

Abstract
Despite advances in understanding tumor biology, malignant gliomas remain incurable. While immunotherapy has improved outcomes in other cancer types, comparable efficacy has not yet been demonstrated for primary cancers of the central nervous system (CNS). T-cell exhaustion, defined as a progressive decrease in effector function, sustained expression of inhibitory receptors, metabolic dysfunction, and distinct epigenetic and transcriptional alterations, contributes to the failure of immunotherapy in the CNS. Herein, we describe recent advances in understanding the drivers of T-cell exhaustion in the glioma microenvironment. We discuss the extrinsic and intrinsic factors that contribute to exhaustion and highlight potential avenues for reversing this phenotype. Our ability to directly target specific immunosuppressive drivers in brain cancers would be a major advance in immunotherapy.
Keywords: Neuro-oncology, immunology, T cells, exhaustion, treatment modalities
T Cell Exhaustion: A Barrier for Effective Immunotherapy
Tumors of the central nervous system (CNS) are some of the most aggressive types of cancer. Annually, an average of 86,355 cases of primary brain tumors occur in the United States, with gliomas accounting for roughly 24.5% of total brain tumors and 80.9% of all malignant ones [1]. Gliomas are thought to develop from mutated progenitor or neuroglial stem cells and are notoriously difficult to treat [2]. Even with the newest lines of treatment, patient survival rates have not changed significantly over the past decade (Box 1).
Box 1. Current Definition and Care of Gliomas.
The World Health Organization (WHO) relies on histological and molecular characteristics in defining tumors of the central nervous system (CNS). Adult diffuse gliomas represent a major class of CNS tumors and are classified as astrocytoma, IDH-mutant; oligodendroglioma, IDH-mutant and 1p/19q-codeleted; and Glioblastoma, IDH-wildtype in the most recent WHO edition. [61]. The standard of care varies based on the clinical presentation of these CNS tumors but is often multifaceted, involving surgery, chemotherapy, radiation, and immunotherapy. High-grade glioma standard of care relies on surgery to debulk and remove all accessible tumor tissue, followed by radiation and chemotherapy to target residual, inaccessible tumor [138,139]. Their location within the brain, their often-fast-growing nature, and their tendency to diffuse into healthy tissue with unclear delineations limit their accessibility and treatment via surgical resection. Radiation, despite producing a survival benefit and improving anti-tumor immunity when combined with immunotherapy agents, has also been linked to lymphopenia and damage to T cells with irradiated T cells displaying decreased proliferative capacity, cytokine production, and disrupted metabolic function through impairments towards glycolysis and energy production [140–149]. Notably, these are characteristics present in exhausted T cell phenotypes, yet it remains unclear exactly how radiation may impact global and intratumoral T cell exhaustion, as opposed to general dysfunction, in the context of gliomas. Treatment using chemotherapy is also challenging given the drug delivery limitations caused by the blood-brain barrier and the difficulty identifying treatment targets that are both specific and adequate to evoke tumor response despite the myriad of signaling pathway overlaps and redundancy. Although exact relationships between chemotherapy and T cell exhaustion in gliomas remain understudied, there is evidence that chemotherapy agents may impart lasting effects on T cells, inhibiting their proliferative capacity, increasing expression of markers associated with exhaustion, and altering metabolic function [150,151]. The impact likely varies depending on the chemotherapy agents used, cancer type, and disease progression, but it will be crucial to further study as this immunologic alteration may have implications for immunotherapeutic treatment modalities and anti-tumor responses. Additionally, corticosteroids are often prescribed to patients with gliomas suffering from treatment-associated toxicities, especially immune-related adverse events (irAEs) in response to immunotherapies as they induce immunosuppression to counteract irAEs [152]. Corticosteroids inhibit T cell functionality – notably dexamethasone, a common corticosteroid prescribed for patients with gliomas, impairing T cell proliferation – and dexamethasone has been shown to limit survival benefit by immunotherapy agents in preclinical and clinical models of glioblastomas [153–155]. Additional research is needed to distinguish between the influence of these corticosteroid-induced immunologic alterations on T cell exhaustion versus general dysfunction in order to enhance the anti-tumor response.
Immunotherapy has revolutionized cancer care, as it relies on enhancing the ability of the patient’s immune cells to eradicate cancer cells. Because the immunological response can target infiltrating and persisting cancer cells, it is a promising approach for treating gliomas. However, patients’ responses vary greatly, and the use of immunotherapy to treat CNS cancers has been far less successful than in other cancer types. To advance the fields of immunotherapy and cancer care, a better understanding of the processes by which immune cell function and anti-tumor responses are suppressed in brain cancers is needed.
The most common form of immunotherapy using immune checkpoint inhibitors, unleashes or redirects the function of T-lymphocytes, which carry out cell-mediated immune responses that directly kill cancer cells and enhance the antitumor capabilities of other immune cells [3]. For full antitumor immunity, T-cells must have both effector functionality and the ability to infiltrate the tumor microenvironment (TME). The discovery that T-cells can undergo exhaustion in certain contexts, including the TME [4] has changed the view of T-cell biology. T-cell exhaustion is defined as a progressive decrease in effector function, sustained expression of inhibitory receptors, loss of cytokine production, metabolic dysfunction, and distinct epigenetic and transcriptional alterations [5,6]. T-cell exhaustion was first identified in murine models of chronic infection, but it has since been shown to occur in other animals and in humans suffering from chronic infection or cancer [7]. Although the delineation between T-cell exhaustion and general dysfunction is not entirely clear, it is becoming evident that T-cell exhaustion is a distinct cellular process accompanied by specific metabolic, transcriptional, and epigenetic changes.
To elucidate potential drivers of T-cell exhaustion and to unravel the cellular mechanisms by which it is induced in gliomas specifically, we reviewed the current knowledge of the microenvironment of gliomas. The efficacy of T-cells that infiltrate gliomas is significantly reduced, and they become exhausted due to both intrinsic and extrinsic factors within the glioma microenvironment. Here, we describe known drivers of T-cell exhaustion that have been identified in gliomas and, where relevant, in other cancers and diseases, with the goal of providing a comprehensive picture of the exhausted state and potential ways to reverse it. We discuss the interplay of metabolism, epigenetic, and transcriptional factors in exhausted T-cells found in gliomas, in order to propose ways to reverse it. Our ability to target immunosuppressive drivers specific to brain cancers, thereby reducing or reversing T-cell exhaustion, has the potential to improve patients’ responses and outcomes in this deadly disease.
Known Markers of Exhausted T Cells
Although the definition of T-cell exhaustion is evolving, currently we define this state by reduced proliferative capacity, reduced production of effector cytokines and cytotoxicity, elevated and sustained expression of multiple inhibitory receptors, altered expression and function of key transcription factors, dysregulation of epigenetic programs, and impaired metabolic activity (Figure 1). T-cells can vary in their extent of exhaustion, with some being capable of re-invigoration but others being terminally dysfunctional [8] (Figure 1). To enhance immune functionality, it is essential to identify the drivers of exhaustion in order to slow and/or reverse it. However, their identification is highly complex because the drivers of exhaustion are often intertwined and varied, depending on extrinsic environments and specific phenotypes.
Figure 1.

Characteristics of functional and terminally exhausted T-cells. Chronic antigen exposure and other intrinsic and extrinsic factors drive T-cell exhaustion in cancer. Expression of inhibitory receptors CTLA-4, PD-1, TIM-3, TIGIT, and LAG-3 increases in exhausted T-cells compared to functional T-cells; transition to an exhausted state is also marked by a decrease in TCF-1 expression and increase in TOX expression. Disturbed TCR signaling, Glut1 upregulation, elevation of inhibitory receptors, and transcriptional and epigenetic changes drive metabolic dysfunction, impairing glycolytic abilities and metabolic flexibility in exhausted T-cells. Cytokines, including IL-2, TNF-α, and IFN-γ, and effector protease granzyme B production also decrease in exhausted T-cells compared to functional ones. These alterations, along with reduced proliferative capacity, results in diminished cytotoxicity as T-cells progress to terminal exhaustion.
T-cell exhaustion occurs as a sequential, sustained reduction in effector function, including alterations in cytokine production and proliferative capability. Loss of production of interleukin-2 (IL-2), a cytokine involved in initiating immune responses and mediating T-cell proliferation, is considered an early sign of exhaustion [9–11]. Loss of IL-2 is often followed by loss of the ability to produce tumor necrosis factor-α (TNF-α), a pro-inflammatory cytokine that helps regulate and direct proper immune responses; loss of production of interferon-γ (IFN-γ), a critical cytokine that drives innate and adaptive immune responses; and decreased granzyme B, a protease secreted by cytotoxic T-cells during immune responses [10,11]. IFN-γ production is more resistant to inhibition and its loss is limited primarily to severely exhausted T-cell subsets [6,9,11] (Figure 1). Although loss of cytokine production was first documented in murine models of chronic infection, similar findings have been observed in cancer models [12–14]. Thus, in both chronic infection and cancer, loss of cytokine production, importantly IL-2, TNF-α, and IFN-γ, is recognized as a defining trait of exhausted T-cells.
At the cell surface, one of the initial markers and drivers of exhaustion is the expression of programmed cell death protein-1 (PD-1); however, roles for PD-1 are complex as PD-1 is induced during T-cell activation and exhausted phenotypes with low/minimal PD-1 expression have been identified [15]. PD-1 largely serves as a down-regulator of the immune system to guard against autoimmunity, but in the context of T-cell exhaustion, it is considered a canonical marker of exhaustion, with sustained expression of PD-1 driving an inhibitory pathway that limits effector functionality [5]. In addition to PD-1, other surface markers of exhaustion include cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), T-cell immunoglobulin and mucin domain-3 (TIM-3), T-cell immunoglobulin and ITIM domain (TIGIT), and lymphocyte activation gene 3 protein (LAG-3). Moreover, key transcription factors mark exhaustion phenotypes, including B lymphocyte–induced maturation protein-1 (Blimp-1), thymocyte selection–associated high-mobility group box protein (TOX), T-bet, and T-cell factor 1 (TCF1)[15]. Expression of these inhibitory receptors and transcription factors often drives metabolic alterations that enhance the exhausted status, such as mitochondrial dysfunction and diminished metabolic flexibility.
The metabolic programs of naïve, activated, and exhausted T cells are very different. While naïve T cells are mostly dependent on oxidative phosphorylation (OXPHOS) for ATP generation, activated T cells switch their metabolic programs to depend on aerobic glycolysis for growth, proliferation, and effector functions such as the production of interferon-gamma. In the study by Chang et al, activated T cells were forced to use only OXPHOS, by removing glucose from the culture medium. The authors found that activated T cells could support proliferation and survival, but the production of interferon-gamma was compromised in the absence of glucose. This finding demonstrates the role of glycolysis in the effector function of activated T cells and the potential association between this pathway and the onset of exhaustion [16] (Figure 1). Indeed, exhausted T cells have a compromised ability to utilize aerobic glycolysis and OXPHOS due to both external factors as well as intrinsic signaling coming from the expression of PD-1. They demonstrated that PD-1 regulated early glycolytic and mitochondrial alterations and repressed transcriptional coactivator (PGC-1) which controls mitochondria biogenesis. Exhausted T-cells appear to compensate for the lack of glucose by upregulating glucose transporter (Glut1) in chronic infection models [17], however, this mechanism has not been yet validated in gliomas.
The proliferative ability of exhausted T-cells is largely modulated by inhibitory receptors such as PD-1, TIM-3, and CTLA-4, that become upregulated during exhaustion. However, the metabolic, transcriptional, and epigenomic alterations that occur in exhausted T-cells further impair their proper proliferation. Nonetheless, loss of cytokine production and of proliferative ability are two core components of T-cell exhaustion and represent two alterations that limit T-cell effector function and anti-tumor immunity.
These characteristics of T-cell exhaustion have been documented in patients with malignant gliomas including glioblastomas [18,19]. Tumor-infiltrating lymphocytes (TILs) in patients with glioblastoma express markers of exhaustion including increased amounts of PD-1, TIM-3, and TIGIT, compared with peripheral blood T-cells and control populations [19]. Additionally, TILs from glioma patients show lower functional capabilities, with decreased production of effector cytokines [19]. To distinguish exhaustion from dysfunction, gene expression profiles of TILs from patients with gliomas were compared with profiles of exhausted T-cells isolated from subjects with chronic viral infection; similar genomic and transcriptional patterns were observed in both samples, with changes in T-bet, PD-1, Eomes, and Blimp-1 detected as exhaustion progressed [19]. In murine SMA-560 glioma models, T-cell exhaustion occurred preferentially in tumor-specific T-cells, suggesting that this was also the case in human gliomas, but the heterogeneity of patient samples limited direct comparison [19].
Finally, it is proposed that persistent antigen exposure and chronic T-cell activation in cancers drive T-cell exhaustion, and this is highly relevant to the exhaustion of gliomal TILs as they are exposed to sustained high levels of antigens in the TME[5,18,20]. The complex nature of the TME also poses many other challenges that have been hypothesized to impact or mediate T-cell function. The glioma microenvironment is notoriously immunosuppressive, and glioma-specific characteristics further impair T-cell efficacy and drive exhaustion.
Extrinsic factors in the glioma microenvironment drive T-cell exhaustion
To deduce the extrinsic factors that promote T-cell exhaustion in gliomas, we reviewed studies that evaluated the environment that contains the tumor-infiltrating lymphocytes. The glioma TME is a heterogeneous environment comprising tumor cells, endothelial cells, immune cells, and fibroblasts. These cells not only interact with one another but are surrounded by an extracellular matrix containing influential factors, such as cytokines, growth factors, enzymes, and metabolites. Moreover, in the glioma TME, hypoxia, nutrient deprivation, and suppressive molecules are prevalent. Cancer cells adapt and overcome these difficulties by altering their metabolic states. Therefore, alterations that occur in the TME can result in metabolic, transcriptomic, and epigenetic changes that affect immune cell function and, in certain cases, drive T-cell exhaustion and dysfunction.
The impact of hypoxia on T-cell exhaustion
Microenvironments of solid tumors are known to be hypoxic, that is, the cells within them do not receive adequate oxygen supply. This has been extensively documented for gliomas (Figure 2) [21–23]. As cancer progresses and tumor density increases, the existing brain vasculature is insufficient to fuel their oxygen supply, which results in both an increase in hypoxic regions as well as the promotion of angiogenesis in the TME [23]. Poor blood supply, as well as the high metabolic activity in actively proliferating or infiltrating tumor cells, depletes available oxygen within the TME, variably resulting in regions of oxygen starvation [24].
Figure 2.
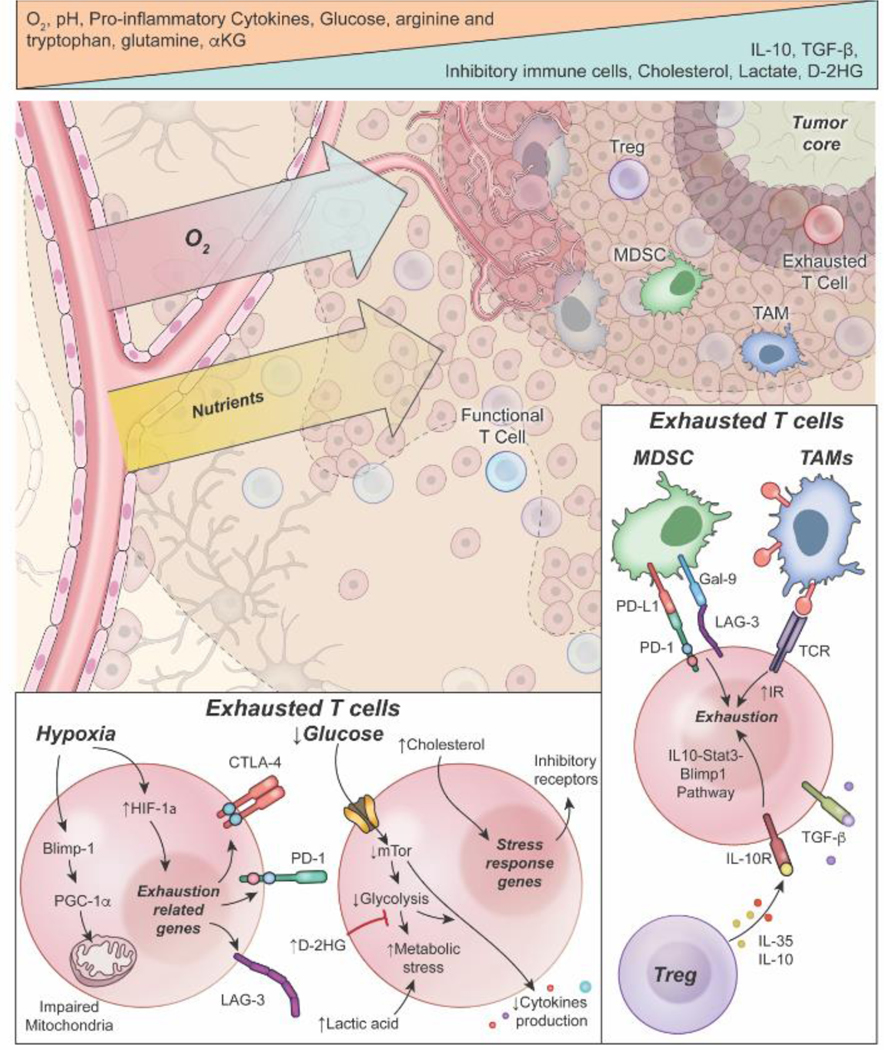
Impacts of glioma microenvironment-specific factors on T-cell exhaustion. Oxygen and nutrient deprivation occur in proximity to the tumor core due to insufficient blood supply and competition by cancer cells. Insufficient oxygen and nutrient levels, notably glucose and amino acid deficiencies, drive progression to exhausted phenotypes in T-cells. Increasing acidity in the tumor microenvironment, as well as increased metabolic by-products (e.g., lactate dehydrogenase, cholesterol, and D-2HG), are implicated in metabolic and transcriptional alterations that promote exhaustion in T-cells. Bottom inset: Hypoxia promotes the expression of transcription factors (e.g. HIF-1α, Blimp-1, etc.) that impair metabolic function and promote the expression of exhaustion-related genes, which leads to the upregulation of inhibitory immune receptors. Glucose deprivation can impair downstream metabolic and signaling pathways that lead to metabolic stress and decreased cytokine production. Increased cholesterol, D-2HG, and lactic acid can enhance this metabolic and epigenetic dysfunction to further promote exhaustion. Bottom right inset: The increased presence of regulatory T cells (Tregs), tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs), along with increased IL-10 and TGF-β, bind to and interact with tumor-infiltrating lymphocytes and promote signaling pathways that can enhance an exhaustive phenotype and suppression of anti-tumor immunity.
Hypoxic stress in the TME has been linked to decreased antitumor immunity and is considered central to promoting cancer aggressiveness [25]. In glioma patients, a high level of hypoxia is linked to poor prognosis and increased immunosuppression in the TME [21]. This hypoxic state drives malignant behavior in gliomas by triggering angiogenesis, immunosuppression, and tumor cell growth largely through pathways mediated by hypoxia-inducible factor HIF1-α [21,26–28]. Hypoxia promotes the expression of HIF-1α and/or HIF-2α, which serve as the primary regulators of hypoxia-related genes and allow for adaptive cellular responses in hypoxic environments [25]. Both hypoxia and the presence of HIF have been shown to be involved in reducing antitumor immunity specifically by inducing T-cell exhaustion.
Scharping et al. noted a strong correlation between severe hypoxia and exhausted intratumoral T-cells [29]. They classified exhausted cells as those that express high sustained levels of inhibitory receptors, such as PD-1 and TIM-3, and that are unable to secrete functional cytokines upon restimulation [29]. Blimp-1, a transcriptional repressor of metabolic reprogramming, was highly upregulated under chronic stimulation in hypoxic conditions, and it repressed the transcriptional coactivator, peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1α, which resulted in decreased mitochondrial reprogramming [29]. Mitochondrial dysfunction and hypoxia were both shown to promote formation of reactive-oxygen species (ROS) in T-cells, which further drove dysfunction and promoted progression to an exhausted state [29].
Notably, these authors observed that exhaustion under hypoxia and chronic antigen stimulation occurred regardless of HIF expression. This indicates that, at least in in vitro conditions, hypoxia drives exhaustion beyond the HIF-1α axis [29]. A meta-analysis of glioma patients showed that high HIF-1α expression was associated with significantly higher displays of exhaustion markers PD-L1, FOXO1, and PRDM1 compared to low HIF-1α expression [30]. However, HIF-1α is likely not the sole mediator of exhaustion in hypoxic conditions [29,31,32].
In addition to HIF-1α, expression of vascular endothelial growth factor (VEGF-A) is triggered by hypoxia and is involved in promoting in vitro differentiation of terminally exhausted CD8+ T-cell populations [33]. Hypoxia also upregulates glioma cell surface expression of PD-L1, which inhibits T-cell functionality through the PD-L1/PD-1 axis [34]. This is a canonical pathway for T-cell exhaustion and results in decreased cytotoxic T-lymphocyte-mediated lysis to allow for glioma cell immune evasion [34].
Hypoxia shapes the transcriptome and epigenome of T-cells leading to exhaustion
Hypoxia and HIF-1α have been shown to alter histone methylation, histone acetylation, and DNA methylation patterns via numerous pathways, and HIF-1α has been linked to downregulation of S-adenosyl methionine (SAM), a substrate required for methylation reactions [35]. Ford et al. were among the first to profile epigenetic changes in TILs in response to hypoxia. They found that terminally exhausted T-cells have distinct chromatin structures that limits their transcriptional ability, and these alterations to chromatin accessibility were driven by altered histone modifications linked to hypoxia [36]. They also compared the transcriptomes of CD8+ T-cells under hypoxic and normoxic conditions and identified several immune-related genes that were downregulated in hypoxic conditions [36]. Reducing hypoxia in tumors restored the expression of many of the genes normally suppressed in terminally exhausted T-cells, which led to the conclusion that the hypoxic environment of tumors drives epigenetic changes that underlie T-cell exhaustion [36]. These results are intriguing as they suggest potential approaches to reverse T-cell exhaustion, and it is evident that more work is needed to understand the multifaceted impacts that hypoxia has on promoting T-cell exhaustion.
Nutrient deprivation contributes to exhausted T-cells
In addition to hypoxia, nutrient deprivation is common in many solid cancers and has been documented in gliomas [37,38]. Poor infiltration of capillaries into the tumor coupled with high demand for nutrients by rapidly growing cancer cells restricts the body’s ability to supply needed nutrients to normal tissue (Figure 2). Carbohydrates and amino acids are often depleted in the glioma microenvironment, resulting in impaired immune cell function. Glucose availability is critical for proper function, proliferation and growth of T-cells. It has been shown that brain tumor cells can outcompete surrounding normal cells for available glucose by upregulating and co-opting neural glucose transporters [39]. This competition, coupled with decreased vascularization of the tumor core, depletes free glucose available to infiltrating immune cells. The lack of glucose is detrimental to T-cells and might be contributing to the exhausted phenotype by forcing T-cells to reprogram their metabolism toward the use of alternative pathways such as one-carbon metabolism, OXPHOS, or to undergo authophagy which allows them to survive but at the cost of altering their function (Figure 2) [37,38,40]. While T-cells upregulate their transport of glucose via Glut1 potentially to overcome this starvation, it is unknown whether this is sufficient to provide enough flux through glycolysis in order to support proper effector function.
Hypoxia has been shown to induce Glut1 upregulation and this response is hypothesized to indicate dependency on glycolysis in exhausted T-cell subsets [17]. Nonetheless, this has yet to be fully studied in glioma models and in microenvironments experiencing glucose restriction, yet it may indicate an additional way by which exhausted T-cells are more susceptible to the detrimental effects of impaired glucose availability.
In murine sarcoma models, limited glucose availability was shown to reduce T-cell antitumor immunity through metabolic restriction, which resulted in decreased mammalian target of rapamycin (mTOR) activity, glycolysis, and cytokine production [41]. In B cell leukemia, impaired glycolysis and mTOR activity has been linked to T-cell exhaustion and expression of exhaustion-related receptors PD-1 and TIM-3 [42]. This situation, however, is complicated, as persistent mTOR signaling is also implicated in metabolic suppression, as it has been shown to drive mitochondrial dysfunction in chronic viral infection [43]. Exhaustion was reversed by PD1 blockade or mTOR inhibition; these findings may indicate that abnormal mTOR signaling, whether high or low, is a driver of exhaustion. mTOR dysregulation may occur in T-cells infiltrating the glioma TME as a result of diminished glucose metabolism due to tumor-specific glucose deprivation [41,42,44]. Lastly, glucose deprivation under hypoxic conditions also results in HIF-1α-mediated expression of LAG3, an immune checkpoint protein often associated with exhaustion [45].
The amino acids arginine and tryptophan are also commonly depleted in the TME due to elevated expression of arginase and indoleamine 2,3 dioxygenase 1 (IDO1) in cancer cells and other immune populations, such as myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) [46,47]. IDO1 becomes aberrantly expressed upon increased signaling in neuro-inflammatory pathways, and its upregulation results in tryptophan depletion [48]. IDO1 was found to be expressed at higher than normal levels in roughly 96% of malignant gliomas, and its expression level is negatively correlated with overall patient survival [48]. Direct ties between IDO1 and exhaustion remain poorly elucidated, but both tryptophan and arginine deprivation results in metabolic impairments that drive T-cell dysfunction[47]. These metabolic impairments may contribute to driving an exhausted T-cell phenotype in glioma, but more research is needed to evaluate these mechanisms.
T cell metabolic reprogramming and mitochondrial fitness impact exhaustion
Nutrient deprivation and hypoxia alter mitochondrial fitness, which is an important determinant of overall T-cell metabolic function (Figure 1) [49]. Damaged mitochondria are typically cleared through mitophagy, which is mediated by hypoxia, nutrient availability, and activation of the phosphatase and tensin homolog (PTEN)-induced putative kinase1 (PINK1)-Parkin pathway [50–52]. In murine melanoma models, TILs have been shown to have reduced mitophagic activity, which leads to the accumulation of dysfunctional mitochondria and decreased metabolic fitness [49]. Such mitochondrial dysregulation drives T-cell exhaustion in TIL populations [49]. T-cell exhaustion is enhanced by microenvironmental stressors, such as hypoxia and glucose deprivation, and when these extrinsic factors were alleviated, mitochondrial fitness was restored with subsequent reduction of exhaustion [49].
TILs in the TME also display impaired oxidative phosphorylation as a result of chronic antigen stimulation [53]. Impaired oxidative phosphorylation suppresses TILs’ proliferative capacity and upregulates exhaustion-linked genes, including PD-1 and CTLA-4 [53]. Schurich et al. documented these mechanisms in viral models, showing that exhausted CD8+ T-cells and impaired mitochondrial dynamics occurred simultaneously [17]. Mitochondrial impairments were associated with T-cells that were more dependent on glucose availability and could not switch to oxidative phosphorylation during periods of glucose depletion, limiting their functional capacity. Vardhana et al. showed that mitochondrial dysfunction in TILs resulted in a heightened nuclear factor of activated T-cell (NFAT) activity as a result of increased intracellular calcium and mitochondrial ROS production, which drove transcriptional changes that promote exhaustion-associated gene expression [53]. The importance of metabolic fitness in relation to Tcell exhaustion was underscored by Bengsch et al., who showed that PD-1 signaling regulated early glycolytic and mitochondrial alterations [43]. Oxidative phosphorylation and glycolysis were suppressed in early exhausted T-cell populations exposed to chronic infection [43]. These findings provide a link by which exhaustion may reduce metabolic fitness and thus cause further dysfunction and suppression of antitumor immunity. This link might be pertinent to glioma TILs experiencing metabolic dysregulation and subsequent exhaustion.
The impact of metabolic by-products and altered cancer metabolism
Hypoxia, nutrient starvation, and the resulting altered cancer cell metabolism promote the production of metabolites that in normal tissue would be removed for excretion but in the TME build up and damage tissue (Figure 2). Hypoxia- and nutrient starvation-induced metabolic shifts in cancer cells result in energy produced through glycolysis instead of the typical citric acid cycle and oxidative phosphorylation [54]. Importantly, this leads to increased production of lactate, which has been shown to promote tumor cell growth, migration, and angiogenesis and to impair the immune response [55].
Lactate exported from glioma cells becomes abundant in the microenvironment of gliomas and glioblastomas [56,57] where it confers immunosuppressive effects. It has been hypothesized that lactate disrupts T-cell functionality by lowering the pH of the TME [58,59]. Lactate accumulation in the TME might also result in end-point inhibition of glycolysis in T-cells, which is exacerbated in low-glucose environments like the TME and drives metabolic stress and decreased effector functionality [58]. Fischer et al. showed that high lactic acid levels in the TME suppressed CD8+ T cell proliferation and cytokine production, impacting T-cell function. They speculated that the high TME lactic acid levels hinder its export from T-cells resulting in subsequent disruption to T-cell metabolism [60]. Whether such immunosuppression occurs via exhaustion pathways and whether it is limited to specific conditions in the TME (such as hypoxia and glucose deprivation) remains unclear. More research is needed to determine whether excess lactic acid is a driver of T-cell exhaustion in gliomas.
D-2-hydroxyglutarate (D-2HG) is an oncometabolite very abundant in glioma microenvironment specifically in tumors with an isocitrate dehydrogenase (IDH) mutation. D-2HG accumulates in gliomas in mM quantities, as a result of mutant IDH1 enzyme, which produce it as a neomorphic activity from α-ketoglutaric acid. IDH mutations now define grade 2 and 3 gliomas and there is a distinct grade 4 IDH-mutated glioma recognized by the 2021 WHO guideline [61]. Importantly the single-amino-acid substitutions that result from the IDH mutation influence glioma development and metabolism [62–64]. In gliomas, D-2HG suppresses antitumor T-cell immunity by disrupting calcium influx, which alters the regulation of T-cell effector function genes [65]. Interestingly, PD-1 levels were reported lower in IDH-mutant gliomas compared to wildtype [65], which suggests that immunosuppression by D-2HG may occur beyond the canonical PD-1-mediated exhaustion pathway. One potential mechanistic link between D-2HG and T-cell exhaustion was reported very recently in a study by Notarangelo et al [66]. The authors showed that D-2HG acts as a direct inhibitor of lactate dehydrogenase alters the glucose metabolism of T-cells, inhibits T-cell proliferation, and their downstream functionality as defined by cytokine production and killing ability. More research is needed to evaluate the role of D-2HG in altering T-cell functionality and inducing exhaustion.
The presence of high levels of cholesterol in the TME has also been correlated with CD8+ T-cell expression of exhaustion-associated receptors PD-1, TIM-3, and LAG-3. Adoptively transferred T-cells acquired increased expression of these receptors and an exhaustion phenotype after exposure to a high-cholesterol TME [67]. Cholesterol reduces the efficacy of lipid metabolism pathways and upregulates stress-response genes, including XBP1, in the endoplasmic reticulum (ER), which has been shown to induce T-cell inhibitory receptor expression, thus delineating a mechanistic link between cholesterol and induction of T-cell dysfunction [67]. Cholesterol is abundant in the TME of the brain, and its availability and metabolism have been linked to glioma and glioblastoma progression [68,69]. Thus, the finding that cholesterol might drive T-cell dysfunction through lipid metabolism and ER-stress pathways may be important for understanding how TILs develop exhaustion in gliomas and glioblastomas.
The impact of other immune cells and cytokines on T-cell exhaustion
The presence of immune cells in the TME helps shape the glioma environment and immune function. Two abundant immune cell subsets that have immunosuppressive effects in the TME of gliomas are tumor-associated macrophages (TAMs) and T-regulatory cells (Tregs) (Figure 2) [70,71]. TAMs have been found to exist in a co-dependent cycle with exhausted CD8+ T-cells [72]. Studies using melanoma models found that, unlike other T-cell subsets, exhausted T-cells contribute to the active recruitment and maturation of macrophages by secreting stimulatory factors, such as colony-stimulating factor 1 (CSF1) [72]. TAMs engage the T-cell receptor (TCR) of CD8+ T cells and increase expression of PD-1 and TOX, which results in failure to produce effector cytokines and exhaustion [72]. Notably, these results were more pronounced in hypoxic environments [72]. Transcriptional and epigenetic profiling indicates that when TAMs are depleted in the TME, exhaustion pathways in TILs are reduced, which allows their effector potential to be restored [72]. Some studies indicated that tumor-associated Tregs induce immunosuppression and T-cell senescence through pathways distinct from exhaustion, as Tregs did not induce significant expression of canonical exhaustion markers on CD4+ and CD8+ T cells [73]. Other studies, however, found that intratumoral Tregs impact exhaustion much more than their peripheral subsets [74]. In non-small cell lung cancer models, Tregs expressing interleukin (IL)-35 and IL-10 directly upregulated inhibitory receptor expression on CD8+ T cells by modulating the Prdm1/BLIMP1 axis [74].
Recently, MDSCs were identified as an additional immune subset capable of driving T-cell exhaustion in chronic infection and cancer. MDSCs highly express Gal-9 and PD-L1, ligands of the inhibitory receptors TIM-3 and PD-1, respectively, and, through these axes, induce T-cell exhaustion [75–77]. Pretreatment of MDSCs with NF-κB inhibitors suppressed the TIM-3/Gal-9 axis and impaired functionality, which diminished the severity of exhaustion in exposed T-cells, restoring proliferation and effector cytokine production [76,77]. The pervasiveness of MDSCs, macrophages, and other myeloid populations has been documented in the glioma microenvironment, both in human patient samples and in genetically engineered murine models [78–80]. In gliomas, these immune cells are implicated in driving immunosuppression, but more research is needed to confirm their involvement in exhaustion and to elucidate their impact on glioma microenvironments.
In addition to infiltrative, suppressive immune cells, cytokines released in the glioma microenvironment likely regulate T-cell exhaustion. Two cytokines that negatively impact T-cell function and have been associated with exhaustion are interleukin-10 (IL-10) and transforming growth factor-β (TGF-β). IL-10 is a pleotropic, anti-inflammatory cytokine that primarily inhibits activation and effector functionality in T-cells [81–84]. Blockade of the IL-10/IL-10R axis prevented functional exhaustion in T-cell subsets and restored effector T-cell activity, as shown by an increase in proliferative and cytokine-producing capabilities [82–84]. Ravi et al. recently identified a correlation between IL-10 and tumor-associated T-cell exhaustion among patients with glioblastomas [85]. RNA-sequencing after IL-10 exposure confirmed this connection, indicating that IL-10 mediated downstream suppressive T-cell pathways and increased expression of exhaustive pathways via the IL-10-STAT3-BLIMP-1 signaling axis, confirming a previous study that found that IL-10 secreted from Treg populations in the TME promoted BLIMP1-mediated exhaustion [74,85].
IL-10 involvement in the T-cell effector function is complex. In contrast to its immunosuppressive effect, IL-10 also helps promote anti-tumor immune responses and maintains CD8+ T-cell effector function in murine models of melanoma and adenocarcinoma [86,87]. Guo et al. documented that stimulation of IL-10-associated pathways metabolically reprogrammed terminally exhausted CD8+ T-cells and promoted oxidative phosphorylation [43,53,87]. The upregulation of oxidative phosphorylation revitalized terminally exhausted T-cells, improved expansion and effector function abilities, and enhanced anti-tumor immunity [87]. In light of earlier work indicating that the IL-10/IL-10R axis promotes T-cell exhaustion, these findings suggest the complex nature of IL-10 roles in exhaustion and variability depending on the context [82]. These studies further document the need for additional research of exhaustion and its many drivers in gliomas and may be clinically important, especially when coupled with other treatments aimed at restoring or improving anti-tumor immune function.
TGF-β, a multifunctional cytokine involved in cell proliferation and differentiation, has been implicated in promoting T-cell exhaustion. Investigations in murine models showed that TGF-β stimulation in precursors to exhausted T cells inhibited mTOR signaling and strongly suppressed cytokine production, thereby promoting exhaustion, metabolic and mitochondrial dysregulation, and reduction in immune response [88]. Attenuation of TGF-β signaling reduced sustained PD-1 expression in T-cells of chronically infected mice models [89]. In patients with B-cell non-Hodgkin lymphoma, treatment with TGF-β resulted in upregulation of exhaustion markers PD-1 and TIM-3 as well as CD70, a co-stimulatory molecule, in intratumoral T-cells [90]. These CD70+ T cells had reduced proliferative capacity and diminished ability to produce IFN-γ, implying that these intratumoral cells became exhausted in response to TGF-β [90]. In gliomas, TGF-β is found in the microenvironment and is implicated in tumor growth and immunosuppression [91,92]. Systemic inhibition of TGF-β in murine models of glioma improved peptide vaccine efficacy and is a focus of therapeutic intervention in human cancer [91,93,94]. More research is needed to elucidate the complex, pleiotropic effect of this cytokine and the potential for these treatment approaches to benefit patients with gliomas.
Understanding the epigenetic and transcriptomic status of exhausted T cells in the glioma microenvironment
Metabolic alterations and microenvironmental factors both have implications in the epigenetic and transcriptional adaptations that cells experience as they progress to exhaustion (Box 2). Specific gene expression patterns–or lack thereof–have been linked to exhaustion (Figure 3) highlighting the importance of understanding the underlying alterations to the epigenome. Difficulty in reinvigorating exhausted T-cells by immune checkpoint blockade or through other therapeutic avenues has also prompted interest in the epigenetic landscape of exhausted T-cells, as epigenetic stability may be a factor limiting exhaustion reversal.
Box 2. Crosstalk between the Metabolome and Epigenome.
Epigenetics is the study of mechanisms by which the intra- and extracellular environments modulate and alter gene activity and expression without changing the nucleotide sequence of genomic DNA. Examples of these alterations include modifications to histone-associated proteins and DNA through acetylation, lactylation, and methylation statuses, which can impact gene expression. Control over the epigenome of cells is a tightly regulated activity in normal cells, as dysregulation and resulting variations in gene expression can result in functional impairments, oncogenesis, or cell death. Products of cellular metabolism are key components of epigenetic regulation (e.g., acetyl-CoA produced during catabolism of acetate, citrate, or pyruvate participates in histone acetylation, D-2hydroxyglutarate produced by IDH1 mutation), and, conversely, control of metabolism is governed by the distinct expression of gene patterns [47,156–159]. Studies of crosstalk between mitochondria, gene alterations, and nuclear reprogramming have found linkages between the metabolomic and epigenetic landscapes of cells [47,156,160–162]. Understanding the role of the epigenome in T-cell exhaustion induction and enhancement will be crucial to identifying mechanisms to reverse it.
Figure 3.

Epigenetic and transcriptional alterations in T-cell exhaustion. Alterations in extrinsic factors and metabolic dysfunction results in disturbed epigenetic profiles among exhausted T cells, which are distinct and have limited reversibility. (a) Impairments to histone acetyltransferase (HAT) and the NAD+-mediated deacetylation axis disrupt histone acetylation and are linked to exhaustion. Mitochondrial dysfunction and subsequent alterations to SAM availability impact histone and DNA methylation patterns to further dysfunction within exhausted T-cells. D-2HG, the product of IDH1 mutant inhibits JMJD3 and TET enzymes leading to a hypermethylated phenotype, and this could induce an exhausted T-cell. (b) NFAT induces the expression of TOX, which results in further epigenetic and transcriptional profiles associated with exhausted phenotypes. These epigenetic and transcriptomic changes result in key traits of T-cell exhaustion: the upregulation of inhibitory receptors (e.g. PD-1, CTLA-4, Tim-3, TIGIT, and Lag-3), decreased production of effector molecules (e.g. IL-2, TNF-α, IFN-γ, and Granzyme B), impaired proliferation, and, ultimately, anti-tumor immune response.
The epigenetic and transcriptomic profiles of exhaustion
Exhausted T-cells have been associated with the expression of specific transcription factors, including NFAT, TOX, NR4A, T-cell factor-1 (TCF1), Eomes, T-bet, Basic leucine zipper transcription factor ATF-like (BATF), and Blimp-1. Open chromatin regions and specific methylation patterns have also been identified for exhausted T-cell populations [95,96] (Figure 3). Notably, chromatin-accessible regions in exhausted murine T-cell populations were identified near the Pdcd1 (PD-1) gene locus, in areas that correspond with enhancer activity [97]. Substantial remodeling of enhancer and transcription factor binding regions has been documented in exhausted T-cells that were not seen in effector T-cells, thereby providing support for the idea that epigenetic and transcriptomic alterations might drive exhaustion and limit reinvigoration of T-cells [96,97]. Belk et al. discovered that chromatin-remodeling proteins, such as INO80 and switch/sucrose non-fermentable (SWI/SNF) chromatin remodeling complex, can regulate exhaustion [98]. Depletion of SWI/SNF protein complexes and INO80 chromatin-remodeling complex reduced the acquisition of exhaustion-associated chromatin remodeling and improved anti-tumor effector capabilities of T-cells, which increased the survival of tumor-bearing mice [98].
Beltra et al. identified four distinct T-cell subsets as they worked to define the epigenetic and transcriptional profiles of exhausted populations [8]. Terminally exhausted T-cells had upregulated expression of inhibitory receptors (Pdcd1, Lag3, Tigit, Cd244) and other genes associated with terminal exhaustion (Entpd1, Cd101, Cd38)[8]. Terminal populations were also enriched for negative regulatory pathways and displayed signs of TCR activation [8]. The translation of these findings to gliomas has not yet been done, but it is likely that similar patterns of altered chromatin-accessible regions exist and are important to identify.
The importance of TOX in driving exhaustion
TOX plays a necessary role in T-cell development and has recently gained attention for its role in driving T-cell exhaustion. TOX expression is driven by NFAT transcription factors following T-cell activation. Sustained expression of TOX was found to drive exhaustion, which was not induced in the absence of TOX [95,99,100]. In TOX-deficient cells, expression of genes associated with exhaustion, such as Pdcd1 and Cd160, and of exhaustion-associated transcription factors, TCF-1, T-bet, and Eomes, was reduced [99]. T cells with a low TOX/T-bet ratio were found in intermediate T-cell exhaustion subsets, but when the ratio of TOX/T-bet was increased to favor TOX, T cells were driven to terminally exhaustive states [8]
A transcriptional meta-analysis of glioma samples from The Cancer Genome Atlas (TCGA) and the Chinese Glioma Genome Atlas showed that TOX was enriched in low-grade and IDH-mutant gliomas and was highly expressed at the leading edge of tumors, indicating the potential for its involvement in oncogenesis, possibly via immunosuppression and promotion of exhaustion [101]. It was also reported that high expression of TOX in glioma cells was associated with less infiltration of immune cells, beyond solely T-cells, into the tumor microenvironment [101]. It has been suggested that TOX could be an important target for immunotherapy in hematological malignancies, and it has been shown for bladder cancer that immune checkpoint blockade can reinvigorate TOX-expressing exhausted TILs [102,103]. Thus, in glioma TILs it may be important to examine the role of TOX and whether it drives exhaustion, as this may have clinical relevancy as a prospective therapeutic target.
The involvement of histone acetylation and deacetylation in exhaustion
Access to DNA and genetic information is regulated in part by histone acetylation and deacetylation. Acetyl-CoA, which is generated during the tricarboxylic acid cycle, serves as a primary source for histone acetylation by histone acetyltransferases (HATs) (Figure 3). Direct connections between glucose availability, glycolysis levels, histone acetylation, and acetyl-CoA have been documented [104–107]. Thus, impaired glycolysis that results from nutrient restriction, inhibitory receptor expression, or other factors specific to TILs in the TME might alter histone acetylation in T-cells by reducing adequate acetyl-CoA levels. Additionally, increasing the concentration of acetate, a key compound in acetyl-CoA synthesis, in vitro was shown to restore histone acetylation, chromatin accessibility, and T-cell effector functions in T-cell populations under chronic glucose restriction, further supporting the roles of acetyl-CoA and metabolic influences in driving epigenetic regulation of T-cell effector functions [108]. The role of histone acetylation in promoting exhaustion is complex and likely specific to the environmental and phenotypic conditions of T-cells, yet is crucial to study as a potential mechanism to reinvigorate exhausted T-cells.
Alterations in nicotinamide adenine dinucleotide (NAD) levels are also implicated in histone modification and lead to downstream deacetylation that reduces effector function (Figure 3). Increased NAD levels and NAD/NADH ratios were shown to increase sirtuin (SIRT) activity, resulting in increased histone deacetylation and upregulated expression of T-bet and its target genes, which have been linked with T-cell exhaustion [47,109]. Production of IFN-γ, a key cytokine in T-cell immune responses, is regulated by SIRT1-mediated H3 lysine 9 acetylation status [47,110]. Therefore, alterations in cellular NAD levels and subsequent SIRT activity may interrupt expression of IFN-γ, a potential mechanism for limiting the effector function of exhausted T-cells [47].
NAD depletion in activated T lymphocytes resulted in impaired proliferation and decreased cytokine production, two key characteristics of T-cell exhaustion [111]. Whether this occurs in glioma remains unclear, although there is evidence that IDH-mutant cancers are vulnerable to NAD depletion through downregulation of the NAD salvage pathway enzyme nicotinate phosphoribosyl transferase (Naprt1)[63,64,112]. In murine glioblastoma models, NAD depletion was shown to upregulate PD-L1, a key molecule implicated in the T-cell exhaustion [113]. More insight into the diversity and impact of NAD availability on T-cell exhaustion in gliomas is needed, as NAD is likely impacted by metabolic regulations that take place and may play an important role in disruptive epigenetic changes.
The role of DNA and histone methylation in exhaustion
DNA methylation patterns are essential for regulating tissue-specific gene expression, recruiting and blocking transcription factors, and imposing genomic imprints. S-adenyl methionine (SAM) is the source of methyl groups in both histone and DNA methylation and is produced via one-carbon metabolism, which has been shown to play important roles in regulating the proliferation of immune cells [104]. Since SAM is produced in mitochondria, its availability may be altered by mitochondrial dysfunction in TILs. Therefore, it will be crucial to investigate the link between mitochondrial dysfunction, SAM levels, and induction of T-cell exhaustion by epigenetic regulation (Figure 3). Demethylation of histones and DNA is controlled by the demethylases Jmjd3 and ten-eleven translocation (TET) protein, which utilize α-ketoglutaric acid (α-KG) as a substrate. Suzuki et al. found that the glutamine–α-KG axis induces CD8 T-cell dysfunction by demethylating a specific histone residue, H3K27, and activating mTORC1 signaling, indicating that α-KG’s impact on the epigenome may be critical for understanding T-cell exhaustion [47,111,114].
Molecular analyses of both tumor-infiltrating CD4+ T cells and peripheral CD4+ T cells from patients with glioblastoma showed distinct changes in their methylation status and transcription profile, confirming the influence that the TME has on shaping the epigenome and transcriptome[115]. Connections to exhaustion were not specifically examined, but, in TILs, there was distinct dysfunction of several genes and ligands related to T-cell function, which further indicated the influence of the glioblastoma on immunosuppression [115]. More directed research is needed to elucidate the mechanisms by which gliomas and glioblastomas affect specific genomic and epigenomic changes in TILs, which is important for reversing exhausted T-cell populations and reinvigorating anti-tumor immunity.
Ability to reverse exhaustion and future directions
Reversal of T-cell exhaustion and enhancement of anti-tumor immunity is likely a critical component of efficacy for immunotherapies for malignant gliomas. We reviewed, from the perspective of immunosuppression, the factors in the microenvironment of glioma that promote tumor growth to potentially identify therapies to ameliorate T-cell exhaustion (Figure 4). Factors such as immunosuppressive cytokines, specific immune cells, upregulation of inhibitory receptors, chronic antigen stimulation, and the difficult nature of the hypoxic, nutrient-deficient tumor microenvironment all drive an exhausted T-cell phenotype, which results in decreased anti-tumor immunity and subsequent tumor growth (Figure 4). Tumor growth in gliomas is also promoted in large part by angiogenesis and immune evasion (Figure 4) [116–120]. Neutralizing the factors that drive tumor growth is critical to tip the scale toward tumor suppression, that can occur by reinvigorating T-cell function and prompting an anti-tumor immune response. Increased availability or recognition of tumor antigens may decrease the likelihood of tumor immune evasion and prompt tumor suppression (Figure 4) [118]. Additional mechanisms that drive sustainable, effective immune responses, such as co-stimulatory molecules, increased involvement of T-cells, and M1-like macrophages, as well as abrogating immunosuppressive activity in the TME by disrupting inhibitory cytokines, angiogenesis, and hypoxic and nutrient-insufficient conditions require further investigation, both individually and in combination (Figure 4) [116,120–123].
Figure 4.

Potential restoration avenues to combat T-cell exhaustion. The balance between exhaustion and functionality in T cells is driven by a complex set of factors, which results in either anti-tumor immunity or tumor growth. Factors that tip T cells towards an exhaustive phenotype include, but are not limited to, expression of negative immune checkpoints, chronic antigen stimulation, resulting epigenetic remodeling and transcriptional changes, hypoxia, metabolic dysfunction, and nutrient deprivation. These factors can be exploited to tip the scale back towards restoration and T cell functionality through approaches including immune checkpoint blockade, normoxia, nutrient restoration, blocking immunosuppressive cytokines, and metabolic reprogramming. Additionally, anti-angiogenetic agents and increasing the abundance of IFN-γ-secreting T cells and M1-like macrophages can promote T cell functionality and tumor suppression. Metabolic modulators, immune checkpoint stimulators and inhibitors, and combinatory therapies (e.g. anti-PD-1 immunotherapy alongside other treatment modalities) can be used to extrinsically induce these factors that promote T cell restoration. (Abbreviation List: IDH1, Isocitrate dehydrogenase 1; IDO, Indoleamine-2,3-dioxygenase; ROS, reactive oxygen species; PGC-1α, peroxisome proliferator-activated receptor-gamma coactivator; A2aR, adenosine A2a receptor; PD-1, programmed cell death protein-1; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; Tim-3, T-cell immunoglobulin and mucin domain-3; LAG-3, lymphocyte activation gene 3 protein; IL-10, interleukin-10; IL-2, interleukin-2; VEGF, vascular endothelial growth factor; TAMs, tumor-associated macrophages; Treg, T-regulatory cells; TGF-β, transforming growth factor-β; IFNg, interferon-γ)
Current strategies to restore exhausted T-cell function have had varied success. Terminally exhausted T-cell populations are characterized by the acquisition of irreversible epigenetic and transcriptional changes, driven heavily by elevated TOX expression and its influence on T-bet [8]. Beltra et al. proposed that this subset is incapable of being restored to full effector functionality and must be replaced by new non-exhausted T-cells to restore anti-tumor immunity.
One way to improve the T-cell anti-tumor immunity is using immune checkpoint blockade, which inhibits checkpoint proteins such as PD-1 and CTLA-4 and prolongs T cell activity. The FDA has approved checkpoint inhibitors that target CTLA-4, PD-1, and PDL-1; others targeting the recently identified checkpoint proteins LAG-3 and TIM-3 are being developed [120,124] (Figure 4, Table 1). Immune checkpoint inhibitors have had limited success in gliomas, and their inability to reverse exhaustion is still unknown. In murine models of chronic infection, PD-1 blockade reactivated some functions of exhausted T-cells, but analysis of accompanying cellular, transcriptional, and epigenetic changes showed scant large-scale remodeling[125]. The epigenetic landscape of the exhausted T-cell subset remained relatively stable throughout PD-1 blockade, and when high antigen exposure was sustained, reinvigorated cells became re-exhausted [125]. Chromatin restructuring that occurs during progression to exhaustion becomes fixed, and epigenetic ‘scars’ are sustained near genes associated with exhaustion, indicating that approaches to reversing exhaustion cannot rely solely on inducing effector function but must also increase epigenetic plasticity [98,126]. Blockade of CTLA-4 has also shown little efficacy within gliomas, however, clinical trials testing checkpoint blockade combinations are currently underway (see Outstanding questions, Figure 4, Table 1). The highly immunosuppressive nature of gliomas is thought to limit the efficacy of immune checkpoint blockade through mechanisms that promote exhaustion and suppress anti-tumor immunity, but the stability of exhausted T-cells also explains their resistance to restoration of function. Other novel methods of immunotherapy include dendritic cell vaccination and CAR T-cell therapy, both of which have proven successful in other cancer types and are being tested in CNS cancer-focused clinical trials (Box 3, see Outstanding questions, Table 1) [120,127,128].
Table 1.
Ongoing clinical trials investigating glioma immunologya
| NCT NUMBER | PHASE | TARGET | NAME OF TRIAL | STATUS | PATIENT ENROLLMENT |
|---|---|---|---|---|---|
| NCT02649582 | I/II | DC-vaccine + Chemotherapy | Adjuvant Dendritic Cell-immunotherapy Plus Temozolomide in Glioblastoma Patients | Recruiting | 20 |
| NCT02465268 | II | DC-vaccine (pp65 DC) + Chemotherapy | Vaccine Therapy for the Treatment of Newly Diagnosed Glioblastoma Multiforme | Recruiting | 175 |
| NCT02208362 | I | Genetically modified T-cell immunotherapy | Genetically Modified T-cells in Treating Patients With Recurrent or Refractory Malignant Glioma | Active, not recruiting | 82 |
| NCT03347097 | I | Genetically modified T-cell immunotherapy | Adoptive Cell Therapy of Autologous TIL and PD1-TIL Cells for Patients With Glioblastoma Multiforme | Active, not recruiting | 40 |
| NCT03696030 | I | HER2-CAR T Cell Therapy | HER2-CAR T Cells in Treating Patients With Recurrent Brain or Leptomeningeal Metastases | Recruiting | 39 |
| NCT02924038 | I | Peptide Vaccination + CD27 MoAb (IMA950, poly-ICLC + Valilumab) | A Study of Varlilumab and IMA950 Vaccine Plus Poly-ICLC in Patients With WHO Grade II Low-Grade Glioma (LGG) | Active, not recruiting | 14 |
| NCT04943718 | I | Peptide Vaccination | Personalized Vaccine for Patients With Recurrent Malignant Glioma | Recruiting | 10 |
| NCT03779230 | I/II | Immunocytokine (L19TNF) | Safety and Efficacy of L19TNF in Patients With Isocitrate Dehydrogenase (IDH) Wildtype WHO Grade III / IV Glioma at First Relapse (GLIOMOON) | Active, not recruiting | 20 |
| NCT04729959 | II | IL-6 + PD-L1 Blockade (Tocilizumab, Atezolizumab) + Radiatior | Testing the Addition of the Immune Therapy Drugs, Tocilizumab and Atezolizumab, to Radiation Therapy for Recurrent Glioblastoma | Suspended (Scheduled Interim Monitoring) | 12 |
| NCT02337686 | II | Anti-PD-1 (Pembrolizumab) | Pembrolizumab in Treating Patients With Recurrent Glioblastoma | Active, not recruiting | 20 |
| NCT04145115 | II | Anti-PD-1 + Anti-CTLA-4 (Nivolumab, Ipilimumab) | A Study Testing the Effect of Immunotherapy (Ipilimumab and Nivolumab) in Patients With Recurrent Glioma With Elevated Mutational Burden | Recruiting | 37 |
| NCT04817254 | II | Anti-PD-1, Anti-CTLA-4 + Chemotherapy (Nivolumab, Ipilimumab + Temozolomide) | Association of Peripheral Blood Immunologic Response to Therapeutic Response to Adjuvant Treatment With Immune Checkpoint Inhibition (ICI) in Patients With Newly Diagnosed Glioblastoma or Gliosarcoma | Recruiting | 48 |
| NCT04606316 | I | Anti-PD-1, Anti-CTLA-4 (Nivolumab, Ipilimumab) + Surgery | Surgical Nivolumab And Ipilimumab For Recurrent GBM | Recruiting | 60 |
| NCT03493932 | I | Anti-PD-1 + Anti-LAG-3 (Nivolumab, BMS-986016) | Cytokine Microdialysis for Real-Time Immune Monitoring in Glioblastoma Patients Undergoing Checkpoint Blockade | Active, not recruiting | 20 |
| NCT03576612 | I | Anti-PD-1 + Immunostimulator (Nivolumab, GMCI) + Radiation + Chemotherapy | GMCI, Nivolumab, and Radiation Therapy in Treating Patients With Newly Diagnosed High-Grade Gliomas (GMCI) | Active, not recruiting | 36 |
| NCT02022384 | IV | Chemoradiation | Immunophenotyping From Blood of Patients With Malignant Gliomas | Active, not recruiting | 50 |
| NCT04065776 | Interventional | Radiation (Hippocampal-avoidance proton therapy) | Evaluation of Hippocampal-Avoidance Using Proton Therapy in Low-Grade Glioma | Recruiting | 74 |
| NCT04781764 | IV | Surgical/Tumor Biopsy | The Study of Microglia/Macrophages Involved Dynamic Evolution of Glioma Microenvironment and the Function and Visualization of Targeted Molecules of Glioma | Recruiting | 60 |
| NCT03189420 | IV | Surgical/Tumor Biopsy | Glioma Microenvironment an Exploratory Study | Active, not recruiting | 30 |
| NCT04461938 | Interventional | Surgical/Tumor Biopsy | Characterization of Metabolic Changes in the Glioma Tumor Tissue Induced by Transient Fasting (ERGO3) | Recruiting | 30 |
| NCT04792437 | IV | Surgical/Tumor Biopsy | Research on Precise Immune Prevention and Treatment of Glioma Based on Multi-omics Sequencing Data | Recruiting | 120 |
| NCT04865315 | IV | Surgical/Tumor Biopsy | A Living Tissue Bank of Patient-Derived Organoids From Glioma Tumors (HiLoGlio) | Active, not recruiting | 50 |
| NCT01246869 | I | MRI and PET Scan Visualization (18F-FDG, 18F-FMISO, H2O) | Assessment of Primary and Metastatic Brain Tumor Hypoxia With Fluoromisonidazole, FDG and Water | Recruiting | 30 |
| NCT05047913 | I | MRI and PET Scan Visualization (18F-FTX) | Comprehensive Evaluation of Tumor Oxygenation, Metabolism and Blood Supply of High Grade Glioma and Cervical Cancers Using Dynamic FAZA PET and Multiparametric MRI | Recruiting | 20 |
| NCT04309552 | I | PET Scan Visualization (18F-FMISO, 18F-FLT PET) | Tumor Hypoxia and Proliferation in Patients With High-Grade Glioma | Recruiting | 30 |
All trials were found on clinicaltrials.gov and were active at time of publication. Trial phases range from I – IV.
Outstanding Questions:
How do current standards of care for CNS cancers impact T cell exhaustion, either synergistically or distinctly from the impact of the cancer microenvironment itself?
As our insight into the immune response to glioma improves, what additional metabolic, epigenetic, and transcriptional alterations are required to drive an exhausted phenotype of T cells? How will these factors interplay with one another?
At what point in the progression of exhaustion do T cells become terminally exhausted and incapable of having functionality restored in gliomas?
How does exhaustion of T cells in gliomas impact the ability of novel therapeutic approaches, such as CAR T cell therapy and dendritic cell vaccination, that are becoming prominent in glioma treatment?
How can combinatorial, multifaceted treatment modalities improve the reversal of T cell exhaustion and enhance anti-tumor immunity?
How can the glioma microenvironment be directly targeted to reduce its immunosuppressive nature and ‘tilt the scales’ in favor of anti-tumor responses rather than cancer progression?
Box 3. The Effect of CAR T Therapy on T-cell Exhaustion.
Following success in hematologic cancers, the use of T cells redirected with Chimeric Antigen Receptors (CAR-T) has gained popularity in malignant brain tumors. Such therapies are still being developed, and few preliminary results from the clinic are available. Many CAR T cell therapies are directed towards epidermal growth factor receptor variant III (EGFRvIII), a gene fusion observed in solid tumors, including gliomas. Early clinical data suggest these CAR T-EGFRvIII cells are both limited by the immunosuppressive microenvironment of gliomas and further induce immunosuppression by prompting upregulation of inhibitory molecules within the TME (e.g. PD-L1, IDO1, and TGF-β) [163,164]. Interestingly, it has been documented that CAR T cells acquire characteristics of exhaustion themselves, including upregulation of inhibitory receptors and transcriptional alterations associated with exhaustion, and it is likely this induction of exhaustion occurs via similar mechanisms as in other T cell subsets [165–171]. In addition to CAR T cell exhaustion, CAR T cell therapy drives further exhaustion of circulating patient T cells, both of which may limit the efficacy of such treatment. Genetic modifications to knock out known exhaustion-related genes among CAR T cells, as well as combining CAR T cell therapies with other forms of immunotherapy, are important avenues to pursue in the future. More research on both T-cell exhaustion and CAR T cell therapies’ involvement in gliomas will be necessary to advance our care of CNS malignancies.
Additional ways to reverse T-cell exhaustion include radioimmunotherapy and chemoimmunotherapy, as well as drugs that target specific drivers of exhaustion, such as hypoxia, metabolic dysregulation, and transcriptional and epigenetic alterations (Figure 4). Some of these approaches are being tested in clinical trials, independently and in combination with immunotherapies (Table 1). Because the glioma microenvironment poses specific threats to T-cell effector function, and because intrinsic factors drive and enhance exhaustion of TILs in gliomas, targeting multiple mechanisms simultaneously could produce the most impactful therapies for restoring anti-tumor immunity and ultimately generating a therapeutic response (see Outstanding questions).
Restoring normoxia has been shown to decrease levels of CD8+ T-cell exhaustion and enhance the efficacy of anti-cancer therapies in cancer models, including gliomas and glioblastomas [29,129,130]. This finding links hypoxia in the glioma microenvironment to immune exhaustion, and thus suggests that modulating the microenvironment could improve treatment (Figure 4). Evidence of reversing the expression of exhaustion-associated transcription factors came from experiments where TOX was deleted [99]. Although inhibitors of TOX have not yet been identified, a potential therapeutic avenue could focus on reducing TOX expression with gene therapy or decreasing its downstream function via small molecule antagonists. Involvement of immunostimulants and treatments to promote cytokine-driven immune activation, such as administration of IL-2 or inhibition of exhaustion-related cytokines, could reverse exhaustion and support other mechanisms by which the immune system can drive tumor suppression (Figure 4, Table 1) [120,131–133]. Use of cytokines to treat gliomas is an exciting avenue that has shown antitumor activity in murine and human soft-tissue sarcoma models [134]. Applications to gliomas and other CNS cancers are being tested in clinical trials (Table 1).
Metabolic alterations of either cancer cells or T-cells aimed at reversing the metabolic phenotype associated with exhaustion are another attractive way to be explored (see Outstanding questions). Acting directly on T-cell metabolism, one strategy is to improve mitochondrial function. This could be achieved by targeting PGC-1α, with bezafibrate, or using antioxidants specific for ROS located in mitochondria are attractive novel therapies. For example, mitoquinone and MitoTempo deceased ROS in mitochondria of exhausted HBV-specific T cells thus restoring the anti-viral activity of these cells [135]. Induction of mitochondrial oxidative stress was enough to limt the ability of T-cells to use OXPHOS for ATP generation and blocked their proliferation. Therefore the use of N-acetyl-cysteine, a known antioxidant, a source of cysteine needed for glutathione, which is a ROS scavenger, restored T-cell function in vitro and in vivo [53]. Preventing the antigen-driven accumulation of mitochondrial ROS has been shown to restore T cell proliferation, effector function, and their killing activity, further validating this strategy. Interestingly, the addition of IL-10, IL-12, and IL-21 has also been reported to improve the mitochondrial function of exhausted T-cells [17,87,136] (Figure 4).
Modulating the extrinsic factors present in the glioma microenvironment coming from the activity of tumor cells by altering tumor-specific pathways is an indirect strategy (see Outstanding questions). This can be done by modulating tumor-specific uptake and utilization of glucose, tryptophan, lactate formation and secretion, D-2HG formation and secretion or cholesterol formation and subsequent secretion into the TME [137]. Tumor specific metabolic modulators such as IDH-mutant inhibitors, IDO inhibitors, or lactate formation inhibitor could be utilized (Figure 4).
Finally, combinatory treatments that decrease the immunosuppressive factors in the glioma microenvironment and reverse T-cell exhaustion via targeting of cancer cells and T-cells symultaheously hold promise (Figure 4). An example of this approach involves combining IDH1 inhibitors, which decrease D-2HG concentration in the microenvironment thus decreasing its immunosuppressive effects and reinvigorating T-cells through checkpoint blockade (Figure 4). It is possible that such combinatorial approaches will prevent T-cells from becoming exhausted or re-exhausted by eliminating the extrinsic factors that lead to exhaustion.
Concluding remarks
Because the glioma microenvironment is unique, complex and is likely a major determinant of tumor immunogenicity, more focus needs to be placed on the interplay between this microenvironment and its direct impact on T-cell function and exhaustion. Additional studies analyzing tumor tissues, particularly after treatments designed to modulate the TME and T-cell function will help elucidate the complex nature of the glioma microenvironment and the role of immune cells (Table 1). Reversal of T-cell exhaustion through cell-specific modulation combined with alteration of the immunosuppressive TME may have profound impacts on the efficacy of immunotherapy for patients with malignant glioma. Ongoing clinical trials and research are showing exciting progress in the ability to treat gliomas and CNS tumors, yet it is likely that no singular approach will be the answer to glioma occurrence and progression. Rather, multi-pronged, combinatorial treatments with an eye towards the involvement of the TME and functionality of immune cells hold the most promise in revolutionizing cancer care.
Highlights:
Gliomas remain notoriously difficult to treat, and the use of immunotherapy to treat CNS cancers has been far less successful than in other cancer types.
T-cell exhaustion is defined by reduced proliferative capacity, reduced production of effector cytokines, sustained expression of inhibitory receptors, altered transcriptional and epigenetic states, and impaired metabolic activity.
The glioma microenvironment contains distinct factors that drive T cell exhaustion and dysfunctional metabolic states.
Metabolic and microenvironmental alterations have unique impacts on the epigenetic and transcriptional changes that drive T cell exhaustion in gliomas.
Reversal of T-cell exhaustion has the potential to enhance anti-tumor immunity and treatment efficacy in malignant gliomas.
Acknowledgments
This work was supported by funding from the National Institutes of Health; National Cancer Institute Center for Cancer Research; and the NIH Cancer Research Training Award. We thank Erina He for the development and illustration of the figures, and Lisa Maroski for scientific editing and review of the manuscript. We acknowledge the support of the National Institutes of Health for providing resources that have contributed to the development of this manuscript.
Footnotes
Declaration of interests
None are declared by the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ostrom QT et al. (2021) CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol 23, iii1-iii105. 10.1093/neuonc/noab200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weller M.et al. (2015) Glioma. Nat Rev Dis Primers 1, 15017. 10.1038/nrdp.2015.17 [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y.and Zhang Z.(2020) The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol 17, 807–821. 10.1038/s41423-020-0488-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moskophidis D.et al. (1993) Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 362, 758–761. 10.1038/362758a0 [DOI] [PubMed] [Google Scholar]
- 5.McLane LM et al. (2019) CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annual Review of Immunology, Vol 37, 2019 37, 457–495. 10.1146/annurev-immunol-041015-055318 [DOI] [PubMed] [Google Scholar]
- 6.Yi JS et al. (2010) T-cell exhaustion: characteristics, causes and conversion. Immunology 129, 474–481. 10.1111/j.1365-2567.2010.03255.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wherry EJ (2011) T cell exhaustion. Nat Immunol 12, 492–499. 10.1038/ni.2035 [DOI] [PubMed] [Google Scholar]
- 8.Beltra JC et al. (2020) Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 52, 825–841 e828. 10.1016/j.immuni.2020.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saeidi A.et al. (2018) T-Cell Exhaustion in Chronic Infections: Reversing the State of Exhaustion and Reinvigorating Optimal Protective Immune Responses. Front Immunol 9, 2569. 10.3389/fimmu.2018.02569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuller MJ et al. (2004) Maintenance, loss, and resurgence of T cell responses during acute, protracted, and chronic viral infections. J Immunol 172, 4204–4214. 10.4049/jimmunol.172.7.4204 [DOI] [PubMed] [Google Scholar]
- 11.Wherry EJ et al. (2003) Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol 77, 4911–4927. 10.1128/jvi.77.8.4911-4927.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmadzadeh M.et al. (2009) Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 114, 1537–1544. 10.1182/blood-2008-12-195792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gehring AJ et al. (2009) Profile of tumor antigen-specific CD8 T cells in patients with hepatitis B virus-related hepatocellular carcinoma. Gastroenterology 137, 682–690. 10.1053/j.gastro.2009.04.045 [DOI] [PubMed] [Google Scholar]
- 14.Jiang Y.et al. (2015) T-cell exhaustion in the tumor microenvironment. Cell Death Dis 6, e1792. 10.1038/cddis.2015.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dolina JS et al. (2021) CD8(+) T Cell Exhaustion in Cancer. Front Immunol 12, 715234. 10.3389/fimmu.2021.715234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang CH et al. (2013) Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. 10.1016/j.cell.2013.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schurich A.et al. (2016) Distinct Metabolic Requirements of Exhausted and Functional Virus-Specific CD8 T Cells in the Same Host. Cell Rep 16, 1243–1252. 10.1016/j.celrep.2016.06.078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davidson TB et al. (2019) Expression of PD-1 by T Cells in Malignant Glioma Patients Reflects Exhaustion and Activation. Clin Cancer Res 25, 1913–1922. 10.1158/1078-0432.CCR-18-1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woroniecka K.et al. (2018) T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin Cancer Res 24, 4175–4186. 10.1158/1078-0432.CCR-17-1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mirzaei R.et al. (2017) T Cell Exhaustion in Glioblastoma: Intricacies of Immune Checkpoints. Trends Immunol 38, 104–115. 10.1016/j.it.2016.11.005 [DOI] [PubMed] [Google Scholar]
- 21.Lin W.et al. (2020) Characterization of Hypoxia Signature to Evaluate the Tumor Immune Microenvironment and Predict Prognosis in Glioma Groups. Front Oncol 10, 796. 10.3389/fonc.2020.00796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei J.et al. (2011) Hypoxia potentiates glioma-mediated immunosuppression. PLoS One 6, e16195. 10.1371/journal.pone.0016195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaur B.et al. (2005) Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol 7, 134–153. 10.1215/S1152851704001115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petrova V.et al. (2018) The hypoxic tumour microenvironment. Oncogenesis 7, 10. 10.1038/s41389-0170011-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chouaib S.et al. (2017) Hypoxic stress: obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 36, 439–445. 10.1038/onc.2016.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brat DJ and Van Meir EG (2001) Glomeruloid microvascular proliferation orchestrated by VPF/VEGF: a new world of angiogenesis research. Am J Pathol 158, 789–796. 10.1016/S0002-9440(10)64025-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensen RL et al. (2014) Preoperative dynamic contrast-enhanced MRI correlates with molecular markers of hypoxia and vascularity in specific areas of intratumoral microenvironment and is predictive of patient outcome. Neuro Oncol 16, 280–291. 10.1093/neuonc/not148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monteiro AR et al. (2017) The Role of Hypoxia in Glioblastoma Invasion. Cells 6. 10.3390/cells6040045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scharping NE et al. (2021) Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol 22, 205–215. 10.1038/s41590-020-00834-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu S.et al. (2021) T-Cell Exhaustion Status Under High and Low Levels of Hypoxia-Inducible Factor 1alpha Expression in Glioma. Front Pharmacol 12, 711772. 10.3389/fphar.2021.711772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tao JH et al. (2015) Hypoxia-inducible factors in T lymphocyte differentiation and function. A Review in the Theme: Cellular Responses to Hypoxia. Am J Physiol Cell Physiol 309, C580–589. 10.1152/ajpcell.00204.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lukashev D.et al. (2006) Cutting edge: hypoxia-inducible factor 1alpha and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J Immunol 177, 4962–4965. 10.4049/jimmunol.177.8.4962 [DOI] [PubMed] [Google Scholar]
- 33.Bannoud N.et al. (2021) Hypoxia Supports Differentiation of Terminally Exhausted CD8 T Cells. Front Immunol 12, 660944. 10.3389/fimmu.2021.660944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barsoum IB et al. (2014) A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res 74, 665–674. 10.1158/0008-5472.CAN-13-0992 [DOI] [PubMed] [Google Scholar]
- 35.Camuzi D.et al. (2019) Regulation Is in the Air: The Relationship between Hypoxia and Epigenetics in Cancer. Cells 8. 10.3390/cells8040300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ford BR et al. (2021) Altered costimulatory signals and hypoxia support chromatin landscapes limiting the functional potential of exhausted T cells in cancer. bioRxiv, 2021.2007.2011.451947. 10.1101/2021.07.11.451947 [DOI] [Google Scholar]
- 37.Lin TW et al. (2017) TDP-43/HDAC6 axis promoted tumor progression and regulated nutrient deprivation-induced autophagy in glioblastoma. Oncotarget 8, 56612–56625. 10.18632/oncotarget.17979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu YQ et al. (2019) Amino acid metabolism-related gene expression-based risk signature can better predict overall survival for glioma. Cancer Sci 110, 321–333. 10.1111/cas.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flavahan WA et al. (2013) Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci 16, 1373–1382. 10.1038/nn.3510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanaka K.et al. (2021) Glioma cells require one-carbon metabolism to survive glutamine starvation. Acta Neuropathol Commun 9, 16. 10.1186/s40478-020-01114-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang CH et al. (2015) Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 162, 1229–1241. 10.1016/j.cell.2015.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siska PJ et al. (2016) Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J Immunol 197, 2532–2540. 10.4049/jimmunol.1502464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bengsch B.et al. (2016) Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity 45, 358–373. 10.1016/j.immuni.2016.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hernandez A.et al. (2021) Glioblastoma: Relationship between Metabolism and Immunosuppressive Microenvironment. Cells 10. 10.3390/cells10123529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Ianni N.et al. (2021) Altered Metabolism in Glioblastoma: Myeloid-Derived Suppressor Cell (MDSC) Fitness and Tumor-Infiltrating Lymphocyte (TIL) Dysfunction. Int J Mol Sci 22. 10.3390/ijms22094460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Speiser DE et al. (2016) Regulatory circuits of T cell function in cancer. Nat Rev Immunol 16, 599–611. 10.1038/nri.2016.80 [DOI] [PubMed] [Google Scholar]
- 47.Franco F.et al. (2020) Metabolic and epigenetic regulation of T-cell exhaustion. Nat Metab 2, 1001–1012. 10.1038/s42255-020-00280-9 [DOI] [PubMed] [Google Scholar]
- 48.Zhai L.et al. (2015) The role of IDO in brain tumor immunotherapy. J Neurooncol 123, 395–403. 10.1007/s11060-014-1687-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu YR et al. (2020) Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat Immunol 21, 1540–1551. 10.1038/s41590-020-0793-3 [DOI] [PubMed] [Google Scholar]
- 50.Webster BR et al. (2014) Regulation of autophagy and mitophagy by nutrient availability and acetylation. Biochim Biophys Acta 1841, 525–534. 10.1016/j.bbalip.2014.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Daskalaki I.et al. (2018) Hypoxia and Selective Autophagy in Cancer Development and Therapy. Front Cell Dev Biol 6, 104. 10.3389/fcell.2018.00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lazarou M.(2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. 10.1038/nature14893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vardhana SA et al. (2020) Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat Immunol 21, 1022–1033. 10.1038/s41590-020-0725-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bartrons R.and Caro J.(2007) Hypoxia, glucose metabolism and the Warburg’s effect. J Bioenerg Biomembr 39, 223–229. 10.1007/s10863-007-9080-3 [DOI] [PubMed] [Google Scholar]
- 55.Agnihotri S.and Zadeh G.(2016) Metabolic reprogramming in glioblastoma: the influence of cancer metabolism on epigenetics and unanswered questions. Neuro Oncol 18, 160–172. 10.1093/neuonc/nov125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reuss AM et al. (2021) The Acidic Brain-Glycolytic Switch in the Microenvironment of Malignant Glioma. Int J Mol Sci 22. 10.3390/ijms22115518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Branco M.et al. (2019) Serum lactate levels are associated with glioma malignancy grade. Clin Neurol Neurosurg 186, 105546. 10.1016/j.clineuro.2019.105546 [DOI] [PubMed] [Google Scholar]
- 58.Tu VY et al. (2021) Beyond the Lactate Paradox: How Lactate and Acidity Impact T Cell Therapies against Cancer. Antibodies (Basel) 10. 10.3390/antib10030025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Romero-Garcia S.et al. (2016) Lactate Contribution to the Tumor Microenvironment: Mechanisms, Effects on Immune Cells and Therapeutic Relevance. Front Immunol 7, 52. 10.3389/fimmu.2016.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fischer K.et al. (2007) Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109, 3812–3819. 10.1182/blood-2006-07-035972 [DOI] [PubMed] [Google Scholar]
- 61.Louis DN et al. (2021) The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23, 1231–1251. 10.1093/neuonc/noab106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han S.et al. (2020) IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. Br J Cancer 122, 1580–1589. 10.1038/s41416-020-0814-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Balss J.et al. (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116, 597–602. 10.1007/s00401-008-0455-2 [DOI] [PubMed] [Google Scholar]
- 64.Yan H.et al. (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360, 765–773. 10.1056/NEJMoa0808710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bunse L.et al. (2018) Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med 24, 1192–1203. 10.1038/s41591-018-0095-6 [DOI] [PubMed] [Google Scholar]
- 66.Notarangelo G.et al. (2022) Oncometabolite d-2HG alters T cell metabolism to impair CD8(+) T cell function. Science 377, 1519–1529. 10.1126/science.abj5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ma X.et al. (2019) Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab 30, 143–156 e145. 10.1016/j.cmet.2019.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ahmad F.et al. (2019) Cholesterol Metabolism: A Potential Therapeutic Target in Glioblastoma. Cancers (Basel) 11. 10.3390/cancers11020146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jin MZ and Jin WL (2020) The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct Target Ther 5, 166. 10.1038/s41392-020-00280-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roesch S.et al. (2018) When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int J Mol Sci 19. 10.3390/ijms19020436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jacobs JF et al. (2009) Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro Oncol 11, 394–402. 10.1215/15228517-2008-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kersten K.et al. (2022) Spatiotemporal co-dependency between macrophages and exhausted CD8(+) T cells in cancer. Cancer Cell 40, 624–638 e629. 10.1016/j.ccell.2022.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu X.et al. (2018) Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat Commun 9, 249. 10.1038/s41467-017-02689-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sawant DV et al. (2019) Adaptive plasticity of IL-10(+) and IL-35(+) Treg cells cooperatively promotes tumor T cell exhaustion. Nat Immunol 20, 724–735. 10.1038/s41590-019-0346-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weber R.et al. (2018) Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front Immunol 9, 1310. 10.3389/fimmu.2018.01310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu S.et al. (2022) TIM3/CEACAM1 pathway involves in myeloid-derived suppressor cells induced CD8(+) T cells exhaustion and bone marrow inflammatory microenvironment in myelodysplastic syndrome. Immunology. 10.1111/imm.13488 [DOI] [PubMed] [Google Scholar]
- 77.Tao J.et al. (2020) CD8(+) T cells exhaustion induced by myeloid-derived suppressor cells in myelodysplastic syndromes patients might be through TIM3/Gal-9 pathway. J Cell Mol Med 24, 1046–1058. 10.1111/jcmm.14825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alban TJ et al. (2020) Glioblastoma Myeloid-Derived Suppressor Cell Subsets Express Differential Macrophage Migration Inhibitory Factor Receptor Profiles That Can Be Targeted to Reduce Immune Suppression. Front Immunol 11, 1191. 10.3389/fimmu.2020.01191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mi Y.et al. (2020) The Emerging Role of Myeloid-Derived Suppressor Cells in the Glioma Immune Suppressive Microenvironment. Front Immunol 11, 737. 10.3389/fimmu.2020.00737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zamler DB et al. (2022) Immune landscape of a genetically engineered murine model of glioma compared with human glioma. JCI Insight 7. 10.1172/jci.insight.148990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moore KW et al. (2001) Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 19, 683–765. 10.1146/annurev.immunol.19.1.683 [DOI] [PubMed] [Google Scholar]
- 82.Blackburn SD and Wherry EJ (2007) IL-10, T cell exhaustion and viral persistence. Trends Microbiol 15, 143–146. 10.1016/j.tim.2007.02.006 [DOI] [PubMed] [Google Scholar]
- 83.Ni G.et al. (2015) Manipulating IL-10 signalling blockade for better immunotherapy. Cell Immunol 293, 126–129. 10.1016/j.cellimm.2014.12.012 [DOI] [PubMed] [Google Scholar]
- 84.Brockman MA et al. (2009) IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood 114, 346–356. 10.1182/blood-2008-12-191296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ravi VM et al. (2022) T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat Commun 13, 925. 10.1038/s41467-022-28523-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fujii S.et al. (2001) Interleukin-10 promotes the maintenance of antitumor CD8(+) T-cell effector function in situ. Blood 98, 2143–2151. 10.1182/blood.v98.7.2143 [DOI] [PubMed] [Google Scholar]
- 87.Guo Y.et al. (2021) Metabolic reprogramming of terminally exhausted CD8(+) T cells by IL-10 enhances anti-tumor immunity. Nat Immunol 22, 746–756. 10.1038/s41590-021-00940-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gabriel SS et al. (2021) Transforming growth factor-beta-regulated mTOR activity preserves cellular metabolism to maintain long-term T cell responses in chronic infection. Immunity 54, 1698–1714 e1695. 10.1016/j.immuni.2021.06.007 [DOI] [PubMed] [Google Scholar]
- 89.Tinoco R.et al. (2009) Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity 31, 145–157. 10.1016/j.immuni.2009.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang ZZ et al. (2014) TGF-beta upregulates CD70 expression and induces exhaustion of effector memory T cells in B-cell non-Hodgkin’s lymphoma. Leukemia 28, 1872–1884. 10.1038/leu.2014.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Han J.et al. (2015) TGF-beta signaling and its targeting for glioma treatment. Am J Cancer Res 5, 945–955 [PMC free article] [PubMed] [Google Scholar]
- 92.Joseph JV et al. (2022) TGF-beta promotes microtube formation in glioblastoma through thrombospondin 1. Neuro Oncol 24, 541–553. 10.1093/neuonc/noab212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ueda R.et al. (2009) Systemic inhibition of transforming growth factor-beta in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin Cancer Res 15, 6551–6559. 10.1158/1078-0432.CCR-09-1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nagaraj NS and Datta PK (2010) Targeting the transforming growth factor-beta signaling pathway in human cancer. Expert Opin Investig Drugs 19, 77–91. 10.1517/13543780903382609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zeng Z.et al. (2020) Exhausted T cells and epigenetic status. Cancer Biol Med 17, 923–936. 10.20892/j.issn.2095-3941.2020.0338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Belk JA et al. (2022) Epigenetic regulation of T cell exhaustion. Nat Immunol 23, 848–860. 10.1038/s41590-022-01224-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sen DR et al. (2016) The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169. 10.1126/science.aae0491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Belk JA et al. (2022) Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell. 10.1016/j.ccell.2022.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Khan O.et al. (2019) TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 571, 211–218. 10.1038/s41586-019-1325-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang R.et al. (2019) Distinct epigenetic features of tumor-reactive CD8+ T cells in colorectal cancer patients revealed by genome-wide DNA methylation analysis. Genome Biol 21, 2. 10.1186/s13059-0191921-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang H.et al. (2020) Correction to: Clinical characterization, genetic profiling, and immune infiltration of TOX in diffuse gliomas. J Transl Med 18, 368. 10.1186/s12967-020-02514-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Han HS et al. (2021) TOX-expressing terminally exhausted tumor-infiltrating CD8(+) T cells are reinvigorated by co-blockade of PD-1 and TIGIT in bladder cancer. Cancer Lett 499, 137–147. 10.1016/j.canlet.2020.11.035 [DOI] [PubMed] [Google Scholar]
- 103.Liang C.et al. (2021) TOX as a potential target for immunotherapy in lymphocytic malignancies. Biomark Res 9, 20. 10.1186/s40364-021-00275-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yerinde C.et al. (2019) Metabolic Control of Epigenetics and Its Role in CD8(+) T Cell Differentiation and Function. Front Immunol 10, 2718. 10.3389/fimmu.2019.02718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lu J.et al. (2021) Metabolic Controls on Epigenetic Reprogramming in Regulatory T Cells. Front Immunol 12, 728783. 10.3389/fimmu.2021.728783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wellen KE et al. (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. 10.1126/science.1164097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cluntun AA et al. (2015) The rate of glycolysis quantitatively mediates specific histone acetylation sites. Cancer Metab 3, 10. 10.1186/s40170-015-0135-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Qiu J.et al. (2019) Acetate Promotes T Cell Effector Function during Glucose Restriction. Cell Rep 27, 2063–2074 e2065. 10.1016/j.celrep.2019.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Buggert M.et al. (2014) T-bet and Eomes are differentially linked to the exhausted phenotype of CD8+ T cells in HIV infection. PLoS Pathog 10, e1004251. 10.1371/journal.ppat.1004251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chang S.and Aune TM (2007) Dynamic changes in histone-methylation ‘marks’ across the locus encoding interferon-gamma during the differentiation of T helper type 2 cells. Nat Immunol 8, 723–731. 10.1038/ni1473 [DOI] [PubMed] [Google Scholar]
- 111.Bruzzone S.et al. (2009) Catastrophic NAD+ depletion in activated T lymphocytes through Nampt inhibition reduces demyelination and disability in EAE. PLoS One 4, e7897. 10.1371/journal.pone.0007897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tateishi K.et al. (2015) Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell 28, 773–784. 10.1016/j.ccell.2015.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li M.et al. (2021) Local Targeting of NAD(+) Salvage Pathway Alters the Immune Tumor Microenvironment and Enhances Checkpoint Immunotherapy in Glioblastoma. Cancer Res 81, 1922. 10.1158/0008-5472.CAN-21-0525 [DOI] [PubMed] [Google Scholar]
- 114.Suzuki J.et al. (2018) The tumor suppressor menin prevents effector CD8 T-cell dysfunction by targeting mTORC1-dependent metabolic activation. Nat Commun 9, 3296. 10.1038/s41467-018-05854-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bam M.et al. (2021) Genome wide DNA methylation landscape reveals glioblastoma’s influence on epigenetic changes in tumor infiltrating CD4+ T cells. Oncotarget 12, 967–981. 10.18632/oncotarget.27955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ahir BK et al. (2020) Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol Neurobiol 57, 2461–2478. 10.1007/s12035-020-01892-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Plate KH et al. (1994) Vascular endothelial growth factor and glioma angiogenesis: coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms. Int J Cancer 59, 520–529. 10.1002/ijc.2910590415 [DOI] [PubMed] [Google Scholar]
- 118.Pearson JRD et al. (2020) Immune Escape in Glioblastoma Multiforme and the Adaptation of Immunotherapies for Treatment. Front Immunol 11, 582106. 10.3389/fimmu.2020.582106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Razavi SM et al. (2016) Immune Evasion Strategies of Glioblastoma. Front Surg 3, 11. 10.3389/fsurg.2016.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tabana Y.et al. (2021) Reversing T-cell exhaustion in immunotherapy: a review on current approaches and limitations. Expert Opin Ther Targets 25, 347–363. 10.1080/14728222.2021.1937123 [DOI] [PubMed] [Google Scholar]
- 121.Vega EA et al. (2008) Combating immunosuppression in glioma. Future Oncol 4, 433–442. 10.2217/14796694.4.3.433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pan Y.et al. (2020) Tumor-Associated Macrophages in Tumor Immunity. Front Immunol 11, 583084. 10.3389/fimmu.2020.583084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Himes BT et al. (2021) Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front Oncol 11, 770561. 10.3389/fonc.2021.770561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Marin-Acevedo JA et al. (2021) Next generation of immune checkpoint inhibitors and beyond. J Hematol Oncol 14, 45. 10.1186/s13045-021-01056-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pauken KE et al. (2016) Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165. 10.1126/science.aaf2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yates KB et al. (2021) Epigenetic scars of CD8(+) T cell exhaustion persist after cure of chronic infection in humans. Nat Immunol 22, 1020–1029. 10.1038/s41590-021-00979-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Miliotou AN and Papadopoulou LC (2018) CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr Pharm Biotechnol 19, 5–18. 10.2174/1389201019666180418095526 [DOI] [PubMed] [Google Scholar]
- 128.Calmeiro J.et al. (2020) Dendritic Cell Vaccines for Cancer Immunotherapy: The Role of Human Conventional Type 1 Dendritic Cells. Pharmaceutics 12. 10.3390/pharmaceutics12020158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mudassar F.et al. (2020) Targeting tumor hypoxia and mitochondrial metabolism with anti-parasitic drugs to improve radiation response in high-grade gliomas. J Exp Clin Cancer Res 39, 208. 10.1186/s13046-02001724-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jayaprakash P.et al. (2018) Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest 128, 5137–5149. 10.1172/JCI96268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Butowski N.(2010) Immunostimulants for malignant gliomas. Neurosurg Clin N Am 21, 53–65. 10.1016/j.nec.2009.08.007 [DOI] [PubMed] [Google Scholar]
- 132.McGranahan T.et al. (2017) History and current state of immunotherapy in glioma and brain metastasis. Ther Adv Med Oncol 9, 347–368. 10.1177/1758834017693750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hashimoto M.et al. (2022) PD-1 combination therapy with IL-2 modifies CD8(+) T cell exhaustion program. Nature. 10.1038/s41586-022-05257-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schliemann C.et al. (2021) Dose escalation and expansion phase I studies with the tumour-targeting antibody-tumour necrosis factor fusion protein L19TNF plus doxorubicin in patients with advanced tumours, including sarcomas. Eur J Cancer 150, 143–154. 10.1016/j.ejca.2021.03.038 [DOI] [PubMed] [Google Scholar]
- 135.Fisicaro P.et al. (2017) Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat Med 23, 327–336. 10.1038/nm.4275 [DOI] [PubMed] [Google Scholar]
- 136.Loschinski R.et al. (2018) IL-21 modulates memory and exhaustion phenotype of T-cells in a fatty acid oxidation-dependent manner. Oncotarget 9, 13125–13138. 10.18632/oncotarget.24442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li R.et al. (2009) IDO inhibits T-cell function through suppressing Vav1 expression and activation. Cancer Biol Ther 8, 1402–1408. 10.4161/cbt.8.14.8882 [DOI] [PubMed] [Google Scholar]
- 138.Fisher JP and Adamson DC (2021) Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 9. 10.3390/biomedicines9030324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Davis ME (2016) Glioblastoma: Overview of Disease and Treatment. Clin J Oncol Nurs 20, S2–8. 10.1188/16.CJON.S1.2-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fuks Z.et al. (1976) Long term effects of radiation of T and B lymphocytes in peripheral blood of patients with Hodgkin’s disease. J Clin Invest 58, 803–814. 10.1172/JCI108532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gray WC et al. (1985) Effects of radiation therapy on T-lymphocyte subpopulations in patients with head and neck cancer. Otolaryngol Head Neck Surg 93, 650–660. 10.1177/019459988509300515 [DOI] [PubMed] [Google Scholar]
- 142.Gough MJ and Crittenden MR (2022) The paradox of radiation and T cells in tumors. Neoplasia 31, 100808. 10.1016/j.neo.2022.100808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Qu Y.et al. (2010) Gamma-ray resistance of regulatory CD4+CD25+Foxp3+ T cells in mice. Radiat Res 173, 148–157. 10.1667/RR0978.1 [DOI] [PubMed] [Google Scholar]
- 144.Li HH et al. (2015) Ionizing Radiation Impairs T Cell Activation by Affecting Metabolic Reprogramming. Int J Biol Sci 11, 726–736. 10.7150/ijbs.12009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Walker MD et al. (1980) Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery. N Engl J Med 303, 1323–1329. 10.1056/NEJM198012043032303 [DOI] [PubMed] [Google Scholar]
- 146.Walker MD et al. (1979) An analysis of dose-effect relationship in the radiotherapy of malignant gliomas. Int J Radiat Oncol Biol Phys 5, 1725–1731. 10.1016/0360-3016(79)90553-4 [DOI] [PubMed] [Google Scholar]
- 147.Sulman EP et al. (2017) Radiation Therapy for Glioblastoma: American Society of Clinical Oncology Clinical Practice Guideline Endorsement of the American Society for Radiation Oncology Guideline. J Oncol Pract 13, 123–127. 10.1200/JOP.2016.018937 [DOI] [PubMed] [Google Scholar]
- 148.Dovedi SJ et al. (2017) Fractionated Radiation Therapy Stimulates Antitumor Immunity Mediated by Both Resident and Infiltrating Polyclonal T-cell Populations when Combined with PD-1 Blockade. Clin Cancer Res 23, 5514–5526. 10.1158/1078-0432.CCR-16-1673 [DOI] [PubMed] [Google Scholar]
- 149.Twyman-Saint Victor C.et al. (2015) Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520, 373–377. 10.1038/nature14292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gassner FJ et al. (2014) Chemotherapy-induced augmentation of T cells expressing inhibitory receptors is reversed by treatment with lenalidomide in chronic lymphocytic leukemia. Haematologica 99, 67–69. 10.3324/haematol.2013.098459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Das RK et al. (2020) Lingering effects of chemotherapy on mature T cells impair proliferation. Blood Adv 4, 4653–4664. 10.1182/bloodadvances.2020001797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dietrich J.et al. (2011) Corticosteroids in brain cancer patients: benefits and pitfalls. Expert Rev Clin Pharmacol 4, 233–242. 10.1586/ecp.11.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tokunaga A.et al. (2019) Selective inhibition of low-affinity memory CD8(+) T cells by corticosteroids. J Exp Med 216, 2701–2713. 10.1084/jem.20190738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Giles AJ et al. (2018) Dexamethasone-induced immunosuppression: mechanisms and implications for immunotherapy. J Immunother Cancer 6, 51. 10.1186/s40425-018-0371-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Iorgulescu JB et al. (2021) Concurrent Dexamethasone Limits the Clinical Benefit of Immune Checkpoint Blockade in Glioblastoma. Clin Cancer Res 27, 276–287. 10.1158/1078-0432.CCR-20-2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hino S.et al. (2013) Metabolism-epigenome crosstalk in physiology and diseases. J Hum Genet 58, 410–415. 10.1038/jhg.2013.57 [DOI] [PubMed] [Google Scholar]
- 157.Xu W.et al. (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30. 10.1016/j.ccr.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Turcan S.et al. (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483. 10.1038/nature10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lu C.et al. (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478. 10.1038/nature10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Quiros PM et al. (2016) Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol 17, 213–226. 10.1038/nrm.2016.23 [DOI] [PubMed] [Google Scholar]
- 161.Matilainen O.et al. (2017) Mitochondria and Epigenetics - Crosstalk in Homeostasis and Stress. Trends Cell Biol 27, 453–463. 10.1016/j.tcb.2017.02.004 [DOI] [PubMed] [Google Scholar]
- 162.Ge T.et al. (2022) Crosstalk between metabolic reprogramming and epigenetics in cancer: updates on mechanisms and therapeutic opportunities. Cancer Commun (Lond) 42, 1049–1082. 10.1002/cac2.12374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Maggs L.et al. (2021) CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front Neurosci 15, 662064. 10.3389/fnins.2021.662064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.O’Rourke DM et al. (2017) A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 9. 10.1126/scitranslmed.aaa0984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Land CA et al. (2020) Chimeric antigen receptor T-cell therapy in glioblastoma: charging the T cells to fight. J Transl Med 18, 428. 10.1186/s12967-020-02598-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Poorebrahim M.et al. (2021) Counteracting CAR T cell dysfunction. Oncogene 40, 421–435. 10.1038/s41388-020-01501-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kouro T.et al. (2022) Exhaustion of CAR T cells: potential causes and solutions. J Transl Med 20, 239. 10.1186/s12967-022-03442-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Moon EK et al. (2014) Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res 20, 4262–4273. 10.1158/1078-0432.CCR-13-2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chen J.et al. (2019) NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534. 10.1038/s41586-019-0985-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Delgoffe GM et al. (2021) The role of exhaustion in CAR T cell therapy. Cancer Cell 39, 885–888. 10.1016/j.ccell.2021.06.012 [DOI] [PubMed] [Google Scholar]
- 171.Tang L.et al. (2021) T Cell Exhaustion and CAR-T Immunotherapy in Hematological Malignancies. Biomed Res Int 2021, 6616391. 10.1155/2021/6616391 [DOI] [PMC free article] [PubMed] [Google Scholar]