

Abstract
Identifying ancestry-specific molecular profiles of late-onset Alzheimer’s Disease (LOAD) in brain tissue is crucial to understand novel mechanisms and develop effective interventions in non-European, high-risk populations. We performed gene differential expression (DE) and consensus network-based analyses in RNA-sequencing data of postmortem brain tissue from 39 Caribbean Hispanics (CH). To identify ancestry-concordant and -discordant expression profiles, we compared our results to those from two independent non-Hispanic White (NHW) samples (n = 731). In CH, we identified 2802 significant DE genes, including several LOAD known-loci. DE effects were highly concordant across ethnicities, with 373 genes transcriptome-wide significant in all three cohorts. Cross-ancestry meta-analysis found NPNT to be the top DE gene. We replicated over 82% of meta-analyses genome-wide signals in single-nucleus RNA-seq data (including NPNT and LOAD known-genes SORL1, FBXL7, CLU, ABCA7). Increasing representation in genetic studies will allow for deeper understanding of ancestry-specific mechanisms and improving precision treatment options in understudied groups.
Keywords: Caribbean-Hispanic, Brain gene expression, Alzheimer’s disease, Bulk tissue, Single cell
1. Introduction
Genetic and environmental factors conferring risk for late-onset Alzheimer’s disease (LOAD) are known to differ across ancestral and ethnic populations (Reitz and Mayeux, 2014), though non-Hispanic White (NHW) cohorts still dominate genomic studies (Sirugo et al., 2019), particularly for neurodegenerative diseases. This work has been foundational for building our understanding of the pathological mechanisms of late-onset Alzheimer’s disease (LOAD), and points toward perturbations in a broad set of biological pathways most often including neuroimmune responses and inflammation, synaptic regulation, oxidative stress and glucose metabolism, and fatty acid metabolism (Calabrò et al., 2020; Morgan et al., 2022). In fact, based on existing literature, the functional reach of LOAD-related pathways is nearly universal; a recent text-mining study of over 200,000 abstracts identified that 91% of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways show LOAD associations within at least 5 published studies (Morgan et al., 2022). Direct bulk and single-cell transcriptomic analyses have also found consistent changes in excitatory and inhibitory neuronal, microglial, and astrocytic cellular populations in LOAD (Consens et al., 2022; Grubman et al., 2019; Mathys et al., 2019). However, with respect to identifying potentially targetable molecular risk mechanisms, the relative lack of data from populations other than NHW has the potential to exacerbate existing disparities in global health equity (Jooma et al., 2019; Weinberger et al., 2020).
Recent work has shown important differences in the genes and molecular pathways involved in risk for and progression of LOAD among different populations, though the scarcity of tissue samples outside of NHW populations has led to a strong bias in the literature toward the least invasive modalities. For example, multi-ancestry genetic analyses how include tens of thousands of individuals from African, African American (AA), Asian, and Hispanic populations (Bellenguez et al., 2022; Kunkle et al., 2021; Lake et al., 2022). Fewer studies have evaluated gene expression profiles in minority groups, with some evidence of differences in cell-free, protein coding messenger RNA (cf-mRNA) abundance in blood between LOAD and controls in AA individuals (Reddy et al., 2022). Transcriptomic analyses of brain tissue in LOAD outside of NHW populations are rarer yet, though single-nucleus RNA sequencing (RNAseq) analysis of frontal cortex from six African American (AA) and NHW individuals found that expression of LOAD-implicated genes (i.e. APOE in ε4 carriers) and gene clusters (neuronal and astrocytic) are differentially expressed between populations (Celis et al., 2020). Additionally, multi-ancestry expression analyses have demonstrated that the inclusion of diverse ancestral populations in expression quantitative trait loci (eQTL) experiments reveals previously unknown mechanistic explanations for known LOAD risk variants (e.g. rs117618017 and expression of APHB1) (Zeng et al., 2022).
Specifically, Caribbean-Hispanics represent an important population of individuals for the study of late-life neurodegenerative disease, as they are uniquely genetically admixed (Moreno-Estrada et al., 2013) and have undergone the most rapid demographic aging within Latin America over the last ten years (Acosta et al., 2021). Recent RNA sequencing analyses of blood in CH individuals (including LOAD and cognitively normal elderly) from Puerto Rico found that approximately 30% of gene expression regulatory variants were not shared between CH and either NHW or African American (AA) populations (Griswold et al., 2021). Despite this, there have been no postmortem brain gene expression studies of LOAD in Hispanic/Latinos populations to date, largely due to the scarcity of available tissue samples and the historical deprioritization of molecular-genetic research in this group. Here we present the first RNA-sequencing analysis of cortical brain tissue from Caribbean-Hispanics (CH) with and without a pathological diagnosis of LOAD. We aimed to identify genes and pathways with ancestry-specific (i.e. those identified in CH only) and ancestry-independent (i.e. those replicating across groups) differential gene expression by comparing transcriptome-wide association differential expression (DE) analyses and co-expression network-based analyses performed in CH and two independent NHW cohorts, processed with identical pipelines.
2. Results
Fig. 1 provides a schematic of our overall study design and Table 1 reports demographic and RNA sequencing depth measures for each cohort included in our study. For the CH cohort, global admixture analysis (Tosto et al., 2015b) (see Supplementary file, Fig. S1) revealed that six individuals did not show all three ancestral components (European, African and Native-American), resulting in 39 genetically-confirmed three-way admixed subjects (nLOAD = 23, nnon-LOAD = 16); five carried a PSEN1 mutation, the G206A substitution - a known mutation associated with familial LOAD in Caribbean Hispanic populations (Athan et al., 2001).
Fig. 1.
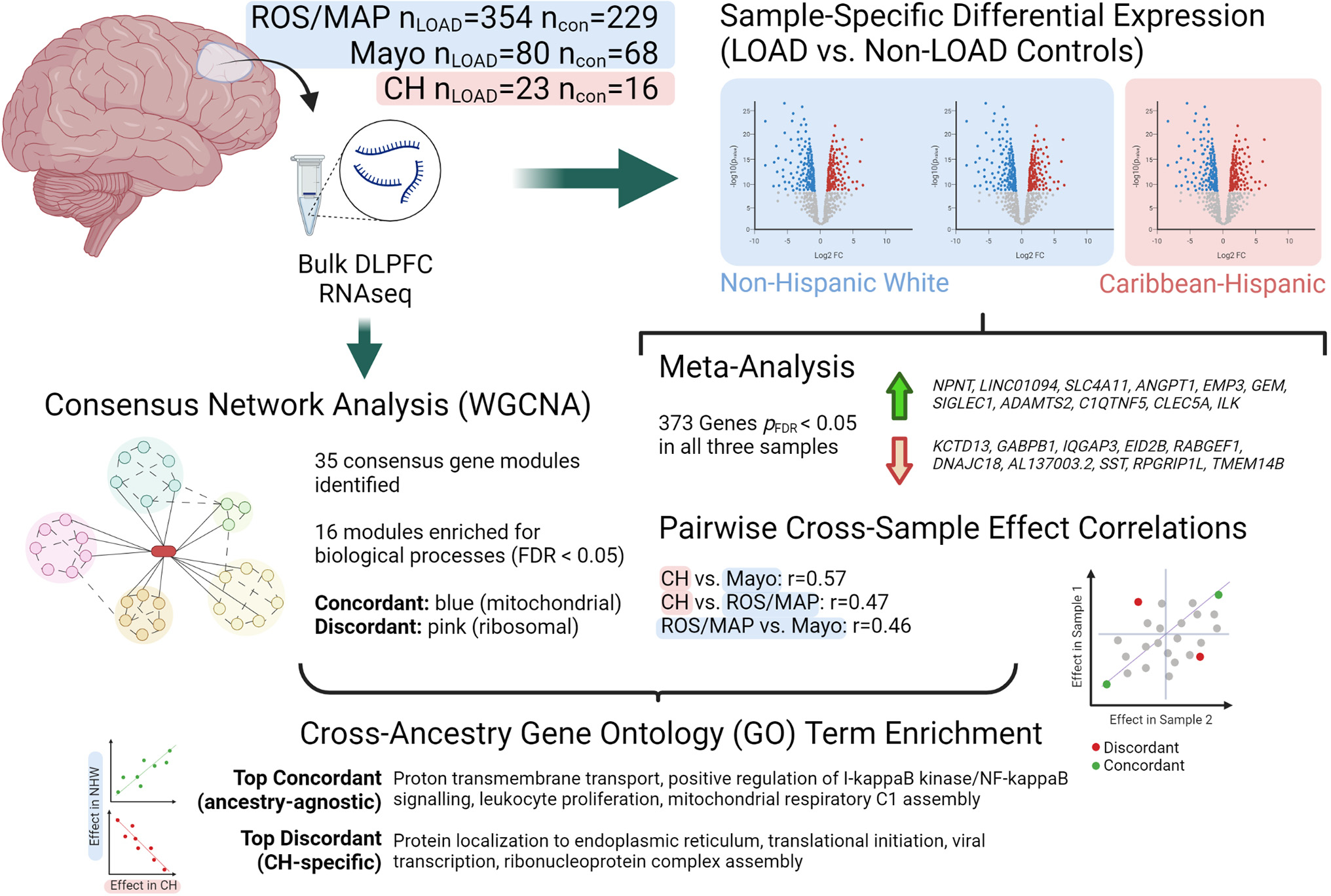
Schematic showing study design and top results. CH = Caribbean Hispanic; con = non-LOAD control; DLPFC = dorsolateral prefrontal cortex; FDR = false discovery rate; LOAD = late-onset Alzheimer’s disease; logFC = log fold-change; ROS/MAP = Religious Orders Study / Memory and Aging Project; WGCNA = weighted gene correlation network analysis. Created with BioRender.com.
Table 1.
Sample Demographics for Caribbean-Hispanic and Non-Hispanic White RNAseq Samples.
| Sample | Non-LOAD | LOAD | P-value |
|---|---|---|---|
|
| |||
| CH | (n = 16) | (n = 23) | |
| sex (M/F) | 8 M, 8F | 7M, 16F | 0.32 |
| age | 66.9 (11.8) | 79 (10.9) | 0.0026 |
| RIN | 5.8 (1.5) | 4.2 (1.5) | 0.0024 |
| PSEN1 carriers (+/−) | 16− | 5+/18− | 0.07 |
| Library size, millions | 16.2 (4.1) | 15.6 (3.8) | 0.65 |
| ROS/MAP (NHW) | (n = 229) | (n = 354) | |
| sex (M/F) | 88 M, 141F | 113 M, 241F | 0.11 |
| age | 87 (7.2) | 90.1 (5.9) | 7.7 × 10−8 |
| RIN | 7.2 (1.1) | 7 (0.9) | 0.015 |
| PMI | 7.1 (4.1) | 7.6 (5.2) | 0.18 |
| Library size, millions | 30.1 (13.5) | 26.5 (8.8) | 4.9 × 10−4 |
| Mayo (NHW) | (n = 68) | (n = 80) | |
| sex (M/F) | 33 M, 35F | 31 M, 49F | 0.25 |
| age | 82.9 (8.4) | 82.6 (7.7) | 0.84 |
| RIN | 7.6 (1) | 8.6 (0.6) | 1.4 × 10−9 |
| PMI | 6.5 (6.5) | 6.7 (4.3) | 0.84 |
| Library size, millions | 40.9 (11.1) | 45.4 (10) | 0.013 |
Note:
p-values correspond to those from two-sided, two-sample t-tests for continuous measures (age, RIN, library size, and PMI) and from two-sided Fisher’s exact tests for dichotomous measures (sex and PSEN1 G206A mutation status).
Summary measures for continuous outcomes are means (standard deviations in brackets). RIN = RNA integrity number; LOAD = late-onset Alzheimer’s disease; M = male; F = female; PMI = postmortem interval; CH = Caribbean-Hispanic (all genetically-confirmed, three-way admixed subjects); NHW = non-Hispanic white; ROS/MAP = Religious Orders Study / Memory and Aging Project; PMI = postmortem interval.
2.1. Caribbean-Hispanics LOAD transcriptome-wide differential expression analysis
We first sought to identify LOAD-differentially expressed genes in CH brain. Robust regression modelling of LOAD status on gene transcript abundance (log2(CPM)) was adjusted for demographic and technical covariates, including a term for PSEN1 mutation carrier status. To further ensure that PSEN1 status would not bias our results, we performed principal components analyses on the CH expression dataset with and without the removal of co-variate effects, confirming that mutation carriers did not cluster separately from the rest of the sample (Fig. S2). Differential expression analysis found a total of 2802 differentially-expressed genes at an FDR significant threshold of q < 0.05 (Table S1). Among the most significantly differentially expressed genes were many with known roles in LOAD, including CD33, TREM2, CR1, CD2AP, HLA-DRB1, MS4A4A, FBXL7, among additional genes identified by the most recent LOAD GWAS meta-analysis (Bellenguez et al., 2022). To functionally characterize differentially expressed genes, we analyzed those significantly up- (n = 1335) and down-regulated (n = 1467) separately, using the FUMA-GWAS tool. Up-regulated genes were significantly enriched for the “BLALOCK_ALZHEIMERS_DISEASE_UP” gene set (pFDR = 9.4 × 10−29) as well as 2578 additional gene sets primarily related to immune cell activation, vascular function, cancers, and components of the extracellular matrix (full results are reported in Table S2). In contrast, the LOAD down-regulated gene set was enriched for the “BLALOCK_ALZHEIMERS_DISEASE_DN” gene set (pFDR = 6.2 × 10−21) and showed fewer additional significant enrichments (152), mostly corresponding to neuronal signaling components, consistent with synaptic loss observed in LOAD.
2.2. Non-Hispanic differential expression analyses and cross-ancestry gene expression comparison
In the Religious Orders Study and Memory and Aging Project (ROS/MAP) and Mayo Clinic non-Hispanic samples, differential expression analyses found 2066/17,665 and 10,878/19,380 significant genes, respectively (Fig. 2D–E; summary statistics in Tables S3 and S4). Validation of these results against published differential expression analyses in these cohorts (Canchi et al., 2019; Logsdon et al., 2019; Mostafavi et al., 2018) demonstrated substantial coherence of transcriptome-wide effects (Supplementary file; Figs. S3 and S4). Comparing between populations, 373 genes reached genome-wide significance in all three differential expression analyses (nuniverse = 15,834, expected overlap = 184; overlap hypergeometric p = 6.6 × 10−43); 364 (98%) with concordant directions of effect (Fig. 2F). Among these, many were of known significance to LOAD, including TREM2, ICA1, VASP, MTMR3, GFAP, and GRIK1 (cross-sample effects of top up- and down-regulated genes shown in Fig. 2G). At uncorrected p < 0.05, 1441 genes were significant in all three analyses, 1360 (94%) with concordant directions of effect. Cross-ancestry meta-analysis found NPNT to be the top DE gene (p = 1.04 × 10−19). Among the LOAD-known genes, several were significantly DE, including FBXL7, TREM2, IQCK, RIN3, CD33, INPP5D, PTK2B, CLU, AD2AP, MEF2C and WWOX (full list in Table S5), with 43 LOAD-known genes at least nominally DE.
Fig. 2.
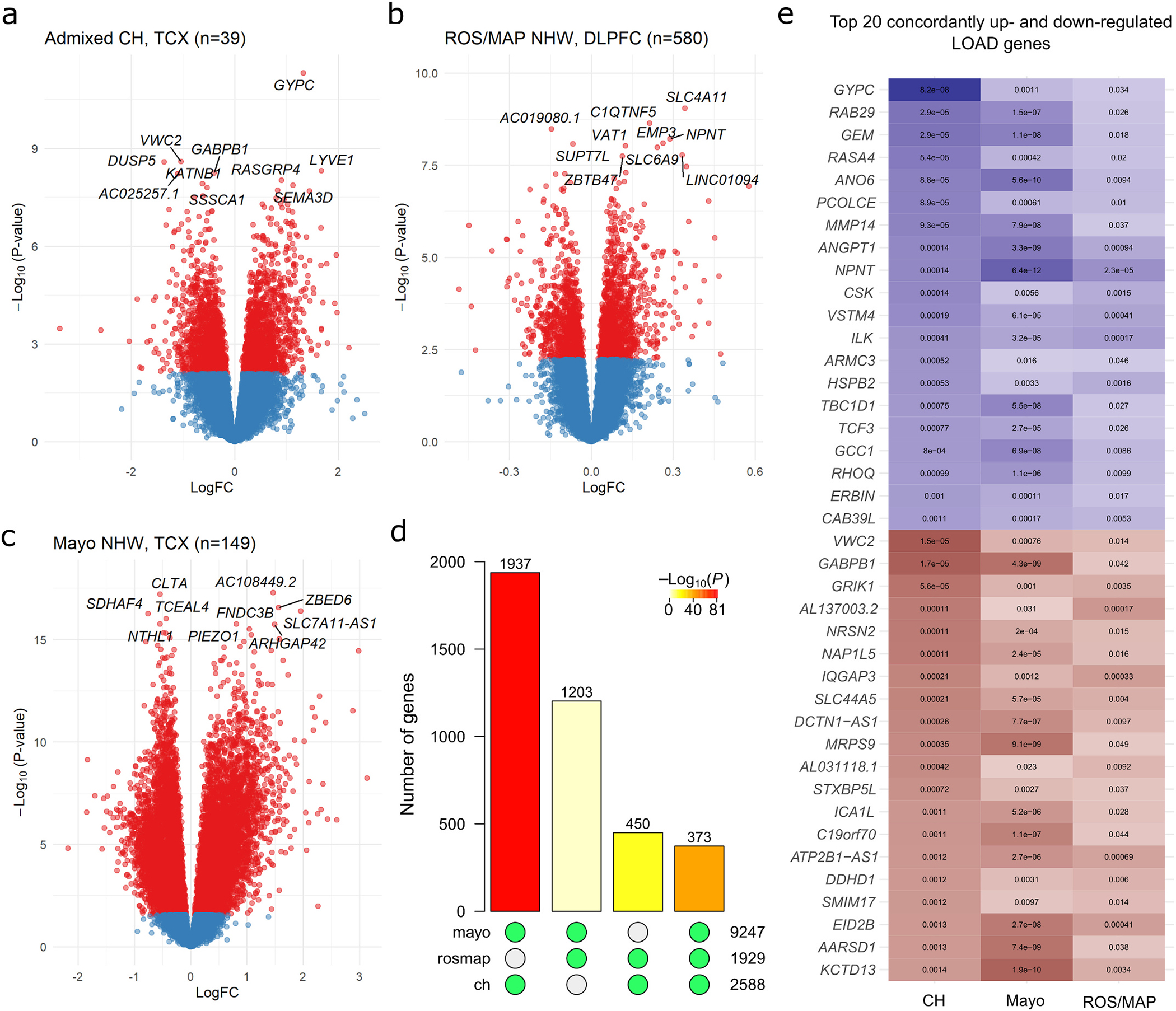
Results of single-gene transcriptome-wide associations with LOAD in Caribbean-Hispanics and non-Hispanic Whites. Gene-level volcano plots for association of genes with LOAD status in A) genetically-confirmed CH subjects (n = 39), and two independent samples of non-Hispanic whites: B) ROS/MAP and C) the Mayo RNAseq cohort. The top ten significantly differentially expressed genes in each analysis are labeled. The Y-axes indicate two-sided −log10(p-values) for robust regression testing differential expression The X-axes indicate log2 fold-change in expression. D) Barplot showing the number of transcriptome-wide FDR-significant (q < 0.05) differentially expressed genes in common between each pair of samples and across all three datasets. Green dots below each bar indicate which sets of significant genes are intersecting. Bar color indicates the p-value for hypergeometric testing of overlap probability (scale shown). E) The top 20 up- and top 20 down-regulated LOAD genes with significant, concordant effects across all three samples, ranked by significance in the CH sample. The color is proportional to moderated t-statistics (blue for positive effect, or higher expression in LOAD, and red for negative effect, or lower expression in LOAD), with FDR-adjusted two-sided p-values labeled inside of tiles. DLPFC = dorsolateral prefrontal cortex source tissue; LogFC = log2 fold-change in expression; NHW = non-Hispanic White; TCX = temporal cortex source tissue.
We then aimed to identify genes with ancestry-specific effects by comparing differential expression statistics between 1) CH vs. ROS/MAP, 2) CH vs. Mayo, and 3) ROS/MAP vs. Mayo, among the set of genes analyzed in all three studies (ngenes = 15,834). We observed a greater proportion of replicated signals between CH and Mayo (1937 in common out of 2588 in CH = 75% replication; expected overlap = 1511, overlap p = 7.8 × 10−81) than between CH and ROS/MAP (450/2588 = 17% replication; expected overlap = 315, overlap p = 1.3 × 10−17). Consistently, pairwise Pearson correlations of the three study’s t-statistics revealed moderate positive relationships (CH vs. Mayo r = 0.57, C. I.99% = [0.56,0.59]; CH vs. ROS/MAP r = 0.47, C.I.99% = [0.46,0.49]; ROS/MAP vs. Mayo r = 0.46, C.I.99% = [0.45,0.48]) (Fig. S5). There were 118 individual genes with significant effects in CH (pFDR < 0.05) and an opposite direction of effect in both Mayo and ROS/MAP (top 10 over- and under-expressed shown in Table S6).
2.3. Identification of ancestry-discordant and –concordant LOAD-related biological processes
To harness the full sets of ranked summary statistics for differential expression in all three analyses, we calculated the pairwise product of t-statistics (“tprod”) for each gene between each pair of analyses. This metric provides a transcriptome-wide ranked measure of similarity in expression magnitude and direction between samples, and was used as input for rank-based gene set enrichment tests. Using this approach, we identified 20 unique, semantically non-redundant biological processes with significant ancestry-discordant or -concordant LOAD-related effects in at least one cross-sample comparison (Fig. 3A). CH vs. Mayo (the comparison with the greatest number of discordant (n = 5) and concordant (n = 11) gene sets): discordant processes included those related to transcription and translation, whereas concordant processes included those related to cellular metabolism, immune response, synaptic transmission, and biomineralization. Notably, the “response to beta-amyloid” (pFDR = 0.013) process was also concordantly downregulated in both datasets, though it was not assigned a dispensability score of 0 by REVIGO (disp = 0.07). Similar themes emerged for both the CH vs. ROS/MAP and ROS/MAP vs. Mayo comparisons, where ribosomal genes were again found to be enriched for discordant effects and neuronal genes for concordant effect, though concordant signals between ROS/MAP and Mayo also included exocrine system development (Fig. 3A,B). Importantly, several of the identified GO groups across sample comparisons had strong overlap in gene identity (S7 Figure), with a core set of 78 genes highly overrepresented in protein localization, viral transcription, and translational initiation categories driving discordant effects.
Fig. 3.
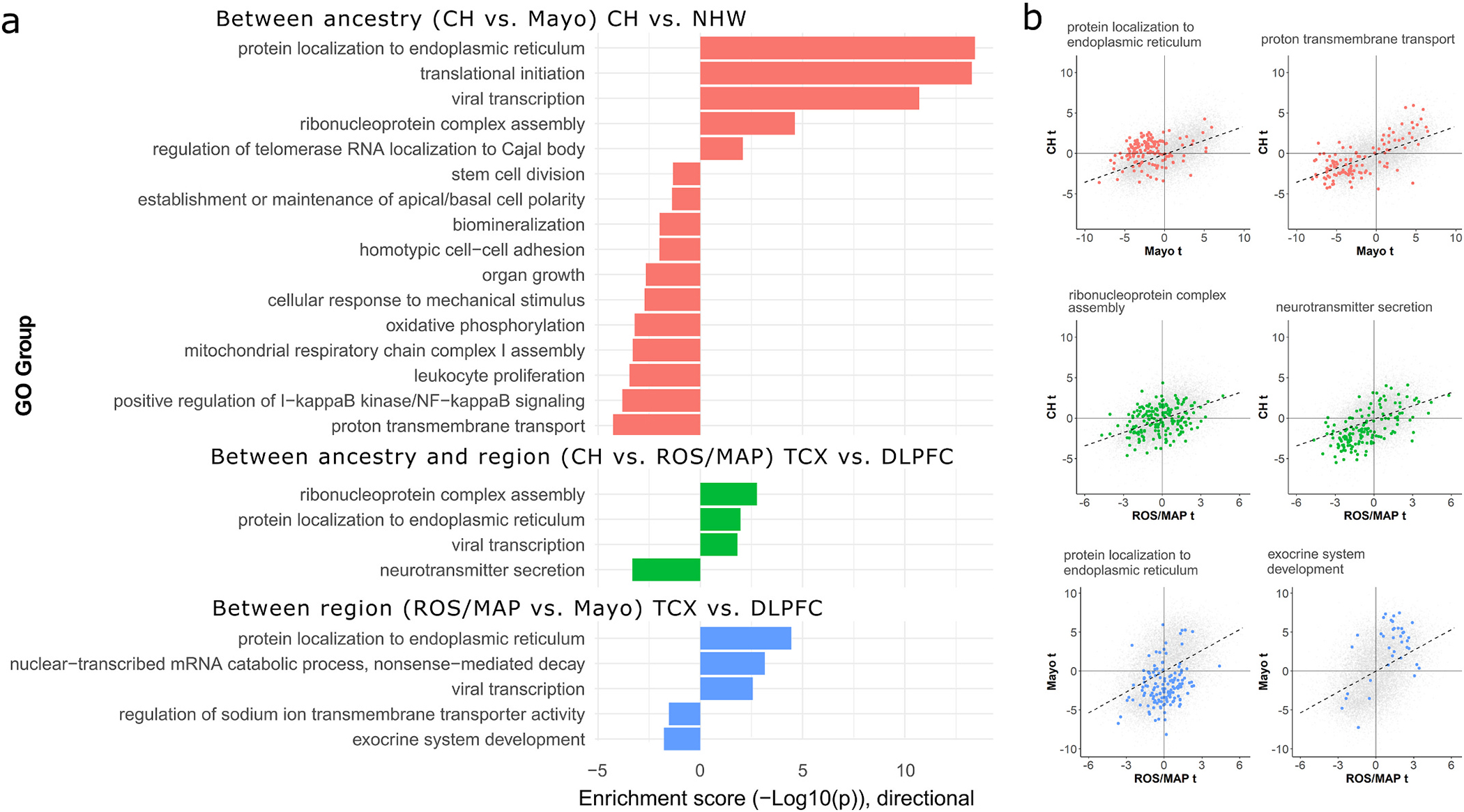
Enrichment analyses for genes with concordant and discordant LOAD effects between ancestry and brain region tissue source. A) Barplot summarizing AUC-based GO enrichment analysis using tprod ranks for all FDR-significant GO terms with a REVIGO dispensability score of 0. The x-axes indicate the enrichment −log10 (two-sided p-values), with values above 0 indicating enrichment toward higher ranks (greater discordance of between-sample effect) and values below 0 indicating enrichment toward lower tanks (greater concordance of between-sample effect). B) Scatterplots for the top discordant (left column) and concordant (right column) groups from panel A for each sample contrast, showing the LOAD differential expression t-values for genes belonging to each enriched gene set within the context of all genes. Genes belonging to labeled GO sets in panel B are colored to match the barplot in panel A. CH = Caribbean-Hispanic; FDR = false discovery rate; GO = gene ontology; DLPFC = dorsolateral prefrontal cortex source tissue; ROS/MAP = Religious Orders Study and Memory and Aging Project; TCX = temporal cortex source tissue.
2.4. Weighted Gene Co-Expression Network Analysis (WGCNA)
Moving beyond the individual gene level to gene networks, we performed consensus co-expression network analyses on all three datasets to understand the degree of shared and distinct network architecture among CH and NHW transcriptomes. A total of 35 discrete consensus gene modules were identified across CH and NHW samples, ranging from 51 to 2715 genes in size (Fig. 4A; Table S7), with 16 modules significantly enriched for at least one biological process (Table S8). Association tests of module eigengenes with LOAD in each dataset revealed largely conserved network-level effects, where CH vs. ROS/MAP effects were most cohesive (r = 0.81, C.I.95% = [0.65,0.90]), followed by Mayo vs. CH (r = 0.68, C.I.95% = [0.45,0.83]) and Mayo vs. ROS/MAP (r = 0.56, C.I.95% = [0.28,0.75]). Several modules with concordant and discordant LOAD associations were identified (Fig. 4B–E; full results in Table S9), among which the blue concordant module was enriched for mitochondrial genes. Further, the discordant pink module (significantly over-expressed in CH, but under-expressed in Mayo) largely represented ribosomal genes (“SRP-dependent cotranslational protein targeting to membrane” pFDR = 7.0 × 10−80).
Fig. 4.
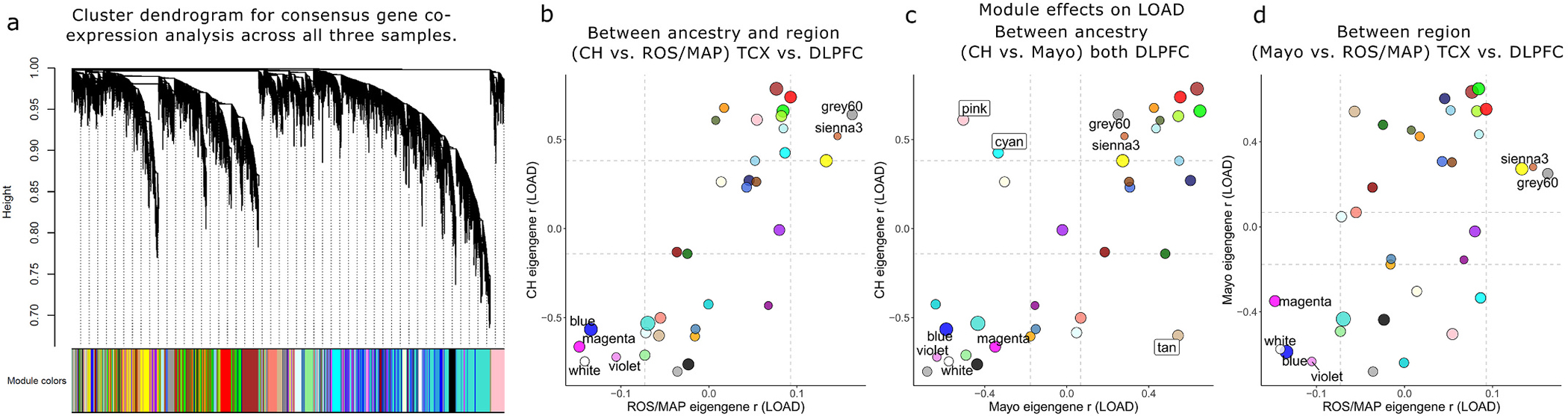
Consensus gene co-expression network module analysis with LOAD. A) Clustered dendrogram showing the common gene set hierarchical structure and consensus gene module definitions. Panels B–D show consensus module eigengene associations with LOAD (Pearson r) for all modules compared between pairwise sample combinations. Modules with concordant effects (pFDR < 0.05 with same direction of effect) in all three samples, or significantly discordant (pFDR < 0.05 in at least two samples but with opposite direction of effect), are labeled; six modules were significantly associated with LOAD in all three samples; greenyellow, brown, and royalblue modules were consistently downregulated, while the red, purple, and lightyellow modules were upregulated. In contrast, four modules showed significant but directionally discordant effects on LOAD when comparing CH to Mayo; salmon and lightcyan modules were significantly upregulated in CH but downregulated in Mayo, whereas black and cyan modules were downregulated in CH but upregulated in Mayo. Colors correspond to module definitions. CH = Caribbean-Hispanic; DLPFC = dorsolateral prefrontal cortex; ROS/MAP = Religious Orders Study and Memory and Aging Project; TCX = temporal cortex.
2.5. Comparison of bulk RNAseq to single cell transcriptomics
To localize our previous findings to specific brain cell types, we analyzed high-depth single-nucleus RNAseq data (snRNAseq) from entorhinal cortex (EC) and superior frontal gyrus (SFG) (Leng et al., 2021). Following clustering, we determined seven major cell types: astroglia, microglia, inhibitory neurons, excitatory neurons, oligodendrocytes, oligodendrocyte progenitors and endothelia (Fig. 5A, B). We then performed differential expression analysis between AD cases and controls (Braak stage 6 versus Braak stage 0, full results for EC per cell type in Table S10) within each cell type cluster separately and cross-referenced these results with our bulk brain-based findings. Regarding the cross-ancestry meta-analysis, 1050 genes were gene-wide significant (p < 1 × 10−6; Table S8): 938 of these genes were expressed in at least 1 cell type in the scRNA-seq, while 829 genes were differentially expressed (DEGs) in at least one cell type in the single cell (Fig. 5D). Excitatory neurons (ExN) showed 679 DEGs (64.7% of the GWA genes in Table S8), followed by astroglia (AG, 636 DEGs, 60.1%), inhibitory neurons (InN, 564 DEGs, 53.7%), oligodendrocytes (OD, 551 DEGs, 52.5%), oligodendrocyte progenitors (OPC, 543 DEGs, 51.7%), microglia (MG, 341 DEGs, 32.5%), and endothelia (EN, 93 DEGs, 8.9%) (Fig. 5D). These genes included the top hit NPNT (AG, InN, OPC, ExN). In addition, 69 out of 75 known LOAD-associated genes were also differentially expressed in at least one cell type in our single-nucleus analysis (Fig. 5C), including SORL1, ABCA7, PLCG2, CLU, FBXL7, UNC5C. For instance, ABCA7 was found DE in neurons only while FBXL7 and IQCK were differently expressed in all cell types but microglia. Finally, GYPC (top DE gene in CH transcriptome) was also found upregulated in astroglia (p = 0.012), microglia (p = 0,009), OPC (p = 3.9 × 10−6), excitatory neurons (p = 2.2 × 10−8).
Fig. 5.
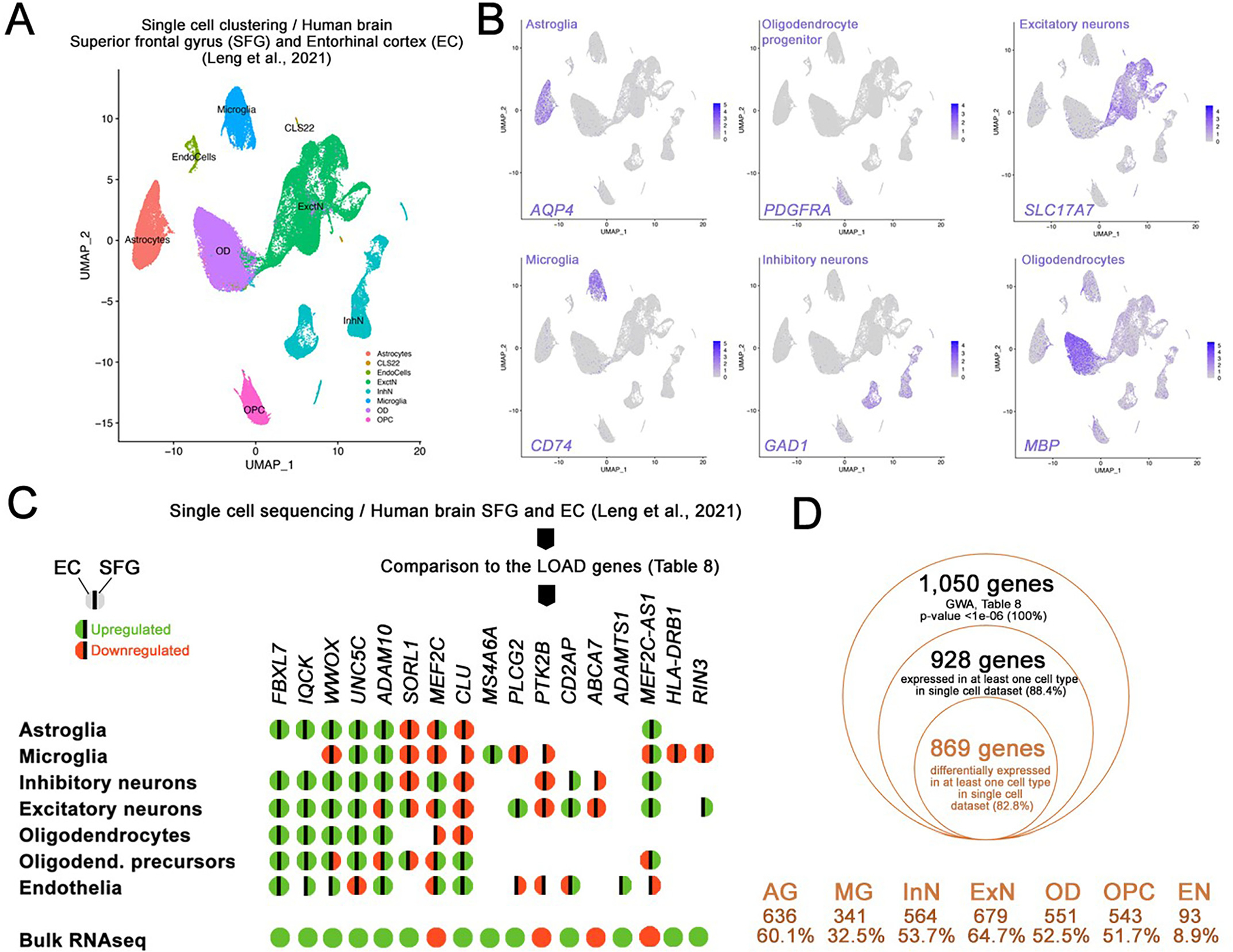
Secondary independent validation of the bulk RNA sequencing with single cell transcriptomics from human brains. (A) Clustering of single cell dataset in Leng et al., 2021 (human brains, EC and SFG, AD cases versus controls; Braak stage 6 versus 0). 7 major cell types were identified as indicated. (B) tSNE plots for major cell type markers (only one out of many were shown) AQP4 for astroglia, PDGFRA for oligodendrocyte progenitors, CD74 for microglia, GAD1 for inhibitory neurons, MBP for oligodendrocytes, SLC17A7 for excitatory neurons. (C) Genes in Table 8 were run against the single cell datasets and differentially expressed genes in various cell types in entorhinal cortex (EC) and superior frontal gyrus (SFG) of human brains with and without 17 CE genes are shown for their differential gene expression in major cell types. Green: upregulation, red: downregulation, left hemisphere: EC, right hemisphere: SFG. No color indicates no significant differential expression. (D) Comparison of GWA genes (Table 8) in single cell dataset (human EC, Braak stage 6 vs 0). 869 genes (82.6% of the initially selected GWA genes) were validated for their differential expression in human EC in at least 1 cell type. Cell types and the number/percentage of the initially selected GWA genes are shown for astroglia (AG), microglia (MG), inhibitory neurons (InN), excitatory neurons (ExN), oligodendrocytes (OD), oligodendrocyte progenitors (OPC), and endothelia (EN).
3. Discussion
To our knowledge, this is the first investigation of gene expression from postmortem LOAD brain in CH individuals. Understanding ancestry-specific molecular profiles of LOAD brain tissue is a first step toward developing interventions effective in non-European Caucasian high-risk populations. We report over 2000 genes differentially expressed in LOAD brain vs. controls within the CH cohort, many with known relevance to LOAD pathophysiology. For example, the top up-regulated gene was GYPC, or glycophorin C, which is found upregulated in human LOAD microglia (Srinivasan et al., 2020) (notably, this gene is also nominally significant in both ROS/MAP and Mayo; p = 0.003, p = 0.0003 respectively). In our single-cell analyses, we consistently found this gene upregulated in astroglia, microglia, oligodendrocyte progenitors and excitatory neurons (Table S9). Up-regulated genes in CH were found significantly enriched for several GWAS catalog traits, including “late onset Alzheimer’s disease” (pFDR = 0.03) and “intraocular pressure” (pFDR = 5.6 × 10−3). The latter is a condition associated with open-angle glaucoma, which shows higher prevalence in Hispanics than NHW (Grosvenor and Hennis, 2011; Quigley et al., 2001) as well as overlaps with genetic risk for LOAD across multiple populations (Gharahkhani et al., 2020).
Our results show substantial overlap in LOAD-related genes between CH and NHW - particularly when comparing same brain region (i.e. in temporal cortex; CH vs. Mayo). This higher concordance suggests that differences in LOAD-related gene expression patterns are greater between sampled brain regions than between ancestries. This degree of inter-regional concordance mirrors recent findings of differential gene expression across multiple brain regions in LOAD (Patel et al., 2019) and Schizophrenia (Collado-Torres et al., 2019). Several genes were consistently differentially expressed across cohorts: among the most significant, many were LOAD known-loci. Notably, BACE2, the lesser known homolog (Wang et al., 2019, p. 2) of the LOAD-associated β-secretase enzyme BACE1, was the eighth most strongly overexpressed gene in our study (pFDR = 4 × 10−5). Most importantly, we found the known CH GWAS-implicated LOAD risk gene, FBXL7 (Tosto et al., 2015a), overexpressed in LOAD at FDR-corrected pFDR < 0.05 in all three samples. At the pathways level, those related to immune cell proliferation, cell killing, and neurotransmitter secretion were found overlapping.
In contrast, among the strongest ancestry-specific genes was CSPG4 (see Table S6), a chondroitin sulfate proteoglycan (also known as neuron-glial antigen 2) which is expressed in oligodendrocyte precursor cells and a subpopulation of NG2+ astrocytes, has been strongly linked to LOAD pathology in humans and mice (Nielsen et al., 2013; Vanzulli et al., 2020), and harbors rare genetic variants in Central American/Hispanic families linked to schizophrenia (de Vrij et al., 2019). Core genes that were increased in LOAD in CH, but decreased in NHW, suggested ancestry-driven differences in the activation of translational machinery in LOAD. Ancestry-specific enrichment analyses highlighted roles of ribosomal genes and those involved in protein synthesis and trafficking, which are emerging as robust and important markers of LOAD pathogenesis (Garcia-Esparcia et al., 2017; Ghosh et al., 2020; Grothe et al., 2018; Hernández-Ortega et al., 2016; Patel et al., 2020; Rasmussen et al., 2015; Shigemizu et al., 2020). We speculate that (SRP)-dependent protein targeting may be disproportionately perturbed in CH patients, given the strong population-specific enrichment in both the single gene and network co-expression analyses. Intriguingly, SRP-dependent genes are strongly dysregulated in periodontitis-affected periodontal tissue (Lundmark et al., 2018). The main driver of periodontitis pathogenesis, P. gingivalis, is known to secrete gingipains which have been linked to LOAD pathogenesis in mice (Ilievski et al., 2018) and humans (Dominy et al., 2019). This connection is re-enforced by the significant enrichment of the GWAS catalog periodontitis gene set in our CH LOAD-upregulated genes. Notably, nephronectin, coded by the NPNT gene (the top DE gene in our cross-ancestral meta-analysis) has been shown to have a critical role in human dental pulp stem cells (Tang and Saito, 2017) and is a top ranking gene in large AD proteomic studies (Bai et al., 2020).
Among our top results, the “protein localization to endoplasmic reticulum” GO group was unanimously enriched in discordantly LOAD-related genes, whether comparing between ancestry, between sampled brain regions, or both ancestry and region (Fig. 3A). Notably, this enrichment was most significant in the between ancestry comparison (same brain region; CH vs. Mayo), highlighting a hierarchy of effects for protein trafficking and related biological processes (e.g. “translational initiation” and “viral transcription; see Fig. S7 for overlap) between the three cohorts. In CH, these genes are largely up-regulated, whereas in Mayo (NHW), these genes are downregulated. While there is moderate trend toward downregulation in ROS/MAP (also NHW), the overall signal driving the strongest enrichment appears to be driven by the unique increases in protein trafficking genes in the CH cohort. While this result is difficult to contextualize given the lack of comparable CH samples, previous work has demonstrated that the effects of LOAD biomarkers (e.g. CSF tau, CSF IL-9, MRI-based functional connectivity) may be strongly attenuated or even reversed in direction between NHW and African American populations (Babulal et al., 2019; Howell et al., 2017; Wharton et al., 2019). Pilot analyses (n = 9 AA; n = 11 NHW) from postmortem brain has also showed over 180 proteins with significant race x LOAD diagnosis interactions, pointing again to ancestry discordant molecular signatures of LOAD and motivating future multi-ancestry ‘omic investigations (Stepler et al., 2020).
Bulk RNA sequencing is a powerful method for determining tissue-level differences in gene abundance. However, is incapable of determining cell type-specific changes in a definitive manner. In our single-nucleus analyses, we validated over 82% of the genes identified by our cross-ancestry meta-analysis, including the top hit, NPNT. This gene was found upregulated in astroglia, oligodendrocyte progenitors and neurons. Several genes with known significant to LOAD showed high consistency between our bulk and scRNAseq data. For example, ABCA7 was found downregulated in neurons only and MS4A6A was found differentially expressed in microglia. UNC5C was found upregulated in all major cell types, with the exception of endothelial cells, where it was found to be downregulated. CLU (found to be upregulated in bulk analyses) was upregulated in endothelial cells and oligodendrocyte precursors, but downregulated in other cell types. This suggests that these genes may have different roles in cell-specific physiology. In contrast, FBXL7 – which has a genome-wide significant risk variant for LOAD in CH individuals (Tosto et al., 2015a) - was found upregulated in nearly all cell types. Our results demonstrate that comparison between bulk and single cell sequencing data can provide useful information on cell types that differentially express genes of interest. This approach can refine biological hypotheses on the functional relevance of these genes to LOAD pathophysiology.
Key limitations in this study include different methods of tissue ascertainment and study design that ultimately impact the results; ROS and MAP are community-based, prospective cohort studies, whereas the CH and Mayo samples are case-control designs selected for diagnosis. Additionally, CH samples were not scanned for rare LOAD mutations using whole genome sequencing, though PSEN1 carriers were identified by imputation or brain-bank record (see Supplementary file). CH specifically are a relatively small brains sample collected over several decades; the difficulties of ascertaining brains in this specific population have mired the availability of large sample sizes of tissue collected over shorter periods (i.e. higher quality). Hispanics tend to not participate in either organ donation in general, or brain donation more specifically, to the same extent as non-Hispanic Whites (NHW) (Frates and Garcia Bohrer, 2002). This resulted in samples with relatively low RNA quality (median RIN = 4.5). Nevertheless, by performing ribosomal RNA depletion prior to sequencing, we ensured that the impact of low RNA quality was mitigated. In fact, ribosomal RNA depletion has been shown to perform very well (Kumar et al., 2017; Schuierer et al., 2017) at amounts far below recommendation and over a wide range of intact and degraded material. Despite these challenges, our analyses proved reliable by showing a moderate concordance between results from all three samples at the genome-wide scale. We were also able to identify many well-known LOAD-associated loci from previous GWAS and sequencing studies such as FBXL7 (previously identified by a CH-specific GWAS from our group (Tosto et al., 2015a)). Finally, we note that differences in age at death between LOAD and non-LOAD participants in the ROS/MAP and CH cohorts were observed (Table 1), meaning that correction for age at death in linear modelling may have partially accounted for a small portion of LOAD-specific differences between groups.
In sum, we performed RNA-sequencing on postmortem brain from a small sample of CH elderly and two large independent NHW cohorts, ultimately identifying numerous candidate genes and biological processes that are consistently dysregulated in LOAD across ancestry or show ancestry-specific dysregulation. Among those genes showing the strongest ancestry-specific effects, we found significant enrichment for ribosomal pathways. Further work in large admixed cohorts will permit a deeper understanding of ancestry-specific mechanisms that may be used to predict risk, onset of pathology, and potentially provide precision treatment options.
4. Materials and methods
4.1. Caribbean Hispanic cohort
We processed postmortem temporal cortex (TCX) tissue from 45 unrelated self-reported CH individuals ascertained from the New York Brain Bank (NYBB) at Columbia University (New York, NY, USA). This research clinic referral-based brain bank consists of over 600 brains from autopsies performed between 1989 and 2016. Cases were selected if age > 50 years old, and neuropathologically defined LOAD or control without any neuropathological diagnosis. The protocol was approved by the institutional review boards of the New York State Psychiatric Institute and Columbia University. Details of the tissue preparation and neuropathological assessment are reported in the Supplementary file and extensively described elsewhere (Vonsattel et al., 1995, 2008). RNA-sequencing methods, expression quantification, and initial quality control are reported in the Supplementary file. Whole-genotyping and admixture estimation has been extensively described in a recent publication(Sariya et al., 2021) from our group and also reported in the Supplementary file.
4.2. Non-Hispanic White (NHW) cohorts
We included in this study two NHW cohorts: 1) The Religious Orders Study and Memory and Aging Project (ROS/MAP) and 2) the Mayo Clinic RNAseq cohort. The sample characteristics of the ROS/MAP cohort subset studied here have been published in detail elsewhere (Bennett et al., 2018). Briefly, RNA sequencing data was analyzed from postmortem dorsolateral prefrontal cortex (DLPFC) of 595 subjects (Bennett et al., 2018). After quality control (QC; see Supplementary file), 583 subjects remained (nAD = 354, nnon-AD = 229). The Mayo RNAseq cohort has also been described in detail (Allen et al., 2018; Allen et al., 2016). After QC (see Supplementary file), 148 subjects remained (nAD = 80, nnon-AD = 68). Protocols were approved by the Mayo Clinic Institutional Review Board and all subjects or next of kin provided informed consent. Neuropathological evaluation details and RNAseq methods are reported in the Supplementary file.
4.3. Ethics approval and consent to participate
For the Caribbean-Hispanic Alzheimer’s Disease Research Centre (ADRC) New York Brain Bank dataset, the protocol was approved by the institutional review boards of the New York State Psychiatric Institute and Columbia University. For the Religious Orders Study and Memory and Aging Project, all study participants provided informed consent and both studies were approved by the Rush University Institutional Review Board. For the Mayo dataset, protocols were approved by the Mayo Clinic Institutional Review Board and all subjects or next of kin provided informed consent.
4.4. RNAseq processing and LOAD Transcriptome-wide differential expression analysis
Gene counts for all three cohorts were post-processed identically. Counts were read into R (v3.6.3) for processing with edgeR (Robinson et al., 2010) and limma/voom (Law et al., 2014; Ritchie et al., 2015). TMM normalization values (using edgeR calcNormFactors) and mean-variance derived observational-level weights were then calculated for linear modelling using limma/voom. For differential expression analyses, the limma package ‘lmFit’ function was used to model log2(expected counts) as a linear function of pathological LOAD status, sequencing batch (brain bank source and flowcell included for Mayo), age, sex, RIN, postmortem interval (not available for the CH sample), percent of usable bases (not included for CH, as it was highly co-linear with RIN, and additional parameters were detrimental to model performance given small sample size), percent duplicated reads, median 3′ bias, and percent of mapped ribosomal bases. In CH models that included PSEN1 mutation carriers, a parameter was added to model its effect. Significance of the contrast between LOAD and non-LOAD status (LOAD vs. control for Mayo) was performed using empirical Bayes moderation (eBayes function).
For ROS/MAP, a stratified sensitivity analysis was performed to ensure that differences in the age range between ROS/MAP and CH samples did not impact our comparisons (Supplementary file). Further validation analyses were performed to ensure consistency of our differential expression analyses of ROS/MAP and Mayo cohorts with previously published work (Supplementary file).
4.5. Differential expression cross-ancestry meta-analysis
We meta-analyzed results from the three transcriptome-wide differential expression analyses employing a fixed-effect model and using logFC and its standard error (SE) as inputs, consistently with previous studies of transcriptomics in LOAD (Patel et al., 2019). The SE was calculated from the logFC values for each gene, separately in each cohort, and used for standard meta-analysis using the R package “rmeta”. LogFC and se ensured that direction of effect was taken into consideration when performing the meta-analysis. Multiple testing correction was applied using the false discovery rate (FDR) method.
4.6. Cross-ancestry comparison of differential expression
To compare differential expression results from our CH and NHW sample analyses (i.e. ROS/MAP and Mayo), we identified genes with 1) ancestry-independent effects - i.e. those with replicated associations with LOAD across CH and NHW samples, and 2) ancestry-specific effects - i.e. those significant in CH and neither NHW samples. To incorporate directionality of effect, magnitude of effect, and effect standard error into our assessment of effect concordance and discordance across samples, we ranked genes by the product of their t-statistics (“tprod”) for association with AD, assigning the highest ranks to genes with the largest discrepancy in direction and effect size between ancestries (i.e. CH vs. Mayo and CH vs. ROS/MAP, separately). Thus, higher ranks indicated sample-specific (“ancestry-discordant”) effects, whereas lower ranks indicated sample-nonspecific (“ancestry-concordant”) effects. To test the impact of gene inclusion criteria, effect statistic, and effect direction on inter-sample correlations, we performed a series of sensitivity analyses (Supplementary file and S6 Figure).
4.7. Functional enrichment analyses
To functionally characterize AD-associated genes in our CH DE analysis we used the Functional Mapping and Annotation of Genome-Wide Association Studies (FUMA-GWAS) GENE2FUNC online utility (https://fuma.ctglab.nl/gene2func/). We performed hypergeometric tests for gene set enrichment separately for genes significantly (transcriptome-wide FDR-adjusted p-value <0.05) up- and down-regulated in LOAD. For rank-based enrichment analyses of cross-sample concordant and discordant gene lists, an area under the curve (AUC) rank-based method was used, as implemented in the tmod R package (Weiner, 2020). Resulting significant GO groups (pFDR < 0.05) were consolidated to minimize semantic redundancy using the REVIGO tool (Supek et al., 2011). For the presentation of results, we chose to select only GO terms with a REVIGO dispensability score of 0; this decision was made to provide the most succinct summary of an otherwise large number of enrichment results while retaining all major relevant pathways. Annotations were limited to those including between 10 and 200 tested genes. GO annotations were extracted from the org.Hs.eg.db and GO.db (version 3.10.0) R packages (Carlson, 2016a, 2016b). For gene co-expression modules, hypergeometric testing was used to evaluate functional enrichment, using the background universe of genes passing QC for consensus module analysis. For all three samples, genes assigned to each consensus module were tested for GO term enrichment including categories of biological processes, cellular components, and molecular functions (Ashburner et al., 2000). FDR correction was used (q < 0.05) to determine significant enrichment.
4.8. Consensus Weighted Gene Co-Expression Network Analysis (WGCNA)
Consensus gene co-expression modules were defined across all three samples using Weighted Gene Co-Expression Analysis (WGCNA) (Zhang and Horvath, 2005) applied to batch- and covariate-corrected partial gene expression residuals (partial with respect to LOAD diagnosis). Detailed description of the method and non-default parameters used in the Supplementary file.
4.9. Comparison of bulk tissue results with single-nucleus RNA sequencing data from human brain
single-nucleus RNA sequencing data (n = 10) for human superior frontal gyrus (SFG) and entorhinal cortex (EC) samples were downloaded from the Gene Expression Omnibus repository under the following GEO ID; GSE147528 (Leng et al., 2021). After extensive QC, we used Seurat V3.1.5 (Hao et al., 2021) for further analyses as described in the Supplementary file. In total, 26 cell clusters (numbered from 0 to 25) were identified. We then generated heat maps and cell types were inferred based on the characteristic gene expression patterns. Major cell types were identified using the following markers (based on Leng et al., 2021): GFAP, SLC1A2 and AQP4 (astroglia); MBP and MOG (oligodendrocytes or OD); PDGFRA and SOX10 (oligondendrocyte precursors or OPC); CD74 and CX3CR1 (microglia); SLC17A7 and CAMK2A (excitatory Neurons, or ExctN); GAD1 and GAD2 (inhibitory Neurons or OD); CLND5 and FLT1 (endothelial cells or EndoCells). Details are provided in the Supplementary file. To identify the differentially expressed genes (DEGs), we compared cases (samples with Braak stage 6) vs. controls (Braak Stage 0) within each main cell type, across Entorhinal Cortex (EC) and superior frontal gyrus (SFG), using the FindMarker function in Seurat. To identify which cell types exhibited overlapping DEGs with our findings from bulk RNAseq, we selected experiment-wide significant (p-value <1.0 × 10−6) from our cross-ancestry meta-analysis, and searched for differentially expressed genes (p < 0.05) in the single-nucleus experiment. We also perform the same investigation for LOAD known-genes.
Supplementary Material
Funding
This work was supported by grants from the National Institutes of Health: R56AG069118, R56AG066889, R21AG054832, R56AG059756, R01AG056531, P50AG008702, P30AG10161, R01AG15819, R01AG17917, U01AG61356, R01AG015473, R56AG063908, P30AG066462, R01NS095922 and P50AG0008702. Funding support for DF was provided by The Koerner Family Foundation New Scientist Program and The Krembil Family Foundation. Taub Institute Schaefer Research Scholars Award (CK) and German Center for Neurodegenerative Diseases (DZNE) (MIC, CK) provided support.
Footnotes
Declaration of Competing Interest
Leon French owns shares in Cortexyme Inc., a company that is developing a gingipain inhibitor to treat Alzheimer’s Disease. The other authors declare no conflict of interest. Funders did not play any role in the design, analysis, or writing or this study.
Data and materials availability
The datasets supporting the conclusions of this article (ROS/MAP and Mayo) are available via approved access at the Synapse AMP-AD Knowledge Portal (https://adknowledgeportal.synapse.org/, doi: https://doi.org/10.7303/syn2580853). All RNA sequencing data for the Caribbean-Hispanic sample will be made available via GEO repository following publication. All analyses were performed using open-source software. No custom algorithms or software were used that are central to the research or not yet described in published literature. Open-sourced code for running all analyses and generating figures presented in the manuscript can be found on github at https://www.github.com/dfelsky/CH_LOAD_TWAS.
CRediT authorship contribution statement
Daniel Felsky: Conceptualization, Methodology, Data curation, Formal analysis, Software, Data curation, Writing – original draft, Writing – review & editing. Ismael Santa-Maria: Data curation, Writing – review & editing. Mehmet Ilyas Cosacak: Investigation, Formal analysis, Validation, Writing – review & editing. Leon French: Conceptualization, Formal analysis, Software. Julie A. Schneider: Investigation. David A. Bennett: Funding acquisition, Investigation. Philip L. De Jager: Funding acquisition, Investigation. Caghan Kizil: Investigation, Formal analysis, Validation, Writing – review & editing. Giuseppe Tosto: Conceptualization, Data curation, Formal analysis, Funding acquisition, Writing – review & editing.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2022.105938.
Data availability
Some data used is currently available within online repositories, and some will be made available online at time of publication (as described in article).
References
- Acosta D, Llibre-Guerra JJ, Jiménez-Velázquez IZ, Llibre-Rodríguez JJ, 2021. Dementia research in the Caribbean Hispanic Islands: present findings and future trends. Front. Public Health 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M, Carrasquillo MM, Funk C, Heavner BD, Zou F, Younkin CS, Burgess JD, Chai H-S, Crook J, Eddy JA, Li H, Logsdon B, Peters MA, Dang KK, Wang X, Serie D, Wang C, Nguyen T, Lincoln S, Malphrus K, Bisceglio G, Li M, Golde TE, Mangravite LM, Asmann Y, Price ND, Petersen RC, Graff-Radford NR, Dickson DW, Younkin SG, Ertekin-Taner N, 2016. Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci. Data 3, 160089. 10.1038/sdata.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M, Wang X, Burgess JD, Watzlawik J, Serie DJ, Younkin CS, Nguyen T, Malphrus KG, Lincoln S, Carrasquillo MM, Ho C, Chakrabarty P, Strickland S, Murray ME, Swarup V, Geschwind DH, Seyfried NT, Dammer EB, Lah JJ, Levey AI, Golde TE, Funk C, Li H, Price ND, Petersen RC, Graff-Radford NR, Younkin SG, Dickson DW, Crook JR, Asmann YW, Ertekin-Taner N, 2018. Conserved brain myelination networks are altered in Alzheimer’s and other neurodegenerative diseases. Alzheimers Dement. 14, 352–366. 10.1016/j.jalz.2017.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G, 2000. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athan ES, Williamson J, Ciappa A, Santana V, Romas SN, Lee JH, Rondon H, Lantigua RA, Medrano M, Torres M, Arawaka S, Rogaeva E, Song YQ, Sato C, Kawarai T, Fafel KC, Boss MA, Seltzer WK, Stern Y, St George-Hyslop P, Tycko B, Mayeux R, 2001. A founder mutation in presenilin 1 causing early-onset Alzheimer disease in unrelated Caribbean Hispanic families. JAMA 286, 2257–2263. 10.1001/jama.286.18.2257. [DOI] [PubMed] [Google Scholar]
- Babulal GM, Quiroz YT, Albensi BC, Arenaza-Urquijo E, Astell AJ, Babiloni C, Bahar-Fuchs A, Bell J, Bowman GL, Brickman AM, Chételat G, Ciro C, Cohen AD, Dilworth-Anderson P, Dodge HH, Dreux S, Edland S, Esbensen A, Evered L, Ewers M, Fargo KN, Fortea J, Gonzalez H, Gustafson DR, Head E, Hendrix JA, Hofer SM, Johnson LA, Jutten R, Kilborn K, Lanctôt KL, Manly JJ, Martins RN, Mielke MM, Morris MC, Murray ME, Oh ES, Parra MA, Rissman RA, Roe CM, Santos OA, Scarmeas N, Schneider LS, Schupf N, Sikkes S, Snyder HM, Sohrabi HR, Stern Y, Strydom A, Tang Y, Terrera GM, Teunissen C, van Lent DM, Weinborn M, Wesselman L, Wilcock DM, Zetterberg H, O’Bryant SE, 2019. Perspectives on ethnic and racial disparities in Alzheimer’s disease and related dementias: update and areas of immediate need. Alzheimers Dement. J. Alzheimers Assoc. 15, 292–312. 10.1016/j.jalz.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B, Wang X, Li Y, Chen P-C, Yu K, Dey KK, Yarbro JM, Han X, Lutz BM, Rao S, Jiao Y, Sifford JM, Han J, Wang M, Tan H, Shaw TI, Cho J-H, Zhou S, Wang H, Niu M, Mancieri A, Messler KA, Sun X, Wu Z, Pagala V, High AA, Bi W, Zhang H, Chi H, Haroutunian V, Zhang B, Beach TG, Yu G, Peng J, 2020. Deep multilayer brain proteomics identifies molecular networks in Alzheimer’s disease progression. Neuron 105, 975–991.e7. 10.1016/j.neuron.2019.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, Naj AC, Campos-Martin R, Grenier-Boley B, Andrade V, Holmans PA, Boland A, Damotte V, van der Lee SJ, Costa MR, Kuulasmaa T, Yang Q, de Rojas I, Bis JC, Yaqub A, Prokic I, Chapuis J, Ahmad S, Giedraitis V, Aarsland D, Garcia-Gonzalez P, Abdelnour C, Alarcón-Martín E, Alcolea D, Alegret M, Alvarez I, Álvarez V, Armstrong NJ, Tsolaki A, Antúnez C, Appollonio I, Arcaro M, Archetti S, Pastor AA, Arosio B, Athanasiu L, Bailly H, Banaj N, Baquero M, Barral S, Beiser A, Pastor AB, Below JE, Benchek P, Benussi L, Berr C, Besse C, Bessi V, Binetti G, Bizarro A, Blesa R, Boada M, Boerwinkle E, Borroni B, Boschi S, Bossù P, Bråthen G, Bressler J, Bresner C, Brodaty H, Brookes KJ, Brusco LI, Buiza-Rueda D, Bûrger K, Burholt V, Bush WS, Calero M, Cantwell LB, Chene G, Chung J, Cuccaro ML, Carracedo Á, Cecchetti R, Cervera-Carles L, Charbonnier C, Chen H-H, Chillotti C, Ciccone S, Claassen JAHR, Clark C, Conti E, Corma-Gómez A, Costantini E, Custodero C, Daian D, Dalmasso MC, Daniele A, Dardiotis E, Dartigues J-F, de Deyn PP, de Paiva Lopes K, de Witte LD, Debette S, Deckert J, del Ser T, Denning N, DeStefano A, Dichgans M, Diehl-Schmid J, Diez-Fairen M, Rossi PD, Djurovic S, Duron E, Düzel E, Dufouil C, Eiriksdottir G, Engelborghs S, Escott-Price V, Espinosa A, Ewers M, Faber KM, Fabrizio T, Nielsen SF, Fardo DW, Farotti L, Fenoglio C, Fernández-Fuertes M, Ferrari R, Ferreira CB, Ferri E, Fin B, Fischer P, Fladby T, Fließbach K, Fongang B, Fornage M, Fortea J, Foroud TM, Fostinelli S, Fox NC, Franco-Macías E, Bullido MJ, Frank-García A, Froelich L, Fulton-Howard B, Galimberti D, García-Alberca JM, García-González P, Garcia-Madrona S, Garcia-Ribas G, Ghidoni R, Giegling I, Giorgio G, Goate AM, Goldhardt O, Gomez-Fonseca D, González-Pérez A, Graff C, Grande G, Green E, Grimmer T, Grünblatt E, Grunin M, Gudnason V, Guetta-Baranes T, Haapasalo A, Hadjigeorgiou G, Haines JL, Hamilton-Nelson KL, Hampel H, Hanon O, Hardy J, Hartmann AM, Hausner L, Harwood J, Heilmann-Heimbach S, Helisalmi S, Heneka MT, Hernández I, Herrmann MJ, Hoffmann P, Holmes C, Holstege H, Vilas RH, Hulsman M, Humphrey J, Biessels GJ, Jian X, Johansson C, Jun GR, Kastumata Y, Kauwe J, Kehoe PG, Kilander L, Ståhlbom AK, Kivipelto M, Koivisto A, Kornhuber J, Kosmidis MH, Kukull WA, Kuksa PP, Kunkle BW, Kuzma AB, Lage C, Laukka EJ, Launer L, Lauria A, Lee C-Y, Lehtisalo J, Lerch O, Lleó A, Longstreth W, Lopez O, de Munain AL, Love S, Löwemark M, Luckcuck L, Lunetta KL, Ma Y, Macías J, MacLeod CA, Maier W, Mangialasche F, Spallazzi M, Marquié M, Marshall R, Martin ER, Montes AM, Rodríguez CM, Masullo C, Mayeux R, Mead S, Mecocci P, Medina M, Meggy A, Mehrabian S, Mendoza S, Menéndez-González M, Mir P, Moebus S, Mol M, Molina-Porcel L, Montrreal L, Morelli L, Moreno F, Morgan K, Mosley T, Nöthen MM, Muchnik C, Mukherjee S, Nacmias B, Ngandu T, Nicolas G, Nordestgaard BG, Olaso R, Orellana A, Orsini M, Ortega G, Padovani A, Paolo C, Papenberg G, Parnetti L, Pasquier F, Pastor P, Peloso G, Pérez-Cordón A, Pérez-Tur J, Pericard P, Peters O, Pijnenburg YAL, Pineda JA, Piñol-Ripoll G, Pisanu C, Polak T, Popp J, Posthuma D, Priller J, Puerta R, Quenez O, Quintela I, Thomassen JQ, Rábano A, Rainero I, Rajabli F, Ramakers I, Real LM, Reinders MJT, Reitz C, Reyes-Dumeyer D, Ridge P, Riedel-Heller S, Riederer P, Roberto N, Rodriguez-Rodriguez E, Rongve A, Allende IR, Rosende-Roca M, Royo JL, Rubino E, Rujescu D, Sáez ME, Sakka P, Saltvedt I, Sanabria Á, Sánchez-Arjona MB, Sanchez-Garcia F, Juan PS, Sánchez-Valle R, Sando SB, Sarnowski C, Satizabal CL, Scamosci M, Scarmeas N, Scarpini E, Scheltens P, Scherbaum N, Scherer M, Schmid M, Schneider A, Schott JM, Selbæk G, Seripa D, Serrano M, Sha J, Shadrin AA, Skrobot O, Slifer S, Snijders GJL, Soininen H, Solfrizzi V, Solomon A, Song Y, Sorbi S, Sotolongo-Grau O, Spalletta G, Spottke A, Squassina A, Stordal E, Tartan JP, Tárraga L, Tesí N, Thalamuthu A, Thomas T, Tosto G, Traykov L, Tremolizzo L, Tybjærg-Hansen A, Uitterlinden A, Ullgren A, Ulstein I, Valero S, Valladares O, Broeckhoven CV, Vance J, Vardarajan BN, van der Lugt A, Dongen JV, van Rooij J, van Swieten J, Vandenberghe R, Verhey F, Vidal J-S, Vogelgsang J, Vyhnalek M, Wagner M, Wallon D, Wang L-S, Wang R, Weinhold L, Wiltfang J, Windle G, Woods B, Yannakoulia M, Zare H, Zhao Y, Zhang X, Zhu C, Zulaica M, Farrer LA, Psaty BM, Ghanbari M, Raj T, Sachdev P, Mather K, Jessen F, Ikram MA, de Mendonça A, Hort J, Tsolaki M, Pericak-Vance MA, Amouyel P, Williams J, Frikke-Schmidt R, Clarimon J, Deleuze J-F, Rossi G, Seshadri S, Andreassen OA, Ingelsson M, Hiltunen M, Sleegers K, Schellenberg GD, van Duijn CM, Sims R, van der Flier WM, Ruiz A, Ramirez A, Lambert J-C, 2022. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436. 10.1038/s41588-022-01024-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA, 2018. Religious orders study and rush memory and aging project. J. Alzheimers Dis. JAD 64, S161–S189. 10.3233/JAD-179939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrò M, Rinaldi C, Santoro G, Crisafulli C, 2020. The biological pathways of Alzheimer disease: a review. AIMS Neurosci. 8, 86–132. 10.3934/Neuroscience.2021005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canchi S, Raao B, Masliah D, Rosenthal SB, Sasik R, Fisch KM, De Jager PL, Bennett DA, Rissman RA, 2019. Integrating gene and Protein expression reveals perturbed functional networks in Alzheimer’s disease. Cell Rep. 28, 1103–1116.e4. 10.1016/j.celrep.2019.06.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson M, 2016a. org.Hs.eg.db.
- Carlson M, 2016b. GO.db.
- Celis K, Griswold AJ, Bussies PL, Rajabli F, Whitehead P, Dorfsman D, Hamilton-Nelson K, Bigio E, Mesulam M, Geula C, Weintraub S, Gearing M, Beecham G, Scott W, Pericak-Vance M, Young J, Vance J, 2020. Transcriptome analysis of single nucleus RNA-seq from Alzheimer disease APOE4 carrier brains in African American (AA) and non-Hispanic whites (NHW) reveals differences in APOE expression (4626). Neurology 94. [Google Scholar]
- Collado-Torres L, Burke EE, Peterson A, Shin J, Straub RE, Rajpurohit A, Semick SA, Ulrich WS, Price AJ, Valencia C, Tao R, Deep-Soboslay A, Hyde TM, Kleinman JE, Weinberger DR, Jaffe AE, 2019. Regional heterogeneity in gene expression, regulation, and coherence in the frontal cortex and Hippocampus across development and schizophrenia. Neuron 103, 203–216.e8. 10.1016/j.neuron.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consens ME, Chen Y, Menon V, Wang Y, Schneider JA, De Jager PL, Bennett DA, Tripathy SJ, Felsky D, 2022. Bulk and single-nucleus transcriptomics highlight intra-telencephalic and somatostatin neurons in Alzheimer’s disease. Front. Mol. Neurosci. 15, 903175 10.3389/fnmol.2022.903175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vrij FM, Bouwkamp CG, Gunhanlar N, Shpak G, Lendemeijer B, Baghdadi M, Gopalakrishna S, Ghazvini M, Li TM, Quadri M, Olgiati S, Breedveld GJ, Coesmans M, Mientjes E, de Wit T, Verheijen FW, Beverloo HB, Cohen D, Kok RM, Bakker PR, Nijburg A, Spijker AT, Haffmans PMJ, Hoencamp E, Bergink V, Vorstman JA, Wu T, Olde Loohuis LM, Amin N, Langen CD, Hofman A, Hoogendijk WJ, van Duijn CM, Ikram MA, Vernooij MW, Tiemeier H, Uitterlinden AG, Elgersma Y, Distel B, Gribnau J, White T, Bonifati V, Kushner SA, 2019. Candidate CSPG4 mutations and induced pluripotent stem cell modeling implicate oligodendrocyte progenitor cell dysfunction in familial schizophrenia. Mol. Psychiatry 24, 757–771. 10.1038/s41380-017-0004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, Nguyen M, Haditsch U, Raha D, Griffin C, Holsinger LJ, Arastu-Kapur S, Kaba S, Lee A, Ryder MI, Potempa B, Mydel P, Hellvard A, Adamowicz K, Hasturk H, Walker GD, Reynolds EC, Faull RLM, Curtis MA, Dragunow M, Potempa J, 2019. Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 5, eaau3333. 10.1126/sciadv.aau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frates J, Garcia Bohrer G, 2002. Hispanic perceptions of organ donation. Prog. Transplant. Aliso Viejo Calif 12, 169–175. 10.7182/prtr.12.3.fn70x07413742531. [DOI] [PubMed] [Google Scholar]
- Garcia-Esparcia P, Sideris-Lampretsas G, Hernandez-Ortega K, Grau-Rivera O, Sklaviadis T, Gelpi E, Ferrer I, 2017. Altered mechanisms of protein synthesis in frontal cortex in Alzheimer disease and a mouse model. Am. J. Neurodegener. Dis. 6, 15–25. [PMC free article] [PubMed] [Google Scholar]
- Gharahkhani P, Jorgenson E, Hysi P, Khawaja AP, Pendergrass S, Han X, Ong JS, Hewitt AW, Segre A, Igo RP, Choquet H, Qassim A, Josyula NS, Bailey JNC, Bonnemaijer P, Iglesias A, Siggs OM, Young T, Vitart V, Thiadens AAHJ, Karjalainen J, Uebe S, Melles RB, Nair KS, Luben R, Simcoe M, Amersinghe N, Cree AJ, Hohn R, Poplawski A, Chen LJ, Cheng C-Y, Vithana EN, Consortium N, Consortium A, Project BJ, Study F, Consortium UBE and Group V, study G, Team, 23andMe Research, Tamiya G, Shiga Y, Yamamoto M, Nakazawa T, Rouhana J, Currant H, Birney E, Wang X, Auton A, Ashaye A, Olawoye O, Williams SE, Akafo S, Ramsay M, Hashimoto K, Kamatani Y, Akiama M, Momozawa Y, Foster PJ, Khaw PT, Morgan JE, Strouthidis NG, Kraft P, Kang JH, Pang CCP, Pasutto F, Mitchell P, Lotery AJ, Palotie A, van Duijn C, Haines J, Hammond C, Pasquale LR, Klaver CCW, Hauser M, Khor CC, Mackey DA, Kubo M, Aung T, Craig J, MacGregor S, Wiggs J, 2020. A large cross-ancestry meta-analysis of genome-wide association studies identifies 69 novel risk loci for primary open-angle glaucoma and includes a genetic link with Alzheimer’s disease. bioRxiv. 10.1101/2020.01.30.927822, 2020.01.30.927822. [DOI] [Google Scholar]
- Ghosh A, Mizuno K, Tiwari SS, Proitsi P, Gomez Perez-Nievas B, Glennon E, Martinez-Nunez RT, Giese KP, 2020. Alzheimer’s disease-related dysregulation of mRNA translation causes key pathological features with ageing. Transl. Psychiatry 10, 1–18. 10.1038/s41398-020-00882-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griswold AJ, Gardner OK, Feliciano-Astacio BE, Van Booven D, Hamilton-Nelson KL, Whitehead PL, Adams LD, Starks TD, Acosta H, Cuccaro ML, Vance JM, Byrd GS, Haines JL, Bush WS, Beecham GW, Pericak-Vance MA, 2021. Expression quantitative trait loci (eQTL) analysis in a diverse Alzheimer disease cohort reveals ancestry-specific regulatory architectures. Alzheimers Dement. 17, e056211 10.1002/alz.056211. [DOI] [Google Scholar]
- Grosvenor D, Hennis A, 2011. Glaucoma in the English-speaking Caribbean. West Indian Med. J. 60, 459–463. [PubMed] [Google Scholar]
- Grothe MJ, Sepulcre J, Gonzalez-Escamilla G, Jelistratova I, Schöll M, Hansson O, Teipel SJ, Alzheimer’s Disease Neuroimaging Initiative, 2018. Molecular properties underlying regional vulnerability to Alzheimer’s disease pathology. Brain J. Neurol. 141, 2755–2771. 10.1093/brain/awy189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, Simmons RK, Buckberry S, Vargas-Landin DB, Poppe D, Pflueger J, Lister R, Rackham OJL, Petretto E, Polo JM, 2019. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 22, 2087–2097. 10.1038/s41593-019-0539-4. [DOI] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R, 2021. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Ortega K, Garcia-Esparcia P, Gil L, Lucas JJ, Ferrer I, 2016. Altered machinery of protein synthesis in Alzheimer’s: from the nucleolus to the ribosome. Brain Pathol. Zurich Switz. 26, 593–605. 10.1111/bpa.12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell JC, Watts KD, Parker MW, Wu J, Kollhoff A, Wingo TS, Dorbin CD, Qiu D, Hu WT, 2017. Race modifies the relationship between cognition and Alzheimer’s disease cerebrospinal fluid biomarkers. Alzheimers Res. Ther. 9, 88. 10.1186/s13195-017-0315-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilievski V, Zuchowska PK, Green SJ, Toth PT, Ragozzino ME, Le K, Aljewari HW, O’Brien-Simpson NM, Reynolds EC, Watanabe K, 2018. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS One 13, e0204941. 10.1371/journal.pone.0204941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jooma S, Hahn MJ, Hindorff LA, Bonham VL, 2019. Defining and achieving health equity in genomic medicine. Ethn. Dis. 29, 173–178. 10.18865/ed.29.S1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Kankainen M, Parsons A, Kallioniemi O, Mattila P, Heckman CA, 2017. The impact of RNA sequence library construction protocols on transcriptomic profiling of leukemia. BMC Genomics 18, 629. 10.1186/s12864-017-4039-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle BW, Schmidt M, Klein H-U, Naj AC, Hamilton-Nelson KL, Larson EB, Evans DA, De Jager PL, Crane PK, Buxbaum JD, Ertekin-Taner N, Barnes LL, Fallin MD, Manly JJ, Go RCP, Obisesan TO, Kamboh MI, Bennett DA, Hall KS, Goate AM, Foroud TM, Martin ER, Wang L-S, Byrd GS, Farrer LA, Haines JL, Schellenberg GD, Mayeux R, Pericak-Vance MA, Reitz C, Writing Group for the Alzheimer’s Disease Genetics Consortium (ADGC), Graff-Radford NR, Martinez I, Ayodele T, Logue MW, Cantwell LB, Jean-Francois M, Kuzma AB, Adams LD, Vance JM, Cuccaro ML, Chung J, Mez J, Lunetta KL, Jun GR, Lopez OL, Hendrie HC, Reiman EM, Kowall NW, Leverenz JB, Small SA, Levey AI, Golde TE, Saykin AJ, Starks TD, Albert MS, Hyman BT, Petersen RC, Sano M, Wisniewski T, Vassar R, Kaye JA, Henderson VW, DeCarli C, LaFerla FM, Brewer JB, Miller BL, Swerdlow RH, Van Eldik LJ, Paulson HL, Trojanowski JQ, Chui HC, Rosenberg RN, Craft S, Grabowski TJ, Asthana S, Morris JC, Strittmatter SM, Kukull WA, 2021. Novel Alzheimer disease risk loci and pathways in African American individuals using the African genome resources panel: a meta-analysis. JAMA Neurol. 78, 102–113. 10.1001/jamaneurol.2020.3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake J, Solsberg CW, Kim JJ, Acosta-Uribe J, Makarious MB, Li Z, Levine K, Heutink P, Alvarado C, Vitale D, Kang S, Gim J, Lee KH, Pina-Escudero SD, Ferrucci L, Singleton AB, Blauwendraat C, Nalls MA, Yokoyama JS, Leonard HL, 2022. Multi-Ancestry Meta-Analysis and Fine-Mapping in Alzheimer’s Disease. 10.1101/2022.08.04.22278442. [DOI] [PMC free article] [PubMed]
- Law CW, Chen Y, Shi W, Smyth GK, 2014. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, Neff N, Li SH, Rodriguez RD, Suemoto CK, Leite REP, Ehrenberg AJ, Pasqualucci CA, Seeley WW, Spina S, Heinsen H, Grinberg LT, Kampmann M, 2021. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci. 24, 276–287. 10.1038/s41593-020-00764-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon BA, Perumal TM, Swarup V, Wang M, Funk C, Gaiteri C, Allen M, Wang X, Dammer E, Srivastava G, Mukherjee S, Sieberts SK, Omberg L, Dang KD, Eddy JA, Snyder P, Chae Y, Amberkar S, Wei W, Hide W, Preuss C, Ergun A, Ebert PJ, Airey DC, Carter GW, Mostafavi S, Yu L, Klein H-U, Consortium the A.-A, Collier DA, Golde T, Levey A, Bennett DA, Estrada K, Decker M, Liu Z, Shulman JM, Zhang B, Schadt E, Jager PLD, Price ND, Ertekin-Taner N, Mangravite LM, 2019. Meta-analysis of the human brain transcriptome identifies heterogeneity across human AD coexpression modules robust to sample collection and methodological approach. bioRxiv 510420. 10.1101/510420. [DOI] [Google Scholar]
- Lundmark A, Gerasimcik N, Båge T, Jemt A, Mollbrink A, Salmén F, Lundeberg J, Yucel-Lindberg T, 2018. Gene expression profiling of periodontitis-affected gingival tissue by spatial transcriptomics. Sci. Rep. 8, 9370. 10.1038/s41598-018-27627-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai L-H, 2019. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Estrada A, Gravel S, Zakharia F, McCauley JL, Byrnes JK, Gignoux CR, Ortiz-Tello PA, Martínez RJ, Hedges DJ, Morris RW, Eng C, Sandoval K, Acevedo-Acevedo S, Norman PJ, Layrisse Z, Parham P, Martínez-Cruzado JC, Burchard EG, Cuccaro ML, Martin ER, Bustamante CD, 2013. Reconstructing the population genetic history of the Caribbean. PLoS Genet. 9, e1003925 10.1371/journal.pgen.1003925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan SL, Naderi P, Koler K, Pita-Juarez Y, Prokopenko D, Vlachos IS, Tanzi RE, Bertram L, Hide WA, 2022. Most pathways can be related to the pathogenesis of Alzheimer‘s Disease. Front. Aging Neurosci. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafavi S, Gaiteri C, Sullivan SE, White CC, Tasaki S, Xu J, Taga M, Klein H-U, Patrick E, Komashko V, McCabe C, Smith R, Bradshaw EM, Root DE, Regev A, Yu L, Chibnik LB, Schneider JA, Young-Pearse TL, Bennett DA, Jager PLD, 2018. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci. 21, 811–819. 10.1038/s41593-018-0154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen HM, Ek D, Avdic U, Orbjörn C, Hansson O, Netherlands Brain Bank, Veerhuis R, Rozemuller AJ, Brun A, Minthon L, Wennström M, 2013. NG2 cells, a new trail for Alzheimer’s disease mechanisms? Acta Neuropathol. Commun. 1, 7. 10.1186/2051-5960-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel H, Dobson RJB, Newhouse SJ, 2019. A Meta-analysis of Alzheimer’s disease brain transcriptomic data. J. Alzheimers Dis. JAD 68, 1635–1656. 10.3233/JAD-181085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Howard D, Man A, Schwartz D, Jee J, Felsky D, Pausova Z, Paus T, French L, 2020. Donor-specific transcriptomic analysis of Alzheimer’s disease-associated Hypometabolism highlights a unique donor, ribosomal proteins and microglia. eNeuro 7. 10.1523/ENEURO.0255-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, West SK, Rodriguez J, Munoz B, Klein R, Snyder R, 2001. The prevalence of glaucoma in a population-based study of Hispanic subjects: Proyecto VER. Arch. Ophthalmol. Chic. Ill 1960 (119), 1819–1826. 10.1001/archopht.119.12.1819. [DOI] [PubMed] [Google Scholar]
- Rasmussen L, de Labio RW, Viani GA, Chen E, Villares J, Bertolucci P-H, Minett TS, Turecki G, Cecyre D, Drigo SA, Smith MC, Payao SLM, 2015. Differential expression of ribosomal genes in brain and blood of Alzheimer’s disease patients. Curr. Alzheimer Res. 12, 984–989. 10.2174/1567205012666151027124017. [DOI] [PubMed] [Google Scholar]
- Reddy JS, Jin J, Lincoln SJ, Ho CCG, Crook JE, Wang X, Malphrus KG, Nguyen T, Tamvaka N, Greig-Custo MT, Lucas JA, Graff-Radford NR, Ertekin-Taner N, Carrasquillo MM, 2022. Transcript levels in plasma contribute substantial predictive value as potential Alzheimer’s disease biomarkers in African Americans. eBioMedicine 78. 10.1016/j.ebiom.2022.103929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C, Mayeux R, 2014. Genetics of Alzheimer’s disease in Caribbean Hispanic and African American populations. Biol. Psychiatry 75, 534–541. 10.1016/j.biopsych.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, 2015. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK, 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinforma. Oxf. Engl. 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariya S, Felsky D, Reyes-Dumeyer D, Lali R, Lantigua RA, Vardarajan B, Jiménez-Velázquez IZ, Haines JL, Shellenberg GD, Pericak-Vance MA, Paré G, Mayeux R, Tosto G, 2021. Polygenic risk score for Alzheimer’s disease in Caribbean Hispanics. Ann. Neurol. 10.1002/ana.26131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuierer S, Carbone W, Knehr J, Petitjean V, Fernandez A, Sultan M, Roma G, 2017. A comprehensive assessment of RNA-seq protocols for degraded and low-quantity samples. BMC Genomics 18, 442. 10.1186/s12864-017-3827-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemizu D, Mori T, Akiyama S, Higaki S, Watanabe H, Sakurai T, Niida S, Ozaki K, 2020. Identification of potential blood biomarkers for early diagnosis of Alzheimer’s disease through RNA sequencing analysis. Alzheimers Res. Ther. 12, 87. 10.1186/s13195-020-00654-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirugo G, Williams SM, Tishkoff SA, 2019. The missing diversity in human genetic studies. Cell 177, 26–31. 10.1016/j.cell.2019.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan K, Friedman BA, Etxeberria A, Huntley MA, van der Brug MP, Foreman O, Paw JS, Modrusan Z, Beach TG, Serrano GE, Hansen DV, 2020. Alzheimer’s patient microglia exhibit enhanced aging and unique transcriptional activation. Cell Rep. 31, 107843 10.1016/j.celrep.2020.107843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepler KE, Mahoney ER, Kofler J, Hohman TJ, Lopez OL, Robinson RAS, 2020. Inclusion of African American/black adults in a pilot brain proteomics study of Alzheimer’s disease. Neurobiol. Dis. 146, 105129 10.1016/j.nbd.2020.105129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, Šmuc T, 2011. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6, e21800. 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Saito T, 2017. The role of Nephronectin on proliferation and differentiation in human dental pulp stem cells. Stem Cells Int. 2017, e2546261 10.1155/2017/2546261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosto G, Fu H, Vardarajan BN, Lee JH, Cheng R, Reyes-Dumeyer D, Lantigua R, Medrano M, Jimenez-Velazquez IZ, Elkind MSV, Wright CB, Sacco RL, Pericak-Vance M, Farrer L, Rogaeva E, St George-Hyslop P, Reitz C, Mayeux R, 2015a. F-box/LRR-repeat protein 7 is genetically associated with Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2, 810–820. 10.1002/acn3.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosto G, Lee JH, Vardarajan BN, Cheng R, Reyes-Dumeyer D, Barral S, Reitz C, Mayeux R, 2015b. O3–05-04: admixture analysis of Alzheimer’s disease in caribbean hispanics. Alzheimers Dement. 11, P229. 10.1016/j.jalz.2015.07.264. [DOI] [Google Scholar]
- Vanzulli I, Papanikolaou M, De-La-Rocha IC, Pieropan F, Rivera AD, Gomez-Nicola D, Verkhratsky A, Rodríguez JJ, Butt AM, 2020. Disruption of oligodendrocyte progenitor cells is an early sign of pathology in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 94, 130–139. 10.1016/j.neurobiolaging.2020.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, Aizawa H, Ge P, DiFiglia M, McKee AC, MacDonald M, Gusella JF, Landwehrmeyer GB, Bird ED, Richardson EP, 1995. An improved approach to prepare human brains for research. J. Neuropathol. Exp. Neurol. 54, 42–56. 10.1097/00005072-199501000-00006. [DOI] [PubMed] [Google Scholar]
- Vonsattel JPG, Del Amaya MP, Keller CE, 2008. Twenty-first century brain banking. Processing brains for research: the Columbia University methods. Acta Neuropathol. (Berl.) 115, 509–532. 10.1007/s00401-007-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Xu Q, Cai F, Liu X, Wu Y, Song W, 2019. BACE2, a conditional β-secretase, contributes to Alzheimer’s disease pathogenesis. JCI Insight 4, 123431. 10.1172/jci.insight.123431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger DR, Dzirasa K, Crumpton-Young LL, 2020. Missing in action: African ancestry brain research. Neuron 107, 407–411. 10.1016/j.neuron.2020.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner J, 2020. tmod: Feature Set Enrichment Analysis for Metabolomics and Transcriptomics.
- Wharton W, Kollhoff AL, Gangishetti U, Verble DD, Upadhya S, Zetterberg H, Kumar V, Watts KD, Kippels AJ, Gearing M, Howell JC, Parker MW, Hu WT, 2019. Interleukin 9 alterations linked to alzheimer disease in african americans. Ann. Neurol. 86, 407–418. 10.1002/ana.25543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng B, Bendl J, Kosoy R, Fullard JF, Hoffman GE, Roussos P, 2022. Multi-ancestry eQTL meta-analysis of human brain identifies candidate causal variants for brain-related traits. Nat. Genet. 54, 161–169. 10.1038/s41588-021-00987-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S, 2005. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, Article17. 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Some data used is currently available within online repositories, and some will be made available online at time of publication (as described in article).