

Summary
Using machine learning (ML), we interrogated the function of all human-chimpanzee variants in 2,645 Human Accelerated Regions (HARs), finding 43% of HARs have variants with large opposing effects on chromatin state and 14% on neurodevelopmental enhancer activity. This pattern, consistent with compensatory evolution, was confirmed using massively parallel reporter assays in chimpanzee and human neural progenitor cells. The species-specific enhancer activity of HARs was accurately predicted from the presence and absence of transcription factor footprints in each species. Despite these striking cis effects, activity of a given HAR sequence was nearly identical in human and chimpanzee cells. This suggests that HARs did not evolve to compensate for changes in the trans environment but instead altered their ability to bind factors present in both species. Thus, ML prioritized variants with functional effects on human neurodevelopment and revealed an unexpected reason why HARs may have evolved so rapidly.
eTOC Blurb
Whalen et al. couple deep learning with functional assays in chimpanzee and human cells to interrogate the neurodevelopmental enhancer potential of 2645 Human Accelerated Regions (HARs). Activity is dominated by cis rather than trans effects, and compensatory changes are identified as a driver of rapid HAR evolution.
Introduction
Human accelerated regions (HARs) are highly conserved sequences that acquired many nucleotide substitutions in humans since we diverged from chimpanzees and, more recently, from archaic hominins1,2. This accelerated substitution rate suggests that HARs are important and that their functions changed during human evolution, perhaps altering traits that distinguish us from chimpanzees and other animals such as differences in morphology, diet, reproduction, and cognition2. HARs and other uniquely human genomic regions could be responsible for our high rates of psychiatric disorders, such as schizophrenia and autism spectrum disorder (ASD), which might be maladaptive by-products of the same changes in the human brain that enabled our unique linguistic and cognitive skills3,4. Indeed, HARs are enriched in disease-associated loci and nearby genes expressed during embryonic development, especially neurodevelopment5–10. Therefore, HARs are exciting candidates for understanding human-specific traits, including our unique disease susceptibilities.
The majority of HARs (96%) reside in noncoding regions. We previously used machine learning (ML) to predict that at least 30% are developmental enhancers based on their epigenetic and sequence features6. Fifty-two prioritized HARs have been analyzed for regulatory activity in transgenic mouse embryos2,6,7,11,12, with 31 (60%) functioning as enhancers in any tissue, 19 in the nervous system, and 14 in telencephalon. The human sequences of nine HARs–31% of 29 where human and chimpanzee sequences were tested–alter evolutionarily conserved enhancer activity. Several differentially regulate transcription factors (TFs) that control uniquely human features of the limb (HAR2/HACNS1: GBX2 target gene)13, testes (2xHAR.238: GLI2)14, skin (2xHAR.20: EN1)15, and brain (HARE5/ANC516: FZD8)11. Thus, sequence changes in HARs during human evolution can alter developmental gene regulation and phenotypes.
The forces that drove accelerated evolution in HARs remain largely unknown. Most HARs appear to have undergone positive selection on the human lineage prior to our divergence from archaic hominins, while ~20% have substitution patterns that are consistent with GC-biased gene conversion and a few show population genetic signatures of ongoing adaptation8,16. But it is unknown how much of the accelerated substitution rate in HARs can be attributed to genetic hitchhiking, recurrent positive selection, compensatory evolution to maintain ancestral functions, or other forces. Suppose it was adaptive for a HAR to evolve 50% lower enhancer activity in neural progenitor cells. With hitchhiking, one of its human-specific variants would have decreased enhancer activity by ~50%, while the others (the hitchhikers) had little effect on enhancer activity. In contrast, under recurrent positive selection, each of the variants would have incrementally decreased enhancer activity summing to a 50% reduction. In compensatory evolution, the HAR would contain some variants that increase and others that decrease enhancer activity. Thus, learning the contributions of individual variants within a HAR to its function could reveal how it evolved to have so many human-specific variants.
We hypothesized that recently developed ML methods that model the gene regulatory activity of non-coding sequences17–19 are capable of predicting how sequence changes alter HAR function. ML has the advantage of being able to leverage massive amounts of epigenetic data and learn complex sequence grammars, while being relatively scalable and cost-effective compared to experimental strategies. Massively parallel reporter assays (MPRAs) are a complementary approach. They measure enhancer function en masse with a quantitative readout based on RNA sequencing20 and can be applied to real or synthetic sequences, including detection of interactions between variants21. In recent years, episomal and lentivirus based MPRAs have been applied to human polymorphisms10, human-chimpanzee fixed differences21,22, and modern human-specific substitutions in HARs23. They have also been used to study human-chimpanzee variants in human-gained enhancers21 and introgressed Neanderthal variants24. However, none of these MPRAs tested HAR enhancers in non-human primate cells to evaluate how the trans environment25 interacts with cis regulatory changes.
We therefore saw an opportunity to combine ML and MPRAs in chimpanzee and human neural progenitor cells (NPCs) to quantify the enhancer activity of human, chimpanzee, and ancestral HAR sequences and investigate their evolutionary histories. We discovered that human-chimpanzee differences in HAR enhancer activity are primarily determined by nucleotide changes rather than differences in the cellular environment and can be predicted from the presence/absence of TF footprints in each species. We also found striking computational and experimental evidence for compensatory evolution to maintain ancestral enhancer activity. This new functional understanding is important because almost all HARs showing enhancer activity in NPCs are genetically and physically linked to neurodevelopmental genes and/or neuropsychiatric disease.
Results
Chimpanzee and human neural progenitor cells model gene regulation in early forebrain development
In order to establish an in vitro system to produce data for modeling the cis effects of variants on HAR enhancer function, we generated NPCs from induced pluripotent stem cell (iPSC) lines of two human and two chimpanzee individuals. Though general species-specific regulatory regions have been identified in organoids26, chimpanzee NPCs have not been used in prior HAR research and are essential for quantifying trans effects of the cellular environment. Neural induction was initiated with noggin, a BMP inhibitor, and cells were cultured in retinoic acid-free media supplemented with growth factors FGF and EGF in order to generate early (N2; 12–18 passages) and late (N3: 20–28 passages) telencephalon-fated neural progenitors (Figure 1). All lines exhibited normal cell morphology (Figure 1A,G) and normal karyotypes (Figure 1D,J), as well as neural rosette morphology at an early induction stage and neural progenitor cell morphology at later stages of differentiation (Figure 1B–C,H–I). Characterization through immunohistochemistry assays showed that both human and chimpanzee NPCs express neural and glial progenitor proteins such as PAX6 and GFAP (Figure 1E–F,K–L). We assessed cell heterogeneity through single-cell RNA-sequencing (scRNA-seq; Table S1) and observed comparable patterns of telencephalon and radial glia marker expression in human and chimpanzee NPCs (Figure 1M). Next, we performed chromatin immunoprecipitation sequencing (ChIP-seq) for the active enhancer-associated histone H3K27ac in N2 and N3 cells from both species, observing high genome-wide concordance between human and chimpanzee NPCs (R2 = 0.862 in N2, 0.712 in N3) and a majority of peaks overlapping published H3K27ac peaks from developing human (54.9%) and adult chimpanzee (54.3%) cortical tissues27,28. This indicates the relevance of our NPCs to in vivo biology while also suggesting that we could discover substantial numbers of novel enhancers in this model of early neurodevelopment.
Figure 1. Characterization of chimpanzee and human neural progenitor cells.
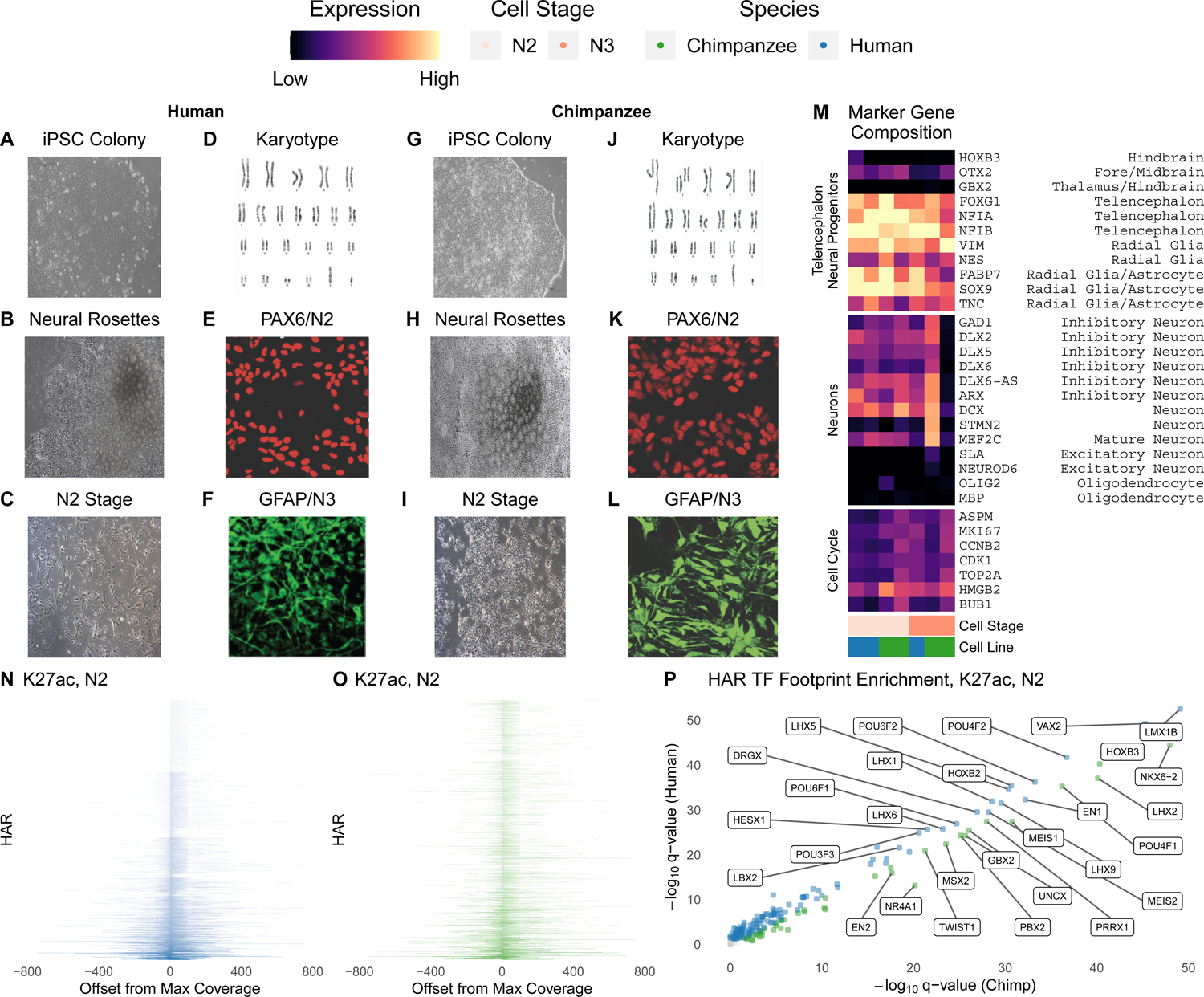
(A-C) Brightfield images of human iPSCs (A). iPSC differentiated into neural rosettes (B) and N2 cells (C) demonstrating typical morphology. (D) Human iPSCs demonstrate normal karyotypes. (E) Human N2 cells express Paired Box 6 (PAX6), a neural marker. (F) Human N3 cells express Glial Fibrillary Acidic Protein (GFAP), a glial marker. (G-I) Brightfield images of chimpanzee iPSCs (G). iPSC differentiated into neural rosettes (H) and N2 cells (I) demonstrating typical morphology. (J) Chimpanzee iPSCs demonstrate normal karyotypes. (K) Chimpanzee N2 cells express PAX6. (L) Chimpanzee N3 cells express GFAP. (M) Percentage of cells in scRNA-seq expressing genes that are markers for the cell cycle or telencephalon and neuronal cell types. Human and chimpanzee N2 and N3 cells show comparable marker expression for radial glia and telencephalon. For example, 50–90% of cells expressed FOXG1, a marker of the telencephalon. (N-O) Coverage (CPM) of H3K27ac ChIP-seq reads at HARs, sorted by maximum CPM, in human (N) and chimpanzee (O) N2 cells. (P) Human and chimpanzee N2 H3K27ac TF footprints are largely concordant, but some TF families with LIM, POU and homeodomains show species-biased enrichment. Select TFs expressed in NPCs59 with large differences in q-value between species are labeled.
Machine learning delineation of HAR enhancers using hundreds of epigenetic features
To epigenetically profile 2645 noncoding HARs from prior studies1, we performed the assay for transposase-accessible chromatin (ATAC-seq) and H3K27me3 ChIP-seq for repressed chromatin in human N2 and N3 cells, an early NPC stage (N1; eleven days after initiating neural induction, passage 1), and astrocyte progenitors (Table 1). Epigenetic marks were generally concordant across species and cell types (Figure S1). The majority of HARs overlap H3K27ac marks in human (Figure 1N) and chimpanzee (Figure 1O) NPCs, although some exhibit species-biased H3K27ac signals. HARs with high H3K27ac mostly lie in open chromatin, whereas those with low H3K27ac tend to overlap peaks of the repression-associated histone H3K27me3, though some HARs have both marks (Figure S1). To directly assess how divergent sequences and epigenetic states alter TF binding in human versus chimpanzee HARs, we ran the histone module of Hmm-based IdentificatioN of Transcription factor footprints (HINT)29 with our NPC H3K27ac ChIP-seq. Footprints of many TFs are differentially enriched between species (Figure 1P; Table S2). EN1, VAX2, and several other homeobox TFs are human-biased, while NKX6–2 is the most chimpanzee-biased. Altogether, this first epigenetic characterization of early chimpanzee neurodevelopment revealed important differences in regulatory potential between human and chimpanzee HARs.
Table 1.
Datasets generated in human and chimpanzee NPCs.
| Human Cell Lines (N) | Chimpanzee Cell Lines (N) | |
|---|---|---|
| LentiMPRA | HS1 N2 (3), N3 (3) WTC N2 (3), N3 (3) | Pt2A N2 (3), N3 (3) Pt5C N2 (3), N3 (3)# |
| scRNA-seq | N2 (1), N3 (1) | N2 (1), N3 (1) |
| ATAC-seq | H1-ESC N1 (2), N2 (2), H9-ESC AP (2) | |
| Hi-C | H1-ESC N2 (2), N3 (2) | |
| H3K27ac ChlP-seq | HS1 N2 (2), N3 (2), H1-ESC N1 (1), H9-ESC AP (1) | Pt2A N2 (2), N3 (2) |
| H3K27me3 ChIP-seq | H1-ESC N1 (1), N2 (1), H9-ESC AP (1) |
Pt5C N2 and N3 lentiMPRA did not pass quality control, and it was not used for modeling.
N1 = Early neural progenitor cells
N2 = Neural progenitor cells
N3 = Glial progenitor cells
AP = Astrocyte progenitor cells
(N) = Number of replicates
To relate this in vitro epigenetic characterization of HARs to in vivo enhancer activity, we first leveraged ML to integrate our 19 ChIP-seq and ATAC-seq experiments with 254 epigenetic studies in primary tissue (Table S3). When considering all developmental stages and regions, 70% of HARs (1846/2645) overlap open chromatin and/or active marks in the human brain (Figure 2A). Significantly fewer HARs (935/2645) have these marks of active regulatory elements in other tissues (Kolmogorov-Smirnov p < 2e-16; Figure 2B), despite similar numbers of datasets. Consistent with multi-tissue enhancer function, 808 HARs have both neural and non-neural marks (Figure S2). These results emphasize that HARs likely function as enhancers in many contexts beyond neurodevelopment, although brain enhancer-associated epigenetic marks are particularly enriched in HARs, as previously reported in smaller datasets6,10,12,21,22,30,31.
Figure 2. The in vivo epigenetic landscape of HARs.
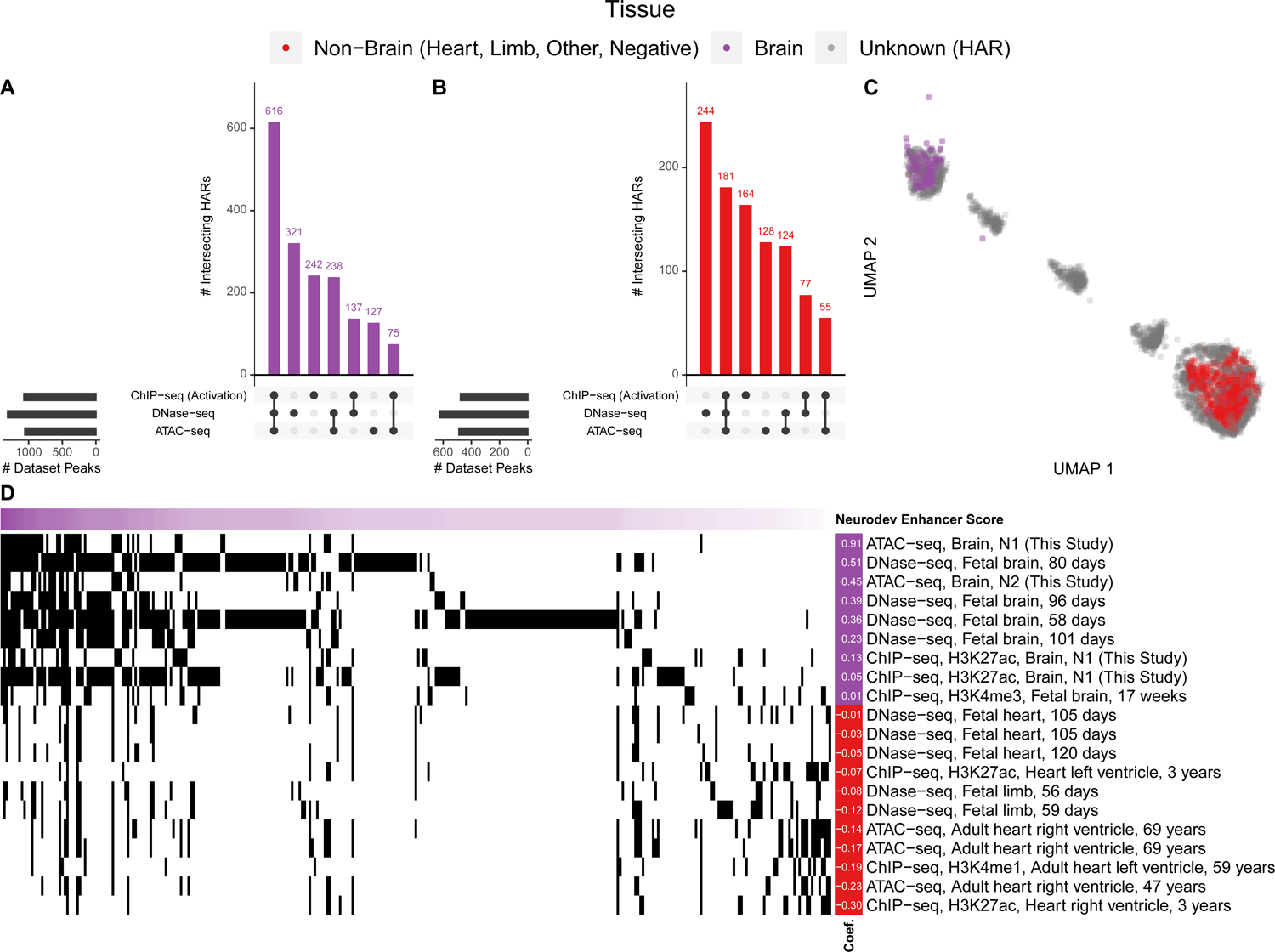
A large collection of open chromatin (ATAC-seq, DNase-seq) and ChIP-seq (TF, histone) datasets from human primary tissues (49% brain, 48% heart, 2% limb; Table S3) were intersected with HARs. (A) Upset plot showing that 1846/2645 HARs overlap at least one type of open chromatin (ATAC-seq, DNase-seq) or activating (H3K4me1, H3K4me3, H3K9ac, H3K27ac, or H3K36me3) mark, while 616/2645 have overlap all three (i.e., ATAC-seq, DNase-seq, and an activating histone). The purple histogram shows the number of HARs with the denoted combination of marks, while the black bars to the left show the number of marks that overlap a HAR. (B) HAR overlaps with activating marks and open chromatin in other tissues. There are significantly more overlaps for the brain compared to non-brain tissues (p-value < 2e-16). Joint heart and brain overlaps are shown in Figure S2. (C) Two-dimensional UMAP projection of HARs (grey) with VISTA heart (red) and brain (purple) enhancers32 showing that some HARs cluster with in vivo validated enhancers. (D) HARs (horizontal axis, sorted so those most similar to VISTA brain enhancers are on the left) with their epigenetic profiles (vertical axis; black indicates overlapping epigenetic features). Shown are the epigenetic features most predictive in a ML model of VISTA brain enhancers (purple) versus non-brain enhancers (VISTA negatives plus enhancers active in other tissues; red), along with their model coefficients (left).
Next, we compared HARs to validated developmental enhancers from the VISTA Enhancer Browser32. We chose VISTA because it measures tissue-specific enhancer activity during embryonic development, and most of the tested sequences are evolutionarily conserved, similar to HARs. After annotating each VISTA sequence with binary vectors denoting overlap with peaks in our epigenetic compendium and embedding these high-dimensional vectors in two dimensions, we observed that neurodevelopmental enhancers have distinct signatures compared to sequences active only in non-brain tissues or without enhancer activity. Then, we annotated and co-embedded HARs in this same epigenetic space. Many HARs cluster with in vivo validated neurodevelopmental enhancers, while others appear not to function as enhancers or to be active in other tissues and developmental stages (Figure 2C).
Motivated by this clear partitioning, we trained a supervised ML model using epigenetic signatures to distinguish VISTA brain enhancers from enhancers that are inactive or active in other tissues. This is a difficult classification problem given the large number of multi-tissue enhancers with overlapping epigenetic signatures. Nonetheless, a L1-penalized logistic regression model can distinguish neurodevelopmental enhancers in held-out VISTA data (median cross-chromosome auPR 0.69, auROC 0.8). Using this model, we scored HARs based on how consistent their epigenetic profiles are with neurodevelopmental enhancer function (Figure 2D). As expected, HARs with higher scores overlap more neurodevelopmental epigenetic marks and have similar co-embedding coordinates to VISTA brain enhancers (Table S7). Thus, ML models are able to integrate hundreds of epigenetic signals to prioritize HARs likely to function as enhancers during neurodevelopment.
2xHAR.183 is a ROCK2 neuronal enhancer
Next, we sought to validate a novel HAR enhancer prediction. We generated chromatin capture (Hi-C) data in our human N2 and N3 cells and used it along with Hi-C from primary fetal brain tissue33–35 to associate HARs with genes they may regulate. This analysis confirmed known regulatory relationships between HARs and developmental genes, including 2xHAR.20 with EN115 and 2xHAR.238 with GLI214. Based on chromatin contacts with the neurodevelopmental gene ROCK2 in NPCs, a PLAC-seq loop to ROCK2 in excitatory neurons35, enhancer annotations (ChromHMM36, FANTOM537), and a high neurodevelopmental enhancer score, we selected 2xHAR.183 for functional characterization (Figure 3A). Consistent with ROCK2’s increasing expression in later stages of embryonic development and at postnatal time points in mice38 (mid-gestation in humans), we observed progressively more open chromatin and greater H3K27ac signal at 2xHAR.183 over developmental stages, with a slightly larger activation signature in chimpanzee compared to human (Figure 3A). Supporting the hypothesis that 2xHAR.183 is part of a neurodevelopmental enhancer, footprint analysis in our human NPCs and ENCODE fetal brain tissue39 identified binding sites for C/EBPBeta, RFX2, PRDM1, and BCL11A (Figure 3B). To test if 2xHAR.183 indeed regulates ROCK2 or the nearby gene E2F6 in an adjacent chromatin domain, we performed CRISPR activation (CRISPRa) in human NGN2-induced iPSC-derived neurons40 and observed increased expression of ROCK2 but not E2F6 (Figure 3C). These findings indicate that 2xHAR.183 is a ROCK2 enhancer in developing neurons.
Figure 3. Validation of an active HAR enhancer regulating ROCK2.
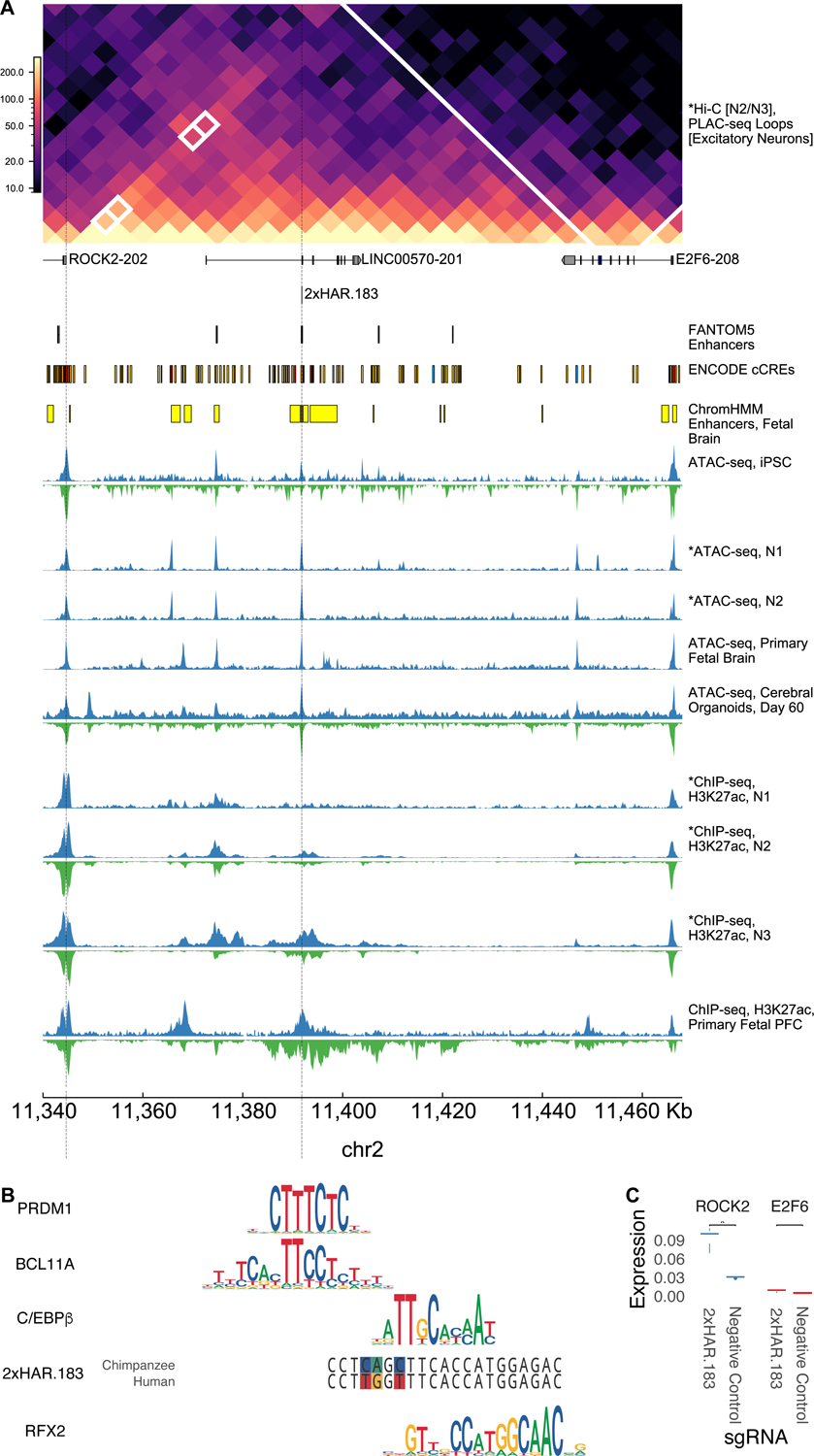
2xHAR.183 was selected for further validation due to its high enhancer score (Figure 2). (A) 2xHAR.183 has a significant chromatin loop with the ROCK2 gene in excitatory neuron PLAC-seq data (5kb resolution binary loop call)35 and contacts ROCK2 in our N2/N3 Hi-C. The gene E2F6 is nearby on the linear genome but has fewer chromatin contacts. 2xHAR.183 overlaps multiple annotations from fetal brain datasets. Chimpanzee and human epigenetic datasets across early neurodevelopment suggest 2xHAR.183 starts and remains accessible in both species, while gaining acetylation beginning at the N2 stage. The activation signature appears later and stronger in chimpanzee versus human cells. (B) Footprints of known neurodevelopmental TFs, C/EBPBeta and RFX2, are contained within 2xHAR.183 and overlap human:chimpanzee variants (colored sites in chimpanzee and human sequences). Additional footprints for PRDM1 and BCL11A were detected adjacent to and partially overlapping the HAR. Height of the nucleotides in each motif indicates information content (0 to 2 bits). (C) CRISPRa validation (3 replicates per target, 4 per control) shows 2xHAR.183 drives strong expression of ROCK2, but not the proximal gene E2F6. Variability between replicates is small for low expression values.
Deep learning predicts that most individual HAR variants alter enhancer activity
To comprehensively test how all individual HAR variants affect enhancer activity, we utilized the deep-learning model Sei41 that predicts how human polymorphisms alter tissue-specific regulatory activity including ten enhancer states. By instead presenting human-chimpanzee fixed differences within HARs to Sei, we could predict if they alter chromatin states (Table S5). This revealed that most HAR variants shift enhancer activity in at least one tissue-specific enhancer state (Figure 4A). Chromatin state changes are generally correlated across different tissues. However, some HAR variants have tissue-specific effects such as trade-offs between brain and B-cell enhancer activity in HAR3 and HAR166, as well as a 2xHAR.170 variant predicted to decrease enhancer activity in brain tissue while increasing activity in all other tissues (Figure 4E). These results demonstrate that deep learning can generate testable hypotheses about HAR variant function.
Figure 4. Human-specific variants shift HAR enhancer profiles in a deep-learning model.
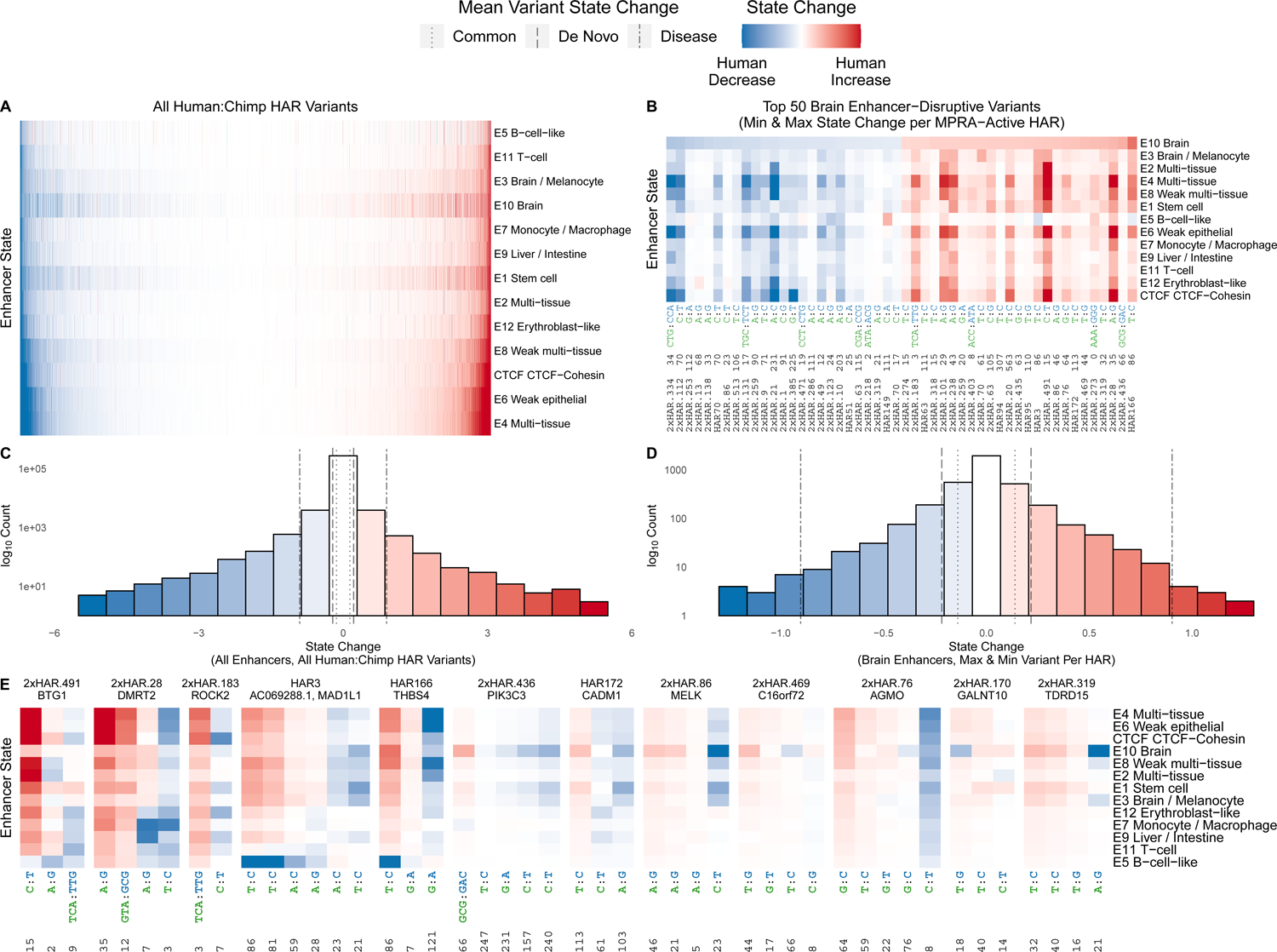
Every human-specific variant in each HAR was evaluated using the deep-learning model Sei41. Variants where the human nucleotide decreases the chromatin state are blue (shade denotes amount of decrease), variants where the human nucleotide increases the chromatin state are red, and complex variants that cannot be scored by Sei are white. (A) The landscape of chromatin state changes (y-axis) induced by all human:chimpanzee variants across all HARs (x-axis), sorted by predicted impact on brain enhancer state. (B) The 50 HAR variants that most increase or decrease brain enhancer state for all HARs that were active in our MPRA. The x-axis shows the HAR name, the offset of the variant from the HAR’s start position, and the human and chimpanzee alleles colored by species and separated by a colon. (C) Histogram of predicted enhancer state changes for all HAR variants from (A). Mean state changes for different classes of variants41 are shown via vertical lines: 1000 Genomes common variants, de novo mutations in healthy individuals, disease-causing mutations (from smallest to largest mean change). Many HAR variants have effects that exceed those of phenotype-associated human polymorphisms. (D) Histogram of predicted brain enhancer state changes for the most disruptive HAR variants in active HARs. Mean state changes for different classes of variants41 as in (C). (E) For 12 HARs containing variants with the largest effects on brain enhancer activity in our Sei analysis, we observed a mix of variants predicted by Sei to increase and decrease enhancer activity. Variants (x-axis) are annotated with their offset from the start of the HAR plus the human and chimpanzee alleles separated by a colon. HARs are annotated with the closest protein-coding gene.
To contextualize these results, we quantified Sei enhancer state changes for HAR variants versus different functional classes of single nucleotide polymorphisms (SNPs). The mean of the largest tissue-specific shift per HAR variant (0.54) is nearly four times higher than common SNPs from the 1000 Genomes Project (0.139), lying between de novo mutations in healthy humans (0.217) and disease mutations in the Human Gene Mutation Database (0.903)41. Using these averages as thresholds, we identified 2121 HAR variants (16%) with absolute effects on brain enhancer activity (Sei state E10) greater than expected compared to common SNPs, 1226 (9%) compared to de novo SNPs, and 61 (< 1%) compared to disease SNPs (Figure 4B & D; Table S5). Variants predicted to increase activity are more common than those predicted to decrease activity, though effect sizes are slightly larger for decreases (Figure 4C–D). Thus, we predict that a substantial number of HAR variants changed enhancer activity during human evolution.
Many HARs contain variants predicted to have opposing effects on enhancer activity
Examining Sei predictions for all variants within the same HAR, we discovered that 43% of HARs contain a mix of variants predicted to increase and decrease enhancer activity beyond the average effect of common variants genome-wide (Figure 4E), and 14% of HARs contain variants with opposing effects on neurodevelopmental enhancer activity. This is significantly more than expected by chance (bootstrap p=0.03). Limiting this analysis to variants whose effects exceed the mean of de novo or disease-causing variants, we observe two or more strongly opposing variants in 30% and 3% of HARs, respectively. Furthermore, many HARs contain individual variants whose effect on enhancer activity is greater than the net effect of all that HAR’s variants. This signature led us to hypothesize that compensatory evolution to fine tune enhancer activity and possibly maintain ancestral activity levels, rather than recurrent selection to successively increase or decrease activity, drove rapid evolution of some HAR enhancers. It is not currently possible to test for variant interactions in the Sei framework, motivating us to move from in silico to in vitro characterization of HAR variants.
MPRA characterization of HAR variants in primate NPCs
Performing a massive ML integration of data from epigenetic assays, transgenic mice, TF motifs, and human genetic variants generated the following testable hypotheses about HAR enhancer function: (i) many HARs function as enhancers in the developing brain, (ii) many human and chimpanzee HAR sequences are differentially active, and (iii) variants within the same HAR interact non-additively to tune activity. To quantitatively test these hypotheses, we used MPRAs to compare the activity of homologous human and chimpanzee sequences (cis effects) in the trans environments of chimpanzee and human NPCs. Interrogating different permutations of HAR variants in cells from both species distinguish this experiment from prior MPRA studies.
We designed an oligonucleotide (oligo) library containing the human and chimpanzee sequences of 714 HARs from our prior studies8,30,42, all potential evolutionary intermediates between the human and chimpanzee sequences (“permutations”) of three HARs (2xHAR.164, 2xHAR.170, 2xHAR.238) with evidence of differences in neurodevelopmental enhancer activity between human and chimpanzee sequences6, 118 positive controls, and 142 negative controls (Methods). We performed lentivirus-based MPRA (lentiMPRA) with this library in N2 and N3 cell lines derived from two humans and two chimpanzees (Figure S3). For each condition, we generated three technical replicates, yielding 18 measurements of enhancer activity for each sequence after quality control. We observed high correlation (median R2 = 0.91) between replicates (Figure 5A).
Figure 5. Species-biased HAR enhancers identified in chimpanzee and human NPCs.
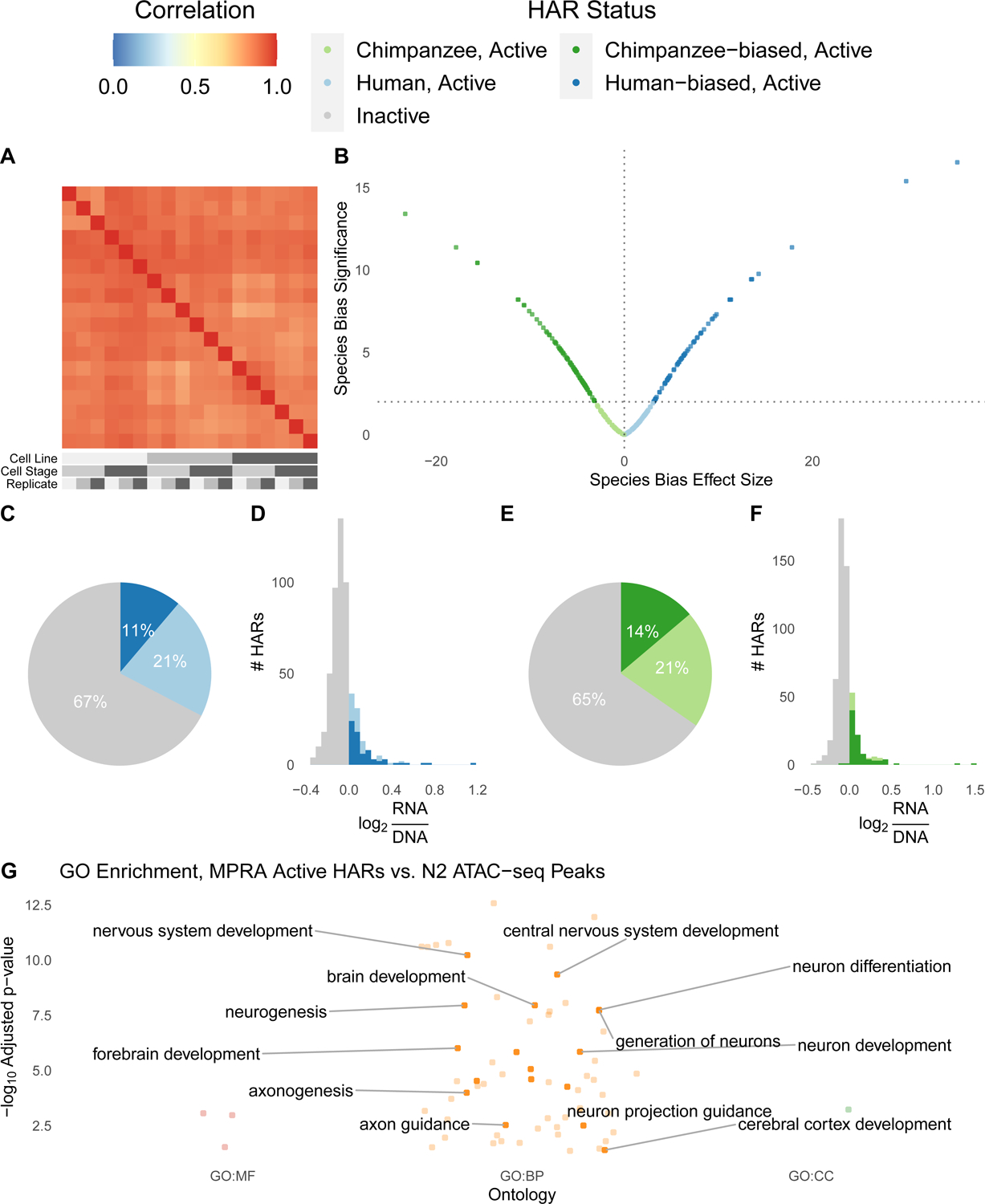
We performed lentiMPRAs in chimpanzee and human cell lines at the N2 and N3 stages of differentiation. (A) Enhancer activity (RNA/DNA ratios batch corrected and normalized for sequencing depth) was highly correlated between technical and biological replicates for eighteen samples passing quality control: 3 replicates (shades of grey) of Pt2a (chimpanzee; dark grey), WTC (human; medium grey), and HS1–11 (human; light grey) iPSC lines differentiated into N2 (medium grey) and N3 (dark grey) cells. (B) Effect size (t-statistic) vs significance (−log10 q-value) for the ratio of human and chimpanzee HAR sequence activity for active HAR enhancers. HARs with species-biased activity are plotted in dark green (chimpanzee sequence more active) or dark blue (human sequence more active). (C) Roughly a third of human HAR sequences are active across samples (log RNA/DNA > median of positive controls in at least 9/18 replicates), and 11% are human-biased (differentially active with human:chimpanzee ratio > 1). (D) Distribution of human HAR sequence enhancer activity for inactive (grey) or active HARs, with active split into human-biased (dark blue) versus not (light blue). (E) Roughly a third of chimpanzee HAR sequences are active across samples, and 14% are chimpanzee-biased. (F) Histogram of chimpanzee sequence activity as in (D). (G) HARs active in lentiMPRA are enriched for many neurodevelopmental GO terms. Colors indicate the type of term: red = molecular function (MF), orange = biological process (BP), green: cellular compartment (CC).
Before comparing human and chimpanzee alleles, we first identified a subset of 293 HARs with activity above the median of positive controls in at least 50% of samples for either the human or chimpanzee sequence. These constitute about one-third of both human and chimpanzee HAR sequences (Figure 5C–F) and include 2xHAR.183. The majority of active HARs (233/293) are in a chromatin domain or loop with a neurodevelopmental gene (Table S7), and these loci are enriched for roles in neurodevelopment, transcription, cell adhesion, axon guidance and neurogenesis (Figure 5G, Table S6).
To validate our lentiMPRA, we compared active HARs to published mouse transgenic reporter assays, mostly performed at embryonic day 11.5, a stage similar to N2 (Table S4). We found significant concordance with in vivo expression for embryonic brain (odds ratio = 3.79, Fisher’s exact test p=0.005) and telencephalon (odds ratio = 7.44, p = 0.00012). We performed mouse reporter experiments for an additional four HARs (HAR152, 2xHAR.133, 2xHAR.518, 2xHAR.548) at developmental stages chosen based on expression of nearby genes and observed enhancer activity for all four (Figure S4). Next, we performed luciferase assays in one human and one chimpanzee cell line for nine active HARs and observed that six were more active than an empty vector (Figure S5). Finally, we quantified activity of H3K27ac versus H3K27me3 peaks included as controls in our lentiMPRA, and we confirmed significantly higher activity for H3K27ac in all samples (Figure S3). These data indicate that our lentiMPRA identified bona fide neuronal enhancers.
Nonetheless, we observed only moderate correlation between neurodevelopmental enhancer scores from our ML model trained on VISTA (Figure 2D) and activity levels in NPC lentiMPRA. To investigate this expected difference43,44, we trained a classifier to distinguish HARs with discordant ML and lentiMPRA scores based on their epigenetic profiles (auPR 0.96; Methods). Analyzing predictive features revealed that lentiMPRA is more permissive, allowing some sequences with closed chromatin in the brain or activating marks outside the brain to show activity in NPCs. Conversely, the ML model is more brain-specific and prioritizes HARs with active marks in whole brain, astrocytes, and hippocampal neurons alongside those with marks in forebrain NPCs. We conclude that it is important to consider the complementary sets of HAR enhancers identified via each approach, as we have done here, with the 48 HARs in the top quartile of both ML and lentiMPRA being particularly high-confidence neurodevelopmental enhancers.
HAR sequence variants alter enhancer activity while the cellular environment does not
We next assessed evidence that lentiMPRA activity levels differed between chimpanzee and human NPCs. We observed strikingly similar activity of HAR enhancers across not only technical but also biological replicates, including different cell species and stages (Figures 5A and S3). In contrast to these limited trans effects, many HARs show consistent differences in activity between human and chimpanzee sequences (Figure S6). These cis effects were significant for 159 HARs (54% of active HARs) at a false discovery rate < 1% (Figure 5B, Table S7). HARs where the human sequence has increased activity (70 human-biased HARs; Figure 5C–D) are slightly less common than those with decreased activity (89 chimpanzee-biased; Figure 5E–F), though effect sizes are similar (Figure 5B). 2xHAR.548, located in a chromatin domain with the neurodevelopmental regulator and disease gene FOXP1, was the most species-biased HAR, showing much higher activity for the human compared to chimpanzee sequence. These results quantitatively demonstrate that cis regulatory features are stronger drivers of HAR enhancer activity than the cellular environment. This observation was possible because we performed lentiMPRA in chimpanzee and human cells.
To validate species-biased HARs, we tested nine homologous chimpanzee and human HAR sequences with luciferase and confirmed statistically significant bias in the expected direction for six (Figure S5). Furthermore, out of six active HARs that have previously shown differential activity between the human and chimpanzee sequence in mouse transgenics (2xHAR.20, 2xHAR.114, 2xHAR.164, 2xHAR.170, 2xHAR.238)6,12,14,15, all except 2xHAR.164 were also species-biased in our lentiMPRA (Table S7). These results are strong evidence that our lentiMPRA accurately detected species-biased HAR enhancer activity.
Species differences in HAR enhancer activity can be predicted from TF footprints
We next sought to use species differences in TF footprints within orthologous HARs (Figure 1P) to better understand how sequence and epigenetic changes in HARs relate to species-biased enhancer activity in lentiMPRA. Most brain-expressed TFs have footprints overlapping multiple HAR variants (Table S2), and some of these also have large Sei brain enhancer activity decreases (Figure 6A) or increases (Figure 6B). A supervised gradient boosting regressor (Methods) was able to use the human and chimpanzee footprints of each HAR to predict their human:chimpanzee lentiMPRA log ratios with very low error (R2=0.8, RMSE=0.04), indicating that loss and gain of TF binding sites is a plausible mechanism through which HAR enhancer activity changed during human evolution. This cis mechanism is consistent with our observing similar activity for HAR sequences in human versus chimpanzee NPCs.
Figure 6. Variants in TF footprints predict HAR species bias.
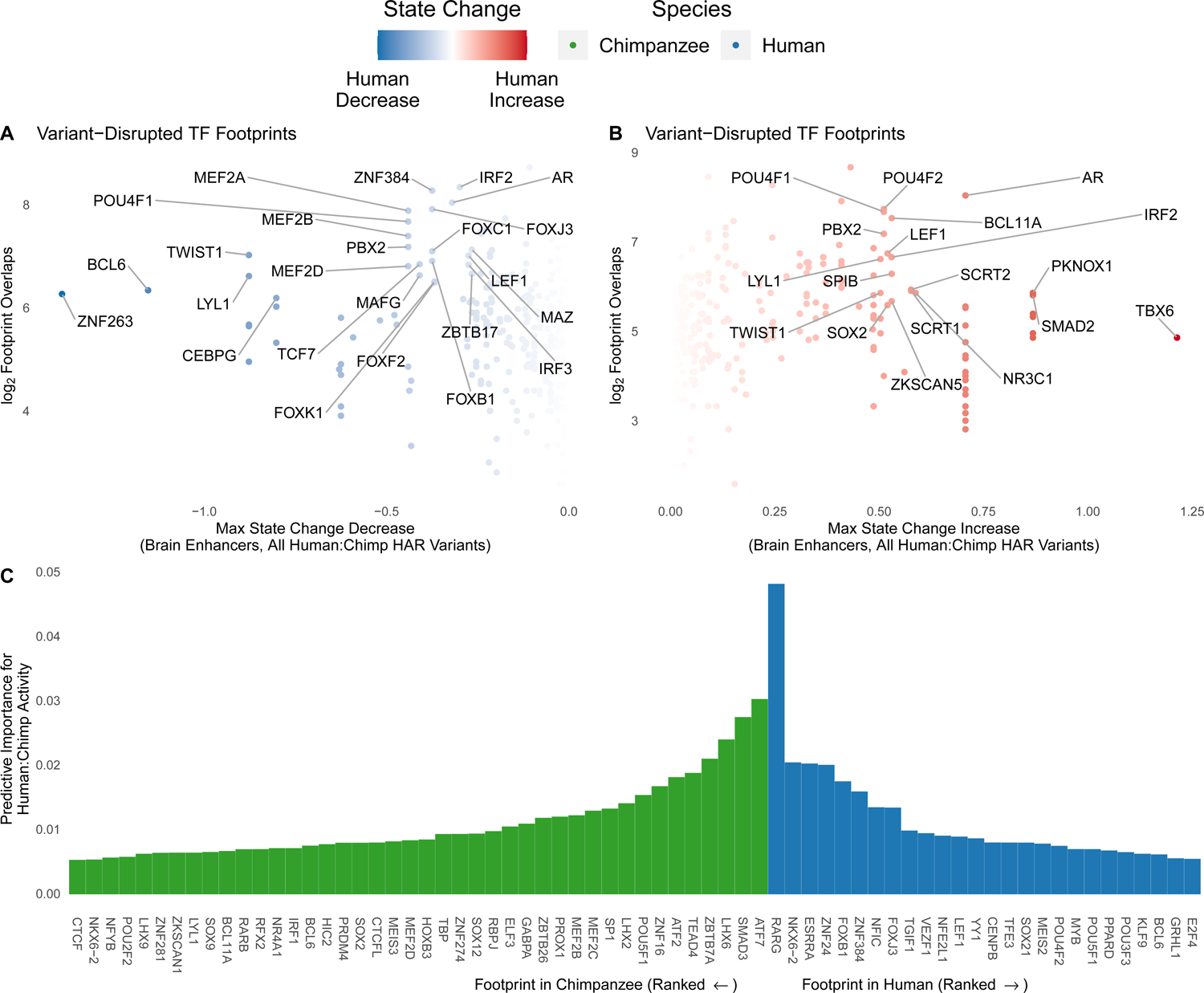
(A) The effects of HAR variants in TF footprints (in human N2 H3K27ac ChIP-seq) on brain enhancer activity were predicted using Sei41. For each TF, the largest decrease in brain enhancer state over all variants (x-axis) is shown against the number of variant-containing footprints (y-axis). Select TFs expressed in NPCs (TPM > 1) and scoring high on one or both metrics are labeled. (B) TFs with the largest predicted increase in brain enhancer activity in the analysis from (A). (C) The species-bias of HAR lentiMPRA activity can be predicted accurately using human and chimpanzee N2 H3K27ac footprints for TFs expressed in NPCs as features in a gradient boosting model. The most important TFs for accurate predictions are shown along with their variable importance scores.
Next, we used variable importance to assess which TF footprints contribute most to this predictive accuracy (Figure 6C). A TF can be important due to its human footprints, its chimpanzee footprints, or both. In each case, the model may leverage a positive or negative association with human:chimpanzee lentiMPRA activity. This analysis highlighted genes associated with neurological disease (MEF2C, NKX6–2) and brain development (FOXB1, ZNF24), as well as TEAD4, which regulates organ size. Other functions represented amongst the top TFs were regulation of cell proliferation and differentiation (SMAD3, LHX2, LHX6, ZNF16, ZBTB7A, POU5F1, FOXJ3, SP1, MEF2B), retinoic acid and estrogen dependent regulation (RARG, ESRRA), chromatin regulation (ATF2, ATF7), and extracellular matrix regulation (ZNF384). Collectively, these results show that changes in HAR neurodevelopmental enhancer activity during human evolution can be accurately recapitulated by the losses and gains of TF footprints.
HAR enhancers are linked to neurodevelopmental gene expression and psychiatric disorders
To aid with interpretation of HAR enhancers in the context of neurobiology and disease, we used linkage disequilibrium to associate HAR variants with neuropsychiatric disorder SNPs45 and brain expression and chromatin quantitative trait loci (QTLs)46–49 (Table S7). We found 55 HAR enhancers with genetic associations to psychiatric disease and/or brain gene expression, many of which also have chromatin interactions with neurodevelopmental genes (Figure S7). We discovered that 2xHAR.170 has a long-range chromatin interaction with GALNT10 and harbors a QTL (rs2434531) where the derived allele is associated with higher expression of GALNT1050. This SNP is in linkage disequilibrium with a schizophrenia-associated SNP (rs11740474)51, and GALNT10 is overexpressed in individuals with schizophrenia52, implicating 2xHAR.170 as a GALNT10 enhancer associated with schizophrenia. Other notable examples of disease associated HARs include 2xHAR.502, which lies in an intron of the language and schizophrenia associated gene FOXP2 and contains a SNP associated with attention deficit hyperactivity disorder. 2xHAR.262 lies in a contact domain with CPSF2, RIN3, and SLC24A4 and contains a SNP associated with bipolar disorder. Collectively, we linked the majority of active HARs to neurodevelopmental genes and associated 20 with psychiatric diseases, underscoring the phenotypic consequences of altering the activity of these deeply conserved enhancers.
Variants within individual HARs interact to tune enhancer activity
We used the permutation oligos to dissect HAR lentiMPRA activity at the single-nucleotide level, first considering all oligos with one chimpanzee variant inserted into the human sequence. This parallels our variant interpretation with Sei, enabling direct comparison. Across variants, Sei’s brain enhancer score correlated loosely with lentiMPRA differential activity (Figures 7A and S8). We observed the strongest concordance for 2xHAR.170, a HAR that is species-biased in lentiMPRA and transgenic mice (Figure 7D–E). To evaluate evidence of compensatory evolution (negative interactions), we modeled permutation lentiMPRA activity using the unique combination of human:chimpanzee variants present in each oligo (Methods), finding that all three tested HARs contain opposing variants. To confirm this, we generated a second lentiMPRA library containing only permutation oligos, observing highly correlated activity measurements (Figure S3) and concordant results.
Figure 7. Variants in HARs interact to tune enhancer activity.
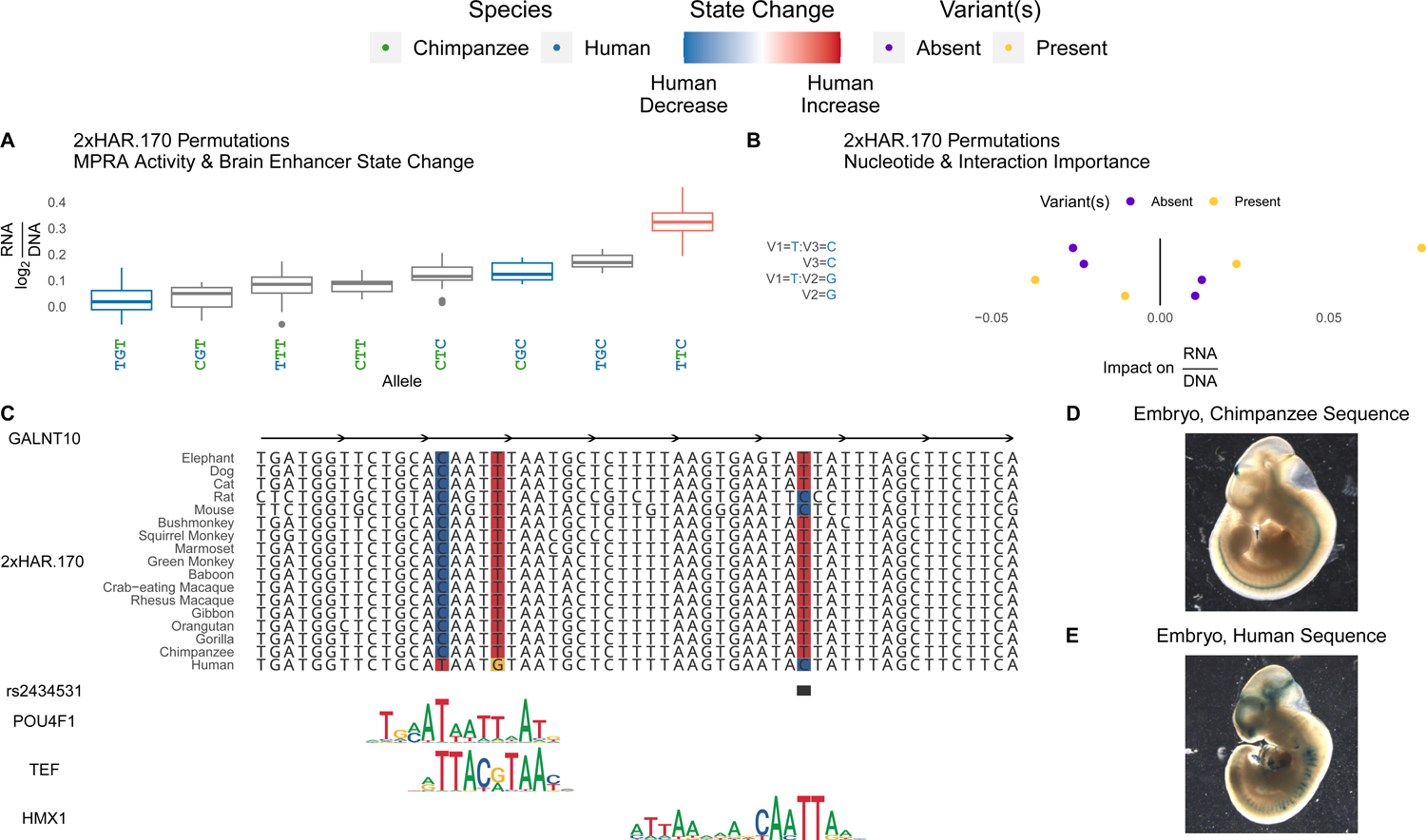
(A) All evolutionary intermediates between chimpanzee and human alleles of 2xHAR.170 were tested via lentiMPRA. Individual variants showed a range of effects on activity (y-axis; log2(RNA/DNA)) that correlated with Sei predicted effects on brain enhancer activity (red = human increase, blue = human decrease). Oligos containing multiple variants revealed interactions between variants that were untestable with Sei (no color). (B) We assessed the importance of each variant using a gradient boosting model that predicts lentiMPRA activity of each permutation using the presence or absence of the human allele at each of the three variants. Interactions between multiple variants (separated by colons on the y-axis) were included as predictors alongside main effects (no colon) to assess their predictive importance. This model confirmed the importance of specific variant interactions (x-axis, positive = higher predicted activity, negative = lower activity). Variant names consist of a V followed by the variant number (1, 2 or 3 ordered from 5’ to 3’), and the allele is shown after the equal sign. Present (yellow) indicates the expected change in enhancer activity for oligos that have the allele denoted on the y-axis, while absent (purple) shows the expected change for oligos that lack the allele. Yellow points at positive impact on RNA/DNA values means that variant or variant combination increases enhancer activity on average across oligos with other variants, while purple points at positive values mean the variant or variant combination decreases activity (i.e., activity is higher when absent). (C) 2xHAR.170 is a candidate intronic enhancer of GALNT10, acquiring a human-specific change from C to T that enhances POU4F1 and TEF binding in our footprint analysis. The human polymorphism rs2434531 is an eQTL for GALNT10, and we predict that the derived allele enhances binding of the repressor HMX1. The HMX1 and TEF footprints were detected in an independent brain footprinting study39. Both results are supported by Sei predictions (4E), lentiMPRA activity (A), and differential activity between the chimpanzee (D) and human (E) sequences in the forebrain and midbrain of transgenic mouse embryos. Adapted from6. The eQTL (rs2434531) is in linkage disequilibrium with a schizophrenia GWAS variant (rs11740474)51. In neuronal cells, 2xHAR.170 is bound by FOXP2, as well as other enhancer-associated proteins (ISL1, HAND2, PHOX2B, FOSL2) and chromatin modifiers (EZH2, SMARCA2, SMARCC1)60.
To determine whether human-specific mutations in HARs interact non-additively, we next dissected variant effects in individual HARs, focusing on the three human variants in 2xHAR.170 (Figure 7B). The derived C allele at the third variant has the largest individual effect in lentiMPRA and Sei analysis (Figure 7A). Footprint analysis (Figure 7C) shows that this C decreases binding affinity of HMX153, a repressor of neural differentiation-driver TLX354, consistent with increased enhancer activity. Since this variant is polymorphic (rs2434531), some humans have the least active permutation, while others have one of the most active, with possible phenotypic consequences. In contrast, the derived alleles at the other two variants individually decrease enhancer activity in lentiMPRA and Sei analysis and have the lowest activity when tested together in combination with the chimpanzee T allele at the third variant. This is consistent with their being proximal and changing high information content positions in a POU4F1 footprint, which is supported by POU4F1 ChIP-seq in fibroblasts55. However, the activity-increasing effect of the derived C allele at the third variant is amplified, not reduced, in the presence of the derived T allele at the first variant. While we do not know the order in which these three variants arose, it is possible that the first two variants compensated for the large, schizophrenia-associated effect of the third variant. Regardless, 2xHAR.170 variants clearly have interacting effects on brain enhancer activity.
Discussion
We used ML models coupled with lentiMPRA and epigenetic experiments in chimpanzee and human NPCs, dozens of which we generated, to functionally profile HARs at single-nucleotide resolution in neurodevelopment. Going beyond prior HAR MPRA studies21,22, we generated data in chimpanzee NPCs, discovering a much greater effect on enhancer activity for HAR sequence variants as compared to species-specific differences in the cellular environment, concordant with greater cis versus trans effects for in vivo enhancers56,57. We showed that species-biased activity can be predicted from TF footprint differences between human and chimpanzee HARs (i.e., differential TF binding). Finally, we dissected the contribution of all variants in each HAR, revealing pervasive interactions between sites, in many cases suggestive of compensatory evolution to maintain ancestral enhancer activity. Altogether, our results prioritize dozens of HARs with evidence of differential neurodevelopmental enhancer activity in humans compared to chimpanzees and other mammals.
A key novelty of our study is the use of ML modeling to efficiently integrate hundreds of epigenetic datasets and screen thousands of variant combinations in silico. We significantly extended prior analyses6,21,22 by (i) using a larger epigenomic compendium, including data we generated in chimpanzee and human NPCs, (ii) analyzing HARs alongside in vivo validated enhancers, revealing a subset of HARs that cluster with tissue-specific enhancers, (iii) showing that differences in chimpanzee versus human sequences and H3K27ac in HARs create species-specific TF footprints that predict differential enhancer activity in lentiMPRA, and (iv) leveraging deep learning to reveal that individual variants within the same HAR often have opposite effects on enhancer activity. ML enabled us to screen many HAR variants with relatively low cost and effort, whereas lentiMPRA and CRISPRa in NPCs provided direct measurements for a smaller number of prioritized sequences.
While our lentiMPRA was highly reproducible in NPCs, it did not perfectly agree with our ML model trained on epigenetic profiles and VISTA brain enhancers. This is expected given observations that only 26% of ENCODE enhancer predictions based on epigenetic marks in K562 cells validated in MPRAs44 and that Drosophila enhancers identified via epigenetic marks and MPRA activity are largely non-overlapping43. MPRAs test sequences outside their native chromatin context, often using insulators, and therefore tend to be permissive (i.e., reporting potential regulatory activity). Conversely, enhancer-associated epigenetic marks do not alone indicate enhancer function. Discordance between lentiMPRA and VISTA also arises from measuring activity in NPCs versus whole embryonic brains, primate versus mouse cells, in vivo versus in vitro reporter assays, and ~100-bp versus ~1,000-bp sequences. Despite these differences, ML and lentiMPRA consistently prioritized dozens of HARs as neurodevelopmental enhancers, and each approach revealed some HAR enhancers missed by the other method. Thus, ML is highly complementary to MPRA, and together they advanced understanding of HAR function.
This iterative combination of ML and experimentation shed light on a major question regarding HARs: why did they acquire so many mutations in the human lineage after being conserved throughout mammalian evolution? This question has been hard to tackle, because most HAR sequence changes occurred before our common ancestor with Neanderthals and other archaic hominins1, which means we cannot directly link sequence changes to phenotypes. We addressed this gap with functional data for individual variants and variant combinations in both human and chimpanzee cells. If a human-specific variant changed enhancer activity relative to the chimpanzee allele in chimpanzee cells but not human cells, we might conclude that it evolved to maintain ancestral activity in the presence of altered trans factors in human cells. We found little evidence for such trans effects and instead concluded that species-biased HARs are driven primarily by sequence changes leading to differential binding of TFs present in NPCs from both species. While investigating effects of individual variants versus variant combinations, we expected that variants within the same HAR would alter enhancer activity in the same direction, potentially interacting synergistically to generate large differences between the human and chimpanzee sequences. In contrast, we found that variants in the same HAR have both positive and negative interactions, and some variants individually increase activity while others decrease it. This suggests that compensatory evolution played a role in the rapid evolution of HARs. Combining these results, we speculate that a typical HAR enhancer may have evolved through initial variants with large changes in activity that were then moderated back towards ancestral levels by subsequent nearby variants.
Genetic and three-dimensional chromatin interactions between HARs and genes provide some insight into why HARs might have evolved in this forward-and-back way. Most HARs with enhancer activity in NPCs interact with neurodevelopmental genes, and many are genetically linked to neuropsychiatric disease SNPs and QTLs. This establishes a connection to differential gene expression and chromatin accessibility, suggesting that differential enhancer activity of the HAR could affect brain development and phenotypes. It has been postulated that changes in the human brain enabling our unique cognitive abilities are “Achilles’ heels” that also contribute to schizophrenia and other psychiatric disorders58. Thus, it is plausible that opposing selection pressures for new cognitive traits and against neurological disease were amongst the evolutionary forces that contributed to interacting, compensatory variants in HAR enhancers and their many sequence differences between humans and chimpanzees.
No doubt the true evolutionary trajectory of HARs is more complex than this one hypothesis. It is also likely that our conclusions are influenced by biases in epigenomic and reporter data. For instance, MPRA, luciferase and transgenic animal enhancer assays test candidate enhancers outside their native chromatin environment. Therefore, it is possible that HAR enhancers in their genomic loci would show trans effects that we could not detect in this study. Another caveat is that our NPCs represent only one cell type and two differentiation time points, whereas our ML analyses predict that HARs function broadly across tissues, cell types, and developmental stages. We addressed the shortcoming that prior HAR MPRA and epigenetic studies used human cells, and we created an in vitro system to assay HARs in chimpanzee neurodevelopment. But HARs may have evolved through selection in other cellular contexts. Sei predicted a limited number of HAR variants with differential effects across tissues. Nonetheless, it will be critical to evaluate how human variants affect HAR enhancer function beyond early neuronal differentiation. The integrated ML and experimental strategy presented here provides a framework for these investigations.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Katherine S. Pollard (katherine.pollard@gladstone.ucsf.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
ATAC-seq, ChIP-seq, Hi-C, and MPRA sequencing data has been deposited to GEO: GSE110760.
Experimental Model and Subject Details
Cell Lines
We performed lentiMPRA in N2 and N3 cells derived from four separate iPSC lines from two human and two chimpanzee males. All lines were reprogrammed from fibroblasts using episomal plasmids according to a recently published protocol61. One iPSC line was previously described (WTC;62), and three were generated from low passage fibroblasts (P3 – P7) from Coriell Cell Repository (Hs1: 2 year old human male, catalog AG07095; Pt2: 6 year old chimpanzee male, Maverick, catalog: S003611; Pt5: 8 year old chimpanzee male, catalog PR00738)25. We electroporated three micrograms of episomal expression plasmid mixture encoding OCT3/4, SOX2, KLF4, L-MYC, LIN28, and shRNA for TP53 into 300,000 fibroblasts from each individual with a Neon Electroporation Device (Invitrogen), using a 100 μL kit, with setting of 1,650V, 10ms, and three pulses25,63. After 5–8 days, cells were detached and seeded onto irradiated SNL feeder cells. The culture medium was replaced the next day with primate ESC medium (Reprocell) containing 5 – 20 ng/mL of βFGF. Colonies were picked after 20 – 30 days, and selected for further cultivation. After three to five passages, colonies were transferred to Matrigel-coated dishes and maintained in mTeSR1 medium (Stem Cell Technologies, 05850) supplemented with Penicillin/Streptomycin/Gentomycin. Further passaging was performed using calcium- and magnesium-free PBS to gently disrupt colonies. Each line showed a normal karyotype, and was recently described25. The UCSF Committee on Human Research and the UCSF GESCR (Gamete, Embryo, and Stem Cell Research) Committee approved all human iPSC experiments. HepG2 cells were cultured in Dulbecco’s Modified Eagle Medium (Corning) supplemented with 10% FBS and Penicillin-Streptomycin, and passaged every 4–5 days using StemPro Accutase (Thermo Fisher Scientific).
Method Details
Neural differentiation of human and chimpanzee iPSCs
Human and chimpanzee iPSCs were cultured in Matrigel-coated plates with mTeSR media in an undifferentiated state. Cells were propagated at a 1:3 ratio by treatment using calcium and magnesium free PBS to gently disrupt colonies by mechanical dissection. To trigger neural induction, iPSCs were split with EDTA at 1:5 ratios in culture dishes coated with matrigel and culture in N2B27 medium (comprised of DMEM/F12 medium (Invitrogen) supplemented with 1% MEM-nonessential amino acids (Invitrogen), 1 mM L-glutamine, 1% penicillin-streptomycin, 50 ng/mL bFGF (FGF-2) (Millipore), 1x N2 supplement, and 1 × B27 supplement (Invitrogen)) supplemented with 100 ng/ml mouse recombinant Noggin (R&D systems). N1 cells were collected eleven days after initiating neural induction. Cells at passages 1–3 were split by collagenase into small clumps, similar to iPSC culture, and continuously cultured in N2B27 medium with Noggin. After passage 3, cells were plated at the density of 5E4 cells/cm2 after disassociation by TrypLE express (Invitrogen) into single-cell suspension, and cultured in N2B27 medium supplemented with 20 ng/mL bFGF and EGF. Cells were maintained under this culture condition for a minimum of three months with a stable proliferative capacity. N2 cells were collected at P12–18 and N3 cells at P20–28.
Validation of N2 and N3 markers through immunostaining
Human and chimpanzee N2 and N3 cells were examined using immunostaining against neural and glial progenitor markers. Cells were cultured in chambered Millipore EZ slides, rinsed with PBS, fixed with 4% paraformaldehyde in PBS for 15 minutes at room temperature, washed three times with ice cold PBS, and permeabilized through incubation for 10 min with PBS containing 0.1% Triton X-100. Cells were washed in PBS three times and incubated with 10% donkey serum for 30 minutes to block nonspecific binding of antibodies. Cells were next incubated with diluted primary antibodies against Nestin (monoclonal mouse, Abcam, AB6142), Pax6 (polyclonal rabbit, Abcam, AB5790), and GFAP (polyclonal rabbit, Chemicon, AB5804) in 10% donkey serum for 1 hour at room temperature. The cells were then washed three times in PBS, 5 minutes each wash, then incubated with a secondary antibody (Alexa 488 donkey anti rabbit, Life technologies; Alexa 546 donkey anti mouse, Life technologies) in donkey serum for 1 hour at room temperature in the dark. Cells were then washed three times with PBS in the dark, then covered with a coverslip in Cytoseal mounting media (Thermo Scientific).
Single Cell RNA-Sequencing
To determine the composition of cell types in human and chimpanzee cell lines used for lentiMPRA, we generated single cell gene expression (scRNA-seq) data and clustered cells from each line based on expression. Cells were captured using the C1TM Single-Cell Auto Prep Integrated Fluidic Circuit (IFC), which uses a microfluidic chip to capture the cells, perform lysis, reverse transcription and cDNA amplification in nanoliter reaction volumes. The details of the protocol are described in PN100-7168 (http://www.fluidigm.com/). Sequencing libraries were prepared after the cDNA was harvested from the C1 microfluidic chip using the Nextera XT Sample Preparation Kit (Illumina), following its protocol with minor modifications. The single cell libraries from each C1 capture were then pooled, cleaned twice with 0.9X Agencourt AMPure XP SPRI beads (Beckman Coulter), eluted in DNA suspension buffer (Teknova) or EB buffer (Qiagen) buffer and quantified using High Sensitivity DNA Chip (Agilent). scRNA-seq paired-end reads were generated for ~50 cells per library (Table S1). Sequencing data is available through the accession number GSE110760 (chimpanzee cells: GSE110759, human cells: GSE110758). We trimmed reads for quality using cutadapt under the Trim Galore! wrapper (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) with the default settings, and Nextera transposase sequences were removed. Reads shorter than 20 bp were discarded. Read level quality control was then assessed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were aligned to the NCBI human reference assembly GRCh38 by HiSat264 using the prefilter-multihits option and a guided alignment via the human Gencode Basic v20 transcriptome. Expression for RefSeq genes was quantified by the featureCounts routine, in the subRead library65, using only uniquely mapping reads and discarding chimeric fragments and unpaired reads. Gene expression values were normalized based on library size as counts per million reads (CPM). We used visual image calls to remove any libraries that originated from C1 chambers with multiple cells. To further identify outlier cells, we removed those with fewer than 1,000 genes detected, or with greater than 20% of reads aligning to mitochondrial or ribosomal genes. Gene expression was analyzed using a threshold of detection for each gene at 2 CPM. We then calculated the percentage of cells expressing regional identity genes (e.g., FOXG1 for telencephalon, DLX6-AS1 for GABAergic neurons, MKI67 for dividing cells, SLC1A3 for radial glia). In both human and chimpanzee cell lines at the NPC and GPC stage, 50–90% of cells expressed telencephalon (FOXG1) and radial glia/astrocyte markers.
Histone ChIP-seq experiments
Ten million human (HS1) and chimpanzee (Pt2a) N2 and N3 cells were crosslinked with 1% formaldehyde for 5 minutes and quenched with 125 mM glycine for 5 minutes. To obtain antibody-beads conjugate, Dynabeads protein A (Invitrogen) and Dynabeads protein G (Invitrogen) were mixed at 1:1 ratio and washed twice with Buffer A (LowCell# ChIP kit, diagenode). 10 μg of H3K27ac antibody (Abcam, ab4729) was added to the beads, and gently agitated at 4°C for 2 hours. ChIP was performed using LowCell# ChIP kit (Diagenode) according to manufacturer’s protocol. Sequencing libraries were generated using Accel-NGS 2S Plus DNA library kit (Swift Biosciences). DNA was quantified with Qubit DNA HS assay kit and Bioanalyzer (Agilent) using the DNA High Sensitivity kit. Sequencing was performed using an Illumina HiSeq 4000 with 50 bp single reads. Two biological replicates were done for each cell type. ChIP-seq was processed by the ENCODE Transcription Factor and Histone ChIP-seq processing pipeline (https://github.com/ENCODE-DCC/chip-seq-pipeline2) using default parameters. The pipeline configuration file was modified to enable alignment of Pt2a cell line data to the panTro6 genome.
ATAC-seq experiments
ATAC-seq was performed according to previously described66. Briefly, 50,000 cells were dissociated using Accutase and precipitated with centrifugation at 500 g for 5 minutes. The cell pellet was washed with PBS, resuspended in 50 mL lysis buffer (10 mM TrisHCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Igepal CA-630), and precipitated with centrifugation at 500 g for 10 minutes. The nuclei pellet was resuspended in 50 mL transposition reaction mixture which includes 25 μL Tagment DNA buffer (Nextera DNA sample preparation kit; Illumina), 2.5 μL Tagment DNA enzyme (Nextera DNA sample preparation kit; Illumina), and 22.5 μL nuclease-free water, and incubated at 37C for 30 minutes. Tagmented DNA was purified with MinElute reaction cleanup kit (QIAGEN). The DNA was size-selected using SPRIselect (Beckman Coulter) according to the man- ufacturer’s protocol. 0.6x and 1.5x volume of SPRIselect was used for right and left side selection, respectively. Library amplification was performed as previously described67. Amplified library was further purified with SPRIselect as described above. DNA was quantified on a Bioanalyzer using the DNA High Sensitivity kit (Agilent). Massively parallel sequencing was performed on an Illumina HiSeq4000 with PE150. ATAC-seq was done in 2 biological replicates for each time point.
Hi-C experiments
Hi-C was performed using the Arima Hi-C kit (Arima Genomics) according to the manufacturer’s instructions. 10 million cells were used. The sequencing library was prepared using Accel-NGS 2S Plus DNA Library Kit (Swift Biosciences) according to the manufacturer’s protocol. Two independent biological replicates were prepared for each cell line. In total eight libraries were pooled and sequenced with paired-end 150-bp reads using two lanes of a NovaSeq6000 S2 (Illumina) at the Chan Zuckerberg Biohub.
LentiMPRA library design
All HARs from our prior studies8,42 that were not fully covered in the human (hg19) and chimpanzee (panTro2) reference genome sequences were included in the library design. These 714 HARs have similar lengths and genomic distributions compared to the larger set of 2645 HARs, so we expect them to be representative. For each HAR, we designed 171-bp oligos representing the orthologous human and chimpanzee sequences. Since HARs have median length 227 bp, most could be synthesized using a single oligo or two highly overlapping oligos. Flanking genomic sequence was added to HARs shorter than 171 bp, and HARs longer than 171 bp were tiled with multiple oligos having variable but considerable overlap depending on the length of the HAR (e.g., 67% overlap between the two oligos for HAR sequences between 171 and 342 bp long, which have mean length 233 bp). A third of HARs could be synthesized using a single oligo and the vast majority required less than three oligos. For the remaining HARs, the multiple oligos were separately quantified for enhancer activity (see below) and assessed for agreement. We observed high correlation between multiple oligos per HAR, likely due to their generally high level of overlap, so we merged all oligos per HAR (summed their reads) for downstream analysis. This produced one activity measurement for each human or chimpanzee HAR sequence.
We additionally synthesized 118 sequences that we expected would show little or no enhancer activity in NPCs (negative controls) and 143 sequences that we expected would drive expression in NPCs (positive controls). Negative controls were comprised of 34 sequences used as negatives by the ENCODE consortium (provided by Rick Meyers) plus 84 human genome sequences located in H3K27me3 ChIP-seq peaks from human N2 and N3 cells (data generated in the Ahituv lab, released with this study). Positive controls included 9 positive enhancer elements from ENCODE (provided by Rick Meyers), 124 human genome sequences located in H3K27ac ChIP-seq peaks from human N2 and N3 cells (data generated in the Ahituv lab, released with this study), and 10 human genome sequences we predicted would function as neurodevelopmental enhancers using our EnhancerFinder algorithm68.
All HAR and control sequences were scanned for restriction sites (for SbfI and EcoRI) and modified to avoid problems in synthesis and cloning. We designed our experiments to ideally have 100 unique 15-bp barcodes per variant to build in robustness to barcode dropout and jackpotting issues, variability in activity across integration sites, and other sources of technical error. These barcodes are not random, but rather designed to be at least two substitutions and one insert-deletion (indel) apart from other barcodes and synthesized with the oligos. The final array design included 2,440 unique 171-bp sequences, each with 100 barcodes, for a total of 244,000 oligos inclusive of HARs and controls.
LentiMPRA library synthesis and cloning
All lentiMPRA sequences were array-synthesized as 230-bp oligos (Agilent Technologies) containing universal priming sites (AGGACCGGATCAACT…CATTGCGTGAACCGA), a 171-bp candidate enhancer sequence, spacer (CCTGCAGGGAATTC), and 15-bp barcode. The amplification and cloning of the enhancers and barcodes into the pLS-mP lentiviral vector was performed as previously described69. Briefly, pLS-mP was cut with SbfI and EcoRI taking out the minimal promoter and EGFP reporter gene. The oligos containing the HAR, spacer, and barcode were amplified with adaptor primers (pLSmP-AG-f and pLSmP-AG-r; Table S8) that have overhangs complementary to the cut vector backbone, and the products were cloned using NEBuilder HiFi DNA Assembly mix (NEB, E2621). The cloning reaction was transformed into electro-competent cells (NEB C3020) and multiple transformations were pooled and midiprepped (Chargeswitch Pro Filter Plasmid Midi Kit, Invitrogen CS31104). The library was then cut using SbfI and EcoRI sites contained within the spacer, so that the minimal promoter and EGFP could be reintroduced via a sticky end ligation (T4 DNA Ligase, NEB M0202). This library was transformed and purified, as previously described, and DNA sequenced to determine complexity.
We estimate that at least 92% of barcodes are correctly synthesized and that per-base substitution errors are about 0.02–0.04% (synthesis and amplification). The observed median number of unique barcodes per variant ranged from 79 to 81 across our 24 replicates, with a 25th percentile of 73 to 76 barcodes and a 75th percentile of 79 to 87 barcodes. The chimpanzee sequence of 2xHAR.335 had particularly low barcode counts, with a median of 17, a minimum of 16, and a maximum of 18. The next worst HAR has a median of 49 barcodes. Thus, barcode count was consistently high for most of the HARs we tested. Also, the number of barcodes was similar across replicates for any given oligo, suggesting synthesis as the source of differential numbers of barcodes. By aiming for 100 barcodes per variant, we generated a library with high numbers of barcodes despite barcode dropout.
Lentivirus library preparation and infection
Lentivirus packaging of the HAR lentiMPRA library was performed by the UCSF Viracore using standard techniques70. Twelve million HEK293T cells were plated in a 15-cm dish and cultured for 24 hours. The cells were co-transfected with 8 μg of the HAR library and 4 μg of packaging vectors using jetPRIME (Polyplus-transfections). The transfected cells were cultured for 3 days and lentiviruses were harvested and concentrated as previously described70. For all human and chimpanzee cell lines and cell stages, about twelve million cells were plated in 15-cm dishes and cultured for 24–48 hours. Cells were infected with a multiplicity of infection (MOI) of 50. When infecting the library into HepG2 cells, 8 μg/mL polybrene was added to the cells. The culture medium was refreshed daily. Infected cells were washed with PBS three times before cell lysis in order to remove any non-integrated lentivirus.
RNA & DNA isolations and sequencing
Genomic DNA and total RNA were extracted using the AllPrep DNA/RNA mini kit (Qiagen). Messenger RNA was purified from the total RNA using Oligotex mRNA mini kit (Qiagen) and treated with Turbo DNAseq to remove contaminating DNA. The RT-PCR, amplification and sequencing of RNA and DNA were performed as previously described69, with some alterations for adding Unique Molecular Identifiers (UMIs) in the process. In brief, mRNA was reverse transcribed with SuperScript II (Invitrogen) using a primer downstream from the barcode (pLSmP-ass-R-UMI-i#; Table S8). The resulting cDNA was split into multiple reactions to reduce PCR jack-potting effects and cDNA amplification performed with Kapa Robust polymerase for three cycles, incorporating unique molecular identifiers (UMIs) of 10 bp length. PCR products were cleaned with AMPure XP beads (Beckman Coulter) to remove primers and concentrate samples. These products underwent a second round of amplification in 8 reactions per replicate for 15 cycles, switching from the UMI-incorporating reverse primer to one containing only the P7 flow cell sequence (P7; Table S8). All reactions were pooled and run on agarose gels for size selection and submitted for sequencing. For DNA, each replicate was amplified for 3 cycles with UMI-incorporating primers, just as the RNA. First round products were cleaned up with AMPure XP beads, and amplified in split reactions, each for 20 cycles. Again, reactions were pooled and gel-purified.
RNA and DNA for all three replicates for all samples were sequenced on an Illumina NextSeq instrument (2×15 bp barcodes + 10bp UMI + 10bp sample index) using custom primers (BARCODE-SEQ-R1-V4, pLSmP-AG-seqIndx, BARCODE-SEQ-R2-V4; Table S8) and are available through the Short Read Archive (SRA) with BioProject accession numbers PRJNA428580 (chimpanzee cells) and PRJNA428579 (human cells). Illumina Paired End reads sequenced the barcodes from the forward and reverse direction and allowed for adapter trimming and consensus calling of tags71. Barcode or UMI sequences containing unresolved bases (N) or not matching the designed length of 15 bp were excluded. In data analysis, each barcode × UMI pair is counted only once and only barcodes matching perfectly to those included in the above oligo design were considered.
ML comparisons with lentiMPRA
Each HAR and VISTA enhancer was described by overlaps with epigenetic datasets in primary brain, heart, and limb tissue and co-embedded in UMAP space. Additionally, a ML model (L1-penalized classifier) was trained to distinguish HARs with high lentiMPRA activity (top 25%) and low ML scores (bottom 25%) from those with low lentiMPRA activity (bottom 25%) and high ML scores (top 25%). The model achieved high accuracy (0.96 auPR), indicating that these groups of discordant HARs have distinct combinations of epigenetic features. HARs with high lentiMPRA activity but low ML scores have marks of active chromatin in non-brain tissues. These are a mix of enhancers with active marks in NPCs and other tissues, plus enhancers inactive in NPCs that nonetheless show high activity in lentiMPRAs due to being tested outside their native chromatin environment that is silent in the embryonic brain. On the other hand, HARs with high ML scores but low lentiMPRA activity overlap open chromatin in samples from the whole fetal brain. These appear to be mostly embryonic brain enhancers active in cell types other than forebrain neurons.
Permutation lentiMPRA
For each of three selected HARs with significant cis effects in our lentiMPRAs and prior evidence of enhancer activity (2xHAR.164, 2xHAR.170, 2xHAR.238), we designed oligos carrying all possible evolutionary intermediates (“permutations”) between homologous human and chimpanzee sequences. Some human HAR sequences differ in length from their homologous chimpanzee sequence due to short insertions and deletions. So to truly isolate the effects of individual human mutations in HARs, permutation oligos were created by mutating these sites and combinations thereof in the chimpanzee sequence. Thus, for HARs with insertions or deletions the oligo containing all human alleles is not the exact human genome sequence, but rather the chimpanzee sequence with all these mutations introduced. Permutation oligos were assayed, quantified and normalized alongside the main lentiMPRA library described above. The one-hot encoded oligo sequence, along with cell species and stage, were used to model the log RNA/DNA ratios for a given HAR using gradient boosting (XGBoost). The model estimates the importance of each nucleotide with a human-chimpanzee sequence difference, interactions between these nucleotides, and interactions between nucleotides and cell species or stage for predicting MPRA activity. We also assayed the permutation oligos as a separate library (“library 2”) in three technical replicates of two human (WTC, HS1–11) and two chimpanzee (Pt2A, Pt5C) cell lines, each at two stages of neural differentiation (N2, N3). For library 2, RNA and DNA count data was quantified as above, including normalizing for sequencing depth and batch correcting for library preparation date using limma.
Luciferase assays
To generate pLS-mP-Luc vector (Addgene 106253), minimal promoter and Luciferase gene fragment was amplified using pGL4.23 (Promega) as a template and inserted into pLS-mP (Addgene 81225) replacing with mP-EGFP. To generate pLS-SV40-mP-Rluc (Addgene106292), renilla luciferase gene was amplified using pGL4.74 (promega) as a template and inserted into pLS-SV40-mP vector (17) replacing with EGFP gene. We used an Agilent array to synthesize human and chimpanzee sequences of 2xHAR.11, 2xHAR.35, 2xHAR.53, 2xHAR.176, 2xHAR.273, 2xHAR.364, 2xHAR.401, 2xHAR.417, 2xHAR.434, and 2xHAR.518, and six negative control sequences (hg19 coordinates): N0 = chr1:10755200–10755371 (astrocyte progenitor H3K27me3 peak), N06 = chr7:27118200–27118371 (N2 H3K27me3 peak), N10 = chr4:8852800–8852971 (N1 H3K27me3 peak), N12 = chr17:46740400–46740571 (N2 H3K27me3 peak), N15 = chr19:1744200–1744371 (N1 H3K27me3 peak), N17 = chr14:37219958–37220129 (ENCODE negative control). These were synthesized along with homology arms on both sides (left: AGCCTGCATTTCTGCCAGGGCCCGCTCTAG, right: CTAGACCTGCAGGCACTAGAGGGTATATA), amplified using Agilent-luc.F and Agilent-luc.R primers (Table S8), and cloned into XbaI site of the pLS-mP-luc using NEBuilder HiFi DNA Assembly Cloning Kit (NEB). Fragments that failed to clone (human 2xHAR.11, chimpanzee 2xHAR.35, human 2xHAR.176, human 2xHAR.273, chimpanzee 2xHAR.364, human and chimpanzee 2xHAR.434, and chimpanzee 2xHAR.518) were synthesized by Twist Bioscience along with homology arms: (left: TGTATATCCGGTCTCTTCTCTGGGTAGTCTCACTCAGCCTGCATTTCTGCCAGGGCCCGCTCTAG, right: CTAGACCTGCAGGCACTAGAGGGTATATAATGGAAGCTCGACTTCCAGCTTGGCAATCCGGTAC), amplified using Twist-luc.F and Twist-luc.R primers (Table S8) and cloned into the pLS-mP-luc. Lentivirus was generated using standard methods70, as described below for the library, individually for each clone with pLS-SV40-mP-Rluc spiked in at 10% of the total amount of plasmid used. 2×104 cells per well (HS1 and Pt2 N3 cells) were seeded in a 96-well plate and were infected with virus 24 hours later. Three independent replicate cultures were transfected per plasmid and two biological replicates were done in different days. Firefly and Renilla luciferase activities were measured on a Synergy 2 microplate reader (BioTek) using the Dual-Luciferase Reporter Assay System (Promega). Enhancer activity was calculated as the fold change of each construct’s firefly luciferase activity normalized to renilla luciferase activity.
Transgenic mouse reporter assays
We selected HAR152, 2xHAR.133, 2xHAR.518, and 2xHAR.548 for in vivo validation with mouse transient transgenic reporter assays based on their lentiMPRA activity, epigenetic profiles, and nearby genes. All HAR sequences were cloned into the Hsp68-LacZ vector (Addgene #37843) and validated by Sanger sequencing. LacZ transgenic mice were generated by Cyagen Biosciences using standard procedures72, harvested and stained for LacZ expression as previously described73. Pictures were taken using an M165FC stereo microscope and a DFC500 12-megapixel camera (Leica).
CRISPR activation experiment
2xHAR.183 was selected for further functional characterization due to its high predicted enhancer score (see Methods: Supervised and Unsupervised Learning Analysis) and overlaps with multiple chromatin interaction datasets. The HAR shares a TAD and has a significant chromatin loop with the ROCK2 gene in excitatory neuron PLAC-seq data35 and contacts ROCK2 in our N2/N3 Hi-C. The gene E2F6 is nearby on the linear genome but has fewer 3D chromatin contacts. Independent from our prediction, 2xHAR.183 overlaps a predicted FANTOM5 enhancer, an ENCODE candidate cis-regulatory element, and a ChromHMM enhancer annotated using fetal brain datasets.
Human excitatory neurons were generated using hiPSCs in the WTC11 background containing a doxycycline inducible neurogenin-2 at the AAVS1 safe harbour locus. In their undifferentiated state, cells were plated in Matrigel-coated plates and cultured with mTeSR media. mTeSR media was changed daily. To induce differentiation, cells were dissociated using Accutase and plated in Matrigel-coated plates. Cells were cultured for 3 days in pre-differentiation media containing KnockOut DMEM/F-12 with 2 ug/mL doxycycline supplemented with 1X N-2 Supplement, 1X NEAA, 10 ng/mL brain-derived neurothrophic factor (BDNF), 10 ng/mL NT-3, and 1ug/mL lamininin. On the first day, ROCK inhibitor was added to the predifferentiation media at a concentration of 10uM. Pre-differentiation media was changed daily for 3 days. To induce maturation, precursor cells were dissociated with Accutase and subplated in poly-D-Lysine coated plates. Cells were cultured in maturation media containing Neurobasal A and DMEM/F12 with 2 ug/mL doxycycline supplemented with 1X N-2 Supplement, 0.5 X B-27 Supplement, 1X NEAA, 0.5X GlutaMax, 10 ng/mL BDNF, 10 ng/mL NT-3, and 1ug/mL lamininin. Cells were maintained in the maturation media for the remaining 14 days. Half media changes were conducted on day 7 and day 14 of differentiation with maturation media minus doxycyline. After 14 days, wells were infected with lentivirus containing dCAS9-VP64_Blast (Addgene Plasmid #61425) and sgRNA targeting 2xHAR.183. The sgRNA sequence ATCATAGGATCAACTCGTTA was selected using CHOPCHOP to target 2xHAR.183 and was cloned into the pLG1 expression vector. Experiments were performed in triplicate and compared to wells infected with dCAS9-VP64 and no sgRNA. RNA was isolated after 5 days infection with lentivirus using the QIAGEN RNeasy kit with gDNA elimination column. RNA quality was investigated using the Agilent 2100 Bioanalyzer system and the RNA 6000 Nano kit, and an RNA integrity number over 9.0 was verified for all samples. cDNA was made from 1 microgram total RNA using SuperScript III First-Strand Synthesis SuperMix. qRT-PCR was performed using Maxima SYBR Green / ROX qPCR master mix and the following oligonucleotides:
| Gapdh_Forward | GTCTCCTCTGACTTCAACAGCG |
| Gapdh_Reverse | ACCACCCTGTTGCTGTAGCCAA |
| Rock2_Forward | CGA GCCGCC AGAGAGAG |
| Rock2_Reverse | CCAAGGAA !MAAGCCATCCAGC |
| E2f6_Forward | TACCCAGTCTCCTCCTGGAC |
| E2f6_Reverse | TATTTTTGATGGCAGCAGGC |
Quantification and Statistical Analysis
Transcription factor footprints
While traditional footprinting methods operate on open chromatin data, the HINT29 method can also compute footprints from histone ChIP-seq data alone, albeit with reduced accuracy. We utilized this strategy, because we have matched H3K27ac ChIP-seq from human and chimpanzee NPCs. Genome-wide footprint analysis was run separately on human N2 H3K27ac ChIP-seq and chimpanzee N2 H3K27ac ChIP-seq, both using HOCOMOCO v11 and JASPAR 2020 TF motifs. Expressed TFs were defined as those with TPM>1 in NPCs59 (Kallisto74 v0.48). HINT (v0.13.2) was used to compute enrichment of footprints within HARs compared to H3K27ac peaks (`rgt-hint footprinting --histonè followed by `rgt-motifanalysis matching`).
RNA/DNA ratios and quantification of enhancer activity
RNA and DNA counts were first normalized per replicate using counts per million reads mapped (CPM). RNA/DNA ratios per HAR per replicate were calculated by taking the sum of RNA counts for all ~80 barcodes assigned to all oligo(s) tiling across each HAR, divided by the sum of all DNA counts for all barcodes across all oligo(s) per HAR, and using only barcodes with >0 counts in DNA. Importantly, we do not compute RNA/DNA for each barcode and average these, but rather use the ratio of the sum of RNA counts and the sum of DNA counts over all detected barcodes, which is more robust to over-represented (PCR “jackpot”) barcodes than first taking the ratio per barcode. We also tried using the ratio of the median RNA and median DNA count, rather than sums (equivalent to means when RNA and DNA have the same number of barcodes), and we observed a correlation of ~98.5%, demonstrating that our quantification method is indeed robust. We summed counts across oligos for HARs tiled using two or more oligos, because we observed generally good agreement between oligos for the same HAR. The resulting RNA/DNA ratios were batch normalized for RNA and DNA library preparation date using limma75.
We focused our differential activity analyses on HARs with the highest and most consistent activity across replicates, specifically, 293 “active” HARs (41%) that drive expression above the median of positive controls in at least 50% of samples for either the human or chimpanzee sequence. These HARs also all have activity above the 75th percentile of the negative controls in at least 50% of samples for either the human or chimpanzee sequence. There is no threshold that perfectly separates positive and negative controls, because their activity distributions overlap despite positives being significantly more active than negatives in all cell lines (Figure S3). This overlap likely represents the permissiveness of MPRAs, which are conducted outside the chromatin environment of the native locus. Importantly, our conclusion that most HARs show small quantitative differential activity between human and chimpanzee sequences is robust to the chosen threshold for active HARs.
Modeling lentiMPRA cis and trans effects
To identify HARs with different enhancer activity between human and chimpanzee sequences (“cis effects”), we used the R limma75 package (3.50.1) to fit a linear model for the mean log2(human [RNA/DNA] / chimpanzee [RNA/DNA]) of each HAR across 18 samples passing QC (human and chimpanzee cells, N2 and N3 stages) with code: lmFit(log2(human_chimp_ratios), model.matrix(~ prep_date)) %>% eBayes(). This fits a linear model for each HAR that adjusts for the library preparation date, which we used to test for mean log-ratios significantly different from zero. P-values were adjusted for multiple testing using the false discovery rate (FDR eBayes q-value <1%), producing 188 differentially active HARs. We also explored using limma with voom or other variance stabilizing transformations but found that these were not needed because the log2(human[RNA/DNA] / chimpanzee [RNA/DNA]) values do not have a strong mean-variance relationship.
Gene Ontology analysis
Gene Ontology (GO) terms associated with the 293 active HAR enhancers were separately compared to those of two background sets: all human N2 ATAC-seq peaks, and a random subset of 20k conserved elements identified with phastCons. Enrichment was computed with g:Profiler76, using the custom statistical domain scope to provide the appropriate background set. The significance threshold was computed using g:SCS, which accounts for the non-independence of terms in the GO hierarchy.
Predicting differential activity from footprints
HARs were intersected with TF footprints detected in human and chimpanzee N2 H3K27ac data (HINT29 v0.13.2), excluding TFs not expressed in NPCs (TPM < 1, Kallisto74 v0.48). Each HAR was then described by a vector of 762 binary features (presence/absence of 381 human and 381 chimpanzee footprints). A supervised gradient boosting regressor (XGBoost) was trained using these features and the log-scale RNA/DNA ratio from the MPRA as a continuous label. 80 percent of HARs were used for training and 20 percent used to detect overfitting during training (early stopping validation set). Variable importance was computed for each feature (TF × species). In-sample R2 and MSE were reported, as HARs are described by sparse and diverse sets of footprints that result in low R2 for out-of-sample data.
Variant-disrupted footprints
The predicted chromatin state changes for all human:chimpanzee HAR variants were computed using Sei and overlapped with predicted footprints for NPC-expressed TFs (TPM > 1). For each TF, the number of overlapping variants and the maximum and minimum state change was computed. For visualization, TFs in the upper quartile of # variant overlaps were labeled if their maximum or minimum state change were in the top or bottom 18th percentile, respectively.
Supervised and unsupervised learning analysis with in vivo epigenetic profiles
A large collection of open chromatin and TF binding datasets from multiple primary tissues (49% brain, 48% heart, 2% limb) were intersected with HARs and shown using Upset plots. The marks H3K4me1, H3K4me3, H3K9ac, H3K27ac, and H3K36me3 were labeled as activating. GEO and ENCODE accessions for these datasets are given in Table S3. Feature vectors encoding the intersection of these datasets with HARs and validated VISTA enhancers32 (all tissues) were projected into two dimensions using UMAP77 (umap-learn v0.5.3, n_neighbors = 8, metric = russellrao). For supervised learning, a logistic regression model with L1 (LASSO) penalty (scikit-learn78 1.0.2) was trained using the feature vectors of validated VISTA enhancers and labeling brain enhancers as positives and non-brain enhancers (candidates that failed to validate or were active in other tissues) as negatives. This model then scored the similarity of HARs to neurodevelopmental enhancers.
Deep learning characterization of HAR variants
All variants between human and chimpanzee HAR alleles were computed by first extracting alignments for each HAR (`mafsInRegion`) from the Zoonomia Consortium79, the largest multi-species alignment (MSA) to date. Human and chimpanzee alignments were then converted from MAF format to FASTA (`msa_view`), and from FASTA to VCF (`jvarkit msa2vcf`)80. The deep learning tool Sei41 then predicted changes in chromatin state for each human:chimpanzee variant across all HARs. Sei utilizes tens of thousands of human datasets which do not exist for chimpanzee; therefore, the predicted score estimates the impact of a chimpanzee variant relative to the human allele. To interpret these changes from an evolutionary perspective, we multiplied predicted scores by −1 so that chimpanzee variants with large negative scores would instead be positive (i.e., the human allele caused an increase) and large positive scores would instead be negative (i.e., the human allele caused a decrease).
Analysis of HAR genetic and physical linkage to genes
Raw Hi-C data was aligned to hg38 and processed with juicer and distiller. Contact domains were called with the arrowhead algorithm in juicer81, while chromatin loops were called using mustache82 on output from distiller83. LD blocks were computed using plink84 using 1000 Genomes85 super-populations. HARs were annotated with multiple data sources including neurodevelopmental enhancer score (machine learning), closest protein coding gene (bedtools), neuropsychiatric variants in the same LD block for all (plink, bedtools), protein coding gene promoters sharing a contact domain in NPC/GPC or CP/GZ33 Hi-C data (juicer, bedtools), variants or protein coding gene promoters interacting with the HAR via chromatin looping in NPC/GPC or CP/GZ33 Hi-C data (mustache, bedtools), and genes interacting with the HAR via chromatin looping in primary tissue PCHi-C34 or PLAC-seq35 data (bedtools).
Variant datasets from the Psychiatric Genomics Consortium included ADHD86, Alzheimer’s Disease87, Autism Spectrum Disorder88, Bipolar Disorder89, Cross-Disorder90, Major Depressive Disorder91, Obsessive-Compulsive Disorder92, Schizophrenia (in review), and Tourette’s Syndrome93. Variant datasets from the PsychENCODE Consortium include expression QTLs (FDR < 0.05, > 1 FPKM in >= 20% of samples) and chromatin QTLs. Other variant datasets included chromatin QTLs in neurons and neural progenitors49, expression QTLs in prefrontal cortex48, and GTEx v8 fine-mapped brain expression QTLs46. Datasets using hg19 coordinates were mapped to hg38 using their rsid in combination with dbSNP build 155.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Experimental Models: Cell Lines | ||
| Human WTC | Miyaoka et al. 2014 | https://doi.org/10.1038/nmeth.2840 |
| Human HS1 | Coriell Cell Repository | AG07095 |
| Chimpanzee Pt2 | Coriell Cell Repository | S003611 |
| Chimpanzee Pt5 | Coriell Cell Repository | PR00738 |
| Antibodies | ||
| monoclonal mouse anti-Nestin | Abcam | Abcam AB6142 |
| polyclonal rabbit anti-Pax6 | Abcam | Abcam AB5790 |
| polyclonal rabbit anti-GFAP | Chemicon | Chemicon AB5804 |
| Recombinant DNA | ||
| pLS-mP | Nadav Ahituv | Addgene 81225 |
| pLS-mP-Luc | Nadav Ahituv | Addgene106253 |
| pLS-SV40-mP-Rluc | Nadav Ahituv | Addgene106292 |
| Hsp68-LacZ vector | Nadav Ahituv | Addgene 37843 |
| dCAS9-VP64_Blast | Feng Zhang | Addgene 61425 |
| Oligonucleotides | ||
| Forward primer for Gapdh | This paper | GTCTCCTCTGACTTCAACAGCG |
| Reverse primer for Gapdh | This paper | ACCACCCTGTTGCTGTAGCCAA |
| Forward primer for Rock2 | This paper | CGAGCCGCCAGAGAGAG |
| Reverse primer for Rock2 | This paper | CCAAGGAAIIIAAGCCATCCAGC |
| Forward primer for E2f6 | This paper | TACCCAGTCTCCTCCTGGAC |
| Reverse primer for E2f6 | This paper | TATTTTTGATGGCAGCAGGC |
| Deposited Data | ||
| Raw and processed MPRA, ChlP-seq, ATAC-seq, and Hi-C sequencing data | This paper | GEO: GSE149268 |
| Human Accelerated Regions | Hubisz and Pollard 2014 | https://doi.org/10.1016/j.gde.2014.07.005 |
| JASPAR 2020 transcription factor binding sites | Castro-Mondragon et al. 2022 | https://jaspar.genereg.net |
| HOCOMOCO v11 transcription factor binding models | Kulakovskiy et al. 2018 | https://hocomoco11.autosome.org |
| Raw and Processed ChlP-seq, ATAC-seq, and DNase-seq sequencing data | ENCODE Consortium | Table S3 |
| Validated human enhancers | Visel et al. 2007 | https://enhancer.lbl.gov |
| TF footprints in ENCODE tissues | Funk et al. 2020 | https://data.nemoarchive.org/other/grant/sament/sament/footprint_atlas |
| Genomes for computing LD blocks by super-population | 1000 Genomes Project Consortium | https://www.internationalgenome.org/data/ |
| Multi-species alignment | Zoonomia Consortium | https://doi.org/10.1038/s41586-020-2876-6 |
| Hi-C chromatin loops | Won et al. 2016 | https://doi.org/10.1038/nature19847 |
| PCHi-C chromatin loops | Song et al. 2019 | https://doi.org/10.1038/s41588-019-0472-1 |
| PLAC-seq chromatin loops | Song et al. 2020 | https://doi.org/10.1038/s41586-020-2825-4 |
| Software and Algorithms | ||
| pandas | McKinney 2012 | https://pandas.pydata.org |
| scikit-learn | Pedregosa et al. 2011 | https://scikit-learn.org |
| xgboost | Chen and Guestrin 2016 | https://github.com/dmlc/xgboost |
| R | R Development Core Team 2018 | https://www.r-project.org |
| bioconductor | Huber et al. 2015 | https://www.bioconductor.org |
| limma | Ritchie et al. 2015 | https://doi.org/10.18129/B9.bioc.limma |
| bedtools2 | Quinlan and Hall 2010 | https://github.com/arq5x/bedtools2 |
| ENCODE ATAC-seq pipeline | Lee et al. 2016 | https://github.com/ENCODE-DCC/atac-seq-pipeline |
| ENCODE ChlP-seq pipeline | Lee et al. 2016 | https://github.com/ENCODE-DCC/chip-seq-pipeline2 |
| HlNT | Gusmao et al. 2014 | https://github.com/CostaLab/reg-gen |
| Sei | Chen et al. 2022 | https://github.com/FunctionLab/sei-framework |
| distiller | Goloborodko et al. 2019 | https://github.com/open2c/distiller-nf |
| juicer | Durand et al. 2016 | https://github.com/aidenlab/juicer |
| mustache | Ardakany et al. 2020 | https://github.com/ay-lab/mustache |
| plink | Purcell et al. 2007 | https://www.cog-genomics.org/plink/1.9 |
| UMAP | McInnes et al. 2018 | https://github.com/lmcinnes/umap |
| jvarkit | Lindenbaum 2021 | https://github.com/lindenb/jvarkit |
| mafslnRegion | Kent et al. 2002 | https://hgdownload.soe.ucsc.edu/admin/exe |
| msa_view | Siepel et al. 2004 | https://github.com/CshlSiepelLab/phast |
| kallisto | Bray et al. 2016 | https://github.com/pachterlab/kallisto |
| g:Profiler | Raudvere et al. 2019 | https://biit.cs.ut.ee/gprofiler/gost |
| Enhancer prediction | This paper | https://doi.org/10.5281/zenodo.7349179 |
eTOC Highlights.
31% of HARs act as enhancers in chimpanzee and human neural progenitor cells
43% of HARs have human variants with large effects on chromatin state
Transcription factor footprints predict human:chimpanzee enhancer activity
Rapid evolution of HAR sequences reflects compensatory tuning of enhancer activity
Acknowledgements
This work was funded by the Gladstone Institutes (K.S.P.), Schmidt Futures Foundation (A.A.P), and NIH grants: DP2MH122400-01 (A.A.P.), R35NS097305 (A.R.K.), FHG011569A (T.F.), R01MH109907 (N.A. and K.S.P.), U01MH116438 (N.A. and K.S.P.), UM1HG009408 (J.S. and N.A.), UM1HG011966 (J.S. and N.A.), and 2R01NS099099 (J.L.R.). K.S.P. is a Chan Zuckerberg Investigator. A.A.P is a New York Stem Cell Foundation Robertson Investigator and member of the UCSF Kavli Institute for Fundamental Neuroscience.
Declaration of Interests
A.K. is a cofounder, consultant, and member of the board of Neurona Therapeutic, a company studying the potential therapeutic use of interneuron transplantation. J.L.R.R. is a cofounder, stockholder, and member of the board of Neurona. N.A. is a cofounder and on the scientific advisory board of Regel Therapeutics and Neomer Diagnostics and receives funding from BioMarin Pharmaceutical Incorporate. K.K. is currently an employee of Fauna Bio.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hubisz MJ, and Pollard KS (2014). Exploring the genesis and functions of Human Accelerated Regions sheds light on their role in human evolution. Curr. Opin. Genet. Dev 29, 15–21. [DOI] [PubMed] [Google Scholar]
- 2.Franchini LF, and Pollard KS (2017). Human evolution: the non-coding revolution. BMC Biol. 15, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burns JK (2004). An evolutionary theory of schizophrenia: cortical connectivity, metarepresentation, and the social brain. Behav. Brain Sci 27, 831–55; discussion 855–85. [DOI] [PubMed] [Google Scholar]
- 4.Crow TJ (1997). Is schizophrenia the price that Homo sapiens pays for language? Schizophrenia Research 28, 127–141. 10.1016/s0920-9964(97)00110-2. [DOI] [PubMed] [Google Scholar]
- 5.Babbitt CC, Warner LR, Fedrigo O, Wall CE, and Wray GA (2011). Genomic signatures of diet-related shifts during human origins. Proceedings of the Royal Society B: Biological Sciences 278, 961–969. 10.1098/rspb.2010.2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Capra JA, Erwin GD, McKinsey G, Rubenstein JLR, and Pollard KS (2013). Many human accelerated regions are developmental enhancers. Philosophical Transactions of the Royal Society B: Biological Sciences 368, 20130025. 10.1098/rstb.2013.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamm GB, Pisciottano F, Kliger R, and Franchini LF (2013). The developmental brain gene NPAS3 contains the largest number of accelerated regulatory sequences in the human genome. Mol. Biol. Evol 30, 1088–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pollard KS, Salama SR, King B, Kern AD, Dreszer T, Katzman S, Siepel A, Pedersen JS, Bejerano G, Baertsch R, et al. (2006). Forces shaping the fastest evolving regions in the human genome. PLoS Genet. 2, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prabhakar S, Noonan JP, Pääbo S, and Rubin EM (2006). Accelerated evolution of conserved noncoding sequences in humans. Science 314, 786. [DOI] [PubMed] [Google Scholar]
- 10.Doan RN, Bae B-I, Cubelos B, Chang C, Hossain AA, Al-Saad S, Mukaddes NM, Oner O, Al-Saffar M, Balkhy S, et al. (2016). Mutations in Human Accelerated Regions Disrupt Cognition and Social Behavior. Cell 167, 341–354.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyd JL, Skove SL, Rouanet JP, Pilaz L-J, Bepler T, Gordân R, Wray GA, and Silver DL (2015). Human-chimpanzee differences in a FZD8 enhancer alter cell-cycle dynamics in the developing neocortex. Curr. Biol 25, 772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prabhakar S, Visel A, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer-Frick I, Morrison H, Fitzpatrick DR, Afzal V, et al. (2008). Human-specific gain of function in a developmental enhancer. Science 321, 1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dutrow EV, Emera D, Yim K, Uebbing S, Kocher AA, Krenzer M, Nottoli T, Burkhardt DB, Krishnaswamy S, Louvi A, et al. (2022). Modeling uniquely human gene regulatory function via targeted humanization of the mouse genome. Nat. Commun 13, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Norman AR, Ryu AH, Jamieson K, Thomas S, Shen Y, Ahituv N, Pollard KS, and Reiter JF (2021). A Human Accelerated Region is a Leydig cell GLI2 Enhancer that Affects Male-Typical Behavior. bioRxiv, 2021.01.27.428524. 10.1101/2021.01.27.428524. [DOI] [Google Scholar]
- 15.Aldea D, Atsuta Y, Kokalari B, Schaffner SF, Prasasya RD, Aharoni A, Dingwall HL, Warder B, and Kamberov YG (2021). Repeated mutation of a developmental enhancer contributed to human thermoregulatory evolution. Proc. Natl. Acad. Sci. U. S. A 118. 10.1073/pnas.2021722118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kostka D, Hubisz MJ, Siepel A, and Pollard KS (2012). The role of GC-biased gene conversion in shaping the fastest evolving regions of the human genome. Mol. Biol. Evol 29, 1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou J, Theesfeld CL, Yao K, Chen KM, Wong AK, and Troyanskaya OG (2018). Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet 50, 1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaishnav ED, de Boer CG, Molinet J, Yassour M, Fan L, Adiconis X, Thompson DA, Levin JZ, Cubillos FA, and Regev A (2022). The evolution, evolvability and engineering of gene regulatory DNA. Nature 603, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Avsec Ž, Agarwal V, Visentin D, Ledsam JR, Grabska-Barwinska A, Taylor KR, Assael Y, Jumper J, Kohli P, and Kelley DR (2021). Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 18, 1196–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inoue F, and Ahituv N (2015). Decoding enhancers using massively parallel reporter assays. Genomics 106, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uebbing S, Gockley J, Reilly SK, Kocher AA, Geller E, Gandotra N, Scharfe C, Cotney J, and Noonan JP (2021). Massively parallel discovery of human-specific substitutions that alter enhancer activity. Proc. Natl. Acad. Sci. U. S. A 118. 10.1073/pnas.2007049118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Girskis KM, Stergachis AB, DeGennaro EM, Doan RN, Qian X, Johnson MB, Wang PP, Sejourne GM, Nagy MA, Pollina EA, et al. (2021). Rewiring of human neurodevelopmental gene regulatory programs by human accelerated regions. Neuron 109, 3239–3251.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weiss CV, Harshman L, Inoue F, Fraser HB, Petrov DA, Ahituv N, and Gokhman D (2021). The cis-regulatory effects of modern human-specific variants. Elife 10. 10.7554/eLife.63713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jagoda E, Xue JR, Reilly SK, Dannemann M, Racimo F, Huerta-Sanchez E, Sankararaman S, Kelso J, Pagani L, Sabeti PC, et al. (2022). Detection of Neanderthal Adaptively Introgressed Genetic Variants That Modulate Reporter Gene Expression in Human Immune Cells. Mol. Biol. Evol 39. 10.1093/molbev/msab304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pollen AA, Bhaduri A, Andrews MG, Nowakowski TJ, Meyerson OS, Mostajo-Radji MA, Di Lullo E, Alvarado B, Bedolli M, Dougherty ML, et al. (2019). Establishing Cerebral Organoids as Models of Human-Specific Brain Evolution. Cell 176, 743–756.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanton S, Boyle MJ, He Z, Santel M, Weigert A, Sanchís-Calleja F, Guijarro P, Sidow L, Fleck JS, Han D, et al. (2019). Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574, 418–422. [DOI] [PubMed] [Google Scholar]
- 27.Markenscoff-Papadimitriou E, Whalen S, Przytycki P, Thomas R, Binyameen F, Nowakowski TJ, Kriegstein AR, Sanders SJ, State MW, Pollard KS, et al. (2020). A Chromatin Accessibility Atlas of the Developing Human Telencephalon. Cell 182, 754–769.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castelijns B, Baak ML, Timpanaro IS, Wiggers CRM, Vermunt MW, Shang P, Kondova I, Geeven G, Bianchi V, de Laat W, et al. (2020). Hominin-specific regulatory elements selectively emerged in oligodendrocytes and are disrupted in autism patients. Nat. Commun 11, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gusmao EG, Dieterich C, Zenke M, and Costa IG (2014). Detection of active transcription factor binding sites with the combination of DNase hypersensitivity and histone modifications. Bioinformatics 30, 3143–3151. [DOI] [PubMed] [Google Scholar]
- 30.Pollard KS, Salama SR, Lambert N, Lambot M-A, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, et al. (2006). An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443, 167–172. [DOI] [PubMed] [Google Scholar]
- 31.Bae B-I, Jayaraman D, and Walsh CA (2015). Genetic changes shaping the human brain. Dev. Cell 32, 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Visel A, Minovitsky S, Dubchak I, and Pennacchio LA (2007). VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Won H, de la Torre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, Gandal MJ, Sutton GJ, Hormozdiari F, Lu D, et al. (2016). Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song M, Yang X, Ren X, Maliskova L, Li B, Jones IR, Wang C, Jacob F, Wu K, Traglia M, et al. (2019). Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat. Genet 51, 1252–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song M, Pebworth M-P, Yang X, Abnousi A, Fan C, Wen J, Rosen JD, Choudhary MNK, Cui X, Jones IR, et al. (2020). Cell-type-specific 3D epigenomes in the developing human cortex. Nature 587, 644–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ernst J, and Kellis M (2017). Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc 12, 2478–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lizio M, Harshbarger J, Shimoji H, Severin J, Kasukawa T, Sahin S, Abugessaisa I, Fukuda S, Hori F, Ishikawa-Kato S, et al. (2015). Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 16, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- 39.Funk CC, Casella AM, Jung S, Richards MA, Rodriguez A, Shannon P, Donovan-Maiye R, Heavner B, Chard K, Xiao Y, et al. (2020). Atlas of Transcription Factor Binding Sites from ENCODE DNase Hypersensitivity Data across 27 Tissue Types. Cell Rep. 32, 108029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang C, Ward ME, Chen R, Liu K, Tracy TE, Chen X, Xie M, Sohn PD, Ludwig C, Meyer-Franke A, et al. (2017). Scalable Production of iPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem Cell Reports 9, 1221–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen KM, Wong AK, Troyanskaya OG, and Zhou J (2022). A sequence-based global map of regulatory activity for deciphering human genetics. Nat. Genet 54, 940–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, et al. (2011). A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindhorst D, and Halfon MS (2022). Reporter gene assays and chromatin-level assays define substantially non-overlapping sets of enhancer sequences. bioRxiv, 2022.04.21.489091. 10.1101/2022.04.21.489091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kwasnieski JC, Fiore C, Chaudhari HG, and Cohen BA (2014). High-throughput functional testing of ENCODE segmentation predictions. Genome Res. 24, 1595–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sullivan PF, Agrawal A, Bulik CM, Andreassen OA, Børglum AD, Breen G, Cichon S, Edenberg HJ, Faraone SV, Gelernter J, et al. (2018). Psychiatric Genomics: An Update and an Agenda. Am. J. Psychiatry 175, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.GTEx Consortium (2013). The Genotype-Tissue Expression (GTEx) project. Nat. Genet 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, Clarke D, Gu M, Emani P, Yang YT, et al. (2018). Comprehensive functional genomic resource and integrative model for the human brain. Science 362. 10.1126/science.aat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Werling DM, Pochareddy S, Choi J, An J-Y, Sheppard B, Peng M, Li Z, Dastmalchi C, Santpere G, Sousa AMM, et al. (2020). Whole-Genome and RNA Sequencing Reveal Variation and Transcriptomic Coordination in the Developing Human Prefrontal Cortex. Cell Rep. 31, 107489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang D, Elwell AL, Aygün N, Krupa O, Wolter JM, Kyere FA, Lafferty MJ, Cheek KE, Courtney KP, Yusupova M, et al. (2021). Cell-type-specific effects of genetic variation on chromatin accessibility during human neuronal differentiation. Nat. Neurosci 24, 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu Y, Li X, Liu J, Luo X-J, and Yao Y-G (2020). SZDB2.0: an updated comprehensive resource for schizophrenia research. Hum. Genet 139, 1285–1297. [DOI] [PubMed] [Google Scholar]
- 51.Hormozdiari F, Zhu A, Kichaev G, Ju CJ-T, Segrè AV, Joo JWJ, Won H, Sankararaman S, Pasaniuc B, Shifman S, et al. (2017). Widespread Allelic Heterogeneity in Complex Traits. Am. J. Hum. Genet 100, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Voisey J, Mehta D, McLeay R, Morris CP, Wockner LF, Noble EP, Lawford BR, and Young RM (2017). Clinically proven drug targets differentially expressed in the prefrontal cortex of schizophrenia patients. Brain Behav. Immun 61, 259–265. [DOI] [PubMed] [Google Scholar]
- 53.Furlan A, Lübke M, Adameyko I, Lallemend F, and Ernfors P (2013). The transcription factor Hmx1 and growth factor receptor activities control sympathetic neurons diversification. EMBO J. 32, 1613–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Divya TS, Lalitha S, Parvathy S, Subashini C, Sanalkumar R, Dhanesh SB, Rasheed VA, Divya MS, Tole S, and James J (2016). Regulation of Tlx3 by Pax6 is required for the restricted expression of Chrnα3 in Cerebellar Granule Neuron progenitors during development. Sci. Rep 6, 30337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hammal F, de Langen P, Bergon A, Lopez F, and Ballester B (2022). ReMap 2022: a database of Human, Mouse, Drosophila and Arabidopsis regulatory regions from an integrative analysis of DNA-binding sequencing experiments. Nucleic Acids Res. 50, D316–D325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ritter DI, Li Q, Kostka D, Pollard KS, Guo S, and Chuang JH (2010). The importance of being cis: evolution of orthologous fish and mammalian enhancer activity. Mol. Biol. Evol 27, 2322–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mattioli K, Oliveros W, Gerhardinger C, Andergassen D, Maass PG, Rinn JL, and Melé M (2020). Cis and trans effects differentially contribute to the evolution of promoters and enhancers. Genome Biol. 21, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crow TJ (2000). Schizophrenia as the price that homo sapiens pays for language: a resolution of the central paradox in the origin of the species. Brain Res. Brain Res. Rev 31, 118–129. [DOI] [PubMed] [Google Scholar]
- 59.Schwartz MP, Hou Z, Propson NE, Zhang J, Engstrom CJ, Santos Costa V, Jiang P, Nguyen BK, Bolin JM, Daly W, et al. (2015). Human pluripotent stem cell-derived neural constructs for predicting neural toxicity. Proc. Natl. Acad. Sci. U. S. A 112, 12516–12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, et al. (2012). Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Okita K, Yamakawa T, Matsumura Y, Sato Y, Amano N, Watanabe A, Goshima N, and Yamanaka S (2013). An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells 31, 458–466. [DOI] [PubMed] [Google Scholar]
- 62.Miyaoka Y, Chan AH, Judge LM, Yoo J, Huang M, Nguyen TD, Lizarraga PP, So P-L, and Conklin BR (2014). Isolation of single-base genome-edited human iPS cells without antibiotic selection. Nature Methods 11, 291–293. 10.1038/nmeth.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bershteyn M, Nowakowski TJ, Pollen AA, Di Lullo E, Nene A, Wynshaw-Boris A, and Kriegstein AR (2017). Human iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia. Cell Stem Cell 20, 435–449.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–360. 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liao Y, Smyth GK, and Shi W (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Research 41, e108–e108. 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Inoue F, Kreimer A, Ashuach T, Ahituv N, and Yosef N (2019). Identification and Massively Parallel Characterization of Regulatory Elements Driving Neural Induction. Cell Stem Cell 25, 713–727.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Erwin GD, Oksenberg N, Truty RM, Kostka D, Murphy KK, Ahituv N, Pollard KS, and Capra JA (2014). Integrating diverse datasets improves developmental enhancer prediction. PLoS Comput. Biol 10, e1003677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Inoue F, Kircher M, Martin B, Cooper GM, Witten DM, McManus MT, Ahituv N, and Shendure J (2017). A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Research 27, 38–52. 10.1101/gr.212092.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang X, and McManus M (2009). Lentivirus Production. Journal of Visualized Experiments. 10.3791/1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kircher M (2012). Analysis of High-Throughput Ancient DNA Sequencing Data. Methods in Molecular Biology, 197–228. 10.1007/978-1-61779-516-9_23. [DOI] [PubMed] [Google Scholar]
- 72.Pu X-A, Young AP, and Kubisch HM (2019). Production of Transgenic Mice by Pronuclear Microinjection. In Microinjection: Methods and Protocols, Liu C and Du Y, eds. (Springer; New York: ), pp. 17–41. [DOI] [PubMed] [Google Scholar]
- 73.Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, et al. (2006). In vivo enhancer analysis of human conserved non-coding sequences. Nature 444, 499–502. [DOI] [PubMed] [Google Scholar]
- 74.Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- 75.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, and Vilo J (2019). g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McInnes L, Healy J, Saul N, and Großberger L (2018). UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw 3, 861. [Google Scholar]
- 78.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research 12, 2825–2830. [Google Scholar]
- 79.Zoonomia Consortium (2020). A comparative genomics multitool for scientific discovery and conservation. Nature 587, 240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lindenbaum P jvarkit: Java utilities for Bioinformatics (Github).
- 81.Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, and Aiden EL (2016). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Systems 3, 95–98. 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roayaei Ardakany A, Gezer HT, Lonardi S, and Ay F (2020). Mustache: multi-scale detection of chromatin loops from Hi-C and Micro-C maps using scale-space representation. Genome Biol. 21, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Goloborodko A, Venev S, Abdennur N, azkalot, and Di Tommaso P (2019). mirnylab/distiller-nf: v0.3.3 10.5281/zenodo.3350937. [DOI] [Google Scholar]
- 84.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, Baldursson G, Belliveau R, Bybjerg-Grauholm J, Bækvad-Hansen M, et al. (2019). Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet 51, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hägg S, Athanasiu L, et al. (2019). Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet 51, 404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium (2017). Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism 8, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mullins N, Forstner AJ, O’Connell KS, Coombes B, Coleman JRI, Qiao Z, Als TD, Bigdeli TB, Børte S, Bryois J, et al. (2021). Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet 53, 817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cross-Disorder Group of the Psychiatric Genomics Consortium (2019). Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell 179, 1469–1482.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, Adams MJ, Agerbo E, Air TM, Andlauer TMF, et al. (2018). Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet 50, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.International Obsessive Compulsive Disorder Foundation Genetics Collaborative (IOCDF-GC) and OCD Collaborative Genetics Association Studies (OCGAS) (2018). Revealing the complex genetic architecture of obsessive-compulsive disorder using meta-analysis. Mol. Psychiatry 23, 1181–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yu D, Sul JH, Tsetsos F, Nawaz MS, Huang AY, Zelaya I, Illmann C, Osiecki L, Darrow SM, Hirschtritt ME, et al. (2019). Interrogating the Genetic Determinants of Tourette’s Syndrome and Other Tic Disorders Through Genome-Wide Association Studies. AJP 176, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ATAC-seq, ChIP-seq, Hi-C, and MPRA sequencing data has been deposited to GEO: GSE110760.