

ABSTRACT
The global coronavirus disease 2019 (COVID-19) pandemic has a detrimental impact on public health. COVID-19 usually manifests as pneumonia, which can progress into acute respiratory distress syndrome (ARDS) related to uncontrolled TH17 immune reaction. Currently, there is no effective therapeutic agent to manage COVID-19 with complications. The currently available anti-viral drug remdesivir has an effectiveness of 30% in SARS-CoV-2–induced severe complications. Thus, there is a need to identify effective agents to treat COVID-19 and the associated acute lung injury and other complications. The host immunological pathway against this virus typically involves the THαβ immune response. THαβ immunity is triggered by type 1 interferon and interleukin-27 (IL-27), and the main effector cells of the THαβ immune response are IL10-CD4 T cells, CD8 T cells, NK cells, and IgG1-producing B cells. In particular, IL-10 exerts a potent immunomodulatory or anti-inflammatory effect and is an anti-fibrotic agent for pulmonary fibrosis. Concurrently, IL-10 can ameliorate acute lung injury or ARDS, especially those caused by viruses. Owing to its anti-viral activity and anti-pro-inflammatory effects, in this review, IL-10 is suggested as a possible treatment agent for COVID-19.
KEYWORDS: Tr1, IL-10, virus, COVID-19, ARDS
Introduction
Recently, the coronavirus Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has caused a severe global outbreak of coronavirus disease 2019 (COVID-19). COVID-19 is a severe respiratory-related disease that usually causes pneumonia. WHO designated the disease as COVID-19. The structure of SARS-CoV-2 is very similar with that of the severe acute respiratory syndrome (SARS) virus; both of them bind to the receptor of angiotensin-converting enzyme 2 (ACE2) to infect pulmonary epithelial cells. SARS-CoV-2 can cause severe complications, including ARDS, with a very high mortality, especially in the elderly population. As of 14 December 2022 650 million COVID-19 cases and more than 6.6 million associated deaths have been reported globally [1] [2].
The clinical manifestations of COVID-19 include cough, fever, dyspnoea, and respiratory pain. Upper respiratory symptoms and diarrhoea are also noted but are less common. The SARS-CoV-2 infection can progress into severe respiratory complications, such as pneumonia or acute lung injury [3]. Progression to acute lung injury or acute respiratory distress syndrome (ARDS) is associated with a high mortality. Comorbidities related to higher mortality include cardiovascular disease, diabetes mellitus, hypertension, obesity, chronic lung disease, chronic liver disease, chronic kidney disease, smoking, and malignancy. Several laboratory findings have been related to worse outcomes, including lymphopenia, increased prothrombin time, and elevated levels of liver enzymes, lactate dehydrogenase, inflammatory markers (e.g. C-reactive protein [CRP] and ferritin), D-dimer, troponin, creatine phosphokinase, and creatinine with acute kidney injury. Some severe COVID-19 patients have laboratory findings of an overt inflammatory response, cytokine release syndrome, continuous fevers, elevated inflammatory markers (e.g. CRP and ferritin), and upregulated levels of pro-inflammatory cytokines (e.g. TNF alpha, IL-1, and IL-6). Thus, the use of IL-6 inhibitors to treat COVID-19 has been attempted, with a success rate of approximately 32% [4]. Chest CT of COVID-19 patients can show a peripheral distribution, ground-glass appearances, fine reticular patterns, vascular thickening, and reverse halo mark. Laboratory diagnostic requires a positive RT-PCR nucleic acid test results.
However, to date, there is no effective therapeutic agent to treat this detrimental infectious disease. An anti-viral agent, remdesivir, has been reported to have a 30% response rate for treating COVID-19. Nevertheless, better treatment strategies for SARS-CoV-2 infection and ARDS are required [5].
Overview of ARDS
ARDS is a syndrome that is related to an overactive innate immune response. In ARDS, hyperactive macrophages and neutrophils release pro-inflammatory cytokines, causing cytokine storm and severe immune cell infiltration in the lung. In the later stage, regulatory cells (Treg) become dominant and mediate pulmonary fibrosis to cause a sequel to ARDS. Because ARDS is a consequence of overactive TH17-like innate immunity, its management should focus on inhibiting the overactive host innate immune response. Immune modulation by suppressing the production of pro-inflammatory cytokines could be a promising strategy. However, there is no successful treatment strategy for ARDS; thus, its prognosis remains very poor. Infectious agents, such as SARS-CoV, can cause ARDS [6–8]. In addition, SARS-CoV can downregulate type 1 interferon signalling, which suppresses the anti-viral adaptive immunity [9,10]. This downregulation is important to the pathophysiology of COVID-19.
ARDS is often a complication of sepsis caused by bacterial infection. However, virus-induced ARDS is also seen. The SARS-CoV or influenza virus can also cause ARDS as a complication of these lethal viral infections [11,12]. ARDS is a major complication of severe COVID-19 disease and can manifest shortly after the onset of dyspnoea. If SARS-CoV2 infection induces ARDS, the mortality rate can become approximate 40%. In a study of 201 hospitalized patients with COVID-19 in Wuhan, 41% developed ARDS, and age >65 years, diabetes mellitus, and hypertension were associated with increased risk of ARDS [13]. In another study of 2741 patients who were hospitalized for COVID-19 in New York City, 24% (n = 665) died or were discharged to hospice care, including 241 patients who were not treated in an intensive care unit [14]. Moreover, among the 749 patients who received intensive care (27% of the total cohort), 647 received mechanical ventilation; of these patients, 60% died, 13% were still ventilated, and 16% were discharged. In a study of 138 patients, ARDS developed in 20% of the patients at an average of eight days after the onset of symptoms; mechanical ventilation was required in 12.3% of the patient [15]. In large studies conducted in the United States, 12–24% of hospitalized patients required mechanical ventilation [14,16].
Host immunological pathways against viral or bacterial pathogens
TH17 immune reaction is the host tolerable immune reaction against extracellular microorganisms, including extracellular bacteria, fungi, and protozoa. The antigen-presenting cells of TH17 immune reaction are type 2 convention dendritic cells (cDC2), and its innate lymphoid cells are type 3 natural cytotoxic receptor-innate lymphoid cells (NCR-ILC3). The component cells for TH17 immune reaction are neutrophils (N2), IL-17 expressing CD4 T cells, iNKT17 cells, and IgA2 B lymphocytes. The antigen-presenting cells are type 1 myeloid dendritic cells, while the innate lymphoid cells are type3 innate lymphoid cells (NCR-ILCs3). The triggering cytokines are IL-6 and TGF-β, and the master transcription factors are STAT3α, and STAT5α/β. TH17 immune response is associated with type 3 immune complex-mediated autoimmunity.
THαβ immunity is the host immune reaction against infectious particles, including viruses and prions [17,18]. Plasmacytoid dendritic cells are the antigen-presenting cells for THαβ immunity, while IL-10 producing innate lymphoid cells (ILC10) are the innate lymphoid cells. The major immune cells are NK cells (NK1), IL-10–producing CD4 T cells, CD8 T cells (Tc2), and IgG1 B cells, and the initiation cytokines are interferons alpha/beta and IL-10. IL-10 is the central effector cytokine in THαβ immune function, and the master transcription factors are STAT1α, STAT1β, and STAT3β. NK cell with IgG1-mediated antibody dependent cellular cytotoxicity is the effector phase of this immune reaction, causing apoptosis of virus- or prion-infected cell. During apoptosis, DNA fragmentation eliminates viral DNA or RNA, and caspase protein digestion destroys prion pathogenic proteins. Type 2 antibody-dependent cellular cytotoxic autoimmunity is associated with the THαβ immunological pathway.
It is worth noting that host protective immunity against extracellular bacteria is TH17 immune response and host protective immunity against viruses is THαβ immune response. Based on my previous study, pathogens can drive wrong host immunological pathways to induce immune evasion. For example, malarial protozoa can drive TH17 and THαβ immune reactions rather than TH1 immune reaction to cause immune evasion to prevent malarial pathogens from the attack of host immunity. TH1 immune reaction is the host immunological pathway against intracellular protozoa and other intracellular micro-organisms. If there is successful initiation of TH1 immunity, the malarial protozoa can be eliminated immediately after malarial infection. SARS-CoV2 infection has also similar strategy by inducing anti-bacterial TH17 immunity and suppressing anti-viral THαβ immunity to cause immune evasion to let the viral pathogens survive in the host. The figure 1 shows the ARDS related anti-bacterial TH17 immunity and anti-viral immunity.
Figure 1.
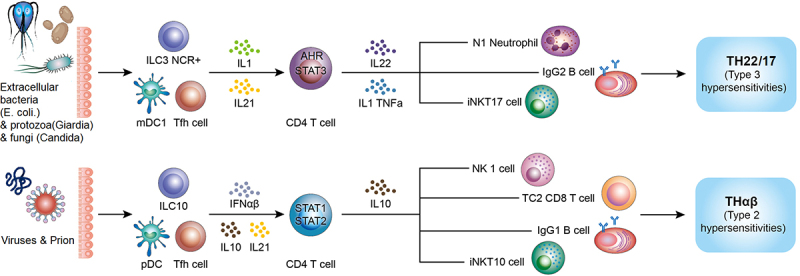
ARDS-related TH17 immunity and anti-viral THαβ immunity. Reprinted from T-H Wen at al. The framework for human host immune responses to four types of parasitic infections and relevant key JAK/STAT signaling IJMS 2021,22:13310.
ARDS is related to anti-extracellular bacterial TH17/TH22 immunity, and virus infection trigger protective THαβ immunity.
Immune pathogenesis of ARDS: Role of TH17 immunity
ARDS is a detrimental respiratory disorder. Septicaemia is the most significant risk factor for developing ARDS. Sepsis is defined as symptomatic bacteraemia due to bacterial infection. Moreover, PMN overactivity is key in the pathophysiology of ARDS. Thus, extracellular microorganism-induced TH17 immune reaction with PMN overactivity should be a master role in the pathogenesis of ARDS.
The duration sequence of ARDS can be categorized into three stages [19]. First, it is the exudative phase. In this stage, damaged alveolar capillary endothelium and type I pneumocytes induce the loss of the alveolar barrier function and accumulation of protein-rich oedema fluid in interstitial alveolar spaces. Pro-inflammatory cytokines including IL-1, IL-6, and TNF-α and chemokines including IL-8, and leukotriene B4 have been detected in the lung in this stage [20,21]. Neutrophils accumulate in the pulmonary tissues [22,23]. Alveolar oedema can contribute to atelectasis, and hyaline-like change begin to form, followed by intrapulmonary shunting and hypoxia. The condition becomes aggravated by microvascular occlusion, leading to increased alveolar dead space. It also leads to pulmonary hypertension. The exudative stage includes the first week of illness after exposure to predominating ARDS risks containing septicaemia, aspiration pneumonia, lung contusion, near-drowning, toxin, or drug inhalation, major trauma, severe burns, multiple transfusions, acute pancreatitis, and post-cardiopulmonary bypass.
The second stage is the proliferative stage. The duration is from days 7 to 21. Although the majority of patients recover after this phase, some patients suffer from progressive lung injury and early lung fibrosis. In histological findings, this stage involves the initiating of lung repairment, organization of pulmonary exudates, and becoming a lymphocyte-dominant pulmonary infiltration. There is also a multiplication of type II pneumocytes, which can produce lung surfactants and trans-differentiate into type I pneumocytes. Besides, the production of type III collagen precursor, a pulmonary fibrosis marker, is initiated.
The third is the fibrotic stage. Although majority of ARDS patients recover lung function 21 days following the initiation of lung injury, several patients suffer from a fibrotic stage that may require mechanical ventilators for maintaining respiratory function. In histological findings, pulmonary oedema and lung exudates in early stages progress to general interstitial fibrosis. Extensive pulmonary fibrosis can contribute to pulmonary hypertension.
Now, we provide a detailed pathophysiology to explicit the three phases of ARDS. In the first exudative phase, PMNs are recruited to the lung owing to chemotactic molecules, such as IL-8, C5, and leukotriene B4. An infection with bacterium in lung tissue can trigger the activation of lung epithelial cells, lung endothelial cells, lung fibroblasts, and alveolar macrophages during sepsis. Toll-like receptors 1, 2, 4, and 5 and heat-shock proteins (HSP60, HSP70) are essential agents that drive TH17 host immunity [24–32]. Heat-shock proteins are essential reactive proteins in situations described above [33–35]. In particular, HSP60 and HSP70 can activate TH17-related Toll-like receptors. Thus, TH17-associated cytokines, such as IL-17, IL-1, TNF-α, and IL-6, as well as TH17-associated chemokines, such as IL-8 are triggered. Bacterial infection can induce pulmonary epithelial cells to release chemokines and cytokines [36–38]. TH17 cytokines activate TH17 immunity, including the PMN effector function fighting against extracellular bacteria. Thus, the ARDS-related cytokine storm is now understood.
However, the CD86 co-stimulation signal, TCR genes, and most MHC genes are downregulated in ARDS. Thus, adaptive T cells, B cell immunity with specific antibody, and TCR responses against bacteria may not be triggered. However, a previous study detected IL-8 autoantibodies in ARDS patients [39]. IL-8 and leukotriene B4 are the main chemo-attractants in pulmonary tissues [40–46]. This chemokine was originally identified in pulmonary giant cell lines [47]. IL-8 has a high affinity for binding to heparin sulphate and chondroitin sulphate, which are abundant in the lung [48,49]. IL-8 in lung tissues can then recruit PMNs to pulmonary. This can provide reasons why IL-8 produced from distant sites, such as the pancreas, during inflammation can lead to ARDS.
Several defects in the IL8 autoantibody were reported. First, the anti-IL-8–IL-8 complex was detected in the sera of 55% of the healthy controls. There was no significant difference in the number of IL-8–anti-IL-8 complex between ARDS patients and the healthy controls. Second, the IL8 autoantibody can suppress IL-8’s binding activity to neutrophils and chemotaxis activity. Thus, the importance of the IL8 autoantibody in ARDS pathogenesis is doubtful. Moreover, several studies support the role ofTH17 immunity in the pathogenesis of ARDS [21,50]. G-CSF, the growth factor of neutrophils, can cause the common symptoms of ARDS [51]. Moreover, suppression of NFkB can attenuate ARDS progression [52,53]. The key THαβ cytokine, IL-10, also reduces the severity of ARDS [54].
Sepsis is the number one important risk factor for ARDS. However, specific pathogens other than bacterium are also risks for ARDS development. For example, malarial infection can cause ARDS complications. P. falciparum can activate HSP60 and HSP70 to trigger TH17 immunity, leading to acute lung injury [55]. SARS-CoV2 and the H1N1 influenza virus can also downregulate anti-viral type 1 interferons and upregulate TH17-like immunity to trigger acute lung injury [7,56]. Thus, TH17 inflammation may be a key pathway in ARDS pathogenesis. It is also noted in burns, major trauma, and acute pancreatic inflammation when TH17-like innate immunity is activated.
In the second proliferative phase, lymphocytes, including TH17-like lymphocytes and Treg lymphocytes, replace PMNs and be the major immune cells in ARDS. TH17-related cytokines, such as IL-17, IL-1, IL-6, and TNF-α, contribute to the inflammation owing to innate immunity activation. However, if bacteria during sepsis or other aetiology suppress the expression of HLA molecules, successful adaptive immune responses with T cell and B cell activation cannot be achieved. One study showed that STAT3, a key transcription factor in TH22/TH17 adaptive immunity, is downregulated. Thus, only the innate immune response is activated in ARDS patients. In addition, activated Treg can secrete TGF-β, which plays a vital role in the pathophysiology of ARDS. Treg cells, the regulatory components of TH17 immunity, are associated with STAT5B activation. So, in the third fibrotic phase, TGF-β-secreting CD4CD25 T helper cells are the dominant immune cells in ARDS [50]. ARDS was induced by bacterial sepsis; thus, failure to induce specific adaptive immunity, such as through TCR and antibodies, cannot successfully overcome bacterial infection. In the early disease stage, overactive innate immunity with PMN activation causes severe lung consolidation, whereas in the later stage, failure of adaptive immunity against bacteria causes abundant regulatory T cells to secrete TGF-β, a powerful fibrosis-promoting agent and the most important and potent stimulant of tissue fibrosis [57,58]. TGF-β promotes the bio-synthesis of collagens [59]; thus, its over-activity in pulmonary tissues causes lung fibrotic change. TGF-β-induced fibrotic change is a process for repairment of cavities caused by bacterial loci, such as abscesses.
The abovementioned pathophysiology can explain the controversial results of many studies. A study found that TLR4 and HSPs can let ARDS become worse [26]. However, other studies reported that TLR4 or HSPs can protect against lung fibrosis after acute lung injury [60–62]. TLR and HSP signals can retain the overactivity of innate inflammatory molecules, such as IL-1. Thus, TGF-β over-activity to promote lung fibrosis is avoided. An animal study reported that neutrophil inhibitors can attenuate the progression of acute lung injury [63]. Thus, TH17 inflammatory processes can fully explain the pathogenesis of ARDS.
Finally, we will mention the two-hit mechanism for Transfusion Related Acute Lung Injury (TRALI). TRALI and ARDS share a common pathogenesis. According to the two-hit hypothesis, TRALI requires the activation of innate immunity, such as CRP, and the production of anti-MHC antibodies. Anti-MHC antibodies may block MHC signalling to prevent the activation of successful adaptive immunity. In addition, downregulated MHC signalling can help trigger the Treg cell components of the TH17 immunological pathway. Thus, overactivated TH17 immunity causes the pathophysiology of ARDS.
Virus induced ARDS
Although ARDS is usually induced by bacterial infection such as sepsis, ARDS can also be induced by virus infection. For example, influenza virus infection can trigger ARDS as its detrimental complication. Influenza virus-induced ARDS has two important components. First, the viral pathogen can infect respiratory epithelial cells. Second, over-activity of innate immune reaction including TH17 immunity is triggered. Influenza virus infection can induce ARDS by fulfilling the above two conditions. H5N1 and H1N1 are the most identified influenza viruses which can induce ARDS. In a previous study, H5N1 is a potent inducer of proinflammatory cytokines including TNF-α in human macrophages. Thus, TH17 over-activity is associated with H5N1 influenza virus infection. Another paper pointed out that the influenza virus targets lung epithelial cells, damaging tight junctions, and killing infected alveolar cells. Infected epitheliums produce pro-inflammatory cytokines that attract neutrophils as well as macrophages and activate adjacent endothelial cells. Activated endothelial cells and infiltrated leucocytes stimulate further immune cell infiltration, and leucocytes induce production of free radicals like reactive oxygen species to damage lung structure. Activated macrophages also cause trigger apoptosis of epithelial cells. Other virus-induced ARDS has also other components. Adenovirus-induced ARDS is similar to bacteria induced ARDS. However, CD3+ CD4+ T cells are reduced by adenovirus-induced ARDS. It suggests that lymphopenia is also an important finding in virus-induced ARDS. As for Herpesviridae, herpes simplex virus (HSV) and cytomegalovirus (CMV) are the two viral pathogens causing nosocomial viral pneumonia that can progress into ARDS. TH17 related innate immunity over-activation is also related to Herpesviridae viruses caused ARDS [64–66].
Coronavirus can also cause virus-induced ARDS including SARS, MERS, and COVID-19, which is caused by SARS-CoV1, MERS-CoV, and SARS-CoV2, respectively. First of all, these coronaviruses can infect lung epithelial cells to cause alveolar damages. In coronavirus infection, defective type 1 interferon response is noted in SARS, MERS, and COVID-19. Suppression of the interferon alpha/beta is commonly noted in coronavirus infection. SARS-CoV infection in vitro failed to activate interferon regulatory factor 3 (IRF3) which is responsible for producing type 1 interferon. In our previous study, failure of interferon alpha production by SARS-CoV1 is also noted in monocytic cell lines. It is known that eight proteins in SARS-CoV antagonize interferon-stimulated gene (ISG) responses, and these proteins have been identified in MERS-CoV with similar functions. NSP14 and NSP10–16 complex can cap viral mRNAs to protect the SARS-CoV mRNAs from being bound by MDA5 and IFIT1 to trigger IRF activity. The SARS-CoV nucleocapsid protein has an inhibitory effect on type 1 interferon induction, too. The SARS-CoV membrane protein represses type I interferon production by stopping the formation of TRAF3/TBK1/IKK complex. The MERS-CoV membrane protein inhibits the nuclear translocation of IRF3 to stop to activate type I interferon promoter. MERS-CoV ORF4a, ORF4b, ORF5 proteins can all inhibit the nuclear trafficking of IRF3 and activation of the interferon promoter. Type 1 interferons are the initiator of anti-viral THαβ immune response. Thus, coronavirus infection cannot successfully trigger the host anti-viral immunological pathway. The second mechanism of coronavirus-induced ARDS is lymphocyte functional impairment. Recent evidence has found that NK cell number reduction, especially perforin+ NK cell reduction, is noted in COVID-19. The absence of cytotoxic granules containing NK cells also means they are functional impairment to lyse virus-infected host cells. Besides, CD8 and CD4 T cell numbers are also reduced in SARS, MERS, or COVID-19 infection. Lymphopenia is a common finding in coronavirus-induced ARDS. Cytotoxic function of CD8 T cells is also impaired in SARS-CoV2 infection. Thus, adaptive immunity is impaired in coronavirus-induced ARDS. The third mechanism of coronavirus-induced ARDS is over-activation of neutrophils and macrophages. Over-activities of neutrophils and macrophages produce dys-regulated pro-inflammatory cytokines in SARS, MERS, or COVID-19 to induce cytokine storm and ARDS. The over-activated neutrophils and macrophages with massive pro-inflammatory cytokines also link to up-regulated TH17 immune reaction. These mechanisms are the pathophysiology of coronavirus-induced ARDS [67,68].
Drugs which may be useful to control COVID-19
Due to the above discussion, we know that over-activation of TH17 immunity is related to the pathogenesis of COVID-19. Thus, therapeutic agents targeting TH17 immune reaction may be useful to manage COVID-19 infection. COVID-19 infection is related to up-regulation of anti-bacterial TH17 immunity and down-regulation of anti-viral THαβ immunity. First, we will talk about drugs, which can suppress TH17 immune reaction including silibinin, baricitinib, ruxolitinib, tocilizumab, risankizumab, brodalumab, secukinumab, and netakimab. STAT3 is the key transcription factor to trigger TH17 immunity. Thus, STAT3 inhibitors such as silibinin was used in a pilot study to see its effects on COVID-19 infection in several cancer patients. In this pilot study, pro-inflammatory biomarkers did reduce after silibinin treatment, so STAT3 inhibitor could be useful to control COVID-19. However, the sample size is small, and we cannot make a conclusion. Besides, JAK molecules also play vital roles in triggering STAT3 transcription factor activation during TH17 immunity. The cooperation of JAK1, JAK2, and TYK2 are responsible of interleukin-6 signal transduction to activate STAT3. Preliminary data also show that Baricitinib, a JAK1 and JAK2 inhibitor, is helpful to control COVID-19 infection. Although it was not statistically significant, Baricitinib can reduce COVID-19 mortality in meta-analysis of randomized clinical trials. Ruxolitinib, a JAK1/JAK2/TYK2 inhibitor, has also been suggested to treat COVID-19 infection. However, the efficacy of ruxolitinib was not confirmed in large clinical trials. Combining IL-6 inhibitor tocilizumab and ruxolitinib has been proposed. The non-helpful results of using this strategy could be due to the overlapping mechanism of blocking interleukin-6 signalling of both inhibitors. Thus, no additional effectiveness was observed. On the other hand, it could cause higher adverse effect by overlapping triggering of interleukin-6 signal transduction pathway. It is worth noting that type 1 interferon and interleukin-10 signal transduction relies on JAK1 and TYK2 to trigger THαβ anti-virus host immune reaction. Thus, blocking JAK1 and TYK2 via ruxolitinib can adversely suppress anti-viral host immunity. This can explain why ruxolitinib is not helpful in controlling COVID-19 infection. This can also explain why JAK2 inhibition plays a key role in control COVID-19 infection. As for the other important transcription factor in TH17 immunity, RAR-related orphan receptor inhibitors, the information is limited in COVID-19 patients. Another TH17-related cytokine, interleukin-23, is an interesting drug target. A case report showing that the IL-23 inhibitor risankizumab used in one psoriasis patient can help to control COVID-19 infection. But, information is limited. Interleukin-17 is the central cytokine in the TH17 immunological pathway. Brodalumab is an IL-17 receptor inhibitor, and secukinumab is an IL-17 inhibitor. Both drugs are suggested to be used to control COVID-19 infection. Nevertheless, we still need randomized clinical trials to confirm the efficacy of the two drugs. The other IL-17 inhibitor netakimab has been tested in a pilot clinical trial. After 3 days of therapy, body temperature, SpO2/FiO2, and CRP improved significantly. However, the ICU transfer rate, need for mechanical ventilation, and one-month mortality rate was statistically non-significantly reduced. A statistically significant decrease in the duration of hospitalization in the netakimab group was observed. These above evidences suggest TH17 immunity inhibition can be helpful in the management in COVID-19 infection. However, the clinical guideline already includes IL-6 inhibitor, a key TH17 initiator, to treat COVID-19 with complications. We will need new drug candidate not only to control TH17 related pro-inflammatory cytokine storm but also to successfully initiate anti-viral THαβ immunity to suppress viral infection. Interleukin-10 can play a vital role [69–72].
IL-10 as host immunity factor against viruses
The host immunological pathway against viruses is the THαβ immunity [18]. The THαβ immune response is driven by type 1 interferon and IL-10. Pegylated IL-10 (pegilodecakin) is the drug form of IL-10 [73]. It has been used in the cancer clinical trial owing to its immune stimulatory effects. The main T helper cells of THαβ immunity are IL-10-CD4 T cells, formerly called Tr1 cells [74]. The major effector cells of THαβ immunity are IL-10-CD4 T cells, NK cells, CD8 T cells, and IgG1-producing B cells. CD8 T cells and NK cells have an antibody-dependent cellular cytotoxicity (ADCC) function, which induces the apoptosis of virus-infected cells, leading to viral and cellular DNA fragmentation and elimination of viruses. Type 1 interferon is the main cytokine that triggers anti-viral immune responses. Because SARS-CoV can downregulate type 1 interferon production to confer the virus survival advantage, type 1 interferons (e.g. interferon-α and interferon-β) have been used to treat SARS infections [75,76].
IL-10 is the key cytokine in the THαβ immune response and a potent anti-viral cytokine [74]. Previous studies have showed that IL-10 can suppress viral infections, including the diseases caused by HSV, alpha virus, HIV, RSV, HCV, Japanese encephalitis virus, influenza virus, vaccinia virus, dengue virus, and coronavirus [77–87]. IL-10 can also reduce coronavirus-induced CNS demyelination or encephalomyelitis [88–92]. Several pox viruses, such as the EBV or CMV, encode viral IL-10 (vIL-10) to mimic cellular IL-10 or serve as a decoy [93,94]. Thus, virus can escape the host immune response through vIL10 [95–98]. These viral encoded IL-10 lack the functions of stimulating thymocyte or PBMC proliferation and B cells class II MHC up-regulation which are normal functions of cellular IL-10 [93–95,97]. Viral encoded IL-10 also suppresses the induction of type 1 interferons from plasmacytoid dendritic cells and prevents the expression of co-stimulating molecules on lymphocytes [99,100]. Type 1 interferons (IFNα & IFNβ) can drive naïve CD4 T cells to become IL-10 producing CD4 T cells, formally called Tr1 cells. IL-10 producing CD4 T cells are the major T helper cells in antiviral THαβ immunity. IL-10 was originally clones as a T cell growth factor. IL-10 can activate the activities of NK cells, B cells, and CD8 T cells [101–104]. IL-10 producing CD8 T cells are the main component cells in anti-viral THαβ immunity. It can also cause B cell antibody isotype switch to IgG1 antibody, which is the anti-viral antibody [18,105]. Lymphocytes including NK cells, CD8 T cells, and IgG1 B cells are main effector cells in anti-viral host immunity. IL-10 is not a pure immunosuppressant because of these functions. IL-10 is also an immunostimulant to trigger host immune reaction against viruses. These findings suggest that IL-10 plays an important role in anti-viral immunity.
TH3 cells, which are detected in chronic virus infection, are characterized by co-secretion of IL-10 and TGF-β [105]. Blackburn and Brooks suggested IL-10 is related to persistent viral infection; however, they neglected the role of TGF-β and IL-10 co-expressing TH3 cells [106,107]. A previous study pointed out that TGF-β signalling contributes to the persistence of lymphocytic choriomenigitis virus (LCMV) infection in mice [108]. In fact, IL-10 exerts a potent anti-viral activity by activating NK cells and CD8 T cells [101]. TGF-β can weaken the anti-viral effect of IL-10 and mildly control chronic virus infections to avoid fulminant viral diseases, such as viral hepatitis [105]. In fact, based on the findings of Blackburn and Brooks, we speculate that the actual cause of persistent virus infection is TGF-β and not IL-10. A figure of showing anti-viral TH
IL-10 as an immune-modulation mediator
IL-10 possesses a strong immune-modulation activity [109] and is considered as an anti-inflammatory cytokine that can suppress pro-inflammatory cytokine production by macrophages [110–113]. IL-10 can also suppress the functions of neutrophils [114,115]. Previous studies have shown IL-10 can suppress the pathogenic TH17-like innate immunity to ameliorate autoimmunity [116–120]. THαβ CD4 T cells can secrete IL-10, and IL-10 can activate CD8 T cells and NK cells, causing immunity against virus infection. Thus, IL-10 has a dual role in anti-viral immunity: suppression of macrophages and neutrophils and activation of CD8 T cells and NK cells for anti-viral immunity [101].
One study reported elevated IL-10 levels in patients with COVID-19 with ARDS; however, whether this serves a protective or harmful role remains unknown [4]. ARDS is related to overactivation of macrophages and neutrophils and manifests as acute lung injury caused by hyperactivity of innate immunity. Indeed, previous studies have showed that IL-10 from Tr1 cells (now called THαβ cells) can ameliorate ARDS in some animal models [54,121]. In addition, SARS-CoV inhibited by CD4 T cells and CD8 T cells in animal models were reported [90,91]. Thus, CD4 T cells secrete IL-10, which subsequently activate CD8 T cells [101]. Overall, IL-10 may suppress coronavirus infection.
TRALI is considered as the same syndrome as ARDS. They have common clinical manifestations. A two-hit hypothesis was proposed for the aetiology of TRALI. First, over-activity of innate immunity is triggered happens. Second, there is the presence of anti-HLA antibodies. In TRALI, the level of IL-10 is usually low, with bad prognosis [122]. A previous study found that TRALI can be treated using IL-10 [123]. Several studies have reported that IL-10 can treat acute lung injury or pulmonary inflammation due to viruses, including influenza virus or respiratory syncytial virus (RSV) [64,82]. Thus, ARDS may also be treated by IL-10 [123]. IL-10 plays a strong protective role in a viral infection of the lung [79]; thus, it may be a good candidate for virus-induced ARDS treatment.
IL-10 as an anti-fibrotic agent
IL-10 has a potent anti-fibrotic activity. It may compete with other pro-fibrotic cytokines, such as TGF-β, to inhibit fibrosis. Several previous studies have found that IL-10 can suppress the activity of fibroblasts [124,125] and ameliorate pulmonary fibrosis [126–129]. The common sequalae of ARDS is pulmonary fibrosis. Overall, we can infer that the use of IL-10 can treat SARS-CoV-2–induced ARDS and pulmonary fibrosis through the anti-viral THαβ immunological pathway, which was previously discussed [105].
Possible overdose and resistance of IL-10 therapy
Previous studies showed that IL-10 injection in healthy voluneters is safe and well tolerated. The administated dosages are 1.0, 2.5, 5.0, 10, 25, 50 ug/kg [130–133]. At high dosage of interleukin-10 administration, adverse effects include flu-like syndrome, mild to moderate decrease of neutrophil counts, mild decrease of lymphocyte count, and mild delayed decrease of platelet counts. Pegylated interleukin-10 can protect itself from metabolizing and prolong the half-life of the drug [134]. Overall, interleukin-10 administration is safe and well tolerated. As for the resistance of interleukin-10, observation found that high glucose in blood circulation is associated with interleukin-10 resistance [135]. However, the detailed mechanism is unclear. In addition, some viruses, such as EBV or CMV encode the viral interleukin-10. Viral interleukin-10 can compete the binding to host interleukin-10 receptor, so it might lead to host interleukin-10 resistance [93–97]. Viral interleukin-10 can still have immuno-suppression effects of interleukin-10, but it abrogates the anti-viral and immuno-stimulating effects of interleukin-10. That is the reason why these viruses can have the phenomenon of immune evasion.
Responses to previous studies
Several previous studies have pointed out that IL-10 levels are elevated after SARS-CoV2 infection. This elevation may be related to the severity of COVID-19. In addition, the levels of pro-inflammatory cytokines, especially IL-6, were more highly elevated in severe cases of COVID-19 [136]. Thus, those authors concluded that IL-10 may be related to worse prognosis of SARS-CoV2 infection [137–139]. Furthermore, those authors suggested that IL-10 may play a role in the pathogenesis of COVID-19. In fact, there is another two papers, which pointed out down-regulation of IL-10 during COVID-19 infection and higher level of IL-10 was seen in recovered patients compared to severe COVID-19 patients [140,141]. Another two studies also had findings against IL-10 elevation in COVID-19 [142,143]. Thus, observations about elevation of IL-10 serum levels during COVID-19 infection are not consistent. A previous study found that IL-10 can be effective to treat solid tumours, suggesting that it is a pro-inflammatory cytokine [73]. However, this conclusion is questionable. IL-10 potently suppresses cytokine storms with elevations of pro-inflammatory cytokines. IL-10 is effective against solid tumours because it is an anti-viral cytokine. Thus, IL-10 has a similar mechanism in combating solid tumours as oncolytic viruses.
Nevertheless, previous studies have some limitations and false interpretations. IL-10 levels can be reactively elevated during COVID-19. Interleukin-10 levels may not be elevated enough to combat severe disease during COVID-19. A study showed that the IL-6/IL-10 ratio is a more ideal prognosis biomarker for COVID-19. Other studies have also suggested that this ratio could be a prognostic indicator for systemic inflammatory diseases [144–146]. Thus, it should be noted that higher levels of IL-6 and lower levels of IL-10 are related to COVID-19 severity and worse prognosis. IL-10 is a key cytokine during THαβ anti-viral immunity, and its production is triggered by type 1 interferons. However, SARS-CoV2 can downregulate type 1 interferons and prevent the induction of IL-10. In fact, IL-10 plays a beneficial role against coronavirus infection. Current clinical guidelines for treating COVID-19 with ARDS is triple therapy: remdesivir, tocilizumab (anti-IL-6 monoclonal antibody), and corticosteroid [147]. Remdesivir is the anti-SARS-CoV2 agent. Tocilizumab is the monoclonal antibody to suppress pro-inflammatory cytokine: IL-6. Corticosteroid is the potent immunosuppressant to suppress ARDS with cytokine storm in COVID-19. Corticosteroid can totally suppress host immune responses, including adaptive immune suppression with lymphocyte inhibition. Thus, it can cause host antiviral immunity to become impaired. Thus, it is not safe to use corticosteroid to treat SARS-CoV2-induced ARDS in COVID-19. IL-10 can inhibit all pro-inflammatory cytokines, including IL-1, IL-6, IL-8, and TNF-α and it can stimulate anti-viral immune reaction with activating CD8 T cells, NK cells, and IgG1 B cells activities. Thus, it is more logical to use IL-10 to treat COVID-19 instead of corticosteroid.
Other THαβ cytokines to control of COVID-19 infection
There are other THαβ cytokines that can also be important to control COVID-19 infection. SARS-CoV can downregulate type 1 interferons. Thus, type 1 interferons could be used to control COVID-19. Additionally, type 3 interferons have a similar anti-viral mechanism as type 1 interferons [148], and SARS-CoV2 can be suppressed by type 3 interferons [149]. Thus, type 3 interferons can also be used to control SARS-CoV2. In addition, IL-27 is a cytokine that upregulates IL-10 [150]. Thus, the anti-viral ability of IL-27 makes it a potential therapeutic agent for COVID-19 [151,152]. These cytokines may serve as possible management strategies to treat COVID-19.
Conclusions
To date, there is no effective therapeutic agent for COVID-19 with ARDS. The success rate of IL-6 inhibitors for COVID-19 treatment is only 32%, which does not satisfy the clinical needs. This review proposes IL-10 as a new therapeutic strategy for COVID-19 with ARDS. We discussed its treatment potential based on the three mechanisms. First, it is a key anti-viral infection cytokine that activates CD8 T cells and NK cells. Second, it exhibits immune modulation activity by suppressing macrophages and neutrophils to produce pro-inflammatory cytokines. Third, IL-10 is a potent anti-fibrotic agent that competes with pro-fibrotic cytokines, such as TGF-β, to inhibit pulmonary fibrosis. Thus, we suggest that IL-10 is a potential therapeutic candidate for COVID-19 with ARDS, especially in SARS-CoV-2 infection complications with ARDS.
Acknowledgements
The authors are very thankful for Taipei Tzu Chi Hospital for providing fundings for this research.
Funding Statement
This research was funded by Taipei Tzu Chi Hospital, grant number TCRD-TPE-111-14
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing not applicable-no new data generated
Institutional review board statement
Not applicable.
Informed consent statement
Not applicable.
References
- [1].Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–14. DOI: 10.1016/s0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wilder-Smith A, Chiew CJ, Lee VJ.. Can we contain the COVID-19 outbreak with the same measures as for SARS? Lancet Infect Dis. 2020;20(5):e102–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mehta P, McAuley DF, Brown M, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rahmati M. Cytokine-targeted therapy in severely ill COVID-19 patients: options and cautions. Eurasian J Med Oncol. 2020. DOI: 10.14744/ejmo.2020.72142 [DOI] [Google Scholar]
- [5].Mason RJ. Pathogenesis of COVID-19 from a cell biology perspective. Eur Respir J. 2020;55(4):2000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jiang Y, Xu J, Zhou C, et al. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am J Respir Crit Care Med. 2005;171(8):850–857. [DOI] [PubMed] [Google Scholar]
- [7].Rockx B, Baas T, Zornetzer GA, et al. Early upregulation of acute respiratory distress syndrome-associated cytokines promotes lethal disease in an aged-mouse model of severe acute respiratory syndrome coronavirus infection. J Virol. 2009;83(14):7062–7074. DOI: 10.1128/JVI.00127-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang Y, Li J, Zhan Y, et al. Analysis of serum cytokines in patients with severe acute respiratory syndrome. Infect Immun. 2004;72(8):4410–4415. DOI: 10.1128/IAI.72.8.4410-4415.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hensley LE, Fritz LE, Jahrling PB, et al. Interferon-β 1a and SARS Coronavirus replication. Emerg Infect Dis. 2004;10(2):317–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hu W, Yen YT, Singh S, et al. SARS-Cov regulates immune function-related gene expression in human monocytic cells. Viral Immunol. 2012;25(4):277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39(5):529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chien JY, Hsueh PR, Cheng WC, et al. Temporal changes in cytokine/chemokine profiles and pulmonary involvement in severe acute respiratory syndrome. Respirology. 2006;11(6):715–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fu Y, Cheng Y, Wu Y. Understanding SARS-CoV-2-mediated inflammatory responses: from mechanisms to potential therapeutic tools. Virol Sin. 2020;35(3):266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Petrilli CM, Jones SA, Yang J, et al. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York city: prospective cohort study. BMJ. 2020;369:m1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–Infected pneumonia in Wuhan, China. JAMA. 2020;323(11):1061–1069. DOI: 10.1001/jama.2020.1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Richardson S, Hirsch JS, Narasimhan M, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York city area. JAMA. 2020;323(20):2052–2059. DOI: 10.1001/jama.2020.6775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tsou A, Chen PJ, Tsai KW, et al. THαβ immunological pathway as protective immune response against prion diseases: an insight for prion infection therapy. Viruses. 2022;14(2):408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hu WC. Human immune responses to Plasmodium falciparum infection: molecular evidence for a suboptimal THαβ and TH17 bias over ideal and effective traditional TH1 immune response. Malar J. 2013;12(1):392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kong SL, Chui P, Lim B, et al. Elucidating the molecular physiopathology of acute respiratory distress syndrome in severe acute respiratory syndrome patients. Virus Res. 2009;145(2):260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lee EJ, Lim JY, Lee SY, et al. The expression of HSPs, anti-oxidants, and cytokines in plasma and bronchoalveolar lavage fluid of patients with acute respiratory distress syndrome. Clin Biochem. 2012;45(6):493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Papadakos PJ. Cytokines, genes, and ARDS. Chest. 2002;121(5):1391–1392. [DOI] [PubMed] [Google Scholar]
- [22].Downey GP, Dong Q, Kruger J, et al. Regulation of neutrophil activation in acute lung injury. Chest. 1999;116:46S–54S. [PubMed] [Google Scholar]
- [23].Lee WL, Downey GP. Neutrophil activation and acute lung injury. Curr Opin Crit Care. 2001;7(1):1–7. [DOI] [PubMed] [Google Scholar]
- [24].Charles PE, Tissieres P, Barbar SD, et al. Mild-stretch mechanical ventilation upregulates toll-like receptor 2 and sensitizes the lung to bacterial lipopeptide. crit care. 2011;15(4):R181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fan J, Li Y, Vodovotz Y, et al. Hemorrhagic shock-activated neutrophils augment TLR4 signaling-induced TLR2 upregulation in alveolar macrophages: role in hemorrhage-primed lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2006;290(4):L738–746. [DOI] [PubMed] [Google Scholar]
- [26].Imai Y, Kuba K, Neely GG, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133(2):235–249. DOI: 10.1016/j.cell.2008.02.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jiang D, Liang J, Fan J, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11(11):1173–1179. DOI: 10.1038/nm1315 [DOI] [PubMed] [Google Scholar]
- [28].Lv T, Shen X, Shi Y, et al. TLR4 is essential in acute lung injury induced by unresuscitated hemorrhagic shock. J Trauma. 2009;66(1):124–131. [DOI] [PubMed] [Google Scholar]
- [29].Sharif R, Dawra R, Wasiluk K, et al. Impact of toll-like receptor 4 on the severity of acute pancreatitis and pancreatitis-associated lung injury in mice. Gut. 2009;58(6):813–819. [DOI] [PubMed] [Google Scholar]
- [30].Togbe D, Schnyder-Candrian S, Schnyder B, et al. TLR4 gene dosage contributes to endotoxin-induced acute respiratory inflammation. J Leukocyte Biol. 2006;80(3):451–457. [DOI] [PubMed] [Google Scholar]
- [31].Wu TT, Chen TL, Loon WS, et al. Lipopolysaccharide stimulates syntheses of toll-like receptor 2 and surfactant protein-A in human alveolar epithelial A549 cells through upregulating phosphorylation of MEK1 and ERK1/2 and sequential activation of NF-κB. Cytokine. 2011;55(1):40–47. [DOI] [PubMed] [Google Scholar]
- [32].Reino DC, Pisarenko V, Palange D, et al. Trauma hemorrhagic shock-induced lung injury involves a gut-lymph-induced TLR4 pathway in mice. PLoS ONE. 2011;6(8):e14829. DOI: 10.1371/journal.pone.0014829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chase MA, Wheeler DS, Lierl KM, et al. Hsp72 induces inflammation and regulates cytokine production in airway epithelium through a TLR4- and NF-κB-Dependent mechanism. J Immunol. 2007;179(9):6318–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ganter MT, Ware LB, Howard M, et al. Extracellular heat shock protein 72 is a marker of the stress protein response in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2006;291(3):L354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Krzyzaniak M, Cheadle G, Peterson C, et al. Burn-induced acute lung injury requires a functional Toll-like receptor 4. Shock. 2011;36(1):24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Khair OA, Davies RJ, Devalia JL. Bacterial-induced release of inflammatory mediators by bronchial epithelial cells. Eur Respir J. 1996;9(9):1913–1922. [DOI] [PubMed] [Google Scholar]
- [37].Smart SJ, Casale TB. Pulmonary epithelial cells facilitate TNF-alpha-induced neutrophil chemotaxis. A role for cytokine networking. J Immunol. 1994;152(8):4087–4094. [PubMed] [Google Scholar]
- [38].Thorley AJ, Ford PA, Giembycz MA, et al. Differential regulation of cytokine release and leukocyte migration by lipopolysaccharide-stimulated primary human lung alveolar type II epithelial cells and macrophages. J Immunol. 2007;178(1):463–473. [DOI] [PubMed] [Google Scholar]
- [39].Kurdowska A, Miller EJ, Noble JM, et al. Anti-IL-8 autoantibodies in alveolar fluid from patients with the adult respiratory distress syndrome. J Immunol. 1996;157(6):2699–2706. [PubMed] [Google Scholar]
- [40].Kunkel SL, Standiford T, Kasahara K, et al. Interleukin-8 (IL-8): the major neutrophil chemotactic factor in the lung. Exp Lung Res. 1991;17(1):17–23. [DOI] [PubMed] [Google Scholar]
- [41].Lin G, Pearson AE, Scamurra RW, et al. Regulation of interleukin-8 expression in porcine alveolar macrophages by bacterial lipopolysaccharide. J Biol Chem. 1994;269(1):77–85. [PubMed] [Google Scholar]
- [42].Nakamura H, Yoshimura K, Jaffe HA, et al. Interleukin-8 gene expression in human bronchial epithelial cells. J Biol Chem. 1991;266(29):19611–19617. [PubMed] [Google Scholar]
- [43].Standiford TJ, Kunkel SL, Basha MA, et al. Interleukin-8 gene expression by a pulmonary epithelial cell line. A model for cytokine networks in the lung. J Clin Invest. 1990;86(6):1945–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Strieter RM, Chensue SW, Basha MA, et al. Human alveolar macrophage gene expression of interleukin-8 by tumor necrosis factor- α , lipopolysaccharide, and interleukin-1 β. Am J Respir Cell Mol Biol. 1990;2(4):321–326. [DOI] [PubMed] [Google Scholar]
- [45].De Luca D, Minucci A, Cogo P, et al. Secretory phospholipase a pathway during pediatric acute respiratory distress syndrome: a preliminary study. Pediatr Crit Care Med. 2011;12(1):e20–24. [DOI] [PubMed] [Google Scholar]
- [46].Donnelly SC, Kunkel RM, Strieter SL, et al. Interleukin-8 and development of adult respiratory distress syndrome in at-risk patient groups. Lancet. 1993;341(8846):643–647. [DOI] [PubMed] [Google Scholar]
- [47].Suzuki K, Miyasaka H, Ota H, et al. Purification and partial primary sequence of a chemotactic protein for polymorphonuclear leukocytes derived from human lung giant cell carcinoma LU65C cells. J Exp Med. 1989;169(6):1895–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Frevert CW, Goodman RB, Kinsella MG, et al. Tissue-specific mechanisms control the retention of IL-8 in lungs and skin. J Immunol. 2002;168(7):3550–3556. [DOI] [PubMed] [Google Scholar]
- [49].Frevert CW, Kinsella MG, Vathanaprida C, et al. Binding of interleukin-8 to heparan sulfate and chondroitin sulfate in lung tissue. Am J Respir Cell Mol Biol. 2003;28(4):464–472. [DOI] [PubMed] [Google Scholar]
- [50].Fahy RJ, Lichtenberger F, McKeegan CB, et al. The acute respiratory distress syndrome: a role for transforming growth factor-beta 1. Am J Respir Cell Mol Biol. 2003;28(4):499–503. [DOI] [PubMed] [Google Scholar]
- [51].Takatsuka H, Takemoto Y, Mori A, et al. Common features in the onset of ARDS after administration of granulocyte colony-stimulating factor. Chest. 2002;121(5):1716–1720. [DOI] [PubMed] [Google Scholar]
- [52].Tanaka S, Nishiumi S, Nishida M, et al. Vitamin K3 attenuates lipopolysaccharide-induced acute lung injury through inhibition of nuclear factor-κB activation. Clin Exp Immunol. 2010;160(2):283–292. DOI: 10.1111/j.1365-2249.2009.04083.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].von Bismarck P, Klemm K, Garcia Wistadt CF, et al. Selective NF-κB inhibition, but not dexamethasone, decreases acute lung injury in a newborn piglet airway inflammation model. Pulm Pharmacol Ther. 2009;22(4):297–304. [DOI] [PubMed] [Google Scholar]
- [54].Wu CL, Lin LY, Yang JS, et al. Attenuation of lipopolysaccharide-induced acute lung injury by treatment with IL-10. Respirology. 2009;14(4):511–521. [DOI] [PubMed] [Google Scholar]
- [55].Van den Steen PE, Geurts N, Deroost K, et al. Immunopathology and dexamethasone therapy in a new model for malaria-associated acute respiratory distress syndrome. Am J Respir Crit Care Med. 2010;181(9):957–968. [DOI] [PubMed] [Google Scholar]
- [56].Zhang Y, Sun H, Fan L, et al. Acute respiratory distress syndrome induced by a swine 2009 H1N1 variant in mice. PLoS ONE. 2012;7:e29347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Daniels CE, Wilkes MC, Edens M, et al. Imatinib mesylate inhibits the profibrogenic activity of TGF-β and prevents bleomycin-mediated lung fibrosis. J Clin Invest. 2004;114(9):1308–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Border WA, Noble NA, Noble NA. Transforming growth factor β in tissue fibrosis. N Engl J Med. 1994;331(19):1286–1292. [DOI] [PubMed] [Google Scholar]
- [59].Kitamura H, Cambier S, Somanath S, et al. Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin αvβ8–mediated activation of TGF-β. J Clin Invest. 2011;121(7):2863–2875. DOI: 10.1172/jci45589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yang HZ, Wang JP, Mi S, et al. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am J Pathol. 2012;180(1):275–292. DOI: 10.1016/j.ajpath.2011.09.019 [DOI] [PubMed] [Google Scholar]
- [61].Hagiwara S, Iwasaka H, Matsumoto S, et al. Association between heat stress protein 70 induction and decreased pulmonary fibrosis in an animal model of acute lung injury. Lung. 2007;185(5):287–293. [DOI] [PubMed] [Google Scholar]
- [62].Hilberath JN, Carlo T, Pfeffer MA, et al. Resolution of Toll-like receptor 4-mediated acute lung injury is linked to eicosanoids and suppressor of cytokine signaling 3. Faseb J. 2011;25(6):1827–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sakashita A, Nishimura Y, Nishiuma T, et al. Neutrophil elastase inhibitor (sivelestat) attenuates subsequent ventilator-induced lung injury in mice. Eur J Pharmacol. 2007;571(1):62–71. [DOI] [PubMed] [Google Scholar]
- [64].Herold S, Becker C, Ridge KM, et al. Influenza virus-induced lung injury: pathogenesis and implications for treatment. Eur Respir J. 2015;45(5):1463–1478. [DOI] [PubMed] [Google Scholar]
- [65].Luyt C, Combes A, Trouillet JL, et al. Virus-induced acute respiratory distress syndrome: epidemiology, management and outcome. La Presse Médicale. 2011;40(12):e561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wu Z, Zhang R, Liu D, et al. Acute respiratory distress syndrome caused by human adenovirus in adults: a prospective observational study in Guangdong, China. Front Med (Lausanne). 2021;8:791163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Aslan A, Aslan C, Zolbanin NM, et al. Acute respiratory distress syndrome in COVID-19: possible mechanisms and therapeutic management. Pneumonia. 2021;13(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Quan C, Li C, Ma H, et al. Immunopathogenesis of coronavirus-induced Acute Respiratory Distress Syndrome (ARDS): potential infection-associated hemophagocytic lymphohistiocytosis. Clinical Microbiology Reviews. 2020;34(1). DOI: 10.1128/cmr.00074-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Selvaraj V, Finn A, Lal A, et al. Baricitinib in hospitalised patients with COVID-19: a meta-analysis of randomised controlled trials. EClinicalMedicine. 2022;49:101489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Spinelli FR, Conti F, Gadina M. HiJAKing SARS-CoV-2? The potential role of JAK inhibitors in the management of COVID-19. Sci Immunol. 2020;5(47). DOI: 10.1126/sciimmunol.abc5367 [DOI] [PubMed] [Google Scholar]
- [71].Wang CJ, Truong AK. COVID-19 infection on IL-23 inhibition. Dermatologic Therapy. 2020;33(6):e13893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zou Y, Meng Z. Literature overview of the IL-17 inhibition from psoriasis to COVID-19. J Inflamm Res. 2021;14:5611–5618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Naing A, Wong DJ, Infante JR, et al. Pegilodecakin combined with pembrolizumab or nivolumab for patients with advanced solid tumours (IVY): a multicentre, multicohort, open-label, phase 1b trial. Lancet Oncol. 2019;20(11):1544–1555. DOI: 10.1016/s1470-2045(19)30514-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Rojas JM, Avia M, Martin V, et al. IL-10: a multifunctional cytokine in viral infections. J Immunol Res. 2017;2017:6104054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Cinatl J, Morgenstern B, Bauer G, et al. Treatment of SARS with human interferons. Lancet. 2003;362(9380):293–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Dandekar S, Perlman AA. Immunopathogenesis of coronavirus infections: implications for SARS. Nat Rev Immunol. 2005;5(12):917–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Cush SS, Reynoso GV, Kamenyeva O, et al. Locally produced IL-10 limits cutaneous vaccinia virus spread. PLOS Pathog. 2016;12(3):e1005493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jiang L, Yao S, Huang S, et al. Type I IFN signaling facilitates the development of IL-10-producing effector CD8 + T cells during murine influenza virus infection. Eur J Immunol. 2016;46(12):2778–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Loebbermann J, Schnoeller C, Thornton H, et al. IL-10 regulates viral lung immunopathology during acute respiratory syncytial virus infection in mice. PLoS ONE. 2012;7(2):e32371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Naicker DD, Werner L, Kormuth E, et al. Interleukin-10 promoter polymorphisms influence HIV-1 susceptibility and primary HIV-1 pathogenesis. J Infect Dis. 2009;200(3):448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Savarin C, Bergmann CC. Fine tuning the cytokine storm by IFN and IL-10 following neurotropic coronavirus encephalomyelitis. Front Immunol. 2018;9:3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Sun J, Madan R, Karp CL, et al. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med. 2009;15(3):277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Sun K, Torres L, Metzger DW. A detrimental effect of interleukin-10 on protective pulmonary humoral immunity during primary influenza a virus infection. J Virol. 2010;84(10):5007–5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Swarup V, Ghosh J, Duseja R, et al. Japanese encephalitis virus infection decrease endogenous IL-10 production: correlation with microglial activation and neuronal death. Neurosci Lett. 2007;420(2):144–149. [DOI] [PubMed] [Google Scholar]
- [85].Tian Y, Seumois G, De-Oliveira-Pinto LM, et al. Molecular signatures of dengue virus-specific IL-10/ifn-γ co-producing CD4 T cells and their association with dengue disease. Cell Rep. 2019;29(13):4482–4495 e4484. DOI: 10.1016/j.celrep.2019.11.098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Tumpey TM, Elner VM, Chen SH, et al. Interleukin-10 treatment can suppress stromal keratitis induced by herpes simplex virus type 1. J Immunol. 1994;153(5):2258–2265. [PubMed] [Google Scholar]
- [87].Wang Y, Rice AP. Interleukin-10 inhibits HIV-1 LTR-directed gene expression in human macrophages through the induction of cyclin T1 proteolysis. Virology. 2006;352(2):485–492. [DOI] [PubMed] [Google Scholar]
- [88].Perlman S, Zhao J. Roles of regulatory T cells and IL-10 in virus-induced demyelination. J Neuroimmunol. 2017;308:6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Puntambekar SS, Hinton DR, Yin X, et al. Interleukin-10 is a critical regulator of white matter lesion containment following viral induced demyelination. Glia. 2015;63(11):2106–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Trandem K, Jin Q, Weiss KA, et al. Virally expressed interleukin-10 ameliorates acute encephalomyelitis and chronic demyelination in coronavirus-infected mice. J Virol. 2011;85(14):6822–6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Trandem K, Zhao J, Fleming E, et al. Highly activated cytotoxic CD8 T cells express protective IL-10 at the peak of coronavirus-induced encephalitis. J Immunol. 2011;186(6):3642–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Zhao J, Zhao J, Fett C, et al. IFN-γ– and IL-10–expressing virus epitope-specific Foxp3+ T reg cells in the central nervous system during encephalomyelitis. J Exp Med. 2011;208(8):1571–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Liu Y, de Waal Malefyt R, Briere F, et al. The EBV IL-10 homologue is a selective agonist with impaired binding to the IL-10 receptor. J Immunol. 1997;158(2):604–613. [PubMed] [Google Scholar]
- [94].Ouyang P, Rakus K, van Beurden SJ, et al. IL-10 encoded by viruses: a remarkable example of independent acquisition of a cellular gene by viruses and its subsequent evolution in the viral genome. J Gen Virol. 2014;95(2):245–262. [DOI] [PubMed] [Google Scholar]
- [95].Avdic S, McSharry BP, Slobedman B. Modulation of dendritic cell functions by viral IL-10 encoded by human cytomegalovirus. Front Microbiol. 2014;5:337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kotenko SV, Saccani S, Izotova LS, et al. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proc Natl Acad Sci U S A. 2000;97(4):1695–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Slobedman B, Barry PA, Spencer JV, et al. Virus-encoded homologs of cellular interleukin-10 and their control of host immune function. J Virol. 2009;83(19):9618–9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Spencer JV, Lockridge KM, Barry PA, et al. Potent immunosuppressive activities of cytomegalovirus-encoded interleukin-10. J Virol. 2002;76(3):1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Chang WL, Barry PA, Szubin R, et al. Human cytomegalovirus suppresses type I interferon secretion by plasmacytoid dendritic cells through its interleukin 10 homolog. Virology. 2009;390(2):330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Chang WL, Baumgarth N, Yu D, et al. Human cytomegalovirus-encoded interleukin-10 homolog inhibits maturation of dendritic cells and alters their functionality. J Virol. 2004;78(16):8720–8731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Mocellin S, Panelli MC, Wang E, et al. The dual role of IL-10. Trends Immunol. 2003;24(1):36–43. [DOI] [PubMed] [Google Scholar]
- [102].Zheng LM, Ojcius DM, Garaud F, et al. Interleukin-10 inhibits tumor metastasis through an NK cell-dependent mechanism. J Exp Med. 1996;184(2):579–584. DOI: 10.1084/jem.184.2.579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Malisan F, Brière F, Bridon JM, et al. Interleukin-10 induces immunoglobulin G isotype switch recombination in human CD40-activated naive B lymphocytes. J Exp Med. 1996;183(3):937–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Fujii S, Shimizu K, Shimizu T, et al. Interleukin-10 promotes the maintenance of antitumor CD8(+) T-cell effector function in situ. Blood. 2001;98(7):2143–2151. [DOI] [PubMed] [Google Scholar]
- [105].Hu WC. A framework of all discovered immunological pathways and their roles for four specific types of pathogens and hypersensitivities. Front Immunol. 2020;11:1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Blackburn SD, Wherry EJ. IL-10, T cell exhaustion and viral persistence. Trends Microbiol. 2007;15(4):143–146. [DOI] [PubMed] [Google Scholar]
- [107].Brooks DG, Trifilo MJ, Edelmann KH, et al. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12(11):1301–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Tinoco R, Alcalde V, Yang Y, et al. Cell-intrinsic transforming growth factor-β signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity. 2009;31(1):145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Murray PJ. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci U S A. 2005;102(24):8686–8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Hutchins AP, Diez D, Miranda-Saavedra D. The IL-10/stat3-mediated anti-inflammatory response: recent developments and future challenges. Brief Funct Genomics. 2013;12(6):489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Kessler B, Rinchai D, Kewcharoenwong C, et al. Interleukin 10 inhibits pro-inflammatory cytokine responses and killing of Burkholderia pseudomallei. Sci Rep. 2017;7(1):42791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Krause P, Morris V, Greenbaum JA, et al. IL-10-producing intestinal macrophages prevent excessive antibacterial innate immunity by limiting IL-23 synthesis. Nat Commun. 2015;6(1):7055. DOI: 10.1038/ncomms8055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Shouval DS, Biswas A, Goettel JA, et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. 2014;40(5):706–719. DOI: 10.1016/j.immuni.2014.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Dang PM, Elbim C, Marie JC, et al. Anti-inflammatory effect of interleukin-10 on human neutrophil respiratory burst involves inhibition of GM-CSF-induced p47phox phosphorylation through a decrease in ERK1/2 activity. Faseb J. 2006;20(9):1504–1506. [DOI] [PubMed] [Google Scholar]
- [115].Laichalk LL, Danforth JM, Standiford TJ. Interleukin-10 inhibits neutrophil phagocytic and bactericidal activity. FEMS Immunol Med Microbiol. 1996;15(4):181–187. [DOI] [PubMed] [Google Scholar]
- [116].Gu Y, Yang J, Ouyang X, et al. Interleukin 10 suppresses Th17 cytokines secreted by macrophages and T cells. Eur J Immunol. 2008;38(7):1807–1813. DOI: 10.1002/eji.200838331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Guo B. IL-10 modulates TH17 pathogenicity during autoimmune diseases. J Clin Cell Immunol. 2016;7(02). DOI: 10.4172/2155-9899.1000400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Kulcsar KA, Baxter VK, Greene IP, et al. Interleukin 10 modulation of pathogenic Th17 cells during fatal alphavirus encephalomyelitis. Proc Natl Acad Sci U S A. 2014;111(45):16053–16058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Li B, Gurung P, Malireddi RK, et al. IL-10 engages macrophages to shift Th17 cytokine dependency and pathogenicity during T-cell-mediated colitis. Nat Commun. 2015;6(1):6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Liu B, Tonkonogy SL, Sartor RB. Antigen-presenting cell production of IL-10 inhibits T-helper 1 and 17 cell responses and suppresses colitis in mice. Gastroenterology. 2011;141(2):653–662. 10.1053/j.gastro.2011.04.053. 662 e651-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Xu S, Xu M, Li GG, et al. Early recruitment of IL-10-producing B cells into alveoli improved the resolution of acute lung injury. Cell Physiol Biochem. 2016;38(5):1752–1760. [DOI] [PubMed] [Google Scholar]
- [122].Kapur R, Kim M, Rebetz J, et al. Low levels of interleukin-10 in patients with transfusion-related acute lung injury. Ann Transl Med. 2017;5(16):339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Kapur R, Kim M, Aslam R, et al. T regulatory cells and dendritic cells protect against transfusion-related acute lung injury via IL-10. Blood. 2017;129(18):2557–2569. DOI: 10.1182/blood-2016-12-758185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Garantziotis S, Brass DM, Savov J, et al. Leukocyte-derived IL-10 reduces subepithelial fibrosis associated with chronically inhaled endotoxin. Am J Respir Cell Mol Biol. 2006;35(6):662–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Shi J, Li J, Guan H, et al. Anti-fibrotic actions of interleukin-10 against hypertrophic scarring by activation of PI3K/AKT and STAT3 signaling pathways in scar-forming fibroblasts. PLoS ONE. 2014;9(5):e98228. DOI: 10.1371/journal.pone.0098228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Ito T, Okada T, Miyashita H, et al. Interleukin-10 expression mediated by an adeno-associated virus vector prevents monocrotaline-induced pulmonary arterial hypertension in rats. Circ Res. 2007;101(7):734–741. DOI: 10.1161/CIRCRESAHA.107.153023 [DOI] [PubMed] [Google Scholar]
- [127].Kurosaki F, Uchibori R, Sehara Y, et al. AAV6-mediated IL-10 expression in the lung ameliorates bleomycin-induced pulmonary fibrosis in mice. Hum Gene Ther. 2018;29(11):1242–1251. [DOI] [PubMed] [Google Scholar]
- [128].Nakagome K, Dohi M, Okunishi K, et al. In vivo IL-10 gene delivery attenuates bleomycin induced pulmonary fibrosis by inhibiting the production and activation of TGF- in the lung. Thorax. 2006;61(10):886–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Spight D, Zhao B, Haas M, et al. Immunoregulatory effects of regulated, lung-targeted expression of IL-10 in vivo. Am J Physiol Lung Cell Mol Physiol. 2005;288(2):L251–265. [DOI] [PubMed] [Google Scholar]
- [130].Asadullah K, Sterry W, Volk HD. Interleukin-10 therapy—Review of a new approach. Pharmacol Rev. 2003;55(2):241–269. [DOI] [PubMed] [Google Scholar]
- [131].Chernoff AE, Granowitz EV, Shapiro L, et al. A randomized, controlled trial of IL-10 in humans. Inhibition of inflammatory cytokine production and immune responses. J Immunol. 1995;154(10):5492–5499. [PubMed] [Google Scholar]
- [132].Huhn RD, Radwanski E, O’connell SM, et al. Pharmacokinetics and immunomodulatory properties of intravenously administered recombinant human interleukin-10 in healthy volunteers. Blood. 1996;87(2):699–705. [PubMed] [Google Scholar]
- [133].Mosser DM, Zhang X. Interleukin-10: new perspectives on an old cytokine. Immunol Rev. 2008;226(1):205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Mattos A, de Jager-Krikken A, de Haan M, et al. Pegylation of interleukin-10 improves the pharmacokinetic profile and enhances the antifibrotic effectivity in CCl(4)-induced fibrogenesis in mice. J Control Release. 2012;162(1):84–91. [DOI] [PubMed] [Google Scholar]
- [135].Barry JC, Shakibakho S, Durrer C, et al. Hyporesponsiveness to the anti-inflammatory action of interleukin-10 in type 2 diabetes. Sci Rep. 2016;6(1):21244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Yang L, Xie X, Tu Z, et al. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct Target Ther. 2021;6(1):255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Zhao Y, Qin L, Zhang P, et al. Longitudinal COVID-19 profiling associates IL-1RA and IL-10 with disease severity and RANTES with mild disease. JCI Insight. 2020;5(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Han H, Ma Q, Li C, et al. Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerg Microbes Infect. 2020;9(1):1123–1130. DOI: 10.1080/22221751.2020.1770129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Lu L, Zhang H, Dauphars DJ, et al. A potential role of interleukin 10 in COVID-19 pathogenesis. Trends Immunol. 2021;42(1):3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Cheng L, Zhu Z, Wang C, et al. COVID-19 induces lower levels of IL-8, IL-10, and MCP-1 than other acute CRS-inducing diseases. Proc Natl Acad Sci U S A. 2021;118(21). DOI: 10.1073/pnas.2102960118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Merza MY, Hwaiz RA, Hamad BK, et al. Analysis of cytokines in SARS-CoV-2 or COVID-19 patients in Erbil city, Kurdistan region of Iraq. PLoS ONE. 2021;16(4):e0250330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Utrero-Rico A, Gonzalez-Cuadrado C, Chivite-Lacaba M, et al. Alterations in circulating monocytes predict COVID-19 severity and include chromatin modifications still detectable six months after recovery. Biomedicines. 2021;9(9):1253. DOI: 10.3390/biomedicines9091253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].McElvaney OJ, McEvoy NL, McElvaney OF, et al. Characterization of the inflammatory response to severe COVID-19 illness. Am J Respir Crit Care Med. 2020;202(6):812–821. DOI: 10.1164/rccm.202005-1583OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Sun J, Su J, Xie Y, et al. Plasma IL-6/IL-10 ratio and IL-8, LDH, and HBDH level predict the severity and the risk of death in AIDS patients with pneumocystis pneumonia. J Immunol Res. 2016;2016:1583951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Taniguchi T, Koido Y, Aiboshi J, et al. Change in the ratio of interleukin-6 to interleukin-10 predicts a poor outcome in patients with systemic inflammatory response syndrome. Crit Care Med. 1999;27(7):1262–1264. [DOI] [PubMed] [Google Scholar]
- [146].McElvaney OJ, Hobbs BD, Qiao D, et al. A linear prognostic score based on the ratio of interleukin-6 to interleukin-10 predicts outcomes in COVID-19. EBioMedicine. 2020;61:103026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Malin JJ, Spinner CD, Janssens U, et al. Key summary of German national treatment guidance for hospitalized COVID-19 patients: key pharmacologic recommendations from a national German living guideline using an evidence to decision framework (last updated 17.05.2021). Infection. 2022;50(1):93–106. DOI: 10.1007/s15010-021-01645-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Sorgeloos F, Kreit M, Hermant P, et al. Antiviral type I and type III interferon responses in the central nervous system. Viruses. 2013;5(3):834–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Felgenhauer U, Schoen A, Gad HH, et al. Inhibition of SARS–CoV-2 by type I and type III interferons. J Biol Chem. 2020;295(41):13958–13964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Zhang H, Madi A, Yosef N, et al. An IL-27-driven transcriptional network identifies regulators of IL-10 expression across T helper cell subsets. Cell Rep. 2020;33(8):108433. DOI: 10.1016/j.celrep.2020.108433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Duan Y, Jia Y, Wang T, et al. Potent therapeutic target of inflammation, virus and tumor: focus on interleukin-27. Int Immunopharmacol. 2015;26(1):139–146. [DOI] [PubMed] [Google Scholar]
- [152].Zhu H, Lou C, Liu P. Interleukin-27 ameliorates coxsackievirus-B3-induced viral myocarditis by inhibiting Th17 cells. Virol J. 2015;12(1):189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable-no new data generated