

Abstract
The complement (C) field has recently experienced a strong resurgence of interest due to, the unexpected discovery of new C functions extending its role beyond immunity and pathogen clearance, a growing list of diseases in which C plays a role, and proliferation of C therapeutics. Importantly, while the majority of C components in circulation are generated by the liver and activated extracellularly, C activation unexpectedly also occurs intracellularly across a broad range of cells. Such cell-autonomous C activation can engage intracellular C receptors, which then drive non-canonical cell-specific effector functions. Thus, much remains to be discovered about C biology. In this short review, we will focus on novel non-canonical activities of C in its ‘classic areas of operation’: kidney and brain biology, infection and autoimmunity – with an outlook on the next generation of C-targeted therapeutics.
Keywords: complement, brain, kidney, autoimmune disease, infections
Introduction
The complement (C) system is an ancient pathogen recognition receptor system (PRR) discovered by Jules Bordet in the late 19th century(1). It consists of over 50 proteins that are mostly generated by the liver and either circulate in the fluid phase or are membrane bound. Most C core proteins exist in a pre-enzymatic form and are activated in a sequential and cascade-like fashion (2–4). There are three activation pathways, the classical (CP), alternative (AP) and lectin pathway (LP) (4). The three pathways are triggered by distinct pathogens or noxious (self) antigens but then cumulate at the activation of the C core proteins C3 (C3a and C3b) and C5 (C5a and C5b) via the formation of C3/C5 convertases. C3 and C5 activation leads to the formation of the membrane attack complex (MAC, C5b-9) that forms pores on the pathogen surface and induces its lytic killing (2, 5). The anaphylatoxins, C3a and C5a signal through G protein-coupled receptors C3aR and C5aR leading to the recruitment and activation of immune cells. C3b (and further break-down products) are strong opsonins and mediate the phagocytic uptake and clearance of pathogens via scavenger cells (6). The C system is regulated by a group of proteins under the umbrella ‘Regulators of C Activation’ that are placed at strategic locations along the pathways (7).
Overall, C is recognized as the surveillance mechanism and the first line of host defense against pathogenic invasion (Figure 1) and C deficiencies are thus often associated with recurrent infections (8). Importantly, because C is central to the detection and removal of self-derived danger (for example, apoptotic cells and immune complexes (ICs)), C deficiencies can also cause autoimmune diseases such as systemic lupus erythematosus (SLE) (9, 10). Further, unwanted, uncontrolled, or prolonged C activation are all connected with a range of highly prevalent inflammatory disease conditions, including arthritis and cardiovascular disease (11, 12). Thus, C is known for decades to be an important therapeutic target and much time and effort has been invested in ‘re-setting’ hyper-complement activation in acute and chronic inflammation.
Figure 1. Visual summary of key insights gained at the AAI/ICS Guest Symposium.
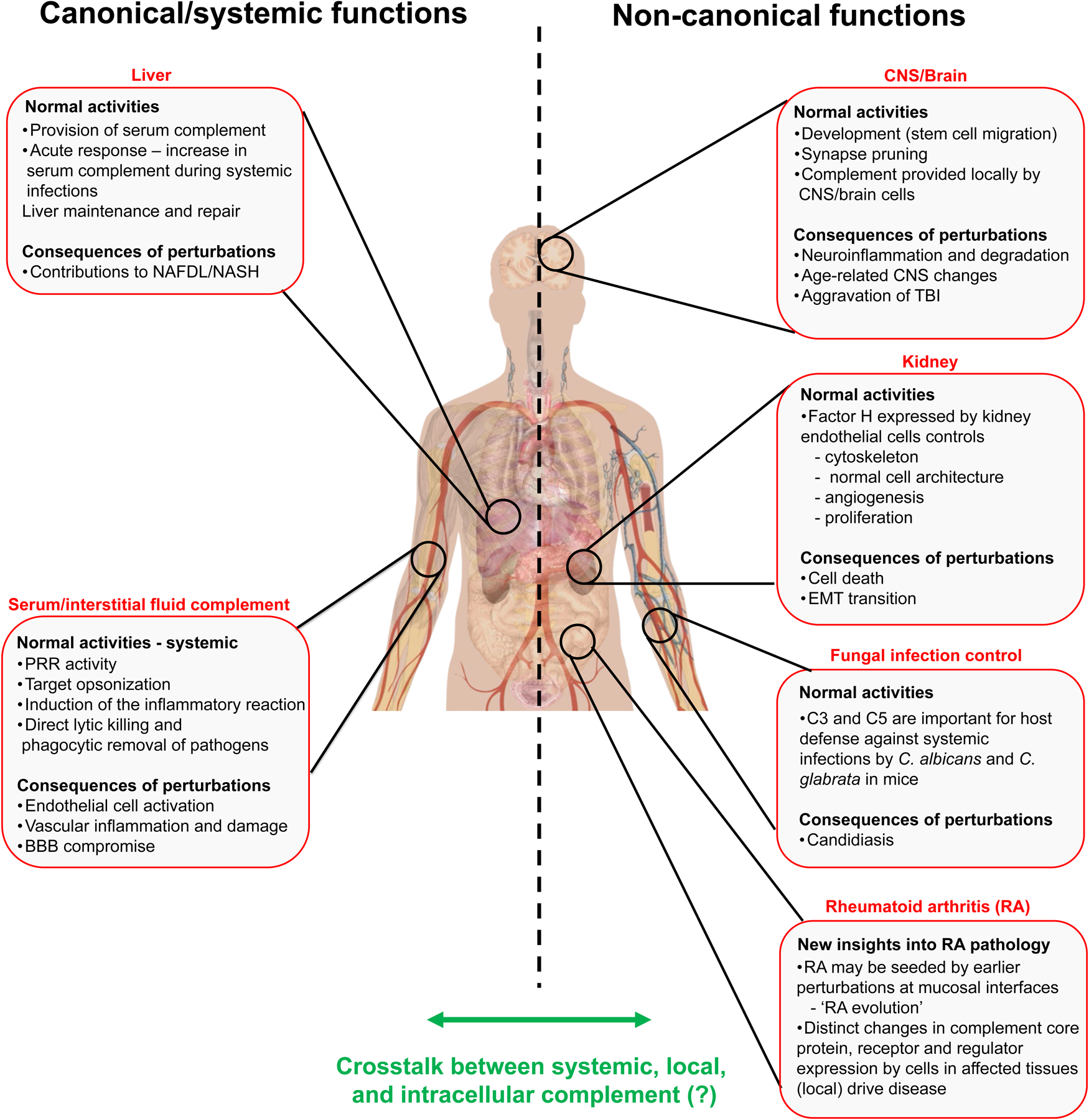
The left side of the graphic summarizes the classic role of liver-derived C and consequences of its perturbations. The right side summarizes the discussed new roles of mostly cell-autonomous C activities in the brain, kidney and during candida infection and new insights gained about local C production during the evolution of rheumatoid arthritis. The exact underlying molecular mechanisms of these new activities remain to be defined, as are the potential cross-talks between the different locations of C activation and function (indicated by green arrow). BBB, blood brain barrier; CNS, central nervous system; EMT, epithelial-mesenchymal transition; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; RA, rheumatoid arthritis; TBI, traumatic brain injury.
Over the last decade, unexpected additional locations of C activation and function have been identified. Liver-derived and serum-circulating C proteins are accepted as key in fighting blood borne pathogens (2, 4, 13). However, studies from as early as the 1980s have indicated that many cell types can synthesize and secrete a range of C components into the close environmental space (14–17) across tissues and organs ranging from brain (18, 19) and eye (20), to kidney (21, 22) and intestine (23). Such local C production is biologically important for protection against infections as, for example, monocyte and macrophage derived C3, C4, and C5 components are required additional drivers in tissue protection against viruses and bacteria (14, 24, 25). An additional layer of complexity with regards to C activity was added by the finding that complement can also be activated within cells and can engage complement activation receptors in subcellular compartments. The activity of intracellular complement (‘the complosome’)(26) is associated with non-canonical C functions as it controls basic cellular processes such as cell metabolism and autophagy. For example, C3a generated intracellularly by human CD4+ T cells engages the lysosomal C3aR and sustains T cell survival via the activation of tonic mammalian target of rapamycin (mTOR) (27). The cell-autonomous engagement of the C regulator/receptor CD46 during T cell activation by T cell generated C3b, induces metabolic reprogramming needed for Th1 induction and cytotoxic T lymphocyte activation (28) (29). Interestingly, such C driven cell-autonomous immunity can also be triggered by C3 fragments that had been carried into the cells’ interior by pathogens that were opsonized with C3 fragments in serum (30).
Thus, the novel insights into C biology across host tissues, in combination with the development of next-generation C therapeutics holds promise to tackle human inflammatory diseases more effectively. The International Complement Society (ICS) organizes an ICS Guest Symposium at the annual conference of The American Association of Immunologists (AAI) with the aim to inform about the progress and particularly the exciting findings in C research, and to engage immunologists across disciplines. At the 2022 AAI conference in Portland, OR (May 6–10), Jessy Alexander (University of Buffalo), and Jeanne Paz (Gladstone Institutes & UCSF) presented unexpected new findings on C activities in the kidney and in the brain, respectively. Further, Michail Lionakis (NIAID, NIH) gave an update on the role of C in fungal infections and hinted towards a new non-canonical C activity in the protection against candidiasis. The ICS Guest Symposium session was concluded by Michael Holers (University of Colorado) who provided an overview about the complex roles played by C during the natural history of rheumatoid arthritis. In this brief review, we will provide a summary about these presentations with appropriate biological background information and discussion on their future implications for C research and beyond.
Intracellular complement controls kidney disease (Jessy J. Alexander)
The kidney is an immunologically active organ (0.5–1% body weight) with 20‐25% (1–1.2 L/min) of cardiac output traversing the kidney, and the glomerulus filtering 90–120 ml/min (31). The kidney becomes a multipronged intersection where the pathogen induces C activation and C evasion strategies, and activated C instructs local innate and adaptive immune system responses. Also, the kidney generates ammonia that interacts with C3 to activate the C alternative pathway (32). The kidney is a major extrahepatic site of C production with different kidney cells synthesizing both C3 and C5 and C regulatory proteins such as CD46, CD55, CD59 and Crry(33) (34). Studies show that along with IC mediated diseases such as lupus nephritis and IgA nephropathy where C levels are a part of the clinical evaluation, C system is also engaged and causes pathology in diseases including anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis(35) and dense deposit disease(36). C inhibitors are effective in diseases such as aHUS, paroxysmal nocturnal hemoglobinuria and ANCA vasculitis but shows only limited benefit in immune complex (IC) mediated diseases such as lupus and rheumatoid arthritis (37–41). ICs are present in 45–65% of patients with glomerular disease(42–44) with varying patterns of glomerular deposition.
As mentioned in the introduction, C activation occurs intracellularly across cells and dictates cellular behavior (26). However, its impact on tissue homeostasis is just beginning to emerge. Dr. Alexander addressed the question whether understanding C compartmentalization, intrinsic complement and its functions better, could be helpful in increasing C therapeutic effectiveness and disease management. To address this, she focused on the central C pathway regulator factor H (FH) (4). Factor H synthesized by the liver restrains the C cascade particularly at the alternative pathway and maintains C3 levels (2, 45). Here, Jessy Alexander presented novel data showing that FH could be serving as the ‘guardian’ (21) from within the kidney endothelial cell (Figure 1). She showed that both human and mouse kidney endothelial cells express FH. Also, reducing endothelial FH levels resulted in altered MASP, C1s, C4 expression, which are initiating proteins of the lectin and classical pathways, suggesting cross-communication or -regulation among the three C pathways (46). Absence of FH also altered the actin cytoskeleton with the formation of stress fibers leading to increased endothelial layer permeability(46). The kidney endothelium is a part of the glomerular filtration apparatus (47). Once the endothelial layer loses its integrity, the glomerular basement membrane that has no C regulators(48) becomes exposed to large C proteins and other toxins. In addition, actin cytoskeletal remodeling influences proliferation and angiogenesis and Jessy Alexander showed data that FH deficiency altered these parameters in kidney endothelial cells. Replenishing intracellular FH by transfection reverted cell proliferation close to normal levels. Importantly, earlier work from her laboratory showed that C5a, the breakdown product of C5, causes similar changes of cytoskeletal remodeling and proliferation of brain endothelial cells (49). Thus, controlled local and/or intracellular complement activation may regulate endothelial cells across tissues. Moreover, a recent publication from another group showed that intracellular FH can drive kidney tubular epithelial cell turn over and epithelial-mesenchymal transition and malignant transformation (50). Jessy Alexander’s laboratory is now focusing on understanding the roles of downstream effectors in FH induced changes of kidney endothelial cells. Some findings include the observation that FH deficiency causes the translocation of nuclear factor kappa B (NF-κB) into the nucleus, where this transcription factor then modulates different innate and adaptive immune responses and plays a critical role in mediating inflammatory responses. Overall, the results presented suggest that modulating FH in kidney endothelial cells could be an effective target for maintaining the glomerular homeostasis.
Of course, these are early insights and understanding the complexities of and potential cross-communications between intracellular, cell-autonomous and serum-derived C will require substantial future work and research efforts. However, this will likely be rewarding as inflammation of the vasculature is common to the broadest range of diseases – unfortunately as recently demonstrated by the SARS-CoV2-induced Coronavirus Disease 2019 (COVID19) (51, 52) with local and/or widespread vessel inflammation being one of its cardinal features.
New roles for complement during brain injury (Jeanne T. Paz)
In her presentation, Dr. Jeanne T. Paz kept with the scheme of dissecting the roles of locally generated complement in tissue biology. The brain is a uniquely placed and immune-privileged organ, separated from systemic effects by the presence of the blood-brain barrier (BBB) (53), in which endothelial cells with intricate junctional formations prevent the influx of immune cells and large proteins. The brain comprises a complex assembly of functionally different cell types such as neurons that transmit impulses, oligodendrocyte cells that generate myelin, astrocytes that maintain homeostasis and microglia, the critical immune cells in the brain (54). Rather unexpectedly, research on the C system in the brain was and is one of the major drivers in our understanding of extra-hepatic and local C activity (55, 56). Since the brain is considered immune-privileged, one might expect any presence of C to be a sign of disease/neuroinflammation. However, C in the brain is actually a driver of normal development and homeostatic tissue activity (Figure 1). So far, most cells assessed in the brain synthesize C proteins. C produced in the brain is involved in neurogenesis (57–59), synaptic pruning (60, 61), scavenging apoptotic debris (62–64) and the response to and removal of inflammatory insults. However, excessive and prolonged C activation does contribute to neurodegenerative disease and to neuropathies during aging (58, 65–67). In addition, C activation in the periphery heavily impacts the brain: the anaphylatoxins C3a and C5a generated during systemic C activation render the BBB leaky (49) and can foster cancer metastasis (68). There is a substantial body of literature on these subjects, and we guide the reader to those for more detailed insights on the exciting and diverse roles of C in the central nervous system (CNS) (61, 65, 69).
Jeanne Paz presented data that further substantiated our understanding that C can indeed play a very sinister role in the brain, specifically during traumatic brain injury (TBI). TBI is a leading cause of disability in children and adults (70). Although TBI acutely disrupts the cortex, the outmost part of the brain, most TBI-related disabilities reflect secondary injuries that accrue over time as consequences of the initial impact. Understanding where, when, and how secondary injuries develop is critical for preventing disability following TBI. The Paz group applied spatial transcriptomics in a mouse model of TBI and found C1q and C4 among the top differentially upregulated genes five weeks after TBI in a subcortical brain region called the thalamus, which is reciprocally connected with the impacted cortex (71). Using immunohistochemistry and in vivo electrophysiology, they noted that increased C1q expression colocalized with neuron loss and chronic inflammation, and correlated with disruption in sleep spindles and emergence of epileptic activities. Mice treated with an antibody that blocks the C-activating effect of C1q ameliorated these pathological features, suggesting that C1q is a disease modifier in TBI. Using single-nucleus RNA sequencing, the Paz group pinpointed microglia, as the source for generating the detrimental C1q production, and suggest overall that the activation of the classical C pathway in the thalamus could be a target for treating TBI-related disabilities (71). The latter is a notion that the lab is now following up with further studies.
Revisiting complement in fungal infections (Michail Lionakis)
The role of serum circulating ‘classic’ C in the protection against fungal infections are mostly understood among complementologists. However, the functions of cell-autonomous and intracellular C activities during pathogen sensing and removal have not yet been explored but is an area of growing interest. Dr. Michail Lionakis gave an update on C in fungal infections and shared some initial insights into new and ongoing work on C and candidiasis in his laboratory. In his presentation, Michail Lionakis first summarized the published work on the role of C in host defense against systemic candidiasis (72–75). Fungal infections during hospitalization are a common occurrence especially in critically ill patients in the intensive care unit. In a 2019 Centers for Disease Control and Prevention (CDC) report, fungi were among the major pathogens that were drug resistant in the United States (76). Along with increased infections by nosocomial pathogens, fungi that are emerging due to climatic and other environmental changes enhance the risk further (77, 78). Improving fungal diagnostic modalities and identifying effective therapeutic targets is an urgent need. The recent introduction in the clinic of the C5-targeted monoclonal antibody eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria and hemolytic uremic syndrome has uncovered a critical contribution of C5a signaling in antifungal host defense as eculizumab-treated patients were reported to develop meningococcal infections and systemic candidiasis with high mortality (79, 80), as well as invasive pulmonary aspergillosis(81). These observations in humans were unexpected because of the lack of reported invasive fungal disease in patients suffering with inherited C5 deficiency (Figure 1). C5 deficiency patients are typically at high risk for invasive infections by encapsulated bacteria (82). This susceptibility prompted the United States Food and Drug Administration (FDA) to update the package insert of eculizumab in 2018 to warn for the risk of invasive fungal infections in addition to pyogenic bacterial disease in vulnerable patients receiving the drug.
Prior work had examined inbred DBA/2 and A/J mouse strains, which are C5-deficient, and had found them to have significantly greater mortality after systemic challenge with C. albicans (72–74), in a model that relies heavily on phagocyte-mediated innate, not lymphocytic, responses for effective host defense (77). These inbred mice had greater fungal burden in several, but not all, examined tissues after infection associated with enhanced pro-inflammatory responses in infected organs (72–74). Yet, the mechanisms of impaired phagocyte-dependent immune responses were not thoroughly examined in those studies and the inbred DBA/2 and A/J mouse strains exhibit additional immunological defects beyond C5 deficiency (83, 84). Another study examined C3-deficient mice after systemic challenge with C. albicans and C. glabrata and found them to exhibit increased mortality after infection with associated enhanced levels of pro-inflammatory cytokines in infected tissues, yet the precise mechanisms of impaired host defense in the absence of C3 were not defined (75). Recent work from the Lionakis lab dissected the mechanisms by which C critically contributes to host defense against invasive fungal disease. This is particularly timely as the rising infections by systemic candidiasis and the multidrug resistant Candida auris (78, 85) infections in response to treatment with eculizumab (79, 86), and other novel complement pathway inhibitors have become a major clinical problem during treatment of various vulnerable patient populations. Michail Lionakis presented unpublished data that shed light on the mechanisms by which C5a-C5aR1 signaling promotes protective phagocyte-dependent host defense against invasive candidiasis in mice and humans – with initial indication that cell-autonomous C5a generation in myeloid cells may be key (Desai et al., under review). The importance of C activation in antifungal immunity was highlighted and underscores the need for careful surveillance of patients treated with various C pathway inhibitors for the development of opportunistic fungal disease.
The three presentations detailed above are examples of changes in our thinking about C biology and C in human disease. Among the key realizations in our field is the understanding that C operates at different and unexpected locations, that is in circulation (liver), locally (cell-derived) and intracellularly (cell-autonomous). In addition, a given role of C in a specific cell manner also changes with the activation state of that cell, and often in a temporal fashion (87, 88). Consequently, we need to understand the distinct roles of C at different locations and over the course of C engagement (onset, progression, resolution) to fully comprehend its role in normal biology and to tackle the system optimally in diseases. This notion was then brought beautifully forward, and applied to the pathogenesis of rheumatoid arthritis by the presentation of Michael Holers.
Stage- and context-dependent roles for complement in rheumatoid arthritis evolution (V. Michael Holers).
Rheumatoid arthritis (RA) is a chronic autoimmune disease whose exact etiology is not completely understood. The triggers include mucosal inflammation and dysbiosis (89). RA affects most often the peripheral small joints causing the synovium to thicken by accumulating and incorporating immune cells to form the pannus that destroys cartilage and bone (90). The LP, CP and AP are likely all activated in RA by diverse triggers such (11, 91) as autoantibodies, necrotic cells or exposed collagen and thus important contributors to the disease pathology in humans and in mouse models of the disease. The activation of major C components C3 and C5 (92) are increased in the synovium in RA indicating perpetuated local C activation. The presentation by Holers provided a somewhat different angle on C and RA: instead of paying, as most do, attention to the symptomatic site, the inflamed joint, his talk focused on the role of the C system throughout the evolution of RA (Figure 1). Holers stressed the fact that RA exhibits a prolonged preclinical period of time that involves the development of chronic mucosal inflammation, especially prominent in the lung and intestine, which is associated with the local production of RA-related autoantibodies designated ACPA (anti-citrullinated protein antibodies)(93) and RF (rheumatoid factor) (94). His team helped spear-head detailed analyses of intact and cleaved C activation components in the sputum of preclinical RA individuals (95). This work detailed the presence of C activation fragments that are associated with neutrophil extracellular trap (NET) formation and elevated cytokines and chemokines (96). Further, the observed C activation during this earlier pre-clinical phase is causally associated with the development of NETs and autoantibody generation. Transition of the disease to involve the synovium was also associated with informative and specific patterns of C gene expression and localization of activation fragments and receptors. As a major outcome, the team noted positive associations in early RA between a subset of synovial, but not peripheral blood cell, C gene expression levels encoding factors such as Factor B and C5aR1, with clinical disease activity. Conversely, and somewhat unexpectedly, C5 gene expression itself is inversely associated with disease activity. Immunohistochemical analyses of the inflamed tissue sites demonstrated regional differences in C factor localization, and inverse relationships between activation fragments and regulatory molecules. Therefore, RA is a disease with substantial opportunities for therapeutic impact, and specific attention to early disease and regulation of the C3/C5 convertases may be particularly worthy.
Conclusions
The C system was thought to be fully functionally defined and well understood for many years. Novel C insights and discoveries over the last two decades, however, have proven us wrong. C is not only a pro-inflammatory pathogen fighter but at the heart of normal cell and tissue development and function. The system not only actively operates body-wide in serum, but also participates on a cellular and sub-cellular level in basic cell physiology and regenerative processes. The exact modes of specifically the latter, non-canonical, C activities and their regulation are incompletely understood and therefore need to be explored further. Much progress has been made in several areas of C therapeutics through the development of drugs such as Eculizumab (41), Avacopan (97), C1 inhibitor, etc., as well as Ravulizumab (40) (a long lasting C5a inhibitor), and sutimlimab (an inhibitor for C1s) that are approved for diseases such as PNH, ANCA vasculitis and cold agglutinin disease, rendering these diseases more manageable. However, a larger number of C therapeutics have not delivered in the clinical and/or in more common disease settings. Understanding the recently discovered non-canonical C activities on a molecular level and their interplay with the classic, circulating components as well as with other PRR systems may provide new opportunities for drug development. Areas that need more in depth exploration include the role of cell-autonomous C in stromal and parenchymal cells in RA (98), the impact of the mucosal microbiota on local C activities in health and disease and the role of C in pain perception and illness behavior. Finally, one of the hottest subjects in the C field is its clear impact on cancer (for good or for worse) and we expect substantial new insights here soon (50, 99–104).
Acknowledgements:
We thank the patients, researchers and clinicians that have added to our understanding of C biology over time. This is a brief review about the recent advances in the C field presented at the AAI and not meant to be an in-depth assessment of complement. Thus, we apologize to the many researchers whose work should be acknowledged but was not cited due to space constraints.
Financial support information
Work in the different laboratories is supported by: in part by the Intramural Research Program of the NIH, the NHLBI (ZIA/hl006223 to CK); R01HL112937 and University of Toledo Medical Research Society Grant (VPF); DoD EP150038 and NIH/NINDS R01 NS078118 grants (JTP); Division of Intramural Research of the NIAID, NIH (ZIA AI001175 to MSL); NIH R01 AR051749 (MVH) and NIH R01 DK111222 (JJA).
Abbreviations
- C
complement
- FH
factor H
- CP
classical
- AP
alternative
- LP
lectin
- MAC, C5b-9
membrane attack complex
- ICs
immune complexes
- ICS
International Complement Society
- AAI
American Association of Immunologists
References
- 1.Cavaillon JM, Sansonetti P, and Goldman M. 2019. 100th Anniversary of Jules Bordet’s Nobel Prize: Tribute to a Founding Father of Immunology. Front Immunol 10: 2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Merle NS, Church SE, Fremeaux-Bacchi V, and Roumenina LT. 2015. Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front Immunol 6: 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walport MJ 2001. Complement. Second of two parts. N Engl J Med 344: 1140–1144. [DOI] [PubMed] [Google Scholar]
- 4.Walport MJ 2001. Complement. First of two parts. N Engl J Med 344: 1058–1066. [DOI] [PubMed] [Google Scholar]
- 5.Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, and Roumenina LT. 2015. Complement System Part II: Role in Immunity. Front Immunol 6: 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carroll MV, and Sim RB. 2011. Complement in health and disease. Adv Drug Deliv Rev 63: 965–975. [DOI] [PubMed] [Google Scholar]
- 7.Noris M, and Remuzzi G. 2013. Overview of complement activation and regulation. Semin Nephrol 33: 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heesterbeek DAC, Angelier ML, Harrison RA, and Rooijakkers SHM. 2018. Complement and Bacterial Infections: From Molecular Mechanisms to Therapeutic Applications. J Innate Immun 10: 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Botto M 2001. Links between complement deficiency and apoptosis. Arthritis Res 3: 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walport MJ, Davies KA, Morley BJ, and Botto M. 1997. Complement deficiency and autoimmunity. Ann N Y Acad Sci 815: 267–281. [DOI] [PubMed] [Google Scholar]
- 11.Holers VM, and Banda NK. 2018. Complement in the Initiation and Evolution of Rheumatoid Arthritis. Front Immunol 9: 1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wenzel UO, Bode M, Kohl J, and Ehmke H. 2017. A pathogenic role of complement in arterial hypertension and hypertensive end organ damage. Am J Physiol Heart Circ Physiol 312: H349–H354. [DOI] [PubMed] [Google Scholar]
- 13.Zhou Z, Xu MJ, and Gao B. 2016. Hepatocytes: a key cell type for innate immunity. Cell Mol Immunol 13: 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lubbers R, van Essen MF, van Kooten C, and Trouw LA. 2017. Production of complement components by cells of the immune system. Clin Exp Immunol 188: 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Portilla D, and Xavier S. 2021. Role of intracellular complement activation in kidney fibrosis. Br J Pharmacol 178: 2880–2891. [DOI] [PubMed] [Google Scholar]
- 16.Eldewi DM, Alhabibi AM, El Sayed HME, Mahmoud SAK, El Sadek SM, Gouda RM, Hassan M, Ibrahim AH, and Abd El Haliem NF. 2019. Expression levels of complement regulatory proteins (CD35, CD55 and CD59) on peripheral blood cells of patients with chronic kidney disease. Int J Gen Med 12: 343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgan BP, and Gasque P. 1997. Extrahepatic complement biosynthesis: where, when and why? Clin Exp Immunol 107: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farber K, Cheung G, Mitchell D, Wallis R, Weihe E, Schwaeble W, and Kettenmann H. 2009. C1q, the recognition subcomponent of the classical pathway of complement, drives microglial activation. J Neurosci Res 87: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gasque P, Singhrao SK, Neal JW, Gotze O, and Morgan BP. 1997. Expression of the receptor for complement C5a (CD88) is up-regulated on reactive astrocytes, microglia, and endothelial cells in the inflamed human central nervous system. Am J Pathol 150: 31–41. [PMC free article] [PubMed] [Google Scholar]
- 20.Mandal MN, and Ayyagari R. 2006. Complement factor H: spatial and temporal expression and localization in the eye. Invest Ophthalmol Vis Sci 47: 4091–4097. [DOI] [PubMed] [Google Scholar]
- 21.Boudhabhay I, and Roumenina LT. 2021. Complement factor H: a guardian within? Kidney Int 100: 747–749. [DOI] [PubMed] [Google Scholar]
- 22.Brooimans RA, Stegmann AP, van Dorp WT, van der Ark AA, van der Woude FJ, van Es LA, and Daha MR. 1991. Interleukin 2 mediates stimulation of complement C3 biosynthesis in human proximal tubular epithelial cells. J Clin Invest 88: 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Ye J, Ren Y, Zuo J, Dai W, He Y, Tan M, Song W, and Yuan Y. 2018. Intracellular activation of complement C3 in Paneth cells improves repair of intestinal epithelia during acute injury. Immunotherapy 10: 1325–1336. [DOI] [PubMed] [Google Scholar]
- 24.Verschoor A, Brockman MA, Knipe DM, and Carroll MC. 2001. Cutting edge: myeloid complement C3 enhances the humoral response to peripheral viral infection. J Immunol 167: 2446–2451. [DOI] [PubMed] [Google Scholar]
- 25.Dunkelberger JR, and Song WC. 2010. Complement and its role in innate and adaptive immune responses. Cell Res 20: 34–50. [DOI] [PubMed] [Google Scholar]
- 26.Arbore G, Kemper C, and Kolev M. 2017. Intracellular complement - the complosome - in immune cell regulation. Mol Immunol 89: 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, Subias M, Pickering MC, Drouet C, Meri S, Arstila TP, Pekkarinen PT, Ma M, Cope A, Reinheckel T, Rodriguez de Cordoba S, Afzali B, Atkinson JP, and Kemper C. 2013. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 39: 1143–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolev M, Dimeloe S, Le Friec G, Navarini A, Arbore G, Povoleri GA, Fischer M, Belle R, Loeliger J, Develioglu L, Bantug GR, Watson J, Couzi L, Afzali B, Lavender P, Hess C, and Kemper C. 2015. Complement Regulates Nutrient Influx and Metabolic Reprogramming during Th1 Cell Responses. Immunity 42: 1033–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niyonzima N, Rahman J, Kunz N, West EE, Freiwald T, Desai JV, Merle NS, Gidon A, Sporsheim B, Lionakis MS, Evensen K, Lindberg B, Skagen K, Skjelland M, Singh P, Haug M, Ruseva MM, Kolev M, Bibby J, Marshall O, O’Brien B, Deeks N, Afzali B, Clark RJ, Woodruff TM, Pryor M, Yang ZH, Remaley AT, Mollnes TE, Hewitt SM, Yan B, Kazemian M, Kiss MG, Binder CJ, Halvorsen B, Espevik T, and Kemper C. 2021. Mitochondrial C5aR1 activity in macrophages controls IL-1beta production underlying sterile inflammation. Sci Immunol 6: eabf2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tam JC, Bidgood SR, McEwan WA, and James LC. 2014. Intracellular sensing of complement C3 activates cell autonomous immunity. Science 345: 1256070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaufman Daniel P., B. H, Knohl Stephen J.. 2022. Physiology, Glomerular Filtration Rate. StatPearls [Internet]. [PubMed] [Google Scholar]
- 32.Nath KA, Hostetter MK, and Hostetter TH. 1985. Pathophysiology of chronic tubulo-interstitial disease in rats. Interactions of dietary acid load, ammonia, and complement component C3. J Clin Invest 76: 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thurman JM, Ljubanovic D, Royer PA, Kraus DM, Molina H, Barry NP, Proctor G, Levi M, and Holers VM. 2006. Altered renal tubular expression of the complement inhibitor Crry permits complement activation after ischemia/reperfusion. J Clin Invest 116: 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nangaku M 1998. Complement regulatory proteins in glomerular diseases. Kidney Int 54: 1419–1428. [DOI] [PubMed] [Google Scholar]
- 35.Jayne DRW, Bruchfeld AN, Harper L, Schaier M, Venning MC, Hamilton P, Burst V, Grundmann F, Jadoul M, Szombati I, Tesar V, Segelmark M, Potarca A, Schall TJ, Bekker P, and Group CS. 2017. Randomized Trial of C5a Receptor Inhibitor Avacopan in ANCA-Associated Vasculitis. J Am Soc Nephrol 28: 2756–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith RJ, Alexander J, Barlow PN, Botto M, Cassavant TL, Cook HT, de Cordoba SR, Hageman GS, Jokiranta TS, Kimberling WJ, Lambris JD, Lanning LD, Levidiotis V, Licht C, Lutz HU, Meri S, Pickering MC, Quigg RJ, Rops AL, Salant DJ, Sethi S, Thurman JM, Tully HF, Tully SP, van der Vlag J, Walker PD, Wurzner R, Zipfel PF, and G. Dense Deposit Disease Focus. 2007. New approaches to the treatment of dense deposit disease. J Am Soc Nephrol 18: 2447–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trouw LA, Pickering MC, and Blom AM. 2017. The complement system as a potential therapeutic target in rheumatic disease. Nat Rev Rheumatol 13: 538–547. [DOI] [PubMed] [Google Scholar]
- 38.Galindo-Izquierdo M, and Pablos Alvarez JL. 2021. Complement as a Therapeutic Target in Systemic Autoimmune Diseases. Cells 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barilla-Labarca ML, Toder K, and Furie R. 2013. Targeting the complement system in systemic lupus erythematosus and other diseases. Clin Immunol 148: 313–321. [DOI] [PubMed] [Google Scholar]
- 40.Kulasekararaj AG, Hill A, Rottinghaus ST, Langemeijer S, Wells R, Gonzalez-Fernandez FA, Gaya A, Lee JW, Gutierrez EO, Piatek CI, Szer J, Risitano A, Nakao S, Bachman E, Shafner L, Damokosh AI, Ortiz S, Roth A, and Peffault de Latour R. 2019. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: the 302 study. Blood 133: 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mache CJ, Acham-Roschitz B, Fremeaux-Bacchi V, Kirschfink M, Zipfel PF, Roedl S, Vester U, and Ring E. 2009. Complement inhibitor eculizumab in atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol 4: 1312–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Briganti EM, Dowling J, Finlay M, Hill PA, Jones CL, Kincaid-Smith PS, Sinclair R, McNeil JJ, and Atkins RC. 2001. The incidence of biopsy-proven glomerulonephritis in Australia. Nephrol Dial Transplant 16: 1364–1367. [DOI] [PubMed] [Google Scholar]
- 43.Swaminathan S, Leung N, Lager DJ, Melton LJ 3rd, Bergstralh EJ, Rohlinger A, and Fervenza FC. 2006. Changing incidence of glomerular disease in Olmsted County, Minnesota: a 30-year renal biopsy study. Clin J Am Soc Nephrol 1: 483–487. [DOI] [PubMed] [Google Scholar]
- 44.National Institutes of Health, N. I. o. D. a. D. a. K. D. B, MD. 2009. U.S. Renal Data System:Incidence of Reported ESRD. [Google Scholar]
- 45.Merle NS, Leon J, Poillerat V, Grunenwald A, Boudhabhay I, Knockaert S, Robe-Rybkine T, Torset C, Pickering MC, Chauvet S, Fremeaux-Bacchi V, and Roumenina LT. 2020. Circulating FH Protects Kidneys From Tubular Injury During Systemic Hemolysis. Front Immunol 11: 1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mahajan S, Jacob A, Kelkar A, Chang A, McSkimming D, Neelamegham S, Quigg RJ, and Alexander JJ. 2021. Local Complement Factor H protects kidney endothelial cell structure and function. Kidney Int. [DOI] [PubMed] [Google Scholar]
- 47.Jourde-Chiche N, Fakhouri F, Dou L, Bellien J, Burtey S, Frimat M, Jarrot PA, Kaplanski G, Le Quintrec M, Pernin V, Rigothier C, Sallee M, Fremeaux-Bacchi V, Guerrot D, and Roumenina LT. 2019. Endothelium structure and function in kidney health and disease. Nat Rev Nephrol 15: 87–108. [DOI] [PubMed] [Google Scholar]
- 48.Thurman JM 2020. Complement and the Kidney: An Overview. Adv Chronic Kidney Dis 27: 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jacob A, Hack B, Chiang E, Garcia JG, Quigg RJ, and Alexander JJ. 2010. C5a alters blood-brain barrier integrity in experimental lupus. FASEB J 24: 1682–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Daugan MV, Revel M, Thouenon R, Dragon-Durey MA, Robe-Rybkine T, Torset C, Merle NS, Noe R, Verkarre V, Oudard SM, Mejean A, Validire P, Cathelineau X, Sanchez-Salas R, Pickering MC, Cremer I, Mansuet-Lupo A, Alifano M, Sautes-Fridman C, Damotte D, Fridman WH, and Roumenina LT. 2021. Intracellular Factor H drives tumor progression independently of the complement cascade. Cancer Immunol Res. [DOI] [PubMed] [Google Scholar]
- 51.Lo MW, Kemper C, and Woodruff TM. 2020. COVID-19: Complement, Coagulation, and Collateral Damage. J Immunol 205: 1488–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Howell MC, Green R, McGill AR, Kahlil RM, Dutta R, Mohapatra SS, and Mohapatra S. 2021. Activation of Intracellular Complement in Lungs of Patients With Severe COVID-19 Disease Decreases T-Cell Activity in the Lungs. Front Immunol 12: 700705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abbott NJ, Ronnback L, and Hansson E. 2006. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7: 41–53. [DOI] [PubMed] [Google Scholar]
- 54.Engelhardt B, and Coisne C. 2011. Fluids and barriers of the CNS establish immune privilege by confining immune surveillance to a two-walled castle moat surrounding the CNS castle. Fluids Barriers CNS 8: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, and Barres BA. 2007. The classical complement cascade mediates CNS synapse elimination. Cell 131: 1164–1178. [DOI] [PubMed] [Google Scholar]
- 56.Alexander JJ, Anderson AJ, Barnum SR, Stevens B, and Tenner AJ. 2008. The complement cascade: Yin-Yang in neuroinflammation--neuro-protection and -degeneration. J Neurochem 107: 1169–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rahpeymai Y, Hietala MA, Wilhelmsson U, Fotheringham A, Davies I, Nilsson AK, Zwirner J, Wetsel RA, Gerard C, Pekny M, and Pekna M. 2006. Complement: a novel factor in basal and ischemia-induced neurogenesis. EMBO J 25: 1364–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coulthard LG, Hawksworth OA, Li R, Balachandran A, Lee JD, Sepehrband F, Kurniawan N, Jeanes A, Simmons DG, Wolvetang E, and Woodruff TM. 2017. Complement C5aR1 Signaling Promotes Polarization and Proliferation of Embryonic Neural Progenitor Cells through PKCzeta. J Neurosci 37: 5395–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gorelik A, Sapir T, Haffner-Krausz R, Olender T, Woodruff TM, and Reiner O. 2017. Developmental activities of the complement pathway in migrating neurons. Nat Commun 8: 15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, and Stevens B. 2012. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74: 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stephan AH, Barres BA, and Stevens B. 2012. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci 35: 369–389. [DOI] [PubMed] [Google Scholar]
- 62.Alawieh A, Elvington A, and Tomlinson S. 2015. Complement in the Homeostatic and Ischemic Brain. Front Immunol 6: 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alawieh A, Elvington A, Zhu H, Yu J, Kindy MS, Atkinson C, and Tomlinson S. 2015. Modulation of post-stroke degenerative and regenerative processes and subacute protection by site-targeted inhibition of the alternative pathway of complement. J Neuroinflammation 12: 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hammad A, Westacott L, and Zaben M. 2018. The role of the complement system in traumatic brain injury: a review. J Neuroinflammation 15: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dalakas MC, Alexopoulos H, and Spaeth PJ. 2020. Complement in neurological disorders and emerging complement-targeted therapeutics. Nat Rev Neurol 16: 601–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Coulthard LG, Hawksworth OA, Conroy J, Lee JD, and Woodruff TM. 2018. Complement C3a receptor modulates embryonic neural progenitor cell proliferation and cognitive performance. Mol Immunol 101: 176–181. [DOI] [PubMed] [Google Scholar]
- 67.Coulthard LG, Hawksworth OA, and Woodruff TM. 2018. Complement: The Emerging Architect of the Developing Brain. Trends Neurosci 41: 373–384. [DOI] [PubMed] [Google Scholar]
- 68.Bouwens van der Vlis TAM, Kros JM, Mustafa DAM, van Wijck RTA, Ackermans L, van Hagen PM, and van der Spek PJ. 2018. The complement system in glioblastoma multiforme. Acta Neuropathol Commun 6: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Magdalon J, Mansur F, Teles ESAL, de Goes VA, Reiner O, and Sertie AL. 2020. Complement System in Brain Architecture and Neurodevelopmental Disorders. Front Neurosci 14: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sariaslan A, Sharp DJ, D’Onofrio BM, Larsson H, and Fazel S. 2016. Long-Term Outcomes Associated with Traumatic Brain Injury in Childhood and Adolescence: A Nationwide Swedish Cohort Study of a Wide Range of Medical and Social Outcomes. PLoS Med 13: e1002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Holden SS, Grandi FC, Aboubakr O, Higashikubo B, Cho FS, Chang AH, Forero AO, Morningstar AR, Mathur V, Kuhn LJ, Suri P, Sankaranarayanan S, Andrews-Zwilling Y, Tenner AJ, Luthi A, Aronica E, Corces MR, Yednock T, and Paz JT. 2021. Complement factor C1q mediates sleep spindle loss and epileptic spikes after mild brain injury. Science 373: eabj2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ashman RB, Bolitho EM, and Papadimitriou JM. 1993. Patterns of resistance to Candida albicans in inbred mouse strains. Immunol Cell Biol 71 (Pt 3): 221–225. [DOI] [PubMed] [Google Scholar]
- 73.Mullick A, Elias M, Picard S, Bourget L, Jovcevski O, Gauthier S, Tuite A, Harakidas P, Bihun C, Massie B, and Gros P. 2004. Dysregulated inflammatory response to Candida albicans in a C5-deficient mouse strain. Infect Immun 72: 5868–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mullick A, Leon Z, Min-Oo G, Berghout J, Lo R, Daniels E, and Gros P. 2006. Cardiac failure in C5-deficient A/J mice after Candida albicans infection. Infect Immun 74: 4439–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsoni SV, Kerrigan AM, Marakalala MJ, Srinivasan N, Duffield M, Taylor PR, Botto M, Steele C, and Brown GD. 2009. Complement C3 plays an essential role in the control of opportunistic fungal infections. Infect Immun 77: 3679–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.(CDC)., C. f. D. C. a. P. 2019. Antibiotic Resistance Threats in the United States, . Atlanta: CDC; https://www.cdc.gov/drugresistance/pdf/threats-report/2019-arthreats-report-508.pdf. [Google Scholar]. [Google Scholar]
- 77.Lionakis MS, and Levitz SM. 2018. Host Control of Fungal Infections: Lessons from Basic Studies and Human Cohorts. Annu Rev Immunol 36: 157–191. [DOI] [PubMed] [Google Scholar]
- 78.Lionakis MS, and Hohl TM. 2020. Call to Action: How to Tackle Emerging Nosocomial Fungal Infections. Cell Host Microbe 27: 859–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Socie G, Caby-Tosi MP, Marantz JL, Cole A, Bedrosian CL, Gasteyger C, Mujeebuddin A, Hillmen P, Vande Walle J, and Haller H. 2019. Eculizumab in paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome: 10-year pharmacovigilance analysis. Br J Haematol 185: 297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Han H, Alagusundaramoorthy S, Swanson K, Gardezi AI, and Chan MR. 2019. Acute Candida albicans Peritonitis in a Patient with Atypical Hemolytic Uremic Syndrome Treated with Eculizumab. Perit Dial Int 39: 575–576. [DOI] [PubMed] [Google Scholar]
- 81.Han X, Su X, Li Z, Liu Y, Wang S, Zhu M, Zhang C, Yang F, Zhao J, Li X, Chen F, and Han L. 2021. Complement receptor 3 mediates Aspergillus fumigatus internalization into alveolar epithelial cells with the increase of intracellular phosphatidic acid by activating FAK. Virulence 12: 1980–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lionakis MS, Netea MG, and Holland SM. 2014. Mendelian genetics of human susceptibility to fungal infection. Cold Spring Harb Perspect Med 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Diez E, Lee SH, Gauthier S, Yaraghi Z, Tremblay M, Vidal S, and Gros P. 2003. Birc1e is the gene within the Lgn1 locus associated with resistance to Legionella pneumophila. Nat Genet 33: 55–60. [DOI] [PubMed] [Google Scholar]
- 84.Mitsos LM, Cardon LR, Ryan L, LaCourse R, North RJ, and Gros P. 2003. Susceptibility to tuberculosis: a locus on mouse chromosome 19 (Trl-4) regulates Mycobacterium tuberculosis replication in the lungs. Proc Natl Acad Sci U S A 100: 6610–6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang X, Hurabielle C, Drummond RA, Bouladoux N, Desai JV, Sim CK, Belkaid Y, Lionakis MS, and Segre JA. 2020. Murine model of colonization with fungal pathogen Candida auris to explore skin tropism, host risk factors and therapeutic strategies. Cell Host Microbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jodele S, Dandoy CE, Lane A, Laskin BL, Teusink-Cross A, Myers KC, Wallace G, Nelson A, Bleesing J, Chima RS, Hirsch R, Ryan TD, Benoit S, Mizuno K, Warren M, and Davies SM. 2020. Complement blockade for TA-TMA: lessons learned from a large pediatric cohort treated with eculizumab. Blood 135: 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rey R, Suaud-Chagny MF, Bohec AL, Dorey JM, d’Amato T, Tamouza R, and Leboyer M. 2020. Overexpression of complement component C4 in the dorsolateral prefrontal cortex, parietal cortex, superior temporal gyrus and associative striatum of patients with schizophrenia. Brain Behav Immun 90: 216–225. [DOI] [PubMed] [Google Scholar]
- 88.Mannes M, Schmidt CQ, Nilsson B, Ekdahl KN, and Huber-Lang M. 2021. Complement as driver of systemic inflammation and organ failure in trauma, burn, and sepsis. Semin Immunopathol 43: 773–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jubair WK, Hendrickson JD, Severs EL, Schulz HM, Adhikari S, Ir D, Pagan JD, Anthony RM, Robertson CE, Frank DN, Banda NK, and Kuhn KA. 2018. Modulation of Inflammatory Arthritis in Mice by Gut Microbiota Through Mucosal Inflammation and Autoantibody Generation. Arthritis Rheumatol 70: 1220–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hitchon CA, and El-Gabalawy HS. 2011. The synovium in rheumatoid arthritis. Open Rheumatol J 5: 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Okroj M, Heinegard D, Holmdahl R, and Blom AM. 2007. Rheumatoid arthritis and the complement system. Ann Med 39: 517–530. [DOI] [PubMed] [Google Scholar]
- 92.Ruddy S, and Colten HR. 1974. Rheumatoid arthritis. Biosynthesis of complement proteins by synovial tissues. N Engl J Med 290: 1284–1288. [DOI] [PubMed] [Google Scholar]
- 93.Kurowska W, Kuca-Warnawin EH, Radzikowska A, and Maslinski W. 2017. The role of anti-citrullinated protein antibodies (ACPA) in the pathogenesis of rheumatoid arthritis. Cent Eur J Immunol 42: 390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wilson D 2006. Rheumatoid factors in patients with rheumatoid arthritis. Can Fam Physician 52: 180–181. [PMC free article] [PubMed] [Google Scholar]
- 95.Willis VC, Demoruelle MK, Derber LA, Chartier-Logan CJ, Parish MC, Pedraza IF, Weisman MH, Norris JM, Holers VM, and Deane KD. 2013. Sputum autoantibodies in patients with established rheumatoid arthritis and subjects at risk of future clinically apparent disease. Arthritis Rheum 65: 2545–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Okamoto Y, Devoe S, Seto N, Minarchick V, Wilson T, Rothfuss HM, Mohning MP, Arbet J, Kroehl M, Visser A, August J, Thomas SM, Charry LL, Fleischer C, Feser ML, Frazer-Abel AA, Norris JM, Cherrington BD, Janssen WJ, Kaplan MJ, Deane KD, Holers VM, and Demoruelle MK. 2022. Association of Sputum Neutrophil Extracellular Trap Subsets With IgA Anti-Citrullinated Protein Antibodies in Subjects at Risk for Rheumatoid Arthritis. Arthritis Rheumatol 74: 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Merkel PA, Niles J, Jimenez R, Spiera RF, Rovin BH, Bomback A, Pagnoux C, Potarca A, Schall TJ, Bekker P, and Investigators C. 2020. Adjunctive Treatment With Avacopan, an Oral C5a Receptor Inhibitor, in Patients With Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. ACR Open Rheumatol 2: 662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Friscic J, Bottcher M, Reinwald C, Bruns H, Wirth B, Popp SJ, Walker KI, Ackermann JA, Chen X, Turner J, Zhu H, Seyler L, Euler M, Kirchner P, Kruger R, Ekici AB, Major T, Aust O, Weidner D, Fischer A, Andes FT, Stanojevic Z, Trajkovic V, Herrmann M, Korb-Pap A, Wank I, Hess A, Winter J, Wixler V, Distler J, Steiner G, Kiener HP, Frey B, Kling L, Raza K, Frey S, Kleyer A, Bauerle T, Hughes TR, Gruneboom A, Steffen U, Kronke G, Croft AP, Filer A, Kohl J, Klein K, Buckley CD, Schett G, Mougiakakos D, and Hoffmann MH. 2021. The complement system drives local inflammatory tissue priming by metabolic reprogramming of synovial fibroblasts. Immunity 54: 1002–1021 e1010. [DOI] [PubMed] [Google Scholar]
- 99.Fridman WH, Petitprez F, Meylan M, Chen TW, Sun CM, Roumenina LT, and Sautes-Fridman C. 2021. B cells and cancer: To B or not to B? J Exp Med 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Galmiche A, Rak J, Roumenina LT, and Saidak Z. 2022. Coagulome and the tumor microenvironment: an actionable interplay. Trends Cancer 8: 369–383. [DOI] [PubMed] [Google Scholar]
- 101.Revel M, Daugan MV, Sautes-Fridman C, Fridman WH, and Roumenina LT. 2020. Complement System: Promoter or Suppressor of Cancer Progression? Antibodies (Basel) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Revel M, Sautes-Fridman C, Fridman WH, and Roumenina LT. 2022. C1q+ macrophages: passengers or drivers of cancer progression. Trends Cancer. [DOI] [PubMed] [Google Scholar]
- 103.Roumenina LT, Daugan MV, Noe R, Petitprez F, Vano YA, Sanchez-Salas R, Becht E, Meilleroux J, Clec’h BL, Giraldo NA, Merle NS, Sun CM, Verkarre V, Validire P, Selves J, Lacroix L, Delfour O, Vandenberghe I, Thuilliez C, Keddani S, Sakhi IB, Barret E, Ferre P, Corvaia N, Passioukov A, Chetaille E, Botto M, de Reynies A, Oudard SM, Mejean A, Cathelineau X, Sautes-Fridman C, and Fridman WH. 2019. Tumor Cells Hijack Macrophage-Produced Complement C1q to Promote Tumor Growth. Cancer Immunol Res 7: 1091–1105. [DOI] [PubMed] [Google Scholar]
- 104.Roumenina LT, Daugan MV, Petitprez F, Sautes-Fridman C, and Fridman WH. 2019. Context-dependent roles of complement in cancer. Nat Rev Cancer 19: 698–715. [DOI] [PubMed] [Google Scholar]