

SUMMARY
Methyl-based methanogenesis is one of three broad categories of archaeal anaerobic methanogenesis, including both the methyl dismutation (methylotrophic) pathway and the methyl-reducing (also known as hydrogen-dependent methylotrophic) pathway. Methyl-based methanogenesis is increasingly recognized as an important source of methane in a variety of environments. Here, we provide an overview of methyl-based methanogenesis research, including the conditions under which methyl-based methanogenesis can be a dominant source of methane emissions, experimental methods for distinguishing different pathways of methane production, molecular details of the biochemical pathways involved, and the genes and organisms involved in these processes. We also identify the current gaps in knowledge and present a genomic and metagenomic survey of methyl-based methanogenesis genes, highlighting the diversity of methyl-based methanogens at multiple taxonomic levels and the widespread distribution of known methyl-based methanogenesis genes and families across different environments.
KEYWORDS: Archaea, anaerobic catabolic pathways, methane, methanogenesis, methanogens, methylated compounds, methylotrophs
INTRODUCTION
Methanogenesis (the production of methane gas [CH4] as a result of energy conservation) is an ancient microbial metabolism that likely played an important role in the evolution of life on Earth (1–3). Methanogenesis occurs as the final step in decomposition of organic matter in anaerobic environments and contributes most of the biotically produced methane, which makes up 70 to 90% of the methane produced on Earth today (the remainder is produced abiotically) (4–7). Today, methane (and consequently methanogenesis) is the second largest contributor to the greenhouse gas effect causing global climate change but at the same time is an important source of energy for human societies as the principal ingredient of natural gas (8, 9). Atmospheric methane concentrations have increased by about 2.5-fold from ~729 ppb in 1750 to ~1,857 ppb in 2018, which is the highest level reached in the past 800,000 years (7). The resulting radiative forcing of this increase is linked to an increase in temperature of 0.5°C when comparing 1850 to 1900 levels to 2010 to 2019 levels (8).
We have a keen interest in understanding the methane cycle from both basic and applied perspectives. Studies of the fundamental biology involved in methanogenesis are yielding new insights into anaerobic metabolism, the function of many poorly understood archaeal and extremophilic taxa, and the evolution of life on Earth as well as discoveries of taxa that can be useful for bioremediation efforts or for improving human health (10–12). We also need an improved and mechanistic understanding of the methane cycle to inform modeling efforts to more accurately understand, predict, and mitigate climate change. While great progress has been made and continues to be made on reconciling global methane sources and sinks, there are still many uncertainties (7, 13, 14). In particular, anthropogenic distortion of the natural methane cycle is a major contributor to climate change, yet new science-based technologies, such as animal feed additives (15) and rice paddy management practices (16), have the potential to reduce emissions. Furthermore, many questions remain regarding positive and negative feedbacks in the climate system, including feedbacks involving methane as a central player (17). For example, will Arctic warming and thawing permafrost contribute to increased methane fluxes? How will increased salinization from sea level rise and/or droughts affect methane fluxes from coastal wetlands? The answers to these questions depend on knowledge of the underlying microbiological processes and how different pathways of methane production will be affected. Another application for methanogenesis research is to study sources of natural gas, a key source of energy for human society. Natural gas sources are diverse, including hydrates, shales, deep aquifers, and coalbeds (9), and contain a substantial amount of biogenic methane from methanogens, including methyl-based methanogens in addition to some thermogenic methane (18, 19). Finally, anaerobic digesters of biomass, manure, or sludge from sewage treatment plants to produce methane to burn as energy rely on microorganisms to convert those products to methane (20). Thus, these two forms of energy production rely on methanogens, and a better understanding of methanogens can help us predict energy sources and produce a greater quantity of energy more efficiently.
There are three major types of methanogenesis: acetoclastic, hydrogenotrophic, and methyl-based methanogenesis (including methyl dismutation and methyl reduction). The relative contributions of these three broad categories vary among ecosystems and are influenced by environmental conditions such as temperature, the extent to which organic matter is degraded, and the other fermentation processes at work (21). There is also a recently described alternative form of methanogenesis from methylated compounds known as methoxydotrophic methanogenesis, whereby aromatic compounds are demethoxylated in a process suggested to be performed by both methanogens and nonmethanogens (22, 23). Furthermore, there are several pathways that yield methane that are not performed by methanogens, including aerobic methylphosphonate degradation (24), aerobic aspartate aminotransferase (25), cyanobacterial photosynthetic (26), and nitrogen fixation (27) pathways as well as proposed plant, animal, and fungal pathways (28, 29). These alternative pathways have been only recently described and are still poorly understood, both in terms of the biochemistry involved and the overall contribution to the global methane budget. Archaeal methanogenesis has been reviewed elsewhere (4, 20, 21, 30–33), but generally, more focus has been given to acetoclastic and hydrogenotrophic methanogenesis. Here, we specifically review methyl-based methanogenesis, as recent work has increasingly pointed to it as an important source of methane in a variety of environments. Methyl-based methanogenesis includes methanogenesis from the following methylated compounds: tetramethylammonium, trimethylamine (TMA), dimethylamine (DMA), monomethylamine (MMA), methanol (MeOH), glycine betaine (GB), dimethylsulfide (DMS), methanethiol (MT), and methylthiopropanoate (MMPA) (Table 1). We synthesize information from the literature on the environments in which methyl-based methanogenesis is prevalent, review methods for determining the source of methane, integrate and summarize biochemical information from the literature and the KEGG (34), BioCyc (35), and ModelSeed (36) databases for each methylated methanogenic substrate, and analyze the distribution and abundances of methyl-based methanogenesis genes and taxa using publicly available genomes and metagenomes in the Joint Genome Institute’s Integrated Microbial Genomes and Microbiomes (IMG/M) database (37).
TABLE 1.
Methyl-based methanogenic substrates
| Namea | Abbreviation | Formula | Structureb | CH3-Dism. | CH3-Red. | Environmentsc | Genesd |
|---|---|---|---|---|---|---|---|
| Tetramethylammonium | QMA | (CH3)4N |
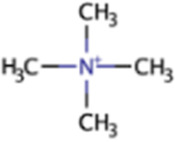
|
X | Marine sediment, industrial wastewater | mtqBCe, mtqAe | |
| Trimethylamine | TMA | (CH3)3N |
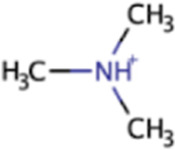
|
X | X | Marine sediment, hypersaline sediment, gut | mttBC, mtbA |
| Dimethylamine | DMA | (CH3)2NH |
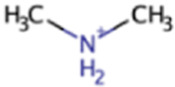
|
X | X | Marine sediment, hypersaline sediment, gut | mtbBC, mtbA |
| Monomethylamine | MMA | CH3NH2 |

|
X | X | Marine sediment, hypersaline sediment, gut | mtmBC, mtbA |
| Methanol | MeOH | CH3OH |

|
X | X | Marine sediment, freshwater sediment | mtaBC, mtaA |
| Glycine betaine | GB | C5H11NO2 |
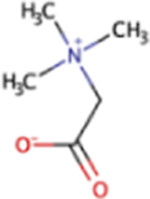
|
X | Marine sediment, hypersaline sediment | mtgBC, mtgA | |
| Dimethyl sulfide | DMS | (CH3)2S |
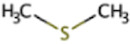
|
X | Marine sediment, hypersaline sediment | mtsAB, mtpC, mtsDEF | |
| Methanethiol | MT | CH3SH |

|
X | Marine sediment, hypersaline sediment, freshwater sediment | mtsAB, mtpC, mtsF | |
| Methylthiopropanoate | MMPA | C5H10O2S |
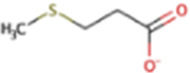
|
X | Marine sediment, hypersaline sediment | mtpP, mtsA, mtpA, mtpCAP |
Substrate names, abbreviations, formulas, structures, use in methyl dismutation (CH3-Dism.), use in methyl reduction (CH3-Red.), environments in which the substrates are present, and genes involved in the demethylation and methyl transfer to coenzyme M are shown.
Structure diagrams from ModelSEED.
Marine sediments here include ocean sediments as well as coastal and estuarine sediments, which are influenced by seawater.
Pathway-specific genes. All pathways would additionally need mcrABG. Some pathways would additionally require the Wood-Ljungdahl methyl branch and hydrogenases and membrane-bound proteins (Fig. 2).
Hypothesized but only demonstrated by one study.
CONDITIONS FAVORABLE FOR METHYL-BASED METHANOGENESIS
To understand when and where methyl-based methanogenesis represents an important microbial metabolic strategy, we must consider both abiotic conditions and biotic interactions. Important abiotic factors are substrate availability and any other environmental filters on the methanogenic archaea, such as temperature, pH, salinity, carbon availability, and nutrient concentrations (38). Biotic interactions include positive syntrophic associations and negative competitive interactions for the same limited resources. Methyl-based methanogenesis is performed by a variety of archaeal genera and species across multiple phyla, including Euryarchaeota, Halobacteriota, Thermoplasmatota, and potentially Crenarchaeota (suggested by genomic data but not confirmed experimentally) (39, 40). Environments that favor these organisms will also favor methyl-based methanogenesis.
As the energy yield of methanogenesis is typically low at only −33 to −131 ΔG° kJ/mol CH4 depending on the pathway (but could also reach a maximum of −241 ΔG° kJ/mol CH4 with glycine betaine), methanogens can be easily outcompeted by other organisms in most conditions (30). More specifically, the energy yield of the acetoclastic pathway is −33 to −36 ΔG° kJ/mol CH4, that of the hydrogenotrophic pathway is −37 to −131 ΔG° kJ/mol CH4 (depending on whether the electron donor is hydrogen, ethanol, or isopropanol and assuming abundant electron donors), that of the methyl dismutation pathway is −31 to −241 ΔG° kJ/mol CH4 (depending on the substrate), and that of the methyl-reducing pathway with methanol is −100 to −113 ΔG° kJ/mol CH4 (depending on whether hydrogen or ethanol is used as the electron donor) (30, 33). In fact, it has been proposed that methanogenesis occurs only when other more energetically favorable electron acceptors, such as nitrate (NO3–), iron (Fe3+), manganese (Mn4+), and sulfate (SO42–), have been depleted (41). For each of the aforementioned electron acceptors, experimental manipulations have shown decreased methane production when those molecules were added (42, 43). One key line of work on methanogenesis has demonstrated that sulfate reducers can outcompete methanogens when sulfate concentrations are high. Importantly, this only applies to the “competitive substrates” acetate and hydrogen, which are also used by sulfate reducers (42, 44). In key early experiments, hydrogenotrophic and acetoclastic methanogenesis were inhibited by sulfate-reducing bacteria, while methyl-based methanogens avoided this competition and remained active even under high sulfate conditions (44–50). More recent studies have built on this work by pointing to the prevalence of methyl-based methanogens and substrates in the sulfate-reducing zone in the upper layer of marine sediments (51–59). While many studies tend to label all methylated methanogenic substrates as “noncompetitive” with sulfate reducers, it is important to note that methanol is used by some sulfate reducers just like H2 and acetate (60). Some studies have still shown methanol-driven increases in methanogenesis concurrent with sulfate reduction (55, 58), but it is also possible that there could be competition between sulfate reducers and methanogens for methanol (61).
Methyl-based methanogenesis can be the dominant methanogenesis pathway in anaerobic ocean environments (e.g., sediments and hydrothermal vents), coastal environments (e.g., seagrass meadows and coastal wetlands), other environments with high salt and/or sulfate concentrations (e.g., solar salterns, hypersaline lakes, and microbial mats) (30, 51–59, 61–68), and even insect guts (69). In some cases, the high salt/high sulfate conditions go hand in hand, and the prevalence of the methyl-based pathway is likely due to other methanogens being outcompeted by sulfate reducers. In other cases where hypersaline environments have low sulfate concentrations or low sulfate reducer populations (70), the prevalence of methyl-based methanogenesis is not due to other methanogens being outcompeted by sulfate reducers but to the environmental conditions not being favorable for survival by other methanogens. This is because some methylotrophic methanogens such as Methanolobus, Methanosalsum, Methanohalophilus, Methanohalobium, and a potential new Methanosarcinaceae genus identified by metagenomics, are halotolerant or halophilic organisms; while they group phylogenetically with other methanogens, they contain the biochemical mechanisms necessary to cope with high salt via production of compatible solutes or by having an acidic proteome (39, 62, 71). Methyl-based methanogens are also favored in saline environments because the compatible solute glycine betaine produced by halophilic bacteria is fermented to TMA or used directly, which fuels methyl-based methanogens. Another potential reason why these methyl-based methanogenic taxa persist in hypersaline environments while other types of methanogens do not is that the relative energy yield of the methyl-based pathway is higher, which enables them to divert more energy to compatible solute production (72).
Such results have implications for predicting the changes in methane fluxes that will occur as many coastal and inland ecosystems are increasingly impacted by sea level rise and seawater intrusion, decreases in freshwater inputs, or human activities (e.g., irrigation and salting roads) (73, 74) that increase salinity or sulfate or both salinity and sulfate together. Ecosystems influenced by seawater will experience increases in both salinity and sulfate. Crop irrigation with groundwater or river water can also lead to increases in both salinity and sulfate, especially in arid to semiarid regions (75, 76). The salting of roads in winter increases salinity but not sulfate, as typically NaCl rather than sulfate salts are used for this purpose. However, some human activities lead to increases in sulfate and not salinity. Commonly used nitrogen, phosphorus, and potassium fertilizers are a major source of sulfates in ecosystems affected by agricultural runoff (75, 77, 78). Additionally, fossil fuel-fired power plants are associated with increases in sulfate but not salinity due to wet and dry deposition of sulfur emissions to the atmosphere (79). This source of sulfate has decreased in the United States and Europe (80) but still contributes a substantial amount of sulfate in some parts of the world (81).
METHODS FOR ASSESSING METHYL-BASED METHANOGENESIS
There are several different ways in which researchers can shed light on the relative contributions of different pathways, including but not limited to methyl-based pathways, to methanogenesis. These can be broadly grouped into four categories: isotope-based methods, geochemical measurements, nucleic acid sequencing, and substrate addition experiments. Each category has its own set of advantages and disadvantages (Table 2). The most convincing and conclusive claims about dominant methane sources will use a combination of these methods, as has been done recently by several studies on methyl-based methanogenesis (59, 64, 65).
TABLE 2.
Summary of the advantages and disadvantages of different methods to determine the importance of different methanogenesis pathways
| Method | Advantages | Disadvantages |
|---|---|---|
| Isotope-based methods | Can clearly distinguish some pathways based on natural 13C abundance | Can be ambiguous, as some substrates have overlapping signatures |
| Can clearly track carbon from substrate to product with 14C label | Can be influenced by other biochemical processes/pathways such as methanotrophy that affect natural 13C abundances | |
| Can be ambiguous if different CH4 pools in the environment mix physically before measurement | ||
| Substrate quantification | Identifies which substrates are present | Can be difficult for some substrates and require specialized analytical methods |
| Quantifies concentration of substrate to help interpret its relevance | Measures substrate pools rather than production and consumption rates | |
| Nucleic acid sequencing | Enables high-throughput processing of many samples | Can be affected by organisms and genes that are present but not active or expressed (DNA) |
| Provides a deeper understanding of the ecology and taxonomy | Can be affected by relic DNA | |
| Facilitates comparisons to databases and other studies | Can be difficult for RNA, which is easily degraded | |
| Can assess which genes are actively expressed (RNA) | ||
| Provides a clear relationship between substrate concentration and CH4 produced | ||
| Substrate addition experiments | Enable manipulating other environmental variables of interest | Are performed under laboratory conditions that cannot completely mimic field conditions |
| Can be performed on microcosms or isolates | Test potential rates of methanogenesis, not the actual field rates | |
| Can utilize inhibitors to help indicate importance of a pathway | ||
| Can be combined with isotope-based methods to clearly link substrate to methane | ||
| Can be combined with microscopy-based methods to show the spatial arrangement of microorganisms |
Isotope-Based Methods
Heavy isotopes are a useful tool for tracking the flow of elements through biological systems. In particular, to study methanogenic pathways, heavy isotopes of carbon and/or hydrogen can be used. Due to the activities of enzymes and their preference for certain isotopes, products of enzymatic reactions will have a different isotopic makeup than the background in the environment.
Isotopic profiling to determine methane sources could involve one of three methods: 13C and/or 2H fractionation of methane and substrates, 13C fractionation of the biomass or lipids of the methanogens, or heavy isotope labeling. Isotopic profiling methods can be performed on environmental samples or combined with substrate addition experiments in the laboratory on microcosms or isolates. The most common isotopic method is examination of the natural abundance of 13C and/or 2H in methane from a particular environment; this method has been used since the late 1950s (82, 83) and can be used to distinguish between biotic and abiotic sources of methane and different pathways of biotic and abiotic methanogenesis (5). Early observations of the differences between the δ13C and δ2H values of methane in marine and freshwater sediments suggested that different substrates might yield different isotopic profiles and led to subsequent experimental work with isolates to match signatures to substrates (82). Isotopic profiling of methane is recognized as an important method for understanding sources of methane at regional to global scales (13).
Carbon fractionation values are more frequently used than hydrogen fractionation values and have been specifically determined for many methanogenic substrates (84). For example, the aerobic bacterial methylphosphonate degradation pathway that produces methane as a byproduct of phosphorus acquisition yields methane molecules with only minor depletion in 13C (mean = −1.3‰) (85), while for anaerobic methanogenesis, 13C depletion values range from −9‰ to −95‰ (Table 3). Within the anaerobic methanogenesis pathways, there is also substantial variation in fractionation values, distinguishing hydrogenotrophic, acetoclastic, and methyl-based methanogenesis (84). Notably, there are still many methylated substrates that have not been analyzed for 13C depletion, which is an area for future work (Table 3). 2H fractionation studies are less common than those on 13C, but there is some evidence of differences in 2H fractionation between CO2-derived methane and acetate-derived methane (86, 87). Although isotopic methods can be clear and definitive in some cases (i.e., when there is no overlap in values), or at least enable researchers to eliminate potential pathways (i.e., when values are out of the range of at least one substrate), there are also some drawbacks, including nondefinitive cases in the overlapping ranges of substrates. The carbon isotope fractionation also depends on other variables, most notably temperature but also growth phase, hydrogen supply, substrate concentration, methanogen species, effects of other enzymes involved in carbon acquisition, and methanotrophy, as methanotrophs preferentially consume 12C-CH4 over 13C-CH4 (86, 88–90). Environmental measurements of isotope fractionation in methane can also be impacted by the mixing of multiple gas sources (91).
TABLE 3.
Summary of isotope delta and fractionation value ranges from the literature
| Pathway | Substrate | δ13Csubstratea | δ2HCH4a | δ13CCH4a | Exptl εCH4-substratea | Refs |
|---|---|---|---|---|---|---|
| Bacterial | MPn | −100.7 to −95.86b | −39 | −5.11 to 2.91 | 85, 104 | |
| Methyl based | QMA | |||||
| TMA | −36.9 to −29.5b | −97 to −83 | −83 to −39 | 84, 86, 88, 93, 225 | ||
| DMA | ||||||
| MMA | ||||||
| MeOH | −46.2 to −37.7b | −129.6 to −46.4 | −94 to −68 | 84–86, 88 | ||
| GB | ||||||
| DMS | −24.4 to −18.6c | −54 to −44 | 86, 225 | |||
| MT | ||||||
| MMPA | ||||||
| Hydrogenotrophic | CO2 | −49.7c to 16.7b | −266 to −153 | −108 to −60 | −95 to −23 | 82, 84–87, 94, 226 |
| Acetoclastic | Acetate | −36.4 to −22.1b | −396 to −266 | −70 to −27 | −35 to −6 | 84, 85, 87, 227 |
The δ13C of the substrates (from the environment or experimental reagents), the δ2H of methane, the δ13C of methane, and the 13C fractionation factor (methane-substrate) ε, calculated from experiments as described elsewhere (84) and is commonly reported in the literature, are shown. All numbers reflect per mille (‰) values. δ2H is calculated using the Vienna Standard Mean Ocean Water standard, while δ13C is calculated using the Vienna Pee Dee Belemnite standard. Blank cells indicate no known reports in the literature as of October 2022.
Reagent used in experiments.
From natural environments.
A related but less frequently used stable isotope method involves looking at the carbon fractionation not of the methane itself, but of the biomass or lipids of the microorganisms hypothesized to be performing the methanogenesis. Archaeal lipids are composed of an isoprenoid chain, an ether linkage, and a glycerol-1-phosphate backbone, while bacterial phospholipids consist of a fatty acid chain, an ester linkage, and a glycerol-3-phosphate backbone. The archaeal domain contains substantial variation in the length, composition, and configuration of the isoprenoid chain and modifications to the polar head groups (92). Differences in lipid and biomass 13C fractionation are expected among different methanogens due to differences in their carbon assimilation pathways (88). This method was developed in the versatile Methanosarcina barkeri species but was found to not be as conclusive as studying 13C fractionation of the methane molecule itself, as the fractionation of methane was more pronounced (88). Even so, it can be used in concert with other methods, and several studies have reported the δ13C of biomass or archaeal lipids (59, 93, 94), although this likely includes nonmethanogens and does not distinguish among organisms using similar carbon assimilation pathways but different energy conservation pathways. Lipids were enriched in 13C relative to the substrate and bulk biomass only when methanogens were grown with added acetate and not H2/CO2, methanol, or trimethylamine under conditions of limited substrate availability (88).
A third isotopic method is to use a 14C tracer to track carbon from a labeled substrate to the methane molecule; in such experiments, a suspected methane-producing environmental sample (e.g., sediment) is brought back to the lab, known quantities of 14C-labeled substrates are added, and 14C-CH4 is quantified. An advantage of this method is that it enables quantification of the contribution of different pathways to the overall methane flux. The contributions of acetate, CO2, trimethylamine, monomethylamine, methanol, and glycine betaine have been studied by this method while, to our knowledge, to date, quaternary methylammonium or tetramethylammonium, dimethylamine, dimethylsulfide, methanethiol, and methylthiopropanoate have not. Such experiments have been done using a variety of sediments, including those from freshwater lakes, the deep ocean, coastal salt marshes, and rice fields as well as rumen fluid and feed (59, 61, 63, 65, 95–100).
Finally, stable isotope probing (SIP) can be used to assess microbial function in environmental samples by using isotopic probes to link identity and function. A wide variety of SIP techniques, including DNA-SIP, RNA-SIP, protein-SIP, phospholipid fatty acid analysis-SIP (PLFA-SIP), and metabolite-SIP have been developed to focus on different aspects of biology and biochemistry (101), and new methods are still actively being developed (e.g., flow-SIP) to minimize uncertainties arising from microbial cross-feeding (102). SIP is often combined with secondary ion mass spectrometry (SIMS) for isotopic and elemental analysis and with in situ hybridization methods, such as fluorescence in situ hybridization (FISH), to link microbial identity to isotopic enrichment.
Substrate Quantification
A seemingly simple way to test if a pathway is relevant in an environment is to directly measure the concentration of the substrate. However, this can be challenging in terms of both measurement and interpretation, as such measurements only quantify pools and not active production or uptake. Many of the methylated methanogenic substrates as well as methylated methane precursors in bacterial methane production pathways are notoriously hard to identify and detect, can be strongly sorbed to sediments, and require specific extraction and spectrometry methods (63, 103). For example, clearly identifying a pool of methylphosphonate required 31P nuclear magnetic resonance spectroscopy (104). In addition to the substrate concentrations themselves informing likely methanogenesis pathways, a second step is to take those concentrations and make thermodynamic calculations that can be used in concert with the substrate concentration data and other environmental parameters to assess the likelihood of the occurrence of a pathway. Assuming that −15 kJ mol substrate−1 energy must be available to be usefully harnessed and support basic biochemical integrity and function (105, 106), if certain pathways fall below this cutoff, it is likely that they do not occur (59). Such conclusions, however, depend on accurately measuring the substrate concentrations, which can be challenging (see above).
Nucleic Acid Sequencing
DNA and RNA sequencing methods can be used to identify relevant pathways via the presence, abundance, or expression of key genes and taxa. This works because many of the pathways have genes, enzymes, and proteins isolated, purified, and described, and many taxa are known methanogens with available cultures and experimental evidence. Thus, metagenomic or genomic data (or better metatranscriptomic or transcriptomic data) can show which genes are present or expressed, while 16S rRNA or mcrA marker gene surveys can identify potentially methanogenic archaeal taxa, many of which have been cultured and have known substrates that they can or cannot metabolize. The 16S or mcrA gene is used to identify organisms in the sample; assessment of methanogenesis capabilities would be based on prior experiments done with the identified taxa. The mcrA gene is also present in anaerobic methanotrophic (ANME) archaea so this gene in itself is not necessarily a marker of methanogens, although it is commonly used as such due to its presence in all methanogens (i.e., all methanogens have mcrA but not all mcrA-containing organisms are methanogens). Sequencing data can be an effective method for broad surveys and to answer other biological, ecological, and functional questions at the same time, and there are extensive databases from many environments that can be queried for genes or taxa of interest.
But sequencing data also have disadvantages. For example, metagenomic data could provide support for a pathway but not conclusive evidence, as it would not show if the genes were actively expressed. This can become especially problematic considering that some taxa are versatile methanogens and may harbor genes for multiple methanogenesis pathways but only be metabolizing a specific substrate under the conditions of the particular sample. Likewise, using taxonomy could also be inconclusive depending on the taxa involved. For example, members of the family Methanosarcinaceae can perform all four major archaeal methanogenic pathways, so an operational taxonomic unit (OTU) or amplicon sequence variant (ASV) assigned only to that family would be inconclusive in terms of identifying the methanogenic pathway, while identifying a particular species that has been verified in culture to use only specific substrates would constitute much stronger evidence. Of the sequence-based methods, generating metatranscriptomic or transcriptomic data under methanogenic conditions would be the most conclusive as it would show the actively expressed genes. Yet, even the conclusions from metatranscriptomic or transcriptomic data can be limited when protein or activity quantification are lacking.
Substrate or Inhibitor Addition Experiments
A final method for distinguishing methane pathways is to perform substrate addition experiments, in which known concentrations of substrates are added to microcosms or cultures, and methane production is measured over time and compared to controls. The benefits of this method are that the production of methane can be validated, the rate of production can be quantified, and the methane flux from different potential substrates can be compared. This is commonly done for pure methanogen cultures to characterize the capabilities of specific isolates (107) but is also done at the microcosm level (70).
In addition to testing methane production from different substrates, chemical inhibitors can also be added to specifically inhibit certain pathways and thereby strengthen conclusions about other pathways. 2-Bromoethanesulfonic acid or chloroform is used to inhibit methanogens and confirm biogenic methane origin (108–110). Sodium molybdate inhibits sulfate reduction, and fluoroacetate inhibits acetate metabolism (100). Methyl fluoride specifically inhibits acetoclastic methanogenesis (84, 99).
Lastly, the substrate addition method can be combined with stable and radioactive isotope methods by performing an isotopically labeled substrate addition (see above section). This would add additional clarity to the interpretation of the results, as potential contributions of nonadditive substrates to net methane production in unlabeled enrichments could confound those results. Substrate addition experiments could also be combined with recently developed microscopy-based methods such as the combination of FISH and biorthogonal noncanonical amino acid tagging (BONCAT), which has been demonstrated to be a powerful tool for assessing microbial function as it enables visualization of newly made proteins (111, 112). BONCAT-FISH has been used to study methane-oxidizing microbial consortia (113) and could be used to identify different methanogenesis pathways by linking microbial taxonomy and translational activity in environmental samples with different substrate additions. The downside of substrate addition experiments is that laboratory conditions do not mimic field conditions, and, as such, the experiment provides information only on the capacity of the microorganisms in a sample to produce methane from a substrate and not the field production rates or even that they actively produce methane in the field.
In summary, there are four main types of methods for determining methane sources (Table 2), and the strongest cases about the relative importance of different pathways can be made by combining multiple lines of evidence, as has been done by several studies on methylotrophic methanogenesis (59, 64, 65).
SUBSTRATE SOURCES AND BIOCHEMISTRY
As the methyl dismutation and methyl-reducing pathways of methanogenesis both involve the demethylation of methylated compounds, a variety of compounds can serve as the primary substrates and carbon sources for methanogenesis. Known methylated substrates include QMA, TMA, DMA, MMA, methanol, methanethiol, MMPA, DMS, dimethylsulfoniopropionate (DMSP), glycine betaine (GB), choline, methionine, and dimethylethanolamine (DMEA). An overview of the biochemical reactions and genes involved in these pathways is presented in Fig. 1, with the exception of choline, methionine, and DMEA, which have demonstrated methane-producing potential but may only function as precursors, as the mechanisms have not been fully characterized (114–116).
FIG 1.

Simplified map of methyl dismutation methanogenesis pathways showing ModelSeed compound names and reaction IDs as well as KEGG Orthology (KO) identifiers (when available) for the genes involved in the reactions. This map focuses on the methyl dismutation steps and does not show all of the proteins and pathways for energy conservation. The map was made with Escher based on MetaCyc, KEGG, and ModelSeed annotations and only includes those pathways in at least one of those databases as of the time of writing. Note that some gene names are described but do not yet have KO assignments. Also note that some reactions are not in ModelSeed (e.g., for betaine). The asterisk (*) indicates that methanogenesis from methylthiopropanoate differs in Ms. barkeri (“M. bar”) and Ms. acetivorans (“M. ace”), with Ms. acetivorans using MtsC proteins instead of MtsB proteins. A question mark (?) denotes hypothesized genes in need of further confirmation.
Generally, methanogenesis from methylated compounds using the methyl dismutation pathway follows a two-step process in which the substrate is demethylated, with the methyl group first transferred to a substrate-specific corrinoid protein and then to coenzyme M (CoM) to produce methyl-CoM (35, 117). Methyl-CoM then reacts with coenzyme B (CoB) to yield methane in the reaction catalyzed by methyl-CoM reductase (encoded by mcrABG). This is the same ultimate methane-forming reaction that occurs in the other methanogenesis pathways. Methanogens performing methyl dismutation reduce 75% of methyl groups using electrons obtained by oxidizing 25% of the methyl groups to CO2 with the methyl branch of the Wood-Ljungdahl pathway (33). Energy conservation happens during membrane-bound electron transport (Fig. 2).
FIG 2.

(A and B) Diagram of methyl dismutation (A) versus methyl-reducing (B) pathways from methanol (adapted from Kurth et al. [33]). The pathway in Methanomassiliicoccus luminyensis is shown in B. Note that methyl-reducing taxa may or may not contain the genes for the methyl branch of the Wood-Ljungdahl pathway, and activity/growth experiments are recommended to confirm the methyl-reducing pathway; WL, Wood-Ljungdahl; MFR, methanofuran; H4MPT, tetrahydromethanopterin; Fwd/Fmd, formylmethanofuran dehydrogenase; Ftr, formylmethanofurantetrahydromethanopterin formyltransferase; Mch, methenyltetrahydromethanopterin cyclohydrolase; Mtd, methylenetetrahydromethanopterin dehydrogenase; Mer, 5,10-methylenetetrahydromethanopterin; Mtr, tetrahydromethanopterin S-methyltransferase; Mta = methyl-coenzyme M methyltransferase (methanol/glycine betaine-specific corrinoid protein); Mcr, methyl-coenzyme M reductase; Fpo, F420H2 dehydrogenase; Hdr, membrane-bound heterodisulfide reductase; Ech-H2ase, energy-conserving hydrogenase; Rnf, Na+-translocating ferredoxin:NAD+ oxidoreductase complex; MP, methanophenazine; AH2, hydrogen donor; A, hydrogen acceptor; H4MPT, tetrahydromethanopterin; MTAC, CoI-corrinoid-Fe-S-proteins; CoM, coenzyme M; CoB, coenzyme B; CoM-S-S-Cob, coenzyme B-coenzyme M heterodisulfide; F420, coenzyme F420; Fd, ferredoxin, a two electron carrier; red, reduced; ox, oxidized. Na+/H+ translation stoichiometry is not represented in the figure.
Even though methyl-based methanogenesis differs significantly from hydrogenotrophic methanogenesis, the methyl reduction pathway is dependent on hydrogen (or formate or ethanol) for reducing electrons (33, 39, 118, 119). Genes for the methyl reduction pathway have been found in the Korarchaeota phylum (120), the Methanonatronarchaeia class (121), the Methanomassiliicoccales order (39, 122), the Methanosphaera genus (Methanobacteriaceae), and the Methanosarcinales order (69), the latter of which also contains members performing methyl dismutation methanogenesis without hydrogen as well as acetoclastic and hydrogenotrophic methanogenesis. Taxa performing methyl-reducing methanogenesis lack the methyl branch of the Wood-Ljungdahl pathway and instead use H2 (or formate or ethanol) as the electron donor. Genomes of organisms that perform the methyl-reducing pathway may completely or partially lack the genes of the methyl branch of the Wood-Ljungdahl pathway (123, 124). This is a key difference between the methyl dismutation and methyl-reducing pathways (Fig. 2). The methyl-reducing pathway may involve one of several different systems of membrane-bound electron transport depending on the species (Fig. S1 in the supplemental material) (33). The methyl-reducing pathway can be confirmed by activity and growth experiments that demonstrate a lack of methane production in the absence of H2 or other electron donors.
There are key differences in how energy is conserved among the hydrogenotrophic, acetoclastic, methyl dismutation, and methyl reduction methanogenesis pathways. In hydrogenotrophic methanogenesis, energy conservation happens exclusively during a methyl transfer reaction involving the membrane-bound Mtr methyltransferase, which transports sodium ions across the membrane, building up a sodium motive force that can be used by ATP synthase (125). In acetoclastic methanogenesis, energy conservation happens at the Mtr step as well as in a membrane-bound ferredoxin-heterodisulfide electron transport chain (33, 126). In contrast to hydrogenotrophic and acetoclastic methanogenesis, energy conservation in methyl-based methanogenesis does not occur at the Mtr step because it operates in reverse (Fig. 2). In methanogenesis via methyl dismutation, energy conservation occurs during membrane-bound electron transport involving ferredoxin, heterodisulfide, and methanophenazine, although there are differences among taxa, such as the model organisms Methanosarcina acetivorans and Methanosarcina barkeri. The hydrogenase-proficient Ms. barkeri uses the Frh, Ech, and Vht hydrogenases, while the hydrogenase-deficient Ms. acetivorans uses the Rnf enzyme complex and the dehydrogenase Fpo (125). In methanogenesis via methyl reduction, energy conservation occurs during membrane-bound electron transport involving ferredoxin and heterodisulfide; whether methanophenazine is used or not and which protein is used varies among taxa (Fig. 2; Fig. S1) (33, 126). Furthermore, in certain instances of methyl reduction, energy conservation may also occur with a sodium motive force, such as that generated by EhbA-Q in Methanosphaera stadtmanae (33, 126, 127).
In the following sections, we will provide an overview of the sources and sinks of each substrate, including biosynthesis and nonmethanogenic degradation pathways that both affect the available pools of the substrates and then describe the methanogenesis pathway from each substrate (Fig. 1).
Tetramethylammonium
Tetramethylammonium or quaternary methylammonium (QMA) is commonly present in a variety of marine animals, including the phyla Cnidaria, Mollusca, and Bryozoa (128). QMA often forms salts of chloride or hydroxide, the latter of which is toxic and is a constituent of industrial wastewater (129). QMA hydroxide is produced for several manufacturing industries for uses that include electronic chips, semiconductors, liquid crystal displays, and light-emitting diodes. QMA is also abundant in wastewater from these industries, which has sparked interest in QMA-degrading microbes (12). The hypothesized methanogenesis pathway from QMA begins when it reacts with H+ and a Co(I) QMA-specific corrinoid protein (MtqC) to form TMA and a methyl-Co(III) QMA-specific corrinoid protein in a reaction catalyzed by a methyltransferase enzyme encoded by mtqB. Next, a second methyltransferase enzyme encoded by mtqA catalyzes the reaction to methylate coenzyme M with the methyl-Co(III) QMA-specific corrinoid protein, yielding methyl-CoM, H+, and the Co(I) QMA-specific corrinoid protein to be recycled back into the first reaction (130) (Fig. 1).
Trimethylamine
Trimethylamine (TMA) is produced via the degradation of several precursors, including QMA (see above), choline, glycine betaine, trimethylamine-N-oxide (TMAO), carnitine, and diacylglyceryl hydroxymethyl N,N,N-trimethyl-β-alanine (DGTA) (131). TMA appears to be released by the cordgrass Spartina alterniflora in salt marshes, is present in benthic animals and phytoplankton in marine ecosystems where it can contribute to methanogenesis (132), and is also present in many plant and fungal species in marine and terrestrial ecosystems (133). TMA is also present in ruminants as a product of betaine degradation (96). In humans, TMA is produced from l-carnitine and choline by gut microflora, is excreted in urine, and has been associated with disease, effects on the circulatory system, and other negative effects (134–136). Because of this, there is interest in the role of methanogenic archaea in the gut to potentially remove TMA (and DMA and MMA) locally (11).
TMA has gained a lot of attention as an important methylated methanogenic substrate due to the ubiquity and abundance of the aforementioned precursors. Choline, glycine betaine, and TMAO are produced abundantly and ubiquitously by both prokaryotic and eukaryotic organisms, particularly in marine environments (93, 137), and genes encoding proteins involved in their degradation to TMA are similarly ubiquitous and abundant (138, 139). TMA dominated the exchangeable pool of amines in a salt marsh (132). Open water and sediment porewater concentrations of TMA range from 0.05 nM to 50 μM (131). In hypersaline environments, TMA can be formed primarily by breaking down glycine betaine, which is an abundant compatible solute in those environments (137). TMA itself has also been suggested to be a compatible solute, and its concentrations vary seasonally and with salinity as a function of benthic invertebrate concentrations (140). TMA has been found to be more abundant in the solid phase than in the dissolved pool, suggesting strong adsorption to sediments, a factor that must be taken into account when considering TMA sinks (103). In shallow marine sediments, TMA and DMA were both detected while MMA was not, suggesting some differences in the cycling of these three methylated amines. In the same study, TMA and DMA concentrations increased as organic matter content increased (141).
TMA can be degraded aerobically and anaerobically via several nonmethanogenic pathways, which thereby decrease pools available for methanogenesis (131, 142–144). Aerobic marine bacteria, particularly those in the Roseobacteria clade, use TMA monooxygenase (encoded by the tmm gene) to use TMA as a carbon and nitrogen source, with some specialized methylotrophs capable of growing on TMA as their sole source of carbon and energy (145). Anaerobic denitrifying bacteria can use TMA as a carbon source for growth with nitrate (142, 146). A second anaerobic pathway is the TMA dehydrogenase pathway, which produces DMA, MMA, formaldehyde, and ammonia (131). TMA can also be used to form acetate by Acetohalobium, a versatile halophilic bacterium (147). In methanogenesis from TMA, TMA is first degraded to DMA in a two-step reaction where a methyl group, catalyzed by the MttB methyltransferase enzyme, is first transferred to the MttC corrinoid protein to generate H+ and methylated MttC (148). The methyl group is then transferred from methyl-MttC to CoM to form methyl-CoM in a reaction catalyzed by a second methyltransferase enzyme (encoded by mtbA), which is the same enzyme and gene used for methyl-CoM production from DMA and MMA (Fig. 1, see below) (144).
An important aspect of methyl group transfer reactions is that they depend on the redox state of the corrinoid protein (e.g., highly reducing Co[I], inactive Co[II], and methylated Co[III]). The iron-sulfur RamA protein, which is often encoded near the methyltransferases in methanogen genomes, is necessary to activate the methyltransferase reactions in the TMA, DMA, and MMA pathways (149, 150). Without RamA, adventitious oxidation of the corrinoid protein to the Co(II) state would inactivate the methyltransferase reactions. RamA returns the corrinoid protein to the Co(I) state via ATP-dependent reduction (150). Recent work on this protein has demonstrated its dependence on ions such as potassium and ammonium and described the steady state kinetics of ATP dependence (151).
Dimethylamine
Environmental dimethylamine (DMA) is likely primarily produced as a product of trimethylamine degradation (see above) and degradation of trimethylamine N-oxide (143). It follows that DMA is ubiquitous in marine waters and sediments just as these two precursors and their precursors are. DMA is also found in human and rat guts, where it is produced from choline, lecithin, methylamine, and methionine and is excreted in urine (134). DMA can be found in plants and fungi; while it was found in only 2 of 28 marine plant species, which is much fewer than TMA (23 of 28 species), DMA was similarly widespread in many species of Basidiomycete fungi (133). DMA (and also MMA) increased seasonally in a salt marsh as a result of new organic matter inputs from senescing marsh grasses (103).
Besides serving as a substrate for methanogenesis, DMA can be broken down photochemically to form N-nitrosodimethylamine (152) as well as by aerobic marine bacteria that use DMA monooxygenase (encoded by dmmABC genes) to form MMA (143). Anaerobic bacteria can also use DMA dehydrogenase to produce MMA and formaldehyde (131). While denitrification involving TMA and MMA has been demonstrated, it is unclear whether denitrification involving DMA can occur or if DMA is only an intermediate in TMA-dependent denitrification (142, 146, 153). Methanogenesis from DMA follows a similar pathway as the pathway for TMA, but MtbB and MtbC proteins are involved instead of MttB and MttC proteins (Fig. 1) (144).
Monomethylamine
Monomethylamine (MMA) is produced in marine environments from microbial degradation of DMA and glycine betaine (154) (see above and below), in animal guts from sarcosine, glycine, creatine, and epinephrine (155, 156), and in terrestrial ecosystems by flowering plants and fungi (133). MMA is present in ruminant guts, where it makes up a substantial portion of the nitrogen content along with ammonia (157), and in human and rat guts, where it is produced primarily from sarcosine and oxidized to CO2 and NH3 (156). Human adults can excrete several milligrams of MMA per day in their urine (158). MMA is a smaller component of animal tissue than TMA, but worms may excrete MMA and contribute to MMA pools in salt marshes where methanogenesis is known to occur in sediments (132, 159).
Like TMA and DMA, aerobic marine bacteria can use MMA as a source of carbon, nitrogen, and energy, in some instances as the sole source (145). MMA can also be used as an electron donor in anaerobic denitrification (153). In the last step of the dehydrogenase pathway originating with TMA, MMA dehydrogenase is used by anaerobic and aerobic bacteria to form ammonia and formaldehyde from MMA (131). Methanogenesis from MMA follows the same general pathway as TMA and DMA except that MtmB and MtmC proteins are used (Fig. 1) (144).
Methanol
Globally, the primary source of methanol (MeOH) is production from plants and subsequent release as a volatile organic compound (160). Dissolved methanol can also be present in coastal and freshwater wetlands as a product of pectin, xylan, lignin, or aromatic acid degradation (98, 161–164). Although these precursors constitute a large fraction of plant-derived organic matter, methanol typically does not contribute as much methane flux as acetate or H2/CO2, perhaps only 1 to 10% (46, 165). Even in saline environments where methanol might be predicted to play a larger role, TMA appears to contribute more to methane flux (46). Methanol is also released by chemical and enzymatic methylation of methoxy groups. In marine environments, in situ production and external deposition from terrestrial ecosystems and the atmosphere contribute to methanol concentrations in ocean sediments ranging from 0.3 μM to 111.7 μM depending on the depth (60).
Methanol can be oxidized aerobically to CO2 by bacteria and fungi that use it as a carbon and energy source (166). Anaerobically, methanol serves as a carbon source and electron donor for denitrification, acetogenesis, and sulfate reduction, all of which consume methanol and could lower the available methanol for methanogenesis (60, 167, 168). Methanogenesis from methanol follows a similar path as TMA, DMA, and MMA but with MtaB and MtaC proteins in the initial step (169) and MtaA instead of MtbA to transfer the methyl group from the corrinoid protein to CoM (170). Similar to RamA in the TMA, DMA, and MMA methyltransferase reactions, the RamM protein is required to reduce the corrinoid-binding proteins in the methanol pathway (149).
Glycine Betaine
Glycine betaine (GB) is an important osmolyte that can be found in high concentrations in saline to hypersaline environments, both inside and outside cells (137, 171). A wide variety of organisms, including plants, archaea, cyanobacteria, and mammals, have been reported to accumulate GB as a compatible solute (137). As it is present in plants, GB consequently is present in the diets of ruminants and other herbivores (96). Five pathways of glycine betaine biosynthesis have been described, encompassing those from choline performed by Gram-positive bacteria, Gram-negative bacteria, and plants and two pathways from glycine (via sarcosine) performed by archaea and bacteria (35). Genes for GB biosynthesis and transport have been found in hypothesized halotolerant methanogenic archaea and methylphosphonate-degrading bacteria (71, 172).
In addition to serving as a substrate for methanogenesis, GB can be degraded via three other pathways, which would consume GB and decrease pools available for methanogenesis. GB degradation forms many other metabolites, including N,N-dimethylglycine, sarcosine, l-serine, pyruvate, and acetate (35, 173). In the ruminant gut, trimethylamine, dimethylglycine, and methionine can be produced from GB (96). Members of the sulfur-reducing archaeal genus Halalkaliarchaeum are able to use GB as a carbon source and electron donor for sulfur reduction (174). The pathway for methanogenesis from GB has been recently described and involves a methyltransferase enzyme (encoded by the mtgB gene) in a reaction between GB and H+ to produce N,N-dimethylglycine and a methyl-Co(III) glycine betaine-specific corrinoid protein MtgC. Then, a methyltransferase enzyme encoded by mtgA, which is homologous to the one involved in methyl-CoM formation from methanol, is used to methylate CoM to produce methyl-CoM and H+ and MtgC, which can be recycled back into the first reaction (175, 176). Additionally, as was the case for methanol, the RamM protein is required to reduce the bound corrinoid of MtgC (176).
Dimethylsulfide
Dimethylsulfide (DMS) is a volatile organic sulfur compound estimated to constitute up to 90% of reduced sulfur in surface seawater. DMS concentrations in seawater follow a clear seasonal pattern of low concentrations during winter and high concentrations during summer, suggesting a biological origin (177). The most likely primary source of DMS in marine ecosystems is the multifunctional compound dimethylsulfoniopropionate (DMSP), which is produced in large quantities by marine plants, algae, phytoplankton, and other organisms and may account for up to 10% of the carbon fixed by marine phytoplankton (178–180). DMSP has been described as an osmoprotectant, cryoprotectant, predator deterrent, and antioxidant (181–183). DMSP can be degraded via several different pathways that occur in both marine and freshwater environments involving bacterial and algal lyase enzymes (184). Two such cleavage pathways directly form DMS as a product, while a third forms methylthiopropanoate (MMPA; see below) (180, 185, 186). Secondary sources of DMS include methionine, methanethiol methylation, dimethyl sulfoxide reduction, and sulfonium salts other than DMSP, such as S-methylmethionine or trimethylsulfonium (35, 181, 187). DMS levels may increase with salinity, as a positive relationship between salinity and DMS levels was reported in algal cultures (188).
DMS can be degraded with DMS monooxygenase (encoded by dmoAB) to form methanethiol (MT) and formaldehyde (189), oxidized to dimethyl sulfoxide with DMS oxygenase (encoded by dsoBCDEF) (190), or oxidized by sulfate- and nitrate-reducing bacteria (191). Methane production from DMS, MT, and MMPA all follow similar pathways (192). A key difference between methanogenesis from DMS, MT, and MMPA and the other six substrates (described above and shown in Fig. 1) is that only one methyltransferase enzyme is present, and it catalyzes both the methylation and demethylation of the corrinoid protein. There are also some differences that have been described between Methanosarcina barkeri and Methanosarcina acetivorans (117, 192). In methanogenesis from DMS in Ms. barkeri, DMS, H+, and MtsB proteins react to form methylated MtsB proteins, a reaction catalyzed by a methyltransferase encoded by mtsA. Then, MtsA combines CoM and the methylated MtsB proteins to form methyl-CoM, H+, and free MtsB proteins to be recycled back into the first reaction (Fig. 1) (192, 193). It has been suggested that in Ms. acetivorans, MtsDFH may be used as the methyltransferase instead of MtsA, and MtsB is not needed (117, 194).
Methanethiol
MT is a volatile sulfur compound with a single methyl group (CH3) and an active thiol (-SH) group that appears as a gas or as a labile acid. As with DMS, MT is produced via degradation of DMSP and methionine, perhaps in even greater levels than DMS (179, 187). In this pathway, which differs from the two DMS-producing cleavage pathways, MMPA is produced as an intermediate, which is transformed through three subsequent reactions to MT (180). Bacteria were shown to take up 15 to 40% of [35S]DMSP sulfur. Alphaproteobacteria degraded DMSP to MT and rapidly incorporated it into macromolecules (195). In addition to these bacteria, algae and plants, particularly in marine environments, can also generate MT. For example, one survey found that MT was produced by 87 of the 118 herbaceous plants studied (196). MT is also produced from DMS with DMS monooxygenase or methanogenesis (see above), from H2S via thiol transmethylation, and from MMPA (179, 181, 193). Substantial rates of MT formation have been observed in Sphagnum peat bogs, salt marsh sediments, and freshwater habitats. MT in the atmosphere is oxidized to dimethyl disulfide (DMDS), while in marine environments, MT could be converted to carbonyl sulfide (181). While some MT is taken up by microorganisms, some MT is also likely sorbed to dissolved organic matter (179).
The methanogenesis pathway from MT is similar to DMS. In Ms. barkeri, MtsB proteins and MT react to produce H2S and methylated MtsB proteins in a reaction catalyzed by a methyltransferase enzyme encoded by mtsA. Subsequently, MtsA catalyzes the reaction to form methyl-CoM as above (Fig. 1) (192). In Methanosarcina acetivorans, MtsF may be used instead of MtsA, with no need for MtsB (117).
Methylthiopropanoate
Methylthiopropanoate (MMPA) is the third of the methylated sulfur compounds that can be produced from DMSP. MMPA can also be produced from methionine or via oxidation of 1,2-dihydroxy-3-keto-5-methylthiopentene in bacteria and plants (35, 197, 198). Besides methanogenesis, it can be demethylated into 3-mercaptopropanoic acid or cleaved into MT, acetaldehyde, and CO2 (184).
Methanogenesis from MMPA was first shown to follow a similar path as DMS and MT in Ms. barkeri (192, 193). In the first reaction, MtsA methylates the MtsB proteins with MMPA and H+, forming 3-mercaptopropanoic acid. In the second reaction, MtsA demethylates the MtsB proteins to methylate CoM. Subsequent work suggested additional mechanisms involving other MMPA-specific genes in Ms. acetivorans (117). In particular, the mtpCAP suite of genes was implicated in growth specifically with MMPA but not with DMS or MT. While not necessary under laboratory conditions, mtpP was suggested as a transporter for MMPA in nature. MtpC proteins were suggested to take the place of MtbB proteins, and MtpA was suggested to be the methyltransferase of the two reactions (117). It was later shown that MtpA only catalyzes the first reaction (methylation of MtpC proteins) but not the second reaction (methylation of CoM). MtsDFH were suggested to play a role, but the second methyltransferase remains unidentified (199). Also, of note for methanogenesis from MMPA in Ms. acetivorans is that the msrH gene is required for transcription of mtpCAP (117).
GENE AND TAXA ENVIRONMENTAL ABUNDANCES
We used the Joint Genome Institute’s Integrated Microbial Genomes and Microbiomes Database (IMG/M) (37) to conduct several different analyses of the distribution of methanogenesis genes and taxa using publicly available genomes (isolate genomes and high-quality metagenome-assembled genomes [MAGs]) and metagenomes. The goal of the analysis was to assess the diversity of organisms containing some of the methyl-based methanogenesis genes, compare the abundance of certain genes involved in methyl-based, hydrogenotrophic, and acetoclastic methanogenesis across a variety of methanogenic environments, and compare the abundance of methanogen families across those same environments. The environments include landfills, sewage treatment plants, wetlands (freshwater and coastal), rice fields, ocean (sediment), human guts, ruminant (cow and sheep) guts, termite guts, hydrothermal vents (plume, sediment, seawater, microbial mat, and host-associated), and hypersaline environments. These environments are hypothesized to have different dominant methanogenic substrates (21, 30, 62). Human and livestock guts, termite guts, and hydrothermal vents are rich in H2, while ocean, coastal wetland, and hypersaline sediments are thought to be rich in methylated compounds. Landfills, sewage treatment, freshwater wetlands, and rice fields are rich in both H2 and acetate, although for simplicity, we will classify these to be hypothesized acetoclastic dominated because the majority of methane is produced by acetoclastic methanogenesis where acetate is abundant (21).
Genome Survey
The isolate and metagenome-assembled genome (MAG) genome survey demonstrated that while the pairs of genes for initial demethylation of TMA, DMA, MMA, and MeOH are present in more archaeal genomes than bacterial genomes, they are present in many bacteria too, with the exception of MMA demethylation genes (Fig. 3). This highlights that both of these domains may perform initial degradation of methylated carbon compounds in the same manner in addition to other degradation pathways that are known to be performed by bacteria (131, 166). While fermentation and acetogenesis performed by bacteria have long been implicated as generating important precursors to archaeal methanogenesis (200), it remains to be seen how bacteria with these particular demethylation genes might interact with or otherwise affect (positively or negatively) methanogenic archaea. The analysis also shows that more genomes contain methanol degradation genes than genes for other pathways, while at the genus level, TMA degradation genes are the most prevalent among methanogenic archaeal genera, followed by genes for degradation of DMA and then MMA. This is generally in line with the ecology of these substrates, with TMA and methanol being produced from more sources than DMA and MMA.
FIG 3.

Number of genera and genomes of archaea and bacteria containing genes for the first step of demethylation for each of the four methylated compounds with complete KO annotations. Archaeal genomes are separated into those containing mcrA and those without mcrA; trimethylamine = mttB and mttC; dimethylamine = mtbB and mtbC; methylamine = mtmB and mtmC; methanol = mtaB and mtaC. Only four bacterial genomes contained mtbA encoding the second enzyme to produce methyl-CoM from TMA/DMA/MMA, but none of these four contained mttBC, mtbBC, or mtmBC. Twenty-six bacterial genomes contained mtaA, encoding the second enzyme to produce methyl-CoM from methanol, and 13 of these contained mtaBC. However, the vast majority (>90%) of archaeal genomes shown here also contained mtbA (TMA/DMA/MMA) or mtaA (methanol). Note that only archaea perform the subsequent step of reducing methyl-CoM to methane (with McrABG) and that this is just for demethylation for methanogenesis; other pathways also degrade these compounds. Also note that the x axis scale is different on each graph. We searched for mttB, mttC, mtbB, mtbC, mtmB, mtmC, mtaB, and mtaC genes on IMG/M on May 11, 2022. The table of genomes containing each gene was downloaded and filtered to include only isolate genomes or high-quality metagenome assembled genomes (224). Only genomes containing both of the genes in each pair were counted.
There was a total of 117 unique genomes with the complete suite of genes for at least one of the methylated amines (TMA, DMA, or MMA) pathways (i.e., containing the pair of genes for the initial demethylation, mtbA for methyl-CoM production, and mcrA for CH4 production). Most of the genomes that contained TMA-processing genes (n = 115) also contained genes to process DMA (n = 113) or MMA (n = 103), with 97 genomes containing the complete suite for all three methylated amines. No single genome examined here contained only TMA-, only DMA-, or only MMA-degrading genes. Overall, this supports the hypothesis that most methanogens that produce methane from TMA can also produce methane from DMA and MMA, which would enable them to generate additional energy from the products of the reactions (i.e., DMA from TMA and MMA from DMA). This is partially supported by isolate culture experiments that test methane production from a suite of substrates and often show methane production for TMA, DMA, and MMA (201–207), although more substrate specificity has also been shown (208).
Metagenome Gene Survey
We analyzed the metagenomes for certain genes that are present in different methanogenesis pathways, which provides information on the potential of different environments to harbor taxa containing those genes. Certain environments were previously hypothesized to have relatively greater contributions of certain methanogenic pathways based on substrate availability and previous work on methanogenic community composition (30). Gene counts were separated into those harbored by archaeal taxa, bacterial taxa, and all other taxa/unknown taxa based on the taxonomic assignment of metagenomic scaffolds by the IMG/M annotation pipeline, since only archaeal genes are expected to participate in methanogenesis pathways. The gene abundance survey of metagenomes demonstrated significant differences in all eight of the genes analyzed, and this held true for the total gene count across all taxa, the gene counts from just the archaeal assigned scaffolds, and the gene counts of scaffolds not assigned to bacteria or archaea (Kruskal-Wallis, P < 0.05; Fig. 4; Table S2 in the supplemental material). For gene counts from bacterial-assigned scaffolds, six of the eight gene abundances were significantly different among the ecosystems, while mcrA (not present in bacteria) and mtsA (only present in very few bacterial scaffolds) were not (Fig. 4; Table S2). This is not surprising due to the known differences in environmental conditions and known taxonomic preferences across these habitats.
FIG 4.

Counts per million (CPM) assembled reads normalized abundance of genes for all core methanogenesis pathways (methyl-CoM reduction, mcrA), acetoclastic methanogenesis (cdhD), hydrogenotrophic methanogenesis (frhA), and methyl-based methanogenesis from methanol (mtaA), trimethylamine (mttC), dimethylamine (mtbC), methylamine (mtmC), or the methylated sulfides (mtsA), which include dimethylsulfide (DMS), methanethiol (MeSH), and methylthiopropanoate (MMPA). Gene counts are split taxonomically between archaea, bacteria, and other (eukaryotes and unassigned) according to the scaffold taxonomic assignments from the IMG/M annotation pipeline. Also shown are the number of copies of the genes in genomes containing the mcrA gene (methanogens), the number of archaeal genera containing the genes, and the number of bacterial genera containing the genes based on isolate genomes and high-quality MAGs in the IMG/M database. Note that “Humans and livestock” and “Termites” refer to gut samples. The following words were searched in IMG/M for each habitat type (the search was performed on February 5, 2022): landfill = “landfill”, sewage treatment = “sewage”, wetlands = “wetland”, rice fields = “rice”, ocean = “ocean”, humans and livestock = “human gut” or “ruminant” or “cow”, termites = “termite”, hydrothermal vents = “hydrothermal”, and hypersaline = “hypersaline.” The results were first filtered to only metagenomes (i.e., metatranscriptomes and isolate genomes were removed). Results were then further filtered based on the information in the study name or genome name to ensure that the metagenomes were actually from the targeted habitats, and only metagenomes containing the mcrA gene were retained. Coastal wetlands were separated from freshwater wetlands using “grep” to extract metagenomes containing the word “coastal” or based on our knowledge of the metagenomic study. Furthermore, only metagenomes with either unrestricted public use status or explicit permission from the principal investigators in the case of restricted use status for metagenomes sequenced at Joint Genome Institute (JGI) (JGI Data Utilization Status = “Restricted” in IMG/M) were used (Table S1 in the supplemental material). The KEGG Orthology (KO) gene counts were downloaded using the “Statistical Analysis” tool on IMG/M, which uses lastal 983 and KEGG Genes v77.1 to assign KO terms. To acquire KO gene counts of just the archaeal portion of the metagenomes, the KO profiles were filtered to include only those found on scaffolds assigned to the domain Archaea. The same was performed for the domain Bacteria. Archaeal and bacterial counts were subtracted from the total counts to yield the counts for all eukaryotic taxa as well as scaffolds with no taxonomic assignment. This was performed using a custom Python script to process three of the output files generated by the IMG/M annotation pipeline, (i) the KO terms of the genes, (ii) the taxonomic assignments of the scaffolds, and (iii) the gene to scaffold mapping. Metagenomes that contained fewer than 1,000 reads with family-level taxonomic information were removed from the data set. The final sample size was 465, including samples from all over the world (Fig. S2; landfill, n = 12; sewage treatment, n = 27; rice field, n = 11; wetlands (freshwater), n = 90; humans and livestock, n = 50; termites, n = 62; hydrothermal vent, n = 130; wetlands (coastal), n = 52; ocean, n = 13; hypersaline, n = 18). Correlations between the selected gene and the other genes in each pathway are shown in Fig. S3. Tables of all genomes containing each of these genes were downloaded from IMG/M to acquire counts of genera per domain that contain each gene. To aid in interpretation of abundances, we also downloaded a KO profile of all IMG/M isolate genomes and high-quality metagenome assembled genomes containing the mcrA gene (the search was performed May 11, 2022; n = 282 genomes), selected the genes of interest by their KOs, and examined the maximum and minimum number of copies across all of the genomes.
The total abundance of the cdhD gene involved in acetoclastic methanogenesis as well as other pathways found in bacteria was significantly greater than hydrogenotrophic and methyl-based methanogenesis genes in five of the six habitats where those two pathways are hypothesized to be dominant and was also greater than mcrA. Indeed, many of the cdhD counts were derived from bacteria, especially in freshwater wetlands, termite guts, and ocean sediments (Fig. 4). When analyzing just the archaeal-assigned scaffolds, cdhD abundance was greatest in hydrothermal vents, followed by freshwater wetland and ocean sediments (Fig. 4). In two instances (ocean sediments and freshwater wetlands), archaeal cdhD abundance was greater than mcrA abundance, suggesting multiple copies per genome and/or presence of cdhD in nonmethanogenic archaea. The total abundance and archaeal abundance of the frhA gene involved in hydrogenotrophic methanogenesis was greatest in hydrothermal vents and was least abundant in ocean and hypersaline sediments (Fig. 4).
In terms of the methyl-based methanogenesis genes, their total abundances generally followed a pattern of genes for utilization of methanol or TMA being the most abundant, followed by DMA, MMA, and DMS (Fig. 4). This pattern was also true for abundance in the archaeal-assigned scaffolds, except that mtbC (for DMA utilization) was the most abundant methyl-based gene in termite guts (Fig. 4). Abundance in the three ecosystems hypothesized to be dominated by methyl-based methanogenesis was not significantly greater than in the other ecosystems except for mtaA (methanol) and mttC (TMA) in ocean sediments.
These metagenomic gene abundances can be confounded by several factors, which could obscure the true patterns of the activity levels of each methanogenic pathway. Most importantly, some of the genes are not strictly specific to each pathway, and most of the genes are not specific even to archaea (Fig. 4). To aid in the interpretation of abundances, genes from archaeal and bacterial scaffolds were counted separately; however, this step still has limitations, as there are many scaffolds that are not taxonomically assigned. In particular, the cdhD gene, while required for acetoclastic methanogenesis and thus present in all acetoclastic methanogens, is not an exclusive marker of acetoclastic methanogenesis, as it is also present in some hydrogenotrophs and other bacterial taxa (acetogens, sulfate-reducers) that use the Wood-Ljungdahl pathway, and this inflated the cdhD counts. However, a low abundance of cdhD, and in particular of archaeal cdhD, does mean a low abundance of acetoclastic taxa (and other taxa with cdhD). This interpretation applies to all of the genes, in fact, as most of them (except mcrA and mtsA) are found in bacteria as well (Fig. 4). Similarly, the TMA methyltransferases have homologs in bacteria that are involved in glycine betaine metabolism (209).
The estimates are further complicated by the fact that some taxa contain genes for multiple pathways, and metagenomic data do not distinguish which of them are actively expressed. This is a well-known issue with the versatile methanogens in the Methanosarcinales order, as discussed elsewhere (70). In the case of distinguishing between methyl dismutation and methyl reduction, a complete or partial absence of the methyl branch of the Wood-Ljungdahl pathway would suggest the methyl reduction pathway, but it is also possible that methyl-reducing taxa have those genes but do not express them (33). Still, we suggest that researchers working with genomic data assess the genomes or MAGs for multiple suites of genes to evaluate the presence of pathways more effectively. For metagenomic data, this is more difficult, as multiple organisms are represented in the metagenome. Furthermore, some genes can be present in multiple copies (Fig. 4), which can affect their overall abundances in metagenomes. More metatranscriptomic surveys are necessary to complement this analysis and more effectively assess the dominant active pathways. Another confounding factor for ocean sediments is depth, which was not available in the IMG/M metadata. Methyl-based methanogenesis is expected to be more abundant in the top layer of sediment, while hydrogenotrophic and acetoclastic methanogenesis are expected to increase further down the sediment profile below the sulfate reduction zone (55).
Metagenome Taxonomic Survey
The metagenome-based taxonomic survey showed major differences at the family level of methanogens across the different habitat types, with all families exhibiting significant differences in abundance among the different environments (Fig. 5; Table S2). Hydrothermal vents had the highest overall abundance of methanogens in terms of counts per million assembled metagenomic reads and were dominated by Methanocaldococcaceae, Methanococcaceae, and, to a lesser extent, Methanosarcinaceae. Methanococcaceae and Methanocaldococcaceae were dominant methanogenic families only in hydrothermal vents, likely due to the extreme thermal adaptations present in some members of these families. Methanosarcinaceae was the most abundant family across the whole data set, with mean counts per million (CPM) >1 in all ecosystems but human and livestock gastrointestinal tracts. These ecosystems were dominated by hydrogenotrophic Methanobacteriaceae, although 14 other methanogen families were present at very low abundances. Methanoregulaceae was the second most abundant family overall; this was driven by their abundance in sewage treatment samples, wetlands, landfills, and rice fields (Fig. 5).
FIG 5.

Counts per million normalized abundance of different methanogen families across different methanogenesis habitats. Numbers above the columns are the number of methanogenic families present (the total number of methanogen families in the whole data set was 18). Also shown are the known substrates/pathways that can be used as published previously (39) but with the methyl-reducing pathway added to Methanosarcinaceae (69). Note that only the top 12 families in the data set are shown here (these families had greater than 1.6 CPM in at least one habitat type). Families are sorted by pathway and then by overall abundance. The data set was assembled as in Fig. 3, except a family-level taxonomic profile was downloaded with the “Statistical Analysis” tool on IMG/M, which uses lastal 983 and the IMG-NR reference database to assign the taxonomy. Methanogenic families were selected as those containing the string “Methano,” followed by filtering out any methanotrophs based on the literature. Known methanogenic pathways performed by the families were taken from the literature (39, 69).
These taxonomic data provide mixed support for some of the proposed dominant pathways, although again it is difficult to know where members of the versatile Methanosarcinaceae family are performing more of the acetoclastic, hydrogenotrophic, methyl dismutation, or methyl-reducing pathways with these data. However, we can use the other two families that perform exclusively acetoclastic methanogenesis (Methanosaetaceae and Methanotrichaceae) as markers; they are indeed more abundant in the hypothesized acetoclastic-dominant habitats with perhaps the exception of rice fields, which had an even distribution of family abundances. Note that Methanosaetaceae was renamed Methanotrichaceae, and these families represent the same group of organisms, but older taxonomic databases and older annotations by the IMG/M pipeline contain both families. Methanosaetaceae/Methanotrichaceae were most abundant in landfills, sewage treatment plants, and freshwater wetlands, but were also abundant in ocean sediments, hydrothermal vents, and coastal wetlands. Human and livestock guts and hydrothermal vents were dominated by hydrogenotrophic taxa, in support of the hypothesized dominance of the hydrogenotrophic pathway there (Fig. S4) even though the hydrogenotrophic gene frhA was not as abundant as cdhD in either of those habitats or in termite guts. Termite guts had about equal mean abundances of hydrogenotrophic (most Methanobacteriaceae) and methyl-reducing methanogens (Methanomassiliicoccaceae). Ocean sediments, which are predicted to be dominated by methylotrophs, were dominated by Methanosarcinaceae, which could indeed be performing methyl-based methanogenesis. Hydrogenotrophic and acetoclastic taxa were present in ocean sediments as well, although to a lesser extent (Fig. 5).
CONCLUSIONS AND FUTURE DIRECTIONS
Methanogens were first cultured in the 1920s. Since then, hundreds of methanogens have been isolated and sequenced, and many new methanogenic substrates have been identified. We now know that archaeal methanogens are more diverse taxonomically and functionally than previously predicted and that biological methane production is not limited to the archaeal domain. Methanogenesis as a mode of growth and energy conservation, however, is still limited to the archaeal domain. Many new studies have come out in recent years demonstrating that methyl-based methanogenesis is the dominant pathway in some environments. These studies use multiple methods, including isotopic profiling, molecular sequencing, substrate quantification, and microcosm incubation methods to draw robust conclusions (59, 63). Genes involved in methyl-based methanogenesis are also widely distributed across taxa and ecosystems, suggesting that it may contribute to methane production in a broader range of environments than previously thought (Fig. 1, 4, and 5) (39).
In the context of two key ongoing global change processes (drought and sea level rise), it is possible that the relative contribution of methyl-based methanogenesis to methane production may increase due to the increasingly saline conditions in ecosystems affected by these processes (210), even if overall methane emissions might decrease (211, 212). Salinity is expected not only to increase but also to become more variable (74). As noted earlier, climate change has already and will continue to increase salinity, especially in estuarine and coastal wetlands, as has been shown by many studies in many places (74, 213, 214). This is due to a combination of sea level rise and declining freshwater inputs as well as other human activities, such as salting roads in winter, irrigation, and vegetation clearing (74). These factors combined with decreasing precipitation in some areas mean that methyl-based methanogenesis could also become more relevant in inland freshwater wetlands.
There is still much work to be done to fully characterize some pathways as well as their isotope fractionations (Fig. 1; Table 3) and to quantify the relative contributions of each substrate to the overall methane production in different environments. We also call for improved computational methods for metabolic modeling of methyl-based methanogenesis as well as methanogenesis in general. Automated genome-scale metabolic modeling methods are continuously making excellent and important methodological improvements. However, it still takes a high degree of manual curation to build accurate metabolic models for methanogens (215–222). This is an active area of research that we expect will soon enable model development for a greater number and broader diversity of methanogens. Research on methyl-based methanogenesis and methanogenesis in general is an exciting and challenging field of research that will continue to capitalize on improving molecular and computational methods, enabling discoveries with significant implications for climate change mitigation and natural gas energy production.
ACKNOWLEDGMENTS
The work conducted by the US Department of Energy (DOE) Joint Genome Institute (https://ror.org/04xm1d337), a DOE Office of Science User Facility, is supported by the Office of Science of the US Department of Energy operated under contract number DE-AC02-05CH11231. We thank Harry Beller for permission to use restricted metagenomes from sewage treatment plants that were part of a project done at Lawrence Berkeley National Laboratory (223). We thank Christa Pennacchio and Tanja Woyke for assistance in checking the data utilization status (restricted versus unrestricted) of metagenomes on IMG/M. We thank three anonymous reviewers for valuable contributions to the manuscript.
Biographies

Clifton P. Bueno de Mesquita is a postdoctoral scholar at the DOE Joint Genome Institute. He received his B.A. in Environmental Studies/Conservation Biology from Middlebury College and his Ph.D. in Ecology and Evolutionary Biology from the University of Colorado Boulder. He has studied the microbiomes of air, snow, soils, sediments, roots, leaves, seeds, and guts. He has previously studied methane production in coarse woody debris and is currently studying methane cycling in estuarine wetlands and industrial salterns. He is fascinated by the taxonomic and functional diversity of microorganisms and has a particular interest in managing greenhouse gas emissions to mitigate climate change.

Dongying Wu is a bioinformatic engineer at the DOE Joint Genome Institute. He received his B.A. in Genetics and Genetic Engineering from Fudan University and his Ph.D. in Genetics from Iowa State University. His major interests are novel lineage discovery and gene function prediction based on large-scale metagenome and metatranscriptome data mining. His contributions are exemplified by his important roles in milestone genome and metagenome projects, such as the sequencing of Arabidopsis thaliana, the Global Ocean Survey, and The Genomic Encyclopedia of Bacteria and Archaea (GEBA) project at the DOE Joint Genome Institute.

Susannah G. Tringe is the Director of the Environmental Genomics and Systems Biology division at Lawrence Berkeley National Laboratory. She received her undergraduate degree in Physics from Harvard University and then went on to a Ph.D. in Biophysics from Stanford University and joined Berkeley Lab as a postdoc at the Joint Genome Institute in 2003. There she developed techniques for using DNA sequence data for comparative analysis of whole microbial communities rather than individual organisms. Her current research focuses on using nucleic acid sequence data to study communities of microbes from diverse environmental niches and understand how they can potentially be harnessed for improved environmental and agricultural outcomes. She has a particular interest in how microbes influence greenhouse gas uptake and release in wetlands, especially as it relates to the methane cycle.
Footnotes
[This article was published on 24 January 2023 with an error in Fig. 3. The figure was updated in the current version, posted on 31 January 2023.]
Supplemental material is available online only.
REFERENCES
- 1.Ueno Y, Yamada K, Yoshida N, Maruyama S, Isozaki Y. 2006. Evidence from fluid inclusions for microbial methanogenesis in the early Archaean era. Nature 440:516–519. doi: 10.1038/nature04584. [DOI] [PubMed] [Google Scholar]
- 2.Kasting JF, Catling D. 2003. Evolution of a habitable planet. Annu Rev Astron Astrophys 41:429–463. doi: 10.1146/annurev.astro.41.071601.170049. [DOI] [Google Scholar]
- 3.Sauterey B, Charnay B, Affholder A, Mazevet S, Ferrière R. 2020. Co-evolution of primitive methane-cycling ecosystems and early Earth’s atmosphere and climate. Nat Commun 11:2705. doi: 10.1038/s41467-020-16374-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Costa KC, Leigh JA. 2014. Metabolic versatility in methanogens. Curr Opin Biotechnol 29:70–75. doi: 10.1016/j.copbio.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 5.Etiope G, Sherwood Lollar B. 2013. Abiotic methane on Earth. Rev Geophys 51:276–299. doi: 10.1002/rog.20011. [DOI] [Google Scholar]
- 6.Conrad R. 2009. The global methane cycle: recent advances in understanding the microbial processes involved. Environ Microbiol Rep 1:285–292. doi: 10.1111/j.1758-2229.2009.00038.x. [DOI] [PubMed] [Google Scholar]
- 7.Saunois M, Stavert AR, Poulter B, Bousquet P, Canadell JG, Jackson RB, Raymond PA, Dlugokencky EJ, Houweling S, Patra PK, Ciais P, Arora VK, Bastviken D, Bergamaschi P, Blake DR, Brailsford G, Bruhwiler L, Carlson KM, Carrol M, Castaldi S, Chandra N, Crevoisier C, Crill PM, Covey K, Curry CL, Etiope G, Frankenberg C, Gedney N, Hegglin MI, Höglund-Isaksson L, Hugelius G, Ishizawa M, Ito A, Janssens-Maenhout G, Jensen KM, Joos F, Kleinen T, Krummel PB, Langenfelds RL, Laruelle GG, Liu L, Machida T, Maksyutov S, McDonald KC, McNorton J, Miller PA, Melton JR, Morino I, Müller J, Murguia-Flores F, et al. 2020. The global methane budget 2000–2017. Earth Syst Sci Data 12:1561–1623. doi: 10.5194/essd-12-1561-2020. [DOI] [Google Scholar]
- 8.IPCC. 2021. Climate Change 2021: the physical science basis. https://www.ipcc.ch/report/sixth-assessment-report-working-group-i/. Accessed April 1, 2022.
- 9.Faramawy S, Zaki T, Sakr AA-E. 2016. Natural gas origin, composition, and processing: a review. J Nat Gas Sci Eng 34:34–54. doi: 10.1016/j.jngse.2016.06.030. [DOI] [Google Scholar]
- 10.Buan NR. 2018. Methanogens: pushing the boundaries of biology. Emerg Top Life Sci 2:629–646. doi: 10.1042/ETLS20180031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaci N, Borrel G, Tottey W, O'Toole PW, Brugère J-F. 2014. Archaea and the human gut: new beginning of an old story. World J Gastroenterol 20:16062–16078. doi: 10.3748/wjg.v20.i43.16062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen W-Y, Kraková L, Wu J-H, Pangallo D, Jeszeová L, Liu B, Yasui H. 2017. Community and proteomic analysis of anaerobic consortia converting tetramethylammonium to methane. Archaea 2017:e2170535. doi: 10.1155/2017/2170535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dlugokencky EJ, Nisbet EG, Fisher R, Lowry D. 2011. Global atmospheric methane: budget, changes and dangers. Philos Trans A Math Phys Eng Sci 369:2058–2072. doi: 10.1098/rsta.2010.0341. [DOI] [PubMed] [Google Scholar]
- 14.Heimann M. 2011. Atmospheric science: enigma of the recent methane budget. Nature 476:157–158. doi: 10.1038/476157a. [DOI] [PubMed] [Google Scholar]
- 15.Roque BM, Salwen JK, Kinley R, Kebreab E. 2019. Inclusion of Asparagopsis armata in lactating dairy cows’ diet reduces enteric methane emission by over 50 percent. J Clean Prod 234:132–138. doi: 10.1016/j.jclepro.2019.06.193. [DOI] [Google Scholar]
- 16.Zou J, Huang Y, Jiang J, Zheng X, Sass RL. 2005. A 3-year field measurement of methane and nitrous oxide emissions from rice paddies in China: effects of water regime, crop residue, and fertilizer application. Glob Biogeochem Cycles doi: 10.1029/2004GB002401. [DOI] [Google Scholar]
- 17.Dean JF, Middelburg JJ, Röckmann T, Aerts R, Blauw LG, Egger M, Jetten MSM, de Jong AEE, Meisel OH, Rasigraf O, Slomp CP, In't Zandt MH, Dolman AJ. 2018. Methane feedbacks to the global climate system in a warmer world. Rev Geophys 56:207–250. doi: 10.1002/2017RG000559. [DOI] [Google Scholar]
- 18.Moore MT, Vinson DS, Whyte CJ, Eymold WK, Walsh TB, Darrah TH. 2018. Differentiating between biogenic and thermogenic sources of natural gas in coalbed methane reservoirs from the Illinois Basin using noble gas and hydrocarbon geochemistry. Geol Soc Spec Publ 468:151–188. doi: 10.1144/SP468.8. [DOI] [Google Scholar]
- 19.Guo H, Yu Z, Liu R, Zhang H, Zhong Q, Xiong Z. 2012. Methylotrophic methanogenesis governs the biogenic coal bed methane formation in Eastern Ordos Basin, China. Appl Microbiol Biotechnol 96:1587–1597. doi: 10.1007/s00253-012-3889-3. [DOI] [PubMed] [Google Scholar]
- 20.Demirel B, Scherer P. 2008. The roles of acetotrophic and hydrogenotrophic methanogens during anaerobic conversion of biomass to methane: a review. Rev Environ Sci Biotechnol 7:173–190. doi: 10.1007/s11157-008-9131-1. [DOI] [Google Scholar]
- 21.Conrad R. 2020. Importance of hydrogenotrophic, aceticlastic and methylotrophic methanogenesis for methane production in terrestrial, aquatic and other anoxic environments: a mini review. Pedosphere 30:25–39. doi: 10.1016/S1002-0160(18)60052-9. [DOI] [Google Scholar]
- 22.Kurth JM, Nobu MK, Tamaki H, de Jonge N, Berger S, Jetten MSM, Yamamoto K, Mayumi D, Sakata S, Bai L, Cheng L, Nielsen JL, Kamagata Y, Wagner T, Welte CU. 2021. Methanogenic archaea use a bacteria-like methyltransferase system to demethoxylate aromatic compounds. ISME J 15:3549–3565. doi: 10.1038/s41396-021-01025-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welte CU, de Graaf R, Dalcin Martins P, Jansen RS, Jetten MSM, Kurth JM. 2021. A novel methoxydotrophic metabolism discovered in the hyperthermophilic archaeon Archaeoglobus fulgidus. Environ Microbiol 23:4017–4033. doi: 10.1111/1462-2920.15546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karl DM, Beversdorf L, Björkman KM, Church MJ, Martinez A, Delong EF. 2008. Aerobic production of methane in the sea. Nature Geosci 1:473–478. doi: 10.1038/ngeo234. [DOI] [Google Scholar]
- 25.Wang Q, Alowaifeer A, Kerner P, Balasubramanian N, Patterson A, Christian W, Tarver A, Dore JE, Hatzenpichler R, Bothner B, McDermott TR. 2021. Aerobic bacterial methane synthesis. Proc Natl Acad Sci USA 118:e2019229118. doi: 10.1073/pnas.2019229118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bižić M, Klintzsch T, Ionescu D, Hindiyeh MY, Günthel M, Muro-Pastor AM, Eckert W, Urich T, Keppler F, Grossart H-P. 2020. Aquatic and terrestrial cyanobacteria produce methane. Sci Adv 6:eaax5343. doi: 10.1126/sciadv.aax5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng Y, Harris DF, Yu Z, Fu Y, Poudel S, Ledbetter RN, Fixen KR, Yang Z-Y, Boyd ES, Lidstrom ME, Seefeldt LC, Harwood CS. 2018. A pathway for biological methane production using bacterial iron-only nitrogenase. Nat Microbiol 3:281–286. doi: 10.1038/s41564-017-0091-5. [DOI] [PubMed] [Google Scholar]
- 28.Lenhart K, Bunge M, Ratering S, Neu TR, Schüttmann I, Greule M, Kammann C, Schnell S, Müller C, Zorn H, Keppler F. 2012. Evidence for methane production by saprotrophic fungi. Nat Commun 3:1046. doi: 10.1038/ncomms2049. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Chen H, Zhu Q, Shen Y, Wang X, Wang M, Peng C. 2015. A novel pathway of direct methane production and emission by eukaryotes including plants, animals and fungi: an overview. Atmos Environ 115:26–35. doi: 10.1016/j.atmosenv.2015.05.019. [DOI] [Google Scholar]
- 30.Lyu Z, Shao N, Akinyemi T, Whitman WB. 2018. Methanogenesis. Curr Biol 28:R727–R732. doi: 10.1016/j.cub.2018.05.021. [DOI] [PubMed] [Google Scholar]
- 31.Lessner DJ. 2009. Methanogenesis biochemistry. Wiley, Hoboken, NJ. [Google Scholar]
- 32.Deppenmeier U. 2002. The unique biochemistry of methanogenesis. Prog Nucleic Acid Res Mol Biol 71:223–283. doi: 10.1016/s0079-6603(02)71045-3. [DOI] [PubMed] [Google Scholar]
- 33.Kurth JM, Op den Camp HJM, Welte CU. 2020. Several ways one goal—methanogenesis from unconventional substrates. Appl Microbiol Biotechnol 104:6839–6854. doi: 10.1007/s00253-020-10724-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanehisa M, Goto S. 2000. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karp PD, Billington R, Caspi R, Fulcher CA, Latendresse M, Kothari A, Keseler IM, Krummenacker M, Midford PE, Ong Q, Ong WK, Paley SM, Subhraveti P. 2019. The BioCyc collection of microbial genomes and metabolic pathways. Brief Bioinform 20:1085–1093. doi: 10.1093/bib/bbx085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seaver SMD, Liu F, Zhang Q, Jeffryes J, Faria JP, Edirisinghe JN, Mundy M, Chia N, Noor E, Beber ME, Best AA, DeJongh M, Kimbrel JA, D'haeseleer P, McCorkle SR, Bolton JR, Pearson E, Canon S, Wood-Charlson EM, Cottingham RW, Arkin AP, Henry CS. 2021. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res 49:D575–D588. doi: 10.1093/nar/gkaa746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen I-MA, Chu K, Palaniappan K, Ratner A, Huang J, Huntemann M, Hajek P, Ritter S, Varghese N, Seshadri R, Roux S, Woyke T, Eloe-Fadrosh EA, Ivanova NN, Kyrpides NC. 2021. The IMG/M data management and analysis system v.6.0: new tools and advanced capabilities. Nucleic Acids Res 49:D751–D763. doi: 10.1093/nar/gkaa939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malyan SK, Bhatia A, Kumar A, Gupta DK, Singh R, Kumar SS, Tomer R, Kumar O, Jain N. 2016. Methane production, oxidation and mitigation: a mechanistic understanding and comprehensive evaluation of influencing factors. Sci Total Environ 572:874–896. doi: 10.1016/j.scitotenv.2016.07.182. [DOI] [PubMed] [Google Scholar]
- 39.Söllinger A, Urich T. 2019. Methylotrophic methanogens everywhere—physiology and ecology of novel players in global methane cycling. Biochem Soc Trans 47:1895–1907. doi: 10.1042/BST20180565. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, Wegener G, Williams TA, Xie R, Hou J, Tian C, Zhang Y, Wang F, Xiao X. 2021. A methylotrophic origin of methanogenesis and early divergence of anaerobic multicarbon alkane metabolism. Sci Adv 7:eabj1453. doi: 10.1126/sciadv.abj1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlesinger WH, Bernhardt ES. 2013. Biogeochemistry: an analysis of global change, 3rd ed. Academic Press, Waltham, MA. [Google Scholar]
- 42.Achtnich C, Bak F, Conrad R. 1995. Competition for electron donors among nitrate reducers, ferric iron reducers, sulfate reducers, and methanogens in anoxic paddy soil. Biol Fertil Soils 19:65–72. doi: 10.1007/BF00336349. [DOI] [Google Scholar]
- 43.Sutton-Grier Ariana E, Keller JK, Koch R, Gilmour C, Megonigal JP. 2011. Electron donors and acceptors influence anaerobic soil organic matter mineralization in tidal marshes. Soil Biol Biochem 43:1576–1583. doi: 10.1016/j.soilbio.2011.04.008. [DOI] [Google Scholar]
- 44.Lovley DR, Klug MJ. 1983. Sulfate reducers can outcompete methanogens at freshwater sulfate concentrations. Appl Environ Microbiol 45:187–192. doi: 10.1128/aem.45.1.187-192.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oremland RS, Polcin S. 1982. Methanogenesis and sulfate reduction: competitive and noncompetitive substrates in estuarine sediments. Appl Environ Microbiol 44:1270–1276. doi: 10.1128/aem.44.6.1270-1276.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oremland RS, Marsh LM, Polcin S. 1982. Methane production and simultaneous sulphate reduction in anoxic, salt marsh sediments. Nature 296:143–145. doi: 10.1038/296143a0. [DOI] [Google Scholar]
- 47.Kristjansson JK, Schönheit P. 1983. Why do sulfate-reducing bacteria outcompete methanogenic bacteria for substrates? Oecologia 60:264–266. doi: 10.1007/BF00379530. [DOI] [PubMed] [Google Scholar]
- 48.Lovley DR, Dwyer DF, Klug MJ. 1982. Kinetic analysis of competition between sulfate reducers and methanogens for hydrogen in sediments. Appl Environ Microbiol 43:1373–1379. doi: 10.1128/aem.43.6.1373-1379.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schönheit P, Kristjansson JK, Thauer RK. 1982. Kinetic mechanism for the ability of sulfate reducers to out-compete methanogens for acetate. Arch Microbiol 132:285–288. doi: 10.1007/BF00407967. [DOI] [Google Scholar]
- 50.Winfrey MR, Zeikus JG. 1977. Effect of sulfate on carbon and electron flow during microbial methanogenesis in freshwater sediments. Appl Environ Microbiol 33:275–281. doi: 10.1128/aem.33.2.275-281.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pimenov N, Davidova I, Belyaev S, Lein A, Ivanov M. 1993. Microbiological processes in marine sediments in the Zaire River Delta and the Benguela upwelling region. Geomicrobiol J 11:157–174. doi: 10.1080/01490459309377948. [DOI] [Google Scholar]
- 52.Xiao K-Q, Beulig F, Kjeldsen KU, Jørgensen BB, Risgaard-Petersen N. 2017. Concurrent methane production and oxidation in surface sediment from Aarhus Bay, Denmark. Front Microbiol 8:1198. doi: 10.3389/fmicb.2017.01198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhuang G-C, Montgomery A, Sibert RJ, Rogener M-K, Samarkin VA, Joye SB. 2018. Effects of pressure, methane concentration, sulfate reduction activity, and temperature on methane production in surface sediments of the Gulf of Mexico. Limnol Oceanogr 63:2080–2092. doi: 10.1002/lno.10925. [DOI] [Google Scholar]
- 54.Zhuang G-C, Xu L, Liang Q, Fan X, Xia Z, Joye SB, Wang F. 2019. Biogeochemistry, microbial activity, and diversity in surface and subsurface deep-sea sediments of South China Sea. Limnol Oceanogr 64:2252–2270. doi: 10.1002/lno.11182. [DOI] [Google Scholar]
- 55.Xu L, Zhuang G-C, Montgomery A, Liang Q, Joye SB, Wang F. 2021. Methyl-compounds driven benthic carbon cycling in the sulfate-reducing sediments of South China Sea. Environ Microbiol 23:641–651. doi: 10.1111/1462-2920.15110. [DOI] [PubMed] [Google Scholar]
- 56.Mitterer RM, Malone MJ, Goodfriend GA, Swart PK, Wortmann UG, Logan GA, Feary DA, Hine AC. 2001. Co-generation of hydrogen sulfide and methane in marine carbonate sediments. Geophys Res Lett 28:3931–3934. doi: 10.1029/2001GL013320. [DOI] [Google Scholar]
- 57.Mitterer RM. 2010. Methanogenesis and sulfate reduction in marine sediments: a new model. Earth Planet Sci Lett 295:358–366. doi: 10.1016/j.epsl.2010.04.009. [DOI] [Google Scholar]
- 58.Maltby J, Sommer S, Dale AW, Treude T. 2016. Microbial methanogenesis in the sulfate-reducing zone of surface sediments traversing the Peruvian margin. Biogeosciences 13:283–299. doi: 10.5194/bg-13-283-2016. [DOI] [Google Scholar]
- 59.Zhuang G-C, Elling FJ, Nigro LM, Samarkin V, Joye SB, Teske A, Hinrichs K-U. 2016. Multiple evidence for methylotrophic methanogenesis as the dominant methanogenic pathway in hypersaline sediments from the Orca Basin, Gulf of Mexico. Geochim Cosmochim Acta 187:1–20. doi: 10.1016/j.gca.2016.05.005. [DOI] [Google Scholar]
- 60.Fischer PQ, Sánchez-Andrea I, Stams AJM, Villanueva L, Sousa DZ. 2021. Anaerobic microbial methanol conversion in marine sediments. Environ Microbiol 23:1348–1362. doi: 10.1111/1462-2920.15434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.King GM, Klug MJ, Lovley DR. 1983. Metabolism of acetate, methanol, and methylated amines in intertidal sediments of Lowes Cove, Maine. Appl Environ Microbiol 45:1848–1853. doi: 10.1128/aem.45.6.1848-1853.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mcgenity T, Sorokin D. 2018. Methanogens and methanogenesis in hypersaline environments, p 283–309. In Stams A, Sousa D (ed), Biogenesis of hydrocarbons. Springer International Publishing, Cham, Switzerland. [Google Scholar]
- 63.Zhuang G-C, Heuer VB, Lazar CS, Goldhammer T, Wendt J, Samarkin VA, Elvert M, Teske AP, Joye SB, Hinrichs K-U. 2018. Relative importance of methylotrophic methanogenesis in sediments of the Western Mediterranean Sea. Geochim Cosmochim Acta 224:171–186. doi: 10.1016/j.gca.2017.12.024. [DOI] [Google Scholar]
- 64.Schorn S, Ahmerkamp S, Bullock E, Weber M, Lott C, Liebeke M, Lavik G, Kuypers MMM, Graf JS, Milucka J. 2022. Diverse methylotrophic methanogenic archaea cause high methane emissions from seagrass meadows. Proc Natl Acad Sci USA 119:e2106628119. doi: 10.1073/pnas.2106628119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiao K-Q, Beulig F, Røy H, Jørgensen BB, Risgaard-Petersen N. 2018. Methylotrophic methanogenesis fuels cryptic methane cycling in marine surface sediment. Limnol Oceanogr 63:1519–1527. doi: 10.1002/lno.10788. [DOI] [Google Scholar]
- 66.King GM. 1988. Methanogenesis from methylated amines in a hypersaline algal mat. Appl Environ Microbiol 54:130–136. doi: 10.1128/aem.54.1.130-136.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Antony CP, Murrell JC, Shouche YS. 2012. Molecular diversity of methanogens and identification of Methanolobus sp. as active methylotrophic Archaea in Lonar crater lake sediments. FEMS Microbiol Ecol 81:43–51. doi: 10.1111/j.1574-6941.2011.01274.x. [DOI] [PubMed] [Google Scholar]
- 68.Cadena S, García-Maldonado JQ, López-Lozano NE, Cervantes FJ. 2018. Methanogenic and sulfate-reducing activities in a hypersaline microbial mat and associated microbial diversity. Microb Ecol 75:930–940. doi: 10.1007/s00248-017-1104-x. [DOI] [PubMed] [Google Scholar]
- 69.Sprenger WW, Hackstein JHP, Keltjens JT. 2007. The competitive success of Methanomicrococcus blatticola, a dominant methylotrophic methanogen in the cockroach hindgut, is supported by high substrate affinities and favorable thermodynamics. FEMS Microbiol Ecol 60:266–275. doi: 10.1111/j.1574-6941.2007.00287.x. [DOI] [PubMed] [Google Scholar]
- 70.Zhou J, Theroux SM, Bueno de Mesquita CP, Hartman WH, Tian Y, Tringe SG. 2022. Microbial drivers of methane emissions from unrestored industrial salt ponds. ISME J 16:284–295. doi: 10.1038/s41396-021-01067-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bueno de Mesquita CP, Zhou J, Theroux SM, Tringe SG. 2021. Methanogenesis and salt tolerance genes of a novel halophilic Methanosarcinaceae metagenome-assembled genome from a former solar saltern. Genes 12:1609. doi: 10.3390/genes12101609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oren A. 1999. Bioenergetic aspects of halophilism. Microbiol Mol Biol Rev 63:334–348. doi: 10.1128/MMBR.63.2.334-348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cao T, Han D, Song X. 2021. Past, present, and future of global seawater intrusion research: a bibliometric analysis. J Hydrol 603:126844. doi: 10.1016/j.jhydrol.2021.126844. [DOI] [Google Scholar]
- 74.Herbert ER, Boon P, Burgin AJ, Neubauer SC, Franklin RB, Ardón M, Hopfensperger KN, Lamers LPM, Gell P. 2015. A global perspective on wetland salinization: ecological consequences of a growing threat to freshwater wetlands. Ecosphere 6:art206. doi: 10.1890/ES14-00534.1. [DOI] [Google Scholar]
- 75.Szynkiewicz A, Witcher JC, Modelska M, Borrok DM, Pratt LM. 2011. Anthropogenic sulfate loads in the Rio Grande, New Mexico (USA). Chem Geol 283:194–209. doi: 10.1016/j.chemgeo.2011.01.017. [DOI] [Google Scholar]
- 76.Papadopoulos I. 1986. Effect of high sulfate irrigation waters on soil salinity and yields. Agron J 78:429–432. doi: 10.2134/agronj1986.00021962007800030006x. [DOI] [Google Scholar]
- 77.Ekholm P, Lehtoranta J, Taka M, Sallantaus T, Riihimäki J. 2020. Diffuse sources dominate the sulfate load into Finnish surface waters. Sci Total Environ 748:141297. doi: 10.1016/j.scitotenv.2020.141297. [DOI] [PubMed] [Google Scholar]
- 78.Orem W, Gilmour C, Axelrad D, Krabbenhoft D, Scheidt D, Kalla P, McCormick P, Gabriel M, Aiken G. 2011. Sulfur in the south Florida ecosystem: distribution, sources, biogeochemistry, impacts, and management for restoration. Crit Rev Environ Sci Technol 41:249–288. doi: 10.1080/10643389.2010.531201. [DOI] [Google Scholar]
- 79.Zhang Y, Mathur R, Bash JO, Hogrefe C, Xing J, Roselle SJ. 2018. Long-term trends in total inorganic nitrogen and sulfur deposition in the US from 1990 to 2010. Atmos Chem Phys 18:9091–9106. doi: 10.5194/acp-18-9091-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lajtha K, Jones J. 2013. Trends in cation, nitrogen, sulfate and hydrogen ion concentrations in precipitation in the United States and Europe from 1978 to 2010: a new look at an old problem. Biogeochemistry 116:303–334. doi: 10.1007/s10533-013-9860-2. [DOI] [Google Scholar]
- 81.Gao Y, Ma M, Yang T, Chen W, Yang T. 2018. Global atmospheric sulfur deposition and associated impaction on nitrogen cycling in ecosystems. J Clean Prod 195:1–9. doi: 10.1016/j.jclepro.2018.05.166. [DOI] [Google Scholar]
- 82.Whiticar MJ, Faber E, Schoell M. 1986. Biogenic methane formation in marine and freshwater environments: CO2 reduction vs. acetate fermentation—isotope evidence. Geochim Cosmochim Acta 50:693–709. doi: 10.1016/0016-7037(86)90346-7. [DOI] [Google Scholar]
- 83.Rosenfeld WD, Silverman SR. 1959. Carbon isotope fractionation in bacterial production of methane. Science 130:1658–1659. doi: 10.1126/science.130.3389.1658.b. [DOI] [PubMed] [Google Scholar]
- 84.Penger J, Conrad R, Blaser M. 2012. Stable carbon isotope fractionation by methylotrophic methanogenic archaea. Appl Environ Microbiol 78:7596–7602. doi: 10.1128/AEM.01773-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Taenzer L, Carini PC, Masterson AM, Bourque B, Gaube JH, Leavitt WD. 2020. Microbial methane from methylphosphonate isotopically records source. Geophys Res Lett 47:e2019GL085872. doi: 10.1029/2019GL085872. [DOI] [Google Scholar]
- 86.Whiticar MJ. 1999. Carbon and hydrogen isotope systematics of bacterial formation and oxidation of methane. Chem Geol 161:291–314. doi: 10.1016/S0009-2541(99)00092-3. [DOI] [Google Scholar]
- 87.Etiope G, Ehlmann BL, Schoell M. 2013. Low temperature production and exhalation of methane from serpentinized rocks on Earth: a potential analog for methane production on Mars. Icarus 224:276–285. doi: 10.1016/j.icarus.2012.05.009. [DOI] [Google Scholar]
- 88.Londry KL, Dawson KG, Grover HD, Summons RE, Bradley AS. 2008. Stable carbon isotope fractionation between substrates and products of Methanosarcina barkeri. Org Geochem 39:608–621. doi: 10.1016/j.orggeochem.2008.03.002. [DOI] [Google Scholar]
- 89.Goevert D, Conrad R. 2009. Effect of substrate concentration on carbon isotope fractionation during acetoclastic methanogenesis by Methanosarcina barkeri and M. acetivorans and in rice field soil. Appl Environ Microbiol 75:2605–2612. doi: 10.1128/AEM.02680-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schubert CJ, Coolen MJL, Neretin LN, Schippers A, Abbas B, Durisch-Kaiser E, Wehrli B, Hopmans EC, Damsté JSS, Wakeham S, Kuypers MMM. 2006. Aerobic and anaerobic methanotrophs in the Black Sea water column. Environ Microbiol 8:1844–1856. doi: 10.1111/j.1462-2920.2006.01079.x. [DOI] [PubMed] [Google Scholar]
- 91.Etiope G, Baciu CL, Schoell M. 2011. Extreme methane deuterium, nitrogen and helium enrichment in natural gas from the Homorod seep (Romania). Chem Geol 280:89–96. doi: 10.1016/j.chemgeo.2010.10.019. [DOI] [Google Scholar]
- 92.Caforio A, Driessen AJM. 2017. Archaeal phospholipids: structural properties and biosynthesis. Biochim Biophys Acta Mol Cell Biol Lipids 1862:1325–1339. doi: 10.1016/j.bbalip.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 93.Summons RE, Franzmann PD, Nichols PD. 1998. Carbon isotopic fractionation associated with methylotrophic methanogenesis. Org Geochem 28:465–475. doi: 10.1016/S0146-6380(98)00011-4. [DOI] [Google Scholar]
- 94.Fuchs G, Thauer R, Ziegler H, Stichler W. 1979. Carbon isotope fractionation by Methanobacterium thermoautotrophicum. Arch Microbiol 120:135–139. doi: 10.1007/BF00409099. [DOI] [Google Scholar]
- 95.Takai Y. 1970. The mechanism of methane fermentation in flooded paddy soil. Soil Sci Plant Nutr 16:238–244. doi: 10.1080/00380768.1970.10433371. [DOI] [Google Scholar]
- 96.Mitchell AD, Chappell A, Knox KL. 1979. Metabolism of betaine in the ruminant. J Anim Sci 49:764–774. doi: 10.2527/jas1979.493764x. [DOI] [PubMed] [Google Scholar]
- 97.Lovley DR, Klug MJ. 1983. Methanogenesis from methanol and methylamines and acetogenesis from hydrogen and carbon dioxide in the sediments of a eutrophic lake. Appl Environ Microbiol 45:1310–1315. doi: 10.1128/aem.45.4.1310-1315.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schink B, Zeikus JGY. 1982. Microbial ecology of pectin decomposition in anoxic lake sediments. Microbiology 128:393–404. doi: 10.1099/00221287-128-2-393. [DOI] [Google Scholar]
- 99.Krüger M, Frenzel P, Conrad R. 2001. Microbial processes influencing methane emission from rice fields. Glob Change Biol 7:49–63. doi: 10.1046/j.1365-2486.2001.00395.x. [DOI] [Google Scholar]
- 100.Winfrey MR, Ward DM. 1983. Substrates for sulfate reduction and methane production in intertidal sediments. Appl Environ Microbiol 45:193–199. doi: 10.1128/aem.45.1.193-199.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Musat N, Musat F, Weber PK, Pett-Ridge J. 2016. Tracking microbial interactions with NanoSIMS. Curr Opin Biotechnol 41:114–121. doi: 10.1016/j.copbio.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 102.Mooshammer M, Kitzinger K, Schintlmeister A, Ahmerkamp S, Nielsen JL, Nielsen PH, Wagner M. 2021. Flow-through stable isotope probing (flow-SIP) minimizes cross-feeding in complex microbial communities. ISME J 15:348–353. doi: 10.1038/s41396-020-00761-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang X, Lee C. 1990. The distribution and adsorption behavior of aliphatic amines in marine and lacustrine sediments. Geochim Cosmochim Acta 54:2759–2774. doi: 10.1016/0016-7037(90)90010-I. [DOI] [Google Scholar]
- 104.Repeta DJ, Ferrón S, Sosa OA, Johnson CG, Repeta LD, Acker M, DeLong EF, Karl DM. 2016. Marine methane paradox explained by bacterial degradation of dissolved organic matter. Nature Geosci 9:884–887. doi: 10.1038/ngeo2837. [DOI] [Google Scholar]
- 105.Hoehler TM. 2004. Biological energy requirements as quantitative boundary conditions for life in the subsurface. Geobiology 2:205–215. doi: 10.1111/j.1472-4677.2004.00033.x. [DOI] [Google Scholar]
- 106.Schink B, Stams AJM. 2006. Syntrophism among prokaryotes, p 309–335. In Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E (ed), The prokaryotes, 3rd ed. Springer Science & Business Media, New York, NY. [Google Scholar]
- 107.Zhang G, Jiang N, Liu X, Dong X. 2008. Methanogenesis from methanol at low temperatures by a novel psychrophilic methanogen, “Methanolobus psychrophilus” sp. nov., prevalent in Zoige Wetland of the Tibetan Plateau. Appl Environ Microbiol 74:6114–6120. doi: 10.1128/AEM.01146-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Oremland RS, Marsh L, DesMarais DJ. 1982. Methanogenesis in Big Soda Lake, Nevada: an alkaline, moderately hypersaline desert lake. Appl Environ Microbiol 43:462–468. doi: 10.1128/aem.43.2.462-468.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chidthaisong A, Conrad R. 2000. Specificity of chloroform, 2-bromoethanesulfonate and fluoroacetate to inhibit methanogenesis and other anaerobic processes in anoxic rice field soil. Soil Biol Biochem 32:977–988. doi: 10.1016/S0038-0717(00)00006-7. [DOI] [Google Scholar]
- 110.Scholten JCM, Conrad R, Stams AJM. 2000. Effect of 2-bromo-ethane sulfonate, molybdate and chloroform on acetate consumption by methanogenic and sulfate-reducing populations in freshwater sediment. FEMS Microbiol Ecol 32:35–42. doi: 10.1111/j.1574-6941.2000.tb00696.x. [DOI] [PubMed] [Google Scholar]
- 111.Hatzenpichler R, Scheller S, Tavormina PL, Babin BM, Tirrell DA, Orphan VJ. 2014. In situ visualization of newly synthesized proteins in environmental microbes using amino acid tagging and click chemistry. Environ Microbiol 16:2568–2590. doi: 10.1111/1462-2920.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hatzenpichler R, Krukenberg V, Spietz RL, Jay ZJ. 2020. Next-generation physiology approaches to study microbiome function at single cell level. Nat Rev Microbiol 18:241–256. doi: 10.1038/s41579-020-0323-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hatzenpichler R, Connon SA, Goudeau D, Malmstrom RR, Woyke T, Orphan VJ. 2016. Visualizing in situ translational activity for identifying and sorting slow-growing archaeal−bacterial consortia. Proc Natl Acad Sci USA 113:E4069–E4078. doi: 10.1073/pnas.1603757113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Watkins AJ, Roussel EG, Webster G, Parkes RJ, Sass H. 2012. Choline and N,N-dimethylethanolamine as direct substrates for methanogens. Appl Environ Microbiol 78:8298–8303. doi: 10.1128/AEM.01941-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Neill AR, Grime DW, Dawson RMC. 1978. Conversion of choline methyl groups through trimethylamine into methane in the rumen. Biochem J 170:529–535. doi: 10.1042/bj1700529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.L’Haridon S, Chalopin M, Colombo D, Toffin L. 2014. 2014. Methanococcoides vulcani sp. nov., a marine methylotrophic methanogen that uses betaine, choline and N,N-dimethylethanolamine for methanogenesis, isolated from a mud volcano, and emended description of the genus Methanococcoides. Int J Syst Evol Microbiol 64:1978–1983. doi: 10.1099/ijs.0.058289-0. [DOI] [PubMed] [Google Scholar]
- 117.Fu H, Metcalf WW. 2015. Genetic basis for metabolism of methylated sulfur compounds in Methanosarcina species. J Bacteriol 197:1515–1524. doi: 10.1128/JB.02605-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sorokin DY, Makarova KS, Abbas B, Ferrer M, Golyshin PN, Galinski EA, Ciordia S, Mena MC, Merkel AY, Wolf YI, van Loosdrecht MCM, Koonin EV. 2017. Discovery of extremely halophilic, methyl-reducing euryarchaea provides insights into the evolutionary origin of methanogenesis. Nat Microbiol 2:17081. doi: 10.1038/nmicrobiol.2017.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hoedt EC, Cuív PÓ, Evans PN, Smith WJM, McSweeney CS, Denman SE, Morrison M. 2016. Differences down-under: alcohol-fueled methanogenesis by archaea present in Australian macropodids. ISME J 10:2376–2388. doi: 10.1038/ismej.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.McKay LJ, Dlakić M, Fields MW, Delmont TO, Eren AM, Jay ZJ, Klingelsmith KB, Rusch DB, Inskeep WP. 2019. Co-occurring genomic capacity for anaerobic methane and dissimilatory sulfur metabolisms discovered in the Korarchaeota. Nat Microbiol 4:614–622. doi: 10.1038/s41564-019-0362-4. [DOI] [PubMed] [Google Scholar]
- 121.Sorokin DY, Merkel AY, Abbas B, Makarova KS, Rijpstra WIC, Koenen M, Sinninghe Damsté JS, Galinski EA, Koonin EV, van Loosdrecht MCMY. 2018. Methanonatronarchaeum thermophilum gen. nov., sp. nov. and “Candidatus Methanohalarchaeum thermophilum”, extremely halo(natrono)philic methyl-reducing methanogens from hypersaline lakes comprising a new euryarchaeal class Methanonatronarchaeia classis nov. Int J Syst Evol Microbiol 68:2199–2208. doi: 10.1099/ijsem.0.002810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kröninger L, Gottschling J, Deppenmeier U. 2017. Growth characteristics of Methanomassiliicoccus luminyensis and expression of methyltransferase encoding genes. Archaea 2017:e2756573. doi: 10.1155/2017/2756573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Thomas CM, Taib N, Gribaldo S, Borrel G. 2021. Comparative genomic analysis of Methanimicrococcus blatticola provides insights into host adaptation in archaea and the evolution of methanogenesis. ISME Commun 1:47. doi: 10.1038/s43705-021-00050-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ou Y-F, Dong H-P, McIlroy SJ, Crowe SA, Hallam SJ, Han P, Kallmeyer J, Simister RL, Vuillemin A, Leu AO, Liu Z, Zheng Y-L, Sun Q-L, Liu M, Tyson GW, Hou L-J. 2022. Expanding the phylogenetic distribution of cytochrome b-containing methanogenic archaea sheds light on the evolution of methanogenesis. ISME J 16:2373–2387. doi: 10.1038/s41396-022-01281-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mand TD, Metcalf WW. 2019. Energy conservation and hydrogenase function in methanogenic archaea, in particular in the genus Methanosarcina. Microbiol Mol Biol Rev 83:e00020-19. doi: 10.1128/MMBR.00020-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Thauer RK, Kaster A-K, Seedorf H, Buckel W, Hedderich R. 2008. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol 6:579–591. doi: 10.1038/nrmicro1931. [DOI] [PubMed] [Google Scholar]
- 127.Fricke WF, Seedorf H, Henne A, Krüer M, Liesegang H, Hedderich R, Gottschalk G, Thauer RK. 2006. The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. J Bacteriol 188:642–658. doi: 10.1128/JB.188.2.642-658.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Anthoni U, Bohlin L, Larsen C, Nielsen P, Nielsen NH, Christophersen C. 1989. Tetramine: occurrence in marine organisms and pharmacology. Toxicon 27:707–716. doi: 10.1016/0041-0101(89)90037-8. [DOI] [PubMed] [Google Scholar]
- 129.Wang C, Lin C-K. 2020. Degradation of tetramethylammonium hydroxide through the coupling of ozonation with other advanced oxidation processes. Ozone Sci Eng 42:439–449. doi: 10.1080/01919512.2019.1695099. [DOI] [Google Scholar]
- 130.Asakawa S, Sauer K, Liesack W, Thauer RK. 1998. Tetramethylammonium:coenzyme M methyltransferase system from Methanococcoides sp. Arch Microbiol 170:220–226. doi: 10.1007/s002030050636. [DOI] [PubMed] [Google Scholar]
- 131.Sun J, Mausz MA, Chen Y, Giovannoni SJ. 2019. Microbial trimethylamine metabolism in marine environments. Environ Microbiol 21:513–520. doi: 10.1111/1462-2920.14461. [DOI] [PubMed] [Google Scholar]
- 132.Wang X-C, Lee C. 1994. Sources and distribution of aliphatic amines in salt marsh sediment. Org Geochem 22:1005–1021. doi: 10.1016/0146-6380(94)90034-5. [DOI] [Google Scholar]
- 133.Smith TA. 1971. The occurrence, metabolism and functions of amines in plants. Biol Rev Camb Philos Soc 46:201–241. doi: 10.1111/j.1469-185x.1971.tb01182.x. [DOI] [PubMed] [Google Scholar]
- 134.Asatoor AM, Simeshoff ML. 1965. The origin of urinary dimethylamine. Biochim Biophys Acta Gen Subj 111:384–392. doi: 10.1016/0304-4165(65)90048-6. [DOI] [PubMed] [Google Scholar]
- 135.Abdul Rahim MBH, Chilloux J, Martinez-Gili L, Neves AL, Myridakis A, Gooderham N, Dumas M-E. 2019. Diet-induced metabolic changes of the human gut microbiome: importance of short-chain fatty acids, methylamines and indoles. Acta Diabetol 56:493–500. doi: 10.1007/s00592-019-01312-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Onyszkiewicz M, Jaworska K, Ufnal M. 2020. Short chain fatty acids and methylamines produced by gut microbiota as mediators and markers in the circulatory system. Exp Biol Med (Maywood) 245:166–175. doi: 10.1177/1535370219900898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Oren A. 1990. Formation and breakdown of glycine betaine and trimethylamine in hypersaline environments. Antonie Van Leeuwenhoek 58:291–298. doi: 10.1007/BF00399342. [DOI] [PubMed] [Google Scholar]
- 138.Zhu Y, Jameson E, Parslow RA, Lidbury I, Fu T, Dafforn TR, Schäfer H, Chen Y. 2014. Identification and characterization of trimethylamine N-oxide (TMAO) demethylase and TMAO permease in Methylocella silvestris BL2. Environ Microbiol 16:3318–3330. doi: 10.1111/1462-2920.12585. [DOI] [PubMed] [Google Scholar]
- 139.Lidbury I, Murrell JC, Chen Y. 2014. Trimethylamine N-oxide metabolism by abundant marine heterotrophic bacteria. Proc Natl Acad Sci USA 111:2710–2715. doi: 10.1073/pnas.1317834111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sørensen J, Glob E. 1987. Influence of benthic fauna on trimethylamine concentrations in coastal marine sediments. Mar Ecol Prog Ser 39:15–21. doi: 10.3354/meps039015. [DOI] [Google Scholar]
- 141.Lee C, Olson BL. 1984. Dissolved, exchangeable and bound aliphatic amines in marine sediments: initial results. Org Geochem 6:259–263. doi: 10.1016/0146-6380(84)90047-0. [DOI] [Google Scholar]
- 142.Kim SG, Bae HS, Lee ST. 2001. A novel denitrifying bacterial isolate that degrades trimethylamine both aerobically and anaerobically via two different pathways. Arch Microbiol 176:271–277. doi: 10.1007/s002030100319. [DOI] [PubMed] [Google Scholar]
- 143.Lidbury I, Mausz MA, Scanlan DJ, Chen Y. 2017. Identification of dimethylamine monooxygenase in marine bacteria reveals a metabolic bottleneck in the methylated amine degradation pathway. ISME J 11:1592–1601. doi: 10.1038/ismej.2017.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Soares JA, Zhang L, Pitsch RL, Kleinholz NM, Jones RB, Wolff JJ, Amster J, Green-Church KB, Krzycki JA. 2005. The residue mass of l-pyrrolysine in three distinct methylamine methyltransferases. J Biol Chem 280:36962–36969. doi: 10.1074/jbc.M506402200. [DOI] [PubMed] [Google Scholar]
- 145.Chen Y. 2012. Comparative genomics of methylated amine utilization by marine Roseobacter clade bacteria and development of functional gene markers (tmm, gmaS). Environ Microbiol 14:2308–2322. doi: 10.1111/j.1462-2920.2012.02765.x. [DOI] [PubMed] [Google Scholar]
- 146.Kim S-G, Bae H-S, Oh H-M, Lee S-T. 2003. Isolation and characterization of novel halotolerant and/or halophilic denitrifying bacteria with versatile metabolic pathways for the degradation of trimethylamine. FEMS Microbiol Lett 225:263–269. doi: 10.1016/S0378-1097(03)00530-5. [DOI] [PubMed] [Google Scholar]
- 147.Zhilina TN, Zavarzin GA. 1990. Extremely halophilic, methylotrophic, anaerobic bacteria. FEMS Microbiol Rev 7:315–321. doi: 10.1111/j.1574-6968.1990.tb04930.x. [DOI] [Google Scholar]
- 148.Ferguson DJ, Krzycki JA. 1997. Reconstitution of trimethylamine-dependent coenzyme M methylation with the trimethylamine corrinoid protein and the isozymes of methyltransferase II from Methanosarcina barkeri. J Bacteriol 179:846–852. doi: 10.1128/jb.179.3.846-852.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ferguson T, Soares JA, Lienard T, Gottschalk G, Krzycki JA. 2009. RamA, a protein required for reductive activation of corrinoid-dependent methylamine methyltransferase reactions in methanogenic archaea. J Biol Chem 284:2285–2295. doi: 10.1074/jbc.M807392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gaston MA, Jiang R, Krzycki JA. 2011. Functional context, biosynthesis, and genetic encoding of pyrrolysine. Curr Opin Microbiol 14:342–349. doi: 10.1016/j.mib.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Huening KA, Jiang R, Krzycki JA. 2020. Kinetic and substrate complex characterization of RamA, a corrinoid protein reductive activase from Methanosarcina barkeri. FEMS Microbiol Lett 367:fnaa128. doi: 10.1093/femsle/fnaa128. [DOI] [PubMed] [Google Scholar]
- 152.Lee C, Yoon J. 2007. UV-A induced photochemical formation of N-nitrosodimethylamine (NDMA) in the presence of nitrite and dimethylamine. J Photochem Photobiol Chem 189:128–134. doi: 10.1016/j.jphotochem.2007.01.022. [DOI] [Google Scholar]
- 153.Mustakhimov I, Kalyuzhnaya MG, Lidstrom ME, Chistoserdova L. 2013. Insights into denitrification in Methylotenera mobilis from denitrification pathway and methanol metabolism mutants. J Bacteriol 195:2207–2211. doi: 10.1128/JB.00069-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tate RL, Alexander M. 1976. Microbial formation and degradation of dimethylamine. Appl Environ Microbiol 31:399–403. doi: 10.1128/aem.31.3.399-403.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Davis EJ, De Ropp RS. 1961. Metabolic origin of urinary methylamine in the rat. Nature 190:636–637. doi: 10.1038/190636a0. [DOI] [PubMed] [Google Scholar]
- 156.Schayer RW, Smiley RL, Kaplan EH. 1952. The metabolism of epinephrine containing isotopic carbon. II. J Biol Chem 198:545–551. doi: 10.1016/S0021-9258(18)55509-5. [DOI] [PubMed] [Google Scholar]
- 157.Hill KJ, Mangan JL. 1964. The formation and distribution of methylamine in the ruminant digestive tract. Biochem J 93:39–45. doi: 10.1042/bj0930039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mitchell SC, Zhang AQ. 2001. Methylamine in human urine. Clin Chim Acta 312:107–114. doi: 10.1016/s0009-8981(01)00608-8. [DOI] [PubMed] [Google Scholar]
- 159.Fitzsimons MF, Jemmett AW, Wolff GA. 1997. A preliminary study of the geochemistry of methylamines in a salt marsh. Org Geochem 27:15–24. doi: 10.1016/S0146-6380(97)00062-4. [DOI] [Google Scholar]
- 160.Millet DB, Jacob DJ, Custer TG, de Gouw JA, Goldstein AH, Karl T, Singh HB, Sive BC, Talbot RW, Warneke C, Williams J. 2008. New constraints on terrestrial and oceanic sources of atmospheric methanol. Atmos Chem Phys 8:6887–6905. doi: 10.5194/acp-8-6887-2008. [DOI] [Google Scholar]
- 161.Schink B, Zeikus JG. 1980. Microbial methanol formation: a major end product of pectin metabolism. Curr Microbiol 4:387–389. doi: 10.1007/BF02605383. [DOI] [Google Scholar]
- 162.Rosell K-G, Svensson S. 1975. Studies of the distribution of the 4-O-methyl-d-glucuronic acid residues in birch xylan. Carbohydr Res 42:297–304. doi: 10.1016/S0008-6215(00)84271-8. [DOI] [Google Scholar]
- 163.Ander P, Eriksson MER, Eriksson K-E. 1985. Methanol production from lignin-related substances by Phanerochaete chrysosporium. Physiol Plant 65:317–321. doi: 10.1111/j.1399-3054.1985.tb02402.x. [DOI] [Google Scholar]
- 164.Donnelly MI, Dagley S. 1980. Production of methanol from aromatic acids by Pseudomonas putida. J Bacteriol 142:916–924. doi: 10.1128/jb.142.3.916-924.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Conrad R, Claus P. 2005. Contribution of methanol to the production of methane and its 13C-isotopic signature in anoxic rice field soil. Biogeochemistry 73:381–393. doi: 10.1007/s10533-004-0366-9. [DOI] [Google Scholar]
- 166.Kolb S. 2009. Aerobic methanol-oxidizing bacteria in soil. FEMS Microbiol Lett 300:1–10. doi: 10.1111/j.1574-6968.2009.01681.x. [DOI] [PubMed] [Google Scholar]
- 167.Christensson M, Lie E, Welander T. 1994. A comparison between ethanol and methanol as carbon sources for denitrification. Water Sci Technol 30:83–90. doi: 10.2166/wst.1994.0255. [DOI] [Google Scholar]
- 168.Litty D, Kremp F, Müller V. 2022. One substrate, many fates: different ways of methanol utilization in the acetogen Acetobacterium woodii. Environ Microbiol 24:3124–3133. doi: 10.1111/1462-2920.16011. [DOI] [PubMed] [Google Scholar]
- 169.Sauer K, Thauer RK. 1998. Methanol: coenzyme M methyltransferase from Methanosarcina barkeri. Eur J Biochem 253:698–705. doi: 10.1046/j.1432-1327.1998.2530698.x. [DOI] [PubMed] [Google Scholar]
- 170.Harms U, Thauer RK. 1996. Methylcobalamin:coenzyme M methyltransferase isoenzymes MtaA and MtbA from Methanosarcina barkeri. Eur J Biochem 235:653–659. doi: 10.1111/j.1432-1033.1996.00653.x. [DOI] [PubMed] [Google Scholar]
- 171.Oren A. 2013. Life at high salt concentrations, p 421–440. In Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (ed), The prokaryotes: prokaryotic communities and ecophysiology. Springer, Berlin, Heidelberg. [Google Scholar]
- 172.Bueno de Mesquita CP, Zhou J, Theroux S, Tringe SG. 2022. Methylphosphonate degradation and salt-tolerance genes of two novel halophilic Marivita metagenome-assembled genomes from unrestored solar salterns. Genes 13:148. doi: 10.3390/genes13010148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Müller E, Fahlbusch K, Walther R, Gottschalk G. 1981. Formation of N,N-dimethylglycine, acetic acid, and butyric acid from betaine by Eubacterium limosum. Appl Environ Microbiol 42:439–445. doi: 10.1128/aem.42.3.439-445.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sorokin DY. 2021. Microbial utilization of glycine betain in hypersaline Soda Lakes. Microbiology 90:569–577. doi: 10.1134/S0026261721050143. [DOI] [Google Scholar]
- 175.Ticak T, Hariraju D, Arcelay MB, Arivett BA, Fiester SE, Ferguson DJ. 2015. Isolation and characterization of a tetramethylammonium-degrading Methanococcoides strain and a novel glycine betaine-utilizing Methanolobus strain. Arch Microbiol 197:197–209. doi: 10.1007/s00203-014-1043-6. [DOI] [PubMed] [Google Scholar]
- 176.Creighbaum AJ, Ticak T, Shinde S, Wang X, Ferguson DJJ. 2019. Examination of the glycine betaine-dependent methylotrophic methanogenesis pathway: insights into anaerobic quaternary amine methylotrophy. Front Microbiol 10:2572. doi: 10.3389/fmicb.2019.02572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Leck C, Larsson U, Bågander LE, Johansson S, Hajdu S. 1990. Dimethyl sulfide in the Baltic Sea: annual variability in relation to biological activity. J Geophys Res 95:3353–3363. doi: 10.1029/JC095iC03p03353. [DOI] [Google Scholar]
- 178.van der Maarel MJEC, Hansen TA. 1997. Dimethylsulfoniopropionate in anoxic intertidal sediments: a precursor of methanogenesis via dimethyl sulfide, methanethiol, and methiolpropionate. Mar Geol 137:5–12. doi: 10.1016/S0025-3227(96)00074-6. [DOI] [Google Scholar]
- 179.Kiene RP. 1996. Production of methanethiol from dimethylsulfoniopropionate in marine surface waters. Mar Chem 54:69–83. doi: 10.1016/0304-4203(96)00006-0. [DOI] [Google Scholar]
- 180.Reisch CR, Stoudemayer MJ, Varaljay VA, Amster IJ, Moran MA, Whitman WB. 2011. Novel pathway for assimilation of dimethylsulphoniopropionate widespread in marine bacteria. Nature 473:208–211. doi: 10.1038/nature10078. [DOI] [PubMed] [Google Scholar]
- 181.Bentley R, Chasteen TG. 2004. Environmental VOSCs—formation and degradation of dimethyl sulfide, methanethiol and related materials. Chemosphere 55:291–317. doi: 10.1016/j.chemosphere.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 182.Sunda W, Kieber DJ, Kiene RP, Huntsman S. 2002. An antioxidant function for DMSP and DMS in marine algae. Nature 418:317–320. doi: 10.1038/nature00851. [DOI] [PubMed] [Google Scholar]
- 183.Alstyne KLV, Wolfe GV, Freidenburg TL, Neill A, Hicken C. 2001. Activated defense systems in marine macroalgae: evidence for an ecological role for DMSP cleavage. Mar Ecol Prog Ser 213:53–65. doi: 10.3354/meps213053. [DOI] [Google Scholar]
- 184.Taylor BF, Gilchrist DC. 1991. New routes for aerobic biodegradation of dimethylsulfoniopropionate. Appl Environ Microbiol 57:3581–3584. doi: 10.1128/aem.57.12.3581-3584.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Curson ARJ, Sullivan MJ, Todd JD, Johnston AWB. 2011. DddY, a periplasmic dimethylsulfoniopropionate lyase found in taxonomically diverse species of Proteobacteria. ISME J 5:1191–1200. doi: 10.1038/ismej.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Alcolombri U, Laurino P, Lara-Astiaso P, Vardi A, Tawfik DS. 2014. DddD is a CoA-transferase/lyase producing dimethyl sulfide in the marine environment. Biochemistry 53:5473–5475. doi: 10.1021/bi500853s. [DOI] [PubMed] [Google Scholar]
- 187.Kiene RP, Visscher PT. 1987. Production and fate of methylated sulfur compounds from methionine and dimethylsulfoniopropionate in anoxic salt marsh sediments. Appl Environ Microbiol 53:2426–2434. doi: 10.1128/aem.53.10.2426-2434.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zhuang G, Yang G, Yu J, Gao Y. 2011. Production of DMS and DMSP in different physiological stages and salinity conditions in two marine algae. Chin J Ocean Limnol 29:369–377. doi: 10.1007/s00343-011-0046-2. [DOI] [Google Scholar]
- 189.Boden R, Borodina E, Wood AP, Kelly DP, Murrell JC, Schäfer H. 2011. Purification and characterization of dimethylsulfide monooxygenase from Hyphomicrobium sulfonivorans. J Bacteriol 193:1250–1258. doi: 10.1128/JB.00977-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Horinouchi M, Kasuga K, Nojiri H, Yamane H, Omori T. 1997. Cloning and characterization of genes encoding an enzyme which oxidizes dimethyl sulfide in Acinetobacter sp. strain 20B. FEMS Microbiol Lett 155:99–105. doi: 10.1111/j.1574-6968.1997.tb12692.x. [DOI] [PubMed] [Google Scholar]
- 191.Tanimoto Y, Bak F. 1994. Anaerobic degradation of methylmercaptan and dimethyl sulfide by newly isolated thermophilic sulfate-reducing bacteria. Appl Environ Microbiol 60:2450–2455. doi: 10.1128/aem.60.7.2450-2455.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tallant TC, Paul L, Krzycki JA. 2001. The MtsA subunit of the methylthiol:coenzyme M methyltransferase of Methanosarcina barkeri catalyses both half-reactions of corrinoid-dependent dimethylsulfide: coenzyme M methyl transfer. J Biol Chem 276:4485–4493. doi: 10.1074/jbc.M007514200. [DOI] [PubMed] [Google Scholar]
- 193.Tallant TC, Krzycki JA. 1997. Methylthiol:coenzyme M methyltransferase from Methanosarcina barkeri, an enzyme of methanogenesis from dimethylsulfide and methylmercaptopropionate. J Bacteriol 179:6902–6911. doi: 10.1128/jb.179.22.6902-6911.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Oelgeschläger E, Rother M. 2009. In vivo role of three fused corrinoid/methyl transfer proteins in Methanosarcina acetivorans. Mol Microbiol 72:1260–1272. doi: 10.1111/j.1365-2958.2009.06723.x. [DOI] [PubMed] [Google Scholar]
- 195.Kiene RP, Linn LJ, González J, Moran MA, Bruton JA. 1999. Dimethylsulfoniopropionate and methanethiol are important precursors of methionine and protein-sulfur in marine bacterioplankton. Appl Environ Microbiol 65:4549–4558. doi: 10.1128/AEM.65.10.4549-4558.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Saini HS, Attieh JM, Hanson AD. 1995. Biosynthesis of halomethanes and methanethiol by higher plants via a novel methyltransferase reaction. Plant Cell Environ 18:1027–1033. doi: 10.1111/j.1365-3040.1995.tb00613.x. [DOI] [Google Scholar]
- 197.Steele RD, Benevenga NJ. 1979. The metabolism of 3-methylthiopropionate in rat liver homogenates. J Biol Chem 254:8885–8890. doi: 10.1016/S0021-9258(19)86782-0. [DOI] [PubMed] [Google Scholar]
- 198.Myers RW, Wray JW, Fish S, Abeles RH. 1993. Purification and characterization of an enzyme involved in oxidative carbon-carbon bond cleavage reactions in the methionine salvage pathway of Klebsiella pneumoniae. J Biol Chem 268:24785–24791. doi: 10.1016/S0021-9258(19)74533-5. [DOI] [PubMed] [Google Scholar]
- 199.Fu H, Goettge MN, Metcalf WW. 2019. Biochemical characterization of the methylmercaptopropionate:Cob(I)alamin methyltransferase from Methanosarcina acetivorans. J Bacteriol 201:e00130-19. doi: 10.1128/JB.00130-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Stams AJM. 1994. Metabolic interactions between anaerobic bacteria in methanogenic environments. Antonie Van Leeuwenhoek 66:271–294. doi: 10.1007/BF00871644. [DOI] [PubMed] [Google Scholar]
- 201.Lomans BP, Maas R, Luderer R, Op den Camp HJM, Pol A, van der Drift C, Vogels GD. 1999. Isolation and characterization of Methanomethylovorans hollandica gen. nov., sp. nov., isolated from freshwater sediment, a methylotrophic methanogen able to grow on dimethyl sulfide and methanethiol. Appl Environ Microbiol 65:3641–3650. doi: 10.1128/AEM.65.8.3641-3650.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Borton MA, Daly RA, O'Banion B, Hoyt DW, Marcus DN, Welch S, Hastings SS, Meulia T, Wolfe RA, Booker AE, Sharma S, Cole DR, Wunch K, Moore JD, Darrah TH, Wilkins MJ, Wrighton KC. 2018. Comparative genomics and physiology of the genus Methanohalophilus, a prevalent methanogen in hydraulically fractured shale. Environ Microbiol 20:4596–4611. doi: 10.1111/1462-2920.14467. [DOI] [PubMed] [Google Scholar]
- 203.Hippe H, Caspari D, Fiebig K, Gottschalk G. 1979. Utilization of trimethylamine and other N-methyl compounds for growth and methane formation by Methanosarcina barkeri. Proc Natl Acad Sci USA 76:494–498. doi: 10.1073/pnas.76.1.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Paterek JR, Smith PH. 1985. Isolation and characterization of a halophilic methanogen from Great Salt Lake. Appl Environ Microbiol 50:877–881. doi: 10.1128/aem.50.4.877-881.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sowers KR, Ferry JG. 1983. Isolation and characterization of a methylotrophic marine methanogen, Methanococcoides methylutens gen. nov., sp. nov. Appl Environ Microbiol 45:684–690. doi: 10.1128/aem.45.2.684-690.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Zhilina TN, Merkel AY. 2019. Methanohalobium, p 1–6. In Whitman WB (ed), Bergey’s Manual of Systematics of Archaea and Bacteria. John Wiley & Sons, Ltd, New York, NY. doi: 10.1002/9781118960608.gbm00515.pub2. [DOI] [Google Scholar]
- 207.Mathrani IM, Boone DR, Mah RA, Fox GE, Lau PPY. 1988. Methanohalophilus zhilinae sp. nov., an alkaliphilic, halophilic, methylotrophic methanogen. Int J Syst Bacteriol 38:139–142. doi: 10.1099/00207713-38-2-139. [DOI] [PubMed] [Google Scholar]
- 208.Shen Y, Chen S-C, Lai M-C, Huang H-H, Chiu H-H, Tang S-L, Rogozin DY, Degermendzhy AG. 2020. Methanolobus halotolerans sp. nov., isolated from the saline Lake Tus in Siberia. Int J Syst Evol Microbiol 70:5586–5593. doi: 10.1099/ijsem.0.004453. [DOI] [PubMed] [Google Scholar]
- 209.Ticak T, Kountz DJ, Girosky KE, Krzycki JA, Ferguson DJ. 2014. A nonpyrrolysine member of the widely distributed trimethylamine methyltransferase family is a glycine betaine methyltransferase. Proc Natl Acad Sci USA 111:E4668–E4676. doi: 10.1073/pnas.1409642111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Jeffries TC, Seymour JR, Newton K, Smith RJ, Seuront L, Mitchell JG. 2012. Increases in the abundance of microbial genes encoding halotolerance and photosynthesis along a sediment salinity gradient. Biogeosciences 9:815–825. doi: 10.5194/bg-9-815-2012. [DOI] [Google Scholar]
- 211.Poffenbarger HJ, Needelman BA, Megonigal JP. 2011. Salinity influence on methane emissions from tidal marshes. Wetlands 31:831–842. doi: 10.1007/s13157-011-0197-0. [DOI] [Google Scholar]
- 212.Al-Haj AN, Fulweiler RW. 2020. A synthesis of methane emissions from shallow vegetated coastal ecosystems. Glob Chang Biol 26:2988–3005. doi: 10.1111/gcb.15046. [DOI] [PubMed] [Google Scholar]
- 213.Dasgupta S, Hossain M, Huq M, Wheeler D. 2015. Climate change and soil salinity: the case of coastal Bangladesh. Ambio 44:815–826. doi: 10.1007/s13280-015-0681-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Nielsen DL, Brock MA. 2009. Modified water regime and salinity as a consequence of climate change: prospects for wetlands of Southern Australia. Clim Change 95:523–533. doi: 10.1007/s10584-009-9564-8. [DOI] [Google Scholar]
- 215.Richards MA, Lie TJ, Zhang J, Ragsdale SW, Leigh JA, Price ND. 2016. Exploring hydrogenotrophic methanogenesis: a genome scale metabolic reconstruction of Methanococcus maripaludis. J Bacteriol 198:3379–3390. doi: 10.1128/JB.00571-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kumar VS, Ferry JG, Maranas CD. 2011. Metabolic reconstruction of the archaeon methanogen Methanosarcina Acetivorans. BMC Syst Biol 5:28. doi: 10.1186/1752-0509-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Nazem-Bokaee H, Gopalakrishnan S, Ferry JG, Wood TK, Maranas CD. 2016. Assessing methanotrophy and carbon fixation for biofuel production by Methanosarcina acetivorans. Microb Cell Fact 15:10. doi: 10.1186/s12934-015-0404-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Goyal N, Widiastuti H, Karimi I, Zhou Z. 2014. A genome-scale metabolic model of Methanococcus maripaludis S2 for CO2 capture and conversion to methane. Mol Biosyst 10:1043–1054. doi: 10.1039/c3mb70421a. [DOI] [PubMed] [Google Scholar]
- 219.Stolyar S, Van Dien S, Hillesland KL, Pinel N, Lie TJ, Leigh JA, Stahl DA. 2007. Metabolic modeling of a mutualistic microbial community. Mol Syst Biol 3:92. doi: 10.1038/msb4100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Benedict MN, Gonnerman MC, Metcalf WW, Price ND. 2012. Genome-scale metabolic reconstruction and hypothesis testing in the methanogenic archaeon Methanosarcina acetivorans C2A. J Bacteriol 194:855–865. doi: 10.1128/JB.06040-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Gonnerman MC, Benedict MN, Feist AM, Metcalf WW, Price ND. 2013. Genomically and biochemically accurate metabolic reconstruction of Methanosarcina barkeri Fusaro, iMG746. Biotechnol J 8:1070–1079. doi: 10.1002/biot.201200266. [DOI] [PubMed] [Google Scholar]
- 222.Feist AM, Scholten JCM, Palsson BØ, Brockman FJ, Ideker T. 2006. Modeling methanogenesis with a genome-scale metabolic reconstruction of Methanosarcina barkeri. Mol Syst Biol 2:2006.0004. doi: 10.1038/msb4100046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Beller HR, Rodrigues AV, Zargar K, Wu Y-W, Saini AK, Saville RM, Pereira JH, Adams PD, Tringe SG, Petzold CJ, Keasling JD. 2018. Discovery of enzymes for toluene synthesis from anoxic microbial communities. Nat Chem Biol 14:451–457. doi: 10.1038/s41589-018-0017-4. [DOI] [PubMed] [Google Scholar]
- 224.Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe-Fadrosh EA, Tringe SG, Ivanova NN, Copeland A, Clum A, Becraft ED, Malmstrom RR, Birren B, Podar M, Bork P, Weinstock GM, Garrity GM, Dodsworth JA, Yooseph S, Sutton G, Glöckner FO, Gilbert JA, Nelson WC, Hallam SJ, Jungbluth SP, Ettema TJG, Tighe S, Konstantinidis KT, Liu W-T, Baker BJ, Rattei T, Eisen JA, Hedlund B, McMahon KD, Fierer N, Knight R, Finn R, Cochrane G, Karsch-Mizrachi I, Tyson GW, Rinke C, Lapidus A, Meyer F, Yilmaz P, Parks DH, Eren AM. Genome Standards Consortium, et al. 2017. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol 35:725–731. doi: 10.1038/nbt.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Zhuang G-C, Lin Y-S, Bowles MW, Heuer VB, Lever MA, Elvert M, Hinrichs K-U. 2017. Distribution and isotopic composition of trimethylamine, dimethylsulfide and dimethylsulfoniopropionate in marine sediments. Mar Chem 196:35–46. doi: 10.1016/j.marchem.2017.07.007. [DOI] [Google Scholar]
- 226.Botz R, Pokojski H-D, Schmitt M, Thomm M. 1996. Carbon isotope fractionation during bacterial methanogenesis by CO2 reduction. Org Geochem 25:255–262. doi: 10.1016/S0146-6380(96)00129-5. [DOI] [Google Scholar]
- 227.Penning H, Claus P, Casper P, Conrad R. 2006. Carbon isotope fractionation during acetoclastic methanogenesis by Methanosaeta concilii in culture and a Lake Sediment. Appl Environ Microbiol 72:5648–5652. doi: 10.1128/AEM.00727-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Download mmbr.00024-22-s0001.xlsx, XLSX file, 0.1 MB (81.3KB, xlsx)
Tables S2 and S3, Fig. S1 to S4. Download mmbr.00024-22-s0002.pdf, PDF file, 0.6 MB (631.6KB, pdf)