

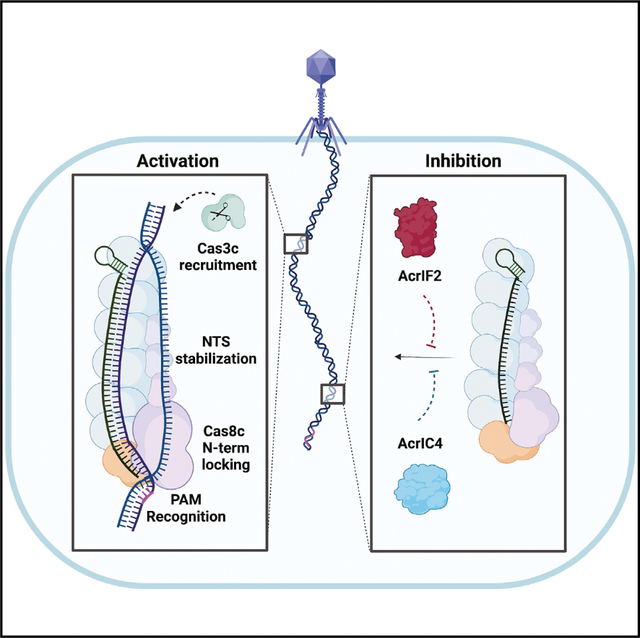
SUMMARY
Type I CRISPR-Cas systems employ multi-subunit Cascade effector complexes to target foreign nucleic acids for destruction. Here, we present structures of D. vulgaris type I-C Cascade at various stages of double-stranded (ds)DNA target capture, revealing mechanisms that underpin PAM recognition and Cascade allosteric activation. We uncover an interesting mechanism of non-target strand (NTS) DNA stabilization via stacking interactions with the “belly” subunits, securing the NTS in place. This “molecular seatbelt” mechanism facilitates efficient R-loop formation and prevents dsDNA reannealing. Additionally, we provide structural insights into how two anti-CRISPR (Acr) proteins utilize distinct strategies to achieve a shared mechanism of type I-C Cascade inhibition by blocking PAM scanning. These observations form a structural basis for directional R-loop formation and reveal how different Acr proteins have converged upon common molecular mechanisms to efficiently shut down CRISPR immunity.
In brief
O’Brien et al. show how the type I-C CRISPR-Cascade complex selectively recognizes and unwinds DNA targets, and how the evicted non-target strand is stabilized via aromatic clamp residues. Furthermore, the mechanisms by which two distinct anti-CRISPR proteins target this process is revealed.
Graphical Abstract
INTRODUCTION
CRISPR and CRISPR-associated (Cas) genes together form a prokaryotic adaptive immune response against foreign genetic elements, such as plasmids and phages.1,2 CRISPR-Cas immunity is established through three major stages: adaptation, maturation, and interference.3,4 Immunological memory is first acquired through the incorporation of short genetic fragments from invading phage or plasmids into the bacterial genome at CRISPR loci.3,4 These short fragments are then transcribed into pre-CRISPR RNAs (crRNAs) and processed into mature crRNA transcripts.5 Cas proteins assemble around the mature crRNA to form either multi-subunit or single subunit crRNA-guided surveillance complexes, utilizing the crRNA as a guide for target recognition.6 In double-stranded (ds)DNA-targeting systems, once a target-flanking protospacer adjacent motif (PAM) is identified and R-loop formation is complete, associated nucleases and/or nuclease domains degrade the designated DNA.7 However, phages have evolved mechanisms to evade CRISPR-Cas surveillance by counter attacking with small inhibitory proteins known as anti-CRISPRs (Acrs).8,9 The interplay between Acrs and CRISPR-Cas systems is one component of the molecular arms race between phage and bacteria.10
CRISPR-Cas systems are highly diverse and can be divided into two major classes.11,12 Class I uses multi-subunit crRNA-guided surveillance complexes, whereas class II relies on a single effector nuclease.13,14 Although the simplicity of class II nucleases enables their use as tools for genome engineering,15 these systems are present in less than 10% of bacteria and archaea,12 whereas the multi-subunit class I systems are far more abundant. This class can be further divided into types I, III, and IV based on effector complex subunit composition, with type I being the most prevalent.12 Type I systems are characterized by the presence of a trans-acting helicase-nuclease, Cas3, and are divided into multiple subtypes (A–G).12,14
The type I-C CRISPR-Cas system contains only three Cas genes in its operon—cas5c, cas7c, and cas8c12—and is therefore considered a minimal Cascade.14 The type I-C Cascade uses Cas5c to process the crRNA instead of the typical RNA endonuclease Cas612,16–18 and does not encode a separate open reading frame for the small subunit (Cas11c) within its operon.12 Recent studies revealed that the D. vulgaris large subunit Cas8c includes an internal ribosome binding site at the C terminus, which encodes a separate small subunit Cas11c.19 We have previously confirmed that this non-canonical Cas11c is identical to the C-terminal domain of Cas8c in both sequence and structure, adopting a helical bundle topology typical of other small subunits.18 The discovery and inclusion of the Cas11c small subunit has been a crucial component for effectively utilizing type I-C systems for genome engineering applications.20
Previous structures and biochemical analysis of the type I-C Cascade have provided insights into complex assembly.17,18 However, as the type I-C CRISPR system emerges as a popular tool for genome engineering,20,21 it will likely be beneficial to understand the mechanisms behind PAM recognition, dsDNA unwinding, and R-loop stabilization in order to provide structural blueprints to guide rational Cascade re-engineering. Additionally, modulating type I-C in vivo functionality through the incorporation of Acr inhibitory proteins is also of interest.20,21 The lack of sequence similarity and structural motifs among Acr proteins has hindered their identification, including those targeting the I-C effector complex.9 However, a method for discovering Acrs recently revealed multiple type I-C Acr proteins that inactivated the I-C effector complex in vivo, including AcrIC4.22 Surprisingly, a previously characterized type I-F inhibitory protein, AcrIF2, also demonstrated the in vivo inhibition of the type I-C Cascade.22–24 How AcrIF2 can efficiently inhibit the structurally distinct type I-F and I-C Cascades remains enigmatic.
Here, we use cryoelectron microscopy (cryo-EM) to determine structures of the type I-C Cascade bound to a dsDNA target. This structure reveals dramatic conformational changes that are crucial for R-loop formation and important insights into non-target strand (NTS) stabilization and PAM recognition. Additionally, we determined structures of the type I-C Cascade bound to AcrIF2 and AcrIC4. These structures reveal that AcrIF2 and AcrIC4 use different strategies to inhibit PAM recognition and subsequent dsDNA target binding by type I-C Cascade. Collectively, this study expands our mechanistic understanding of the type I-C effector complex and provides insights into the modes of inhibition that Acrs use to help phage evade type I CRISPR-Cas bacterial immune systems.
RESULTS
DNA binding induces conformational changes in the type I-C Cascade
We purified the D. vulgaris type I-C Cascade as previously described, assembled the complex bound to a 75-bp dsDNA target containing a complimentary protospacer and 5′-TTC PAM (Figures 1A–1C and S1–S4), and determined a 2.9 Å-resolution cryo-EM structure, allowing us to build a complete atomic model. Incidentally, we observed a subset of particles within our cryo-EM dataset that formed a partial R-loop but lacked visible density for the NTS, which resulted in an additional structure at 2.9 Å resolution (Figures 1D and S2–S4). Additionally, we improved our previously determined18 apo-Cascade cryo-EM structure from 3.1 to 2.7 Å, enabling us to unambiguously model several regions of the complex that were poorly resolved in our previous structure (Figures 1E left, S2, and S3). Due to domain flexibility, residues 290–348 within the Cas8c subunit are missing in both the apo and DNA-bound structures. In the dsDNA-bound structure, a combination of density subtraction, focused three-dimensional (3D) classification, and local refinement significantly improved the resolution of the flexible Cas8c N terminus (Cas8c N-term), and AlphaFold2 was used to further assist with modeling of a full R-loop structure (Figures 1C, S2, and S3).25–27
Figure 1. Structural rearrangements in type I-C Cascade required for R-loop formation.
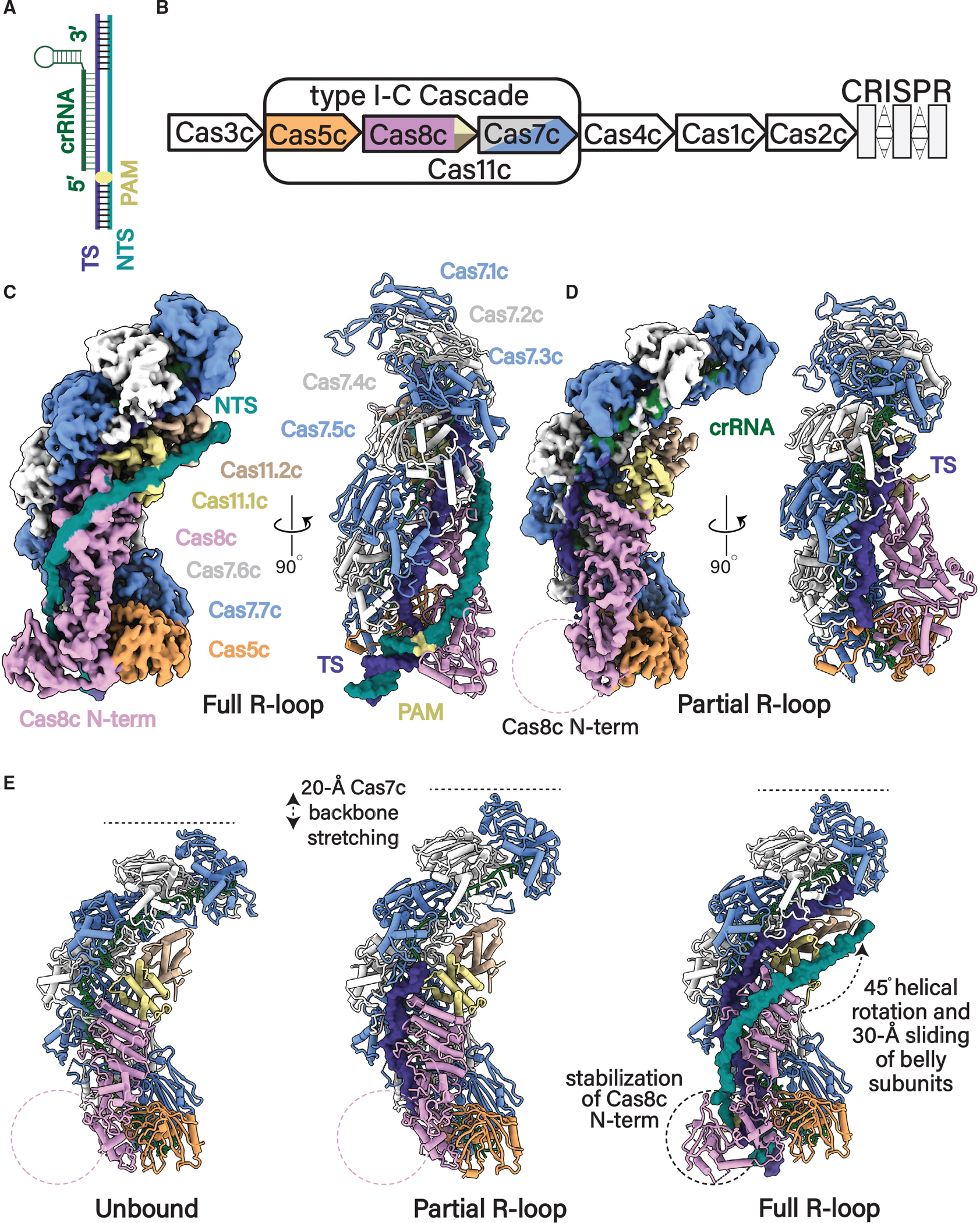
(A) Schematic of R-loop formed by Cascade (only the nucleic acid components are shown). Nucleic acids are colored as follows: TS, dark blue; NTS, cyan; crRNA, green.
(B) Organization of the D. vulgaris type I-C operon. Subunits are colored as follows: Cas7, alternative blue and gray; Cas8c, plum; Cas5c, light orange; Cas11c, yellow and tan.
(C) 2.9 Å cryoelectron microscopy (cryo-EM) structure and atomic model of the type I-C Cascade in full R-loop formation.
(D) 2.8 Å cryoelectron microscopy (cryo-EM) structure and atomic model of the type I-C Cascade in a partial R-loop.
(E) Conformational changes of type I-C Cascade induced upon full and partial R-loop formation when compared with our improved 2.7 Å resolution apo-Cascade cryo-EM model. Subunits and nucleic acid are colored as in (A) and (B).
The overall architecture of type I-C Cascade with or without target DNA resembles a caterpillar, composed of four different Cas proteins with a stoichiometry of Cas7c7Cas5c1Cas8c1Cas11c2 and a single 35-nt crRNA (Figures 1C–1E). The complex first assembles with the crRNA-processing subunit, Cas5c, sitting at the base of the complex and cradling the 5′ end of the crRNA handle. Seven Cas7s stack on top of the Cas5c, oligomerizing along the crRNA in a helical filament until they cap the 3′ end of the crRNA. Finally, the large Cas8c subunit and two non-canonical Cas11 small subunits nestle inside the belly of the complex, completing the arrangement of subunits in this effector.17,18 In both the partial and full R-loop structures, the target strand (TS) is wedged inside the complex, hybridized to the crRNA (Figures 1C and 1D). In the full R-loop structure, the NTS is guided across the belly of the complex by the Cas8c and Cas11c belly subunits (Figure 1C) (explained in detail later).
Although maintaining the same overall caterpillar architecture, the type I-C Cascade undergoes multiple conformational rearrangements to achieve full R-loop propagation (Figure 1E). The Cas7-crRNA backbone becomes extended in the DNA-bound structures, stretching by ~20 Å to accommodate the base pairing of the crRNA to the TS protospacer (Figure 1E middle; Video S1). Additionally, the Cas8c N-term is stabilized upon dsDNA binding, interacting with the NTS at the base of the complex exclusively when bound to dsDNA (Figures 1C and 1E, right). However, the most dramatic conformational change involves a largely uniform rigid-body rearrangement of the belly subunits. dsDNA binding causes the Cas8c C-terminal domain and the two Cas11c subunits to shift upward by 30 Å and rotate by ~45° (Figure 1E, right; Video S2). Since these conformational changes are exclusive to the dsDNA-bound structure, it is likely that they occur by allosteric signaling within Cascade upon recognition of a suitable PAM and accompany R-loop propagation (Figure 1E).
Cas8c-mediated NTS stabilization
Previous type I Cascade structures captured with complete R-loops had limited resolution of the NTS, preventing direct visualization of how the NTS is stabilized during R-loop propagation.28–31 This mechanism is critical for kinetically partitioning the forward reaction, preventing dsDNA reannealing and favoring R-loop completion.28–31 Since charge-swap mutations introduced at this site resulted in a substantial DNA-binding defect for type I-E and I-F Cascades, it has been proposed that the NTS is stabilized predominantly through non-specific electrostatic interactions with the surface of the small and large subunits.28–30 Our dsDNA-bound Cascade structure exhibited well-resolved cryo-EM density for the displaced NTS, enabling us to confidently model 41 nt of the NTS and provide a structural snapshot of the interactions between the NTS and type I-C Cascade (Figures 2 and S4). As previously hypothesized, positively charged residues (R205, R408, K561, K607, and K608) on the surface of the C terminus of Cas8c participate in non-specific electrostatic interactions with the negatively charged NTS backbone28–30 (Figures 2A–2C) of which K607 and K608 are highly conserved (Figure S5). Because Cas11c is identical in sequence and structure to the C terminus of Cas8c, the analogous positively charged residues in the Cas11 subunits (K73 and K119) interact with the phosphodiester backbone until the NTS is reunited with the TS at the top of the complex (Figure 2B).
Figure 2. Cas8c utilizes aromatic base stacking for NTS clamping.
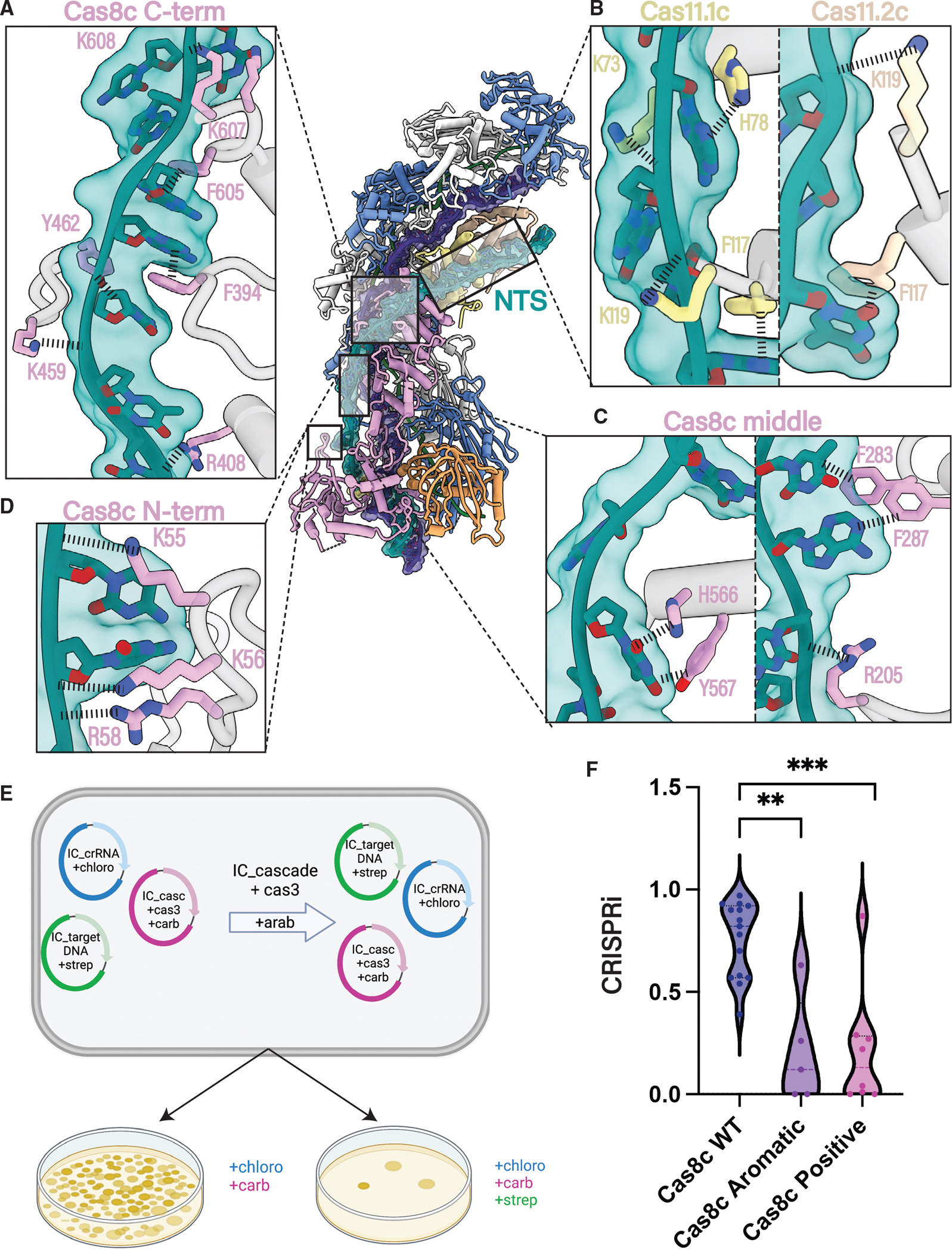
(A–C) Specific residues in Cas8c and Cas11 small subunits are involved in NTS stabilization. Positively charged and aromatic residues across the surface of the Cas8c and Cas11 subunits form non-specific interactions with the NTS backbone and bases, respectively.
(D) Cas8c N-term acts as a dsDNA vice that clamps around the dsDNA helix, facilitating the initial melting of the duplex using highly conserved, positively charged residues to hold the NTS in place and to prevent reannealing with the TS.
(E) Type I-C Cascade-Cas3 in vivo plasmid interference assay design.
(F) Quantification of the in vivo assay demonstrated in (E). Each data point is an independent replicate. All aromatic residues exhibited in (A)–(D) (F283, F287, F394, H566, Y462, Y567, and F605) were mutated to alanine in Cas8c aromatic mutant. All positive residues exhibited in (A)–(D) (K607, K608, R408, R205, K459, R205, R58, K56, and K55) were mutated to alanine in the Cas8c positive mutant. The interference efficiency of mutants was significantly decreased compared with WT. Mutants are statistically significant to WT (**p = 0.006, ***p = 0.0008).
Akin to the type I-F Cascade, the Cas8c N-term acts as a dsDNA “vice” that clamps around the dsDNA helix and triggers the initial melting of the duplex (Figure 2D).23,24,28 Highly conserved, positively charged residues (K55, K56, and R58) within the Cas8c vice make sequence-independent contacts with the negatively charged NTS backbone (Figures 2D and S4–S5). Since the N terminus of Cas8c could not be resolved in the apo or R-loop intermediate structures, these charge-charge contacts between the Cas8c N-term and the NTS likely play a role in stabilizing the NTS during R-loop formation. This is analogous to the stabilization of the Cas8a N-term by PAM recognition in the type I-A Cascade system.32
In addition to electrostatic contacts, we identified multiple interactions between aromatic residues and NTS nucleobases, stabilizing the single-stranded region of the NTS. Aromatic residues on the surface of the Cas8c C terminus (F283, F287, F394, H566, Y462, Y567, and F605) participate in stacking interactions with NTS bases (Figures 2A–2C and S4). Analogous Cas11c residues H78 and F117 are also involved in base stacking toward the PAM-distal end of the NTS (Figure 2B), and residues Y462 and F605 are highly conserved among type I-C Cascades (Figure S5). To test the functional significance of these aromatic and positively charged residues, we developed an in vivo type I-C Cascade-Cas3 interference assay (Figure 2E). Plasmid interference efficiency was compared for wild-type (WT) I-C Cascade-Cas3, type I-C Cascade containing an aromatic mutant cluster in Cas8c (F605A, Y462A, F394A, H566A, Y567A, F287A, and F283A), and a type I-C Cascade containing a positive charge mutant cluster in Cas8c (K607A, K608A, R408A, R205A, K459A, R205A, R58A, K56A, and K55A). Both mutants demonstrated a significant decrease in interference efficiency compared with the WT, likely due to significant destabilization of the NTS hindering optimal recruitment of Cas3c and subsequent dsDNA cleavage (Figure 2F). Further analysis of the NTS path in the type I-E Cascade revealed multiple aromatic residues similarly positioned along the putative NTS path29 (Figure S5), suggesting that this mechanism of NTS stabilization may occur within multiple type I systems.
Cas8c N-term is responsible for PAM recognition
The PAM is a short nucleotide motif on the NTS that is upstream of the corresponding protospacer sequence on the TS (Figure 1A). The recognition of this sequence allows CRISPR effector complexes to distinguish self from non-self DNA and initiate R-loop formation.33 In the type I-C Cascade, the 5′ TTC PAM is recognized from the minor grove by the N-term of the Cas8c large subunit (Figure 3A). The N-term of Cas8c clamps around the dsDNA helix and positions four loops to facilitate PAM recognition and strand separation (Figures 3B and 3C). N72 contacts the PAM, protruding from a glycine loop that wedges between the AT-3:TNT-3 base pairs (Figures 3C, 3D, and S4). N72 is within hydrogen bonding distance to both the TS AT-3 and the NTS TNT-3, creating a dual contact with both strands (Figure 3D). Thus, it is not surprising that N72A is highly conserved across type I-C Cascades and supports the strict tolerance for only pyrimidines at PAM NT−3 position for some type I-C Cascades (Figure S5).34 Although a glycine loop is a PAM-recognition feature present in type I-E Cascades, the use of asparagine for PAM recognition bears more of a resemblance to type I-F Cascade.28–30 Additionally, we observe a highly conserved glutamine wedge that stacks above the PAM and intercalates between the two DNA strands (Figures 3C, 3E, S4, and S5). A glutamine wedge is a common PAM-recognition feature and is found in other class I and class II systems. For instance, the Cas12 complex utilizes a pair of Gln residues that stack over the terminal base pair of the PAM, thereby triggering R-loop formation.29–31,35,36 Q212 hydrogen bonds with GT-1, sterically displacing the first two nucleotides of the protospacer and forcing them to rotate outward (Figure 3E). The final two loops involved in PAM recognition contain three positively charged residues (K92, K94, and R363) that non-specifically stabilize the PAM duplex phosphodiester backbone (Figure 3C). The mutation of either N72 or Q212 residue abrogated dsDNA binding in vitro, confirming their importance (Figures 3F and 3G). Similarly, N72A and Q212A demonstrated a severe interference defect in vivo when compared with WT, where the three positively charged PAM backbone stabilizing residues, K92A/K94A/R363A, only slightly reduced interference (Figure 3H). These results, along with the complimentary binding data, suggest that Q212 and N72 are essential for type I-C Cascade PAM recognition (Figure 3I).
Figure 3. Cas8c N-term has a minimal PAM-recognition requirement.
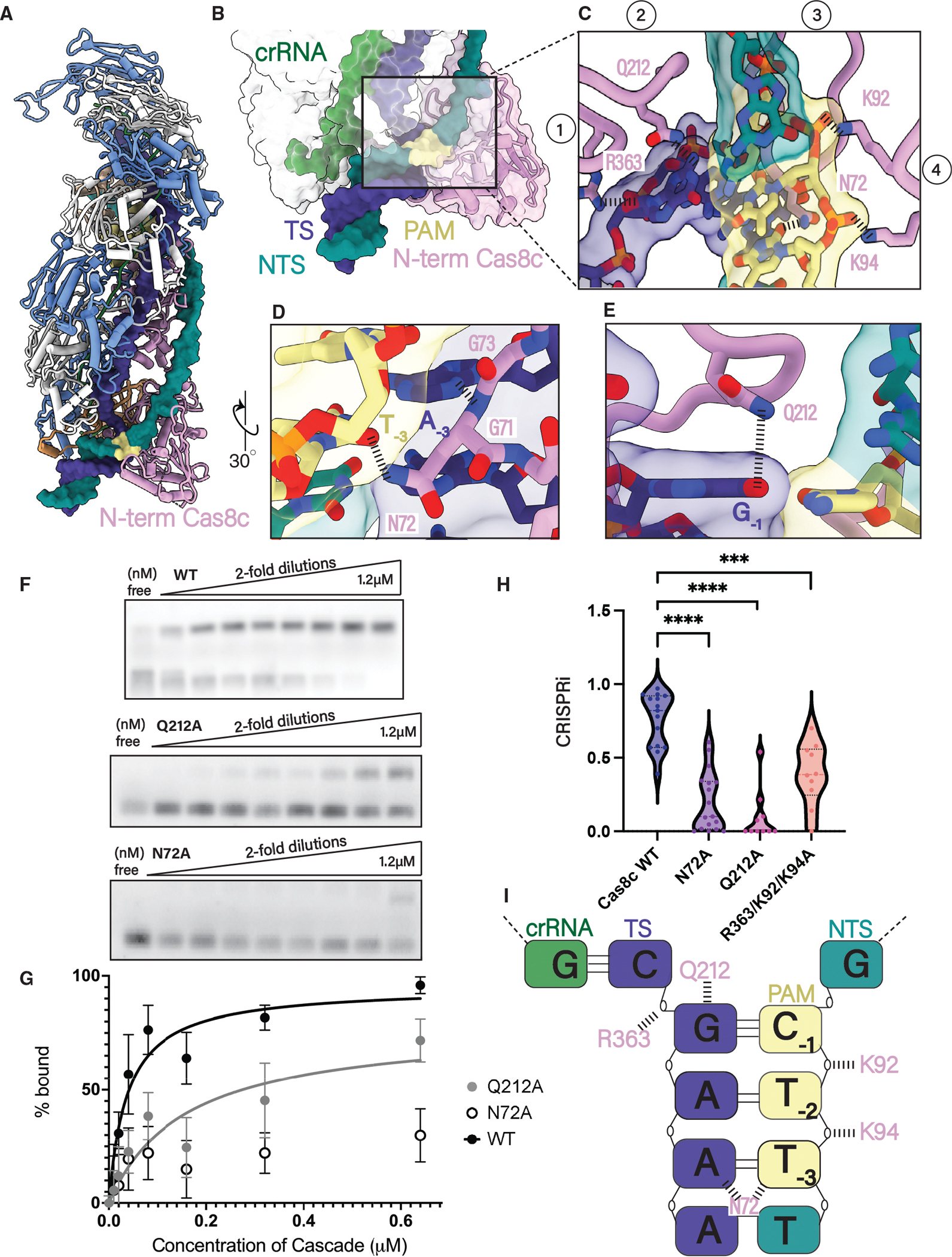
(A) 5′-TTC PAM motif is recognized from the minor grove by the N-term of the Cas8c large subunit.
(B and C) (B) The N-term of Cas8c clamps around the dsDNA helix and (C) positions four loops to facilitate PAM recognition and strand separation.
(D) N72 makes one contact with the PAM, protruding from a glycine loop that wedges into the duplex.
(E) dsDNA splitting is facilitated by a glutamine wedge that stacks above the PAM and intercalates between the two DNA strands.
(F) Electrophoretic mobility shift assays (EMSAs) measuring the binding of increasing concentrations of Cascade to a fluorescently labeled dsDNA target with a 5′-TTC PAM. Experiments were done in triplicate and representative results are shown.
(G) Quantification of EMSA data. Each point is the average of at least three independent replicates. WT Cascade Kd(app) is 37 nM, and Q212A mutant Kd(app) is 155 μM. The binding curve of the N72A mutant could not be fit and a Kd could not be determined.
(H) Quantification of type I-C Cascade in vivo plasmid interference assay with PAM mutants. Each data point is an independent replicate. PAM mutants Q212A and N72A demonstrated the most severe defect in interference efficiency compared with WT and the R363A/K92A/K94A mutant. **** demonstrates mutants are statistically significant to WT (N72A: p < 0.0001, Q212A: p < 0.0001, R363A/K92A/K94A: p = 0.0003).
(I) Schematic of the five residues involved in PAM recognition for type I-C Cascade. Two residues (Q212 and N72) make specific contact with the PAM bases and three residues (K92, K94, and R363) stabilize the phosphate backbone.
AcrIF2 blocks PAM recognition and induces an inactive Cas8c conformation
Phages can utilize Acr proteins to deactivate CRISPR-Cas effectors and escape detection.8,9 AcrIF2 has demonstrated high levels of CRISPR suppression in vivo across multiple type I subtypes.22 To determine the mechanism of AcrIF2 inhibition toward the type I-C Cascade, we determined a 3.0 Å cryo-EM structure of AcrIF2 bound to the type I-C Cascade (Figures 4A and S1–S3). Although the binding of AcrIF2 does not induce any conformational changes within the Cascade structure relative to the apo model, AcrIF2 does interact with and stabilize the Cas8c N-term, which is absent in the apo structure due to high flexibility (Figure 4A). The Cas8c N-term is otherwise only observed in the full R-loop structure, forming a network of contacts with the PAM-adjacent dsDNA (Figures 1C and 4A inset). AcrIF2 is positioned at the hinge-like interface that tethers the Cas8c N-term to the complex, making several electrostatic interactions with the electropositive DNA-binding surface of Cas8c (Figures 4B and 4C). It has been previously shown that AcrIF2 and AcrIF10 both target the PAM site in the type I-F Cascade using slightly different positioning on the Cas8f subunit (Figure S6).24 Compared with cryo-EM structures of the type I-F Cascade bound to AcrIF2 and AcrIF10, AcrIF2 uses the same acidic interface to bind both Cas8f and Cas8c at the same PAM-recognition site23,24 (Figures 4C and S6). However, AcrIF2 is oriented parallel rather than perpendicular to Cas8c (i.e., is rotated by 90°), which provides additional contacts with Cas8c than Cas8f23,24 (Figures 4B and S6). The structural superposition of the type I-C dsDNA-bound model shows severe clashing between the PAM and AcrIF2 (Figure 4D). AcrIF2 is positioned directly at the PAM-recognition site, both sterically blocking PAM binding and interacting with Q212 (Figure 4D). Additionally, AcrIF2 binding induces an extended, inactive formation of the Cas8c N-term (Figures 4D and 4E). Thus, AcrIF2 engages in a two-pronged attack to prevent dsDNA binding of the type I-C Cascade by simultaneously blocking PAM recognition and jamming the vice-like Cas8c N-term in a non-productive conformation (Figures 4D and 4E; Video S3). To support this, we observed efficient inhibition of type I-C CRISPRi by AcrIF2 in vivo (Figure 4F).
Figure 4. AcrIF2 blocks both PAM recognition and domain rearrangements required for R-loop formation.
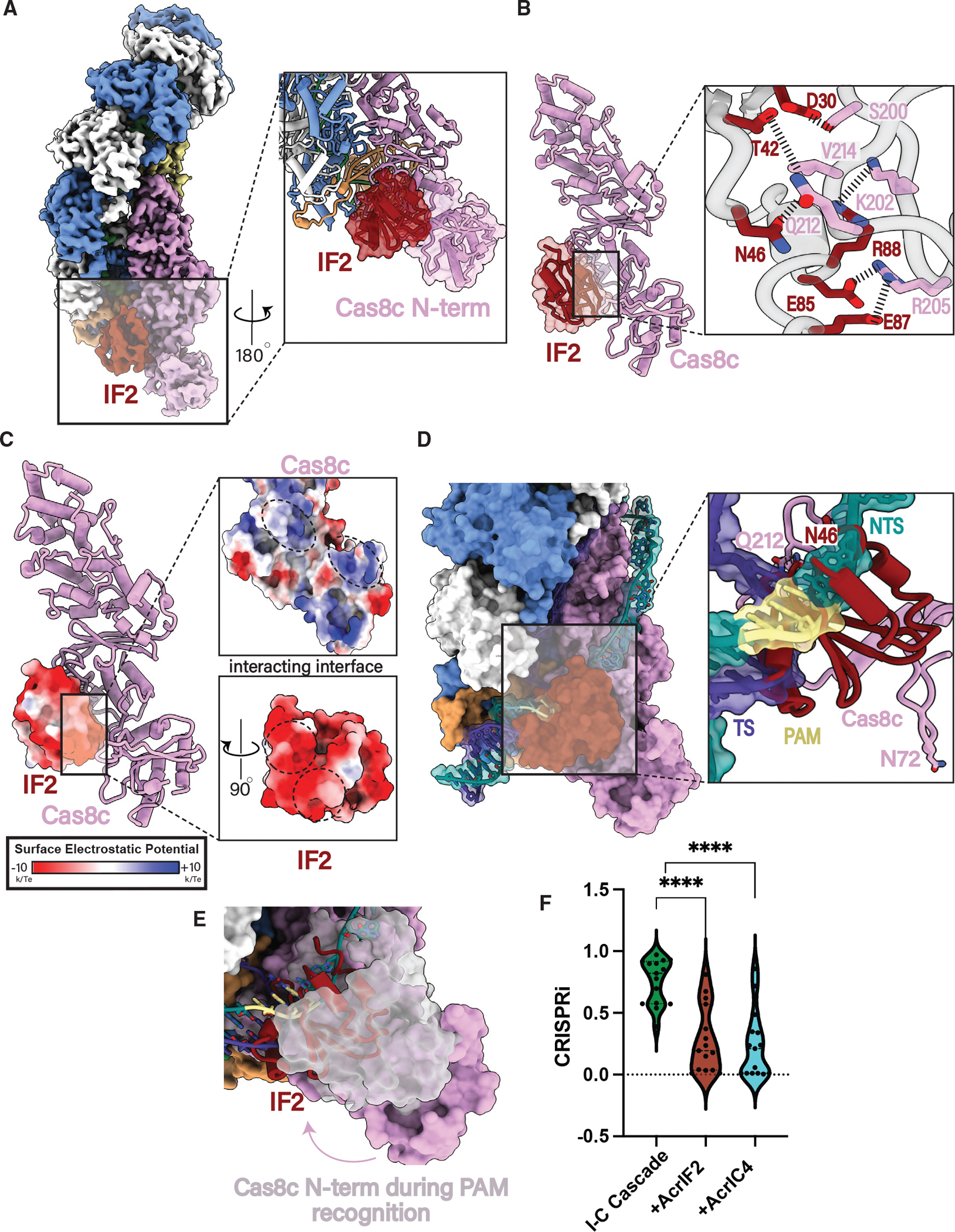
(A) 3.0 Å resolution cryoelectron structure of type I-C Cascade bound to AcrIF2 and atomic model demonstrating the presence of Cas8c N-term (inset).
(B) AcrIF2 exclusively interacts with Cas8c through a large network of non-specific interactions.
(C) The surface of AcrIF2 is negatively charged and sits within two positively charged surfaces between the Cas8c N-term and the rest of the Cas8c subunit.
(D) Structural superposition of the type I-C dsDNA-bound model shows severe clashing between PAM residues and AcrIF2. AcrIF2 additionally hinders the rearrangement of Cas8c N-term containing important residues involved in PAM recognition (N72).
(E) AcrIF2 occludes PAM recognition by Cas8c N-terminal domain. Pink, AcrIF2-bound structure; gray, full R-loop structure.
(F) Type I-C Cascade-Cas3 in vivo plasmid interference assays demonstrate AcrsIF2 and AcrIC4 inhibit DNA degradation. **** demonstrate AcrIF2 and AcrIC4 are statistically significant (AcrIF2: p < 0.0001, AcrC4: p < 0.0001).
AcrIC4 occludes the PAM site yet makes different Cascade contacts from AcrIF2
Unlike AcrIF2, AcrIC4 demonstrated high levels of CRISPR suppression in vivo, exclusively targeting the type I-C subtype.22 To understand the mechanism of this type I-C Cascade-specific Acr, we solved a 3.1 Å cryo-EM structure of the type I-C Cascade bound to AcrIC4 (Figures 5A and S1–S3). AcrIC4 contacts Cas7.6c and Cas8c (Figures 5A and 5B) and uses an extensive negatively charged surface to block PAM recognition, as shown by superposition with the full R-loop structure (Figures 5B–5D). The distinct architecture of type I-C Cascade provides the spatial arrangement of Cas7.6c and Cas8c to provide a binding interface for AcrIC4 that is absent on type I-F Cascade, providing a structural rationale for why (unlike AcrIF2) AcrIC4 is not a dual Cascade inhibitor. We observed similar CRISPR inhibition by AcrIC4 in vivo compared with AcrIF2 (Figure 4F). Although AcrIC4 uses a strategy similar to AcrIF2 to prevent CRISPRi, the Acr proteins do so by targeting slightly different (but partially overlapping) surfaces of the complex (Figure 5E). This indicates that although PAM blocking is an effective Acr tactic, there are many structural solutions to achieve this common goal.
Figure 5. AcrIC4 occludes the PAM site but makes different Cascade contacts than AcrIF2.
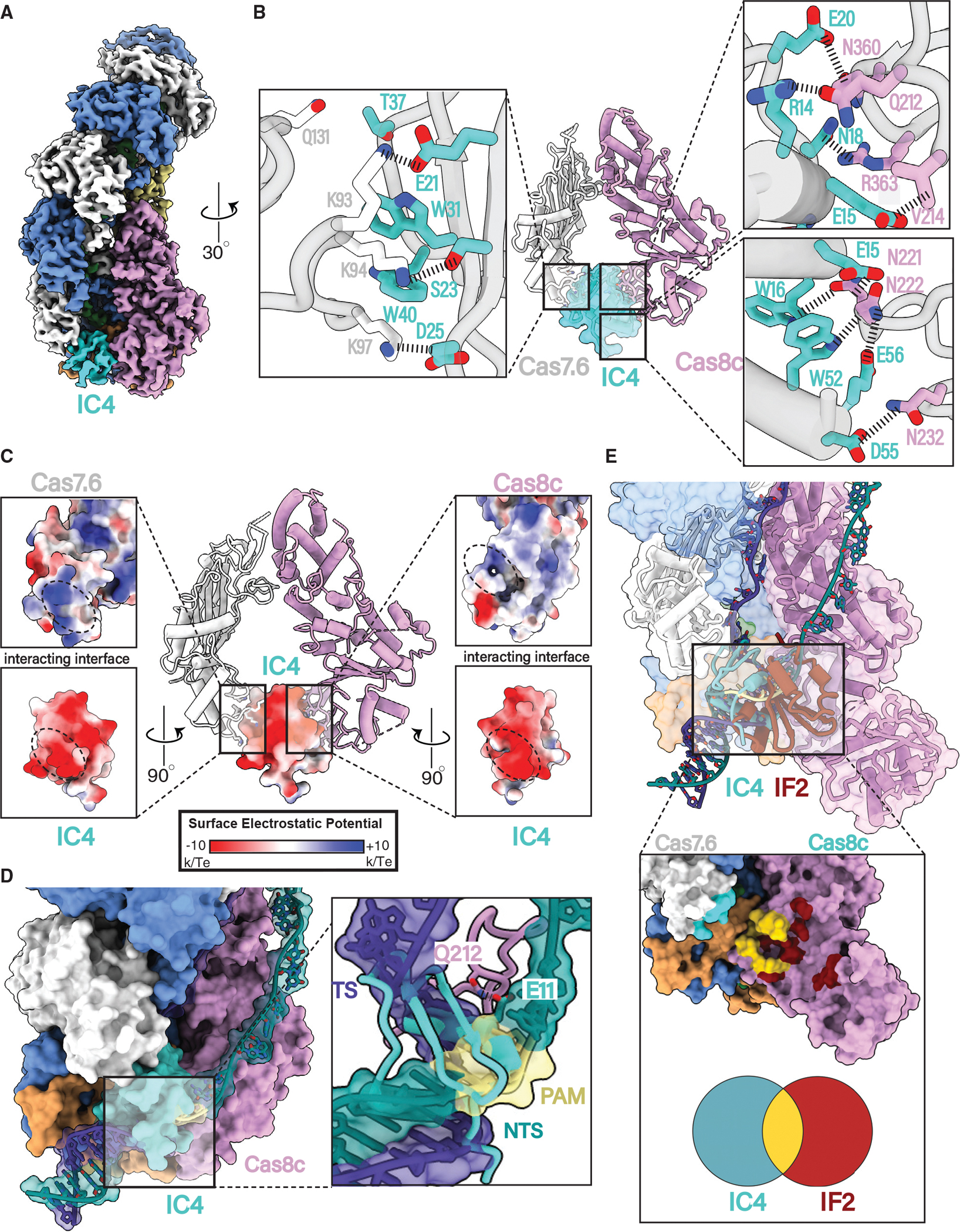
(A) 3.1 Å resolution cryoelectron structure of the type I-C Cascade bound to AcrIC4.
(B) AcrIC4 interacts with both Cas8c and Cas7c through a large network of non-specific interactions.
(C) AcrIC4 is entirely negatively charged and wedged between positively charged Cas8c and Cas7.6 surfaces.
(D) Structural superposition of the type I-C dsDNA-bound model shows severe clashing between PAM residues and AcrIC4.
(E) Overlay of the IF2 and IC4 binding sites demonstrates a partially overlapping interface at the PAM site.
DISCUSSION
Here, we describe the mechanisms by which the type I-C Cascade is activated for dsDNA targeting and how two Acr proteins can deactivate the effector (Figure 6). Three high-resolution cryo-EM structures of Cascade at different stages of activation reveal critical details about how target DNA binding triggers major conformational rearrangements for R-loop formation (Figure 1). Although the binding of the DNA target induces complex elongation, the recognition of a cognate PAM sequence and R-loop propagation are essential to trigger conformational changes in the C terminus of Cas8c and Cas11c subunits, locking the complex into a complete R-loop. Similar phenomena have been observed for the type I-F and type I-A Cascade.28,32 Thus, our structures highlight the importance of the duplex vs. ssDNA binding for type I systems in triggering R-loop formation28–31 and establishing conformational control mechanisms to ensure correct target recognition for Cas3 recruitment and activation.37,38
Figure 6. Model for type I-C Cascade-dsDNA activation and inhibition by Acrs.
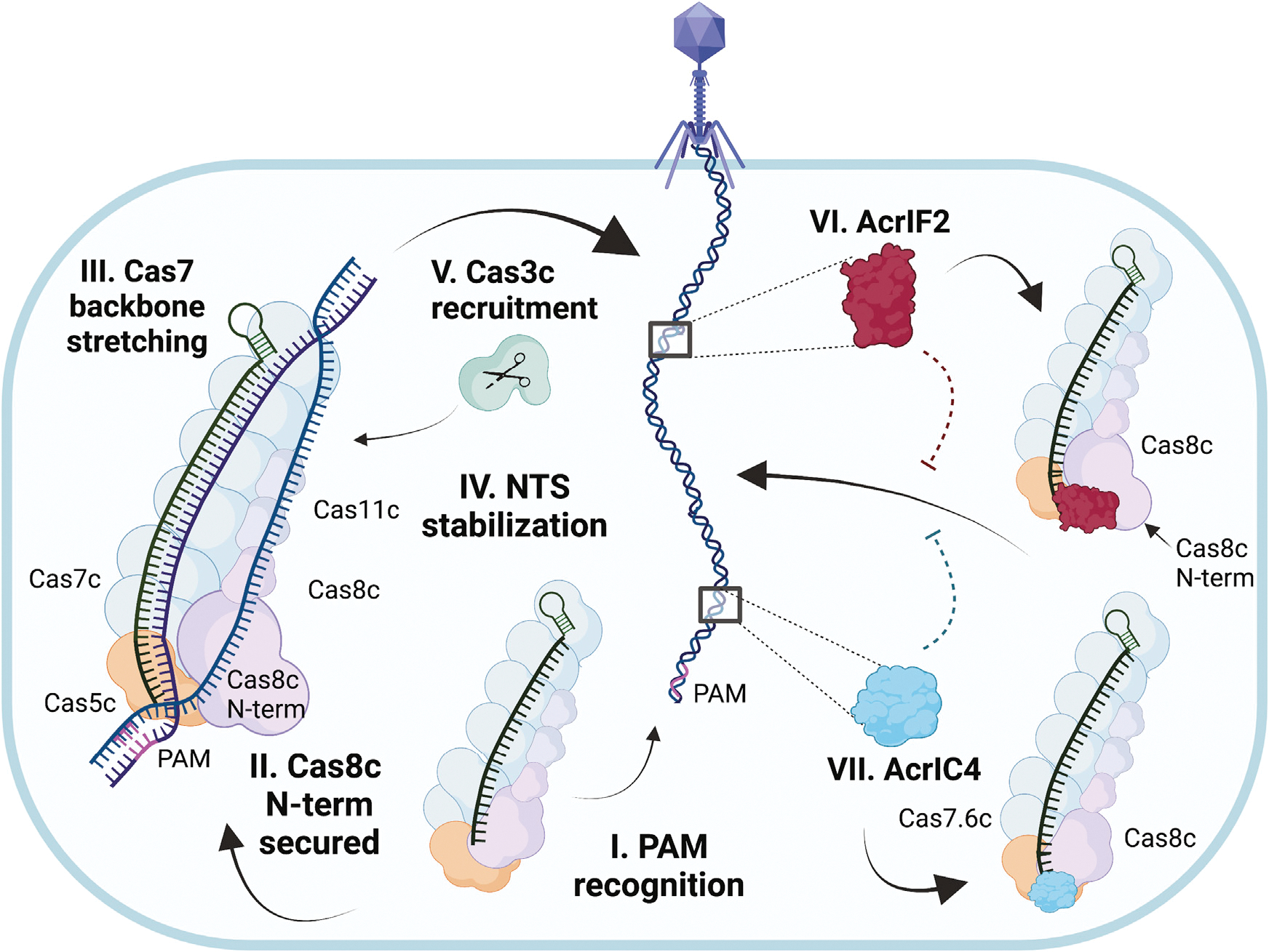
(I) The type I-C Cascade Cas8c has a minimal PAM-recognition requirement. (II) Cas8c N-term is stabilized upon dsDNA binding and responsible for the initial melting of the duplex. (III) Cas7 backbone stretches to accommodate the annealing of TS to crRNA. (IV) Cas8c and Cas11 subunits undergo conformational rotation to support NTS stabilization through an intriguing mechanism of NTS-base clamping between aromatic residues. (V) Once complete R-loop conformation has been established, Cas3c is recruited to degrade the foreign DNA. (VI) AcrIF2 crowds the Cas8c N-term PAM-recognition region and jams the vice-like Cas8c C terminus into a non-productive configuration. (VII) AcrIC4 makes specific contacts with Cas7.6 and Cas8c causing steric hindrance at the PAM-recognition site. This cartoon was created with Biorender.com.
Despite the importance of the NTS in creating a stable R-loop, a thorough understanding of the structural mechanisms behind NTS stabilization was previously hindered by limited resolution in this region in other structures.28–31 Our full R-loop structure reveals a path for the entirety of the NTS, allowing visualization of interactions between the NTS and the Cas8c and Cas11c belly subunits (Figure 2) through both electrostatic and stacking interactions. When alanine substitutions were introduced at this site, the mutants demonstrated a significant decrease in interference efficiency, suggesting the importance of both aromatic and positively charged residues involved in NTS stabilization and Cas3 recruitment. Interestingly, the type I-E Cascade contains similar aromatic residues located along the NTS pathway that would be ideal for providing similar contacts. Thus, the combination of charge-charge and base-stacking interactions underpins the importance of securing the NTS during R-loop formation across type I CRISPR-Cas surveillance complexes.
Our structural and biochemical data provide valuable insights into PAM recognition (Figure 3). Although clear shared characteristics with other Cascades are evident, type I-C Cascade demonstrates a more minimal PAM-recognition scheme compared with its other type I counterparts. It requires only two residues (N72 and Q212) to interact with PAM bases and is a surprising mechanism for type I Cascades28–31. Type I-C CRISPR systems have consistently demonstrated a strong preference for the 5′-TTC-3′ PAM in vivo.34 Yet, for such a strict tolerance, fewer interactions are required to recognize the PAM by the Cas8c subunit, compared with those of other Cascades. Recent structural studies on the type I-C acquisition complex showed that Cas4 incorporates several PAM-recognition residues, which results in a particularly stringent PAM-recognition mechanism.39 Thus, we hypothesize that the strong preference of a type I-C 5′-TTC PAM is likely perpetuated during acquisition, rather than the interference stage of CRISPR-Cas immunity. However, future studies will be necessary to fully elucidate these mechanisms. It may emerge that the promiscuous PAM-recognition mechanism of type I-C Cascade could be exploited to engineer versatile Cascade-Cas3 genome engineering tools, as has been done for Cas9.40
Finally, we present the first structures of the type I-C Cascade bound to AcrIF2 and AcrIC4. Through our structural analysis, we were able to demonstrate the different strategies Acrs use to achieve the same goal of blocking PAM binding (Figures 4 and 5). AcrIC4 interacts with both the Cas7c and Cas8c subunits and inhibits dsDNA binding by acting as a negatively charged structural blockade at the PAM-recognition site. By contrast, AcrIF2 wedges into the positively charged hinge of the Cas8c subunit, creating bipartite steric hindrance of both PAM and the N-term of Cas8c. Previous structural analysis of AcrIF2 proposed that this Acr was representative of a “DNA mimic”; however, we believe that the mechanism of AcrIF2 is more complex in type I-C Cascade.23 The majority of AcrIF2 is negatively charged, which enables it to wedge into the positively charged hinges of Cas8 subunits across both type I-C and type I-F subtypes. Additionally, AcrIF2 creates a second level of inhibition by holding the Cas8c N-term away from stable PAM clamping and R-loop propagation. It has recently been demonstrated for the type I-A Cascade that the conformation of Cas8 N-term regulates Cas3 recruitment and activation, raising the possibility that the Cas8c N-term conformation induced by AcrIF2 binding may also inhibit Cas3c association with the complex.32 Recent studies on the type I-A Cascade-Cas3 complex revealed how this system can be adapted as a sensitive dsDNA detection and bi-directional deletion tool in human cells.32 By contrast, the type I-C Cascade-Cas3 complex represents a unidirectional editor that requires fewer cas genes to be delivered for in vivo applications. Additionally, this system does not require Cas3 binding for target recognition. The Nla type I-C system has performed remarkably well in vivo, reaching up to 95% targeting in HAP1 cells and 50% editing in hESCs, outperforming other unidirectional editors.20 Collectively, these structures could provide the basis for further improvement of type I-C Cascade as a genome engineering tool and regulation by Acrs for these applications.
Limitations of the study
This work describes the structural and biochemical mechanisms of a purified CRISPR-Cascade. Although we also tested some of these conclusions in vivo in Escherichia coli (E. coli), we did not test in the native organism, D. vulgaris, or in mammalian cells, which will further inform how this complex could be repurposed for genome editing purposes. Furthermore, it remains to be seen how these studies will inform genome editing in mammalian cells. Invariably, we only solved structures of certain states of R-loop formation. Other states possibly exist that provide further information into the full mechanism of R-loop formation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, David W. Taylor (dtaylor@utexas.edu).
Materials availability
This study did not generate any new unique reagents.
Data and code availability
The cryo-EM map and associated atomic models of the structures of apo type I-C Cascade, Cascade with a partial R-loop, a full R-loop, and bound to AcrIF2 and AcrIC4 have been deposited into the Electron Microscopy Data Bank with accession codes EMD-27402, EMD-27403, EMD-27393, 27412, and EMD-27409 and the Protein Data Bank with associated accession codes PDB: 8DEX, PDB: 8DFA, PDB: 3DEJ, PDB: 8DFS, and PDB: 2DFO, respectively. Accession numbers are listed in the key resources table. Raw data were deposited on Mendeley at https://doi.org/10.17632/spp853ngyh.1 and are publicly available as of the date of publication. DOIs are listed in the key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Bacterial and Virus Strains | ||
|
| ||
| BL21-AI competent cells | ThermoFisher | C607003 |
| DH5a | ThermoFisher | 18258012 |
| NiCo21(DE3) Competent E. coli | NEB | C2529H |
| BL21(DE3) cells | NEB | C25257H |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Type IC Cas recombinant proteins | This study | N/A |
| Anti-CRISPR IF2 recombinant protein | This study | N/A |
| Anti-CRISPR IC4 recombinant protein | This study | N/A |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Q5 Site-Directed Mutagenesis Kit | NEB | E0554S |
| Gibson Assembly Cloning Kit | NEB | E5510S |
| Q5 High Fidelity 2X Master MIx | NEB | M0492S |
| Q1Aprep Spin Miniprep Kit | Qiagen | 27104 |
|
| ||
| Deposited Data | ||
|
| ||
| Type I-C Cascade Apo Structure | This study | EMD-27402PDB 8DEX |
| Type I-C Cascade Partial R-loop Structure | This study | EMD-27403PDB 8DFA |
| Type I-C Cascade Full R-loop Structure | This study | EMD-27939PDB 8DEJ |
| Type I-C Cascade bound to AcrIF2 | This study | EMD-27412PDB 8DFS |
| Type I-C Cascade bound to AcrIC4 | This study | EMD-27409PDB 8DFO |
| Type I-C Cascade-Cas3 plasmid used for in vivo assay | This study | addgene #196400 |
| Mendeley data | This study | https://doi.org/10.17632/spp853ngyh.1 |
| AcrIC4 plasmid used for type I-C Cascade in vivo assay | This study | addgene #196403 |
| AcrIF2 plasmid used for type I-C Cascade in vivo assay | This study | addgene #196402 |
| type I-C Cascade target plasmid used for in vivo assay | This study | addgene #196399 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers and DNA substrates | Table S3 | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| Plasmids | Table S2 | N/A |
|
| ||
| Software and Algorithms | ||
|
| ||
| cryoSPARC | Punjani et al.41 | https://cryosparc.com/ |
| LEGINON | Suloway et al., 200542 | https://nramm.nysbc.org/software/ |
| WARP | Tegunov et al.43 | http://www.warpem.com/warp/ |
| CHIMERAX | Petterson et al.44 | http://plato.cgl.ucsf.edu/chimerax/download.html |
| COOT | Emsley et al.45 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| ISOLDE | Croll et al.46 | https://isolde.cimr.cam.ac.uk/ |
| PHENIX | Adams et al.47 | https://phenix-online.org/ |
| SerialEM | Mastronarde48 | https://bio3d.colorado.edu/SerialEM/ |
| cryoSPARC Live | Punjani et al.27 | https://cryosparc.com/live |
| AlphaFold | Varadi et al.26 | https://github.com/deepmind/alphafold |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
BL21-AI competent cells (ThermoFisher, C607003), DH5a cells (ThermoFisher, 18258012), NiCo21(DE3) competent cells (NEB, C2529H), and BL21(DE3) cells (EB, C25257H) were used in this study.
METHOD DETAILS
Generation of AcrIC4 Plasmid
The AcrIC4 protein sequence (WP_153575361.1)22 was codon optimized and cloned into the pET His6 SUMO vector (addgene #48313) using ligation independent using the primers listed in Table S3. Fusion tags are added to the 5’ end of each primer. All associated plasmids and vectors can be found in Tables S2 and S3, respectively.
Oligonucleotide Preparation
DNA oligonucleotides used in cleavage assays, gel shifts, and electron microscopy were purchased from Integrated DNA Technologies. The dsDNA duplex was formed by mixing equimolar TS and NTS (Table S3) in 40 mM Tris (pH 8.0) and 38 mM MgCl2, heating at 95°C for 2 min, and slow cooling at room temperature for at least 10 min.17
Protein Purification
The WT and PAM mutants of D. vulgaris type I-C Cascade (addgene #81185) were co-expressed with crRNA (addgene #81186) in NiCo21(DE3) E. coli cells.17,18 AcrIF2 (addgene #89234)23 and AcrIC422 were also co-expressed in Nico21(DE3) E. Coli cells and follow the same following purification protocol as I-C Cascade. Cells were grown at 37°C to an OD600 of 0.6–0.8 and induced by the addition of 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG). After overnight growth at 18°C, the cells were harvested and lysed by sonication in a buffer containing 50 mM HEPES–NaOH (pH 7.5), 500 mM KCl, 5% (v/v) glycerol, 1 mM tris(2-carboxyethyl)phosphine (TCEP), 0.01% Triton X-100, 0.5 mM PMSF, and complete Roche mini protease inhibitor tablets. The lysate was centrifuged at 27,000 × g and applied to a HisTrap Ni-NTA affinity column, pre-equilibrated in lysate 50 mM HEPES–NaOH (pH 7.5), 500 mM KCl, 5% (v/v) glycerol, 1 mM tris(2-carboxyethyl)phosphine (TCEP). The protein-bound resin was washed with buffer containing 50 mM HEPES–NaOH (pH 7.5), 150 mM KCl, 5% (v/v) glycerol, and 1 mM TCEP and a second buffer containing 20 mM imidazole, 50 mM HEPES–NaOH (pH 7.5), 150 mM KCl, 5% (v/v) glycerol, and 1 mM TCEP. Protein was eluted with 50 mM HEPES–NaOH (pH 7.5), 150 mM KCl, 5% (v/v) glycerol, 1 mM TCEP, and an imidazole gradient up to 500mM. Approximately 1 mg of TEV protease was added per 25 mg of protein and the protein-TEV mixture was dialyzed at 4°C overnight against size-exclusion buffer. The protein was then concentrated to approximately 20mg/ml and run over a Superdex 200 Increase 10/300 GL size-exclusion column in a buffer containing 50 mM HEPES–NaOH (pH 7.5), 150 mM KCl, 5% (v/v) glycerol, and 1 mM TCEP. Protein was analyzed for purity by 10–20% SDS-Page (Figure S1) and then dialyzed overnight into the storage buffer containing 20 mM HEPES–NaOH (pH 7.5), 100 mM KCl, 5% (v/v) glycerol, and 1 mM TCEP. All proteins were finally concentrated, flash frozen in liquid nitrogen, and stored at −80°C. All associated plasmids and vectors can be found in Tables S2 and S3, respectively.
Cryo-EM Preparation, Data Collection, and Data Processing
The type I-C Cascade was mixed with dsDNA target at a 1:2 molar ratio (complex:dsDNA). Target binding was facilitated by incubating the mixture at 30° C for 30 min. CF-2/2 grids were first glow discharged for 60s and then a layer graphene oxide was added.49,50 3uL of protein was deposited on the grid in a chamber kept at 4 °C in 100% humidity, and excess protein was blotted away for 4s with a force of 0 after a 0.5s incubation time for 4s using filter paper at. The grid was then plunge frozen into liquid ethane using a FEI Vitrobot Mark IV. Frozen-hydrated samples of type I-C Cascade were directly visualized using a FEI Titan Krios microscope equipped with a Gatan K3 direct electron detector. Using the automated data-collection software LEGINON,51 we acquired 5,399 movies at a nominal magnification of 22,500x, corresponding to a calibrated pixel size of 1.1 Å/pixel, a dosage of 15 e−/pixel/s, and a defocus range of −1.2 μm to −2.2 μm. Movies were collected on a Gatan K3 in 20 frames over an exposure time of 3 s (150 ms/frame), giving a total exposure of 45 e−/pixel. Data collected from the FEI Titan Krios were loaded in the real-time pre-processing tool WARP43 for motion correction, CTF-estimation, and non-templated particle picking. 1.2 million particles and 5,399 micrographs were uploaded to cryoSPARC v3.2.41
For the Cascade-dsDNA dataset, particles extracted in WARP were imported into cryoSPARC v3.241 for 2D classification and 778,052 particles were selected for ab-initio reconstruction, subsequent hetero-refinement, and non-uniform refinement. After multiple rounds of hetero and non-uniform refinement, 381,497 particles were selected for density subtraction, local classification, and focused 3D classification to separate out the full R-loop from the partial R-loop particles. Two distinct classes containing full and partial R-loop states resulted from the 3D classification, likely resulting from the incomplete hybridization of the TS and NTS during duplex synthesis. The dsDNA bound particle set independently went through local refinement to improve the N-terminus of Cas8c and the NTS. The particles from the full and partial R-loop classes were then selected for CTF refinement and a final non-uniform refinement. 174,004 particles resulted in a final reconstruction at 2.80-Å resolution cryo-EM map of the type I-C Cascade in the partial R-loop formation, and 96,964 particles resulted in a final reconstruction at 2.86-Å resolution cryo-EM map of the type I-C Cascade bound to dsDNA. Both reconstructions were determined using the 0.143 gold standard Fourier Shell Correlation calculated from two independent half-sets criterion. The apo model was docked into both maps in ChimeraX44 and used to assist de-novo building in Coot45 and refined in PHENIX47 and ISOLDE.46 Alpha Fold was used to assist de-novo building of the N-term Cas8c, TS, and NTS in Coot.45
To generate the type I-C Acr bound structures, type I-C Cascade was mixed independently with AcrIC4 and AcrIF2 at a 1:10 molar ratio (Cascade:Acr) and diluted to a concentration of 0.3mg/ml. Acr binding was facilitated by incubating the mixture at 30° C for 30 min. The CF-1.2/1.3 grids were first plasma cleaned for 30s in a Solarus 950 plasma cleaner (Gatan) and 2.5uL of sample was deposited on the grid. Excess protein was blotted away after a 0.5s incubation time for 6s with a force of 0 using filter paper at 4 °C in 100% humidity. The grid was then vitrified in liquid ethane using a FEI Vitrobot Mark IV. Frozen-hydrated samples of type I-C Cascade bound to AcrIC4 and AcrIF2 were directly visualized using a FEI Glacios cryo-EM microscope operating at 200 kV equipped with a FEI Falcon 4 direct electron detector. Data were collected in SerialEM,48 with a pixel size of 0.94 Å/pixel, a defocus range of −1.5 to −2.5 μm, a total exposure time of 15s for a total accumulated final dosage of 40.5 e−/Å2 split into 60 EER fractions. 4,078 and 3,520 movies were collected from the Falcon 4 detector of type I-C Cascade bound to IF2 and IC4, respectively, and uploaded to cryoSPARC Live v4.0.0-privatebeta.227 for on-the-fly motion correction, CTF estimation and particle picking.
After motion and CTF correction, templates for template-based picking were generated using the apo-model in cryoSPARC Live v4.0.0-privatebeta.2.27 Template-based particle picking of the type I-C Cascade bound to AcrIF2 and AcrIC4 resulted in 942,324 and 1.4 million particles, respectively. All subsequent data processing was performed in cryoSPARC v3.2.41 The particles were subjected to 2D classification, of which 500,650 and 654,965 particles were selected, respectively. Multiple rounds of ab initio reconstruction and heterogeneous refinement preceded a non-uniform refinement composed of 160,716 and 128,780 particles, respectively. This subset of particles was then re-extracted and exposed to CTF refinement. A mask surrounding the cryo-EM density of the Acrs was generated in ChimeraX44 before 3D classification to improve the quality of this region of the maps. After 3D classification, a final round of non-uniform refinement and CTF refinement resulted in final cryo-EM maps composed of 21,625 particles at a 3.0-Å resolution at for the type I-C Cascade-AcrIF2 complex and 21,651 particles at a 3.1-Å resolution for type I-C Cascade-AcrIC4 complex. Both reconstructions were determined using the 0.143 gold standard Fourier Shell Correlation – calculated from two independent half-sets – criterion. The apo type I-C model was docked into both cryo-EM maps and a previous model of AcrIF2 was docked into the IF2-bound cryo-EM map (5UZ9). AlphaFold 226 was used to assist de-novo building of AcrIC4 in Coot.45 Both final structures were refined in PHENIX47 and ISOLDE.46
To improve our original 3.1-Å resolution structure of the apo type I-C Cascade, the type I-C Cascade was diluted to a concentration of 0.3mg/ml. The CF-1.2/1.3 grids were first plasma cleaned for 30s in a Solarus 950 plasma cleaner (Gatan) and 2.5uL of sample was deposited on the grid. Excess protein was blotted away after a 0.5s incubation time for 6s with a force of 0 using filter paper at 4 °C in 100% humidity. The grid was then plunge frozen into liquid ethane using a FEI Vitrobot Mark IV. Frozen-hydrated samples of type I-C Cascade were directly visualized using a FEI Glacios cryo-EM microscope operating at 200 kV equipped with a Gatan Falcon 4 direct electron detector. Data were collected in SerialEM,48 with a pixel size of 0.94 Å/pixel, a defocus range of −1.5 to −2.5 μm, a total exposure time of 15s for a total accumulated final dosage of 40.5 e−/Å2 which was split into 60 EER fractions. 1,782 movies were collected from the Falcon 4 detector of type I-C Cascade and uploaded to cryoSPARC Live v4.0.0-privatebeta.227 for on-the-fly motion correction, CTF estimation and particle picking.
After motion and CTF correction, templates for template-based picking were generated using the apo-model in cryoSPARC Live v4.0.0-privatebeta.2.27 Template-based particle picking of the type I-C Cascade resulted in 617,365 extracted particles. All subsequent data processing was performed in cryoSPARC v3.2.41 The extracted apo type I-C Cascade particles were then combined with the 942,324 and 1.4 million particles extracted from the Acr bound datasets mentioned above to get a total particle count of 3 million particles. The total 3 million particles were subjected to a round of 2D classification, of which 1.1 million particles were selected. Multiple rounds of ab initio reconstruction and heterogeneous refinement resulted in a final non-uniform refinement composed of 619,957 particles. The reconstructions were determined using the 0.143 gold standard Fourier Shell Correlation – calculated from two independent half-sets – criterion. The previous type I-C model was docked into the cryo-EM map and used to assist de-novo building in Coot45 and unambiguously model several regions of the complex that were poorly resolved previously. The final structure was refined in PHENIX47 and ISOLDE.46
Electrophoretic Mobility Shift Assays
Gel shift assays were performed in 1× binding buffer [50 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 0.03% tween]. WT and mutant Cascades were diluted into 1× binding buffer to concentrations of 10nM, 20nM, 40nM, 80nM, 160nM, 320nM, 640nM, and1280nm. An assembled dsDNA duplex containing a 5’FAM-TS and complimentary NTS was added to a final concentration of 10 nM. Varying concentrations of Cascade were incubated with dsDNA at 37°C for 30 min and resolved at 4°C on 1% agarose containing 1× TBE. DNA was visualized by fluorescence imaging and images were quantified using ImageJ software. The fraction of DNA bound (amount of bound DNA divided by the sum of free and bound DNA) was plotted versus the concentration of type I-C Cascade and fit to standard one-site binding isotherm (all R-square values ≥ 0.98) using Prism (GraphPad). Reported Kd(app) values are the average of at least three independent experiments, and error bars represent the standard deviation.
In Vivo Interference Assays
The in vivo interference assay was adapted from the plasmid system from Dillard et al.37 All genes necessary for the formation and assembly of the D. vulgaris type I-C Cascade (Cas7c-Cas8c-Cas5c from addgene #81185)17 and associated cas3 (AAS94335.1) were cloned into a pBAD-based vector with ampicillin antibiotic resistance (addgene #196400). The primers used to generate the Cas8c PAM Q212A and N72A mutants can be found in Table S4. Gene blocks of Cas8c aromatic mutants (F605A, Y462A, F394A, H566A, Y567A, F287A, F283A), positively charged mutants (K607A, K608A, R408A, R205A, K459A, R205A, R58A, K56A, K55A), and PAM non-specific mutants (R363A, K92A, and K94A) were ordered Integrated DNA Technologies and ligated into the pBAD-based vector using Gibson assembly. The primers to generate these plasmids can be found in Tables S4 and S5. Plasmid #81185 was used as associated type I-C Cascade crRNA containing chloramphenicol resistance. The 35 base pair target sequence with a 5’ TTC PAM site were cloned into a pCDF-Duet1 vector with streptomycin resistance (addgene #196399). AcrIF2 (from addgene #89234) and AcrIC4 were cloned into a pET28b vector with kanamycin resistance, respectively (addgene #196403 and #196403 respectively). LB agar plates were prepared with the following antibiotic concentrations: 50 μg/ml kanamycin, 100 μg/ml carbenicillin (or ampicillin), 50 μg/ml streptomycin, and 34 μg/ml chloramphenicol. Vectors harboring the crRNA and target sequenced were co-transformed into BL21-AI cells and were made electrocompetent with a series of glycerol washes. The remaining vectors were transformed using electroporation in order to obtain various E. Coli strains containing: crRNA+target+IC_cascade_cas3, crRNA+target+IC_cascade_cas3_Q212A, crRNA+target+IC_cascade_cas3_N2A, crRNA+target+IC_cascade_cas3_R363A_K92A_K94A, crRNA+target+IC_cascade_cas3__Cas8c_aromatic_mutant, and crRNA+target+IC_cascade_cas3_Cas8c_positive_mutant. crRNA+target+IC_cascade_cas3+AcrIF2, or crRNA+target+IC_cascade_cas3+AcrIC4. Single colonies of E. coli with WT crRNA+target+IC_cascade_cas3 and strains of Cas8c mutants were inoculated into 5ml of LB containing the appropriate antibiotics and grown overnight at 37° shaking at 225 rpm. The following day, the cells were centrifuged at room temperature at 3000 rpm for 10 minutes. The cells were decanted and resuspended with 5ml of LB with no antibiotics. A 1:100 dilution of the resuspended overnight cell cultures was inoculated into fresh LB with no antibiotics and grown to an OD 0.5 at 37° shaking. Cells were induced with a final concentration of 0.5% L-arabinose and grown for an additional 4 hours at 37°. Cell cultures were then serially 10-fold diluted and plated on LB agar plates containing chloramphenicol and carbenicillin, and chloramphenicol, carbenicillin, and streptomycin. Cleavage efficiency was calculated by the ratio of colony forming units (CFUs) on plates without streptomycin and with streptomycin. This was repeated for single colonies of E. coli harboring the crRNA+target_IC_cascade_cas3+AcrIF2 and crRNA+target+IC_cascade_cas3+AcrIC4. The induced cultures from these strains were serially 10-fold diluted and plated on LB agar plates containing chloramphenicol, carbenicillin and kanamycin, and chloramphenicol, carbenicillin, kanamycin, and streptomycin. Cleavage interference of the anti-crispr was calculated by the ratio of colony forming units (CFUs) on plates without streptomycin and with streptomycin. All associated plasmids and vectors can be found in Tables S2 and S3, respectively. Each sample was comprised of 3–15 biological replicates. Welch’s t-test was used to calculate statistical significance of the cleavage efficiency of cell lines harboring each anti-crispr, respectively, compared to the cleavage efficiency of cell lines harboring IC-cascade and cas3 without the addition of an anti-crispr. **** demonstrates statistically significant.
QUANTIFICATION AND STATISTICAL ANALYSIS
In vivo interference assays
Cleavage efficiency was calculated by the ratio of colony forming units (CFUs) on plates without streptomycin and with streptomycin. This was repeated for single colonies of E. coli harboring the crRNA+target_IC_cascade_cas3+AcrIF2 and crRNA+target+IC_cascade_cas3+AcrIC4. The induced cultures from these strains were serially 10-fold diluted and plated on LB agar plates containing chloramphenicol, carbenicillin and kanamycin, and chloramphenicol, carbenicillin, kanamycin, and streptomycin. Cleavage interference of the anti-crispr was calculated by the ratio of colony forming units (CFUs) on plates without streptomycin and with streptomycin. All associated plasmids and vectors can be found in Tables S2 and S3, respectively. Each sample was comprised of 3–15 biological replicates. Welch’s t-test was used to calculate statistical significance of the cleavage efficiency of cell lines harboring each anti-crispr, respectively, compared to the cleavage efficiency of cell lines harboring IC-cascade and cas3 without the addition of an anti-crispr. **** demonstrates statistical significance compared to WT (N72A: P < 0.0001, Q212A: P < 0.0001, R363A/K92A/K94A: P = 0.0003, Cas8c aromatic mutant: P = 0.006, Cas8c positive mutant: P = 0.0008, AcrIF2: P < 0.0001, AcrC4: P < 0.0001).
Supplementary Material
Highlights.
Three structures revealing various stages of Cascade-dsDNA target capture
Unique mechanism of non-target strand stabilization via stacking interactions
Cas8c N-term rearranges for PAM recognition
Distinct strategies used by two anti-CRISPR proteins to evade Cascade
ACKNOWLEDGMENTS
We thank Axel Brilot for expert cryo-EM assistance and members of the Taylor lab for helpful discussions. We especially thank Evan Schwartz and Isabel Strohkendl for their insightful feedback on the manuscript. We also thank Ailong Ke and Ilya Finkelstein for the plasmids used to create our in vivo interference assays. Data were collected at the Sauer Structural Biology Laboratory at the University of Texas at Austin. This work was supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) R35GM138348 (to D.W.T.), Welch Foundation Research Grant F-1938 (to D.W.T.), and a Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation Medical Research Grant (to D.W.T.). D.W.T. is a CPRIT Scholar supported by the Cancer Prevention and Research Institute of Texas (RR160088) and an American Cancer Society Research Scholar supported by the American Cancer Society (RSG-21–050-01-DMC).
Footnotes
DECLARATION ON INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as living with a disability.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2023.01.024.
REFERENCES
- 1.Wiedenheft B, Sternberg SH, and Doudna JA (2012). RNA-guided genetic silencing systems in bacteria and archaea. Nature 482, 331–338. 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 2.Marraffini LA (2015). CRISPR-Cas immunity in prokaryotes. Nature 526, 55–61. [DOI] [PubMed] [Google Scholar]
- 3.Nuñez JK, Bai L, Harrington LB, Hinder TL, and Doudna JA (2016). CRISPR immunological memory requires a Host Factor for Specificity. Mol. Cell 62, 824–833. 10.1016/j.molcel.2016.04.027. [DOI] [PubMed] [Google Scholar]
- 4.Nuñez JK, Kranzusch PJ, Noeske J, Wright AV, Davies CW, and Doudna JA (2014). Cas1–Cas2 complex formation mediates spacer acquisition during CRISPR–Cas adaptive immunity. Nat. Struct. Mol. Biol. 21, 528–534. 10.1038/nsmb.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, and Oost J. van der (2008). Small crispr RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrangou R, and Marraffini LA (2014). CRISPR-Cas systems: prokaryotes upgrade to adaptive immunity. Mol. Cell 54, 234–244. 10.1016/j.molcel.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Datsenko KA, Pougach K, Tikhonov A, Wanner BL, Severinov K, and Semenova E (2012). Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 3, 945. 10.1038/ncomms1937. [DOI] [PubMed] [Google Scholar]
- 8.The discovery, mechanisms, and evolutionary impact of anti-CRISPRs | Annual Review of Virology. 10.1146/annurev-virology-101416-041616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pawluk A, Davidson AR, and Maxwell KL (2018). Anti-CRISPR: discovery, mechanism and function. Nat. Rev. Microbiol. 16, 12–17. 10.1038/nrmicro.2017.120. [DOI] [PubMed] [Google Scholar]
- 10.Hampton HG, Watson BNJ, and Fineran PC (2020). The arms race between bacteria and their phage foes. Nature 577, 327–336. 10.1038/s41586-019-1894-8. [DOI] [PubMed] [Google Scholar]
- 11.Makarova KS, Wolf YI, and Koonin EV (2018). Classification and nomenclature of CRISPR-Cas systems: where from here? CRISPR J. 1, 325–336. 10.1089/crispr.2018.0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, Charpentier E, Cheng D, Haft DH, Horvath P, et al. (2020). Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 18, 67–83. 10.1038/s41579-019-0299-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Makarova KS, Haft DH, Barrangou R, Brouns SJJ, Charpentier E, Horvath P, Moineau S, Mojica FJM, Wolf YI, Yakunin AF, et al. (2011). Evolution and classification of the CRISPR–Cas systems. Nat. Rev. Microbiol. 9, 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJJ, Charpentier E, Haft DH, et al. (2015). An updated evolutionary classification of CRISPRCas systems. Nat. Rev. Microbiol. 13, 722–736. 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bravo JPK, Liu MS, Hibshman GN, Dangerfield TL, Jung K, McCool RS, Johnson KA, and Taylor DW (2022). Structural basis for mismatch surveillance by CRISPR–Cas9. Nature 603, 343–347. 10.1038/s41586-022-04470-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nam KH, Haitjema C, Liu X, Ding F, Wang H, DeLisa MP, and Ke A (2012). Cas5d protein processes Pre-crRNA and assembles into a cascade-like interference complex in subtype I-C/Dvulg CRISPR-Cas system. Structure 20, 1574–1584. 10.1016/j.str.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hochstrasser ML, Taylor DW, Kornfeld JE, Nogales E, and Doudna JA (2016). DNA targeting by a minimal CRISPR RNA-guided cascade. Mol. Cell 63, 840–851. 10.1016/j.molcel.2016.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Brien RE, Santos IC, Wrapp D, Bravo JPK, Schwartz EA, Brodbelt JS, and Taylor DW (2020). Structural basis for assembly of non-canonical small subunits into type I-C Cascade. Nat. Commun. 11, 5931. 10.1038/s41467-020-19785-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McBride TM, Schwartz EA, Kumar A, Taylor DW, Fineran PC, and Fagerlund RD (2020). Diverse CRISPR-Cas complexes require independent translation of small and large subunits from a single gene. Mol. Cell 80, 971–979.e7. 10.1016/j.molcel.2020.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Tan R, Krueger RK, Gramelspacher MJ, Zhou X, Xiao Y, Ke A, Hou Z, and Zhang Y (2022). Cas11 enables genome engineering in human cells with compact CRISPR-Cas3 systems. Mol. Cell 82, 852–867.e5. 10.1016/j.molcel.2021.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Csörgő B, León LM, Chau-Ly IJ, Vasquez-Rifo A, Berry JD, Mahendra C, Crawford ED, Lewis JD, and Bondy-Denomy J (2020). A compact Cascade–Cas3 system for targeted genome engineering. Nat. Methods 17, 1183–1190. 10.1038/s41592-020-00980-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.León LM, Park AE, Borges AL, Zhang JY, and Bondy-Denomy J (2021). Mobile element warfare via CRISPR and anti-CRISPR in Pseudomonas aeruginosa. Nucleic Acids Res. 49, 2114–2125. 10.1093/nar/gkab006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chowdhury S, Carter J, Rollins MF, Golden SM, Jackson RN, Hoffmann C, Nosaka L, Bondy-Denomy J, Maxwell KL, Davidson AR, et al. (2017). Structure reveals mechanisms of viral suppressors that intercept a CRISPR RNA-guided surveillance complex. Cell 169, 47–57.e11. 10.1016/j.cell.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo TW, Bartesaghi A, Yang H, Falconieri V, Rao P, Merk A, Eng ET, Raczkowski AM, Fox T, Earl LA, et al. (2017). Cryo-EM structures reveal mechanism and inhibition of DNA targeting by a CRISPR-Cas surveillance complex. Cell 171, 414–426.e12. 10.1016/j.cell.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, et al. (2022). AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444. 10.1093/nar/gkab1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Punjani A (2021). Real-time cryo-EM structure determination. Microsc. Microanal. 27, 1156–1157. 10.1017/S1431927621004360. [DOI] [Google Scholar]
- 28.Rollins MF, Chowdhury S, Carter J, Golden SM, Miettinen HM, Santiago-Frangos A, Faith D, Lawrence CM, Lander GC, and Wiedenheft B (2019). Structure reveals a mechanism of CRISPR-RNAguided nuclease recruitment and anti-CRISPR viral mimicry. Mol. Cell 74, 132–142.e5. 10.1016/j.molcel.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayes RP, Xiao Y, Ding F, van Erp PBG, Rajashankar K, Bailey S, Wiedenheft B, and Ke A (2016). Structural basis for promiscuous PAM recognition in type I-E Cascade from E. coli. Nature 530, 499–503. 10.1038/nature16995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao Y, Luo M, Hayes RP, Kim J, Ng S, Ding F, Liao M, and Ke A (2017). Structure basis for directional R-loop formation and substrate handover mechanisms in Type I CRISPR-Cas system. Cell 170, 48–60.e11. 10.1016/j.cell.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartz EA, McBride TM, Bravo JPK, Wrapp D, Fineran PC, Fagerlund RD, and Taylor DW (2022). Structural rearrangements allow nucleic acid discrimination by type I-D Cascade. Nat. Commun. 13, 2829. 10.1038/s41467-022-30402-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu C, Ni D, Nam KH, Majumdar S, McLean J, Stahlberg H, Terns MP, and Ke A (2022). Allosteric control of type I-A CRISPR-Cas3 complexes and establishment as effective nucleic acid detection and human genome editing tools. Mol. Cell 82, 2754–2768.e5. 10.1016/j.molcel.2022.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mojica FJM, Díez-Villaseñor C, García-Martínez J, and Almendros C (2009). Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology (Reading) 155, 733–740. 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- 34.Leenay RT, Maksimchuk KR, Slotkowski RA, Agrawal RN, Gomaa AA, Briner AE, Barrangou R, and Beisel CL (2016). Identifying and visualizing functional PAM diversity across CRISPR-Cas systems. Mol. Cell 62, 137–147. 10.1016/j.molcel.2016.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stella S, Alcón P, and Montoya G (2017). Structure of the Cpf1 endonuclease R-loop complex after target DNA cleavage. Nature 546, 559–563. 10.1038/nature22398. [DOI] [PubMed] [Google Scholar]
- 36.Swarts DC, van der Oost J, and Jinek M (2017). Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-Cas12a. Mol. Cell 66, 221–233.e4. 10.1016/j.molcel.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dillard KE, Brown MW, Johnson NV, Xiao Y, Dolan A, Hernandez E, Dahlhauser SD, Kim Y, Myler LR, Anslyn EV, et al. (2018). Assembly and translocation of a CRISPR-Cas primed acquisition complex. Cell 175, 934–946.e15. 10.1016/j.cell.2018.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao Y, Luo M, Dolan AE, Liao M, and Ke A (2018). Structure basis for RNA-guided DNA degradation by Cascade and Cas3. Science 361, eaat0839. 10.1126/science.aat0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dhingra Y, Suresh SK, Juneja P, and Sashital DG (2022). PAM binding ensures orientational integration during Cas4-Cas1-Cas2 mediated CRISPR adaptation. Preprint at bioRxiv. 10.1101/2022.05.30.494039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walton RT, Christie KA, Whittaker MN, and Kleinstiver BP (2020). Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 368, 290–296. 10.1126/science.aba8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 42.Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, and Carragher B (2005). Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60. 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 43.Tegunov D, and Cramer P (2019). Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 16, 1146–1152. 10.1038/s41592-019-0580-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, and Ferrin TE (2021). UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 46.Croll TI (2018). Isolde: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol. 74, 519–530. 10.1107/S2059798318002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). Phenix: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51. 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 49.Palovcak E, Wang F, Zheng SQ, Yu Z, Li S, Betegon M, Bulkley D, Agard DA, and Cheng Y (2018). A simple and robust procedure for preparing graphene-oxide cryo-EM grids. J. Struct. Biol. 204, 80–84. 10.1016/j.jsb.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martin TG, Boland A, Fitzpatrick AWP, and Scheres SHW (2016). Graphene oxide grid preparation. 10.6084/m9.figshare.3178669.v1. [DOI]
- 51.Potter CS, Chu H, Frey B, Green C, Kisseberth N, Madden TJ, Miller KL, Nahrstedt K, Pulokas J, Reilein A, et al. (1999). Leginon: a system for fully automated acquisition of 1000 electron micrographs a day. Ultramicroscopy 77, 153–161. 10.1016/S0304-3991(99)00043-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM map and associated atomic models of the structures of apo type I-C Cascade, Cascade with a partial R-loop, a full R-loop, and bound to AcrIF2 and AcrIC4 have been deposited into the Electron Microscopy Data Bank with accession codes EMD-27402, EMD-27403, EMD-27393, 27412, and EMD-27409 and the Protein Data Bank with associated accession codes PDB: 8DEX, PDB: 8DFA, PDB: 3DEJ, PDB: 8DFS, and PDB: 2DFO, respectively. Accession numbers are listed in the key resources table. Raw data were deposited on Mendeley at https://doi.org/10.17632/spp853ngyh.1 and are publicly available as of the date of publication. DOIs are listed in the key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Bacterial and Virus Strains | ||
|
| ||
| BL21-AI competent cells | ThermoFisher | C607003 |
| DH5a | ThermoFisher | 18258012 |
| NiCo21(DE3) Competent E. coli | NEB | C2529H |
| BL21(DE3) cells | NEB | C25257H |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Type IC Cas recombinant proteins | This study | N/A |
| Anti-CRISPR IF2 recombinant protein | This study | N/A |
| Anti-CRISPR IC4 recombinant protein | This study | N/A |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Q5 Site-Directed Mutagenesis Kit | NEB | E0554S |
| Gibson Assembly Cloning Kit | NEB | E5510S |
| Q5 High Fidelity 2X Master MIx | NEB | M0492S |
| Q1Aprep Spin Miniprep Kit | Qiagen | 27104 |
|
| ||
| Deposited Data | ||
|
| ||
| Type I-C Cascade Apo Structure | This study | EMD-27402PDB 8DEX |
| Type I-C Cascade Partial R-loop Structure | This study | EMD-27403PDB 8DFA |
| Type I-C Cascade Full R-loop Structure | This study | EMD-27939PDB 8DEJ |
| Type I-C Cascade bound to AcrIF2 | This study | EMD-27412PDB 8DFS |
| Type I-C Cascade bound to AcrIC4 | This study | EMD-27409PDB 8DFO |
| Type I-C Cascade-Cas3 plasmid used for in vivo assay | This study | addgene #196400 |
| Mendeley data | This study | https://doi.org/10.17632/spp853ngyh.1 |
| AcrIC4 plasmid used for type I-C Cascade in vivo assay | This study | addgene #196403 |
| AcrIF2 plasmid used for type I-C Cascade in vivo assay | This study | addgene #196402 |
| type I-C Cascade target plasmid used for in vivo assay | This study | addgene #196399 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers and DNA substrates | Table S3 | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| Plasmids | Table S2 | N/A |
|
| ||
| Software and Algorithms | ||
|
| ||
| cryoSPARC | Punjani et al.41 | https://cryosparc.com/ |
| LEGINON | Suloway et al., 200542 | https://nramm.nysbc.org/software/ |
| WARP | Tegunov et al.43 | http://www.warpem.com/warp/ |
| CHIMERAX | Petterson et al.44 | http://plato.cgl.ucsf.edu/chimerax/download.html |
| COOT | Emsley et al.45 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| ISOLDE | Croll et al.46 | https://isolde.cimr.cam.ac.uk/ |
| PHENIX | Adams et al.47 | https://phenix-online.org/ |
| SerialEM | Mastronarde48 | https://bio3d.colorado.edu/SerialEM/ |
| cryoSPARC Live | Punjani et al.27 | https://cryosparc.com/live |
| AlphaFold | Varadi et al.26 | https://github.com/deepmind/alphafold |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.