potential of Asymmetric Hydrogenation at Merck. Acc. Chem. Res. 2007, 40, 1320–1326. [DOI] [PubMed] [Google Scholar]
- 387.Krska SW; DiRocco DA; Dreher SD Shevlin M The Evolution of Chemical High-Throughput Experimentation To Address Challenging Problems in Pharmaceutical Synthesis. Acc. Chem. Res. 2017, 50, 2976–2985 [DOI] [PubMed] [Google Scholar]
- 388.DiRocco D; Dykstra K; Krska S; Vachal P; Conway DV Tudge, M. Late-Stage Functionalization of Biologically Active Heterocycles Through Photoredox Catalysis. Angew. Chem. Int. Ed. 2014, 53, 4802–4806. [DOI] [PubMed] [Google Scholar]
- 389.Rein J Annand JR; Wismer MK; Fu J; Siu JC; Klapars A; Strotman NA; Kalyani D; Lehnherr D; Lin S Unlocking the Potential of High-Throughput Experimentation for Electrochemistry with a Standardized Microscale Reactor. ACS Cent. Sci. 2021, DOI: 10.1021/acscentsci.1c00328x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.V&P Scientific, https://www.vp-scientific.com/index.html (accessed April 4, 2021).
- 391.Lin S; Dikler S; Blincoe WD; Ferguson RD; Sheridan RP; Peng Z; Conway DV; Zawatzky K; Wang H; Cernak T; Davies IW; DiRocco DA; Sheng H; Welch CJ; Dreher SD Mapping the Dark Space of Chemical Reactions with Extended Nanomole Synthesis and MALDI-TOF MS. Science 2018, 361, eaar6236. [DOI] [PubMed] [Google Scholar]
- 392.Santanilla AB; Regalado EL; Pereira T; Shevlin M; Bateman K; Campeau L-C; Schneeweis J; Beritt S; Shi Z-C; Nantermet P; et al.Nanomole-Scale High-Throughput Chemistry for the Synthesis of Complex Molecules. Science 2015, 347, 49–53. [DOI] [PubMed] [Google Scholar]
- 393.For contour light intensity maps illustrating this effect for Kessil lamps, see: https://www.kessil.com/science/resources.php (accessed April 4, 2020).
- 394.Penn PhD Photoreactor homepage, https://www.pennphd.com/#home (accessed April 4, 2021).
- 395.Le CC; Wismer MK; Shi Z-C; Conway DV; Li G; Vachal P; Davies IW; MacMillan DWC A General Small-Scale Reactor To Enable Standardization and Acceleration of Photocatalytic Reactions. ACS Cent. Sci. 2017, 3, 647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.HeptaoChem homepage, https://www.hepatochem.com/ (accessed April 4, 2021).
- 397.Photoreactor.de homepage, http://wppr.photoreactor.de/ (accessed April 4, 2021).
- 398.IKA Electrochemistry webpage https://www.ika.com/en/Products-Lab-Eq/Electrochemistry-Kit-csp-516/ElectraSyn-20-Package-cpdt-20008980/ (accessed April 4, 2021).
- 399.Siu T; Li W; Yudin AK Parallel Electrosynthesis of α-Alkoxycarbamates, α-Alkoxyamides, and α-Alkoxysulfonamides Using the Spatially Addressable Electrolysis Platform (SAEP). J. Comb. Chem. 2000, 2, 545–549. [DOI] [PubMed] [Google Scholar]
- 400.Tajima T; Nakajima A Parallel Electrosynthesis of N-Acyliminium Ion Equivalents Using Silica Gel-Supported Piperidine. Chem. Lett. 2009, 38, 160–161. [Google Scholar]
- 401.Gütz C; Klöckner B; Waldvogel SR Electrochemical Screening for Electroorganic Synthesis. Org. Process Res. Dev. 2016, 20, 26–32. [Google Scholar]
- 402.Wills AG; Charvet S; Battilocchio C; Scarborough CC; Wheelhouse KMP; Poole DL; Carson N; Vantourout JC High-Throughput Electrochemistry: State of the Art, Challenges, and Perspective. Org. Process Res. Dev. 2021, DOI: 10.1021/acs.oprd.1c00167. [DOI] [Google Scholar]
- 403.Martin VI; Goodell JR; Ingham OJ; Porco JA Jr.; Beeler AB Multidimensional Reaction Screening for Photochemical Transformations as a Tool for Discovering New Chemotypes. J. Org. Chem. 2014, 79, 3838–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Sun AC; Steyer DJ; Allen AR; Payne EM; Kennedy RT; Stephenson CRJ A Droplet Microfluidic Platform for High-Throughput Photochemical Reaction Discovery. Nat. Commun. 2020, 11, 6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Lalevée J; Fouassier JP (2012) Overview of Radical Initiation. In Encyclopedia of Radicals in Chemistry, Biology and Materials, John Wiley & Sons, Inc., Hoboken, NJ, USA, DOI: 10.1002/9781119953678. [DOI] [Google Scholar]
- 406.Davies J; Morcillo SP; Douglas JJ; Leonori D Hydroxylamine Derivatives as Nitrogen-Radical Precursors in Visible-Light Photochemistry. Chem. Eur. J. 2018, 24, 12154–12163. [DOI] [PubMed] [Google Scholar]
- 407.Smith JM; Dixon JA; deGruyter JN; Baran PS Alkyl Sulfinates: Radical Precursors Enabling Drug Discovery. J. Med Chem. 2019, 62, 2256–2264. [DOI] [PubMed] [Google Scholar]
- 408.Benzophenone oximes: Hasebe M; Tsuchiya T Photodecarboxylative Chlorination of Carboxylic Acids via Their Benzophenone Oxime Esters. Tetrahedron Lett. 1988, 29, 6287–6290. [Google Scholar]
- 409.Kvasovs N; Gevorgyan V Contemporary Methods for Generation of Aryl Radicals. Chem. Soc. Rev. 2021, 50, 2244–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Chatgilialoglu C Structural and Chemical Properties of Silyl Radicals. Chem. Rev. 1995, 95, 1229–1251. [Google Scholar]
- 411.Dénès F; Pichowicz M; Povie G; Renaud P Thiyl Radicals in Organic Synthesis. Chem. Rev. 2014, 114, 2587–2693. [DOI] [PubMed] [Google Scholar]
- 412.Chatgilialoglu C; Ferreri C; Landais Y; Timokhin VI Thirty Years of (TMS)3SiH: A Milestone in Radical-Based Synthetic Chemistry. Chem. Rev. 2018, 118, 6516–6572. [DOI] [PubMed] [Google Scholar]
- 413.For recent examples of using metal centered radicals (e.g. •Mn(CO)5 for XAT), see: Nuhant P; Oderinde MS; Genovino J; Juneau A; Gagné Y; Allais C; Chinigo GM; Choi C; Sach NW; Bernier L; et al. Visible-Light-Initiated Manganese Catalysis for C−H Alkylation of Heteroarenes: Applications and Mechanistic Studies. Angew. Chem. Int. Ed. 2017, 56, 15309–15313.
- 414.Wang L; Lear JM; Rafferty SM; Fosu SC; Nagib DA Ketyl Radical Reactivity via Atom Transfer Catalysis. Science 2018, 362, 225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Rafferty SM; Rutherford JE; Zhang L; Wang L; Nagib DA Cross-Selective Aza-Pinacol Coupling via Atom Transfer Catalysis. J. Am. Chem. Soc. 2021, 143, 5622–5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Wang X; Zhu B; Dong J; Tian H; Liu Y; Song H; Wang Q Visible-Light-Mediated Multicomponent Reaction for Secondary Amine Synthesis. Chem. Commun. 2021. 57, 5028–5031. [DOI] [PubMed] [Google Scholar]
- 417.For an example of using alpha-amino radicals for XAT, see: Constantin T; Zanini M; Regni A; Sheikh NS; Juliá F; Leonori D Aminoalkyl Radicals as Halogen-Atom Transfer Agents for Activation of Alkyl and Aryl Halides. Science 2020, 367, 1021–1026.
- 418.Morcillo SP Radical-Promoted C−C Bond Cleavage: A Deconstructive Approach for Selective Functionalization. Angew.Chem. Int. Ed. 2019, 58, 14044–14054. [DOI] [PubMed] [Google Scholar]
- 419.Photodecarboxylation: Budac D; Wan P Photodecarboxylation: Mechanism and Synthetic Utility. J. Photochem. Photobiology A, 1992, 67, 135–166. [Google Scholar]
- 420.Pratsch G; Lackner GL; Overman LE Constructing Quaternary Carbons from N-(Acyloxy)Phthalimide Precursors of Tertiary Radicals Using Visible-Light Photocatalysis. J. Org. Chem. 2015, 80, 6025–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.For methods utilizing UV light, see: Okada K; Okamoto K; Oda M A New and Practical Method of Decarboxylation: Photosensitized Decarboxylation of N-Acyloxyphthalimides via Electron-Transfer Mechanism. J. Am. Chem. Soc. 1988, 110, 8736–8738.
- 422.Okada K; Okamoto K; Oda M Photochemical Chlorodecarboxylation via an Electron Transfer Mechanism. J. Chem. Soc., Chem. Commun. 1989, 1636−1637. [Google Scholar]
- 423.For methods utilizing visible light, see: Okada K; Okamoto K; Morita N; Okubo K; Oda M Photosensitized Decarboxylative Michael Addition Through N-(Acyloxy)phthalimides via an Electron-Transfer Mechanism. J. Am. Chem. Soc. 1991, 113, 9401–9402.
- 424.Okada K; Okubo K; Morita N; Oda M Reductive Decarboxylation of N-(Acyloxy)phthalimides via Redox-Initiated Radical Chain Mechanism. Tetrahedron Lett. 1992, 33, 7377–7380. [Google Scholar]
- 425.Okada K; Okubo K; Morita N; Oda M Redox-Mediated Decarboxylative Photo-Phenylselenenylation of N-Acyloxyphthalimides. Chem. Lett. 1993, 22, 2021–2024. [Google Scholar]
- 426.Zhang X; MacMillan DWC Alcohols as Latent Coupling Fragments for Metallaphotoredox Catalysis: sp3–sp2 Cross-Coupling of Oxalates with Aryl Halides. J. Am. Chem. Soc. 2016, 138, 13862–13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Nawrat CC; Jamison CR; Slutskyy Y; MacMillan DWC; Overman LE Oxalates as Activating Groups for Alcohols in Visible Light Photoredox Catalysis: Formation of Quaternary Centers by Redox-Neutral Fragment Coupling. J. Am. Chem. Soc. 2015, 137, 11270–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Tellis JC; Kelly CB; Primer DN; Jouffroy M; Patel NR; Molander GA Single-Electron Transmetalation via Photoredox/Nickel Dual Catalysis: Unlocking a New Paradigm for sp3–sp2 Cross-Coupling. Acc. Chem. Res. 2016, 49, 1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Matsuoka D; Nishigaichi Y Photosubstitution of Dicyanoarenes by Hypervalent Allylsilicon Reagents via Photoinduced Electron Transfer. Chem. Lett. 2014, 43, 559–561. [Google Scholar]
- 430.Matsuoka D; Nishigaichi Y Allyl-transfer Reaction from Photoexcited Hypervalent Allylsilicon Reagent toward Dicyanobenzenes. Chem. Lett. 2015, 44, 163–165. [Google Scholar]
- 431.Corcé V; Chamoreau L-M; Derat E; Goddard J-P; Ollivier C; Fensterbank L Silicates as Latent Alkyl Radical Precursors: Visible-Light Photocatalytic Oxidation of Hypervalent Bis-Catecholato Silicon Compounds. Angew. Chem. Int. Ed. 2015, 54, 11414–11418. [DOI] [PubMed] [Google Scholar]
- 432.Jouffroy M; Primer DN; Molander GA Base-Free Photoredox/Nickel Dual-Catalytic Cross-Coupling of Ammonium Alkylsilicates. J. Am. Chem. Soc. 2016, 138, 475–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Vasudevan D Direct and Indirect Electrochemical Reduction of Organic Halides in Aprotic Media. Russ. J. Electrochem. 2005, 41, 310–314. [Google Scholar]
- 434.Lambert FL; Kobayashi K Polarography of Organic Halogen Compounds. I. Steric Hindrance and the Half-Wave Potential in Alicyclic and Aliphatic Halides. J. Am. Chem. Soc. 1960, 82, 5324–5328. [Google Scholar]
- 435.Lambert FL; Ingall GB Voltammetry of Organic Halogen Compounds. IV. The Reduction of Organic Chlorides at the Vitreous (Glassy) Carbon Electrode. Tetrahedron Lett. 1974, 15, 3231–3234. [Google Scholar]
- 436.Welin ER; Warkentin AA; Conrad JC; MacMillan DWC Enantioselective α-Alkylation of Aldehydes by Photoredox Organocatalysis: Rapid Access to Pharmacophore Fragments from β-Cyanoaldehydes. Angew. Chem. Int. Ed. 2015, 54, 9668–9672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Nicewicz DA; MacMillan DWC Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Roth H; Romero N; Nicewicz D Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. [Google Scholar]
- 439.For deamination strategies using Katritzky salts as alkyl radical precursors, see: Correia JTM; Fernandes VA; Matsuo BT; Delgado JAC; de Souza WC; Paixão MW Photoinduced Deaminative Strategies: Katritzky Salts As Alkyl Radical Precursors. Chem. Commun. 2020, 56, 503–514.
- 440.Grimshaw J; Moore S; Thompson N; Trocha-Grimshaw J Carbon–nitrogen bond cleavage in π-radicals derived by reduction of N-benzyl- and N-allyl-pyridinium salts. J. Chem. Soc., Chem. Commun. 1983, 783–784. [Google Scholar]
- 441.Ashley MA; Rovis T Photoredox-Catalyzed Deaminative Alkylation via C–N Bond Activation of Primary Amines. J. Am. Chem. Soc. 2020, 142, 18310–18316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Nakajima K; Miyake Y; Nishibayashi Y Synthetic Utilization of α-Aminoalkyl Radicals and Related Species in Visible Light Photoredox Catalysis. Acc. Chem. Res. 2016, 49, 1946–1956. [DOI] [PubMed] [Google Scholar]
- 443.Fundamentals and Applications of Organic Electrochemistry: Synthesis, Materials, Devices, Fuchigami T, Atobe M, Inagi S, Eds.; John Wiley & Sons, Ltd: New York, 2015; pp. 56–63. [Google Scholar]
- 444.Yoshida J.-i.; Kataoka K; Horcajada R; Nagaki A Modern Strategies in Electroorganic Synthesis. Chem. Rev. 2008, 108, 2265–2299 [DOI] [PubMed] [Google Scholar]
- 445.Yoshida J Electroauxiliary. In Encyclopedia of Applied Electrochemistry; Kreysa G, Ota K, Savinell RF, Eds.; Springer: New York, 2014; pp 386–392. [Google Scholar]
- 446.Stateman LM; Nakafuku KM; Nagib DA Remote C–H Functionalization via Selective Hydrogen Atom Transfer. Synthesis 2018, 50, 1569–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Capaldo L; Ravelli D Hydrogen Atom Transfer (HAT): A Versatile Strategy for Substrate Activation in Photocatalyzed Organic Synthesis. Eur. J. Org. Chem. 2017, 2056–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Revathi L; Ravindar L; Fang W-Y; Rakesh KP; Qin H-L Visible Light-Induced C−H Bond Functionalization: A Critical Review. Adv. Synth. Catal. 2018, 360, 4652–4698. [Google Scholar]
- 449.Capaldo L; Quadri LL; Ravelli D Photocatalytic Hydrogen Atom Transfer: The Philosopher’s Stone for Late-Stage Functionalization? Green Chem. 2020, 22, 3376–3396. [Google Scholar]
- 450.Hoffmann N Proton-Coupled Electron Transfer in Photoredox Catalytic Reactions. Eur. J. Org. Chem. 2017, 1982–1992. [Google Scholar]
- 451.Gentry EC; Knowles RR Synthetic Applications of Proton-Coupled Electron Transfer. Acc. Chem. Res. 2016, 49, 1546–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Weinberg DR; Gagliardi CJ; Hull JF; Murphy CF; Kent CA; Westlake BC; Paul A; Ess DH; McCafferty DG; Meyer TJ Proton-Coupled Electron Transfer. Chem. Rev. 2012, 112, 4016–4093. [DOI] [PubMed] [Google Scholar]
- 453.Tyburski R; Liu T; Glover SD; Hammarström L Proton-Coupled Electron Transfer Guidelines, Fair and Square. J. Am. Chem. Soc. 2021, 143, 560–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Chu L; Ohta C; Zuo Z; MacMillan DWC Carboxylic Acids as A Traceless Activation Group for Conjugate Additions: A Three-Step Synthesis of (±)-Pregabalin. J. Am. Chem. Soc. 2014, 136, 10886–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Zuo Z; MacMillan DWC Decarboxylative Arylation of α-Amino Acids via Photoredox Catalysis: A One-Step Conversion of Biomass to Drug Pharmacophore. J. Am. Chem. Soc. 2014, 136, 5257–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Noble A; McCarver SJ; MacMillan DWC Merging Photoredox and Nickel Catalysis: Decarboxylative Cross-Coupling of Carboxylic Acids with Vinyl Halides. J. Am. Chem. Soc. 2015, 137, 624–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Liu J; Liu Q; Yi H; Qin C; Bai R; Qi X; Lan Y; Lei A Visible-Light-Mediated Decarboxylation/Oxidative Amidation of α-Keto Acids with Amines under Mild Reaction Conditions Using O2. Angew. Chem. Int. Ed. 2013, 53, 502–506. [DOI] [PubMed] [Google Scholar]
- 458.Gutiérrez-Bonet A; Tellis JC; Matsui JK Vara BA; Molander GA 1,4-Dihydropyridines as Alkyl Radical Precursors: Introducing the Aldehyde Feedstock to Nickel/Photoredox Dual Catalysis. ACS Catal. 2016, 6, 8004–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Goddard J-P; Ollivier C; Fensterbank L Photoredox Catalysis for the Generation of Carbon Centered Radicals. Acc. Chem. Res. 2016, 49, 1924–1936. [DOI] [PubMed] [Google Scholar]
- 460.Yasu Y; Koike T; Akita M Visible Light-Induced Selective Generation of Radicals from Organoborates by Photoredox Catalysis. Adv. Synth. Catal. 2012, 354, 3414–3420. [Google Scholar]
- 461.Matsui JK; Lang SB; Molander GA Photoredox-Mediated Routes to Radicals: The Value of Catalytic Radical Generation in Synthetic Methods Development. ACS Catal. 2017, 7, 2563–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Miyazawa K; Yasu Y; Koike T; Akita M Visible-light-induced hydroalkoxymethylation of electron-deficient alkenes by photoredox catalysis. Chem. Commun. 2013, 49, 7249–7251. [DOI] [PubMed] [Google Scholar]
- 463.Shu C; Noble A; Aggarwal VK Photoredox-Catalyzed Cyclobutane Synthesis by a Deboronative Radical Addition−Polar Cyclization Cascade. Angew. Chem. Int. Ed. 2019, 58, 3870–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Wilger DJ; Gesmundo NJ; Nicewicz DA Catalytic Hydrotrifluoromethylation of Styrenes and Unactivated Aliphatic Alkenes via an Organic Photoredox System. Chem. Sci. 2013, 4, 3160–3165. [Google Scholar]
- 465.Knauber T; Chandrasekaran R; Tucker JW; Chen JM; Reese M; Rankic DA; Sach N; Helal C Ru/Ni Dual Catalytic Desulfinative Photoredox Csp2-Csp3 Cross-Coupling of Alkyl Sulfinate Salts and Aryl Halides. Org. Lett. 2017, 19, 6566–6569. [DOI] [PubMed] [Google Scholar]
- 466.Yoshida J; Nishiwaki K Redox Selective Reactions of Organo-Silicon and -Tin Compounds. J. Chem. Soc., Dalton Trans. 1998, 16, 2589–2596. [Google Scholar]
- 467.Remeru C; Kelly CB; Patel NR; Molander GA Aminomethylation of Aryl Halides Using α-Silylamines Enabled by Ni/Photoredox Dual Catalysis. ACS Catal. 2017, 7, 6065–6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Lackner GL; Quasdorf KW; Pratsch G; Overman LE Fragment Coupling and the Construction of Quaternary Carbons Using Tertiary Radicals Generated From tert-Alkyl N-Phthalimidoyl Oxalates By Visible-Light Photocatalysis. J. Org. Chem. 2015, 80, 6012–6024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Grimshaw J; Moore S; Grimshaw JT Electrochemical Reactions. Part 26. Radicals Derived by Reduction of N-Alkylpyridinium Salts and Homologous N,N'-Polymethylenebispyridinium Salts. Cleavage of the Carbon−Nitrogen Bond. Acta Chem. Scand. Ser. B 1983, 37, 485–489. [Google Scholar]
- 470.Sun AC; McClain EJ; Beatty JW; Stephenson CRJ Visible Light-Mediated Decarboxylative Alkylation of Pharmaceutically Relevant Heterocycles. Org. Lett. 2018, 20, 3487–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Staveness D; Irene Bosque I; Stephenson CRJ Free Radical Chemistry Enabled by Visible Light-Induced Electron Transfer. Acc. Chem. Res. 2016, 49, 2295–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Liao L-L; Cao GM; Ye J-H; Sun G-Q; Zhou W-J; Gui Y-Y; Yan S-S; Shen G; Yu D-G Visible-Light-Driven External-Reductant-Free Cross-Electrophile Couplings of Tetraalkyl Ammonium Salts. J. Am. Chem. Soc. 2018, 140, 17338–17342. [DOI] [PubMed] [Google Scholar]
- 473.Elofson RM; Gadallah FF, Substituent FF Effects in the Polarography of Aromatic Diazonium Salts. J. Org. Chem. 1969, 34, 854–857. [Google Scholar]
- 474.Pause L; Robert M; Savéant J-M Can Single-Electron Transfer Break an Aromatic Carbon−Heteroatom Bond in One Step? A Novel Example of Transition between Stepwise and Concerted Mechanisms in the Reduction of Aromatic Iodides. J. Am. Chem. Soc. 1999, 121, 7158–7159. [Google Scholar]
- 475.Nagib DA; Scott ME; MacMillan DWC Enantioselective α-Trifluoromethylation of Aldehydes via Photoredox Organocatalysis. J. Am. Chem. Soc. 2009, 131, 10875–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Nagib DA; MacMillan DWC Trifluoromethylation of Arenes and Heteroarenes by Means of Photoredox Catalysis. Nature 2011, 480, 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Yasu Y; Koike T; Akita M Three-component Oxytrifluoromethylation of Alkenes: Highly Efficient and Regioselective Difunctionalization of C–C Bonds Mediated by Photoredox Catalysts. Angew. Chem. Int. Ed. 2012, 51, 9567–9571 [DOI] [PubMed] [Google Scholar]
- 478.Kabatc J; Ortylb J; Kostrzewska K New Kinetic and Mechanistic Aspects of Photosensitization of Iodonium Salts in Photopolymerization of Acrylates. RSC Adv. 2017, 7, 41619–41629. [Google Scholar]
- 479.Magdesieva TV; Kravchuk DN; Butin KP Electrochemical Arylation of Cobalt and Nickel Chelates With Diphenyliodonium Salts. Russ. Chem. Bull. 2002, 51, 85–89. [Google Scholar]
- 480.Vaillant FL; Wodrich MD; Waser J Room Temperature Decarboxylative Cyanation of Carboxylic Acids Using Photoredox Catalysis and Cyanobenziodoxolones: A Divergent Mechanism Compared to Alkynylation. Chem. Sci. 2017, 8, 1790–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Pandey G; Rao KSSP; Sekhar BBV Photosensitized One-Electron Reductive Cleavage of a Carbon–Selenium Bond: A Novel Chemoselective Deselenenylation and Phenylselenenyl Group Transfer Radical Chain Reaction. J. Chem. Soc., Chem. Commun. 1993, 1636–1638. [Google Scholar]
- 482.Gray HB; Winkler JR Long-range electron transfer. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 3534–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.The depth, path length (L), to which light can penetrate into the solution achieving a certain level of transmittance (T), is governed by the concentration (c) and molar absorptivity (ε) of the photocatalyst is limited as illustrated in Figure 10 and by the following equation:
- 484.Huang H; Jia K; Chen Y Radical Decarboxylative Functionalizations Enabled by Dual Photoredox Catalysis. ACS Catal. 2016, 6, 4983–4988. [Google Scholar]
- 485.Ramadoss V; Zheng Y; Shao X; Tian L; Wang Y Advances in Electrochemical Decarboxylative Transformation Reactions. Chem. Eur. J. 2020, 27, 3213–3228. [DOI] [PubMed] [Google Scholar]
- 486.Roughley SD; Jordan AM The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem. 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]
- 487.Xiang J; Shang M; Kawamata Y; Lundberg H; Reisberg SH; Chen M; Mykhailiuk P; Beutner G; Collins MR; Davies A; et al. Hindered Dialkyl Ether Synthesis With Electrogenerated Carbocations. Nature 2019, 573, 398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Mao R; Balon J; Hu X Decarboxylative C(sp3)−O Cross-Coupling. Angew. Chem. Int. Ed. 2018, 57, 13624–13628. [DOI] [PubMed] [Google Scholar]
- 489.Shibutani S; Kodo T; Takeda M; Nagao K; Tokunaga N; Sasaki Y; Ohmiya H Organophotoredox-Catalyzed Decarboxylative C(sp3)–O Bond Formation. J. Am. Chem. Soc. 2020, 142, 1211–1216. [DOI] [PubMed] [Google Scholar]
- 490.Shibutani S; Nagao K; Ohmiya H Organophotoredox-Catalyzed Three-Component Coupling of Heteroatom Nucleophiles, Alkenes, and Aliphatic Redox Active Esters. Org. Lett. 2021, 23, 1798–1803. [DOI] [PubMed] [Google Scholar]
- 491.Proctor RSJ; Phipps RJ Recent Advances in Minisci-Type Reactions. Angew. Chem. Int. Ed. 2019, 58, 13666–13699. [DOI] [PubMed] [Google Scholar]
- 492.Jin J; MacMillan DWC Alcohols As Alkylating Agents in Heteroarene C–H Functionalization. Nature, 2015, 525, 87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.DiRocco DA; Dykstra K; Krska S; Vachal P; Conway DV; Tudge M Late-Stage Functionalization of Biologically Active Heterocycles Through Photoredox Catalysis. Angew. Chem. Int. Ed. 2014, 53, 4802–4806. [DOI] [PubMed] [Google Scholar]
- 494.Garza-Sanchez RA; Tlahuext-Aca A; Tavakoli G; Glorius F Visible Light-Mediated Direct Decarboxylative C–H Functionalization of Heteroarenes. ACS Catal. 2017, 7, 4057–4061. [Google Scholar]
- 495.Cheng W-M; Shang R; Fu M-C; Fu Y Photoredox-Catalysed Decarboxylative Alkylation of N-Heteroarenes with N-(Acyloxy)Phthalimides. Chem. Eur. J. 2017, 23, 2537–2541. [DOI] [PubMed] [Google Scholar]
- 496.Liu Y; Xue L; Shi B; Bu F; Wang D; Lu L; Shi R; Lei A Catalyst-Free Electrochemical Decarboxylative Cross-Coupling of N-Hydroxyphthalimide Esters and N-Heteroarenes towards C(sp3)–C(sp2) Bond Formation. Chem. Commun. 2019, 55, 14922–14925. [DOI] [PubMed] [Google Scholar]
- 497.Lian F; Xu K; Meng W; Zhang H; Tana Z; Zeng C Nickel-Catalyzed Electrochemical Reductive Decarboxylative Coupling of N-Hydroxyphthalimide Esters With Quinoxalinones.Chem. Commun. 2019, 55, 14685–14688. [DOI] [PubMed] [Google Scholar]
- 498.Kawabata J-I; Nozawa K; Kawai K-I; Nakajima S Electrochemical Hydroxylation, Methoxylation, and Hydroxymethylation of Active Methine Compounds. Liebigs Ann. Chem. 1990, 181–183. [Google Scholar]
- 499.Huff CA; Cohen RD; Dykstra KD; Streckfuss E; DiRocco DA; Krska SW Photoredox-Catalyzed Hydroxymethylation of Heteroaromatic Bases. J. Org. Chem. 2016, 81, 6980–6987. [DOI] [PubMed] [Google Scholar]
- 500.Biezczad B; Perego LA; Melchiorre P Photochemical C−H Hydroxyalkylation of Quinolines and Isoquinolines. Angew. Chem. Int. Ed. 2019, 58, 16878–16883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Xu K; Jiang Y-Y; Liu Y-G; Sun B-G; Zheng C-C Electrocatalytic Minisci Acylation Reaction of N-Heteroarenes Mediated by NH4I. Org. Lett. 2017, 19, 5517–5520. [DOI] [PubMed] [Google Scholar]
- 502.Jouffroy M; Kong J Direct C−H Carbamoylation of Nitrogen-Containing Heterocycles. Chem. Eur. J. 2019, 25, 2217–2221. [DOI] [PubMed] [Google Scholar]
- 503.Proctor RSJ; Davis HJ; Phipps RJ Catalytic Enantioselective Minisci-Type Addition to Heteroarenes. Science 2018, 360, 419–422. [DOI] [PubMed] [Google Scholar]
- 504.Fu M-C; Shang R; Zhao B; Wang B; Fu Y Photocatalytic Decarboxylative Alkylations Mediated by Triphenylphosphine and Sodium Iodide. Science 2019, 363, 1429–1434. [DOI] [PubMed] [Google Scholar]
- 505.Zheng D; Studer A Asymmetric Synthesis of Heterocyclic γ-Amino-Acid and Diamine Derivatives by Three-Component Radical Cascade Reactions. Angew. Chem. Int. Ed. 2019, 58, 15803–15807. [DOI] [PubMed] [Google Scholar]
- 506.Proctor RSJ; Chuentragool R Colgan AC; Phipps RJ Hydrogen Atom Transfer-Driven Enantioselective Minisci Reaction of Amides. J. Am. Chem. Soc. 2021, 143, 4928–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Yan H; Hou Z-W; Xu H-C Photoelectrochemical C−H Alkylation of Heteroarenes with Organotrifluoroborates. Angew. Chem. Int. Ed. 2019, 58, 4592–4595. [DOI] [PubMed] [Google Scholar]
- 508.Xu P; Chen P-Y; Xu H-C Scalable Photoelectrochemical Dehydrogenative Cross-Coupling of Heteroarenes with Aliphatic C−H Bonds. Angew. Chem. Int. Ed. 2020, 59, 14275–14280. [DOI] [PubMed] [Google Scholar]
- 509.Capaldo L; Quadri LL; Merli D; Ravelli D Photoelectrochemical Cross-Dehydrogenative Coupling of Benzothiazoles With Strong Aliphatic C–H Bonds. Chem. Commun. 2021, DOI: 10.1039/d1cc01012c. [DOI] [PubMed] [Google Scholar]
- 510.Purser S; Moore PR; Swallow S; Gouverneur V Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]
- 511.Mondal R; Agbaria M; Nairoukh Z Fluorinated Rings: Conformation and Application. Chem. Eur. J. 2021, 27, 7193–7213.. [DOI] [PubMed] [Google Scholar]
- 512.Lai C; Mallouk TE A New Approach to the Photochemical Trifluoromethylation of Aromatic Compounds. J. Chem. Soc., Chem. Commun. 1993, 1359–1361. [Google Scholar]
- 513.Beatty JW; Douglas JJ; Cole KP; Stephenson CRJ A Scalable and Operationally Simple Radical Trifluoromethylation. Nat. Commun. 2015, 6, 7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Beatty JW; Douglas JJ; Miller R; McAtee RC; Cole KP; Stephenson CRJ Photochemical Perfluoroalkylation with Pyridine N-Oxides: Mechanistic Insights and Performance on a Kilogram Scale. Chem 2016, 1, 456–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.O'Brien AG; Maruyama A; Inokuma Y; Fujita M; Baran PS; Blackmond DG Radical C–H Functionalization of Heteroarenes under Electrochemical Control. Angew. Chem. Int. Ed. 2014, 53, 11868–11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Dou G-Y; Jiang Y-Y; Xu K; Zeng C-C Electrochemical Minisci-Type Trifluoromethylation of Electron-Deficient Heterocycles Mediated by Bromide Ions. Org. Chem. Front. 2019, 6, 2392–2397. [Google Scholar]
- 517.Qiu Y; Scheremetjew A; Finger LH; Ackermann L Electrophotocatalytic Undirected C−H Trifluoromethylations of (Het)Arenes. Chem. Eur. J. 2020, 26, 3241–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Shi L, Xia W Photoredox Functionalization of C–H Bonds Adjacent to a Nitrogen Atom. Chem. Soc. Rev. 2012, 41, 7687–7697. [DOI] [PubMed] [Google Scholar]
- 519.Beatty JW; Stephenson CRJ Amine Functionalization via Oxidative Photoredox Catalysis: Methodology Development and Complex Molecule Synthesis. Acc. Chem. Res. 2015, 48, 1474–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Hu J; Wang J; Nguyen TH; Zheng N The Chemistry of Amine Radical Cations Produced by Visible Light Photoredox Catalysis. Beilstein J. Org. Chem. 2013, 9, 1977–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521.Mitchell EA; Peschiulli A; Lefevre N; Meerpoel L; Maes BUW Direct a-Functionalization of Saturated Cyclic Amines. Chem. Eur. J. 2012, 18, 10092–10142 [DOI] [PubMed] [Google Scholar]
- 522.Shono T; Hamaguchi H; Matsumura Y Electroorganic chemistry. XX. Anodic Oxidation of Carbamates. J. Am. Chem. Soc. 1975, 97, 4264–4268. [Google Scholar]
- 523.Shono T; Matsumura Y; Tsubata K Electroorganic Chemistry. 46. A New Carbon-Carbon Bond Forming Reaction at the α-Position of Amines Utilizing Anodic Oxidation as a Key Step. J. Am. Chem. Soc. 1981, 103, 1172–1176. [Google Scholar]
- 524.Ross S; Finkelstein D; Peterson RC Anodic Oxidations. III. The Reaction Mechanism in the Electrochemical Acetoxylation and Alkoxylation of N,N-Dimethylamides. J. Am. Chem. Soc. 1966, 88, 4657–4660. [Google Scholar]
- 525.Nyberg K; Servin R Large-Scale Laboratory Electrolysis in Organic Systems. III. The Synthesis of Alpha-Methoxyalkylamides. Cyclic Acylimmonium Precursors. Acta Chem. Scand. Ser. B 1976, 30, 640–642. [Google Scholar]
- 526.Shono T; Matsumura Y; Tsubata K Anodic Oxidation of N-Carbomethoxypyrrolidine: 2-Methoxy-N-Carbomethoxypyrrolidine. Org. Synth. 1985, 63, 206–213. [Google Scholar]
- 527.Yoshida J; Suga S; Suzuki S; Kinomura N; Yamamoto A Fujiwara, K. Direct Oxidative Carbon−Carbon Bond Formation Using the “Cation Pool” Method. 1. Generation of Iminium Cation Pools and Their Reaction with Carbon Nucleophiles. J. Am. Chem. Soc. 1999, 121, 9546–9549. [Google Scholar]
- 528.Yoshida J.-i.; Suga S Basic Concepts of “Cation Pool” and “Cation Flow” Methods and Their Applications in Conventional and Combinatorial Organic Synthesis. Chem. Eur. J. 2002, 8, 2650–2658. [DOI] [PubMed] [Google Scholar]
- 529.Wang F; Rafiee M; Stahl SS Electrochemical Functional-Group-Tolerant Shono-type Oxidation of Cyclic Carbamates Enabled by Aminoxyl Mediators. Angew. Chem. Int. Ed. 2018, 57, 6686–6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530.Morisaki K; Morimoto H; Ohshima T Recent Progress on Catalytic Addition Reactions to N-Unsubstituted Imines. ACS Catal. 2020, 10, 6924–6951. [Google Scholar]
- 531.Leitch JA; Rossolini T; Rogova T; Maitland JAP; Dixon DJ α-Amino Radicals via Photocatalytic Single-Electron Reduction of Imine Derivatives. ACS Catal. 2020, 10, 2009–2025. [Google Scholar]
- 532.Garrido-Castro AF; Maestro MC; Alemán J α-Functionalization of Imines via Visible Light Photoredox Catalysis. Catalysts 2020, 10, 562. [Google Scholar]
- 533.Maruyama T; Suga S; Yoshida J Radical Addition to “Cation Pool”. Reverse Process of Radical Cation Fragmentation. J. Am. Chem. Soc. 2005, 127, 7324–7325. [DOI] [PubMed] [Google Scholar]
- 534.Maruyama T; Suga S; Yoshida J Distannane Mediated Reaction of N-Acyliminium Ion Pools With Alkyl Halides. A Chain Mechanism Involving Radical Addition Followed by Electron Transfer. Tetrahedron 2006, 62, 6519–6525. [Google Scholar]
- 535.Maruyama T; Mizuno Y; Shimizu I; Suga S; Yoshida J Reactions of a N-Acyliminium Ion Pool with Benzylsilanes. Implication of a Radical/Cation/Radical Cation Chain Mechanism Involving Oxidative C−Si Bond Cleavage. J. Am. Chem. Soc. 2007, 129, 1902–1903. [DOI] [PubMed] [Google Scholar]
- 536.Kim S; Hayashi K; Kitano Y; Tada M; Chiba K Anodic Modification of Proline Derivatives Using a Lithium Perchlorate/Nitromethane Electrolyte Solution. Org. Lett. 2002, 4, 3735–3737. [DOI] [PubMed] [Google Scholar]
- 537.Suga S; Suzuki S; Yoshida J Reduction of a “Cation Pool”: A New Approach to Radical Mediated C−C Bond Formation. J. Am. Chem. Soc. 2002, 124, 30–31. [DOI] [PubMed] [Google Scholar]
- 538.Suga S; Suzuki S; Maruyama T; Yoshida, Generation of Carbon Free Radicals by Reduction of the Cation Pool. J. Bull. Chem. Soc. Jpn. 2004, 77, 1545–1554. [Google Scholar]
- 539.Chiba T; Takata Y Anodic Cyanation of Tertiary Aliphatic and Heterocyclic Amines. J. Org. Chem. 1977, 42, 2973–2977. [Google Scholar]
- 540.Konno A; Fuchigami T; Fujita Y; Nonaka T Electrolytic Transformation of Fluoroorganic Compounds. 5. Anodic Cyanation of 2,2, 2-Trifluoroethylamines. J. Org. Chem. 1990, 55, 1952–1954. [Google Scholar]
- 541.Gall EL; Hurvois J-P; Sinbanbhit S Regio- and Diastereoselective Synthesis of α-Cyanoamines by Anodic Oxidation of 6-Membered α-Silylamines. Eur. J. Org. Chem. 1999, 2645–2653. [Google Scholar]
- 542.Girard N; Hurvois J-P Anodic Cyanation of C-4 Hydroxylated Piperidines: Total Synthesis of (±)-Alkaloid 241D. Tetrahedron Lett. 2007, 48, 4097–4099. [Google Scholar]
- 543.Libendi SS; Demizua Y; Onomura O Direct Electrochemical α-Cyanation of N-Protected Cyclic Amines. Org. Biomol. Chem. 2009, 7, 351–356. [DOI] [PubMed] [Google Scholar]
- 544.Tajima T; Nakajima A Direct Oxidative Cyanation Based on the Concept of Site Isolation. J. Am. Chem. Soc. 2008, 130, 10496–10497 [DOI] [PubMed] [Google Scholar]
- 545.Lennox AJJ; Goes SL; Webster MP; Koolman HF; Djuric SW; Stahl SS Electrochemical Aminoxyl-Mediated α-Cyanation of Secondary Piperidines for Pharmaceutical Building Block Diversification. J. Am. Chem. Soc. 2018, 140, 11227–11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Pan Y; Wang S; Kee CW; Dubuisson E; Yang Y; Loh KP; Tan C-H Graphene Oxide and Rose Bengal: Oxidative C–H Functionalisation of Tertiary Amines Using Visible Light. Green Chem. 2011, 13, 3341–3344. [Google Scholar]
- 547.Yilmaz O; Oderinde MS; Emmert MH Photoredox-Catalyzed Cα–H Cyanation of Unactivated Secondary and Tertiary Aliphatic Amines: Late-Stage Functionalization and Mechanistic Studies J. Org. Chem. 2018, 83, 11089–11100. [DOI] [PubMed] [Google Scholar]
- 548.Heitz DR; Rizwan K; Molander GA Visible-Light-Mediated Alkenylation, Allylation, and Cyanation of Potassium Alkyltrifluoroborates with Organic Photoredox Catalysts. J. Org. Chem. 2016, 81, 7308–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Quanquan Z; Dan L; Wenjing X; Liangqiu L Visible-Light Photoredox Catalytic α-Cyanation Reactions of Tertiary Amines. Acta Chim. Sinica 2020, 75, 110–114. [Google Scholar]
- 550.Ramirez NP; König B; Gonzalez-Gomez JC Decarboxylative Cyanation of Aliphatic Carboxylic Acids via Visible-Light Flavin Photocatalysis. Org. Lett. 2019, 21, 1368–1373. [DOI] [PubMed] [Google Scholar]
- 551.Luo Y-R Handbook of Bond Dissociation Energies in Organic Compounds, 1st ed.; CRC Press, 2003. [Google Scholar]
- 552.HAT: Hoshikawa T, Yoshioka S, Kamijo S, Inoue M Photoinduced Direct Cyanation of C(sp3)–H Bonds. Synlett 2013, 45, 874–887. [Google Scholar]
- 553.Wakaki T; Sakai K; Enomoto T; Kondo M; Masaoka S; Oisaki K; Kanai M C(sp3)–H Cyanation Promoted by Visible-Light Photoredox/ Phosphate Hybrid Catalysis. Chem. Eur. J. 2018, 24, 8051–8055. [DOI] [PubMed] [Google Scholar]
- 554.To W-P; Liu Y; Lau T-C; Che C-M A Robust Palladium(II)-Porphyrin Complex as Catalyst for Visible Light Induced Oxidative C–H Functionalization Chem. Eur. J. 2013, 19, 5654–5664. [DOI] [PubMed] [Google Scholar]
- 555.Rueping M; Zhu S; Koenigs RM Visible-Light Photoredox Catalyzed Oxidative Strecker Reaction. Chem. Commun. 2011, 47, 12709–12711. [DOI] [PubMed] [Google Scholar]
- 556.Rueping M; Zoller J; Fabry DC; Poscharny K; Koenigs RM Weirich TE; Mayer J Light-Mediated Heterogeneous Cross Dehydrogenative Coupling Reactions: Metal Oxides as Efficient, Recyclable, Photoredox Catalysts in C–C Bond-Forming Reactions. Chem. Eur. J. 2012, 18, 3478–3481. [DOI] [PubMed] [Google Scholar]
- 557.Kumar P; Varma S; Jain SL A TiO2 Immobilized Ru(II) Polyazine Complex: A Visible-Light Active Photoredox Catalyst for Oxidative Cyanation of Tertiary Amines. J. Mater. Chem. A 2014, 2, 4514–4519. [Google Scholar]
- 558.Rueping M; Vila C; Bootwicha T Continuous Flow Organocatalytic C−H Functionalization and CrossDehydrogenative Coupling Reactions: Visible Light Organophotocatalysis for Multicomponent Reactions and C−C, C−P Bond Formations. ACS Catal. 2013, 3, 1676–1680. [Google Scholar]
- 559.Tucker JW; Zhang Y; Jaimison TF; Stephenson CRJ Visible-Light Photoredox Catalysis in Flow. Angew. Chem. Int. Ed. 2012, 51, 4144–4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.To W-P; Tong GS-M; Lu W; Ma C; Liu J; Chow AL-F; Che C-M Luminescent Organogold(III) Complexes with Long-Lived Triplet Excited States for Light-Induced Oxidative C-H Bond Functionalization and Hydrogen Production. Angew. Chem. Int. Ed. 2012, 51, 2654–2657. [DOI] [PubMed] [Google Scholar]
- 561.Wu Y; Yi H; Lei A Electrochemical Acceptorless Dehydrogenation of N-Heterocycles Utilizing TEMPO as Organo-Electrocatalyst. ACS Catal. 2018, 8, 1192–1196. [Google Scholar]
- 562.Balayeva NO, Zheng N, Dillert R, Bahnemann DW Visible-Light-Mediated Photocatalytic Aerobic Dehydrogenation of N-heterocycles by Surface-Grafted TiO2 and 4-amino-TEMPO. ACS Catal. 2019, 9, 10694–10704. [Google Scholar]
- 563.Bergonzini G; Schindler CS; Wallentin C-J; Jacobsen EN; Stephenson CRJ Photoredox Activation and Anion Binding Catalysis in the Dual Catalytic Enantioselective Synthesis of β-Amino Esters. Chem. Sci. 2014, 5, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.Diccianni JB; Diao T Mechanisms of Nickel-Catalyzed Cross-Coupling Reactions. Trends in Chemistry 2019, 1, 830–844. [Google Scholar]
- 565.Ruiz-Castillo P; Buchwald SL Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Corcoran EB; Pirnot MT; Lin S; Dreher SD; DiRocco DA; Davies IW; Buchwald SL; MacMillan DWC Aryl Amination Using Ligand-Free Ni(II) Salts and Photoredox Catalysis. Science 2016, 353, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Kudisch M; Lim C-H; Thordarson P; Miyake GM Energy Transfer to Ni-Amine Complexes in Dual Catalytic, Light-Driven C–N Cross-Coupling Reactions. J. Am. Chem. Soc. 2019, 141, 19479–19486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Park BY; Pirnot MT; Buchwald SL Visible Light-Mediated (Hetero)Aryl Amination Using Ni(II) Salts and Photoredox Catalysis in Flow: A Synthesis of Tetracaine. J. Org. Chem. 2020, 85, 3234–3244. [DOI] [PubMed] [Google Scholar]
- 569.Oderinde MS; Jones NH; Juneau A; Frenette M; Aquila B; Tentarelli S; Robbins DW; Johannes JW Highly Chemoselective Iridium Photoredox and Nickel Catalysis for the Cross-Coupling of Primary Aryl Amines with Aryl Halides. Angew. Chem. Int. Ed. 2016, 55, 13219–13223. [DOI] [PubMed] [Google Scholar]
- 570.Lim C-H; Kudisch M; Liu B; Miyake GM C–N Cross-Coupling via Photoexcitation of Nickel–Amine Complexes. J. Am. Chem. Soc. 2018, 140, 7667–7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Till NA; Tian L; Dong Z; Scholes GD; MacMillan DWC Mechanistic Analysis of Metallaphotoredox C–N Coupling: Photocatalysis Initiates and Perpetuates Ni(I)/Ni(III) Coupling Activity. J. Am. Chem. Soc. 2020, 142, 15830–15841. [DOI] [PubMed] [Google Scholar]
- 572.Sun R; Qin Y; Nocera DG General Paradigm in Photoredox Nickel-Catalyzed Cross-Coupling Allows for Light-Free Access to Reactivity. Angew. Chem. Int. Ed. 2020, 59, 9527–9533. [DOI] [PubMed] [Google Scholar]
- 573.Li C; Kawamata Y; Nakamura H; Vantourout JC; Liu Z; Hou Q; Bao D; Starr JT; Chen J; Yan M; et al. Electrochemically Enabled, Nickel-Catalyzed Amination. Angew. Chem. Int. Ed. 2017, 56, 13088–13093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Kawamata Y; Vantourout JC; Hickey DP; Bai P; Chen L; Hou Q; Qiao W; Barman K; Edwards MA; Garrido-Castro AF; et al. Electrochemically Driven, Ni-Catalyzed Aryl Amination: Scope, Mechanism, and Applications. J. Am. Chem. Soc. 2019, 141, 6392–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Eichman CC; Stambuli JP Transition Metal Catalyzed Synthesis of Aryl Sulfides. Molecules 2011, 16, 590–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Oderinde MS; Frenette M; Robbins DW; Aquila B; Johannes JW Photoredox Mediated Nickel-Catalyzed Cross-Coupling of Thiols With Aryl and Heteroaryl Iodides via Thiyl Radicals. J. Am. Chem. Soc. 2016, 138, 1760–1763. [DOI] [PubMed] [Google Scholar]
- 577.Wang Y; Deng L; Wang X; Wu Z; Wang Y; Pan Y Electrochemically Promoted Nickel-Catalyzed Carbon–Sulfur Bond Formation. ACS Catal. 2019, 9, 1630–1634. [Google Scholar]
- 578.Liu D; Ma H-X; Fang P; Mei T-S Nickel-Catalyzed Thiolation of Aryl Halides and Heteroaryl Halides through Electrochemistry. Angew. Chem. Int. Ed. 2019, 58, 5033–5037. [DOI] [PubMed] [Google Scholar]
- 579.Dombrowski AW; Gesmundo NJ; Aguirre AL; Sarris KA; Young JM; Bogdan AR; Martin MC; Gedeon S; Wang Y Expanding the Medicinal Chemist Toolbox: Comparing Seven C(sp2)–C(sp3) Cross-Coupling Methods by Library Synthesis. ACS Med. Chem. Lett. 2020, 11, 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580.Littke AF; Dai C; Fu GC Versatile Catalysts for the Suzuki Cross-Coupling of Arylboronic Acids with Aryl and Vinyl Halides and Triflates under Mild Conditions. J. Am. Chem. Soc. 2000, 122, 4020–4028. [Google Scholar]
- 581.Kataoka N; Shelby Q; Stambuli JP; Hartwig JF Air Stable, Sterically Hindered Ferrocenyl Dialkylphosphines for Palladium-Catalyzed C−C, C−N, and C−O Bond-Forming Cross-Couplings. J. Org. Chem. 2002, 67, 5553–5566. [DOI] [PubMed] [Google Scholar]
- 582.For a minireview, see Doucet H Suzuki–Miyaura Cross-Coupling Reactions of Alkylboronic Acid Derivatives or Alkyltrifluoroborates with Aryl, Alkenyl or Alkyl Halides and Triflates. Eur. J. Org. Chem. 2008, 2008, 2013–2030.
- 583.Leonori D; Aggarwal VK Stereospecific Couplings of Secondary and Tertiary Boronic Esters. Angew. Chem. Int. Ed. 2015, 54, 1082–1096. [DOI] [PubMed] [Google Scholar]
- 584.Ma X; Murray B; Biscoe MR Stereoselectivity in Pd-Catalysed Cross-Coupling Reactions of Enantioenriched Nucleophiles. Nat. Rev. Chem. 2020, 4, 584–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Molander GA; Ellis N Organotrifluoroborates: Protected Boronic Acids That Expand the Versatility of the Suzuki Coupling Reaction. Acc. Chem. Res. 2007, 40, 275–286. [DOI] [PubMed] [Google Scholar]
- 586.Darses S; Genet J-P Potassium Organotrifluoroborates: New Perspectives in Organic Synthesis. Chem. Rev. 2008, 108, 288–325. [DOI] [PubMed] [Google Scholar]
- 587.Jahn U Radicals in Transition Metal Catalyzed Reactions? Transition Metal Catalyzed Radical Reactions?: A Fruitful Interplay Anyway. In Radicals in Synthesis III; Heinrich M, Gansäuer A, Eds.; Topics in Current Chemistry; Springer: Berlin, Heidelberg, 2012; pp 323–451. [DOI] [PubMed] [Google Scholar]
- 588.Kwiatkowski MR; Alexanian EJ Transition-Metal (Pd, Ni, Mn)-Catalyzed C–C Bond Constructions Involving Unactivated Alkyl Halides and Fundamental Synthetic Building Blocks. Acc. Chem. Res. 2019, 52, 1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Tellis JC; Primer DN; Molander GA Single-Electron Transmetalation in Organoboron Cross-Coupling by Photoredox/Nickel Dual Catalysis. Science 2014, 345, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Primer DN; Karakaya I; Tellis JC; Molander GA Single-Electron Transmetalation: An Enabling Technology for Secondary Alkylboron Cross-Coupling. J. Am. Chem. Soc. 2015, 137, 2195–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Tellis JC; Amani J; Molander GA Single-Electron Transmetalation: Photoredox/Nickel Dual Catalytic Cross-Coupling of Secondary Alkyl β-Trifluoroboratoketones and -Esters with Aryl Bromides. Org. Lett. 2016, 18, 2994–2997. [DOI] [PubMed] [Google Scholar]
- 592.Karakaya I; Primer DN; Molander GA Photoredox Cross-Coupling: Ir/Ni Dual Catalysis for the Synthesis of Benzylic Ethers. Org. Lett. 2015, 17, 3294–3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Karimi-Nami R; Tellis JC; Molander GA Single-Electron Transmetalation: Protecting-Group-Independent Synthesis of Secondary Benzylic Alcohol Derivatives via Photoredox/Nickel Dual Catalysis. Org. Lett. 2016, 18, 2572–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594.Matsui JK; Molander GA Direct α-Arylation/Heteroarylation of 2-Trifluoroboratochromanones via Photoredox/Nickel Dual Catalysis. Org. Lett. 2017, 19, 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595.El Khatib M; Serafim RAM; Molander GA α-Arylation/Heteroarylation of Chiral α-Aminomethyltrifluoroborates by Synergistic Iridium Photoredox/Nickel Cross-Coupling Catalysis. Angew. Chem. Int. Ed. 2016, 55, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Alam R; Molander GA Direct Synthesis of Secondary Benzylic Alcohols Enabled by Photoredox/Ni Dual-Catalyzed Cross-Coupling. J. Org. Chem. 2017, 82, 13728–13734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Ryu D; Primer DN; Tellis JC; Molander GA Single-Electron Transmetalation: Synthesis of 1,1-Diaryl-2,2,2-Trifluoroethanes by Photoredox/Nickel Dual Catalytic Cross-Coupling. Chem. Eur. J. 2016, 22, 120–123. [DOI] [PubMed] [Google Scholar]
- 598.Yamashita Y; Tellis JC; Molander GA Protecting Group-Free, Selective Cross-Coupling of Alkyltrifluoroborates with Borylated Aryl Bromides via Photoredox/Nickel Dual Catalysis. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 12026–12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599.Gutierrez O; Tellis JC; Primer DN; Molander GA; Kozlowski MC Nickel-Catalyzed Cross-Coupling of Photoredox-Generated Radicals: Uncovering a General Manifold for Stereoconvergence in Nickel-Catalyzed Cross-Couplings. J. Am. Chem. Soc. 2015, 137, 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Biswas S; Weix DJ Mechanism and Selectivity in Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Halides with Alkyl Halides. J. Am. Chem. Soc. 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Schley ND; Fu GC Nickel-Catalyzed Negishi Arylations of Propargylic Bromides: A Mechanistic Investigation. J. Am. Chem. Soc. 2014, 136, 16588–16593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Breitenfeld J; Ruiz J; Wodrich MD; Hu X Bimetallic Oxidative Addition Involving Radical Intermediates in Nickel-Catalyzed Alkyl–Alkyl Kumada Coupling Reactions. J. Am. Chem. Soc. 2013, 135, 12004–12012. [DOI] [PubMed] [Google Scholar]
- 603.Yin H; Fu GC Mechanistic Investigation of Enantioconvergent Kumada Reactions of Racemic α-Bromoketones Catalyzed by a Nickel/Bis(Oxazoline) Complex. J. Am. Chem. Soc. 2019, 141, 15433–15440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604.Primer DN; Molander GA Enabling the Cross-Coupling of Tertiary Organoboron Nucleophiles through Radical-Mediated Alkyl Transfer. J. Am. Chem. Soc. 2017, 139, 9847–9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Yuan M; Song Z; Badir SO; Molander GA; Gutierrez O On the Nature of C(sp3)–C(sp2) Bond Formation in Nickel-Catalyzed Tertiary Radical Cross-Couplings: A Case Study of Ni/Photoredox Catalytic Cross-Coupling of Alkyl Radicals and Aryl Halides. J. Am. Chem. Soc. 2020, 142, 7225–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606.Milligan JA; Phelan JP; Badir SO; Molander GA Alkyl Carbon–Carbon Bond Formation by Nickel/Photoredox Cross-Coupling. Angew. Chem. Int. Ed. 2019, 58, 6152–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Luo J; Hu B; Wu W; Hu M; Liu TL Nickel-Catalyzed Electrochemical C(sp3)−C(sp2) Cross-Coupling Reactions of Benzyl Trifluoroborate and Organic Halides. Angew. Chem. Int. Ed. 2021, 60, 6107–6116. [DOI] [PubMed] [Google Scholar]
- 608.Everson DA; Weix DJ Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J. Org. Chem. 2014, 79, 4793–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609.Duan Z; Li W; Lei A Nickel-Catalyzed Reductive Cross-Coupling of Aryl Bromides with Alkyl Bromides: Et3N as the Terminal Reductant. Org. Lett. 2016, 18, 4012–4015. [DOI] [PubMed] [Google Scholar]
- 610.Everson DA; Jones BA; Weix DJ Replacing Conventional Carbon Nucleophiles with Electrophiles: Nickel-Catalyzed Reductive Alkylation of Aryl Bromides and Chlorides. J. Am. Chem. Soc. 2012, 134, 6146–6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.Zhang P; Le C. “Chip”; MacMillan DWC Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway for Cross-Electrophile Coupling. J. Am. Chem. Soc. 2016, 138, 8084–8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Chen TQ; MacMillan DWC A Metallaphotoredox Strategy for the Cross-Electrophile Coupling of α-Chloro Carbonyls with Aryl Halides. Angew. Chem. Int. Ed. 2019, 58, 14584–14588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613.Bacauanu V; Cardinal S; Yamauchi M; Kondo M; Fernández DF; Remy R; MacMillan DWC Metallaphotoredox Difluoromethylation of Aryl Bromides. Angew. Chem. Int. Ed. 2018, 57, 12543–12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614.Sakai HA; Liu W; Le C “Chip”; MacMillan, D. W. C. Cross-Electrophile Coupling of Unactivated Alkyl Chlorides. J. Am. Chem. Soc. 2020, 142, 11691–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Shou-Kun Zhang S-K; Samanta RC; Del Vecchio A Ackermann L Chem. Eur. J. 2020, 26, 10936–10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Durandetti M; Nédélec J-Y; Périchon J Nickel-Catalyzed Direct Electrochemical Cross-Coupling between Aryl Halides and Activated Alkyl Halides. J. Org. Chem. 1996, 61, 1748–1755. [DOI] [PubMed] [Google Scholar]
- 617.Durandetti M; Périchon J; Nédélec J-Y Asymmetric Induction in the Electrochemical Cross-Coupling of Aryl Halides with α-Chloropropionic Acid Derivatives Catalyzed by Nickel Complexes. J. Org. Chem. 1997, 62, 7914–7915. [DOI] [PubMed] [Google Scholar]
- 618.Perkins RJ; Pedro DJ; Hansen EC Electrochemical Nickel Catalysis for Sp2-Sp3 Cross-Electrophile Coupling Reactions of Unactivated Alkyl Halides. Org. Lett. 2017, 19, 3755–3758. [DOI] [PubMed] [Google Scholar]
- 619.Truesdell BL; Hamby TB; Sevov CS General C(sp2)–C(sp3) Cross-Electrophile Coupling Reactions Enabled by Overcharge Protection of Homogeneous Electrocatalysts. J. Am. Chem. Soc. 2020, 142, 5884–5893. [DOI] [PubMed] [Google Scholar]
- 620.Perkins RJ; Hughes AJ; Weix DJ; Hansen EC Metal-Reductant-Free Electrochemical Nickel-Catalyzed Couplings of Aryl and Alkyl Bromides in Acetonitrile. Org. Process Res. Dev. 2019, 23, 1746–1751. [Google Scholar]
- 621.DeLano TJ; Reisman SE Enantioselective Electroreductive Coupling of Alkenyl and Benzyl Halides via Nickel Catalysis. ACS Catal. 2019, 9, 6751–6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Li Z; Sun W; Wang X; Li L; Zhang Y; Li C Electrochemically Enabled, Nickel-Catalyzed Dehydroxylative Cross-Coupling of Alcohols with Aryl Halides. J. Am. Chem. Soc. 2021, 143, 3536–3543. [DOI] [PubMed] [Google Scholar]
- 623.Mucha A; Kafarski P; Berlicki Ł Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [DOI] [PubMed] [Google Scholar]
- 624.Demmer CS; Krogsgaard-Larsen N; Bunch L Review on Modern Advances of Chemical Methods for the Introduction of a Phosphonic Acid Group. Chem. Rev. 2011, 111, 7981–8006. [DOI] [PubMed] [Google Scholar]
- 625.Montchamp J-L Phosphinate Chemistry in the 21st Century: A Viable Alternative to the Use of Phosphorus Trichloride in Organophosphorus Synthesis. Acc. Chem. Res. 2014, 47, 77–87. [DOI] [PubMed] [Google Scholar]
- 626.Chen L; Liu X-Y; Zou Y-X Recent Advances in the Construction of Phosphorus-Substituted Heterocycles, 2009–2019. Adv.Synth. Catal. 2020, 362, 1724–1818. [Google Scholar]
- 627.Han C; Zhu L; Zhao F; Zhang Z; Wang J; Deng Z; Xu H; Li J; Ma D; Yan P Suppressing Triplet State Extension for Highly Efficient Ambipolar Phosphine Oxide Host Materials in Blue PHOLEDs. Chem. Commun. 2014, 50, 2670–2672. [DOI] [PubMed] [Google Scholar]
- 628.Kotani S; Nakajima M Recent Advances in Asymmetric Phosphine Oxide Catalysis. Tetrahedron Lett. 2020, 61, 151421. [Google Scholar]
- 629.Zhao W; Yan PK; Radosevich AT A Phosphetane Catalyzes Deoxygenative Condensation of α-Keto Esters and Carboxylic Acids via PIII/PV═O Redox Cycling. J. Am. Chem. Soc. 2015, 137, 616–619. [DOI] [PubMed] [Google Scholar]
- 630.Nykaza TV; Harrison TS; Ghosh A; Putnik RA; Radosevich AT A Biphilic Phosphetane Catalyzes N–N Bond-Forming Cadogan Heterocyclization via PIII/PV═O Redox Cycling. J. Am. Chem. Soc. 2017, 139, 6839–6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Nykaza TV; Ramirez A; Harrison TS; Luzung MR; Radosevich AT Biphilic Organophosphorus-Catalyzed Intramolecular Csp2–H Amination: Evidence for a Nitrenoid in Catalytic Cadogan Cyclizations. J. Am. Chem. Soc. 2018, 140, 3103–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Li G; Nykaza TV; Cooper JC; Ramirez A; Luzung MR; Radosevich AT An Improved PIII/PV═O-Catalyzed Reductive C–N Coupling of Nitroaromatics and Boronic Acids by Mechanistic Differentiation of Rate- and Product-Determining Steps. J. Am. Chem. Soc. 2020, 142, 6786–6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Li G; Qin Z; Radosevich ATP (III)/P(V)-Catalyzed Methylamination of Arylboronic Acids and Esters: Reductive C–N Coupling with Nitromethane as a Methylamine Surrogate. J. Am. Chem. Soc. 2020, 142, 16205–16210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634.(a) Jockusch S; Turro NJ Phosphinoyl Radicals: Structure and Reactivity. A Laser Flash Photolysis and Time-Resolved ESR Investigation. J. Am. Chem. Soc. 1998, 120, 11773–11777. [Google Scholar]; (b) Leca D; Fensterbank L; Lacôte E; Malacria M Recent Advances in the Use of Phosphorus-Centered Radicals in Organic Chemistry. Chem. Soc. Rev. 2005, 34, 858–865. [DOI] [PubMed] [Google Scholar]
- 635.Luo K; Yang W-C; Wu L Photoredox Catalysis in Organophosphorus Chemistry. Asian J. Org. Chem. 2017, 6, 350–367. [Google Scholar]
- 636.Ung SP-M; Mechrouk VA; Li C-J Shining Light on the Light-Bearing Element: A Brief Review of Photomediated C–H Phosphorylation Reactions. Synthesis 2020, 53, 1003–1022. [Google Scholar]
- 637.Sbei N; Martins GM; Shirinfar B; Ahmed N Electrochemical Phosphorylation of Organic Molecules. Chem. Rec. 2020, 20, 1530–1552. [DOI] [PubMed] [Google Scholar]
- 638.Xuan J; Zeng T-T; Chen J-R; Lu L-Q; Xiao W-J Room Temperature C–P Bond Formation Enabled by Merging Nickel Catalysis and Visible-Light-Induced Photoredox Catalysis. Chem. Eur. J. 2015, 21, 4962–4965. [DOI] [PubMed] [Google Scholar]
- 639.Christiansen A; Li C; Garland M; Selent D; Ludwig R; Spannenberg A; Baumann W; Franke R; Börner A On the Tautomerism of Secondary Phosphane Oxides. Eur. J. Org. Chem. 2010, 2010, 2733–2741. [Google Scholar]
- 640.Liao L-L; Gui Y-Y; Zhang X-B; Shen G; Liu H-D; Zhou W-J; Li J; Yu D-G Phosphorylation of Alkenyl and Aryl C–O Bonds via Photoredox/Nickel Dual Catalysis. Org. Lett. 2017, 19, 3735–3738. [DOI] [PubMed] [Google Scholar]
- 641.Sengmany S; Ollivier A; Le Gall E; Léonel E A Mild Electroassisted Synthesis of (Hetero)Arylphosphonates. Org. Biomol. Chem. 2018, 16, 4495–4500. [DOI] [PubMed] [Google Scholar]
- 642.Daili F; Ouarti A; Pinaud M; Kribii I; Sengmany S; Le Gall E; Léonel E Nickel-Catalyzed Electrosynthesis of Aryl and Vinyl Phosphinates: Nickel-Catalyzed Electrosynthesis of Aryl and Vinyl Phosphinates. Eur. J. Org. Chem. 2020, 3452–3455. [Google Scholar]
- 643.Bai Y; Liu N; Wang S; Wang S; Ning S; Shi L; Cui L; Zhang Z; Xiang J Nickel-Catalyzed Electrochemical Phosphorylation of Aryl Bromides. Org. Lett. 2019, 21, 6835–6838. [DOI] [PubMed] [Google Scholar]
- 644.Zhu C; Yue H; Nikolaienko P; Rueping M Merging Electrolysis and Nickel Catalysis in Redox Neutral Cross-Coupling Reactions: Experiment and Computation for Electrochemically Induced C–P and C–Se Bonds Formation. CCS Chem. 2020, 2, 179–190. [Google Scholar]
- 645.Zhao C; Rakesh KP; Ravidar L; Fang W-Y; Qin H-L Pharmaceutical and Medicinal Significance of Sulfur (SVI)-Containing Motifs for Drug Discovery: A Critical Review. Eur. J. Med. Chem. 2019, 162, 679–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 646.Meanwell NA Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 2011, 54, 2529–2591. [DOI] [PubMed] [Google Scholar]
- 647.Dong J; Krasnova L; Finn MG; Sharpless KB Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry. Angew. Chem. Int. Ed. 2014, 53, 9430–9448. [DOI] [PubMed] [Google Scholar]
- 648.Shannon DA; Gu C; McLaughlin CJ; Kaiser M; Hoorn R. A. L. van der; Weerapana E Sulfonyl Fluoride Analogues as Activity-Based Probes for Serine Proteases. ChemBioChem 2012, 13, 2327–2330. [DOI] [PubMed] [Google Scholar]
- 649.Narayanan A; Jones LH Sulfonyl Fluorides as Privileged Warheads in Chemical Biology. Chem. Sci. 2015, 6, 2650–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 650.Zhao Q; Ouyang X; Wan X; Gajiwala KS; Kath JC; Jones LH; Burlingame AL; Taunton J Broad-Spectrum Kinase Profiling in Live Cells with Lysine-Targeted Sulfonyl Fluoride Probes. J. Am. Chem. Soc. 2017, 139, 680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651.Ye S; Li X; Xie W; Wu J Photoinduced Sulfonylation Reactions Through the Insertion of Sulfur Dioxide. Eur. J. Org. Chem. 2020, 2020, 1274–1287. [Google Scholar]
- 652.Ai C; Shen H; Song D; Li Y; Yi X; Wang Z; Ling F; Zhong W Metal- and Oxidant-Free Electrochemical Synthesis of Sulfinic Esters From Thiols and Alcohols. Green. Chem. 2019, 21, 5528–5531. [Google Scholar]
- 653.Tan KYD; Teng GF; Fan WY Photocatalytic Transformation of Organic and Water-Soluble Thiols into Disulfides and Hydrogen under Aerobic Conditions Using Mn(CO)5Br. Organometallics 2011, 30, 4136–4143. [Google Scholar]
- 654.Li XB; Li ZJ; Gao YJ; Meng QY; Yu S; Weiss RG; Tung CH; Wu LZ Mechanistic Insights into the Interface-Directed Transformation of Thiols into Disulfides and Molecular Hydrogen by Visible-Light Irradiation of Quantum Dots. Angew. Chem. Int. Ed. 2014, 53, 2085–2089. [DOI] [PubMed] [Google Scholar]
- 655.Kumar P; Singh G; Tripathi D; Jain SL Visible Light Driven Photocatalytic Oxidation of Thiols to Disulfides Using Iron Phthalocyanine Immobilized on Graphene Oxide as a Catalyst Under Alkali Free Conditions. RSC Adv. 2014, 4, 50331–50337. [Google Scholar]
- 656.Tankam T; Poochampa K; Vilaivan T; Sukwattanasinitt M; Wacharasindhu S Organocatalytic Visible Light Induced S–S Bond Formation for Oxidative Coupling of Thiols to disulfides. Tetrahedron 2016, 72, 788–793. [Google Scholar]
- 657.Liu W; Wang C; Huang Y; Chen Q; Wang L; He M Visible-Light-Mediated Facile Synthesis of Disulfides Using Reusable TiO2/MoS2 Nanocomposite Photocatalyst. Synth. Commun. 2016, 46, 1268–1274. [Google Scholar]
- 658.Talla A; Driessen B; Straathof NJW; Milroy L-G; Brunsveld L; Hessel V; Noël T Metal-Free Photocatalytic Aerobic Oxidation of Thiols to Disulfides in Batch and Continuous-Flow. Adv. Synth. Catal. 2015, 357, 2180–2186. [Google Scholar]
- 659.Dethe DH; Srivastava A; Dherange BD; Kumar BV Unsymmetrical Disulfide Synthesis through Photoredox Catalysis. Adv. Synth. Catal. 2018, 360, 3020–3025. [Google Scholar]
- 660.Ai C; Shen H; Song D; Li Y; Yi X; Wang Z; Ling F; Zhong W Metal- and Oxidant-Free Electrochemical Synthesis of Sulfinic Esters From Thiols and Alcohols. Green. Chem. 2019, 21, 5528–5531. [Google Scholar]
- 661.Laudadio G; Straathof NJW; Lanting MD; Knoops B; Hessel V; Noël T An Environmentally Benign and Selective Electrochemical Oxidation of Sulfides and Thiols in a Continuous-Flow Microreactor. Green Chem. 2017, 19, 4061–4066. [Google Scholar]
- 662.Liua S; Chen B; Yang Y; Yang Y; Chen Q; Zeng X; Xu B Electrochemical Oxidations of Thioethers: Modulation of Oxidation Potential Using a Hydrogen Bonding Network. Electrochem. Commun. 2019, 109, 106583–1065. [Google Scholar]
- 663.Lutz S; Steckhan E Liese A. First Asymmetric Electroenzymatic Oxidation Catalyzed by a Peroxidase. Electrochem. Commun. 2004, 6, 583–587. [Google Scholar]
- 664.Li B; Liu A-H; He L-N; Yang Z-Z; Gao J; Chen K-H Selectivity Switch in the Aerobic Oxygenation of Sulfides Photocatalysed by Visible-Light-Responsive Decavanadate. Green Chem. 2020, 22, 3896–3905. [Google Scholar]
- 665.Li Y; Rizvi S. A.-e.-A.; Hu D; Sun D; Gao A; Zhou Y; Li J; Jiang X Selective Late-Stage Oxygenation of Sulfides with Ground-State Oxygen by Uranyl Photocatalysis. Angew. Chem. Int. Ed. 2019, 58, 13499–13506. [DOI] [PubMed] [Google Scholar]
- 666.Ma L; Zhou H; Xu M; Hao P; Kong X; Duan H Integrating Hydrogen Production With Anodic Selective Oxidation of Sulfides Over a CoFe Layered Double Hydroxide Electrode. Chem. Sci. 2020, 12, 938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 667.Meadows DC; Gervay-Hague J Vinyl Sulfones: Synthetic Preparations and Medicinal Chemistry Applications. Med. Res. Rev. 2006, 26, 793–814. [DOI] [PubMed] [Google Scholar]
- 668.El-Awa A; Noshi MN; du Jourdin XM; Fuchs PL Evolving Organic Synthesis Fostered by the Pluripotent Phenylsulfone Moiety. Chem. Rev. 2009, 109, 2315–2349. [DOI] [PubMed] [Google Scholar]
- 669.Palmer JT; Rasnick D; Klaus JL; Bromme D Vinyl Sulfones as Mechanism-Based Cysteine Protease Inhibitors. J. Med. Chem. 1995, 38, 3193–3196. [DOI] [PubMed] [Google Scholar]
- 670.Liu S; Zhou B; Yang H; He Y; Jiang Z-X; Kumar S; Wu L; Zhang Z-Y Aryl Vinyl Sulfonates and Sulfones as Active Site-Directed and Mechanism-Based Probes for Protein Tyrosine Phosphatases. J. Am. Chem. Soc. 2008, 130, 8251–8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 671.Morales-Sanfrutos J; Lopez-Jaramillo J; Ortega-Muñoz M; Megia-Fernandez A; Perez-Balderas F; Hernandez-Mateo F; Santoyo-Gonzalez F Vinyl Sulfone: A Versatile Function for Simple Bioconjugation and Immobilization. Org. Biomol. Chem. 2010, 8, 667–675. [DOI] [PubMed] [Google Scholar]
- 672.Söveges B; Imre T; Szende T; Póti ÁL; Cserép GB; Hegedűs T; Kele P; Németh K A Systematic Study of Protein Labeling by Fluorogenic Probes Using Cysteine Targeting Vinyl Sulfone-Cyclooctyne Tags. Org. Biomol. Chem. 2016, 14, 6071–6078. [DOI] [PubMed] [Google Scholar]
- 673.Meyer AU; Jäger S; Prasad Hari D; König B Visible Light-Mediated Metal-Free Synthesis of Vinyl Sulfones from Aryl Sulfinates. Adv. Synth. Catal. 2015, 357, 2050–2054. [Google Scholar]
- 674.Meyer AU; Straková K; Slanina T; König B Eosin Y (EY) Photoredox-Catalyzed Sulfonylation of Alkenes: Scope and Mechanism. Chem. Eur. J. 2016, 22, 8694–8699. [DOI] [PubMed] [Google Scholar]
- 675.Ji Y; Brueckl T; Baxter RD; Fujiwara Y; Seiple IB; Su S; Blackmond DG; Baran PS Innate C-H Trifluoromethylation of Heterocycles. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14411–14415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676.Zhou Q; Gui J; Pan C-M; Albone E; Cheng X; Suh EM; Grasso L; Ishihara Y; Baran PS Bioconjugation by Native Chemical Tagging of C–H Bonds. J. Am. Chem. Soc. 2013, 135, 12994–12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 677.Zhou Q; Ruffoni A; Gianatassio R; Fujiwara Y; Sella E; Shabat D; Baran PS Direct Synthesis of Fluorinated Heteroarylether Bioisosteres. Angew. Chem. Int. Ed. 2013, 52, 3949–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 678.Gianatassio R; Kawamura S; Eprile CL; Foo K; Ge J; Burns AC; Collins MR; Baran PS Simple Sulfinate Synthesis Enables C–H Trifluoromethylcyclopropanation. Angew. Chem. Int. Ed. 2014, 53, 9851–9855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 679.Luo Y-C; Pan X-J; Yuan G-Q An Efficient Electrochemical Synthesis of Vinyl Sulfones from Sodium Sulfinates and Olefins. Tetrahedron 2015, 71, 2119–2123. [Google Scholar]
- 680.Terent’ev AO; Mulina OM; Pirgach DA; Ilovaisky AI; Syroeshkin MA; Kapustina NI; Nikishin GI Electrosynthesis of Vinyl Sulfones from Alkenes and Sulfonyl Hydrazides Mediated by KI: Аn Electrochemical Mechanistic Study. Tetrahedron 2017, 73, 6871–6879. [Google Scholar]
- 681.Wang H; Lu Q; Chiang C-W; Luo Y; Zhou J; Wang G; Lei A Markovnikov-Selective Radical Addition of S-Nucleophiles to Terminal Alkynes through a Photoredox Process. Angew. Chem. Int. Ed. 2017, 56, 595–599. [DOI] [PubMed] [Google Scholar]
- 682.Mo Z-Y; Zhang Y-Z; Huang G-B; Wang X-Y; Pan Y-M; Tang H-T Electrochemical Sulfonylation of Alkynes with Sulfonyl Hydrazides: A Metal- and Oxidant-Free Protocol for the Synthesis of Alkynyl Sulfones. Adv. Synth. Catal. 2020, 362, 2160–2167. [Google Scholar]
- 683.Markitanov YM; Timoshenko VM; Shermolovich YG β-Keto Sulfones: Preparation and Application in Organic Synthesis. Journal of Sulfur Chemistry 2014, 35, 188–236. [Google Scholar]
- 684.Nájera C; Yus M Desulfonylation Reactions: Recent Developments. Tetrahedron 1999, 55, 10547–10658. [Google Scholar]
- 685.Yang H; Carter RG; Zakharov LN Enantioselective Total Synthesis of Lycopodine. J. Am. Chem. Soc. 2008, 130, 9238–9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 686.Cai S; Chen D; Xu Y; Weng W; Li L; Zhang R; Huang M Visible-Light-Promoted Syntheses of β-Keto Sulfones from Alkynes and Sulfonylhydrazides. Org. Biomol. Chem. 2016, 14, 4205–4209. [DOI] [PubMed] [Google Scholar]
- 687.Yang D; Huang B; Wei W; Li J; Lin G; Liu Y; Ding J; Sun P; Wang H Visible-Light Initiated Direct Oxysulfonylation of Alkenes with Sulfinic Acids Leading to β-Ketosulfones. Green Chem. 2016, 18, 5630–5634. [Google Scholar]
- 688.Yavari I; Shaabanzadeh S Electrochemical Synthesis of β-Ketosulfones from Switchable Starting Materials. Org. Lett. 2020, 22, 464–467. [DOI] [PubMed] [Google Scholar]
- 689.Pagire SK; Paria S; Reiser O Synthesis of β-Hydroxysulfones from Sulfonyl Chlorides and Alkenes Utilizing Visible Light Photocatalytic Sequences. Org. Lett. 2016, 18, 2106–2109. [DOI] [PubMed] [Google Scholar]
- 690.Chan C-K; Lo N-C; Chen P-Y; Chang M-Y An Efficient Organic Electrosynthesis of β-Hydroxysulfones. Synthesis 2017, 49, 4469–4477. [Google Scholar]
- 691.Liu N-W; Liang S; Manolikakes G Recent Advances in the Synthesis of Sulfones. Synthesis 2016, 48, 1939–1973. [Google Scholar]
- 692.Liu N-W; Liang S; Margraf N; Shaaban S; Luciano V; Drost M; Manolikakes G Nickel-Catalyzed Synthesis of Diaryl Sulfones from Aryl Halides and Sodium Sulfinates. Eur. J. Org. Chem. 2018, 2018, 1208–1210. [Google Scholar]
- 693.Yue H; Zhu C; Rueping M Cross-Coupling of Sodium Sulfinates with Aryl, Heteroaryl, and Vinyl Halides by Nickel/Photoredox Dual Catalysis. Angew. Chem. Int. Ed. 2018, 57, 1371–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 694.Liu N-W; Hofman K; Herbert A; Manolikakes G Visible-Light Photoredox/Nickel Dual Catalysis for the Cross-Coupling of Sulfinic Acid Salts with Aryl Iodides. Org. Lett. 2018, 20, 760–763. [DOI] [PubMed] [Google Scholar]
- 695.Cabrera-Afonso MJ; Lu Z-P; Kelly CB; Lang SB; Dykstra R; Gutierrez O; Molander GA Engaging Sulfinate Salts via Ni/Photoredox Dual Catalysis Enables Facile Csp2–SO2R Coupling. Chem. Sci. 2018, 9, 3186–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 696.Chen L; Liang J; Chen Z; Chen J; Yan M; Zhang X A Convenient Synthesis of Sulfones via Light Promoted Coupling of Sodium Sulfinates and Aryl Halides. Adv. Synth. Catal. 2019, 361, 956–960. [Google Scholar]
- 697.Crisenza GEM; Mazzarella D; Melchiorre P Synthetic Methods Driven by the Photoactivity of Electron Donor–Acceptor Complexes. J. Am. Chem. Soc. 2020, 142, 5461–5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 698.Johnson TC; Elbert BL; Farley AJM; Gorman TW; Genicot C; Lallemand B; Pasau P; Flasz J; Castro JL; MacCoss M; et al. Direct Sulfonylation of Anilines Mediated by Visible Light. Chem. Sci. 2018, 9, 629–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 699.Ye S; Li Y; Wu J; Li Z Thiourea Dioxide as a Source of Sulfonyl Groups: Photoredox Generation of Sulfones and Sulfonamides from Heteroaryl/Aryl Halides. Chem. Commun. 2019, 55, 2489–2492. [DOI] [PubMed] [Google Scholar]
- 700.Yuan Y; Yu Y; Qiao J; Liu P; Yu B; Zhang W; Liu H; He M; Huang Z; Lei A Exogenous-Oxidant-Free Electrochemical Oxidative C–H Sulfonylation of Arenes/Heteroarenes with Hydrogen Evolution. Chem. Commun. 2018, 54, 11471–11474. [DOI] [PubMed] [Google Scholar]
- 701.Nematollahi D; Varmaghani F Paired Electrochemical Synthesis of New Organosulfone Derivatives. Electrochim. Acta 2008, 53, 3350–3355. [Google Scholar]
- 702.Beiginejad H; Nematollahi D; Varmaghani F; Bayat M; Salehzadeh H Efficient Factors on the Reaction Rate and Site-Selectivity in Sulfonylation of Catechol and Hydroquinone Derivatives: Experimental and Theoretical Studies. J. Electrochem. Soc. 2013, 160, G3001. [Google Scholar]
- 703.Nematollahi D; Baniardalan M; Khazalpour S; Pajohi-Alamoti MR Product Diversity by Changing the Electrode Potential. Synthesis, Kinetic Evaluation and Antibacterial Activity of Arylsulfonyl-4,4’-Biphenol and Bis-Arylsulfonyl-4,4’-Biphenol Derivatives. Electrochim. Acta 2016, 191, 98–105. [Google Scholar]
- 704.Nikl J; Lips S; Schollmeyer D; Franke R; Waldvogel SR Direct Metal- and Reagent-Free Sulfonylation of Phenols with Sodium Sulfinates by Electrosynthesis. Chem. Eur. J. 2019, 25, 6891–6895. [DOI] [PubMed] [Google Scholar]
- 705.Nikl J; Ravelli D; Schollmeyer D; Waldvogel SR Straightforward Electrochemical Sulfonylation of Arenes and Aniline Derivatives Using Sodium Sulfinates. ChemElectroChem 2019, 6, 4450–4455. [Google Scholar]
- 706.Wu Y-C; Jiang S-S; Luo S-Z; Song R-J; Li J-H Transition-Metal- and Oxidant-Free Directed Anodic C–H Sulfonylation of N,N-Disubstituted Anilines with Sulfinates. Chem. Commun. 2019, 55, 8995–8998. [DOI] [PubMed] [Google Scholar]
- 707.Kim W; Kim HY; Oh K Electrochemical Radical–Radical Cross-Coupling Approach between Sodium Sulfinates and 2 H -Indazoles to 3-Sulfonylated 2H -Indazoles. Org. Lett. 2020, 22, 6319–6323. [DOI] [PubMed] [Google Scholar]
- 708.Smith BR; Eastman CM; Njardarson JT Beyond C, H, O, and N! Analysis of the Elemental Composition of U.S. FDA Approved Drug Architectures. J. Med. Chem. 2014, 57, 9764–9773. [DOI] [PubMed] [Google Scholar]
- 709.Ilardi EA; Vitaku E; Njardarson JT Data-Mining for Sulfur and Fluorine: An Evaluation of Pharmaceuticals To Reveal Opportunities for Drug Design and Discovery. J. Med. Chem. 2014, 57, 2832–2842. [DOI] [PubMed] [Google Scholar]
- 710.Bag S; Tulsan R; Sood A; Cho H; Redjeb H; Zhou W; LeVine H; Török B; Török M Sulfonamides as Multifunctional Agents for Alzheimer’s Disease. Bioorg. Med. Chem. Lett. 2015, 25, 626–630. [DOI] [PubMed] [Google Scholar]
- 711.Apaydın S; Török M Sulfonamide Derivatives as Multi-Target Agents for Complex Diseases. Bioorg. Med. Chem. Lett. 2019, 29, 2042–2050. [DOI] [PubMed] [Google Scholar]
- 712.Ballatore C; Huryn DM; Smith AB Carboxylic Acid (Bio)Isosteres in Drug Design. ChemMedChem 2013, 8, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 713.Lassalas P; Gay B; Lasfargeas C; James MJ; Tran V; Vijayendran KG; Brunden KR; Kozlowski MC; Thomas CJ; Smith AB; et al. Structure Property Relationships of Carboxylic Acid Isosteres. J. Med. Chem. 2016, 59, 3183–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 714.Bahrami K; Khodaei MM; Soheilizad M Direct Conversion of Thiols to Sulfonyl Chlorides and Sulfonamides. J. Org. Chem. 2009, 74, 9287–9291. [DOI] [PubMed] [Google Scholar]
- 715.DeBergh JR; Niljianskul N; Buchwald SL Synthesis of Aryl Sulfonamides via Palladium-Catalyzed Chlorosulfonylation of Arylboronic Acids. J. Am. Chem. Soc. 2013, 135, 10638–10641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 716.Chen Y; Murray PRD; Davies AT; Willis MC Direct Copper-Catalyzed Three-Component Synthesis of Sulfonamides. J. Am. Chem. Soc. 2018, 140, 8781–8787. [DOI] [PubMed] [Google Scholar]
- 717.Jiang Y; Wang Q-Q; Liang S; Hu L-M; Little RD; Zeng C-C Electrochemical Oxidative Amination of Sodium Sulfinates: Synthesis of Sulfonamides Mediated by NH4I as a Redox Catalyst. J. Org. Chem. 2016, 81, 4713–4719. [DOI] [PubMed] [Google Scholar]
- 718.Zhao J; Xu J; Chen J; Wang X; He M Metal-Free Oxidative Coupling of Amines with Sodium Sulfinates: A Mild Access to Sulfonamides. RSC Adv. 2014, 4, 64698–64701. [Google Scholar]
- 719.Pan X; Gao J; Liu J; Lai J; Jiang H; Yuan G Synthesis of Sulfonamides via I2-Mediated Reaction of Sodium Sulfinates with Amines in an Aqueous Medium at Room Temperature. Green Chem. 2015, 17, 1400–1403. [Google Scholar]
- 720.Yang K; Ke M; Lin Y; Song Q Sulfonamide Formation from Sodium Sulfinates and Amines or Ammonia under Metal-Free Conditions at Ambient Temperature. Green Chem. 2015, 17, 1395–1399. [Google Scholar]
- 721.Wei W; Liu C; Yang D; Wen J; You J; Wang H Metal-Free Direct Construction of Sulfonamides via Iodine- Mediated Coupling Reaction of Sodium Sulfinates and Amines at Room Temperature. Adv. Synth. Catal. 2015, 357, 987–992. [Google Scholar]
- 722.Buathongjan C; Beukeaw D; Yotphan S Iodine-Catalyzed Oxidative Amination of Sodium Sulfinates: A Convenient Approach to the Synthesis of Sulfonamides under Mild Conditions. Eur. J. Org. Chem. 2015, 2015, 1575–1582. [Google Scholar]
- 723.Blum SP; Karakaya T; Schollmeyer D; Klapars A; Waldvogel SR Metal-Free Electrochemical Synthesis of Sulfonamides Directly from (Hetero)Arenes, SO2, and Amines. Angew. Chem. Int. Ed. 2021, 60, 5056–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 724.Hell SM; Meyer CF; Laudadio G; Misale A; Willis MC; Noël T; Trabanco AA; Gouverneur V Silyl Radical-Mediated Activation of Sulfamoyl Chlorides Enables Direct Access to Aliphatic Sulfonamides from Alkenes. J. Am. Chem. Soc. 2020, 142, 720–725. [DOI] [PubMed] [Google Scholar]
- 725.Kim T; McCarver SJ; Lee C; MacMillan DWC Sulfonamidation of Aryl and Heteroaryl Halides through Photosensitized Nickel Catalysis. Angew. Chem. Int. Ed. 2018, 57, 3488–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 726.Davies W; Dick JH CCLXXXVI.—Aromatic Sulphonyl Fluorides. A Convenient Method of Preparation. J. Chem. Soc. 1931, 2104–2109. [Google Scholar]
- 727.Bianchi TA; Cate LA Phase Transfer Catalysis. Preparation of Aliphatic and Aromatic Sulfonyl Fluorides. J. Org. Chem. 1977, 42, 2031–2032. [Google Scholar]
- 728.Talko A; Barbasiewicz M Nucleophilic Fluorination with Aqueous Bifluoride Solution: Effect of the Phase-Transfer Catalyst. ACS Sustainable Chem. Eng. 2018, 6, 6693–6701. [Google Scholar]
- 729.Tribby AL; Rodríguez I; Shariffudin S; Ball ND Pd-Catalyzed Conversion of Aryl Iodides to Sulfonyl Fluorides Using SO2 Surrogate DABSO and Selectfluor. J. Org. Chem. 2017, 82, 2294–2299. [DOI] [PubMed] [Google Scholar]
- 730.Davies AT; Curto JM; Bagley SW; Willis MC One-Pot Palladium-Catalyzed Synthesis of Sulfonyl Fluorides from Aryl Bromides. Chem. Sci. 2017, 8, 1233–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 731.Laudadio G; Bartolomeu A de A; Verwijlen LMHM; Cao Y; de Oliveira KT; Noël T Sulfonyl Fluoride Synthesis through Electrochemical Oxidative Coupling of Thiols and Potassium Fluoride. J. Am. Chem. Soc. 2019, 141, 11832–11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 732.Kirihara M; Naito S; Nishimura Y; Ishizuka Y; Iwai T; Takeuchi H; Ogata T; Hanai H; Kinoshita Y; Kishida M; et al. Oxidation of Disulfides with Electrophilic Halogenating Reagents: Concise Methods for Preparation of Thiosulfonates and Sulfonyl Halides. Tetrahedron 2014, 70, 2464–2471. [Google Scholar]
- 733.Zheng Q; Dong J; Sharpless KB Ethenesulfonyl Fluoride (ESF): An On-Water Procedure for the Kilogram-Scale Preparation. J. Org. Chem. 2016, 81, 11360–11362. [DOI] [PubMed] [Google Scholar]
- 734.Xu R; Xu T; Yang M; Cao T; Liao S A Rapid Access to Aliphatic Sulfonyl Fluorides. Nat. Commun. 2019, 10, 3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 735.Martins GM; Shirinfar B; Hardwick T; Ahmed N A Green Approach: Vicinal Oxidative Electrochemical Alkene Difunctionalization. ChemElectroChem 2019, 6, 1300–1315. [Google Scholar]
- 736.Abou-Hamdan H; Kouklovsky C; Vincent G Dearomatization Reactions of Indoles to Access 3D Indoline Structures. Synlett 2020, 31, 1775–1788. [Google Scholar]
- 737.Siu JC; Parry JB; Lin S Aminoxyl-Catalyzed Electrochemical Diazidation of Alkenes Mediated by a Metastable Charge-Transfer Complex. J. Am. Chem. Soc. 2019, 141, 2825–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 738.Wu J; Dou Y; Guillot R; Kouklovsky C; Vincent G Electrochemical Dearomative 2,3-Difunctionalization of Indoles. J. Am. Chem. Soc. 2019, 141, 2832–2837. [DOI] [PubMed] [Google Scholar]
- 739.Shen T; Lambert TH Electrophotocatalytic diamination of Vicinal C–H bonds. Science 2021, 371, 620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 740.Fu N; Sauer GS; Lin S Electrocatalytic Radical Dichlorination of Alkenes with Nucleophilic Chlorine Sources. J. Am. Chem. Soc. 2017, 139, 15548–15553. [DOI] [PubMed] [Google Scholar]
- 741.Dong X; Roeckl JL; Waldvogel SR; Morandi B Merging Shuttle Reactions and Paired Electrolysis for Reversible Vicinal Dihalogenations. Science 2021, 371, 507–514. [DOI] [PubMed] [Google Scholar]
- 742.Lian P; Long W; Li J; Zheng Y; Wan X Visible-Light-Induced Vicinal Dichlorination of Alkenes through LMCT Excitation of CuCl2. Angew Chem Int. Ed. 2020, 59, 23603–23608. [DOI] [PubMed] [Google Scholar]
- 743.Doobary S; Sedikides AT; Caldora HP; Poole DL; Lennox AJJ Electrochemical Vicinal Difluorination of Alkenes: Scalable and Amenable to Electron-Rich Substrates. Angew. Chem. Int. Ed. 2020, 59, 1155–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 744.Wu J; Guillot R; Kouklovsky C; Vincent G Electrochemical Dearomative Dihydroxylation and Hydroxycyclization of Indoles. Advanced Synthesis & Catalysis 2020, 362, 1712–1719. [Google Scholar]
- 745.Cai C-Y; Shu X-M; Xu H-C Practical and Stereoselective Electrocatalytic 1,2-Diamination of Alkenes. Nat. Commun. 2019, 10, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 746.Govaerts S; Angelini L; Hampton C; Malet-Sanz L; Ruffoni A; Leonori D Photoinduced Olefin Diamination with Alkylamines. Angew. Chem. Int. Ed. 2020, 59, 15021–15028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 747.Qin Q; Han Y-Y; Jiao Y-Y; He Y; Yu S Photoredox-Catalyzed Diamidation and Oxidative Amidation of Alkenes: Solvent-Enabled Synthesis of 1,2-Diamides and α-Amino Ketones. Org. Lett. 2017, 19, 2909–29012. [DOI] [PubMed] [Google Scholar]
- 748.Reed NL; Herman MI; Miltchev VP; Yoon TP Tandem Copper and Photoredox Catalysis in Photocatalytic Alkene Difunctionalization Reactions. Beilstein J. Org. Chem. 2019, 15, 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 749.Fu N; Song L; Liu J; Shen Y; Siu JC; Lin S New Bisoxazoline Ligands Enable Enantioselective Electrocatalytic Cyanofunctionalization of Vinylarenes. J. Am. Chem. Soc. 2019, 141, 14480–14485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 750.Guo Q; Wang M; Peng Q; Huo Y; Liu Q; Wang R; Xu Z Dual-Functional Chiral Cu-Catalyst-Induced Photoredox Asymmetric Cyanofluoroalkylation of Alkenes. ACS Catal. 2019, 9, 4470–4476. [Google Scholar]
- 751.Ilchenko NO; Janson PG; Szabo KJ Copper-Mediated Cyanotrifluoromethylation of Styrenes Using the Togni Reagent. J. Org. Chem. 2013, 78, 11087–11091. [DOI] [PubMed] [Google Scholar]
- 752.He Y-T; Li L-H; Yang Y-F; Zhou ZZ; Hua H-L; Liu X-Y; Liang Y-M Copper-Catalyzed Intermolecular Cyanotrifluoromethylation of Alkenes. Org. Lett. 2014, 16, 270–273. [DOI] [PubMed] [Google Scholar]
- 753.Wang F; Wang D; Wan X; Wu L; Chen P; Liu G Enantioselective Copper-Catalyzed Intermolecular Cyanotrifluoromethylation of Alkenes via Radical Process. J. Am. Chem. Soc. 2016, 138, 15547–15550. [DOI] [PubMed] [Google Scholar]
- 754.Zhang W; Lin S Electroreductive Carbofunctionalization of Alkenes with Alkyl Bromides via a Radical-Polar Crossover Mechanism. J. Am. Chem. Soc. 2020, 142, 20661–20670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 755.For anti-Markovnikov hydroformylation, see: Huang H; Yu C; Zhang Y; Zhang Y; Mariano PS; Wang W Chemo- and Regioselective Organo-Photoredox Catalyzed Hydroformylation of Styrenes via a Radical Pathway. J. Am. Chem. Soc. 2017, 139, 9799–9802.
- 756.For anti-Markovnikov carbamoylation, see: Prieto A; Taillefer M Visible-Light Decatungstate/Disulfide Dual Catalysis for the Hydro-Functionalization of Styrenes. Org. Lett. 2021, 23, 1484–1488.
- 757.For anti-Markovnikov hydrotrifluoromethylation see: Wu X; Chu L; Qing F-L Silver-Catalyzed Hydrotrifluoromethylation of Unactivated Alkenes with CF3SiMe3. Angew. Chem. Int. Ed. 2013, 52, 2198–2202.
- 758.Mizuta S; Verhoog S; Engle KM; Khotavivattana T; O’Duill M; Wheelhouse K; Rassias G; Médebielle M; Gouverneur V Catalytic Hydrotrifluoromethylation of Unactivated Alkenes. J. Am. Chem. Soc. 2013, 135, 2505–2508. [DOI] [PubMed] [Google Scholar]
- 759.For anti-Markovnikov hydrodifluoromethylation, see: Sumino S; Uno M; Fukuyama T; Ryu I; Matsuura M; Yamamoto A; Kishikawa Y Photoredox-Catalyzed Hydrodifluoroalkylation of Alkenes Using Difluorohaloalkyl Compounds and a Hantzsch Ester. J. Org. Chem. 2017, 82, 5469–5474.
- 760.For a minireview on nickel-centered photoredox methods towards alkene dicarbofunctionalization, see: Zhu C; Yue H; Chu L; Rueping M Recent Advances in Photoredox and Nickel Dual-Catalyzed Cascade Reactions: Pushing the Boundaries of Complexity. Chemical Science 2020, 11, 4051–4064.
- 761.Phelan JP; Lang SB; Compton JS; Kelly CB; Dykstra R; Gutierrez O; Molander GA Redox-Neutral Photocatalytic Cyclopropanation via Radical/Polar Crossover. J. Am. Chem. Soc. 2018, 140, 8037–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 762.For anti-Markovnikov hydrocarboxylation, see: Seo H; Liu A; Jamison TF Direct β-Selective Hydrocarboxylation of Styrenes with CO2 Enabled by Continuous Flow Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 13969–13972.
- 763.For unactivated alkene dicarbofunctionalization with internal carboxylation, see: Yatham VR; Shen Y; Martin R Catalytic Intermolecular Dicarbofunctionalization of Styrenes with CO2 and Radical Precursors. Angew. Chem. Int. Ed. 2017, 56, 10915–10919.
- 764.Treacy SR; Rovis T Copper Catalyzed C(sp3)–H Bond Alkylation via Photoinduced Ligand-to-Metal Charge Transfer. J. Am. Chem. Soc. 2021, 143, 2729–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 765.Bhaskaran RP; Babu BP Progress in Electrochemical Trifluoromethylation Reactions. Adv. Synth. Catal. 2020, 362, 5219–5237. [Google Scholar]
- 766.Koike T; Akita M New Horizons of Photocatalytic Fluoromethylative Difunctionalization of Alkenes. Chem. 2018, 4, 409–437. [Google Scholar]
- 767.Koike T; Akita M Fine Design of Photoredox Systems for Catalytic Fluoromethylation of Carbon−Carbon Multiple Bonds. Acc. Chem. Res. 2016, 49, 1937–1944. [DOI] [PubMed] [Google Scholar]
- 768.Oh SH; Malpani YR; Ha N; Jung Y-S; Han SB Vicinal Difunctionalization of Alkenes: Chlorotrifluoromethylation with CF3SO2Cl by Photoredox Catalysis. Org. Lett. 2014, 16, 1310–1313. [DOI] [PubMed] [Google Scholar]
- 769.Huang Y; Hong H; Zou Z; Liao C; Lu J; Qin Y; Li Y; Chen L Electrochemical Vicinal Aminotrifluoromethylation of Alkenes: High Regioselective Acquisition of β-Trifluoromethylamines. Org. Biomol. Chem. 2019, 17, 5014–5020. [DOI] [PubMed] [Google Scholar]
- 770.Yasu Y; Koike T; Akita M Intermolecular Aminotrifluoromethylation of Alkenes by Visible-Light-Driven Photoredox Catalysis. Org. Lett. 2013, 15, 2136–2139. [DOI] [PubMed] [Google Scholar]
- 771.Noto N; Koike T; Akita M Metal-free di- and tri-fluoromethylation of alkenes realized by visible-light-induced perylene photoredox catalysis. Chem. Sci. 2017, 8, 6375–6379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 772.Jud W; Kappe CO; Cantillo D Catalyst-Free Oxytrifluoromethylation of Alkenes through Paired Electrolysis in Organic-Aqueous Media. Chem. Eur. J. 2018, 24, 17234–17238. [DOI] [PubMed] [Google Scholar]
- 773.Yasu Y Koike, T.; Akita, M. Three-component Oxytrifluoromethylation of Alkenes: Highly Efficient and Regioselective Difunctionalization of C=C Bonds Mediated by Photoredox Catalysts. Angew. Chem. Int. Ed. 2012, 51, 9567–9571. [DOI] [PubMed] [Google Scholar]
- 774.Margrey KA; Nicewicz DA A General Approach to Catalytic Alkene Anti-Markovnikov Hydrofunctionalization Reactions via Acridinium Photoredox Catalysis. Acc. Chem. Res. 2016, 49, 1997–2006. [DOI] [PubMed] [Google Scholar]
- 775.Onuska NPR; Schutzbach-Horton ME; Collazo JLR; Nicewicz DA Anti-Markovnikov Hydroazidation of Activated Olefins via Organic Photoredox Catalysis. Synlett 2020, 31, 55–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 776.Song L; Fu N; Ernst BG; Lee W-H; Frederick MO; DiStasio RA Jr.; Lin S Dual Electrocatalysis Enables Enantioselective Hydrocyanation of Conjugated Alkenes. Nat. Chem. 2020, 12, 747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 777.Wang D; Zhu N; Chen P; Lin Z; Liu G Enantioselective Decarboxylative Cyanation Employing Cooperative Photoredox Catalysis and Copper Catalysis. J. Am. Chem. Soc. 2017, 139, 15632–15635. [DOI] [PubMed] [Google Scholar]
- 778.Sha W; Deng L; Ni S; Mei H; Han J; Pan Y Merging Photoredox and Copper Catalysis: Enantioselective Radical Cyanoalkylation of Styrenes. ACS Catal. 2018, 8, 7489–7494. [Google Scholar]
- 779.Zhang G; Hu X; Chiang C-W; Yi H; Pei P; Singh AK; Lei A Anti-Markovnikov Oxidation of β-Alkyl Styrenes with H2O as the Terminal Oxidant. J. Am. Chem. Soc. 2016, 138, 12037–12040. [DOI] [PubMed] [Google Scholar]
- 780.Hu X; Zhang G; Bu F; Lei A Visible-Light-Mediated Anti-Markovnikov Hydration of Olefins. ACS Catal. 2017, 7, 1432–1437. [Google Scholar]
- 781.Xiong P; Long H; Song J; Wang Y; Li J-F; Xu H-C Electrochemically Enabled Carbohydroxylation of Alkenes with H2O and Organotrifluoroborates. J. Am. Chem. Soc. 2018, 140, 16387–16391. [DOI] [PubMed] [Google Scholar]
- 782.Yang D-T; Zhu M; Schiffer ZJ; Williams K; Song X; Liu X; Manthiram K Direct Electrochemical Carboxylation of Benzylic C–N Bonds with Carbon Dioxide. ACS Catal. 2019, 9, 4699–4705. [Google Scholar]
- 783.Liao L-L; Cao G-M; Ye J-H; Sun G-Q; Zhou W-J; Gui Y-Y; Yan S-S; Shen G; Yu D-G Direct Electrochemical Carboxylation of Benzylic C–N Bonds with Carbon Dioxide. J. Am. Chem. Soc. 2018, 140, 17338–17342. [DOI] [PubMed] [Google Scholar]
- 784.Isse AA; Ferlin MG; Gennaro A Electrocatalytic Reduction of Arylethyl Chlorides at Silver Cathodes in the Presence of Carbon Dioxide: Synthesis of 2-Arylpropanoic Acids. J. Electroanal. Chem. 2005, 581, 38–45. [Google Scholar]
- 785.Isse AA; De Giusti A; Gennaro A; Falciola L; Mussini PR Electrochemical Reduction of Benzyl Halides at a Silver Electrode. Electrochim. Acta 2006, 51, 4956–4964. [Google Scholar]
- 786.Isse AA; Gennaro A Electrocatalytic Carboxylation of Benzyl Chlorides at Silver Cathodes in Acetonitrile. Chem. Commun. 2002, 2798–2799. [DOI] [PubMed] [Google Scholar]
- 787.Nicastri MC; Lehnherr D; Lam Y.-h.; DiRocco DA; Rovis T Synthesis of Sterically Hindered Primary Amines by Concurrent Tandem Photoredox Catalysis. J. Am. Chem. Soc. 2020, 142, 987–998. [DOI] [PubMed] [Google Scholar]
- 788.Rong J; Seeberger PH; Gilmore K Chemoselective Photoredox Synthesis of Unprotected Primary Amines Using Ammonia. Org. Lett. 2018, 20, 4081–4085. [DOI] [PubMed] [Google Scholar]
- 789.It is not known whether PC1 can facilitate a E- to Z-isomerization of these oxime substrates (or the reverse process), a scenario where this isomerization is slow (or not occurring) with respect to N–O bond homolysis could explain the incomplete consumption of the ortho-methyl substituted oxime observed under cross-coupling conditions. It is conceivable that the triplet sensitization of that E-oxime substrate by PC1 could even result in forming the related Z-oxime in its S0 configuration whereby this Z-oxime would no longer be sensitizable by PC1 due to the T1 to S0 energy gap (ca. 74 kcal•mol−1) being outside the triplet sensitization range of PC1 (ca. 56 kcal•mol−1).
- 790.The electrochemical window of the solvent/electrolyte may limit how reducing of a substrate one can activate as decomposition of the solvent/electrolyte may dominate at potential at or beyond the solvent/electrolyte limit.
- 791.For application of Bordwell’s equation in PCET, see: Bordwell FG; Cheng J-P; Harrelson JA Homolytic Bond Dissociation Energies in Solution From Equilibrium Acidity and Electrochemical Data. J. Am. Chem. Soc. 1988, 110, 1229–1231.
- 792.Bi-directional building blocks refer to compounds with two or more functional groups that can be utilized to further structurally elaborate the building block – in essence having two or more exit-vectors for building complexity and exploring chemical space.
- 793.For discussion of direct-to-biology and parallel library synthesis, see: Santanilla AB; Regalado ER; Pereira T; Shevlin M; Bateman K; Campeau LC; Schneeweis J; Berritt S; Shi Z-C; Nantermet P; Liu Y; Helmy R; Welch CJ; Vachal P; Davies IW; Cernak T; Dreher SD Nanomole-Scale High-Throughput Chemistry for the Synthesis of Complex Molecules. Science 2015, 347, 49–53.
- 794.Musgrave OC Oxidation of alkyl aryl ethers. Chem. Rev. 1969, 69, 499–531. [Google Scholar]
- 795.Osa T; Yildiz A; Kuwana T Electrooxidation of Benzene. J. Am. Chem. Soc. 1969, 91, 3994–3995. [Google Scholar]
- 796.Morofuji T; Shimizu A; Yoshida J Electrochemical C–H Amination: Synthesis of Aromatic Primary Amines via N-Arylpyridinium Ions. J. Am. Chem. Soc. 2013, 135, 5000–5003. [DOI] [PubMed] [Google Scholar]
- 797.Morofuji T; Shimizu A; Yoshida J Direct C–N Coupling of Imidazoles with Aromatic and Benzylic Compounds via Electrooxidative C–H Functionalization. J. Am. Chem. Soc. 2014, 136, 4496–4499. [DOI] [PubMed] [Google Scholar]
- 798.Morofuji T; Shimizu A; Yoshida J Heterocyclization Approach for Electrooxidative Coupling of Functional Primary Alkylamines with Aromatics. J. Am. Chem. Soc. 2015, 137, 9816–9819. [DOI] [PubMed] [Google Scholar]
- 799.Romero NA; Margrey KA; Tay NE; Nicewicz DA Site-Selective Arene C-H Amination via Photoredox Catalysis. Science 2015, 349, 1326–1330. [DOI] [PubMed] [Google Scholar]
- 800.Margrey KA; Levens A; Nicewicz DA Direct Aryl C−H Amination with Primary Amines Using Organic Photoredox Catalysis. Angew. Chem. Int. Ed. 2017, 56, 15644–15648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 801.McManus JB; Nicewicz DA Direct C–H Cyanation of Arenes via Organic Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 2880–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 802.Hayrapetyan D; Rit RK; Kratz M; Tschulik K; Gooßen LJ TEMPO-Catalyzed Electrochemical C–H Thiolation: Synthesis of Benzothiazoles and Thiazolopyridines from Thioamides. Chem. Eur. J. 2018, 24, 11288–11291. [DOI] [PubMed] [Google Scholar]
- 803.Lebedev A; Jiao J; Lee J; Yang F; Allison N; Herschman H; Sadeghi S Radiochemistry on Electrodes: Synthesis of an 18F-Labelled and in vivo Stable COX-2 Inhibitor. PLOS ONE 2017, 12, e0176606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 804.Chen W; Huang Z; Tay NES; Giglio B; Wang M; Wang H; Wu Z; Nicewicz DA; Li Z Direct Arene C–H Fluorination with 18F− via Organic Photoredox Catalysis. Science 2019, 364, 1170–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 805.Qian X-Y; Li S-Q; Song J; Xu H-C TEMPO-Catalyzed Electrochemical C–H Thiolation: Synthesis of Benzothiazoles and Thiazolopyridines from Thioamides. ACS Catal. 2017, 7, 2730–2734. [Google Scholar]
- 806.Tabaković I; Trkovnik M; Batušić M; Tabaković K Electrochemical Synthesis of Heterocyclic Compounds; VII1. Anodic Synthesis of 1,3-Thiazole Derivatives. Synthesis 1979, 8, 590–592. [Google Scholar]
- 807.Zhang G; Liu C; Yi H; Meng Q; Bian C; Chen H; Jian J-X; Wu L-Z Lei, A. External Oxidant-Free Oxidative Cross-Coupling: A Photoredox Cobalt-Catalyzed Aromatic C–H Thiolation for Constructing C–S Bonds. J. Am. Chem. Soc. 2015, 137, 9273–9280. [DOI] [PubMed] [Google Scholar]
- 808.Hein SJ; Lehnherr D; Arslan H; Uribe-Romo FJ; Dichtel WR Alkyne Benzannulation Reactions for the Synthesis of Novel Aromatic Architectures. Acc. Chem. Res. 2017, 50, 2776–2788. [DOI] [PubMed] [Google Scholar]
- 809.Senese AD; Chalifoux WA Nanographene and Graphene Nanoribbon Synthesis via Alkyne Benzannulations. Molecules 2019, 24, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 810.Narita A; Wang X-Y; Feng X; Müllen K New Advances in Nanographene Chemistry. Chem. Soc. Rev. 2015, 44, 6616–6643. [DOI] [PubMed] [Google Scholar]
- 811.Jolly A; Miao D; Daigle M; Morin J-F Emerging Bottom-Up Strategies for the Synthesis of Graphene Nanoribbons and Related Structures. Angew. Chem. Int. Ed. 2019, 59, 4624–4633. [DOI] [PubMed] [Google Scholar]
- 812.Lehnherr D; Chen C; Pedramrazi Z; DeBlase CR; Alzola JM; Keresztes I; Lobkovsky EB; Crommie MF; Dichtel WR Sequence-Defined oligo(ortho-Arylene) Foldamers Derived from the Benzannulation of ortho(Arylene Ethynylene)s. Chem. Sci. 2016, 7, 6357–6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 813.Zhai L; Shukla R; Rathore R Oxidative C−C Bond Formation (Scholl Reaction) with DDQ as an Efficient and Easily Recyclable Oxidant. Org. Lett. 2009, 11, 3474–3477. [DOI] [PubMed] [Google Scholar]
- 814.Grzybowski M; Skonieczny K; Butenschön H; Gryko DT Comparison of Oxidative Aromatic Coupling and the Scholl Reaction. Angew. Chem. Int. Ed. 2013, 52, 9900–9930. [DOI] [PubMed] [Google Scholar]
- 815.King BT; Kroulík J; Robertson CR; Rempala P; Hilton CL; Korinek JD; Gortari LM Controlling the Scholl Reaction. J. Org. Chem. 2007, 72, 2279–2288. [DOI] [PubMed] [Google Scholar]
- 816.Crawford PW; Carlos E; Ellegood JC; Cheng CC; Dong Q; Liu DF; Luo YL The Electrochemistry of Antineoplastic Furanquinones: Electrochemical Properties of Benzo[b]naphtho[2,3-d]furan-6,11-dione Derivatives. Electrochim. Acta 1996, 41, 2399–2403. [Google Scholar]
- 817.Mallory FB; Mallory CW Chapter 30: Photocyclization of Stilbenes and Related Molecules. In Organic Reactions; Dauben WG, Ed.; John Wiley & Sons: New York, 1984, 1–456. DOI: 10.1002/0471264180.or030.01. [DOI] [Google Scholar]
- 818.Molloy MS; Snyder JA; Bragg AE Structural and Solvent Control of Nonadiabatic Photochemical Bond Formation: Photocyclization of o-Terphenyl in Solution. J. Phys. Chem. A 2014, 118, 3913–3925. [DOI] [PubMed] [Google Scholar]
- 819.Bushby RJ; Hardy C Regiospecific Synthesis of 2,3,6,7,10,11-Hexasubstituted Triphenylenes by Oxidative Photocyclisation of 3,3",4,4',4",5'-Hexasubstituted 1,1':2'1"-TerphenyIs. J. Chem. Soc., Perkin Trans. 1 1986, 721–723. [Google Scholar]
- 820.Liu L; Yang B; Katz TJ; Poindexter MK J. Org. Chem. 1991, 56, 3769–3775. [Google Scholar]
- 821.Mueller A; Amsharov KY Synthesis of Robust Precursors for the Controlled Fabrication of (6,6), (8,8), (10,10), and (12,12) Armchair Single-Walled Carbon Nanotubes. Eur. J. Org. Chem. 2015, 3053–3056. [Google Scholar]
- 822.Röckl JL; Pollok D; Franke R; Siegfried R Waldvogel, S. R. A Decade of Electrochemical Dehydrogenative C,C-Coupling of Aryls. Acc. Chem. Res. 2020, 53, 45–61. [DOI] [PubMed] [Google Scholar]
- 823.Huang Z; Lumb J-P Phenol-Directed C–H Functionalization. ACS Catal. 2019, 9, 521–555. [Google Scholar]
- 824.Kirste A; Schnakenburg G; Stecker F; Fischer A; Waldvogel SR Anodic Phenol–Arene Cross-Coupling Reaction on Boron-Doped Diamond Electrodes. Angew. Chem. Int. Ed. 2010, 49, 971–975. [DOI] [PubMed] [Google Scholar]
- 825.Kirste A; Schnakenburg G; Waldvogel SR Anodic Coupling of Guaiacol Derivatives on Boron-Doped Diamond Electrodes. Org. Lett. 2011, 13, 3226–2129. [DOI] [PubMed] [Google Scholar]
- 826.Kirste A; Nieger M; Malkowsky IM; Stecker F; Fischer A; Waldvogel SR ortho-Selective Phenol-Coupling Reaction by Anodic Treatment on Boron-Doped Diamond Electrode Using Fluorinated Alcohols. Chem. Eur. J. 2009, 15, 2273–2277. [DOI] [PubMed] [Google Scholar]
- 827.Dahms B; Kohlpaintner PJ; Wiebe A; Breinbauer R; Schollmeyer D; Waldvogel SR Selective Formation of 4,4’-Biphenols by Anodic Dehydrogenative Cross- and Homo-Coupling Reaction. Chem. Eur. J. 2019, 25, 2713–2716. [DOI] [PubMed] [Google Scholar]
- 828.Malkowsky IM; Rommel CE; Fröhlich R; Griesbach U; Putter H; Waldvogel SR Novel Template-Directed Anodic Phenol-Coupling Reaction. Chem. Eur. J. 2006, 12, 7482–7488. [DOI] [PubMed] [Google Scholar]
- 829.Selt M; Franke R; Waldvogel SR Supporting-Electrolyte-Free and Scalable Flow Process for the Electrochemical Synthesis of 3,3′,5,5′-Tetramethyl-2,2′-biphenol. Org. Process Res. Dev. 2020, 24, 2347–2355. [Google Scholar]
- 830.Niederer KA; Gilmartin PH; Kozlowski MC Oxidative Photocatalytic Homo- and Cross-Coupling of Phenols: Nonenzymatic, Catalytic Method for Coupling Tyrosine. ACS Catal. 2020, 10, 14615–14623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 831.Kirste A; Elser B; Schnakenburg G; Waldvogel SR Efficient Anodic and Direct Phenol-Arene C,C Cross-Coupling: The Benign Role of Water or Methanol. J. Am. Chem. Soc. 2012, 134, 3571–3576. [DOI] [PubMed] [Google Scholar]
- 832.Zeng C; Wang B; Zhang H; Sun M; Huang L; Gu Y; Qiu Z; Müllen, Gu C; Ma Y Electrochemical Synthesis, Deposition, and Doping of Polycyclic Aromatic Hydrocarbon Films. J. Am. Chem. Soc. 2021, 143, 2682–2687. [DOI] [PubMed] [Google Scholar]
- 833.Daigle M; Picard-Lafon A; Soligo E; Morin J-F Regioselective Synthesis of Nanographenes by Photochemical Cyclodehydrochlorination. Angew. Chem. Int. Ed. 2016, 55, 2042–2047. [DOI] [PubMed] [Google Scholar]
- 834.Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- 835.For a recent example, see: Hong CM; Xu Y; Chung JYL; Schultz DM; Weisel M; Varsolona RJ; Zhong Y-L; Purohit AK; He CQ; Gauthier DR; et al. Development of a Commercial Manufacturing Route to 2-Fluoroadenine, The Key Unnatural Nucleobase of Islatravir. Org. Process Res. Dev. 2020. 25, 395–404.
- 836.For a review on fluorinated pharmaceuticals in clinical trials, see: Zhou Y; Wang J; Gu Z; Wang S; Zhu W; Aceña JL; Soloshonok VA; Izawa K; Liu H Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518.
- 837.Kirsch P Modern Fluoroorganic Chemistry, 1st ed.; John Wiley & Sons, Ltd: Weinheim, Germany, 2004. [Google Scholar]
- 838.Senaweera SM; Singh A; Weaver JD Photocatalytic Hydrodefluorination: Facile Access to Partially Fluorinated Aromatics. J. Am. Chem. Soc. 2014, 136, 3002–3005. [DOI] [PubMed] [Google Scholar]
- 839.Selivanova GA; Reshetov AV; Beregovaya IV; Vasil’eva NV; Bagryanskaya I. Yu.; Shteingarts VD Hydrodefluorination of Polyfluoro-2-Naphthylamines by Zn in Aqueous NH3: A Correlation of the Product Distribution and the Computationally Predicted Regioselectivity of the Substrate Radical Anion Fragmentation. Journal of Fluorine Chemistry 2012, 137, 64–72. [Google Scholar]
- 840.Singh A; Kubik JJ; Weaver JD Photocatalytic C–F Alkylation; Facile Access to Multifluorinated Arenes. Chem. Sci. 2015, 6, 7206–7212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 841.Senaweera S; Weaver JD Dual C–F, C–H Functionalization via Photocatalysis: Access to Multifluorinated Biaryls. J. Am. Chem. Soc. 2016, 138, 2520–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 842.Singh A; Fennell CJ; Weaver JD Photocatalyst Size Controls Electron and Energy Transfer: Selectable E/Z Isomer Synthesis via C–F Alkenylation. Chem. Sci. 2016, 7, 6796–6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 843.Teegardin KA; Weaver JD Polyfluoroarylation of Oxazolones: Access to Non-Natural Fluorinated Amino Acids. Chem. Commun. 2017, 53, 4771–4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 844.Day JI; Weaver JD Selective and Scalable Perfluoroarylation of Nitroalkanes. J. Org. Chem. 2017, 82, 6801–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 845.Priya S; Weaver JD Prenyl Praxis: A Method for Direct Photocatalytic Defluoroprenylation. J. Am. Chem. Soc. 2018, 140, 16020–16025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 846.Qin J; Zhu S; Chu L Dual Photoredox-/Palladium-Catalyzed Cross-Electrophile Couplings of Polyfluoroarenes with Aryl Halides and Triflates. Organometallics 2021, 40, 2246–2252. [Google Scholar]
- 847.Drakesmith FG Electrochemical Reduction of Fluorinated Aromatic Carboxylic Acids. J. Chem. Soc., Perkin Trans. 1 1972, 184–189. [Google Scholar]
- 848.Kariv-Miller E; Vajtner Z Electroreductive Dehalogenation of Fluorobenzenes. J. Org. Chem. 1985, 50, 1394–1399. [Google Scholar]
- 849.Wu W-B; Li M-L; Huang J-M Electrochemical Hydrodefluorination of Fluoroaromatic Compounds. Tetrahedron Lett. 2015, 56, 1520–1523. [Google Scholar]
- 850.Xie S-L; Gao X-T; Wu H-H; Zhou F; Zhou J Direct Electrochemical Defluorinative Carboxylation of gem-Difluoroalkenes with Carbon Dioxide. Org. Lett. 2020, 22, 8424–8429. [DOI] [PubMed] [Google Scholar]
- 851.Zhu C; Zhang Y-F; Liu Z-Y; Zhou L; Liu H; Feng C Selective C–F Bond Carboxylation of gem-Difluoroalkenes with CO2 by Photoredox/Palladium Dual catalysis. Chem. Sci. 2019, 10, 6721–6726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 852.Metternich JB; Gilmour R One Photocatalyst, n Activation Modes Strategy for Cascade Catalysis: Emulating Coumarin Biosynthesis with (−)-Riboflavin. J. Am. Chem. Soc. 2016, 138, 1040–1045. [DOI] [PubMed] [Google Scholar]
- 853.Zafrani Y; Yeffet D; Sod-Moriah G; Berliner A; Amir D; Marciano D; Gershonov E; Saphier S Difluoromethyl Bioisostere: Examining the “Lipophilic Hydrogen Bond Donor” Concept. J. Med. Chem. 2017, 60, 797–804. [DOI] [PubMed] [Google Scholar]
- 854.Sessler CD; Rahm M; Becker S; Goldberg JM; Wang F; Lippard SJ CF2H, a Hydrogen Bond Donor. J. Am. Chem. Soc. 2017, 139, 9325–9332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 855.Feng Z; Xiao Y-L; Zhang X Transition-Metal (Cu, Pd, Ni)-Catalyzed Difluoroalkylation via Cross-Coupling with Difluoroalkyl Halides. Acc. Chem. Res. 2018, 51, 2264–2278. [DOI] [PubMed] [Google Scholar]
- 856.Meanwell NA Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- 857.Lund H; Jensen NJ; Almqvist S-O; Enzell CR; Taticchi A; Mannervik B Electroörganic Preparations. XXXVI. Stepwise Reduction of Benzotrifluoride. Acta Chem. Scand. 1974, 28b, 263–265. [Google Scholar]
- 858.Fuchigami T Electrochemical Reactions of Fluoro Organic Compounds. In Electrochemistry V; Steckhan E, Ed.; Topics in Current Chemistry; Springer: Berlin, Heidelberg, 1994; pp 1–37. [Google Scholar]
- 859.Andrieux CP; Combellas C; Kanoufi F; Savéant J-M; Thiébault A Dynamics of Bond Breaking in Ion Radicals. Mechanisms and Reactivity in the Reductive Cleavage of Carbon−Fluorine Bonds of Fluoromethylarenes. J. Am. Chem. Soc. 1997, 119, 9527–9540. [Google Scholar]
- 860.Saboureau C; Troupel M; Sibille S; Périchon J Electroreductive Coupling of Trifluoromethylarenes with Electrophiles: Synthetic Applications. J. Chem. Soc., Chem. Commun. 1989, 1138–1139. [Google Scholar]
- 861.Clavel P; Lessene G; Biran C; Bordeau M; Roques N; Trévin S; Montauzon D de. Selective Electrochemical Synthesis and Reactivity of Functional Benzylic Fluorosilylsynthons. J. Fluorine Chem. 2001, 107, 301–310. [Google Scholar]
- 862.Chen K; Berg N; Gschwind R; König B Selective Single C(sp3)–F Bond Cleavage in Trifluoromethylarenes: Merging Visible-Light Catalysis with Lewis Acid Activation. J. Am. Chem. Soc. 2017, 139, 18444–18447. [DOI] [PubMed] [Google Scholar]
- 863.Wang H; Jui NT Catalytic Defluoroalkylation of Trifluoromethylaromatics with Unactivated Alkenes. J. Am. Chem. Soc. 2018, 140, 163–166. [DOI] [PubMed] [Google Scholar]
- 864.Vogt DB; Seath CP; Wang H; Jui NT Selective C–F Functionalization of Unactivated Trifluoromethylarenes. J. Am. Chem. Soc. 2019, 141, 13203–13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 865.Sap JBI; Straathof NJW; Knauber T; Meyer CF; Médebielle M; Buglioni L; Genicot C; Trabanco AA; Noël T; am Ende CW; Gouverneur V Organophotoredox Hydrodefluorination of Trifluoromethylarenes with Translational Applicability to Drug Discovery. J. Am. Chem. Soc. 2020, 142, 9181–9187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 866.Zimmerman HE A Mechanistic Analysis of the Birch Reduction. Acc. Chem. Res. 2012, 45, 164–170. [DOI] [PubMed] [Google Scholar]
- 867.Peters BK, Rodriguez KX, Reisberg SH, Beil SB, Hickey DP, Kawamata Y, Collins M, Starr J, Chen L, Udyavara S, et al. Scalable and Safe Synthetic Organic Electroreduction Inspired by Li-ion Battery chemistry. Science 2019, 363, 838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 868.Chatterjee A; König B Birch-Type Photoreduction of Arenes and Heteroarenes by Sensitized Electron Transfer. Angew. Chem. Int. Ed. 2019, 58, 14289–14294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 869.Cole JP; Chen D-F; Kudisch M; Pearson RM; Lim C-H; Miyake G; Organocatalyzed Birch Reduction Driven by Visible Light. J. Am. Chem. Soc. 2020, 142, 13573–13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 870.Masui M; Hara S; Ueshima T; Kawaguchi T Anodix Oxidation of Compounds Having Benzylic or Allylic Carbon and α-Carbon to Hetero Atom Using N-Hydroxyphthalimide as a Mediator. Chem. Pham. Bull. 1983, 31, 4209–4211. [Google Scholar]
- 871.Masui M; Hosomi K; Tsuchida K, Ozaki S Electrochemical Oxidation of Olefins Using N-Hydroxyphthalimide as a Mediator. Chem. Pharm. Bull. 1985, 33, 4798–4802. [Google Scholar]
- 872.Horn EJ; Rosen BR; Chen Y; Tang J; Chen K; Eastgate MD; Baran PS Scalable and Sustainable Electrochemical Allylic C–H Oxidation. Nature 2016, 533, 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 873.Saito M; Kawamata Y; Meanwell M; Navratil R; Chiodi D; Carlson E; Hu P; Chen L; Udyavara S; Kingston C; et al. N-Ammonium Ylide Mediators for Electrochemical C–H Oxidation. J. Am. Chem. Soc. 2021, 143, 7859–7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 874.Laudadio G; Govaerts S; Wang Y; Ravelli D; Koolman HF; Fagnoni M; Djuric SW; Noël T Selective C(sp3)−H Aerobic Oxidation Enabled by Decatungstate Photocatalysis in Flow. Angew. Chem. Int. Ed. 2018, 57, 4078–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 875.Shultz DM; Lévesque F; DiRocco DA; Reibarkh M; Ji Y; Joyce LA; Dropinski JF; Sheng H; Sherry BD; Davies IW Oxyfunctionalization of the Remote C−H Bonds of Aliphatic Amines by Decatungstate Photocatalysis. Angew. Chem. Int. Ed. 2017, 56, 15274–15278. [DOI] [PubMed] [Google Scholar]
- 876.Feng X; Pi Y; Song Y; Xu Z; Li Z; Lin W Integration of Earth-Abundant Photosensitizers and Catalysts in Metal–Organic Frameworks Enhances Photocatalytic Aerobic Oxidation. ACS Catal. 2021, 11 (3), 1024–1032. [Google Scholar]
- 877.Tateno H; Iguchi S; Miseki Y; Sayama K Photo-Electrochemical C−H Bond Activation of Cyclohexane Using a WO3 Photoanode and Visible Light. Angew. Chem. Int. Ed. 2018, 57, 11238–11241. [DOI] [PubMed] [Google Scholar]
- 878.Nicholas AM de P; Arnold DR Thermochemical Parameters for Organic Radicals and Radical Ions. Part 1. The Estimation of the pKa of Radical Cations Based on Thermochemical Calculations. Can. J. Chem. 1982, 60, 2165–2179. [Google Scholar]
- 879.Oliva M; Coppola GA; Eycken E. V. V. der; Sharma UK Photochemical and Electrochemical Strategies Towards Benzylic C−H Functionalization: A Recent Update. Adv. Synth. Catal. 2021, 363, 1810–1834. [Google Scholar]
- 880.Lee BJ; DeGlopper KS; Yoon TP Angew. Chem. Int. Ed. 2020, 59, 197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 881.Wang H; Liang K; Xiong W; Samanta S; Li W; Lei A Electrochemical Oxidation-Induced Etherification via C(sp3)─H/O─H Cross-Coupling. Sci. Adv. 2020, 6, eaaz0590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 882.Lv S; Zhang G; Chen J; Gao W Electrochemical Dearomatization: Evolution from Chemicals to Traceless Electrons. Adv. Synth. Catal. 2020, 362, 462–477. [Google Scholar]
- 883.Deng X; Rong J; Wang L; Vasdev N; Zhang L; Josephson L; Liang SH Chemistry for Positron Emission Tomography: Recent Advances in 11C-, 18F-, 13N-, and 15O-Labeling Reactions. Angew. Chem. Int. Ed. 2019, 58, 2580–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 884.Bloom S; Knippel JL; Lectka T A photocatalyzed Aliphatic Fluorination. Chem. Sci. 2014, 5, 1175–1178. [Google Scholar]
- 885.Bloom S; McCann M; Lectka T Photocatalyzed Benzylic Fluorination: Shedding “Light” on the Involvement of Electron Transfer. Org. Lett. 2014, 16, 6338–6341. [DOI] [PubMed] [Google Scholar]
- 886.Halperin SD; Fan H; Chang S; Martin RE; Britton R A Convenient Photocatalytic Fluorination of Unactivated C–H Bonds. Angew. Chem. Int. Ed. 2014, 53, 4690–4693. [DOI] [PubMed] [Google Scholar]
- 887.West JG; Bedell TA; Sorensen EJ The Uranyl Cation as a Visible-Light Photocatalyst for C(sp3)−H Fluorination. Angew. Chem. Int. Ed. 2016, 55, 8923–8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 888.Nodwell MB; Yang H; Čolović M; Yuan Z; Merkens H; Martin RE; Bénard F; Schaffer P; Britton R 18F-Fluorination of Unactivated C–H Bonds in Branched Aliphatic Amino Acids: Direct Synthesis of Oncological Positron Emission Tomography Imaging Agents. J. Am. Chem. Soc. 2017, 139, 3595–3598. [DOI] [PubMed] [Google Scholar]
- 889.Yuan Z; Nodwell MB; Yang H; Malik N; Merkens H; Bénard F; Martin RE; Schaffer P; Britton R Site-Selective, Late-Stage C−H 18F-Fluorination on Unprotected Peptides for Positron Emission Tomography Imaging. Angew. Chem. Int. Ed. 2018, 57, 12733–12736. [DOI] [PubMed] [Google Scholar]
- 890.Takahira Y; Chen M; Kawamata Y; Mykhailiuk P; Nakamura H; Peters BK; Reisberg SH; Li C; Chen L; Hoshikawa T; et al. Electrochemical C(sp3)–H Fluorination. Synlett 2019, 30, 1178–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 891.Leibler I; Tekle-Smith MA; Doyle A A General Strategy for C(sp3)–H Functionalization with Nucleophiles Using Methyl Radical as a Hydrogen Atom Abstractor. ChemRxiv 2021. DOI: 10.26434/chemrxiv.13644218.v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 892.Zhang Y; Fitzpatrick NA; Bedre I; Yayla HG; Lall MS; Musacchio P A Photoredox-Catalyzed Approach for Formal Hydride Abstraction to Enable a General Csp3–H Fluorination with HF. ChemRxiv 2021. DOI: 10.26434/chemrxiv.14109797.v1. [DOI] [Google Scholar]
- 893.Chu JCK; Rovis T Amide-Directed Photoredox-catalysed C–C Bond Formation at Unactivated sp3 C–H bonds. Nature, 2016, 539, 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 894.Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Nature 2016, 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 895.For a light-initiated HLF reaction see: Wappes EA; Fosu SC; Chopko TC; Nagib DA Triiodide-Mediated δ-Amination of Secondary C−H Bonds. Angew. Chem. Int. Ed. 2016, 55, 9974–9978.
- 896.Shono T; Matsumura Y; Katoh S; Takeuchi K; Sasaki K; Kamada T; Shimizu R Electroorganic Chemistry. 120. New Patterns of Anodic Oxidation of Amides. Synthesis of α-Amino Aldehyde Acetals and Pyrrolidines from Amines. J. Am. Chem. Soc. 1990, 112, 2368–2372. [Google Scholar]
- 897.Hu X; Zhang G; Bu F; Nie L; Lei A Electrochemical-Oxidation-Induced Site-Selective Intramolecular C(sp3)–H Amination. ACS Catal. 2018, 8, 9370–9375. [Google Scholar]
- 898.Wang F; Stahl SS Merging Photochemistry with Electrochemistry: Functional-Group Tolerant Electrochemical Amination of C(sp3)−H Bonds. Angew. Chem. Int. Ed. 2019, 58, 6385–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 899.For a history of alcohol oxidation via chemical methods with oxoammoniums and nitroxides, see: Bobbitt JM; Brückner C; Merbouh N Oxoammonium- and Nitroxide-Catalyzed Oxidation of Alcohols. In Organic Reactions; Denmark SE, Ed.; Wiley: Hoboken, NJ, 2010; Vol. 74, pp 103–424.
- 900.For a summary of redox potentials for aminoxyl species, see: (a) Reference 212 (b) Reference 227
- 901.Taitt BJ; Bender MT; Choi K-S Impacts of the Regeneration Pathways of the Oxoammonium Cation on Electrochemical Nitroxyl Radical-Mediated Alcohol Oxidation. ACS Catal. 2020, 10, 265–275. [Google Scholar]
- 902.Semmelhack MF; Chou CS; Cortes DA Nitroxyl-Mediated Electrooxidation of Alcohols to Aldehydes and Ketones. J. Am. Chem. Soc. 1983, 105, 4492–4494. [Google Scholar]
- 903.Yang X; Fan Z; Shen Z; Li M Electrocatalytic Synthesis of Nitriles from Aldehydes with Ammonium Acetate as the Nitrogen Source. Electrochim. Acta 2017, 226, 53–59. [Google Scholar]
- 904.Chen Q; Fang C; Shen Z; Li M Electrochemical Synthesis of Nitriles from Aldehydes Using TEMPO as a Mediator. Electrochem. Commun. 2016, 64, 51–55. [Google Scholar]
- 905.Schnatbaum K; Schäfer HJ Electroorganic Synthesis 66: Selective Anodic Oxidation of Carbohydrates Mediated by TEMPO. Synthesis 1999, 1999, 864–872. [Google Scholar]
- 906.Schämann M; Schäfer HJ TEMPO-Mediated Anodic Oxidation of Methyl Glycosides and 1-Methyl and 1-Azido Disaccharides. Eur. J. Org. Chem. 2003, 2003, 351–358. [Google Scholar]
- 907.Rafiee M; Konz ZM; Graaf MD; Koolman HF; Stahl SS Electrochemical Oxidation of Alcohols and Aldehydes to Carboxylic Acids Catalyzed by 4-Acetamido-TEMPO: An Alternative to “Anelli” and “Pinnick” Oxidations. ACS Catal. 2018, 6738–6744. [Google Scholar]
- 908.Rafiee M; Miles KC; Stahl SS Electrocatalytic Alcohol Oxidation with TEMPO and Bicyclic Nitroxyl Derivatives: Driving Force Trumps Steric Effects. J. Am. Chem. Soc. 2015, 137, 14751–14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 909.Shibuya M; Tomizawa M; Suzuki I; Iwabuchi Y 2-Azaadamantane N-Oxyl (AZADO) and 1-Me-AZADO: Highly Efficient Organocatalysts for Oxidation of Alcohols. J. Am. Chem. Soc. 2006, 128, 8412–8413. [DOI] [PubMed] [Google Scholar]
- 910.Nandi J; Ovian JM; Kelly CB; Leadbeater NE Oxidative Functionalisation of Alcohols and Aldehydes via the Merger of Oxoammonium Cations and Photoredox Catalysis. Org. Biomol. Chem. 2017, 15, 8295–8301. [DOI] [PubMed] [Google Scholar]
- 911.Pistritto VA; Paolillo JM; Bisset KA; Leadbeater NE Oxidation of α-Trifluoromethyl and Non-Fluorinated Alcohols via the Merger of Oxoammonium Cations and Photoredox Catalysis. Org. Biomol. Chem. 2018, 16, 4715–4719. [DOI] [PubMed] [Google Scholar]
- 912.Nandi J; Witko ML; Leadbeater NE Combining Oxoammonium Cation Mediated Oxidation and Photoredox Catalysis for the Conversion of Aldehydes into Nitriles. Synlett 2018, 29, 2185–2190. [Google Scholar]
- 913.Nandi J; Hutcheson EL; Leadbeater NE Combining Photoredox Catalysis and Oxoammonium Cations for the Oxidation of Aromatic Alcohols to Carboxylic Acids. Tet. Lett. 2021, 63, 152632. [Google Scholar]
- 914.Perutz RN; Procacci B Photochemistry of Transition Metal Hydrides. Chem. Rev. 2016, 116, 8506–8544. [DOI] [PubMed] [Google Scholar]
- 915.Franke R; Little RD Electrons and Holes as Catalysts in Organic Electrosynthesis. ChemElectroChem 2019, 6, 4373–4382. [Google Scholar]
- 916.Okada Y; Yamaguchi Y; Ozakia A; Chiba K Aromatic “Redox Tag”-Assisted Diels–Alder Reactions by Electrocatalysis. Chem. Sci. 2016, 7, 6387–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 917.Nakayama K; Maeta N; Horiguchi G; Kamiya H; Okada Y Radical Cation Diels–Alder Reactions by TiO2 Photocatalysis. Org. Lett. 2019, 21, 2246–22250. [DOI] [PubMed] [Google Scholar]
- 918.Lin S; Ischay MA; Fry CG; Yoon TP Radical Cation Diels–Alder Cycloadditions by Visible Light Photocatalysis. J. Am. Chem. Soc. 2011, 133, 19350–19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 919.For an early example of a photochemically triggered cycloaddition, see: Mlcoch J; Steckhan E Photochemically Initiated, Electron-Transfer-Catalyzed Diels-Alder Reactions of Electron-Rich Dienophiles. Angew. Chem. Int. Ed. 1985, 24, 412–414.
- 920.For an early example of an electrochemically triggered cycloaddition, see: Mlcoch J; Steckhan E Electrochemically Induced [4+2]-cycloadditions - a Mechanistic Interpretation of the Cation Radical Diels-Alder Reaction Based on Preparative Results. Tetrahedon Lett. 1985, 28, 1081–1084.
- 921.For intramolecular [2+2] cyclizations that proceed with chemical reductants, see: Baik T-G; Luis AL; Wang L-C; Krische MJ A Diastereoselective Metal-Catalyzed [2 + 2] Cycloaddition of Bis-Enones. J. Am. Chem. Soc. 2001, 123, 6716–6717.
- 922.Wang L-C; Jang H-Y; Roh Y; Lynch V; Schultz AJ; Wang X; Krische MJ Diastereoselective Cycloreductions and Cycloadditions Catalyzed by Co(dpm)2-Silane (dpm = 2,2,6,6-Tetramethylheptane-3,5-Dionate): Mechanism and Partitioning of Hydrometallative versus Anion Radical Pathways. J. Am. Chem. Soc. 2002, 124, 9448–9453. [DOI] [PubMed] [Google Scholar]
- 923.Yang J; Felton GAN; Bauld NL; Krische MJ Chemically Induced Anion Radical Cycloadditions: Intramolecular Cyclobutanation of Bis(Enones) via Homogeneous Electron Transfer. J. Am. Chem. Soc. 2004, 126, 1634–1635. [DOI] [PubMed] [Google Scholar]
- 924.Yang J; Cauble DF; Berro AJ; Bauld NL; Krische MJ Anion Radical [2 + 2] Cycloaddition as a Mechanistic Probe: Stoichiometry- and Concentration-Dependent Partitioning of Electron-Transfer and Alkylation Pathways in the Reaction of the Gilman Reagent Me2CuLi·LiI with Bis(Enones). J. Org. Chem. 2004, 69, 7979–7984. [DOI] [PubMed] [Google Scholar]
- 925.Roh Y; Jang H-Y; Lynch V; Bauld NL; Krische MJ Anion Radical Chain Cycloaddition of Tethered Enones: Intramolecular Cyclobutanation and Diels−Alder Cycloaddition. Org. Lett. 2002, 4, 611–613. [DOI] [PubMed] [Google Scholar]
- 926.Ischay MA; Anzovino ME; Du J; Yoon TP Efficient Visible Light Photocatalysis of [2+2] Enone Cycloadditions. J. Am. Chem. Soc. 2008, 130, 12886–12887. [DOI] [PubMed] [Google Scholar]
- 927.Wang Q; Wang Q; Zhang Y; Mohamer YM; Pacheco C; Zheng N; Zare RN; Chen H Electrocatalytic Redox Neutral [3 + 2] Annulation of N-Cyclopropylanilines and Alkenes. Chem. Sci. 2021, 12, 969–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 928.Muller P Glossary of Terms Used in Physical Organic Chemistry (IUPAC Recommendations 1994). Pure Appl. Chem. 1994, 66, 1077–1184. [Google Scholar]
- 929.Cai Y; Wang J; Zhang Y; Li Z; Hu D; Zheng N; Chen H Detection of Fleeting Amine Radical Cations and Elucidation of Chain Processes in Visible-Light-Mediated [3 + 2] Annulation by Online Mass Spectrometric Techniques. J. Am. Chem. Soc. 2017, 139, 12259–12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 930.White DH; Noble A; Booker-Milburn KI; Aggarwal VK Diastereoselective Photoredox-Catalyzed [3 + 2] Cycloadditions of N-Sulfonyl Cyclopropylamines with Electron-Deficient Olefins. Org. Lett. 2021, 23, 3038–3042. [DOI] [PubMed] [Google Scholar]
- 931.Newman MS; Karnes HA The Conversion of Phenols to Thiophenols via Dialkylthiocarbamates. J. Org. Chem. 1966, 31, 3980–3984. [Google Scholar]
- 932.Kwart H; Evans ER The Vapor Phase Rearrangement of Thioncarbonates and Thioncarbamates. J. Org. Chem. 1966, 31, 410–413. [Google Scholar]
- 933.Roesel AF; Ugandi M; Huyen NTT; Májek M; Broese T; Roemelt M; Francke R Electrochemically Catalyzed Newman−Kwart Rearrangement: Mechanism, Structure−Reactivity Relationship, and Parallels to Photoredox Catalysis. J. Org. Chem. 2020, 85, 8029–8044. [DOI] [PubMed] [Google Scholar]
- 934.Francke R; Cericola D; Kötz R; Weingarth D; Waldvogel SR Novel Electrolytes for Electrochemical Double Layer Capacitors Based on 1,1,1,3,3,3-Hexafluoropropan-2-ol. Electrochim. Acta 2012, 62, 372–380. [Google Scholar]
- 935.Perkowski AJ; Cruz CL; Nicewicz DA Ambient-Temperature Newman−Kwart Rearrangement Mediated by Organic Photoredox Catalysis. J. Am. Chem. Soc. 2015, 137, 15684–15687. [DOI] [PubMed] [Google Scholar]
- 936.Cruz CL; Nicewicz DA Mechanistic Investigations into the Cation Radical Newman−Kwart Rearrangement. ACS Catal. 2019, 9, 3926–3935. [Google Scholar]
- 937.Pedersen SK; Ulfkjær A; Newman MN; Yogarasa S; Petersen AU; Sølling TI; Pittelkow M Inverting the Selectivity of the Newman−Kwart Rearrangement via One Electron Oxidation at Room Temperature. J. Org. Chem. 2018, 83, 12000–12006. [DOI] [PubMed] [Google Scholar]
- 938.Gendron T; Pereira R; Abdi HY; Witney TH; Årstad E Iron(II)/Persulfate Mediated Newman–Kwart Rearrangement. Org. Lett. 2020, 22, 274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 939.Boyer C; Bulmus V; Davis TP; Ladmiral V; Liu J; Perrier S Bioapplications of RAFT Polymerization. Chem. Rev. 2009, 109, 5402–5436. [DOI] [PubMed] [Google Scholar]
- 940.Moad G; Rizzardo E; Thang SH Toward Living Radical Polymerization. Acc. Chem. Res. 2008, 41, 1133–1142. [DOI] [PubMed] [Google Scholar]
- 941.Chen M; Zhong M; Johnson JA Light-Controlled Radical Polymerization: Mechanisms, Methods, and Applications. Chem. Rev. 2016, 116, 10167–10211. [DOI] [PubMed] [Google Scholar]
- 942.Xu J; Jung K; Atme A; Shanmugam S; Boyer C A Robust and Versatile Photoinduced Living Polymerization of Conjugated and Unconjugated Monomers and Its Oxygen Tolerance. J. Am. Chem. Soc. 2014, 136, 5508–5519. [DOI] [PubMed] [Google Scholar]
- 943.Note: we use dispersity instead of polydispersity to describe the “numerical attribute of the dispersion of a distribution” as suggested by new IUPAC conventions, see: Stepto RFT Dispersity in Polymer Science (IUPAC Recommendations 2009). Pure Appl. Chem. 2009, 81, 351–353.
- 944.Xu J; Jung K; Boyer C Oxygen Tolerance Study of Photoinduced Electron Transfer–Reversible Addition–Fragmentation Chain Transfer (PET-RAFT) Polymerization Mediated by Ru(bpy)3Cl2. Macromolecules 2014, 47, 4217–4229. [Google Scholar]
- 945.Shanmugam S; Xu J; Boyer C Exploiting Metalloporphyrins for Selective Living Radical Polymerization Tunable over Visible Wavelengths. J. Am. Chem. Soc. 2015, 137, 9174–9185. [DOI] [PubMed] [Google Scholar]
- 946.Xu J; Shanmugam S; Duong HT; Boyer C Organo-Photocatalysts for Photoinduced Electron Transfer-Reversible Addition–Fragmentation Chain Transfer (PET-RAFT) Polymerization. Polym. Chem. 2015, 6, 5615–5624. [Google Scholar]
- 947.Shanmugam S; Xu J; Boyer C Light-Regulated Polymerization under Near-Infrared/Far-Red Irradiation Catalyzed by Bacteriochlorophyll a. Angew. Chem. Int. Ed. 2016, 55, 1036–1040. [DOI] [PubMed] [Google Scholar]
- 948.Shanmugam S; Boyer C Stereo-, Temporal and Chemical Control through Photoactivation of Living Radical Polymerization: Synthesis of Block and Gradient Copolymers. J. Am. Chem. Soc. 2015, 137, 9988–9999. [DOI] [PubMed] [Google Scholar]
- 949.Xu J; Shanmugam S; Fu C; Aguey-Zinsou K-F; Boyer C Selective Photoactivation: From a Single Unit Monomer Insertion Reaction to Controlled Polymer Architectures. J. Am. Chem. Soc. 2016, 138, 3094–3106. [DOI] [PubMed] [Google Scholar]
- 950.Xu J; Fu C; Shanmugam S; Hawker CJ; Moad G; Boyer C Synthesis of Discrete Oligomers by Sequential PET-RAFT Single-Unit Monomer Insertion. Angew. Chem. Int. Ed. 2017, 56, 8376–8383. [DOI] [PubMed] [Google Scholar]
- 951.Bünsow J; Mänz M; Vana P; Johannsmann D Electrochemically Induced RAFT Polymerization of Thermoresponsive Hydrogel Films: Impact on Film Thickness and Surface Morphology. Macromol. Chem. Phys. 2010, 211, 761–767. [Google Scholar]
- 952.Wang Y; Fantin M; Park S; Gottlieb E; Fu L; Matyjaszewski K Electrochemically Mediated Reversible Addition–Fragmentation Chain-Transfer Polymerization. Macromolecules 2017, 50, 7872–7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 953.Lorandi F; Fantin M; Shanmugam S; Wang Y; Isse AA; Gennaro A; Matyjaszewski K Toward Electrochemically Mediated Reversible Addition–Fragmentation Chain-Transfer (eRAFT) Polymerization: Can Propagating Radicals Be Efficiently Electrogenerated from RAFT Agents? Macromolecules 2019, 52, 1479–1488. [Google Scholar]
- 954.Sang W; Xu M; Yan Q Coenzyme-Catalyzed Electro-RAFT Polymerization. ACS Macro Lett. 2017, 6, 1337–1341. [DOI] [PubMed] [Google Scholar]
- 955.Min K; Gao H; Matyjaszewski K Preparation of Homopolymers and Block Copolymers in Miniemulsion by ATRP Using Activators Generated by Electron Transfer (AGET). J. Am. Chem. Soc. 2005, 127, 3825–3830. [DOI] [PubMed] [Google Scholar]
- 956.Magenau AJD; Strandwitz NC; Gennaro A; Matyjaszewski K Electrochemically Mediated Atom Transfer Radical Polymerization. Science 2011, 332, 81–84. [DOI] [PubMed] [Google Scholar]
- 957.Fantin M; Isse AA; Venzo A; Gennaro A; Matyjaszewski K Atom Transfer Radical Polymerization of Methacrylic Acid: A Won Challenge. J. Am. Chem. Soc. 2016, 138, 7216–7219. [DOI] [PubMed] [Google Scholar]
- 958.Chmielarz P; Fantin M; Park S; Isse AA; Gennaro A; Magenau AJD; Sobkowiak A; Matyjaszewski K Electrochemically Mediated Atom Transfer Radical Polymerization (eATRP). Prog. Polym. Sci. 2017, 69, 47–78. [Google Scholar]
- 959.Zhang G; Song IY; Ahn KH; Park T; Choi W Free Radical Polymerization Initiated and Controlled by Visible Light Photocatalysis at Ambient Temperature. Macromolecules 2011, 44, 7594–7599. [Google Scholar]
- 960.Fors BP; Hawker CJ Control of a Living Radical Polymerization of Methacrylates by Light. Angew. Chem. Int. Ed. 2012, 51, 8850–8853. [DOI] [PubMed] [Google Scholar]
- 961.Treat NJ; Fors BP; Kramer JW; Christianson M; Chiu C-Y; Read de Alaniz J; Hawker CJ Controlled Radical Polymerization of Acrylates Regulated by Visible Light. ACS Macro Lett. 2014, 3, 580–584. [DOI] [PubMed] [Google Scholar]
- 962.Treat NJ; Sprafke H; Kramer JW; Clark PG; Barton BE; Read de Alaniz J; Fors BP; Hawker CJ Metal-Free Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2014, 136, 16096–16101. [DOI] [PubMed] [Google Scholar]
- 963.Theriot JC; Lim C-H; Yang H; Ryan MD; Musgrave CB; Miyake GM Organocatalyzed atom transfer radical polymerization driven by visible light. Science 2016, 352, 1082–1086. [DOI] [PubMed] [Google Scholar]
- 964.McCarthy BG; Pearson RM; Lim C-H; Sartor SM; Damrauer NH; Miyake GM Structure–Property Relationships for Tailoring Phenoxazines as Reducing Photoredox Catalysts. J. Am. Chem. Soc. 2018, 140, 5088–5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 965.Pearson RM; Lim C-H; McCarthy BG; Musgrave CB; Miyake GM Organocatalyzed Atom Transfer Radical Polymerization Using N-Aryl Phenoxazines as Photoredox Catalysts. J. Am. Chem. Soc. 2016, 138, 11399–11407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 966.Yang F; Zhu X; Li C; Yang J; Stansbury JW; Nie J Electro-Initiated Cationic Polymerization in the Presence of Potassium Hexafluoroantimonate. RSC Adv. 2014, 4, 22224–22229. [Google Scholar]
- 967.Yang F; Yang J; Zheng K; Stansbury JW; Nie J Electro-Induced Cationic Polymerization of Vinyl Ethers by Using Ionic Liquid 1-Butyl-3-Methylimidazolium Tetrafluoroborate as Initiator. Macromol. Chem. Phys. 2015, 216, 380–385. [Google Scholar]
- 968.Crivello JV The Discovery and Development of Onium Salt Cationic Photoinitiators. J. Polym. Sci., Part A: Polym. Chem. 1999, 37, 4241–4254. [Google Scholar]
- 969.Yagci Y; Durmaz YY; Aydogan B Phenacyl Onium Salt Photoinitiators: Synthesis, Photolysis, and Applications. Chem. Rec. 2007, 7, 78–90. [DOI] [PubMed] [Google Scholar]
- 970.Sangermano M Advances in Cationic Photopolymerization. Pure Appl. Chem. 2012, 84, 2089–2101. [Google Scholar]
- 971.Dumur F; Gigmes D; Fouassier J-P; Lalevée J Organic Electronics: An El Dorado in the Quest of New Photocatalysts for Polymerization Reactions. Acc. Chem. Res. 2016, 49, 1980–1989. [DOI] [PubMed] [Google Scholar]
- 972.Messina MS; Axtell JC; Wang Y; Chong P; Wixtrom AI; Kirlikovali KO; Upton BM; Hunter BM; Shafaat OS; Khan SI; et al. Visible-Light-Induced Olefin Activation Using 3D Aromatic Boron-Rich Cluster Photooxidants. J. Am. Chem. Soc. 2016, 138, 6952–6955. [DOI] [PubMed] [Google Scholar]
- 973.Perkowski AJ; You W; Nicewicz DA Visible Light Photoinitiated Metal-Free Living Cationic Polymerization of 4-Methoxystyrene. J. Am. Chem. Soc. 2015, 137, 7580–7583. [DOI] [PubMed] [Google Scholar]
- 974.Ciftci M; Yoshikawa Y; Yagci Y Living Cationic Polymerization of Vinyl Ethers through a Photoinduced Radical Oxidation/Addition/Deactivation Sequence. Angew. Chem. Int. Ed. 2017, 56, 519–523. [DOI] [PubMed] [Google Scholar]
- 975.Kottisch V; Michaudel Q; Fors BP Cationic Polymerization of Vinyl Ethers Controlled by Visible Light. J. Am. Chem. Soc. 2016, 138, 15535–15538. [DOI] [PubMed] [Google Scholar]
- 976.Michaudel Q; Chauviré T; Kottisch V; Supej MJ; Stawiasz KJ; Shen L; Zipfel WR; Abruña HD; Freed JH; Fors BP Mechanistic Insight into the Photocontrolled Cationic Polymerization of Vinyl Ethers. J. Am. Chem. Soc. 2017, 139, 15530–15538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 977.Peterson B; Lin S; Fors BP Electrochemically Controlled Cationic Polymerization of Vinyl Ethers. J. Am. Chem. Soc. 2018, 140, 2076–2079. [DOI] [PubMed] [Google Scholar]
- 978.Sang W; Yan Q Electro-Controlled Living Cationic Polymerization. Angew. Chem. Int. Ed. 2018, 57, 4907–4911. [DOI] [PubMed] [Google Scholar]
- 979.Schrock RR; Hoveyda AH Molybdenum and Tungsten Imido Alkylidene Complexes as Efficient Olefin-Metathesis Catalysts. Angew. Chem. Int. Ed. 2003, 42, 4592–4633. [DOI] [PubMed] [Google Scholar]
- 980.Ogba OM; Warner NC; O’Leary DJ; Grubbs RH Recent advances in ruthenium-based olefin metathesis. Chem. Soc. Rev. 2018, 47, 4510–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 981.Miura T; Kim S; Kitano Y; Tada M; Chiba K Electrochemical Enol Ether/Olefin Cross-Metathesis in a Lithium Perchlorate/Nitromethane Electrolyte Solution. Angew. Chem. Int. Ed. 2006, 45, 1461–1463. [DOI] [PubMed] [Google Scholar]
- 982.Ogawa KA; Goetz AE; Boydston AJ Metal-Free Ring-Opening Metathesis Polymerization. J. Am. Chem. Soc. 2015, 137, 1400–1403. [DOI] [PubMed] [Google Scholar]
- 983.Goetz AE; Boydston AJ Metal-Free Preparation of Linear and Cross-Linked Polydicyclopentadiene. J. Am. Chem. Soc. 2015, 137, 7572–7575. [DOI] [PubMed] [Google Scholar]
- 984.Gilet M; Mortreux A; Nicole J; Petit F New Efficient Route to Alkene Metathesis Catalysts. Electrogeneration of Catalytically Active Species From Tungsten Hexachloride. J. Chem. Soc., Chem. Commun. 1979, 12, 521–522. [Google Scholar]
- 985.Gilet M; Mortreux A; Folest JC; Petit F Electrochemically Induced Generation of Catalytic Species for Olefin Metathesis From Molybdenum and Tungsten Chlorides. J. Am. Chem. Soc. 1983, 105, 3876–3881. [Google Scholar]
- 986.Ahumada G; Ryu Y; Bielawski CW Potentiostatically Controlled Olefin Metathesis. Organometallics 2020, 39, 1744–1750. [Google Scholar]
- 987.Theunissen C; Ashley MA; Rovis T Visible-Light-Controlled Ruthenium-Catalyzed Olefin Metathesis. J. Am. Chem. Soc. 2019, 141, 6791–6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 988.Kärkäs MD; Matsuura BS; Monos TM; Magallanes G; Stephenson CRJ Transition-metal catalyzed valorization of lignin: the key to a sustainable carbon-neutral future. Org. Biomol. Chem. 2016, 14, 1853–1914. [DOI] [PubMed] [Google Scholar]
- 989.Nguyen JD; Matsuura BS; Stephenson CRJ A Photochemical Strategy for Lignin Degradation at Room Temperature. J. Am. Chem. Soc. 2014, 136, 1218–1221. [DOI] [PubMed] [Google Scholar]
- 990.Kärkäs MD; Bosque I; Matsuura BS; Stephenson CR J. Org. Lett. 2016, 18, 5166–5169. [DOI] [PubMed] [Google Scholar]
- 991.Magallanes G; Kärkäs MD; Bosque I; Lee S; Maldonado S; Stephenson CR J. ACS Catal. 2019, 9, 2252–2260. [Google Scholar]
- 992.Yang C; Kärkäs MD; Magallanes G; Chan K; Stephenson CRJ Organocatalytic Approach to Photochemical Lignin Fragmentation. Org. Lett. 2020, 22, 8082–8085. [DOI] [PubMed] [Google Scholar]
- 993.Bosque I; Magallanes G; Rigoulet M; Kärkäs MD; Stephenson CRJ Redox Catalysis Facilitates Lignin Depolymerization. ACS Cent. Sci. 2017, 3, 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 994.Tay NES; Nicewicz DA Cation Radical Accelerated Nucleophilic Aromatic Substitution via Organic Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 16100–16104. [DOI] [PubMed] [Google Scholar]
- 995.Nguyen ST; Murray PRD; Knowles RR Light-Driven Depolymerization of Native Lignin Enabled by Proton-Coupled Electron Transfer. ACS Catal. 2020, 10, 800–805. [Google Scholar]
- 996.Rafiee M; Alherech M; Karlen SD; Stahl SS Cation Radical Accelerated Nucleophilic Aromatic Substitution via Organic Photoredox Catalysis. Electrochemical Aminoxyl-Mediated Oxidation of Primary Alcohols in Lignin to Carboxylic Acids: Polymer Modification and Depolymerization. J. Am. Chem. Soc. 2019, 141, 15266–15276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 997.Li S; Kim S; Davis AH; Zhuang J; Shuler EW; Willinger D; Lee J-J; Zheng W; Sherman BD; Yoo CG; et al. Photocatalytic Chemoselective C−C Bond Cleavage at Room Temperature in Dye-Sensitized Photoelectrochemical Cells. ACS Catalysis, 2021, 11, 3771–3781. [Google Scholar]
- 998.Mairanovsky VG Electro-Deprotection—Electrochemical Removal of Protecting Groups. Angew. Chem. Int. Ed. 1976, 15, 281–292. [Google Scholar]
- 999.Montenegro MI The Electrochemical Cleavage of Protecting Groups. Electrochim. Acta, 1986, 31, 607–620. [Google Scholar]
- 1000.(a) Luo X; Ma X; Lebreux F; Markó IE; Lam K Chem. Commun. 2018, 54, 9969–9972. [DOI] [PubMed] [Google Scholar]; (b) Lam K; Markó IE Chemoselective Chemical and Electrochemical Deprotections of Aromatic Esters. Org. Lett. 2009, 11, 2752–2755. [DOI] [PubMed] [Google Scholar]
- 1001.Lam K; Markó IE Chemoselective Chemical and Electrochemical Deprotections of Aromatic Esters. Org. Lett. 2009, 11, 2752–2755. [DOI] [PubMed] [Google Scholar]
- 1002.Shi S-H; Liang Y; Jiao N Electrochemical Oxidation Induced Selective C–C Bond Cleavage. Chem. Rev. 2021, 121, 485–505. [DOI] [PubMed] [Google Scholar]
- 1003.Platen M; Steckhan E Mild and Effective Removal of Dithioketal Protecting Groups by Triarylamine Cation Radicals as Homogeneous Electron Transfer Agents. Chem. Ber. 1984, 117, 1679–1694. [Google Scholar]
- 1004.Glotz G; Kappe CO; Cantillo D Electrochemical N-Demethylation of 14-Hydroxy Morphinans: Sustainable Access to Opioid Antagonists. Org. Lett. 2020, 22, 6891–6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1005.Ripper JA; Tiekink ERT; Scammells PJ Photochemical N-Demethylation of Alkaloids. Bioorg. Med. Chem. Lett. 2001, 11, 443–445. [DOI] [PubMed] [Google Scholar]
- 1006.Chen Y; Glotz G; Cantillo D Kappe CO Chem. Eur. J. 2020, 26, 2973–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1007.Bal MK; Banks CE; Jones AJ Metabolism Mimicry: An Electrosynthetic Method for the Selective Deethylation of Tertiary Benzamides. ChemElectroChem 2019, 6, 4284–4291. [Google Scholar]
- 1008.Electrically Driven N(sp2)–C(sp2/3) Bond Cleavage of Sulfonamides. ACS Sustainable Chem. Eng. 2020, 8, 3487–3493. [Google Scholar]
- 1009.Weinreb SM; Epling GA; Comi R; Reitano M Efficacious Cleavage of the Benzyl Ether Protecting Group by Electrochemical Oxidation. J. Org. Chem. 1975, 40, 1356–1358. [Google Scholar]
- 1010.Schmidt W; Steckhan E Mild Oxidative Removal of the p-Methoxybenzyl Ether Protecting Group by Homogeneous Electron Transfer. Angew. Chem. Int. Ed. 1978, 17, 673–674. [Google Scholar]
- 1011.Zeng C.-c.; Zhang N.-t.; Lam CM; Little DR Novel Triarylimidazole Redox Catalysts: Synthesis, Electrochemical Properties, and Applicability to Electrooxidative C–H Activation. Org. Lett. 2012, 14, 1314–1317. [DOI] [PubMed] [Google Scholar]
- 1012.Green RA; Jolley KE; Al-Hadedi AAM; Pletcher D; Harrowven DC; De Frutos O; Mateos C; Klauber DJ; Rincón JA; Brown RCD Electrochemical Deprotection of para-Methoxybenzyl Ethers in a Flow Electrolysis Cell. Org. Lett. 2017, 19, 2050–2053. [DOI] [PubMed] [Google Scholar]
- 1013.Tucker JW; Narayanam JMR; Shaha PS; Stephenson CRJ Oxidative Photoredox Catalysis: Mild and Selective deprotection of PMB Ethers Mediated by Visible Light. Chem. Commun. 2011, 47, 5040–5042. [DOI] [PubMed] [Google Scholar]
- 1014.Ahn DK; Kang YW; Woo SK Oxidative Deprotection of p-Methoxybenzyl Ethers via Metal-Free Photoredox Catalysis. J. Org. Chem. 2019, 84, 3612–3623. [DOI] [PubMed] [Google Scholar]
- 1015.Boyd JW; Schmalzl PW; Miller LL Mechanism of Anodic Cleavage of Benzyl Ethers. J. Am. Chem. 1980, 102, 3856–3862. [Google Scholar]
- 1016.Lacobucci S; Filippova N; d'Alarcao M; Deprotection of p-Methoxyphenyl Pyranosides by Anodic Oxidation. Carbohydr. Res. 1995, 277, 321–325. [DOI] [PubMed] [Google Scholar]
- 1017.Platen M; Steckhan E Oxidative Deblocking of the 4-Methoxybenzyl Thioether Protecting Group: Application to the Directed Synthesis of Poly-Cystinyl Peptides. Liebigs Ann. Chem. 1984, 1563–1576. [Google Scholar]
- 1018.Otake Y; Williams JD; Rincon JA; de Frutos O Mateos, C.; Kappe, C. O. Photochemical Benzylic Bromination in Continuous Flow Using BrCCl3 and Its Application to Telescoped p-Methoxybenzyl Protection. Org. Biomol. Chem. 2019, 17, 1384–1388. [DOI] [PubMed] [Google Scholar]
- 1019.Mairanovsky VG Electro-Deprotection—Electrochemical Removal of Protecting Groups. Angew. Chem. Int. Ed. 1976, 15, 281–292. [Google Scholar]
- 1020.Horner L; Neumann H Studien zum Vorgang der Wasserstoffübertragung, XII: Hydrierende Spaltung von Sulfonen mit Tetramethylammonium als Elektronenüberträger. Chem. Ber. 1965, 98, 1715–1721. [Google Scholar]
- 1021.Horner L; Neumann H Studien zum Vorgang der Wasserstoffübertragung, XIII. Reduktive Spaltung von Säureamiden und Estern mit Tetramethylammonium (TMA). Benzoyl- und Tosylrest als Schutzgruppe bei Peptidsynthesen. Chem. Ber. 1965, 98, 3462–3469. [Google Scholar]
- 1022.Iwasaki T; Matsumoto K; Matsuoka M; Takahashi T; Okumura K Detosylation of N-Tosyl Amino Acids and Peptides by Electrolytic Reduction. Bull. Chem. Soc. Jpn. 1973, 46, 852–855. [Google Scholar]
- 1023.Kossai R; Jeminet G; Simonet J Cathodic Behaviour of p-Toluenesulfonamides in Organic Solvents. Electrochim. Acta 1977, 22, 1395–1402. [Google Scholar]
- 1024.Kossai R; Simonet J; Jeminet G Electrochemical Reduction of Complex Sulfonamides: A Cathodic Synthesis of Aza and Aza-oxa Ligands. Tetrahedron Lett. 1979, 20, 1059–1062. [Google Scholar]
- 1025.Lebouc A; Martigny P; Carlier R; Simonet J La Deprotection Electrochimique des Amines (II) - Problèmes Posés pas la Coupure Cathodique des p.Toluène Sulfonamides. Tetrahedron 1985, 41, 1251–1258. [Google Scholar]
- 1026.Kossai R Catalyse Redox Homogene de Reduction des p-Toluenesulfonamides. Determination des Parametres Cinetiques et Thermodynammiques. Electrochim. Acta 1986, 31, 1643–1651. [Google Scholar]
- 1027.Roemmele RC; Rapoport H Removal of N-arylsulfonyl groups from hydroxy α-amino acids. J. Org. Chem. 1988, 53, 2367–2371. [Google Scholar]
- 1028.Kossai R; Emir B; Simonet J Mousset G The Cathodic Cleavage of Aromatic Sulphonamides: An Elegant Way To Produce Amino Radicals Themselves Fully Characterized by Means of the Spin Marking Technique. J. Electroanal. Chem. Interfacial Electrochem. 1989, 270, 253–260. [Google Scholar]
- 1029.Goulaouic-Dubois C; Guggisberg A; Hesse M Protection of Amines by the Pyridine-2-sulfonyl Group and Its Cleavage under Mild Conditions (SmI2 or Electrolysis). J. Org. Chem. 1995, 60, 5969–5972. [Google Scholar]
- 1030.Electrodic Cleavage of the N–S Bond in N-Tosylcarboxamides. A New Entry to N-Unsubstituted Lactams. J. Chem. Soc. Perkin Trans 1 1992, 2001–2004. [Google Scholar]
- 1031.Khanjiin NA; Hesse M Synthesis of the Macrocyclic Spermidine Alkaloid (±)-(2R*,3R*)-3-Hydroxycelacinnine Helv. Chim. Acta 2001, 84, 1253–1267. [Google Scholar]
- 1032.Coeffard V; Thobie-Gautier C; Beaudet I; Le Grognec E; Quintard J-P Mild Electrochemical Deprotection of N-Phenylsulfonyl N-Substituted Amines Derived from (R)-Phenylglycinol. Eur. J. Org. Chem. 2008, 383–391. [Google Scholar]
- 1033.Coeffard V; Thobie-Gautier C; Beaudet I; Le Grognec E; Quintard J-P Mild Electrochemical Deprotection of N-Phenylsulfonyl N-Substituted Amines Derived from (R)-Phenylglycinol. Eur. J. Org. Chem. 2012, 383–391. [Google Scholar]
- 1034.Viaud P; Coeffard V; Thobie-Gautier C; Beaudet I; Galland N; Quintard J-P; Le Grognec E Electrochemical Cleavage of Sulfonamides: An Efficient and Tunable Strategy to Prevent β-Fragmentation and Epimerization. Org. Lett. 2012, 14, 942–945. [DOI] [PubMed] [Google Scholar]
- 1035.Scialdone O; Belfiore C; Filardo G; Galia A; Sabatino MA; Silvestri G Direct Electrochemical Detosylation of Tetratosylcyclen to Cyclen With Carbon Cathodes. Electrochim. Acta 2005, 51, 598–604. [Google Scholar]
- 1036.Oda K; Ohnuma T; Ban Y A Facile Removal of the Arenesulfonyl Group by Electrochemical Reduction of Sulfonamides in a New Cooperative System of Anthracene and Ascorbic Acid: The Control of Crisscross Annulation. J. Org. Chem. 1984, 49, 953–959. [Google Scholar]
- 1037.Senboku H; Nakahara K; Fukuhara T; Hara S Hg Cathode-Free Electrochemical Detosylation of N,N-Disubstituted p-Toluenesulfonamides: Mild, Efficient, and Selective Removal of N-tosyl group. Tetrahedron Lett. 2010, 51, 435–438. [Google Scholar]
- 1038.Nyasse B; Grehn L; Maia HLS; Monteiro LS; Ragnarsson U 2-Naphthalenesulfonyl as a Tosyl Substitute for Protection of Amino Functions. Cyclic Voltammetry Studies on Model Sulfonamides and Their Preparative Cleavage by Reduction. J. Org. Chem. 1999, 64, 7135–7139. [Google Scholar]
- 1039.Maia HLS Monteiro, L. S.; Sebastião, J. Mild Reductive Cleavage of Tryptophan and Histidine Side-Chain Protecting Groups. Eur. J. Org. Chem. 2001, 1967–1970. [Google Scholar]
- 1040.Xuan J; Li B-J; Feng Z-J; Sun G-D; Ma H-H; Yuan Z-W; Chen J-R; Lu L-Q; Xiao W-J Desulfonylation of Tosyl Amides through Catalytic Photoredox Cleavage of N–S Bond Under Visible-Light Irradiation. Chem. Asian J. 2013, 8, 1090–1094. [DOI] [PubMed] [Google Scholar]
- 1041.Liu Q; Liu Y-X; Song H-J; Wanga Q-M Electron Transfer Photoredox Catalysis: Development of a Photoactivated Reductive Desulfonylation of an Aza-Heteroaromatic Ring. Adv. Synth. Catal. 2020, 362, 3110–3115. [Google Scholar]
- 1042.MacKenzie IA; Wang L; Onuska NPR; Williams OF; Begam K; Moran AM; Dunietz BD; Nicewicz DA Discovery and Characterization of an Acridine Radical Photoreductant. Nature 2020, 580, 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1043.Hasegawa E; Tanaka T; Izumiya N; Kiuchi T; Ooe Y; Iwamoto H; Takizawa S.-y.; Murata S Protocol for Visible-Light-Promoted Desulfonylation Reactions Utilizing Catalytic Benzimidazolium Aryloxide Betaines and Stoichiometric Hydride Donor Reagents. J. Org. Chem. 2020, 85, 4344–4353. [DOI] [PubMed] [Google Scholar]
- 1044.Hasegawa E; Izumiya N; Miura T; Ikoma T; Iwamoto H; Takizawa S; Murata S Benzimidazolium Naphthoxide Betaine Is a Visible Light Promoted Organic Photoredox Catalyst. J. Org. Chem. 2018, 83, 3921–3927. [DOI] [PubMed] [Google Scholar]
- 1045.Hasegawa E; Nagakura Y; Izumiya N; Matsumoto K; Tanaka T; Miura T; Ikoma T; Iwamoto H; Wakamatsu K Visible Light and Hydroxynaphthylbenzimidazoline Promoted Transition-Metal-Catalyst-Free Desulfonylation of N-Sulfonylamides and N-Sulfonylamines. J. Org. Chem. 2018, 83, 10813–10825 [DOI] [PubMed] [Google Scholar]
- 1046.Saboureau C; Troupel M; Sibille S; D'Incan E; Perichon J Electroformylation of Organic Halides in N, N-Dimethylformamide: An Efficient Electrosynthesis of Aldehydes. J. Chem. Soc., Chem. Commun. 1989, 895–896. [Google Scholar]
- 1047.Carelli I; Chiarotto I; Cacchi S; Pace P; Amatore C; Jutand A; Meyer G Electrosynthesis of Aromatic Aldehydes by Palladium-Catalyzed Carbonylation of Aryl Iodides in the Presence of Formic Acid. Eur. J. Org. Chem. 1999, 1471–1473. [Google Scholar]
- 1048.Huang H; Li X; Yu C; Zhang Y; Mariano PS; Wang W Visible-Light-Promoted Nickel- and Organic-Dye-Cocatalyzed Formylation Reaction of Aryl Halides and Triflates and Vinyl Bromides with Diethoxyacetic Acid as a Formyl Equivalent. Angew. Chem. Int. Ed. 2017, 56, 1500–1505. [DOI] [PubMed] [Google Scholar]
- 1049.Nielsen MK; Shields BJ; Liu J Williams MJ; Zacuto MJ; Doyle AG Mild, Redox-Neutral Formylation of Aryl Chlorides Through the Photocatalytic Generation of Chlorine Radicals. Angew. Chem. Int. Ed. 2017, 56, 7191–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1050.Zhao B; Shang R; Cheng W-M; Fu Y Decarboxylative Formylation of Aryl Halides With Glyoxylic Acid by Merging Organophotoredox With Palladium Catalysis. Org. Chem. Front. 2018, 5, 1782–1786. [Google Scholar]
- 1051.Siu T; Yudin AK Practical Olefin Aziridination with a Broad Substrate Scope. J. Am. Chem. Soc. 2002, 124, 530–531. [DOI] [PubMed] [Google Scholar]
- 1052.Siu T; Picard CJ; Yudin AK Development of Electrochemical Processes for Nitrene Generation and Transfer J. Org. Chem. 2005, 70, 932–937. [DOI] [PubMed] [Google Scholar]
- 1053.Chen J; Yan W-Q; Lam CM; Zheng C-C; Hu L-M; Little RD Electrocatalytic Aziridination of Alkenes Mediated by n-Bu4NI: A Radical Pathway. Org. Lett. 2015, 17, 986–989. [DOI] [PubMed] [Google Scholar]
- 1054.Li J; Huang W; Chem J; He L; Cheng X; Li G Electrochemical Aziridination by Alkene Activation Using a Sulfamate as the Nitrogen Source. Angew. Chem. Int. Ed. 2019, 57, 5695–5698. [DOI] [PubMed] [Google Scholar]
- 1055.Yu W-L; Chen J-Q; Wei Y-L; Wang Z-Y; Xu. P.-F. Alkene Functionalization for the Stereospecific Synthesis of Substituted Aziridines by Visible-Light Photoredox Catalysis. Chem. Commun. 2018, 54, 1948–1951. [DOI] [PubMed] [Google Scholar]
- 1056.Elinson MN; Dorofeeva EO; Vereshchagin AN; Nikishi GI Electrochemical Synthesis of Cyclopropanes. Russ. Chem. Rev. 1995, 84, 485. [Google Scholar]
- 1057.Herraiza AG; Suero MG New Alkene Cyclopropanation Reactions Enabled by Photoredox Catalysis via Radical Carbenoids. Synthesis 2019, 51, 2821–2828. [Google Scholar]
- 1058.Corey EJ; Bauld NL; La Londe RT; Casanova J; Kaiser ET Generation of Cationic Carbon by Anodic Oxidation of Carboxylic Acids. J. Am. Chem. Soc. 1960, 82, 2645–2646. [Google Scholar]
- 1059.Bours J; Morton M; Fry AJ Electrochemical Oxidation of Cyclooctatetraene in the Presence of Allyltrimethylsilane. Anodic Trialkylation with Bicyclization. Tetrahedron Letters 2012, 53, 1015–1017. [Google Scholar]
- 1060.Chaussard J; Folest J-C; Nedelec J-Y; Perichon J; Sibille S; Troupel M Use of Sacrificial Anodes in Electrochemical Functionalization of Organic Halides. Synthesis 1990, 369–381. [Google Scholar]
- 1061.Casanova J; Reddy VP Electrochemistry of the Carbon–Halogen Bond. PATAI'S Chemistry of Functional Groups. 10.1002/9780470682531.pat0018 [DOI] [Google Scholar]
- 1062.Périchon J; Gosmini C; Buriez C Electrochemical Generation and Reaction of Zinc Reagents. PATAI'S Chemistry of Functional Groups. 10.1002/9780470682531.pat0380 [DOI] [Google Scholar]
- 1063.Peters DG; Oxidation and Reduction of Halogen-containing Compounds. In Encyclopedia of Electrochemistry: Online. 10.1002/9783527610426.bard080008 [DOI] [Google Scholar]
- 1064.Freund A Ueber Trimethylen. J. Prakt. Chem. 1882, 26, 367–377. [Google Scholar]
- 1065.Rifi MR Electrochemical Preparation of Bicyclobutanes and Other Strained Cycloalkanes. J. Am. Chem. Soc. 1967, 89, 4442–4445. [Google Scholar]
- 1066.Shono T; Matsumura Y; Tsubata K; Sugihara Y Electroorganic Chemistry. Part 58. New Synthesis of Cyclopropanes From 1,3-Dicarbonyl Compounds Utilizing Electroreduction of 1,3-Dimethanesulfonates. J. Org. Chem. 1982, 47, 3090–3094. [Google Scholar]
- 1067.For a minireview, see: Petrosyan VA; Niyazymbetov ME Electrochemical Methods for the Generation of Carbenes and Their Analogues. Russ. Chem. Rev. 1989, 58, 644.
- 1068.Fritz HP; Kornrumpf W Elektrochemische Synthesen, XV. Eine Verbesserte Kathodische Erzeugung von Dichlorcarben. Justus Liebigs Ann. Chem. 1978, 1416–1423. [Google Scholar]
- 1069.Fritz HP; Kornrumpf W Electrochemical Syntheses: Part XVI. Electrochemical Synthesis of Difluorocarbene. J. Electroanal. Chem. Interfacial Electrochem. 1979, 100, 217–223. [Google Scholar]
- 1070.Fritz HP; Kornrumpf W Die Reduktion von CBr2F2 Durch Blei - Ein Neuartiger Weg Zum Difluorcarben / The Reduction of CBr2F2 by Lead -a Novel Pathway to Difluorocarbene. Z. Naturforsch. B 1981, 36, 1375–1380. [Google Scholar]
- 1071.Baizer MM; Chruma JL Electrolytic Reductive Coupling. XXI. Reduction of Organic Halides in the Presence of Electrophiles. J. Org. Chem. 1972, 37, 1951–1960. [Google Scholar]
- 1072.Petrosyan VA; Niyazymbetov ME; Baryshnikova TK Synthesis Based on Electrogenerated Carbenes Communication 1. Some Reactions with the Participation of Dichlorocarbene. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1988, 37, 81–85. [Google Scholar]
- 1073.Durandetti S; Sibille S; Perichon J Electrochemical cyclopropanation of alkenes using dibromomethane and zinc in dichloromethane/DMF mixture. J. Org. Chem. 1991, 56, 3255–3258. [Google Scholar]
- 1074.For a minireview, see: Niyazymbetov ME; Evans DH The Utility of Carbanions and Heteroatom-Anions in Electroorganic Synthesis. Tetrahedron 1993, 49, 9627–9687.
- 1075.Le Menn JC; Sarrazin J; Tallec A Cyclocondensation Reactions from Electrogenerated Halomalonate Carbanions—Compared Reactivity of Bromo and Chloro Anions and Influence of the Supporting-Electrolyte Cation. Electrochim. Acta 1990, 35, 563–566. [Google Scholar]
- 1076.Goumain S; Jubault P; Feasson C; Collignon N A New and Efficient Electrosynthesis of 2-Substituted 1,1-Cyclopropanediylbis(Phosphonates). Synthesis 1999, 1903–1906. [Google Scholar]
- 1077.Duquenne C; Goumain S; Jubault P; Feasson C; Quirion J-C Electrosynthesis of α-Arylated β-Substituted Cyclopropylphosphonates. Synthesis of a Phosphonic Analogue of Minalcipran. Org. Lett. 2000, 2, 453–455. [DOI] [PubMed] [Google Scholar]
- 1078.Petrosyan VA; Vasil’ev AA; Tatarinova VI Electrosynthesis of Cyclopropane Derivatives by a Perkin-Type Reaction. Russ. Chem. Bull. 1994, 43, 84–88. [Google Scholar]
- 1079.Sengmany S; Léonel E; Paugam JP; Nédélec J-Y Cyclopropane Formation by Copper-Catalysed Indirect Electroreductive Coupling of Activated Olefins and Activated α,α,α-Trichloro or Gem-Dichloro Compounds. Synthesis 2002, 0533–0537. [Google Scholar]
- 1080.Oudeyer S; Léonel E; Paugam JP; Nédélec J-Y Copper-Catalyzed Electrosynthesis of 1-Acyl-2,2-Diphenylcyclopropanes and Their Behaviour in Acidic Medium. Tetrahedron 2003, 59, 1073–1081. [Google Scholar]
- 1081.Oudeyer S; Léonel E; Paugam JP; Sulpice-Gaillet C; Nédélec J-Y Formation of Polysubstituted Chlorocyclopropanes from Electrophilic Olefins and Activated Trichloromethyl Compounds. Tetrahedron 2006, 62, 1583–1589. [Google Scholar]
- 1082.Saegusa T; Yonezawa K; Ito Y Synthetic Reactions by Complex Catalysts. XXVII. Cyclopropane Synthesis Using a Copper Carbenoid from Trichloromethyl Compound, Copper and Isonitrile. Synthetic Communications 1972, 2, 431–439. [Google Scholar]
- 1083.Saegusa T; Yonezawa K; Murase I; Konoike T; Tomita S; Ito Y Synthetic Reactions by Complex Catalysts. XXX. Synthesis of Cyclic Compounds by Means of Copper-Isonitrile Complex. Copper(I) Carbenoid Intermediate. J. Org. Chem. 1973, 38, 2319–2328. [Google Scholar]
- 1084.Ito Y; Yonezawa K; Saegusa T Synthetic Reactions by Complex Catalysts. XXXIII. Synthesis of Vinylcyclopropane Derivatives by Copper Isonitrile Complexes. Copper Vinylcarbenoid Intermediates. J. Org. Chem. 1974, 39, 1763–1765. [Google Scholar]
- 1085.Saegusa T; Ito Y Synthesis of Cyclic Compounds via Copper-Isonitrile Complexes. Synthesis 1975, 291–300. [Google Scholar]
- 1086.Sengmany S; Léonel E; Paugam JP; Nédélec J-Y Cyclopropane Formation by Nickel-Catalysed Electroreductive Coupling of Activated Olefins and Unactivated Gem-Dibromo Compounds. Tetrahedron 2002, 58, 271–277. [Google Scholar]
- 1087.Ozaki S; Matsui E; Waku J; Ohmori H Cyclopropanation by Tandem Radical [2 + 1] Cycloaddition Conducted by Nickel Complexes Catalyzed Electroreduction. Tetrahedron Lett. 1997, 38, 2705–2708. [Google Scholar]
- 1088.White DA Syntheses with Electrogenerated Halogens: Part II. Oxidation of Malonate Esters. J. Electrochem. Soc. 1977, 124, 1177. [Google Scholar]
- 1089.Elinson MN; Fedukovich SK; Lindeman SV; Aleksanyan MS; Struchkov Yu. T.; Nikishin GI Electrochemical Cyclization of the Tetramethyl Ester of Propane-1,1,3,3-Tetracarboxylic Acid in the Presence of Salts of Hydrogen Halides. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1989, 38, 1467–1471. [Google Scholar]
- 1090.Okimoto M; Takahashi Y; Kakuchi T Electrooxidative Formation of 1,2-Diaroylcyclopropanes from 1,3-Diaroylpropanes in the Presence of KI. Bull. Chem. Soc. Jpn. 2003, 76, 207–208. [Google Scholar]
- 1091.Nikishin GI; Élinson MN; Fedukovich SK Electrochemical Dehydrotrimerization of Dimethyl Malonate to the Hexamethyl Ester of Cyclopropanehexacarboxylic Acid. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1986, 35, 1749–1749. [Google Scholar]
- 1092.Elinson MN; Lizunova TL; Dekaprilevich MO; Struchkov YT; Nikishin GI Electrochemical Cyclotrimerization of Cyanoacetic Ester Into Trans-1,2,3-Tricyanocyclopropane-1,2,3-Tricarboxylate. Mendeleev Commun. 1993, 3, 192–193. [Google Scholar]
- 1093.Elinson MN; Feducovich SK; Bushuev SG; Zakharenkov AA; Pashchenko DV; Nikishin GI Electrochemical Transformation of Malonate and Alkylidenemalonates into 3-Substituted Cyclopropane-1,1,2,2-Tetracarboxylates. Mendeleev Commun. 1998, 8, 15–16. [Google Scholar]
- 1094.Elinson MN; Feducovich SK; Bushuev SG; Pashchenko DV; Nikishin GI Electrochemical Transformation of Cyanoacetic Ester and Alkylidenecyanoacetic Esters into 3-Substituted 1,2-Dicyanocyclopropane-1,2-Dicarboxylates. Russ. Chem. Bull. 1998, 47, 1133–1136. [Google Scholar]
- 1095.Elinson MN; Feducovich SK; Lizunova TL; Nikishin GI Electrochemical Conversions of Alkyl Alkylidenecyanoacetates into 3-Substituted Dialkyl 1,2-Dicyanocyclopropane-1,2-Dicarboxylates. Russ. Chem. Bull. 1999, 48, 1526–1529. [Google Scholar]
- 1096.Elinson MN; Fedukovich SK; Zakharenkov AA; Nikishin GI Electrochemical Transformations of Alkylidenemalonates into Substituted Cyclopropanecarboxylates. Mendeleev Commun. 1999, 9, 20–22. [Google Scholar]
- 1097.Elinson MN; Feducovich SK; Starikova ZA; Olessova OS; Vereshchagin AN; Nikishin GI Stereoselective Electrochemical Transformation of Alkylidenecyanoacetates and Malonate into (E)-3-Substituted-2-Cyanocyclopropane-1,1,2-Tricarboxylates. Tetrahedron Letters 2000, 41, 4937–4941. [Google Scholar]
- 1098.Elinson MN; Fedukovich SK; Zaimovskaya TA; Vereshchagin AN; Nikishin GI Electrocatalytic Transformation of Malononitrile and Cycloalkylidenemalononitriles into Spirotricyclic and Spirotetracyclic Compounds Containing Cyclopropane and Pyrroline Fragments. Russ. Chem. Bull. 2003, 52, 2241–2246. [Google Scholar]
- 1099.Elinson MN; Fedukovich SK; Vereshchagin AN; Dorofeev AS; Dmitriev DE; Nikishin GI Electrocatalytic Transformation of Malononitrile and Cycloalkylidenemalononitriles into Spirobicyclic and Spirotricyclic Compounds Containing 1,1,2,2-Tetracyanocyclopropane Fragment. Russ. Chem. Bull. 2003, 52, 2235–2240. [Google Scholar]
- 1100.Elinson MN; Feducovich SK; Zaimovskaya TA; Vereshchagin AN; Gorbunov SV; Nikishin GI Electrocatalytic Transformation of Dialkyl Malonates and Arylidene- or Alkylidenemalononitriles into Dialkyl Esters of 3-Substituted 2,2-Dicyanocyclopropane-1,1-Dicarboxylic Acids. Russ. Chem. Bull. 2005, 54, 1593–1598. [Google Scholar]
- 1101.Elinson MN; Feducovich SK; Starikova ZA; Vereshchagin AN; Belyakov PA; Nikishin GI Stereoselective Electrocatalytic Transformation of Malonate and Alkylidenecyanoacetates into (E)-3-Substituted 2-Cyanocyclopropane-1,1,2-Tricarboxylates. Tetrahedron 2006, 62, 3989–3996. [Google Scholar]
- 1102.O. Dorofeeva E; N. Elinson M; N. Vereshchagin A; O. Stepanov N; S. Bushmarinov I; A. Belyakov P; O. Sokolova O; I. Nikishin G Electrocatalysis in MIRC Reaction Strategy: Facile Stereoselective Approach to Medicinally Relevant Spirocyclopropylbarbiturates from Barbituric Acids and Activated Olefins. RSC Adv. 2012, 2, 4444–4452. [Google Scholar]
- 1103.Elinson MN; Feducovich SK; Vereshchagin AN; Gorbunov SV; Belyakov PA; Nikishin GI Electrocatalytic Multicomponent Cyclization of an Aldehyde, Malononitrile and a Malonate into 3-Substituted-2,2-Dicyanocyclopropane-1,1-Dicarboxylate—the First One-Pot Synthesis of a Cyclopropane Ring from Three Different Molecules. Tetrahedron Lett. 2006, 47, 9129–9133. [Google Scholar]
- 1104.Vereshchagin AN; Elinson MN; Dorofeeva EO; Stepanov NO; Zaimovskaya TA; Nikishin GI Electrocatalytic and Chemical Methods in MHIRC Reactions: The First Example of the Multicomponent Assembly of Medicinally Relevant Spirocyclopropylbarbiturates from Three Different Molecules. Tetrahedron 2013, 69, 1945–1952. [Google Scholar]
- 1105.Zhang Y; Qian R; Zheng X; Zeng Y; Sun J; Chen Y; Ding A; Guo H Visible Light Induced Cyclopropanation of Dibromomalonates With Alkenes via Double-SET by Photoredox Catalysis. Chem. Commun. 2015, 51, 54–57. [DOI] [PubMed] [Google Scholar]
- 1106.del Hoyo AM; Herraiz AG; Suero MG A Stereoconvergent Cyclopropanation Reaction of Styrenes. Angew. Chem. Int. Ed. 2017, 56, 1610–1613. [DOI] [PubMed] [Google Scholar]
- 1107.del Hoyo AM; Suero MG Photoredox-Catalyzed Cyclopropanation of Michael Acceptors. Eur. J. Org. Chem. 2017, 2122–2125. [Google Scholar]
- 1108.Sayes M; Benoit G; Charette AB Borocyclopropanation of Styrenes Mediated by UV-light Under Continuous Flow Conditions. Angew. Chem. Int. Ed. 2018, 57, 13514–13518. [DOI] [PubMed] [Google Scholar]
- 1109.Guo T; Zhang L; Liu X; Fang Y; Jin X; Yang Y; Li Y; Chen B; Ouyang M Visible-Light-Promoted Redox-Neutral Cyclopropanation Reactions of α-Substituted Vinylphosphonates and Other Michael Acceptors with Chloromethyl Silicate as Methylene Transfer Reagent. Adv. Synth. Catal. 2018, 360, 4459–4463. [Google Scholar]
- 1110.Luo W; Fang Y; Zhang L; Xu T; Liu Y; Li Y; Jin X; Bao J; Wu X; Zhang Z Bromomethyl Silicate: A Robust Methylene Transfer Reagent for Radical-Polar Crossover Cyclopropanation of Alkenes. Eur. J. Org. Chem. 2020, 1778–1781. [Google Scholar]
- 1111.Davies HML; Antoulinakis EG Intermolecular Metal-Catalyzed Carbenoid Cyclopropanations. In Organic Reactions; American Cancer Society, 2004; pp 1–326. [Google Scholar]
- 1112.Wu W; Lin Z; Jiang H Recent Advances in the Synthesis of Cyclopropanes. Org. Biomol. Chem. 2018, 16, 7315–7329. [DOI] [PubMed] [Google Scholar]
- 1113.Pastor-Pérez L; Wiebe C; Pérez-Prieto J; Stiriba S-E A Tetramethoxybenzophenone as Efficient Triplet Photocatalyst for the Transformation of Diazo Compounds. J. Org. Chem. 2007, 72, 1541–1544. [DOI] [PubMed] [Google Scholar]
- 1114.Pastor-Pérez L; Barriau E; Frey H; Pérez-Prieto J; Stiriba S-E Photocatalysis within Hyperbranched Polyethers with a Benzophenone Core. J. Org. Chem. 2008, 73, 4680–4683. [DOI] [PubMed] [Google Scholar]
- 1115.Sarabia FJ; Ferreira EM Radical Cation Cyclopropanations via Chromium Photooxidative Catalysis. Org. Lett. 2017, 19, 2865–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1116.Li P; Zhao J; Shi L; Wang J; Shi X; Li F Iodine-catalyzed diazo activation to access radical reactivity Nat. Commun. 2018, 9, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1117.Shu C; Mega RS; Andreassen BJ; Noble A; Aggarwal VK Synthesis of Functionalized Cyclopropanes from Carboxylic Acids by a Radical Addition–Polar Cyclization Cascade. Angew. Chem. Int. Ed. 2018, 57, 15430–15434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1118.Milligan JA; Phelan JP; Polites VC; Kelly CB; Molander GA Radical/Polar Annulation Reactions (RPARs) Enable the Modular Construction of Cyclopropanes. Org. Lett. 2018, 20, 6840–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1119.Milligan JA; Burns KL; Le AV; Polites VC; Wang Z-J; Molander GA; Kelly CB Radical-Polar Crossover Annulation: A Platform for Accessing Polycyclic Cyclopropanes. Adv. Synth. Catal. 2020, 362, 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1120.Luo W; Yang Y; Fang Y; Zhang X; Jin X; Zhao G; Zhang L; Li Y; Zhou W; Xia T; et al. Photoredox-Catalyzed Cyclopropanation of 1,1-Disubstituted Alkenes via Radical-Polar Crossover Process. Adv. Synth. Catal. 2019, 361, 4215–4221. [Google Scholar]
- 1121.Jurberg ID; Davies HML Blue Light-Promoted Photolysis of Aryldiazoacetates. Chem. Sci. 2018, 9, 5112–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1122.Herraiz AG; Suero MG A Transition-Metal-Free & Diazo-Free Styrene Cyclopropanation. Chem. Sci. 2019, 10, 9374–9379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1123.Shono T; Kise N Uematsu N; Morimoto S; Okazaki E Electroorganic Chemistry. 124. Electroreductive Intramolecular Coupling of α,ω-Bromoalkyl) β-Keto Esters. J. Org. Chem. 1990, 55, 5037–5041. [Google Scholar]
- 1124.Inokuchi T; Tsuji M; Kawafuchi H; Torii S Indirect Electroreduction of 2-Alkyl-2-(bromomethyl)cycloalkanones with Cobaloxime to Form 3-Alkyl-2-alkenones via 1,2-Acyl Migration. J. Org. Chem. 1991, 56, 5945–5948. [Google Scholar]
- 1125.Petti A; Natho P; Lam, Parson PJ Regioselective Electrochemical Cyclobutanol Ring Expansion to 1-Tetralones. Eur. J. Org. Chem. 2021, 854–858. [Google Scholar]
- 1126.Zhao K; Yamashita K; Carpenter JE; Sherwood TC; Ewing WR; Cheng PTW; Knowles RR Catalytic Ring Expansions of Cyclic Alcohols Enabled by Proton Coupled Electron Transfer. J. Am. Chem. Soc. 2019, 141, 8752–8757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1127.Kang J-C; Tu Y-Q; Dong J-W; Chen C; Zhou J; Ding T-M; Zai J-T; Chen Z-M; Zhang S-Y Electrochemical Semipinacol Rearrangements of Allylic Alcohols: Construction of All-Carbon Quaternary Stereocenters. Org. Lett. 2019, 21, 2536–2540. [DOI] [PubMed] [Google Scholar]
- 1128.Jung HI; Kim Y; Kim DY Electrochemical Trifluoromethylation/Semipinacol Rearrangement Sequences of Alkenyl Alcohols: Synthesis of β-CF3-Substituted Ketones. Org. Biomol. Chem. 2019, 17, 3319–3323. [DOI] [PubMed] [Google Scholar]
- 1129.Guan Z; Wang H; Huang Y; Wang K; Wang S; Lei A Electrochemical Oxidative Aryl(alkyl)trifluoromethylation of Allyl Alcohols via 1,2-Migration. Org. Lett. 2019, 21, 4619–4622. [DOI] [PubMed] [Google Scholar]
- 1130.Sahoo B; Li J -Long, Glorius, F. Visible-Light Photoredox-Catalyzed Semipinacol-Type Rearrangement: Trifluoromethylation/Ring Expansion by a Radical–Polar Mechanism. Angew. Chem. Int. Ed. 2015, 54, 11577–11580. [DOI] [PubMed] [Google Scholar]
- 1131.Byeol S; Kim DY Visible-Light-Induced Photocatalytic Trifluoromethylation/1,2-Carbon Migration Sequences for the Synthesis of CF3)-Substituted Cyclic Ketones. J. Fluor. Chem. 2015, 178, 214–218. [Google Scholar]
- 1132.Kim YJ; Kim DY Electrochemical Radical Arylsulfonylation/Semipinacol Rearrangement Sequences of Alkenylcyclobutanols: Synthesis of β-Sulfonated Cyclic Ketones. Tetrahedron Lett. 2019, 60, 1287–1290. [Google Scholar]
- 1133.Kim YJ; Kim DY Electrochemical Radical Selenylation/1,2-Carbon Migration and Dowd–Beckwith-Type Ring-Expansion Sequences of Alkenylcyclobutanols. Org. Lett. 2019, 21, 1021–1025. [DOI] [PubMed] [Google Scholar]
- 1134.Kim JH; Kim DH Photocatalytic Reductive Sulfonylation and 1,2-Alkyl Migration Cascades of Vinyl Cyclobutanols: A Synthesis of β-Sulfonated Cyclopentanones. Synth. Commun. 2020, 50, 207–216. [Google Scholar]
- 1135.He F-S; Wu Y; Li W; Xia H; Wu J Photoredox-Catalyzed Sulfonylation of Alkenylcyclobutanols with the Insertion of Sulfur Dioxide Through Semipinacol Rearrangement. Org. Chem. Front. 2019, 6, 1873–1878. [Google Scholar]
- 1136.Shu X.-z. Zhang M; He Y; Frei H; Toste FD Dual Visible Light Photoredox and Gold-Catalyzed Arylative Ring Expansion. J. Am. Chem. Soc. 2014, 136, 5844–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1137.Xioa F; Guo Y; Zeng Y-F Recent Developments in Radical Cross-Coupling of Redox-Active Cycloketone Oximes. Adv. Synth. Catal. 2021, 363, 120–143. [Google Scholar]
- 1138.Steiman TJ; Liu J; Mengiste A; Doyle AG Synthesis of β-Phenethylamines via Ni/Photoredox CrossElectrophile Coupling of Aliphatic Aziridines and Aryl Iodides. J. Am. Chem. Soc. 2020, 142, 7598–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1139.Parasram M; Shields BJ; Ahmad O; Knauber T; Doyle AG Regioselective Cross-Electrophile Coupling of Epoxides and (Hetero)aryl Iodides via Ni/Ti/Photoredox Catalysis. ACS Catal. 2020, 10, 5821–5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1140.Sun H; Yang C; Lin R; Xia W Regioselective Ring-Opening Nucleophilic Addition of Aziridines through Photoredox Catalyst. Adv. Synth. Catal. 2014, 356, 2775–2780. [Google Scholar]
- 1141.Yu X-Y; Zhou Q-Q; Wang P-Z; Liao C-M; Chen J-R; Xiao W-J Dual. Photoredox/Nickel-Catalyzed Regioselective Cross-Coupling of 2-Arylaziridines and Potassium Benzyltrifluoroborates: Synthesis of β-Substitued Amines. Org. Lett. 2018, 20, 421–424. [DOI] [PubMed] [Google Scholar]
- 1142.Xu C-H; Li J-H; Xian J-N; Deng W Merging Photoredox/Nickel Catalysis for Cross-Electrophile Coupling of Aziridines with Pyridin-1-ium Salts via Dearomatization. Org. Lett. 2021, DOI: 10.1021/acs.orglett.1c01077. [DOI] [PubMed] [Google Scholar]
- 1143.Larraufie M-H; Pellet R; Fensterbank L; Goddard J-P; Lacôte E; Malacria M; Ollivier C Visible-Light-Induced Photoreductive Generation of Radicals from Epoxides and Aziridines. Angew. Chem. Int. Ed. 2011, 50, 4463–4466. [DOI] [PubMed] [Google Scholar]
- 1144.Hartman RL; McMullen JP; Jensen KF Deciding Whether To Go with the Flow: Evaluating the Merits of Flow Reactors for Synthesis. Angew. Chem. Int. Ed. 2011, 50, 7502–7519. [DOI] [PubMed] [Google Scholar]
- 1145.Amatore C Basic Concepts. In Organic Electrochemistry, 5th Edition; Hammerich O, Speiser B Eds.; CRC Press: New York, 2016, p. 71. [Google Scholar]
- 1146.Mo Y; Lu Z; Rughoobur G; Patil P; Gershenfeld N; Akinwande AI; Buchwald SL; Jensen KF Microfluidic Electrochemistry for Single-Electron Transfer Redox-Neutral Reactions. Science 2020, 368, 1352–1357. [DOI] [PubMed] [Google Scholar]
- 1147.McNally A; Prier CK; MacMillan DWC Discovery of an α-Amino C–H Arylation Reaction Using the Strategy of Accelerated Serendipity. Science 2011, 334, 1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1148.Lan JY; Schuster GB Photoalkylation of Dicyanoarenes with Alkyltriphenylborate Salts. J. Am. Chem. Soc. 1985, 107, 6710–6711. [Google Scholar]
- 1149.Lima F; Kabeshov MA; Tran DN; Battilocchio C; Sedelmeier J; Sedelmeier G; Schenkel B; Ley SV Visible Light Activation of Boronic Esters Enables Efficient Photoredox C(sp2)–C(sp3) Cross-Couplings in Flow. Angew. Chem. Int. Ed. 2016, 55, 14085–14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1150.For an analogous reaction using 4-alkyl-1,4-dihydropyridines, see: Buzzetti L; Prieto A; Roy SR; Melchiorre P Radical-Based C−C Bond-Forming Processes Enabled by the Photoexcitation of 4-Alkyl-1,4-Dihydropyridines. Angew. Chem. Int. Ed. 2017, 56, 15039–15043.
- 1151.Cuthbertson JD; MacMillan DWC The Direct Arylation of Allylic sp3 C–H Bonds via Organic and Photoredox Catalysis. Nature 2015, 519, 74–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1152.Cheng W-M; Shang R; Fu Y Photoredox/Brønsted Acid Co-Catalysis Enabling Decarboxylative Coupling of Amino Acid and Peptide Redox-Active Esters with N-Heteroarenes. ACS Catal. 2017, 7, 907–911. [Google Scholar]
- 1153.Welin ER; Le C; Arias-Rotondo DM; McCusker JK; MacMillan DWC Photosensitized, Energy Transfer-Mediated Organometallic Catalysis Through Electronically Excited Nickel(II). Science 2017, 355, 380–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1154.Shipley JW; Rogers MT The Electrolysis of Some Organic Compounds with Alternating Current. Can. J. Res. 1939, 17b, 147–158. [Google Scholar]
- 1155.Kunkely H; Merz A; Vogler A Alternating Current Electrolysis of Transition-Metal Carbonyl Complexes: Electrochemically Induced Photochemistry. J. Am. Chem. Soc. 1983, 105, 7241–7243. [Google Scholar]
- 1156.Alkire RC; Tsai JE Electrosynthesis of Propylene Oxide with Alternating Current. J. Electrochem. Soc. 1982, 129, 1157–1158. [Google Scholar]
- 1157.Lee B; Naito H; Nagao M; Hibino T Alternating-Current Electrolysis for the Production of Phenol from Benzene. Angew. Chem. Int. Ed. 2012, 51, 6961–6965. [DOI] [PubMed] [Google Scholar]
- 1158.Rodrigo S; Guansekera D; Mahajan JP; Luo L Alternating Current Electrolysis for Organic Synthesis. Curr. Opin. Electrochem. 2021, 28, 100712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1159.Pesco A m.; Cheh HY Mass Transfer in Alternating Current Electrolysis. J. Electrochem. Soc. 1984, 131, 2259–2266. [Google Scholar]
- 1160.Remick AE; Marcus RA Studies on Alternating Current Electrolysis: IV. Mathematical Treatment of Reversible Electron Transfer With Alternating Voltage Control and Distorted Current. J. Electrochem. Soc. 1962, 109, 628–634. [Google Scholar]
- 1161.Blanco DE; Lee B; Modestino MA Optimizing Organic Electrosynthesis Through Controlled Voltage Dosing and Artificial Intelligence. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 17683–17689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1162.Sattler LE; Otten CJ; Hilt G Alternating Current Electrolysis for the Electrocatalytic Synthesis of Mixed Disulfide via Sulfur–Sulfur Bond Metathesis towards Dynamic Disulfide Libraries. Chem. Eur. J. 2020, 26, 3129–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1163.Bortnikov EO; Semenov SN Coupling of Alternating Current to Transition-Metal Catalysis: Examples of Nickel-Catalyzed Cross-Coupling. J. Org. Chem. 2021, 86, 782–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1164.Fedkiw PS; Scott WD Jr. Selectivity Changes in Electrochemical Reaction Sequences by Modulated Potential Control. J. Electrochem. Soc. 1984, 131, 1304. [Google Scholar]
- 1165.Wills AG; Poole DL; Alder CM; Reid M A Mechanistic and Cautionary Case Study on the Use of Alternating Potential in Electrochemical Reactions. ChemElectroChem 2020, 7, 2771–2776. [Google Scholar]
- 1166.Kawamata Y; Hayashi Y; Carlson E; Shaji S; Waldmann D; Simmons BJ; Edwards J; Zapf CW; Saito M; Baran PS Chemoselective Electrosynthesis Using Rapid Alternating Polarity. ChemRxiv 2021. DOI: 10.26434/chemrxiv.14394602.v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1167.Schotten C; Bourne RA; Kapur N; Nguyen BN; Willans CE Electrochemical Generation of N-Heterocyclic Carbenes for Use in Synthesis and Catalysis. Adv. Synth. Catal. 2021, 363, 3189–3200. [Google Scholar]
- 1168.Liu B, Zhang Y, Xu D, Chen W, Chem. Commun. 2011, 47, 2883–2885. [DOI] [PubMed] [Google Scholar]
- 1169.Lake BRM; Bullough EK; Williams TJ; Whitwood AC; Little MA; Willans CE Simple and Versatile Selective Synthesis of Neutral and Cationic Copper(I) N-Heterocyclic Carbene Complexes Using an Electrochemical Procedure. Chem. Commun. 2012, 48, 4887–4889. [DOI] [PubMed] [Google Scholar]
- 1170.Bullough EK; Little MA; Willans CE Electrochemical Synthesis of a Tetradentate Copper N-Heterocyclic Carbene Calix[4]arene and Its Transmetalation to Palladium: Activity of the Palladium Complex in Suzuki–Miyaura Cross-Coupling. Organometallics 2013, 32, 570–577. [Google Scholar]
- 1171.Stephen HR, Schotten C, Nicholls TP, Woodward M, Bourne RA, Kapur N, Willans CE, A Versatile Electrochemical Batch Reactor for Synthetic Organic and Inorganic Transformations and Analytical Electrochemistry. Org. Process Res. Dev. 2020, 24, 1084–1089. [Google Scholar]
- 1172.Galuppo C; Alvarenga J Queiroz AC; Messias I; Nagao R; Abbehausen C The Electrosynthesis of Gold(I) Complexes: A Clean, One-Pot Method. Electrochem. Commun. 2020, 110, 106620. [Google Scholar]
- 1173.Schotten C; Taylor CJ; Bourne RA; Chamberlain TW; Nguyen BN; Kapur N; Willans CE Alternating Polarity for Enhanced Electrochemical Synthesis. React. Chem. Eng. 2021, 6, 147–151. [Google Scholar]
- 1174.Karimian N; Hashemi P; Afkhami A; Bagheri A The Principles of Bipolar Electrochemistry and Its Electroanalysis Applications. Curr. Opin. Electrochem. 2019, 17, 30–37. [Google Scholar]
- 1175.Crooks RM Principles of Bipolar Electrochemistry. ChemElectroChem 2016, 3, 357–359. [Google Scholar]
- 1176.Koefoed L; Pedersen SU; Daasbjerg K Bipolar Electrochemistry—A Wireless Approach for Electrode Reactions. Curr. Opin. Electrochem. 2017, 2, 13–17. [Google Scholar]
- 1177.Gamero-Quijano A; Molina-Osorio AF; Peljo P; Scanlon MD Closed Bipolar Electrochemistry in a Four-Electrode Configuration. Phys. Chem. Chem. Phys. 2019, 21, 9627–9640. [DOI] [PubMed] [Google Scholar]
- 1178.Fosdick SE; Knust KN; Scida K; Crooks RM Bipolar Electrochemistry. Angew. Chem Int. Ed. 2013, 52, 10438–10456. [DOI] [PubMed] [Google Scholar]
- 1179.Shida N; Zhou Y; Inagi S Bipolar Electrochemistry: A Powerful Tool for Electrifying Functional Material Synthesis. Acc. Chem. Res. 2019, 52, 2598–2608. [DOI] [PubMed] [Google Scholar]
- 1180.Plana D; Shul G; Stephenson MJ; Dryfe RAW The Voltammetric Response of Bipolar Cells: Mechanistic Investigations of Electroless Deposition. Electrochem. Commun. 2009, 11, 61–64. [Google Scholar]
- 1181.Manji A; Oloman CW Electrosynthesis of Propylene Oxide in a Bipolar Trickle-Bed Reactor. J. Appl. Electrochem. 1987, 17, 532–544. [Google Scholar]
- 1182.Fosdick SE; Berglund SP; Mullins CB; Crooks RM Parallel Screening of Electrocatalyst Candidates Using Bipolar Electrochemistry. Anal. Chem. 2013, 85, 2493–2499. [DOI] [PubMed] [Google Scholar]
- 1183.Miyamoto K; Nishiyama H; Tomita I; Inagi S Development of a Split Bipolar Electrode System for Electrochemical Fluorination of Triphenylmethane. ChemElectroChem 2019, 6, 97–100. [Google Scholar]
- 1184.Glaser F; Kerzig C; Wenger OS Multi-Photon Excitation in Photoredox Catalysis: Concepts, Applications, Methods. Angew. Chem. Int. Ed. 2020, 59, 10266–10284. [DOI] [PubMed] [Google Scholar]
- 1185.Nguyen JD; D’Amato EM; Narayanam JMR; Stephenson CRJ Engaging Unactivated Alkyl, Alkenyl and Aryl Iodides in Visible-Light-Mediated Free Radical Reactions. Nature Chemistry 2012, 4, 854–859. [DOI] [PubMed] [Google Scholar]
- 1186.Shon J-H; Kim D; Rathnayake MD; Sittel S; Weaver J; Teets TS Photoredox Catalysis on Unactivated Substrates With Strongly Reducing Iridium Photosensitizers. Chem. Sci. 2021, 12, 4069–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1187.Li H; Tang X; Pang JH; Wu X; Yeow EKL; Chiba S Polysulfide Anions as Visible Light Photoredox Catalysts for Aryl Cross-Couplings. J. Am. Chem. Soc. 2021, 143, 481–487. [DOI] [PubMed] [Google Scholar]
- 1188.Ghosh I; Ghoh T; Bardagi JI; König B Reduction of Aryl Halides by Consecutive Visible Light-Induced Electron Transfer Processes. Science 2014, 346, 725–728. [DOI] [PubMed] [Google Scholar]
- 1189.Ghosh I; König B Chromoselective Photocatalysis: Controlled Bond Activation through Light-Color Regulation of Redox Potentials. Angew. Chem. Int. Ed. 2016, 55, 7676–7679. [DOI] [PubMed] [Google Scholar]
- 1190.Kerzig C; Guo X; Wenger OS Unexpected Hydrated Electron Source for Preparative Visible-Light Driven Photoredox Catalysis. J. Am. Chem. Soc. 2019, 141, 2122–2127. [DOI] [PubMed] [Google Scholar]
- 1191.Connell TU; Fraser CL; Czyz ML; Smith ZM; Hayne DJ; Doeven EH; Agugiaro J; Wilson DJD; Adcock JL; Scully AD; et al. The Tandem Photoredox Catalysis Mechanism of [Ir(ppy)2(dtb-bpy)]+ Enabling Access to Energy Demanding Organic Substrates. J. Am. Chem. Soc. 2019, 141, 17646–17658. [DOI] [PubMed] [Google Scholar]
- 1192.Czyz ML; Taylor MS; Horngren TH; Polyzos A Reductive Activation and Hydrofunctionalization of Olefins by Multiphoton Tandem Photoredox Catalysis. ACS Catal. 2021, 11, 5472–5480. [Google Scholar]
- 1193.Barham JP; König B Synthetic Photoelectrochemistry. Angew. Chem. Int. Ed. 2020, 59, 11732–11747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1194.Yu Y; Guo P; Zhong J-S; Yuan Y; Ye K-Y Merging Photochemistry With Electrochemistry in Organic Synthesis. Org. Chem. Front. 2020, 7, 131–135. [Google Scholar]
- 1195.Capaldo L; Quadri LL; Ravelli D Merging Photocatalysis with Electrochemistry: The Dawn of a new Alliance in Organic Synthesis. Angew. Chem. Int. Ed. 2019, 58, 17508–17510. [DOI] [PubMed] [Google Scholar]
- 1196.Sivula K; van de Krol R Semiconducting Materials for Photoelectrochemical Energy Conversion. Nat. Rev. Mater. 2016, 1, 15010. [Google Scholar]
- 1197.Gouda A; Liu T; Byers JC Augustynksi J; Santato C Best Practices in Photoelectrochemistry. J. Power Sources 2021, 482, 228958. [Google Scholar]
- 1198.Zhang L, Liardet L, Luo J; Ren D; Grätzel M; Hu X Photoelectrocatalytic arene C–H amination. Nat. Catal. 2019, 2, 366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1199.Scheffold R; Orlinski RJ Am. Chem. Soc. 1983, 105, 7200–7202. [Google Scholar]
- 1200.Huang H; Strater ZM; Rauch M; Shee J; Sisto TJ; Nuckolls C; Lambert TH Electrophotocatalysis with a Trisaminocyclopropenium Radical Dication. Angew. Chem. Int. Ed. 2019, 58, 13318–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1201.Huang H; Lambert TH Electrophotocatalytic SNAr Reactions of Unactivated Aryl Fluorides at Ambient Temperature and Without Base. Angew. Chem. Int. Ed. 2020, 59, 658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1202.Pistritto VA; Schutzbach-Horton ME; Nicewicz DA Nucleophilic Aromatic Substitution of Unactivated Fluoroarenes Enabled by Organic Photoredox Catalysis. J. Am. Chem. Soc. 2020, 142, 17187–17194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1203.Shi W; Zhang J; Zhao F; Wei W; Liang F; Zhang Y; Zhou S Nucleophilic Aromatic Substitution of Unactivated Aryl Fluorides with Primary Aliphatic Amines by Organic Photoredox Catalysis. Chem. Eur. J. 2020, 26, 14823–14827. [DOI] [PubMed] [Google Scholar]
- 1204.Targos K; Williams OP; Wickens ZK Unveiling Potent Photooxidation Behavior of Catalytic Photoreductants. J. Am. Chem. Soc. 2021, 143, 4125–4132. [DOI] [PubMed] [Google Scholar]
- 1205.Huang H; Lambert TH Electrophotocatalytic C–H Heterofunctionalization of Arenes. Angew. Chem. Int. Ed. 2021, 60, 11163–11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1206.Kim H; Kim H; Lambert T; Lin S Reductive Electrophotocatalysis: Merging Electricity and Light To Achieve Extreme Reduction Potentials. J. Am. Chem. Soc. 2020, 142, 2087–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1207.Laissue PP; Alghamdi RA; Tomancak P; Reynaud EG; Shroff H Assessing Phototoxicity in Live Fluorescence Imaging. Nat. Methods 2017, 14, 657–661. [DOI] [PubMed] [Google Scholar]
- 1208.Wäldchen S; Lehmann J; Klein T; van de Linde S; Sauer M Light-Induced Cell Damage in Live-Cell Super-Resolution Microscopy. Scientific Reports 2015, 5, 15348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1209.For exceptions, see reference 18
- 1210.For a recent study on polynuclear photoredox catalysts with bathochromic absorption shifts, see Cerfontaine S; Wehlin SAM; Elias B; Troian-Gautier L Photostable Polynuclear Ruthenium(II) Photosensitizers Competent for Dehalogenation Photoredox Catalysis at 590 nm. J. Am. Chem. Soc. 2020, 142, 5549–5555.
- 1211.Yanai N; Kimizuka N New Triplet Sensitization Routes for Photon Upconversion: Thermally Activated Delayed Fluorescence Molecules, Inorganic Nanocrystals, and Singlet-to-Triplet Absorption. Acc. Chem. Res. 2017, 50, 2487–2495. [DOI] [PubMed] [Google Scholar]
- 1212.Ravetz BD; Pun AB; Churchill EM; Congreve DN; Rovis T; Campos LM Photoredox Catalysis Using Infrared Light via Triplet Fusion Upconversion. Nature 2019, 565, 343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1213.Singh-Rachford TN; Castellano FN J. Phys. Chem. A. 2008. 112, 3550–3556. [DOI] [PubMed] [Google Scholar]
- 1214.Sommer JR; Yang Y; Sommer JR; Shelton AH; Schanze KS; Xue J; Reynolds JR Chem. Mater. 2011. 23, 5296–5304. [Google Scholar]
- 1215.Deng F; Sommer JR; Myahkostupov M; Schanze KS; Castellano FN Near-IR Phosphorescent Metalloporphyrin as a Photochemical Upconversion Sensitizer. Chem. Commun. 2013, 49, 7406–7408. [DOI] [PubMed] [Google Scholar]
- 1216.Mashraqui SH; Kellogg RM Tetrahedron Lett. 1985, 26, 1453–1456. [Google Scholar]
- 1217.Natarajan P; Kumar N; Sharma M Org. Chem. Front. 2016, 3, 1265–1270. [Google Scholar]
- 1218.Miyake GM; Theriot JC Perylene as an Organic Photocatalyst for the Radical Polymerization of Functionalized Vinyl Monomers through Oxidative Quenching with Alkyl Bromides and Visible Light. Macromolecules 2014, 47, 8255–8261. [Google Scholar]
- 1219.Kerzig C; Wenger OS Sensitized Triplet–Triplet Annihilation Upconversion in Water and Its Application to Photochemical Transformations. Chem. Sci. 2018, 9, 6670–6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1220.Montalti M; Credi A; Prodi L; Gandolfi MT Handbook of Photochemistry, 3rd. ed.; CRC Press: Boca Raton, 2006. DOI: 10.1201/9781420015195. [DOI] [Google Scholar]
- 1221.Pfund B; Steffen DM; Schreier MR; Bertrams M-S; Ye C; Börjesson K; Wenger OS; Kerzig C UV Light Generation and Challenging Photoreactions Enabled by Upconversion in Water. J. Am. Chem. Soc. 2020, 142, 10468–10476. [DOI] [PubMed] [Google Scholar]
- 1222.Whittemore TJ; Xue C; Huang J; Gallucci JC; Turro C Single-Chromophore Single-Molecule Photocatalyst for the Production of Dihydrogen Using Low-Energy Light. Nat. Chem. 2020, 12, 180–185. [DOI] [PubMed] [Google Scholar]
- 1223.Ravetz BD; Tay NES; Joe C; Sezen-Edmonds M; Schmidt MA; Tan Y; Janey JA; Eastgate MD; Rovis T Development of a Platform for Near-Infrared Photoredox Catalysis. ACS Cent. Sci. 2020, 6, 2053–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1224.Mei L; Veleta JM; Gianetti TL Helical Carbenium Ion: A Versatile Organic Photoredox Catalyst for Red-Light-Mediated Reactions. J. Am. Chem. Soc. 2020, 142, 12056–12061. [DOI] [PubMed] [Google Scholar]
- 1225.Smith MB; Michl J Singlet Fission. Chem. Rev. 2010, 110, 6891–6936. [DOI] [PubMed] [Google Scholar]
- 1226.Casanova D Chem. Rev. 2018, 118, 7164–7207. [DOI] [PubMed] [Google Scholar]
- 1227.Miyata K; Conrad-Burton FS; Geyer FL; Zhu X-Y Triplet Pair States in Singlet Fission. Chem. Rev. 2019, 119, 4261–4292. [DOI] [PubMed] [Google Scholar]
- 1228.Tykwinski RR Synthesis of Unsymmetrical Derivatives of Pentacene for Materials Applications. Acc. Chem. Res. 2019, 52, 2056–2069. [DOI] [PubMed] [Google Scholar]
- 1229.Zirzlmeier J; Lehnherr D; Coto PB; Chernick ET; Casillas R; Basel BS; Thoss M; Tykwinski RR; Guldi DM Singlet Fission in Pentacene Dimers. Proc. Natl. Acad. Sci. USA 2015, 112, 5325–5330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1230.Zirzlmeier J; Casillas R; Reddy SR; Coto PB; Lehnherr D; Chernick ET; Papadopoulos I; Thoss M; Tykwinski RR; Guldi DM Solution-Based Intramolecular Singlet Fission in Cross-Conjugated Pentacene Dimers. Nanoscale 2016, 8, 10113–10123. [DOI] [PubMed] [Google Scholar]
- 1231.Hetzer C; Guldi DM; Tykwinski RR Pentacene Dimers as a Critical Tool for the Investigation of Intramolecular Singlet Fission. Chem. Eur. J. 2018, 24, 8245–8257.. [DOI] [PubMed] [Google Scholar]
- 1232.Sanders SN; Pun AB; Parenti KR; Kumarasamy E; Yablon LM; Sfeir MY; Campos LM Understanding the Bound Triplet-Pair State in Singlet Fission. Chem 2019, 5, 1988–2005. [Google Scholar]
- 1233.Sanders SN; Kumarasamy E; Pun AB; Trinh MT; Choi B; Xia J; Taffet EJ; Low JZ; Miller JR; Roy X; et al. Quantitative Intramolecular Singlet Fission in Bipentacenes. J. Am. Chem. Soc. 2015, 137, 8965–8972. [DOI] [PubMed] [Google Scholar]
- 1234.Fuemmeler EG; Sanders SN; Pun AB; Kumarasamy E; Zeng T; Miyata K; Steigerwald ML; Zhu X-Y; Sfeir MY; Campos LM; Ananth N A Direct Mechanism of Ultrafast Intramolecular Singlet Fission in Pentacene Dimers. ACS Cent. Sci. 2016, 2, 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1235.Gromski PS; Granda JM; Cronin L Universal Chemical Synthesis and Discovery with ‘The Chemputer’. Trends Chem. 2020, 2, 4–12. [Google Scholar]
- 1236.Shields BJ; Stevens J; Li J; Parasram M; Damani F; Alvarado JIM; Janey JM; Adams RP; Doyle AG Bayesian Reaction Optimization as a Tool for Chemical Synthesis. Nature 2021, 590, 89–96. [DOI] [PubMed] [Google Scholar]
- 1237.DiLuzio S; Mdluli V; Connell TU; Lewis J; VanBenschoten V; Bernhard S High-Throughput Screening and Automated Data-Driven Analysis of the Triplet Photophysical Properties of Structurally Diverse, Heteroleptic Iridium(III) Complexes. J. Am. Chem. Soc. 2021, 143, 1179–1194. [DOI] [PubMed] [Google Scholar]
- 1238.Lowry MS; Hudson WR; Pascal RA Jr., Bernhard S Accelerated Luminophore Discovery through Combinatorial Synthesis. J. Am. Chem. Soc. 2004, 126, 14129–14135. [DOI] [PubMed] [Google Scholar]
- 1239.Juneau A; Hope TO; Malenfant J; Mesko M; McNeill J Frenette M Method to Predict Reagents in Iridium-Based Photoredox Catalysis. ChemRxiv 2020. DOI: 10.26434/chemrxiv.13058024.v1. [DOI] [Google Scholar]
- 1240.Robinson SG; Sigman MS Integrating Electrochemical and Statistical Analysis Tools for Molecular Design and Mechanistic Understanding. Acc. Chem. Res. 2020, 53, 289–299. [DOI] [PubMed] [Google Scholar]
- 1241.Reid JP; Proctor RSJ; Sigman MS; Phipps RJ Predictive Multivariate Linear Regression Analysis Guides Successful Catalytic Enantioselective Minisci Reactions of Diazines. J. Am. Chem. Soc. 2019, 141, 19178–19185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1242.Sandford C; Fries LR; Ball TE; Minteer SD; Sigman MS Mechanistic Studies Into the Oxidative Addition of Co(I) Complexes: Combining Electroanalytical Techniques with Parameterization J. Am. Chem. Soc. 2019, 141, 18877–18889. [DOI] [PubMed] [Google Scholar]
- 1243.Dörr M; Röckl JL; Rein J; Schollmeyer D; Waldvogel SR Electrochemical C–H Functionalization of (Hetero)Arenes—Optimized by DoE. Chem. Eur. J. 2020, 26, 10195–10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1244.Liu L; Chen W; Liu T; Kong X; Zheng J; Li Y Rational Design of Hydrocarbon-Based Sulfonated Copolymers for Proton Exchange Membranes. J. Mater. Chem. A 2019, 7, 11847–11857. [Google Scholar]
- 1245.Min K; Choi B; Park K; Cho E Machine Learning Assisted Optimization of Electrochemical Properties for Ni-Rich Cathode Materials. Sci Rep. 2018, 8, 15778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1246.Whitacre JF; Mitchell J; Dave A; Wu W; Burke S Viswanathan, V. An Autonomous Electrochemical Test Stand for Machine Learning Informed Electrolyte Optimization. J. Electrochem. Soc. 2019, 166, A4181. [Google Scholar]
- 1247.Chen Y; Tian B; Cheng Z; Li X; Huang M; Sun Y; Liu S; Cheng X; Li S; Ding M Electro-Descriptors for the Performance Prediction of Electroorganic Synthesis. Angew. Chem. Int. Ed. 2021, 60, 4199–4207. [DOI] [PubMed] [Google Scholar]
- 1248.Demissie TB; Rudd K; Hansen JH DFT as a Powerful Predictive Tool in Photoredox Catalysis: Redox Potentials and Mechanistic Analysis. Organometallics 2015, 34, 4218–4228. [Google Scholar]
- 1249.Strieth-Kalthoff F; Glorius F Triplet Energy Transfer Photocatalysis: Unlocking the Next Level. Chem 2020, 6, 1888–1903. [Google Scholar]
- 1250.Litman ZC; Wang Y; Zhao H; Hartwig JF Cooperative Asymmetric Reactions Combining Photocatalysis and Enzymatic Catalysis. Nature 2018, 560, 355–359. [DOI] [PubMed] [Google Scholar]
- 1251.Molloy JJ; Schafer M; Wienhold M; Morack T; Daniliuc CG; Gilmore R Science 2020, 369, 302–306. [DOI] [PubMed] [Google Scholar]
- 1252.Hölzl-Hobmeier A, Bauer A, Silva AV, Huber SM, Bannwarth C, Bach T Catalytic Deracemization of Chiral Allenes by Sensitized Excitation With Visible Light. Nature 2018, 564, 240–243. [DOI] [PubMed] [Google Scholar]
- 1253.Scholtz SO; Farney EP; Kim S; Bates DM; Yoon TP Spin-Selective Generation of Triplet Nitrenes: Olefin Aziridination through Visible-Light Photosensitization of Azidoformates. Angew. Chem. Int. Ed. 2016, 55, 2239–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1254.Parasram M, and Gevorgyan V Visible Light-Induced Transition Metal-Catalyzed Transformations: Beyond Conventional Photosensitizers. Chem. Soc. Rev. 2017, 46, 6227–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1255.Yoo SJ; Li L-J; Zeng C-C; Little DR Polymeric Ionic Liquid and Carbon Black Composite as a Reusable Supporting Electrolyte: Modification of the Electrode Surface. Angew. Chem. Int. Ed. 2015, 54, 3744–3747. [DOI] [PubMed] [Google Scholar]
- 1256.Artz J; Müller TE; Thenert K Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [DOI] [PubMed] [Google Scholar]
- 1257.Wei X; Li Y; Chen L; Shi J Formic Acid Electro-Synthesis by Concurrent Cathodic CO2 Reduction and Anodic CH3OH Oxidation. Angew. Chem. Int. Ed. 2021, 60, 3148–3155. [DOI] [PubMed] [Google Scholar]
- 1258.Mikkelsen M; Jørgensen M; Krebs FC The Teraton Challenge. A Review of Fixation and Transformation of Carbon Dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar]
- 1259.Liu Q; Wu L; Jackstell R; Beller M Using Carbon Dioxide as a Building Block in Organic Synthesis. Nat. Commun. 2016, 6, 5933. [DOI] [PubMed] [Google Scholar]
- 1260.Kanan MK; Nocera DG In Situ Formation of an Oxygen-Evolving Catalyst in Neutral Water Containing Phosphate and Co2+. Science, 2008, 321, 1072–1075. [DOI] [PubMed] [Google Scholar]
- 1261.Zou XX; Zhang YN Noble Metal-Free Hydrogen Evolution Catalysts for Water Splitting. Chem. Soc. Rev. 2015, 44, 5148–5180. [DOI] [PubMed] [Google Scholar]
- 1262.Jin HY; Guo CX; Liu X; Liu JL; Vasileff A; Jiao Y; Zheng Y; Qiao SZ Emerging Two-Dimensional Nanomaterials for Electrocatalysis. Chem. Rev. 2018, 118, 6337–6408. [DOI] [PubMed] [Google Scholar]
- 1263.Song J; Wei C; Huang Z-F; Liu C; Zeng L; Wang X; Xu ZJ A Review on Fundamentals for Designing Oxygen Evolution Electrocatalysts. Chem. Soc. Rev. 2020, 49, 2196–2214. [DOI] [PubMed] [Google Scholar]
- 1264.Jiao Y; Zheng Y; Jaroniec M; Qiao SZ Design of Electrocatalysts for Oxygen- and Hydrogen-Involving Energy Conversion Reactions. Chem. Soc. Rev. 2015, 44, 2060–2086. [DOI] [PubMed] [Google Scholar]
- 1265.Spitler MT; Modestino MA; Deutsch TG; Xiang CX; Durrant JR; Esposito DV; Haussener S; Maldonado S; Sharp ID; Parkinson BA; et al. Practical Challenges in the Development of Photoelectrochemical Solar Fuels Production. Sustain. Energy Fuels 2020, 4, 985–995. [Google Scholar]
- 1266.Neri G; Forser M; Cowan AJ; Photochemical Energy Storage. In Energy Storage Option and Their Environmental Impact, Hester RE, Harrison RM, Eds.; Royal Society of Chemistry: London, 2019, pp.184–209. [Google Scholar]
- 1267.(a) DuBois DL Development of Molecular Electrocatalysts for Energy Storage. Inorg. Chem. 2014, 53, 3935–3960. [DOI] [PubMed] [Google Scholar]; (b) Concepcion JJ; Jurss JW; Brennaman MK; Hoertz PG; Patrocinio AOT; Iha NYM; Templeton JL; Meyer TJ Making Oxygen with Ruthenium Complexes. Acc. Chem. Res. 2009, 42, 1954–1965. [DOI] [PubMed] [Google Scholar]
- 1268.Koohi-Fayegh S; Rosen MA A Review of Energy Storage Types, Applications and Recent Developments. Journal of Energy Storage 2020, 27, 101047. [Google Scholar]
- 1269.Kwabi DG; Ji Y; Aziz MJ Electrolyte Lifetime in Aqueous Organic Redox Flow Batteries: A Critical Review. Chem. Rev. 2020, 120, 6467–6489. [DOI] [PubMed] [Google Scholar]
- 1270.Electrochemistry for the Environment; Comninellis C, Guohua C, Eds.; Springer: New York, 2010. [Google Scholar]
- 1271.Deng Y; Zhu X; Chen N; Feng C; Wang H; Kuang P; Hu W Review on Electrochemical System for Landfill Leachate Treatment: Performance, Mechanism, Application, Shortcoming, and Improvement Scheme. Sci. Total Environ. 2020, 745, 140768. [DOI] [PubMed] [Google Scholar]
- 1272.Deng Y; Englehardt JD; Electrochemical Oxidation for Landfill Leachate Treatment. Waste Manag. 2007, 27, 380–388. [DOI] [PubMed] [Google Scholar]
- 1273.Sharma A; Ahmad J; Flora SJS Application of Advanced Oxidation Processes and Toxicity Assessment of Transformation Products. Environ. Res. 2018, 167, 223–233. [DOI] [PubMed] [Google Scholar]
- 1274.Curto D; Franzitta V; Guercio A A Review of the Water Desalination Technologies. Appl. Sci. 2021. 11, 670. [Google Scholar]
- 1275.Knust KN; Hlushkou D; Anand RK; Tallarek U; Crooks RM Electrochemically Mediated Seawater Desalination. Angew. Chem. Int. Ed. 2013, 52, 8107–8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1276.Witt S; Rancis N; Maldonado V Electrochemical Destruction of 'Forever Chemicals': The Right Solution at the Right Time. Electrochem. Soc. Interface 2020, 29, 73–75. [Google Scholar]