

Abstract
Background:
Proprotein convertase subtilisin-kexin type 9 (PCSK9) chaperones the hepatic low-density lipoprotein receptor (LDLR) for lysosomal degradation, elevating serum LDL cholesterol and promoting atherosclerotic heart disease. Though the major effect on the hepatic LDLR comes from secreted PCSK9, the details of PCSK9 reuptake into the hepatocyte remain unclear. In both tissue culture and animal models, heparan sulfate proteoglycans (HSPGs) on hepatocytes act as co-receptors to promote PCSK9 reuptake. We hypothesized that if this PCSK9:HSPG interaction is important in humans, disrupting it with unfractionated heparin (UFH) would acutely displace PCSK9 from the liver and increase plasma PCSK9.
Methods:
We obtained remnant plasma samples from 160 subjects undergoing cardiac catheterization before and after administration of intravenous UFH. PCSK9 levels were determined using a commercial ELISA (enzyme-linked immunosorbent assay).
Results:
Median plasma PCSK9 was 113 ng/ml prior to UFH and 119 ng/ml afterwards. This difference was not significantly different (p = 0.83, 95% CI: −6.23 to 6.31 ng/ml). Equivalence testing provided 95% confidence that UFH would not raise plasma PCSK9 by more than 4.7%. Among all subgroups, only subjects with the lowest baseline PCSK9 concentrations exhibited a response to UFH (8.8% increase, adj. p = 0.044). A modest correlation was observed between baseline plasma PCSK9 and the change in plasma PCSK9 due to UFH (RS = −0.3634, p < 0.0001).
Conclusion:
Administration of UFH does not result in a clinically meaningful effect on circulating PCSK9 among an unselected population of humans. The results cast doubt on the clinical utility of disrupting the PCSK9:HSPG interaction as a general therapeutic strategy for PCSK9 inhibition. However, the observations suggest that in selected populations, disrupting the PCSK9:HSPG interaction could still affect PCSK9 reuptake and offer a therapeutic benefit.
Graphical Abstract
Introduction
Atherosclerotic heart disease remains the leading cause of death in industrialized countries1, with low-density lipoprotein (LDL) its main reversible risk factor2. The hepatic LDL receptor (LDLR) clears LDL from the bloodstream, and thus its upregulation protects against cardiovascular disease3. The LDLR is controlled by several homeostatic mechanisms including proprotein convertase subtilisin/kexin type 9 (PCSK9)4, which chaperones the hepatic LDLR5 for lysosomal degradation. Accordingly, therapeutic disruption of PCSK9 lowers LDL and improves cardiovascular outcomes6.
Circulating PCSK9 drives hepatic LDLR degradation6,7. As such, PCSK9 reinternalization and lysosomal delivery are important mechanisms to understand. Though PCSK9 binds the LDLR8, its affinity at physiological pH (KD = 170–628 nM)9,10 is much lower than circulating concentrations (1–6 nM)11, suggesting that another factor aids PCSK9 reuptake. Both clathrin-mediated PCSK9 internalization12 and a separate caveolin-mediated pathway13 have been described. Further, while the LDLR itself is required for PCSK9 reentry10, the binding of PCSK9 to the LDLR is not14.
Heparan sulfate proteoglycans (HSPGs) participate in lipoprotein metabolism in multiple ways. First, HSPGs capture triglyceride-rich lipoprotein remnants, promoting their breakdown independent of the LDLR15. Second, HSPGs affect PCSK9 entry and clearance in tissue culture and animal models16,17. An arginine-rich region on the PCSK9 prodomain binds to both HSPGs and heparin16, a heterogenous species of sulfated oligosaccharides used clinically as an anticoagulant. Previously, we have shown that the binding sites on PCSK9 for both HSPGs and LDL overlap and that LDL attenuates the effect of HSPGs on PCSK9 reentry17. The gain-of-function S127R mutation4, whose mechanism remains elusive, introduces a positively charged arginine on PCSK9 near the HSPG binding site, increasing dependence on the HSPGs17. In mice, stripping oligosaccharides from hepatic HSPGs inhibits PCSK9 function and raises PCSK9 levels by displacing the liver-bound PCSK9 not yet internalized16. Additionally, proteinuric renal failure increases sulfation of HSPGs, raising their affinity for PCSK9 and driving dyslipidemia18. Last, PCSK9 concentrations rise in patients with acute coronary syndrome (ACS)19, but it is unclear whether this results from ACS itself or the heparin treatment that typically accompanies it20.
To date, the relationship between HSPGs and PCSK9 has not been directly tested in humans. We hypothesized that if HSPGs are critical co-receptors for PCSK9 in humans, administering a competitive inhibitor would displace PCSK9 from the liver and raise circulating PCSK9, mimicking the animal models16. We therefore leveraged remnant blood samples from cardiac catheterization patients undergoing short term anticoagulation with unfractionated heparin (UFH) in an investigation we named the Heparin Blockade of PCSK9 (“HepBlock9”) study. To our surprise, we observed no difference in circulating PCSK9 in our overall cohort after UFH administration, suggesting that heparin is unlikely to be successfully repurposed as a general strategy for PCSK9 inhibition.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Study Subjects and Samples
All protocols were approved by the Institutional Review Board of the University of California, San Francisco (UCSF) as a no subject contact study with a waiver of informed consent (CHR #18–26734). Adults presenting to the cardiac catheterization laboratory at the Zuckerberg San Francisco General Hospital (ZSFG) receiving intravenous (IV) UFH were considered. Subjects were fasted at least 6 hrs prior to the procedure, except for those in whom clinical urgency precluded the fast (n = 6). Remnants from blood routinely obtained during the procedure were transferred to sterile heparinized tubes and promptly placed on ice. Within 8 hrs, the plasma was isolated by centrifugation (1000 × g for 10 min at 4 °C), deidentified, and stored at −80 °C. Based on plasma PCSK9 levels in the general population11, we estimated that 150 subjects would provide 80% power (1 − β) at 95% confidence (two-tailed α = 0.05) to detect a 4 ng/ml change in PCSK9 concentrations (7% of total) after UFH administration. This is a conservative estimate compared to the 25% increase in circulating murine PCSK9 observed after heparinase treatment16, yet similar to the 7.4% increase in PCSK9 concentrations observed with statins21.
In a separate exploratory cohort, we obtained samples from inpatients receiving a continuous infusion of IV UFH. Remnants from citrated blood samples, used to determine the partial thromboplastin time (PTT) before and every 6 hrs after initiation of UFH, were stored at room temperature in the clinical laboratory for up to 24 hrs. Plasma was then isolated, re-labeled, deidentified, and stored at −80 °C as noted above.
Clinical data, including demographics, laboratory values, medication records, medical histories, diagnosis codes, and cardiac catheterization reports, were obtained from the longitudinal electronic medical record and reviewed by study investigators remote from the time of clinical presentation.
Inclusion and Exclusion Criteria
In the main cohort, we included all adults ≥ 18 years of age presenting to the cardiac catheterization laboratory who had blood samples obtained immediately before and approximately 5 to 20 minutes after receiving IV UFH. Exclusion criteria were limited to 1) active use of heparin or heparinoids, defined as IV UFH within 4 hrs, subcutaneous (SC) UFH or prophylactic dosing of SC low-molecular weight heparin (LMWH) within 24 hrs, or therapeutic dosing of SC LMWH within 48 hrs, all chosen to ensure adequate clearance (at least 4 half-lives) prior to the procedure22, or 2) active liver disease, defined by plasma alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (AP), or total bilirubin (TB) greater than three times the upper limit of normal. For subjects in whom liver tests were unavailable (8 total), we reviewed the medical records to exclude active liver disease.
Pre-defined strata included the following: 1) sex, defined as male, female, or nonbinary, with the latter encompassing all subjects with any discordance between legal sex, sex at birth, or sex identity; 2) age, defined as <55, 55–64, 65–74, or ≥75 years; 3) race, self-identified as White, Black or African American (AA), Asian, American Indian or Native Alaskan, Native Hawaiian or other Pacific Islander, Other, or Unknown; 4) ethnicity, self-defined as Hispanic or not Hispanic; 5) obstructive coronary artery disease (CAD), defined as a ≥70% stenosis in any coronary artery, a ≥50% stenosis in the left main coronary artery, or positive physiologic testing such as fractional-flow reserve (FFR) or instant wave-free ratio (iFR); 6) atherosclerosis, defined as any atherosclerotic plaque observed on coronary angiography; 7) acute coronary syndrome (ACS), defined as evidence of an acute plaque rupture, thrombus, or culprit lesion on coronary angiography; 8) percutaneous coronary intervention (PCI) performed during cardiac catheterization; 9) active statin use at time of cardiac catheterization; 10) non-high density lipoprotein (non-HDL) cholesterol; 11) creatinine; and 12) dose of the IV UFH bolus. All clinical definitions in the record were adjudicated by a board-certified cardiologist.
Enzyme-Linked Immunosorbent Assays (ELISAs)
Samples were analyzed via a commercial PCSK9 ELISA kit (Abcam) compatible with heparinized plasma according to the manufacturer’s instructions with minor modifications. Samples were diluted 50-fold in sample diluent and assayed in duplicate; all measurements resulted within the manufacturer’s reference range. End point absorbance at λ = 450 nm was recorded on a Spark plate reader (Tecan). Concentrations were interpolated from a 4-parameter logistic regression curve (Prism 9, GraphPad Software) fit to the background-subtracted standard run in each plate. For the exploratory cohort of subjects treated with UFH for 6 to 24 hrs, commercial ELISAs compatible with citrated samples were used for PCSK9 (RayBiotech) and apolipoprotein B (ApoB, MabTech).
Statistical Analysis
Interpolated biomarker concentrations from pre- and post-heparin samples were tested for normality using the D’Agostino and Pearson test. Nonparametric tests were used for non-Gaussian data: the Wilcoxon matched-pairs signed rank or Mann-Whitney tests between two groups, as appropriate, and the Kruskal-Wallis and Dunn’s multiple comparisons tests for three or more groups. The Wilcoxon signed rank test was also used to compare the change in PCSK9 to the hypothetical median of 0. When appropriate, the Holm-Sidak correction was applied to account for multiple strata. For Gaussian data, pairwise comparisons were performed with paired t tests and adjusted for multiple hypothesis testing with the Holm-Sidak method when appropriate. Statistical significance was set at adjusted p = 0.05.
Results
A Diverse Cohort of Patient Samples
We obtained a total of 180 paired pre- and post-UFH samples from subjects undergoing cardiac catheterization (Fig. 1A). We excluded two subjects with unclear records of heparin administration, four subjects with delays in sample processing, and twelve subjects that received heparin products within a predetermined window prior to the procedure. As we hypothesized that preserved hepatocyte function is required for PCSK9 internalization, we also excluded two subjects with active liver disease. In total, this left 160 subjects available for analysis (Fig. S1).
Figure 1: HepBlock9 Study.
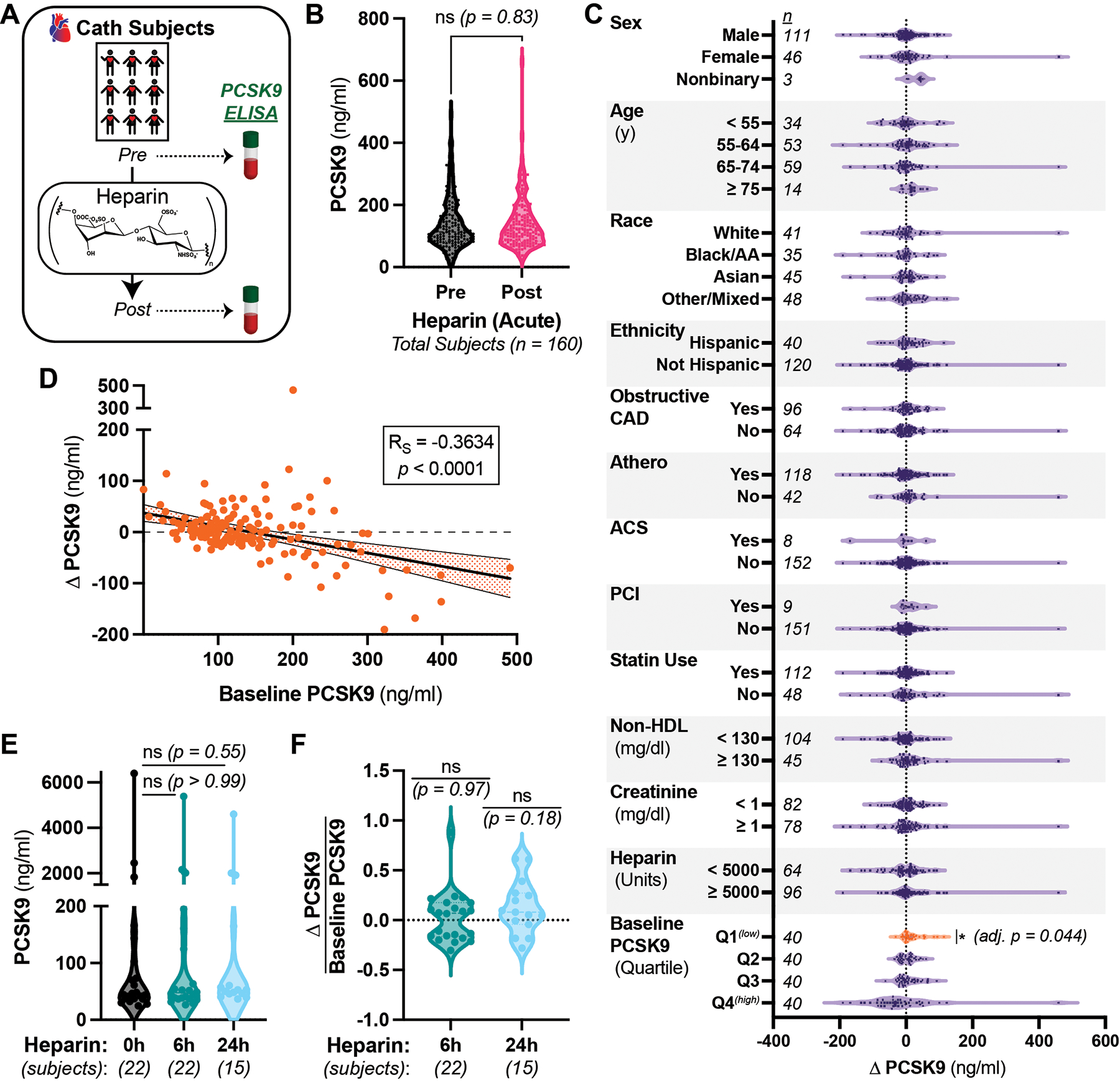
A) Schematic of study. Remnant blood samples before and after heparin in subjects undergoing cardiac catheterization were analyzed for plasma PCSK9. B) Plasma PCSK9 in study subjects before (black) and after (pink) IV UFH. C) Difference in plasma PCSK9 before and after IV UFH in subjects stratified by the indicated variables. Data from the first (lowest) quartile (Q1) of baseline PCSK9 concentrations highlighted in orange. Non-significant p values omitted for clarity. D) Correlation between baseline PCSK9 and change in PCSK9 from the main cohort, with each subject an orange circle. Spearman’s coefficient (RS) shown in box and linear trendline with 95% confidence interval displayed. E) Plasma PCSK9 before (black) or after 6 (teal) or 24 (light blue) hr infusions of IV UFH. F) Fractional difference in plasma PCSK9 before and after IV UFH infusions. Source data is the same as from E. All panels) Unless otherwise noted, Wilcoxon signed-rank tests shown, comparing matched-pairs between groups or to a hypothetical median of 0, with Holm-Sidak corrections for multiple hypothesis testing when appropriate. Note the discontinuous Y axis in D and E. n = number of subjects in each group, ns = non-significant at adjusted p > 0.05.
The subjects were 69% male, 29% female, and 2% nonbinary, with a mean age of 62 (Table 1). Our cohort was ethnically diverse, as 74% were non-White and 25% were Hispanic, consistent with the safety-net population of our institution. As expected from a cohort undergoing coronary angiography, both atherosclerosis (74%) and obstructive coronary disease (40%) were highly prevalent. Additionally, 70% were taking a statin, and the mean non-HDL cholesterol was 117 mg/dl among those with available values (n = 149).
Table 1: Subject Characteristics.
Clinical characteristics of the main cohort of HepBlock9 study subjects (n = 160).
| Age – mean (SD) | 62.1 (10.2) |
| Nonbinary | 3 (1.8) |
| Other/Unknown | 40 (25.0) |
| Hispanic Ethnicity – no. (%) | 40 (25.0) |
| Obstructive Coronary Artery Disease – no. (%) | 64 (40) |
| Any Atherosclerosis – no. (%) | 118 (73.8) |
| Acute Coronary Syndrome – no. (%) | 8 (5.0) |
| Percutaneous Coronary Intervention Performed – no. (%) | 9 (5.6) |
| Statin – no. (%) | 112 (70.0) |
| Triglycerides (n = 146) | 140/118 (84.9) |
| ≥1 – no. (%) | 78 (48.8) |
| ≥5000 – no. (%) | 96 (60.0) |
Intravenous Heparin Does Not Acutely Raise Circulating PCSK9 Concentrations
We then compared plasma PCSK9 in the paired pre- and post-UFH samples. The mean and median baseline PCSK9 concentrations were 137 and 113 ng/ml, respectively, and the mean and median post-UFH PCSK9 concentrations were 139 and 119 ng/ml, respectively. The differences between the matched samples were not significant (p = 0.83, Fig. 1B). Equivalence testing provided 95% confidence that the rise in median PCSK9 levels due to UFH was no more than 5.35 ng/ml, or 4.7% of the baseline. We conclude from these data that IV UFH does not raise PCSK9 levels acutely in a general population.
Subgroup Analyses of Effects from Heparin on Plasma PCSK9
PCSK9 concentrations are affected by several homeostatic mechanisms11,21,23, so we explored the interaction between UFH and circulating PCSK9 after stratifying by pre-defined variables. Neither male nor female subjects showed a significant change in plasma PCSK9 after UFH (Fig. 1C). Consistent with prior literature11, baseline PCSK9 concentrations were 25% higher in women (median 135 ng/ml) than men (median 108 ng/ml), though this did not reach statistical significance (p = 0.19, Fig. S2). We found no significant effects of UFH when stratifying by age, race, ethnicity, manifestations of CAD or its treatment, statin therapy, non-HDL cholesterol, renal function, or heparin dose (Fig. 1C). The baseline PCSK9 values among these strata were also not significantly different (Fig. S2). Additionally, we observed no correlation between any baseline lipid parameter and the effect of UFH on circulating PCSK9 (Fig. S3A–F). By contrast, upon stratifying by baseline PCSK9, we observed a significant increase (8.8%) in PCSK9 from UFH only among the subjects in the lowest quartile of baseline PCSK9 (< 83.9 ng/ml, orange, adj. p = 0.044, Fig. 1C, Table S1). Among all subjects, we also observed a significant negative correlation between baseline PCSK9 and the change in PCSK9 after UFH (RS = −0.3634, p < 0.0001, Fig. 1D). These data suggest that a subpopulation of patients, particularly those with low plasma PCSK9, may be susceptible to disrupting the PCSK9:HSPG interaction by UFH.
Continuous Intravenous Heparin for 24 Hours Does Not Affect PCSK9 Concentrations
We evaluated the acute administration of UFH because the reported half-life of circulating PCSK9 is only 5 minutes24. To assess whether longer UFH treatments are required to disrupt hepatic PCSK9 uptake, we compared baseline PCSK9 concentrations in the heparin-naïve cohort to the 12 subjects excluded due to pre-procedural heparin exposure. We observed no significant difference (p = 0.24, Fig. S4A). As expected, pre- and post-UFH PCSK9 concentrations in these 12 excluded subjects also did not significantly differ (p = 0.30, Fig. S4B).
We also measured PCSK9 concentrations in 22 separate patients anticoagulated with continuous IV UFH for at least 6 hrs. In these subjects, mean and median PCSK9 concentrations were 536 and 45 ng/ml before UFH and 487 and 47 ng/ml after UFH, respectively, but were not significantly different (p > 0.99, Fig. 1E). We also observed no significant effect after 24 hrs of continuous UFH for the 15 of these subjects so treated (p = 0.55, Fig. 1E). There was also no significant difference in the fractional change in PCSK9 concentrations after UFH at either timepoint (Fig. 1F). Consistent with the prior literature25,26, we also observed no effect of either 6 or 24 hrs of UFH administration on plasma ApoB levels (Fig. S5A&B). Together, these data suggest against heparin-based treatments inhibiting PCSK9 reuptake or function in an unselected population.
Discussion
In this study, we mechanistically studied a critical regulator of lipoprotein homeostasis directly in humans. Our goal was to interrogate whether the clinical use of UFH could be repurposed to inhibit PCSK9, either for cholesterol lowering or improved pathogen clearance27. Our study exhibits multiple strengths. First, we used the ideal physiologic model system: human patients. Second, our question is clinically relevant, since UFH is ubiquitously used as an anticoagulant. UFH is attractive to repurpose for an alternative indication, so our results, which reflect therapeutic doses of UFH, remain relevant even though they support the null hypothesis. Third, our temporal observations, before and after UFH, improve our ability to draw causal inferences. Fourth, our ethnically diverse subject population (74% non-White) increases the generalizability of our findings. Last, the use of subjects as their own controls improves the power of our study, as it minimizes confounding from the variability in PCSK9 concentrations observed between individuals11,21,23.
Our study also has several limitations. First, our observations are limited to static plasma measurements and therefore provide only an indirect glimpse into PCSK9 homeostasis. We cannot assess PCSK9 synthesis or degradation rates, shifts in PCSK9 from one body compartment to another, or directly confirm displacement of PCSK9 from hepatic HSPGs. Second, subjects were in a fasted state, with PCSK9 levels expected to fall during the study28. We observed no correlation between the change in PCSK9 levels and the time between blood draws (RS = −0.027, p = 0.81, n = 78). This makes it unlikely, though not impossible, for the homeostatic reduction in PCSK9 over the time course of our experiment to obscure a small true result. Third, while our study was adequately powered for our primary hypothesis, it was not necessarily powered to answer each subgroup analysis. Thus, the effect of heparin in raising PCSK9 levels in the subjects with low baseline PCSK9 should be considered exploratory. The findings may be biologically plausible, as low circulating PCSK9 from increased sequestration on the liver surface could result in more displaced PCSK9 upon treatment with UFH. However, low baseline PCSK9 could result from other mechanisms, such as the transcriptional effects of prolonged fasting29. We observed no differences in subgroups expected to be more sensitive to HSPG blockade, including those with low non-HDL, which disrupts the PCSK9:HSPG interaction17, or those on statins, which drive transcription of30 and increase circulating PCSK931. Thus, further investigations are needed to probe whether a mechanistic relationship between circulating PCSK9 and the PCSK9:HSPG interaction truly exists, as well as whether the baseline PCSK9, or any other biomarker, could identify a subpopulation responsive to a PCSK9:HSPG targeting therapy. Fourth, our measurements do not differentiate between different circulating forms of PCSK9, such as the less-active furin-cleaved species32 or PCSK9 complexed with LDL or Lipoprotein(a)33,34. Though we did not observe a correlation between the effects of UFH and any lipoprotein species, the functional consequences of these various forms of PCSK9 are still being explored, and thus it is possible our results could be clarified by considering these different PCSK9 species in the future. Fifth, we assessed the acute administration of UFH, not the chronic use of heparins. Thus, we can make no definitive conclusions regarding long-term HSPG blockade. Last, we used UFH due to its practicality, but UFH is a heterogenous preparation with pleiotropic effects. It is possible that a more specific inhibitor of the PCSK9:HSPG interaction could make this strategy more successful.
Several explanations could account for the discrepancy between our observations and those from prior models16,17. First, minor differences in PCSK9 regulation exist between the mouse and the human35,36. The murine LDLR may be more sensitive to PCSK9 than the human counterpart35,37, and it is tantalizing to speculate that the PCSK9:HSPG interaction might drive this difference. Second, the clinical dosing of UFH differs from prior animal studies. Though the approximate ratio between the amounts of UFH administered and PCSK9 targeted is slightly higher in our study, more UFH was used in the mouse model to demonstrate UFH-mediated blockade of exogenous, supraphysiologic PCSK916. Third, in vitro and tissue culture models may not recapitulate the complexities of in vivo physiology. Last, we did not perform the study under specific disease conditions that could affect potential regulators of the PCSK9:HSPG interaction, such as extreme hyperlipidemia or proteinuric renal disease18. This does not alter our overall conclusions, but we also cannot extrapolate our findings to such conditions. Studying these disease states in the future may be informative.
In summary, we assessed whether heparin could acutely interfere with PCSK9 uptake into the liver by measuring PCSK9 concentrations in subjects receiving IV UFH for a routine clinical procedure. Our overall results support the null hypothesis, as we found no evidence that UFH could act as a potential strategy to inhibit PCSK9 function across a general, unselected population. However, our results do suggest that this strategy may be feasible in certain subpopulations, particularly those with low levels of circulating PCSK9, and therefore warrants further study.
Supplementary Material
Highlights.
Prior studies suggest that PCSK9 interacts with hepatic heparan sulfate proteoglycans (HSGPs) to enter the liver and raise cholesterol levels
Heparin, a competitive inhibitor of HSPGs, does not affect PCSK9 concentrations across an unselected population of humans, suggesting it is unlikely to be successfully repurposed as a general inhibitor of PCSK9
In a subgroup of patients with low baseline PCSK9 concentrations, PCSK9 levels rise after acute heparin administration, suggesting that this population might benefit therapeutically
Understanding the mechanistic relationship between circulating PCSK9 and the PCSK9:HSPG interaction may help identify a subpopulation that could benefit from PCSK9:HSPG disruption to inhibit PCSK9
Acknowledgments:
We thank the UCSF cardiology fellows and nursing staff of the ZSFG cardiac catheterization laboratory for preservation of remnant samples.
Sources of Funding:
This work was supported by the NIH/NHLBI (K08 HL124068, R03 HL145259, R01 HL146404, and R01 HL159457), a Pfizer ASPIRE Cardiovascular Award, the Harris Fund, and the Research Evaluation and Allocation Committee of the UCSF School of Medicine, all to JSC.
Non-standard Abbreviations and Acronyms
- AA
African American
- ACS
acute coronary syndrome
- ALT
alanine aminotransferase
- AP
alkaline phosphatase
- ApoB
apolipoprotein B
- AST
aspartate aminotransferase
- CAD
coronary artery disease
- ELISA
enzyme-linked immunosorbent assay
- FFR
fractional-flow reserve
- HDL
high density lipoprotein
- HepBlock9
Heparin Blockade of PCSK9 Study
- HSPG
heparan sulfate proteoglycan
- iFR
instant wave-free ratio
- IV
intravenous
- LDL
low density lipoprotein
- LDLR
low density lipoprotein receptor
- LMWH
low molecular weight heparin
- PCI
percutaneous coronary intervention
- PCSK9
proprotein convertase subtilisin-kexin type 9
- PTT
partial thromboplastin time
- SC
subcutaneous
- TB
total bilirubin
- UCSF
University of California San Francisco
- UFH
unfractionated heparin
- ZSFG
Zuckerberg San Francisco General Hospital
Footnotes
Disclosures: JSC has received consulting fees from Gilde Healthcare and Eko.
References
- 1.Ahmad FB, Cisewski JA, Anderson RN. Provisional Mortality Data - United States, 2021. MMWR Morb Mortal Wkly Rep. 2022;71:597–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Cheng S, Delling FN, et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation. 2021;143:e254–e743. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. [DOI] [PubMed] [Google Scholar]
- 5.Maxwell KN, Fisher EA, Breslow JL. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci U S A. 2005;102:2069–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376:1713–1722. [DOI] [PubMed] [Google Scholar]
- 7.Lagace TA, Curtis DE, Garuti R, McNutt MC, Park SW, Prather HB, Anderson NN, Ho YK, Hammer RE, Horton JD. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J Clin Invest. 2006;116:2995–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwon HJ, Lagace TA, McNutt MC, Horton JD, Deisenhofer J. Molecular basis for LDL receptor recognition by PCSK9. Proc Natl Acad Sci U S A. 2008;105:1820–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cunningham D, Danley DE, Geoghegan KF, Griffor MC, Hawkins JL, Subashi TA, Varghese AH, Ammirati MJ, Culp JS, Hoth LR, et al. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat Struct Mol Biol. 2007;14:413–419. [DOI] [PubMed] [Google Scholar]
- 10.Fisher TS, Lo Surdo P, Pandit S, Mattu M, Santoro JC, Wisniewski D, Cummings RT, Calzetta A, Cubbon RM, Fischer PA, et al. Effects of pH and low density lipoprotein (LDL) on PCSK9-dependent LDL receptor regulation. J Biol Chem. 2007;282:20502–20512. [DOI] [PubMed] [Google Scholar]
- 11.Lakoski SG, Lagace TA, Cohen JC, Horton JD, Hobbs HH. Genetic and metabolic determinants of plasma PCSK9 levels. J Clin Endocrinol Metab. 2009;94:2537–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, Cohen JC, Hobbs HH. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 2007;282:18602–18612. [DOI] [PubMed] [Google Scholar]
- 13.Jang HD, Lee SE, Yang J, Lee HC, Shin D, Lee H, Lee J, Jin S, Kim S, Lee SJ, et al. Cyclase-associated protein 1 is a binding partner of proprotein convertase subtilisin/kexin type-9 and is required for the degradation of low-density lipoprotein receptors by proprotein convertase subtilisin/kexin type-9. Eur Heart J. 2020;41;239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeVay RM, Shelton DL, Liang H. Characterization of proprotein convertase subtilisin/kexin type 9 (PCSK9) trafficking reveals a novel lysosomal targeting mechanism via amyloid precursor-like protein 2 (APLP2). J Biol Chem. 2013;288:10805–10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foley EM, Gordts PLSM, Stanford KI, Gonzales JC, Lawrence R, Stoddard N, Esko JD. Hepatic remnant lipoprotein clearance by heparan sulfate proteoglycans and low-density lipoprotein receptors depend on dietary conditions in mice. Arterioscler Thromb Vasc Biol. 2013;33:2065–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gustafsen C, Olsen D, Vilstrup J, Lund S, Reinhardt A, Wellner N, Larsen T, Andersen CBF, Weyer K, Li J-P, et al. Heparan sulfate proteoglycans present PCSK9 to the LDL receptor. Nat Commun. 2017;8:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galvan AM, Chorba JS. Cell-associated heparin-like molecules modulate the ability of LDL to regulate PCSK9 uptake. J Lipid Res. 2019;60:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shrestha P, Yazdani S, Vivès RR, El Masri R, Dam W, van de Sluis B, van den Born J. Proteinuria converts hepatic heparan sulfate to an effective proprotein convertase subtilisin kexin type 9 enzyme binding partner. Kidney Int. 2021;99:1369–1381. [DOI] [PubMed] [Google Scholar]
- 19.Gencer B, Montecucco F, Nanchen D, Carbone F, Klingenberg R, Vuilleumier N, Aghlmandi S, Heg D, Räber L, Auer R, et al. Prognostic value of PCSK9 levels in patients with acute coronary syndromes. Eur Heart J. 2016;37(6):546–553. [DOI] [PubMed] [Google Scholar]
- 20.Amsterdam EA, Wenger NK, Brindis RG, Casey DEJ, Ganiats TG, Holmes DRJ, Jaffe AS, Jneid H, Kelly RF, Kontos MC, et al. 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;130:e344–426. [DOI] [PubMed] [Google Scholar]
- 21.Mayne J, Dewpura T, Raymond A, Cousins M, Chaplin A, Lahey KA, LaHaye SA, Mbikay M, Ooi TC, Chrétien M. Plasma PCSK9 levels are significantly modified by statins and fibrates in humans. Lipids Health Dis. 2008;7:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirsh J, Anand SS, Halperin JL, Fuster V. Guide to anticoagulant therapy: Heparin: A statement for healthcare professionals from the American Heart Association. Circulation. 2001;103:2994–3018. [DOI] [PubMed] [Google Scholar]
- 23.Persson L, Cao G, Stahle L, Sjoberg BG, Troutt JS, Konrad RJ, Galman C, Wallen H, Eriksson M, Hafstrom I, et al. Circulating Proprotein Convertase Subtilisin Kexin Type 9 Has a Diurnal Rhythm Synchronous With Cholesterol Synthesis and Is Reduced by Fasting in Humans. Arterioscler Thromb Vasc Biol. 2010;30:2666–2672. [DOI] [PubMed] [Google Scholar]
- 24.Grefhorst A, McNutt MC, Lagace TA, Horton JD. Plasma PCSK9 preferentially reduces liver LDL receptors in mice. J Lipid Res. 2008;49:1303–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lever WF, Smith PA, Hurley NA. Effects of intravenous heparin on the plasma lipoproteins in primary hypercholesteremic xanthomatosis and idiopathic hyperlipemia. Science. 1953;118:653–654. [DOI] [PubMed] [Google Scholar]
- 26.Herzstein J, Wang CI, Adlersberg D. Effect of heparin on plasma lipid partition in man: Studies in normal persons and in patients with coronary atherosclerosis, nephrosis and primary hyperlipemia. Ann Intern Med. 1954;40:290–306. [DOI] [PubMed] [Google Scholar]
- 27.Walley KR, Thain KR, Russell JA, Reilly MP, Meyer NJ, Ferguson JF, Christie JD, Nakada TA, Fjell CD, Thair SA, et al. PCSK9 is a critical regulator of the innate immune response and septic shock outcome. Sci Transl Med. 2014;6:258ra143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Browning JD, Horton JD. Fasting reduces plasma proprotein convertase, subtilisin/kexin type 9 and cholesterol biosynthesis in humans. J Lipid Res. 2010;51:3359–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krysa JA, Ooi TC, Proctor SD, Vine DF. Nutritional and Lipid Modulation of PCSK9: Effects on Cardiometabolic Risk Factors. J Nutr. 2017;147:473–481. [DOI] [PubMed] [Google Scholar]
- 30.Dubuc G, Chamberland A, Wassef H, Davignon J, Seidah NG, Bernier L, Prat A. Statins Upregulate PCSK9, the Gene Encoding the Proprotein Convertase Neural Apoptosis-Regulated Convertase-1 Implicated in Familial Hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24:1454–1459. [DOI] [PubMed] [Google Scholar]
- 31.Careskey HE, Davis RA, Alborn WE, Troutt JS, Cao G, Konrad RJ. Atorvastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9. J Lipid Res. 2008;49:394–398. [DOI] [PubMed] [Google Scholar]
- 32.Essalmani R, Susan-Resiga D, Chamberland A, Abifadel M, Creemers JW, Boileau C, Seidah NG, Prat A. In vivo evidence that furin from hepatocytes inactivates PCSK9. J Biol Chem. 2011;286:4257–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lagace TA. PCSK9 and LDLR degradation: regulatory mechanisms in circulation and in cells. Curr Opin Lipidol. 2014;25:387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shapiro MD, Tavori H, Fazio S. PCSK9: From Basic Science Discoveries to Clinical Trials. Circ Res. 2018;122:1420–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith GA, Padmanabhan A, Lau BH, Pampana A, Li L, Lee CY, Pelonero A, Nishino T, Sadagopan N, Xia VQ, et al. Cold shock domain–containing protein E1 is a posttranscriptional regulator of the LDL receptor. Sci Transl Med. 2022;14:eabj8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dandan M, Han J, Mann S, Kim R, Mohammed H, Nyangau E, Hellerstein M. Turnover Rates of the Low-Density Lipoprotein Receptor and PCSK9: Added Dimension to the Cholesterol Homeostasis Model. Arterioscler Thromb Vasc Biol. 2021;41:2866–2876. [DOI] [PubMed] [Google Scholar]
- 37.Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, Hammer RE, Moon YA, Horton JD. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci U S A. 2005;102:5374–5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.