

Abstract
Data analysis pipelines are now established as an effective means for specifying and executing bioinformatics data analysis and experiments. While scripting languages, particularly Python, R and notebooks, are popular and sufficient for developing small-scale pipelines that are often intended for a single user, it is now widely recognized that they are by no means enough to support the development of large-scale, shareable, maintainable and reusable pipelines capable of handling large volumes of data and running on high performance computing clusters. This review outlines the key requirements for building large-scale data pipelines and provides a mapping of existing solutions that fulfill them. We then highlight the benefits of using scientific workflow systems to get modular, reproducible and reusable bioinformatics data analysis pipelines. We finally discuss current workflow reuse practices based on an empirical study we performed on a large collection of workflows.
Keywords: Scientific workflows, Bioinformatics, Reuse, Reproducibility
Graphical Abstract
Highlights
-
•
Elicitation of the problems faced when designing large-scale bioinformatics pipelines.
-
•
Review of existing solutions for developing reuseable bioinformatics pipelines.
-
•
Quantitative and qualitative study on current reuse of bioinformatics workflow systems.
1. Introduction
The availability of large dataset along with data transformation and analysis tools, has revolutionized how bioinformaticians conduct computational experiments [1]. These data analyses are increasingly performed through pipelines implemented using scripting languages or notebooks [2]. These pipelines link and intertwine data transformation and analysis tools to convert raw input data into results that allow scientists to gain insights and draw conclusions about the validity of a hypothesis or known fact.
The above approach, mainly involving custom scripts, has been extensively used for small-scale experiments involving a small number of datasets and a small number of analysis and transformation tools [3]. For instance, in the case a single user (bioinformatician) is responsible for the design and the execution of the pipeline as well as the analysis of its results, she or he has prior knowledge of the datasets involved and the analysis tools being used. It is also fair to assume that the pipeline is being run on a single local and homogeneous computing environment.
However, with the above approach, developing large and complex pipelines handling massive amounts of data becomes challenging [4], [5], especially when teams with different expertise and operating in a distributed (and potentially heterogeneous) computing execution environment are involved. In addition, it does not facilitate pipeline sharing and reuse [3]. A moderately sized pipeline can quickly become difficult to understand and maintain, and even more difficult to reuse by third parties. This has become a major concern as bioinformaticians (and scientists in general) are expected to share their resources, including datasets, analysis tools, and also pipelines, in a form that can be readily understood and reused by peers.
The objective of this review article is threefold.
Firstly, we highlight the barriers that need to be overcome to enable the development, sharing and reuse of large-scale pipelines.
Secondly, we show that scientific workflows (e.g., [6], [7], [8]), in combination with other auxiliary technologies, such as tool registries [9], address some of these barriers. Even though scientific workflow systems are not new, they have been around for over two decades, and are now mature enough to be used routinely. The first generation of scientific workflow systems (e.g., Taverna [10]) was designed for users with a low level of programming skills and featured a workbench to compose workflows by graphically dragging and dropping “modules” and linking them together. Thus, these systems had little acceptance among proficient bioinformatics developers. In recent years, however, there has been an emergence of a new generation of scientific workflow systems, notably Nextflow [7] and Snakemake [6]. These script-based systems offer developers significant control over workflow design, configuration and execution. This second generation of workflow systems has gained wider acceptance among bioinformaticians as they address many of the challenges faced by pipeline developers and users. Thirdly, we focus on reuse, one of the major benefits that can be drawn from scientific workflows. In particular, we examine the practical impact of workflow reuse by thoroughly examining a pool of real-world workflows.
Accordingly, the article is structured as follows. We begin by outlining the challenges that need to be overcome to enable the management of large-scale pipelines in section 2. We then present elements of solutions that can be adopted to overcome some of these hurdles, particularly scientific workflows, in section 3. We report on an empirical analysis that examines the state of workflow reuse in practice in section 4, before concluding and discussing the paper in section 5.
2. Difficulties in the development of bioinformatics pipelines
This section outlines why managing large-scale pipelines throughout all stages of their life cycle, from development to sharing and reuse (see Fig. 1) is a difficult task.
Fig. 1.

Life cycle of a bioinformatics pipeline.
To illustrate such difficulties, we use a few real examples encountered during the large scale biomedical project “understanding the pathophysiology of IntraCranial ANeurysm” (ICAN). ICAN aims to better understand and predict the development and rupture of intracranial aneurysms [11], [12]. For this study, biologists and physicians have set up a large collection of biological samples from a population of 3000 individuals. This bio-collection has been used to generate and analyse various types of data, including genomic sequencing data, cerebral vascular organ neuroimaging data, and clinical data (like family history and lifestyle). This data is used to create predictive models for assessing the risk of development and rupture of intracranial aneurysms. Integrating and analysing such heterogeneous data requires specific expertise and a wide variety of software tools, which makes the implementation, execution and sharing among the project stakeholders (and ultimately the community) a difficult task.
2.1. Pipeline development
The first type of problem we can define is related to the actual development of the pipeline, which usually involves multiple programming languages and software environments.
In the ICAN project, sequencing data is analyzed with multiple tools (e.g., BWA [13], GATK[14], Picard Tools [15]), and statistical analyses are performed using Python and R librairies1 in Jupyter Notebooks [2]. The identification of the right tools to use is a first barrier to overcome. The combination of these multiple tools, languages and environments adds a layer of complexity to the development of the pipeline.
2.2. Pipeline testing
Code testing constitutes the next difficulty. Whether it is functional unit testing (testing one part of the program in isolation), integration testing (testing the combination of multiple parts of the program), or environment testing (testing how the program can be run on various platforms), requires significant effort from the pipeline developer(s) to delineate the boundaries of a test (which part of a pipeline to test), and to specify the test cases (selecting example inputs and defining expected outputs).
2.3. Deployment
Most pipelines cannot be run locally, on a single server containing all the necessary data. They usually need to be deployed on a large-scale High Performance Computing (HPC) infrastructure. This deployment step can be tedious, time-consuming, and requires a technical expertise beyond programming (see [16]). Executing a pipeline on a HPC cluster requires setting the computing tasks in the right order, this is called a job submission. For example, the challenges lie in executing the right commands to submit jobs on the right waiting queue, with the right amount of CPUs and memory, task success or failure (e.g., manually check whether the jobs finished successfully, and re-submit them if necessary), availability of computing nodes and deployed workloads (e.g., choosing the right queue depending on the planned run time or memory requirements).
2.4. Maintenance
Maintaining consistency and robustness of the pipeline over time is crucial. However, given the complexity of the pipelines, a previously functioning pipeline may experience failure or produce unexpected results due to one or more of the following causes: changes in hardware environment, changes in software environment (e.g., tools are no longer available or have been updated) or changes in datasets (for example, reference genome sequences are constantly evolving). This may require regular updates of the pipeline itself.
2.5. Reproducing
Ensuring a pipeline can be run over time (maintenance) and over site (deployment) is directly related to the ability to reproduce its results. A pipeline that runs successfully at one site may not work at another site, or may yield unexpected results, due to a different cluster scheduler, hardware, or software environment.
2.6. Reuse
Reusing an analysis pipeline cannot be performed if the pipeline is not reproducible. In other words, Reproducibility is the basis of cumulative science: if the pipeline has been designed to be reproducible then there is hope for it to be more easily shared and reused (in part or in whole) by third parties.
Three needs should be met for reuse.
First, for pipelines to be shared and reused, they must be easily understandable to determine their relevance and how they can be reused in different analyses. Here both pipeline documentation and pipeline modularity (were pipelines are not written as linear code/scripts) play a key role.
Second, once developed, pipelines should be made available in registries that enable developers and curators to document, share with a team or community, or publish pipelines. Pipeline registries must have the capability to track a pipeline’s history over time, various versions and modifications of a pipeline may be developed (to adapt to changes in the execution environment and/or to changes in functional needs).
Third, in addition to sharing pipelines, sharing their constituting “modules”, that are called processors, can help developers save valuable time during development. These processors should be documented with dedicated metadata and annotations in order to be easily found.
3. Landscape of available solutions
In this section, we present elements of solutions to the problems described in the previous section. We will first focus on generic solutions for managing tools, environments, and code. These elements of solutions are generic, meaning they do not rely on the specific language or technology used to develop the pipelines: pipelines can be developed as scripts, notebooks, or scientific workflows.
We then describe a specific solution to manage pipelines, namely using scientific workflow systems, and show how this solves several of the major problems encountered.
3.1. Generic elements of solutions
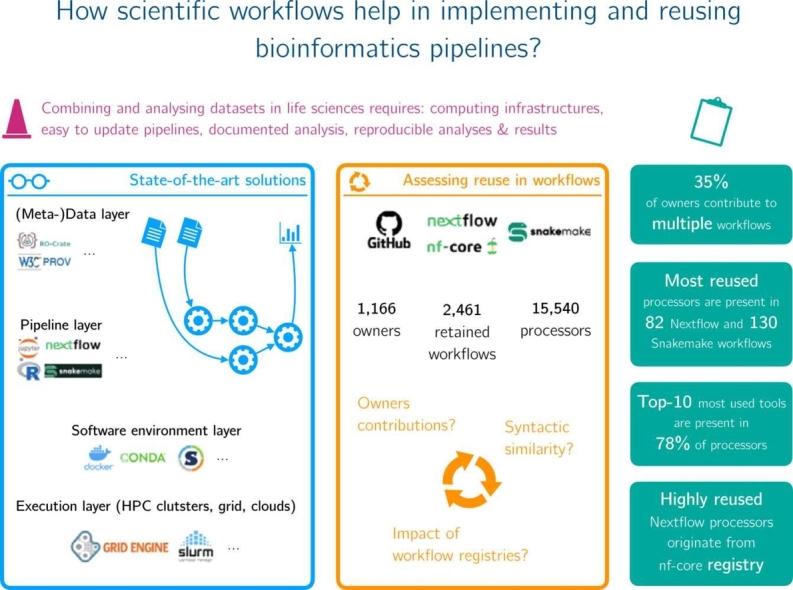
Available solutions can be used to support different facets of pipeline management. Fig. 2 summarizes generic solutions to deal with the multiple layers of pipeline management: data, pipeline, environment, and execution layers.
Fig. 2.

Generic elements of solutions. Analysis can be defined with four layers: Data, pipeline, software environment, and execution layers. On each layer some elements of solutions which are widely used nowadays.
3.1.1. Supporting pipeline development
3.1.1.1. Identifying the right tools to use
As mentioned earlier, bioinformatics pipelines are rarely made of purely new pieces of code. Rather, they make calls to existing tools. In this regard, bio.tools [17] provides an element of solution by offering a large repository of bioinformatics tools. As of January 2023, bio.tools provides 27,538 entries. bio.tools is the successor of Biocatalogue [18], the pioneer tool repository.
3.1.1.2. Developing pipelines
The “Pipeline layer” defined in Fig. 2 provides elements of solution for pipeline implementation. They include Notebooks (e.g., Jupyter) and scientific workflows (e.g., Snakemake, Nextflow). Notebooks are increasingly used as a mean to share and display source code while interactively providing a visualization of the results [2]. Workflow systems will be described in section 3.2.
On the left hand side of Fig. 2, a few examples of software able to support collaborative development are presented. Bioinformatics developers use various IDE (integrated development environments) equipped with plugins; they work in collaborative platforms like GitHub or GitLab to share and review code. In this domain, Git has become the universal reference to manage code version and collaboration.
3.1.2. Tracing data for testing and reuse
Once a first version of the pipeline has been developed, the testing phase may start and needs dedicated datasets. The “Data layer” defined in Fig. 2 represents the datasets consumed and produced during an execution of a given bioinformatics pipeline. Representing and tracing such data provenance in a uniform way is key, both for testing and documenting pipelines which will allow reuse.
Elements of solutions include the W3C PROV recommendation2 that offers a highly generic model for exchanging provenance data on the Web. As PROV does not explicitly provide all the concepts necessary for modeling pipelines or their executions, several PROV-compliant extension languages co-exist: wfprov,3 prov-wf [19] and ProvONE.4
A recent initiative to annotate scientific datasets with lightweight provenance metadata has been introduced, namely, RO-crate.5 It benefits from Schema.org annotations [20], a controlled vocabulary initially proposed to increase the findability of digital objects on the web.
3.1.3. Ensuring pipelines to stand over time and place
3.1.3.1. Deployment
The “Execution layer” defined in Fig. 2 represents the various execution infrastructures, from cloud to cluster or grid. Developers must ensure their pipelines are able to run in such various configurations. For example, in HPC cluster environments, different schedulers may be available (e.g., SLURM [21], PBS [22], LSF or SGE). Even if they solve some difficulties (job scheduling, parallel execution, queue management, etc.), users still have to interact with potentially heterogeneous schedulers, to submit the right jobs in the right order, and to deal with re-submission of failed job executions.
Many tips and solutions have been suggested to handle pipeline job submissions in these environments (see [16]). However, workflow systems, described in section 3.2, currently constitute one of the easiest solution.
3.1.3.2. Maintenance, reproducibility and reuse
The “Environment layer”, defined in Fig. 2, displays several approaches that have been proposed to describe, store and share the execution environment of the pipeline and its scientific context. Capturing this information is key to ensure the pipeline maintenance and thus reproducibility, its ability to continue running over time, and therefore make it reuseable by third parties.
Three families of solutions co-exist.
First, virtualization technologies such as VMware,6 KVM,7 VirtualBox,8 and Vagrant9 can be used (and have been widely used in the past) to package or “freeze” pipeline software environments. As they require storing and executing the entire runtime environment (including the operating system), these solutions are particularly expensive.
The second kind of solutions are based on containers and represent remarkable and lightweight alternatives. They only capture specific dependencies required by applications, and share low-level components provided by the operating system. The containers are built from recipes, simple text files describing how they are constructed, and facilitating their composition, management and sharing. Examples include OpenVZ,10 LXC,11 and more widely used in bioinformatics: Docker [23], and Singularity [24].
The third kind of solutions are based on package management systems, such as Conda.12 They facilitate tool and dependency installation, environment management and sharing (to some extent), but do not completely solve the heterogeneity of execution machines and operating systems. They are also often used to easily create containers (e.g., BioContainers [25], [26]).
These tools are a huge step forward for maintenance, reproducibility and reuse. However, they are still difficult to integrate in pipelines, which is the responsibility of the developer. Again, workflows systems are a great help in orchestrating all these solutions together.
3.2. Scientific workflow systems
Scientific workflow systems[1] have been designed to help bioinformatics scientists to design and execute workflows at multiple levels [27] throughout pipelines life cycle. Scientific workflow systems define workflows as a chain of processors, each performing specific bioinformatics operations by encapsulating a tool or a script. These processors are chained together by data flow: the input of a processor is connected to the output of the previous one, which determines the order in which they are executed.
Pioneer systems include Taverna [10], Kepler [28] and VisTrails [29] have been excellent research prototypes, but are no longer maintained. Over the past decade, three systems - Galaxy [8], Snakemake [6], and Nextflow [30] - have reached a good level of maturity and are now frequently used by bioinformaticians for managing their data analysis.
The main difference between Galaxy and the other two is the targeted users. While Galaxy targets end-users without programming skills (workflow development is made by drag-and-drop actions on predefined steps), Nextflow and Snakemake target bioinformaticians who are proficient in scripting languages, making them increasingly popular in a community with growing development skills.
We will now review scientific workflow systems, especially Snakemake and Nextflow, in light of their ability to provide solutions at each stage of the pipeline life cycle.
3.2.1. Supporting pipelines life cycle with scientific workflows
Scientific workflow systems play a central role in the orchestration of all the layers described in Fig. 2, and act at all stages of pipelines life cycle.
3.2.1.1. Development
Workflows propose an abstract representation of pipelines, allowing easily mixing multiple programming languages and tools. In workflow implementations, scripts are encapsulated: each step of the analysis is described in a unified form, a processor, in a language specific to the workflow system. A processor contains a script (e.g., Python, R, Shell) or a call to a bioinformatics tool (available in the software environment). The encapsulation defines an interface (in the programming point-of-view) as simple as possible: inputs (data types and parameters), outputs, and executions are specified in a language and a format recognized by the workflow system.
3.2.1.2. Pipeline testing
The abstract representation of pipelines provided by workflow systems facilitates testing, which can be then performed within a single development environment. Integration tests and deployment tests are then performed within workflow systems, still in the same development environment.
Snakemake proposes a dedicated unit test framework, it is also possible in Nextflow to implement unit tests via nf-core [31] (described in next paragraphs).
3.2.1.3. Deployment
As orchestrators of all the analysis layers (processing, execution, tools and environment, see section 2 and Fig. 2), workflows schedule job execution using workflow structure, monitor job execution and re-submission (success and failure), and manage job submissions on a large diversity of HPC infrastructures with almost no effort from the developer.
More precisely, workflow systems allow the underlying execution machines (local, clustered, cloud, etc.) to be completely decoupled from the workflow implementation, by separating the workflow logic from its configuration (which machine and scheduler it runs on). As for the optimization of execution, workflow systems add a layer of task scheduling on top of the operating system that executes the task and the HPC scheduler (e.g., SLURM), therefore allowing to fully leverage the parallel nature of the workflow to execute the tasks in the right order and distribute them on all the available computing resources. Workflow systems supporting distributed scheduling have been discussed in detail in [32].
3.2.1.4. Maintenance and reproducibility
Workflow systems make use of containers (Docker/Singularity) or environments (Conda). In doing so, they decouple the implementation of each step from its environment configuration (which determines the container it runs in). They are able to manage both the software environment (via Docker, Singularity, Conda) and the execution environment. Maintaining and updating the software and execution environments used by a workflow, thus making it reproducible, becomes simpler as a result.
3.2.1.5. Sharing/Reuse
To increase their reusability, workflows can be composed of independent processors that can be reused and chained to form new workflows. With encapsulation and modularity, sub-workflows can be created, allowing designers to partially hide workflow complexity and facilitate the reuse of its individual units.
For example, Snakemake proposes several levels of encapsulation and modularity: “wrappers” encapsulate steps, “includes” include an external workflow in the current one, and “modules” define external workflows. Since the transition to the second version of its language (DSL2), Nextflow also offers increased encapsulation and modularity. In particular, DSL2 defines “modules” and “sub-workflows” that allow individual processes (i.e., steps) and sub-workflows to be used across several workflows.
A second element of solution for sharing and reusing workflows is also facilitated by workflow repositories. A few of them have been developed in the past, such as myExperiment [33] (pioneer system), CrowdLabs [34] or SHIWA [35] to name a few. These repositories are no longer maintained as they were associated to Taverna, Kepler and VisTrails systems. Currently active repositories include the Galaxy repository [36] and WorkflowHub [37] (the successor of myExperiment). Other initiatives propose databases of curated workflows, such as nf-core [31] (42 workflows) and sequana [38] (11 workflows) for Nextflow and Snakemake respectively. However, GitHub remains the most important source of workflows, with several thousands of bioinformatics workflows available to users. We shall get back to this point in the next section.
3.2.2. Workflow example from the ICAN project
The ICAN project is a very good illustration of the needs met in large-scale multidisciplinary health projects. In particular in such projects, due to legal constraints regarding the protection of personal data, it is not possible to relocate data from one partner’s site to another. Pipelines must be deployed at the site of the data-holding partner. It is thus crucial to follow good practices during the entire pipeline life cycle. This facilitates the pipeline sharing among partners, ensuring the pipeline runs properly and delivers the expected results, even in HPC environments differing from the development and testing environment. Importantly, the use of standards over proprietary languages for specifying the pipeline and its configuration, as well as for specifying software and execution environments is crucial to achieve a good level of reproducibility.
As highlighted previously, scientific workflow systems provide concrete answers to such needs. We introduce one example of a workflow developed in the context of the ICAN project which performs data analysis (quality control). Our workflow has been developed in Snakemake, it is called BAM_QC 13 and is depicted in Fig. 3. Such a workflow assesses the quality of raw sequencing data by computing several useful metrics (e.g. sequence length, level of contamination or percentage of sequences mapped onto the reference genome) on all the samples. The results of this workflow are used to decide whether to accept or reject the samples.
Fig. 3.

Schematic representation of BAM_QC, the workflow performing sample quality control. It is made of 7 steps: i) InsertCompute and StatsInserts estimate the length of the sequence fragments, ii) SamRun and catStats compute several statistics about mapping, and iii) somalier_extract and somalier_relate compute relatedeness between samples.
Each step of the workflow is implemented as a rule in Snakemake (see Fig. 4), a rule specifies i) its inputs and outputs, ii) the script to execute, and iii) its configuration (software environment and execution machines to use), usually documented in an independent file. While this workflow is rather simple, it would have been more difficult to satisfy all the requirements described in section 2 (in terms of reuse, maintenance, etc.) without a scientific workflow system.
Fig. 4.

Example of a Snakemake rule that estimates the length of sequenced fragments. It specifies its inputs (two cram files), its outputs (one pdf and one txt file), its parameters (one reference fasta file), its environment (Conda, defined in a yaml file alongside), the number of threads that it will use, and the script to execute (involving the call to two bioinformatics tools namely Samtools and Picard).
By using a workflow system, the pipeline code (which implements the logic of the experiment or data analysis) is decoupled from the input datasets and the configuration setup. This allows the same workflow (code) to be portable and executable using different input datasets and across different environments/platforms.
4. Workflow systems and bioinformatics pipelines reuse
In this section, we present the results of a comprehensive study we conducted on the reuse of scientific workflows. Our aim is to show how these workflow systems are being used in practice by the bioinformatics community and gain a better understanding of reuse practices. While previous studies (e.g., [39]) focused on Taverna [10] and Galaxy [8] workflow systems, our study is the first to consider the increasingly popular Snakemake [6] and Nextflow [30] workflow systems.
To do so, we studied the reuse of Nextflow and Snakemake workflows available on GitHub.
After presenting how we have collected workflows, we will introduce our results through three main points. First, workflow reuse through the perspective of repository owners and contributors is studied. Second, reuse practices are examined by comparing workflow code (copy paste, forks, etc.). Third, reuse of individual workflow steps between users is analyzed. While these aspects are mainly quantitative, we will then shift to a qualitative perspective about reuse by examining the bioinformatics operations executed in the collected workflows.
The source code for the data collection and the reuse analyses is available on three GitHub repositories: extraction of information from Snakemake workflows,14 extraction of information from Nextflow workflows15 and experiments and Figures of the papers.16
4.1. Collection of workflows
Figure 5 illustrates our approach for extracting and collecting information about the workflows used in our study. We implemented and executed the three software components (CrawlWF, ParsWF, and WF2BT) to search, extract, parse and annotate Snakemake and Nextflow workflows from GitHub.
Fig. 5.

Workflow extraction and annotation process. It is made of three main software components: i) CrawlWF searches and extracts workflows from GitHub, ii) ParsWF parses the workflows to extract meaningful informations, and iii) WF2BT matches workflow processors to bio.tools annotations.
After workflow collection and extraction from GitHub, CrawlWF harvests GitHub metadata to associate each workflow with its owner (who owns the repository) and contributors (who contributed to the repository). ParsWF then examines the content of the repository, and retains only the well-formed workflow files by syntactically checking Nextflow and Snakemake specifications. It then extracts information about the workflow steps (workflow processors). Finally, WF2BT annotates workflow steps with metadata retrieved from the bio.tools [9] registry. WF2BT proceeds in three steps: (i) extracts the shell script from the processor implementation; (ii) extracts the command names from the shell script to constitute a set of candidate tools; (iii) matches the candidate tools to the list of tool names in bio.tools. Candidate tools that could not be matched to the bio.tools registry (no match with any bio.tool name) were compared to biocontainers [25] metadata, as each container is associated with bio.tools identifiers and comes with a list of shell commands it provides. That way, WF2BT lists the set of tools used by each processor and which have a bio.tools entry.
Using the above approach, as of May 2022, CrawlWF was able to extract 1790 Nextflow and 3866 Snakemake workflows. Of these, ParsWF retained 1675 well-formed Nextflow and 2946 well-formed Snakemake workflows (Fig. 6 provides the evolution of the number of Snakemake and Nextflow workflows available on GitHub in the past eight years). From these, WF2BT retained only 1186 Nextflow and 1257 Snakemake matched-tools workflows (containing at least one processor associated with at least one tool in bio.tools), which represents a total of 2443 retained workflows. Such workflows contain respectively 9652 and 5888 matched-tools processors (individual processors for which at least one tool has a bio.tools entry).
Fig. 6.

Evolution of the monthly and cumulative number of Nextflow and Snakemake workflows available on GitHub since 2014.
Matched-tools processors and matched-tools workflows are the objects of interest in the rest of the study.
4.2. Workflow owners and contributors
We first focus on how the workflows are distributed among repository/workflows owners and contributors. To do so, we have extracted this information from GitHub using CrawlWF. Each workflow is associated to exactly one owner (the owner of the GitHub repository), and potentially several contributors (the GitHub users that have at least contributed once to the repository).
We found that the 1186 Nextflow and 1257 Snakemake matched-tools workflows were owned by 650 and 535 owners respectively. This represents a total of 1166 workflow owners (19 owners published both Nextflow and Snakemake workflows). Considering the ten owners who have provided the largest number of workflows in Nextflow and Snakemake (top ten owners), they have actually published 15% of all Nextflow and of all Snakemake workflows. We are thus not in a situation where workflows are systematically uploaded by a restricted number of owners. Interestingly, 31% (Snakemake) vs. 42% (Nextflow) authors have published at least two workflows. There exists a set of owners who provide several workflows to the community, and overall, workflow authors in general produce several workflows and are willing to share their workflows.
While these results have been obtained on workflow owners, similar trends are observed on workflow contributors (in Nextflow, 37% of contributors have contributed to two workflows or more, and this figure reaches 44% for Snakemake).
4.3. Identifying reused workflows
The question of workflow reuse is now explored, considering complete (whole workflow) rather than partial (workflow parts) reuse.
To do so, two complementary metrics are used. First, we looked for exact matches among texts of the workflow codes to detect copy and paste between workflows. Second, we examined GitHub’s project forking to identify workflow reuse. Forking allows a Git user to explicitly create a new project starting from the code of a parent project, by copying its full content and history. For each workflow project, we have access to its number of forks, and so to the number of times it has been taken as a source of a new workflow.
As a result, we did not find any pair of workflows with identical source code. Then looking at the number of forks provided more interesting insights. Table 1 provides the number of Nextflow and Snakemake workflows forked more than n times (n = 3, 5, 10, 50). As a reminder, the total number of workflows is 1, 186 and 1, 257, respectively in Nextflow and Snakemake. This means, for instance, that there are 45 Nextflow and 57 Snakemake workflows which have been forked more than 10 times.
Table 1.
Percentage of Nextflow and Snakemake workflows that have been forked more than 3, 5, 10 and 50 times.
| Threshold | 3 | 5 | 10 | 50 |
|---|---|---|---|---|
| Nextflow | 13.7% | 7.8% | 3.8% | 0.8% |
| Snakemake | 14.5% | 7.9% | 4.6% | 0.2% |
A close look at workflow forking revealed two predominant practices: (i) a workflow is forked, the copy then undergoes modifications to re-purpose the workflow to a new context of use, (ii) a popular workflow is forked to create a stable, unchanged snapshot for its current users, allowing the original workflow to continue to evolve to meet new users’ needs. In both cases, the original and forked workflows are deemed not to be identical. In view of the above, it is assumed in this paper that, while workflow reuse was identified via the presence of forking, the workflows were modified sufficiently to not be considered identical by the other metric.
4.4. Identifying reused processors
Another objective of this study is to identify fine-grain reuse, notably by detecting copy-paste followed by slight modifications of processors by users. Detecting such similar processors can be performed using plagiarism tools [40] or following the methods also used in other studies [41], [42]. Plagiarism tools are more suited to longer code than processor code, we thus chose to follow the same direction as previous reuse studies [40] by using the Levenshtein’s distance (as implemented in jellyfish17) classically normalized by the size of the processor. As a result the normalized distance lies between 0 (no similarity) and 1 (identical codes).
In order to identify reuse (as copy-paste and slight changes), we thus deemed two processors similar if their Levenshtein’s similarity score was higher than 0.85. This threshold has been chosen based on an inspection of the similar processors obtained considering four values of threshold (0.80, 0.85, 0.90 and 0.95). As a result, 0.85 appeared to be the best value to account for the action of copy-pasting followed by slight changes. We then formed “groups” of processor occurrences using this threshold. Each group represents a unique processor gathering all the highly similar processor occurrences.
4.4.1. Comparative processor reuse study between nextflow and snakemake
Barplots in Fig. 7 represent the distribution of the number of workflows a processor is reused in (reuse across workflows) in Nextflow and Snakemake. As Nextflow and Snakemake have approximately the same amount of workflows, we do not normalize the quantity of workflows.
Fig. 7.

Distribution of processors reuse across workflows in Nextflow and Snakemake.
The barplot of Fig. 7 shows that the reuse profile is similar between Nextflow and Snakemake. Interestingly, the most reused processors are more frequently reused in Nextflow than in Snakemake. Looking at the ”top-x processors” (the x processors that are found in the most workflows), Nextflow processors are more reused across workflows: the top-25 processors are reused in 3.34% (resp. 1.85%) of Nextflow (resp. Snakemake) processors; the top-30 processors are reused in 2.42% of Nextflow processors and 0.93% of Snakemake processors.
4.4.2. Comparative nextflow processor reuse study: role of nf-core
nf-core [31] being a well-known curated repository of Nextflow workflows, the next experiment aims to investigate whether nf-core has an impact on reuse of processors in Nextflow workflows.
In this experiment.
-
•
an nf-core workflow is a workflow whose owner is nf-core.
-
•
a non-nf-core workflow has an owner different from nf-core.
-
•
an nf-core processor is a processor that is used in at least one nf-core workflow.
-
•
a non-nf-core processor is a processor which is never used in an nf-core workflow.
Considering only non-nf-core workflows, the question is: is there a difference between nf-core processors reuse and non-nf-core processors reuse?
Figure 8 represents the distribution of nf-core processor reuse (top) versus the distribution non-nf-core processors reused (bottom) in non-nf-core workflows. It can be observed that nf-core processors reuse is slightly higher than the reuse of non-nf-core processors.
Fig. 8.

Distribution of processors reuse across non nf-core workflows. Both nf-core and non nf-core processors are considered.
4.5. Identifying reused tools
We now focus on identifying the kind of operations users implemented in their workflows using the bioinformatics tools used as a proxy. In order to identify reuse of tools, we count the number of times each tool is used considering all workflow processors.
When studying the tools used in processors, we found that some of them were widely used, as much as 2841 times, indicating that many users performed very similar tasks.
When looking at the list of the most used tools (top tools) for Nextflow and Snakemake, we notice that 9 out of 10 top tools are common between Nextflow and Snakemake. In the following, we will focus on the 14 tools which are common between the top 20 tools of Nextflow and Snakemake. Such tools are presented in Table 2.
Table 2.
14 tools in common between the top 20 Nextflow tools and the top 20 Snakemake tools. # NF: Number of Nextflow processors in which they appear, # SM: Number of Snakemake processors in which they appear. Category: (i) genomic toolkits (GT), (ii) sequence mappers (MAP), (iii) quality control (QC) tools, and (iv) domain-specific tools (DST).
| Tool | # NF | # SM | Category |
|---|---|---|---|
| Samtools | 2841 | 2045 | GT |
| BEDTools[43] | 384 | 603 | GT |
| BCFtools[44] | 929 | 360 | GT |
| BWA | 412 | 356 | MAP |
| GATK | 1067 | 269 | GT |
| FastQC[45] | 770 | 236 | QC |
| Bowtie[46] | 243 | 177 | MAP |
| STAR[47] | 234 | 152 | MAP |
| MultiQC[48] | 707 | 137 | QC |
| Minimap2[49] | 149 | 137 | MAP |
| Picard | 269 | 137 | GT |
| seqtk | 96 | 94 | GT |
| Cutadapt[50] | 80 | 81 | GT |
| QIIME[51] | 207 | 90 | DST |
While some tools are used for generic bioinformatics data processing (e.g. Samtools), a number of them are domain-specific tools performing specific bioinformatic tasks and reflecting trends of tool usage in bioinformatics workflows (e.g. QIIME for microbiome analysis). Not surprisingly, toolkits, mappers and quality control tools are widely used, as they are involved in many workflows (even several times in some workflows) for different types of analysis: BWA and FastQC are used prior to most sequence data analyses, and Samtools is used for multiple tasks (e.g., converting between mapping formats, filtering reads) involved in different kinds of analyses (e.g., RNA-Seq, SNP calling).
Generally, we find that (i) a few tools are widely re-used, and (ii) widely used tools depend little on the workflow system used, as the top used tools are almost the same in Nextflow and Snakemake.
The next section discusses the paper and in particular the results obtained in this study of reuse.
5. Discussion and perspectives
An increasing number of papers highlight the benefits of using scientific workflow systems to develop complex pipelines instead of considering only (python, R or bash) scripts. In [52], authors emphasize how Nextflow, Snakemake and Galaxy are helpful solutions to design, execute and share large-scale multi-omics pipelines. In [1], authors focus on the ability of workflow systems to design reproducible experiments. Good practices papers have also emerged: [53] provides guidelines to develop scientific workflows in the context of high-throughput sequencing data analysis; [54] guides developers of research software in developing computational tools fully utilized in workflow management systems.
The originality of the present paper is to consider all the stages of the bioinformatics pipeline life cycle and for each stage both elicit the problems encountered and provide an overview of a series of solutions. More precisely, we have first presented generic elements of solutions, that can be used in the development of any kind of pipeline. We have then focused on scientific workflow systems and especially on the new generation of workflow systems - Snakemake and Nextflow - that provide significant technical advancements: native support of containers help capturing the execution environment, making pipelines more easily reproducible thus easier to share and re-execute. Such workflow management systems also help integrating all components of a pipeline, such as tools, scripts and bash commands in a seamless way. Furthermore, the modularity of workflow languages makes it easier to isolate single steps, not only making the pipelines easier for others to understand but also facilitating reuse of the workflow steps.
Another major contribution of this paper is a quantitative and qualitative study of reuse of in-use Snakemake and Nextflow workflows. Our study reveals that there is already effective reuse and provides three main conclusions. First, between a third and half of the workflow owners have implemented more than one workflow. Second, we found evidence of reuse in the source code of processors. Third, we have highlighted a set of tools that are regularly used among pipelines implemented as workflows. Previous reuse studies were performed ten years ago on the earliest systems. In particular, [39] considered a set of 898 workflows from the Taverna system. The conclusions of [39] differ from ours. Regarding authorship, we showed that the top 10 workflow owners published 15% of the workflows (compared to 62% previously), which indicates that workflow system usage is no longer limited to a restricted community of experts, but to a growing range of scientists. As a consequence, authors are now more willing to share and reuse workflows with each other. Our study finally reveals that the existence of dedicated curated repositories such as nf-core helps to promote reuse practices.
For an even broader adoption of workflow systems, the main perspectives are to provide ways to discover, retrieve and compare workflows. This point has been considered in the previous generation of workflow systems [41] and is still pointed out by editorials [55], very recent community papers [56] and review papers [3]. Earlier approaches based on the Taverna workflow systems (finding workflows [57], [42], indexing workflows [58], recommending workflows [59], [33], [60]) are unfortunately not well adapted to the new generation of systems and repositories where workflows are more heterogeneous in terms of specification and widely spread on the Web. Technical and algorithmic challenges remain to deal with the distributed and continuously growing and evolving nature of (git based) workflow repositories [55], [56].
Last but not least, a key aspect to achieve in the following years for a wider adoption of workflows by users and for an increase of the workflow reuse practices is directly related to the ability to identify high-quality, namely, FAIR workflows. The ’FAIR Guiding Principles for scientific data management and stewardship’ [61] provides guidelines to improve the Findability, Accessibility, Interoperability, and Reuse of digital assets. Applying FAIR principles to scientific workflows [62], [63] is particularly important as it includes defining metrics e.g. to assess the ability of a workflow to be reproduced or reused, thus providing key quality information to workflow (re)users.
CRediT authorship contribution statement
Marine Djaffardjy: Investigation, Software, Writing – original draft. George Marchment: Software. Clémence Sebe: Software. Raphael Blanchet: Resources, Writing – review & editing. Khalid Bellajhame: Writing – review & editing. Alban Gaignard: Supervision, Investigation, Writing – original draft, Writing – review & editing. Frédéric Lemoine: Writing – review & editing. Sarah Cohen-Boulakia: Supervision, Investigation, Writing – original draft, Writing – review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Code available at https://github.com/r-blanchet/BAM_QC
References
- 1.Cohen-Boulakia S., Belhajjame K., Collin O., Chopard J., Froidevaux C., Gaignard A., Hinsen K., Larmande P., LeBras Y., Lemoine F., Mareuil F., Ménager H., Pradal C., Blanchet C. Scientific workflows for computational reproducibility in the life sciences: status, challenges and opportunities. Fut Gen Comput Syst. 2017;75:284–298. [Google Scholar]
- 2.A. Rule, A. Birmingham, C. Zuniga, I. Altintas, S.-C. Huang, R. Knight, N. Moshiri, M.H. Nguyen, S.B. Rosenthal, F. Pérez, et al., Ten simple rules for writing and sharing computational analyses in jupyter notebooks (2019). [DOI] [PMC free article] [PubMed]
- 3.Wratten L., Wilm A., Göke J. Reproducible, scalable, and shareable analysis pipelines with bioinformatics workflow managers. Nat Methods. 2021;18(10):1161–1168. doi: 10.1038/s41592-021-01254-9. [DOI] [PubMed] [Google Scholar]
- 4.Van Vliet M. Seven quick tips for analysis scripts in neuroimaging. PLoS Comput Biol. 2020;16(3) doi: 10.1371/journal.pcbi.1007358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spjuth O., Bongcam-Rudloff E., Hernández G.C., Forer L., Giovacchini M., Guimera R.V., Kallio A., Korpelainen E., Kańduła M.M., Krachunov M., et al. Experiences with workflows for automating data-intensive bioinformatics. Biol Direct. 2015;10(1):1–12. doi: 10.1186/s13062-015-0071-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Köster J., Rahmann S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics. 2012;28(19):2520–2522. doi: 10.1093/bioinformatics/bts480. [DOI] [PubMed] [Google Scholar]
- 7.J.P. Kurs, M. Simi, F. Campagne, Nextflowworkbench: Reproducible and reusable workflows for beginners and experts, bioRxiv (2016). 10.1101/041236. [DOI]
- 8.Afgan E., Nekrutenko A., Grüning B.A., Blankenberg D., Goecks J., Schatz M.C., Ostrovsky A.E., Mahmoud A., Lonie A.J., Syme A., Fouilloux A., Bretaudeau A., Nekrutenko A., Kumar A., Eschenlauer A.C., DeSanto A.D., Guerler A., Serrano-Solano B., Batut B., Grüning B.A., Langhorst B.W., Carr B., Raubenolt B.A., Hyde C.J., Bromhead C.J., Barnett C.B., Royaux C., Gallardo C., Blankenberg D., Fornika D.J., Baker D., Bouvier D., Clements D., de Lima Morais D.A., Tabernero D.L., Lariviere D., Nasr E., Afgan E., Zambelli F., Heyl F., Psomopoulos F., Coppens F., Price G.R., Cuccuru G., Corguillé G.L., Kuster G.V., Akbulut G.G., Rasche H., Hotz H.-R., Eguinoa I., Makunin I., Ranawaka I.J., Taylor J.P., Joshi J., Hillman-Jackson J., Goecks J., Chilton J.M., Kamali K., Suderman K., Poterlowicz K., Yvan L.B., Lopez-Delisle L., Sargent L., Bassetti M.E., Tangaro M.A., van den Beek M., Čech M., Bernt M., Fahrner M., Tekman M., Föll M.C., Schatz M.C., Crusoe M.R., Roncoroni M., Kucher N., Coraor N., Stoler N., Rhodes N., Soranzo N., Pinter N., Goonasekera N.A., Moreno P.A., Videm P., Melanie P., Mandreoli P., Jagtap P.D., Gu Q., Weber R.J.M., Lazarus R., Vorderman R.H.P., Hiltemann S., Golitsynskiy S., Garg S., Bray S.A., Gladman S.L., Leo S., Mehta S.P., Griffin T.J., Jalili V., Yves V., Wen V., Nagampalli V.K., Bacon W.A., de Koning W., Maier W., Briggs P.J. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022;50(W1):W345–W351. doi: 10.1093/nar/gkac247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ison J., Ienasescu H., Chmura P., Rydza E., Menager H., Kalaš M., Schwammle V., Gruning B., Beard N., Lopez R., Duvaud S., Stockinger H., Persson B., Var^ekova R.S., Raček T., Vondrašek J., Peterson H., Salumets A., Jonassen I., Hooft R., Nyronen T., Valencia A., Capella S., Gelpi J., Zambelli F., Savakis B., Leskošek B., Rapacki K., Blanchet C., Jimenez R., Oliveira A., Vriend G., Collin O., Van Helden J., Løngreen P., Brunak S. The bio.tools registry of software tools and data resources for the life sciences. Genome Biol. 2019;20(1):1–4. doi: 10.1186/s13059-019-1772-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolstencroft K., Haines R., Fellows D., Williams A., Withers D., Owen S., Soiland-Reyes S., Dunlop I., Nenadic A., Fisher P., et al. The taverna workflow suite: designing and executing workflows of web services on the desktop, web or in the cloud. Nucleic Acids Res. 2013:gkt328. doi: 10.1093/nar/gkt328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bourcier R., Scouarnec S.L., Bonnaud S., Karakachoff M., Bourcereau E., Heurtebise-Chrétien S., Menguy C., Dina C., Simonet F., Moles A., Lenoble C., Lindenbaum P., Chatel S., Isidor B., Génin E., Deleuze J.-F., Schott J.-J., Marec H.L., Loirand G., Desal H., Redon R. Rare coding variants in angptl6 are associated with familial forms of intracranial aneurysm. Am J Hum Genet. 2018;102(1):133–141. doi: 10.1016/j.ajhg.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rousseau O., Karakachoff M., Gaignard A., Bellanger L., Bijlenga P., ConstantDitBeaufils P., L’Allinec V., Levrier O., Aguettaz P., Desilles J.-P., Michelozzi C., Marnat G., Vion A.-C., Loirand G., Desal H., Redon R., Gourraud P.-A., Bourcier R. Location of intracranial aneurysms is the main factor associated with rupture in the ican population. J Neurol, Neurosurg Psychiatry. 2021;92(2):122–128. doi: 10.1136/jnnp-2020-324371. [DOI] [PubMed] [Google Scholar]
- 13.Li H., Durbin R. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., et al. The genome analysis toolkit: a mapreduce framework for analyzing next-generation dna sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broad Institute, Picard tools, 〈http://broadinstitute.github.io/picard/〉 (Accessed: 2022/10/24; version 2.27.4).
- 16.Alnasir J.J. Fifteen quick tips for success with hpc, ie, responsibly bashing that linux cluster. PLOS Comput Biol. 2021;17(8) doi: 10.1371/journal.pcbi.1009207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ison J., Rapacki K., Ménager H., Kalaš M., Rydza E., Chmura P., Anthon C., Beard N., Berka K., Bolser D., et al. Tools and data services registry: a community effort to document bioinformatics resources. Nucleic Acids Res. 2016;44(D1):D38–D47. doi: 10.1093/nar/gkv1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhagat J., Tanoh F., Nzuobontane E., Laurent T., Orlowski J., Roos M., Wolstencroft K., Aleksejevs S., Stevens R., Pettifer S., et al. Biocatalogue: a universal catalogue of web services for the life sciences. Nucleic Acids Res. 2010:gkq394. doi: 10.1093/nar/gkq394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.F. Costa, V. Silva, D. de Oliveira, K.A. C.S. Ocaña, E.S. Ogasawara, J. Dias, M. Mattoso, Capturing and querying workflow runtime provenance with PROV: a practical approach, in: Joint 2013 EDBT/ICDT Conferences, EDBT/ICDT ’13, Genoa, Italy, March 22, 2013, Workshop Proceedings, 2013, pp. 282–289.
- 20.Guha R.V., Brickley D., Macbeth S. Schema. org: evolution of structured data on the web. Commun ACM. 2016;59(2):44–51. [Google Scholar]
- 21.M.A. Jette, A.B. Yoo, M. Grondona, Slurm: Simple linux utility for resource management, in: In Lecture Notes in Computer Science: Proceedings of Job Scheduling Strategies for Parallel Processing (JSSPP) 2003, Springer-Verlag, 2002, 44–60.
- 22.H. Feng, V. Misra, D. Rubenstein, Pbs: a unified priority-based scheduler, in: Proceedings of the 2007 ACM SIGMETRICS international conference on Measurement and modeling of computer systems, 2007, pp. 203–214.
- 23.Boettiger C. An introduction to docker for reproducible research. ACM SIGOPS Oper Syst Rev. 2015;49(1):71–79. [Google Scholar]
- 24.Kurtzer G.M., Sochat V., Bauer M.W. Singularity: scientific containers for mobility of compute. PloS One. 2017;12(5) doi: 10.1371/journal.pone.0177459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.daVeigaLeprevost F., Grüning B.A., AlvesAflitos S., Röst H.L., Uszkoreit J., Barsnes H., Vaudel M., Moreno P., Gatto L., Weber J., et al. Biocontainers: an open-source and community-driven framework for software standardization. Bioinformatics. 2017;33(16):2580–2582. doi: 10.1093/bioinformatics/btx192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grüning B., Dale R., Sjödin A., Chapman B.A., Rowe J., Tomkins-Tinch C.H., Valieris R., Köster J. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat Methods. 2018;15(7):475–476. doi: 10.1038/s41592-018-0046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pradal C., Artzet S., Chopard J., Dupuis D., Fournier C., Mielewczik M., Nègre V., Neveu P., Parigot D., Valduriez P., Boulakia S.C. Infraphenogrid: a scientific workflow infrastructure for plant phenomics on the grid. Future Gener Comp Syst. 2017;67:341–353. [Google Scholar]
- 28.Ludäscher B., Altintas I., Berkley C., Higgins D., Jaeger E., Jones M., Lee E.A., Tao J., Zhao Y. Scientific workflow management and the kepler system. Concurr Comput: Pract Exp. 2006;18(10):1039–1065. [Google Scholar]
- 29.Callahan S.P., Freire J., Santos E., Scheidegger C.E., Silva C.T., Vo H.T. In: Proceedings of the ACM SIGMOD International Conference on Management of Data, Chicago, Illinois, USA, June 27–29, 2006. Chaudhuri S., Hristidis V., Polyzotis N., editors. ACM; 2006. Vistrails: visualization meets data management; pp. 745–747. (pp.) [Google Scholar]
- 30.DiTommaso P., Chatzou M., Floden E.W., Barja P.P., Palumbo E., Notredame C. Nextflow enables reproducible computational workflows. Nat Biotechnol. 2017;35(4):316. doi: 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- 31.Ewels P.A., Peltzer A., Fillinger S., Patel H., Alneberg J., Wilm A., Garcia M.U., DiTommaso P., Nahnsen S. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol 2020 38:3. 2020;38(3):276–278. doi: 10.1038/s41587-020-0439-x. 〈https://www.nature.com/articles/s41587-020-0439-x〉 [DOI] [PubMed] [Google Scholar]
- 32.Juve G., Chervenak A.L., Deelman E., Bharathi S., Mehta G., Vahi K. Characterizing and profiling scientific workflows. Future Gener Comp Syst. 2013;29(3):682–692. [Google Scholar]
- 33.Goble C.A., Bhagat J., Aleksejevs S., Cruickshank D., Michaelides D., Newman D., Borkum M., Bechhofer S., Roos M., Li P., et al. myexperiment: a repository and social network for the sharing of bioinformatics workflows. Nucleic Acids Res. 2010;38(suppl 2):W677–W682. doi: 10.1093/nar/gkq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mates P., Santos E., Freire J., Silva C.T. Scientific and Statistical Database Management. Springer; 2011. Crowdlabs: Social analysis and visualization for the sciences; pp. 555–564. (pp.) [Google Scholar]
- 35.Korkhov V., Krefting D., Montagnat J., Huu T.T., Kukla T., Terstyanszky G., Manset D., Caan M., Olabarriaga S. Shiwa workflow interoperability solutions for neuroimaging data analysis. Stud Health Technol Inf. 2012;175 [PubMed] [Google Scholar]
- 36.Blankenberg D., VonKuster G., Bouvier E., Baker D., Afgan E., Stoler N., Taylor J., Nekrutenko A., et al. Dissemination of scientific software with galaxy toolshed. Genome Biol. 2014;15(2):403. doi: 10.1186/gb4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goble C., Soiland-Reyes S., Bacall F., Owen S., Williams A., Eguinoa I., Droesbeke B., Leo S., Pireddu L., Rodríguez-Navas L., et al. Implementing fair digital objects in the eosc-life workflow collaboratory. Zenodo. 2021 [Google Scholar]
- 38.Cokelaer T., Desvillechabrol D., Legendre R., Cardon M. ‘sequana’: a set of snakemake ngs pipelines. J Open Source Softw. 2017;2(16):352. [Google Scholar]
- 39.J. Starlinger, S.C. Boulakia, U. Leser, (re)use in public scientific workflow repositories, in: Scientific and Statistical Database Management - 24th International Conference, SSDBM 2012, Chania, Crete, Greece, June 25–27, 2012, 2012, Proceedings, pp. 361–378.
- 40.Novak M., Joy M., Kermek D. Source-code similarity detection and detection tools used in academia: a systematic review. ACM Trans Comput Educ. 2019;19(3) doi: 10.1145/3313290. (may) (may) [DOI] [Google Scholar]
- 41.Cohen-Boulakia S., Leser U. Search, adapt, and reuse: the future of scientific workflows. ACM SIGMOD Rec. 2011;40(2):6–16. [Google Scholar]
- 42.Starlinger J., Brancotte B., Cohen-Boulakia S., Leser U. Similarity search for scientific workflows. Proc VLDB Endow. 2014;7(12):1143–1154. [Google Scholar]
- 43.Quinlan A.R., Hall I.M. Bedtools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. The sequence alignment/map format and samtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fastqc(Jun 2015). 〈https://qubeshub.org/resources/fastqc〉.
- 46.Langmead B., Salzberg S.L. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. Star: ultrafast universal rna-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ewels P., Magnusson M., Lundin S., Käller M. Multiqc: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32(19):3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–12. [Google Scholar]
- 51.Caporaso J.G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F.D., Costello E.K., Fierer N., Peña A.G., Goodrich J.K., Gordon J.I., et al. Qiime allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krassowski M., Das V., Sahu S.K., Misra B.B. State of the field in multi-omics research: from computational needs to data mining and sharing. Front Genet. 2020;11 doi: 10.3389/fgene.2020.610798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reiter T., Brooks† P.T., Irber† L., Joslin† S.E., Reid† C.M., Scott† C., Brown C.T., Pierce-Ward N.T. Streamlining data-intensive biology with workflow systems. GigaScience. 2021;10(1):1–19. doi: 10.1093/gigascience/giaa140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brack P., Crowther P., Soiland-Reyes S., Owen S., Lowe D., Williams A.R., Groom Q., Dillen M., Coppens F., Gruning B., Eguinoa I., Ewels P., Goble C. Ten simple rules for making a software tool workflow-ready. PLoS Comput Biol. 2022;18(3):1–11. doi: 10.1371/journal.pcbi.1009823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Atkinson M., Gesing S., Montagnat J., Taylor I. Scientific workflows: past, present and future. Future Gener Comput Syst. 2017;75:216–227. doi: 10.1016/j.future.2017.05.041. [DOI] [Google Scholar]
- 56.R.F. DaSilva, H. Casanova, K. Chard, I. Altintas, R.M. Badia, B. Balis, T. Coleman, F. Coppens, F. DiNatale, B. Enders, et al., A community roadmap for scientific workflows research and development, in: 2021 IEEE Workshop on Workflows in Support of Large-Scale Science (WORKS), IEEE, 2021, pp. 81–90.
- 57.A. Goderis, D.D. Roure, C. Goble, J. Bhagat, D. Cruickshank, P. Fisher, D. Michaelides, F. Tanoh, Discovering scientific workflows: The myexperiment benchmarks, Project report (April 2008). 〈https://eprints.soton.ac.uk/265662/〉.
- 58.J. Stoyanovich, B. Taskar, S. Davidson, Exploring repositories of scientific workflows, Proceedings of the ACM SIGMOD International Conference on Management of Data (2010). 10.1145/1833398.1833405. [DOI]
- 59.D. De Roure, C. Goble, Lessons from myExperiment: Two insights into emerging e-Research practice, UK eScience All Hands Meeting 2009 (2009)6–8. 〈http://eprints.ecs.soton.ac.uk/17662/〉.
- 60.A. Halioui, T. Martin, P. Valtchev, A.B. Diallo, Ontology-based workflow pattern mining: Application to bioinformatics expertise acquisition, Proceedings of the ACM Symposium on Applied Computing Part F128005 (2017)824–827. 10.1145/3019612.3019866. [DOI]
- 61.Wilkinson M.D., Dumontier M., Aalbersberg I.J., Appleton G., Axton M., Baak A., Blomberg N., Boiten J.-W., da SilvaSantos L.B., Bourne P.E., et al. The fair guiding principles for scientific data management and stewardship. Sci data. 2016;3 doi: 10.1038/sdata.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goble C., Cohen-Boulakia S., Soiland-Reyes S., Garijo D., Gil Y., Crusoe M.R., Peters K., Schober D. Fair computational workflows. Data Intell. 2020;2(1–2):108–121. doi: 10.1162/dint_a_00033. [DOI] [Google Scholar]
- 63.Celebi R., Moreira J.R., Hassan A.A., Ayyar S., Ridder L., Kuhn T., Dumontier M. Towards FAIR protocols and workflows: the OpenPREDICT use case. PeerJ Comput Sci. 2020;6:1–29. doi: 10.7717/PEERJ-CS.281. [DOI] [PMC free article] [PubMed] [Google Scholar]