

Abstract
Conformational dynamics contribute importantly to enzyme catalysis, such that targeted conformational constraint may affect catalysis. Firefly luciferases undergo extensive structural change during catalysis; key residues form a hydrophobic pocket, excluding water and enabling maximally energetic light production. Point mutants almost always luminesce at longer wavelengths (lower energy) than the wild type. Conformational constraint, using dipeptide analogue 3 at a position critical for optimized excited state structure, produced luciferase emission at a shorter wavelength by ~10 nm. In comparison, introduction of conformationally constrained analogues 4, 5, or 7 afforded luciferases emitting at longer wavelengths, while a related unconstrained luciferase (analogue 6) exhibited wild-type emission. The constrained luciferases tested were more stable than the wild type. Protein modeling demonstrated that the “inside” or “outside” orientation of the conformationally constrained dipeptide led to the shorter or longer emission wavelength, respectively. More broadly, these results suggest that local conformational constraint can control specific elements of enzyme behavior, both in vitro and in vivo. This represents the first example of studying enzyme function by introducing conformationally constrained dipeptides at a specific protein position. The principles discovered here in luciferase modification will enable studies to control the wavelength emission and photophysical properties of modified luciferases.
Keywords: genetic code reprogramming, modified ribosomes, conformational constraint, luciferase bioluminescence and stability, protein modeling
Graphical Abstract
Introduction
At present bioluminescence, especially in the context of luciferase-based reporter assays, is employed broadly. Applications include measuring the infectivity of coronaviruses, detecting protein–protein interactions, high-throughput screening in drug discovery, and bioluminescence resonance energy transfer.1,2 Firefly luciferase produces bioluminescence by a stepwise process1–6 that includes the oxygenation of its luciferin substrate in the presence of O2 and adenosine 5′-triphosphate (ATP), affording a dioxetanone intermediate. Cleavage of the dioxetanone results in an excited state of oxyluciferin that emits visible light (Supporting Information Figure S1).3–7 Interestingly, different firefly species emit light of diverse wavelengths, although all are believed to use the same basic chemistry in doing so. Several mechanisms have been suggested for the differences in emission wavelength, although the actual source(s) is an issue that has persisted for some decades without definitive resolution.7–16
Modified luciferins and engineered luciferases have improved the properties and utility of the luciferases. Nonetheless, further improvements in stability and cell compatibility are still required. Several point mutants of Luciola cruciata luciferase emitted over a range of >50 nm, suggesting that the color differences could be due to changes in the tertiary structure of this luciferase.7,13,14 Notably, the mutant luciferases virtually always emitted at longer wavelengths (lower energy) than the wild type, consistent with the suggestion that the native enzymes have been optimized through evolution.7,13,14,17,18 The development of new strategies for luciferase synthesis that alter emission wavelength and stability should further expand the scope of their applications.
In recent years, genetic code expansion19–21 has emerged as a powerful tool for protein elaboration with a large number of noncanonical amino acids, enabling the rapid development of several areas, such as cell therapy,22 immunosuppression enhancement,23 and in cellulo protein labeling with strongly fluorescent amino acids.24 Biologically active peptides have long been modified with conformationally constrained motifs to improve their binding affinity and metabolic stability.25–27 In comparison, there has been no reported study of enzyme function by incorporating a conformationally constrained dipeptide at a specific protein position. Our laboratory has developed a dual selection procedure to obtain a set of modified ribosomes able to carry out protein synthesis with a variety of noncanonical amino acids, including conformationally constrained dipeptides.28–30 We employed this technique to prepare analogues of firefly luciferase with conformationally constrained dipeptides at a position known to be critical for light emission.
The crystal structure of Photinus pyralis luciferase features a large N-terminal domain comprising ~430 amino acids and is connected to a smaller C-terminal domain of ~100 amino acids.31 Based on structural and mechanistic similarities with acyl-CoA ligases and peptide synthetases within the same superfamily, the putative active site residues of the luciferase were identified as groups of conserved residues on the surfaces of the N- and C-terminal domains. These residues faced each other, across the large cleft between the domains, suggesting that the domains might come together during light production, excluding water from the active site, and thus decreasing quenching of the excited state product.31 A subsequent crystallographic study employed L. cruciata luciferase, bound either to AMP + oxyluciferin or to the high energy intermediate 5′-O-[N-(dehydroluciferyl)sulfamoyl]adenosine (DLSA).14 The latter complex was characterized by much closer association with both Cα and the side chain of Ile288 and was linked to an altered H-bond network involving the side chain of Ser286. This resulted in a closed form of the luciferase–DLSA complex, in which the DLSA benzothia-zole ring was tightly contained within a hydrophobic pocket formed in part by the Ile288 side chain. Comparatively, the structure of the luciferase–DLSA complex involving a S286N mutation afforded a complex in which the Ile288 side chain in the DLSA complex was essentially unchanged from that of wild-type luciferase in the presence of AMP + oxyluciferin, indicating a less rigid and hydrophobic open-form complex.
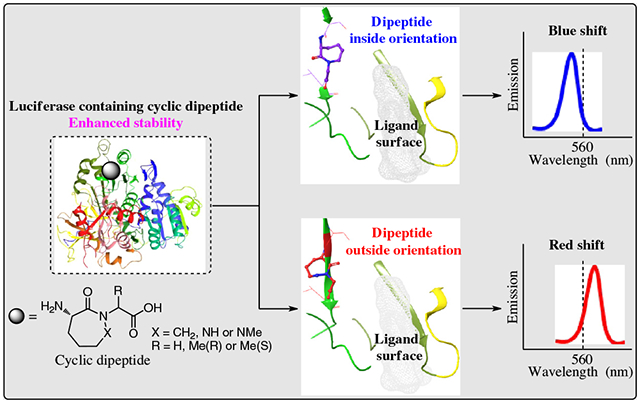
Presently, we have focused on alterations of the region of firefly luciferase suggested to be important for defining the geometry of the closed form of luciferase during production of bioluminescence (residues 286–288 for L. cruciata luciferase, corresponding to residues 284–286 for P. pyralis luciferase). It seemed logical to think that if this region of firefly luciferase was responsible for defining the geometry of the closed form of the light-producing complex in luciferases, both luciferases should be altered similarly when analogous point mutations were introduced. Experimentally, we altered the luciferase region of interest with conformationally constrained cyclic dipeptides and observed the effects on bioluminescence emission wavelength and other properties (Figure 1). Additionally, the modified luciferases were modeled on a putative active site structure to facilitate a better understanding of the experimental results.
Figure 1 |.
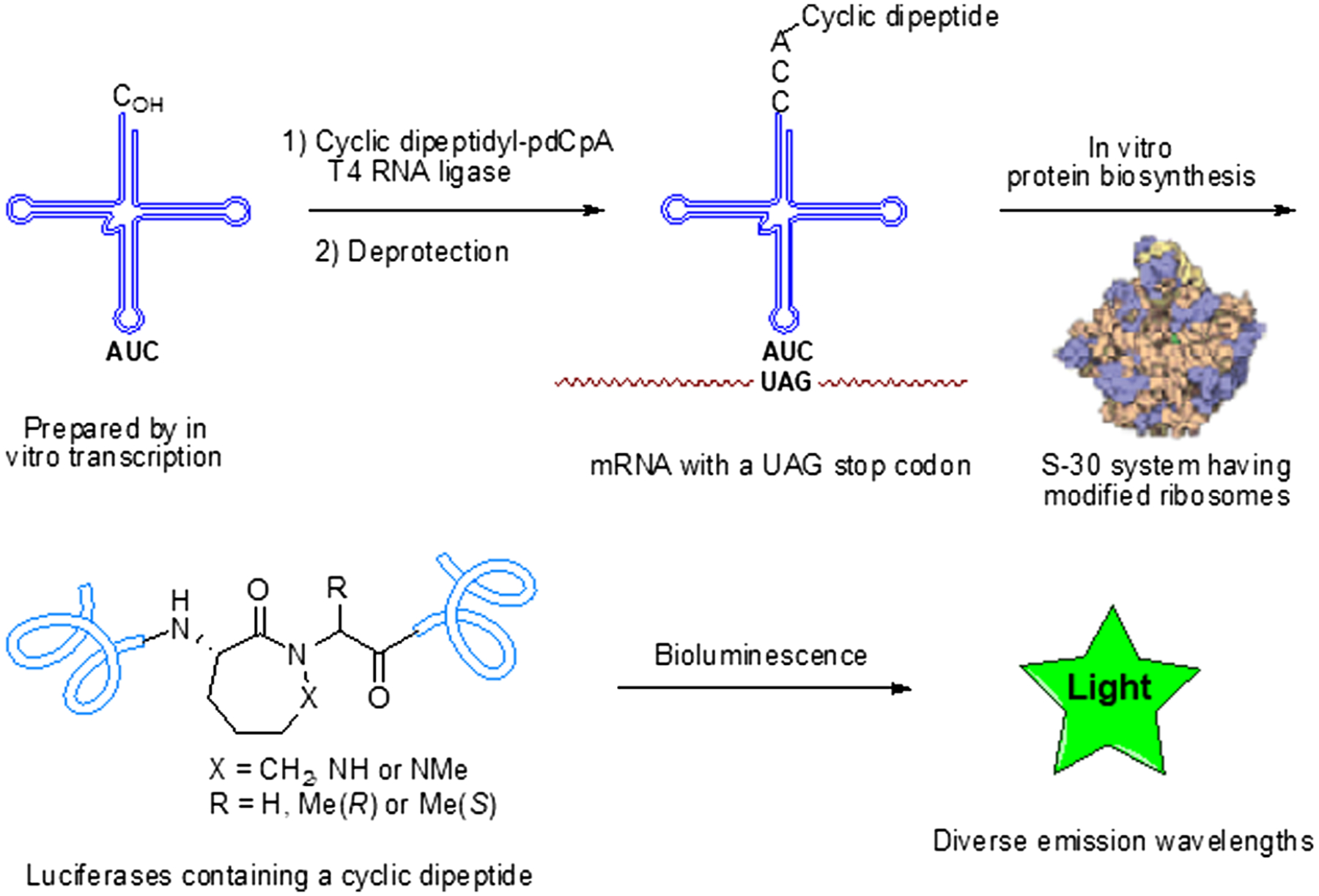
In vitro biosynthesis of firefly luciferase containing conformationally constrained dipeptides. The activated suppressor tRNA was prepared by T4 RNA ligase-mediated condensation of a dipeptidyl-pdCpA derivative with an abbreviated suppressor tRNA transcript (tRNA-COH). As wild-type ribosomes do not utilize dipeptidyl-tRNAs, ribosomes modified in their 23S ribosomal RNA were prepared, and clone 010328R2 was found to be capable of incorporating dipeptides into proteins.29,32 The several modified luciferases containing these cyclic dipeptides exhibited a variety of bioluminescence emission spectra.
Experimental Methods
Preparation of suppressor tRNAs activated with cyclic dipeptides 1–5, 2R-7, and 2S-7
The activation of suppressor tRNACUA-COH in vitro by cyclic dipeptides 1–5, 2R-7, and 2S-7 was carried out essentially as described previously for single amino acids.17,18 Briefly, a 100-μL reaction mixture (total volume) in 100 mM Na Hepes, pH 7.5, contained 1 mM ATP, 15 mM MgCl2, 100 μg of suppressor tRNACUA-COH, 0.5 A260 unit of N-pentenoyl-protected cyclic dipeptidyl-pdCpA, 15% dimethyl sulfoxide (DMSO), and 100 units of T4 RNA ligase. (We have determined that 1 mg of salt-free aminoacyl-pdCpA in 100 mL of DMSO affords a value of ~0.1 A260 unit/mL.) The reaction mixture was incubated at 37 °C for 2 h, and the reaction was quenched by the addition of 0.1 volume of 3 M NaOAc, pH 5.2, and precipitated with 3 volumes of cold ethanol. The pellet obtained after centrifugation (15,000 × g, 15 min, 4 °C) was dissolved in 80 μL of autoclaved water. The efficiency of ligation was estimated by 8% polyacrylamide-7 M urea gel electrophoresis, which was run for ~2–3 h in 0.1 M NaOAc buffer, pH 5.2. The gel was stained with 0.25% methylene blue, which was dissolved in 10% aqueous acetic acid. The N-pentenoyl-protected cyclic dipeptidyl-tRNACUAs were deprotected by treatment with 5 mM aqueous I2 at 25 °C for 15 min. The solution was centrifuged. The supernatant was adjusted to 0.3 M NaOAc and then treated with 3 volumes of cold EtOH to precipitate the cyclic dipeptidyl-tRNACUA, which was isolated by centrifugation (15,000 × g, 15 min, 4 °C). The pellet of activated tRNA was washed with 70% ethanol, air-dried, and dissolved in 20 μL of RNase-free water.
Preparation of S-30 extracts from cells having modified ribosomes
Ten-microliter aliquots from glycerol stocks of BL-21 (DE-3) cells, harboring plasmids with a modified rrnB gene (010328R2) selected previously,29,32 were placed on LB agar supplemented with 100 μg/mL of ampicillin and grown at 37 °C for 16–18 h. A single colony was picked and transferred into 3 mL of LB medium supplemented with 100 μg/mL ampicillin and grown at 37 °C for 3 h in a thermostatic shaker until OD600 was ~0.3. This culture was then diluted with 100 mL of LB medium, pH 8.0, supplemented with 100 μg/mL ampicillin, 1 mM isopropyl-β-d-thiogalactoside (IPTG), and 3 μg/mL of erythromycin and grown at 37 °C until OD600 ~0.5–1.0 was reached. The cells were harvested by centrifugation (5000 × g at 4 °C for 10 min) and washed three times with cold S-30 buffer (1 mM Tris-OAc, pH 8.2, 1.4 mM Mg(OAc)2, 6 mM KOAc, and 0.1 mM dithiothreitol (DTT)) supplemented with β-mercaptoethanol (0.5 mL/L), and once with the same buffer, supplemented with a lower concentration of β-mercaptoethanol (0.05 mL/L). After the final centrifugation, the wet pellet was weighed and 1.27 mL of S-30 buffer without β-mercaptoethanol was added to suspend 1 g of cells. The volume of the suspension was measured and used for estimation of the amount of other components of the lysis mixture. The preincubation mixture (0.29 M Tris-OAc, pH 8.2, 9 mM Mg(OAc)2, 13 mM ATP, 84 mM phosphoenolpyruvate, 4.4 mM DTT, and 5 μM canonical amino acid mixture) in a 0.3 mL volume, as well as 15 units of pyruvate kinase and 10 μg of lysozyme were added per 1 mL of cell suspension, and the final mixture was incubated at 37 °C for 30 min. The incubation mixture was then frozen at −80 °C (30 min), thawed at 37 °C (~30 min), then again frozen at −80 °C, and thawed at room temperature. Ethylene glycol tetraacetic acid was then added to a final concentration of 2.5 mM, and the cells were incubated at 37 °C for 30 min. The same molar amount of CaCl2 was added, mixed well, and frozen (−80 °C for 30 min). The frozen mixture was centrifuged (15,000 × g at 4 °C for 1 h), and the supernatant was collected with autoclaved tubes and stored in aliquots (~0.1 mL) at −80 °C.
Site-directed mutagenesis of the P. pyralis luciferase gene
Polymerase chain reaction (PCR) mutagenesis was done to prepare a gene with codon TAG in lieu of the two codons corresponding to Ala285 and Leu286. A modified Quik-ChangeTN site-directed mutagenesis protocol was used with plasmids pETLucwt as templates and 5′ GGATTACAAGATTCAAAGTTAGCTGGTGCCAACCC3′ as primer. This plasmid was used for the synthesis of luciferases containing modified dipeptides 1–6, 2R-7, and 2S-7.
For the preparation of modified luciferases containing modifications at positions 284 (Ser→Asn) and 286 (Leu→Ala and Leu→Val), plasmids pETLucwt and pETLuc285286, respectively, were mutagenized as above with the following primers:
5′ GGATTACAAGATTCAAAACGCGCTGGTGCCAACCC 3’ (for Asn284), 5′ GGATTACAAGATTCAAAGTGCGGCGCTGGTGCCAA CCC 3’ (for Ala285Ala286), and 5′ GGATTACAAGATTCAAAGTGCGGTCCTGGTGCCAA CCC 3’ (for Ala285Val286).
For oligonucleotide primer phosphorylation, reaction mixtures (20 μL total volume) containing 100 pmol of primer, 1 mM ATP, 70 mM Tris buffer, pH 7.6, 10 mM MgCl2, 5 mM DTT, and 1 unit of T4 polynucleotide kinase were incubated at 37 °C for 1 h and then chilled in ice.
The PCR was carried out in 50 μL (total volume) of 35 mM Tris–HCl, pH 8.0, containing 300 ng of template, 14 pmol of primer, 10 nmol of dNTPs, 12 mM KOAc, 5 mM DTT, 0.05% Triton X-100, 0.05 mM EDTA, 2.5 U of Pfu polymerase and 20 U of Taq DNA ligase. The thermal cycler was programmed as follows: preincubation at 95 °C for 2 min, 18 cycles at 95 °C for 1 min, 43 °C for 1 min, and 65 °C for 12 min. Then, samples were incubated for 7 min at 72 °C and cooled to room temperature. One microliter of restriction endonuclease DpnI was added, and the reaction mixture was incubated at 37 °C for 2 h. Then the sample was subjected to denaturation at 95 °C for 1 min, followed by two cycles at 95 °C for 1 min, 50 °C for 1 min, and 70 °C for 12 min.
DH5α high-efficiency competent cells (>107 cfu/μg) were transformed using 5 μL of the PCR products per 50 μL of cell suspension, and the transformants were placed on the LB agar, prepared with 60 μg/mL of kanamycin and incubated at 37 °C for 18–24 h. Plasmids from single colonies were isolated and utilized in in vitro translation to determine their ability to support the synthesis of full-size protein. Selected plasmids were sequenced to verify the presence of the desired mutations.
Plasmid pETΔLuc, containing the truncated P. pyralis luciferase gene (amino acids 249–450, full length gene numbering), was prepared by Synbio Technology, LLC and was used as a template for PCR mutagenesis for preparation of a gene with TAG in lieu of the two codons corresponding to Ala285 and Leu286, using the same primer described above for full size luciferase. The plasmid so obtained, pETΔLuc285286, was used for preparation of protein containing cyclic peptide 3 for mass spectrometric analysis.
In vitro protein translation
P. pyralis luciferase translation reactions were conducted in 15 μL of incubation mixture containing 0.3 μL/μL of an S-30 system, 170 ng/μL plasmid, 35 mM Tris-acetate, pH 7.4, 190 mM potassium glutamate, 30 mM ammonium acetate, 2 mM DTT, 0.2 μg/μL total Escherichia coli tRNA, 3.5% poly(ethylene glycol) 6000, 20 μg/mL folinic acid, 20 mM ATP and guanosine 5′-triphosphate, 5 mM cytidine 5′-triphosphate and uridine 5′-triphosphate, 100 μM amino acid mixture, 0.5 μCi/μL of [35S]methionine, and 1 μg/μL rifampicin. Plasmids having wild-type and modified luciferase genes were added to a final concentration of 1 μg plasmid/15 μL reaction mixture. Samples containing plasmids having a modified gene (one TAG codon corresponding to luciferase protein positions 285/286) were treated with ~2 μg of the appropriate misacylated suppressor tRNACUA. The protein synthesis reactions were conducted 1 h at 37 °C for 1 h and the reactions were terminated by adding 2× electrophoresis loading buffer (2% SDS (w/v), 5% β-mercaptoethanol (v/v), 20% glycerol (w/v), 0.05% bromophenol blue, 0.15 M Tris-HCl, pH 7.0), and then incubated at 100 °C for 2 min. The analysis of samples after in vitro translation was done by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 5–10 μL of final samples per well.
For larger scale preparation of proteins having cyclic dipeptides, in vitro translation was done in reaction mixtures (2–5 mL total volume) as described above, but without [35S] methionine and using increased amounts of amino acids (~300 μM).
Purification of truncated luciferases, wild type and modified containing cyclic peptide 3
Samples of truncated luciferases were obtained by in vitro translation in 0.5- and 2-mL reaction mixtures (vide supra), respectively, for wild-type and modified proteins, using an S-30 system prepared from clone 010328R2, having a modified ribosome and plasmids pETΔLucwt and pETΔLuc285286, respectively. In the case of the reaction mixture with pETΔLuc285286 plasmid, a suppressor tRNA activated with cyclic peptide 3 was also present. Final reaction mixtures were diluted three fold with 100 mM Tris–HCl, pH 8.3 (wash buffer) and loaded on Strep-Tactin Sepharose (100 μL volume, IBA Life-sciences, Gottingen, Germany), equilibrated with the same buffer, then washed with 2 mL of the same buffer. Bound proteins were then eluted with 0.5 mL of the same buffer, supplemented with 2.5 mM desthiobiotin, and concentrated/desalted by Amicon Ultra 10k centrifugal filters (Merck Millipore Ltd.). The final volume of samples was about 30 μL. Samples were analyzed by SDS-PAGE with Coomassie R-250 staining; the protein concentration was estimated by comparison to known concentrations of a commercial sample of lysozyme.
In vivo expression of luciferases containing noncanonical amino acids
Plasmid pETLuc285286, having a modified P. pyralis luciferase gene with TAG codon in lieu of codons GCG (Ala, position 285) and CTG (Leu, position 286), were cotransformed with pTECH-Pyl-OP33–35 in BL-21(DE-3) competent cells. Cultures in 2YT medium, supplemented with 60 μg/mL kanamycin and 30 μg/mL chloramphenicol, were prepared from single colonies and kept at −80 °C as glycerol (15%) stocks until use. Before in vivo expression started, the corresponding cultures were grown in 5 mL of 2YT medium, supplemented with the two antibiotics, until OD600 ~0.5 was reached. Samples were transferred to a flask with 30 mL of 2YT medium, supplemented with the two antibiotics, and were incubated at 37 °C until OD600 was ~0.4. Then IPTG was added to 1 mM concentration to activate protein production. Simultaneously, the corresponding cyclic dipeptides were added to all cultures to 2 mM final concentration. A culture prepared without the addition of any cyclic dipeptide was prepared as a control using the same procedure. Cultures were incubated overnight at 37 °C and transferred to 50-mL sterile centrifuge tubes. Pellets of cells were prepared by centrifugation (3500 × g, 15 min, 4 °C) and resuspended in lysis buffer (50 mM Tris–HCl, pH 8.0, 10 μg/mL of lysozyme). Three freeze/thaw cycles were carried out and after the last frozen step samples were centrifuged at 4 °C and 15,000 × g for 40 min. The samples were placed on ice, and the supernatants were transferred to clean tubes. Proteins from the lysates were purified by Strep-Tactin chromatography, concentrated/desalted using 30K Amicon Ultra centrifugal filters, and analyzed by SDS- PAGE.
Measurement of bioluminescence emission spectra
Bioluminescence spectra were measured from 450 to 670 nm using a Varian Cary-Eclipse spectrometer in 50 mM Tris–HCl, pH 7.4, supplemented with 10 mM MgCl2 and 2 mM ATP. Forty μL of 2 mM luciferin solution, in 50 mM Tris–HCl, pH 7.4, was mixed with 10 μL of luciferase and diluted to about 50 nM (~3.3 ng/μL). The final concentration of luciferase in solution was about 10 nM (~0.6 ng/μL). Spectra were recorded immediately after the enzyme samples were added to the luciferin solution.
Thermal stability study
Each luciferase sample, diluted to about 50 nM concentration with 50 mM Tris–HCl, pH 7.4, was divided into several 15-μL aliquots, which were incubated at 37 °C for 20, 40, 60, and 80 min and then kept on ice until their bioluminescence measurements. The bioluminescence of all samples was measured immediately after mixing 5 μL of each sample with 45 μL of 2 mM luciferin in 50 mM Tris–HCl, pH 7.4, supplemented with 10 mM MgCl2 and 2 mM ATP. The data was expressed as the percent of bioluminescence relative to a control sample that was incubated on ice throughout the experiment.
Circular dichroism melting curve
Circular dichroism (CD) spectra were collected using a Jasco J-815 instrument. The melting curves of wild-type luciferase (Luc-WT, blue) and Luc-3 (orange) were determined over the temperature range of 20–60 °C (heating rate, 1 °C/min; detection wavelength, 222 nm). Luc-WT was a truncated version of P. pyralis luciferase containing residues 249–450 of wild-type luciferase. Luc-3 was a truncated version of P. pyralis luciferase containing residues 249–450 of wild-type luciferase with dipeptide 3 in lieu of Ala285 and Leu286. The Tm values were estimated based on the maximum slope.
Protein modeling
The protein modeling was performed with Schrodinger (2018–1 release). Step 1, imported Protein Data Bank (PDB) file 4G37 (downloaded from the PDB) into Maestro Elements; Step 2, modified residues 285 and 286 of each mutant luciferase to the corresponding noncanonical dipeptides with 3D Builder; Step 3, preprocessed the protein structure with Protein Preparation Wizard using default parameters, which enabled the bond order assignments and hydrogen additions. Step 4, used PROPKA to optimize the H-bond assignments; Step 5, removed waters having less than 3 H-bonds to non-waters. Step 6, minimized the protein structure with OPLS3 force field. Finally, displayed the binding pocket area and exported the structure.
Results and Discussion
Point mutations in P. pyralis and L. cruciata luciferases produced analogous effects
Consistent with the findings discussed above, wild-type L. cruciata luciferase had its emission maximum at 560 nm, while that of the S286N mutant was at 605 nm.14 The apparent importance of Ile 288 to formation of the closed form was also studied. L. cruciata luciferase mutant I288V emitted maximally at 560 nm with a second emission maximum at 613 nm, suggesting a second excited state of lower energy. Less lipophilic mutant I288A also had a (single) fluorescence emission maximum at 613 nm.14 Although not studied previously, we now report that analogous structural changes in P. pyralis luciferase had a very similar effect. Alteration of Ser284 to Asn altered the emission wavelength from 561 to 603 nm, while alteration of Leu286 successively to Ala and Val altered the resulting emission wavelengths to 590 and 600 nm, respectively (Figures 2a and 2b). Interestingly, wild-type P. pyralis luciferase also exhibited a second emission as a shoulder at ~605–610 nm (vide infra). Thus, parallel changes in these two luciferases had similar effects.
Figure 2 |.
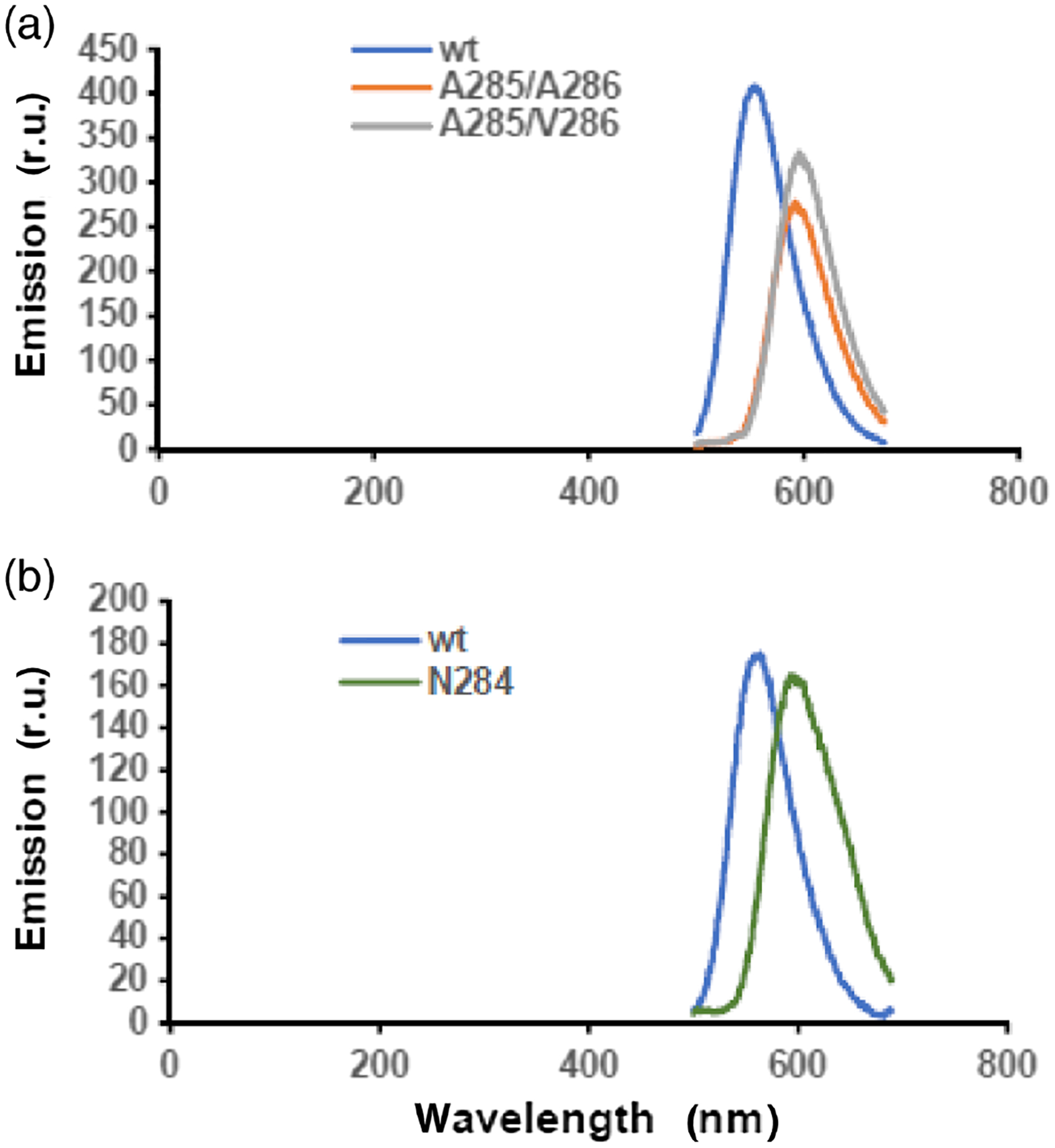
Emission spectra of four types of P. pyralis luciferases. The average λmax for wild-type luciferase was 560 ± 3 nm. (a) For mutant A285/A286, the average λmax was 590 ± 5 nm; for mutant A285/V286, the average λmax was 600 ± 3 nm. (b) For mutant N284, the average λmax was 603 ± 4 nm.
Conformationally constrained dipeptides were prepared and incorporated into P. pyralis luciferase biosynthetically using modified ribosomes selected for that purpose
In this study, we explored the effects of constraining the conformational flexibility of amino acids 285 and 286 in P. pyralis luciferase (analogous to positions 287 and 288 in L. cruciata luciferase) by the introduction of conformationally constrained dipeptides. Eight dipeptides were studied (Figure 3). Dipeptides 1–5 were prepared as described previously.32 Also prepared were dipeptides 6, 2R-7, and 2S-7. The synthesis of S-lysylglycine (6) was straightforward, involving the condensation of di-N-Boc-S-lysine with glycine tert-butyl ester in the presence of 1-hydroxybenzotriazole and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride. Deprotection was accomplished using trifluoroacetic acid (TFA) (Supporting Information Figure S2).
Figure 3 |.
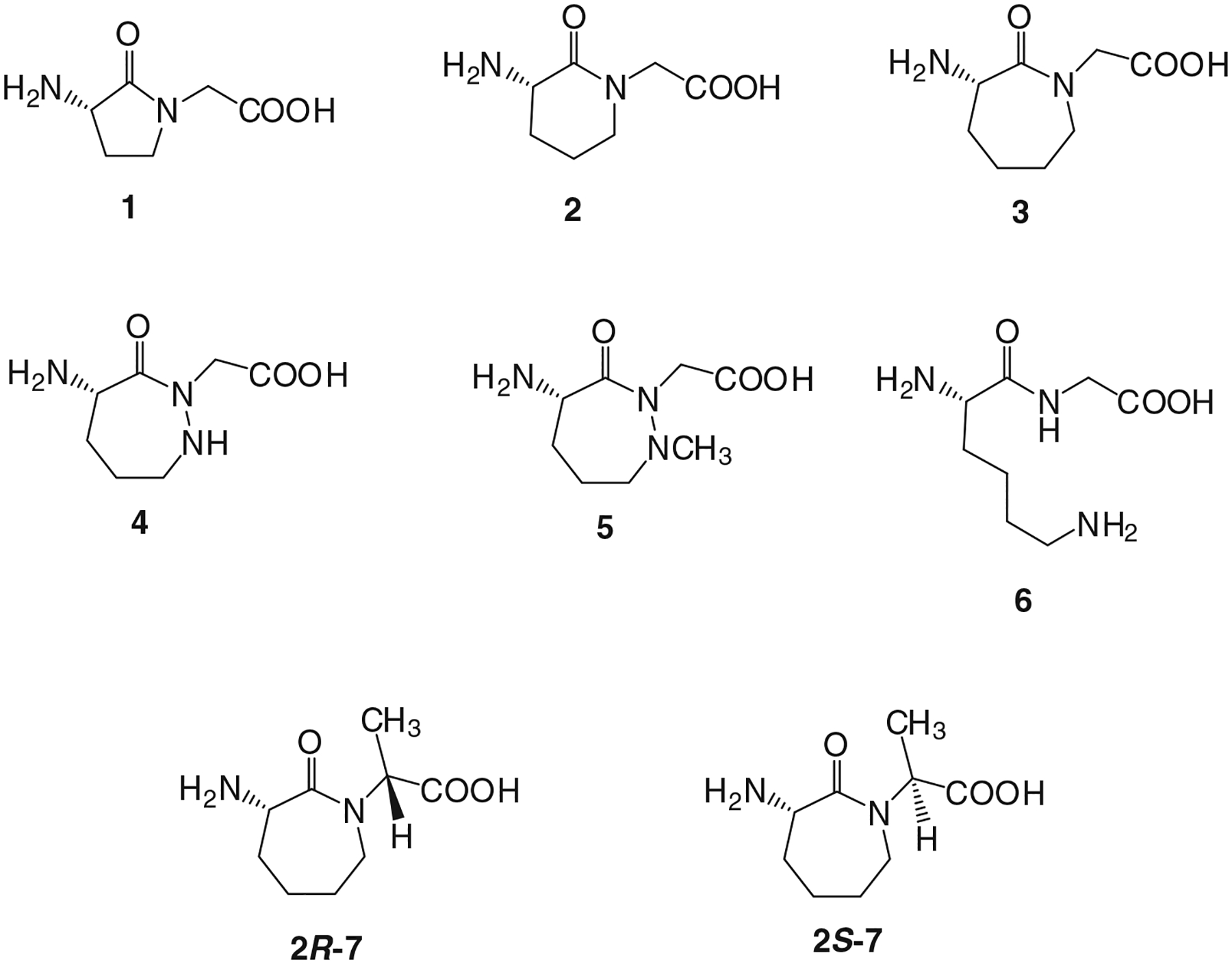
Conformationally constrained dipeptides introduced into P. pyralis luciferase in lieu of Ala285 and Leu286. The dipeptides were each used to aminoacylate a suppressor tRNACUA transcript, and the activated tRNAs were employed in an in vitro protein synthesizing system containing an S-30 extract prepared from ribosomal clone 010328R2.29,32
The synthesis of dipeptide N-protected 2R-7 began with the N-alkylation of tert-butyl (S)-(2-oxoazepan-3-yl)carbamate (9) with (S)-2-bromopropanoic acid, followed by conversion of intermediate 10 to methyl ester 11 in 80% overall yield (Supporting Information Figure S3). Lactam 11 was treated with TFA to remove the Boc protecting group, which was replaced with an N-pentenoyl group, affording 13 in 92% overall yield for two steps. Deprotection of this intermediate successively with LiOH and HCl afforded N-pentenoyl dipeptide 2R-7. Diastereomeric 2S-7 was prepared from 9 and (R)-2-bromopropanoic acid using the same overall procedure (Supporting Information Figure S4).
The deprotected products were used to introduce the synthetic dipeptides into firefly luciferase in E. coli by their inclusion in the culture medium for the bacteria (vide infra). For in vitro synthesis of P. pyralis luciferases containing the conformationally constrained dipeptides, each of the dipeptides was converted to its Nα-protected cyanomethyl ester and then to the respective Nα-protected aminoacyl-pdCpA derivative, as illustrated in Supporting Information Figures S3 and S4. As noted in the figures, the activated pdCpA derivatives were ligated to abbreviated suppressor tRNA-COHs lacking the pCpA sequence present at the 3′-end of functional tRNAs. These misacylated tRNAs were then employed in the biosynthesis of luciferases containing a cyclic dipeptide (Figure 1).
Because native bacterial ribosomes do not utilize dipeptidyl-tRNAs, we employed a strategy of our design29,30 to identify ribosomes that would recognize dipeptidyl-tRNAs and incorporate the dipeptide moiety into nascent proteins. Using a plasmid borne E. coli rrnB gene, we mutagenized DNA regions corresponding to the 23S rRNA that had been reported to be important for peptide bond formation.30 The library of clones so obtained was tested for sensitivity to dipeptide analogues of puromycin, a functional analogue of aminoacyl-tRNA able to interact with the ribosomal A-site.29,30 Clone 010328R2 was sensitive to a dipeptidyl-puromycin derivative containing the dipeptide p-meth-oxyphenylalanylglycine; an S-30 preparation prepared from this clone utilized suppressor tRNAs activated with the cyclic dipeptides and incorporated the cyclic dipeptides into firefly luciferase.
The activated suppressor tRNAs were employed for in vitro protein biosynthesis to incorporate the dipeptides into positions 285 and 286 of P. pyralis luciferase. While constrained dipeptides 1 and 2 were incorporated only minimally, and were not studied further, the remainder of the dipeptides were incorporated in modest yields (~3–5%) sufficient to permit study of light emission by the modified luciferases (Supporting Information Table S1 and Figure S5). The incorporation of dipeptide 3 into a modified luciferase was verified by mass spectrometry (Supporting Information Table S2).
Effects of local conformational constraint on the properties of P. pyralis luciferase
Initial activation/emission experiments were carried out with modified luciferases containing dipeptides 3, 4, and 5 in comparison with wild type (Figure 4a). As shown, wild type emitted at 560 nm with a shoulder at 605–610 nm. The luciferases containing 1,2-diazepanes 4 and 5 at positions 285/286 each contained two emission maxima of roughly equal intensities at 560 and 605–610 nm, reinforcing the interpretation of the two peaks observed for wild type as due to different excited states. Interestingly, while the luciferase containing 1,2-diazepane 4 exhibited slightly more intense emission at 560 nm than at 605–610 nm, the reverse was true for the luciferase containing methylated 1,2-diazepane 5. Plausibly, the N-methyl group may have further distorted the structure of the complex required for light production. In comparison, the luciferase containing conformationally constrained dipeptide 3 at positions 285/286 emitted predominantly as a single peak in experiments at a wavelength in the range 547–552 nm, that is, at higher energy than the wild type. Thus, conformational constraint of a region of luciferase known14 to be critical to the energy of emitted light resulted in higher energy emission. This seems consistent with the interpretation that the (constrained) conformation of luciferase containing 3 is closer to the reaction coordinate for light production than that of wild type.36,37
Figure 4 |.
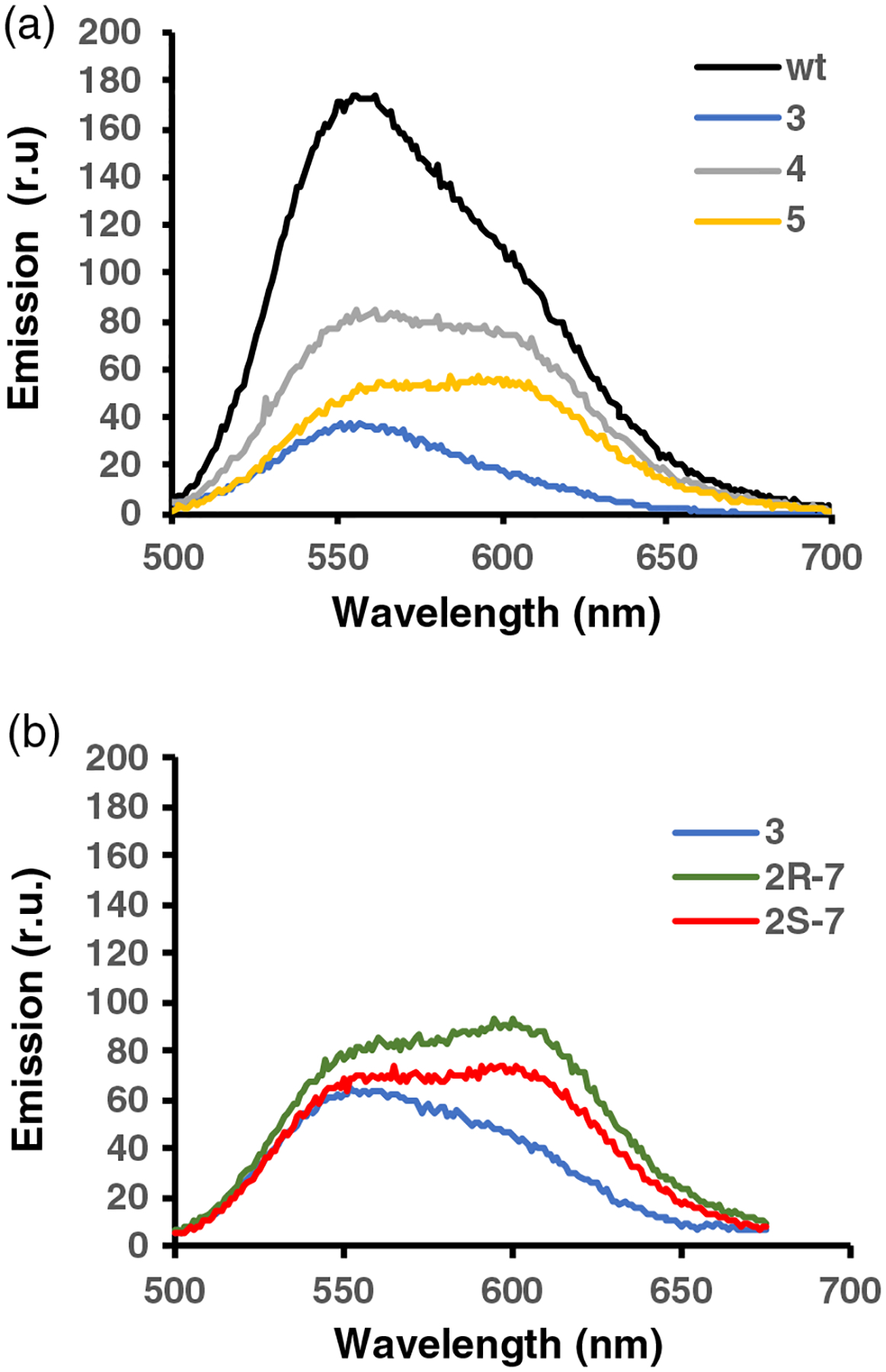
Emission spectra of P. pyralis luciferases. (a) These included wild-type luciferase, and modified luciferases containing 3, 4, and 5, and (b) modified luciferases containing 3, 2R-7, and 2S-7 in lieu of amino acids Ala285 and Leu286. The data are representative of three independent experiments.
Luciferases containing 4 and 5 displayed similar bioluminescence properties, implying the lack of significant effect of the additional N-methyl substituent in 5. Nonetheless, in the belief that methylation of the peptide backbone might be informative, we also studied analogues of 3 C-methylated on the embedded glycine moiety. The individual diastereomers 2R-7 and 2S-7 were used to activate suppressor tRNAs, and then for synthesis of the modified luciferases containing 2R-7 and 2S-7 at positions 285 and 286 of P. pyralis luciferase. As shown in Figure 4b, comparison of the bioluminescence spectra of the luciferases containing dipeptides 3, 2R-7, and 2S-7 demonstrated that the latter two proteins afforded quite similar spectra. Each exhibited two peaks, one at 556 nm and a second, larger peak at 613 nm. These two peaks were putatively due to different excited states, as noted for the luciferases in Figure 4a.
The same emission characteristics were noted when these luciferases were expressed in cellulo in E. coli (Figure 5). Cellular tRNACUA activation was carried out using pyrrololysyl-tRNA synthetase and suppressor tRNAPyl; these were expressed from pTECH-Pyl-OP33–35 and have been used for the incorporation of amino acid analogues into proteins; they have been found to have a relaxed specificity for the position of what is normally the α-amino group of the activated amino acid moiety.24,33–35 Modified P. pyralis luciferase mRNAs were expressed from plasmid pETLuc285286, having a TAG codon in lieu of codons GCG (Ala, position 285) and CTG (Leu, position 286). These two plasmids were cotransformed into E. coli, harboring a plasmid for the modified 23S ribosomal RNA.
Figure 5 |.
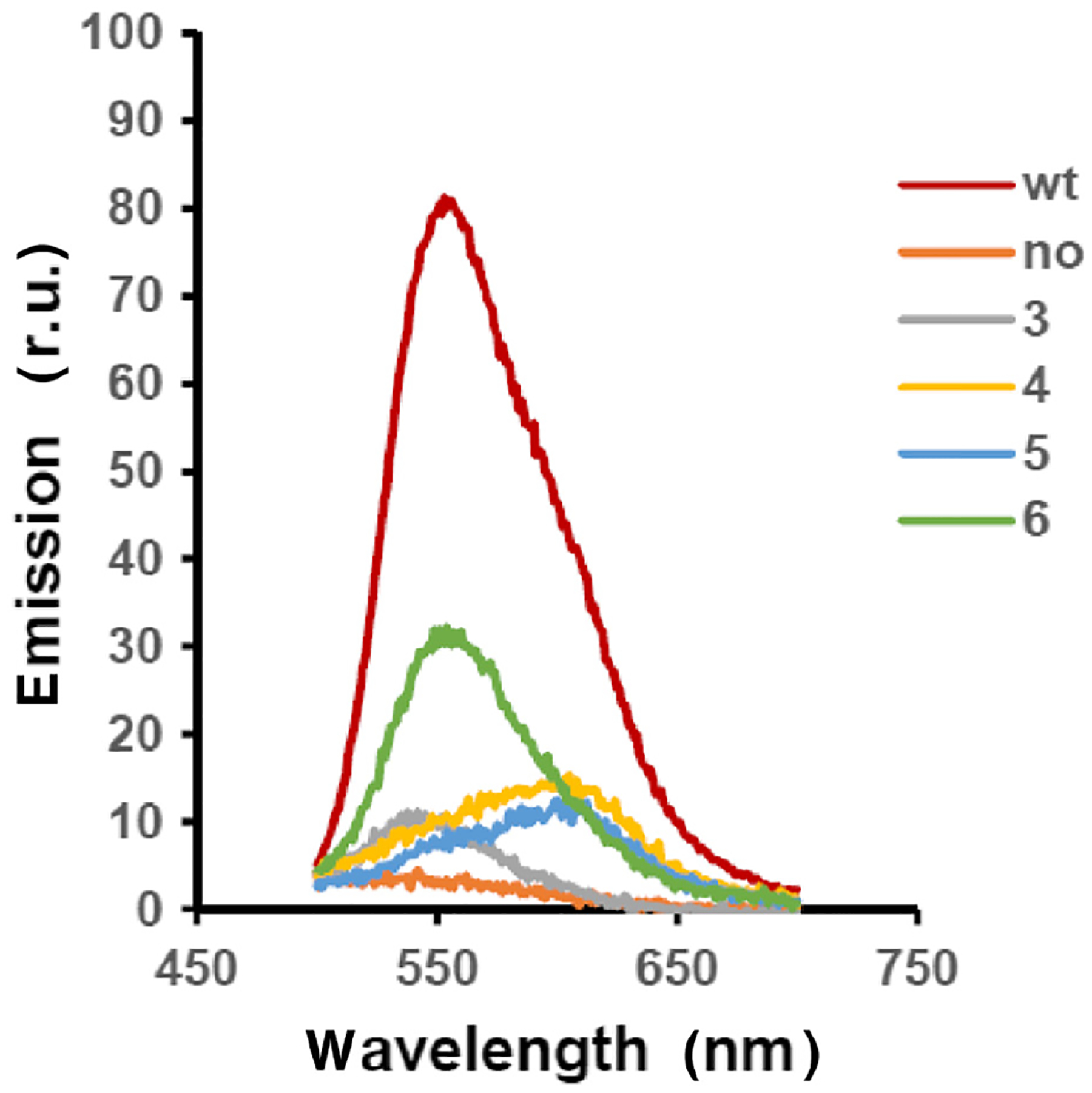
Bioluminescence spectra of wild-type and modified P. pyralis luciferases prepared in cellulo. Wild-type P. pyralis luciferase was expressed in E. coli from plasmid pETLuc. Modified P. pyralis luciferase mRNAs were expressed from plasmid pETLuc285286, having a TAG codon in lieu of codons GCG (Ala, position 285) and CTG (Leu, position 286). This plasmid was cotransformed with pTECH-Pyl-OP33–35 which expressed suppressor tRNAPyl and its corresponding pyrrolysyltRNA synthetase. Growth of the bacteria on 2YT medium in the presence of 2 mM cyclic dipeptides 3, 4, 5, or 6 afforded E. coli containing the corresponding luciferases. A control experiment (no) lacked any cyclic dipeptide. The emission spectra are representative from three independent bioluminescence measurements.
Also studied in the in cellulo experiment was a luciferase containing dipeptide 6 (Supporting Information Figure S2), a ring-opened (conformationally unconstrained) version of dipeptide 3. In the absence of conformational constraint, this luciferase exhibited the same emission characteristics as wild type (Figure 5). It may be noted that this is the first report of inclusion of conformationally constrained dipeptides in a protein expressed in cellulo, and the approach should find further utility in studying protein function both in vitro and in vivo.
To study the effect of local conformational constraint on the biochemical properties of luciferase, we investigated the thermal stability of representative luciferases, which has been related to the dynamic behavior of firefly luciferase.38 As shown in Figure 6a, incubation of wild-type luciferase and luciferase containing 6 at 37 °C in 50 mM Tris–HCl, pH 7.4, for 80 min resulted in a steady loss in their ability to produce light, as aliquots were taken as a function of time and then treated with luciferin, ATP, and Mg2+. Wild-type bioluminescence diminished from 100% to 27 ± 3% over a period of 80 min, while luciferase containing 6 retained 22.4% of its initial activity. In contrast, the luciferases containing constrained dipeptides 3 and 5 retained 64 ± 9% and 74 ± 5%, respectively, of their original light-emission properties following 80 min of incubation. The enhanced thermostability of the enzyme mutants was further confirmed by CD (Figure 6b).39
Figure 6 |.
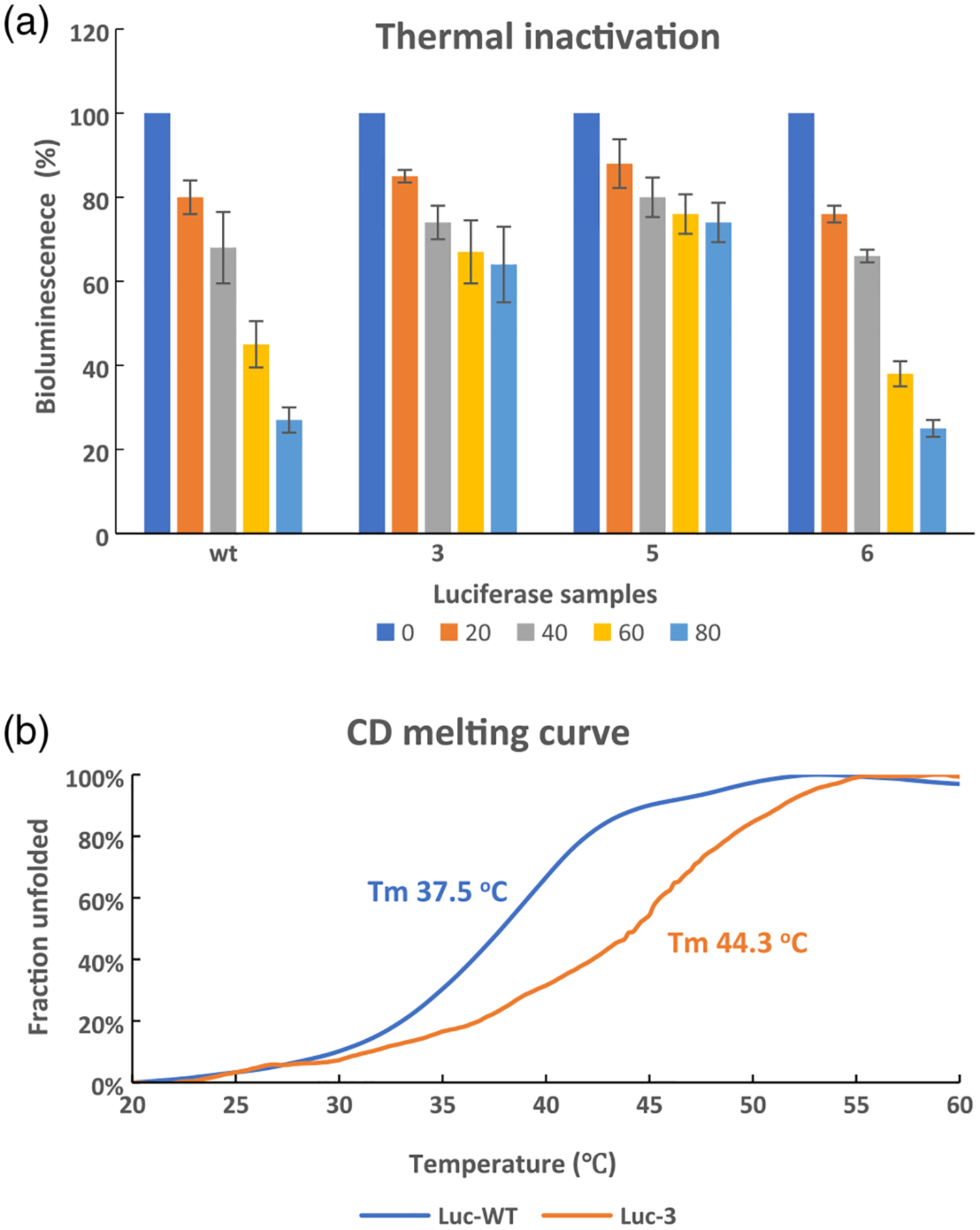
Thermal stability tests. (a) Thermal inactivation. Wild-type P. pyralis luciferase and modified luciferases were incubated at 37 °C for 20, 40, 60, and 80 min, and then tested for their bioluminescence after the addition of ATP + luciferin. The data are representative of three independent experiments. (b) CD melting curves of Luc-WT (blue) and Luc-3 (orange) in 50 mM Tris-HCl, pH 7.4. The Tm values were estimated based on the maximum slope. Luc-WT or Luc-3 was a truncated version of P. pyralis luciferase containing residues 249–450 of wild-type luciferase or wild-type luciferase with dipeptide 3 in lieu of Ala285 and Leu286.
Finally, to better understand the experimental results, luciferases containing conformationally constrained dipeptides (3, 4, or 2S-7) were modeled on the basis of an oxidative catalytic conformation.40,41 Luciferase containing conformationally unconstrained dipeptide 6 was also modeled (Figures 7 and 8). We found two types of orientation (“inside” and “outside”) of the conformationally constrained dipeptides. Compared with the structure of wild-type luciferase, the cyclic seven-membered ring of Luc-3 orientates into the binding pocket; Leu 287 and Val 288 also have some changes in orientation and are closer to the binding pockets (Figure 7a). The distance of key residues (amino acids 284–289) of Luc-3 is much closer to the binding pocket (Figures 8a and 8b). Thus, Luc-3 could bind the excited state of oxyluciferin in a more rigid and hydrophobic microenvironment, which results in emission at a shorter wavelength (higher energy).
Figure 7 |.
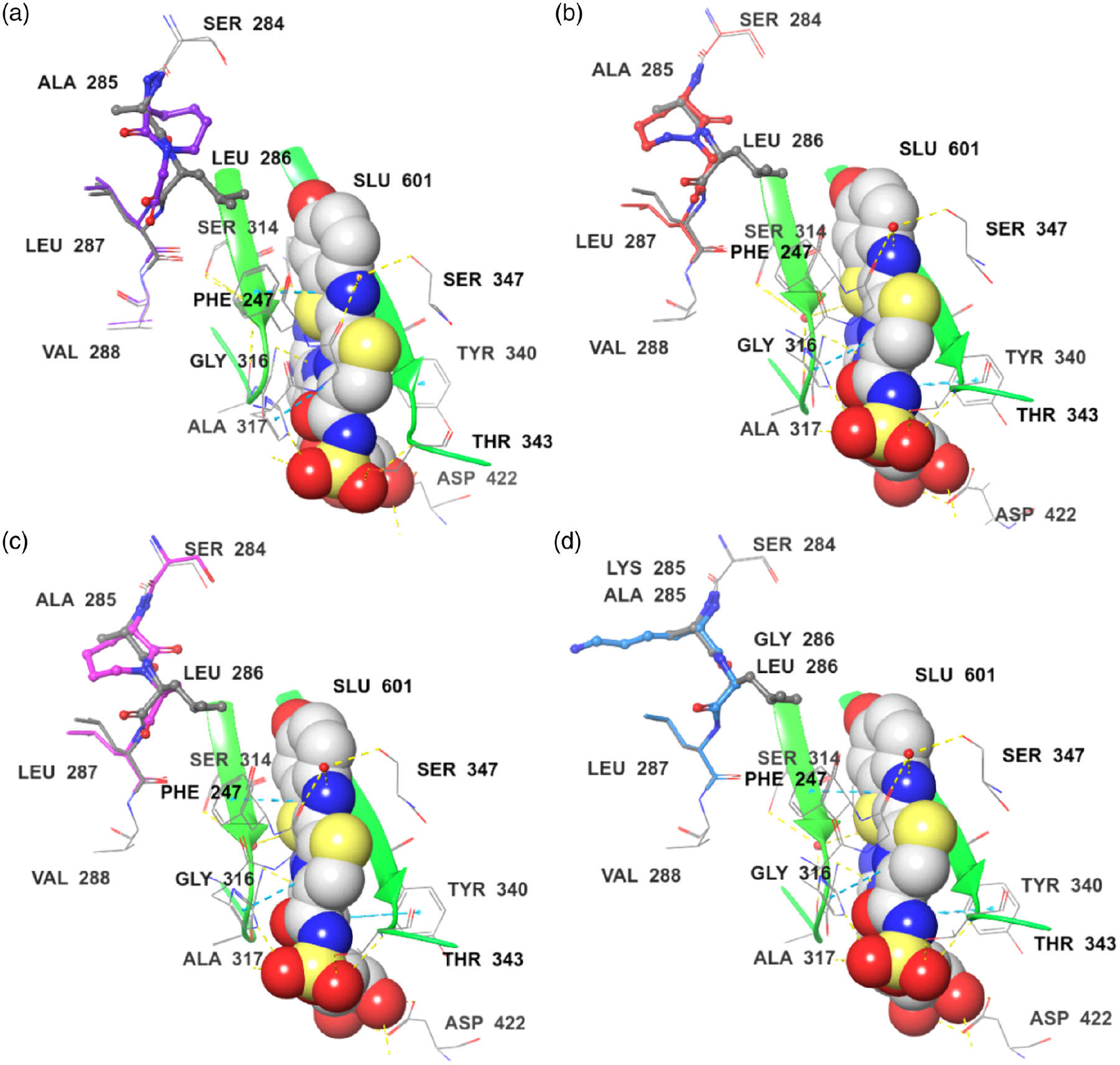
Modeling of comparison of key residues in luciferin binding pocket. The ligand, DLSA (SLU601), was displayed as spheroidal structure. (a) Superposition of the Luc (WT, PDB 4G37) (gray) and Luc-3 (purple) complexes. (b) Superposition of the Luc (WT, PDB 4G37) (gray) and Luc-4 (red) complexes. (c) Superposition of the Luc (WT, PDB 4G37) (gray) and Luc-(2S)-7 (pink) complexes. (d) Superposition of the Luc (WT, PDB 4G37) (gray) and Luc-6 (light blue) complexes.
Figure 8 |.
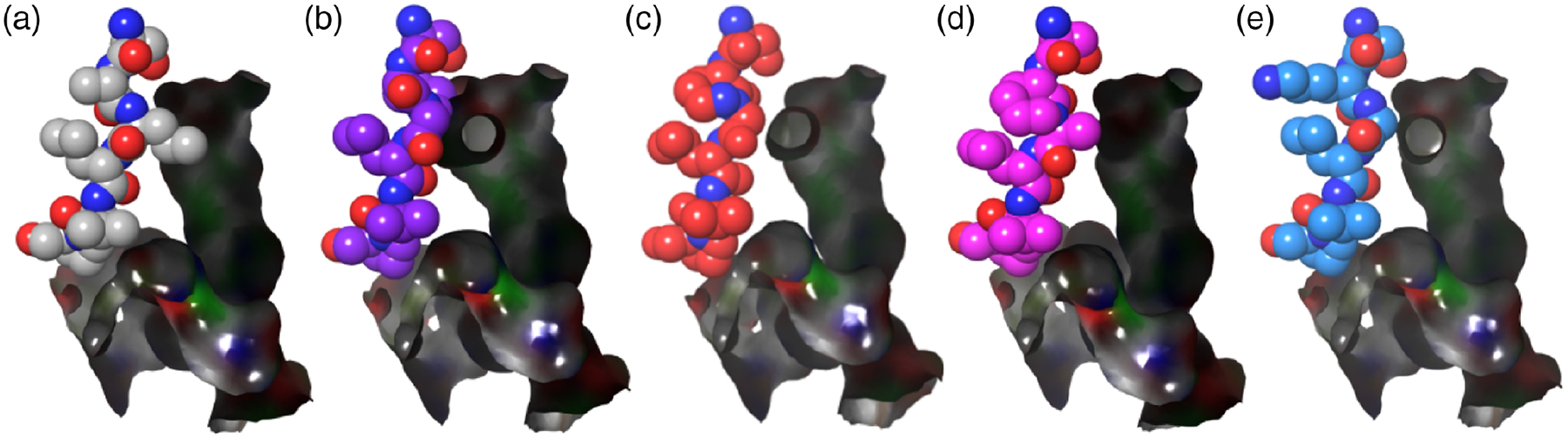
Modeling of interaction between key residues (amino acids 284–289, spheroidal structure) and luciferin binding pocket (surface, black). (a) Luc (WT, PDB 4G37). (b) Luc-3. (c) Luc-4. (d) Luc-(2S)-7. (e) Luc-6.
In comparison, for Luc-4 or Luc-(2S)-7, the cyclic seven-member ring orientates outside the binding pocket, and Ser 284 and Leu 287 are farther away from the binding pocket in comparison with the structure of wild-type luciferase (Figures 7b and 7c). The key residues (amino acids 284–289) of Luc-4 or Luc-(2S)-7 make the binding pocket less rigid and more hydrophilic (Figures 8a, 8c, and 8d). Accordingly, the emission wavelengths of Luc-4 and Luc-(2S)-7 are red-shifted (lower energy). For Luc-6, all of the residues exhibit almost the same orientation with wild-type luciferase (Figure 7d). Luc-6 also forms a binding pocket similar to that of wild-type luciferase (Figures 8a and 8e). As would be expected, the emission spectra of 6 and wild type are similar. These results provide compelling data that rationalize the alteration of emission observed upon introduction of the conformationally constrained dipeptides.
We note that conformational constraint can result in a loss of degrees of freedom and thereby increase protein stability. There are many examples of altered protein stability, including that of firefly luciferase, and increased protein stability can result in alteration of emission wavelength. To the best of our knowledge, examples of point mutations that result in variants emitting at shorter wavelength are quite limited (see, e.g., refs 13 and 42). We believe that the shorter emission wavelength observed for luciferase containing conformationally constrained dipeptide 3 results not from increased thermostability per se, but from the formation of a conformation that more closely approximates the reaction coordinate leading to light production than that of the wild type.
Conclusion
Local conformational constraints have been introduced into peptides for many years by straightforward chemical approaches and constitute a productive strategy for increasing the target affinity and stability of peptides of medicinal interest.25–27 In comparison, the preparation of proteins containing localized residues that are constrained conformationally is more problematic from a technical perspective. In the past few years, we have described a new method for modifying bacterial ribosomes and selecting ribosomes that recognize suppressor tRNAs activated with dipeptides and dipeptidomimetics, enabling the introduction of modified residues within proteins.24,28–30 A recent extension of this strategy has enabled the introduction of two types of conformationally constrained dipeptides.32
In this study, this technique has been applied to the study of the energy of bioluminescence produced by firefly luciferase. Based on an X-ray crystallographic analysis of amino acid residues thought to be important for producing bioluminescence in firefly luciferase, we replaced residues Ala285 and Leu286 with each of several conformationally constrained cyclic dipeptides (Figure 3). Dipeptide 3 was designed as a protein constituent potentially capable of stabilizing the closed complex14 believed to form the basis for the high-energy bioluminescence of wild-type luciferase. In fact, introduction of this dipeptide in lieu of Ala-Leu resulted in a shift to shorter wavelength by ~10 nm both in vitro and in vivo. Structural alteration of the 2-oxoazepane moiety of 3 in luciferase to 3-oxo-1,2-diazepane 4 was expected to further rigidify this structural element of the protein and resulted in emission maxima at 560 nm and 605–610 nm (Figure 3). The further N-methylation of this cyclic dipeptide to afford 5 resulted in a luciferase having the same two emission maxima, but with relatively enhanced emission at longer wavelength, perhaps reflecting further distortion of the closed complex by the N-methyl group. Further, the luciferases having local conformational constraint, that is, 3 and 5, exhibited enhanced thermal stability compared with wild-type luciferase.
Further probing of the geometry of the closed complex believed to be responsible for bioluminescence was carried out by methylation of the embedded glycine moiety, that is, by introduction of 2R-7 and 2S-7 into luciferase. As shown in Figure 4b, these methylated analogues also resulted in enhancement of the longer wavelength emission. While the two bioluminescence spectra were similar, the spectrum of 2R-7 had relatively stronger emission at longer wavelength, plausibly indicating greater distortion of the closed complex by the orientation of the added C-methyl group in this analogue.
Based on the oxidation mechanism40,41 proposed for light production, our protein modeling results demonstrated that the unconstrained analogue (Luc 6) associates with the binding pocket in a fashion similar to the wild type. The luciferase having conformationally constrained dipeptide 3 (Luc 3) that emits at shorter wavelength was found to have the cyclic 7-membered ring oriented into the binding pocket with key residues (amino acids 284–289), increasing the hydrophobicity of the binding pocket, consistent with the observed wavelength change. Two analogues that produced shifts to longer wavelength (Luc 4 and Luc 2S-7) had their cyclic 7-membered rings oriented outside the binding pocket, and the orientation of amino acids 284–289 produced less hydrophobic-binding pockets.
Protein conformational dynamics have been studied extensively as an important element of the catalytic competence of enzymes36,37 and have been shown to be essential to catalytic light production by firefly luciferase.43 While the magnitude of such dynamic behavior likely varies from one enzyme to another, the existence of enzymes believed to undergo large conformational changes along their reaction coordinate would seem to offer an opportunity to understand and influence the catalytic properties of such enzymes. Firefly luciferase might be thought to represent one such enzyme, and the results described above suggest that purposeful alteration of conformational dynamics can be exploited to control specific facets of enzyme behavior.
The several naturally occurring types of luciferases have been employed extensively to monitor protein expression. The diversity of species that bioluminesce at different wavelengths has enabled their combined use in experiments that monitored multiple proteins. The present insights regarding changes in photophysical behavior as structure is altered by conformational constraint should facilitate the development of luciferases that emit at wavelengths sufficiently distinct to enable additional applications to be realized.44
Supplementary Material
Acknowledgments
We thank Prof. Dieter Söll, Yale University, for plasmid pTECH-Pyl-OP. We thank Prof. Yu Rao, and his laboratory at Tsinghua University, for their help with the protein modeling.
Funding Information
This work was supported by research grants R01GM12367 and R35GM140819 from the National Institutes of General Medical Sciences, NIH.
Footnotes
Supporting Information
Supporting Information is available and includes all characterization data related to this study.
Conflict of Interest
The authors declare no competing interests.
References
- 1.Syed AJ; Anderson JC Applications of Bioluminescence in Biotechnology and Beyond. Chem. Soc. Rev 2021, 50, 5668–5705. [DOI] [PubMed] [Google Scholar]
- 2.Kahlke T; Umbers KDL Bioluminescence. Curr. Biol 2016, 26, 313–314. [DOI] [PubMed] [Google Scholar]
- 3.DeLuca M Firefly Luciferase. Adv. Enzymol 1976, 44, 37–68. [DOI] [PubMed] [Google Scholar]
- 4.Ugarova NN Luciferase of Luciola mingrelica Fireflies. Kinetics and Regulation Mechanism. J. Biolumin. Chemilumin 1989, 4, 406–418. [DOI] [PubMed] [Google Scholar]
- 5.Wood KV The Chemical Mechanism and Evolutionary Development of Beetle Bioluminescence. Photochem. Photobiol 1995, 62, 662–673. [Google Scholar]
- 6.Branchini BR; Behney CE; Southworth TL; Fontaine DM; Gulick AM; Vinyard DJ; Brudvig GW Experimental Support for a Single Electron-Transfer Oxidation Mechanism in Firefly Bioluminescence. J. Am. Chem Soc 2015, 137, 7592–7595. [DOI] [PubMed] [Google Scholar]
- 7.Baldwin TO Firefly Luciferase: The Structure Is Known but the Mystery Remains. Structure 1996, 4, 223–228. [DOI] [PubMed] [Google Scholar]
- 8.Seliger HH; McElroy WD The Colors of Firefly Bioluminescence: Enzyme Configuration and Species Specificity. Proc. Natl. Acad. Sci. U. S. A 1964, 52, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White EH; Rapaport E; Hopkins TA; Seliger HH Chemi- and Bioluminesence of Firefly Luciferin. J. Am. Chem. Soc 1969, 91, 2178–2180. [DOI] [PubMed] [Google Scholar]
- 10.White EH; Rapaport E; Seliger HH; Hopkins TA The Chemi- and Bioluminescence of Firefly Luciferin: An Efficient Chemical Production of Electronically Excited States. Bioorg. Chem 1971, 1, 92–122. [Google Scholar]
- 11.Wood KV; Lam YA; Seliger HH; McElroy WD Complementary DNA Coding Click Beetle Luciferases Can Elicit Bioluminescence of Different Colors. Science 1989, 244, 700–702. [DOI] [PubMed] [Google Scholar]
- 12.Wood KV; Lam YA; McElroy WD Introduction to Beetle Luciferases and Their Applications. J. Biolumin. Chemilumin 1989, 4, 289–301. [DOI] [PubMed] [Google Scholar]
- 13.Kajiyama N; Nakano E Isolation and Characterization of Mutants of Firefly Luciferase Which Produce Different Colors of Light. Protein Eng. 1991, 4, 691–693. [DOI] [PubMed] [Google Scholar]
- 14.Nakatsu T; Ichiyama S; Hiratake J; Saldanha A; Kobashi N; Sakata K; Kato H Structural Basis for the Spectral Difference in Luciferase Bioluminescence. Nature 2006, 440, 372–376. [DOI] [PubMed] [Google Scholar]
- 15.Mao Y Dynamics Studies of Luciferase Using Elastic Network Model: How the Sequence Distribution of Luciferase Determines Its Color. Prot. Eng. Des. Select 2011, 24, 341–349. [DOI] [PubMed] [Google Scholar]
- 16.Branchini BR; Southworth TL; Fontaine DM; Murtiashaw MH; McGurk A; Talukder MH; Qureshi R; Yetil D; Sundlov JA; Gulick AM Cloning of the Orange Light-Producing Luciferase from Photinus scintillans—A New Proposal on How Bioluminescence Color Is Determined. Photochem. Photobiol 2017, 93, 479–485. [DOI] [PubMed] [Google Scholar]
- 17.Mamaev SV; Laikhter AL; Arslan T; Hecht SM Firefly Luciferase: Alteration of the Color of Emitted Light Resulting from Substitutions at Position 286. J. Am. Chem. Soc 1996, 118, 7243–7244. [Google Scholar]
- 18.Arslan T; Mamaev SV; Mamaeva NV; Hecht SM Structurally Modified Firefly Luciferase. Effects of Amino Acid Substitution at Position 286. J. Am. Chem. Soc 1997, 119, 10877–10887. [Google Scholar]
- 19.Torre DDL; Chin JW Reprogramming the Genetic Code. Nat. Rev. Genet 2021, 22, 169–184. [DOI] [PubMed] [Google Scholar]
- 20.Manandhar M; Chun E; Romesberg FE Genetic Code Expansion: Inception, Development, Commercialization. J. Am. Chem. Soc 2021, 143, 4859–4878. [DOI] [PubMed] [Google Scholar]
- 21.Cui ZL; Johnston WA; Alexandrov K Cell-Free Approach for Non-Canonical Amino Acids Incorporation into Polypeptides. Front. Bioeng. Biotechnol 2020, 8, 1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen C; Yu GL; Huang YJ; Cheng WH; Li YX; Sun Y; Ye HF; Liu T Genetic-Code-Expanded Cell-Based Therapy for Treating Diabetes in Mice. Nat. Chem. Biol 2022, 18, 47–55. [DOI] [PubMed] [Google Scholar]
- 23.Zhang B; Sun J; Wang Y; Ji D; Yuan Y; Li S; Sun Y; Hou Y; Li P; Zhao L; Yu F; Ma W; Cheng B; Wu L; Hu J; Wang M; Song W; Li X; Li H; Fei Y; Chen H; Zhang L; Tsokos GC; Zhou D; Zhang X Site-Specific PEGylation of Interleukin-2 Enhances Immunosuppression via the Sustained Activation of Regulatory T Cells. Nat. Biomed. Eng 2021, 5, 1288–1305. [DOI] [PubMed] [Google Scholar]
- 24.Chen S; Ji X; Gao M; Dedkova LM; Hecht SM In Cellulo Synthesis of Proteins Containing a Fluorescent Amino Acid. J. Am Chem. Soc 2019, 141, 5597–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bozovičar K; Bratkovič T Small and Simple, Yet Sturdy: Conformationally Constrained Peptides with Remarkable Properties. Int. J. Mol. Sci 2021, 22, 1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng N; Christensen SB; Blakely A; Dowell C; Purushottam L; McIntosh JM; Chou DHC Development of Conformationally Constrained α–RgIA Analogues as Stable Peptide Antagonists of Human α9α10 Nicotinic Acetylcholine Receptors. J. Med. Chem 2020, 63, 8380–8387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gisemba SA; Ferracane MJ; Murray TF; Aldrich JV Conformational Constraint Between Aromatic Residue Side Chains in the “Message” Sequence of the Peptide Arodyn Using Ring Closing Metathesis Results in a Potent and Selective Kappa Opioid Receptor Antagonist. J. Med. Chem 2021, 64, 3153–3164. [DOI] [PubMed] [Google Scholar]
- 28.Dedkova LM; Fahmi NE; Paul R; del Rosario M; Zhang L; Chen S; Feder G; Hecht SM β-Puromycin Selection of Modified Ribosomes for in Vitro Incorporation of β-Amino Acids. Biochemistry 2012, 51, 401–415. [DOI] [PubMed] [Google Scholar]
- 29.Maini R; Dedkova LM; Paul R; Madathil MM; Roy Chowdhury S; Chen S; Hecht SM Ribosome-Mediated Incorporation of Dipeptides and Dipeptide Analogues into Proteins in Vitro. J. Am. Chem. Soc 2015, 137, 11206–11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dedkova LM; Hecht SM Expanding the Scope of Protein Synthesis Using Modified Ribosomes. J. Am. Chem. Soc 2019, 141, 6430–6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conti E; Franks NP; Brick P Crystal Structure of Firefly Luciferase Throws Light on a Superfamily of Adenylate-Forming Enzymes. Structure 1996, 4, 287–298. [DOI] [PubMed] [Google Scholar]
- 32.Zhang C; Bai X; Dedkova LM; Hecht SM Protein Synthesis with Conformationally Constrained Cyclic Dipeptides. Bioorg. Med. Chem 2020, 28, 115780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nozawa K; O’Donoghue P; Gundllapalli S; Araiso Y; Ishitani R; Umehara T; Söll D; Nureki O Pyrrolysyl-tRNA Synthetase-tRNA(Pyl) Structure Reveals the Molecular Basis of Orthogonality. Nature 2009, 457, 1163–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobayashi T; Yanagisawa T; Sakamoto K; Yokoyama S Recognition of Non-Alpha-Amino Substrates by Pyrrolysyl-tRNA Synthetase. J. Mol. Biol 2009, 385, 1352–1360. [DOI] [PubMed] [Google Scholar]
- 35.Wan W; Tharp JM; Liu WR Pyrrolysyl-tRNA Synthetase: An Ordinary Enzyme but an Outstanding Genetic Code Expansion Tool. Biochim. Biophys. Acta 2014, 1844, 1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henzler-Wildman KA; Thai V; Lei M; Ott M; Wolf-Watz M; Fenn T; Pozharski E; Wilson MA; Petsko GA; Karplus M; Hübner CG; Kern D Intrinsic Motions Along an Enzymatic Reaction Trajectory. Nature 2007, 450, 838–844. [DOI] [PubMed] [Google Scholar]
- 37.Hammes GG; Benkovic SJ; Hammes-Schiffer S Flexibility, Diversity, and Cooperativity: Pillars of Enzyme Catalysis. Biochemistry 2011, 50, 10422–10430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jazayeri FS; Amininasab M; Hosseinkhani S Structural and Dynamical Insight into Thermally Induced Functional Inactivation of Firefly Luciferase. PLoS ONE 2017, 12, e0180667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li JC; Nastertorabi F; Xuan WM; Han GW; Stevens RC; Schultz PG A Single Reactive Noncanonical Amino Acid Is Able to Dramatically Stabilize Protein Structure. ACS Chem. Biol 2019, 14, 1150–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Branchini BR; Rosenberg JC; Fontaine DM; Southworth TL; Behney CE; Uzasci L Bioluminescence Is Produced from a Trapped Firefly Luciferase Conformation Predicted by the Domain Alternation Mechanism. J. Am. Chem. Soc 2011, 133, 11088–11091. [DOI] [PubMed] [Google Scholar]
- 41.Sundlov JA; Fontaine DM; Southworth TL; Branchini BR; Gulick AM Crystal Structure of Firefly Luciferase in a Second Catalytic Conformation Supports a Domain Alternation Mechanism. Biochemistry 2012, 51, 6493–6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Branchini BR; Southworth TL; Khattak NF; Michelini E; Roda A Red- and Green-Emitting Firefly Luciferase Mutants for Bioluminescent Reporter Applications. Anal. Biochem 2005, 345, 140–148. [DOI] [PubMed] [Google Scholar]
- 43.Szarecka A; Xu Y; Tang P Dynamics of Firefly Luciferase Inhibition by General Anesthetics: Gaussian and Anisotropic Network Analyses. Biophys. J 2007, 93, 1895–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weihs F; Wang J; Pfleger KDG; Dacres H Experimental Determination of the Bioluminescence Resonance Energy Transfer (BRET) Förster Distances of nanoBRET and Red-Shifted BRET Pairs. Anal. Chim. Acta X 2020, 6, 100059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.