

Abstract
Inherited retinal dystrophies comprise a broad group of genetic eye diseases without effective treatment. Among them, Stargardt disease is the second most prevalent pathology. This pathology triggers progressive retinal degeneration and vision loss in children and adults. In recent years, the evolution of several genome editing technologies, such as the CRISPR-Cas9 system, has revolutionized disease modeling and personalized medicine. Human induced pluripotent stem cells also provide a valuable tool for in vitro disease studies and therapeutic applications. Here, we show precise correction of two ABCA4 pathogenic variants in human induced pluripotent stem cells from two unrelated patients affected with Stargardt disease. Gene editing was achieved with no detectable off-target genomic alterations, demonstrating efficient ABCA4 gene correction without deleterious effects. These results will contribute to the development of emerging gene and cell therapies for inherited retinal dystrophies.
Keywords: MT: RNA/DNA Editing, CRISPR-Cas9, gene editing, human induced pluripotent stem cells, Stargardt disease, inherited retinal dystrophies, ABCA4 gene
Graphical abstract
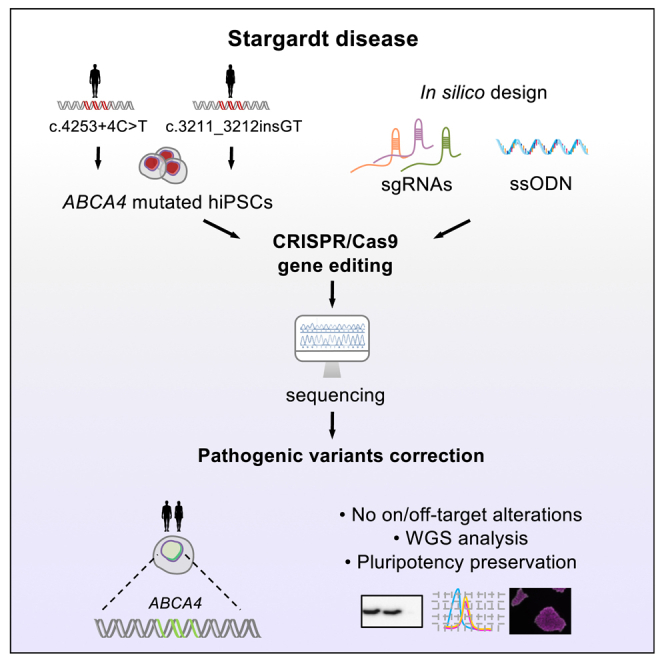
Stargardt disease is an inherited retinal dystrophy that causes progressive retinal degeneration with no treatment. Gene editing technologies allow precise correction of pathogenic variants for personalized medicine. We have reversed damaging variants in two Stargardt patient-derived iPSC lines by CRISPR-Cas9 without undesired genomic alterations and preserving their pluripotency.
Introduction
Stargardt disease (STGD1; OMIM: 248200) is an autosomal recessive inherited retinal dystrophy (IRD) caused by biallelic mutations in the ATP-binding cassette (ABC) transporter subfamily A4 gene (ABCA4; OMIM: 601691).1,2 STGD1 is the most prevalent inherited macular dystrophy and accounts for 12% of IRD-related blindness, with 1 in 8,000–10,000 individuals affected.3,4 The 7,328 bp ABCA4 gene encodes a 2,273 amino acid protein that is mainly expressed in photoreceptors.5 The gene is composed of 50 exons with seven transcript variants, of which only two are protein coding. ABC transporters are transmembrane transport proteins that hydrolyze ATP for substrate pumping across cell membranes.6 Pathogenic variants of this gene can also result in other inherited retinal disorders, such as cone-rod dystrophy and fundus flavimaculatus.2,7
The number of identified ABCA4 mutations has continuously increased with the advent of high-throughput next-generation sequencing. A total of 1,780 ABCA4 variants have been defined in the Human Gene Mutation Database (HGMD Professional 2022.4, released December 2022), of which 1,519 are identified as pathogenic.8 The vast majority correspond to missense and nonsense variants,9 while splicing substitutions and small deletions, insertions, duplications, and indels account for approximately 35%.8 Of note, according to the HGMD, gross deletions and insertions or duplications comprise only 2.25% of ABCA4 alterations.8 Moreover, mutations located in non-coding regions, especially deep intronic ones, are gaining attention because of their multiple effects on transcriptomic and proteomic complexity, such as the modification or generation of splice sites.9,10 Nonetheless, barely 2.47% of ABCA4 variants have been reported in non-codifying locations.8
In the past few decades, several therapeutic approximations have been developed to modulate STGD1 clinical features.9,11,12,13 Because the majority of STGD1 cases are associated with ABCA4, gene therapy provides a powerful approach to halt retinopathy progression. Gene or cell replacement therapies have attracted great interest in recent years, instead of drug-based ones, and are considered one of the most promising therapeutic strategies. Nevertheless, gene therapy is still emerging and has several problems to overcome, such as the limited cargo capacity of adenoviruses for large genes like ABCA4.14,15 Nanoparticles are one of the non-viral-mediated gene therapy approaches that are being explored.16 Antisense oligonucleotides (AONs) have also been developed as an alternative approximation for curative modulation of splicing mutations.12
Therapeutics relying on gene editing enable permanent gene correction, avoiding continuous re-administration and treatment.17 Different genome editing technologies have been developed recently, including transcription activator-like effector nucleases (TALENs), zinc-finger nucleases (ZFNs), and clustered regularly interspaced short palindromic repeats (CRISPR)-associated nuclease Cas9.18 Specifically, recent advances in the CRISPR-Cas9 technology have considerably benefited the biotechnology and biomedicine fields.19,20,21,22 CRISPR-Cas9 gene editing relies on cell-based repair mechanisms to restore DNA double-strand breaks (DSBs) produced by Cas9. The genomic region of interest is targeted by using a single guide RNA (sgRNA), and it needs to be adjacent to the protospacer-adjacent motif (PAM) “NGG” in the case of SpCas9 used in this study.20,23 DNA repair can be performed through two major pathways: non-homologous end-joining (NHEJ) and homology-directed repair (HDR). In NHEJ the two ends of the DSB are randomly re-ligated, generating indels, whereas in the case of HDR, a donor template—such as a single-stranded oligodeoxynucleotide (ssODN)—is used, allowing precise DNA edition.20,21,22
Several registered clinical trials of cell-based therapies for STGD1 are in progress. However, a clinical trial success similar to that accomplished for other IRDs remains to be achieved.9,11 Many trials rely on the potential of stem cells, such as human embryonic stem cells (hESCs) and bone marrow-derived stem cells, in procedures like the subretinal transplantation of differentiated retinal pigment epithelium (RPE) cells and the evaluation of safety and functionality.9 In addition, human induced pluripotent stem cells (hiPSCs) constitute a readily available source of patient-derived cells that could potentially be used for curative strategies, which is crucial because of the lack of cell resources and cell transplant rejection.23,24
The combination of hiPSCs and CRISPR-Cas9 technology enables in vitro gene editing in patient-derived cells to correct their specific mutations and allow their differentiation into retina cells for autologous transplantation, constituting a powerful tool for personalized medicine.25 Nonetheless, low gene editing efficiency, potential adverse effects, and off-target effects must be considered and evaluated.24
In this study, we aimed to correct two ABCA4 variants from two STGD1 patients carrying compound heterozygous mutations. The c.4253+4C>T variant was predicted to cause a splicing defect, and the c.3211_3212insGT probably generates a frameshift (p.Ser1071Cysfs∗14) (Table 1). We tested CRISPR-Cas9 and TALEN genome-editing strategies to correct the ABCA4 sequence in patient-derived iPSCs. We successfully edited both pathogenic variants using CRISPR-Cas9 technology without genomic alterations in the predicted off-targets, which was confirmed by Sanger and whole-genome sequencing (WGS). In addition, we achieved a significant increase in the number of edited clones by modifying the hiPSC culture and transfection conditions, thus optimizing the CRISPR-Cas9 assay. Moreover, gene editing did not compromise the expression of pluripotency markers in corrected clones compared with parental ones.
Table 1.
In silico analysis of patient variant pathogenicity according to the ENSEMBL and ALAMUT predictions
| Patient ID | hiPSC line | Allele | Zygosis | Variant | Amino acid change | Predictorsa | dbSNP | MAF TopMed | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Fi22/01 | FRIMOi003-A | 1 | Het | c.4253+4C>T | – | 3P/1N | rs61754044 | 0.000004 (1/264690) | Özgül et al.26 |
| 2 | Het | c.6089G>A | p.Arg2030Gln | 14P/2N | rs61750641 | 0.000423 (112/264690) | Lewis et al.27 | ||
| Fi15/32 | FRIMOi004-A | 1 | Het | c.3211_3212insGT | p.Ser1071Cysfs∗14 | – | rs61750064 | none | Allikmets et al.5 |
| 2 | Het | c.514G>A | p.Gly172Ser | 6P/10N | rs61748532 | 0.000344 (91/264690) | Jaakson et al.28 | ||
| – | Het | c.2023G>A | p.Val675Ile | 13P/3N | rs575453437 | 0.000008 (2/264690) | Fujinami et al.29 | ||
| – | Het | c.6148G>C | p.Val2050Leu | 11P/5N | rs41292677 | 0.003876 (1026/264690) | Allikmets et al.5 |
MAF, minor allele frequency.
Predictions are expressed as “P” for pathogenic and “N” for neutral. See materials and methods for predictor details.
The results obtained in this study demonstrate for the first time efficient ssODN-mediated CRISPR-Cas9 mutation repair in hiPSCs derived from STGD1 patients. These data encourage investigation of CRISPR-Cas9 gene editing to reverse pathogenic variants as a promising tool for STGD1 research and as a potential therapeutic strategy for this IRD.
Results
sgRNA and TALEN screening for targeting STGD1-related ABCA4 mutations
Two hiPSC lines from two unrelated STGD1 patients carrying compound heterozygous mutations in the ABCA4 gene were obtained as previously described.1 One of the patients (hereafter referred to as Fi22/01 and whose hiPSC line was named FRIMOi003-A) harbors one ABCA4 pathogenic variant in each allele (Figure 1A). One variant probably causes a splicing defect (c.4253+4C>T), and the other one is a widely described missense mutation (c.6089G>A, p.Arg2030Gln) (Table 1). The other patient (Fi15/32, and hiPSC line named FRIMOi004-A) carries a dinucleotide insertion that generates a frameshift in allele 1 (c.3211_3212insGT) and three missense mutations in allele 2 (c.514G>A p.Gly172Ser, c.2023G>A p.Val675Ile, and c.6148G>C p.Val2050Leu) (Figure 1A and Table 1).
Figure 1.

sgRNA and TALEN screening for targeting STGD1-related ABCA4 mutations
(A) Schematic overview of the ABCA4 gene and diagnosed variants of Fi22/01 and Fi15/32 from both alleles. Scale bar represents 1,000 bp. Scheme was designed using the Exon-Intron Graphic Maker software (www.wormweb.org) based on the UCSC Genome Browser ABCA4 sequence. (B) Cleavage efficiency chart of the different sgRNA and TALEN designs screened in wild-type hiPSCs. Relative cleavage efficiency was calculated relative to parental (not cut) bands. (C) Schematic representation of the locus of pathogenic variants selected for gene editing with sgRNAs and TALEN sequences for patient Fi22/01. The sgRNA targeted sequence used for CRISPR-Cas9 appears color highlighted with the patient’s mutation site in red. The PAM is underlined and exons are marked in a box. (D) As in (C) but for patient Fi15/32.
To better characterize these ABCA4 mutations we performed in silico analysis of their prevalence and pathogenicity. The ENSEMBL-derived results showed that all missense ABCA4 variants, except c.514G>A from patient Fi15/32, were predicted to be pathogenic (Table 1). However, all of them have been previously described in STGD1 cases (Table 1). Also, TopMed database information pointed out that c.3211_3212insGT (allele 1) and c.2023G>A (allele 2) variants in patient Fi15/32 could be the most relevant contributing to STGD1, as they appear in a low or null frequency in the population (Table 1).
We designed different sgRNA and TALEN mRNA pair sequences near the variants (Table 2). We chose sgRNAs depending on the presence of the canonical NGG PAM sequence—needed by Cas9 to cleave the DNA—and the TALENs according to the maximum cleavage efficiency score. Notably, we did not design sgRNAs for the c.514G>A variant because the predictors suggested neutrality (Table 1).
Table 2.
List of sgRNA and TALEN sequences designed to target ABCA4 pathogenic variants
| hiPSC line | Variant | Sequence ID | Technology | Distancea | Sequenceb,c | Strand | PAM |
|---|---|---|---|---|---|---|---|
| FRIMOi003-A | c.4253+4C>T | 1 | CRISPR | −5 | TTCTTCAGGTGCGCGGACTC | + | GGG |
| 2 | CRISPR | −5 | TTCTTCAGGTGTGCGGACTC | + | GGG | ||
| T1 | TALEN | 0 | AGCAGTACACCTTCTTCA, ACAGAGGAGAATGGTGAC | +, − | – | ||
| c.6089G>A p.Arg2030Gln | 3 | CRISPR | −9 | GCAATTGATGAGCTGCTCAC | + | AGG | |
| T2 | TALEN | 0 | GCAATTGATGAGCTGCTC, GCCGGGCATAAAGGTAAA | +, − | – | ||
| FRIMOi004-A | c.3211_3212insGT p.Ser1071Cysfs∗14 | 4 | CRISPR | −45 | TAACTCTCCCGTCCTTCTTG | − | AGG |
| 5 | CRISPR | +87 | CTTACTCGAGACGCTCAATC | + | TGG | ||
| 6 | CRISPR | 1 | GCATGCAGAGAAAGCTGTGT | + | CGG | ||
| T3 | TALEN | 0 | GCCTCCAGGTGGCATGCA, TCCCACAAAGGCAATGGC | +, − | – | ||
| c.2023G>A p.Val675Ile | 7 | CRISPR | 0 | GACTGTGAAGAGCATCGTCT | + | TGG | |
| c.6148G>C p.Val2050Leu | 8 | CRISPR | +35 | GGCAGTCGGCGTAGACAGTC | − | AGG | |
| 9 | CRISPR | 0 | ATACTCCAGTTTGCAACCTA | − | GGG |
Distance is expressed in base pairs from cut to edit site.
TALEN sequences are expressed as forward and reverse sequences separated by a comma.
The nucleotides modified from wild-type sequence for targeting the patient’s variant are in italic.
To examine the cleavage efficiency of all these guides, we transfected them into wild-type hiPSCs, and DSBs were detected using the endonuclease T7 I-mediated system. We obtained an overall DNA cleavage efficiency between 15% and 45% with sgRNA/Cas9, as previously reported (Figure 1B).30 However, using the TALEN approximation, we detected only a small proportion of cleaved DNA in the case of the T2 mRNA pair targeting the c.6089G>A variant (Figure 1B). No DNA cut was observed with the other two TALEN pairs (T1 and T3, targeting variants c.4253+4C>T in patient Fi22/01 and c.3211_3212insGT in Fi15/32, respectively) testing two different electroporation conditions compared with the positive control (Figures S1A and S1B). Since TALEN-mediated cleavage was low or undetectable in our hands, we decided to correct STGD1 pathogenic variants by using CRISPR-Cas9 technology.
sgRNAs harboring patient mutations show increased cleavage efficiency in patient-derived hiPSCs
STGD1 is an autosomal recessive retinal dystrophy caused by compound heterozygous mutations. Thus, we aimed to reverse one of the alleles of each patient-derived hiPSC line to stop the disease progression. To determine which of the ABCA4 variants was the most suitable for correction in each patient, we took advantage of the prediction analysis performed before to study the degree of pathogenicity for each one (Table 1).
In the case of patient Fi22/01, both variants were predicted to be pathogenic. Hence, considering that all sgRNAs explored exhibited good cleavage efficiency (Figure 1B), we decided to use all sequences (sgRNAs 1 to 3). Regarding patient Fi15/32—who carries four likely pathogenic variants (Table 1)—we selected the c.3211_3212insGT in allele 1 (sgRNAs 4 to 6), because targeting only one mutation is simpler for experimental design than targeting allele 2, which carries three variants.
Then, we performed a first experiment for setting up conditions of gene editing assay. We chose sgRNA4 to correct c.3211_3212insGT in FRIMOi004-A, due to its higher DNA cleavage efficiency (Figure 1B). We obtained a gene editing efficiency of almost 15% but also a high percentage of genomic alterations, in both edited and unedited clones (Figures S2A and S2B). Notably, all the clones contained on-target deletions (small or large) or indels close to the DSB (Figure S2A).
These results prompted us to explore whether we could improve sgRNA and ssODN designs to avoid this huge number of on-target defects. The use of sgRNA guides designed with the shortest possible distance between the DSB and the editing site is, reportedly, the most efficient for single-base substitutions, since they minimize undesired genomic alterations.31 Importantly, sgRNA4 used in this first assay cut DNA at −45 bp from the targeted edit site (Table 2). Accordingly, we decided to continue editing assays with sgRNA1 and sgRNA2 to correct the c.4253+4C>T variant in patient Fi22/01 (Figure 1C) and sgRNA6 for c.3211_3212insGT in patient Fi15/32 (Figure 1D).
Next, we examined whether inclusion of the patient’s mutation in the sgRNA sequence could enhance DNA cleavage specificity by targeting only the mutated allele. For this purpose, we previously designed sgRNA1 and sgRNA2, which share the same sequence with the exception of the single-nucleotide change corresponding to the patient’s ABCA4 mutation in sgRNA2 (nucleotide in italic in Table 2). When transfected into wild-type hiPSCs, sgRNA1 almost doubled the ability of sgRNA2 to induce Cas9-mediated DSB (Figures 2A and 2B). In contrast, in the FRIMOi003-A cell line, sgRNA2 displayed increased cleavage compared with sgRNA1 (Figures 2A and 2B).
Figure 2.

sgRNA and ssODN design optimization to effectively correct STGD1-related variants
(A) Representative gel of comparison between sgRNA1 and sgRNA2 in wild-type hiPSCs and in FRIMOi003-A hiPSCs. hiPSCs were transfected with or without sgRNA, and PCR amplification of the desired locus was run in a 2% agarose gel for cleaved band visualization and (in B) quantification. Asterisks indicate gel lanes with detectable fragments with the expected size resulting from T7E1 cutting. Untreated genomic DNA from the same hiPSC line used for transfection was used as a negative control showing the intact parental band. (B) Quantification of DNA bands from gel in (A). At least two experiments for each sgRNA were done. Bar graphs show the mean and the error bar the standard error of the mean. (C) ssODN design for CRISPR-Cas9-mediated gene editing of c.4253+4C>T ABCA4 pathogenic variant. Sequence highlighted is the template used for repair. Phosphorothioate modifications are represented in yellow, PAM modification is in green, and the patient’s mutation correction is either in red or in blue. (D) As in (C) but for correcting the c.3211_3212insGT variant. (E) Schematic representation of assay conditions followed for CRISPR-Cas9-mediated genome editing in hiPSCs. sgRNA design targets the mutated allele and ssODN repair template harbors the Cas9-blocking mutation and corrects the patient variant. After 72–96 h culture, single colonies were picked and cultured for further analysis. The figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
Because of the intrinsic variability and transfection effectiveness across hiPSC lines, we could not compare sgRNA cleavage efficiencies between different cell lines. Nevertheless, the results obtained here show that sgRNA2 displayed better cleavage efficiency in FRIMOi003-A than in sgRNA1. This suggests an increase in specificity when using an sgRNA harboring the patient’s variant to target the mutated allele in patient-derived hiPSCs compared with wild-type ones. Subsequently, we decided to continue our study with sgRNA2 for the c.4253+4C>T variant in FRIMOi003-A and sgRNA6 for c.3211_3212insGT in FRIMOi004-A, which was also designed carrying the dinucleotide insertion.
ssODN design optimization to effectively correct STGD1-related variants
ssODNs have been extensively used as donor repair templates for DNA editing and have been reported to increase HDR-mediated repair in single-nucleotide substitutions.32 As we have seen, the sgRNA sequence is important to improve the CRISPR-Cas9 system. Likewise, ssODN design is also critical for efficient HDR-mediated DNA correction or modification.31,33,34 In the editing assay performed with sgRNA4, we obtained on-target genomic aberrations in all clones analyzed, probably due to the distance between the DSB and the editing site and also to the ssODN template, which was used without design optimization (Figures S2A and S2C).
To further enhance the CRISPR-Cas9 gene editing assay, we designed one new ssODN for each variant to correct (Figures 2C and 2D), considering several recently described important parameters for on-target single-base editing.34,35 First, ssODNs must be 70–85 nt long. Second, the predicted DSB site should be at the center of the template and with homologous left and right arms to the targeted sequence of approximately 30 nt each. Third, in addition to the specific disease-causing mutation correction, ssODNs also incorporate a nucleotide change in the PAM (silent mutation) to avoid re-cleavage of the target DNA by Cas9 after HDR repair.31,33,34,36 Last, phosphorothioate bases at the edges of the template were added to increase ssODN stability (nucleotides in yellow in Figures 2C and 2D). Of note, we took into account the preservation of the amino acid sequence—in the case of codifying regions—when introducing Cas9-blocking mutations. Also, we considered that these changes do not modify splicing patterns, which was confirmed with ALAMUT software, and evolutionary conservation was examined to introduce the most frequent alternative nucleotide.
ssODN-mediated DNA editing relies on HDR, which is less effective than the NHEJ pathway in mammalian cells.21,37 Therefore, we used an HDR activator (L755507) and an NHEJ inhibitor (M3814) to improve HDR-mediated DNA editing, as previously reported.37 In addition, Cas9 delivery type has been found to be significant in on-target gene editing efficiency and in avoiding undesirable genomic modifications.21,31 In this sense, plasmid-based Cas9 overexpression in edited cells has been related to lower knockin efficiencies and to a dramatic increase in the re-cutting of edited sites.31 Hence, we decided to transfect hiPSCs with ribonucleoprotein (RNP) complexes comprising sgRNA, ssODN, and Cas9 protein for our assays.
To summarize, to precisely correct our patient-derived ABCA4 mutated hiPSC lines, we used high fidelity (HiFi) Cas9 protein with optimal concentrations and design of the sgRNA and ssODN and delivered them as RNP complexes. Moreover, we used an HDR activator and an NHEJ inhibitor to improve donor template-mediated DNA repair (Figure 2E).
Efficient correction of STGD1-related variants by CRISPR-Cas9 in hiPSCs
hiPSC lines from both STGD1 patients were subjected to CRISPR-Cas9-mediated gene editing to correct ABCA4 mutations. Briefly, a single-cell suspension of hiPSCs was electroporated with the Neon transfection system together with HiFi Cas9, sgRNA, and ssODN (Figure 2E). Control clones of hiPSCs transfected without sgRNA and ssODN were also obtained in parallel in each experiment (parental clones).
We screened more than 50 single isolated edited clones in the region of interest, which was approximately 300–600 bp in length, using Sanger sequencing (Figures 3A and 3C). The primer sequences are listed in Table S1. We obtained a correction efficiency of around 5% for the c.4253+4C>T variant (a total of 3 edited clones out of 57 clones screened; Figures 3B and 3E) and approximately 11% for the c.3211_3212insGT variant (6 of 53 clones; Figures 3D and 3E).
Figure 3.

Efficient correction of STGD1-related variants by CRISPR-Cas9 in hiPSCs
(A) FRIMO003-A hiPSC clones were subjected to Sanger sequencing to analyze gene editing in the locus specified after CRISPR-Cas9. Representative captures of chromatograms from Sanger results of parental and one edited clone are shown. (B) Pie chart of the percentage of edited clones in FRIMOi003-A cells. (C) As in (A) but with FRIMOi004-A. Representative capture of edited clones with PAM modification is also shown. (D) Pie chart of the percentage of edited clones according to variant correction and PAM modification in FRIMOi004-A cells. (E) Summary of the on-target events in total screened clones for each hiPSC line after CRISPR-Cas9-mediated gene editing. (F) Pie chart of the percentage of edited clones in FRIMOi004-A according to variant correction and PAM modification after cell culture conditions optimization. (G) Comparison between CRISPR-Cas9-mediated gene editing efficiency assays after normal and optimized conditions in the FRIMOi004-A hiPSC line.
PAM silent mutation was detected in only three of the FRIMOi004-A corrected clones, but no clones incorporated it in FRIMOi003-A cells (Figure 3E). Notably, the Cas9-blocking mutation was found in heterozygosis in all cases, presumably because the wild-type allele was not recognized by our editing strategy. Consistently, the parental clones screened showed the presence of the patient’s variant (Figures 3A and 3C). All edited clones were also analyzed for genomic alterations, and no insertions, deletions, indels, or single-nucleotide changes were observed in the on-target region (Figure 3E).
These results demonstrate efficient gene correction of the c.4253+4C>T and c.3211_3212insGT ABCA4 variants by the CRISPR-Cas9 system without on-target genomic alterations. The use of L755507 (HDR activator) and M3814 (NHEJ inhibitor) together with ssODN and sgRNA designs significantly contributed to precise gene editing for single-base modifications.
Increase in gene editing efficiency in hiPSCs after CRISPR-Cas9 system optimization
CRISPR-Cas9-mediated genome editing typically has a low correction efficiency (between 2% and 20%).25,31,34,38,39 Moreover, many CRISPR-Cas9 experiments to correct disease-related mutations in hiPSCs have been performed with Cas9-overexpressing plasmids and not by directly transfecting Cas9 protein as an RNP.34,38,40 In an attempt to increase gene editing efficiency, we tested if we could optimize the CRISPR-Cas9 assay by modifying cell culture conditions before and after transfection.
For that purpose, we used the FRIMOi004-A cell line to correct the c.3211_3212insGT variant because it yielded a higher editing percentage. Compared with the previous experiment, this assay was performed when cells exhibited exponential proliferation—that is, at the highest growth rate—and the cells were maintained with ROCK inhibitor during all single-cell suspension preparations. Then, FRIMOi004-A cells were electroporated with the RNP complex comprising the sgRNA, ssODN, and HiFi Cas9 and immediately seeded and cultured with HDR activator (L755507) and NHEJ inhibitor (M3814) for 48 h, instead of 24 h.
Screening of transfected cells revealed a considerable increase in the number of edited clones compared with the previous assay (Figures 3F and 3G). After conditions optimization, we obtained a total of 30 ABCA4 edited clones out of 42 clones screened, resulting in a 70% success rate (Figure 3G). Of these, 17 clones had incorporated only the variant correction and 13 had modified the pathogenic variant and also the PAM sequence (Figure 3F). In addition, sequencing analysis showed no undesirable genomic modifications at the on-target locus (data not shown), as in the previous assay.
Collectively, these results indicate that gene editing efficiency is influenced by many factors that should be finely tuned. The above results demonstrate that hiPSC culture and electroporation conditions, Cas9 delivery method, and use of L755507 and M3814 are decisive points for successful CRISPR-Cas9-mediated gene editing.
hiPSC clones preserve the expression of pluripotency markers after gene editing
To study whether hiPSCs compromised their pluripotency after editing assays, cell clones were cultured in parallel to the parental ones. Edited clones conserved hiPSC colony-like morphology in culture, similar to parental controls, and no differences in proliferation or morphology were observed during cell culture (Figure 4A). Of note, few clones were lost after the first passage because of the low number of cells. Immunofluorescence analysis of these clones showed similar expression of NANOG, SOX2, SSEA4, and TRA-160 pluripotency markers in parental and edited cells (Figures 4B and S3A).
Figure 4.

hiPSC clones preserve the expression of pluripotency markers after gene editing
(A) Bright-field and (B) immunofluorescence pictures of parental and edited clone colonies after performing CRISPR-Cas9 in FRIMOi003-A and FRIMOi003-A. Expression of pluripotency markers NANOG, SOX2, SSEA4, and TRA-160 was assessed by immunofluorescence. Representative captures are shown. Scale bar represents 200 or 100 μm in bright-field or immunofluorescence pictures, respectively. (C) Parental and edited clones were assessed for the expression of the indicated genes by qRT-PCR in FRIMOi003-A and FRIMOi004-A. Data are represented as the mean of at least two different clones for each hiPSC line. The error bar represents the standard error of the mean. (D) As in (C), but data are represented as a heatmap of gene expression. “P” indicates parental and “E” edited clones. (E) Protein lysates of parental (P) and edited (E) clones from FRIMOi003-A and FRIMOi004-A were blotted for SOX2, NANOG, OCT4, and α-tubulin as loading control. See Figure S3B for full unedited blots. (F) Immunofluorescence staining of OTX2, BRACHYURY, and SOX17 in parental and edited clones after differentiation into ectodermal, mesodermal, and endodermal lineages, respectively. Representative captures are shown. Scale bar indicates 50 μm.
hiPSC clones were also assessed for pluripotency marker expression at the mRNA and protein levels. mRNA expression analysis of pluripotency markers revealed no significant differences after gene editing (Figures 4C and 4D). In addition, similar protein levels of NANOG, SOX2, SSEA4, and TRA-160 were observed in edited clones compared with parental hiPSCs, indicating preservation of pluripotency (Figures 4E and S3B).
One of the characteristics of hiPSCs is their ability to differentiate into the three germ layers, being able to generate virtually every committed cell type in human tissues. To assess if gene editing had compromised this capacity, we subjected edited clones to ectodermal, endodermal, and mesodermal lineage differentiation and analyzed them for the expression of OTX2, SOX17, and BRACHYURY markers, respectively. Immunofluorescence evaluation demonstrated the capability of these cells to differentiate into the three germ layers, and no differences were found compared with parental clones (Figure 4F). Notably, none of these markers were expressed in undifferentiated clones, confirming their pluripotency conservation (Figure S3C).
Correction of the ABCA4 variants does not result in off-target alterations after CRISPR-Cas9-mediated gene editing
One of the main problems of gene editing is the generation and detection of genomic alterations in off-target regions, which can occur frequently due to the presence of similar regions throughout the genome. To further analyze the genomic conservation of edited hiPSC clones and evaluate the specificity of the CRISPR-Cas9 strategy, we first used Sanger sequencing to screen the genomic regions homologous to sgRNA2 and sgRNA6.
First, we predicted the off-targets, allowing a maximum of three mismatches in the DNA sequence compared with the sgRNA. We found seven potential off-targets for sgRNA2 and 57 for sgRNA6 (Table S2). Notably, most of them correspond to intergenic or deep-intronic regions. All off-targets with two mismatches and those with three mismatches corresponding to exons and intronic sequences in close proximity to exons were selected for the screening (off-targets and primers are listed in Tables 3 and S3, respectively). We analyzed by Sanger sequencing approximately 400 bp surrounding the homologous sequence and adjacent PAM in these seven off-targets (Figures 5A and 5B). Accurate analysis of these loci revealed that there were no genomic alterations (insertions, deletions, indels, or single-base modifications) in any of the 35 edited clones screened, compared with parental clones (Figures 5A and 5B).
Table 3.
Sequencing results from selected sgRNA2 and sgRNA6 off-targets
| sgRNA | Off-target ID | DNA sequencea,b | Chr | Position | Strand | Mismatches | Gene | Location | Del/ins/indel | Single-nt mod. |
|---|---|---|---|---|---|---|---|---|---|---|
| sgRNA2 | OT-1 | TTCTTCAGtTGTaCtGACTCTGG | chr12 | 80,295,911 | − | 3 | OTOGL | intron 31 | 0/2 | 0/2 |
| OT-2 | TcaTTCAGGTGTGaGGACTCTGG | chr22 | 46,361,593 | + | 3 | CELSR1 | exon 35 | 0/2 | 0/2 | |
| sgRNA6 | OT-3 | GCATcCAGAGAAAGCTaTGTAGG | chr1 | 112,903,490 | − | 2 | – | intergenic | 0/33 | 0/33 |
| OT-4 | cCATGCAGAGAAAGCTtTGaAGG | chr8 | 79,763,025 | + | 3 | HEY1 | exon 5 | 0/33 | 0/33 | |
| OT-5 | GCATGCAGAGgAgGCTtTGTAGG | chr1 | 170,663,322 | − | 3 | PRRX1 | exon 1 | 0/33 | 0/33 | |
| OT-6 | GaATGCAGAGAAgGCTtTGTGGG | chr1 | 183,127,284 | + | 3 | LAMC1 | exon 17 | 0/33 | 0/33 | |
| OT-7 | aCATGCAGtGAAAGCTGTGgAGG | chr6 | 72,272,584 | − | 3 | RIMS1 | intron 22 | 0/33 | 0/33 |
Nucleotides with mismatch compared with the sgRNA reference sequence are in lowercase.
PAM sequence is in italic.
Figure 5.

Correction of the ABCA4 variants does not result in off-target events after CRISPR-Cas9-mediated gene editing
(A) Representative captures of chromatograms showing Sanger sequencing reads of PCR products from predicted off-targets for sgRNA2. On top are reference and consensus sequences and on the bottom the sequence of one edited clone. The sequence homologous to sgRNA2 and the adjacent PAM motif are highlighted (in red). (B) As in (A) but for sgRNA6.
In addition, we sought to evaluate the potential pathogenicity and splice site alteration of deep intronic off-targets if nucleotide changes from our ssODN were introduced into these regions. For this, all intronic off-target regions were subjected to in silico analysis prediction in the unlikely and hypothetical scenario that, after DNA cut, the cell repair machinery would have used the ssODN template for repair. Four predictors were consulted for this analysis using ALAMUT software. The results obtained predicted a potential splicing alteration in a minority of cases (last columns in Table S2). Regarding sgRNA2 off-targets, two intronic regions were predicted to alter a donor splice site. In contrast, in sgRNA6 off-targets we found only four of all the intronic regions with more than two predictors positive for donor and/or acceptor splicing site alteration (Table S2).
It is worth noting that other undesired genomic events across the genome could escape the Sanger sequencing assessment in the predicted off-targets. Thus, we decided to perform WGS in FRIMOi004-A edited cells under optimized conditions to search for genomic variants induced by the CRISPR-Cas9 system. For the parental clone we obtained 689,126,811 high-quality reads, with a mean coverage of 30.55, and 601,239,281 in the edited clone, with a mean coverage of 26.64.
To compare both genomes, we searched for variants with high/moderate predicted pathogenicity impact or with a potential modifier effect. WGS analysis revealed 7,081 putative pathogenic or modifier variants, of which approximately 3% appeared in a different zygosity between parental and edited clones. However, all of them arose due to an incorrect assignation derived from the Var/Depth estimation by the bioinformatic platform in low balanced values. Finally, the only difference between the parental and the edited clone in the total 7,081 variants corresponded to the patient’s mutation corrected after CRISPR-Cas9 gene editing. Last, we observed no differences in the detected copy number variations (CNVs), indicating the absence of gross deletions or insertions generated after gene editing.
Based on these data, we can conclude that the use of a mutated sgRNA with a low number of predicted off-targets—at least with no mismatches—together with ssODN-mediated repair and HiFi Cas9 used in this study provides a promising strategy for hiPSC gene editing for ABCA4 pathogenic variants (single-base substitutions or small insertions) without deleterious effects. Off-target analysis by Sanger sequencing and WGS displayed no undesired genomic alterations, suggesting high specificity of the gene editing assay.
Discussion
STGD1 is an IRD that results in macular degeneration and visual loss and has yet no cure. Thus, there is an unmet need to develop therapeutic approaches to prevent disease progression. The use of CRISPR-Cas9-mediated gene editing has exponentially increased in recent years. Regarding eye diseases, research resulting in several advances has been conducted with special interest in gene therapy strategies.41,42,43 However, therapeutic application remains controversial owing to technical limitations.23,41 There are many concerns regarding the potential risks associated with an in vivo CRISPR-Cas9 system, such as oncogenicity related to DSBs,44 undesired off-target effects, and immune and inflammatory responses.43,45,46
hiPSCs have revolutionized basic research on human diseases and have generated interest as a potential cellular resource for treating pathologies.24,47 The combination of CRISPR and hiPSCs is an important tool for disease study, modeling, and therapeutic applications.24 Nonetheless, precise genome-editing in hiPSCs remains inefficient.48
In the past decade, numerous studies on gene editing in inherited eye disorders have been performed using different cell types and CRISPR-Cas9 approximations.33,36,49,50 Many of these assays use Cas9-overexpressing plasmids.34,36,49,51 This type of delivery is commonly associated with a high number of genomic aberrations and re-cutting events due to uncontrolled Cas9 production and activity,31,34 and sequencing analysis of predicted off-targets is not routinely done.33,49 In addition, plasmid-based Cas9 transfection has been reported to be less efficient in knockin and single-base substitutions than in delivering Cas9 as a purified protein in an RNP complex.31,38 Cas9 transfection together with sgRNA and ssODN as an RNP complex results in a remarkable reduction in Cas9 re-cutting of edited sites because of its rapid degradation.31,38,52
In the present study, efficient gene editing in two STGD1-related ABCA4 pathogenic variants was achieved through ssODN-mediated repair. One of the STGD1-related variants corresponds to an intronic single-base substitution between exons 28 and 29 (c.4253+4C>T). The other corresponds to an insertion of a GT in exon 22 of the ABCA4 gene (c.3211_3212insGT). De Angeli et al. recently published their results on a deep-intronic ABCA4 variant causing a splicing defect in cone photoreceptor precursor cells via plasmid-mediated overexpression of Cas9.49 The authors described the successful restoration of ABCA4 transcript levels in minigene splicing assays. Nonetheless, the recovery of ABCA4 transcription is not done through ssODN-mediated repair for permanent gene editing, and the assessment of potential off-targets could not be done.49
Importantly, deep-intronic mutations account for only 2.47% of ABCA4 described variants. Here we show precise ssODN-mediated single-nucleotide gene editing in the ABCA4 sequence without detected genomic alterations. Hence, the CRISPR-Cas9 assay performed in this study is a promising approximation for the potential treatment of STGD1 disease.
Previous work in our lab and the results published here recommend a fine balance between sgRNA selection and ssODN design together with the CRISPR-Cas9 approximation used, to achieve precise gene editing without undesired re-cutting and off-target effects. These results indicate that there are several key points for accurate single-base gene editing in hiPSCs. For example, the use of an HDR activator and NHEJ inhibitor significantly contributed to efficient gene editing comparing both assay conditions (Figure 3G). In addition, these treatments markedly changed the number of correctly edited clones without alterations at the on-target site (Figures 3C and S2A). Also, we and others have found that the base-pair distance between the cutting and the editing site is important for increasing the on-target editing ratio without undesired genomic events.31 Of note, TALEN technology was not effective in performing DNA cleavage in our hands, possibly due to poor DNA cutting efficiency or the targeted locus itself.
Notably, Cas9-blocking mutations have been demonstrated to reduce re-cutting events.31,33,34 However, we found a significant number of edited clones that did not incorporate this silent mutation and showed no signs of re-cutting or DNA alterations. We speculate that these clones could arise from cells that could have used the wild-type allele as a repair template. Because of that, and considering that our hiPSCs are derived from patients carrying heterozygous mutations, the sgRNA design harboring patient variants could be important to discriminate between corrected and non-corrected alleles, avoiding Cas9 re-cutting of on-target sites.
Off-target abnormality detection is one of the main problems after gene editing. Several online tools predict potential regions homologous to the sgRNA that may be recognized and cut.53,54,55 However, WGS allows detection of variants and other alterations across the whole genome.56 Nevertheless, only events with high readability are well covered and therefore detected, and short read mapability or software limitations can also hamper data analysis.56,57 Because of that, we performed Sanger sequencing of selected off-targets from in silico prediction. From more than 150 off-target sequences analyzed, no genomic modifications were found in the edited clones studied for either sgRNA. Homologous regions with more than three mismatches were not considered because of the absence of genomic abnormalities in off-target regions with fewer than four mismatches. In addition, WGS analysis showed no differences in edited clones compared with parental ones, supporting the absence of off-target aberrations.
Importantly, it is possible that the well-known Cas9 off-target activity could have produced undesired abnormalities, including gross indels, deletions, or insertions, but they were not detected in the screening performed here. However, it is worth noting that a heterozygous SNP was maintained in FRIMOi003-A edited clones (data not shown) and that FRIMOi004-A corrected clones carrying PAM modification were found in a heterozygous state, indicating proper on-target edition and conservation of both alleles.
The eye is an advantageous organ for therapy development because it is very accessible, its anatomy is well studied, and it is easy to image and monitor disease progression. Moreover, its relative isolation from the rest of the body reduces the impact of any systemic adverse effects of therapy.58 There is no effective treatment for retinal dystrophies caused by gene variants yet, but many efforts are being made to develop gene replacement therapies, which have demonstrated good efficacy and safety in ongoing clinical trials.45
Genome editing in hiPSCs provides a tool with tremendous value for disease investigation and molecular and cellular research, avoiding the use of viral vectors to introduce exogenous material. In addition, it is very useful for genotype-phenotype correlation studies and the cells can serve as a, theoretically, unlimited cell source for potential autologous cellular therapy. Nevertheless, in vivo CRISPR-Cas9 gene editing is still in the early stages and there are many concerns regarding undesired effects.23,46 A clinical trial with hESC-derived RPE cells to treat STGD1 is ongoing to evaluate the safety of subretinal transplantation of these stem cell-derived differentiated cells.9,59 Research on both gene therapy and cellular therapy approaches is crucial for the immediate future of regenerative and personalized medicine and for the treatment of STGD1 retinal dystrophy.58,59
Materials and methods
hiPSC culture and transfection
hiPSC lines FRIMOi003-A and FRIMOi004-A derived from STGD1 patients (Fi22/01 and Fi15/32, respectively) carrying ABCA4 heterozygous mutations were obtained as previously described in Riera et al.1 For some experiments, wild-type hiPSCs were used from a patient without any ophthalmologic disease and no genetic variants related to retinal dystrophies. hiPSC colonies were maintained in StemFlex medium (Thermo Fisher Scientific, Waltham, MA, USA) and cultured on Matrigel-coated dishes (Merck, Bedford, MA, USA). To obtain hiPSC single-cell suspensions, hiPSC colonies were detached with TrypLE (Thermo Fisher Scientific), centrifuged, and counted before Neon-mediated transfection (Thermo Fisher Scientific). For hiPSC differentiation, clones were subjected to ectodermal, mesodermal, and endodermal lineage differentiation analysis using the Human Pluripotent Stem Cell Functional Identification Kit (R&D Systems, Minneapolis, MN, USA), according to the manufacturer’s instructions.
The study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of the Institut de Microcirurgia Ocular (protocol code 170505_117; date of approval June 2, 2017).
In silico analysis of patient variants
The damaging variants diagnosed in patients were subjected to in silico analysis for pathogenicity prediction using various tools. Missense mutations were analyzed with ENSEMBL Variant Effect Predictor (VEP),60 which provides results from a range of algorithms to assess the potential pathogenicity of a variant. Predictors used by VEP were LRT, MutationTaster, FATHMM, PROVEAN, MetaSVM, MetaLR, MetaRNN, PRIMATEAI, DEOGEN2, BayesDel_addAF, ClinPred, fathmm_MFL_coding, fathmm_XF_coding, SIFT, PolyPhen, and Loftool. Intronic and synonymous mutations were studied using ALAMUT software (version 1.4; Sophia Genetics, Switzerland) with the following predictors: Splice Site Analysis (SFF), MaxEnt, Splice Site Prediction by Neural Network (NNSPLICE), and GeneSplicer. The TopMed database was used to assess a mutation’s prevalence through minor allele frequency (MAF). dbSNP refers to the variant ID.
sgRNA, TALEN, and ssODN design
sgRNAs and TALENs were designed using the Invitrogen TrueDesign genome editor (Thermo Fisher Scientific) and are listed in Table 2. sgRNAs were selected according to their predicted efficiency and lowest number of potential off-targets. Mutated sgRNAs were modified to harbor the STGD1-related variant. HTR2A TALEN pairs were used as positive controls for TALEN cleavage assessment. The target locus was amplified using specific forward (5′-AGAAAATTACACAGCAATAAAATATAGCGG-3′) and reverse (5′-CCAATATTAATATGTAGCAAAAAGAGGGAG-3′) primers.
ssODNs were designed as follows: the cutting site was centered, and ssODNs were designed with a total length of 75–85 nt ensuring 30–35 nt left and right arms with perfect sequence homology. Phosphorothioate nucleotide modifications were added to the ends of the ssODNs to increase their stability and were synthesized using a PAGE purification method. ssODN sequences are shown in Figures 2C, 2D, and S2C. PAM sequence modification to induce Cas9-blocking mutation was incorporated in the ssODN repair template with a mutation in the second or third nucleotide of the PAM (NGG). Conservation of the reading frame, amino acid change, and SNP prevalence of the nucleotide modification was performed using ALAMUT software (version 1.4; Sophia Genetics). Nucleotide changes were analyzed using PhyloP and the UCSC Genome Browser.
Genomic cleavage detection assay
To detect locus-specific cleavage of genomic DNA by CRISPR-Cas9 and TALEN, we used the GeneArt Genomic Cleavage Detection Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Briefly, hiPSCs were transfected with sgRNA or TALEN and, 4 days later, PCR amplification of the desired locus was performed. A single band was confirmed by agarose gel electrophoresis. Next, the PCR product was subjected to several rounds of denaturation and re-annealing to generate mismatches that were detected and cleaved by the detection enzyme. The resultant bands were visualized by agarose gel electrophoresis with an iBrightCL1000 (Thermo Fisher Scientific). Quantification of band intensity for correlation with Cas9 activity was done using iBright analysis software (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Human iPSC CRISPR-Cas9-mediated genome editing
For genome editing, 1 × 105 hiPSCs were electroporated with 10 pmol of sgRNA, 15 pmol of ssODN, and 10 pmol of HiFi Cas9 protein (Thermo Fisher Scientific). In parallel, hiPSCs without sgRNA and ssODN were transfected as controls. Electroporation was performed using the Neon transfection system (Thermo Fisher Scientific). The electroporation conditions that were optimal in our cells were two pulses of 1,200 V and 20 ms (referred to as v2), instead of one pulse of 1,000 V and 30 ms (referred to as v1). Immediately after electroporation, hiPSCs were seeded onto Matrigel-coated dishes and cultured in StemFlex medium supplemented with 10 μM ROCK inhibitor (Merck, Bedford, MA, USA), 10 μM HDR activator L755507 (Merck), and 0.5 μM NHEJ inhibitor M3814 (Selleckchem, Houston, TX, USA) for 24 h. Then, the cell culture medium was replaced with fresh StemFlex medium, and the cells were cultured until colonies formed from the single-cell suspension. When colonies had developed but were still small enough to ensure individual clones, more than 50 clones were picked and cultured. After approximately 1 week, individual clones were expanded, and a fraction of cells from each clone was collected for genotyping analysis by Sanger sequencing (Macrogen, Madrid, Spain). The positive and parental clones were further expanded and sequenced again to confirm the desired genotypes.
PCR amplification, Sanger sequencing, and data analysis
PCR amplification of the desired genomic region was performed and run on a gel to ensure a single DNA band and negative control. PCR products were purified using 96-well Acroprep Advance plates (Pall, Ann Arbor, MI, USA) with a vacuum manifold (Pall). The products were Sanger sequenced with forward and/or reverse primers (Macrogen). All primer sequences used in this study are listed in Tables S1 and S3. Sanger sequencing results were downloaded from the manufacturer’s platform and the data were aligned and analyzed.
Off-target prediction and analysis
Off-target prediction was performed using the online tool Cas-OFFinder.61 Three or fewer mismatches were allowed for the algorithm to run the prediction. First, all predicted off-targets with fewer than three mismatches were analyzed. Second, all exonic regions and the intronic ones that were in close proximity to exons were covered by Sanger sequencing. In addition, potential splicing effects in deep-intronic off-targets were predicted using ALAMUT software (Sophia Genetics). Intronic regions likely to be recognized by sgRNAs were subjected to potential splicing analysis in the case of DNA sequence modification according to our ssODN in the entire sgRNA sequence (which includes mismatch substitutions, patient variant modification, and Cas9-blocking mutation in PAM). Intergenic regions were not analyzed in this study. PCR amplification of selected off-targets (summarized in Table S3) was performed in three parental clones and all edited clones and then subjected to Sanger sequencing.
Whole-genome sequencing
WGS was performed in collaboration with Macrogen (Seoul, Korea). Briefly, the latest version of the Ilumina Miseq (Illumina, San Diego, CA, USA) sequencing platform was used. TruSeq Nano was used for library design and preparation. Libraries were then sequenced with NovaSeq6000 (Illumina) according to the manufacturer’s instructions. Paired end reads of 101 nt length were generated and sorted into amplicons.
Data analysis
A WGS data analysis report was performed using GeneSystems software (Sistemas Genómicos, Valencia, Spain), and BAM, BAI, VCF, TSV, and BED files were obtained. WGS reads were aligned against the human reference genome version GRCh38/hg38 using Burrows-Wheeler Aligner62 and “in-house” scripts. After read mapping, low-quality reads and PCR duplicates were removed, and variant calling was done using the GATK algorithm,63 CNVKit64 for CNVs, and Manta65 for the rest of the structural variants. Data were filtered with a minimum coverage of 20×, MAFs lower than 1/10,000, and an allelic fraction for heterozygosity above 0.38. Frequent variants in the patient population of origin (Spanish) were rejected. Identified variants were annotated using the Ensembl database or AnnotSV tool in the case of structural variants.66 CNV data were analyzed considering the following parameters: bin number ≥4; cnid score ≥5; copy number ≤1.5 and ≥2.5.
Immunofluorescence staining
For immunofluorescence analysis of pluripotency markers, iPSC clones were seeded onto Matrigel-coated ibidi slides (ibidi, Gräfeling, Germany) and cultured in StemFlex medium. When colonies formed, the ibidi slides were fixed in 4% paraformaldehyde (Thermo Fisher Scientific) for 15 min at room temperature. Next, the cells were permeabilized with 0.25% Triton X-100 in PBS and incubated for 1 h in a blocking solution (5% fetal bovine serum, 4% bovine serum albumin, and 0.5% Tween in PBS) at room temperature. hiPSC clones were then incubated overnight at 4°C with NANOG (D73G4; Cell Signaling Technology, Beverly, MA, USA), SOX2-AlexaFluor488 (E−4, Santa Cruz Biotechnology, Dallas, TX, USA), SSEA4-AlexaFluor488 (BD Pharmingen, Franklin Lakes, NJ, USA), or TRA-160-AlexaFluor488 (BD Pharmingen) antibodies. Anti-rabbit AlexaFluor488-conjugated secondary antibody (Invitrogen) was used for NANOG staining. Immunofluorescence visualization and imaging were performed with Zeiss Axiovert and Axiocam 503 mono (Carl Zeiss, Jena, Germany). Fluorescence images were processed using ImageJ software (NIH, Bethesda, MD, USA).
RNA extraction and quantitative real-time PCR
To assess the gene expression of pluripotency markers, RNA was extracted using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. cDNA was obtained using a Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics, Basel, Switzerland) and analyzed by real-time PCR using QuantStudio and TaqMan probes (Thermo Fisher Scientific).
Protein extraction and western blotting
Protein from hiPSC cultures was extracted with Pierce RIPA lysis buffer supplemented with a Halt protease inhibitor single-use cocktail (Thermo Fisher Scientific). Lysates were clarified by centrifugation and quantified by Bradford assay. Samples were then boiled and loaded onto 10% polyacrylamide gels and transferred to a polyvinylidene fluoride (PVDF) membrane (Roche). Membranes were blocked with 5% non-fat milk and blotted with the corresponding primary antibodies overnight at 4°C: α-tubulin (Proteintech, Rosemont, IL, USA), OCT4 (9B7; Thermo Fisher Scientific), NANOG (D73G4; Cell Signaling Technology), or SOX2 (D6D9; Cell Signaling Technology). Blots were then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies, and the luminescence reaction was developed with SuperSignal West Pico PLUS chemiluminescent substrate (Thermo Fisher Scientific). Blots were re-probed after incubation with Restore Plus western blot stripping buffer (Thermo Fisher Scientific). Full unedited blot images are included as supplementary figures (Figure S3B).
Statistical analysis
Statistical analysis was performed using Prism 9.3.1 (GraphPad Software, La Jolla, CA, USA). Statistical significance was assessed using the non-parametric Mann-Whitney U test and set at values of p > 0.05. Bar graphs throughout the article show the mean and standard error of the mean.
Acknowledgments
We are indebted to the patients for their participation in the study. Informed consent was obtained from the subjects involved in the study. The authors also thank Bernard Faure for his contribution. This work was supported by a private donation (grant Fi-201401), by a grant from Fundació Bancària “la Caixa” (LCF/PR/PR17/11120006), Barcelona, Spain, and by the Fundació de Recerca de l’Institut de Microcirurgia Ocular de Barcelona, Spain.
Author contributions
L.S. performed the experimental work in the study; S.R.-N. and P.M.-V. performed the validation and methodology analysis; L.S., A.N.-F., and E.P. contributed to project conceptualization and methodology; E.P. conceived and supervised the study; L.S. wrote the manuscript; and E.P. supervised the manuscript. All the authors have provided critical comments on the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2023.02.032.
Supplemental information
Data availability
The authors declare that all relevant data supporting the findings of this study are available within the article and its supplementary information. Raw data and additional results not shown are available from the corresponding author (E.P.) upon reasonable request.
References
- 1.Riera M., Patel A., Burés-Jelstrup A., Corcostegui B., Chang S., Pomares E., Corneo B., Sparrow J.R. Generation of two iPS cell lines (FRIMOi003-A and FRIMOi004-A) derived from Stargardt patients carrying ABCA4 compound heterozygous mutations. Stem Cell Res. 2019;36:101389. doi: 10.1016/j.scr.2019.101389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koenekoop R.K. The gene for Stargardt disease, ABCA4, is a major retinal gene: a mini-review. Ophthalmic Genet. 2003;24:75–80. doi: 10.1076/opge.24.2.75.13996. [DOI] [PubMed] [Google Scholar]
- 3.Tsang S.H., Sharma T. Stargardt disease. Adv. Exp. Med. Biol. 2018:139–151. doi: 10.1007/978-3-319-95046-4_27. [DOI] [PubMed] [Google Scholar]
- 4.Runhart E.H., Dhooge P., Meester-Smoor M., Pas J., Pott J.W.R., van Leeuwen R., Kroes H.Y., Bergen A.A., de Jong-Hesse Y., Thiadens A.A., et al. Stargardt disease: monitoring incidence and diagnostic trends in The Netherlands using a nationwide disease registry. Acta Ophthalmol. 2022;100:395–402. doi: 10.1111/aos.14996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allikmets R., Singh N., Sun H., Shroyer N.F., Hutchinson A., Chidambaram A., Gerrard B., Baird L., Stauffer D., Peiffer A., et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997;15:236–246. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 6.Dahl S.G., Sylte I., Ravna A.W. Structures and models of transporter proteins. J. Pharmacol. Exp. Ther. 2004;309:853–860. doi: 10.1124/jpet.103.059972. [DOI] [PubMed] [Google Scholar]
- 7.Cremers F.P., van de Pol D.J., Van Driel M., den Hollander A.I., van Haren F.J., Knoers N.V., Tijmes N., Bergen A.A., Rohrschneider K., Blankenagel A., et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum. Mol. Genet. 1998;7:355–362. doi: 10.1093/hmg/7.3.355. [DOI] [PubMed] [Google Scholar]
- 8.Stenson P.D., Mort M., Ball E.V., Chapman M., Evans K., Azevedo L., Hayden M., Heywood S., Millar D.S., Phillips A.D., Cooper D.N. The Human Gene Mutation Database (HGMD(®)): optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020;139:1197–1207. doi: 10.1007/s00439-020-02199-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang D., Heath Jeffery R.C., Aung-Htut M.T., McLenachan S., Fletcher S., Wilton S.D., Chen F.K. Stargardt disease and progress in therapeutic strategies. Ophthalmic Genet. 2022;43:1–26. doi: 10.1080/13816810.2021.1966053. [DOI] [PubMed] [Google Scholar]
- 10.Albert S., Garanto A., Sangermano R., Khan M., Bax N.M., Hoyng C.B., Zernant J., Lee W., Allikmets R., Collin R.W.J., Cremers F.P.M. Identification and rescue of splice defects caused by two neighboring deep-intronic ABCA4 mutations underlying stargardt disease. Am. J. Hum. Genet. 2018;102:517–527. doi: 10.1016/j.ajhg.2018.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piotter E., McClements M.E., MacLaren R.E. Therapy approaches for stargardt disease. Biomolecules. 2021;11:1179. doi: 10.3390/biom11081179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cremers F.P.M., Lee W., Collin R.W.J., Allikmets R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020;79:100861. doi: 10.1016/j.preteyeres.2020.100861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu L.J., Liu J., Adelman R.A. Novel therapeutics for Stargardt disease. Graefes Arch. Clin. Exp. Ophthalmol. 2017;255:1057–1062. doi: 10.1007/s00417-017-3619-8. [DOI] [PubMed] [Google Scholar]
- 14.Trapani I. Vol. 1715. 2018. Dual AAV vectors for stargardt disease. (Methods in Molecular Biology). [DOI] [PubMed] [Google Scholar]
- 15.Tornabene P., Trapani I., Centrulo M., Marrocco E., Minopoli R., Lupo M., Iodice C., Gesualdo C., Simonelli F., Surace E.M., Auricchio A. Inclusion of a degron reduces levelsof undesired inteins after AAV-mediated proteintrans-splicing in the retina. Mol. Ther. Methods Clin. Dev. 2021;23:448–459. doi: 10.1016/j.omtm.2021.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun D., Schur R.M., Sears A.E., Gao S.-Q., Vaidya A., Sun W., Maeda A., Kern T., Palczewski K., Lu Z.-R. Non-viral gene therapy for stargardt disease with ECO/pRHO-ABCA4 self-assembled Nanoparticles. Mol. Ther. 2020;28:293–303. doi: 10.1016/j.ymthe.2019.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bansal M., Acharya S., Sharma S., Phutela R., Rauthan R., Maiti S., Chakraborty D. CRISPR Cas9 based genome editing in inherited retinal dystrophies. Ophthalmic Genet. 2021;42:365–374. doi: 10.1080/13816810.2021.1904421. [DOI] [PubMed] [Google Scholar]
- 18.Gaj T., Gersbach C.A., Barbas C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaminski M.M., Abudayyeh O.O., Gootenberg J.S., Zhang F., Collins J.J. CRISPR-based diagnostics. Nat. Biomed. Eng. 2021;5:643–656. doi: 10.1038/s41551-021-00760-7. [DOI] [PubMed] [Google Scholar]
- 20.Lander E.S. The heroes of CRISPR. Cell. 2016;164:18–28. doi: 10.1016/j.cell.2015.12.041. [DOI] [PubMed] [Google Scholar]
- 21.Sander J.D., Joung J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J.Y., Doudna J.A. CRISPR technology: a decade of genome editing is only the beginning. Science. 2023;379:eadd8643. doi: 10.1126/science.add8643. [DOI] [PubMed] [Google Scholar]
- 23.Javaid D., Ganie S.Y., Hajam Y.A., Reshi M.S. CRISPR/Cas9 system: a reliable and facile genome editing tool in modern biology. Mol. Biol. Rep. 2022;49:12133–12150. doi: 10.1007/s11033-022-07880-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sen T., Thummer R.P. CRISPR and iPSCs: recent developments and future perspectives in neurodegenerative disease modelling, research, and therapeutics. Neurotox. Res. 2022;40:1597–1623. doi: 10.1007/s12640-022-00564-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park C.Y., Sung J.J., Choi S.H., Lee D.R., Park I.H., Kim D.W. Modeling and correction of structural variations in patient-derived iPSCs using CRISPR/Cas9. Nat. Protoc. 2016;11:2154–2169. doi: 10.1038/nprot.2016.129. [DOI] [PubMed] [Google Scholar]
- 26.Ozgül R.K., Durukan H., Turan A., Öner C., Ogüs A., Farber D.B. Molecular analysis of the ABCA4 gene in Turkish patients with Stargardt disease and retinitis pigmentosa. Hum. Mutat. 2004;23:523. doi: 10.1002/humu.9236. [DOI] [PubMed] [Google Scholar]
- 27.Lewis R.A., Shroyer N.F., Singh N., Allikmets R., Hutchinson A., Li Y., Lupski J.R., Leppert M., Dean M. Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am. J. Hum. Genet. 1999;64:422–434. doi: 10.1086/302251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaakson K., Zernant J., Külm M., Hutchinson A., Tonisson N., Glavač D., Ravnik-Glavač M., Hawlina M., Meltzer M.R., Caruso R.C., et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum. Mutat. 2003;22:395–403. doi: 10.1002/humu.10263. [DOI] [PubMed] [Google Scholar]
- 29.Fujinami K., Lois N., Davidson A.E., Mackay D.S., Hogg C.R., Stone E.M., Tsunoda K., Tsubota K., Bunce C., Robson A.G., et al. A longitudinal study of Stargardt disease: clinical and electrophysiologic assessment, progression, and genotype correlations. Am. J. Ophthalmol. 2013;155:1075–1088.e13. doi: 10.1016/j.ajo.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 30.Sentmanat M.F., Peters S.T., Florian C.P., Connelly J.P., Pruett-Miller S.M. A survey of validation strategies for CRISPR-cas9 editing. Sci. Rep. 2018;8:888. doi: 10.1038/s41598-018-19441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okamoto S., Amaishi Y., Maki I., Enoki T., Mineno J. Highly efficient genome editing for single-base substitutions using optimized ssODNs with Cas9-RNPs. Sci. Rep. 2019;9:4811. doi: 10.1038/s41598-019-41121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richardson C.D., Ray G.J., DeWitt M.A., Curie G.L., Corn J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016;34:339–344. doi: 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- 33.Fuster-García C., García-García G., González-Romero E., Jaijo T., Sequedo M.D., Ayuso C., Vázquez-Manrique R.P., Millán J.M., Aller E. USH2A gene editing using the CRISPR system. Mol. Ther. Nucleic Acids. 2017;8:529–541. doi: 10.1016/j.omtn.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simkin D., Papakis V., Bustos B.I., Ambrosi C.M., Ryan S.J., Baru V., Williams L.A., Dempsey G.T., McManus O.B., Landers J.E., et al. Homozygous might be hemizygous: CRISPR/Cas9 editing in iPSCs results in detrimental on-target defects that escape standard quality controls. Stem Cell Rep. 2022;17:993–1008. doi: 10.1016/j.stemcr.2022.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kong J., Kim S.R., Binley K., Pata I., Doi K., Mannik J., Zernant-Rajang J., Kan O., Iqball S., Naylor S., et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Ther. 2008;15:1311–1320. doi: 10.1038/gt.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanjurjo-Soriano C., Erkilic N., Baux D., Mamaeva D., Hamel C.P., Meunier I., Roux A.F., Kalatzis V. Genome editing in patient iPSCs corrects the most prevalent USH2A mutations and reveals intriguing mutant mRNA expression profiles. Mol. Ther. Methods Clin. Dev. 2020;17:156–173. doi: 10.1016/j.omtm.2019.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shams F., Bayat H., Mohammadian O., Mahboudi S., Vahidnezhad H., Soosanabadi M., Rahimpour A. Advance trends in targeting homology-directed repair for accurate gene editing: an inclusive review of small molecules and modified CRISPR-Cas9 systems. Bioimpacts. 2022;12:371–391. doi: 10.34172/bi.2022.23871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park C.Y., Kim D.H., Son J.S., Sung J.J., Lee J., Bae S., Kim J.H., Kim D.W., Kim J.S. Functional correction of large factor VIII gene chromosomal inversions in hemophilia A patient-derived iPSCs using CRISPR-cas9. Cell Stem Cell. 2015;17:213–220. doi: 10.1016/j.stem.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 39.Park C.Y., Halevy T., Lee D.R., Sung J.J., Lee J.S., Yanuka O., Benvenisty N., Kim D.W. Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived neurons. Cell Rep. 2015;13:234–241. doi: 10.1016/j.celrep.2015.08.084. [DOI] [PubMed] [Google Scholar]
- 40.Bin Moon S., Lee J.M., Kang J.G., Lee N.E., Ha D.I., Kim D.Y., Kim S.H., Yoo K., Kim D., Ko J.H., Kim Y.S. Highly efficient genome editing by CRISPR-Cpf1 using CRISPR RNA with a uridinylate-rich 3′-overhang. Nat. Commun. 2018;9:3651. doi: 10.1038/s41467-018-06129-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cring M.R., Sheffield V.C. Gene therapy and gene correction: targets, progress, and challenges for treating human diseases. Gene Ther. 2022;29:3–12. doi: 10.1038/s41434-020-00197-8. [DOI] [PubMed] [Google Scholar]
- 42.Garafalo A.V., Cideciyan A.V., Héon E., Sheplock R., Pearson A., WeiYang Yu C., Sumaroka A., Aguirre G.D., Jacobson S.G. Progress in treating inherited retinal diseases: early subretinal gene therapy clinical trials and candidates for future initiatives. Prog. Retin. Eye Res. 2020;77:100827. doi: 10.1016/j.preteyeres.2019.100827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodrigues G.A., Shalaev E., Karami T.K., Cunningham J., Slater N.K.H., Rivers H.M. Pharmaceutical development of AAV-based gene therapy products for the eye. Pharm. Res. (N. Y.) 2018;36:29. doi: 10.1007/s11095-018-2554-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Urnov F.D. CRISPR–Cas9 can cause chromothripsis. Nat. Genet. 2021;53:768–769. doi: 10.1038/s41588-021-00881-4. [DOI] [PubMed] [Google Scholar]
- 45.Hu X., Zhang B., Li X., Li M., Wang Y., Dan H., Zhou J., Wei Y., Ge K., Li P., Song Z. The application and progression of CRISPR/Cas9 technology in ophthalmological diseases. Eye. 2022 doi: 10.1038/s41433-022-02169-1. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeballos C M.A., Gaj T. Next-generation CRISPR technologies and their applications in gene and cell therapy. Trends Biotechnol. 2021;39:692–705. doi: 10.1016/j.tibtech.2020.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salas A., Duarri A., Fontrodona L., Ramírez D.M., Badia A., Isla-Magrané H., Ferreira-de-Souza B., Zapata M.Á., Raya Á., Veiga A., García-Arumí J. Cell therapy with hiPSC-derived RPE cells and RPCs prevents visual function loss in a rat model of retinal degeneration. Mol. Ther. Methods Clin. Dev. 2021;20:688–702. doi: 10.1016/j.omtm.2021.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhargava N., Thakur P., Muruganandam T.P., Jaitly S., Gupta P., Lohani N., Goswami S.G., Saravanakumar V., Bhattacharya S.K., Jain S., Ramalingam S. Development of an efficient single-cell cloning and expansion strategy for genome edited induced pluripotent stem cells. Mol. Biol. Rep. 2022;49:7887–7898. doi: 10.1007/s11033-022-07621-9. [DOI] [PubMed] [Google Scholar]
- 49.De Angeli P., Reuter P., Hauser S., Schöls L., Stingl K., Wissinger B., Kohl S. Effective splicing restoration of a deep-intronic ABCA4 variant in cone photoreceptor precursor cells by CRISPR/SpCas9 approaches. Mol. Ther. Nucleic Acids. 2022;29:511–524. doi: 10.1016/j.omtn.2022.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jo D.H., Song D.W., Cho C.S., Kim U.G., Lee K.J., Lee K., Park S.W., Kim D., Kim J.H., Kim J.S., et al. CRISPR-Cas9–mediated therapeutic editing of Rpe65 ameliorates the disease phenotypes in a mouse model of Leber congenital amaurosis. Sci. Adv. 2019;5:eaax1210. doi: 10.1126/sciadv.aax1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roux L.N., Petit I., Domart R., Concordet J.P., Qu J., Zhou H., Joliot A., Ferrigno O., Aberdam D. Modeling of aniridia-related keratopathy by CRISPR/Cas9 genome editing of human limbal epithelial cells and rescue by recombinant PAX6 protein. Stem Cell. 2018;36:1421–1429. doi: 10.1002/stem.2858. [DOI] [PubMed] [Google Scholar]
- 52.Kantor A., McClements M.E., Maclaren R.E. Crispr-cas9 dna base-editing and prime-editing. Int. J. Mol. Sci. 2020;21:6240. doi: 10.3390/ijms21176240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stemmer M., Thumberger T., Del Sol Keyer M., Wittbrodt J., Mateo J.L. CCTop: an intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS One. 2015;10:e0124633. doi: 10.1371/journal.pone.0124633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh R., Kuscu C., Quinlan A., Qi Y., Adli M. Cas9-chromatin binding information enables more accurate CRISPR off-target prediction. Nucleic Acids Res. 2015;43:e118. doi: 10.1093/nar/gkv575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Montague T.G., Cruz J.M., Gagnon J.A., Church G.M., Valen E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014;42:W401–W407. doi: 10.1093/nar/gku410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cromer M.K., Barsan V.V., Jaeger E., Wang M., Hampton J.P., Chen F., Kennedy D., Xiao J., Khrebtukova I., Granat A., et al. Ultra-deep sequencing validates safety of CRISPR/Cas9 genome editing in human hematopoietic stem and progenitor cells. Nat. Commun. 2022;13:4724. doi: 10.1038/s41467-022-32233-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barbitoff Y.A., Abasov R., Tvorogova V.E., Glotov A.S., Predeus A.V. Systematic benchmark of state-of-the-art variant calling pipelines identifies major factors affecting accuracy of coding sequence variant discovery. BMC Genom. 2022;23:155. doi: 10.1186/s12864-022-08365-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jayakody S.A., Gonzalez-Cordero A., Ali R.R., Pearson R.A. Cellular strategies for retinal repair by photoreceptor replacement. Prog. Retin. Eye Res. 2015;46:31–66. doi: 10.1016/j.preteyeres.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 59.Sanie-Jahromi F., Nowroozzadeh M.H. RPE based gene and cell therapy for inherited retinal diseases: a review. Exp. Eye Res. 2022;217:108961. doi: 10.1016/j.exer.2022.108961. [DOI] [PubMed] [Google Scholar]
- 60.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. The Ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Talevich E., Shain A.H., Botton T., Bastian B.C. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 2016;12:e1004873. doi: 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen X., Schulz-Trieglaff O., Shaw R., Barnes B., Schlesinger F., Källberg M., Cox A.J., Kruglyak S., Saunders C.T. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–1222. doi: 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 66.Geoffroy V., Herenger Y., Kress A., Stoetzel C., Piton A., Dollfus H., Muller J. An integrated tool for structural variations annotation. Bioinformatics. 2018;34:3572–3574. doi: 10.1093/bioinformatics/bty304. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all relevant data supporting the findings of this study are available within the article and its supplementary information. Raw data and additional results not shown are available from the corresponding author (E.P.) upon reasonable request.