

Abstract
Mucosal tissues are constitutively colonized by a wide assortment of host-adapted microbes. This includes the polymorphic fungus Candida albicans which is a primary target of human adaptive responses. Immunogenicity is replicated after intestinal colonization in preclinical models with a surprising array of protective benefits for most hosts, but harmful consequences for a few. The interaction between fungus and host is complex, and traditionally, the masking of antigenic fungal ligands has been viewed as a tactic for fungal immune evasion during invasive infection. However, we propose that dynamic expression of cell wall moieties, host cell lysins, and other antigenic C. albicans determinants is necessary during the more ubiquitous context of intestinal colonization to prime immunogenicity and optimize mammalian host symbiosis.
Living with microbial mavens of immunology
Mucosal barriers constitute a critical interface between our own tissues and the external world filled with microbes. The enormous surface area of the intestine and lung make them common sites of microbial infection. It is therefore amazing to consider that most barrier tissues are not sterile, but naturally colonized by a wide variety of microbes [1–4]. Beneficial effects primed by microbial colonization are becoming increasingly recognized with regard to digestion, development and obesity, behavior and socialization, as well in response to circadian changes [5–12]. Many of these benefits are not limited to the commensal niche, and involve non-mucosal and even systemic responses. Mucosal tissues therefore constitute dynamic boundaries where a mutually beneficial symbiosis between microbe and host occurs.
Classical experiments revealed that mice housed under gnotobiotic germ-free conditions (see Glossary) were highly susceptible to intestinal inflammation [13], while mice eradicated of colonizing bacteria (through administration of multiple antibiotics) showed increased susceptibility to localized and systemic viral infections [14, 15]. Complementary studies demonstrated that adding microbes back into the intestines of germ-free or antibiotic-treated mice provided beneficial effects, even when utilizing host-adapted viral or fungal commensals [16, 17]. Our understanding of how commensal microbes exert these effects remains rudimentary, and we have much to learn from these microbial immunology mavens. Fungi are eukaryotes and appear to be especially important for stimulating host immunity, which may reflect their ability to adopt multiple morphologies with expression of unique microbial ligands and their significantly increased mass relative to bacteria [18–20]. Until recently, however, their contribution was often overlooked given their “invisibility” to traditional 16S ribosomal sequencing approaches.
Commensalism in this context can be mutually beneficial. Microbes gain a hospitable niche, whereas mammalian hosts can reap the benefits of tonic immune stimulation. While most commensal microbes are innocuous for the vast majority of individuals, cohabitation with microbes creates the potential for opportunistic infection. Infection from the “inside-out” by commensal-pathobiont microbes such as Staphylococcus aureus, Escherichia coli, and other Enterobacteriaceae species are important causes of human infection [21–26]. However, despite ubiquitous colonization by these microbes with the potential to cause invasive infection, only a very small fraction of individuals experience disease. Thus, mucosal tissues contain a variety of microbes that are neutral or even beneficial to most individuals, while sometimes causing debilitating infections in a few. Dissecting the host-microbe factors that stimulate protective host benefits, and those that erode this delicate symbiosis, represent exciting windows for investigating how our bodies naturally work in concert with commensal microbes.
C. albicans is a leading cause of invasive infection, and consistently, the number one cause of fungemia [27–32]. The annual incidence of invasive candidiasis ranges from 7 to 29 cases per 100,000 individuals, with an estimated 90 cases per 100,000 hospitalizations associated with exceptionally high mortality. Primary risk factors for invasive candidiasis include colonization, the presence of a central venous catheter, recent abdominal surgery, broad-spectrum antibiotic use, and neutropenia [33–40]. However, aside from the importance of these risk factors, one also needs to consider their relative prevalence. An estimated 40–60% of healthy individuals are intestinally colonized with C. albicans [41]. Comparatively, central venous catheter use and neutropenia are each relatively rare, estimated at 0.015% and <0.5% incidence, respectively [42–45]. This epidemiological discordance indicates that most C. albicans-colonized individuals do not develop invasive infection (see Clinician’s Corner). In this Opinion, we propose that resistance to fungal infection amongst the majority of colonized individuals reflects, at least in part, colonization-induced activation of protective immune responses. Reciprocally, transplantation, cancer and other immune compromising conditions [25, 26, 29, 37, 40] that override these colonization-induced benefits allow C. albicans to transition from beneficial commensal to invasive pathogen. We discuss novel, recently described host benefits arising from C. albicans gut colonization, including both local and systemic effects, as well as the negative consequences of fungal dysbiosis.
Clinician’s corner.
Clinicians primarily view fungi negatively focusing on invasive infections caused by C. albicans and other fungal species. However, recent studies show fungal commensals provide multiple benefits including stimulating immune cells that protects against invasive infection.
Most people never develop invasive fungal infection, despite harboring potentially pathogenic fungi in the intestine and other mucosal tissues. The very small subset of individuals with invasive fungal infection have defects in the protective immune components (e.g. neutrophils) stimulated by commensal fungi, and therefore do not reap colonization- induced protective benefits.
Fungi are generally good and fungal colonization benefits the vast majority of individuals across populations, but with negative consequences such as infection, allergies and disorders associated with pathological inflammation for a few.
Instead of focusing on therapies to eradicate C. albicans and other fungi, we can also learn a lot from these microbial immunology mavens. Understanding how commensal fungi boost immunity can be exploited to develop improved therapies to optimize antimicrobial treatments against C. albicans and other difficult to treat infections.
Colonization-induced protection against invasive infection
C. albicans colonization has been recently shown to stimulate a wide variety of host responses (Table 1), which are summarized in several outstanding recent reviews [20, 46–51]. This fungal species is the primary target of human antifungal antibodies, including IgG in the serum and IgA in the intestines [52, 53], as well as circulating antifungal Th17 CD4+ cells [54]. Systemic immunogenicity primed by colonization is recapitulated in mice, especially those reared under specific pathogen-free (SPF) conditions. For example, activation of circulating granulocytes occurs after experimental C. albicans murine intestinal colonization [55–57]. These responses mimic those observed in SPF mice after “re-wilding”, in which animals are released into an outdoor enclosure to acquire environmental microbes [58]. Using recombinant C. albicans engineered to express defined model antigens to establish colonization in mice shows that fungal commensalism primes the accumulation of CD4+ T cells with surrogate fungal-specificity poised for production of the neutrophil-activating cytokine IL-17 [55, 59].
Table 1.
Recently described impacts of C. albicans commensal colonization
| Phenotype | Species | Notes | References |
|---|---|---|---|
| Protection against C. albicans invasive infection | Mouse | Gut adapted strains, trained immunity | [56] |
| Th17 mediated, requires tonic stimulation | [55] | ||
| Antibody mediated | [52] | ||
| B cell independent | [59] | ||
| Protection against infection by other fungal pathogens | Mouse | Aspergillus fumigatus | [56] |
| Candida auris | [52] | ||
| Protection against infection by bacterial pathogens | Mouse | P. aeruginosa after C. albicans systemic priming | [56] |
| S. aureus after C. albicans systemic priming | |||
| S. aureus, requires tonic stimulation | [55] | ||
| C. rodentium | [65] | ||
| Protection against intestinal inflammation | Mouse | Fluconazole treatment (DSS) | [66] |
| Mono-colonization (DSS) | [17] | ||
| Mucosal vs. luminal (DSS) | [65] | ||
| Protection against airway inflammation | Mouse | Fluconazole treatment (HDM) | [66] |
| Fluconazole treatment, CX3CR1 cell dependent | [67] | ||
| Socialization behavior | Mouse | IL-17 dependent | [65] |
| Increased circulating granulocytes | Mouse | Rewilding with fungal acquisition | [58] |
| Promotes intestinal inflammation | Mouse/Human | CX3CR1-cell defects (Crohn’s disease) | [75] |
| Mouse | Hyphae-locked | [53] | |
| Mouse/Human | Candidalysin-dependent high cell-damaging strains (ulcerative colitis) | [69] | |
| Human | Increased IgA hyphae targets (Crohn’s disease) | [99] | |
| Human | Th17 cells | [54] | |
| Human | Fecal transplant (ulcerative colitis) | [100] | |
| Mouse/Human | Malassezia (Crohn’s disease) | [73] | |
| Promotes airway inflammation | Human | COPD, Cystic fibrosis, ABPA | [54] |
| Mouse | Requires tonic stimulation (HDM) | [55] | |
| Mouse | Candidalysin-dependent | [81] | |
| Promotes oral inflammation | Mouse | Innate IL-17, candidalysin-dependent | [70] |
COPD: chronic obstructive pulmonary disease
Several groups have recently shown that mice intestinally colonized with C. albicans are highly resistant to a subsequent intravenous infection with the same species, exhibiting increased survival and reduced fungal burden in the kidney, compared to controls [52, 55, 56]. C. albicans colonization-induced protection in mice occurs in parallel with the activation of circulating granulocytes [58], and these cells are essential since depletion of such Ly6G+ or Gr1+ myeloid cells overrides colonization-induced protection [55, 57]. These findings dovetail closely with neutropenia being the primary risk factor for invasive fungal infections in humans [34, 37, 39, 40]. Teleologically, avoiding harm to their host makes sense for commensal C. albicans. Reciprocally, individuals with defects in protective immune components, such as neutrophils stimulated by commensal colonization, do not reap the benefits of colonization-induced protection. Colonization is not only non-protective for these individuals, but can also represent an important risk factor for invasive infection [25, 26, 29, 37, 40]. Thus, we propose that the co-evolution of the fungus and host has optimized protective benefits associated with gut colonization for the majority of individuals. Prior studies showed that C. albicans colonization in mice devoid of commensal bacteria (via multiple antibiotics) had increased survival compared with non-colonized control mice following dextran sulfate sodium (DSS)-induced colitis or infection by respiratory viral pathogens such as influenza A virus (IAV), indicating that commensal fungi can replace the tonic stimulatory effects of commensal bacteria [17]. More recent studies establishing colonization without antibiotics or using antibiotics with narrow scope to facilitate colonization [52,55,56] suggest that commensal fungi can not only replace the tonic stimulatory effects of commensal bacteria but can also stimulate more fine-tuned adaptations, one of which is the protection against invasive C. albicans infection.
While neutrophils appear to be essential for colonization-induced protection, the necessity for adaptive immune components, including antibody-producing B cells and fungal-specific T cells, differs between studies. For example, C. albicans cells of the standard strain (SC5314) that were passaged in the gut of standard laboratory mice lost their ability to undergo filamentous growth, a key morphological change in response to host adaptation [60, 61], yet provided increased colonization-induced protection in both C57BL/6 wild-type and Rag1−/− mice (the latter being deficient in both B and T cells) [56]. This would suggest that such protection might be due to innate immune cells or trained immunity [56, 62]. However, other studies indicate that adaptive immunity is important for colonized-induced protection. For example, one study showed that C. albicans colonized mice produce antifungal IgG that protects against disseminated fungal infection [52]. Here, donor B cells from SC5314-colonized mice overturned the susceptibility of B cell-deficient μMT−/− mice to a normally lethal intravenous dose of C. albicans, and IgG from C. albicans-colonized mice also bypassed susceptibility caused by the myeloablative drug cyclophosphamide [52]. Additional studies highlighted the importance of Th17 cells since administration of anti-CD4 cell depleting IgG or anti-IL-17A and IL-17F cytokine neutralizing antibodies each overturned C. albicans colonization-induced protection against systemic infection, which persisted even in μMT−/− mice [55, 59]. Of note, priming C. albicans-specific CD4+ T cells requires specialized antigen-presenting cells which express the autoimmune regulator (AIRE) transcriptional regulator, which may explain the increased susceptibility of individuals with autoimmune polyendocrine syndrome type 1 caused by spontaneous AIRE loss-of-function mutations [63, 64]. Mice with conditional loss of Aire in peripheral immune cells that express the Th17 associated-transcription factor Rorc (RorcCre mice intercrossed with Airefl/fl mice) show increased C. albicans titers in the intestine and tongue after oral inoculation, and reduced survival associated with increased C. albicans titers in the kidney after intravenous infection [63]. Thus, C. albicans colonization primed the activation of the protective Th17-neutrophil axis in humans is faithfully modeled in mice.
These studies find differing requirements for B and T cells in mediating colonization-induced protection, which we speculate might relate to differences in the composition of commensal microbes present and/or C. albicans strain-specific features, which could be addressed by performing the identical experiment using mice from different animal facilities. Nonetheless, commensal C. albicans immunogenicity shown by expansion of fungal-specific CD4+ T cells and accumulation of anti-fungal IgG, along with colonization-induced protection against invasive infection has been consistently demonstrated based on improved survival and reduced fungal pathogen burden in normally sterile target tissues [52, 55, 56]. Thus, C. albicans colonization is associated with resistance to C. albicans invasive infection, and important areas for future investigation include further dissecting the microbial and immunological basis for how commensal C. albicans stimulates systemic immunity.
Immunological benefits of C. albicans commensalism
Recent studies further show that the benefits of C. albicans colonization are not restricted only to protection against C. albicans, but extend to infection by a variety of other pathogens. Indeed, mice colonized with the aforementioned afilamentous C. albicans are more resistant to subsequent systemic challenge with the distantly related fungus Aspergillus fumigatus, as evidenced by increased survival rates after infection [56]. Moreover, protection against systemic A. fumigatus does not require B and T cells since Candida-induced protection is still present in Rag1−/− mice [56]. Protection against antigenically unrelated bacterial pathogens including Staphylococcus aureus and Pseudomonas aeruginosa also occurs for mice systemically primed or intestinally colonized with C. albicans [55, 56]. The scope of protection is likely restricted to extracellular pathogens susceptible to neutrophil-mediated immunity because C. albicans colonization has not been found to impact the susceptibility of mice to IAV infection [55]. In this study, a single antibiotic (ampicillin) facilitated fungal colonization, without causing large-scale reductions in the intestinal bacterial load since bacterial genomic equivalents in the feces quantified by shotgun se- quencing were similar between ampicillin treated and control mice [55]. In contrast, C. albicans mono-colonization enhanced protection against IAV infection if the mice were first depleted of nearly all bacteria using multiple antibiotics [17, 55]. Thus, the impact of C. albicans gut colonization on viral infection is dependent on the bacterial microbiome; fungal cells can effectively substitute for large-scale quantitative reductions in gut bacteria to promote systemic anti-viral immunity, but do not provide additive anti-viral protection when the bacterial microbiome is largely intact.
Spatial heterogeneity represents another distinguishing feature for fungal commensals (Figure 1). Certain fungal species, including those of the genus Candida, are found naturally adjacent to the intestinal mucosa, whereas other species primarily occupy the intestinal lumen [65]. Oral gavage to establish intestinal colonization with a consortium of fungi including C. albicans, Saccharomyces cerevisiae, and Saccharomycopsis fibulifera that share mucosal proximity, protected mice against DSS-induced colitis, shown by prolonged animal survival and reduced intestinal permeability [65]. Mice selectively reconstituted with these mucosa-associated fungi were also more resistant to intestinal infection by the mucosal attaching-effacing bacterium Citrobacter rodentium, as evidenced from reduced weight loss and decreased fecal pathogen burden [65]. Accordingly, C. albicans-colonization can activate the Th17 cell-neutrophil axis which broadly protects against antigenically unrelated pathogens [56, 57]. Interestingly, not all colonization-induced protection appears to be antigen-independent. Protection against the emerging fungal pathogen Candida auris in mice can be mediated by cross-reactive anti-fungal IgG antibodies primed by C. albicans-intestinal colonization [52]. Nonetheless, because C. albicans can protect against infection by antigenically unrelated pathogens (such as S. aureus and C. rodentium), we posit that in the larger context of fungal-host symbiosis, these results suggest that commensal fungi not only protect their hosts against auto-infection from the “inside-out”, but also from infection by a variety of other fungal and bacterial pathogens.
Figure 1. Model depicting how spatial dynamics may govern the immunogenicity of commensal fungi.
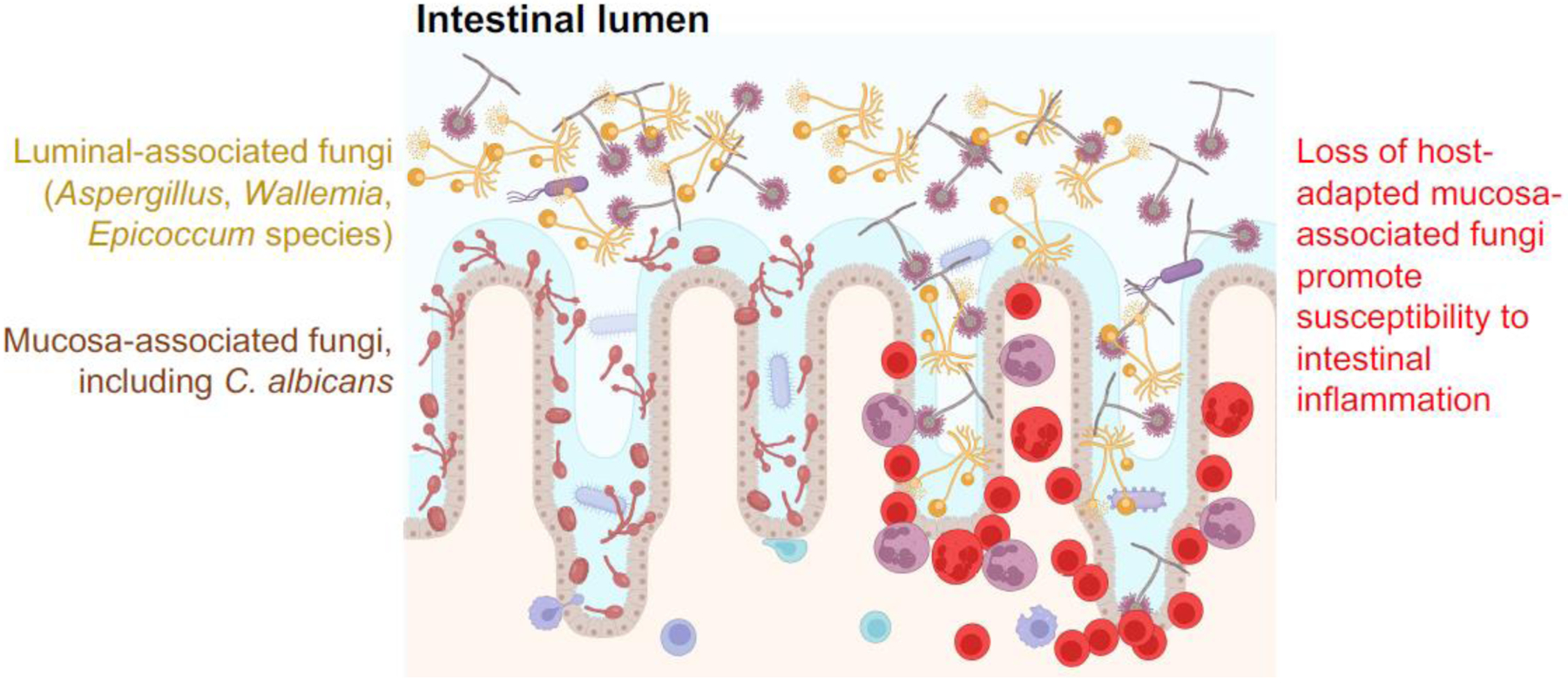
C. albicans and other fungal species lie in close approximation with the intestinal mucosa, whereas other fungi including Aspergillus, Wallemia, and Epicoccum species are more restricted to the intestinal lumen. Recent studies [65] show that administration of fluconazole to mice to selectively eradicate C. albicans promotes their susceptibility to DSS-induced intestinal inflammation which may involve less-host adapted luminal fungi species gaining access to the intestinal mucosa, and thereby causing local accumulation of acute inflammatory cells.
Commensal fungi have also been shown to promote an increasingly wide array of non-infectious physiological processes. These include socialization amongst mice colonized with the aforementioned consortium of mucosa-associated fungi, including C. albicans [65]. These behavioral changes require IL-17 receptor expression by neuronal cells, since enhanced socialization is overturned in mice with conditional loss of IL-17 receptor in Baf53+ neurons (BAF53bCre mice intercrossed with IL17rafl/fl mice) [65]. Likewise, mice harboring a diversity of commensal fungi have increased susceptibility to DSS-induced colitis and house dust mite (HDM)-induced lung inflammation upon commensal C. albicans eradication using the antifungal agent fluconazole [66, 67]. Therefore, we speculate that proximity to the intestinal mucosa is likely to play important roles in distinguishing C. albicans from other fungi such as Aspergillus, Wallemia and Epicoccum species, given the more luminal location of the latter species [65] (Figure 1). Collectively, we argue that immunogenicity of commensal fungi might contribute to the control of behavior and mucosal inflammation, with distinctions between fungal species, based in part on their relative proximity to the intestinal mucosa. However, this remains conjectural and warrants robust investigation.
Immunological consequences of fungal colonization
Multiple consequences associated with colonization-induced immunogenicity have been described. In particular, there is strong evidence that fungal dysbiosis plays a contributing role to human inflammatory bowel disease (IBD), as changes in the composition of the mycobiome often accompany IBD onset or worsening symptom severity [49, 50]. Moreover, patients with Crohn’s disease have been reported to harbor increased accumulation of C. albicans-specific Th17 CD4+ T cells in peripheral blood mononuclear cells [54]. This response might be protective, as treatment with the anti-IL-17A monoclonal antibody, eculizumab, has caused increased disease symptoms compared with placebo controls [68]. Also, recent studies have highlighted C. albicans strain-to-strain heterogeneity in driving intestinal inflammation, suggesting that additional analysis of immunogenic and toxic protein expression by C. albicans in the commensal state is warranted [69]. For instance, C. albicans strains with high macrophage-damaging capacity are more enriched amongst ulcerative colitis patients compared with individuals without IBD, and reconstituting mice with these strains enhances their susceptibility to DSS-induced intestinal injury [69]. Therefore, macrophage damaging capability may be a major determinant of strain-specific disease induction by C. albicans.
Interestingly, the macrophage-damaging capacity of C. albicans clinical isolates has also been linked to filamentation and, in particular, to the peptide toxin candidalysin; indeed, targeted deletion of ECE1 encoding candidalysin (ece1−/−) in C. albicans clinical isolates with high damaging capacity could reduce their ability to prime accumulation of IL-17A-producing CD4+ T cells in mice after intestinal colonization, whereas low concentrations of Th17 inflammation in mice colonized with clinical isolates with low damaging capacity were not further reduced by ECE1 deletion [69, 70] (Figure 2). The importance of filamentation has been further shown by a similarly increased susceptibility to DSS-induced colitis in mice colonized with wildtype C. albicans or hyphal-locked strains compared with reduced inflammation severity in mice colonized with yeast-locked strains (based on histological scoring) [53]. Thus, we posit that a likely consequence of C. albicans immunogenicity is an increased susceptibility to aberrant intestinal inflammation, especially when colonized by strains predisposed to filamentation and candidalysin production [20, 71], or amongst individuals with genetic risk factors for IBD, such as mutations in the following proteins, CARD9, CX3CR1 or CLEC7A (human Dectin-1) [50, 52, 72–75].
Figure 2. Host-adapted C. albicans shows a range of host cell-damaging capacities and expression of host cell toxins linked with filamentation.
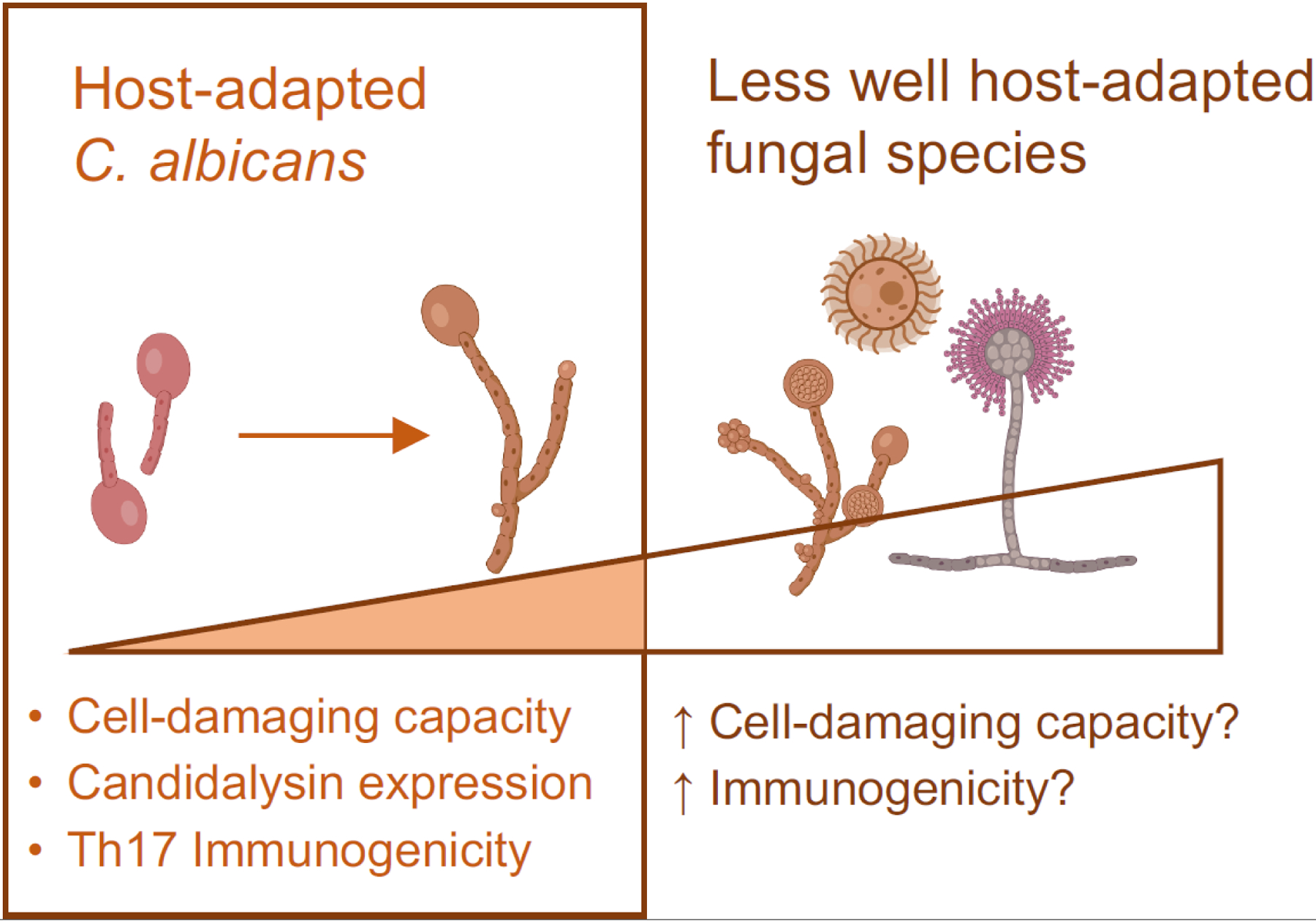
Recent studies show that C. albicans filamentation is associated with enhanced host cell-damaging capacity and immunogenicity [53, 69]. Expression of the fungal toxin candidalysin is linked to filamentation and contributes to the immunogenicity of C. albicans human clinical isolates with high macrophage cell-damaging capacity [69]. These results highlighting the heterogeneity in C. albicans suggest that colonization by fungal strains predisposed to filamentation and candidalysin production may increase susceptibility to aberrant inflammation consequences. By extension, colonization with fungal species that are likely to be even less host-adapted, and their access to the intestinal mucosa with fungal dysbiosis, might help explain the susceptibility to aberrant inflammation associated with the presence of non-Candida fungal species in the intestine of mice and humans [49, 50, 65–67, 73].
Aberrant airway inflammation is another potential consequence of commensal C. albicans colonization, reflecting a shared gut-lung immunological axis [76, 77]. A variety of respiratory disorders including asthma, chronic obstructive pulmonary disease (COPD), and cystic fibrosis have been associated with fungal colonization and the peripheral accumulation of C. albicans-specific Th17 CD4+ T cells [54]. C. albicans-primed Th17 CD4+ T cells are also broadly cross-reactive with other fungi, including Aspergillus, thus contributing to the manifestation of allergic disorders, including allergic bronchopulmonary aspergillosis [54]. Indeed, C. albicans elicits an extremely strong Th17 response [54], and given its ubiquity in the human gut, it is likely to represent a major evolutionary force in promoting Th17 responses. These phenotypes are faithfully modeled in C. albicans-colonized mice with increased susceptibility to airway inflammation induced by HDM or Aspergillus spores [55, 78, 79], and likely not limited only to C. albicans, since colonization of mice with other non-albicans Candida species (C. glabrata, C. tropicalis, or C. parapsilosis) can also exacerbate airway inflammation [80]. Airway inflammation induced by C. albicans shares with intestinal inflammation the requirement for candidalysin since intranasal administration of candidalysin-deficient (ece1−/−), compared with wildtype C. albicans, led to reduced IL-13 plus IL-17 concentrations, and to diminished airway accumulation of eosinophils, neutrophils, and macrophages [81]. Thus, the consequences of C. albicans immunogenicity during intestinal colonization can include aberrant inflammation in the lung, intestine, and potentially other mucosal tissues.
Reconciling whether C. albicans colonization stimulates or suppresses inflammation.
As discussed above, C. albicans colonization can either promote or protect against pathological inflammation. One way to reconcile these somewhat discordant results is to consider C. albicans as part of the larger mycobiome in each experimental context. Mice housed under SPF conditions in several institutions are devoid of commensal fungi [55, 58, 82]. Establishing fungal colonization in this context can stimulate systemic Th17 immunity associated with increased inflammation in the intestine and other mucosal barrier tissues [55, 58]. Thus, compared to mice without fungal colonization, commensal C. albicans can be immunogenic and drive local and systemic Th17 inflammation. In turn, administration of antifungal agents can eradicate fungal colonization and eliminate both the protective and harmful effects of C. albicans colonization, when compared with mice devoid of other fungal commensals [55].
Comparatively, mice housed in other SPF facilities are naturally colonized with a variety of fungal species including Candida, Aspergillus, Wallemia and Epicoccum [66, 67, 83]. Instead of fungal eradication, fluconazole treatment (targeting Candida and other yeast) likely causes fungal dysbiosis, associated with increased accumulation of Aspergillus, Wallemia, and Epicoccum species [66, 67]. Enriched amounts of these non-Candida species is associated with enhanced susceptibility of mice to DSS-induced weight loss and intestinal inflammation, as well as HDM-induced lung inflammation, as shown by the increased accumulation of neutrophils, macrophages, and eosinophils in the airway of fluconazole-treated mice, compared with controls [66, 67]. These considerations suggest that C. albicans might not only be immunogenic during commensal colonization, but might also limit the accumulation of other commensal fungi species that could be more immunogenic; this in turn, could stimulate pathological inflammation (Figure 2).
Differences in immunogenicity might also reflect the proximity of C. albicans to the mucosa, compared with other fungal species that are more confined to the intestinal lumen [65] (Figure 1). Immunogenicity distinctions between relatively friendly fungi such as C. albicans, and other potentially less well host-adapted fungal species are supported by the increased susceptibility of Malassezia-colonized mice to intestinal inflammation, and the correlation in humans between intestinal Malassezia colonization and the severity of Crohn’s disease [73]. Thus, we propose that not all fungi are equally immunogenic in the commensal state, and symbiosis between C. albicans and mammalian hosts might include fine-tuning to simultaneously optimize protection against invasive infection and averting aberrant inflammation triggered by more immunogenic fungi. Additional studies comparing immune responses after mono-fungal colonization, and investigating the dynamic competition between C. albicans and other fungi for the commensal niche, are therefore a priority.
Perspectives on C. albicans immunogenicity
C. albicans immunogenicity in the commensal state, whether beneficial or harmful, involves host cell stimulation. A diverse repertoire of fungal ligands stimulate host cells through a variety of host pattern recognition receptors (PRRs) [39, 48, 84–86]. These include mannans, β-glucan, chitin, and fungal nucleic acids, which stimulate through Toll-like receptors, C-type lectin receptors, NOD-like receptors, and RIG1-like receptors. A variety of other C. albicans proteins including Als1, Als3, and Mp65 are the targets of adaptive T cell and humoral antibody responses [53, 54, 86, 87]. Reciprocally, masking these immunogenic moieties is thought to promote immune evasion during infection or to avert inflammation during commensal growth [88, 89]. This makes sense in the context of commensal colonization if immune stimulation is primarily detrimental. However, if the protective benefits of commensal C. albicans colonization are more universally advantageous, we argue that immunogenicity via the unmasking of fungal antigens is likely to be more favorable, resulting in coordinated stimulation of local intestinal immune and epithelial cells [90].
Teleologically, the accelerated replication of fungi (minutes to hours) compared with mammalian hosts (months to decades), should select for non-immunogenic commensals if immunogenicity uniformly has negative consequences for fungal fitness. Instead, fungal commensals actively stimulate immune cells via a variety of host PRRs, and the degree of C. albicans colonization-induced protection against invasive infection is significantly reduced in mice with functional defects in TLR2, TLR4, Dectin-1, and Dectin-2 [59]. These antifungal receptors are expressed by multiple cell types including CX3CR1 mononuclear phagocyte cells, and conditional depletion of CX3CR1 in CD11c+ antigen-presenting cells (diphtheria toxin administration to CD11cCre CX3CR1DTR mice) overrides the accumulation of IL-17-producing CD4+ T cells in the intestine of C. albicans colonized mice [75]. Moreover, missense mutations in human CX3CR1 have been associated with impaired antifungal responses in Crohn’s disease [75]. These considerations suggest that mammalian co-evolution with fungal commensals is intricately fine-tuned, allowing immunogenic fungal moieties to be selectively expressed when beneficial, but with safeguards in place for masking exposure to limit inflammation. For example, while masked β-glucan exposure by C. albicans might help evade detection by innate immune cells during invasive infection [88, 89], exposing antigenic fungal moieties to prime the host’s local and systemic effects in the more ubiquitous context of C. albicans colonization is likely to be more mutually beneficial to both host and fungus.
This newfound appreciation that fungal immunogenicity might be more uniformly beneficial than detrimental underscores the need for more focused investigation on how exposure of antigenic moieties is regulated, especially in the context of commensal colonization. Important clues come from in vitro studies showing that changes in pH and oxygen concentrations that mimic host tissues, can influence C. albicans expression of immunogenic cell wall components such as mannans, chitin, and β-glucan [85, 89, 91]. For example, hypoxic conditions reduce C. albicans β-glucan exposure, with paralleled reduced susceptibility to phagocytosis by primary murine macrophage cells and diminished production of proinflammatory cytokines such as TNFα and RANTES, compared with cells stimulated with C. albicans grown under normal oxygen conditions [92]. Reciprocally, suppression of C. albicans respiration via exposure to oxidase inhibitors salicylhydroxamic acid and sodium nitroprusside can promote surface chitin and β-glucan exposure which is associated with enhanced immunogenicity, as evidenced by increased phagocytosis from the J774 murine macrophage cell line [93]. Acidic conditions can also stimulate exposure of immunogenic β-glucan and chitin by C. albicans through pH regulated expression of the CHT2 chitinase [94]. Thus, surface expression and exposure of immunogenic cell wall components by C. albicans is not static, but dynamically regulated by environmental cues.
Links between C. albicans dynamic gene expression and immunogenicity during intestinal colonization were recently investigated by varying the expression of UME6, a transcription factor responsible for promoting filamentation in response to environmental signals, since UME6-deficient (ume6−/−) C. albicans cells are defective for filamentation when grown under conditions (serum supplementation, 37°C, or nitrogen and carbon starvation) that do promote filamentation in wildtype C. albicans [95–97]. The results showed that immunogenicity was lost in mice colonized by either UME6-deficient or UME6-overexpressing C. albicans, but restored in mice colonized by C. albicans forced to oscillate between UME6-on and UME6-off states, as evidenced by the relative accumulation of Th17 CD4+ T cells and the resistance of mice to wild-type C. albicans intravenous infection [59]. Moreover, β-glucan exposure, along with expression of GSC1 and MNT2 required for β-glucan and mannan biosynthesis, respectively, were sharply reduced in both UME6-on and UME6-off locked states, with the amounts of each rebounding in C. albicans fungi transitioning between the two states; this indicated that oscillating, instead of static, UME6 expression promoted the expression of these antigenic cell-wall components. In turn, intestinal reconstitution with β-glucan and mannan restored immunogenicity to mice colonized by UME6-on or UME6-off locked commensal C. albicans, further demonstrating that the functional absence of these cell wall components was responsible for the loss of immunogenicity by C. albicans locked into UME6-on or UME6-off states [59]. Thus, the expression of β-glucan and mannan during intestinal colonization appeared to be dynamically regulated by oscillating expression of UME6, and only indirectly linked with fungal morphology since similar proportions of yeast and hyphae were present during colonization with wild-type or ume6−/− cells [97]. We propose that this fluid immunogenicity by C. albicans is a consequence of long-term coevolution of a commensal with its mammalian host. Potentially, the expression of UME6 and other fungal virulence factors associated with filamentation such as candidalysin might undergo continuous refinement to achieve optimal symbiosis, maximizing the benefits of this balance for the majority of hosts. In this scenario of active refining selection, colonization by C. albicans outliers in immunogenicity or with dysregulated expression of antigenic components may cause aberrant inflammation in some individuals.
Concluding remarks
A scientific renaissance in appreciation of commensal fungi has begun. These eukaryotic cells can adopt a variety of different morphologies, are much larger than bacteria, and express antigens which are distinct from bacteria. It is therefore not surprising that the outcomes of colonization with C. albicans and other fungal species are fundamentally unique from commensal bacteria. The emerging theme reporting that C. albicans colonization can confer a variety of benefits to mammalian hosts is highlighted in recent studies [52, 55, 56, 58, 65–67]. Here, we have argued that colonization can protect us not only from invasive fungal infection, but also from infection by antigenically unrelated bacterial pathogens by stimulating the activation of innate immune cells, [55, 56, 65]. Beyond host antimicrobial immunity, commensal C. albicans might also control how we interact with one another by modulating the responsiveness of our neurons to cytokine stimulation, and therefore influence social behaviors, although this remains conjectural [65]. Overall, the results discussed here challenge the generally negative perception of fungi as microbes that only cause difficult-to-treat infections or stimulate allergic-type reactions. These negative consequences for a few individuals are likely offset by far greater host benefits of colonization with C. albicans and other host-adapted fungal species. Limitations to current studies include the near-exclusive focus on C. albicans, and the reliance on rodent colonization and disease models that may not reproduce all aspects of human-microbiota interactions and the pathogenesis of clinical infection, autoinflammatory and allergic disorders. These limitations open several new questions (see Outstanding questions), with answers that will allow improved appreciation of the putative beneficial effects of commensal fungi, which might also be therapeutically dissociated from pathogenic and inflammatory consequences.
Outstanding questions.
Which commensal fungi, in addition to C. albicans, can also prime Th17 immunogenicity, and how is expression of antigenic moieties by fungal species regulated? This information can broaden our understanding of the potential development of fungal probiotics and/or immunogenic fungal components to fine-tune immune responses.
What controls the proximity of commensal fungi to intestinal mucosa, and is mucosal proximity linked with immunogenicity? Do fungi confined to the intestinal lumen gain proximity to the intestinal mucosa during pathological inflammation or when mucosa-associated fungi are eliminated?
What are the links between morphological changes and immunogenicity for C. albicans and other commensal fungi species in the commensal state? Are fungal-specific toxins such as candidalysin associated with pathological inflammation also required for stimulating protective Th17 immunogenicity? This information can help us better understand the molecular basis of C. albicans immunogenicity in the commensal state.
How does acquisition of fungal colonization during unique developmental windows (e.g., in neonates) impact the priming of Th17 immunogenicity? This information can help us understand the long-term health consequences of C. albicans colonization that is naturally initiated in humans shortly after birth.
Is priming protective Th17 systemic responses unique to fungal colonization of the intestine, or are similar responses primed in other barrier tissues (oral mucosa, vaginal mucosal, upper airway, skin)? This information can help us develop improved therapies that exploit the systemic immunomodulatory effects of commensal microbes.
How is immunological tolerance to commensal fungi mediated, and are there specialized immune cells dedicated to suppressing aberrant inflammation stimulated by commensal C. albicans and other fungal species? This information can help us better understand how the balance between immunostimulation and immunosuppression signals are integrated in complex tissues such as the intestine, and which must tolerate self-antigens as well as genetically foreign antigens expressed by commensal microbes.
Significance.
Fungi including Candida albicans colonize the human intestine with local and systemic host benefits, which require fungal immunogenicity in the commensal state. Investigating the molecular and cellular mechanisms sustaining this immunological symbiosis can generate approaches to therapeutically fine-tune immunity, optimize host protection, yet limit invasion.
Highlights Box.
Fungi that colonize the intestine convey unique symbiotic host benefits including tonic calibration of systemic immunity, and protection against invasive infection in rodents with potentially direct translational relevance to human immunity.
C. albicans colonization in mice protects against infection by antigenically unrelated pathogens, aberrant intestinal inflammation, and injury, and can promote social behavior changes through systemic immunological influence.
Immunogenicity during commensal colonization is beneficial to mice, and occurs with dynamically regulated exposure of antigenic fungal cell wall ligands.
Commensal C. albicans in mice are mucosa-associated and can protect against intestinal injury induced by other commensal fungal species that primarily reside within the intestinal lumen.
Commensal C. albicans are heterogeneous in humans and mice, with variations in immune cell-damaging capacity associated with filamentation and production of the toxin candidalysin.
Acknowledgments
This work was supported by NIH-NIAID through grants DP1AI131080 (SSW), R01AI141893 (RJB), R01AI081704 (RJB).SSW is supported by the HHMI Faculty Scholar’s program (#55108587) and Burroughs Wellcome Fund Investigator in the Pathogenesis Award (#1011031).
GLOSSARY
- Autoimmune regulator (AIRE)
Transcription-factor primarily expressed in medullar thymic epithelial cells; promotes the expression of proteins elsewhere in the body, allowing maturing T cells with self-reactivity to be deleted during development.
- Dextran sulfate sodium (DSS)-induced colitis
Experimental model of intestinal injury induced colitis. Animals are administered sulfated polysaccharides that are toxic to colonic epithelial cells, resulting in compromised mucosal barrier function and intestinal inflammation.
- Dysbiosis
Deviance in the composition of the microbial communities found in healthy individuals.
- Extracellular pathogens
Microbes that cause infection without a need to gain access to grow inside host cells. This contrasts with intracellular pathogens which efficiently gain access and cause infection inside cells.
- Filamentous growth
Multiple fungi can form filaments in response to environmental stimuli. C. albicans can grow as yeast which are rounded single cells, or as filamentous hyphae or pseudohyphae; yeast-locked cells exhibit increased intestinal colonization fitness in antibiotic-treated mice.
- Gnotobiotic germ-free conditions
Completely lack commensal or pathogenic microbes. Mice are introduced into these artificial conditions through rederivation, and raised under sterile conditions.
- House dust mite (HDM)-induced airway inflammation
Experimental respiratory tract exposure to the host dust mite – an ubiquitous environmental allergen. Exposure induces an inflammatory response, reactivity, and remodeling of airway cells.
- Hyphal-locked strains
C. albicans strains engineered to ensure that the NRG1 gene is turned off to maintain cells in the hyphal morphology.
- Mycobiome
Abundance and diversity of fungi species present within individuals or a broader subset of mammalian hosts.
- Pattern recognition receptors (PRRs)
Host cellular receptors that recognize and respond to stimulation by evolutionarily conserved macromolecular structures expressed by microbes, e.g.Toll-like receptor 4 that allow host cells to recognize and respond to stimulation by lipopolysaccharide.
- Pathobiont microbes
Potentially pathological microbes that can either colonize innocuously without causing harm or cause infection after first establishing colonization.
- Re-wilding
Relocation of mice or other mammalian hosts from more controlled laboratory settings to less artificial outdoor environments containing more diverse and heterogenous microbes.
- Specific pathogen-free (SPF) conditions
Lack defined pathogenic microbes. While there are general guidelines, there are no currently universally accepted definitions of SPF conditions for biomedical research facilities given the difficulties distinguishing between pathogenic, pathobiont, and more innocuous commensal microbes.
- Symbiosis
Mutually beneficial to two individual parties. In the context of microbial colonization of mammalian hosts, symbiosis refers to interactions that are beneficial to both microbe and host.
- Th17 CD4+ cells
Subset of CD4+ T helper cells; express the transcriptional regulator RORγt and selectively produce the proinflammatory cytokine IL-1; mediate host defense against a wide variety of bacterial and fungal pathogens that primarily reside in the extracellular compartment of host cells.
- Th17-neutrophil axis
Coordinated activation of neutrophils after stimulation by CD4+ T cells that differentiate under the specialized T-helper 17 lineage.
- Trained immunity
Ability of non-antigen-specific innate immune cells (e.g., macrophages, neutrophils, and granulocytes) to sustain activation long-term after stimulation.
- Yeast-locked cells
C. albicans cells engineered to adopt only the yeast morphology due to constitutive expression of the NRG1 gene.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests The authors declare no competing financial interests.
References
- 1.O’Hara AM and Shanahan F (2006) The gut flora as a forgotten organ. EMBO Rep 7 (7), 688–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Backhed F et al. (2005) Host-bacterial mutualism in the human intestine. Science 307 (5717), 1915–20. [DOI] [PubMed] [Google Scholar]
- 3.Eckburg PB et al. (2005) Diversity of the human intestinal microbial flora. Science 308 (5728), 1635–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qin J et al. (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464 (7285), 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blander JM et al. (2017) Regulation of inflammation by microbiota interactions with the host. Nat Immunol 18 (8), 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ansaldo E et al. (2021) Control of immunity by the microbiota. Annu Rev Immunol 39, 449–479. [DOI] [PubMed] [Google Scholar]
- 7.Littman DR and Pamer EG (2011) Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe 10 (4), 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brooks JF 2nd and Hooper LV (2020) Interactions among microbes, the immune system, and the circadian clock. Semin Immunopathol 42 (6), 697–708. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y and Hooper LV (2019) Immune control of the microbiota prevents obesity. Science 365 (6451), 316–317. [DOI] [PubMed] [Google Scholar]
- 10.Morais LH et al. (2021) The gut microbiota-brain axis in behaviour and brain disorders. Nat Rev Microbiol 19 (4), 241–255. [DOI] [PubMed] [Google Scholar]
- 11.Foster JA (2022) Modulating brain function with microbiota. Science 376 (6596), 936–937. [DOI] [PubMed] [Google Scholar]
- 12.Brodin P (2022) Immune-microbe interactions early in life: A determinant of health and disease long term. Science 376 (6596), 945–950. [DOI] [PubMed] [Google Scholar]
- 13.Rakoff-Nahoum S et al. (2004) Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118 (2), 229–241. [DOI] [PubMed] [Google Scholar]
- 14.Abt MC et al. (2012) Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37 (1), 158–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ichinohe T et al. (2011) Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A 108 (13), 5354–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kernbauer E et al. (2014) An enteric virus can replace the beneficial function of commensal bacteria. Nature 516 (7529), 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang TT et al. (2017) Commensal fungi recapitulate the protective benefits of intestinal bacteria. Cell Host Microbe 22 (6), 809–816 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Underhill DM and Iliev ID (2014) The mycobiota: interactions between commensal fungi and the host immune system. Nat Rev Immunol 14 (6), 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neville BA et al. (2015) Candida albicans commensalism in the gastrointestinal tract. FEMS Yeast Res 15 (7), fov081. [DOI] [PubMed] [Google Scholar]
- 20.Iliev ID (2022) Mycobiota-host immune interactions in IBD: coming out of the shadows. Nat Rev Gastroenterol Hepatol 19 (2), 91–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paling FP et al. (2020) Association of Staphylococcus aureus colonization and pneumonia in the intensive care unit. JAMA Netw Open 3 (9), e2012741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tamburini FB et al. (2018) Precision identification of diverse bloodstream pathogens in the gut microbiome. Nat Med 24 (12), 1809–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adelman MW et al. (2020) The gut microbiome’s role in the development, maintenance, and outcomes of sepsis. Crit Care 24 (1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelly MS et al. (2019) Gut colonization preceding mucosal barrier Injury bloodstream infection in pediatric hematopoietic stem cell transplantation recipients. Biol Blood Marrow Transplant 25 (11), 2274–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhai B et al. (2020) High-resolution mycobiota analysis reveals dynamic intestinal translocation preceding invasive candidiasis. Nat Med 26 (1), 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marco F et al. (1999) Elucidating the origins of nosocomial infections with Candida albicans by DNA fingerprinting with the complex probe Ca3. J Clin Microbiol 37 (9), 2817–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pfaller MA and Diekema DJ (2007) Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev 20 (1), 133–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ricotta EE et al. (2021) Invasive candidiasis species distribution and trends, United States, 2009–2017. J Infect Dis 223 (7), 1295–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown GD et al. (2012) Hidden killers: human fungal infections. Sci Transl Med 4 (165), 165rv13. [DOI] [PubMed] [Google Scholar]
- 30.Arendrup MC et al. (2011) National surveillance of fungemia in Denmark (2004 to 2009). J Clin Microbiol 49 (1), 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cleveland AA et al. (2012) Changes in incidence and antifungal drug resistance in candidemia: results from population-based laboratory surveillance in Atlanta and Baltimore, 2008–2011. Clin Infect Dis 55 (10), 1352–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edmond MB et al. (1999) Nosocomial bloodstream infections in United States hospitals: a three-year analysis. Clin Infect Dis 29 (2), 239–244. [DOI] [PubMed] [Google Scholar]
- 33.Mishra AA and Koh AY (2018) Adaptation of Candida albicans during gastrointestinal tract colonization. Curr Clin Microbiol Rep 5 (3), 165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uzun O et al. (2001) Risk factors and predictors of outcome in patients with cancer and breakthrough candidemia. Clin Infect Dis 32 (12), 1713–1717. [DOI] [PubMed] [Google Scholar]
- 35.Yapar N (2014) Epidemiology and risk factors for invasive candidiasis. Ther Clin Risk Manag 10, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poissy J et al. (2020) Risk factors for candidemia: a prospective matched case-control study. Crit Care 24 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castagnola E et al. (2006) Fungal infections in children with cancer: a prospective, multicenter surveillance study. Pediatr Infect Dis J 25 (7), 634–639. [DOI] [PubMed] [Google Scholar]
- 38.Thomas-Ruddel DO et al. (2022) Risk factors for invasive Candida infection in critically ill patients: A Systematic Review and Meta-analysis. Chest 161 (2), 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lionakis MS and Netea MG (2013) Candida and host determinants of susceptibility to invasive candidiasis. PLoS Pathog 9 (1), e1003079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Desai JV and Lionakis MS (2018) The role of neutrophils in host defense against invasive fungal infections. Curr Clin Microbiol Rep 5 (3), 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nash AK et al. (2017) The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 5 (1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kornbau C et al. (2015) Central line complications. Int J Crit Illn Inj Sci 5 (3), 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGee DC and Gould MK (2003) Preventing complications of central venous catheterization. N Engl J Med 348 (12), 1123–1133. [DOI] [PubMed] [Google Scholar]
- 44.O’Grady NP et al. (2011) Summary of recommendations: Guidelines for the prevention of intravascular catheter-related infections. Clin Infect Dis 52 (9), 1087–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsieh MM et al. (2007) Prevalence of neutropenia in the U.S. population: age, sex, smoking status, and ethnic differences. Ann Intern Med 146 (7), 486–492. [DOI] [PubMed] [Google Scholar]
- 46.Swidergall M and LeibundGut-Landmann S (2022) Immunosurveillance of Candida albicans commensalism by the adaptive immune system. Mucosal Immunol, online ahead of print. 10.1038/s41385-022-00536-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gutierrez MW et al. (2022) “Molding” immunity-modulation of mucosal and systemic immunity by the intestinal mycobiome in health and disease. Mucosal Immunol 15 (4) 573–583. [DOI] [PubMed] [Google Scholar]
- 48.Zhou Y et al. (2021) The interactions between Candida albicans and mucosal immunity. Front Microbiol 12, 652725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iliev ID and Cadwell K (2021) Effects of intestinal fungi and viruses on immune responses and inflammatory bowel diseases. Gastroenterology 160 (4), 1050–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Underhill DM and Braun J (2022) Fungal microbiome in inflammatory bowel disease: a critical assessment. J Clin Invest 132 (5), e155786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gaffen SL and Biswas PS (2022) Fungi make fun guys. Cell Host Microbe 30 (3), 277–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doron I et al. (2021) Human gut mycobiota tune immunity via CARD9-dependent induction of anti-fungal IgG antibodies. Cell 184 (4), 1017–1031 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ost KS et al. (2021) Adaptive immunity induces mutualism between commensal eukaryotes. Nature 596 (7870), 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bacher P et al. (2019) Human Anti-fungal Th17 immunity and pathology rely on cross-reactivity against Candida albicans. Cell 176 (6), 1340–1355 e15. [DOI] [PubMed] [Google Scholar]
- 55.Shao TY et al. (2019) Commensal Candida albicans positively calibrates systemic Th17 immunological responses. Cell Host Microbe 25 (3), 404–417 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tso GHW et al. (2018) Experimental evolution of a fungal pathogen into a gut symbiont. Science 362 (6414), 589–595. [DOI] [PubMed] [Google Scholar]
- 57.Reales-Calderon JA et al. (2021) Gut-evolved Candida albicans induces metabolic changes in neutrophils. Front Cell Infect Microbiol 11, 743735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yeung F et al. (2020) Altered immunity of laboratory mice in the natural environment Is associated with fungal colonization. Cell Host Microbe 27 (5), 809–822 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shao TY et al. (2022) Candida albicans oscillating UME6 expression during intestinal colonization primes systemic Th17 protective immunity. Cell Rep 39 (7), 110837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu Y et al. (2013) Synergistic regulation of hyphal elongation by hypoxia, CO(2), and nutrient conditions controls the virulence of Candida albicans. Cell Host Microbe 14 (5), 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kadosh D and Lopez-Ribot JL (2013) Candida albicans: adapting to succeed. Cell Host Microbe 14 (5), 483–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Netea MG et al. (2020) Defining trained immunity and its role in health and disease. Nat Rev Immunol 20 (6), 375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dobes J et al. (2022) Extrathymic expression of Aire controls the induction of effective Th17 cell-mediated immune response to Candida albicans. Nat Immunol, online ahead of print. 10.1038/s41590-022-01247-6 [DOI] [PubMed] [Google Scholar]
- 64.Oikonomou V and Lionakis MS (2022) Extrathymic Aire primes Candida-specific Th17 cells. Nat Immunol, online ahead of print. 10.1038/s41590-022-01252-9 [DOI] [PubMed] [Google Scholar]
- 65.Leonardi I et al. (2022) Mucosal fungi promote gut barrier function and social behavior via Type 17 immunity. Cell 185 (5), 831–846 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wheeler ML et al. (2016) Immunological consequences of intestinal fungal dysbiosis. Cell host & microbe 19 (6), 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li X et al. (2018) Response to fungal dysbiosis by gut-resident CX3CR1(+) mononuclear phagocytes aggravates allergic airway disease. Cell Host Microbe 24 (6), 847–856 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hueber W et al. (2012) Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 61 (12), 1693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li XV et al. (2022) Immune regulation by fungal strain diversity in inflammatory bowel disease. Nature 603 (7902), 672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Verma AH et al. (2017) Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Sci Immunol 2 (17), eaam8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Iliev ID and Leonardi I (2017) Fungal dysbiosis: immunity and interactions at mucosal barriers. Nat Rev Immunol 17 (10), 635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Iliev ID et al. (2012) Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336 (6086), 1314–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Limon JJ et al. (2019) Malassezia Is associated with Crohn’s disease and exacerbates colitis in mouse models. Cell Host Microbe 25 (3), 377–388 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mahmoudi E et al. (2021) The role of mycobiota-genotype association in inflammatory bowel diseases: a narrative review. Gut Pathog 13 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Leonardi I et al. (2018) CX3CR1(+) mononuclear phagocytes control immunity to intestinal fungi. Science 359 (6372), 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ma Y et al. (2021) The gut-lung axis in systemic inflammation. Role of mesenteric lymph as a conduit. Am J Respir Cell Mol Biol 64 (1), 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Raftery AL et al. (2020) Links between inflammatory bowel disease and chronic obstructive pulmonary disease. Front Immunol 11, 2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Noverr MC et al. (2005) Development of allergic airway disease in mice following antibiotic therapy and fungal microbiota increase: role of host genetics, antigen, and interleukin-13. Infection and immunity 73 (1), 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Noverr MC et al. (2004) Role of antibiotics and fungal microbiota in driving pulmonary allergic responses. Infection and immunity 72 (9), 4996–5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim YG et al. (2014) Gut dysbiosis promotes M2 macrophage polarization and allergic airway inflammation via fungi-induced PGE(2). Cell Host Microbe 15 (1), 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wu Y et al. (2021) Candida albicans elicits protective allergic responses via platelet mediated T helper 2 and T helper 17 cell polarization. Immunity 54 (11), 2595–2610 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McDonough LD et al. (2021) Candida albicans isolates 529L and CHN1 exhibit stable colonization of the murine gastrointestinal tract. mBio 12 (6), e0287821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Iliev ID et al. (2012) Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science, 1221789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Romani L (2011) Immunity to fungal infections. Nat Rev Immunol 11 (4), 275–88. [DOI] [PubMed] [Google Scholar]
- 85.Garcia-Rubio R et al. (2019) The fungal cell wall: Candida, Cryptococcus, and Aspergillus species. Front Microbiol 10, 2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Speakman EA et al. (2020) T cell antifungal immunity and the role of C-type lectin receptors. Trends Immunol 41 (1), 61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Drummond RA et al. (2014) Cutting edge: Failure of antigen-specific CD4+ T cell recruitment to the kidney during systemic candidiasis. J Immunol 193 (11), 5381–5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen T et al. (2022) When is it appropriate to take off the mask? Signaling pathways that regulate ß(1,3)-glucan exposure in Candida albicans. Frontiers in Fungal Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cottier F and Hall RA (2019) Face/Off: the interchangeable side of Candida albicans. Front Cell Infect Microbiol 9, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cohen-Kedar S et al. (2021) Commensal fungi and their cell-wall beta-glucans direct differential responses in human intestinal epithelial cells. Eur J Immunol 51 (4), 864–878. [DOI] [PubMed] [Google Scholar]
- 91.Braunsdorf C et al. (2016) Fungal sensing of host environment. Cell Microbiol 18 (9), 1188–1200. [DOI] [PubMed] [Google Scholar]
- 92.Pradhan A et al. (2018) Hypoxia promotes immune evasion by triggering beta-glucan masking on the Candida albicans cell surface via mitochondrial and cAMP-protein kinase A Signaling. mBio 9 (6), e01318–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Duvenage L et al. (2019) Inhibition of classical and alternative modes of respiration in Candida albicans leads to cell wall remodeling and increased macrophage recognition. mBio 10 (1), e02535–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sherrington SL et al. (2017) Adaptation of Candida albicans to environmental pH induces cell wall remodelling and enhances innate immune recognition. PLoS Pathog 13 (5), e1006403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Banerjee M et al. (2008) UME6, a novel filament-specific regulator of Candida albicans hyphal extension and virulence. Mol Biol Cell 19 (4), 1354–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Banerjee M et al. (2013) Expression of UME6, a key regulator of Candida albicans hyphal development, enhances biofilm formation via Hgc1- and Sun41-dependent mechanisms. Eukaryot Cell 12 (2), 224–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Carlisle PL et al. (2009) Expression levels of a filament-specific transcriptional regulator are sufficient to determine Candida albicans morphology and virulence. Proc Natl Acad Sci U S A 106 (2), 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Witchley JN et al. (2019) Candida albicans morphogenesis programs control the balance between gut commensalism and invasive infection. Cell Host Microbe 25 (3), 432–443 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Doron I et al. (2021) Mycobiota-induced IgA antibodies regulate fungal commensalism in the gut and are dysregulated in Crohn’s disease. Nat Microbiol 6 (12), 1493–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Leonardi I et al. (2020) Fungal trans-kingdom dynamics linked to responsiveness to fecal microbiota transplantation (FMT) therapy in ulcerative colitis. Cell Host Microbe 27 (5), 823–829 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]