

Abstract
Heart failure with preserved ejection fraction (HFpEF) represents one of the greatest challenges facing cardiovascular medicine today. Despite being the most common form of HF worldwide, there has been limited success in developing therapeutics for this syndrome. This is largely due to our incomplete understanding of the biology driving its systemic pathophysiology and the heterogeneity of clinical phenotypes, which are increasingly being recognized as distinct HFpEF phenogroups. Development of efficacious therapeutics fundamentally relies on robust preclinical models that not only faithfully recapitulate key features of the clinical syndrome but also enable rigorous investigation of putative mechanisms of disease in the context of clinically relevant phenotypes. In this review, we propose a preclinical research strategy that is conceptually grounded in model diversification and aims to better align with our evolving understanding of the heterogeneity of clinical HFpEF. Although heterogeneity is often viewed as a major obstacle in preclinical HFpEF research, we challenge this notion and argue that embracing it may be the key to demystifying its pathobiology. Here we first provide an overarching guideline for developing HFpEF models through a stepwise approach of comprehensive cardiac and extra-cardiac phenotyping. We then present an overview of currently available models, focused on the three leading phenogroups, which are primarily based on aging, cardiometabolic stress, and/or chronic hypertension. We discuss how well these models reflect their clinically relevant phenogroup and highlight some of the more recent mechanistic insights they are providing into the complex pathophysiology underlying HFpEF.
Subject Terms: Basic Science Research, Animal Models of Human Disease, Cardiovascular Disease, Heart Failure
Keywords: HFpEF, preclinical models, aging, cardiometabolic, hypertension
Introduction
The global prevalence of heart failure (HF) is estimated at >30 million individuals. Rapidly increasing in incidence and prevalence, HF is thought to have contributed to 1 in 8 deaths in 20171,2. Two major phenotypes of HF are recognized: HF with reduced ejection fraction (HFrEF) and HF with preserved ejection fraction (HFpEF)3,4. HFpEF is a syndrome marked by substantial morbidity and mortality, including a 35% two-year rate of HF hospitalization and 14% two-year mortality5. Importantly, HFpEF has been rising over the past decade by 10% relative to HFrEF, and this gap is projected to increase owing to the aging of the population and the increasing prevalence of conditions that predispose to its development, particularly hypertension, obesity, metabolic syndrome, and diabetes5–8. Already, HFpEF is the most common form of HF.
Whereas HFrEF and HFpEF present similarly as HF (shortness of breath, edema, exercise intolerance), recent evidence, both preclinical and clinical, supports a model in which the two syndromes are mechanistically distinct pathophysiological entities. In addition to evidence emerging from analyses of gene expression patterns in both preclinical9–12 and human specimens13, the fact that neurohormonal therapies established to be effective in HFrEF have failed to improve clinical outcomes in HFpEF supports this concept. Also, transition from HFpEF to HFrEF in patients is rare5,14. In aggregate, these data lend considerable credence to the notion that HFrEF and HFpEF are mechanistically dissimilar.
Despite the increasing recognition of distinct pathophysiology, the biological mechanisms underlying HFpEF remain largely unclear. It is important to recognize that HFpEF is a complex clinical syndrome, not a single disease, one characterized by heterogeneous clinical manifestations, high comorbidity burden, and multiorgan systemic pathophysiology. Abnormalities in the heart (such as concentric left ventricular hypertrophy, left atrial remodeling, diastolic dysfunction, impaired cardiac reserves) are frequently present in HFpEF patients15. Similarly, non-cardiac features, such as systemic and pulmonary vascular dysfunction, skeletal muscle impairments, body composition changes, are also widespread in HFpEF and highly variable among patients16,17. Indeed, within the syndrome of HFpEF, distinct phenogroups can be identified stemming from different predisposing conditions and unique responses to treatment6,14,18,19. Also, different HFpEF phenogroups may have dissimilar geographic distributions. For example, hypertensive, stiff, hypertrophic hearts in lean HFpEF patients (“skinny HFpEF”) are encountered more commonly in Asia than in the West20.
Our limited understanding of the molecular mechanisms responsible for the phenotypic heterogeneity in HFpEF has led not only to a lack of consensus on how to define this clinical entity but has also created a major obstacle to generating effective therapeutics. If drug discovery and development in HFpEF are to accelerate and treatment strategies improve, it is critical that we develop a better understanding of specific mechanisms driving the complexity and variability in HFpEF pathophysiology. This will inevitably require an integrated approach among basic, translational, clinical, and epidemiological scientists. In clinical research, the “one-size-fits-all” approach previously used in HFrEF has evolved into a more phenotype-specific research approach. This strategy focuses on classifying the heterogenous HFpEF population into sub-types that are more homogenous based on distinct characteristics observed clinically, largely driven by associated comorbidities, clinical features, myocardial phenotypes, or phenomapping21. Arguably the HFpEF field has been led astray numerous times owing to over-reliance on models that purport to reflect HFpEF but do not recapitulate the clinical realities of the syndrome or manifest major features rarely seen in clinical HFpEF. For example, models marked by transition to HFrEF and termed “temporary HFpEF”, or the presumption that diastolic dysfunction is tantamount to “diastolic heart failure” or HFpEF, are errors found too frequently in the published literature.
As elucidation of pathophysiological mechanisms of disease relies on the availability and veracity of animal and cellular models, the existence of multiple phenotypes of clinical HFpEF necessitates that preclinical researchers similarly adapt to the evolving field and fully embrace the heterogeneity of the syndrome. Instead of expecting a single animal model to reproduce all aspects of the diverse clinical manifestations of the HFpEF syndrome, we propose that model development should be multifold and focus on capturing the hallmark features of specific phenogroups in clinical HFpEF. This strategy would not only be more faithful to the heterogeneity of the clinical syndrome, but diversifying our preclinical models better aligns with emerging clinical strategies to personalize treatment approaches in HFpEF.
To that end, this review begins by presenting a generalized strategy for developing animal models of HFpEF that is fundamentally rooted in capturing clinically relevant phenotypes of this syndrome. The goal of this review is not to present every proposed model of HFpEF, which are not only rapidly increasing in number but have also been reviewed in depth in prior references22–25. Rather, we highlight the currently available models that we collectively feel best represent the three leading HFpEF phenogroups, those primarily based on the leading risk factors of advanced age, cardiometabolic stress, and chronic hypertension. We discuss how these models capture the key features of their respective phenogroup and HFpEF, in general. We then highlight some of the recent mechanistic insights they have provided, and comment on what we believe are some important next steps to better understanding the pathobiology underlying these major phenogroups.
Developing HFpEF Models: A Stepwise Phenotype-based Approach
Assessing whether an animal model accurately recapitulates the clinical syndrome of HFpEF is undeniably challenging. The inability of animals to express the cardinal symptoms of HF, breathlessness and fatigue, make modeling HF in animals inherently difficult. In HFpEF, the varying degrees by which multiple cardiac and extra-cardiac factors contribute to the underlying pathophysiology further complicates our ability to model this complex syndrome. Moreover, unlike HFrEF, which can develop rapidly after an acute cardiac injury (e.g., myocardial infarction), HFpEF is a more chronic HF syndrome that generally develops after years of insidious myocardial and extra-cardiac tissue remodeling. The emphasis placed on developing “quick” models to shorten the experimental timeframe in preclinical research is thus at direct odds with the chronicity that underlies clinical HFpEF.
Despite these challenges, it is incumbent on basic and translational scientists to develop models that are not only faithful to the clinical syndrome but can also be easily reproduced and utilized by others in our collective quest to unveil molecular mechanisms driving HFpEF pathophysiology. Indeed, this has been recognized as a critical shortcoming in the field that needs to be addressed in order for meaningful advances in therapeutic development to emerge26,27.
In an attempt to provide some consensus to this rapidly evolving field, we propose the following stepwise phenotyping strategy in developing animal models of HFpEF. This recognizes that no single model will fully recapitulate the multitude of pathophysiological features associated with the notoriously heterogeneous syndrome of clinical HFpEF. Later in this review, we highlight animal models that we believe best represent the leading HFpEF phenogroups, and in which this generalized phenotyping approach can be tailored to capture the phenotypes most relevant to that specific phenogroup. Notably, the general algorithm presented here emphasizes small animal models, but much of this strategy can and should be translated to large animals. Figure 1: Flowchart for developing HFpEF models.
Figure 1: A stepwise phenotype-based approach to developing HFpEF models.
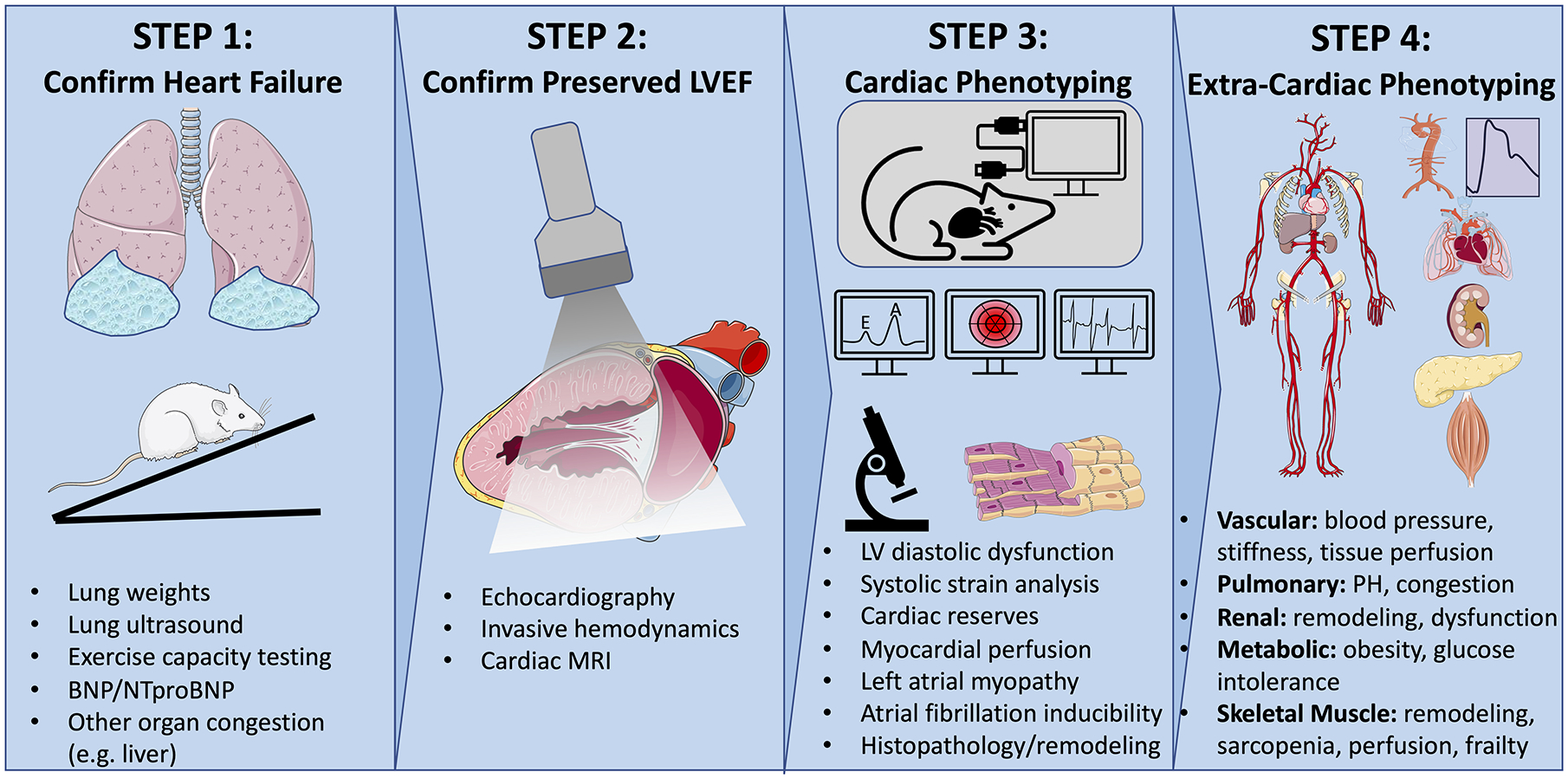
HFpEF is a clinical syndrome with complex systemic pathophysiology that includes functional impairments in both cardiac and extra-cardiac tissues. Developing reliable animal models of HFpEF requires that investigators rigorously assess and validate multiple clinically relevant phenotypes, starting with the hallmark features of HF, congestion and exercise intolerance (1st panel). Next, preserved left ventricular ejection fraction, a defining feature of HFpEF, should be confirmed (2nd panel). Additional cardiac and extra-cardiac phenotyping should then be assessed, and tailored to the specific phenogroup, being modeled (3rd and 4th panels). The illustration was made using images from smart.servier.com. Illustration credit: Ben Smith
Step 1: Confirm the presence of hallmark HF phenotypes
Before even proposing a model of HFpEF (or HFrEF), it is critical that one is confident that the model exhibits key features of HF pathophysiology, which includes congestion and/or exercise intolerance. Clinically, HF is diagnosed by the development of symptomatic dyspnea or fatigue, which is then corroborated by signs of volume overload, elevated filling pressures, impaired exercise capacity, and/or altered tissue perfusion from reduced cardiac output. Of course, symptoms cannot be elicited from an animal, but pulmonary congestion and exercise capacity can be objectively measured and should be among the first phenotypes evaluated.
Congestion can be assessed directly via lung weights (either wet weight or wet:dry weight ratio) or invasive hemodynamics. As obesity and aging are increasingly incorporated into HFpEF models, we encourage investigators to report lung weights normalized not only to tibia length but also to body weight. These comorbid processes are associated with overall physiological increases in organ weights, and thus, increased lung weights may not necessarily indicate pathology or congestion when compared to leaner or younger controls. Notably, a major limitation of these methods is that they can only be performed either post-mortem or during terminal procedures. To circumvent this shortcoming, recent advances in lung ultrasound herald promise in noninvasively assessing pulmonary edema in animals28, which could allow for serial assessments throughout a study. Additionally, circulating BNP, an established clinical HF biomarker that serves as an indicator of increased myocardial wall stress, can be measured in serum or plasma and has been associated with pathologic cardiac remodeling and dysfunction in mice29.
Similarly, we recommend that exercise capacity be thoroughly assessed in every animal model of HFpEF. Exercise intolerance is a hallmark feature of HF, and exercise capacity is an objective metric than can be accurately assessed with thorough methodological training and rigorous compliance to established protocols. The details of animal exercise testing are beyond the scope of this review, but we point the interested reader to several references that provide detailed protocols30,31. Notably, there is currently no consensus in the field regarding standard exercise testing protocols in preclinical HFpEF studies. In an effort to mimic what is done clinically32, we suggest that investigators use graded treadmill protocols, which not only enable fairly controlled assessment of peak exercise capacity, but can be paired with other technologies to assess cardiac reserves33–35. These protocols usually run mice at 10–20° inclines with progressive increases in treadmill speed until animal exhaustion. Similar to advocating for normalization of lung weight to body weight, in addition to tibia length, we recommend that investigators report exercise capacity readouts that appropriately adjust for the animal’s body weight. Weight is a critical factor in determining the amount of work an animal can accomplish during an exercise test and is markedly influenced by both obesity and aging, which are increasingly being incorporated into HFpEF model development. Work achieved on a treadmill exercise test is essentially the product of the animal weight, acceleration due to gravity, and the vertical height traveled, the latter of which incorporates the treadmill grade, velocity profile, and total time run.
Step 2: Confirm preserved left ventricular ejection fraction
Once a HF phenotype is confirmed in the animal model, via congestion and/or exercise intolerance (preferably both), a thorough assessment of cardiac contractile performance should be performed. This can be done via echocardiography, cardiac MRI, and/or invasive hemodynamics. Detailed methodology for these techniques is beyond the scope of this review, but we refer the interested reader to the following references for more information36–38.
In generating HFpEF models, robust validation of preserved left ventricular ejection fraction (LVEF) is critical, as this is a defining feature of the syndrome. With its wide availability and relatively low cost, echocardiography has become the most common method for assessing cardiac function in animals and can be performed with or without sedation. If the former is used, careful attention to anesthesia selection and depth, and their potential confounding effects on myocardial function, should be addressed36,39. The recommended method for quantifying LVEF in clinical echocardiography is Simpson’s biplane method of discs, which integrates multiple apical views to measure LV volumes at end-diastole and end-systole. In small animals, apical views are generally not of sufficient quality to accurately measure LV volumes, and thus LVEF is usually calculated from volumes acquired from parasternal long axis views or extrapolated from linear dimensions from the parasternal short axis view. Both of these methods have limitations, including multiple assumptions regarding the symmetry and uniformity of LV geometry. Careful attention should be focused on ensuring that parasternal long axis views are not foreshortened, and if using linear measurements, we recommend that investigators also report fractional shortening. If feasible, further validation of LVEF preservation with cardiac MRI or invasive hemodynamics is often useful and can provide additional confidence in this core feature of HFpEF.
Although LVEF is preserved in HFpEF, it is important to note that cardiac contractile performance is not perfectly normal. The ability of the heart to augment its output under physiologic or pathologic stress is markedly impaired in HFpEF due to deficiencies in cardiac reserves, which include inotropic, chronotropic, and lusitropic properties40,41. Whereas this hallmark cardiac phenotype of HFpEF is well established in the clinical community, it has been largely ignored in preclinical HFpEF research. Given the clinical relevance of impaired cardiac reserve in HFpEF pathophysiology, we recommend that this cardiac phenotype not only be incorporated in model development and validation, but also in the assessment of therapeutic efficacy of experimental interventions. Both implantable telemetry units33,34 and echocardiography35,41 have been paired with exercise-induced stress in rodents, and can be used reliably assess cardiac reserves in small animal models of HFpEF. Additionally, hemodynamic stress with dobutamine, isoproterenol, phenylephrine, or volume challenges can be paired with invasive hemodynamics42,43 or cardiac MRI44 to assess cardiac reserve, but this requires obligate anesthesia, which in itself, can alter the myocardial response to stress.
Step 3: Evaluate additional common cardiac phenotypes
In addition to impaired cardiac reserve, many HFpEF patients manifest evidence of subclinical systolic and diastolic dysfunction at rest, reflective of abnormal mechanical properties of the myocardium41,45,46. Subclinical systolic dysfunction, also referred to as impaired myocardial strain or deformation, and diastolic dysfunction, which is more reflective of the lusitropic properties of the heart, are most often assessed by echocardiography or invasive hemodynamics. Technical details for these methods are provided in the following references36,42,46. Whereas these cardiac functional phenotypes can provide additional credence to a given model, it is important to note that although impaired systolic strain and diastolic dysfunction are common in HFpEF, these metrics are also seen in many individuals without HFpEF (e.g., untreated hypertensive individuals or healthy older adults without HF), and thus have limited specificity and sensitivity in the diagnosis of the syndrome.
Histopathologic studies have afforded valuable insights into the adverse myocardial structural remodeling associated with HFpEF in humans. Manifestations of modest cardiomyocyte hypertrophy, myocardial fibrosis, and microvascular rarefaction are commonly observed in HFpEF patients47, and should be thoroughly evaluated in all HFpEF models with standard (immuno)histological techniques. Noninvasive approaches to assess cardiac remodeling and myocardial perfusion without sacrificing the animal can also be accomplished, but often require advanced imaging with cardiac MRI, contrast echocardiography, and/or PET36. In rodents, transcriptional profiles associated with pathologic cardiac hypertrophy, which include upregulation of the MYH7 isoform and natriuretic peptides (NPPA and NPPB), can provide additional molecular support for a pathological, as opposed to physiological, growth process in the heart35,48.
Lastly, although it is not commonly included in HFpEF model characterization at this time, we suggest that investigators consider incorporating arrhythmogenic potential, specifically atrial fibrillation (AF) inducibility, in the initial cardiac phenotyping of their models. AF is highly prevalent in HFpEF, occurring in 30–50% of these patients49,50. Whether AF drives HFpEF or vice versa is unclear, but their coexistence strongly suggests that they share similar pathophysiology. Indeed, many of the interventions used to induce HFpEF in rodents, including obesity and increased afterload, also increase AF inducibility51, further highlighting that the mechanisms inducing HFpEF phenotypes in animals likely contribute to AF pathophysiology.
Step 4: Evaluate common extra-cardiac phenotypes
As highlighted throughout this review, multiple cardiac, but also noncardiac impairments, contribute to the systemic pathophysiology underlying HFpEF. Whereas rigorous cardiac phenotyping has been a mainstay in HFpEF model characterization to date, it is equally important to address the non-cardiac phenotypes. Details regarding the relevance of non-cardiac phenotypes in specific HFpEF subgroup models will be highlighted in the following sections and should be prioritized accordingly. However, first we provide a more general overview of how one might approach extra-cardiac phenotyping in HFpEF models.
Systemic and pulmonary vascular remodeling are both major contributors to HFpEF pathophysiology. Systemic arterial hypertension is the most common risk factor in HFpEF41, and blood pressure (BP) should be reported in every model. BP can be measured via invasive hemodynamics but can also be assessed using non-terminal tail cuff systems or telemetry units52. It is important to note that it is not only the absolute difference in arterial blood pressure that matters, but ventricular-vascular mismatch that occurs with arterial stiffening also plays an important role in certain subgroups of HFpEF, especially older patients53. Similar to humans, arterial stiffness can be assessed in animals using noninvasive ultrasound techniques that measure aortic pulse wave velocity54. Pulmonary artery pressures, pulmonary vascular resistance, and right ventricular (RV) function are more challenging to assess in animals, and often require terminal invasive hemodynamic techniques55. However, noninvasive methods using echocardiography and cardiac MRI are available and can provide direct measurements of RV structure and function and pulmonary artery size, along with some indirect assessments of pulmonary hemodynamics56,57.
Impairments in peripheral oxygen extraction in skeletal muscle are increasingly recognized as major contributors to exercise intolerance in HFpEF41,58,59. Like the heart, skeletal muscle in HFpEF patients manifests changes in myocyte fiber type distribution and reduced capillary density60, which can be assessed with standard (immuno)histological methods. Contrast ultrasonography, MRI, and laser Doppler methods can also be used for functional assessment of skeletal muscle perfusion61–63.
Chronic kidney disease (CKD) is highly prevalent in HFpEF, present in nearly 50% of patients64. Similar to the striking co-existence of AF with HFpEF, the frequency with which CKD is observed in HFpEF suggests that common pathobiology drives both processes and that the relationship between these two entities is not just associative but rather causal. Assessing kidney function in HFpEF models is rarely performed, but we propose that given its prominent role in HFpEF pathophysiology, it should be measured regularly. In addition to histopathologic assessments of structural remodeling in the kidney, renal function can be reliably evaluated in rodents by measuring serum creatinine (preferably via enzymatic or HPLC methods) or cystatin C levels65,66.
All the aforementioned non-cardiac processes may have direct influences on cardiac physiology and hemodynamics, but multiple other comorbidities, including diabetes, obesity, anemia, coronary artery disease, and chronic pulmonary disease are also highly prevalent in clinical HFpEF. Whereas hemodynamically significant coronary artery disease and chronic pulmonary disease are not commonly present in small animals, even with metabolic, hypertensive, or aging interventions, glucose intolerance and obesity are associated with some of these interventions, and should be assessed with standard laboratory and biomorphic measurements.
Current Models of the Major HFpEF Phenogroups:
AGE-ASSOCIATED HFpEF:
HFpEF is inextricably linked to aging. Even before its formal recognition as a distinct HF phenotype, some of the first case reports describing the syndrome suggested it might represent a hypertensive cardiomyopathy exclusive to the elderly67. Indeed, HFpEF constitutes the vast majority of incident HF in older adults68, and the rising prevalence of HFpEF is largely attributed to the aging of populations worldwide5. Although age-related HFpEF inevitably shares many features with other HFpEF phenogroups, this specific phenogroup is not only associated with more adverse myocardial and electrical remodeling, but also more diffuse extra-cardiac impairments in the vascular, renal, pulmonary, and skeletal muscle systems, suggesting a more systemic process may underlie HFpEF pathophysiology in older patients69–71.
Elegant studies in human cardiovascular physiology over the past 50+ years have provided important insights into how cardiovascular aging potentially contributes to HFpEF pathophysiology72–74. Normal aging, itself, leads to many of the hallmark phenotypes observed in HFpEF. Exercise capacity progressively declines by ~10% per decade in healthy ambulatory individuals, but accelerates after the seventh decade75. Similar to HFpEF, exercise intolerance in older adults derives largely from reductions in cardiac reserve, but impairments in peripheral oxygen extraction also contribute72–74,76. Additionally, progressive age-related arterial stiffening contributes to the development of cardiac hypertrophy, adverse myocardial remodeling, and the subclinical systolic and diastolic dysfunction observed in the elderly72,73.
It is important to note that despite the strikingly high prevalence of HFpEF in the elderly, the majority of older adults do not have HFpEF despite possessing many of its hallmark phenotypes. Although the culmination of structural and functional changes that occur with aging of the cardiovascular system does not necessarily equate to age-related HFpEF, a longstanding hypothesis has been that acceleration or dysregulation of mechanisms associated with this aging process may be the trigger that transitions it to a more pathologic HF phenotype73,77; however, underlying mechanisms remain unclear. Yet, based on this hypothesis, a strategy of discovering causal mechanisms of cardiovascular aging in animals and then testing their therapeutic relevance in other HF models has become a common experimental approach to glean mechanistic insights into age-related HFpEF.
Models:
Aged C57BL/6 Mouse
Cardiovascular aging has been studied in multiple species, but our understanding of the molecular underpinnings driving this process have been derived largely from rodent models that display many of the features found in human cardiovascular aging78 (Table 1). The aged C57BL/6 mouse (generally >24 months old) is by far the most common rodent model used in cardiac aging studies. Extensive cardiovascular phenotyping of the aged C57BL/6 mouse has consistently shown that these animals display many of the hallmark features of HFpEF, including exercise intolerance, congestion, preserved LVEF, impaired systolic strain, diminished cardiac reserve, diastolic dysfunction, LV pathologic remodeling, and arterial stiffness35,79–81. Importantly, these HFpEF phenotypes develop in the absence of overt hypertension, pulmonary vascular disease, glucose intolerance, or obesity, making this model uniquely suited to study age-specific mechanisms that do not necessarily overlap with other HFpEF phenogroups. Interestingly, cardiac HFpEF phenotypes are more pronounced in aged males, compared to similarly aged females82, which is somewhat discordant with the female predominance in age-related HFpEF69.
Table 1:
Phenotype-based characterization of proposed age-associated HFpEF models.
| Aged C57BL/6 Mouse | Aged F344 Rat | SAMP8 Mouse | C57BL/6 + HFD (3mo) +ANGII (1mo) | C57BL/6 +HFD (13mo) +DOCP (1mo) | SAMP8 +WD (4mo) | |
|---|---|---|---|---|---|---|
| Age (months) | 24–30 | 26–36 | 6–12 | 21–25 | 16 | 6 |
| Exercise intolerance | Y | Y | N | N/A | Y | Y |
| Pulmonary congestion | Y | Y | Y | Y | Y | Y |
| Preserved LVEF | Y/N | Y/N | Y | Y | Y | Y |
| Diastolic dysfunction | Y | Y | Y/N | Y | Y | Y |
| Impaired systolic strain | Y | N/A | NA | N/A | N/A | Y |
| Impaired cardiac reserve | Y | N/A | N | N/A | N/A | NA |
| Cardiac hypertrophy | Y | Y | Y/N | Y | Y | Y |
| Hypertension | N | N | N | Y | Y | N |
| Vascular stiffness | Y | Y | N | Y | N/A | N |
| Sarcopenia | Y | Y | N/A | N | N/A | NA |
| Renal dysfunction | Y | Y | Y | N | N/A | N/A |
| AF inducibility | Y | Y | N/A | N/A | N/A | N/A |
Y = yes. N = no. N/A = not available.
There is some disagreement in the field regarding whether the aged C57BL/6 mouse truly represents a model of HFpEF as opposed to physiological cardiovascular aging. Nevertheless, the consistent observation that it recapitulates many of the hallmark HFpEF phenotypes has led investigators to use it as a platform to discover cardiac aging-related mechanisms that can be targeted for HF therapeutics83. A detailed discussion of the rapidly growing number of putative cardiac aging mechanisms that have been discovered in the aged C57BL/6 mouse is beyond the scope of this review, but includes mitochondrial dysfunction79,84, alterations in autophagy80, cellular senescence85,86, TGFβ family signaling41,87, epigenetic modulation88–90, and inflammation91.
Aged Fisher 344 Rat
Like the aged C57BL/6 mouse, the aged Fisher 344 (F344) rat also recapitulates many of the phenotypes seen in human cardiac aging and HFpEF. These changes develop progressively with age and start to manifest around 24–30 months, becoming more prominent at 36 months. At these later timepoints, animals display evidence of exercise intolerance, pathologic cardiac hypertrophy, and diastolic dysfunction92–96. Early studies showed that both aged male and female F344 rats also manifest evidence of systolic dysfunction, which has diminished enthusiasm for using this cardiac aging model to study mechanisms of age-related HFpEF93–95. However, a more recent echocardiographic study of aged F344 rats has suggested that LVEF may be preserved in aged females (at 26 and 30 months)96. These conflicting data will need to be reconciled if this model is to be utilized in age-related HFpEF research.
SAMP8 Mouse
Although the shorter average lifespan of rodents (around 2–3 years) has made them a powerful tool for studying mechanisms of cardiovascular aging and age-related HFpEF, the substantial length of time needed to acquire clinically relevant phenotypes in these animals nevertheless presents a major challenge. The increasing availability of aged rodents through commercial and government-sponsored rodent colonies has alleviated some of this burden, but these resources are often limited by financial and access issues. To circumvent the longer aging period necessary in wild-type rodents, an accelerated model of cardiac aging that has been proposed as a reasonable alternative is the senescence accelerated-prone mouse (SAMP). SAMP mice were generated in the 1970s from selective inbreeding of the AKR/J strain and display variable degrees of accelerated aging phenotypes97. Two studies have undertaken cardiovascular phenotyping in the SAMP8 strain and have reported a modest degree of diastolic dysfunction in these mice within 6 months in the absence of LV systolic dysfunction98,99. However, there is some discordance in cardiac functional phenotypes (e.g. diastolic dysfunction) and myocardial fibrosis. Moreover, neither study showed convincing evidence of cardiac hypertrophy or significant changes in blood pressure or vascular stiffness. Additionally, although six-month-old SAMP8 mice ran slightly less during treadmill exercise testing compared with two month-old SAMP8 mice, this was not adjusted for the increased body weight in older animals, and the modest deficit in exercise capacity was even smaller than that observed in wild-type controls99. As with the aged F344 rat model, more definitive phenotyping of this model will need to be completed to determine whether it can be used to study age-related HFpEF.
Multiple-Hit Aging Models
Although our success in modeling age-related HFpEF in animals has lagged behind rigorous clinical phenotyping and recognition of this older HFpEF phenogroup, the field has begun to address some of the shortcomings and limitations of current models and strategies. More recent efforts are employing a strategy of incorporating prevalent comorbidities, including obesity and hypertension, in aged animals. The rationale behind this strategy is strongly grounded in clinical data that report high prevalence of both these comorbidities in older adults with HFpEF69–71. This strategy essentially proposes that comorbidities may be the necessary trigger that tips an already compromised and vulnerable, aged cardiovascular system into HF.
Three recent studies suggest that this may indeed be a promising approach to modeling age-related HFpEF99–101. Exposure to a high-fat diet (HFD) and renin-angiotensin-aldosterone system (RAAS) stimulants, either angiotensin II (ANGII) or deoxycorticosterone pivalate (DOCP), amplified many of the cardiac and extra-cardiac HFpEF phenotypes observed in age-matched controls (Table 1)100,101. Moreover, exposure of SAMP8 mice to a high-salt and high-fat Western diet (WD) similarly elicited multiple HFpEF phenotypes, including exercise intolerance, pulmonary congestion, pathologic cardiac hypertrophy, and vascular dysfunction, that were not present in age-matched SAMP8 mice.
Recent Mechanistic Insights and Knowledge Gaps:
These more recent studies not only highlight the current trend and rationale for multiple-hit approaches to model development in age-related HFpEF, but importantly are also providing new insights into the pathobiology of this phenogroup. It is notable that both studies in which metabolic and hypertensive stress were incorporated in aged C57BL/6 mice similarly identified mechanisms of metabolic dysregulation and cardiac inflammation as potentially key mediators of HFpEF pathophysiology100,101.
In the aging+HFD+DOCP model101, aberrant ketone metabolism induced mitochondrial dysfunction and NLRP3 inflammasome activation, which interestingly was ameliorated by increasing circulating β-hydroxybutyrate levels. The authors found that increased circulating β-hydroxybutyrate rescued HFpEF phenotypes, not by increasing myocardial energy supplies (as has been proposed in HFrEF), but by inhibiting mitochondrial protein hyperacetylation and inflammation through suppression of fatty acid uptake and citrate synthase activation. These data further support the notion that HFpEF is likely mechanistically distinct from HFrEF and resonates with mechanisms emerging from cardiometabolic HFpEF models (see following section). The lack of HFpEF phenotypes in their age-matched control (16-month-old C57BL/6), however, raises the question of whether metabolic inflammation is specific to age-related HFpEF or more reflective of the underlying mechanisms driving cardiometabolic HFpEF.
The aging+HFD+ANGII model, which incorporates dietary/hypertensive interventions much later in life (starting at 18–22 months) and is likely more consistent with the clinical scenario leading up to age-related HFpEF, also suggests that metabolic inflammation is likely relevant to the pathobiology contributing to age-related HFpEF100. Metabolic, mitochondrial, inflammatory, and TGFβ family signaling pathways were all enriched in the cardiac transcriptomic profiles of these mice compared to age-matched controls. Although young controls were not included in this study for additional comparison to normal physiological aging, all of these biological processes have previously been implicated in cardiac aging83, suggesting that the introduction of HFD+ANGII in late life amplifies these processes beyond what occurs with normal aging.
Ultimately, how these added comorbidities alter intrinsic cardiovascular aging mechanisms that contribute to HFpEF in older adults and how this pathobiology potentially differs from other HFpEF phenogroups needs to be explored further. Exposure of SAMP8 mice to WD is one recent example of how we can start to address these questions99. Although the SAMP8 mouse only recapitulates some features of age-related HFpEF, it does display evidence of endothelial senescence and dysfunction that is thought to contribute to some of its HFpEF phenotypes. The introduction of WD further amplified the accelerated age-related endothelial senescence, inflammation, and dysfunction seen in the SAMP8 HFpEF model. Whether specifically targeting endothelial senescence in these mice rescues HFpEF phenotypes is currently unknown.
Lastly, as alluded to earlier in this section, a key criticism of studying age-related HFpEF pathobiology in naturally aging rodents is the difficulty in discerning whether findings from these studies are reflective of normal cardiovascular aging as opposed to pathologic HFpEF. Validation of putative aging mechanisms in non-age-related HF models can provide support for its relevance in HF pathophysiology but does not necessarily confirm a causal role in age-related HFpEF. Although undoubtedly challenging, we propose that if a cardiovascular aging mechanism is to be interpreted as a causal mediator in age-related HFpEF, gain- and loss-of-function experiments must also be performed in aged animals with emphasis on HFpEF phenotyping. Moreover, given the more systemic pathophysiology appreciated in this phenogroup69–71, additional attention should be paid to many of the extra-cardiac phenotypes, including atrial fibrillation susceptibility, frailty, and skeletal muscle impairments.
CARDIOMETABOLIC HFpEF:
The world is experiencing a global rise in obesity and associated metabolic stress. In 2016, more than 1.9 billion adults worldwide were overweight and, of these, over 650 million were obese (13% of the world’s adult population). In the US, the average body mass index of HFpEF patients is >35 kg/m2 102. Coupled with this is a worsening global epidemic of hypertension. Whereas HFpEF is a heterogenous syndrome presenting as diverse clinical phenotypes, arguably HFpEF elicited by metabolic alterations – i.e., cardiometabolic HFpEF – is emerging as the most prevalent form of HFpEF.
A preponderance of patients with HFpEF are obese with hypertension, and the worsening global spectrum of these two disorders very likely contributes to the increasing burden of HFpEF, now outstripping HFrEF in prevalence. Obese HFpEF subjects often present with metabolic syndrome or type 2 diabetes and hypertension. Furthermore, obese HFpEF patients, when compared with lean HFpEF patients, often manifest worse functional parameters and increased risk of poor outcomes103. Of note, amelioration of cardiometabolic parameters through behavioral or nutritional strategies (e.g., exercise and weight loss) favorably impact HFpEF clinical outcomes; nevertheless, a dearth of targeted pharmacological, weight loss therapies remains a major challenge, although two Phase 3 clinical trials are underway to test reduction of fat mass in obesity-related HFpEF (NCT04847557 and NCT04916470).
Multiple lines of evidence, both clinical and epidemiological, support the role of cardiometabolic stress as a major driver of cardiac hypertrophy and dysfunction in HFpEF pathogenesis. However, it is important to recognize that cardiometabolic alterations in HFpEF subjects have effects beyond the heart, impacting a majority of organ systems including the vasculature and skeletal muscle104, which highlights the importance of comprehensive phenotyping in model development for this phenogroup.
Models:
Here we point to a number of animals that have recently emerged as potential preclinical models of cardiometabolic HFpEF based on their ability to recapitulate many of the phenotypic and molecular features of this phenogroup (Table 2):
Table 2:
Phenotype-based characterization of proposed cardiometabolic HFpEF models.
| Obese ZSF1 Rat | Goto Kakizaki Rat | C57BL/6 + HFD + L-NAME | Miniswine + WD + DOCA | |
|---|---|---|---|---|
| Obesity | Y | N | Y | Y |
| Glucose intolerance | Y | Y | Y | Y |
| Hypertension | Y | Y | Y | Y |
| Exercise intolerance | Y | N/A | Y | N/A |
| Pulmonary congestion | Y | Y | Y | Y |
| Preserved LVEF | Y | Y | Y | Y |
| Diastolic dysfunction | Y | Y | Y | Y |
| Impaired systolic strain | N/A | NA | Y | N/A |
| Impaired cardiac reserve | Y | N/A | N/A | N/A |
| Pathologic cardiac hypertrophy | Y | Y | Y | Y |
| Vascular dysfunction | Y | Y | Y | Y |
| Sarcopenia | Y | N/A | N/A | N/A |
| Renal dysfunction | Y | Y | N/A | Y |
Y = yes. N = no. N/A = not available.
Goto Kakizaki rat
The non-obese Goto-Kakizaki rat has traditionally been used as a model of type 2 diabetes and was generated by selective breeding of Wistar rats based on glucose intolerance. However, in addition to spontaneous diabetes, these rats display modest hypertension, which is associated with vascular and renal dysfunction, along with cardiac hypertrophy105. Given these phenotypes, which are commonly observed in HFpEF, there has been interest in exploring whether this animal may represent a reasonable model for studying cardiometabolic HFpEF. More extensive phenotyping of these rats has shown that by 48 weeks of age these animals exhibit concentric LV hypertrophy associated with adverse myocardial remodeling, pulmonary congestion, and diastolic dysfunction106. Whereas their LVEF at 48 weeks is within the normal range for rats in general, it is important to note that LVEF significantly declines from 24 to 48 weeks in these animals, which is not entirely consistent with clinical HFpEF.
Obese ZSF1 rat
Fairly consistent HFpEF phenotyping of the obese diabetic Zucker fatty spontaneously hypertensive F1 hybrid (ZSF1) rat has led the field to generally accept this animal as a faithful model of cardiometabolic HFpEF43,84,107–109. By 10–20 weeks, obese ZSF1 rats display multiple cardiac phenotypes consistent with HFpEF, including concentric LV hypertrophy, preserved LVEF, diastolic dysfunction, increased LV filling pressures with impaired lusitropic reserves, lung congestion, and exercise intolerance43,108. The myocardium of these animals also displays features of adverse remodeling commonly seen in HFpEF patients, including cardiomyocyte hypertrophy, interstitial fibrosis, and microvascular fibrotic changes, the latter of which is also observed in the kidneys110. Equally important is the remarkable recapitulation of extra-cardiac HFpEF phenotypes seen in this model. Consistent with cardiometabolic stress, there is insulin resistance and obesity, but also modest hypertension, increased vascular stiffness, renal dysfunction, and adverse skeletal muscle remodeling associated with impaired perfusion and function107,110–113. Importantly, many of the HFpEF phenotypes are observed in both male and female obese ZSF1 rats, suggesting that both sexes can and should be used in preclinical studies114.
High fat diet + L-NAME mouse
Similar to recent trends in age-related HFpEF model development, a multi-hit approach to cardiometabolic HFpEF has been pursued in mice, which has led to an impressive recapitulation of clinical HFpEF phenotypes9. In this model, animals are exposed to two hits: a HFD that triggers obesity and metabolic syndrome and L-NAME (L-NG-Nitro arginine methyl ester), an inhibitor of constitutive nitric oxide synthases that elicits a modest increase in blood pressure (≈40mmHg). The rationale for this model stems from convergence of the two major comorbidities seen in HFpEF patients: mechanical loading (hypertension) and metabolic/inflammatory stress (obesity, metabolic syndrome, diabetes). Either “hit” in isolation is insufficient to trigger HFpEF; rather, it is the convergence of the two that elicits a phenotype consistent with cardiometabolic HFpEF. These animals manifest diastolic dysfunction, preserved LVEF, impaired systolic strain, exercise intolerance, elevated LV filling pressures, microvascular dysfunction, modest cardiomyocyte hypertrophy, capillary rarefaction, predisposition to atrial fibrillation, and modest myocardial fibrosis9. Importantly, these animals do not develop HFrEF over time. However, it appears that female mice have an attenuated cardiac phenotype compared with their male counterparts, and this protective effect is not mediated by female sex hormones115.
Western diet + DOCA miniswine
Recently, a similar multi-hit approach in Göttingen miniswine has been pursued as a novel large animal model of HFpEF. Although the larger size and associated costs make mechanistic investigations challenging in the pig, it notably has more similar cardiovascular structure and physiology to humans, which has been a major impetus for using this animal in cardiovascular therapeutic testing116. In this model, HFpEF phenotypes were induced in 14-month-old female Göttingen minipigs (17–20 kg) using a combination of DOCA and a customized WD high in fat, fructose, cholesterol, and salt.117 These minipigs develop hypertension, morbid obesity, glucose intolerance, and hyperlipidemia within 20 weeks consistent with a metabolic syndrome. Importantly, they demonstrate marked concentric LV hypertrophy associated with preserved LVEF, adverse left atrial remodeling, diastolic dysfunction, and increased LV filling pressures. Moreover, coronary endothelial dysfunction and increased renal fibrosis were also observed, suggesting systemic, multiorgan pathophysiology likely occurs in these animals, similar to clinical HFpEF. Exercise capacity has not yet been assessed in this model, thus more studies are warranted in order to verify its suitability.
Recent Mechanistic Insights and Knowledge Gaps:
The relatively recent development of the obese ZSF1 rat and the two-hit (HFD/L-NAME) murine models of HFpEF are rapidly advancing our understanding of the molecular underpinnings of cardiometabolic HFpEF. The obese ZSF1 rat was essentially validated as a model of HFpEF less than a decade ago and has repeatedly proven to be faithful to the functional phenotypes of the clinical syndrome, as well as to the underlying biology observed in humans with HFpEF. The obese ZSF1 rat notably displays severe diastolic dysfunction and myocardial stiffness which has largely been attributed to titin hypophosphorylation108, a finding also observed in human HFpEF118. Moreover, it recapitulates much of the pathobiology underlying the coronary microvascular inflammation hypothesis introduced by Paulus and Tschope in 2013119,120. However, beyond validating reported molecular findings from human HFpEF specimens, more recent systems-based, -omics approaches in this model have begun to shed light on novel putative mechanisms driving cardiometabolic HFpEF. Cardiome-directed network analysis of bulk RNAseq datasets in these rats identified an overarching theme of dysregulated mitochondrial oxidative metabolism interconnected with endothelial dysfunction, inflammation, and alterations in apoptosis, autophagy, sarcomere/cytoskeleton, and extracellular matrix121. Interestingly, although only hypothesis-generating, these findings in the obese ZSF1 rat largely resonate with the mechanistic work emerging from the two-hit murine model of cardiometabolic HFpEF.
Recent mechanistic studies in the HFD/L-NAME murine model have uncovered a causal link between the combination of obesity and hypertension in the pathophysiology of HFpEF9–12. In these animals, obesity and metabolic stress induce a systemic, low-grade pro-inflammatory state that has been termed metabolic inflammation or meta-inflammation. Downstream of the meta-inflammatory events, cardiomyocytes manifest a significant steatotic response, and consequent lipotoxicity, stemming from impaired myocardial utilization of lipids as an energy source10. These events occur in concert with alterations in mitochondrial fatty acid oxidation and impaired myocardial energetics12.
We and others have proposed that visceral adiposity, common in cardiometabolic HFpEF, is a core mediator of the meta-inflammation in this phenogroup and likely contributes to HFpEF pathophysiology via multiple mechanisms, including promotion of insulin resistance and hypertension, but also via direct influences on cardiac metabolism and immune activation11,122,123. Obesity is associated with polarization of macrophages toward a pro-inflammatory phenotype as well as release of chemokines that initiates recruitment of immune cells. Lipids can also act as inflammatory molecules themselves and promote recruitment of immune cells to HFpEF myocardium and well as directly alter cardiomyocyte metabolism10. Moreover, epicardial adipose tissue with associated local secretion of cytokines has been proposed as a mechanism contributing to meta-inflammation in HFpEF124, although underlying mechanisms remain obscure.
Concomitant with accumulating evidence implicating metabolic stress in HFpEF pathogenesis, a pivotal and related role of immune mechanisms is unfolding123. Clinical data suggest that pro-inflammatory molecules play an important role in HFpEF development and progression125–127. Several adipokines promote endothelial dysfunction and reduce vascular compliance in obese HFpEF patients124. Myocardial infiltration by inflammatory cells, a phenomenon detected in endomyocardial biopsies from HFpEF patients128, promotes HFpEF pathogenesis9,10,116,123,129. T cells have been identified in human myocardium in diverse forms of HF130, and myeloid cells and innate macrophages have also been implicated in the development of diastolic dysfunction91. These data, and others, lend strong credence to a model in which inflammatory events, downstream of metabolic stress, contribute to the pathogenesis of cardiometabolic HFpEF.
Work in the HFD/L-NAME model is revealing how metabolic stress and inflammation are intertwined in HFpEF, and recently identified activation of the inflammatory molecule iNOS (inducible nitric oxide synthase) as a key mechanism triggering nitrosative stress in cardiometabolic HFpEF131. Similar biology has not only been observed in myocardial samples from HFpEF patients, but pharmacological inhibition or genetic silencing of iNOS ameliorates the HFpEF phenotype in HFD/L-NAME challenged mice. However, clinical trials targeting immune mechanisms, such as IL-6 or TNFα, have disappointed126,127,132,133. Therefore, immune modulation as a therapy for HF remains debated. Based on emerging data from the obese ZSF1 and HFD/L-NAME models, we propose that an approach involving a two-pronged attack combining strategies targeting both metabolism and inflammation, is of interest and should be further explored.
In aggregate, findings from preclinical models of cardiometabolic HFpEF, in parallel with clinical data, suggest that HF in this particular phenogroup stems from a chronic inflammatory syndrome that arises in the setting of multiple pro-inflammatory comorbidities. Accumulating evidence point to bidirectional crosstalk between metabolic stress and inflammation, in which adipose tissue, a metabolically active tissue, influences both cardiac metabolism and immune activation. However, the roles of specific immune cells and mediators remain to be elucidated and need to be further explored in these models and validated in patients.
HYPERTENSION-ASSOCIATED HFpEF:
Hypertension remains the most common comorbidity in patients with HFpEF27,134, presenting alone or associated with other comorbidities135. Overall, the pathophysiology of HFpEF in hypertensive patients is complex and multifactorial. The classic paradigm is that poorly controlled hypertension results in LV hypertrophy, left atrial remodeling, diastolic dysfunction and eventually HF136. Whereas LV hypertrophy and myocardial overload can lead to cardiac dysfunction137, it is important to note that some patients with isolated hypertension do not manifest adverse myocardial remodeling, such as increased stiffness due to higher collagen deposition or titin hypophosphorylation, which are characteristically seen in the myocardium of patients with hypertension-induced HFpEF138,139. Despite the high prevalence of hypertension in HFpEF clinical trials, analysis from I-PRESERVE, CHARM-Preserved, and TOPCAT found that approximately 1/3 to 2/3rds of patients with HFpEF did not have LV hypertrophy140–143. Hypertension induces vascular changes, including increases in arterial stiffness that can also lead to ventricular-vascular uncoupling, afterload mismatch, and cardiac dysfunction, in the absence of LV hypertrophy144. The variable contribution of these pathophysiological processes such as the degree of diastolic dysfunction, type of cardiac remodeling, and extent of neurohormonal activation, may in turn also lead to different hypertension-associated HFpEF phenotypes, possibly requiring distinct therapeutic approaches135.
Models:
The effects of increased afterload on cardiac remodeling and function have been thoroughly studied over the years, and several animal models of hypertension-associated HFpEF have been reported that range from malignant hypertension to mild-moderate hypertension. We propose that the latter is more clinically relevant as that is the phenotype more commonly seen in HFpEF patients. It is often accompanied by concentric LV hypertrophy and remodeling, along with diastolic dysfunction22, which should both be rigorously assessed in models of this phenogroup. Moreover, in attempt to be faithful to the clinical syndrome of HFpEF, LVEF must remain preserved without a later decrement in LVEF, which is a major reason why we generally favor the following models of the hypertension-associated HFpEF phenogroup. In addition to an elevated LV end-diastolic pressure (LVEDP), other signs of HF, such as congestion (e.g., lung and liver) and exercise intolerance, should also be evident to fulfill HFpEF criteria23. See Table 3 for details.
Table 3:
Phenotype-based characterization of proposed hypertension-associated HFpEF models.
| Angiotensin II | DOCA/UNX/Salt | SAUNA | |
|---|---|---|---|
| Systemic hypertension | Y (dose dependent) | Y (species dependent) | Y |
| Exercise intolerance | Y (strain dependent) | N/A | Y |
| Pulmonary congestion | Y (strain dependent) | N | Y |
| Diastolic dysfunction | Y | Y | Y |
| LV concentric hypertrophy | Y (strain dependent) | Y | Y |
Y = yes. N = no. N/A = not available.
Angiotensin II model:
Chronic stimulation with angiotensin II (ANGII, in a dose-dependent manner) in mice leads to cardiac hypertrophy and pathologic myocardial remodeling, both in the presence145–147 and absence148,149 of hypertension. This is accompanied by diastolic dysfunction, including worsening LV isovolumetric relaxation time and increased LVEDP146,147,149–151. However, both preserved148,149 and reduced151 LVEF have been documented with chronic ANGII stimulation and as such is both a model of HFpEF and HFrEF depending on the dose and duration of ANGII. Notably, although C57BL/6J mice develop compensatory concentric hypertrophy and fibrosis in response to ANGII145,150, Balb/c mice manifest severe LV chamber dilatation152 which is infrequently seen in HFpEF. C57BL/6J mice exposed to ANGII manifest pulmonary congestion, as well as exercise intolerance145,150,152. In these mice, the impaired exercise capacity appears to be related to skeletal muscle abnormalities, including impaired mitochondrial function and skeletal muscle atrophy153. Thus, although special attention should be paid to the background strain of the animal used, as well as to the dosage and duration of angiotensin chosen, this may be a valid model of hypertension-associated HFpEF.
DOCA + UNX + SALT model:
Another model of hypertension-associated HFpEF in rats is the administration of deoxycortisone acetate (DOCA), accompanied by unilateral nephrectomy (UNX, one week before DOCA, at 6–10 weeks of age) and 1% NaCl drinking water154,155. This typically results in moderate hypertension, cardiac hypertrophy and fibrosis156 accompanied by severe diastolic dysfunction, manifesting a restrictive pattern, while LVEF is preserved157,158. Although cardiac hypertrophy and diastolic dysfunction are consistent, blood pressure responses are variable and have been reported to be unchanged or mildly increased in DOCA mice157 159,160. Importantly, there is evidence of exercise intolerance, but not pulmonary congestion160,161.
SAUNA model:
More recent studies have used a combination of salted drinking water, unilateral nephrectomy, and chronic aldosterone (SAUNA162,163) to induce HFpEF in both rats and mice (Figure 3). Moderate hypertension in this model is accompanied by concentric LV hypertrophy and remodeling164–168, with evidence of diastolic dysfunction, including elevated LVEDP, reduced diastolic relaxation, and prolonged time constant of pressure decay162,169 while LVEF remains preserved. Importantly, lung congestion168,170,171 and exercise intolerance163,172 are both present in this model. In addition to increased myocardial fibrosis, LV tissue from SAUNA mice display increased expression of the shorter stiffer N2B titin transcript isoform171, similar to the human HFpEF phenotype in which changes in diastolic stiffness due to titin modifications have also been described139,173. Inflammation is associated with the adverse cardiac remodeling and diastolic dysfunction seen in hypertension associated-HFpEF, and in SAUNA mice, increased macrophage density is observed in the myocardium, consistent with findings in HFpEF patients162. It has been proposed that cardiac macrophages activate fibroblasts stimulating cardiac remodeling and leading to impaired myocardial relaxation and increased stiffness.
Figure 3: SAUNA model of hypertension-associated HFpEF.
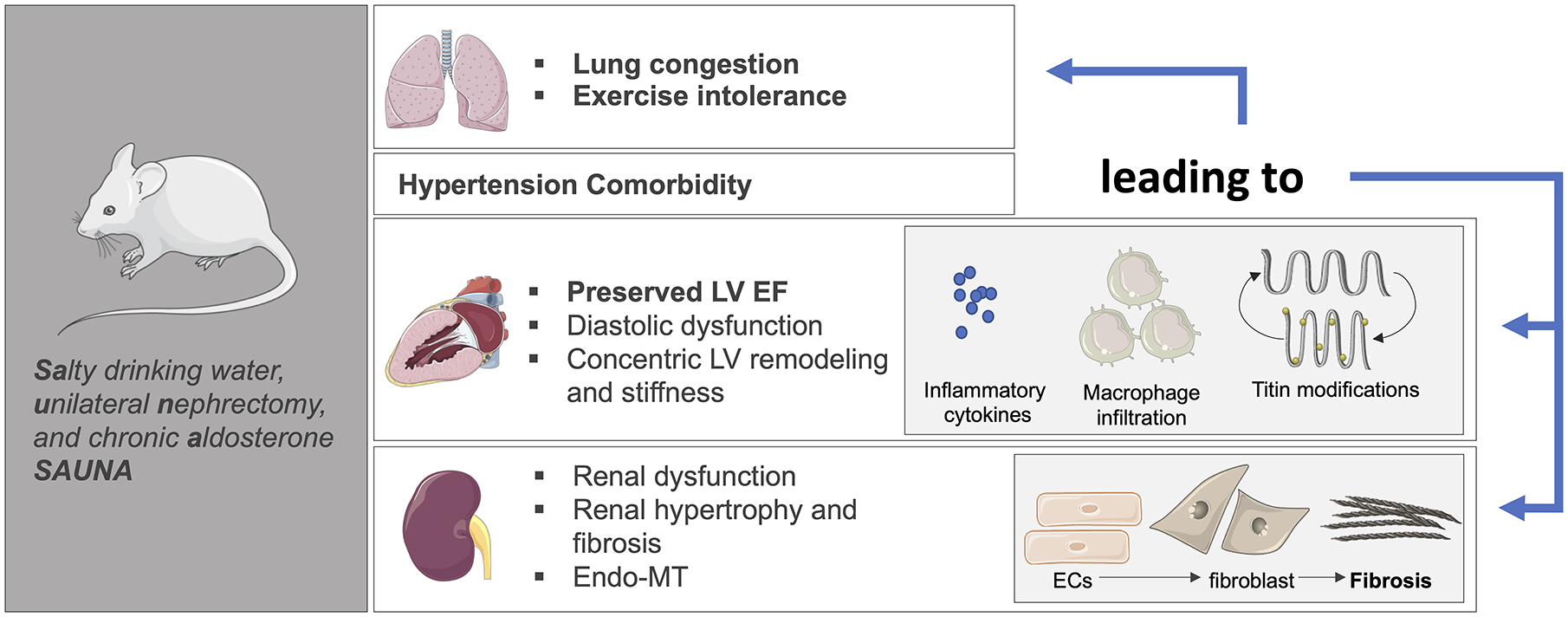
The SAUNA model using a combination of SAlty drinking water, Unilateral Nephrectomy, and chronic Aldosterone to induce a model of hypertension-associated HFpEF in rodents. The schematic displays some of the key cardiac and extra-cardiac (e.g. chronic kidney disease) observed in this model. Recent mechanistic work in this model has implicated inflammation, endo-MT, and titin modifications as underlying pathogenic processes in the pathophysiology of hypertension-associated HFpEF. The illustration was made using images from smart.servier.com.
Additional models:
Other proposed models of hypertension-associated HFpEF that are described in the literature include the Dahl salt-sensitive rat, the spontaneously hypertensive rat, the transverse aortic constriction-induced pressure overload murine model22,174, or transverse aortic constriction in cats175. In addition to the first two models demonstrating severely elevated or malignant hypertension, all four demonstrate progression to eccentric/dilated LV hypertrophy and ultimately a reduction in LVEF176. Although characterized by an initial compensatory phase, with signs consistent with HFpEF, such as congestion or exercise intolerance, this phase is usually followed by a decompensated phase consistent with LV chamber enlargement, eccentric remodeling and further deterioration of systolic function and LVEF177–181. Such progression from HFpEF to HFrEF is rarely observed in patients with HFpEF182,183, which would suggest that these models differ from the clinical syndrome.
Recent Mechanistic Insights and Knowledge Gaps:
The effects of increased afterload or chronic hypertension on LV hypertrophy, remodeling, and dysfunction are well established136. However, recent studies in the above models are deepening our understanding of the molecular mechanisms – e.g. titin isoform switches, increased macrophage content, epigenetic regulation91,171,184 – by which hypertension-associated myocardial remodeling transitions to the clinical syndrome of HFpEF. Similar to model development for age-associated and cardiometabolic HFpEF, more recent strategies for generating models of this phenogroup are also incorporating multiple hits, but ones that are more focused on comorbidities that are highly prevalent in this phenogroup (e.g. CKD).
In particular, HFpEF associated with chronic hypertension is often accompanied by renal dysfunction (commonly referred to as cardiorenal HFpEF)6,21, which is characterized by greater LV hypertrophy and worse cardiac mechanics, that lead to RV dysfunction and poorer prognosis185,186. Conventional thinking supports a cardiocentric origin for cardiorenal syndrome, driven by hemodynamic dysfunction and poor forward flow or renal venous congestion leading to renal hypoperfusion, activation of the renin-angiotensin-aldosterone system, and arginine and vasopressin hypersecretion187–189. However, renal dysfunction in HFpEF (including metabolic, electrolyte, and systemic impairments) contributes to a systemic pro-inflammatory state, diminished nitric oxide bioavailability and endothelial dysfunction, which can promote cardiomyocyte stiffening, cardiac hypertrophy, and interstitial fibrosis by crosstalk between the endothelium and cardiomyocyte compartments137,190.
The more recent models of hypertension-associated HFpEF, specifically those that include unilateral nephrectomy in addition to high salt loads and pharmacological neurohormonal activation, have not only captured this inextricable link between the heart and kidneys in HFpEF, but are also unveiling new insights into the pathophysiology of this phenogroup that are likely mediated through organ crosstalk and mechanisms that are interestingly independent of BP. Recent work in the SAUNA model has implicated a potentially causal role of endothelial-mesenchymal transition (endo-MT) in the renal fibrosis and cardiorenal syndrome of HFpEF191. The moderate hypertensive SAUNA HFpEF model notably demonstrates LV hypertrophy and fibrosis, but also features of renal dysfunction including kidney remodeling, characterized by glomerular hypertrophy and increased intraglomerular cells, accompanied by an elevation of serum creatinine and albuminuria192. Renal fibrosis in SAUNA HFpEF is strongly associated with an increase in the pool of myofibroblasts, consistent with the activation of endo-MT, suggesting that endo-MT is likely an important source of myofibroblasts that contribute to the renal fibrosis underlying cardiorenal HFpEF191. Interestingly, serum samples from HFpEF patients induced similar endo-MT readouts in cultured human aortic endothelial cells, suggesting that circulating factors in HFpEF can promote this pro-fibrotic process. Moreover, recent findings from the DOCA/UNX/Salt model of hypertension-associated HFpEF have also shown that myocardial fibrosis in this phenogroup is at least partially regulated by epigenetic mechanisms, independent of blood pressure184. Histone deacetylase (HDAC) inhibition was effective in reversing established diastolic dysfunction in this model, suggesting that it might be a promising targeted therapy in this phenogroup. Whether epigenetic modifications are also related endo-MT or whether they lead to increased fibrosis in other organs in HFpEF, are unclear, as is the source and identity of the circulating factors that trigger endo-MT in this phenogroup.
Lastly, similar to heart-kidney crosstalk, adipose tissue also communicates with the heart, a process that has interestingly been found in the SAUNA model, which does not incorporate overt metabolic stress. Metabolic, endocrine and humoral signaling between the heart and the adipose tissue has been proposed to be a critical player in HFpEF pathophysiology170. Natriuretic peptides, which are increased in HFpEF, are known to modulate adipose tissue, altering energy expenditure and metabolism, and induce “browning” of white adipose tissue, which has been observed in the SAUNA model170,193,194. To date, whether browning of adipose tissue in HFpEF is deleterious or an adaptive stress response remains largely unknown.
Ultimately, further investigations into the molecular mechanisms driving cardiac and extra-cardiac fibrosis and dysfunction in hypertension-associated HFpEF is needed. The crosstalk between organs is undoubtedly important to the underlying pathophysiology of this phenogroup, and likely in HFpEF in general, and should be further explored.
CONCLUSION:
Recent progress in clinical phenotyping of HFpEF has led to a paradigm shift in the field, which now recognizes that HFpEF is not a single disease entity but rather a systemic syndrome marked by a multitude of different phenogroups with varying pathophysiology. As our knowledge of the extent and variability of cardiac and extra-cardiac phenotypes in clinical HFpEF continues to broaden, it is imperative that we incorporate the same rigor and completeness in preclinical model characterization to ensure that the most important and clinically relevant phenotypes of this syndrome are reliably captured. However, expanding our phenotyping requirements for these models is not enough. As our understanding of HFpEF rapidly evolves, it is paramount that preclinical investigators embrace these changes. At the center of this is the now widely recognized clinical concept of HFpEF heterogeneity, which will likely require that we similarly diversify our approach to model development. Preclinical models of HFpEF that are employing this strategy – by selectively incorporating metabolic stress, mineralocorticoid signaling/hypertension, and/or ageing to model HFpEF phenogroups – are not only unveiling biology unique to specific phenogroups, but also identifying mechanisms (such as inflammation) that could be universally causal in HFpEF. No animal model is perfect. Despite the complex nature of HFpEF, a reductionist approach focusing on the contribution of a dominant comorbidity (e.g., hypertension or obesity) to the HFpEF phenotype and pathogenesis remains as important as multiple comorbidity models (e.g., obesity and aging or hypertension and obesity). Equally important is incorporating both sexes in preclinical investigations, which is particularly relevant in HFpEF, given the sex differences observed in this clinical syndrome. Finally, preclinical models of HFpEF with other comorbidities such as HFpEF associated with sleep apnea, atrial fibrillation, pulmonary hypertension, chronic kidney disease, etc, alone or in combination, are necessary to further elucidate pathogenic mechanisms and inform drug discovery for this undertreated clinical syndrome.
Figure 2: Animal models of the aging, cardiometabolic, and hypertension-associated HFpEF phenogroups.
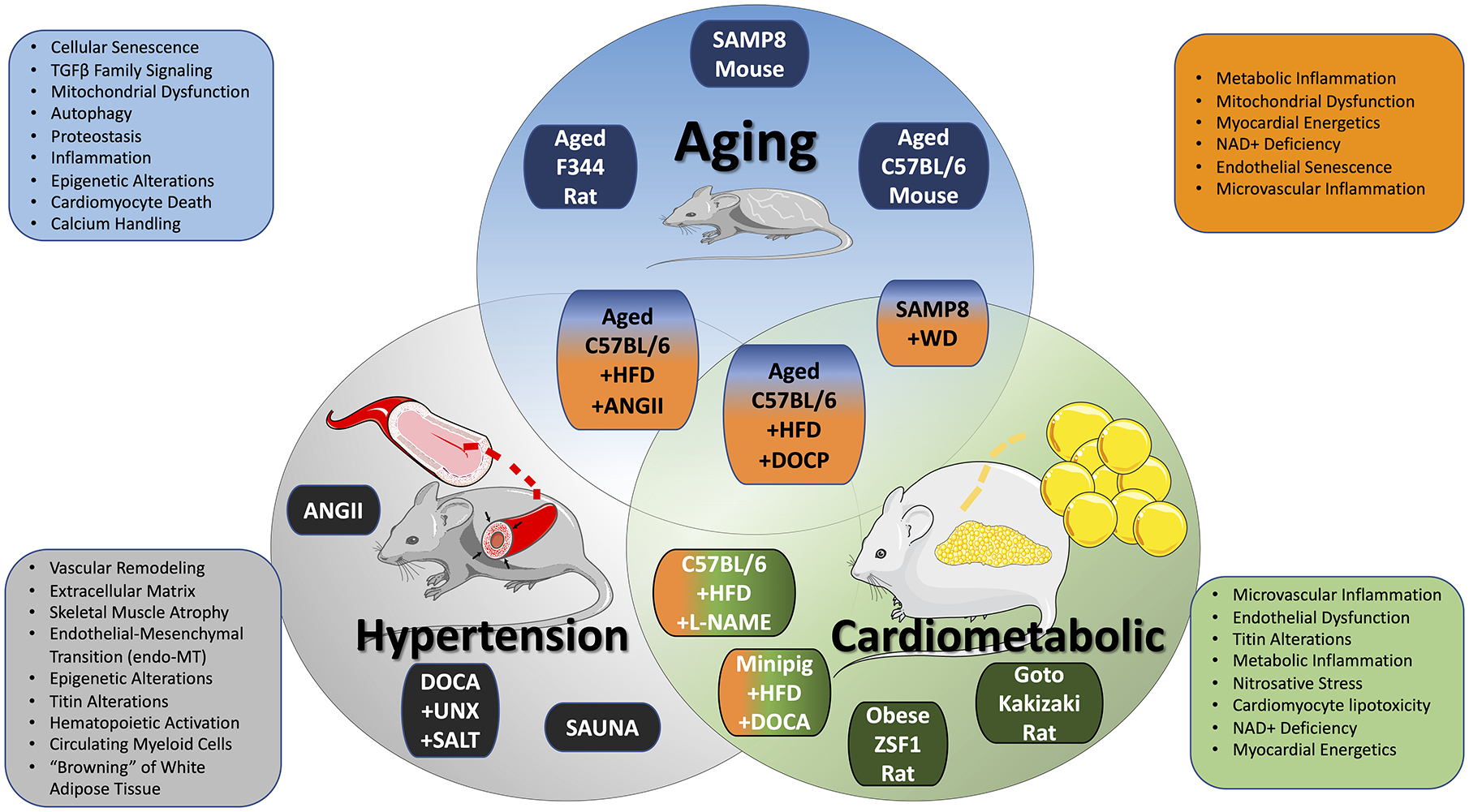
Advanced age, metabolic syndrome, and chronic hypertension represent the dominant comorbidities contributing to HFpEF pathogenesis. The Venn diagram displays animal models that are relatively unique to these primary HFpEF phenogroups, along with newer multi-hit strategies being used to integrate these primary comorbidities in model development. Recent mechanistic insights into the underlying pathobiology of these phenogroups, and HFpEF in general, that have been derived from these models are displayed. Blue = age-associated HFpEF. Green = cardiometabolic HFpEF. Gray = hypertension-associated HFpEF. Orange = multi-hit, integrated HFpEF. The illustration was made using images from smart.servier.com. Illustration credit: Ben Smith.
Source of Funding
This work was supported by grants from the National Institutes of Health: HL126012 (JAH), HL128215 (JAH), HL147933 (JAH), HL155765 (JAH), HL145985 (FS), AG064328 (JR); American Heart Association: 14SFRN20510023 (JAH), Fred and Ines Yeatts Fund for Innovative Research (JR).
Non-standard Abbreviations and Acronyms:
- HFpEF
Heart Failure with Preserved Ejection Fraction
- LVEF
Left Ventricular Ejection Fraction
- AF
Atrial Fibrillation
- BP
Blood Pressure
- CKD
Chronic Kidney Disease
- SAMP
Senescence Accelerated Prone Mouse
- HFD
High Fat Diet
- WD
Western Diet
- DOCP
Deoxycorticosterone Pivalate
- ZSF1
Zucker Fatty Spontaneously Hypertensive F1 Hybrid
- L-NAME
L-NG-Nitro Arginine Methyl Ester
- DOCA
Deoxycorticosterone Acetate
- UNX
Unilateral Nephrectomy
- SAUNA
Salty Drinking Water - Unilateral Nephrectomy - Aldosterone
Footnotes
Disclosures
Dr. Hill is a coinventor on a patent application (PCT/ US/2017/037019) that was filed in June 2017 (provisional application filed in June 2016). The patent relates to the diet used for modeling HFpEF. The other authors report no conflicts.
REFERENCES:
- 1.Timmis A, Townsend N, Gale C, Grobbee R, Maniadakis N, Flather M, Wilkins E, Wright L, Vos R, Bax J, et al. European Society of Cardiology: Cardiovascular Disease Statistics 2017. Eur Heart J. 2018;39:508–579. doi: 10.1093/eurheartj/ehx628 [DOI] [PubMed] [Google Scholar]
- 2.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019;139:e56–e528. doi: 10.1161/CIR.0000000000000659 [DOI] [PubMed] [Google Scholar]
- 3.Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, Gonzalez-Juanatey JR, Harjola VP, Jankowska EA, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37:2129–2200. doi: 10.1093/eurheartj/ehw128 [DOI] [PubMed] [Google Scholar]
- 4.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr., Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, et al. 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128:1810–1852. doi: 10.1161/CIR.0b013e31829e8807 [DOI] [PubMed] [Google Scholar]
- 5.Dunlay SM, Roger VL, Redfield MM. Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2017;14:591–602. doi: 10.1038/nrcardio.2017.65 [DOI] [PubMed] [Google Scholar]
- 6.Shah SJ, Kitzman DW, Borlaug BA, van Heerebeek L, Zile MR, Kass DA, Paulus WJ. Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation. 2016;134:73–90. doi: 10.1161/CIRCULATIONAHA.116.021884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitzman DW, Shah SJ. The HFpEF Obesity Phenotype: The Elephant in the Room. J Am Coll Cardiol. 2016;68:200–203. doi: 10.1016/j.jacc.2016.05.019 [DOI] [PubMed] [Google Scholar]
- 8.Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA. Evidence Supporting the Existence of a Distinct Obese Phenotype of Heart Failure With Preserved Ejection Fraction. Circulation. 2017;136:6–19. doi: 10.1161/CIRCULATIONAHA.116.026807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. 2019;568:351–356. doi: 10.1038/s41586-019-1100-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schiattarella GG, Altamirano F, Kim SY, Tong D, Ferdous A, Piristine H, Dasgupta S, Wang X, French KM, Villalobos E, et al. Xbp1s-FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat Commun. 2021;12:1684. doi: 10.1038/s41467-021-21931-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schiattarella GG, Rodolico D, Hill JA. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovascular research. 2021;117:423–434. doi: 10.1093/cvr/cvaa217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tong D, Schiattarella GG, Jiang N, Altamirano F, Szweda PA, Elnwasany A, Lee DI, Yoo H, Kass DA, Szweda LI, et al. NAD(+) Repletion Reverses Heart Failure With Preserved Ejection Fraction. Circulation research. 2021;128:1629–1641. doi: 10.1161/CIRCRESAHA.120.317046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hahn VS, Knutsdottir H, Luo X, Bedi K, Margulies KB, Haldar SM, Stolina M, Yin J, Khakoo AY, Vaishnav J, et al. Myocardial Gene Expression Signatures in Human Heart Failure with Preserved Ejection Fraction. Circulation. 2020; Published online October 29, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borlaug BA, Redfield MM. Diastolic and systolic heart failure are distinct phenotypes within the heart failure spectrum. Circulation. 2011;123:2006–2013; discussion 2014. doi: 10.1161/CIRCULATIONAHA.110.954388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagueh SF. Heart failure with preserved ejection fraction: insights into diagnosis and pathophysiology. Cardiovasc Res. 2021;117:999–1014. doi: 10.1093/cvr/cvaa228 [DOI] [PubMed] [Google Scholar]
- 16.Parikh KS, Sharma K, Fiuzat M, Surks HK, George JT, Honarpour N, Depre C, Desvigne-Nickens P, Nkulikiyinka R, Lewis GD, et al. Heart Failure With Preserved Ejection Fraction Expert Panel Report: Current Controversies and Implications for Clinical Trials. JACC Heart Fail. 2018;6:619–632. doi: S2213–1779(18)30464–5 [pii]; 10.1016/j.jchf.2018.06.008 [doi] [DOI] [PubMed] [Google Scholar]
- 17.Borlaug BA. The pathophysiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2014;11:507–515. doi: nrcardio.2014.83 [pii]; 10.1038/nrcardio.2014.83 [doi] [DOI] [PubMed] [Google Scholar]
- 18.Borlaug BA. Evaluation and management of heart failure with preserved ejection fraction. Nature reviews Cardiology. 2020;17:559–573. doi: 10.1038/s41569-020-0363-2 [DOI] [PubMed] [Google Scholar]
- 19.Hahn VS, Knutsdottir H, Luo X, Bedi K, Margulies KB, Haldar SM, Stolina M, Yin J, Khakoo AY, Vaishnav J, et al. Myocardial Gene Expression Signatures in Human Heart Failure With Preserved Ejection Fraction. Circulation. 2021;143:120–134. doi: 10.1161/CIRCULATIONAHA.120.050498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tromp J, Teng TH, Tay WT, Hung CL, Narasimhan C, Shimizu W, Park SW, Liew HB, Ngarmukos T, Reyes EB, et al. Heart failure with preserved ejection fraction in Asia. Eur J Heart Fail. 2019;21:23–36. doi: 10.1002/ejhf.1227 [DOI] [PubMed] [Google Scholar]
- 21.Shah SJ. Precision Medicine for Heart Failure with Preserved Ejection Fraction: An Overview. J Cardiovasc Transl Res. 2017;10:233–244. doi: 10.1007/s12265-017-9756-y [doi];10.1007/s12265-017-9756-y [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Valero-Munoz M, Backman W, Sam F. Murine Models of Heart Failure with Preserved Ejection Fraction: a “Fishing Expedition”. JACC Basic Transl Sci. 2017;2:770–789. doi: 10.1016/j.jacbts.2017.07.013 [doi];S2452-302X(17)30216-4 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Withaar C, Lam CSP, Schiattarella GG, de Boer RA, Meems LMG. Heart failure with preserved ejection fraction in humans and mice: embracing clinical complexity in mouse models. Eur Heart J. 2021;42:4420–4430. doi: 6355308 [pii]; 10.1093/eurheartj/ehab389 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lourenco AP, Leite-Moreira AF, Balligand JL, Bauersachs J, Dawson D, de Boer RA, de Windt LJ, Falcao-Pires I, Fontes-Carvalho R, Franz S, et al. An integrative translational approach to study heart failure with preserved ejection fraction: a position paper from the Working Group on Myocardial Function of the European Society of Cardiology. Eur J Heart Fail. 2018;20:216–227. doi: 10.1002/ejhf.1059 [DOI] [PubMed] [Google Scholar]
- 25.Barandiaran Aizpurua A, Schroen B, van Bilsen M, van Empel V. Targeted HFpEF therapy based on matchmaking of human and animal models. Am J Physiol Heart Circ Physiol. 2018;315:H1670–H1683. doi: 10.1152/ajpheart.00024.2018 [DOI] [PubMed] [Google Scholar]
- 26.Roh J, Houstis N, Rosenzweig A. Why Don’t We Have Proven Treatments for HFpEF? Circ Res. 2017;120:1243–1245. doi: 10.1161/CIRCRESAHA.116.310119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah SJ, Borlaug BA, Kitzman DW, McCulloch AD, Blaxall BC, Agarwal R, Chirinos JA, Collins S, Deo RC, Gladwin MT, et al. Research Priorities for Heart Failure With Preserved Ejection Fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation. 2020;141:1001–1026. doi: 10.1161/CIRCULATIONAHA.119.041886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Villalba-Orero M, Lopez-Olaneta MM, Gonzalez-Lopez E, Padron-Barthe L, Gomez-Salinero JM, Garcia-Prieto J, Wai T, Garcia-Pavia P, Ibanez B, Jimenez-Borreguero LJ, et al. Lung ultrasound as a translational approach for non-invasive assessment of heart failure with reduced or preserved ejection fraction in mice. Cardiovasc Res. 2017;113:1113–1123. doi: 10.1093/cvr/cvx090 [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Sano S, Yura Y, Ke Z, Sano M, Oshima K, Ogawa H, Horitani K, Min KD, Miura-Yura E, et al. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight. 2020;5. doi: 10.1172/jci.insight.135204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poole DC, Copp SW, Colburn TD, Craig JC, Allen DL, Sturek M, O’Leary DS, Zucker IH, Musch TI. Guidelines for animal exercise and training protocols for cardiovascular studies. Am J Physiol Heart Circ Physiol. 2020;318:H1100–H1138. doi: 10.1152/ajpheart.00697.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Platt C, Houstis N, Rosenzweig A. Using exercise to measure and modify cardiac function. Cell Metab. 2015;21:227–236. doi: 10.1016/j.cmet.2015.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arena R, Myers J, Williams MA, Gulati M, Kligfield P, Balady GJ, Collins E, Fletcher G, American Heart Association Committee on Exercise R, Prevention of the Council on Clinical C, et al. Assessment of functional capacity in clinical and research settings: a scientific statement from the American Heart Association Committee on Exercise, Rehabilitation, and Prevention of the Council on Clinical Cardiology and the Council on Cardiovascular Nursing. Circulation. 2007;116:329–343. doi: 10.1161/CIRCULATIONAHA.106.184461 [DOI] [PubMed] [Google Scholar]
- 33.Desai KH, Sato R, Schauble E, Barsh GS, Kobilka BK, Bernstein D. Cardiovascular indexes in the mouse at rest and with exercise: new tools to study models of cardiac disease. Am J Physiol. 1997;272:H1053–1061. doi: 10.1152/ajpheart.1997.272.2.H1053 [DOI] [PubMed] [Google Scholar]
- 34.Lujan HL, DiCarlo SE. Cardiac output, at rest and during exercise, before and during myocardial ischemia, reperfusion, and infarction in conscious mice. Am J Physiol Regul Integr Comp Physiol. 2013;304:R286–295. doi: 10.1152/ajpregu.00517.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roh JD, Houstis N, Yu A, Chang B, Yeri A, Li H, Hobson R, Lerchenmuller C, Vujic A, Chaudhari V, et al. Exercise training reverses cardiac aging phenotypes associated with heart failure with preserved ejection fraction in male mice. Aging Cell. 2020;19:e13159. doi: 10.1111/acel.13159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindsey ML, Kassiri Z, Virag JAI, de Castro Bras LE, Scherrer-Crosbie M. Guidelines for measuring cardiac physiology in mice. Am J Physiol Heart Circ Physiol. 2018;314:H733–H752. doi: 10.1152/ajpheart.00339.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pacher P, Nagayama T, Mukhopadhyay P, Batkai S, Kass DA. Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat Protoc. 2008;3:1422–1434. doi: 10.1038/nprot.2008.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kass DA, Hare JM, Georgakopoulos D. Murine cardiac function: a cautionary tail. Circ Res. 1998;82:519–522. doi: 10.1161/01.res.82.4.519 [DOI] [PubMed] [Google Scholar]
- 39.Roth DM, Swaney JS, Dalton ND, Gilpin EA, Ross J Jr. Impact of anesthesia on cardiac function during echocardiography in mice. Am J Physiol Heart Circ Physiol. 2002;282:H2134–2140. doi: 10.1152/ajpheart.00845.2001 [DOI] [PubMed] [Google Scholar]
- 40.Borlaug BA, Olson TP, Lam CS, Flood KS, Lerman A, Johnson BD, Redfield MM. Global cardiovascular reserve dysfunction in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2010;56:845–854. doi: 10.1016/j.jacc.2010.03.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roh JD, Hobson R, Chaudhari V, Quintero P, Yeri A, Benson M, Xiao C, Zlotoff D, Bezzerides V, Houstis N, et al. Activin type II receptor signaling in cardiac aging and heart failure. Sci Transl Med. 2019;11. doi: 10.1126/scitranslmed.aau8680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, Prabhu SD, Rockman HA, Kass DA, Molkentin JD, Sussman MA, et al. Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res. 2012;111:131–150. doi: 10.1161/RES.0b013e3182582523 [DOI] [PubMed] [Google Scholar]
- 43.Leite S, Oliveira-Pinto J, Tavares-Silva M, Abdellatif M, Fontoura D, Falcao-Pires I, Leite-Moreira AF, Lourenco AP. Echocardiography and invasive hemodynamics during stress testing for diagnosis of heart failure with preserved ejection fraction: an experimental study. Am J Physiol Heart Circ Physiol. 2015;308:H1556–H1563. doi: ajpheart.00076.2015 [pii]; 10.1152/ajpheart.00076.2015 [doi] [DOI] [PubMed] [Google Scholar]
- 44.Wiesmann F, Ruff J, Engelhardt S, Hein L, Dienesch C, Leupold A, Illinger R, Frydrychowicz A, Hiller KH, Rommel E, et al. Dobutamine-stress magnetic resonance microimaging in mice : acute changes of cardiac geometry and function in normal and failing murine hearts. Circ Res. 2001;88:563–569. doi: 10.1161/01.res.88.6.563 [DOI] [PubMed] [Google Scholar]
- 45.Kraigher-Krainer E, Shah AM, Gupta DK, Santos A, Claggett B, Pieske B, Zile MR, Voors AA, Lefkowitz MP, Packer M, et al. Impaired systolic function by strain imaging in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2014;63:447–456. doi: 10.1016/j.jacc.2013.09.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Obokata M, Reddy YNV, Borlaug BA. Diastolic Dysfunction and Heart Failure With Preserved Ejection Fraction: Understanding Mechanisms by Using Noninvasive Methods. JACC Cardiovasc Imaging. 2020;13:245–257. doi: 10.1016/j.jcmg.2018.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131:550–559. doi: 10.1161/CIRCULATIONAHA.114.009625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bostrom P, Mann N, Wu J, Quintero PA, Plovie ER, Panakova D, Gupta RK, Xiao C, MacRae CA, Rosenzweig A, et al. C/EBPbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143:1072–1083. doi: 10.1016/j.cell.2010.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kotecha D, Lam CS, Van Veldhuisen DJ, Van Gelder IC, Voors AA, Rienstra M. Heart Failure With Preserved Ejection Fraction and Atrial Fibrillation: Vicious Twins. J Am Coll Cardiol. 2016;68:2217–2228. doi: 10.1016/j.jacc.2016.08.048 [DOI] [PubMed] [Google Scholar]
- 50.Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Bohm M, Brunner-La Rocca HP, Choi DJ, Chopra V, Chuquiure-Valenzuela E, et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N Engl J Med. 2021;385:1451–1461. doi: 10.1056/NEJMoa2107038 [DOI] [PubMed] [Google Scholar]
- 51.Schuttler D, Bapat A, Kaab S, Lee K, Tomsits P, Clauss S, Hucker WJ. Animal Models of Atrial Fibrillation. Circ Res. 2020;127:91–110. doi: 10.1161/CIRCRESAHA.120.316366 [DOI] [PubMed] [Google Scholar]
- 52.Zhao X, Ho D, Gao S, Hong C, Vatner DE, Vatner SF. Arterial Pressure Monitoring in Mice. Curr Protoc Mouse Biol. 2011;1:105–122. doi: 10.1002/9780470942390.mo100149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Borlaug BA, Kass DA. Ventricular-vascular interaction in heart failure. Heart Fail Clin. 2008;4:23–36. doi: 10.1016/j.hfc.2007.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Butlin M, Tan I, Spronck B, Avolio AP. Measuring Arterial Stiffness in Animal Experimental Studies. Arterioscler Thromb Vasc Biol. 2020;40:1068–1077. doi: 10.1161/ATVBAHA.119.313861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin-6 overexpression induces pulmonary hypertension. Circ Res. 2009;104:236–244, 228p following 244. doi: 10.1161/CIRCRESAHA.108.182014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thibault HB, Kurtz B, Raher MJ, Shaik RS, Waxman A, Derumeaux G, Halpern EF, Bloch KD, Scherrer-Crosbie M. Noninvasive assessment of murine pulmonary arterial pressure: validation and application to models of pulmonary hypertension. Circ Cardiovasc Imaging. 2010;3:157–163. doi: 10.1161/CIRCIMAGING.109.887109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Nierop BJ, van Assen HC, van Deel ED, Niesen LB, Duncker DJ, Strijkers GJ, Nicolay K. Phenotyping of left and right ventricular function in mouse models of compensated hypertrophy and heart failure with cardiac MRI. PLoS One. 2013;8:e55424. doi: 10.1371/journal.pone.0055424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dhakal BP, Malhotra R, Murphy RM, Pappagianopoulos PP, Baggish AL, Weiner RB, Houstis NE, Eisman AS, Hough SS, Lewis GD. Mechanisms of exercise intolerance in heart failure with preserved ejection fraction: the role of abnormal peripheral oxygen extraction. Circ Heart Fail. 2015;8:286–294. doi: 10.1161/CIRCHEARTFAILURE.114.001825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Houstis NE, Eisman AS, Pappagianopoulos PP, Wooster L, Bailey CS, Wagner PD, Lewis GD. Exercise Intolerance in Heart Failure With Preserved Ejection Fraction: Diagnosing and Ranking Its Causes Using Personalized O2 Pathway Analysis. Circulation. 2018;137:148–161. doi: 10.1161/CIRCULATIONAHA.117.029058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kitzman DW, Nicklas B, Kraus WE, Lyles MF, Eggebeen J, Morgan TM, Haykowsky M. Skeletal muscle abnormalities and exercise intolerance in older patients with heart failure and preserved ejection fraction. Am J Physiol Heart Circ Physiol. 2014;306:H1364–1370. doi: 10.1152/ajpheart.00004.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seol SH, Davidson BP, Belcik JT, Mott BH, Goodman RM, Ammi A, Lindner JR. Real-time contrast ultrasound muscle perfusion imaging with intermediate-power imaging coupled with acoustically durable microbubbles. J Am Soc Echocardiogr. 2015;28:718–726 e712. doi: 10.1016/j.echo.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Streif JU, Hiller KH, Waller C, Nahrendorf M, Wiesmann F, Bauer WR, Rommel E, Haase A. In vivo assessment of absolute perfusion in the murine skeletal muscle with spin labeling MRI. Magnetic resonance imaging. J Magn Reson Imaging. 2003;17:147–152. doi: 10.1002/jmri.10229 [DOI] [PubMed] [Google Scholar]
- 63.Tang GL, Kim KJ. Laser Doppler Perfusion Imaging in the Mouse Hindlimb. J Vis Exp. 2021. doi: 10.3791/62012 [DOI] [PubMed] [Google Scholar]
- 64.Ter Maaten JM, Damman K, Verhaar MC, Paulus WJ, Duncker DJ, Cheng C, van Heerebeek L, Hillege HL, Lam CS, Navis G, et al. Connecting heart failure with preserved ejection fraction and renal dysfunction: the role of endothelial dysfunction and inflammation. Eur J Heart Fail. 2016;18:588–598. doi: 10.1002/ejhf.497 [DOI] [PubMed] [Google Scholar]
- 65.Keppler A, Gretz N, Schmidt R, Kloetzer HM, Groene HJ, Lelongt B, Meyer M, Sadick M, Pill J. Plasma creatinine determination in mice and rats: an enzymatic method compares favorably with a high-performance liquid chromatography assay. Kidney Int. 2007;71:74–78. doi: 10.1038/sj.ki.5001988 [DOI] [PubMed] [Google Scholar]
- 66.Song S, Meyer M, Turk TR, Wilde B, Feldkamp T, Assert R, Wu K, Kribben A, Witzke O. Serum cystatin C in mouse models: a reliable and precise marker for renal function and superior to serum creatinine. Nephrol Dial Transplant. 2009;24:1157–1161. doi: 10.1093/ndt/gfn626 [DOI] [PubMed] [Google Scholar]
- 67.Topol EJ, Traill TA, Fortuin NJ. Hypertensive hypertrophic cardiomyopathy of the elderly. N Engl J Med. 1985;312:277–283. doi: 10.1056/NEJM198501313120504 [DOI] [PubMed] [Google Scholar]
- 68.Gottdiener JS, Arnold AM, Aurigemma GP, Polak JF, Tracy RP, Kitzman DW, Gardin JM, Rutledge JE, Boineau RC. Predictors of congestive heart failure in the elderly: the Cardiovascular Health Study. J Am Coll Cardiol. 2000;35:1628–1637. doi: 10.1016/s0735-1097(00)00582-9 [DOI] [PubMed] [Google Scholar]
- 69.Tromp J, Shen L, Jhund PS, Anand IS, Carson PE, Desai AS, Granger CB, Komajda M, McKelvie RS, Pfeffer MA, et al. Age-Related Characteristics and Outcomes of Patients With Heart Failure With Preserved Ejection Fraction. J Am Coll Cardiol. 2019;74:601–612. doi: 10.1016/j.jacc.2019.05.052 [DOI] [PubMed] [Google Scholar]
- 70.Upadhya B, Kitzman DW. Heart Failure with Preserved Ejection Fraction in Older Adults. Heart Fail Clin. 2017;13:485–502. doi: 10.1016/j.hfc.2017.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shah SJ, Katz DH, Selvaraj S, Burke MA, Yancy CW, Gheorghiade M, Bonow RO, Huang CC, Deo RC. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation. 2015;131:269–279. doi: 10.1161/CIRCULATIONAHA.114.010637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circulation. 2003;107:346–354. doi: 10.1161/01.cir.0000048893.62841.f7 [DOI] [PubMed] [Google Scholar]
- 73.Strait JB, Lakatta EG. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail Clin. 2012;8:143–164. doi: 10.1016/j.hfc.2011.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roh J, Rhee J, Chaudhari V, Rosenzweig A. The Role of Exercise in Cardiac Aging: From Physiology to Molecular Mechanisms. Circ Res. 2016;118:279–295. doi: 10.1161/CIRCRESAHA.115.305250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fleg JL, Morrell CH, Bos AG, Brant LJ, Talbot LA, Wright JG, Lakatta EG. Accelerated longitudinal decline of aerobic capacity in healthy older adults. Circulation. 2005;112:674–682. doi: 10.1161/CIRCULATIONAHA.105.545459 [DOI] [PubMed] [Google Scholar]
- 76.Beere PA, Russell SD, Morey MC, Kitzman DW, Higginbotham MB. Aerobic exercise training can reverse age-related peripheral circulatory changes in healthy older men. Circulation. 1999;100:1085–1094. doi: 10.1161/01.cir.100.10.1085 [DOI] [PubMed] [Google Scholar]
- 77.Cheng S, Fernandes VR, Bluemke DA, McClelland RL, Kronmal RA, Lima JA. Age-related left ventricular remodeling and associated risk for cardiovascular outcomes: the Multi-Ethnic Study of Atherosclerosis. Circ Cardiovasc Imaging. 2009;2:191–198. doi: 10.1161/CIRCIMAGING.108.819938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dai DF, Chen T, Johnson SC, Szeto H, Rabinovitch PS. Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal. 2012;16:1492–1526. doi: 10.1089/ars.2011.4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, Gollahon K, Martin GM, Loeb LA, Ladiges WC, et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, Harger A, Schipke J, Zimmermann A, Schmidt A, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016;22:1428–1438. doi: 10.1038/nm.4222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sindler AL, Fleenor BS, Calvert JW, Marshall KD, Zigler ML, Lefer DJ, Seals DR. Nitrite supplementation reverses vascular endothelial dysfunction and large elastic artery stiffness with aging. Aging Cell. 2011;10:429–437. doi: 10.1111/j.1474-9726.2011.00679.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kane AE, Bisset ES, Heinze-Milne S, Keller KM, Grandy SA, Howlett SE. Maladaptive Changes Associated With Cardiac Aging Are Sex-Specific and Graded by Frailty and Inflammation in C57BL/6 Mice. J Gerontol A Biol Sci Med Sci. 2021;76:233–243. doi: 10.1093/gerona/glaa212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li H, Hastings MH, Rhee J, Trager LE, Roh JD, Rosenzweig A. Targeting Age-Related Pathways in Heart Failure. Circ Res. 2020;126:533–551. doi: 10.1161/CIRCRESAHA.119.315889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Abdellatif M, Trummer-Herbst V, Koser F, Durand S, Adao R, Vasques-Novoa F, Freundt JK, Voglhuber J, Pricolo MR, Kasa M, et al. Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci Transl Med. 2021;13. doi: 10.1126/scitranslmed.abd7064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Anderson R, Lagnado A, Maggiorani D, Walaszczyk A, Dookun E, Chapman J, Birch J, Salmonowicz H, Ogrodnik M, Jurk D, et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019;38. doi: 10.15252/embj.2018100492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lewis-McDougall FC, Ruchaya PJ, Domenjo-Vila E, Shin Teoh T, Prata L, Cottle BJ, Clark JE, Punjabi PP, Awad W, Torella D, et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell. 2019;18:e12931. doi: 10.1111/acel.12931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Loffredo FS, Steinhauser ML, Jay SM, Gannon J, Pancoast JR, Yalamanchi P, Sinha M, Dall’Osso C, Khong D, Shadrach JL, et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell. 2013;153:828–839. doi: 10.1016/j.cell.2013.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lyu G, Guan Y, Zhang C, Zong L, Sun L, Huang X, Huang L, Zhang L, Tian XL, Zhou Z, et al. TGF-beta signaling alters H4K20me3 status via miR-29 and contributes to cellular senescence and cardiac aging. Nat Commun. 2018;9:2560. doi: 10.1038/s41467-018-04994-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jeong MY, Lin YH, Wennersten SA, Demos-Davies KM, Cavasin MA, Mahaffey JH, Monzani V, Saripalli C, Mascagni P, Reece TB, et al. Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci Transl Med. 2018;10. doi: 10.1126/scitranslmed.aao0144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S, Kaluza D, Treguer K, Carmona G, Bonauer A, et al. MicroRNA-34a regulates cardiac ageing and function. Nature. 2013;495:107–110. doi: 10.1038/nature11919 [DOI] [PubMed] [Google Scholar]
- 91.Hulsmans M, Sager HB, Roh JD, Valero-Munoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B, et al. Cardiac macrophages promote diastolic dysfunction. J Exp Med. 2018;215:423–440. doi: 10.1084/jem.20171274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brenner DA, Apstein CS, Saupe KW. Exercise training attenuates age-associated diastolic dysfunction in rats. Circulation. 2001;104:221–226. doi: 10.1161/01.cir.104.2.221 [DOI] [PubMed] [Google Scholar]
- 93.Walker EM Jr., Nillas MS, Mangiarua EI, Cansino S, Morrison RG, Perdue RR, Triest WE, Wright GL, Studeny M, Wehner P, et al. Age-associated changes in hearts of male Fischer 344/Brown Norway F1 rats. Ann Clin Lab Sci. 2006;36:427–438. [PubMed] [Google Scholar]
- 94.Boluyt MO, Converso K, Hwang HS, Mikkor A, Russell MW. Echocardiographic assessment of age-associated changes in systolic and diastolic function of the female F344 rat heart. J Appl Physiol (1985). 2004;96:822–828. doi: 10.1152/japplphysiol.01026.2003 [DOI] [PubMed] [Google Scholar]
- 95.Pacher P, Mabley JG, Liaudet L, Evgenov OV, Marton A, Hasko G, Kollai M, Szabo C. Left ventricular pressure-volume relationship in a rat model of advanced aging-associated heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H2132–2137. doi: 10.1152/ajpheart.00405.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fannin J, Rice KM, Thulluri S, Dornon L, Arvapalli RK, Wehner P, Blough ER. Age-associated alterations of cardiac structure and function in the female F344xBN rat heart. Age (Dordr). 2014;36:9684. doi: 10.1007/s11357-014-9684-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Takeda T, Hosokawa M, Higuchi K. Senescence-accelerated mouse (SAM): a novel murine model of senescence. Exp Gerontol. 1997;32:105–109. doi: 10.1016/s0531-5565(96)00036-8 [DOI] [PubMed] [Google Scholar]
- 98.Reed AL, Tanaka A, Sorescu D, Liu H, Jeong EM, Sturdy M, Walp ER, Dudley SC Jr., Sutliff RL. Diastolic dysfunction is associated with cardiac fibrosis in the senescence-accelerated mouse. Am J Physiol Heart Circ Physiol. 2011;301:H824–831. doi: 10.1152/ajpheart.00407.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gevaert AB, Shakeri H, Leloup AJ, Van Hove CE, De Meyer GRY, Vrints CJ, Lemmens K, Van Craenenbroeck EM. Endothelial Senescence Contributes to Heart Failure With Preserved Ejection Fraction in an Aging Mouse Model. Circ Heart Fail. 2017;10. doi: 10.1161/CIRCHEARTFAILURE.116.003806 [DOI] [PubMed] [Google Scholar]
- 100.Withaar C, Meems LMG, Markousis-Mavrogenis G, Boogerd CJ, Sillje HHW, Schouten EM, Dokter MM, Voors AA, Westenbrink BD, Lam CSP, et al. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc Res. 2021;117:2108–2124. doi: 10.1093/cvr/cvaa256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Deng Y, Xie M, Li Q, Xu X, Ou W, Zhang Y, Xiao H, Yu H, Zheng Y, Liang Y, et al. Targeting Mitochondria-Inflammation Circuit by beta-Hydroxybutyrate Mitigates HFpEF. Circ Res. 2021;128:232–245. doi: 10.1161/CIRCRESAHA.120.317933 [DOI] [PubMed] [Google Scholar]
- 102.Bragazzi NL, Zhong W, Shu J, Abu Much A, Lotan D, Grupper A, Younis A, Dai H. Burden of heart failure and underlying causes in 195 countries and territories from 1990 to 2017. Eur J Prev Cardiol. 2021;28:1682–1690. doi: 10.1093/eurjpc/zwaa147 [DOI] [PubMed] [Google Scholar]
- 103.Packer M, Kitzman DW. Obesity-Related Heart Failure With a Preserved Ejection Fraction: The Mechanistic Rationale for Combining Inhibitors of Aldosterone, Neprilysin, and Sodium-Glucose Cotransporter-2. JACC Heart Fail. 2018;6:633–639. doi: 10.1016/j.jchf.2018.01.009 [DOI] [PubMed] [Google Scholar]
- 104.Mishra S, Kass DA. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2021. doi: 10.1038/s41569-020-00480-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cheng ZJ, Vaskonen T, Tikkanen I, Nurminen K, Ruskoaho H, Vapaatalo H, Muller D, Park JK, Luft FC, Mervaala EM. Endothelial dysfunction and salt-sensitive hypertension in spontaneously diabetic Goto-Kakizaki rats. Hypertension. 2001;37:433–439. doi: 10.1161/01.hyp.37.2.433 [DOI] [PubMed] [Google Scholar]
- 106.Meagher P, Civitarese R, Lee X, Gordon M, Bugyei-Twum A, Desjardins JF, Kabir G, Zhang Y, Kosanam H, Visram A, et al. The Goto Kakizaki rat: Impact of age upon changes in cardiac and renal structure, function. PLoS One. 2021;16:e0252711. doi: 10.1371/journal.pone.0252711 [doi];PONE-D-20-39982 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Leite S, Cerqueira RJ, Ibarrola J, Fontoura D, Fernandez-Celis A, Zannad F, Falcao-Pires I, Paulus WJ, Leite-Moreira AF, Rossignol P, et al. Arterial Remodeling and Dysfunction in the ZSF1 Rat Model of Heart Failure With Preserved Ejection Fraction. Circ Heart Fail. 2019;12:e005596. doi: 10.1161/CIRCHEARTFAILURE.118.005596 [DOI] [PubMed] [Google Scholar]
- 108.Hamdani N, Franssen C, Lourenco A, Falcao-Pires I, Fontoura D, Leite S, Plettig L, Lopez B, Ottenheijm CA, Becher PM, et al. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail. 2013;6:1239–1249. doi: CIRCHEARTFAILURE.113.000539 [pii]; 10.1161/CIRCHEARTFAILURE.113.000539 [doi] [DOI] [PubMed] [Google Scholar]
- 109.Schauer A, Draskowski R, Jannasch A, Kirchhoff V, Goto K, Mannel A, Barthel P, Augstein A, Winzer E, Tugtekin M, et al. ZSF1 rat as animal model for HFpEF: Development of reduced diastolic function and skeletal muscle dysfunction. ESC Heart Fail. 2020;7:2123–2134. doi: 10.1002/ehf2.12915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.van Dijk CG, Oosterhuis NR, Xu YJ, Brandt M, Paulus WJ, van Heerebeek L, Duncker DJ, Verhaar MC, Fontoura D, Lourenco AP, et al. Distinct Endothelial Cell Responses in the Heart and Kidney Microvasculature Characterize the Progression of Heart Failure With Preserved Ejection Fraction in the Obese ZSF1 Rat With Cardiorenal Metabolic Syndrome. Circ Heart Fail. 2016;9:e002760. doi: 10.1161/CIRCHEARTFAILURE.115.002760 [DOI] [PubMed] [Google Scholar]
- 111.Goto K, Schauer A, Augstein A, Methawasin M, Granzier H, Halle M, Craenenbroeck EMV, Rolim N, Gielen S, Pieske B, et al. Muscular changes in animal models of heart failure with preserved ejection fraction: what comes closest to the patient? ESC Heart Fail. 2021;8:139–150. doi: 10.1002/ehf2.13142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tofovic SP, Kusaka H, Kost CK Jr., Bastacky S Renal function and structure in diabetic, hypertensive, obese ZDFxSHHF-hybrid rats. Ren Fail. 2000;22:387–406. doi: 10.1081/jdi-100100882 [DOI] [PubMed] [Google Scholar]
- 113.Espino-Gonzalez E, Tickle PG, Benson AP, Kissane RWP, Askew GN, Egginton S, Bowen TS. Abnormal skeletal muscle blood flow, contractile mechanics and fibre morphology in a rat model of obese-HFpEF. J Physiol. 2021;599:981–1001. doi: 10.1113/JP280899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nguyen ITN, Brandt MM, van de Wouw J, van Drie RWA, Wesseling M, Cramer MJ, de Jager SCA, Merkus D, Duncker DJ, Cheng C, et al. Both male and female obese ZSF1 rats develop cardiac dysfunction in obesity-induced heart failure with preserved ejection fraction. PLoS One. 2020;15:e0232399. doi: 10.1371/journal.pone.0232399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tong D, Schiattarella GG, Jiang N, May HI, Lavandero S, Gillette TG, Hill JA. Female Sex Is Protective in a Preclinical Model of Heart Failure With Preserved Ejection Fraction. Circulation. 2019;140:1769–1771. doi: 10.1161/CIRCULATIONAHA.119.042267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lelovas PP, Kostomitsopoulos NG, Xanthos TT. A comparative anatomic and physiologic overview of the porcine heart. J Am Assoc Lab Anim Sci. 2014;53:432–438. [PMC free article] [PubMed] [Google Scholar]
- 117.Sharp TE 34d, Scarborough AL, Li Z, Polhemus DJ, Hidalgo HA, Shchumacher JD, Matsuura TR, Jenkins JS, Kelly DP, Goodchild TT, et al. Novel Gottingen miniswine model of heart failure with preserved ejection fraction integrating multiple comorbidities. JACC Basic Transl Sci. 2021;6:154-170. doi: 10.1016/j.jactbs.2020.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Borbely A, Falcao-Pires I, van Heerebeek L, Hamdani N, Edes I, Gavina C, Leite-Moreira AF, Bronzwaer JG, Papp Z, van der Velden J, et al. Hypophosphorylation of the Stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res. 2009;104:780–786. doi: 10.1161/CIRCRESAHA.108.193326 [DOI] [PubMed] [Google Scholar]
- 119.Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschope C, Leite-Moreira AF, Musters R, Niessen HW, Linke WA, et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail. 2016;4:312–324. doi: 10.1016/j.jchf.2015.10.007 [DOI] [PubMed] [Google Scholar]
- 120.Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–271. doi: 10.1016/j.jacc.2013.02.092 [DOI] [PubMed] [Google Scholar]
- 121.Summer G, Kuhn AR, Munts C, Miranda-Silva D, Leite-Moreira AF, Lourenco AP, Heymans S, Falcao-Pires I, van Bilsen M. A directed network analysis of the cardiome identifies molecular pathways contributing to the development of HFpEF. J Mol Cell Cardiol. 2020;144:66–75. doi: 10.1016/j.yjmcc.2020.05.008 [DOI] [PubMed] [Google Scholar]
- 122.Schiattarella GG, Sequeira V, Ameri P. Distinctive patterns of inflammation across the heart failure syndrome. Heart Fail Rev. 2021;26:1333–1344. doi: 10.1007/s10741-020-09949-5 [DOI] [PubMed] [Google Scholar]
- 123.Schiattarella GG, Alcaide P, Condorelli G, Gillette TG, Heymans S, Jones EAV, Kallikourdis M, Lichtman AH, Marelli-Berg FM, Shah S, et al. Immunometabolic Mechanisms of Heart Failure with Preserved Ejection Fraction. Nature Cardiovascular Research. 2022;1:211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Packer M Epicardial Adipose Tissue May Mediate Deleterious Effects of Obesity and Inflammation on the Myocardium. J Am Coll Cardiol. 2018;71:2360–2372. doi: 10.1016/j.jacc.2018.03.509 [DOI] [PubMed] [Google Scholar]
- 125.Nishida K, Otsu K. Inflammation and metabolic cardiomyopathy. Cardiovasc Res. 2017;113:389–398. doi: 10.1093/cvr/cvx012 [DOI] [PubMed] [Google Scholar]
- 126.Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation. 2001;103:2055–2059. doi: 10.1161/01.cir.103.16.2055 [DOI] [PubMed] [Google Scholar]
- 127.Briasoulis A, Androulakis E, Christophides T, Tousoulis D. The role of inflammation and cell death in the pathogenesis, progression and treatment of heart failure. Heart Fail Rev. 2016;21:169–176. doi: 10.1007/s10741-016-9533-z [DOI] [PubMed] [Google Scholar]
- 128.Hahn VS, Yanek LR, Vaishnav J, Ying W, Vaidya D, Lee YZJ, Riley SJ, Subramanya V, Brown EE, Hopkins CD, et al. Endomyocardial Biopsy Characterization of Heart Failure With Preserved Ejection Fraction and Prevalence of Cardiac Amyloidosis. JACC Heart Fail. 2020;8:712–724. doi: 10.1016/j.jchf.2020.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Paulus WJ. Unfolding Discoveries in Heart Failure. N Engl J Med. 2020;382:679–682. doi: 10.1056/NEJMcibr1913825 [DOI] [PubMed] [Google Scholar]
- 130.Nevers T, Salvador AM, Grodecki-Pena A, Knapp A, Velazquez F, Aronovitz M, Kapur NK, Karas RH, Blanton RM, Alcaide P. Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure. Circulation Heart failure. 2015;8:776–787. doi: 10.1161/CIRCHEARTFAILURE.115.002225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schiattarella GG, Rodolico D, Hill JA. Metabolic Inflammation in Heart Failure with Preserved Ejection Fraction. Cardiovascular research. 2020. doi: 10.1093/cvr/cvaa217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rai A, Narisawa M, Li P, Piao L, Li Y, Yang G, Cheng XW. Adaptive immune disorders in hypertension and heart failure: focusing on T-cell subset activation and clinical implications. J Hypertens. 2020;38:1878–1889. doi: 10.1097/HJH.0000000000002456 [DOI] [PubMed] [Google Scholar]
- 133.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT, Anti TNFTACHFI. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107:3133–3140. doi: 10.1161/01.CIR.0000077913.60364.D2 [DOI] [PubMed] [Google Scholar]
- 134.Tromp J, Claggett BL, Liu J, Jackson AM, Jhund PS, Kober L, Widimsky J, Boytsov SA, Chopra VK, Anand IS, et al. Global Differences in Heart Failure With Preserved Ejection Fraction: The PARAGON-HF Trial. Circ Heart Fail. 2021;14:e007901. doi: 10.1161/CIRCHEARTFAILURE.120.007901 [doi] [DOI] [PubMed] [Google Scholar]
- 135.Kasiakogias A, Rosei EA, Camafort M, Ehret G, Faconti L, Ferreira JP, Brguljan J, Januszewicz A, Kahan T, Manolis A, et al. Hypertension and heart failure with preserved ejection fraction: position paper by the European Society of Hypertension. J Hypertens. 2021;39:1522–1545. doi: 10.1097/HJH.0000000000002910 [doi];00004872-202108000-00006 [pii] [DOI] [PubMed] [Google Scholar]
- 136.Messerli FH, Rimoldi SF, Bangalore S. The Transition From Hypertension to Heart Failure: Contemporary Update. JACC Heart Fail. 2017;5:543–551. doi: S2213–1779(17)30317–7 [pii]; 10.1016/j.jchf.2017.04.012 [doi] [DOI] [PubMed] [Google Scholar]
- 137.Shah SJ, Kitzman DW, Borlaug BA, van HL, Zile MR, Kass DA, Paulus WJ. Phenotype-Specific Treatment of Heart Failure With Preserved Ejection Fraction: A Multiorgan Roadmap. Circulation. 2016;134:73–90. doi: CIRCULATIONAHA.116.021884 [pii]; 10.1161/CIRCULATIONAHA.116.021884 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee CJ, Park S. Hypertension and Heart Failure with Preserved Ejection Fraction. Heart Fail Clin. 2021;17:337–343. doi: S1551–7136(21)00023–4 [pii]; 10.1016/j.hfc.2021.02.002 [doi] [DOI] [PubMed] [Google Scholar]
- 139.Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, Slater R, Palmer BM, Van BP, Meyer M, et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. 2015;131:1247–1259. doi: CIRCULATIONAHA.114.013215 [pii]; 10.1161/CIRCULATIONAHA.114.013215 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lewis GA, Schelbert EB, Williams SG, Cunnington C, Ahmed F, McDonagh TA, Miller CA. Biological Phenotypes of Heart Failure With Preserved Ejection Fraction. J Am Coll Cardiol. 2017;70:2186–2200. doi: S0735–1097(17)39620–1 [pii]; 10.1016/j.jacc.2017.09.006 [doi] [DOI] [PubMed] [Google Scholar]
- 141.Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJ, Michelson EL, Olofsson B, Ostergren J, Yusuf S, Pocock S. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet. 2003;362:759–766. doi: S0140673603142821 [pii]; 10.1016/s0140-6736(03)14282-1 [doi] [DOI] [PubMed] [Google Scholar]
- 142.Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, Anderson S, Donovan M, Iverson E, Staiger C, et al. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med. 2008;359:2456–2467. doi: NEJMoa0805450 [pii]; 10.1056/NEJMoa0805450 [doi] [DOI] [PubMed] [Google Scholar]
- 143.Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, Clausell N, Desai AS, Diaz R, Fleg JL, et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. 2014;370:1383–1392. doi: 10.1056/NEJMoa1313731 [doi] [DOI] [PubMed] [Google Scholar]
- 144.Samson R, Jaiswal A, Ennezat PV, Cassidy M, Le Jemtel TH. Clinical Phenotypes in Heart Failure With Preserved Ejection Fraction. J Am Heart Assoc. 2016;5. doi: JAHA.115.002477 [pii]; 10.1161/JAHA.115.002477 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Becher PM, Lindner D, Miteva K, Savvatis K, Zietsch C, Schmack B, Van LS, Westermann D, Schultheiss HP, Tschope C. Role of heart rate reduction in the prevention of experimental heart failure: comparison between If-channel blockade and beta-receptor blockade. Hypertension. 2012;59:949–957. doi: HYPERTENSIONAHA.111.183913 [pii]; 10.1161/HYPERTENSIONAHA.111.183913 [doi] [DOI] [PubMed] [Google Scholar]
- 146.Murdoch CE, Chaubey S, Zeng L, Yu B, Ivetic A, Walker SJ, Vanhoutte D, Heymans S, Grieve DJ, Cave AC, et al. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J Am Coll Cardiol. 2014;63:2734–2741. doi: S0735–1097(14)01557–5 [pii]; 10.1016/j.jacc.2014.02.572 [doi] [DOI] [PubMed] [Google Scholar]
- 147.Ichihara S, Senbonmatsu T, P E Jr, Ichiki T, Gaffney FA, Inagami T. Angiotensin II type 2 receptor is essential for left ventricular hypertrophy and cardiac fibrosis in chronic angiotensin II-induced hypertension. Circulation. 2001;104:346–351. doi: 10.1161/01.cir.104.3.346 [doi] [DOI] [PubMed] [Google Scholar]
- 148.Matsumoto E, Sasaki S, Kinoshita H, Kito T, Ohta H, Konishi M, Kuwahara K, Nakao K, Itoh N. Angiotensin II-induced cardiac hypertrophy and fibrosis are promoted in mice lacking Fgf16. Genes Cells. 2013;18:544–553. doi: 10.1111/gtc.12055 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Regan JA, Mauro AG, Carbone S, Marchetti C, Gill R, Mezzaroma E, Valle RJ, Salloum FN, Van Tassell BW, Abbate A, et al. A mouse model of heart failure with preserved ejection fraction due to chronic infusion of a low subpressor dose of angiotensin II. Am J Physiol Heart Circ Physiol. 2015;309:H771–H778. doi: ajpheart.00282.2015 [pii]; 10.1152/ajpheart.00282.2015 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Glenn DJ, Cardema MC, Ni W, Zhang Y, Yeghiazarians Y, Grapov D, Fiehn O, Gardner DG. Cardiac steatosis potentiates angiotensin II effects in the heart. Am J Physiol Heart Circ Physiol. 2015;308:H339–H350. doi: ajpheart.00742.2014 [pii]; 10.1152/ajpheart.00742.2014 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Westermann D, Becher PM, Lindner D, Savvatis K, Xia Y, Frohlich M, Hoffmann S, Schultheiss HP, Tschope C. Selective PDE5A inhibition with sildenafil rescues left ventricular dysfunction, inflammatory immune response and cardiac remodeling in angiotensin II-induced heart failure in vivo. Basic Res Cardiol. 2012;107:308. doi: 10.1007/s00395-012-0308-y [doi] [DOI] [PubMed] [Google Scholar]
- 152.Peng H, Yang XP, Carretero OA, Nakagawa P, D’Ambrosio M, Leung P, Xu J, Peterson EL, Gonzalez GE, Harding P, et al. Angiotensin II-induced dilated cardiomyopathy in Balb/c but not C57BL/6J mice. Exp Physiol. 2011;96:756–764. doi: expphysiol.2011.057612 [pii]; 10.1113/expphysiol.2011.057612 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kadoguchi T, Kinugawa S, Takada S, Fukushima A, Furihata T, Homma T, Masaki Y, Mizushima W, Nishikawa M, Takahashi M, et al. Angiotensin II can directly induce mitochondrial dysfunction, decrease oxidative fibre number and induce atrophy in mouse hindlimb skeletal muscle. Exp Physiol. 2015;100:312–322. doi: 10.1113/expphysiol.2014.084095 [doi] [DOI] [PubMed] [Google Scholar]
- 154.Ogata T, Miyauchi T, Sakai S, Takanashi M, Irukayama-Tomobe Y, Yamaguchi I. Myocardial fibrosis and diastolic dysfunction in deoxycorticosterone acetate-salt hypertensive rats is ameliorated by the peroxisome proliferator-activated receptor-alpha activator fenofibrate, partly by suppressing inflammatory responses associated with the nuclear factor-kappa-B pathway. J Am Coll Cardiol. 2004;43:1481–1488. doi: 10.1016/j.jacc.2003.11.043 [doi];S0735109704001901 [pii] [DOI] [PubMed] [Google Scholar]
- 155.Matsumura Y, Kuro T, Konishi F, Takaoka M, Gariepy CE, Yanagisawa M. Enhanced blood pressure sensitivity to DOCA-salt treatment in endothelin ET(B) receptor-deficient rats. Br J Pharmacol. 2000;129:1060–1062. doi: 10.1038/sj.bjp.0703157 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Grobe JL, Mecca AP, Mao H, Katovich MJ. Chronic angiotensin-(1–7) prevents cardiac fibrosis in DOCA-salt model of hypertension. Am J Physiol Heart Circ Physiol. 2006;290:H2417–H2423. doi: 01170.2005 [pii]; 10.1152/ajpheart.01170.2005 [doi] [DOI] [PubMed] [Google Scholar]
- 157.Jeong EM, Monasky MM, Gu L, Taglieri DM, Patel BG, Liu H, Wang Q, Greener I, Dudley SC, Jr., Solaro RJ. Tetrahydrobiopterin improves diastolic dysfunction by reversing changes in myofilament properties. J Mol Cell Cardiol. 2013;56:44–54. doi: S0022–2828(12)00431–2 [pii]; 10.1016/j.yjmcc.2012.12.003 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Allan A, Fenning A, Levick S, Hoey A, Brown L. Reversal of cardiac dysfunction by selective ET-A receptor antagonism. Br J Pharmacol. 2005;146:846–853. doi: 0706384 [pii]; 10.1038/sj.bjp.0706384 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lovelock JD, Monasky MM, Jeong EM, Lardin HA, Liu H, Patel BG, Taglieri DM, Gu L, Kumar P, Pokhrel N, et al. Ranolazine improves cardiac diastolic dysfunction through modulation of myofilament calcium sensitivity. Circ Res. 2012;110:841–850. doi: CIRCRESAHA.111.258251 [pii]; 10.1161/CIRCRESAHA.111.258251 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mohammed SF, Ohtani T, Korinek J, Lam CS, Larsen K, Simari RD, Valencik ML, Burnett JC Jr., Redfield MM. Mineralocorticoid accelerates transition to heart failure with preserved ejection fraction via “nongenomic effects”. Circulation. 2010;122:370–378. doi: CIRCULATIONAHA.109.915215 [pii]; 10.1161/CIRCULATIONAHA.109.915215 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bowen TS, Eisenkolb S, Drobner J, Fischer T, Werner S, Linke A, Mangner N, Schuler G, Adams V. High-intensity interval training prevents oxidant-mediated diaphragm muscle weakness in hypertensive mice. FASEB J. 2017;31:60–71. doi: fj.201600672R [pii]; 10.1096/fj.201600672R [doi] [DOI] [PubMed] [Google Scholar]
- 162.Hulsmans M, Sager HB, Roh JD, Valero-Munoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B, et al. Cardiac macrophages promote diastolic dysfunction. J Exp Med. 2018;215:423–440. doi: jem.20171274 [pii]; 10.1084/jem.20171274 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yoon S, Kim M, Lee H, Kang G, Bedi K, Margulies KB, Jain R, Nam KI, Kook H, Eom GH. S-Nitrosylation of Histone Deacetylase 2 by Neuronal Nitric Oxide Synthase as a Mechanism of Diastolic Dysfunction. Circulation. 2021;143:1912–1925. doi: 10.1161/CIRCULATIONAHA.119.043578 [doi] [DOI] [PubMed] [Google Scholar]
- 164.Brilla CG, Weber KT. Mineralocorticoid excess, dietary sodium, and myocardial fibrosis. J Lab Clin Med. 1992;120:893–901. doi: 0022–2143(92)90267-O [pii] [PubMed] [Google Scholar]
- 165.Brilla CG, Weber KT. Reactive and reparative myocardial fibrosis in arterial hypertension in the rat. Cardiovasc Res. 1992;26:671–677. doi: 10.1093/cvr/26.7.671 [doi] [DOI] [PubMed] [Google Scholar]
- 166.Young M, Fullerton M, Dilley R, Funder J. Mineralocorticoids, hypertension, and cardiac fibrosis. J Clin Invest. 1994;93:2578–2583. doi: 10.1172/JCI117269 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sam F, Xie Z, Ooi H, Kerstetter DL, Colucci WS, Singh M, Singh K. Mice lacking osteopontin exhibit increased left ventricular dilation and reduced fibrosis after aldosterone infusion. Am J Hypertens. 2004;17:188–193. doi: S0895706103012032 [pii]; 10.1016/j.amjhyper.2003.10.007 [doi] [DOI] [PubMed] [Google Scholar]
- 168.Zhang L, Chen J, Yan L, He Q, Xie H, Chen M. Resveratrol Ameliorates Cardiac Remodeling in a Murine Model of Heart Failure With Preserved Ejection Fraction. Front Pharmacol. 2021;12:646240. doi: 10.3389/fphar.2021.646240 [doi];646240 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yang HJ, Kong B, Shuai W, Zhang JJ, Huang H. MD1 deletion exaggerates cardiomyocyte autophagy induced by heart failure with preserved ejection fraction through ROS/MAPK signalling pathway. J Cell Mol Med. 2020;24:9300–9312. doi: 10.1111/jcmm.15579 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Valero-Munoz M, Li S, Wilson RM, Hulsmans M, Aprahamian T, Fuster JJ, Nahrendorf M, Scherer PE, Sam F. Heart Failure With Preserved Ejection Fraction Induces Beiging in Adipose Tissue. Circ Heart Fail. 2016;9:e002724. doi: CIRCHEARTFAILURE.115.002724 [pii]; 10.1161/CIRCHEARTFAILURE.115.002724 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Valero-Munoz M, Li S, Wilson RM, Boldbaatar B, Iglarz M, Sam F. Dual Endothelin-A/Endothelin-B Receptor Blockade and Cardiac Remodeling in Heart Failure With Preserved Ejection Fraction. Circ Heart Fail. 2016;9. doi: CIRCHEARTFAILURE.116.003381 [pii]; 10.1161/CIRCHEARTFAILURE.116.003381 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wilson RM, De Silva DS, Sato K, Izumiya Y, Sam F. Effects of fixed-dose isosorbide dinitrate/hydralazine on diastolic function and exercise capacity in hypertension-induced diastolic heart failure. Hypertension. 2009;54:583–590. doi: HYPERTENSIONAHA.109.134932 [pii]; 10.1161/HYPERTENSIONAHA.109.134932 [doi] [DOI] [PubMed] [Google Scholar]
- 173.Hamdani N, Paulus WJ. Myocardial titin and collagen in cardiac diastolic dysfunction: partners in crime. Circulation. 2013;128:5–8. doi: CIRCULATIONAHA.113.003437 [pii]; 10.1161/CIRCULATIONAHA.113.003437 [doi] [DOI] [PubMed] [Google Scholar]
- 174.Conceicao G, Heinonen I, Lourenco AP, Duncker DJ, Falcao-Pires I. Animal models of heart failure with preserved ejection fraction. Neth Heart J. 2016;24:275–286. doi: 10.1007/s12471-016-0815-9 [doi];10.1007/s12471-016-0815-9 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wallner M, Eaton DM, Berretta RM, Liesinger L, Schittmayer M, Gindlhuber J, Wu J, Jeong MY, Lin YH, Borghetti G, et al. HDAC inhibition improves cardiopulmonary function in a feline model of diastolic dysfunction. Science translational medicine. 2020;12. doi: 10.1126/scitranslmed.aay7205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Gomes AC, Falcao-Pires I, Pires AL, Bras-Silva C, Leite-Moreira AF. Rodent models of heart failure: an updated review. Heart Fail Rev. 2013;18:219–249. doi: 10.1007/s10741-012-9305-3 [doi] [DOI] [PubMed] [Google Scholar]
- 177.Doi R, Masuyama T, Yamamoto K, Doi Y, Mano T, Sakata Y, Ono K, Kuzuya T, Hirota S, Koyama T, et al. Development of different phenotypes of hypertensive heart failure: systolic versus diastolic failure in Dahl salt-sensitive rats. J Hypertens. 2000;18:111–120. doi: 10.1097/00004872-200018010-00016 [doi] [DOI] [PubMed] [Google Scholar]
- 178.Bing OH, Brooks WW, Robinson KG, Slawsky MT, Hayes JA, Litwin SE, Sen S, Conrad CH. The spontaneously hypertensive rat as a model of the transition from compensated left ventricular hypertrophy to failure. J Mol Cell Cardiol. 1995;27:383–396. doi: 10.1016/s0022-2828(08)80035-1 [doi] [DOI] [PubMed] [Google Scholar]
- 179.Boluyt MO, Robinson KG, Meredith AL, Sen S, Lakatta EG, Crow MT, Brooks WW, Conrad CH, Bing OH. Heart failure after long-term supravalvular aortic constriction in rats. Am J Hypertens. 2005;18:202–212. doi: S0895–7061(04)01017–9 [pii]; 10.1016/j.amjhyper.2004.08.034 [doi] [DOI] [PubMed] [Google Scholar]
- 180.Knight WE, Chen S, Zhang Y, Oikawa M, Wu M, Zhou Q, Miller CL, Cai Y, Mickelsen DM, Moravec C, et al. PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc Natl Acad Sci U S A. 2016;113:E7116–E7125. doi: 1607728113 [pii]; 10.1073/pnas.1607728113 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ma J, Luo T, Zeng Z, Fu H, Asano Y, Liao Y, Minamino T, Kitakaze M. Histone Deacetylase Inhibitor Phenylbutyrate Exaggerates Heart Failure in Pressure Overloaded Mice independently of HDAC inhibition. Sci Rep. 2016;6:34036. doi: srep34036 [pii]; 10.1038/srep34036 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Abbate A, Arena R, Abouzaki N, Van Tassell BW, Canada J, Shah K, Biondi-Zoccai G, Voelkel NF. Heart failure with preserved ejection fraction: refocusing on diastole. Int J Cardiol. 2015;179:430–440. doi: S0167–5273(14)02267–0 [pii]; 10.1016/j.ijcard.2014.11.106 [doi] [DOI] [PubMed] [Google Scholar]
- 183.Dunlay SM, Roger VL, Weston SA, Jiang R, Redfield MM. Longitudinal changes in ejection fraction in heart failure patients with preserved and reduced ejection fraction. Circ Heart Fail. 2012;5:720–726. doi: CIRCHEARTFAILURE.111.966366 [pii]; 10.1161/CIRCHEARTFAILURE.111.966366 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Travers JG, Wennersten SA, Pena B, Bagchi RA, Smith HE, Hirsch RA, Vanderlinden LA, Lin YH, Dobrinskikh E, Demos-Davies KM, et al. HDAC Inhibition Reverses Preexisting Diastolic Dysfunction and Blocks Covert Extracellular Matrix Remodeling. Circulation. 2021;143:1874–1890. doi: 10.1161/CIRCULATIONAHA.120.046462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Gori M, Senni M, Gupta DK, Charytan DM, Kraigher-Krainer E, Pieske B, Claggett B, Shah AM, Santos AB, Zile MR, et al. Association between renal function and cardiovascular structure and function in heart failure with preserved ejection fraction. Eur Heart J. 2014;35:3442–3451. doi: ehu254 [pii]; 10.1093/eurheartj/ehu254 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Unger ED, Dubin RF, Deo R, Daruwalla V, Friedman JL, Medina C, Beussink L, Freed BH, Shah SJ. Association of chronic kidney disease with abnormal cardiac mechanics and adverse outcomes in patients with heart failure and preserved ejection fraction. Eur J Heart Fail. 2016;18:103–112. doi: 10.1002/ejhf.445 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Damman K, Testani JM. The kidney in heart failure: an update. Eur Heart J. 2015;36:1437–1444. doi: ehv010 [pii]; 10.1093/eurheartj/ehv010 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Agrawal A, Naranjo M, Kanjanahattakij N, Rangaswami J, Gupta S. Cardiorenal syndrome in heart failure with preserved ejection fraction-an under-recognized clinical entity. Heart Fail Rev. 2019;24:421–437. doi: 10.1007/s10741-018-09768-9 [doi];10.1007/s10741-018-09768-9 [pii] [DOI] [PubMed] [Google Scholar]
- 189.Ananthram MG, Gottlieb SS. Renal Dysfunction and Heart Failure with Preserved Ejection Fraction. Heart Fail Clin. 2021;17:357–367. doi: S1551–7136(21)00038–6 [pii]; 10.1016/j.hfc.2021.03.005 [doi] [DOI] [PubMed] [Google Scholar]
- 190.Ter Maaten JM, Damman K, Verhaar MC, Paulus WJ, Duncker DJ, Cheng C, van HL, Hillege HL, Lam CS, Navis G, et al. Connecting heart failure with preserved ejection fraction and renal dysfunction: the role of endothelial dysfunction and inflammation. Eur J Heart Fail. 2016;18:588–598. doi: 10.1002/ejhf.497 [doi] [DOI] [PubMed] [Google Scholar]
- 191.Valero-Munoz M, Oh A, Faudoa E, Breton-Romero R, El AF, Bujor A, Sam F. Endothelial-Mesenchymal Transition in Heart Failure With a Preserved Ejection Fraction: Insights Into the Cardiorenal Syndrome. Circ Heart Fail. 2021;14:e008372. doi: 10.1161/CIRCHEARTFAILURE.121.008372 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sam F, Duhaney TA, Sato K, Wilson RM, Ohashi K, Sono-Romanelli S, Higuchi A, De Silva DS, Qin F, Walsh K, et al. Adiponectin deficiency, diastolic dysfunction, and diastolic heart failure. Endocrinology. 2010;151:322–331. doi: en.2009–0806 [pii]; 10.1210/en.2009-0806 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Sengenes C, Berlan M, De Glisezinski I, Lafontan M, Galitzky J. Natriuretic peptides: a new lipolytic pathway in human adipocytes. FASEB J. 2000;14:1345–1351. [PubMed] [Google Scholar]
- 194.Jeanson Y, Carriere A, Casteilla L. A New Role for Browning as a Redox and Stress Adaptive Mechanism? Front Endocrinol (Lausanne). 2015;6:158. doi: 10.3389/fendo.2015.00158 [DOI] [PMC free article] [PubMed] [Google Scholar]