

Abstract
Endothelial cells (ECs) grant access of disseminated cancer cells to distant organs. However, the molecular players regulating the activation of quiescent ECs at the premetastatic niche (PMN) remain elusive. Here, we find that ECs at the PMN coexpress tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) and its cognate death receptor 5 (DR5). Unexpectedly, endothelial TRAIL interacts intracellularly with DR5 to prevent its signaling and preserve a quiescent vascular phenotype. In absence of endothelial TRAIL, DR5 activation induces EC death and nuclear factor κB/p38–dependent EC stickiness, compromising vascular integrity and promoting myeloid cell infiltration, breast cancer cell adhesion, and metastasis. Consistently, both down-regulation of endothelial TRAIL at the PMN by proangiogenic tumor-secreted factors and the presence of the endogenous TRAIL inhibitors decoy receptor 1 (DcR1) and DcR2 favor metastasis. This study discloses an intracrine mechanism whereby TRAIL blocks DR5 signaling in quiescent endothelia, acting as gatekeeper of the vascular barrier that is corrupted by the tumor during cancer cell dissemination.
Endothelial TRAIL at the pre-metastatic niche ensures a quiescent vasculature, and its depletion fosters metastasis seeding.
INTRODUCTION
Despite numerous successful advances in the treatment of localized solid tumors, metastasis remains the Achilles tendon of cancer therapy and the leading cause of death of patients with cancer (1). It is undoubtful that the vascular lining is not a passive by-stander but is actively involved in many aspects of tumor progression and metastasis (2, 3). Besides contributing to the trafficking of oxygen, metabolites, waste, and immune cells within the tumor, endothelial cells (ECs) are the main route of metastasis dissemination and constitute an interactive barrier that needs to be crossed first by intravasating cancer cells at the primary tumor and afterward by circulating cancer cells reaching the metastatic site (3, 4). The access of cancer cells to the vasculature has gained the spotlight in the hallmarks of cancer (5), although the focus so far has been mostly on EC interactions at the primary tumor site, whereas the interplay between cancer cells and ECs at the premetastatic niche (PMN) remains elusive. Primary tumors secrete signals that precondition distant organs to the arrival of metastatic cancer cells, enforcing the formation of the PMN (6). A better understanding of how quiescent ECs in the PMN are hijacked by the tumor and how, in turn, phenotypic changes of the vascular lining affect on the success of metastatic dissemination could help to assess the risk of metastasis and provide novel strategies to prevent or treat their occurrence.
Tumor necrosis factor (TNF)–related apoptosis-inducing ligand (TRAIL), the protein encoded by the gene TNF superfamily member 10 (TNFSF10), was found as an arm of the natural killer (NK) cell armamentarium, able to selectively kill disseminated cancer cells upon binding to its cognate death receptors (TRAIL-R1/DR4 and TRAIL-R2/DR5 in humans and DR5 in mice), thereby limiting tumor initiation and metastasis (7–12). TRAIL receptor agonists have been tested in clinical trials, but despite being well tolerated, their efficacy did not meet the expectations when used as a monotherapy (13–16). Several resistance mechanisms have been proposed, including apoptosis evasion downstream of TRAIL receptor and the engagement of alternative signaling pathways that may even result in tumor-promoting effects (14, 15, 17–22). Moreover, three TRAIL decoy receptors (DcRs) have been described (TRAIL-R3/DcR1, TRAIL-R4/DcR2, and osteoprotegerin) that have affinity for TRAIL but are unable to transduce the proapoptotic signal and, thus, are believed to mediate resistance to TRAIL-induced apoptosis (14, 23–28). However, genetic models to study their physiological role and importance in vivo are still missing.
Recent reports have questioned the unique selectivity of the effect of TRAIL on cancer cells (14, 27, 29–33), and the endogenous TRAIL system has been associated with several pathophysiological conditions besides cancer (22, 34–36). Thus, the role of the TRAIL system in cancer has been, so far, oversimplified under a cancer cell–centric paradigm, probably overlooking diverse and undiscovered roles of TRAIL in cells from the host. For instance, exogenous TRAIL was reported to induce apoptosis in DR5-expressing tumor ECs and vascular disruption and to reduce tumor growth (30). However, the role of endogenous TRAIL in EC biology and its impact in metastasis remains unknown.
Here, we found that both mouse and human ECs in the PMN express high levels of TRAIL, which impedes early metastasis colonization via a mechanism independent of DR5-mediated cancer cell apoptosis. We suggest that this physiologically relevant barrier is overruled by tumor-derived signals, including vascular endothelial growth factor–A (VEGF-A) and placental growth factor (PlGF), which down-regulate endothelial TRAIL at the PMN, while the presence of DcR1 and DcR2 could hinder TRAIL as well. Together, tampering with endothelial TRAIL compromises the integrity of the vascular barrier, promotes inflammatory cell recruitment, and favors metastatic cell lodging.
RESULTS
Endogenous TRAIL restrains cancer cell colonization and metastasis independently of DR5 expression in cancer cells
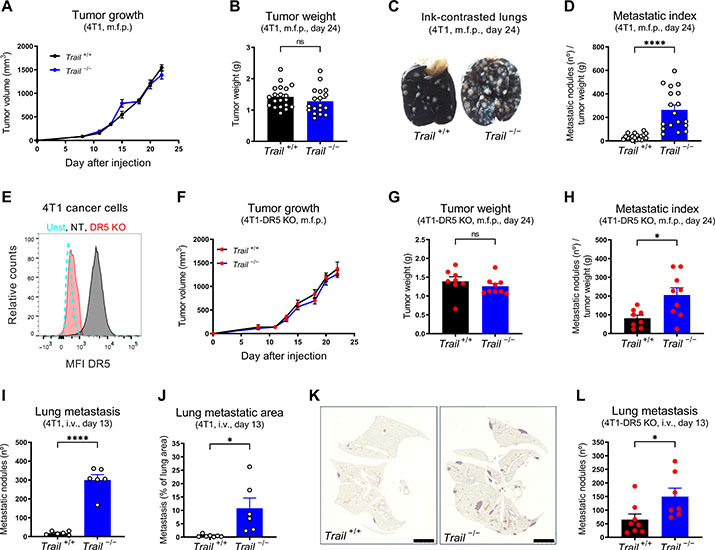
To dissect the effect of TRAIL in nonmalignant cells during cancer progression and metastasis, constitutive and ubiquitous TRAIL wild-type (WT) and knockout (KO) mice (Trail+/+ and Trail−/−, respectively, in a Balb/C background) were orthotopically injected with 4T1 or EMT6.5 cells, two well-established orthotopic and highly metastatic triple-negative breast cancer models (37, 38). In both models, TRAIL deletion did not significantly affect tumor growth (Fig. 1, A and B, and fig. S1, A and B), but it severely increased the number of spontaneous lung metastases (Fig. 1, C and D, and fig. S1, C and D). Moreover, TRAIL deletion did not affect tumor growth (fig. S1E), but it increased metastasis even in the 4T07 nonmetastatic model (fig. S1, F and G). To exclude the direct effect of TRAIL on cancer cells, we generated 4T1-DR5 KO cancer cells (Fig. 1E) and injected them in WT and TRAIL KO mice. In addition, in absence of DR5 in cancer cells, TRAIL deletion did not affect tumor growth (Fig. 1, F and G) but still increased spontaneous metastasis (Fig. 1H), suggesting the existence of a previously unidentified antimetastatic effect of TRAIL that is independent of DR5 in cancer cells.
Fig. 1. Endogenous TRAIL restrains cancer cell colonization and metastasis independently of DR5 expression in cancer cells.

(A to D) Tumor growth (A), tumor weight (B), representative images of ink-contrasted lungs (C), and metastatic index (D) 24 days after orthotopic mammary fat pad (m.f.p.) injection of 4T1 cells in constitutive TRAIL WT (Trail+/+) and KO (Trail −/−) mice. (E) Fluorescence-activated cell sorting (FACS) analysis of cell surface DR5 protein in sorted 4T1-DR5 KO and 4T1–NT [nontargeting guide RNA (gRNA) control] cells. unst, unstained. (F to H) Tumor growth (F), tumor weight (G), and metastatic index (H) 24 days after orthotopic m.f.p. injection of 4T1-DR5 KO cells in Trail+/+ and Trail −/− mice. (I to K) Number of ink-contrasted lung metastasis (I) or metastatic area in hematoxylin and eosin (H&E) staining (J and K) 13 days after intravenous (i.v.) injection of 4T1 cells in Trail+/+ and Trail −/− mice. Scale bars, 2,5 mm. (L and M) Number of ink-contrasted lung metastasis 13 days after i.v. injection of 4T1-DR5 KO cells in Trail+/+ and Trail −/− mice (L) or 12 days after i.v. injection of E0771 cells in induced ubiquitous TRAIL KO (R26ΔT10) versus control (R26W/W) mice (M). (N) FACS analysis of cell surface DR5 protein in E0771-DR5 KO and E0771-NT cells. (O) Number of ink-contrasted lung metastasis 12 days after i.v. injection of E0771-DR5 KO cells in induced R26W/W and R26ΔT10 mice. (P to R) Number of 4T1 cancer cell colony-forming units (CFU) (P), mRNA expression of dLNGFR (Q), or bioluminescent signal (R) in perfused lung tissue after i.v. injection of 4T1 (P), 4T1-dLNGFR (Q), or 4T1-Akaluc cells (R) in Trail+/+ and Trail−/− mice. (S and T) mRNA expression of dLNGFR (S) or bioluminescent signal (T) in perfused lung tissue after i.v. injection of E0771-dLNGFR (S) or E0771-Akaluc cells (T) in induced R26W/W and R26ΔT10 mice. All graphs show means ± SEM. ns, not significant. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Several features of the tumor microenvironment (TME) have been described to foster malignancy and metastatic spread (2, 3, 39). We analyzed multiple traits of the primary tumor, such as hypoxia (fig. S1H), immune cell composition (fig. S1, I to P), and vascularization (fig. S1, Q and R). However, we did not find any significant differences between tumors derived from TRAIL WT and KO mice that could explain our phenotype. To assess the contribution of TRAIL during metastatic dissemination once cancer cells are in the blood stream, we used an experimental metastasis model in which cancer cells are injected in the tail vein. Similar to the orthotopic model, TRAIL deletion resulted in a significant increase in experimental lung metastases both in animals injected with 4T1-WT cancer cells (Fig. 1, I to K) or with 4T1-DR5 KO cancer cells (Fig. 1L). To induce TRAIL deletion in specific cell types, we generated Trail floxed mice that we then intercrossed with different Cre-deleter strains (all in a C57BL/6 background). Ruling out possible developmental defects, inducible ubiquitous deletion of TRAIL before tail vein injection of E0771-WT or E0771-DR5 KO breast cancer cells fueled experimental metastasis (Fig. 1, M to O).
To dissect whether TRAIL impaired early organ colonization or the outgrowth of established lesions, we used either an ex vivo cancer cell colony formation assay (37), cancer cells traceable by the expression of a truncated low-affinity nerve growth factor receptor (dLNGFR), or a single-cell bioluminescence imaging system (40). In all the assays, constitutive or inducible deletion of TRAIL significantly increased lung cancer cell burden at 24 hours after intravenous injection, which was steadily amplified at later time points (Fig. 1, P to T). In sum, these data reveal that TRAIL impairs early metastatic organ colonization via a previously unknown mechanism independent of DR5-mediated cancer cell apoptosis.
EC-derived TRAIL at the PMN restrains early metastatic colonization
To identify which cells express TRAIL, we sorted different cell populations from the lungs of tumor-free mice (fig. S2). Although TRAIL was expressed by several immune cell populations (i.e., NK cells, CD8+ T cells, and neutrophils), ECs expressed 25- to 40-fold higher levels of TRAIL than any of the immune cell types (Fig. 2A). Analysis of publicly available human and murine single-cell RNA sequencing (scRNA-seq) atlases corroborated that ECs are the major source of TRAIL in the lung (Fig. 2B and fig. S3A) (41, 42) and that the lung is the organ with a higher percentage of TRAIL-expressing ECs, followed by the colon, liver, and kidney (fig. S3B) (43). In murine lung ECs, TRAIL protein was detected mostly intracellularly (Fig. 2C) and minimally at the cell surface (Fig. 2D).
Fig. 2. ECs in the premetastatic lung (and liver) are the main source of TRAIL.

(A) mRNA expression of Tnfsf10 in cells sorted from perfused lungs of tumor-free WT mice. (B) T-distributed stochastic neighbor embedding (t-SNE) plots showing the expression of TNFSF10 for different cell subsets (epithelial, endothelial, stromal, and immune) from uninvolved pulmonary tissue of three different patients with lung cancer (P1, P2, and P3). (C and D) FACS analysis of intracellular (in) (C) or extracellular (ex) (D) TRAIL protein on lung ECs from Trail+/+ and Trail−/− mice. ΔMFI = MFIstained − MFIisotype (C) or ΔMFI = MFIstained − MFIFMO (D). All graphs show means ± SEM. ***P < 0.001; ****P < 0.0001.
To assess the effect of EC-derived TRAIL in metastasis, we generated a novel murine strain to induce the deletion of TRAIL specifically in ECs (ECΔT10) (fig. S4, A and B), which resulted in an almost complete depletion of TRAIL in pulmonary tissue (Fig. 3A), underscoring that ECs are the main source of TRAIL in the lung under homeostatic conditions. Phenotypically, EC-specific deletion of TRAIL did not affect primary tumor volume (Fig. 3B), weight (Fig. 3C), hypoxia (fig. S4C), vascularization (fig. S4D), or immune cell composition (fig. S4, E to J). However, it strongly increased spontaneous (Fig. 3, D and E) and experimental lung metastases (Fig. 3, F to H), with an already higher metastatic burden 24 hours after the injection of either E0771-WT cancer cells (Fig. 3I) or E0771-DR5 KO cancer cells (Fig. 3J). Conversely, promoting TRAIL expression with EC-targeting lipid nanoparticles (LNPs) (fig. S4, K and L) (44) significantly rescued the increase in experimental lung metastasis induced by the EC-specific deletion of TRAIL and decreased the metastatic burden in WT mice (Fig. 3K). Liver metastasis also increased upon EC-specific or ubiquitous deletion of TRAIL (Fig. 3, L to N, and fig. S4, M and N), showing that EC-derived TRAIL controls metastasis in several vascular beds. In line with an antimetastatic role of TRAIL expressed by NK cells (7–9, 12), NK cell–specific deletion of TRAIL (fig. S4O) led to more experimental lung metastasis (Fig. 3, O and P). However, this effect was less pronounced (2.2-fold increase at end stage) when compared to the EC-specific deletion of TRAIL (12.4-fold increase; Fig. 3, G and H), and no significant differences were observed at 24 hours (Fig. 3Q).
Fig. 3. EC-derived TRAIL at the PMN restrains early metastatic colonization.

(A) Protein levels of TRAIL in perfused lung tissue lysates of induced EC-specific TRAIL WT (ECW/W) and KO (ECΔT10) mice. (B to E) Tumor growth (B), tumor weight (C), number of ink-contrasted lung metastasis (D), and metastatic index (E) 27 days after orthotopic m.f.p. injection of E0771 cells in induced ECW/W and ECΔT10 mice. (F to H) Number of ink-contrasted lung metastasis (F) or metastatic area in H&E staining (G and H) 12 days after i.v. injection of E0771 cells in induced ECW/W and ECΔT10 mice. Scale bars, 500 μm. (I to K) mRNA expression of dLNGFR in perfused lung tissue 24 hours after i.v. injection of E0771-dLNGFR (I and K) or E0771-dLNGFR-DR5 KO cells (J) in induced ECW/W and ECΔT10 mice. In (K), anti-CD31 (αCD31)–LNP/empty or anti-CD31–LNP/Trail (8 μg per mouse) was injected i.v. 48 and 24 hours before cancer cell injection, and statistical significance was calculated by one-tailed unpaired t test. (L) FACS analysis of the cancer cell burden 14 days after intrasplenic (i.s.) injection of E0771-dLNGFR cells in ECW/W and ECΔT10 mice. (M) Representative images of H&E staining of livers 14 days after i.s. injection of E0771-dLNGFR cells in ECW/W and ECΔT10 mice. Incidence indicates the fraction of mice displaying metastatic lesions upon pathological analysis of liver cross sections. Scale bars, 500 μm. (N) Liver metastatic area in H&E staining 14 days after i.s. injection of 4T1-CD90.1 cells in Trail+/+ and Trail−/− mice. (O and P) Lung metastatic area in H&E staining 12 days after i.v. injection of E0771 cells in NK-specific TRAIL WT (NKW/W) and KO (NKΔT10) mice. Scale bars, 500 μm. (Q) mRNA expression of dLNGFR in perfused lung tissue after i.v. injection of E0771-dLNGFR cells in NKW/W and NKΔT10 mice. All graphs show means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Together, we demonstrate that TRAIL is highly expressed in ECs, where it restrains metastatic colonization. While we confirm that NK cell–derived TRAIL also harnesses metastatic growth to some extent, endothelial TRAIL has a major role in preventing cancer cell dissemination in the early steps of the metastatic process.
TRAIL expression is a trait of EC quiescence and is down-regulated by tumor-derived factors during PMN formation
Given the role of endothelial TRAIL at the metastatic site but the absence of a vascular phenotype in the primary tumor, we further characterized TRAIL expression in these vascular beds. In mice orthotopically injected with E0771 cells, TRAIL expression was about 8.5-fold higher in lung ECs than in primary tumor–associated ECs (Fig. 4A). In postmortem samples from two patients with breast cancer with lung metastases, although other cell types were also positive for TRAIL, expression analysis focused on the vasculature revealed that TRAIL protein colocalized with the parenchymal lung vessels adjacent to the metastatic lesion, whereas it was significantly lower in metastasis-associated ECs (Fig. 4, B and C). In a patient where ECs from uninvolved pulmonary tissue clustered according to their distance from the lung tumor (41), TRAIL expression was markedly higher in distal than in proximal ECs (Fig. 4, D to F). This indicates that TRAIL expression is higher in more quiescent vascular beds distal from the tumor or metastatic lesion and is lower in tumor-associated, neoangiogenic vascular beds.
Fig. 4. TRAIL expression is a trait of EC quiescence.

(A) mRNA expression of Tnfsf10 in ECs sorted from perfused lungs or primary tumors of E0771 tumor–bearing WT mice (day 14). (B and C) Histological analysis of vasculature (CD31; red), TRAIL (green), and autofluorescence (blue) in a metastatic lesion or its adjacent parenchymal tissue in postmortem lung samples from two patients with breast cancer. Dots in (C) represent the average intensity in 10 different fields (5 fields per patient). Arrowheads in (B) show colocalization of CD31 and TRAIL (yellow). Scale bars, 50 μm. (D to F) t-SNE plots showing the origin (distal versus proximal) or expression of indicated genes (D), violin plot showing expression levels (E), or the percentage of expressing (+) and nonexpressing (−) cells (F) in ECs from patient 3. (G) Immunofluorescence costaining of TRAIL (green), Ki-67 (magenta), and Hoechst (blue). Higher-resolution inset of confluent (A) or proliferative (Ki-67+) areas (B). Scale bar, 50 μm. (H to J) Protein levels of TRAIL in whole-cell extract (WCE), intracellular, or surface fractions of quiescent HUVECs (H); in supernatant protein precipitates or WCE of quiescent HUVECs treated with the pan-caspase inhibitor q-VD-OPh (qVD) (50 μM) for 18 hours (I); or in proliferating, dense, or quiescent HUVECs (5-ethynyl-2’-deoxyuridine, EdU) (J). In (H), VE-cadherin is the representative surface protein. (K) mRNA expression of TNFSF10 in proliferating, dense, or quiescent HUVECs. (L) Volcano plot showing TNFSF10 transcript distribution. Positive and negative log2 (fold change) values (x axis) represent up-regulation or down-regulation, respectively, in quiescent compared to proliferative HUVECs. Dots in blue represent significant differentially expressed genes. (M) mRNA expression of TNFSF10 in quiescent HUVECs treated with VEGF-A (50 ng/ml), bFGF (10 ng/ml), or PlGF-1 (50 ng/ml) for 17 hours. All graphs show means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
In human umbilical vein ECs (HUVECs) TRAIL protein was mostly detected intracellularly and minimally on the cell surface (Fig. 4, G and H) and was present in HUVECs’ supernatant (Fig. 4I), suggesting that it may also be shed. TRAIL expression was higher in dense and quiescent than in proliferating (Ki-67+) HUVECs (Fig. 4, G, J, and K, and fig. S5A). Supporting this, TRAIL appeared as the most up-regulated gene in quiescent compared to proliferative HUVECs in a publicly available RNA-seq dataset (Fig. 4L) (45). Treatment of quiescent HUVECs with proangiogenic factors such as VEGF-A, basic fibroblast growth factor (bFGF), or PlGF reduced TRAIL expression (Fig. 4M). In contrast, proinflammatory/antitumoral cytokines such as interferon-γ (IFN-γ) up-regulated TRAIL, as previously reported (7, 12), while other cytokines known to be present in the TME had no significant effect (fig. S5B). The multitargeted receptor tyrosine kinase inhibitor sunitinib [with the highest affinity for both VEGF receptor 1 (VEGFR1), binding to PlGF and VEGF-A, and VEGFR2, binding to VEGF-A] rescued VEGF-A–induced TRAIL down-regulation. Moreover, either in the absence or in the presence of VEGF-A, MAPK (mitogen-activated protein kinase) kinase (MEK) inhibition up-regulated, while phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR)/Akt inhibition down-regulated TRAIL (fig. S5, C and D). This suggests that TRAIL is normally silenced by pathways inducing EC proliferation [i.e., MEK/extracellular signal–regulated kinase (ERK)] (46), while its expression is sustained by pathways important for EC homeostasis and survival (PI3K/mTOR/Akt) (47). Treatment of quiescent HUVECs with cancer cell–conditioned medium reduced TRAIL expression (Fig. 5A), which was rescued by sunitinib (Fig. 5B). In vivo, TRAIL expression was repressed by the administration of a conditioned medium from VEGF-A– or PlGF-overexpressing cancer cells (Fig. 5, C and D, and fig. S6, A to D), which correlated with an increased early metastatic colonization (Fig. 5, E and F).
Fig. 5. TRAIL expression is down-regulated by tumor-derived factors during PMN formation.

(A and B) mRNA expression of TNFSF10 in HUVECs treated for 24 hours with a conditioned medium (CM) of E0771 or 4T1 cancer cells for 48 hours and/or with sunitinib (1 μM). (C and D) FACS analysis of intracellular TRAIL protein in lung ECs from WT mice injected 12 times i.v. with a CM of E0771 cells overexpressing (OE) murine VEGF-A164 (C) or PlGF (D) for 48 hours. ΔMFI = MFIstained − MFIisotype. (E and F) mRNA expression of dLNGFR in perfused lung tissue 24 hours after i.v.injection of E0771-dLNGFR in WT mice injected 12 times i.v. with a CM of E0771 cells overexpressing VEGF-A164 (E) or PlGF (F) for 48 hours. Vehicle represents a mock medium, pseudo-conditioned in absence of cancer cells. (G) mRNA expression of Ly6g, S100a8, S100a9, Cd68, Emr1, and dLNGFR in perfused lung tissue from WT mice after orthotopic m.f.p. injection of E0771-dLNGFR cells. (H to K) Histological analysis of Ly-6G+ neutrophil area (H and I) and F4/80+ macrophage area (J and K) in tumor-free (TF) WT mice or tumor-bearing (TB) WT mice 10 days after orthotopic m.f.p. injection of E0771-dLNGFR cells. Scale bars, 50 μm. (L) mRNA expression of Tnfsf10 and dLNGFR in perfused lung tissue from WT mice after orthotopic m.f.p. injection of E0771-dLNGFR cells. FC, fold change. (M) FACS analysis of intracellular TRAIL protein in lung ECs from tumor-free WT mice or 10 days after orthotopic m.f.p. injection of E0771-dLNGFR cells. ΔMFI = MFIstained − MFIisotype. All graphs show means ± SEM. *P < 0.05; **P < 0.01; ##P < 0.01; ****P < 0.0001.
Given that tumors produce proangiogenic factors that could have effects at the distant metastatic sites, we interrogated whether primary tumors have the ability to alter TRAIL expression at the PMN. One of the hallmarks of the PMN formation is the recruitment of myeloid cells to the lung (6, 48). Neutrophils, but not macrophages, infiltrate the lungs from day 7 onward after orthotopic tumor implantation (Fig. 5, G to K), which is accompanied by a concomitant up-regulation of PMN-associated S100a family proteins (Fig. 5G), together confirming the establishment of a PMN. E0771 cancer cells spontaneously colonize the lung between days 10 and 14 (Fig. 5L and fig. S6E). Compared to tumor-free mice, expression of TRAIL significantly decreased both in pulmonary tissue and specifically in lung ECs before cancer cell arrival, during the PMN formation (Fig. 5, L and M, and fig. S6F).
These data unveil that the mature and quiescent endothelia, but not angiogenic vessels in the tumor or in the metastasis, represent a major source of endogenous TRAIL. We show that tumor-derived soluble factors, such as VEGF-A, bFGF, and PlGF, reduce TRAIL expression in lung ECs. Genetic or pathological down-regulation of TRAIL by tumor-derived factors “destabilizes” the vascular barrier and contributes to the formation of a PMN.
EC-specific depletion of TRAIL triggers apoptosis
As previously reported (34, 49, 50), exogenous TRAIL induced the assembly of the “death-inducing signaling complex” (DISC), constituted by DR5, Fas-associated protein with death domain (FADD), and cleaved caspase-8 (fig. S7A). The consequent apoptosis was equally rescued by both the pan-caspase inhibitor q-VD-OPh (qVD) and the caspase-8–specific inhibitor z-IETD-FMK (zIETD) (Fig. 6A). However, the angiocrine role of endogenous EC-derived TRAIL in EC biology is mostly unknown. To shed light on this, we silenced TRAIL in HUVECs (Fig. 6B and fig. S7, B and C). Upon culture confluency, TRAIL silencing resulted in caspase-8 and caspase-3 cleavage (Fig. 6B) and subsequent apoptosis (Fig. 6, C to E), which could be modestly rescued by zIETD and completely by qVD (Fig. 6, C, E, and F). zIETD inhibited caspase-8 cleavage, as expected, but it failed to revert the cleavage of caspase-3 (fig. S7D), overall arguing that absence of endogenous TRAIL in ECs promotes both caspase-8–dependent and –independent apoptosis. To assess whether TRAIL deletion affected vascular tightness, we measured transendothelial electrical resistance (TEER) in scramble control (shSCR) and shTRAIL HUVECs. In shSCR HUVECs, TEER steadily increased over time as cells grow until confluence and tighten EC junctions, which was paralleled by an increase in TRAIL expression (Fig. 6G). Conversely, TEER significantly dropped down from day 2 onward in shTRAIL HUVECs (Fig. 6G), indicating that deficiency of TRAIL in HUVECs hinders the possibility to make a tight, intact, and confluent vascular layer.
Fig. 6. EC-specific depletion of TRAIL triggers apoptosis.

(A) FACS analysis of viable [annexin V− propidium iodide (PI)−] HUVECs treated with SuperKillerTRAIL (recTRAIL; 100 ng/ml), pan-caspase inhibitor qVD (50 μM), or caspase-8–specific inhibitor zIETD (50 μM) for 5 days. (B) Protein levels of pro–caspase-8, cleaved caspase-8 (clCasp8), pro–caspase-3, cleaved caspase-3 (clCasp3), and TRAIL in shSCR or shTRAIL HUVECs. For each time point, the densitometry fold change of shTRAIL toward shSCR is indicated. (C and D) Time-dependent analysis of cell death by cytotox incorporation in shSCR and shTRAIL HUVECs treated with qVD (50 μM; green line) or zIETD (100 μM; blue line) since day 1. Representative images at 34 hours (D). (E) Analysis of viable (annexin V− PI−) shSCR and shTRAIL HUVECs at day 3 treated with qVD (20 μM) since day 1. (F) Protein levels of pro–caspase-8, cleaved caspase-8, pro–caspase-3, cleaved caspase-3, and TRAIL in shSCR and shTRAIL HUVECs at day 5 treated with qVD (50 μM) since day 0. (G) TEER measurement and mRNA expression of TNFSF10 of shSCR and shTRAIL HUVEC monolayers. (H and I) FACS analysis of the percentage of dead blood vessel ECs (BECs; CD45−CD31+PDPN−) in perfused lungs from tumor-free induced ECW/W and ECΔT10 mice. In (I), anti-CD31 (αCD31)–conjugated LNP/empty or anti-CD31–LNP/Trail (8 μg per mouse) was injected i.v. 48 and 24 hours before cancer cell injection, and statistical significance was calculated by one-tailed unpaired t test. (J) GSEA enrichment plots showing the comparison of the gene expression profiles in lung ECs sorted from tumor-free induced ECW/W and ECΔT10 mice. NES, normalized enrichment score. (K and L) Protein levels of fibronectin and TRAIL in perfused lung tissue lysates of tumor-free induced ECW/W and ECΔT10 mice. (M) mRNA expression of Fn1 (fibronectin) and dLNGFR in perfused lung tissue from WT mice after orthotopic m.f.p. injection of E0771-dLNGFR cells. All graphs show means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To assess whether TRAIL deletion in vivo also affected the integrity of the lung vasculature, we analyzed the lungs of tumor-free mice by flow cytometry. EC-specific deletion of TRAIL increased the percentage of dead blood vessel ECs (BECs) (Fig. 6H and figs. S8 and S9), which was reverted by the reexpression of TRAIL with EC-targeting LNPs (Fig. 6I). Next, we sorted lung ECs from tumor-free WT and EC-specific TRAIL KO mice and performed bulk RNA-seq (fig. S7E). Gene set enrichment analysis (GSEA) confirmed a signature of apoptosis (Fig. 6J). As a consequence of vascular leakage, fibronectin can leak from the serum to the lung parenchyma. We observed that EC-specific deletion of TRAIL resulted in higher fibronectin protein levels in perfused lung tissue of tumor-free animals (Fig. 6, K and L), indicating an increase in vascular leakiness, as its transcripts during PMN formation did not change (Fig. 6M). Thus, these data show that the expression of TRAIL in quiescent ECs is required to maintain the integrity of the vascular barrier.
EC-specific depletion of TRAIL favors a proinflammatory state that facilitates leukocyte and cancer cell adhesion
Adhesion to the vascular wall is a critical event for successful metastatic colonization (4, 51, 52). EC-specific deletion of TRAIL promoted a proadhesion and proinflammatory transcriptional program in lung ECs from tumor-free mice, characterized by the up-regulation of adhesion molecules (namely, Icam1, Sele, and Vcam1) and chemokines (namely, Ccl2 and Ccl5) (Fig. 7, A and B). Consistently, the expression of adhesion molecules (i.e., ICAM1, SELE, and VCAM1) was the lowest in TRAILhigh distal ECs and the highest in TRAILlow proximal ECs in patient 3 from the scRNA-seq atlas of the human lung (Fig. 7, C to I) (41). Upon silencing of TRAIL in HUVECs, the expression of the E-Selectin and intercellular adhesion molecule–1 (ICAM1) increased (Fig. 7, J to L), which could not be reverted by the treatment with the pan-caspase inhibitor qVD (Fig. 7, J and K), proving that the proadhesive signature is independent from caspase activity. Functionally, this translated into an increased cancer cell and leukocyte adhesion to shTRAIL compared to shSCR HUVEC monolayers (Fig. 7, M and N). In tumor-free mice, EC-specific deletion of TRAIL increased the infiltration of immune cells into the lungs (Fig. 7, O and P) and in particular of myeloid cells (Fig. 7, Q and R), such as neutrophils (Fig. 7, S and T), which are known to be recruited to the lung in preparation of the PMN (6, 48). In contrast, lung-resident alveolar macrophages remained unchanged (Fig. 7U). Pretreatment of mice with ICAM1 and E-Selectin blocking antibodies before cancer cell injection abrogated the increase in cancer cell lodging 24 hours after injection observed upon EC-specific deletion of TRAIL (Fig. 7V). This was confirmed by quantification of tdTomato+ E0771 cells in whole lungs by three-dimensional (3D) confocal microscopy (Fig. 7, W and X).
Fig. 7. EC-specific depletion of TRAIL favors a proinflammatory state that facilitates leukocyte and cancer cell adhesion.

(A) mRNA expression of indicated genes in lung ECs sorted from tumor-free induced ECW/W and ECΔT10 mice. (B) FACS analysis of the percentage of adhesive ECs [vascular cell adhesion molecule–1+ (VCAM1+) E-Selectin+ ICAM1high] in perfused lungs from tumor-free induced ECW/W and ECΔT10 mice. (C to I) t-SNE plots showing the origin (distal versus proximal) or expression of indicated genes (C), violin plot showing expression levels (D, F, and H), or the percentage of expressing (+) and nonexpressing (−) cells (E, G, and I) in ECs from patient 3. (J and K) mRNA expression of SELE (J) and ICAM1 (K) in shSCR and shTRAIL HUVECs at day 3 treated with qVD (50 μM) since day 0. (L) Protein levels of ICAM1 and E-Selectin in shSCR and shTRAIL HUVECs. For each time point, the densitometry fold change of shTRAIL toward shSCR is indicated. TRAIL knockdown efficiency is shown in Fig. 6B. (M and N) E0771 or 4T1 cells (M) or white blood cells (WBC) (N) adhered to shSCR and shTRAIL HUVEC monolayers at day 6 after 1 hour of contact. (O to U) FACS analysis of the percentage or number of immune cells (O and P), myeloid cells excluding alveolar macrophages (AMs) (Q and R), neutrophils (S and T), and AM (U) in perfused lungs from tumor-free induced ECW/W and ECΔT10 mice. (V to X) mRNA expression of dLNGFR in perfused lung tissue (V), number of cancer cells (W), and representative images (X) showing tdTomato+ cancer cells (magenta) and 4′,6-diamidino-2-phenylindole (DAPI) (blue) in lungs 24 hours after i.v. injection of E0771-dLNGFR-tdTomato cells in induced ECW/W and ECΔT10 mice. Anti-ICAM1 and anti–E-Selectin blocking antibodies (each 10 mg/kg) or immunoglubulin G (IgG) control (20 mg/kg) were injected intravenously 3 hours before cancer cell injection. Scale bars, 250 μm. All graphs show means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
These data prove that TRAIL expression in quiescent lung ECs is required to keep a proinflammatory program at bay. Upon depletion of TRAIL, the endothelium becomes inflamed and expresses higher levels of adhesion molecules and cytokines that orchestrate leukocyte and cancer cell adhesion, together fostering metastatic colonization.
Activation of p38 and nuclear factor κB underlies the proadhesive state of the endothelium upon TRAIL depletion
Besides triggering the extrinsic apoptosis pathway, TRAIL receptor signaling is upstream of multiple signaling pathways, such as PI3K/Akt, Erk1/2, nuclear factor κB (NF-κB), p38, and c-Jun N-terminal kinase, which are linked to survival, invasiveness, and inflammation (14, 17–20, 22). Silencing of TRAIL increased the phosphorylation of p38 (Fig. 8A), which has been associated with EC stress, apoptosis, and metastasis (53). Moreover, TRAIL silencing activated the NF-κB pathway, as seen by the phosphorylation of IκBα (the inhibitory protein of NF-κB), a subsequent decrease in its total levels (Fig. 8A) and by the nuclear accumulation of NF-κB (Fig. 8B). In this line, gene signatures of NF-κB activation and p38 downstream signaling were up-regulated in lung ECs sorted from tumor-free EC-specific TRAIL KO mice compared to WT mice (Fig. 8, C and D). Upon silencing of TRAIL, DR4 and/or DR5 coimmunoprecipitated with IκB kinase α (IKKα), IKKβ, NF-κB essential modulator (NEMO), receptor-interacting protein 1 (RIP1), caspase-8, and FADD (Fig. 8E). In shTRAIL HUVECs and in absence of exogenous TRAIL, deletion of DR4 strongly rescued the phosphorylation of p38, while deletion of DR5 had a minor effect (fig. S7F). Noteworthy, the pan-caspase inhibitor qVD failed to revert the changes in IκBα and in p38 phosphorylation, suggesting that caspase activity is not upstream to these changes (Fig. 8F). Inhibition of NF-κB or p38 pathways almost completely rescued the up-regulation of E-Selectin and ICAM1 observed upon silencing of TRAIL (Fig. 8, G to L) and reduced the adhesion of E0771 cells to shTRAIL HUVECs (Fig. 8, M and N).
Fig. 8. Activation of p38 and NF-κB underlies the proadhesive state of the endothelium upon TRAIL depletion.

(A) Protein levels of p38 (total and ph-Thr180/Tyr182) and IκBα (total and ph-Ser32/36) in shSCR or shTRAIL HUVECs. For each time point, the densitometry fold change of shTRAIL toward shSCR is indicated. For TRAIL knockdown efficiency, see Fig. 6B. (B) Immunofluorescence staining of NF-κB (green) and Hoechst (blue) in shSCR or shTRAIL HUVECs. Scale bars, 20 μm. (C and D) GSEA enrichment plots showing the comparison of the gene expression profiles in lung ECs sorted from tumor-free induced ECW/W and ECΔT10 mice. (E) Coimmunoprecipitation (co-IP) of DR4, DR5, or IgG control in HUVECs at day 3, showing protein levels of IKKα, ΙΚΚβ, caspase-8, RIP1, NEMO, FADD, DR4, DR5, and TRAIL. Input = WCE. (F) Protein levels of p38 (total and ph-Thr180/Tyr182) and total IκBα in shSCR and shTRAIL HUVECs at day 5 treated with the pan-caspase inhibitor qVD (50 μM) since day 0. For TRAIL knockdown efficiency and caspase cleavage inhibition, see Fig. 6F. (G to J) mRNA expression of ICAM1 (G and I) and SELE (H and J) in shSCR and shTRAIL HUVECs at day 6 treated either with the IκB inhibitor IKK-16 (NF-κBi; 2,5 μM) (G and H) or with the p38 inhibitor SB203580 (p38i; 10 μM) (I and J) since day 4. (K and L) Protein levels of ICAM1, E-Selectin, and TRAIL in shSCR and shTRAIL HUVECs at day 6 treated either with NF-κBi (2.5 or 5 μM) (K) or with p38i (1 or 10 μM) (L) since day 4. (M and N) E0771 cancer cells adhered to shSCR and shTRAIL HUVEC monolayers at day 6 after 1 hour of contact. HUVECs were treated either with NF-κBi (2.5 μM) for 24 hours (M) or with p38i (10 μM) for 48 hours (N). All graphs show means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Overall, this shows that silencing of TRAIL activates a stress and proinflammatory response characterized by the activation of complementary NF-κB and p38 signaling pathways. In turn, these induce the expression of adhesion molecules and promote leukocyte and cancer cell adhesion, which is independent of the concomitant caspase activation.
Endothelial DR5 and TRAIL DcRs favor metastatic spread
Given that the antimetastatic effect of endothelial TRAIL is greatly EC autonomous and independent from the expression of DR5 in cancer cells, we next sought to investigate whether and how TRAIL receptors in nonmalignant cells, and particularly in ECs, could be involved in the control of metastasis. ECs expressed the highest levels of DR5 among all the cells sorted from tumor-free lungs (Fig. 9A and fig. S2). Similar to TRAIL, DR5 protein was mostly found intracellularly in lung ECs (Fig. 9B). Unexpectedly, inducible ubiquitous or EC-specific deletion of DR5 almost completely prevented experimental lung metastases at end stage (Fig. 9, C and D) and at 24 hours after injection (Fig. 9E), indicating that, oppositely to the role of EC-derived TRAIL, EC-derived DR5 promotes metastasis formation. Dual deletion of EC-derived TRAIL and DR5 almost completely rescued the increase in metastasis (Fig. 9F), suggesting that DR5 mediates the increase in metastasis seen in the absence of EC-derived TRAIL. Compared to EC-specific TRAIL KO mice, deletion of EC-derived DR5 and dual deletion of EC-derived TRAIL and DR5 reduced EC demise (Fig. 9, G and H) and abrogated the increase in immune infiltration (both the total immune infiltrate and the individual immune cell subsets) (Fig. 9, I to L, and fig. S10, A to D), as well as the induction of the proadhesive transcriptional program (Fig. 9M). While murine lung ECs express only DR5, which was found almost entirely intracellularly (Fig. 9B), HUVECs express both DR5 and DR4 (fig. S10E) (34, 49), which was detected intracellularly and on the cell surface (Fig. 9N). However, TRAIL and DR5 interacted intracellularly in quiescent HUVECs (Fig. 9, O and P). Upon EC-specific deletion of TRAIL, we observed a significant increase in DR5 exposure at the cell surface, as indicated by extracellular flow cytometry staining (Fig. 9Q), which was rescued by the LNP-mediated reexpression of TRAIL (Fig. 9R). However, shTRAIL HUVECs were equally sensitive to exogenous recombinant TRAIL-mediated apoptosis than shSCR HUVECs (fig. S10F). Overall, this suggests that DR5 exerts a prometastatic role independently of EC-derived TRAIL, which is unleashed in the absence of TRAIL ligand. Notably, our data show that mice lacking TRAIL ubiquitously display the same vascular phenotype, further supporting a ligand-independent mechanism of action.
Fig. 9. Endothelial DR5 favors metastatic spread.

(A) mRNA expression of Tnfrsf10b in cells sorted from perfused lungs of tumor-free WT mice. (B) FACS analysis of DR5 protein on lung BECs from tumor-free induced EC-specific DR5 WT (ECW/W) or KO (ECΔDR5) mice. (C and D) Number of ink-contrasted lung metastasis 12 days after i.v. injection of E0771 cells in induced ubiquitous DR5 WT (R26W/W) or KO (R26ΔDR5) (C) or ECW/W and ECΔDR5 mice (D). (E and F) mRNA expression of dLNGFR in perfused lung tissue after i.v. injection of E0771-dLNGFR cells in induced ECW/W, ECΔDR5, ECΔT10, or EC-specific TRAIL and DR5 double KO mice (ECΔT10ΔDR5) mice. Figure 3I previously showed dataset relative to ECΔT10. (G to L) FACS analysis of the percentage of dead BECs (G and H), immune cells (I and J), and neutrophils (K and L) in perfused lungs from tumor-free induced ECW/W, ECΔDR5, or ECΔT10ΔDR5 mice. Figures 6H and 7 (O, P, S, and T) previously showed datasets relative to ECΔT10. (M) mRNA expression of indicated genes in lung ECs sorted from tumor-free induced ECW/W and ECΔT10ΔDR5 mice. (N) Protein levels of DR4 and DR5 in WCE, intracellular, or surface fractions in quiescent HUVECs. VE-cadherin is the representative surface protein. (O) Co-IP of DR5, TRAIL, or IgG control in quiescent HUVECs. Input = WCE. (P) Proximity ligation assay (PLA) between TRAIL and DR5 (red), VE-cadherin (green), and Hoechst (blue) in HUVECs. Scale bar, 10 μm. (Q and R) FACS analysis of extracellular DR5 protein on lung BECs from induced ECW/W and ECΔT10 mice. In (R), anti-CD31 (αCD31)–conjugated LNP/empty or anti-CD31–LNP/Trail (8 μg per mouse) was injected i.v. 48 and 24 hours before cancer cell injection, and statistical significance was calculated by one-tailed unpaired t test. ΔMFI = MFIstained − MFIFMO. All graphs show means ± SEM. **P < 0.01; ***P < 0.001; ****P < 0.0001.
TRAIL decoy receptors (TRAIL-R3/DcR1 and TRAIL-R4/DcR2) mediate resistance to TRAIL-induced apoptosis (23–26, 28), although genetic models to study their physiological role and importance in vivo have not been reported. To assess a possible contribution of DcR1 and DcR2 in the homeostatic control of TRAIL-related functions, we generated a novel DcR1 and DcR2 double full KO murine strain (fig. S11, A to C). Deletion of DcR1/2 reduced the number of experimental lung metastases (Fig. 10A), which confirms in vivo their role as natural TRAIL inhibitors. Among our sorted cell populations from tumor-free lungs, DcR1 was abundantly expressed by and neutrophils (Fig. 10B), whereas DcR2 was highly expressed by ECs, CD8+ T cells, and neutrophils (Fig. 10C). To dissect which cell(s) were responsible for this phenotype, we generated reciprocal chimeric mice. Deletion of DcR1/2 in the host nonimmune compartment, but not in the hematopoietic compartment, reduced experimental lung metastases (Fig. 10D), indicating that the prometastatic effect of DcR1/2 is carried out by a nonimmune cell.
Fig. 10. Endogenous TRAIL DcRs favor metastatic spread.

(A) Number of ink-contrasted lung metastasis 12 days after i.v. injection of E0771 cells in constitutive DcR1/2 WT (Dcr1/2+/+) and KO (Dcr1/2−/−) mice. (B and C) mRNA expression of Tnfrsf23 (DcR1) (B) and Tnfrsf22 (DcR2) (C) in ECs, NK cells, CD8+ T cells, and neutrophils sorted from perfused lungs of tumor-free WT mice. (D) Number of ink-contrasted lung metastasis 11 days after i.v. injection of E0771 cells in chimeras obtained from the reciprocal bone marrow transplantation of constitutive DcR1/2 WT (Dcr1/2+/+) and KO (Dcr1/2−/−) mice. Arrows indicate genotypes of the bone marrow donor → recipient mice. (E and F) Graphical abstract. (E) Steady-state, high TRAIL in ECs. In steady state, endothelial TRAIL holds DR5 intracellularly and prevents its activation in quiescent ECs, thereby supporting cell survival, quiescence, and a resting anti-inflammatory/antiadhesive state. Together, this ensures the stability of the vascular barrier. (F) PMN, low TRAIL in ECs. Tampering with TRAIL in the PMN by transcriptional down-regulation in response to tumor-derived factors (i.e., VEGF-A, PlGF, and others) or by DcR-mediated entrapment liberates DR5. The increased availability of DR5 at the cell surface is sufficient to trigger its activation in a ligand-independent manner. As a result, vascular integrity is compromised by the occurring EC apoptosis and NF-κB/p38–mediated stickiness, favoring immune cell recruitment and ICAM1/E-Selectin–mediated cancer cell adhesion. Together, this process promotes cancer cell dissemination and metastasis in distant organs. All graphs show means ± SEM. *P < 0.05; **P < 0.01.
In conclusion, these data indicate that while EC-derived TRAIL protects against metastasis, its receptor DR5 in ECs exerts a prometastatic signaling, which is unleashed by the genetic deletion of endothelial TRAIL (Fig. 10, E and F). Physiologically, we unravel at least two possible mechanisms that counter the protective function of endothelial TRAIL, i.e., the transcriptional down-regulation of TRAIL by tumor-derived signals and the entrapment of TRAIL in the PMN by the DcRs (Fig. 10, E and F).
DISCUSSION
It is well accepted that a primary tumor can precondition the metastatic niche to the arrival of disseminated cancer cells. However, the mechanism underlying the activation of quiescent blood vessels at the PMN in response to tumor-derived signals has been understudied so far. Here, we unveil that endothelial TRAIL in a distant organ such as lungs or liver preserves the integrity and functions of the vascular barrier. In a tumor context, this mechanism is hijacked to facilitate breast cancer cell lodging in the PMN and metastasis formation (Fig. 10, E and F).
To date, the antimetastatic effect of TRAIL was ascribed to NK cell–mediated killing of disseminated DR5-expressing cancer cells (7–9, 12). However, the genetic tools available so far were limited to murine strains where TRAIL was constitutively and ubiquitously deleted. Using newly generated, cell-specific TRAIL KO mice, we unveil that EC-derived TRAIL sustains the integrity and quiescence of the endothelium, which precludes early cancer cell lodging in distant organs independently from DR5 expression in cancer cells themselves. Ultimately, deletion of TRAIL in ECs resulted in a marked increase in metastasis, particularly when compared to the milder effect of NK cell–specific TRAIL depletion. We found that ECs are the main source of TRAIL in the PMN, as seen by mining TRAIL expression in different sorted cell types and in tissue extracts, or in publicly available scRNA-seq datasets from murine and human lungs (41, 42, 54). This expression was the highest in ECs from tumor-free or unaffected lungs in mice and humans, respectively, whereas it was reduced in both tumor and metastasis-associated ECs. In line with this, TRAIL expression was low in proliferating ECs and strongly up-regulated by quiescence in vitro. Thus, we propose that TRAIL is required in mature, quiescent vascular beds and dispensable during angiogenesis.
Non-EC derived TRAIL (recombinant or endogenous) has been associated with either proapoptotic or promitogenic and promigratory effects on ECs in vitro (34, 49, 50, 55, 56) and has been shown to protect against vascular oxidative stress and EC dysfunction (57). However, the angiocrine role of endogenous EC-derived TRAIL remained unknown. This is the first report in which absence (rather than presence) of endothelial TRAIL results in caspase-8–dependent and independent EC demise. However, deletion of TRAIL does not cause fatal vascular defects, likely because it does not equally affect all the ECs, but only a fraction of them, and compensatory mechanisms may also be at play. Besides compromising EC survival and vascular stability, TRAIL depletion in ECs results in an increased activation and number of sticky “hotspots” within the endothelial lining, which promote leukocyte and cancer cell adhesion both on cultured ECs and at the PMN. Supporting our results, these adhesion molecules seem to be almost exclusively expressed by a subset of TRAILlow human lung ECs (41). Mechanistically, EC adhesion in absence of TRAIL is at least partially mediated by both p38 and NF-κB signaling, resulting in the up-regulation of ICAM1 and E-Selectin. These results are unexpected since it is reported that the activation of these pathways can be downstream of DR5 upon TRAIL binding in other contexts (20, 22), whereas, in ECs, these pathways are triggered by the absence of the ligand. Other groups have reported that ligand/receptor engagement promotes the formation of a proinflammatory complex (14, 15, 17–22). The data shown in this manuscript suggest that a complex with similar composition may be recruited to TRAIL receptors upon silencing, genetic KO, or pathophysiological down-regulation of endothelial TRAIL ligand. We show that the elimination of endothelial TRAIL elicits a cascade consisting in the liberation of DR5 from the intracellular space to the EC surface. This process is sufficient to engage IKKs (that will activate NF-κB) and to promote downstream p38 phosphorylation. Therefore, it seems that tampering with intracellular endothelial TRAIL leads to increased receptor availability at the cell surface that will initiate a proinflammatory, proadhesive, and proapoptotic endothelial phenotype in a ligand-independent manner as described before in other contexts (19).
Given the important effect of EC-derived TRAIL in the control of vascular homeostasis, we sought to investigate whether and how TRAIL receptors were mechanistically and phenotypically involved. Previous studies have mostly focused on the role of death receptors in cancer cells. Although several nonmalignant cells have been reported to express DR5 (14, 27, 29–33), its expression and function at the PMN were never investigated. Consistent with an EC-autonomous effect, we found that lung ECs express high levels of DR5. Instead of phenocopying the prometastatic effect of EC-specific deletion of TRAIL, EC-specific deletion of DR5 markedly reduced early lung metastases. Furthermore, concomitant EC-specific deletion of TRAIL and DR5 in vivo rescued the phenotype observed in EC-specific TRAIL single KO mice. These findings unveil that, in contrast to the expectation, the expression of TRAIL and its cognate receptor DR5 in ECs exert opposite effects on metastasis formation and that TRAIL-mediated inhibition of DR5 impairs metastasis. We found that in ECs, both TRAIL and its receptor DR5 are mostly detected intracellularly, where they interact with each other. Given this unusual feature, we can speculate that this homeostatic ligand-dependent “blockage” might happen through intracrine interaction between TRAIL and DR5. In contrast, recombinant TRAIL induces EC apoptosis by activating a conventional DR5 signaling. However, we exclude that endothelial TRAIL depletion sensitizes the endothelium to extracellular TRAIL because EC-specific and ubiquitous TRAIL KO mice display a similar phenotype and TRAIL silencing in ECs enhances apoptosis irrespective of increasing concertation of exogenous recombinant TRAIL. It is exciting to see how our findings define DR5 in ECs at the metastatic niche as another dependence receptor pathway (58, 59), whereby the ligand blocks a signaling elicited by its cognate receptor, ultimately preserving vascular homeostasis. This notion raises the question of whether TRAIL receptor antagonists may be better candidates than TRAIL receptor agonists in tumors where cancer cells have acquired resistance to TRAIL-mediated apoptosis or where DR5 promotes prosurvival and proinvasive effects (14–16). Alternatively, we suggest that LNPs delivering a TRAIL mRNA selectively in lung ECs might represent a therapeutic tool to prevent cancer cell extravasation. We prove that the induction of TRAIL in ECs through LNP-mediated delivery “holds” DR5 intracellularly, prevents EC apoptosis, hinders early cancer cell extravasation into the lungs of WT mice, and abrogates almost completely cancer cell dissemination/metastasis in EC-specific TRAIL KO mice.
Overall, we suggest that endothelial TRAIL is required to maintain EC homeostasis and survival and to prevent the activation of a p38 and NF-κB-driven proinflammatory and proadhesion transcriptional program, ultimately ensuring a proper vascular barrier function. Primary tumors are able to shape several features of the distant organs, creating a PMN permissive for incoming circulating cancer cells (6, 48). We have found that primary tumor-derived soluble factors naturally suppress TRAIL expression in ECs, which precedes the arrival of cancer cells themselves. Among the factors responsible for this effect, we have identified the proangiogenic molecules VEGF-A and PlGF, both binding to VEGFRs (and, to a less extent, bFGF) although, given the plethora of signals coming from the tumor, we do not exclude that other factors might contribute too. Mechanistically, we suggest that the activation of receptor tyrosine kinases in ECs, e.g., VEGFRs, is the main trigger for TRAIL suppression, greatly through the proproliferative cascade MEK/MAPK signaling (which is consistent with the observation that Ki-67high ECs did not express TRAIL). This pathophysiological situation is similar to the even more extreme scenario offered by EC-specific TRAIL deletion, in which cancer cell adhesion and immune cell infiltration are both enhanced and vascular integrity is compromised. VEGF-A–targeted therapies have faced important challenges, partly because of the lack of predictive biomarkers (60, 61). If VEGF-A induces permeability of the pulmonary vascular bed through TRAIL down-regulation, then one can hypothesize that those patients where VEGF-A–targeted therapies abrogate this process are virtually those that respond positively to the treatment. Together, our data highlight endothelial TRAIL at the PMN as a physiologically relevant gatekeeper of vascular barrier function that can be overruled by the tumor during cancer cell dissemination.
We define a pathophysiological control of this pathway by disclosing the relevance of naturally occurring TRAIL inhibitors, the TRAIL decoy receptors DcR1 and DcR2 (23–25, 28). Although expressed by several immune cells, only the deletion of DcR1 and/or DcR2 in nonimmune cells (likely by ECs themselves) impairs metastasis. Therefore, given our evidence on the intracrine interaction of TRAIL and DR5, we speculate that EC-derived DcRs could compete intracellularly with DR5, thereby harnessing TRAIL’s antimetastatic effect.
In conclusion, we show that TRAIL in ECs is required to ensure EC survival and a resting anti-inflammatory and antiadhesive state, thus resulting in the formation of a tight and quiescent barrier. In the context of cancer, this mechanism is possibly overruled in a way that the down-regulation of TRAIL and the blockade of TRAIL by DcRs would foster the dysregulation and activation of this barrier, the initiation of inflammation, and cancer cell adhesion and dissemination, all events promoting metastasis.
MATERIALS AND METHODS
Animals
WT C57BL/6NTac and Balb/cAnNCrl mice were purchased from the internal stock of the KU Leuven. Constitutive and ubiquitous Tnfsf10-deficient mice (8) (referred to as Trail+/+ and Trail−/−) in Balb/c background were received from Prof. M. Smyth (Peter MacCallum Cancer Centre, Australia) and Prof. J. Weiss (The National Cancer Institute, USA).
Conditional Tnfsf10 KO mice (Tnfsf10Lox/Lox) were generated in the C57BL/6N background using a targeted ES cell line from KOMP/EUCOMM (EPD0244_2_E08). Positive clones were confirmed by Southern blot. The neomycin resistance cassette was deleted in vivo by using recombinase flippase (FLP)-mediated recombination. The final transgenic line carries two LoxP sites flanking the exon 3 in each Tnfsf10 allele. Tnfsf10Lox/Lox mice were intercrossed with mice expressing the tamoxifen-inducible Cre recombinase under the Rosa26 promoter (Rosa26.iCreERT2xTnfsf10Lox/Lox, for inducible ubiquitous deletion and referred to as R26W/W and R26ΔT10), the VE-cadherin promoter (Cdh5.iCreERT2xTnfsf10Lox/Lox, for inducible EC-specific deletion and referred to as ECW/W and ECΔT10), or a constitutively active Cre recombinase under the NKp46 promoter (62) (Nkp46.iCrexTnfsf10Lox/Lox, for NK cell–specific deletion and referred to as NKW/W and NKΔT10). Nkp46.iCre mice (62) were obtained from Prof. E. Vivier (Centre d’Immunologie de Marseille-Luminy, France).
Prof. H. Walczak (Deutsches Krebsforschungszentrum, Germany) provided the conditional Tnrfsf10b KO mice (DR5Lox/Lox) mice (9). Tnfrsf10bLox/Lox mice were intercrossed with mice expressing the tamoxifen-inducible Cre recombinase under the Rosa26 promoter (Rosa26.iCreERT2xTnfrsf10bLox/Lox, for inducible ubiquitous deletion and referred to as R26W/W and R26ΔDR5) or the VE-cadherin promoter (Cdh5.iCreERT2xTnfrsf10bLox/Lox, for inducible EC-specific deletion and referred to as ECW/W and ECΔDR5) and with Cdh5.iCreERT2xTnfsf10Lox/Lox mice (Cdh5.iCreERT2xTnfsf10Lox/LoxxTnfrsf10bLox/Lox, for inducible EC-specific double deletion of TRAIL and DR5 and referred to as ECW/W and ECΔT10ΔDR5).
In collaboration with Cyagen (USA), we generated constitutive Tnfrsf22/Tnfrsf23-double KO mice (referred to as Dcr1/2+/+ and Dcr1/2−/−) by introducing a point mutation in exon 3 of both genes by CRISPR-Cas9 technology [guide RNA (gRNA) sequence targeting both genes: CACATTGTCCTTGAGTATGGGGG]. The targeted allele of Tnfrsf22 has an insertion of 1 base pair (bp) between nucleotides 135 and 136. The targeted allele of Tnfrsf23 has a deletion of 2 bp corresponding to nucleotides 97 and 98. The mRNAs transcribed from the targeted alleles with frameshift mutations undergo nonsense-mediated decay and result in the absence of the protein. Analysis of potential off-target sites including NGA, NAG, and NGG protospacer adjacent motive (PAM) sequence by polymerase chain reaction (PCR) and next generation sequencing (NGS) confirmed that they were intact.
All mice used were between 8 and 14 weeks old. Deletion of the Tnfsf10 and/or Tnfrsf10b floxed allele in murine strains expressing a tamoxifen-inducible Cre recombinase (iCreERT2) was induced by intraperitoneal injections of tamoxifen (1 mg per mouse per day) for 5 days (Rosa26.iCreERT2) or 10 days (Cdh5.iCreERT2) before the experiment. When necessary, mice were anesthetized with a mix of ketamine (100 mg/kg) and xylazine (10 mg/kg). Housing and all experimental procedures were performed in accordance with the guidelines of the Institutional Animal Care and Research Advisory Committee of the KU Leuven (ECD156/2014, ECD200/2016, and ECD226/2017).
Cell culture
4T1 and E0771 medullary breast adenocarcinoma cell lines were obtained from the American Type Culture Collection and CH3 Biosystems, respectively, and EMT6.5 cells were a gift from R. L. Anderson (Peter MacCallum Cancer Centre, Melbourne, Australia). 4T1 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) high glucose (Gibco, 41965-039) supplemented with 10% fetal bovine serum (FBS) (Gibco), penicillin (100 U/ml), streptomycin (100 μg/ml), and 2 mM l-glutamine (Gibco) (referred to as DMEM complete). EMT6.5 cells were cultured in Alpha minimum essential medium (MEM) Eagle without l-glutamine (Lonza, BE12-169F) supplemented with 10% FBS (Gibco), penicillin (100 U/ml), streptomycin (100 μg/ml), and 2 mM l-glutamine (Gibco). E0771 cells were cultured in Roswell Park Memorial Institute (RPMI)1640 with l-glutamine (Gibco, 21875-034) supplemented with 10% FBS (Gibco), penicillin (100 U/ml), streptomycin (100 μg/ml), and 2 mM l-glutamine (Gibco) (referred to as RPMI complete). Cells were incubated at 37°C in 95% air and 5% CO2. A lentiviral vector encoding a mutated firefly luciferase [akaluciferase (Akaluc)] (40), Renilla luciferase, puromycin resistance, and miRFP670 was introduced by lentiviral transduction in 4T1 and E0771 cells. Transduced cells were sorted according to miRFP670 expression, and miRFP670+ cells are hereby referred to as 4T1-Akaluc or E0771-Akaluc. A lentiviral vector (LentiGuide) expressing a dLNGFR as a surface reporter protein was introduced by lentiviral transduction in 4T1 and E0771 cells. Transduced cells were sorted to obtain a pure dLNGFR+ cell population. A lentiviral vector expressing tdTomato (Addgene, plasmid #27353, provided by Prof. J. Van Rheenen) and blasticidin resistance was introduced by lentiviral transduction in E0771-dLNGFR+ cells. Subsequently, tdTomato+ cells were selected for 5 days with blasticidin (1 μg/ml; InvivoGen, ant-bl-05). Generation of DR5 KO cells was achieved by transfection of 4T1 and E0771 cells with a plasmid encoding for Thy1.1 (as a surface reporter protein), Cas9, and the gRNA of interest (nontargeting gRNA, GAACAGTCGCGTTTGCGACT; DR5 gRNA, TAGAATGTACCTGCTAGACA) (LentiCRISPR v2 Thy1.1) using Lipofectamine3000 (Invitrogen, L3000008, according to the manufacturer’s instructions). The Thy1.1 was used to negatively select the clones where the plasmid might have been stably incorporated. Subsequently, cells were sorted to obtain a pure “Cas9-free” DR5 WT (Thy1.1− DR5+) or KO (Thy1.1− DR5−) populations. A lentiviral vector expressing murine VEGF-A164 (pRRL56 mVEGFA164), murine PlGF (pRRL56 mPlGF), or its corresponding empty vector was introduced by lentiviral transduction in E0771 cells. Overexpression of VEGF-A or mPlGF was confirmed by quantitative reverse transcription PCR (qRT-PCR) and by the quantification of the cytokine levels in cancer cell supernatants with mouse VEGF-A Quantikine ELISA kit (R&D, MMV00) or mouse PlGF-2 Quantikine ELISA kit (R&D, MP200) according to the manufacturer’s instructions.
HUVECs from pooled donors were obtained from PromoCell and cultured in 0.2% gelatin-coated dishes in Endothelial cell growth medium 2 (EGM2) medium supplemented with the provided growth factors (PromoCell, C-22011), penicillin (100 U/ml), and streptomycin (100 μg/ml; Gibco). HUVECs were used between passages 3 and 7, and the medium was refreshed every 48 or 72 hours. To induce EC quiescence by contact inhibition, at day 0, HUVECs were seeded in supplemented EGM2 at a density of 30,000 cells/cm2; at day 3, the medium was replaced for a 1:1 mixture of supplemented EGM2 and Medium 199 (M199) with HEPES (Gibco, 12340-030) supplemented with 10% FBS (Gibco), penicillin (100 U/ml) and streptomycin (100 μg/ml) (referred to as quiescence medium); and at day 6, cells were collected for analysis. For the analysis at different densities, at day 0, HUVECs were seeded in supplemented EGM2 at a density of 30,000 or 2500 cells/cm2 for dense or proliferating conditions, respectively, and analyzed at day 4. For in vitro gene silencing experiments, at day −1, HUVECs were seeded in supplemented EGM2 at a density of 15,000 cells/cm2, transduced overnight with pLKO lentiviral vectors coding for a short hairpin RNA against TNFSF10 (Sigma-Aldrich, TRCN0000005928) and the respective scramble as control vector at a multiplicity of infection of 20, and re-fed with fresh medium the next day.
Chemical inhibitors and cytokines
Inhibitors were, unless indicated otherwise, used in the following concentrations: pan-caspase inhibitor qVD (50 μM; Gentaur, 607-A1901), caspase-8–specific inhibitor zIETD (100 μM; Selleck Chemicals, S7314), sunitinib (1 μM; LC Laboratories, S-8803), IκB inhibitor IKK-16 (NF-κBi, 2.5 and 5 μM; Selleck Chemicals, S2882), p38 inhibitor SB203580 (p38i; 1 and 10 μM; Selleck Chemicals, S1076), MEK inhibitor pimasertib (1 μM; MedChemExpress, HY-12042), PI3K/mTOR inhibitor dactolisib (10 μM; MedChemExpress, HY-50673), and Akt inhibitor ipatasertib (10 μM; MedChemExpress, HY-15186A). Samples treated with vehicle only served as negative controls.
Cytokines were, unless indicated otherwise, used in the following concentrations: murine interleukin-2 (IL-2) (100 ng/ml; PeproTech, 212-12), murine IL-15 (50 ng/ml; PeproTech, 210-15), human VEGF-A (50 ng/ml; PeproTech, 100-20), human PlGF-1 (50 ng/ml; PeproTech, 100-06), human bFGF (10 ng/ml; PromoCell, C-39211), human IFN-γ (10 ng/ml; PeproTech, 300-02), human SuperKillerTRAIL [100 ng/ml for cell death assay and 1 μg/ml for coimmunoprecipitation (co-IP); Adipogen, AG-40 T-0002], human TNFα (20 ng/ml; PeproTech, 300-01A), human IL-1α (5 ng/ml; PeproTech, 200-01A), human IL-3 (20 ng/ml; PeproTech, 200-03), human IL-4 (20 ng/ml; PeproTech, 200-04), human IL-6 (20 ng/ml; PeproTech, 200-06), human IL-10 (150 ng/ml; PeproTech, 200-10), human IL-13 (25 ng/ml; PeproTech, 200-13), transforming growth factor–β (TGFβ) (20 ng/ml; PeproTech, 100-21).
Tnfsf10 mRNA production and formulation into LNPs
Tnfsf10 mRNAs were produced as described (63). Tnfsf10 was codon-optimized and cloned into a 101 adenosine tail (polyadenylate tail) containing in vitro transcription template. Tnfsf10 mRNA was nucleoside-modified (m1Ψ-5′-triphosphate) and cotranscriptionally capped using the trinucleotide CleanCap (TriLink) and precipitated and purified using cellulose chromatography (64). mRNA was analyzed by electrophoresis using native agarose gels and stored at −20°C. Cellulose-purified mRNAs were encapsulated in LNPs using a self-assembly process in which an aqueous solution of mRNA at pH 4.0 is rapidly mixed with a solution of lipids dissolved in ethanol (65). LNPs used in this study were similar in composition to those described previously (65, 66). The nanoparticles contained the commercially available ionizable cationic lipid ALC-0315 (Cayman Chemicals), phosphatidylcholine, cholesterol, and polyethylene glycol (PEG) lipid at molar ratio 50:10:38.5:1.5, respectively, and were encapsulated at an RNA to total lipid ratio of ~0.05 (w/w). The control LNP had no RNA included. LNPs had a diameter between 80 and 90 nm and a polydispersity index (PDI) of <0.2 as measured by dynamic light scattering using a Zetasizer Nano ZS (Malvern Instruments Ltd., Malvern, UK) instrument. mRNA-LNP formulations were stored at −80°C at a concentration of mRNA of ~1 mg/ml.
Preparation and characterization of targeted LNPs
For preparation of targeted LNP antibodies to murine CD31/platelet endothelial cell adhesion molecule 1 (PECAM1) (BD Biosciences, clone MEC13.3) were conjugated to nanoparticles via N-succinimidyl S-acetylthioacetate (SATA)–maleimide conjugation chemistry (44). The mRNA-LNP construct was modified with maleimide functioning group (DSPE-PEG-mal) by postinsertion technique with minor modifications (67). The antibody was functionalized with SATA (Sigma-Aldrich) and deprotected using hydroxylamine, followed by removal of the unreacted components by G-25 Sephadex Quick Spin Protein columns (Roche Applied Science, Indianapolis, IN). The reactive SH groups on the antibody were then conjugated to LNP maleimide. Size analysis of anti-CD31/Tnfsf10 mRNA-LNPs was performed using Malvern Zetasizer Nano ZS (Malvern Instruments Ltd., Malvern, UK). Antibody conjugation enlarged LNP diameter from 78.7 ± 0.4 nm (PDI, 0.016 ± 0.004) to 113.1 ± 1.1 nm (PDI, 0.137 ± 0.019).
Bone marrow transplantation
To generate bone marrow reciprocal chimeras, 5- to 8-week-old recipient mice of the appropriate murine strain were sublethally irradiated with 9.2 gray (Gy). Subsequently, 10 × 106 bone marrow cells from donor mice of the appropriate genotype were injected intravenously in the tail vein of recipient mice. Experimental metastasis experiments were initiated 5 weeks after bone marrow reconstitution.
Tumor experiments
A total of 106 4T1 or EMT6.5 cells in a volume of 50 μl of phosphate-buffered saline (PBS) were injected orthotopically to the right mammary fat pad (m.f.p.) of the second nipple. Tumor size was measured three times a week using a caliper and calculated using the formula: V = (π × d2 × D)/6, where d is the minor tumor axis and D is the major tumor axis. On the last day, tumor weight was registered and samples were collected for histological or FACS analysis. Lung metastatic nodules were visualized after intratracheal injection of 15% India ink solution. Superficial metastatic nodules were assessed under a stereomicroscope.
Experimental lung metastasis
A total of 100,000 4T1 or 250,000 E0771 cells in a volume of 200 μl of PBS were intravenously injected in the tail vein. For analysis of cancer cell burden at early time points, 4T1-Akaluc, 4T1-dLNGFR, E0771-Akaluc, E0771-dLNGFR, or E0771-dLNGFR-tdTomato were used. At the end stage (13 days for 4T1 or 12 days for E0771), lung metastatic nodules were contrasted after intratracheal injection of 15% India ink solution. Alternatively, lung tissue was collected after perfusion of animals with a saline through the right ventricle, and metastasis were assessed by hematoxylin and eosin staining on paraffin sections. For ex vivo quantification of cancer cell burden at early time points, lung tissue was collected after perfusion of animals with a saline through the right ventricle, and bioluminescence was measured in an IVIS Spectrum (detailed below), snap-frozen (to assess dLNGFR expression by qRT-PCR), digested (to perform colony formation assay), or fixed in 2% paraformaldehyde (PFA) (for whole-lung imaging). When indicated, a mixture of anti-ICAM1 (Bio X Cell, BE0020) and anti–E-Selectin (Bio X Cell, BE0294) blocking antibodies (10 mg/kg of each) or its corresponding immunoglubulin G2b (IgG2b) isotype control (Bio X Cell, BE0090) (20 mg/kg) were intravenously injected in the tail vein 3 hours before cancer cell injection. When indicated, anti-CD31–conjugated control LNPs (anti-CD31–LNP/empty) or anti-CD31–conjugated LNPs loaded with murine Trail mRNA (anti-CD31–LNP/Trail) (8 μg per mouse) were intravenously injected 48 and 24 hours before cancer cell injection.
Experimental liver metastasis
Mice were anesthetized and the spleen was exposed via an incision in the lateral flank. A total of 100,000 4T1-CD90.1 cells or 15,000 E0771-dLNGFR cells in a volume of 30 μl of PBS were slowly injected in the spleen, and cancer cells were allowed to circulate to the liver for 5 min. Afterward, the vasculature in the upper part of the spleen was ligated, the spleen was removed by cauterization, and the incisions were stitched. For the E0771 model, only male mice were used.
In vivo bioluminescence
Mice were injected intravenously with 4T1-Akaluc or E0771-Akaluc cells. For imaging at the indicated time points, mice were injected intraperitoneally with TokeOni Akalumine substrate (30 mg/kg; Sigma-Aldrich, 808350) dissolved in PBS (10 mM). After 10 min, the animals were perfused with saline through the right ventricle. Lungs were dissected positioned in the IVIS Spectrum (PerkinElmer), and images were acquired (medium binning, f stop = 1, time = 60 s). Bioluminescence images were analyzed using the LivingImage (PerkinElmer, Waltham, MA) processing software. Regions of interest (ROIs) were drawn around the bioluminescent signals, and measurements were generated as the total flux (in photons per second per square centimeter per steradian) from the selected ROIs. For analysis, ROIs for all lungs were kept identical, and the same ROI was overlaid on all lungs of every group to avoid differences in background signal contributing too much to the overall photon flux. We further calculated the specific Akaluc signal by subtracting the average values obtained for control animals (not receiving 4T1-Akaluc or E0771-Akaluc cells but still receiving Akalumine substrate) to the ROI region of experimental animals.
Colony formation assay
Animals were perfused with saline through the right ventricle. Lungs were dissected, minced, and collected in gentleMACS C tubes containing 5 ml of lung digestion buffer [DMEM high glucose supplemented with 1% penicillin-streptomycin (Gibco), collagenase II (1 mg/ml; Gibco, 17101015), collagenase IV (2.5 mg/ml; Worthington Biochemical, LS004188) and deoxyribonuclease I (DNase I) (40 U/ml; Sigma-Aldrich, D4527)] and automatically dissociated on the gentleMACS Dissociator (Miltenyi Biotec) with the program 37_m_LDK_1. Single-cell suspensions were washed in FACS buffer (PBS supplemented with 2% FBS and 2 mM EDTA), filtered through a 70-μm pore-sized mesh and centrifuged at 360g for 5 min. Cells were resuspended and plated in DMEM complete medium supplemented with 6-thioguanine (60 μM, Sigma-Aldrich, A4882) and plated in 10-cm petri dishes. After 6 days, the colonies were fixed with 4% PFA for 15 min, stained crystal violet (0.1 g/liter) in distilled H2O for 30 min, and counted.
In vivo administration of cancer cell conditioned medium
E0771 cancer cells (empty vector, VEGF164-OE, or PlGF-OE) were seeded in 10-cm petri dishes and allowed to grow for 24 to 72 hours. The medium was replaced by RPMI medium without glutamine and without phenol red (Gibco, 32404014), and cells were incubated for 48 hours. The same medium was incubated in parallel in 10-cm petri dishes without cancer cells to generate vehicle medium. Conditioned medium was collected and centrifuged at 3130g for 10 min. Supernatants were supplemented with Hepes (20 mM) and filtered (0.22 μm). Eight- to 10-week-old WT C57BL/6NTac mice were injected with 300 μl of conditioned medium (or PBS) daily in the tail vein 5 times/week and euthanized 17 days after the first injection. Animals were perfused with saline through the right ventricle. Lungs were dissected, minced, and collected for FACS processing.
Whole-lung clearing and immunostaining
Animals were perfused with saline through the right ventricle. Lungs were dissected and fixed in 2% PFA (pH 7.4) for 24 hours at 4°C. Next, lungs were cleared for 3 to 5 days using the FLASH tissue clearing method (68) with some modifications: Clearing steps took place before immunostaining to preserve tdTomato endogenous fluorescence, and primary antibody tissue penetration and dehydration steps were omitted. DAPI (4′,6-diamidino-2-phenylindole) (Thermo Fisher Scientific, D1306; 1:500) was incubated for 4 to 5 hours using an orbital shaker at room temperature (RT). Whole lungs were kept in PBS for short-term storage at 4°C or in refractive index–matched solution for at least 1 hour before imaging.
Whole-lung imaging
For whole-lung tissue imaging, lung pieces embedded in refractive index–matched solution were placed onto thin microscope coverslips (Duran Group, 235503704) and imaged using an inverted confocal microscope (TCS SP8, Leica Microsystems) with a 25× water objective (long working distance; numerical aperture, 0.95) and equipped spectral hybrid detectors. To localize metastatic cells, a xy overview was generated for each lung piece. Next, metastatic cells were imaged in 3D with more detail by generating detailed xyz tile scans with Z-step size of 0.5 to 1 μm. DAPI signal was excited with a 405-nm laser and collected between 410 and 500 nm, and tdTomato was excited with a 561-nm laser and collected between 570 and 640 nm. All images were collected at 12-bit depth with 1024 by 1024 pixels per tile. Images were processed using the LasX 3D visualization software module (Leica Microsystems), and signal intensities were linearly adjusted for visualization purposes. The number of tdTomato+ metastatic cells was counted using confocal images obtained from lung overviews. Total imaged area of each lung was used to normalize the number of metastatic cells per square millimeter.
Histological analysis and immunostainings of murine tissue
For serial sections cut at 7 μm in thickness, tissue samples were fixed in 2% PFA overnight at 4°C, dehydrated, and embedded in paraffin. Paraffin slides were first rehydrated to further proceed with antigen retrieval in citrate solution (DAKO, S1699) at 100°C for 20 min. If necessary, then 0.1% hydrogen peroxide was added to methanol to block endogenous peroxidases. The sections were blocked with the appropriate serum (DAKO) and incubated overnight with the following antibodies: rat anti-CD31 (BD Pharmingen, 550274; 1:50), rat anti-F4/80 (Serotec, MCA497F; 1:100), rabbit anti-pimonidazole (Hypoxyprobe, PAb2627AP; 1:100). Appropriate secondary antibodies were used: Alexa Fluor 488–, Alexa Fluor 647–, or Alexa Fluor 568–conjugated secondary antibodies (Molecular Probes; 1:200), biotin-labeled antibodies (Jackson ImmunoResearch; 1:300) and, when necessary, tyramide signal amplification (TSA) system amplification [fluorescein, Cy3, or biotin/3,3′-diaminobenzidine (DAB)] (PerkinElmer, Life Sciences) were performed according to the manufacturer’s instructions. Hoechst solution (Thermo Fisher Scientific, H3570; 1:1000) was used to visualize nuclei. Whenever sections were stained in fluorescence, ProLong Gold Antifade Mountant without DAPI (Invitrogen, P36930) was used. Microscopic analysis was done with an Olympus BX41 microscope and CellSense imaging software.
Hypoxia assessment
Tumor hypoxia was detected by injecting intraperitoneally pimonidazole hydrochloride (60 mg/kg; Hypoxyprobe, HP3-100KIT) into tumor-bearing mice 1 hour before mice were euthanized and tumors were harvested. To detect the formation of pimonidazole adducts, tumor formalin-fixed paraffin-embedded (FFPE) sections were immunostained with anti-pimonidazole (Hypoxyprobe, PAb2627AP; 1:100) according to the manufacturer’s instructions.
Patient selection and sample collection
FFPE samples from metastases and normal lung tissue were obtained through the ethically approved UPTIDER program (UZ/KU Leuven Program for Post-mortem Tissue Donation to Enhance Research, NCT04531696, S64410). In this project, patients with metastatic breast cancer that consent to participate undergo a rapid research autopsy in the first 12 hours after death. Briefly, upon death of a participating patient the body is transported to the morgue of the UZ Leuven hospital. Several types of body fluids are collected, as well as extensive malignant and adjacent normal tissue samples from different organs in the body, including the ones that are difficult to reach during the life of the patient. Tissue samples are processed under different conditions (snap-frozen in liquid nitrogen, frozen in optimal cutting temperature compound, collected in neutral-buffered formalin for further processing in FFPE blocks, or fresh in specific medium for in vivo development). For our experiments described in this paper, FFPE samples were used. The UPTIDER project is coordinated and performed by the Laboratory for Translational Breast Cancer Research at our institution, KU Leuven, under the lead of Prof. C. Desmedt and Prof. Dr. G. Floris.
Histological analysis and immunostainings of human tissue
Lung tissue adjacent to breast cancer lung metastasis and the corresponding lung metastasis samples was obtained from a patient from the postmortem tissue donation program UPTIDER (UZ/KU Leuven Post-mortem Tissue Donation program to Enhance Research, https://clinicaltrials.gov/ct2/show/NCT04531696). Immunofluorescent staining of FFPE was performed on a LabSat device (Lunaphore Technologies SA). Preprocessing steps such as dewaxing was performed on Leica ST5010 Autostainer XL device. Antigen retrieval was subsequently performed on LabSat, using the proprietary antigen retrieval buffer of pH 9. TRAIL staining was performed using anti-human TRAIL antibody (R&D, AF375; 60 μg/ml) for 10 min, followed by 2-min incubation with Alexa Fluor 647 AffiniPure donkey anti-goat IgG (H + L) (Jackson ImmunoResearch, 705-605-147; 1:420). CD31 staining was performed using anti-human PECAM-1/CD31 antibody (LifeSpan BioSciences, LS-B16850-50, 1:50) for 10 min, followed by 2-min incubation with Alexa Fluor 555 donkey anti-mouse IgG (H + L) highly cross-adsorbed antibody (Invitrogen, A-31570; 1:300). Scanning was performed with Zeiss Axio scan Z1 with adapted scanning profile.
Immunofluorescent images were analyzed using QuPath (69). Five regions per lesion were selected for quantification of signal. Regions with unspecific staining and debris were avoided. Within the region, the endothelium of the small vessel was delineated by polygon annotation tool. Intensity of TRAIL in Cy5 channel was calculated using the built-in “compute intensity features” module. Settings were selected with a pixel size of 1 μm and a tile diameter of 25 μm. The autofluorescent signal of the respective region was calculated in the exact same way and subtracted from the TRAIL intensity.
Immunofluorescence of HUVECs
Eighteen-millimeter glass coverslips were coated with 0.2% gelatin for 1 hour at RT. Subsequently, gelatin-coated coverslips were exposed to 2.5% glutaraldehyde in PBS for 10 min at RT, followed by 70% ethanol for 30 min at RT. After washing with PBS, active aldehyde groups were neutralized with 2 mM glycine in PBS overnight at 37°C. Glycine was removed and the coverslips were washed with PBS before seeding the HUVECs. HUVECs were fixed in 4% PFA for 10 min at 4°C and blocked and permeabilized with blocking buffer [PBS supplemented with 0.3% Triton X-100 and 3% bovine serum albumin (BSA) fraction V] for 1 hour at RT. The following primary antibodies diluted in blocking buffer were incubated overnight at 4°C: goat anti-human TRAIL (R&D, AF375; 1:25), rabbit anti–Ki-67 (Abcam, ab15580; 1:1000) and mouse anti–NF-κB (Cell Signaling Technology, 6956; 1:400). Appropriate secondary antibodies were used: Alexa Fluor 488–, Alexa Fluor 647–, or Alexa Fluor 568–conjugated secondary antibodies (Molecular Probes; 1:200). Hoechst solution (Thermo Fisher Scientific, H3570; 1:1000) was used to visualize nuclei. Coverslips were mounted with ProLong Gold Antifade Mountant without DAPI (Invitrogen, P36930). Microscopic analysis was done with an Olympus BX41 microscope and CellSense imaging software.
Proximity ligation assay
HUVECs grown in 0.2% gelatin-coated coverslips (described above) were fixed in 4% PFA for 10 min at 4°C and permeabilized with PBS supplemented with 0.3% Triton X-100 for 10 min at 4°C. For the proximity ligation assay (PLA) of TRAIL and DR5, the Duolink In Situ Red Starter Kit Goat/Rabbit (Sigma-Aldrich, DUO92105) was utilized according to the manufacturer’s protocol, which was combined with immunofluorescence of VE-cadherin. The following primary antibodies were incubated at 4°C overnight: goat anti-human TRAIL (R&D, AF375; 1:25), rabbit anti-human DR5 (Cell Signaling Technology, 8074; 1:50), and mouse anti–VE-cadherin (BD Biosciences, 610252; 1:100). Donkey anti-mouse Alexa Fluor 488 (Molecular Probes; 1:200) was incubated together with the anti-goat and anti-rabbit PLA probes. Microscopic analysis was done with an Olympus BX41 microscope and CellSense imaging software.
Flow cytometry (FACS) analysis and cell sorting
Before dissecting the lungs, animals were perfused with saline through the right ventricle. Lungs were dissected, minced, and collected in gentleMACS C tubes containing 5 ml of lung digestion buffer [DMEM high glucose supplemented with 1% penicillin-streptomycin (Gibco), collagenase II (1 mg/ml; Gibco, 17101015), collagenase IV (2.5 mg/ml; Worthington Biochemical, LS004188), and DNase I (40 U/ml; Sigma-Aldrich, D4527)] and automatically dissociated on the gentleMACS Dissociator (Miltenyi Biotec) with the program 37_m_LDK_1. Tumors were minced and collected in gentleMACS C tubes containing 5 ml of tumor digestion buffer [RPMI 1640 supplemented with 10% FBS (Gibco), 1% l-glutamine (Gibco), 1% penicillin-streptomycin (Gibco), collagenase I (1 mg/ml; Gibco, 17100017), Dispase (2 mg/ml; Gibco, 17105041), and DNase I (0.2 mg/ml; Roche, 11284932001)] and automatically dissociated on the gentleMACS Dissociator (Miltenyi Biotec) with the program 37_m_TDK_2 (4T1) or 37_m_TDK_1 (E0771). Livers were dissected, mince, and collected in 50-ml Falcon tubes containing 5 ml of liver digestion buffer [DMEM high glucose supplemented with 1% penicillin-streptomycin (Gibco), 1× sodium pyruvate (Gibco), 1× MEM nonessential amino acid solution (Gibco), collagenase I (1 mg/ml; Gibco, 17100017), collagenase II (1 mg/ml; Gibco, 17101015), Dispase (2.5 U/ml; Gibco, 17105041), and DNase I (20 U/ml; Sigma-Aldrich, D4527)] and placed at 37°C for 30 min with regular vortexing to allow dissociation. Single-cell suspensions were washed in FACS buffer (PBS supplemented with 2% FBS and 2 mM EDTA), filtered through a 70-μm pore-sized mesh, and centrifuged at 360g for 5 min. Red blood cell (RBC) lysis was performed using Hybri-Max (Sigma-Aldrich, R7757) for 3 min at 37°C, subsequently washed in FACS buffer, filtered through a 40-μm pore-sized mesh, and centrifuged at 360g for 5 min. Cells were resuspended in FACS buffer, counted and incubated for 15 min at 4°C with mouse BD Fc block (BD Biosciences, 553142), and stained with the appropriate fluorochrome-conjugated antibodies and viability dye (Invitrogen, 65-0866-14) for 30 min at 4°C, protected from light (details on the FACS panels are available upon request). For intracellular staining, after staining for surface antigens and viability, washed cells were fixed and permeabilized (Invitrogen, 00-5523-00) for 30 min at 4°C and incubated with the fluorochrome-conjugated antibody overnight. Cells were washed and either resuspended in FACS buffer and sorted with a FACSAria III (BD Biosciences) or fixed in a 1:1 mixture of IC fixation buffer (eBioscience, 00-8222-49) and FACS buffer and analyzed with a BD Fortessa X-20 (BD Biosciences) or a FACSCanto II (BD Biosciences). For absolute cell counts, precision count beads (BioLegend, 424902) were added by reverse pipetting before analysis. FMO (fluorescence minus one) controls and isotype controls (where the antibody is substituted with its corresponding fluorochrome-conjugated isotype control, in the presence of all the other antibodies and viability dye) were prepared when required to ensure proper gating of positive populations. Plotted data represent median fluorescence intensity (MFI) of the stained samples after subtracting the appropriate control (FMO or isotype for extracellular or intracellular markers, respectively).
For cell death analysis, supernatants of HUVECs cultures (containing dead cells) were pooled with HUVECs collected with trypsin. Subsequently, cells were washed in annexin V binding buffer (BioLegend, 422201) and stained with allophycocyanine (APC)-conjugated annexin V (BioLegend, 640941; 1:50) and propidium iodide (Sigma-Aldrich, P4864; 1:2000) for 15 min. Stained samples were further diluted with annexin V binding buffer and immediately analyzed with a BD Fortessa X-20 (BD Biosciences). Flow cytometry data were analyzed with FlowJo 10 software (Tree Star).
RNA-seq of lung ECs
RNA from 2 million sorted lung ECs (viability dye− CD45− CD31+) from EC-specific TRAIL WT and KO mice (Cdh5.iCreERT2xTnfsf10Lox/Lox) was isolated, and the concentration, as well as purity, was determined spectrophotometrically using the Nanodrop ND-1000 (Nanodrop Technologies).
Per sample, an amount of 3000 ng of total RNA was used as input. Subsequently, preparation was performed with KAPA stranded mRNA-seq according to the protocol, and samples were pooled equimolar. Sequencing was performed as single end of 50 bp on HiSeq 4000 from Illumina. Thirty-five to 40 million reads were generated per sample.
Gene reads counts were estimated by RNA-Seq by Expectation-Maximization (RSEM) using GENCODE reference annotation of the mouse genome (GRCm38), version M24 (Ensembl 99). Differential expression of EC-specific TRAIL KO (ECΔT10) versus control (ECW/W) was performed using the DESeq2 R package (v1.26.0). The significance was determined by padj < 0.05. The GSEA was performed using the preranked gene list by log2 fold change (ECΔT10 versus ECW/W control) and the MSigDB gene sets. Only gene sets containing between 5 and 500 genes were retained, and the number of permutations was set at 1000.
RNA extraction, cDNA synthesis, and qRT-PCR
Cultured cells were collected in RLT lysis buffer containing 1% β-mercaptoethanol and RNA was isolated using RNeasy Micro or Mini kits (QIAGEN, 74004 and 74106, respectively). Lung tissue was ribolysed in TRIzol, extracted with chloroform, and subsequently isolated using RNeasy Mini kit (QIAGEN, 74004). Reverse transcription to cDNA was performed using SuperScript III First-Strand Synthesis System (Invitrogen, 18080051), for sorted cells, or the Quantitect Reverse Transcription Kit (QIAGEN, 205314), for cultured cells and tissue, according to the manufacturer’s instructions. qRT-PCR amplification was performed using commercial or homemade primers (IDT) in the QuantStudio 12K Flex Real-Time PCR system (Applied Biosystems). Hypoxanthine-guanine phosphoribosyltransferase (HPRT) or β-actin was used as housekeeping genes.
Co-IP of endogenous TRAIL and DR5
Co-IP was performed using the Co-Immunoprecipitation Kit (Thermo Fisher Scientific, 26149) and according to the manufacturer’s instructions. Briefly, AminoLink Plus Coupling Resin was coupled with anti-human TRAIL (R&D, AF375), anti-human DR5 (R&D, AF631), or normal goat IgG (R&D, AB-108-C) antibodies at a proportion of 1.3 mg of antibody/mg of protein according to the manufacturer’s instructions. HUVECs [untreated or treated with SuperKillerTRAIL (1 μg/ml) for 30 min] were lysed for 5 min on ice with IP lysis buffer (provided in the kit) [25 mM tris, 0.15 M NaCl, 1 mM EDTA, 1% NP-40, and 5% glycerol (pH 7.4)] supplemented with Halt Protease and Phosphatase Inhibitor Cocktails (Thermo Fisher Scientific, 78440). After centrifugation at 13,000g for 10 min at 4°C, the supernatant was recovered and protein concentration was measured by the bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific, 23225) according to the manufacturer’s instructions. Protein extracts were precleared with the control agarose resin and subsequently incubated overnight at 4°C with the antibody-coupled AminoLink Plus Coupling Resin. After washing unbound proteins, resin-bound proteins were eluted in elution buffer (pH 2.8). Protein lysates were denatured by adding lane marker sample buffer 5× [0.3 M tris-HCl, 5% SDS, 50% glycerol, lane marker tracking dye, and 5% β-mercaptoethanol (pH 6.8)] and incubating at 95°C for 5 min and analyzed by Western blot.
Cell surface protein isolation
Cell surface protein isolation was performed using the Cell Surface Protein Isolation Kit (Thermo Fisher Scientific, 89881) and according to the manufacturer’s instructions. Briefly, surface HUVEC monolayers were biotinylated for 30 min at 4°C to label surface proteins. Subsequently, the reaction was quenched and HUVECs were harvested, pelleted, and lysed for 30 min on ice in lysis buffer (provided in the kit) supplemented with cOmplete Mini protease inhibitor (Roche, 11836145001) and PhosSTOP phosphatase inhibitor (Roche, 04906837001). After centrifugation at 10,000g for 2 min at 4°C, the supernatant was recovered and incubated for 60 min at RT with NeutrAvidin Agarose. The flow-through containing unbound intracellular proteins was collected by centrifugation, the NeutrAvidin Agarose was washed, and biotinylated surface proteins were eluted in SDS–polyacrylamide gel electrophoresis (PAGE) sample buffer supplemented with 50 mM dithiothreitol (DTT) and analyzed by Western blot.
Protein precipitation from supernatants
Protein precipitation from filtered HUVECs’ supernatants was performed by mixing supernatant:methanol:chloroform (4:4:1). After centrifugation at 18,000g for 5 min at RT, an interphase containing the protein was formed. The upper aqueous phase was removed, and the remaining lower organic phase and interphase were mixed with the same initial volume of methanol. After centrifugation at 18,000g for 5 min at RT, the supernatant was discarded and the protein pellet was dried, dissolved in 1× SDS Laemmli buffer (Bio-Rad, 1610737), and analyzed by Western blot.
Protein extraction and Western blot
Protein extraction from murine lungs was performed by ribolysing the perfused left lobe in radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific, 89900) supplemented with cOmplete Mini protease inhibitor (Roche, 11836145001) and PhosSTOP phosphatase inhibitor (Roche, 04906837001). Lung lysates were incubated on ice for 15 min before centrifuging at 17,000g for 15 min at 4°C to remove cellular debris. Supernatants were subsequently collected. Whole-cell protein extraction from HUVECs was performed using a modified Laemmli extraction buffer [62.5 mM tris-HCl (pH 6.8), 10% glycerol, and 2% SDS] supplemented with cOmplete Mini protease inhibitor (Roche, 11836145001) and PhosSTOP phosphatase inhibitor (Roche, 04906837001). Membranes and DNA were disrupted by passing the protein extracts through a 29-gauge syringe. Protein concentration was measured by the BCA protein assay (Thermo Fisher Scientific, 23225) according to the manufacturer’s instructions. Protein samples (20 to 50 μg) were denatured by adding reducing loading buffer and incubating at 95°C for 5 min. Proteins were resolved by SDS-PAGE and transferred to nitrocellulose membrane Trans-Blot Turbo Midi 0.2 μm Nitrocellulose (Bio-Rad, 1704159) using Trans-Blot Turbo Transfer System (Bio-Rad). Nonspecific binding was blocked in PBS with 0.1% Tween 20 (PBST) containing 5% nonfat dry milk (Cell Signaling Technology, 9999). Subsequently, membranes were incubated overnight at 4°C with the following primary antibodies diluted in PBST containing 5% BSA or nonfat dry milk (according to the manufacturer’s recommendations): human TRAIL (Cell Signaling Technology, 3219; 1:1000), murine TRAIL (R&D, AF1121; 1:500), murine DcR2 (R&D, MAB1816; 1:500), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Cell Signaling Technology, 2218; 1:1000), VE-cadherin (BD Biosciences, 610252; 1:1000), tubulin–horseradish peroxidase (HRP) (Abcam, ab21058; 1:2000), pro–caspase-3 (Cell Signaling Technology, 9668; 1:1000), cleaved caspase-3 (Cell Signaling Technology, 9116; 1:1000), pro–caspase-8 (Cell Signaling Technology, 4790 or 9746; 1:1000), cleaved caspase-8 (Cell Signaling Technology, 9496; 1:1000), vinculin (Sigma-Aldrich, V9131; 1:2000), ICAM1 (Cell Signaling Technology, 67836; 1:1000), E-Selectin (Santa Cruz Biotechnology, sc-137054; 1:800), p38 ph-Thr180/Tyr182 (Cell Signaling Technology, 4511; 1:1000), p38 (Cell Signaling Technology, 8690; 1:1000), IκBα ph-Ser32/36 (Cell Signaling Technology, 9246; 1:1000), IκBα (Cell Signaling Technology, 4814; 1:1000), DR4 (Cell Signaling Technology, 42533; 1:1000), DR5 (Cell Signaling Technology, 8074; 1:1000), IKKα (Cell Signaling Technology, 2682; 1:1000), IKKβ (Cell Signaling Technology, 8943; 1:1000), RIP1 (Cell Signaling Technology, 3493; 1:1000), NEMO (BD Biosciences, 611306; 1:1000), and FADD (Cell Signaling Technology, 2782; 1:1000). The day after, membranes were incubated for 1 hour at RT with appropriate HRP-conjugated secondary antibodies (Cell Signaling Technology, 7074 or 7076; 1:2000; and DAKO, P044901-2; 1:2000). Signal was visualized by Novex ECL Chemiluminescent Substrate (Invitrogen, WP20005) or SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, 34096) and acquired by a LAS 4000 charge-coupled device camera with ImageQuant software (GE Healthcare). Tubulin, GAPDH, or vinculin are shown as loading controls.
Live-cell imaging
To assess cell viability over time, HUVECs were seededin 96-well black plates with clear bottom (VWR, 734-1609) for in vitro gene silencing as described above. At the indicated time, culture medium was replaced by culture medium containing 250 nM cytotox green reagent (Essen BioScience, 4633) in the presence or the absence of the pan-caspase inhibitor qVD (50 μM; Gentaur, 607-A1901) or the caspase-8–specific inhibitor zIETD (100 μM; Selleck Chemicals, S7314). The cells were then incubated and monitored by the IncuCyte live-cell imaging system (Essen BioScience), and images were acquired every 2 hours. Cytotox+ area per well was analyzed over time by the IncuCyte ZOOM software.
Transendothelial electrical resistance
TEER was measured as described in (45). Briefly, 25,000 shSCR or shTRAIL HUVECs were seeded in EGM2 on 0.1% gelatin-coated 6.5-mm polyester transwells with 0.4-mm pore size (Corning, CLS3470). The TEER was measured using the Endohm-6 electrode (World Precisions Instruments) connected to an EVOM2 voltohmmeter (World Precisions Instruments). Gelatin-coated wells without cells were used to measure the intrinsic electrical resistance of the inserts, and these values were then subtracted to the values measured in the presence of cells. Measurements were performed every day, taking three measurements per construct.
Cancer cell adhesion assay
HUVECs were seeded in 96-well plates for in vitro gene silencing as described above, and the adhesion assay was performed on day 6. E0771-dLNGFR-tdTomato or 4T1-tdTomato cells were harvested, counted, and resuspended in M199 medium (Gibco, 12340-030) supplemented with 1% BSA. HUVECs’ supernatant was completely removed and cancer cells were placed on top (40,000 cells per well) and allowed to adhere for 1 hour (37°C and 5%CO2). Nonadherent cells were removed by washing two times with PBS. The adhered cancer cells and HUVECs were collected with trypsin, transferred to a V-bottom 96-well plate, centrifuged at 360g for 5 min at 4°C, and fixed in a 1:1 mixture of IC fixation buffer (eBioscience, 00-8222-49) and FACS buffer by reverse pipetting. Precision count beads (BioLegend, 424902) were added by reverse pipetting before analysis of tdTomato+ cells with a Fortessa LSR-II (BD Biosciences).
Leukocyte adhesion assay
HUVECs were seeded in 96-well plates for in vitro gene silencing as described above, and the adhesion assay was performed on day 6. Blood was collected by cardiac puncture and kept in lithium heparin Microtainer tubes (BD Biosciences, 365965). To isolate white blood cells (WBCs), the blood was diluted in 1.25% dextran in saline to allow the sedimentation of RBCs. After 30 min, the erythrocyte-poor upper layer was collected and washed in PBS supplemented with 0.1% BSA. The remaining RBCs were lysed in a hypotonic solution of 0.2% NaCl for 30 s and brought under isotonic condition with 1.6% NaCl and 0.1% glucose. WBCs were washed in PBS supplemented with 0.1% BSA, counted, and resuspended in DMEM complete. HUVECs’ supernatant was completely removed, and WBCs were placed on top (100,000 cells per well) and allowed to adhere for 1 hour (37°C and 5% CO2). Nonadherent cells were removed by washing two times with PBS. The adhered WBCs and HUVECs were collected with trypsin, transferred to a V-bottom 96-well plate, stained for CD45, centrifuged at 360g for 5 min at 4°C, and fixed in a 1:1 mixture of IC fixation buffer (eBioscience, 00-8222-49) and FACS buffer by reverse pipetting. Precision count beads (BioLegend, 424902) were added by reverse pipetting before analysis of tdTomato+ cells with a Fortessa LSR-II (BD Biosciences).
Statistical analysis
All statistical analysis was performed using GraphPad Prism 9. Unless otherwise stated in the figure legend, statistical significance was calculated by two-tailed unpaired t test on two experimental conditions or two-way analysis of variance (ANOVA) when more than two experimental groups were compared. Outlier test was performed. All data are represented as means ± SEM. P values are indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Acknowledgments
We thank I. Tassaert, S. Appelmans, Á. González Merino, B. Pérez González, J. Verelst, E. Vandeputte, A. Jacquemotte, J. Diender, S. Willox, S. Trusso, E. Achten, R. Kroes, S. Vinckier, M. Declerq, O. Marín-Béjar, and R. Faria for technical help. We thank Prof. M. Smyth (Peter MacCallum Cancer Centre, Australia) and Prof. J. Weiss (The National Cancer Institute, USA) for providing the constitutive Tnfsf10-deficient mice, Prof. E. Vivier (Centre d’Immunologie de Marseille-Luminy, France) for providing the Nkp46.iCre mice (62), and Prof. H. Walczak (Deutsches Krebsforschungszentrum, Germany) for providing the Tnfrsf10bLox/Lox mice (9). We thank Prof. J. Van Rheenen (Netherlands Cancer Institute, Netherlands) for providing the Addgene plasmid #27353 encoding for tdTomato. We thank S. Vlayen for administrative support. We thank the patients, their families and all the multidisciplinary team from KU Leuven and UZ Leuven.
Funding: C.R.-D. was supported by an FWO doctoral fellowship (1108917 N), and R.M.-P. was supported by an FWO postdoctoral fellowship (12N4915N). G.C. was supported by FIRC/AIRC fellowships (grant no. 25254), AIRC Short-Term Fellowships, and FEBS Short-Term Fellowship. The postmortem tissue donation program UPTIDER is supported by a grant from the University Hospitals from Leuven (KOOR 2021), as well as a C1 grant (14/21/114) from KU Leuven. M.D.S. is supported by the Nadine de Beauffort Funds. M.M. received an ERC consolidator grant (773208), FWO grant (G0D1617N), and funds from the VIB TechWatch team.
Author contributions: C.R.-D. performed the experimental design, acquisition, analysis, and interpretation of all data and wrote the manuscript. E.L.-G., I.C., G.C., and F.C. assisted with conducting experiments and data analysis. P.Z. analyzed (sc)RNA-seq data. L.M. performed whole-lung immunostaining, imaging, and quantification. M.D.S., T.G., and E.I. performed the analysis of human samples from UPTIDER. D.L. assisted with FACS sorting and in vivo experiments and provided technical support. J.S. assisted with tail vein injections, FACS sorting, and in vivo experiments and provided technical support. M.-G.A. and V.V.S. conceptualized, planned, and provided LNPs. H.P. provided the data interpretation. D.W. and V.R.M. conceptualized, developed, and provided LNPs. S.S. provided reagents and assisted in the Akaluc bioluminescence experiments. C.D. provided and performed analysis of human samples from UPTIDER and gave scientific input. C.L.G.J.S. performed whole-lung immunostaining, imaging, and quantification and gave scientific input. A.S. gave scientific input on RNA-seq data. M.D.M. performed all the cloning and assisted in generating the constitutive ubiquitous Tnfrsf22/23 double KO mice. R.M.-P. performed the experimental design, acquisition, analysis and interpretation of data, conducted the scientific direction, and wrote the manuscript. M.M. supported financially the work, performed the experimental design and data interpretation, conducted scientific direction, and wrote the manuscript.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) data repository with the accession number GSE199461. The samples and data from human patients with breast cancer under the UPTIDER program can be provided by KU Leuven/C. Desmedt pending scientific review of the proposal, ethical committee approval, and a completed material/data transfer agreement. Requests for these samples should be submitted to C. Desmedt or M. Maetens (https://gbiomed.kuleuven.be/english/research/50488876/54887282). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
Supplementary Materials
This PDF file includes:
Fig. S1 to S11
REFERENCES AND NOTES
- 1.P. S. Steeg, Targeting metastasis. Nat. Rev. Cancer 16, 201–218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.L. Bejarano, M. J. C. Jordāo, J. A. Joyce, Therapeutic targeting of the tumor microenvironment. Cancer Discov. 11, 933–959 (2021). [DOI] [PubMed] [Google Scholar]
- 3.J. Fares, M. Y. Fares, H. H. Khachfe, H. A. Salhab, Y. Fares, Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 5, 28 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.N. Wettschureck, B. Strilic, S. Offermanns, Passing the vascular barrier: Endothelial signaling processes controlling extravasation. Physiol. Rev. 99, 1467–1525 (2019). [DOI] [PubMed] [Google Scholar]
- 5.D. Hanahan, Hallmarks of cancer: New dimensions. Cancer Discov. 12, 31–46 (2022). [DOI] [PubMed] [Google Scholar]
- 6.H. Peinado, H. Zhang, I. R. Matei, B. Costa-Silva, A. Hoshino, G. Rodrigues, B. Psaila, R. N. Kaplan, J. F. Bromberg, Y. Kang, M. J. Bissell, T. R. Cox, A. J. Giaccia, J. T. Erler, S. Hiratsuka, C. M. Ghajar, D. Lyden, Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 17, 302–317 (2017). [DOI] [PubMed] [Google Scholar]
- 7.K. Takeda, Y. Hayakawa, M. J. Smyth, N. Kayagaki, N. Yamaguchi, S. Kakuta, Y. Iwakura, H. Yagita, K. Okumura, Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat. Med. 7, 94–100 (2001). [DOI] [PubMed] [Google Scholar]
- 8.E. Cretney, K. Takeda, H. Yagita, M. Glaccum, J. J. Peschon, M. J. Smyth, Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J. Immunol. 168, 1356–1361 (2002). [DOI] [PubMed] [Google Scholar]
- 9.A. Grosse-Wilde, O. Voloshanenko, S. Lawrence Bailey, G. M. Longton, U. Schaefer, A. I. Csernok, G. Schütz, E. F. Greiner, C. J. Kemp, H. Walczak, TRAIL-R deficiency in mice enhances lymph node metastasis without affecting primary tumor development. J. Clin. Investig. 118, 100–110 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.H. Walczak, R. E. Miller, K. Ariail, B. Gliniak, T. S. Griffith, M. Kubin, W. Chin, J. Jones, A. Woodward, T. Le, C. Smith, P. Smolak, R. G. Goodwin, C. T. Rauch, J. C. Schuh, D. H. Lynch, Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 5, 157–163 (1999). [DOI] [PubMed] [Google Scholar]
- 11.A. Ashkenazi, R. C. Pai, S. Fong, S. Leung, D. A. Lawrence, S. A. Marsters, C. Blackie, L. Chang, A. E. McMurtrey, A. Hebert, L. DeForge, I. L. Koumenis, D. Lewis, L. Harris, J. Bussiere, H. Koeppen, Z. Shahrokh, R. H. Schwall, Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Invest. 104, 155–162 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.M. J. Smyth, E. Cretney, K. Takeda, R. H. Wiltrout, L. M. Sedger, N. Kayagaki, H. Yagita, K. Okumura, Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon γ-dependent natural killer cell protection from tumor metastasis. J. Exp. Med. 193, 661–670 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.J. Lemke, S. von Karstedt, J. Zinngrebe, H. Walczak, Getting TRAIL back on track for cancer therapy. Cell Death Differ. 21, 1350–1364 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.S. von Karstedt, A. Montinaro, H. Walczak, Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 17, 352–366 (2017). [DOI] [PubMed] [Google Scholar]
- 15.S. von Karstedt, H. Walczak, An unexpected turn of fortune: Targeting TRAIL-Rs in KRAS-driven cancer. Cell Death Discov. 6, 14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.M. Snajdauf, K. Havlova, J. Vachtenheim, A. Ozaniak, R. Lischke, J. Bartunkova, D. Smrz, Z. Strizova, The TRAIL in the treatment of human cancer: An update on clinical trials. Front. Mol. Biosci. 8, 628332 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.T. Hartwig, A. Montinaro, S. von Karstedt, A. Sevko, S. Surinova, A. Chakravarthy, L. Taraborrelli, P. Draber, E. Lafont, F. Arce Vargas, M. A. El-Bahrawy, S. A. Quezada, H. Walczak, The TRAIL-induced cancer secretome promotes a tumor-supportive immune microenvironment via CCR2. Mol. Cell 65, 730–742.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.S. Von Karstedt, A. Conti, O. J. Sansom, H. Walczak, Cancer cell-autonomous TRAIL-R signaling promotes KRAS-Driven cancer progression, invasion, and metastasis. Cancer Cell 27, 561–573 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.G. P. Sullivan, H. O’Connor, C. M. Henry, P. Davidovich, D. M. Clancy, M. L. Albert, S. P. Cullen, S. J. Martin, TRAIL receptors serve as stress-associated molecular patterns to promote ER-stress-induced inflammation. Dev. Cell 52, 714–730.e5 (2020). [DOI] [PubMed] [Google Scholar]
- 20.K. Azijli, B. Weyhenmeyer, G. J. Peters, S. De Jong, F. A. E. Kruyt, Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: Discord in the death receptor family. Cell Death Differ. 20, 858–868 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.C. M. Henry, S. J. Martin, Caspase-8 acts in a non-enzymatic role as a scaffold for assembly of a pro-inflammatory “FADDosome” complex upon TRAIL stimulation. Mol. Cell 65, 715–729.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 22.L. Cardoso Alves, N. Corazza, O. Micheau, P. Krebs, The multifaceted role of TRAIL signaling in cancer and immunity. FEBS J. 288, 5530–5554 (2021). [DOI] [PubMed] [Google Scholar]
- 23.P. Schneider, D. Olson, A. Tardivel, B. Browning, A. Lugovskoy, D. Gong, M. Dobles, S. Hertig, K. Hofmann, H. Van Vlijmen, Y.-M. Hsu, L. C. Burkly, J. Tschopp, T. S. Zheng, Identification of a new murine tumor necrosis factor receptor locus that contains two novel murine receptors for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J. Biol. Chem. 278, 5444–5454 (2003). [DOI] [PubMed] [Google Scholar]
- 24.G. Pan, J. Ni, Y. F. Wei, G. I. Yu, R. Gentz, V. M. Dixit, An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 277, 815–818 (1997). [DOI] [PubMed] [Google Scholar]
- 25.J. P. Sheridan, S. A. Marsters, R. M. Pitti, A. Gurney, M. Skubatch, D. Baldwin, L. Ramakrishnan, C. L. Gray, K. Baker, W. I. Wood, A. D. Goddard, P. Godowski, A. Ashkenazi, Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 277, 818–821 (1997). [DOI] [PubMed] [Google Scholar]
- 26.D. Mérino, N. Lalaoui, A. Morizot, P. Schneider, E. Solary, O. Micheau, Differential inhibition of TRAIL-mediated DR5-DISC formation by decoy receptors 1 and 2. Mol. Cell. Biol. 26, 7046–7055 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.M. Liguori, C. Buracchi, F. Pasqualini, F. Bergomas, S. Pesce, M. Sironi, F. Grizzi, A. Mantovani, C. Belgiovine, P. Allavena, Functional TRAIL receptors in monocytes and tumor-associated macrophages: A possible targeting pathway in the tumor microenvironment. Oncotarget 7, 41662–41676 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.L. O’Leary, A. M. van der Sloot, C. R. Reis, S. Deegan, A. E. Ryan, S. P. S. Dhami, L. S. Murillo, R. H. Cool, P. C. de Sampaio, K. Thompson, G. Murphy, W. J. Quax, L. Serrano, A. Samali, E. Szegezdi, Decoy receptors block TRAIL sensitivity at a supracellular level: The role of stromal cells in controlling tumour TRAIL sensitivity. Oncogene 35, 1261–1270 (2016). [DOI] [PubMed] [Google Scholar]
- 29.G. Germano, R. Frapolli, C. Belgiovine, A. Anselmo, S. Pesce, M. Liguori, E. Erba, S. Uboldi, M. Zucchetti, F. Pasqualini, M. Nebuloni, N. van Rooijen, R. Mortarini, L. Beltrame, S. Marchini, I. Fuso Nerini, R. Sanfilippo, P. G. Casali, S. Pilotti, C. M. Galmarini, A. Anichini, A. Mantovani, M. D’Incalci, P. Allavena, Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 23, 249–262 (2013). [DOI] [PubMed] [Google Scholar]
- 30.N. S. Wilson, A. Yang, B. Yang, S. Couto, H. Stern, A. Gogineni, R. Pitti, S. Marsters, R. M. Weimer, M. Singh, A. Ashkenazi, Proapoptotic activation of death receptor 5 on tumor endothelial cells disrupts the vasculature and reduces tumor growth. Cancer Cell 22, 80–90 (2012). [DOI] [PubMed] [Google Scholar]
- 31.S. P. Cullen, S. J. Martin, Fas and TRAIL “death receptors” as initiators of inflammation: Implications for cancer. Semin. Cell Dev. Biol. 39, 26–34 (2015). [DOI] [PubMed] [Google Scholar]
- 32.E. E. McGrath, H. M. Marriott, A. Lawrie, S. E. Francis, I. Sabroe, S. A. Renshaw, D. H. Dockrell, M. K. B. Whyte, TNF-related apoptosis-inducing ligand (TRAIL) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. J. Leukoc. Biol. 90, 855–865 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.T. Condamine, V. Kumar, I. R. Ramachandran, J. I. Youn, E. Celis, N. Finnberg, W. S. El-Deiry, R. Winograd, R. H. Vonderheide, N. R. English, S. C. Knight, H. Yagita, J. C. McCaffrey, S. Antonia, N. Hockstein, R. Witt, G. Masters, T. Bauer, D. I. Gabrilovich, ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J. Clin. Invest. 124, 2626–2639 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.H. Forde, E. Harper, C. Davenport, K. D. Rochfort, R. Wallace, R. P. Murphy, D. Smith, P. M. Cummins, The beneficial pleiotropic effects of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) within the vasculature: A review of the evidence. Atherosclerosis 247, 87–96 (2016). [DOI] [PubMed] [Google Scholar]
- 35.A. T. Braithwaite, H. M. Marriott, A. Lawrie, Divergent roles for TRAIL in lung diseases. Front. Med. 5, 212 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.F. Bossi, S. Bernardi, G. Zauli, P. Secchiero, B. Fabris, TRAIL modulates the immune system and protects against the development of diabetes. J. Immunol. Res. 2015, 680749 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.B. A. Pulaski, S. Ostrand-Rosenberg, Mouse 4T1 breast tumor model. Curr. Protoc. Immunol. 39, 20–22 (2001). [DOI] [PubMed] [Google Scholar]
- 38.S. A. Williams, Y. Harata-Lee, I. Comerford, R. L. Anderson, M. J. Smyth, S. R. McColl, Multiple functions of CXCL12 in a syngeneic model of breast cancer. Mol. Cancer 9, 250 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.C. Riera-Domingo, A. Audigé, S. Granja, W. C. Cheng, P. C. Ho, F. Baltazar, C. Stockmann, M. Mazzone, Immunity, hypoxia, and metabolism–the ménage à trois of cancer: Implications for immunotherapy. Physiol. Rev. 100, 1–102 (2020). [DOI] [PubMed] [Google Scholar]
- 40.S. Iwano, M. Sugiyama, H. Hama, A. Watakabe, N. Hasegawa, T. Kuchimaru, K. Z. Tanaka, M. Takahashi, Y. Ishida, J. Hata, S. Shimozono, K. Namiki, T. Fukano, M. Kiyama, H. Okano, S. Kizaka-Kondoh, T. J. McHugh, T. Yamamori, H. Hioki, S. Maki, A. Miyawaki, Single-cell bioluminescence imaging of deep tissue in freely moving animals. Science 359, 935–939 (2018). [DOI] [PubMed] [Google Scholar]
- 41.K. J. Travaglini, A. N. Nabhan, L. Penland, R. Sinha, A. Gillich, R. V. Sit, S. Chang, S. D. Conley, Y. Mori, J. Seita, G. J. Berry, J. B. Shrager, R. J. Metzger, C. S. Kuo, N. Neff, I. L. Weissman, S. R. Quake, M. A. Krasnow, A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.I. Angelidis, L. M. Simon, I. E. Fernandez, M. Strunz, C. H. Mayr, F. R. Greiffo, G. Tsitsiridis, M. Ansari, E. Graf, T. M. Strom, M. Nagendran, T. Desai, O. Eickelberg, M. Mann, F. J. Theis, H. B. Schiller, An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 10, 963 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.J. Kalucka, L. P. M. H. de Rooij, J. Goveia, K. Rohlenova, S. J. Dumas, E. Meta, N. V. Conchinha, F. Taverna, L. A. Teuwen, K. Veys, M. García-Caballero, S. Khan, V. Geldhof, L. Sokol, R. Chen, L. Treps, M. Borri, P. de Zeeuw, C. Dubois, T. K. Karakach, K. D. Falkenberg, M. Parys, X. Yin, S. Vinckier, Y. Du, R. A. Fenton, L. Schoonjans, M. Dewerchin, G. Eelen, B. Thienpont, L. Lin, L. Bolund, X. Li, Y. Luo, P. Carmeliet, Single-cell transcriptome atlas of murine endothelial cells. Cell 180, 764–779.e20 (2020). [DOI] [PubMed] [Google Scholar]
- 44.H. Parhiz, V. V. Shuvaev, N. Pardi, M. Khoshnejad, R. Y. Kiseleva, J. S. Brenner, T. Uhler, S. Tuyishime, B. L. Mui, Y. K. Tam, T. D. Madden, M. J. Hope, D. Weissman, V. R. Muzykantov, PECAM-1 directed re-targeting of exogenous mRNA providing two orders of magnitude enhancement of vascular delivery and expression in lungs independent of apolipoprotein E-mediated uptake. J. Control. Release 291, 106–115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.J. Kalucka, L. Bierhansl, N. V. Conchinha, R. Missiaen, I. Elia, U. Brüning, S. Scheinok, L. Treps, A. R. Cantelmo, C. Dubois, P. de Zeeuw, J. Goveia, A. Zecchin, F. Taverna, F. Morales-Rodriguez, A. Brajic, L. C. Conradi, S. Schoors, U. Harjes, K. Vriens, G. A. Pilz, R. Chen, R. Cubbon, B. Thienpont, B. Cruys, B. W. Wong, B. Ghesquière, M. Dewerchin, K. De Bock, X. Sagaert, S. Jessberger, E. A. V. Jones, B. Gallez, D. Lambrechts, M. Mazzone, G. Eelen, X. Li, S. M. Fendt, P. Carmeliet, Quiescent endothelial cells upregulate fatty acid β-oxidation for vasculoprotection via redox homeostasis. Cell Metab. 28, 881–894.e13 (2018). [DOI] [PubMed] [Google Scholar]
- 46.M. Chrzanowska-Wodnicka, A. E. Kraus, D. Gale, G. C. White, J. Vansluys, Defective angiogenesis, endothelial migration, proliferation, and MAPK signaling in Rap1b-deficient mice. Blood 111, 2647–2656 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O. Dormond, J. C. Madsen, D. M. Briscoe, The effects of mTOR-Akt interactions on anti-apoptotic signaling in vascular endothelial cells. J. Biol. Chem. 282, 23679–23686 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.J. Jablonska, S. Lang, R. V. Sionov, Z. Granot, The regulation of pre-metastatic niche formation by neutrophils. Oncotarget 8, 112132–112144 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.M. S. Patil, S. P. Cartland, M. M. Kavurma, TRAIL signals, extracellular matrix and vessel remodelling. Vasc. Biol. 2, R73–R84 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.J. H. Li, N. C. Kirkiles-Smith, J. M. McNiff, J. S. Pober, TRAIL induces apoptosis and inflammatory gene expression in human endothelial cells. J. Immunol. 171, 1526–1533 (2003). [DOI] [PubMed] [Google Scholar]
- 51.H. Harjunpää, M. L. Asens, C. Guenther, S. C. Fagerholm, Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front. Immunol. 10, 1078 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.S. Hiratsuka, S. Goel, W. S. Kamoun, Y. Maru, D. Fukumura, D. G. Duda, R. K. Jain, Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc. Natl. Acad. Sci. U.S.A. 108, 3725–3730 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.I. Corre, F. Paris, J. Huot, I. Corre, F. Paris, J. Huot, The p38 pathway, a major pleiotropic cascade that transduces stress and metastatic signals in endothelial cells. Oncotarget 8, 55684–55714 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.L. He, M. Vanlandewijck, M. A. Mäe, J. Andrae, K. Ando, F. Del Gaudio, K. Nahar, T. Lebouvier, B. Laviña, L. Gouveia, Y. Sun, E. Raschperger, Å. Segerstolpe, J. Liu, S. Gustafsson, M. Räsänen, Y. Zarb, N. Mochizuki, A. Keller, U. Lendahl, C. Betsholtz, Single-cell RNA sequencing of mouse brain and lung vascular and vessel-associated cell types. Sci Data 5, 180160 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.G. Cantarella, G. Di Benedetto, D. Ribatti, G. Saccani-Jotti, R. Bernardini, Involvement of caspase 8 and c-FLIPL in the proangiogenic effects of the tumour necrosis factor-related apoptosis-inducing ligand (TRAIL). FEBS J. 281, 1505–1513 (2014). [DOI] [PubMed] [Google Scholar]
- 56.P. Secchiero, A. Gonelli, E. Carnevale, D. Milani, A. Pandolfi, D. Zella, G. Zauli, TRAIL promotes the survival and proliferation of primary human vascular endothelial cells by activating the Akt and ERK pathways. Circulation 107, 2250–2256 (2003). [DOI] [PubMed] [Google Scholar]
- 57.P. Manuneedhi Cholan, S. P. Cartland, L. Dang, B. S. Rayner, S. Patel, S. R. Thomas, M. M. Kavurma, TRAIL protects against endothelial dysfunction in vivo and inhibits angiotensin-II-induced oxidative stress in vascular endothelial cells in vitro. Free Radic. Biol. Med. 126, 341–349 (2018). [DOI] [PubMed] [Google Scholar]
- 58.J. Luchino, M. Hocine, M. C. Amoureux, B. Gibert, A. Bernet, A. Royet, I. Treilleux, P. Lécine, J. P. Borg, P. Mehlen, S. Chauvet, F. Mann, Semaphorin 3E suppresses tumor cell death triggered by the plexin D1 dependence receptor in metastatic breast cancers. Cancer Cell 24, 673–685 (2013). [DOI] [PubMed] [Google Scholar]
- 59.M. Brisset, M. Grandin, A. Bernet, P. Mehlen, F. Hollande, Dependence receptors: New targets for cancer therapy. EMBO Mol. Med. 13, e14495 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.J. M. L. Ebos, C. R. Lee, W. Cruz-Munoz, G. A. Bjarnason, J. G. Christensen, R. S. Kerbel, Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15, 232–239 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.S. Loges, M. Mazzone, P. Hohensinner, P. Carmeliet, Silencing or fueling metastasis with VEGF inhibitors: Antiangiogenesis Revisited. Cancer Cell 15, 167–170 (2009). [DOI] [PubMed] [Google Scholar]
- 62.E. Narni-Mancinelli, J. Chaix, A. Fenis, Y. M. Kerdiles, N. Yessaad, A. Reynders, C. Gregoire, H. Luche, S. Ugolini, E. Tomasello, T. Walzer, E. Vivier, Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc. Natl. Acad. Sci. U.S.A. 108, 18324–18329 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.M. G. Alameh, I. Tombácz, E. Bettini, K. Lederer, C. Sittplangkoon, J. R. Wilmore, B. T. Gaudette, O. Y. Soliman, M. Pine, P. Hicks, T. B. Manzoni, J. J. Knox, J. L. Johnson, D. Laczkó, H. Muramatsu, B. Davis, W. Meng, A. M. Rosenfeld, S. Strohmeier, P. J. C. Lin, B. L. Mui, Y. K. Tam, K. Karikó, A. Jacquet, F. Krammer, P. Bates, M. P. Cancro, D. Weissman, E. T. Luning Prak, D. Allman, M. Locci, N. Pardi, Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity 54, 2877–2892.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.M. Baiersdörfer, G. Boros, H. Muramatsu, A. Mahiny, I. Vlatkovic, U. Sahin, K. Karikó, A facile method for the removal of dsRNA contaminant from in vitro-transcribed mRNA. Mol. Ther. Nucleic Acids 15, 26–35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.M. A. Maier, M. Jayaraman, S. Matsuda, J. Liu, S. Barros, W. Querbes, Y. K. Tam, S. M. Ansell, V. Kumar, J. Qin, X. Zhang, Q. Wang, S. Panesar, R. Hutabarat, M. Carioto, J. Hettinger, P. Kandasamy, D. Butler, K. G. Rajeev, B. Pang, K. Charisse, K. Fitzgerald, B. L. Mui, X. Du, P. Cullis, T. D. Madden, M. J. Hope, M. Manoharan, A. Akinc, Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol. Ther. 21, 1570–1578 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen, X. Du, D. Butler, L. Eltepu, S. Matsuda, J. K. Narayanannair, K. G. Rajeev, I. M. Hafez, A. Akinc, M. A. Maier, M. A. Tracy, P. R. Cullis, T. D. Madden, M. Manoharan, M. J. Hope, Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. 51, 8529–8533 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.T. Ishida, D. L. Iden, T. M. Allen, A combinatorial approach to producing sterically stabilized (Stealth) immunoliposomal drugs. FEBS Lett. 460, 129–133 (1999). [DOI] [PubMed] [Google Scholar]
- 68.H. A. Messal, J. Almagro, M. Zaw Thin, A. Tedeschi, A. Ciccarelli, L. Blackie, K. I. Anderson, I. Miguel-Aliaga, J. van Rheenen, A. Behrens, Antigen retrieval and clearing for whole-organ immunofluorescence by FLASH. Nat. Protoc. 16, 239–262 (2020). [DOI] [PubMed] [Google Scholar]
- 69.P. Bankhead, M. B. Loughrey, J. A. Fernández, Y. Dombrowski, D. G. McArt, P. D. Dunne, S. McQuaid, R. T. Gray, L. J. Murray, H. G. Coleman, J. A. James, M. Salto-Tellez, P. W. Hamilton, QuPath: Open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S11