

Abstract
Regulation of Aberrant Protein Production (RAPP) is a protein quality control in mammalian cells. RAPP degrades mRNAs of nascent proteins not able to associate with their natural interacting partners during synthesis at the ribosome. However, little is known about the molecular mechanism of the pathway, its substrates, or its specificity. The Signal Recognition Particle (SRP) is the first interacting partner for secretory proteins. It recognizes signal sequences of the nascent polypeptides when they are exposed from the ribosomal exit tunnel. Here, we reveal the generality of the RAPP pathway on the whole transcriptome level through depletion of human SRP54, an SRP subunit. This depletion triggers RAPP and leads to decreased expression of the mRNAs encoding a number of secretory and membrane proteins. The loss of SRP54 also leads to the dramatic upregulation of a specific network of HSP70/40/90 chaperones (HSPA1A, DNAJB1, HSP90AA1, and others), increased ribosome associated ubiquitination, and change in expression of RPS27 and RPS27L suggesting ribosome rearrangement. These results demonstrate the complex nature of defects in protein trafficking, mRNA and protein quality control, and provide better understanding of their mechanisms at the ribosome.
Keywords: Signal sequence, Secretory proteins, Protein synthesis and transport, Translational control, Signal Recognition Particle
Graphical Abstract
Introduction
Cells synthesize thousands of proteins that need to be transported to different organelles, secreted outside of the cell, integrated into different membranes, or remain in the cytoplasm. Cells evolved several mechanisms for proper and efficient protein transport and folding. These processes are tightly regulated to eliminate the appearance of incorrect proteins in the wrong subcellular localization that may be harmful for a cell. However, sometimes defects in the transported proteins can occur due to mutations, premature stop codons in mRNA templates, mRNA truncations, or defects in the components of the transport machinery. These proteins are not able to reach their correct subcellular localizations or do not fold properly. Aberrant proteins are potentially dangerous, and in many cases lead to human diseases [1–5]. Therefore, there are several mRNA and protein quality control mechanisms that were evolved to prevent synthesis or remove these undesired defective proteins [6–14]. Many of these processes happen co-translationally when polypeptide nascent chains are being synthesized by the ribosome. Defective mRNAs with premature stop codons, truncations, or with missing stop codons are detected and eliminated by nonsense mediated decay, no-go decay, or non-stop decay, respectively. Truncated polypeptides resulting from incomplete synthesis or because of stress are removed by the ribosome quality control complex (RQC) [8, 14]. The Regulation of Aberrant Protein Production (RAPP) pathway senses protein interactions during their translation and degrades their mRNAs if these interactions are disrupted by mutations in the synthesized proteins or in the interacting factors (see recent reviews for details [1, 6].) Several protein quality control pathways in the cytosol and the endoplasmic reticulum (ER) remove misfolded proteins missed by the quality control mechanisms at the ribosome. However, the precise details of these processes are still poorly understood.
More than a third of all cellular proteins are secretory and membrane proteins [15]. One of the major protein targeting pathways is Signal Recognition Particle (SRP) dependent protein targeting [16–20]. SRP recognizes special N-terminal targeting signals termed signal sequences or signal peptides [21–25]. Although signal sequences do not have strong amino acid homology, they share a similar organization and physicochemical properties. A typical signal sequence contains an N-terminal positively charged n-domain, a hydrophobic core (h-domain), and a C-terminal c-domain [26, 27]. While the h-domain is crucial for protein translocation and the n-domain for its targeting efficiency, the c-domain is important for processing and cleavage of the signal sequence [25, 28–30].
SRP is a ribonucleoprotein complex with multiple subunits. It targets proteins to the endoplasmic reticulum (ER) membrane for translocation, processing, modifications, and maturation in secretory protein biogenesis. In mammals, SRP consists of six proteins (SRP9, SRP14, SRP68, SRP72, SRP19 and SRP54) and one noncoding 7SL RNA [18]. Defects in SRP subunits are associated with multiple human diseases [31]. The SRP54 subunit of the targeting complex recognizes and directly binds the signal sequence of a secretory protein during its synthesis [22, 24, 25, 32]. The integrity of the hydrophobic core of the signal sequence is crucial for its interaction with SRP54 [25]. The failure of SRP54 to bind to a polypeptide nascent chain due to a mutation in the hydrophobic core of the signal sequence triggers the RAPP protein quality control pathway and consequently leads to the mRNA degradation of the aberrant secretory protein [33–36]. RAPP is unique among other protein quality controls – it senses interactions of the polypeptide nascent chains during their synthesis on the ribosome and destroys their mRNA templates if normal interactions are disrupted in order to prevent synthesis of potentially harmful products. We found that the loss of the SRP54 subunit also triggers the RAPP response [33, 34, 36]. However, it is still unknown if the RAPP pathway is a general mechanism controlling the synthesis and targeting of secretory and membrane proteins, or if it is involved in the regulation of a small number of specific proteins.
We hypothesize that the RAPP pathway surveys synthesis of many secretory and membrane proteins, and that its activation leads to specific changes in expression of proteins associated with a cellular stress response. In this study, we activated RAPP by depleting SRP54 and evaluated gene expression at the whole transcriptome level. Our data demonstrate that the loss of the SRP54 subunit results in mRNA depletion of many secretory and membrane proteins. It also leads to a complex stress response that occurs at the ribosome and includes specific chaperones upregulation, ribosome rearrangement, and ubiquitination. Our results support the idea that RAPP is a general mechanism of protein quality control, demonstrate involvement of distinct chaperone networks in the process, and uncover the complex nature of the co-translational events at the ribosome during translation.
Results
Effect of SRP54 depletion on the whole transcriptome in human cells
To evaluate SRP function in the RAPP pathway on the whole transcriptome level in mammalian cells, we depleted SRP54 using RNAi technology, performed Deep RNA sequencing, and compared the results with the transcriptome of the control cells. About 90% of the reads were successfully mapped to the human genome GRCh38.p13, and the sample distance analysis showed excellent reproducibility between three biological replicates (Figure 1A, Supplementary Table S1).
Figure 1. Deep RNA sequencing in Hela Tet-ON cells depleted of SRP54 subunit of the human Signal Recognition Particle.
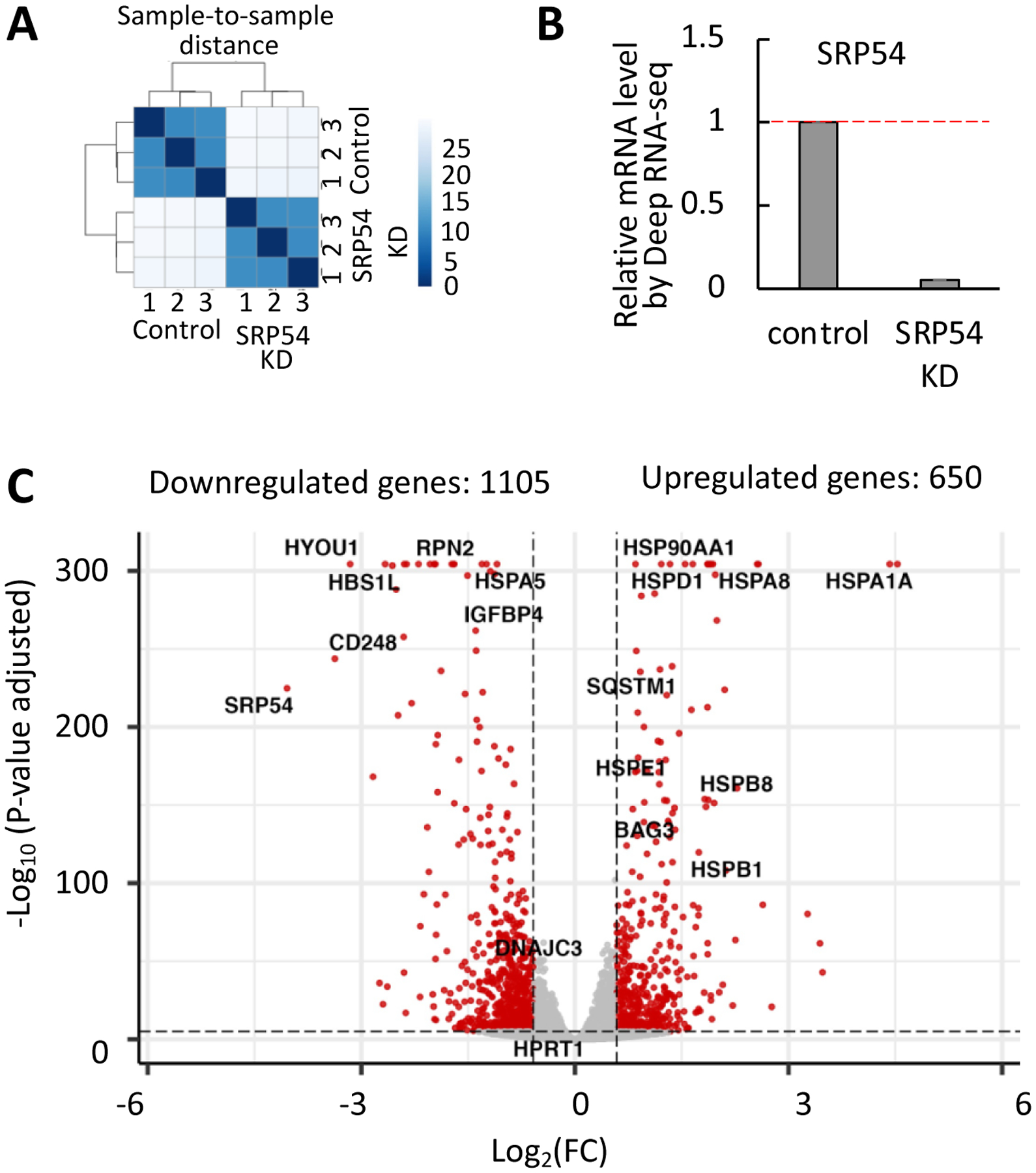
(A) Evaluation of the Deep RNA sequencing data quality. Sample clustering analysis was done to verify reproducibility of the results between biological replicates (n=3). The Euclidean distance between samples (axis on the right from 0 to 25) was calculated using the R function. Bright blue color reflects a close distance and good data quality between the replicates, and light blue shows far distance. Control 1, 2, 3 and SRP54 KD 1, 2, 3 correspond to the samples marked AK51, AK52, AK53, AK54, AK55, AK56, respectively, in the supplementary data analysis files.
(B) Evaluation of the SRP54 knockdown. Gene expression was quantified as a ratio between number of SRP54 transcript counts in SRP54 KD cells and control cells. Red dash line presents mRNA level in control sample.
(C) The Volcano plot of differently expressed genes in SRP54 KD cells. The X-axis represents log2 of the fold change (FC, negative values are downregulated and positive values are upregulated genes), the Y-axis shows statistical significance by representing negative log10 of the adjusted p-values. Gene expression changes with the adjusted p-values ≤ 0.05 and FC ≥ |1.5| were considered significant (marked in red). The examples of the affected genes are labeled.
We were able to generate an efficient depletion of SRP54 as demonstrated by comparing counts of the SRP54 sequences between control and siSRP54-transfected cells; the SRP54 mRNA decreased almost twentyfold (Figure 1B). Using the DESeq2 algorithm [37], a total of 18830 transcripts were detected, and 7215 genes were differentially expressed (Supplementary File 1). Affected transcripts were filtered with a cut-off at ≥1.5 fold change in gene expression. The final number of transcripts affected by the SRP54 depletion is 1755, with 650 and 1105 transcripts upregulated and downregulated, respectively (Supplementary Files 2 and 3). The Volcano plot in Figure 1C shows the magnitude of change in gene expression (X-axis) relative to its statistical significance (Y-axis). Examples of upregulated (top right) and downregulated (top left) transcripts are annotated.
Downregulated transcripts in SRP54-depleted cells show enrichment in molecular processes associated with extracellular proteins, including cell adhesion, response to growth factors, extracellular matrix and structure organization, and others (Supplementary Figure S1). Among upregulated transcripts, enrichment of proteins associated with protein folding modulators and ubiquitination was observed. A diverse group of heat shock proteins is upregulated. Gene ontology analysis indicates that defective SRP may affect targeting of extracellular proteins, and upregulate processes associated with stress response and protein misfolding.
SRP54 plays an important role in modulating protein sorting. To estimate the impact of SRP54 knockdown on protein targeting, we examined the number of differentially expressed transcripts per subcellular localization. For this analysis, proteins were classified in four large groups regarding their localization: cytoplasm, nucleus, mitochondria, and secretome. The largest effect from the loss of SRP54 was observed on the secretome (n=718; 43.1%), but the loss of SRP54 also affects transcripts encoding cytoplasm (n=402; 24.1%), nucleus (n=487; 29.2%), and mitochondria (n=60; 3.6%) proteins (Figure 2A, Supplementary File 4). Comparing the distribution of down and upregulated proteins in the afore mentioned categories, the secretome group was notably downregulated in SRP defective cells with 68% (500 of 718) of all differentially expressed transcripts of secretory proteins. In addition, since many secretory proteins have signal sequences, we also completed analysis of proteins containing signal sequences. Using the Uniprot database, we identified transcripts of 376 proteins with signal sequences that were differentially expressed. In agreement with the results for the secretome group, 83% (313 of 376) of differentially expressed transcripts of proteins containing signal sequences were preferentially downregulated (Figure 2A, Supplementary File 4). Figure 2B shows presentation of the differentially expressed transcripts in the same categories as in Figure 2A using violin plots. This analysis shows that there are no notable differences in the average fold change for all evaluated categories. However, similar to Figure 2A, this analysis illustrates a large difference in the affected numbers of transcripts. The secretome and signal sequence-containing proteins were preferentially downregulated after SRP54 depletion; in contrast, cytoplasmic proteins have almost equal amounts of down and upregulated proteins. The data suggest that downregulated proteins from the secretome and from proteins with signal sequences are subjected to the RAPP regulation in conditions when SRP54 is depleted. We also observed an increase of downregulated transcripts for mitochondrial and nuclear proteins, suggesting that some of these proteins may require SRP for targeting.
Figure 2. Analysis of differentially expressed transcripts regarding their encoded proteins’ localization in the cell and in relation of the signal sequence presence.
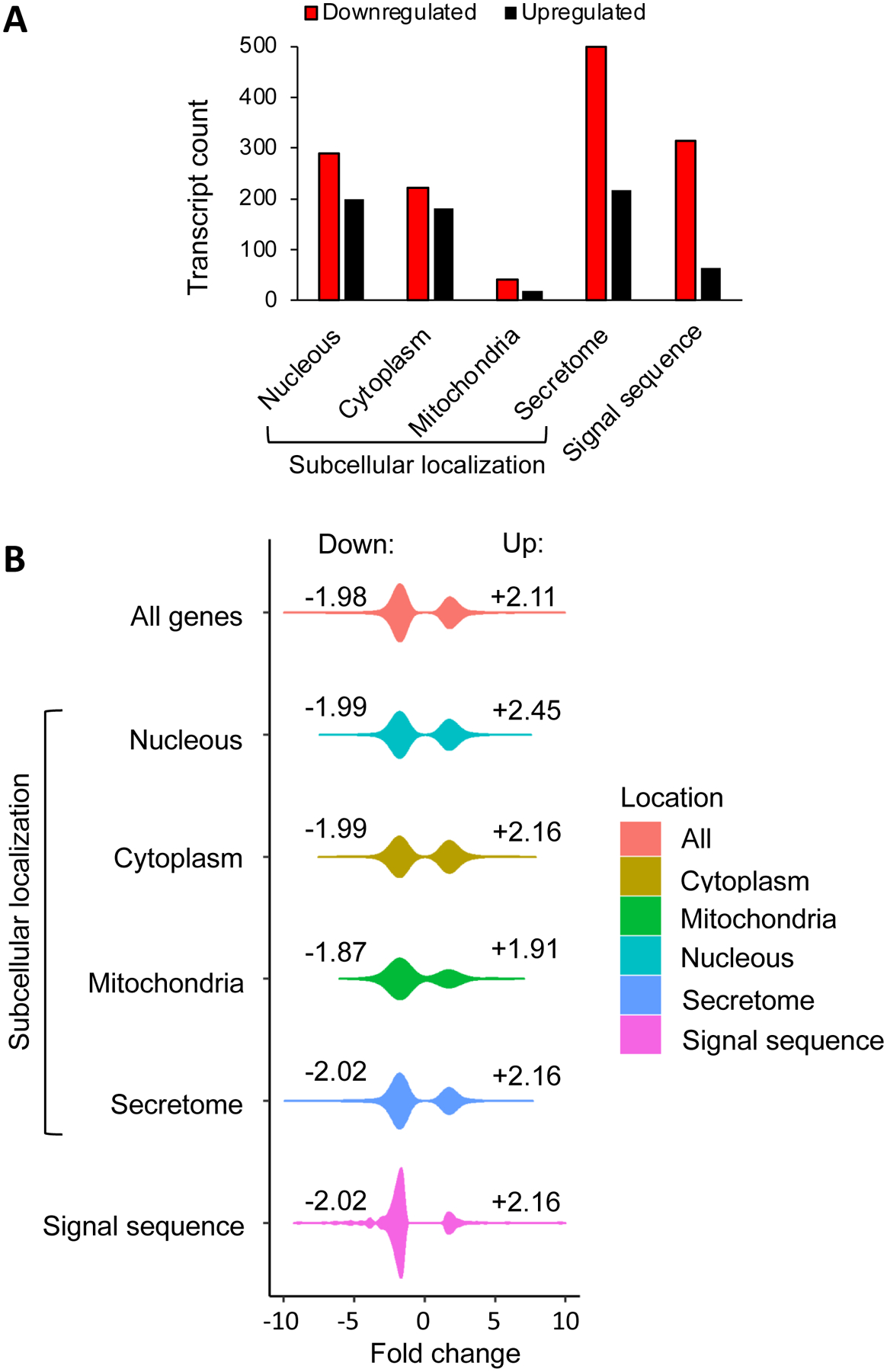
(A) The distribution of down and upregulated transcripts was estimated per subcellular localization and represented as bar plots. Proteins were classified in four large groups regarding their localization: cytoplasm, nucleus, mitochondria, and secretome. Analysis was done on the base of the Human Protein Atlas database. Cytoplasm includes cytosol, actin filaments, centrosome, intermediate filaments, microtubules; nucleus includes proteins localized in nuclear membrane, nucleoli, and nucleoplasm; mitochondria include proteins localized in mitochondria; secretome combines ER, Golgi, plasma membrane, vesicles, and secreted proteins. In addition to subcellular localization the presence of signal sequence was evaluated as an independent category based on UniProtKB/Swiss-Prot repository. Black bars are upregulated transcripts, red bars are downregulated transcripts.
(B) Representation of the data shown in panel (A) as violin plots. The distributions of the linear fold changes (effect size) are shown using kernel density estimation. Negative and positive values correspond to downregulated (Down, left side of distributions) and upregulated (Up, right side of distributions) transcripts. Numbers labeled close to each distribution represent the average fold change for down and upregulated transcripts per category. Classification of proteins in different groups are as in panel (A).
Additionally, 236 of 313 downregulated transcripts (75.4%) with annotated signal sequences are associated with human diseases based on the Genetic Association Database (Supplementary Figure S2, Supplementary File 5). These results highlight that the loss of SRP targeting can contribute to multiple human diseases by affecting expression of important secretory proteins.
While most transcripts of proteins with signal sequences are decreased after SRP depletion, some are upregulated (Figure 2). This suggests that a subset of proteins with signal sequences is not regulated by RAPP, and is not SRP-dependent. To determine possible differences between SRP-dependent and SRP-independent proteins we analyzed overall length of the proteins and the signal sequences’ features. The analysis showed that there is no correlation between the peptide length and differential expression when all proteins were evaluated without bias (Supplementary Figure S3A). However, we observed that upregulated proteins with signal sequences show a wider distribution of the peptides’ lengths in comparison with downregulated proteins with signal sequences (Supplementary Figure S3B). No differences were observed between signal sequences’ hydrophobicity of these two groups of proteins (Supplementary Figure S4). However, our results indicate that the N-region of downregulated proteins trends to show a higher positive net charge (Supplementary Figure S5). Thus, although some small distinctions were found, there are no obvious striking differences between these two groups of proteins. Although many secretory proteins have signal sequences, not all of them are SRP-dependent and are instead transported by SRP-independent mechanisms. It is still unknown what specific features make proteins SRP-dependent or not, and require further testing to answer this question.
Earlier, we demonstrated that SRP54 depletion causes a reduction of mRNA levels of intestinal alkaline phosphatase, calreticulin and BIP (Binding Immunoglobulin Protein, gene name HSPA5, heat shock protein family A (Hsp70) member 5) [33]. All these proteins have signal sequences and are regulated by RAPP. Analysis of the Deep RNA-seq data also clearly demonstrates that mRNAs of the above proteins are affected by SRP54 knockdown (Supplementary Figure S6). These studies corroborate our findings based on the Deep RNA-seq analysis.
Thus, our study emphasizes the crucial role of SRP in the normal physiology of mammalian cells, suggesting an additional role in functional protection of the secretory and membrane protein mRNAs in addition to its role in protein targeting. Our findings also suggest that the RAPP pathway is a general mechanism for SRP-dependent proteins, due to their expression downregulation in the Deep RNA seq.
SRP54 depletion affects secretory and membrane proteins expression on mRNA and protein levels
Our Deep RNA-seq data demonstrate that the mRNA expression of many secretory and membrane proteins decreases, suggesting activation of the RAPP pathway by SRP54 depletion (Figures 1, 2). To verify the data obtained by the Deep RNA-seq, we independently tested the expression on mRNA and protein levels for several proteins that were found downregulated by the RNA-seq. We selected representative proteins from three groups: proteins with cleavable signal sequences and extracellular and ER lumen localization (group 1); lipoproteins and single-pass membrane proteins with cleavable signal sequences (group 2); and multi-pass membrane proteins with and without signal sequences (group 3) (Supplementary Table S2). Selected targets for group 1 include PDIA3 (protein disulfide isomerase family A, member 3), LOX (lysyl oxidase), HYOU1 (hypoxia up-regulated 1), SPOCK1 (SPARC (osteonectin), cwcv and kazal like domains proteoglycan 1), P4HA3 (prolyl 4-hydroxylase subunit alpha 3), and IGFBP4 (insulin like growth factor binding protein 4). Group 2 includes CD24, a protein that is attached to lipid bilayer, and CD248 (endosialin), a protein that has a single transmembrane span. For the group 3, we selected two multi-pass membrane proteins: CHRND, a cholinergic receptor, with 4 transmembrane domains (TMDs) and a cleavable signal sequence; and KCNQ1, a potassium channel, with 7 TMDs and without a signal sequence. Relative mRNA levels of these proteins determined by Deep RNA-seq are shown in Figures 3A (for the groups 1 and 2) and 4A (for the group 3).
Figure 3. SRP54 depletion affects secreted proteins expression.
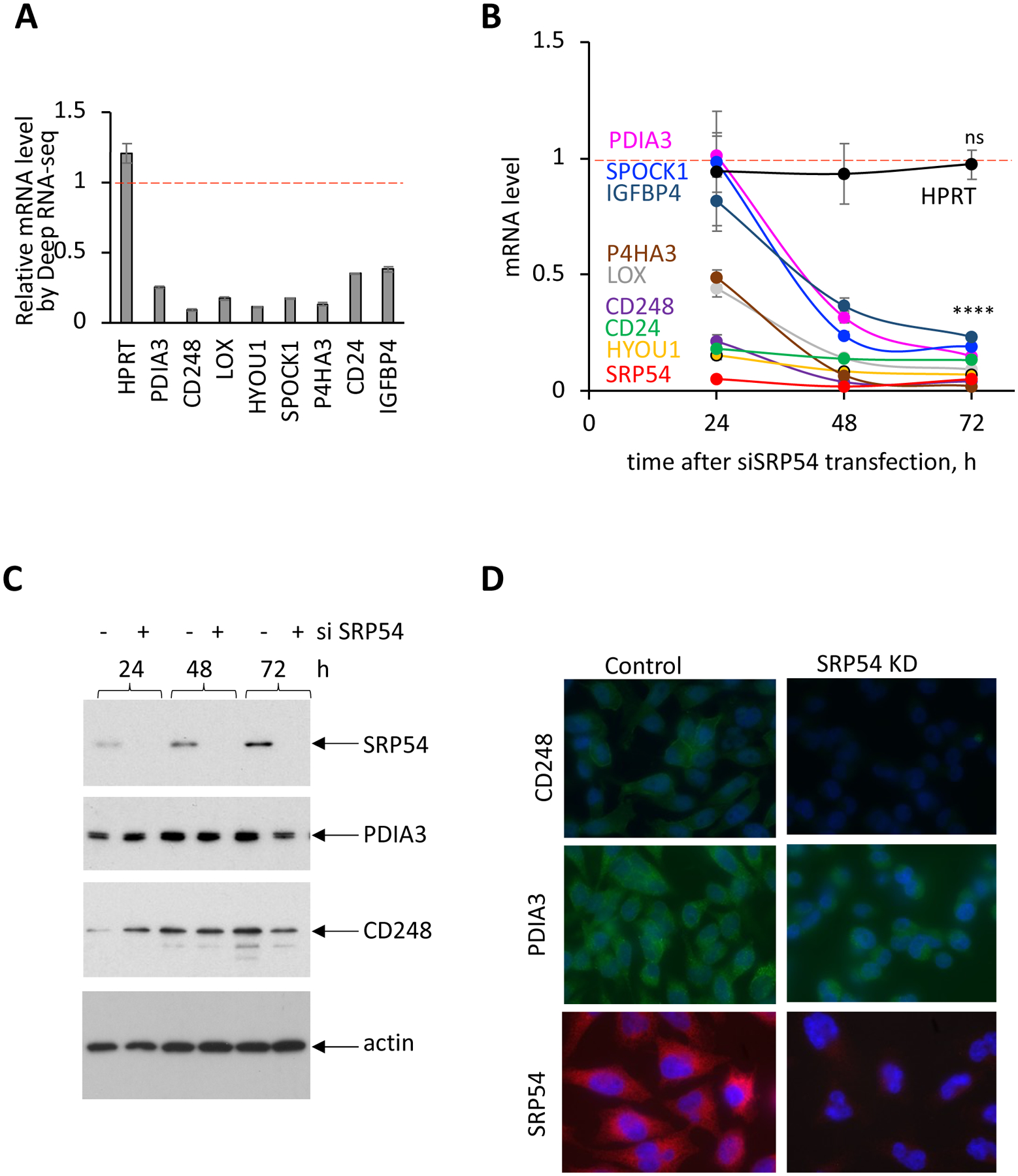
HeLa Tet-ON cells were transfected with SRP54 specific siRNA (siSRP54). Samples for mRNA and protein analysis were collected at 24, 48 and 72 hours after transfection.
(A) Relative mRNA levels determined by Deep RNA-seq analysis, shown as a ratio between transcript counts in SRP54 KD and control cells.
(B) mRNA levels of secreted proteins, SRP54 and the control cytosolic protein HPRT in SRP54 KD cells determined by RT-qPCR in independent from the Deep RNA-seq set of experiments, at 24, 48 and 72 hours after siSRP54 transfection. Data were normalized to actin mRNA and presented relatively to the mRNA levels in control cells (marked by red dashed lines). Standard errors were calculated based on the values from three independent experiments. Two-tailed unpaired t test was applied for evaluating statistical significance for 72 hours-time points: ns, not significant; ****, p<0.0001.
(C) Western blot detection of CD248, PDIA3, SRP54 and actin in control (−) and siSRP54 treated (+) cells at different time points after siRNA transfection as indicated. Actin blot was done as a loading control.
(D) Immunostaining of control and SRP knockdown (SRP54 KD) cells with CD248 (top), PDIA3 (middle) and SRP54 (bottom) specific antibodies. Cells were stained at 48 hours after siSRP54 transfection.
In these experiments we succeeded an efficient SRP54 knockdown as demonstrated by RT-qPCR analysis (mRNA level, Figure 3B) and by western blot (protein level, Figure 3C). SRP54 was not detectable even at 24 h time point after siSRP54 transfection. The transcript of the cytosolic protein HPRT (hypoxanthine phosphoribosyltransferase 1) was used as a negative control since it is not a substrate for SRP [33]. The HPRT transcript level was not affected by SRP54 depletion as demonstrated by the Deep RNA-seq (Figure 3A) and by RT-qPCR in the time-course experiment (Figure 3B). The analysis of transcripts of the proteins with signal sequences from the groups 1 and 2 is shown in Figure 3B. Remarkably, the expression of these mRNAs was dramatically affected in SRP54 knockdown cells. After 72 hours of SRP54 depletion, mRNA levels of all tested proteins with signal sequences were significantly decreased. However, we observed some differences in the rate of the process. While the mRNA levels for CD248, HYOU1, CD24, LOX, and P4HA3 were already decreased at least 50% or more at 24 hours post-siSRP54, the mRNAs of PDIA3, IGFBP4 and SPOCK1 were more stable and still at high levels at this time point even though SRP54 was not detectable. To test how SRP54 depletion affects secretory proteins on the protein level, we selected two proteins, PDIA3 and CD248, and evaluated their expression by western blotting (Figure 3C). We observed that the effect of SRP54 knockdown was slower on the protein level than on the mRNA level, however, a substantial protein decrease was observed at 72 hours. We also tested the effect of the SRP54 knockdown by immunostaining, demonstrating a decrease in the protein expression of CD248 and PDIA3 in conditions when SRP54 was efficiently depleted (Figure 3D).
The Deep RNA-seq analysis of multi-pass membrane proteins (group 3) showed significant decrease in their transcript levels (Figure 4A). These results were further verified by RT-qPCR in samples taken at different time points after siSRP54 transfection. mRNA levels of these proteins are significantly reduced in cells with defective SRP (Figure 4B); and protein expression, evaluated by western blotting, was also decreased in a time-dependent manner (Figure 4C).
Figure 4. SRP54 depletion affects expression of membrane proteins with multiple transmembrane domains.
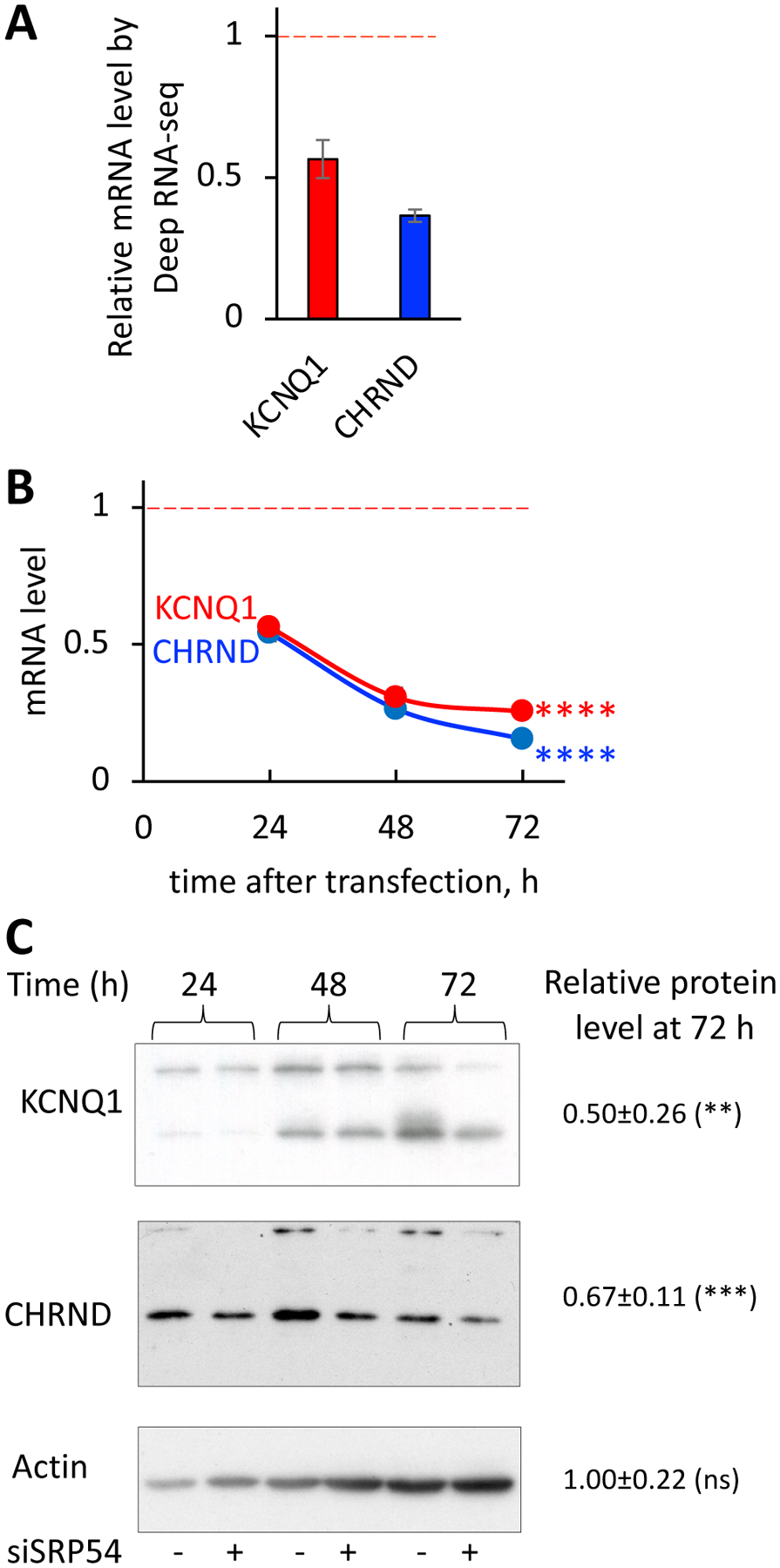
HeLa Tet-ON cells were transfected with SRP54 specific siRNA (SRP54 KD). Samples for mRNA and protein analysis were prepared as described on Figure 3.
(A) Relative mRNA levels of CHRND and KCNQ1 proteins determined by Deep RNA-seq analysis, shown as a ratio between transcript counts in SRP54 KD and control cells (marked by red dashed line).
(B) mRNA levels of multi-pass membrane proteins were determined by RT-qPCR at 24, 48 and 72 hours after siSRP54 transfection. Data were normalized to actin mRNA and presented relatively to the mRNA levels in control cells (marked by red dashed line). Two-tailed unpaired t test was applied for evaluating statistical significance for 72 hours-time points: ns, not significant; ****, p<0.0001.
(C) Western blot detection of CHRND and KCNQ1 in control (−) and siSRP54 treated (+) cells at different time points after siRNA transfection as indicated. Actin blot was done as a loading control. Numbers represent relative protein expression at 72 h-time point calculated by normalizing the signal in SRP54 depleted cells to the one in the control samples. Quantification of 4–5 biological repeats was done using ImageJ. Standard deviations are shown. Statistical analysis for 72 hours-time point was done using unpaired two-tailed t test. P-values are shown: ns, not significant; **, p < 0.01; ***, p < 0.001.
We observed that the effect of SRP54 depletion on protein expression was slower than the effect on mRNA. Our results suggest that mRNA and protein clearance mechanisms have different rates, and the lack of SRP54 has a delayed negative effect on protein expression. As it was demonstrated earlier, on average, mammalian proteins are five times more stable than their mRNAs [38]. Our data are in a good agreement with these observations.
Finally, we conclude that SRP54 depletion affects secreted and membrane protein expression on mRNA and protein levels most likely through the loss of SRP interaction with a nascent chain during translation and activation of the RAPP pathway. The data demonstrate that RAPP is a general mechanism for quality control of secretory and membrane proteins and support the idea of an mRNA protective function of SRP.
Loss of SRP54 induces upregulation of specific chaperones
Further analysis of the Deep RNA-seq data shows that cells undergo global changes in gene expression. We observe that transcripts of several specific heat shock proteins (HSP) or chaperones are upregulated in the SRP54 knockdown cells, implying that the deficiency in protein targeting leads to a comprehensive stress response in human cells (Supplementary File 6). Bioinformatic analysis of Deep RNA-seq data identifies 170 proteins that belong to heat shock protein families/chaperones with transcript counts more than 10. 68 of them have altered transcript levels in SRP54 KD background, while 102 are unaffected. Remarkably, 62 transcripts are upregulated and only 6 are downregulated. From 62 upregulated transcripts, 41 are products of pseudogenes and were not used in further analyses. Therefore, a total of 21 upregulated and 6 downregulated heat shock proteins and chaperones respond to depletion of SRP54 and are shown in Figure 5A. Affected heat shock proteins differ in size, cellular abundance and function, implying the complex nature of cellular response to the stress caused by a defective targeting factor. We observed seven representatives of Heat Shock Protein Family A (Hsp70) (e.g. HSPA1A, HSPA8), four proteins from DNAJ family chaperones (HSP40), and two HSP90 proteins among the upregulated transcripts. Two nucleotide exchange factors from different families, BAG3 and HSPH1, are involved in the ATP hydrolysis cycle by HSP70 ATPases [39]. Among small heat shock proteins upregulated in SRP54 KD cells, HSPB8 and HSPB1 are known to have inducible expression and chaperone properties [40]. Two HSPs, HSPD1 (HSP60) ATP-ase with its cochaperone HSPE1 (HSP10), with connection to mitochondrial proteins folding [41, 42] are also found upregulated when SRP54 is depleted.
Figure 5. Upregulation of chaperones (heat shock proteins, HSPs) in SRP54 depleted cells.
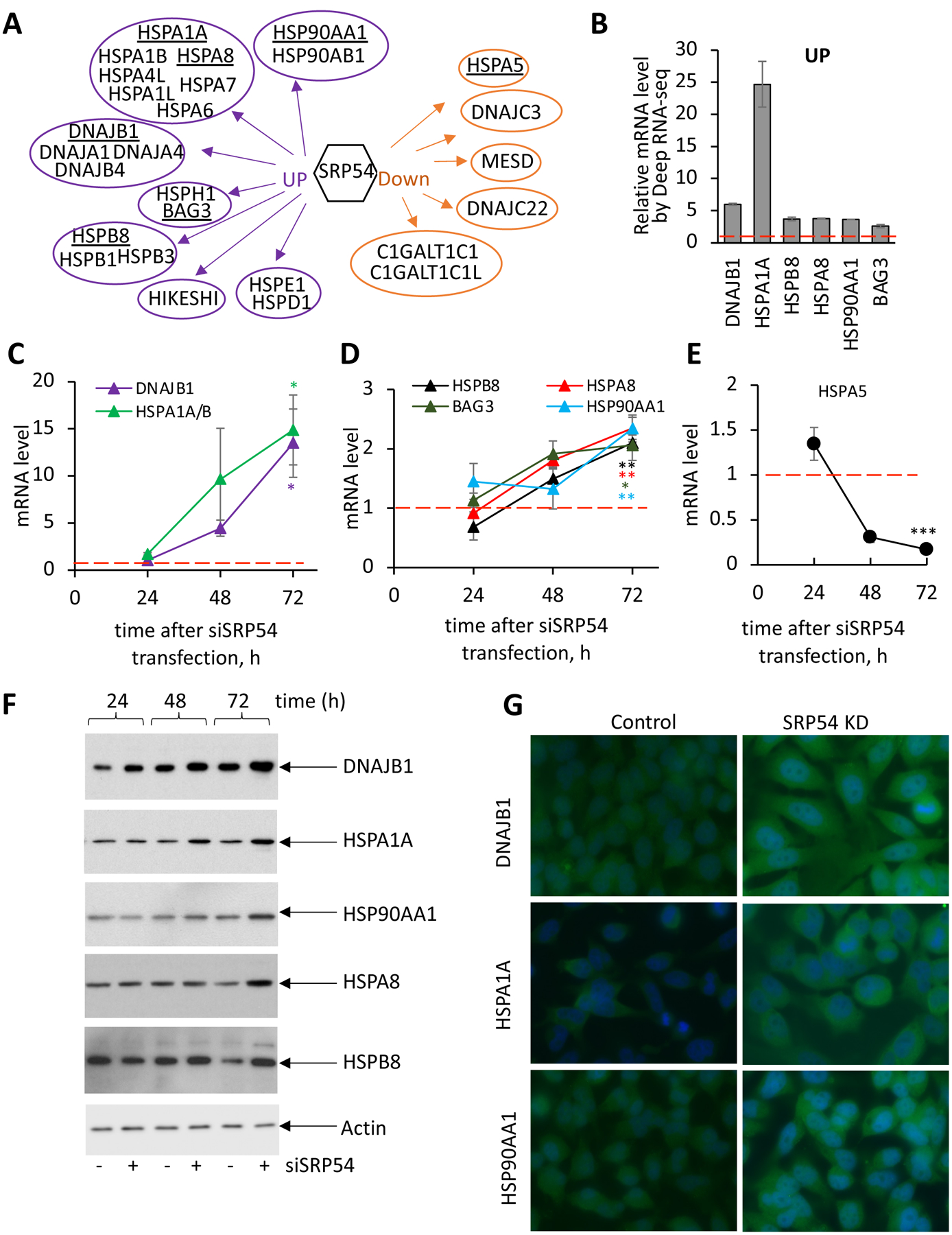
(A) Schematic presentation of heat shock proteins from different families that are affected in SRP54 knockdown cells as determined by Deep RNA-seq (see also Supplementary File 6). 21 upregulated and 6 downregulated HSPs grouped by families are shown. Underlined chaperones/HSPs were chosen for further analysis.
(B) Relative mRNA levels of selected HSPs determined by Deep RNA-seq analysis. mRNA level was calculated as a ratio between transcripts counts in SRP54 knockdown and control cells. Red dash line presents the value in control cells.
(C) mRNA levels for chaperones DNAJB1 and HSPA1A/B at different time points after siSRP54 transfection were evaluated by RT-qPCR in independent from the Deep RNA-seq experiments at 24, 48 and 72 hours-time points after siRNA specific to SRP54 (siSRP54) transfection. Red dash line shows corresponding mRNA levels in control cells. Error bars are standard errors (n=3). Two-tailed unpaired t test was applied for evaluating statistical significance at the 72 hours- time point. P-values are shown in the corresponding colors: *, p < 0.05.
(D) mRNA levels of HSPB8, HSPA8, BAG3, and HSP90AA1 in SRP54 depleted cells analyzed as described for (C). Red dash line shows corresponding mRNA levels in control cells. Error bars are standard errors (n=3). Two-tailed unpaired t test was applied for evaluating statistical significance at the 72 hours- time point. P-values are shown in the corresponding colors: *, p < 0.05; **, p < 0.01.
(E) mRNA levels of HSPA5 in SRP54 depleted cells analyzed as described for (C). Red dash line shows corresponding mRNA levels in control cells. Error bars are standard errors (n=3). Two-tailed unpaired t test was applied for evaluating statistical significance at the 72 hours- time point. P-values are shown in the corresponding colors: ***; p < 0.001.
(F) Western blot assay of the chaperones/HSPs DNAJB1, HSPA1A, HSP90AA1, HSPA8, and HSPB8 in control (−) and siSRP54 treated cells (+) as indicated. Actin was used as loading control.
(G) Immunostaining of HeLa Tet-ON control (left images) and SRP54 KD (right images) cells with antibodies against DNAJB1 (top), HSPA1A (middle), and HSP90AA1 (bottom). Cells were stained at 48 hours after transfection.
While upregulated chaperones may be involved in the RAPP pathway or in a stress response associated with defective protein trafficking, the downregulated chaperones may be consistent with being substrates for the RAPP pathway. Remarkably, all observed downregulated HSPs were found or predicted as proteins with ER localization, secreted, or possessing a signal sequence or transmembrane span [43–47]. This suggests that they are most likely substrates for the RAPP pathway. Among these downregulated HSPs, there are two specific chaperones involved in protein glycosylation (C1GALT1C1 and C1GALT1C1L), two HSP40s of DnaJ family member C (DNAJC22 and C3), and two chaperones (MESD and HSPA5, also known as BIP or GRP78) with a key role in protein folding in the ER. Figure 5B illustrates the mRNA expression levels of the selected HSPs evaluated by Deep RNA-seq analysis. Supplementary Table S3 emphasizes these HSPs’ involvement in the stress response, protein quality control, protein folding and degradation, stress granule disassembly, and even mRNA stability. Two chaperones, HSPA1A and DNAJB1, are dramatically upregulated (by a fold change of about 25 and 6, respectively), while expression of the others, HSBP8, HSP90AA1, HSPA8, and BAG3, is also significantly increased but to a lesser degree by a fold change of 2.6–3.8. HSPA5 (also known as BIP or GRP78), an ER localized chaperone and potential RAPP substrate [33], is downregulated (Supplementary Figure S6) in agreement with previous studies.
To verify Deep RNA-seq data, we tested the chaperone mRNA levels by RT-qPCR at different time points after siSRP54 transfection. mRNA levels of all examined chaperones except HSPA5 gradually increased during the time course validating the changes detected by Deep RNA-seq (Figures 5C, D). As expected, the HSPA5 mRNA level decreased in these conditions (Figure 5E) supporting its regulation by the RAPP pathway. Analysis of selected chaperones by western blot at different time points clearly demonstrates gradually increasing levels of DNAJB1, HSPA1A, HSP90AA1, HSPA8, and HSPB8 proteins upon SRP54 depletion (Figure 5F). Immunostaining of DNAJB1, HSPA1A, and HSP90AA1 in conditions of SRP54 depletion also show significant upregulation of these proteins (Figure 5G). Thus, our data demonstrate specific upregulation of certain chaperones (heat shock proteins) in response to SRP54 depletion.
Is there crosstalk between upregulation of chaperones and specific mRNA degradation during the RAPP pathway?
In this study, we demonstrate that expression of mRNAs of many secretory proteins are decreased when SRP54 protein is lost. At the same time, expression of specific heat shock proteins is upregulated. Some of these chaperones are involved in mRNA stability, protein quality controls, stress granules disassembly and stress (see Supplementary Table S3 for details). Upregulation of multiple chaperones in response to the SRP54 knockdown and the RAPP induction suggest two possible scenarios of their action: first, by association with the mRNA degradation mechanism during RAPP; or, second, by their involvement at the later stages of RAPP as a stress response to aberrant proteins appeared in the cell. For instance, their role may prevent aberrant proteins from aggregation and promote their clearance. There is also possibility that these chaperones do not have direct role in the RAPP mechanism. To answer the question about possible chaperones’ involvement in the modulation of mRNA degradation, we experimentally tested if their knockdowns affect the secretory protein mRNA levels in conditions when SRP54 is still present and when SRP54 is depleted. First, we transfected HeLa Tet-ON cells with siRNAs specific to DNAJB1, HSPA1A, and HSPB8, and siSRP54 as a control for upregulation of chaperones. RT-qPCR showed that knockdown was efficient and the mRNA levels of all these proteins are significantly decreased in cells with corresponding siRNAs (Figure 6A). The expression of DNAJB1 and HSPB8 on the protein level was also significantly depleted, while HSPA1A had only partial knockdown (Figure 6B, compare lanes 1 and 3). Based on the very low mRNA level of HSPA1A in the cells transfected with siHSPA1A and relatively high level of HSPA1A protein that remained in these cells, we conclude that HSPA1A protein is very stable and still detectable even 72 hours after siRNA transfection. As expected, the SRP54 depletion alone leads to significant increase in mRNA and protein levels of all above chaperones (Figure 6A, B, compare lanes 1 and 2 in B). However, these effects were significantly impaired in the cells with double knockdowns with siRNAs specific to HSPs and SRP54 – expression of all chaperones were significantly reduced, and HSPB8 protein was practically undetectable (Figure 6A, B).
Figure 6. Depletion of chaperones does not affect mRNA degradation of the secreted proteins.
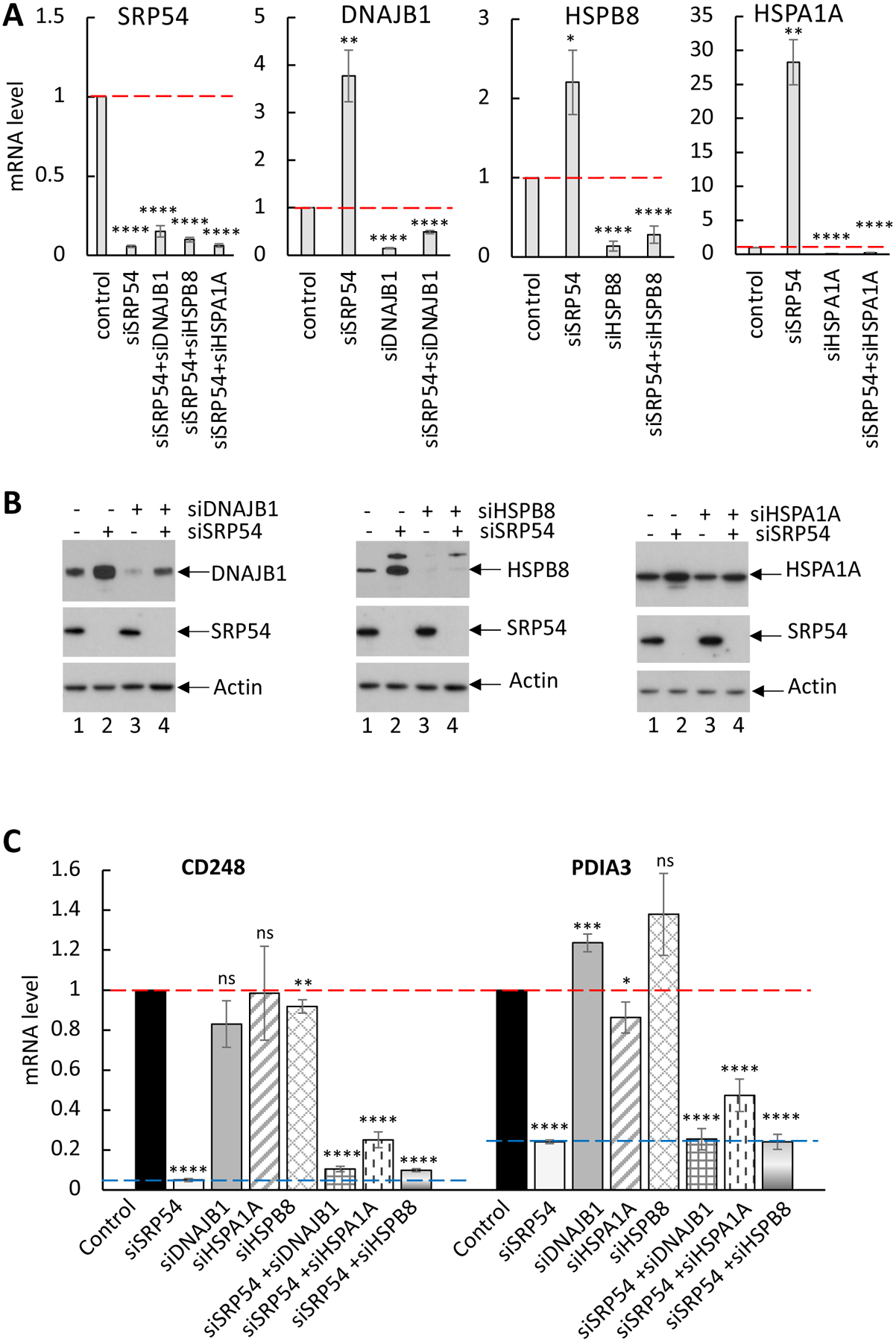
HeLa Tet-ON cells were transfected with the following siRNAs: siDNAJB1, siHSPA1A, siHSP90AA1, siHSPB8 alone or in combination with siSRP54.
(A) Confirmation of knockdowns for all tested chaperones/HSPs and SRP54 by RT-qPCR. Red dash lanes show mRNA level of corresponding genes in control cells. Standard errors are calculated based on 3–4 independent experiments. Two-tailed t-test was applied for pairwise comparisons: *, p < 0.05; **, p < 0.01; ***; p < 0.001; ****, p<0.0001.
(B). Confirmation of knockdowns for all tested chaperones/HSPs and SRP54 by western blot. Actin blot was used as a loading control.
(C) mRNA levels of secreted proteins CD248 and PDIA3 were analyzed by RT-qPCR in cells with a single chaperone/HSP KD or in combination with siSRP54 as indicated. Red dash lanes show mRNA level of the secreted proteins in control cells. Blue dash lane indicates the level of CD248 and PDIA3 mRNA in SRP54 KD cells. Standard errors are calculated based on 3–4 independent experiments. Two-tailed t-test was applied for pairwise comparisons: *, p < 0.05; **, p < 0.01; ***; p < 0.001; ****, p<0.0001; ns, not significant. All data were compared to mRNA level in control cells.
To test if the above chaperones are involved in downregulation of secretory protein mRNA, we analyzed mRNA levels of the RAPP substrates CD248 and PDIA3 in the cells where HSPs or SRP54 were solely knocked down, or HSPs and SRP54 were knocked down together. Analysis of the mRNA levels by RT-qPCR shows that the lack of DNAJB1 (grey symbols) and HSPB8 (diamond grid symbols) or decrease in HSPA1A (diagonal stripes symbol) level did not affect the mRNA levels of CD248 and PDIA3 (Figure 6C). For comparison, samples from SRP54 KD cells (light grey symbol) showed significant decrease in mRNA for CD248 and PDIA3.
If the chaperones upregulated in SRP54 KD cells contribute to mRNA degradation of the secretory proteins, then we would anticipate that the depletion of specific chaperones in the background of an SRP54 knockdown might rescue the mRNAs of these secreted proteins. However, analysis of mRNA levels of secretory proteins in the cells with double knockdowns showed that the combination of SRP54 depletion with knockdowns of DNAJB1, HSP1A1, or HSPB8 have very little effect on mRNA levels of CD248 and PDIA3 (Figure 6C). Thus, the data indicate that HSPA1A, DNAJB1 and HSPB8 do not specifically affect the mRNA degradation of secreted proteins. Therefore, our results suggest that these chaperones might have a different role in the RAPP pathway other than mRNA degradation.
SRP54 depletion leads to ribosomal stress
The current hypothesis on SRP function in protein targeting states that SRP scans ribosomes during protein synthesis until it detects and binds a signal sequence located at the N-terminus of the polypeptide nascent chain that is emerging from the ribosome exit tunnel. Mammalian SRP displays elongation arrest during targeting that prevents the exposure of the unbound nascent chain to the cytosolic environment before its targeting to the ER. A defect in the functional targeting factor, SRP, can generate dramatic stress on the cell and its translation machinery. Stressed ribosomes are known to change composition and recruit other cellular factors to resolve the stress [48, 49]. Therefore, we searched the Deep RNA-seq data for mRNA changes associated with the composition of ribosomes. Generally, ribosomes are relatively conserved ribonucleoprotein complexes and do not substantially change their protein composition. However, there are some examples of ribosome rearrangements and specialization under specific conditions. The human ribosome is composed of two subunits: a small 40S subunit, that contains 33 proteins with an 18S rRNA; and a large 60S subunit with 47 proteins in complex with 28S, 5.8S and 5S rRNA [50]. Strikingly, analysis of the Deep RNA-seq data reveals that the transcript for ribosomal protein S27 (RPS27, eS27) is downregulated by about two times when SRP54 is depleted (Figure 7A). Upregulation up to 27% was also observed for 6 proteins from the 60S subunit and 5 proteins from the 40S subunit. Changes at the mRNA levels of other ribosomal proteins in SRP54 KD cells were smaller; only approximately a 10–20% decrease was observed for about half of the 60S ribosomal protein transcripts and for 4 transcripts for the 40S subunit, while the mRNAs of other ribosomal proteins were relatively unaffected (Supplementary File 1). In comparison with RPS27, transcript levels of 40S small ribosomal protein S6 (RPS6, eS6), and 60S large ribosomal protein L11 (RPL11, uL5 were not significantly affected (Figure 7A). To verify the Deep RNA-seq data we determined mRNA levels by RT-qPCR at different time points of the SRP54 depletion in independent set of experiments. As shown in Figure 7B, RPS27 transcript was significantly downregulated, while mRNA levels of the 60S subunit protein RPL11 and the 40S subunit protein RPS6 were less affected. A similar effect was observed on the protein level as well. A gradual decrease in the level of RPS27 protein with progression of the siSRP54 treatment is clearly shown by western blot, while expression of the RPL11 and RPS6 proteins was less affected even at 72 hours (Figure 7C). Notably, according to the Deep RNA-seq data, the RPS27L (S27-like) transcript was ~70% upregulated in SRP54 KD cells (Figure 7A) suggesting that this variant could replace RPS27 when cells lack SRP54. RPS27L is the paralog of RPS27. Both are 9.5 kDa proteins, and are only different in three amino acid residues. RPS27 and RPS27L are involved in cancer cell survival, and their knockdowns lead to 18S rRNA maturation defects, ribosomal stress and consequently cell apoptosis [51, 52].
Figure 7. SRP54 depletion affects ribosomal proteins and 18S RNA.
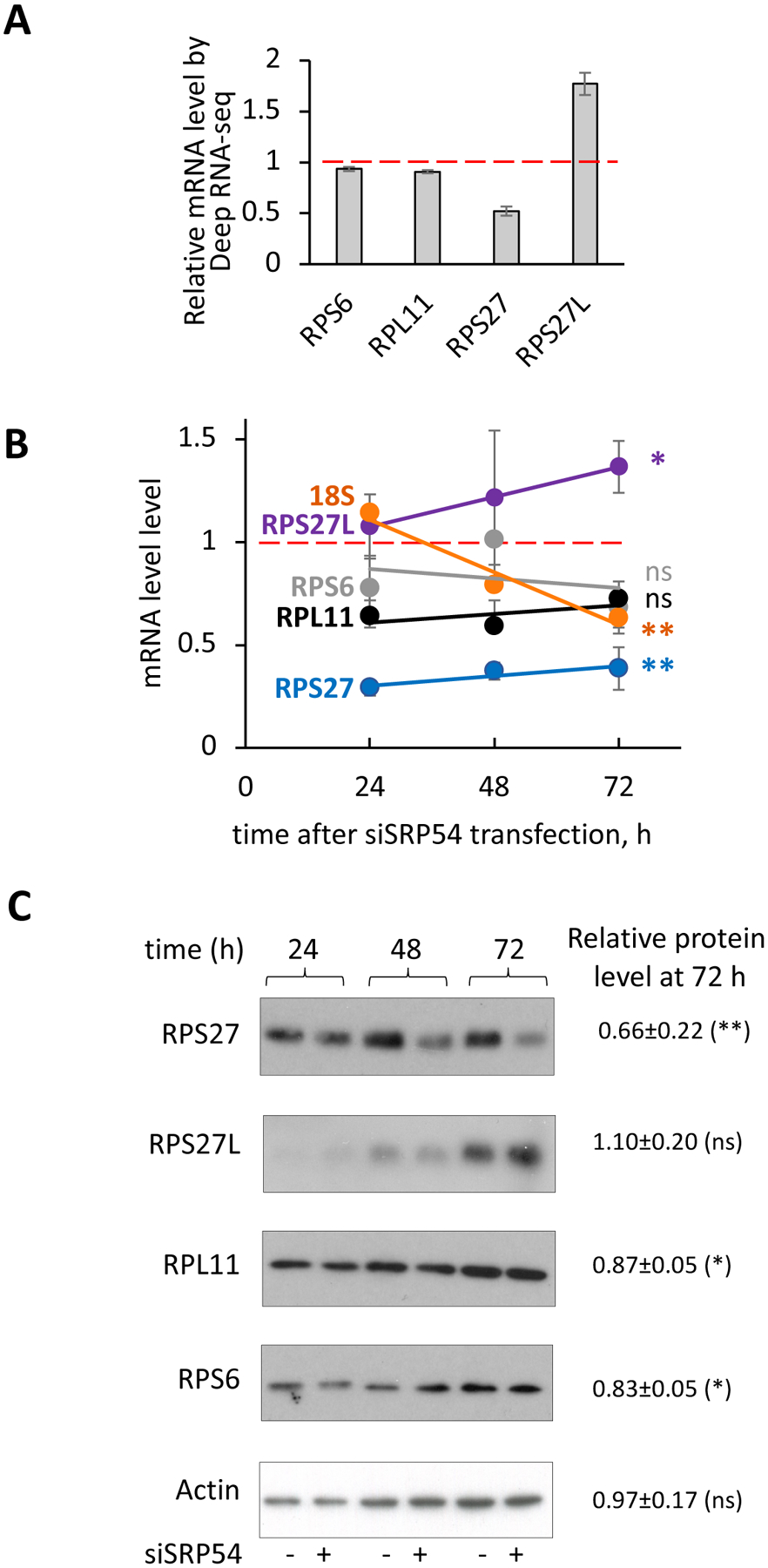
(A) Relative mRNA levels of RPS27, RPS27L, RPS6 and RPL11 determined by Deep RNA-seq analysis. mRNA levels were calculated as a ratio between transcript counts in the SRP54 KD cells and transcripts in the control cells. Red dash line presents mRNA level in the control cells.
(B) mRNA levels of ribosomal proteins RPS27 (blue), RPS27L (purple), RPS6 (grey), RPL11 (black) and 18S (orange) determined by RT-qPCR in independent from the Deep RNA-seq experiments obtained at 24, 48 and 72 hours-time points after siSRP54 transfection. Data were normalized to actin and shown relatively to mRNA level in the control cells (red dash line). Standard errors were calculated based on values from 2–6 independent experiments. Samples for RNA extraction was taken at 24, 48 and 72 hours after siSRP54 transfection. Statistical analysis for 72 hours-time point was done using unpaired two-tailed t test. P-values are shown in corresponding colors: *, p < 0.05; **, p < 0.01; ns, not significant.
(C) Western blotting of RPS27, RPS27L, RPL11, RPS6 and actin. Whole cell lysates protein samples were prepared from control and SRP54 depleted HeLa Tet-on cells at different time points. Representative blots are shown. Actin was used as loading control. Numbers represent relative protein expression at 72 h-time point calculated by normalizing the signal in SRP54 depleted cells to the one in the control samples. Quantification of 2–5 biological repeats was done using ImageJ. Standard deviations are shown. Statistical analysis for 72 hours-time point was done using unpaired two-tailed t test. P-values are shown: ns, not significant; *, p < 0.05; **, p < 0.01.
Despite small difference between RPS27 and RPS27L, we were able to distinguish their expression on mRNA level by RT-qPCR using selective primers and confirm the Deep RNA-seq data. Indeed, a significant increase in RPS27L mRNA level upon SRP54 depletion was observed (Figure 7B). Western blotting also clearly demonstrates different patterns for RPS27 and RPS27L expression (Fig. 7C). While we observed notable time-dependent RPS27 protein expression decrease upon SRP54 depletion, RPS27L showed a trend in moderate expression increase. Although the proteins are different in 3 amino acids only, the difference in the western blot profiles indicate that the antibodies can distinguish between these two proteins. Most likely the real increase in RPS27L protein expression is larger than observed, and may be hidden by potential cross-reactivity to RPS27. All data together (Deep RNA-seq, mRNA levels by RT-qPCR, and protein expression by western blot) demonstrate that SRP54 depletion leads to RPS27 downregulation and RPS27L upregulation suggesting ribosome rearrangement.
To verify if SRP54 depletion affects ribosomes, we performed polysome profiling. Polysome profiling is a technique to separate ribosome subunits (40S and 60S), monosomes (80S), and polysomes (disomes, trisomes, etc.) by ultracentrifugation on a sucrose gradient followed by fractionation. Polysome profiling provides very distinct A260 absorbance spectra with characteristic peaks for the above categories [53]. We used this technique to test for A260 spectra differences in the composition of ribosomal subunits, monosomes, and polysomes prepared from the control and SRP54 knockdown cells. As shown in Figure 8A, A260 absorbance spectra of control (black line) and SRP54 KD (red line) look similar. However, there is a small decrease in absorbance for the small (40S) subunit peak and increase for the large (60S) subunit peak in the SRP54 KD spectrum (Figure 8B). Although, no obvious changes were observed for polysome fractions, the 80S monosome was slightly decreased. Western blot analysis of prepolysome fractions (40S, 60S, 80S) in control and SRP54 KD cells also revealed that the amount of RPS27 in the 40S subunit and 80S ribosome was about 30% and 20% less, respectively, when SRP54 is depleted (Figure 8C, D). Interestingly, a similar pattern was observed for another 40S subunit protein, RPS6, while RPL11 showed a small decrease in 60S and 80S. It was shown that reduced RPS27 levels due to clinical mutations interferes with normal ribosome biogenesis and affects maturation of 18S rRNA, a 40S subunit ribosomal RNA [54]. We hypothesize that the decrease in RPS27 level in SRP54 depleted cells may affect maturation of 18S rRNA. Indeed, as shown on Figure 7B, the level of 18S rRNA is decreasing in a time-dependent manner after siSRP54 transfection. The greatest change in 18S rRNA level (about 30%) was observed at about 72 hours post-siSRP54 transfection. This result can explain decrease in 40S subunit observed during sucrose gradient fractionation above. In summary, the data demonstrate that SRP54 depletion leads to ribosomal stress affecting ribosome stability and ribosome composition through the changed levels of ribosomal proteins RPS27 (down) and RPS27L (up) and decreased level of 18S ribosomal RNA.
Figure 8. Effect of SRP54 depletion on ribosomes determined by polysome profiling.
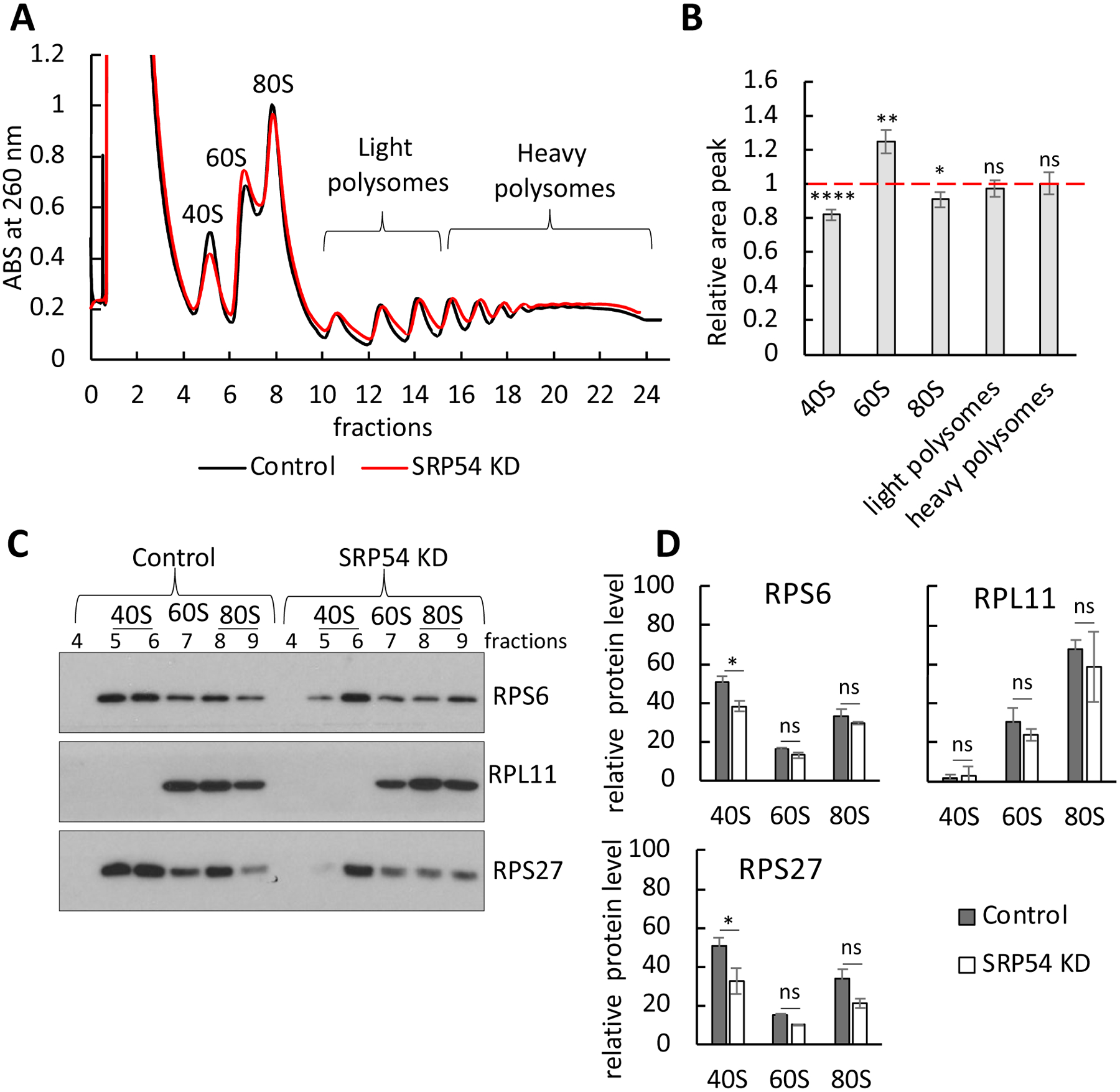
(A) Polysome profile in HeLa-TetON control cells and cells transfected with SRP54 siRNA. Cell lysates were subjected to 10% - 50% sucrose gradient centrifugation to achieve separation of ribosomal fractions. Representative absorbance spectra at 260 nm are shown (black line is control; red line is SRP54 knockdown). The peaks for small (40S), large (60S) subunits, as well as monosome (80S) and polysomes are indicated.
(B) Relative area peaks of ribosomal fractions shown on panel (A) were quantified. Area peak in SRP54 KD cells was normalized to the one in the control cells. The quantification was done for a several independent runs (n=7); standard errors are shown. Red dashed line indicates the area of peaks (taken as 1) in control cells. Statistical analysis was done using unpaired two-tailed t test. P-values are shown: *, p < 0.05; **, p < 0.01; ****, p<0.0001; ns, not significant.
(C) Analysis of the ribosomal proteins from the prepolysomes fractions (40S, 60S, and 80S) in the control and SRP54 KD. Samples were analyzed by western blot for RPS27, RPS6, RPL11 ribosomal proteins. Representative blots are shown.
(D) Quantification of RPS27, RPS6 and RPL11 from blots in the panel C. Quantification of ribosomal proteins in 40S, 60S and 80S fractions from two independent experiments was done by the using of ImageJ software and normalized to a total signal in prepolysomal (40S, 60S and 80S) fractions. Standard deviation is shown. Statistical analysis of ribosomal protein blots quantifications is done using Two-way Anova with Tukey’s multiple comparison test. P-values are shown in corresponding colors: *, p < 0.05; ns, not significant.
SRP54 depletion leads to upregulation of ubiquitination
As demonstrated in Figure 1 – 4, SRP54 depletion activates the RAPP pathway and leads to downregulation of secretory and membrane protein mRNAs most likely through their degradation. The RAPP pathway is initiated when a nascent chain containing a signal sequence is exposed from the ribosomal tunnel but it is unable to interact with SRP [33, 34, 36]. Thus, this event causes accumulation of incompletely synthesized secretory proteins on the ribosome, preventing ribosomes from completing translation and initiating synthesis of new proteins. It is also possible that incompletely synthesized proteins are released from the ribosomes and misfolded in the cytoplasm. To remedy the situation, the ribosomes should be recycled and aberrant peptides should be degraded. Cells label proteins designated for degradation with ubiquitin, a 76-amino acid peptide. There are two major pathways for protein degradation: proteasomal and lysosomal (autophagy) [55]. It is suggested that different polyubiquitination patterns of proteins targeted for degradation will determine which pathway cells will be using. For example, K48-linked ubiquitination targets proteins to the proteasome, while K63-linked ubiquitination directs proteins to autophagy [56–60]. K48- and K63-linked ubiquitination can be distinguished by immunological methods with specific antibodies. Western blotting assays revealed elevated levels of K48-linked ubiquitination in cells depleted of SRP54 at 48 and 72 hours after siSRP54 transfection, while K63-linked ubiquitination was not significantly changed (Figure 9A, B). These data suggest that there is accumulation of K48 ubiquitinated proteins that are substrates for the proteasome, and thus possibly implicate the proteasome in the clearance of partly synthesized proteins.
Figure 9. Ubiquitination profiles in the control and SRP54 knockdown cells.
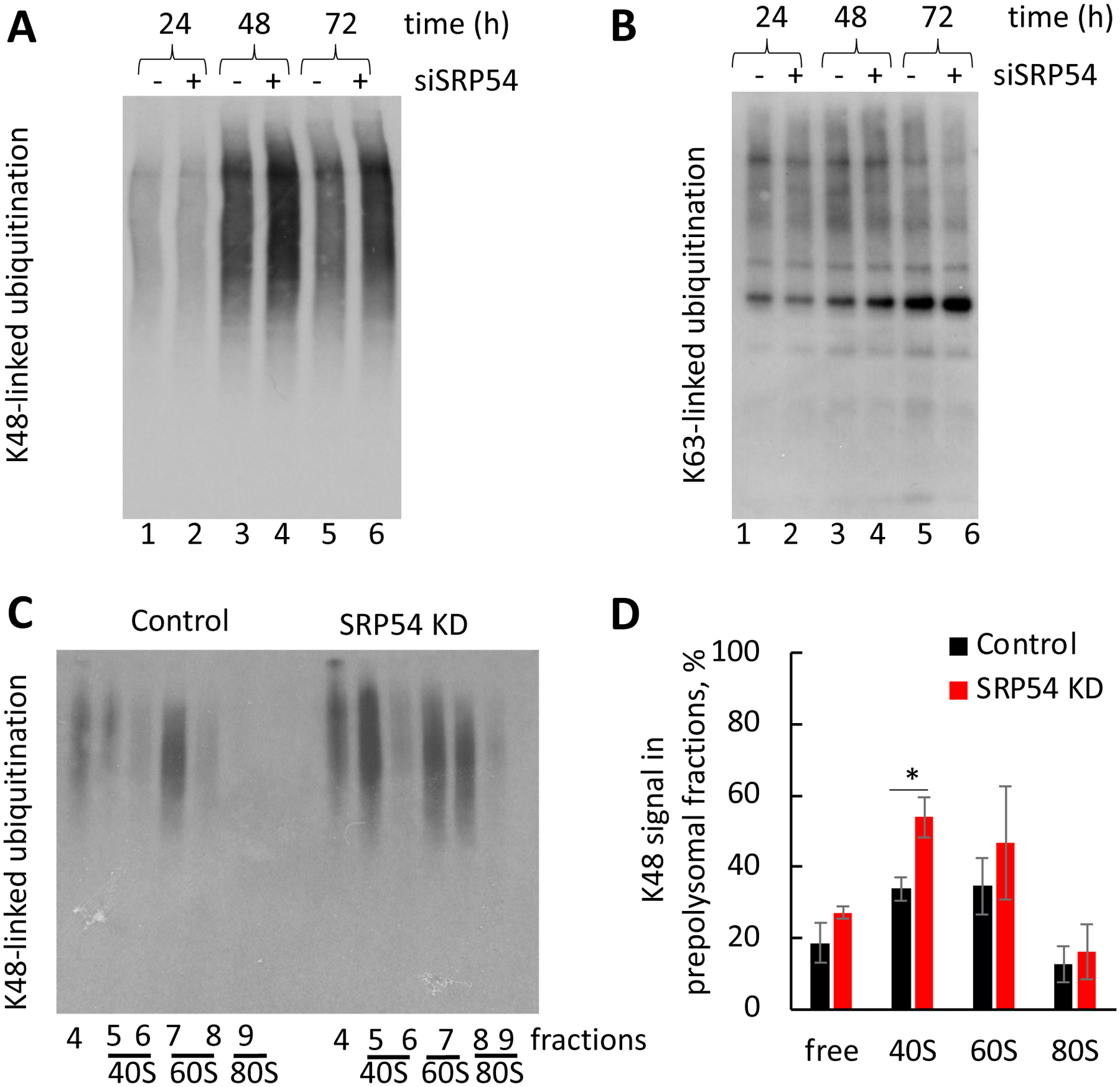
(A) Time course of K48-linked ubiquitination in the control HeLa Tet-On (−) and siSRP54 transfected cells (+). Samples were prepared as described for Figure 3.
(B) Time course of K63-linked ubiquitination in the same samples as from (A).
(C) Western blotting against K48 ubiquitination in ribosome subunits and monosome fractions. Fractions from the polysome fractionation experiments were used for analysis. Representative blot is shown.
(D) The K48 signals from panel (C) were quantified using ImageQuant software. The signal intensity was normalized to the total K48 signal in prepolysomal fractions in the control cells. Average values of three independent repeats are shown. P value shown (*, p < 0.05) was calculated using two-tailed unpaired t-test.
To test if enhanced ubiquitination in SRP54 depleted cells is connected to translational events, we probed the ribosome and polysome fractions, prepared as described above, for colocalization of ubiquitinated proteins with ribosomes and polysomes. Notably, K48 ubiquitination in SRP54 KD ribosomes and their subunits is higher than in control samples (Figure 9C, D). The most noticeable difference in K48 ubiquitination between control and SRP54 KD samples is found in the 40S subunit. Therefore, we conclude that elevated ubiquitination in SRP54 KD cells is associated, at least in part, with ribosomes and their subunits.
Discussion
Regulation of Aberrant Protein Production (RAPP) is a protein quality control pathway in mammalian cells. It surveys proteins during their synthesis and degrades their mRNAs if the nascent proteins are not able to associate with their natural partners at the ribosome polypeptide exit site [33, 34, 36]. This pathway is unique because it senses aberrant proteins and eliminates not only defective proteins but also their mRNAs. The specificity of the pathway and its place among other protein quality control mechanisms are discussed in details in recent publications [1, 6]. The major characteristic of the pathway is downregulation of mRNA when a protein is not able to be recognized by SRP [33, 34, 36]. There is also indication that SRP and RAPP are involved in the biogenesis of alpha-synuclein, the protein without a signal sequence [61]. However, the molecular mechanism of RAPP is still mainly unknown.
In this work we completed a knockdown of the SRP54 subunit of the SRP complex in cultured human cells to model conditions when secretory proteins are unable to interact with SRP, and studied its consequences at the whole transcriptome level. SRP54 is the subunit that is in direct contact with secretory protein signal sequences [22, 24, 62]. Thus, its depletion leads to the inability of the SRP complex to interact with signal sequences or transmembrane spans of nascent chains of the proteins that use SRP as a targeting factor. Our study demonstrates, for the first time, that majority of differentially expressed secretory/membrane protein mRNAs are downregulated when SRP54 is depleted. These data suggest that they are controlled by the RAPP pathway, implementing the pathway as a general mechanism of protein quality control. By analyzing the mRNA and protein expression of several selected secretory and membrane proteins in independent time-course experiments of SRP54 depletion, we verified that their mRNA and protein expression are decreasing in a time-dependent manner. Activation of the RAPP pathway prevents synthesis of aberrant secretory and membrane proteins and clears the cell of potentially harmful products, preventing the accumulation of aberrant proteins in the cytoplasm of mammalian cells. Interestingly, SRP depletion in yeast leads to a different scenario through activation of an alternative SRP-independent targeting mechanism as well as mistargeting of the ER transcripts to mitochondria [63, 64]. Yeast likely do not need a mechanism like RAPP because they have the ability to adapt to SRP loss by upregulation of different protein transport pathways. Thus, the RAPP pathway was developed later evolutionarily and is a characteristic feature of protein quality control for higher eukaryotes.
While SRP54 loss leads to decrease expression of many secretory and membrane protein mRNAs, there are sets of mRNAs of secreted, membrane proteins and proteins with clearly identified signal sequences that were not downregulated. These findings suggest that these proteins are SRP independent and most likely are not regulated by RAPP. However, our analysis of overall length of the proteins and signal sequences’ features did not elucidate specific characteristics for SRP-dependent and SRP-independent proteins. Thus, the mechanisms of protein selection by SRP or by components of other secretory pathways require future studies.
Remarkably, the cellular response to SRP54 depletion is also associated with a dramatic increase of specific chaperones’ expression, ribosomal stress, potential ribosomal rearrangement, and ribosome associated K48 ubiquitination. We hypothesize that all these processes are ancillary to the RAPP pathway (Figure 10). We posit that their functions are distinct from the primary mRNA degradation role during RAPP. Indeed, our experiments showed that these chaperones are not involved specifically in the mRNA downregulation (Figure 6). Most likely these chaperones are engaged in interactions with partly synthesized polypeptides to prevent their misfolding and accumulation in the cytoplasm and facilitate their clearance. It is possible that these chaperones are not directly involved in RAPP, and their upregulation is an indirect result of cellular stress associated with protein transport defects. We cannot also exclude that these chaperones are also associated with modulation of other cellular processes resulted from the loss of some membrane or secretory proteins’ expression. Thus, the determination of the exact role of these chaperones requires further studies.
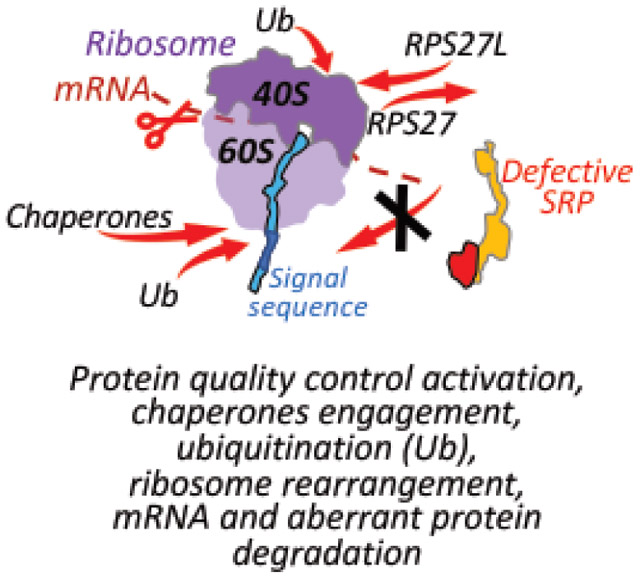
Figure 10. Early events at the ribosome as a response to a lost nascent chain interaction with SRP.
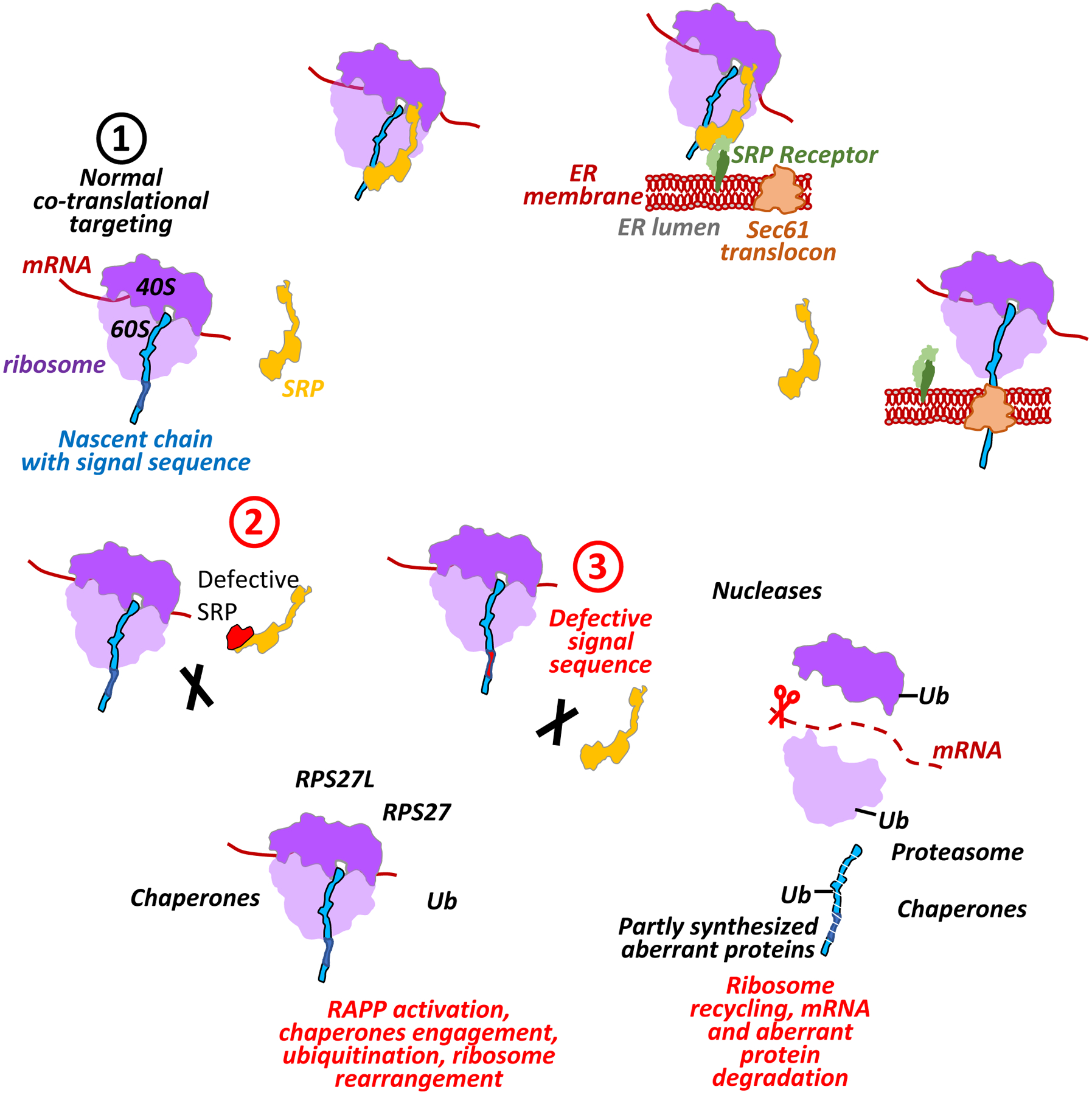
The figure shows a hypothetical model demonstrating RAPP activation and engagement of specific chaperones, ubiquitination and ribosome rearrangement as a result of the lost SRP interactions with polypeptide nascent chains. During normal protein synthesis (scenario 1) SRP co-translationally binds signal sequence of the exposed nascent chain, pauses the translation and targets the ribosome-nascent chain complex to the SRP receptor in the ER membrane. Then ribosome-nascent chain complex is transferred to the Sec61 translocon releasing SRP. Translation is resumed, and the protein is co-translationally translocated into ER lumen, and the signal sequence is cleaved off by a signal peptidase (not shown in the figure). If interactions with SRP are lost due to defects in SRP (scenario 2) or mutations in the signal sequence of the nascent chain (scenario 3), the cells respond to this failure by activation of the RAPP pathway engaging AGO2 (not shown). This triggers upregulation of the distinct chaperones (heat shock proteins), forming specific chaperone network to prevent aggregation of the partly synthesized aberrant peptides and provide help in their clearance. The RAPP activation also triggers K48-linked ubiquitination (Ub). It may affect the ribosomal proteins facilitating the ribosome recycling, as well as label aberrant polypeptides for degradation by proteasome. Ribosomes undergo rearrangement due to decrease of the RPS27 and increase RPS27L proteins during the stress induced by the SRP54 depletion. RPS27L could potentially substitute RPS27 in effort to restore or maintain the translation activity of the ribosomes during stress. Finally, mRNAs of secretory and membrane proteins are degraded by engagement of still unidentified nuclease(s). Partly synthesized nascent polypeptides are degraded by proteasome to prevent their accumulation in the cytoplasm. Ribosome subunits 40S, 60S, and translating mRNA are marked in light purple, dark purple and brown color, correspondingly, SRP is in yellow, nascent chain is in blue, signal sequence is in dark blue, defective signal sequence is shown in red, SRP receptor subunits SRa and SRb are in light and dark green, correspondingly, Sec61 translocon is in orange-brown.
It is very interesting that the SRP54 depletion leads to potential ribosome rearrangement through decreased RPS27 and increased RPS27L expression as compensation for the loss of RPS27. Although involvement of RPS27L needs to be further elucidated, we propose that the RPS27 exchange with RPS27L may provide better fitness for the ribosome and facilitate protein synthesis in conditions of protein targeting failure and RAPP pathway activation. The mechanism by which changes in the expression of ribosomal proteins RPS27/RPS27L could affect mRNA translation is not fully understood. Modulation of ribosome function by individual ribosomal proteins can globally or selectively impact translation by affecting only some mRNAs [65]. Variations in ribosomal protein composition could also introduce changes to gene expression [66]. It has been recently shown that differential expression of ribosomal protein paralogs modifies ribosome composition in response to stress in yeast; however, the functional consequences of this are still not well understood [49]. In agreement with yeast ribosomal stress responses, our data demonstrate changes in mammalian ribosome composition caused by the lack of SRP. Downregulation of RPS27, if not complemented by its paralog RPS27L, could shift the stoichiometry of ribosomal proteins in corresponding subunits and result in fewer functional ribosomes. On the other hand, complementation of depleted ribosomal proteins with its paralogs could modify overall ribosome structure that could affect specific mRNA recognition. Considering that the loss of SRP54 triggers mRNA depletion of secretory proteins, we hypothesize that the ribosome might contribute to RAPP pathway activity. Ribosome heterogeneity was suggested to play a role in selective translation [65, 67, 68]. Some ribosomal proteins are also suggested to have extra-ribosomal functions [69, 70]. Furthermore, changes in availability of functional RPS27 was reported to cause interference with 18S rRNA maturation, which is a component of the 40S ribosomal subunit. Here we show that the peak corresponding to the 40S subunit measured by total RNA absorbance at 260 nm is smaller in SRP54 depleted cells than in the control cells (Figure 8A, B). Changes in total 18S rRNA pool with progression of siSRP54 depletion was also detected (Figure 7B). However, it is not clear if these 20–30% changes in ribosome composition in cells lacking SRP affect translation globally or specifically. Other ribosome associated factors could also influence ribosome heterogeneity include modification of rRNA and ribosomal proteins [65, 68].
The activation of the RAPP pathway leads to elimination of many secretory and membrane protein mRNAs when SRP54 is depleted as shown in Figures 1 – 4. We demonstrated earlier that the mRNA degradation during RAPP is translation dependent [34]. Although the mRNA is degraded, there are remaining nascent chains of partly synthesized proteins that are still attached to the ribosomes. It is also possible that some of these peptides are released in the cytoplasm. These aberrant polypeptides are potentially harmful and need to be degraded while the ribosomes are recycled. E3 ubiquitin ligases mark the peptides and proteins destined for degradation with ubiquitin. It was also demonstrated earlier that specific ribosomal proteins are ubiquitinated during RQC [71, 72] and during the unfolded protein response (UPR) [73]. In contrast to the ubiquitinated aberrant proteins, the ribosomal protein ubiquitination is reversible and has a regulatory role [74]. In the current study, we discovered that SRP54 depletion leads to an increase in K48-type polyubiquitination suggesting that they are substrates for the proteasome. Remarkably, this elevated ubiquitination is associated with ribosomes and ribosomal subunits (Figure 9). Most likely, the maximum ubiquitination signal in the 40S fractions is associated with modification of the ribosomal proteins, and the ubiquitination in the 60S fractions is associated with ubiquitination of the aberrant polypeptides. Although yeast respond differently to SRP depletion by upregulation of an alternative mechanism for protein transport [63, 64] instead of the specific mRNA degradation in RAPP, the lack of SRP in yeast triggers involvement of the Hel2 protein, which is known to ubiquitinate small ribosomal proteins to rescue colliding ribosomes [75]. ZNF598, a mammalian homolog of Hel2, is involved in ubiquitination of small ribosomal proteins in response to stalled ribosomes on premature polyadenylated mRNAs, and it is a ribosome collision sensor [72, 76, 77]. However, it is still unknown if it is involved in response of the SRP54 knockdown and in the RAPP pathway.
Although the E3 ubiquitin ligases and specific substrates need to be determined, our data are consistent with the idea that the nascent chains of partly synthesized proteins as well as ribosomal proteins are ubiquitinated for degradation and/or recycling during RAPP.
We propose a hypothetical scheme of involvement of these specific chaperones, possible ribosome rearrangement, and ubiquitination in the Regulation of Aberrant Production (Figure 10). When a signal sequence or transmembrane span of a nascent chain is exposed from the ribosome exit tunnel in normal conditions, it is recognized by SRP. Then the ribosome-nascent chain-SRP complex is successfully targeted to the SRP receptor (SR) in the ER membrane. Finally, the protein is co-translationally translocated through the Sec61 translocon into the ER lumen for maturation and for further transport through the Golgi to the plasma membrane or secretion outside of the cell. The SRP-nascent chain binding obstructs other ribosome-associated factors to interact with the nascent chain, preventing RAPP activation, and protecting the mRNAs. However, when the nascent chain is unable to interact with SRP because of an absent or defective SRP subunit, or because of the mutations in the polypeptide nascent chain itself, the space that is normally taken by the SRP remains open. This leads to engagement of AGO2, as we found earlier [33], and recruitment of the machinery for mRNA degradation. We demonstrate, for the first time in this study, that this process is followed by the global downregulation of SRP-dependent protein mRNAs and recruitment of specific chaperones. Although the precise role of these chaperones is not established yet, they may help to prevent misfolding and facilitate clearance of partly synthesized polypeptides. Finally, this leads to the engagement of E3 ligases to mark the partly synthesized nascent chains for the proteasomal degradation and label the ribosomal proteins for ribosome recycling. Although many details of the RAPP pathway are still unknown, our model provides a conceptual network of events that may take place during the RAPP pathway activation. This study demonstrates that the RAPP is a general mechanism of protein quality control, and it is not limited to a few secretory proteins. Clearly, our findings open more questions about the finer details of the molecular mechanism to be answered in the future.
Materials and Methods
Cells, growth conditions, siRNA transfections
HeLa Tet-On cells (Clontech) were grown in 6-well plates for Deep RNA-seq and in 12-well plates for other experiments at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium-high glucose (DMEM, Sigma Aldrich) supplied with 10% fetal bovine serum (FBS, Sigma Aldrich) and penicillin and streptomycin mixture (100 units/ml and 100 μg/ml correspondingly) (Sigma Aldrich). Where indicated, siSRP54-transfected HeLa Tet-On cells were treated with proteasome inhibitor MG132 (Sigma Aldrich) or lysosomal inhibitor Bafilomycin (Sigma Aldrich) at growth conditions for 8 h with 10 μM and 0.2 μM final concentrations correspondingly. siRNA transfections were done as described [33, 36]. siRNAs were used at concentrations of 12.0 −13.8 nM for transfections. The sequences of siRNAs used in this study are presented in Supplementary Table S4.
Deep RNA sequencing and bioinformatics analysis
Hela Tet-On cells were grown and transfected with 12 nM siRNA against SRP54 or control siRNA as described earlier [33]. Three biological replicates were used for each experimental condition. Samples were collected 55 hours after transfection, total RNA was purified and used for the whole transcriptome RNA sequencing (Deep RNA-seq) that was completed at the Genomics Core, UT Southwestern Medical Center. Illumina TruSeq Stranded kit was used to generate the cDNA library. Agilent 2100 Bioanalyzer System was used for total RNA and library quality check. Quant-iT PicoGreen dsDNA Assay kit from Invitrogen and PerkinElmer Victor X3 2030 Multilabel Reader to measure the concentration. Samples were sequenced on Illumina HiSeq 2500 sequencer with high through-put single end (SE) flow cell and 50 bases calling. Data obtained from Deep RNA-seq included more than 25 million reads per sample. The reads were analyzed by FastQC (Version 0.11.9) for quality control. Reads processing included the filtering by an average Phred score ≥ 25 and the trimming of Illumina adapters using Trimmomatic algorithm (Galaxy Version 0.38.0) [78]. The processed reads were mapped to the GRCh38 human reference genome (https://useast.ensembl.org), using the algorithm Burrows-Wheeler Aligner (Version 0.7.17.4) in Galaxy project platform (https://usegalaxy.org/) [79]. Samtools algorithm (Galaxy Version 1.9) was used to estimate the mapping quality and generate the coordinate sorted bam files. The gene count matrix was calculated from sorted bam files using the featureCounts algorithm (Galaxy Version 1.6.4) [80].
Differential gene expression analysis was completed using DESeq2 (Version 1.26.0) package of Bioconductor. To analyze the treatment effect and biological replicate distribution, the Euclidean distance between samples was visualized in a heatmap, using the function heatmap.2 from the gplots package (Version 3.0.3). Significantly expressed genes were selected by base mean (mean of normalized counts of all samples) ≥ 10. An absolute fold of change ≥ 1.5 and a Benjamini–Hochberg adjusted P-value ≤ 0.05 were fixed as the cut-off to detect differentially expressed genes (DEG) after SRP54 knockdown. The DEG were visualized in a Volcano plot using the function ggplot2 [81]. The gene names were added using the package MyGene.Info (1.22.0) of Bioconductor [82]. Subcellular localization of differentially expressed gene was evaluated using The Human Proteome: The Cell Atlas repository (https://www.proteinatlas.org/humanproteome/cell). Proteins were classified to four subcellular localizations groups: 1) nucleus, includes proteins localized in nuclear membrane, nucleoli, and nucleoplasm; 2) cytoplasm includes cytosol, actin filaments, centrosome, intermediate filaments, microtubules 3) mitochondria include proteins localized in mitochondria. 4) secretome combines ER, Golgi, plasma membrane, vesicles, and membrane and secreted proteins predicted by majority-based method (Supplementary File 4). Genes coding proteins with signal sequences were annotated by using ‘UniProtKB/Swiss-Prot’ database [83]. The number of DEG and the distribution of fold changes were represented as bar plots and violin plots, respectively. Violin plots were performed using the function ‘ggplot2’ of R software. The signal sequence regions, including the n-terminal region (N-region), the central hydrophobic region (H-region), and the C-terminal region (C-region) that is extended until the cleavage site, were identified using the ‘SignalP 6.0’ slow algorithm [84].
Signal sequences and their regions, were analyzed in terms of hydrophobicity (scale= ‘KyteDoolittle’), hydrophobic moment (angle = 100, window = 5), and net charge (pH = 7, pKscale = ‘Lehninger’) across the ‘Peptides’ package in R software [85]. The distribution of DEG with annotated signal sequence per protein length was explored by using density plots using the function ggplot2 in R that use Kernel smoothing to represent frequency distributions. Both down and upregulated genes were analyzed by gene ontology analysis (GO). Gene ontology over-representation test based on molecular functions was completed by using the ‘ClusterProfiler enrichGO’ function (pvalueCutoff = 0.05, pAdjustMethod = “BH”, qvalueCutoff = 0.2, minGSSize = 10, maxGSSize). All genes significantly expressed were considered as background for Gene Ontology analysis. The set of downregulated proteins with signal sequences were analyzed by their association with human diseases. The analysis was based on the Genetic Association Database (GAD) using the DAVID Bioinformatics Resources 6.8, NIAID/NIH.
RNA purification, cDNA preparation and RT-qPCR analysis
Total RNA was purified using NucleoSpin RNA purification kit (Takara), or Trizol reagent (Life technologies), or RNeasy Mini kit (Qiagen), and quantified using NanoDrop (Thermo Scientific). cDNA samples were prepared using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real time-quantitative polymerase chain reactions (RT-qPCR) were done on the Quant Studio 12 K Flex Real-Time PCR System with using Power SYBR Green PCR Master Mix (Applied Biosystems) according to the manufacturer’s protocol. The Comparative CT method was used to quantify the RT-qPCR results [86]. For analysis of gene expression in polysome fractions synthetic OmpA-RNA was added to the fractions prior to RNA purification and used for normalization of RT-PCR data as described [53]. The following mRNAs were analyzed by RT-qPCR: Signal Recognition Particle 54, (SRP54, NM_003136); CD248 molecule, endosialin (CD248, NM_020404); protein disulfide isomerase family A, member 3 (PDIA3, NM_005313); SPARC (osteonectin), cwcv and kazal like domains proteoglycan 1 (SPOCK1, NM_004598); lysyl oxidase (LOX, NM_002317); prolyl 4-hydroxylase subunit alpha 3 (P4HA3, NM_182904); hypoxia up-regulated 1 (HYOU1, NM_006389); CD24 molecule (CD24, NM_013230); insulin like growth factor binding protein 4 (IGFBP4, NM_001552); hypoxanthine phosphoribosyltransferase 1 (HPRT1, NM_000194), cholinergic receptor nicotinic delta subunit (CHRND, NM_000751.3); potassium voltage-gated channel subfamily Q member 1 (KCNQ1; NM_000218.3); beta actin (ACTB, NM_001101); heat shock protein family B (small) member 8 (HSPB8, NM_014365); shock protein family A (Hsp70) member 8 (HSPA8, NM_006597); heat shock protein 90kDa alpha (cytosolic), class A member 1 (HSP90AA1, NM_001017963); BCL2 associated athanogene 3 (BAG3, NM_004281); DnaJ (Hsp40) homolog, subfamily B, member 1 (DNAJB1, NM_006145); heat shock protein family A (Hsp70) member 1A (HSPA1A, NM_005345); heat shock protein family A (Hsp70) member 1B (HSPA1B, NM_005346); heat shock protein family A (HSP70) member 5 (HSPA5, NM_005347); Homo sapiens ribosomal protein L11 (RPL11, NM_000975); Homo sapiens ribosomal protein S6 (RPS6, NM_001010); Homo sapiens ribosomal protein S27 (RPS27, NM_001030); Homo sapiens ribosomal protein S27-like (RPS27L, NM_015920); and also human 18S ribosomal RNA (X03205).The sequences of RT-qPCR primers used in this study are presented in Supplementary Table S5.
Western blotting
Samples for total cell protein expression analysis were collected 72 h after siRNA transfections or as indicated. HeLa Tet-On Cells were lysed in 1x sample buffer and incubated for 5 min at 95 °C. Western blotting techniques were done according to manufacturer’s protocol with minor modifications. Proteins were separated on 12, 15, or gradient 4–20% SDS-PA gels (as indicated in figure legends) and transferred to PVDF membrane with 0.45 μm pore size (Thermo Scientific). For HSPB8 blotting PVDF membrane with 0.2 μm pore size (Thermo Scientific) was used. Additional treatment of HSPB8 membrane in 0.4% paraformaldehyde (Electron Microscopy Sciences) in Dulbecco’s Phosphate Buffered Saline (DPBS, Sigma) was done for 30 min at room temperature prior to incubation with antibodies. The list of antibodies used in this study is presented in Supplementary Table S4. Protein signals were visualized with western blotting luminol reagent (Santa Cruz Biotechnology, Inc) or SuperSignal West Pico PLUS or Femto Chemiluminescent Substrates (Thermo Scientific).
Immunostaining
HeLa Tet-ON growth and transfection conditions were used as described above with minor modifications. Cells were grown on glass cover slips for 40–46 h after siSRP54 transfection. Cells were fixed with 4% paraformaldehyde solution prepared in Dulbecco’s phosphate-buffered saline (DPBS, Sigma-Aldrich), then washed in DPBS twice and permeabilized with 0.2% Triton X-100, 3% BSA, and DPBS for 20 min at 4°C. Incubation with primary antibodies was done in the same buffer for 1 h at room temperature, incubation with secondary antibodies was done for 30 min in the dark. BSA was omitted from the washing buffer and washing was repeated at least three times for 5 min after each antibody incubation. Cells slips then were mounted on the slides with Prolong Gold antifade reagent supplemented with DAPI (Life Technologies) overnight. Alexa Fluor 488 Goat anti-rabbit IgG (catalog number A11008, Invitrogen) was used as a secondary antibody for PDIA3, CD248, HSPA1A, DNAJB1, and HSP90AA1 immunostaining. Alexa Fluor 555 Goat anti-mouse IgG (catalog number A21422, Invitrogen) was used for SRP54 staining. Images were collected using Zeiss Axiovert 200 M Microscope (TTUHSC Imaging Center).
Polysome profiling
For polysome profiling experiments HeLa Tet-On cells with starting density of 0.6×105 cells/ml were grown in 15 cm dish (Thermo Scientific) for 20 h. The cells were then transfected with 19 nM siRNA SRP54 and grown for 48 hours. The cells were treated with 100 μg/ml cycloheximide and incubated for 15 min at 37°C with 5% CO2. before lysis. The cells were lysed on ice in the buffer containing 20 mM Hepes-KOH (pH 7.5), 0.5% NP-40, 10 mM MgCl2, 2 mM DTT, 1x protease inhibitor cocktail (EDTA-free), 1 mg/mL heparin, and 100 μg/ml cycloheximide. Details of cell lysate preparation are described previously [53]. A260 of cleared by centrifugation lysates was measured (it was about 30 units), and the samples were adjusted to the same A260 before ultracentrifugation. 450 μl of the samples were subjected to ultracentrifugation using Beckman SW 41 rotor through the 10%−50% sucrose gradient, containing 20 mM Hepes-KOH (pH 7.5), 10 mM MgCl2, 1 mM DTT, 1 mg/mL heparin, for 2 h at 260,000xg and 4 °C. After centrifugation, 500 μl fractions were collected using Piston Gradient Fractionator (BioComp Instruments). Protein samples were subjected to 15-folds concentrating by precipitation with trichloroacetic acid (TCA), washed twice with ice-cold acetone and dissolved in 1x SDS-PAGE sample buffer.
Statistical analysis
The data in all figures are averaged from independent biological repeats as indicated in the figure legends. Standard errors or standard deviations are shown. Statistical analysis was done using Prism 9 software. Two-tailed t-test was applied where indicated. Two-way Anova followed by Tukey’s multiple comparison test was applied for quantification of K48-linked ubiquitination blot. P-values indicating statistical significance are shown as ns: not significant; *: p < 0.05; **: p < 0.01; ***: p < 0.001. ****: p<0.0001.
Supplementary Material
Highlights.
Human SRP targets secretory proteins to ER and protects their mRNAs
SRP54 depletion globally downregulates secretory and membrane protein mRNAs
SRP54 knockdown upregulates specific chaperone network and ubiquitination
Loss of SRP54 leads to ribosomal stress and change in expression of RPS27 and RPS27L
Data demonstrate complex nature of SRP function and dramatic consequences of defects
Acknowledgements
The authors thank Sarah C. Miller for critical reading of the manuscript and help in its editing. This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM135167. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CRediT authorship contribution statement
Elena B. Tikhonova: Methodology, Validation, Formal analysis, Investigation, Writing - Original Draft, Writing - Review & Editing, Visualization.
Sneider Alexander Gutierrez Guarnizo: Methodology, Formal analysis, Data Curation, Investigation, Writing - Review & Editing.
Morgana K. Kellogg: Validation, Formal analysis, Investigation, Writing - Review & Editing.
Alexander Karamyshev: Validation, Investigation, Writing - Review & Editing.
Igor M. Dozmorov: Formal analysis, Data Curation, Writing - Review & Editing.
Zemfira N. Karamysheva: Conceptualization, Methodology, Validation, Investigation, Writing - Review & Editing, Visualization.
Andrey L. Karamyshev: Conceptualization, Methodology, Validation, Investigation, Writing - Original Draft, Writing - Review & Editing, Visualization, Supervision, Project administration, Funding acquisition.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
Deep RNA-seq data have been submitted to the GEO database (https://www.ncbi.nlm.nih.gov/geo/) and available under accession number GSE182922.
References
- [1].Karamyshev AL, Tikhonova EB, Karamysheva ZN. Translational Control of Secretory Proteins in Health and Disease. Int J Mol Sci. 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med (Berl). 2003;81:678–99. [DOI] [PubMed] [Google Scholar]
- [3].Zimmermann R, Muller L, Wullich B. Protein transport into the endoplasmic reticulum: mechanisms and pathologies. Trends in molecular medicine. 2006;12:567–73. [DOI] [PubMed] [Google Scholar]
- [4].Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev. 2007;87:1377–408. [DOI] [PubMed] [Google Scholar]
- [5].Lin WJ, Salton SR. The regulated secretory pathway and human disease: insights from gene variants and single nucleotide polymorphisms. Front Endocrinol (Lausanne). 2013;4:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Karamyshev AL, Karamysheva ZN. Lost in Translation: Ribosome-Associated mRNA and Protein Quality Controls. Front Genet. 2018;9:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].D’Orazio KN, Green R. Ribosome states signal RNA quality control. Mol Cell. 2021;81:1372–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Brandman O, Hegde RS. Ribosome-associated protein quality control. Nat Struct Mol Biol. 2016;23:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sontag EM, Samant RS, Frydman J. Mechanisms and Functions of Spatial Protein Quality Control. Annu Rev Biochem. 2017;86:97–122. [DOI] [PubMed] [Google Scholar]
- [10].Joazeiro CAP. Mechanisms and functions of ribosome-associated protein quality control. Nat Rev Mol Cell Biol. 2019;20:368–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Inada T Quality controls induced by aberrant translation. Nucleic Acids Res. 2020;48:1084–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yip MCJ, Shao S. Detecting and Rescuing Stalled Ribosomes. Trends Biochem Sci. 2021;46:731–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Shao S, Hegde RS. Target Selection during Protein Quality Control. Trends Biochem Sci. 2016;41:124–37. [DOI] [PubMed] [Google Scholar]
- [14].Sitron CS, Brandman O. Detection and Degradation of Stalled Nascent Chains via Ribosome-Associated Quality Control. Annu Rev Biochem. 2020;89:417–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- [16].Koch HG, Moser M, Muller M. Signal recognition particle-dependent protein targeting, universal to all kingdoms of life. Rev Physiol Biochem Pharmacol. 2003;146:55–94. [DOI] [PubMed] [Google Scholar]
- [17].Akopian D, Shen K, Zhang X, Shan SO. Signal recognition particle: an essential protein-targeting machine. Annu Rev Biochem. 2013;82:693–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wild K, Halic M, Sinning I, Beckmann R. SRP meets the ribosome. Nat Struct Mol Biol. 2004;11:1049–53. [DOI] [PubMed] [Google Scholar]
- [19].Kellogg MK, Miller SC, Tikhonova EB, Karamyshev AL. SRPassing Co-translational Targeting: The Role of the Signal Recognition Particle in Protein Targeting and mRNA Protection. Int J Mol Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hsieh HH, Shan SO. Fidelity of Cotranslational Protein Targeting to the Endoplasmic Reticulum. Int J Mol Sci. 2021;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Walter P, Ibrahimi I, Blobel G. Translocation of proteins across the endoplasmic reticulum. I. Signal recognition protein (SRP) binds to in-vitro-assembled polysomes synthesizing secretory protein. J Cell Biol. 1981;91:545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Krieg UC, Walter P, Johnson AE. Photocrosslinking of the signal sequence of nascent preprolactin to the 54-kilodalton polypeptide of the signal recognition particle. Proc Natl Acad Sci U S A. 1986;83:8604–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Noriega TR, Tsai A, Elvekrog MM, Petrov A, Neher SB, Chen J, et al. Signal recognition particle-ribosome binding is sensitive to nascent chain length. J Biol Chem. 2014;289:19294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kurzchalia TV, Wiedmann M, Girshovich AS, Bochkareva ES, Bielka H, Rapoport TA. The signal sequence of nascent preprolactin interacts with the 54K polypeptide of the signal recognition particle. Nature. 1986;320:634–6. [DOI] [PubMed] [Google Scholar]
- [25].Nilsson I, Lara P, Hessa T, Johnson AE, von Heijne G, Karamyshev AL. The code for directing proteins for translocation across ER membrane: SRP cotranslationally recognizes specific features of a signal sequence. J Mol Biol. 2015;427:1191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].von Heijne G Signal sequences. The limits of variation. J Mol Biol. 1985;184:99–105. [DOI] [PubMed] [Google Scholar]
- [27].von Heijne G The signal peptide. J Membr Biol. 1990;115:195–201. [DOI] [PubMed] [Google Scholar]
- [28].Nesmeyanova MA, Karamyshev AL, Karamysheva ZN, Kalinin AE, Ksenzenko VN, Kajava AV. Positively charged lysine at the N-terminus of the signal peptide of the Escherichia coli alkaline phosphatase provides the secretion efficiency and is involved in the interaction with anionic phospholipids. FEBS Lett. 1997;403:203–7. [DOI] [PubMed] [Google Scholar]
- [29].Karamyshev AL, Karamysheva ZN, Kajava AV, Ksenzenko VN, Nesmeyanova MA. Processing of Escherichia coli alkaline phosphatase: role of the primary structure of the signal peptide cleavage region. J Mol Biol. 1998;277:859–70. [DOI] [PubMed] [Google Scholar]
- [30].Kalinin AE, Mikhaleva NI, Karamyshev AL, Karamysheva ZN, Nesmeyanova MA. Interaction of mutant alkaline phosphatase precursors with membrane phospholipids in vivo and in vitro. Biochemistry (Mosc). 1999;64:1021–9. [PubMed] [Google Scholar]
- [31].Kellogg MK, Tikhonova EB, Karamyshev AL. Signal Recognition Particle in Human Diseases. Front Genet. 2022;13:898083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jomaa A, Eitzinger S, Zhu Z, Chandrasekar S, Kobayashi K, Shan SO, et al. Molecular mechanism of cargo recognition and handover by the mammalian signal recognition particle. Cell Rep. 2021;36:109350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Karamyshev AL, Patrick AE, Karamysheva ZN, Griesemer DS, Hudson H, Tjon-Kon-Sang S, et al. Inefficient SRP interaction with a nascent chain triggers a mRNA quality control pathway. Cell. 2014;156:146–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pinarbasi ES, Karamyshev AL, Tikhonova EB, Wu IH, Hudson H, Thomas PJ. Pathogenic Signal Sequence Mutations in Progranulin Disrupt SRP Interactions Required for mRNA Stability. Cell Rep. 2018;23:2844–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Karamysheva ZN, Tikhonova EB, Karamyshev AL. Granulin in Frontotemporal Lobar Degeneration: Molecular Mechanisms of the Disease. Front Neurosci. 2019;13:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tikhonova EB, Karamysheva ZN, von Heijne G, Karamyshev AL. Silencing of Aberrant Secretory Protein Expression by Disease-Associated Mutations. J Mol Biol. 2019;431:2567–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, et al. Global quantification of mammalian gene expression control. Nature. 2011;473:337–42. [DOI] [PubMed] [Google Scholar]
- [39].Rauch JN, Gestwicki JE. Binding of human nucleotide exchange factors to heat shock protein 70 (Hsp70) generates functionally distinct complexes in vitro. J Biol Chem. 2014;289:1402–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14:105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bross P, Fernandez-Guerra P. Disease-Associated Mutations in the HSPD1 Gene Encoding the Large Subunit of the Mitochondrial HSP60/HSP10 Chaperonin Complex. Front Mol Biosci. 2016;3:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Vilasi S, Bulone D, Caruso Bavisotto C, Campanella C, Marino Gammazza A, San Biagio PL, et al. Chaperonin of Group I: Oligomeric Spectrum and Biochemical and Biological Implications. Front Mol Biosci. 2017;4:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hsieh JC, Lee L, Zhang L, Wefer S, Brown K, DeRossi C, et al. Mesd encodes an LRP5/6 chaperone essential for specification of mouse embryonic polarity. Cell. 2003;112:355–67. [DOI] [PubMed] [Google Scholar]
- [44].Munro S, Pelham HR. An Hsp70-like protein in the ER: identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell. 1986;46:291–300. [DOI] [PubMed] [Google Scholar]
- [45].Gething MJ. Role and regulation of the ER chaperone BiP. Seminars in cell & developmental biology. 1999;10:465–72. [DOI] [PubMed] [Google Scholar]
- [46].Bole DG, Dowin R, Doriaux M, Jamieson JD. Immunocytochemical localization of BiP to the rough endoplasmic reticulum: evidence for protein sorting by selective retention. J Histochem Cytochem. 1989;37:1817–23. [DOI] [PubMed] [Google Scholar]
- [47].Piette BL, Alerasool N, Lin ZY, Lacoste J, Lam MHY, Qian WW, et al. Comprehensive interactome profiling of the human Hsp70 network highlights functional differentiation of J domains. Mol Cell. 2021. [DOI] [PubMed] [Google Scholar]
- [48].Liu B, Han Y, Qian SB. Cotranslational response to proteotoxic stress by elongation pausing of ribosomes. Mol Cell. 2013;49:453–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ghulam MM, Catala M, Abou Elela S. Differential expression of duplicated ribosomal protein genes modifies ribosome composition in response to stress. Nucleic Acids Res. 2020;48:1954–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. The structure of the eukaryotic ribosome at 3.0 A resolution. Science. 2011;334:1524–9. [DOI] [PubMed] [Google Scholar]
- [51].Xiong X, Cui D, Bi Y, Sun Y, Zhao Y. Neddylation modification of ribosomal protein RPS27L or RPS27 by MDM2 or NEDP1 regulates cancer cell survival. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2020;34:13419–29. [DOI] [PubMed] [Google Scholar]
- [52].O’Donohue MF, Choesmel V, Faubladier M, Fichant G, Gleizes PE. Functional dichotomy of ribosomal proteins during the synthesis of mammalian 40S ribosomal subunits. J Cell Biol. 2010;190:853–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Karamysheva ZN, Tikhonova EB, Grozdanov PN, Huffman JC, Baca KR, Karamyshev A, et al. Polysome Profiling in Leishmania, Human Cells and Mouse Testis. J Vis Exp. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wang R, Yoshida K, Toki T, Sawada T, Uechi T, Okuno Y, et al. Loss of function mutations in RPL27 and RPS27 identified by whole-exome sequencing in Diamond-Blackfan anaemia. Br J Haematol. 2015;168:854–64. [DOI] [PubMed] [Google Scholar]
- [55].Liebl MP, Hoppe T. It’s all about talking: two-way communication between proteasomal and lysosomal degradation pathways via ubiquitin. Am J Physiol Cell Physiol. 2016;311:C166–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hjerpe R, Rodriguez MS. Alternative UPS drug targets upstream the 26S proteasome. The international journal of biochemistry & cell biology. 2008;40:1126–40. [DOI] [PubMed] [Google Scholar]
- [57].Grice GL, Nathan JA. The recognition of ubiquitinated proteins by the proteasome. Cell Mol Life Sci. 2016;73:3497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol. 2016;18:579–86. [DOI] [PubMed] [Google Scholar]
- [59].Erpapazoglou Z, Walker O, Haguenauer-Tsapis R. Versatile roles of k63-linked ubiquitin chains in trafficking. Cells. 2014;3:1027–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Nathan JA, Kim HT, Ting L, Gygi SP, Goldberg AL. Why do cellular proteins linked to K63-polyubiquitin chains not associate with proteasomes? EMBO J. 2013;32:552–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Hernandez SM, Tikhonova EB, Baca KR, Zhao F, Zhu X, Karamyshev AL. Unexpected Implication of SRP and AGO2 in Parkinson’s Disease: Involvement in Alpha-Synuclein Biogenesis. Cells. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Janda CY, Li J, Oubridge C, Hernandez H, Robinson CV, Nagai K. Recognition of a signal peptide by the signal recognition particle. Nature. 2010;465:507–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Costa EA, Subramanian K, Nunnari J, Weissman JS. Defining the physiological role of SRP in protein-targeting efficiency and specificity. Science. 2018;359:689–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Mutka SC, Walter P. Multifaceted physiological response allows yeast to adapt to the loss of the signal recognition particle-dependent protein-targeting pathway. Mol Biol Cell. 2001;12:577–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Guo H Specialized ribosomes and the control of translation. Biochem Soc Trans. 2018;46:855–69. [DOI] [PubMed] [Google Scholar]
- [66].Kondrashov N, Pusic A, Stumpf CR, Shimizu K, Hsieh AC, Ishijima J, et al. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell. 2011;145:383–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Slavov N, Semrau S, Airoldi E, Budnik B, van Oudenaarden A. Differential Stoichiometry among Core Ribosomal Proteins. Cell Rep. 2015;13:865–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Gerst JE. Pimp My Ribosome: Ribosomal Protein Paralogs Specify Translational Control. Trends in genetics : TIG. 2018;34:832–45. [DOI] [PubMed] [Google Scholar]
- [69].Floristan A, Morales L, Hanniford D, Martinez C, Castellano-Sanz E, Dolgalev I, et al. Functional analysis of RPS27 mutations and expression in melanoma. Pigment Cell Melanoma Res. 2020;33:466–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zhou X, Liao WJ, Liao JM, Liao P, Lu H. Ribosomal proteins: functions beyond the ribosome. J Mol Cell Biol. 2015;7:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Matsuo Y, Ikeuchi K, Saeki Y, Iwasaki S, Schmidt C, Udagawa T, et al. Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nat Commun. 2017;8:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Juszkiewicz S, Hegde RS. Initiation of Quality Control during Poly(A) Translation Requires Site-Specific Ribosome Ubiquitination. Mol Cell. 2017;65:743–50 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Higgins R, Gendron JM, Rising L, Mak R, Webb K, Kaiser SE, et al. The Unfolded Protein Response Triggers Site-Specific Regulatory Ubiquitylation of 40S Ribosomal Proteins. Mol Cell. 2015;59:35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Garshott DM, Sundaramoorthy E, Leonard M, Bennett EJ. Distinct regulatory ribosomal ubiquitylation events are reversible and hierarchically organized. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Matsuo Y, Inada T. The ribosome collision sensor Hel2 functions as preventive quality control in the secretory pathway. Cell Rep. 2021;34:108877. [DOI] [PubMed] [Google Scholar]
- [76].Garzia A, Jafarnejad SM, Meyer C, Chapat C, Gogakos T, Morozov P, et al. The E3 ubiquitin ligase and RNA-binding protein ZNF598 orchestrates ribosome quality control of premature polyadenylated mRNAs. Nat Commun. 2017;8:16056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Juszkiewicz S, Chandrasekaran V, Lin Z, Kraatz S, Ramakrishnan V, Hegde RS. ZNF598 Is a Quality Control Sensor of Collided Ribosomes. Mol Cell. 2018;72:469–81 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. [DOI] [PubMed] [Google Scholar]
- [81].Wickham H. ggplot2: Elegant graphics for data analysis. Springer-Verlag; New York; 2016. [Google Scholar]
- [82].Mark A, Thompson R, Afrasiabi C, Wu C. mygene: Access MyGene.Info_ services. R package version 1.22.0. 2019. [Google Scholar]
- [83].UniProt C UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480–D9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Teufel F, Almagro Armenteros JJ, Johansen AR, Gislason MH, Pihl SI, Tsirigos KD, et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature biotechnology. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Osorio D, Rondón-Villarreal P, Torre R. Peptides: A Package for Data Mining of Antimicrobial Peptides. The R Journal. 2015;7:4–14. [Google Scholar]
- [86].Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Deep RNA-seq data have been submitted to the GEO database (https://www.ncbi.nlm.nih.gov/geo/) and available under accession number GSE182922.