

Abstract
Bivalent chemical degraders, otherwise known as Proteolysis Targeting Chimeras (PROTACs), have proven to be an efficient strategy for targeting overexpressed or mutated proteins in cancer. PROTACs provide an alternative approach to small molecule inhibitors, which are restricted by occupancy-driven pharmacology, often resulting in acquired inhibitor resistance via compensatory increases in protein expression. Despite the advantages of bivalent chemical degraders, they often have suboptimal physicochemical properties and optimization for efficient degradation remains highly unpredictable. Herein, we report the development of a potent EED-targeted PRC2 degrader, UNC7700. UNC7700 contains a unique cis-cyclobutane linker and potently degrades PRC2 components EED (DC50 = 111 nM; Dmax = 84%), EZH2WT/EZH2Y641N (DC50 = 275 nM; Dmax = 86%), and to a lesser extent SUZ12 (Dmax = 44%) after 24 hours in a diffuse large B-cell lymphoma (DLBCL) DB cell line. Characterization of UNC7700 and related compounds for ternary complex formation and cellular permeability to provide a rationale for the observed improvement in degradation efficiency remained challenging. Importantly, UNC7700, dramatically reduces H3K27me3 levels and is anti-proliferative in DB cells (EC50 = 0.79 ± 0.53 μM).
Graphical Abstract
INTRODUCTION
Polycomb repressive complex 2 (PRC2) contains three core components, EED, SUZ12, and EZH2, which are responsible for the deposition and propagation of Histone 3 Lysine 27 trimethylation (H3K27me3). An increase in H3K27me3 levels arising from either overexpression or mutation of EZH2 is associated with aberrant transcriptional repression and is a common hallmark of a variety of cancers.1 Specifically, in diffuse large B-cell lymphoma (DLBCL) there are EZH2 gain-of-function mutations in the SET domain which cause PRC2 ‘hyperactivity’ and result in an increase in H3K27me3 levels.2,3 Although numerous potent small molecule EZH2 inhibitors have been reported that are effective at decreasing H3K27me3 levels, they are often met with inhibitor resistance due to the development of secondary point mutations such as the Y641N mutation in the SET domain.4,5 As a result, several allosteric PRC2 inhibitors (e.g. EED226 and A-395) that target the WD40 domain of EED have been developed which similarly decrease H3K27me3 levels.6,7 While catalytic and allosteric PRC2 inhibitors are initially effective at decreasing H3K27me3 levels and are in clinical development, this strategy may be vulnerable to acquired resistance and new strategies are needed to combat PRC2 driven cancers.5
Recent efforts to target PRC2 and circumvent the possibility of acquired inhibitor resistance have focused on the discovery and use of bivalent chemical degraders, or Proteolysis Targeting Chimeras (PROTACs).1,8–11 Bivalent chemical degraders have proven to be effective at degrading challenging targets, eliciting novel pharmacology, and overcoming acquired inhibitor resistance. Bivalent chemical degraders are constructed of a ligand for a protein-of-interest (POI) and for an E3 ligase that are joined by a linker, commonly consisting of alkyl or PEG chains of varying lengths. The ligands bind to their respective targets, bringing the POI and E3 ligase in close proximity to form a ternary complex consisting of POI:degrader:E3 ligase.12,13 In the case of the E3 ligase Von-Hippel Lindau (VHL), it is part of the CRL2VHL complex that also contains a ubiquitylated E2 ligase that then facilitates ubiquitylation of an available lysine residue on the POI.14 The ubiquitin tagged POI is then brought to the proteasome for degradation, and the released degrader molecule can continue to facilitate degradation of the POI in a catalytic fashion.
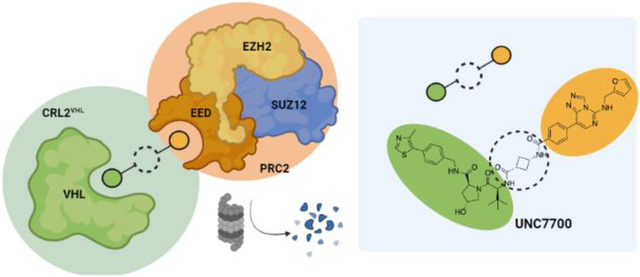
We reported a novel EED-targeted PRC2 bivalent chemical degrader, UNC6852, which recruits VHL to degrade PRC2 components EED, EZH2, and to a lesser extent SUZ12 in DLBCL cell lines (Figure 1).10 UNC6852 also reduces H3K27me3 levels and is anti-proliferative in DB and Pfeiffer DLBCL cells. Other EED- and EZH2-targeted degraders have also recently been reported (Figure 1).8,9 Similar to UNC6852, the EED-targeted degraders PROTAC 1 and 2 contain a related EED ligand (MAK683) and VHL recruiter, while the EZH2-targeted degrader E7 contains an EZH2 ligand and a cereblon (CRBN) recruiter. Additional strategies have also been utilized to degrade PRC2 including hydrophobic tagging (MS1943) and disruption of the EED-EZH2 protein-protein interaction to destabilize the complex (DC-PRC2in-01).11,15 Ma et al. show that MS1943 is anti-proliferative in triple negative breast cancer (TNBC) cells and suppresses tumor growth in vivo with once daily intraperitoneal administration of MS1943 (150 mg/kg).11 Overall, the effects of PRC2-targeted degraders in DLBCL and TNBC cells and in vivo models are encouraging, and these molecules could serve as leads towards novel therapies for cancers with aberrant PRC2 catalytic and non-catalytic functions.
Figure 1.
The current suite of EED- and EZH2-targeted PRC2 degraders and our newly developed EED-targeted degraders UNC7700 and UNC7698 (middle).
The multi-step mechanism of bivalent chemical degraders has led to the pursuit of a rulebook to facilitate the design and optimization of effective degraders such as those reported for PRC2 in a more rational and efficient manner. Degrader development involves consideration of multiple parameters including, but not limited to, cell permeability, binary target engagement, ternary complex formation, lysine availability and ubiquitylation, and proteasomal degradation. Despite a plethora of assays that exist to assess these key parameters, the results of such assays are not necessarily predictive of effective degradation of the POI.16 For example, Donovan et al. recently published their efforts to map the degradable kinome and explore key variables for effective degradation; however, they highlighted a lack of correlation between degradation and binary target engagement, ternary complex formation, and protein expression levels in cellular systems, reinforcing that chemical degrader optimization can be both difficult and unpredictable.16
Herein, we report the optimization of UNC6852 and development of a second generation EED-targeted PRC2 degrader, UNC7700, which has a cis-cyclobutane linker as opposed to the propyl linker of UNC6852 (Figure 1). UNC7700 degrades EED approximately 15-fold more potently than UNC6852 in DB cells and can effectively degrade the core PRC2 components in DLBCL cells after 24 hours (EED DC50 = 111 nM, Dmax = 84%; EZH2WT/EZH2Y641N DC50 = 275 nM, Dmax = 86%; SUZ12 Dmax = 44%). UNC7700 also more potently reduces H3K27me3 levels and demonstrates a more robust anti-proliferative effect than UNC6852. Additionally, we explored the effects of cis- and trans-cyclobutane linkers on binary target engagement, cell permeability, and ternary complex formation to better rationalize the differences in degradation efficiency between UNC6852, UNC7700, and UNC7698.
RESULTS AND DISCUSSION
Linker optimization leads to more effective PRC2 degradation.
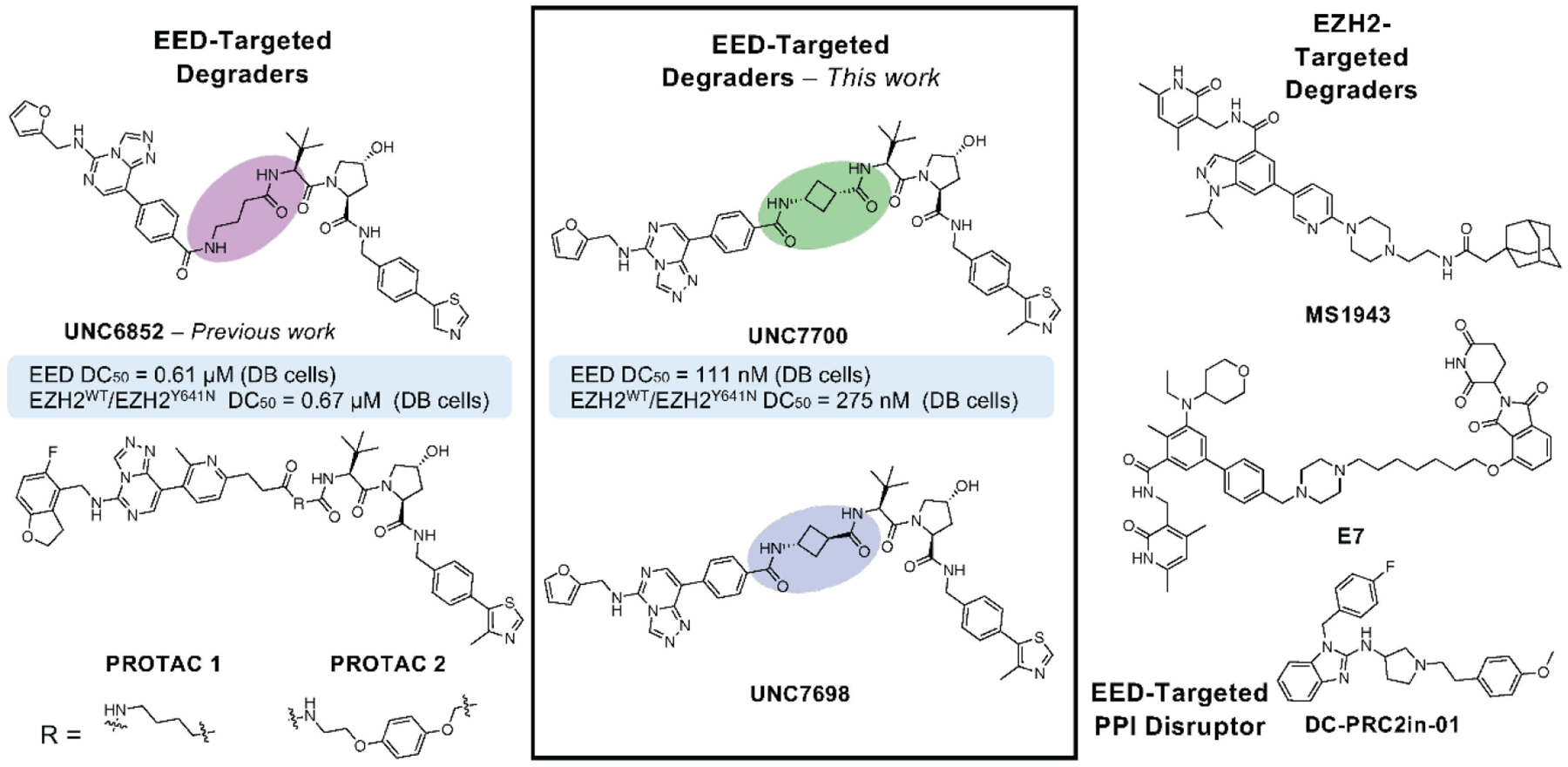
We designed a series of PROTACs varying the linker region of our previously reported degrader, UNC6852, while the ligands for EED and VHL remained unchanged (Figure 2A, Supplemental Table 1). Modifications to the propyl linker primarily included cyclic analogues to restrict flexibility and/or add conformational bias upon ternary complex formation. We synthesized compounds with a cis/trans-cyclobutane (UNC7327), racemic pyrrolidine (UNC7701), 4-piperidine (UNC7326), 4-fluoro-piperidine (UNC7703), and 3-piperidine (UNC7702) linker to explore different ring sizes and trajectories. In some cases, we further modified the amide between EED226 and the linker to give secondary and tertiary amines (X = CH2) resulting in UNC7744, UNC7742, and UNC7743. Additionally, the sulfonamide matched pair of UNC7326 and UNC7743 was synthesized (X = SO2, UNC7741) (Figure 2A). All compounds were synthesized as described in the supplementary Information (Supplemental Information 2. Chemistry).
Figure 2.
Identification of potent PRC2 degraders. A) Structures of EED-targeted degraders and % degradation of EED and EZH2 ± the standard deviation compared to DMSO in DB cells (3 μM, 24 hr). Results were quantified based on western blot and are reported as the average of 2 biological replicates ± standard deviation (*1 biological replicate was performed). EED IC50 values were determined by TR-FRET and are reported as the average of 3 biological replicates ± the standard deviation. Green = EED IC50 <100 nM or degradation >70%; Yellow = EED IC50 100–500 nM or degradation 50–70%; Orange = Degradation 20–50%; Red = Degradation <20%. B-C) Representative western blot analysis of EED and EZH2 degradation reported in A.
To first assess EED target engagement, we determined the IC50 values for all compounds by TR-FRET as previously described.10,17 All of the degraders synthesized displayed potent binding in the nanomolar range to His-tagged EED (Figure 2A and Supplemental Figure 1). Next, the extent of degradation of core PRC2 components EED and EZH2WT/EZH2Y641N was assessed via western blot analysis upon treatment of DB cells with 3 μM compound for 24 hours (Figure 2). UNC7327, which contains a mixture of cis/trans-1,3-substituted cyclobutane ring, was identified as an improved degrader compared to UNC6852 under these conditions (Figure 2 and Supplemental Figure 2). Several other degraders such as UNC7741 and UNC7326 also exhibited robust EED and EZH2WT/EZH2Y641N degradation; however, a follow up dose response study in DB cells indicated that the two degraders were not as effective at degrading the PRC2 components as UNC6852 (EED DC50 = 0.61 ± 0.18 μM, Dmax = 94% at 30 uM; EZH2WT/EZH2Y641N DC50 = 0.67 ± 0.24 μM, Dmax = 96% at 30 uM) (Supplemental Figure 3).10 The degraders with a pyrrolidine and 1,3-substituted piperidine linker induced modest degradation, while the secondary and tertiary amine containing compounds overall showed weak or no significant degradation of the PRC2 components. These differences in degradation efficiencies could be attributed to multiple factors including differences in cell permeability or ternary complex formation.
Stereoisomers UNC7700 and UNC7698 have different degradation profiles.
Due to the improved efficacy of UNC7327, we decided to synthesize the individual stereoisomers to better understand the active species(s). Degraders UNC7700 and UNC7698 were synthesized containing either a cis- or a trans-cyclobutane linker, respectively (Figure 3A). The two compounds were tested in a dose response format (0.01–3 μM) at 24 hr in DB cells via western blot (Figure 3B and C and Supplemental Figure 4). Intriguingly, while UNC7700 showed potent degradation of EED, EZH2WT/EZH2Y641N, and SUZ12 (EED DC50 = 111 nM, Dmax = 84%; EZH2WT/EZH2Y641N DC50 = 275 nM, Dmax = 86%; SUZ12 DC50 = N/A, Dmax = 44%), UNC7698 degraded EED with a more modest Dmax and did not display significant EZH2WT/EZH2Y641N and SUZ12 degradation under these conditions (EED DC50 = 67 nM, Dmax = 64%; EZH2WT/EZH2Y641N DC50 = N/A, Dmax = 15%; SUZ12 DC50 = N/A, Dmax = 29%). This result was further corroborated by quantitative targeted proteomics analysis of the levels of EED, EZH2WT/EZH2Y641N, and SUZ12 in DB cells treated with 3 μM compound for 24 hr (Figure 3D). Analysis of UNC7700 and UNC7698 in our EED TR-FRET assay revealed that both compounds are equally potent binders of EED and they bind with comparable affinities to UNC6852 (Figure 3E). This indicates that the improved degradation of EED with UNC7700 and UNC7698 relative to UNC6852 is not a result of more potent EED binding, and the different degradation profiles of UNC7700 and UNC7698 are also independent of EED binding.
Figure 3.
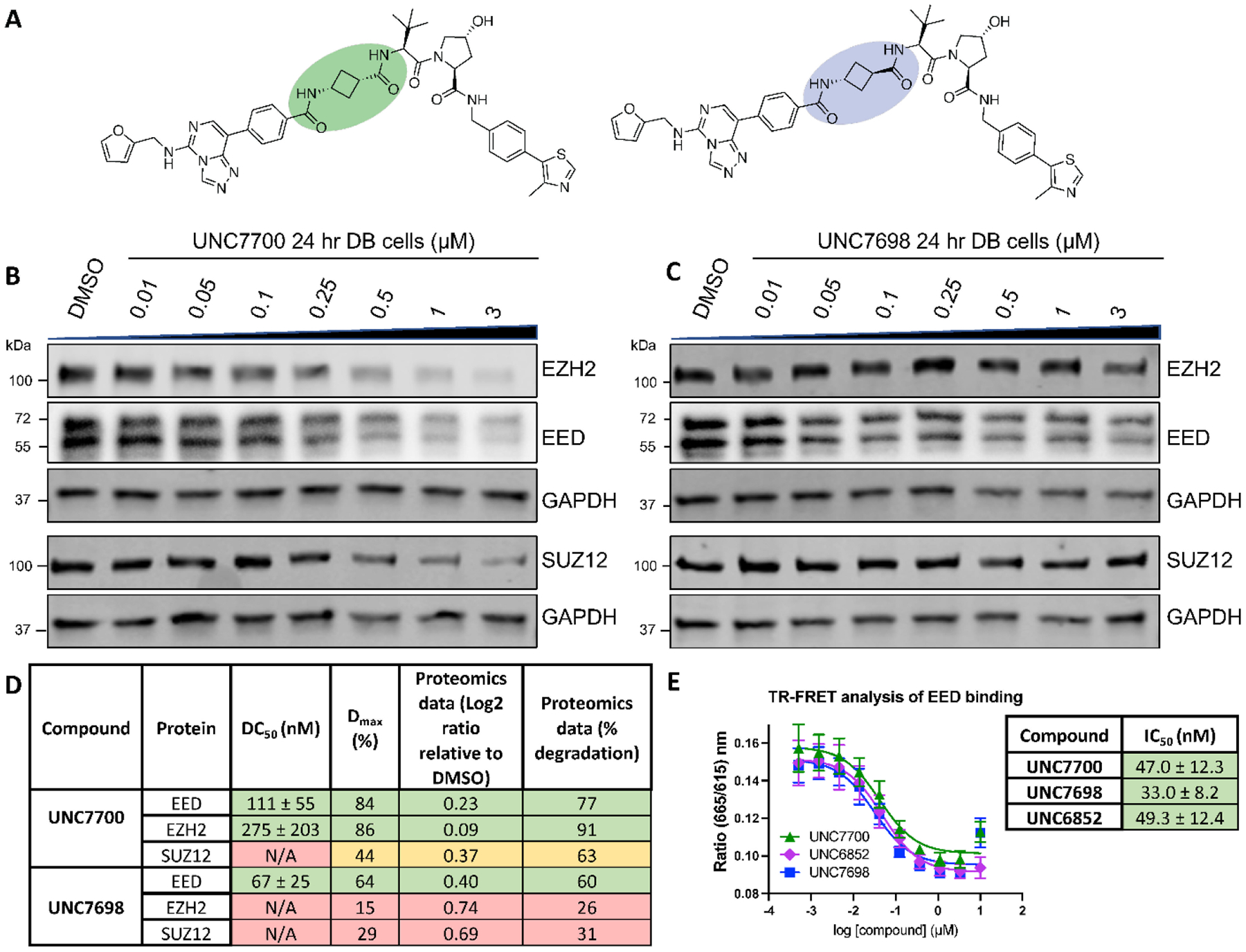
Structurally similar PRC2 degraders, UNC7700 and UNC7698, demonstrate improved degradation compared to UNC6852 and different degradation profiles. A) Structures of UNC7700 (cis-cyclobutane degrader) and UNC7698 (trans-cyclobutane degrader). B) Western blot analysis of PRC2 components following UNC7700 treatment of DB cells in a dose response format (0.01 – 3 μM, 24 hr). GAPDH is a loading control. C) Western blot analysis of PRC2 components following UNC7698 treatment of DB cells in a dose response format (0.01 – 3 μM, 24 hr). GAPDH is a loading control. D) Quantification of western blots in B) and C). Data are representative of 3 biological replicates ± standard error. Quantification of the extent of EED, EZH2, and SUZ12 degradation as assessed by mass spectrometry-based targeted proteomics analysis. E) EED IC50 values determined by TR-FRET. Data are representative of 3 biological replicates ± the standard deviation.
UNC7700 mediated degradation is VHL and proteosome dependent.
To confirm that UNC7700 facilitates PRC2 degradation through the recruitment of VHL, we synthesized a negative control compound, UNC7971, which has a cis-hydroxyproline in the VHL binding region of the molecule (Figure 4A). This compound is unable to bind to VHL and as a result does not elicit degradation of PRC2 components (Figure 4B). Furthermore, addition of the proteasome inhibitor MG132 prior to UNC7700 cell treatment blocks PRC2 degradation, confirming that degradation is proteosome dependent (Figure 4C).
Figure 4.
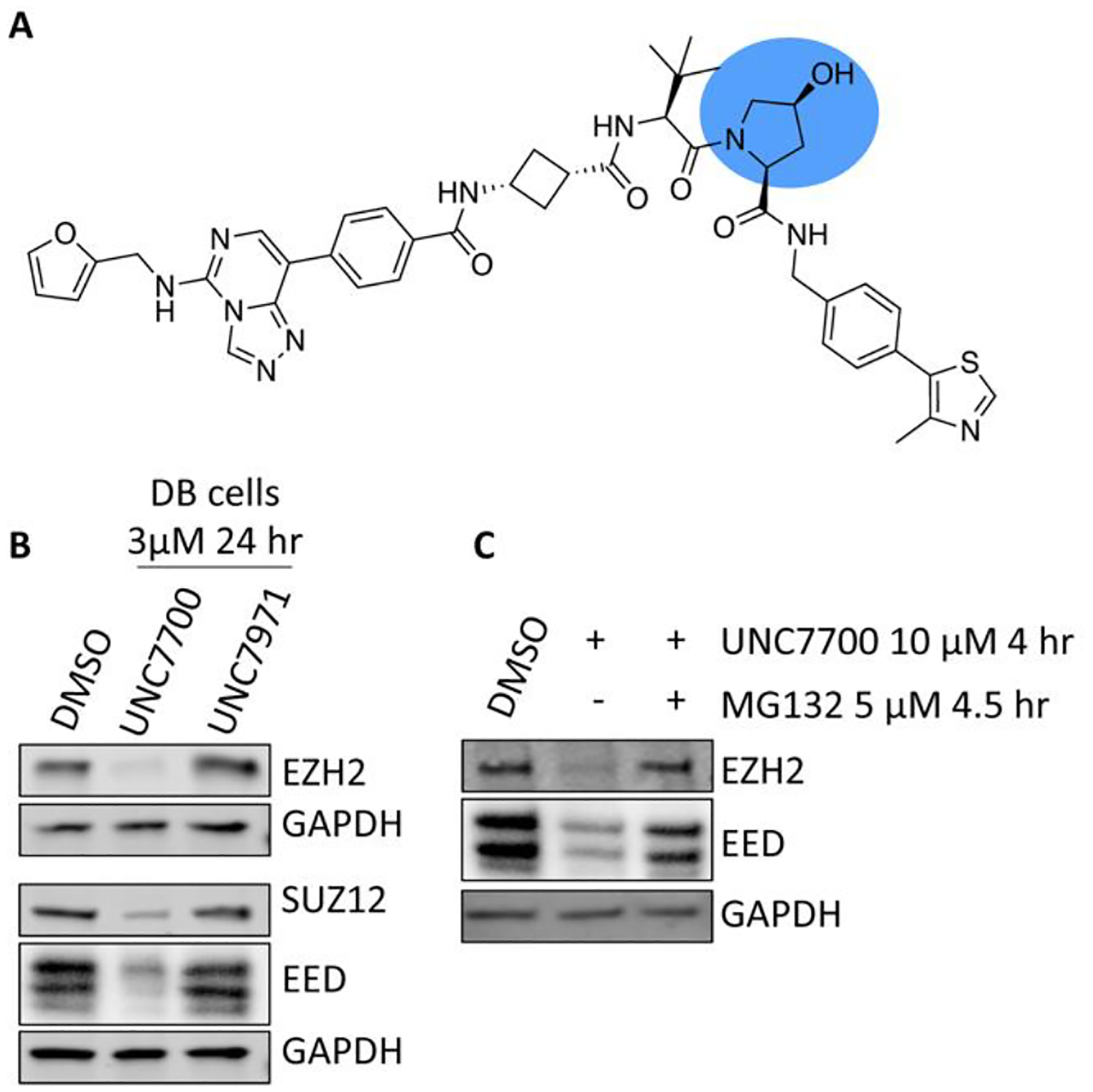
UNC7700 mediated PRC2 degradation is VHL and proteosome dependent. A) Chemical structure of negative control degrader UNC7971. B) Negative control UNC7971 does not bind to VHL and hence does not induce PRC2 degradation. C) Addition of proteosome inhibitor MG132 blocks proteosome mediated degradation.
PROTAC fragments demonstrate a correlation between cell permeability and degradation efficacy.
To better understand the differences in degradation efficacies between our closely related degrader molecules, we sought to quantitatively explore structure-permeability relationships using the chloroalkane penetration assay (CAPA). Appending a chloroalkane PEG tag (ct) to a compound-of-interest enables assessment of relative cytosolic compound concentrations based on covalent reaction with a stably expressing HaloTag-GFP fusion located in the outer mitochondrial membrane.18,19 In this pulse-chase assay, cells are pulsed with the ct-compound and chased with a chloroalkane TAMRA dye to determine the extent of ct-compound that has entered the cytosol of the cell, as the observed fluorescence intensity is inversely proportional to the cellular penetration of the ct-molecule. Critically, the CAPA assay enables rank ordering of permeability for low permeability compounds that cannot be adequately distinguished in standard permeability assays such as Caco-2.20
We first synthesized small molecule fragments of a subset of our degraders containing the EED ligand, linker region, and a chloroalkane PEG tag (ct) (Figure 5A and Supplemental Table 2).18,20 We prepared ct-compounds containing the cis- and trans-cyclobutane linker isomers, EED-cis-ct (UNC7792) and EED-trans-ct (UNC7794), based on UNC7700 and UNC7698, respectively (Figure 5A). EED-linker-ct fragments were also synthesized based on degraders UNC6852, UNC7326, UNC7741, UNC7743, and UNC7744 (Supplemental Figure 5). CAPA analysis for these PROTAC fragments after 4 hours showed that in general, an increase in permeability correlated with an increase in degradation efficiency for the corresponding PROTACs (Supplemental Figure 5). Remarkably, a stark contrast in permeability was observed between EED-cis-ct (UNC7792) and EED-trans-ct (UNC7794), with EED-cis-ct being ~1000-fold more permeable than EED-trans-ct (Figure 5B). EED-propyl-ct (UNC7706) resulted in a CP50 that was about half-way between the two cyclobutyl compounds. Overall, this reinforces that subtle differences in linker structures have the potential to influence compound permeability in a meaningful way.
Figure 5.
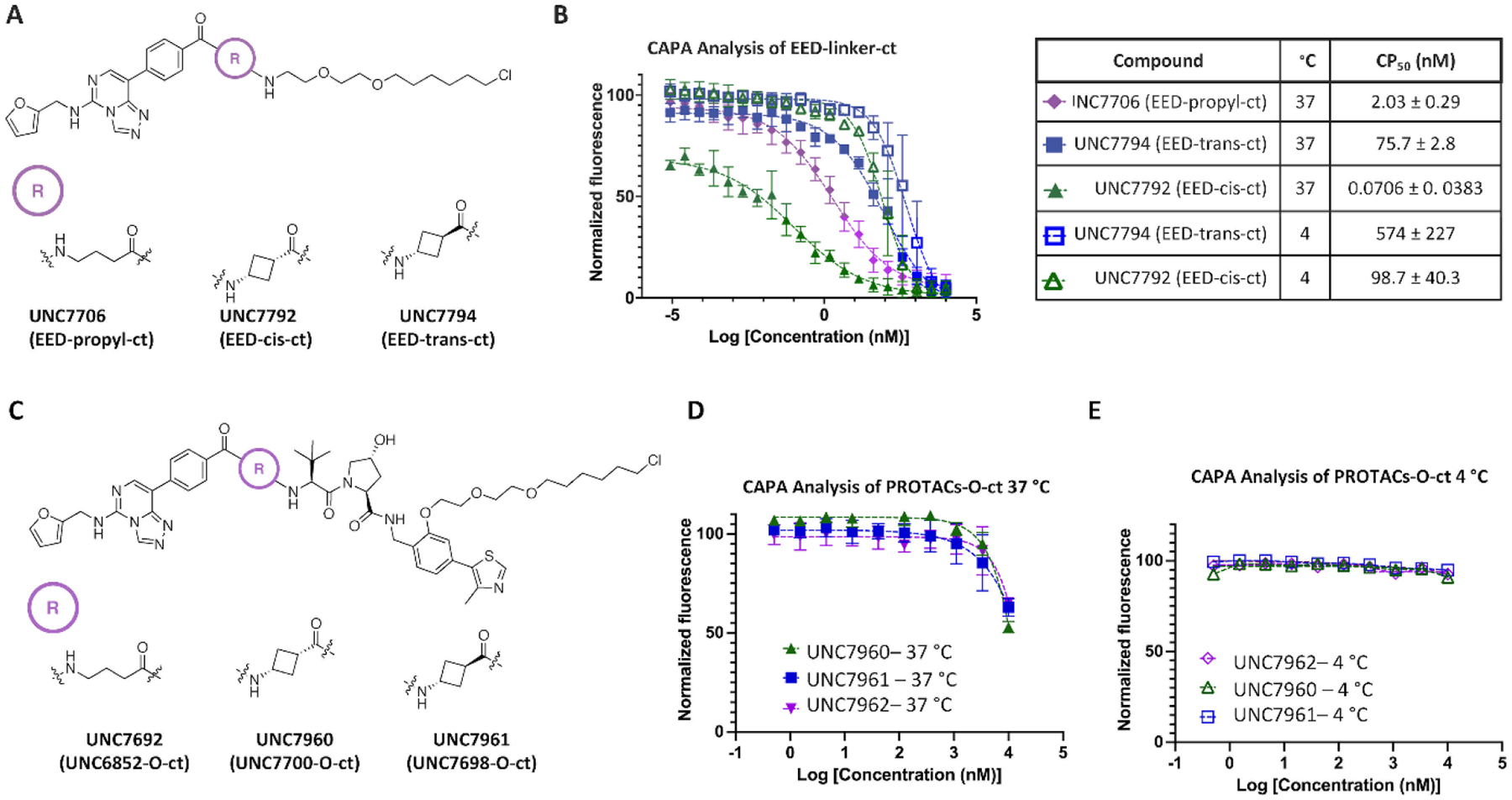
CAPA facilitates evaluation of the permeability of degraders and degrader fragments. A) Chemical structures of fragment chloroalkane tagged compounds EED-propyl-ct, EED-cis-ct, and EED-trans-ct. B) CAPA analysis of select EED-linker-ct fragments in A) at 4 °C and 37 °C and the corresponding CP50 values ± standard deviation. C) Chemical structures of chloroalkane tagged PROTACs (PROTAC-O-ct) UNC6852-O-ct, UNC7700-O-ct, and UNC7698-O-ct. D) CAPA analysis of PROTAC-O-ct compounds in C) at 37 °C indicating no significant differences in permeability. E) CAPA analysis of PROTAC-O-ct compounds at 4 °C shows no cell permeation.
Ct-UNC7700 and ct-UNC7698 have similar permeability via CAPA analysis.
We were next interested in evaluating if the permeability trends that were seen with our degrader fragments were similarly observed in the context of the larger degrader molecules containing the VHL-recruiting ligand. Ct-degrader molecules were synthesized with the ct incorporated within the VHL ligand at a position known to be amenable to functionalization (Supplemental Chemistry Information).21,22 Specifically, an O-linked ct on the phenyl ring of the VHL ligand provided an intermediate to facilitate the synthesis of ct-PROTACs in a convergent fashion (Figure 5C, Supplemental Scheme 7). We synthesized ct-PROTACs based on UNC6852 (UNC6852-O-ct, UNC7692) and our new degraders (UNC7700-O-ct, UNC7960 and UNC7698-O-ct, UNC7961). Intriguingly, comparison of the cellular permeability of these ct-PROTACs resulted in comparable levels of permeability (Figure 5D). All ct-PROTACs were also significantly less permeable than their corresponding ct-fragments. Extending the compound incubation time in this assay from 4 hours to 8, 24, and 48 hours did not result in increased permeability, indicating that the ct-PROTACs do not need increased incubation (or pulse) times to reach the cytosol (Supplemental Figure 6).18,19
Due to the differences in size of the ct-fragments relative to the full ct-PROTACs, it is possible that the two sets of compounds utilize different mechanisms to enter the cell. If the ct-PROTACs utilize an energy-dependent mechanism of uptake as opposed to passive diffusion, this could be determined by incubating them at a decreased temperature of 4 °C.18,19 Active transport mechanisms such as endocytosis rely on energy transfer; therefore, the permeability of the compounds should be impeded at decreased temperatures if they are entering the cytosol via active transport.23,24 At a reduced temperature the membrane should become rigid which limits the effects of compound entry by endocytosis and active transport mechanisms.25 When we performed CAPA at 4 °C, none of the ct-PROTACs entered the cytosol to a measurable extent, suggesting a potential active transport route of entry (Figure 5E). For comparison, we also tested our fragment compounds, EED-cis-ct (UNC7792) and EED-trans-ct (UNC7794), in the CAPA assay at 4 °C. We observed a modest change in permeability with the trans-isomer (8-fold decrease), while the cis- isomer displayed a 1397-fold decrease in observed permeability at 4 °C, suggesting that entry by an active transport mechanism plays a larger role (Figure 5B). We speculate that the observed permeability differences of the fragment compounds EED-cis-ct and EED-trans-ct are indicative of the true permeability differences between UNC7700 and UNC7698, and that adding the ct-label to the full PROTACs obscures this trend, either by changing the mechanism of cell permeability or limiting the overall permeability so that these differences can no longer be discerned. This reveals a limitation present in all studies dependent on labelled compounds, as labels are not free from their own effects on the property measured.
We tested the permeability of UNC7700 in a Caco-2 assay and confirmed that the permeability was below the limit of detection of the assay (Supplemental Table 3). This is in line with our previously published data showing that Caco-2 was unable to assess the permeability of PROTACs such as MZ1.20 This further demonstrates that CAPA has a larger dynamic range for assessing and comparing the permeability of PROTACs. Additionally, the percent recovery of UNC7700 from the donor compartment, cell lysate, and assay well varied from 79–106%, suggesting that the poor Caco-2 permeability observed was not due to non-specific binding, membrane partitioning, or lysosomal trapping, among other mechanisms for decreased recovery.26
UNC7700 has lower conformational mobility compared to UNC7698.
To further explore the physical properties of the cis- and trans-PROTAC isomers, UNC7700 and UNC7698, we investigated whether the compounds’ conformational dynamics may affect their cell permeability. To this end, molecular dynamics (MD) simulations of UNC7700 and UNC7698 were performed and analyzed. The conformational ensembles of the compounds were explored through clustering using the k-means technique.27 Sampled conformations were clustered into groups with root mean square distances (RMSD) between the group members of 1 ångström (Å) or less. The number of clusters can be considered as a proxy to conformational mobility of the degrader and the normalized cluster size as a proxy to the free energy of the corresponding metastable conformational state. The clustering metrics show that UNC7700 and UNC7698 display significant differences in their dynamic behavior. In particular, UNC7700 has lower conformational mobility than UNC7698. UNC7700 shows fewer clusters than UNC7698 (53 and 97, respectively) and the probability that UNC7700 will adopt its dominant conformation (corresponding to the largest cluster) is 15% versus 8% for UNC7698. We also found that the dominant, most stable conformations adopted by UNC7700 represent a peculiar, folded state in which the triazolo-pyrimidine aromatic system of the EED ligand stacks with the phenyl ring of the VHL ligand (Figure 6A). This contrasts with the most stable conformations of UNC7698 which are extended and lack this stacking interaction (Figure 6A). One corollary of the differing dynamic behavior of UNC7698 and UNC7700 is that, despite only minor differences in their chemical structures, they are perceived by their cellular environment as substantially different molecules. In particular, the polar solvent-accessible surface area of UNC7700 averaged over all three trajectories is 255 Å2 versus 314 Å2 for UNC7698 (the average molecular volumes are 976 and 1207 Å3, respectively). Therefore, UNC7700 is perceived by its environment as an overall smaller and more lipophilic molecule than UNC7698, and both of these characteristics are in general predictive of more effective cell penetration. This idea of PROTAC solution conformations influencing cell permeability was recently reported by Atilaw et al., and they confirmed this result by NMR analysis of molecular flexibility in solution (NAMFIS).28 Overall, our MD studies reveal that UNC7700 may have increased permeability due to its more compact and folded conformation relative to UNC7698, and this increase in permeability may be partially responsible for differences in degradation efficacies.
Figure 6.
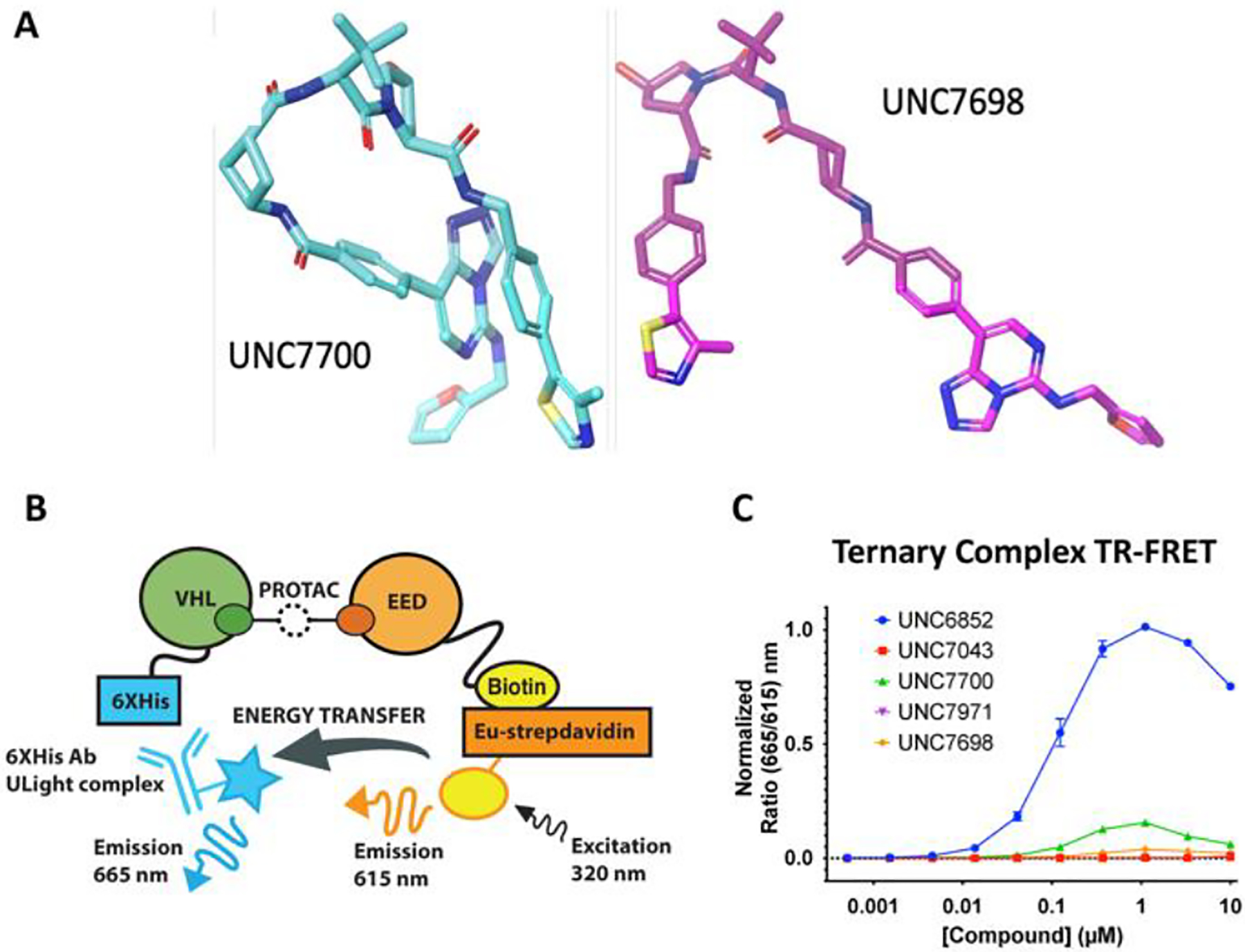
A) Molecular dynamics with UNC7700 and UNC7698 shows that UNC7700 has a compact structure whereas UNC7698 is more extended. B) Ternary complex assay with EED-biotin and His-VHL. A FRET signal is produced when the two proteins are in proximity. C) Ternary complex formation with EED and VHL is observed with UNC6852 and UNC7700.
Evaluation of ternary complex formation.
As the formation of a productive E3-degrader-POI complex can influence degradation kinetics and selectivity, we next assessed ternary complex formation in the presence of UNC7700, the corresponding negative control compound UNC7971, and UNC7698 with a FRET-based proximity assay (Figure 6B and C). We also tested our first generation PROTAC, UNC6852, and the corresponding negative control, UNC7043. Using Avi-tagged biotinylated EED and His-VHL, we first optimized the TR-FRET assay in the presence of UNC6852. The assay was performed in a dose response fashion with up to 10 μM UNC6852 due to the poor solubility observed above this concentration. The ternary complex assay showed signs of a bell-shaped curve in the presence of UNC6852; however, the concentration of UNC6852 was not high enough to generate a full bell curve10. After optimizing the assay conditions for UNC6852, the second generation PROTACs were similarly evaluated. Interestingly, the overall signal intensity for UNC7700 was much weaker compared to UNC6852. The observed decrease in signal intensity is likely the result of a change in orientation between EED-biotin and VHL-His in the presence of UNC7700. If the biotin tag on EED is positioned further away from the His tag on VHL, this could decrease the efficiency of energy transfer and the signal intensity.29 Therefore, the signal intensity in the assay is unique to each PROTAC ternary complex and is not necessarily reflective of degradation efficacy.16 Instead, it has been reported that the PROTAC concentration corresponding to the peak height for each degrader is indicative of the capacity to form a ternary complex and maximum degrader efficacy.30 Both UNC6852 and UNC7700 demonstrate a maximum peak height at around 1 μM, and UNC7698, which is able to partially degrade EED in DB cells, shows a small increase above baseline at a similar concentration (Figure 6B). Consistent with western blot data, negative control compounds UNC7043 and UNC7971 do not show any increase in FRET signal. Overall, these results confirm that a ternary complex is formed in presence of UNC7700, UNC7698, and UNC6852 as expected based on the fact that they are able to elicit some form of degradation; however, this suggests that the more rigid linkers of UNC7700 and UNC7698 result in a significant change in orientation between EED and VHL in this system relative to the more flexible linker of UNC6852 and that the extent of energy transfer is not necessarily related to degradation efficiency.
Computational investigation of the disparate degradation profiles of UNC7700 and UNC7698.
To better understand the differences in degradation profiles between UNC7700 and UNC7698 and elucidate how the dynamic properties of the cyclobutyl linkers affect ternary complex stability, we employed all-atom MD simulations of the ternary PRC2-PROTAC-E3 ligase complexes. First, models of possible EED-VHL complexes were obtained using HADDOCK, a protein-protein docking algorithm.31 The most plausible poses were selected under two constraints: (i) the resulting VHL pose did not overlap with the other proteins of the PRC2 complex (EZH2 and SUZ12), and (ii) the EED-VHL complex enabled connection of the monovalent EED and VHL ligands by the appropriate linkers without perturbing their co-crystal 3D poses (see Methods for more details on how initial 3D ternary complex models were built). The k-means clustering method was then used to depict the conformational free energy landscapes of the ternary complexes.27 The MD snapshots of ternary complexes were clustered in the feature space of Structural Protein-Ligand Interaction Fingerprints (SPLIF) (a threshold RMSD of 1 Å was applied as a cluster inclusion criterion).32 In SPLIF space, clusters can be interpreted as distinct ligand poses or microstates of ligand-protein interaction. Based on the clustering data, EED- and VHL-bound UNC7700 displayed a higher conformational mobility (a total of 3408 ligand poses) than UNC7698 (1402 ligand poses). This is a notable result given that in solvent, UNC7700 is less mobile than UNC7698, suggesting that UNC7698 is likely to be subject to a significantly higher entropic penalty upon ternary complex formation. Visual inspection of representative samples of MD snapshots shows significantly larger “clouds” of cartoon representations for PRC2-UNC7700-VHL complexes compared to the equivalent UNC7698 complexes (Figure 7A and B). Moreover, the VHL “cloud” in the UNC7700-bound complex tends to lean toward EZH2 to a larger extent than it does in the UNC7698-bound complex (Figure 7C and D). A quantitative analysis shows that throughout the simulations, VHL and EZH2 in the UNC7700-bound complex displayed on average 360 interacting atom pairs (defined as atoms closer than 4 Å from each other) and a maximum of 859 interacting atom pairs, while the average and maximum numbers for the UNC7698 complex were 219 and 559, respectively. The most likely reason for this closer EZH2-VHL interaction is that UNC7700, even when embedded in the ternary complex, tends to adopt a bent conformation (Figure 7E) that positions VHL toward EZH2, while the extended conformations adopted by UNC7698 do not promote similar interactions between VHL and EZH2. Overall, the MD simulation data comparing the two PRC2-PROTAC-VHL complexes provided sound structural rationale for our prior observations. The simulations suggest a mechanism by which UNC7700 binds the ternary complex more tightly than UNC7698 due to a less significant entropic penalty on binding. Furthermore, UNC7700 appears to induce more abundant interactions between VHL and EZH2 than UNC7698 which could explain why UNC7700 potently degrades EZH2 while UNC7698 does not.
Figure 7.
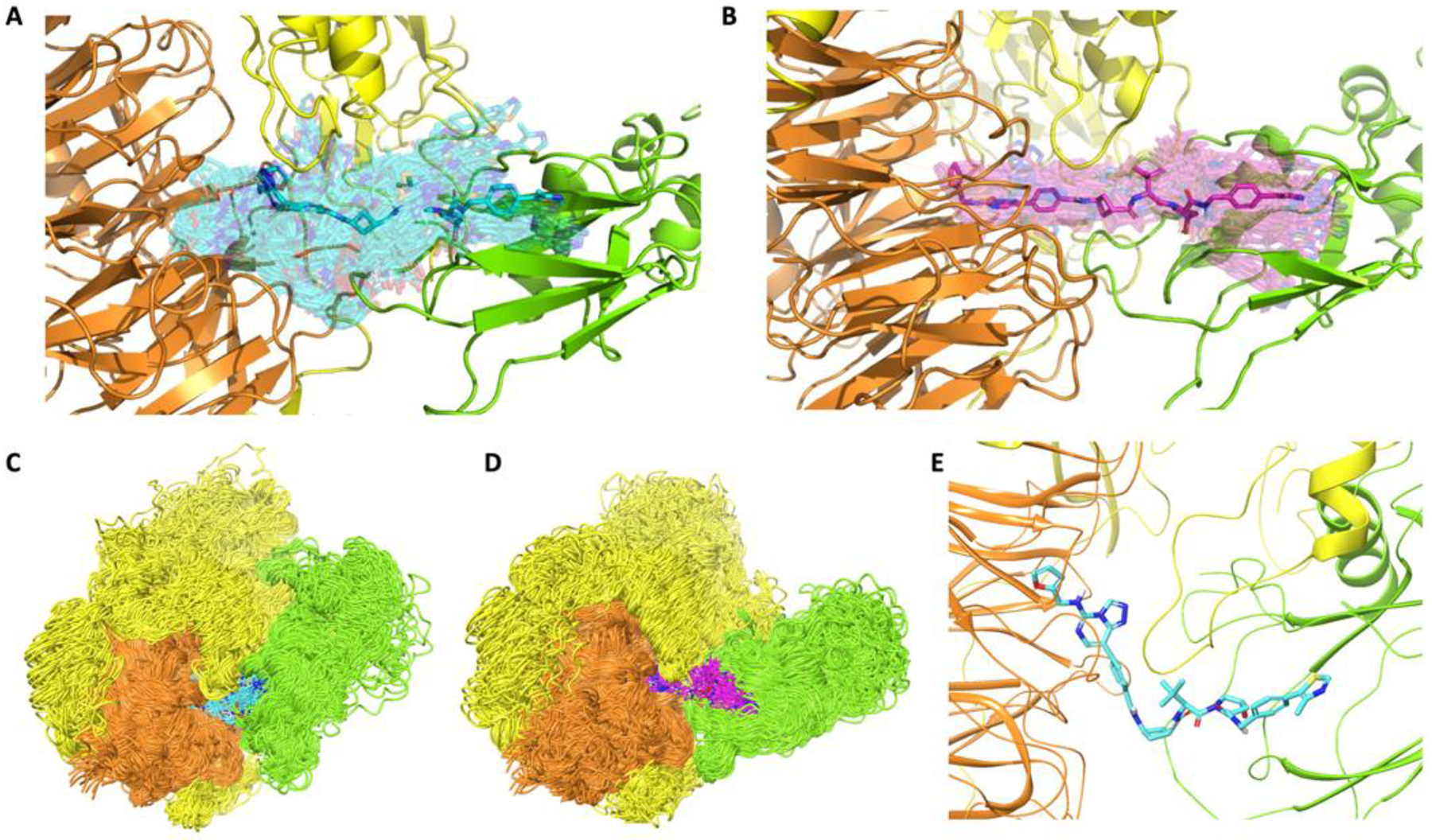
A, B) Representative ensembles of PROTAC conformations (transparent sticks) within ternary PRC2-PROTAC-VHL complexes for UNC7700 (A, cyan) and UNC7698 (B, magenta) sampled from MD trajectories. The most stable conformers in each ensemble are rendered in solid sticks. C, D) Representative ensembles of protein conformations within ternary PRC2-PROTAC-VHL complexes with UNC7700 (C, cyan) and UNC7698 (D, magenta) sampled from MD trajectories. E) A sample stable conformer of UNC7700 extracted from the MD trajectories of the ternary complex. VHL = Green; EED = Orange; EZH2 = Yellow.
UNC7700 decreases H3K27me3 levels and is anti-proliferative in DB cells.
We next sought to compare the effects of UNC7700, which potently degrades all core PRC2 components, on H3K27me3 levels to those of UNC7698, which selectively degrades EED. DB cells were dosed (3 μM for 96 hr) with our first-generation PRC2 degrader UNC6852, our second-generation degraders UNC7700 and UNC7698, the small molecule EED antagonist EED226, and the small molecule EZH2 inhibitor UNC1999 (Figure 8A, Supplemental Figure 7). Despite substantial degradation of EED and EZH2WT/EZH2Y641N with UNC6852 treatment, only a small decrease in H3K27me3 levels at this concentration and time point was observed. In contrast, UNC7700 showed complete degradation of EED and EZH2WT/EZH2Y641N and an 85% decrease in H3K27me3 levels, indicating that UNC7700 is substantially more effective than UNC6852. UNC7698 degrades EED to a much greater extent than EZH2WT/EZH2Y641N, which is consistent with our previous data at 24 hr in DB cells (Figure 3C); however, UNC7698 shows little decrease in H3K27me3 levels at this concentration and time point. Small molecule control compounds EED226 and UNC1999 decrease H3K27me3 levels by 63% and 76%, respectively.
Figure 8.
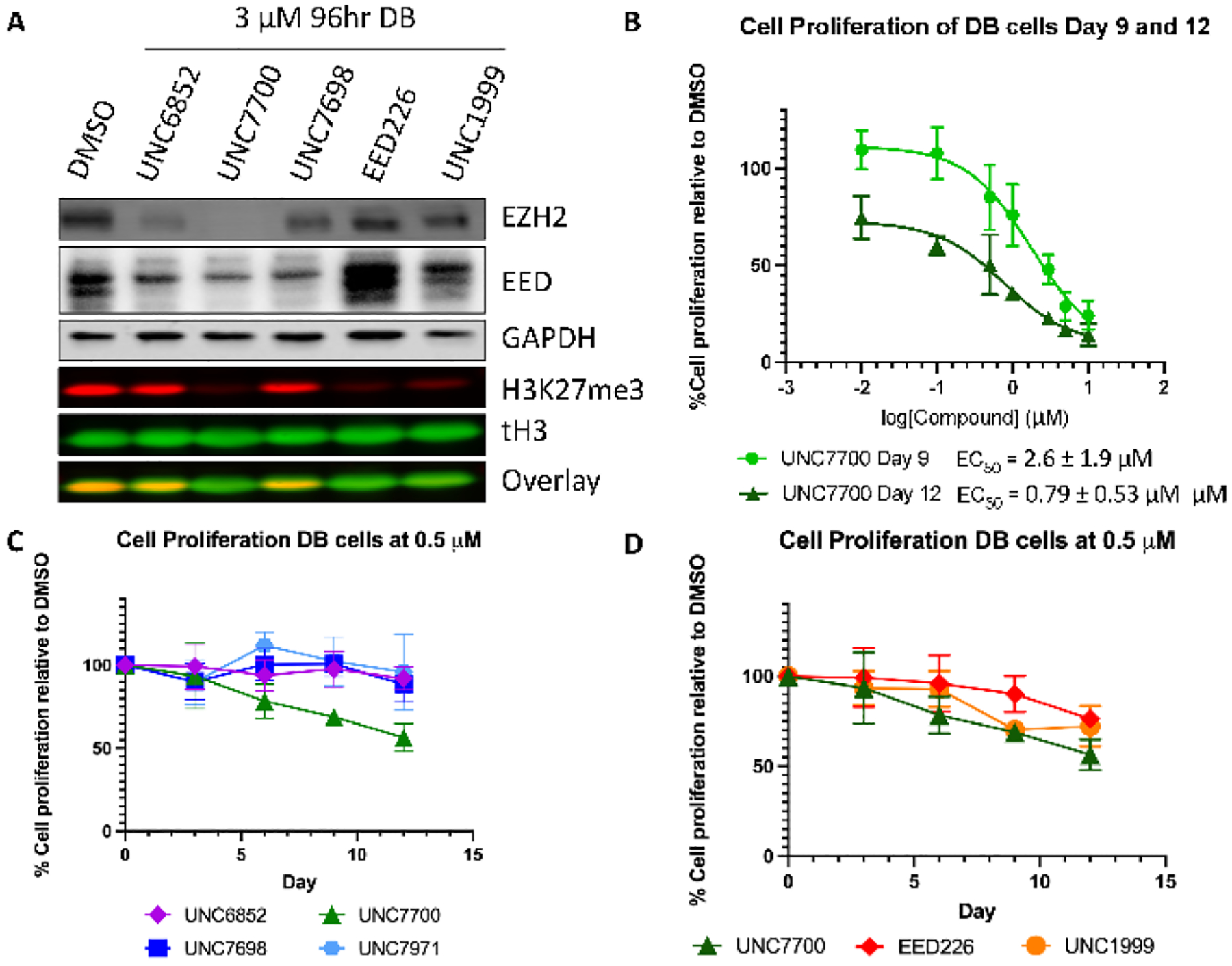
UNC7700 decreases H3K27me3 levels and is anti-proliferative in DB cells. A) Comparison of the degradation profiles of UNC7700, UNC7698, UNC6852, EED226, and UNC1999 and the resulting H3K27me3 levels. B) Treatment of DB cells with UNC7700 results in a decrease in cell proliferation in a dose response fashion. C) Effect of UNC7700 on DB cell proliferation compared to UNC6852, UNC7698, and negative control UNC7971 after 12 days at 500 nM. D) Effect of UNC7700 on DB cell proliferation compared to EED226 and UNC1999 after 12 days at 500 nM.
The effect of our second generation degraders on the proliferation of DB cells was tested in a dose response format (0.01 – 10 μM) for 12 days. UNC7700 had a significant impact on proliferation with an EC50 of 0.79 ± 0.53 μM after 12 days and 2.6 ± 1.9 μM after 9 days (Figure 8B); however, we observed about a 30% loss in cell viability after 12 days at the top concentrations tested (5 – 10 μM) (Supplemental Figure 8A). DB cells were next treated for 12 days with 0.5 μM UNC7700, UNC7698, UNC6852, EED226, UNC1999, and the negative control compound for UNC7700, UNC7971. After 12 days, there was no cell toxicity observed with any of the compounds at this lower concentration (Supplemental Figure 9B). Importantly, UNC7700 demonstrated the most significant anti-proliferative effect of the compounds tested. After 12 days, 0.5 μM UNC7700 reduced cell growth by 46% compared to DMSO and there was no reduction in proliferation with the corresponding negative control degrader UNC7971 (Figure 8C). UNC6852 and UNC7698 had only modest effects on cell growth under these conditions, with an 8% and 12% decrease in proliferation, respectively, compared to DMSO treated cells. Last, UNC7700 elicited a stronger antiproliferative effect than the potent small molecule inhibitors, EED226 (reduced by 24%) and UNC1999 (reduced by 28%) (Figure 8D).
In conclusion, we identified UNC7700, a more potent and efficacious PRC2 degrader than our previously reported degrader, UNC6852, from a screen of second-generation molecules with modifications to the linker region. Interestingly, we observed that UNC7700, which contains a cis-cyclobutane linker, differs from its closely related trans-isomer, UNC7698, in its PRC2 degradation profile. Specifically, UNC7700 potently degrades EZH2 in addition to EED whereas UNC7698 is more selective for EED. As expected, UNC7700 mediated PRC2 degradation was found to be both VHL and proteasome dependent. CAPA analysis with EED-linker-ct fragments suggested a difference in cellular permeability between the cis- and the trans-cyclobutane isomers, with the cis-cyclobutane isomer demonstrating significantly improved permeability. Consistent with this, molecular dynamics indicated that UNC7700 exists predominantly in a folded state with a lower solvent-accessible surface area, likely facilitated by intramolecular hydrogen bonding, while UNC7698 remains in an extended state that could result in decreased permeability. Therefore, these results support the idea that increased permeability of UNC7700 may be one factor that contributes to the improved activity of UNC7700.
Evaluation of ternary complex formation in the presence of UNC6852, UNC7700, and UNC7698 revealed that while all 3 compounds can effectively form an EED-PROTAC-VHL ternary complex, they likely do so with different orientations based on the differences in overall FRET signals observed. Molecular dynamics simulations with the PRC2-PROTAC-VHL complexes suggested that UNC7700 has a lower entropic penalty of binding to the ternary complex and more frequent and abundant interactions between VHL and EZH2 occur in the presence of UNC7700 as compared to UNC7698. These results provide a possible explanation as to why UNC7700 effectively degrades EZH2 while UNC7698 does not and point towards a mechanism whereby EZH2 is ubiquitylated directly in the presence of UNC7700; however, other possible explanations may still exist. For example, it is possible that the two compounds bind different variants of PRC2 or UNC7698 binds EED in the context of non-PRC2 proteins, leading to the different degradation profiles observed. While UNC7698 showed significant EED degradation with minimal loss of EZH2 and SUZ12, this did not result in a substantial decrease in H3K27me3 levels or anti-proliferative effects in DB cells. In contrast, UNC7700 resulted in near complete loss of EED, EZH2, and SUZ12, elicited substantial decreases in H3K27me3 levels, and demonstrated more robust anti-proliferative effects than the small molecule inhibitors EED226 and UNC1999 under the conditions tested.
Overall, this effort served to reinforce the complexity of PROTAC development, while also highlighting the broad potential impacts of subtle changes in the composition of the linker. We employed a variety of assays and techniques including CAPA, MD simulations, and a ternary complex assay to better understand the impacts of subtle linker modifications on degrader efficacy. As PRC2 remains a target of high therapeutic interest, UNC7700 may serve as an improved tool to evaluate the consequences of potent PRC2 degradation.
Methods
Protein Expression and Purification
His-VHL (residues 1–154, N-terminal tag, purchased from Novus Biologicals, Catalog No. NBC1-18497). His-tagged EED protein was purified as previously reported.10
Full length biotinylated EED (reference sequence AAD08714) was generated by cloning the EED coding sequence into a modified pET28 vector that includes an N-terminal hexahistidine tag, thrombin cleavage site, and an Avi tag sequence (GLNDIFEAQKIEWHE). The resulting construct was cotransformed into Rosetta2 BL21(DE3)pLysS competent cells (Novagen, EMD Chemicals, San Diego, CA) along with an expression plasmid for E. coli biotin ligase, BirA (Addgene plasmid # 20857).33 A 1 L culture was grown to mid log phase at 37 °C at which time the temperature was lowered to 16 °C and protein expression was induced by addition of 0.2 mM IPTG. D-biotin was added to the culture at this time to a final concentration of 50 μM. Expression was allowed to continue overnight. Cells were harvested by centrifugation and pellets were stored at −80 °C.
EED was purified by resuspending thawed cell pellets in 15 mL of BugBuster protein extraction reagent (EMD Millipore, Burlington, MA) supplemented with 1X EDTA free protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN), 30 mM imidazole and Benzonase nuclease (EMD Millipore, Burlington, MA). Cells were lysed by incubating at room temperature for 30 minutes with rocking. The cell lysate was clarified by centrifugation and loaded onto a 1 mL HisTrap FF column (Cytiva, Marlborough, MA) that had been preequilibrated with 10 column volumes of binding buffer (50mM sodium phosphate pH 7.2, 500 mM NaCl, 30mM imidazole) using an AKTA FPLC (Cytiva, Marlborough, MA). The column was washed with 15 column volumes of binding buffer and protein was eluted in a linear gradient to 100% elution buffer (50 mM sodium phosphate pH 7.2, 500mM NaCl, 500 mM imidazole) over 20 column volumes. Peak fractions containing the desired protein were pooled and concentrated to 2ml in an Amicon Ultra-15 concentrator 10,000 molecular weight cut-off (Merck Millipore, Carrigtwohill Co. Cork IRL).
The N-terminal hexahistidine tag was removed by thrombin cleavage according to manufacturer’s recommendations (Novagen, EMD Chemicals, San Diego, CA). Briefly, purified protein was incubated with thrombin at a final concentration of 1 unit thrombin per milligram tagged protein for 16 hours at 4 °C. The cleavage reaction was then passed over a 1ml HisTrap FF column (Cytiva, Marlborough, MA) as described above to remove any protein that still retained the tag. The column flow through was collected and concentrated to 1.75 mL using an Amicon Ultra-15 concentrator, 10,000 molecular weight cut-off (Merck Millipore, Carrigtwohill Co. Cork IRL).
Biotinylated EED was isolated by passing the 1.75 mL concentrated protein over a 2 mL monomeric avidin column by gravity flow following manufacturer’s recommendations (Pierce Biotechnology/Thermo Scientific, Rockford, IL). Bound protein was eluted in 12 mL PBS containing 2 mM D-biotin and concentrated to 2 mL in an Amicon Ultra-15 concentrator 10,000 molecular weight cut-off (Merck Millipore, Carrigtwohill Co. Cork IRL). Concentrated protein was loaded onto a HiLoad 26/60 Superdex 200 prep grade column (Cytiva, Marlborough, MA) that had been preequilibrated with 1.2 column volumes of sizing buffer (25mM Tris pH 7.5, 250 mM NaCl, 2 mM DTT, 5% glycerol) using an ATKA FPLC (Cytiva, Marlborough, MA). Protein was eluted isocratically in sizing buffer over 1.3 column volumes at a flow rate of 2 mL/min collecting 3 mL fractions. Peak fractions were analyzed for purity by SDS-PAGE and those containing pure protein were pooled and concentrated in an Amicon Ultra-15 concentrator 10,000 molecular weight cut-off (Merck Millipore, Carrigtwohill Co. Cork IRL). Concentrated protein was aliquoted and stored at −80 °C in sizing buffer.
EED Binding TR-FRET Assay
The EED TR-FRET assay was performed as previously reported.10,17 The assay conditions contain EED-6xHis (15 nM), Perkin Elmer ULight anti6xHis-antibody (10 nM, product code TRF0105-M), Perkin Elmer Lance Eu-W1024 Streptavidin (2 nM), biotinylated EED ligand UNC5390 also named UNC5114-biotin (15 nM). The compounds were tested as 10-point dose response curves with a top final concentration of 10 μM. Data are reported as three biological replicates ± standard deviation.
TR-FRET Ternary Complex Formation Assay
Formation of ternary complexes composed of EED, PROTAC and VHL was monitored by time-resolved fluorescence energy transfer from a europium donor to a ULight™ acceptor. Assays were run using white, low-volume, flat-bottom, nonbinding, 384-well microplates (Greiner Bio-One, Catalog No. 784904) containing a total assay volume of 10 μL per well with a final DMSO concentration of 1% (v/v). The assay buffer was composed of 20 mM Tris (pH 7.5), 150 mM NaCl, 0.05 mM Tween 20, and 2 mM DTT. LANCE Europium-W1024 Streptavidin (2 nM) and LANCE Ultra ULight™-anti-6xHis antibody (100 nM) were used as donor and acceptor fluorophores associated with biotinylated-EED (residues 1–441, N-terminal tag; 160 nM) and His-VHL proteins (residues 1–154, N-terminal tag; 320 nM), respectively. Compounds were evaluated using 10 point, three-fold dose responses beginning at 10 μM.
For every independent experiment, three technical replicates were performed simultaneously for each compound. A 10 point, three-fold serial dilution of each compound at 3X (3% v/v DMSO) final assay concentration was prepared in a separate 384-well plate. 10 no-compound control wells were also prepared to be used as a low signal control. A “master mix” composed of donor and acceptor fluorophores and both proteins was prepared at a 1.5X (0% DMSO) final assay concentration. A 16 channel VIAFLO electronic multichannel pipette (Integra, Part No. 4641) was used to plate the compounds and master mix at a 1X final assay concentration. The plates were sealed with clear covers, mixed gently on a tabletop shaker, and if needed due to the presence of bubbles, centrifuged at 1000 × g for 2 minutes, and allowed to incubate in a dark space. After 4 h, the plate was read on an EnVision 2103 Multilabel Plate Reader (PerkinElmer) using a 320 nm excitation filter and 615 and 665 nm emission filters. Emission signals were measured simultaneously using a dual mirror D400/D630 and 100-μsec delay. The TR-FRET output signal was expressed as a ratio of acceptor/donor (665/615 nm) emission counts. The data was then normalized by subtracting the averaged signal of the no-compound control wells. The normalized TR-FRET signals for each compound technical replicate were averaged and plotted against the dosing concentrations in GraphPad Prism. Two independent experiments were plotted simultaneously with errors bars to display the standard error from the independent experiments.
Cell Culture
DB cells (ATCC® CRL-2289™) were purchased from The Tissue Culture Facility at UNC Chapel Hill and were cultured in RPMI 1640 (Gibco) and 10% FBS (VWR Life Sciences Seradigm). MDA-MB-468 cells (ATCC® HTB-132™) were purchased from The Tissue Culture Facility at UNC Chapel Hill and were cultured in DMEM High Glucose, 10% FBS (VWR Life Sciences Seradigm), 1% Penicillin-Streptomycin (Sigma), 1% Non-essential amino acids (Sigma). HeLa cells stably expressing the HaloTag-GFP-Mitochondria construct were provided by the Kritzer lab.1 Cells were cultured in DMEM high glucose media (Sigma) supplemented with 10% FBS (VWR Life Sciences Seradigm), 1% Penicillin-Streptomycin (Sigma), and 1 μg/mL Puromycin (InvivoGen) to select for Halo-Tag expressing populations. All cell lines were incubated at 37°C with 5% CO2.
Cell Dosing and Lysis
For degradation analysis, cells were cultured in 6 well plates (Olympus Genesee Scientific, 25–105) and dosed with the appropriate concentration of bivalent chemical degrader from a DMSO stock. Adherent cells (MDA-MB-468) were seeded at 600,000 cell/well for 24 hr analysis, and 200,000 cells/well for 96 hr analysis. At the appropriate time point, cells were washed with PBS (2 × 1 mL), scraped in cold PBS (1 mL). centrifuged, aspirated, and lysed in 40–50 μL of Cytobuster lysis buffer (Cytobuster™ (71009), 1X Protease Inhibitor Cocktail (Active Motif, 37490), 2 μL/mL Benzonase® nuclease (Millipore, ≥ 90% SDS page, E1014). Non adherent cells (DB) were seeded at 800,000 cells/well for 24 hr analysis and 50,000 cells/well for 96 hr analysis. At the appropriate time point, cells were centrifuged, aspirated, washed with PBS (1 mL) and this process repeated twice. The cells were lysed in 40–50 μL of Cytobuster lysis buffer.
The protein levels were quantified using Protein Assay Dye Reagent Concentrate (Bio-Rad, 5000006) using a known concentration of BSA standard.
Western Blot Analysis
Cell lysate (20 mg – DB cells; 25–30 mg MDA-MB-468 cells) was combined with Laemmli buffer (Bio-Rad; 2X - 1610737 or 4X - 1610747) containing 2-mercaptoethanol (5%) and samples were boiled at 95 °C prior to gel loading. Gels (15mL, 15 well; 4–15% precast mini-PROTEAN TGX gels, Bio-Rad, 4561046DC; or 4–15% precast mini-PROTEAN TGX Stain Free gels, Bio-Rad 4568086) were placed in a Mini-PROTEAN tetra cell at 200V in 1X Tris/Gycine/SDS running buffer (Bio-Rad, 1610772). Molecular weight ladders used were either Precision Plus Protein Dual Color Standard (Bio-Rad, 161–0374), or PageRuler Plus pre-stained protein ladder (ThermoFisher, 26619). Protein was transferred onto Immobilon-FL PVDF Membranes (Millipore Sigma, IPFL00010), with 1X Tris/Gycine transfer buffer (Bio-Rad, 1610772) and methanol (0.2% volume) at 100V for 1 hr at 4 °C. Membranes were blocked at room temperature for 1 hr with Odyssey blocking buffer (TBS, LI-COR, 926–31099), and the incubated with primary antibodies overnight at 4 °C.
Primary Antibodies
anti-EED (1:500, R&D Systems, AF5827), anti-EZH2 (1:1000, D2C9 XP Cell Signaling Technology, 5246S), anti-SUZ12 (1:500, D39F6 XP Cell Signaling Technology, 3737S), anti-GAPDH-AlexaFluor 680 (1:5000, Abcam, ab184095), anti-GAPDH (1:5000, EMD Millipore, AB2302), anti-H3K27me3 (1:2000, Abcam, ab6002), anti-Histone H3 (1:5000, Abcam, ab1791), anti-HA (Abcam, 1:500, ab9110). Membranes were incubated with the corresponding secondary antibodies for 1hr at room temperature prior to imaging. Fluorescence imaging was performed on a LI-COR Odyssey. For chemiluminescent detection, membranes were activated with ECL Prime western blotting detection reagent (Amersham, RPN2232) and imaged on a Bio-Rad Chemidoc.
Li-COR Fluorescent Antibodies
IR Dye 680RD (1:10000, Goat anti-mouse, LI-COR, 926–68070), IR Dye 800CW (1:10000, Goat anti-rabbit, LI-COR, 926–32211).
Chemi-Doc Chemiluminescent Secondary Antibodies
Goat anti-chicken HRP (1:10000, LifeTech/Novex, A16054), Donkey anti-sheep HRP (1:10000, LifeTech/Novex, A16041), Donkey anti-rabbit HRP (1:10000, LifeTech/Novex, A16035).
Western Blot Quantification
Western blots were analyzed by firstly calculating the densitometry on either ImageStudio software or ImageLab software for LICOR or Chemidoc imaging, respectively. The densitometry of EED was calculated by quantifying the lower band at ~55 kDa based upon our previously reported antibody validation.10 The densitometry of the protein of interest band relative to the densitometry of each corresponding GAPDH band was calculated. The resulting densitometry relative to the DMSO band was calculated to give the % degradation. For the dose response and time study these values were plotted in GraphPad Prism against the corresponding concentration or time of degrader treatment. An inhibitor concentration vs response (three parameters) regression was plotted and the IC50 values were taken from G-raphPad Prism which corresponded to either the apparent half-life (t1/2, when protein levels were plotted against time), or the half maximal degradation concentration (DC50, when protein levels were plotted against concentration). The maximal degradation (Dmax) was calculated based on the % degradation at 3 μM after 24 hr.
Proteomics
Sample Preparation:
DB cells were seeded in T25 cm plates (0.8 million cells/mL in 10 mL total media) and dosed with DMSO, UNC7700, or UNC7698 (3 μM for 24 hr). After 24 hr the cells were centrifuged, media aspirated, and washed 3 × PBS with cells centrifuged at 4 °C. Cell lysis was for 30 mins on ice with 400 μL of lysis buffer (8M urea in 50 mM Tris-HCl pH 7.8, 1X protease inhibitor cocktail and 1X phosphatase inhibitor cocktail). The lysis mixture was centrifuged at 14000 rpm for 15 mins at 4 °C and the supernatant collected. Protein lysates (200 μg; n=3) were reduced with 5 mM DTT at 56 °C for 30 min, then alkylated with 15 mM iodoacetamide at room temperature in the dark for 45 min. The samples were diluted to 1M urea and subjected to digestion with LysC (Wako) for 2 h and trypsin (Promega) overnight at 37 °C at a 1:50 enzyme:protein ratio. The resulting peptide samples were acidified to 0.5% TFA, desalted using Thermo desalting spin columns, then the eluates were dried via vacuum centrifugation. Peptide concentration was determined using Pierce Quantitative Fluorometric Peptide Assay, then all samples were diluted to 0.5 μg/μL concentration prior to LC-MS/MS analysis.
LC-MS/MS Analysis:
The peptide samples (1.5 μg) were analyzed by LC-MS/MS using an Easy nLC 1200 coupled to a QExactive HF mass spectrometer (Thermo Scientific). Samples were injected onto an Easy Spray PepMap C18 column (75 μm id × 25 cm, 2 μm particle size) (Thermo Scientific) and separated over a 2 hr method. The gradient for separation consisted of 5–48% mobile phase B at a 250 nL/min flow rate, where mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in 80% ACN. Peptide identification was confirmed for the peptides of interest using a data-dependent acquisition method (DDA), then a targeted parallel reaction monitoring (PRM) method was developed for specific Suz12, EED and EZH2 peptides (3 peptides per protein). The QExactive HF was operated in MS1 and PRM mode. Resolution for the precursor scan (m/z 400–1200) was set to 60,000 with a AGC target set to 1e6 and a maximum injection time set to 200 ms. PRM scans (15,000 resolution) consisted of higher collision dissociate (HCD) set to 27; AGC target set to 1e6; maximum injection time set to 100 ms; isolation window of 2 Da.
Data Analysis:
For the untargeted DDA analysis, raw data files were processed using Proteome Discoverer version 2.1 (Thermo Scientific). Peak lists were searched against a reviewed Uniprot human database, appended with a common contaminants database, using Sequest. The following parameters were used to identify tryptic peptides for protein identification: 10 ppm precursor ion mass tolerance; 0.02 Da product ion mass tolerance; up to two missed trypsin cleavage sites; (C) carbamidomethylation was set as a fixed modification; (M) oxidation was set as a variable modification. Peptide false discovery rates (FDR) were calculated by the Percolator node using a decoy database search and data were filtered using a 1% FDR cutoff. For the targeted, PRM analysis, spectral libraries of the targeted peptides were generated from the DDA Proteome Discoverer results. Raw data were imported into Skyline and peak areas were extracted from the MS2 data (up to 10 fragment ions per peptide). One quantifiable peptide per protein was used to calculate log2 ratios using extracted peak areas (compound treatment versus DMSO control).
Chloroalkane penetration assay (CAPA)
CAPA was performed as previously described with some modifications.1 HaloTag- GFP HeLa cells were seeded at 5000 cells per well on a 384-well plate and allowed to adhere overnight. For every independent experiment, three technical replicates were performed simultaneously for each compound. On the day of the experiment, compounds were prepared in a separate 384-well plate at final 1X concentration and total volume of 60 μL per well. Twenty concentration points were generated by performing a three-fold serial dilution of 1 mM DMSO compound stocks in HeLa media, ensuring a final DMSO concentration of 1%. Compound-free control wells containing 1% DMSO in media were also prepared to be used as no-pulse (100% signal) and no-pulse/no-chase (0% signal) controls. The media of the 384-well assay plate containing cells was then removed and 50 μL of the compound and control samples from the dilution plate were added to each well. The plate was incubated for 4 h at 37°C with 5% CO2. The media was then removed, and the cells were washed with phenol red-free Opti-MEM (Gibco) and incubated for 30 min. The media was then removed, and compound and no-pulse control wells were chased with 5 μM ct-TAMRA—synthesized according to literature procedures1 and prepared in phenol red-free Opti-MEM, and incubated for 30 min. The no-pulse/no-chase control wells were washed with phenol red-free Opti-MEM instead. The media was then removed, and the wells were washed with phenol red-free Opti-MEM media supplemented with 10% FBS and 1% Penicillin-Streptomycin and incubated for 15 min. The media was then removed, and the wells were washed with PBS (Corning). The media was then removed, and the cells were trypsinized with phenol red-free 0.5% trypsin-EDTA (Gibco) diluted 1:1 with PBS. After a 20 min incubation, the cells were quenched and resuspended with 50% FBS in PBS and analyzed by flow cytometry (iQue Screener PLUS, Intellicyt®).
The normalized fluorescence signals for each compound technical replicate was plotted against the log of the dosing concentration. GraphPad Prism was used to fit the dose response curves using the “log(inhibitor) vs. response -- Variable slope (four parameters)” model. CP50 values generated from the antilog of the fitted LogIC50 values were recorded for each compound for every independent experiment. The CP50 values were averaged across the independent experiments, and the standard error of the mean was determined.
Caco-2
Compounds were evaluated by Sai Life Sciences Ltd. at the DMPK testing facility in India. Methods are as previously reported.20
Computational Docking
Molecular dynamics (MD) simulations for all systems were performed using the Gromacs 2018.2 simulation package with CHARMM27 protein force field.34 The 3D structures of ternary complexes PRC2-ligand-VHL were built in several steps. First, EED and EZH2 – two proteins putatively involved in interactions with the other members of the ternary complex – were extracted from the crystal structure of PRC2 (PDB: 5HYN). Second, the structure of EED-226, a monomeric EED-binding portion of our bifunctional ligands, was docked to EED by 3D alignment of the EED-EZH2 complex with the EED-bound EED-226 (5WUK). Third, we used HADDOCK a protein-protein docking algorithm, to generate structures of EED-VHL complexes (see below the details of the docking procedure).31,35 Next, the structure of “VHL Ligand 1”, a monomeric VHL-binding portion of our bifunctional ligands, was docked to VHL by 3D alignment of the EED-EZH2-VHL with the VHL-bound Ligand 1 (the latter complex was extracted from 6HAX, a VHL-PROTAC-SMARCA2 complex). Finally, EED-226 was connected to VHL Ligand 1 by an appropriate linker to result in a protein-bound bifunctional ligand. All the above steps were done using the Maestro modeling suite (release 2016–2, Schrödinger, LLC: New York, NY). The generated models of the ternary complexes with UNC7700 and UNC7698 served as starting points for MD simulations. CHARMM22 force field parameters for UNC7700 and UNC7698 were generated by Swissparam.36 End caps were added to both termini of each protein. The protein complex was minimized in vacuum using steepest decent algorithm for 5,000 steps or until the maximum force of 1,000 kJ*mol−1*nm−1 was reached. The molecular systems were then solvated in TIP3P water, counterions were added to ensure the systems’ electric neutrality, and NaCl ions (0.15 M) were added by randomly replacing certain water molecules in order to mimic physiological conditions.37 An energy minimization with solvent was then performed, followed by a two-step equilibration: 5 ns in NVT ensemble at 310 K using the modified Berendsen thermostat and 5 ns in NPT ensemble at 1 atm (and 310 K) using the Parinello-Rahman pressure coupling.38 All simulations were conducted using the Leapfrog integrator in periodic boundary conditions. The particle mesh Ewald algorithm controlled the long-range electrostatic interactions.39 Bonds involving hydrogen atoms were constrained using the linear constraint solver algorithm (LINCS).40 The production simulations were performed in NVT ensemble. For each of the three systems, three independent ~5 μs MD simulations were run. Molecular visualizations were produced using Maestro [Schrodinger, LLC]. MD trajectories were clustered and analyzed by means of the Pipeline Pilot data processing environment (v. 18.1.100.11, BIOVIA, 3dsbiovia.com). The input data (sets of the protein’s atomic coordinates and the backbone f and y angles) were generated from the MD trajectories using custom Pipeline Pilot scripts (protein structures were centered and aligned using the Gromacs trjconv tool). The clustering technique used was k-means with Euclidian distance metrics. The cluster aggregation criteria were chosen so that root mean square distances (RMSD) between the cluster members would be on the order of 1 Å.
EED-VHL docking calculations were performed using the HADDOCK web service31,35. Two protein structures were selected for docking – EED extracted from the PRC2 structure (5HYN) and VHL extracted from 6HAX, a VHL-PROTAC-SMARCA2 complex. A set of default HADDOCK parameters was used for all docking simulations. The parameter file and all input and output HADDOCK files are available upon request. Top ten docking poses were visually inspected in the context of PRC2-VHL complex. A VHL pose that did not overlap with EZH2 was selected for the ternary complex model.
Cell Proliferation Analysis
Exponentially growing DB cells were seeded in a 12 well plate (Corning Costar, CLS3513) at a cell density of 100K cells/mL in complete media (2 mL). Every 3 days the media was exchanged, cells were split back to the seeding density, and the compound or DMSO control were re-dosed. At each time point the cells were counted on an automated Bio-Rad TC20 cell counter with Trypan blue (Abcam, ab233465) and cell counting slides (1450015) to give the cell count (cells/mL) and cell viability (%). The % cell proliferation is calculated based on the total cell number relative to the DMSO control at the same time point. To determine an EC50, the % confluency of the cells relative to DMSO were plotted against log[inhibitor concentration] and the results were analyzed in GraphPad Prism with a log(inhibitor) vs response - variable slope (four parameters). EC50 values and are reported as an average of three biological replicates ± standard deviation.m
Supplementary Material
Acknowledgements
Our table of contents graphic was generated by biorender.com. The authors thank M. Uguen and J. Tabor for reviewing the primary chemistry data and R. Johnson and P. Buttery for reviewing the primary biology data. The authors thank B. Hardy for assembly of the screening plate for TR-FRET.
Funding Sources
This work was supported by the National Institute on Drug Abuse, NIH (grants R61DA047023 and R33DA047023) and the University Cancer Research Fund, University of North Carolina at Chapel Hill to L.I.J. and the National Institute of General Medical Sciences, NIH (grant R35GM139514) to S.V.F. This research is based in part upon work conducted using the UNC Proteomics Core Facility, which is supported in part by P30CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center.
Footnotes
Supporting Information
Supplemental Figures 1–9 as referenced above full western blots, full structures of all bivalent degraders and chloroalkane tagged compounds (Tables S1 and S2), Caco-2 data (Table S3) (PDF)
Chemistry: general procedures, synthetic schemes, experimental methods, compound characterization (PDF)
Raw data from whole proteome profiling (XLSX)
This material is available free of charge at http://pubs.acs.org.
References
- 1.Das P & Taube JH Regulating methylation at h3k27: A trick or treat for cancer cell plasticity. Cancers 12, 1–33 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yap DB et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117, 2451–2459 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker T et al. Acquisition of a single EZH2 D1 domain mutation confers acquired resistance to EZH2-targeted inhibitors. Oncotarget 6, 32646–32655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gibaja V et al. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene 35, 558–566 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bisserier M & Wajapeyee N Mechanisms of resistance to ezh2 inhibitors in diffuse large b-cell lymphomas. Blood 131, 2125–2137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qi W et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat Chem Biol 13, 381–388 (2017). [DOI] [PubMed] [Google Scholar]
- 7.He Y et al. The EED protein-protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat Chem Biol 13, 389–395 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Liu Z et al. Design and Synthesis of EZH2-Based PROTACs to Degrade the PRC2 Complex for Targeting the Noncatalytic Activity of EZH2. J Med Chem 64, 2829–2848 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Hsu JHR et al. EED-Targeted PROTACs Degrade EED, EZH2, and SUZ12 in the PRC2 Complex. Cell Chem Biol 27, 41–46.e17 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Potjewyd F et al. Degradation of Polycomb Repressive Complex 2 with an EED-Targeted Bivalent Chemical Degrader. Cell Chem Biol 27, 47–56.e15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma A et al. Discovery of a first-in-class EZH2 selective degrader. Nat Chem Biol 16, 214–222 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neklesa TK, Winkler JD & Crews CM Targeted protein degradation by PROTACs. Pharmacol Therapeut 174, 138–144 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Qi J & Zhang G Proteolysis-targeting chimeras for targeting protein for degradation. 11, 723–741 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Cardote TAF, Gadd MS & Ciulli A Crystal Structure of the Cul2-Rbx1-EloBC-VHL Ubiquitin Ligase Complex. Struct Lond Engl 1993 25, 901–911.e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du D et al. Structure-Guided Development of Small-Molecule PRC2 Inhibitors Targeting EZH2–EED Interaction. J Med Chem 64, 8194–8207 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Donovan KA et al. Mapping the Degradable Kinome Provides a Resource for Expedited Degrader Development. Cell 183, 1714–1731.e10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rectenwald JM et al. A General TR-FRET Assay Platform for High-Throughput Screening and Characterizing Inhibitors of Methyl-Lysine Reader Proteins. Slas Discov 24, 693–700 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peraro L et al. Cell Penetration Profiling Using the Chloroalkane Penetration Assay. J Am Chem Soc 140, 11360–11369 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deprey K & Kritzer JA Quantitative measurement of cytosolic penetration using the chloroalkane penetration assay. Methods Enzymol 641, 277–309 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foley CA, Potjewyd F, Lamb KN, James LI & Frye SV Assessing the Cell Permeability of Bivalent Chemical Degraders Using the Chloroalkane Penetration Assay. Acs Chem Biol 15, 290–295 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buckley DL et al. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. Acs Chem Biol 10, 1831–1837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maniaci C et al. Homo-PROTACs: Bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat Commun 8, 830 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shinoda W Permeability across lipid membranes. Biochimica Et Biophysica Acta Bba - Biomembr 1858, 2254–2265 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Dougherty PG, Sahni A & Pei D Understanding Cell Penetration of Cyclic Peptides. Chem Rev 119, 10241–10287 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chanaday NL & Kavalali ET Time course and temperature dependence of synaptic vesicle endocytosis. Febs Lett 592, 3606–3614 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Bednarczyk D & Sanghvi MV The impact of assay recovery on the apparent permeability, a function of lysosomal trapping. Xenobiotica 50, 1–8 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Kanungo T et al. An efficient k-means clustering algorithm: analysis and implementation. Ieee T Pattern Anal 24, 881–892 (2002). [Google Scholar]
- 28.Atilaw Y et al. Solution Conformations Shed Light on PROTAC Cell Permeability. Acs Med Chem Lett 12, 107–114 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Degorce F et al. HTRF: A Technology Tailored for Drug Discovery-A Review of Theoreti-cal Aspects and Recent Applications. Current Chemical Genomics 3, 22–32 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin W & Chen T General Stepwise Approach to Optimize a TR-FRET Assay for Characterizing the BRD/PROTAC/CRBN Ternary Complex. Acs Pharmacol Transl Sci 4, 941–952 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zundert GCP van et al. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J Mol Biol 428, 720–725 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Da C & Kireev D Structural Protein–Ligand Interaction Fingerprints (SPLIF) for Structure-Based Virtual Screening: Method and Benchmark Study. J Chem Inf Model 54, 2555–2561 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Howarth M, Takao K, Hayashi Y & Ting AY Targeting quantum dots to surface proteins in living cells with biotin ligase. Proc National Acad Sci 102, 7583–7588 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vanommeslaeghe K et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31, 671–90 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wassenaar TA et al. WeNMR: Structural Biology on the Grid. J Grid Comput 10, 743–767 (2012). [Google Scholar]
- 36.Zoete V, Cuendet MA, Grosdidier A & Michielin O SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 32, 2359–68 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Mark P & Nilsson L Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J Phys Chem 105, 9954–9960 (2001). [Google Scholar]
- 38.Nosé S & Klein ML A study of solid and liquid carbon tetrafluoride using the constant pressure molecular dynamics technique. J Chem Phys 78, 6928–6939 (1983). [Google Scholar]
- 39.Essmann U et al. A smooth particle mesh Ewald method. J Chem Phys 103, 8577–8593 (1995). [Google Scholar]
- 40.GRANT JA, GALLARDO MA & PICKUP BT A fast method of molecular shape comparison: A simple application of a Gaussian description of molecular shape. J Comput Chem 17, 1653–1666 (1996). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.