

Abstract
Motivation
Accurate diagnostic classification and biological interpretation are important in biology and medicine, which are data-rich sciences. Thus, integration of different data types is necessary for the high predictive accuracy of clinical phenotypes, and more comprehensive analyses for predicting the prognosis of complex diseases are required.
Results
Here, we propose a novel multi-task attention learning algorithm for multi-omics data, termed MOMA, which captures important biological processes for high diagnostic performance and interpretability. MOMA vectorizes features and modules using a geometric approach and focuses on important modules in multi-omics data via an attention mechanism. Experiments using public data on Alzheimer’s disease and cancer with various classification tasks demonstrated the superior performance of this approach. The utility of MOMA was also verified using a comparison experiment with an attention mechanism that was turned on or off and biological analysis.
Availability and implementation
The source codes are available at https://github.com/dmcb-gist/MOMA.
Supplementary information
Supplementary materials are available at Bioinformatics online.
1 Introduction
Deep neural networks (DNNs) have been developed with diverse variations for increased performance according to different goals and domains; these include convolutional neural networks based on image data, recurrent neural networks based on sequence data, generative adversarial networks based on adversarial training and auto-encoders (AE) for learning representation. However, in biology and medicine, models require not only performance and prediction accuracy but also interpretability (Hanczar et al., 2020): interpretability helps physicians make accurate decisions related to diagnosis and treatment courses, thus impacting patients’ lives. High-performance models also reveal new biological relationships, for which interpretability is indispensable.
Module detection is an essential process for interpreting the heterogeneity between samples, as genetic or transcriptional variation differs with samples, even for the same disease. A module in this context is defined as a gene set with inter-relationships between genes. Many studies, using various machine learning methods, have identified inter-relationships including canonical correlation analysis (Min et al., 2021), clustering (Fu and Medico, 2007), generalization of principal component analysis (Argelaguet et al., 2018) and non-negative matrix factorization (NMF) (Yang and Michailidis, 2016). Recently, Min et al. (2021) constructed a canonical correlation analysis-based model and identified cancer-specific and shared microRNA-gene modules using multi-omics data sets of 33 cancer types from The Cancer Genome Atlas (TCGA). However, these traditional methods miss non-linear relationships between features (Argelaguet et al., 2018). Recently, neural network-based models have been developed (Chen et al., 2018; Dwivedi et al., 2020) that overcome these shortcomings and solve the lack of interpretability. Chen et al. (2018) have used AEs to embed gene-set nodes, include genes in the gene set through the first layer, and combine the gene set with the superset through weights in the second layer. The study by Dwivedi et al. (2020) uses AE and a light-up procedure to associate genes with hidden nodes to derive disease modules. These lines of work focus on unsupervised learning; however, with the accumulation of clinical phenotypes, such as disease presence, disease stage, drug response and medical value, supervised learning with clinical phenotypes is more appropriate for broad and flexible interpretation.
With advances in high-throughput techniques, the large amount of biomedical data, including molecular and medical information, have been accumulated (Bennett et al., 2018; Chen et al., 2021; Hutter and Zenklusen, 2018; Zeng et al., 2021), which can improve our current understanding of various diseases. Integration of these data promises to improve the prognosis and predictive accuracy of clinical phenotypes and facilitate more comprehensive analyses of complex disease prognosis (Hasin et al., 2017; Subramanian et al., 2020). Existing studies have demonstrated the advantage of integrating multi-biomedical data with various approaches, including matrix factorization (Chen and Zhang, 2018; Lock et al., 2013; Zhang et al., 2011, 2012; Zhang and Zhang, 2019), networks (Huang et al., 2019a; Koh et al., 2019) and DNNs (Chaudhary et al., 2018; Huang et al., 2019b; Sharifi-Noghabi et al., 2019). For example, Zhang et al. (2011) developed an NMF-based model to integrate microRNA and mRNA expression data in ovarian cancer. Chen and Zhang (2018) proposed the NMF framework in a network manner (NetNMF) to integrate pairwise genomic data. Recently, Zhang and Zhang (2019) adopted a matrix factorization-based model to reveal common as well as specific patterns from multiple datasets.
However, the integration of multi-biomedical data is one of the major challenges. Early integration, as reported by Chaudhary et al. (2018), is an approach to concatenate multiple datasets into a single dataset. However, early integration has three main disadvantages (Rappoport and Shamir, 2018). First, the weight is focused on a dataset with many features. Second, the distribution difference between datasets is ignored. Third, the input dimension of the model is increased. Late integration models (Sharifi-Noghabi et al., 2019) overcome these problems by separately learning each dataset and then integrating them. This approach preserves the unique data distribution. However, weak signals in each dataset may be lost during the integration phase (Rappoport and Shamir, 2018). Moreover, the biological interactions between features in multiple datasets may be lost (Sharifi-Noghabi et al., 2019).
Motivated by these limitations, we propose a Multi-task Attention Learning Algorithm for Multi-omics Data (MOMA) model, a novel and flexible approach that learns complex biological knowledge from multi-biomedical data to achieve high performance and interpretability. Using a geometrical approach, genes and modules were vectorized, and the vector sum of genes included in the module was interpreted as the corresponding module vector. To the best of our knowledge, this work represents the first attempt to use gene and module vectorization in DNNs. An attention mechanism (Bahdanau et al., 2014) was also designed as a mediator to identify related modules among multiple data. The result is an intermediate integration that overcomes the limitations of early and late integration, maintains a unique distribution of each dataset, considers biological interactions, and does not increase the input dimension of the model. MOMA also improves classification performance by focusing on important modules that are highly related to each other in each dataset.
The main contributions of this article are summarized as follows:
This work introduces a new type of module detection system based on geometric interpretation.
This work demonstrates the application of an attention mechanism for learning with a focus on the relevant modules between multi-omics data.
Experimental results with various public datasets and various classification tasks demonstrate that this model outperforms several strong baselines, including the top-performing systems.
Interpretation of module detection illustrates why the proposed model works well.
The algorithm has been validated by turning on and off the attention mechanism, which is an important part of the MOMA algorithm.
2 Our proposed MOMA model
We propose the MOMA model (Fig. 1) for multi-omics module analysis and classification. Our model is composed of three stages: (i) building a module for each dataset using the module encoder, (ii) focusing on the important modules between each omics data using module attention and (iii) multi-task learning for each dataset in a fully connected layer.
Fig. 1.

Overview of the proposed MOMA algorithm for two omics datasets: gene expression and DNA methylation. Each module encoder generates module vectors for each dataset. The module encoder consists of a fully connected layer. The weights of the fully connected layer represent the association between genes and modules, and the weight vector of each gene represents weights between the gene and each module. The vector of the gene is the product of the gene input value and its weight vectors. Each module vector is generated by the sum of the vectors of all genes in that module. The inner product of a gene vector and a module vector represents the degree of gene contribution to the module vector. The gene expression and DNA methylation module vectors are fed into an attention layer that assigns higher weights to the relevant module vectors for disease prediction. The model is trained via multi-task learning
2.1 Module encoder
The module encoder consists of a fully connected layer that connects features of the omics data to each module, where each module is represented by a vector. Let weights of the fully connected layer represent the association between features and modules of the jth omics data and a weight vector corresponding to each feature and each module represent associations between them. The output nodes of the fully connected layer are normalized to unit vectors, which represent module vectors. Given a training sample , where xj denotes the sample under the jth omics of J omics datasets and y is the corresponding label, let denote the module encoder of jth omics data. The module vectors of jth omics data Mj are defined as follows:
| (1) |
where θmodule denotes the weights of fmodule, Nj is the number of modules of jth omics data, and D is the dimension of the module vector.
2.2 Module attention
A module attention mechanism was devised to focus on modules with high similarity between each omics data module. Cosine similarity was used to measure relevance. Let Att denote the module attention matrix between the module vectors of two omics datasets and Attlk denote the element in row l and column k (that is the (l, k)th element of Att). Attlk stores the relationship information with possible dependence between the lth module from one omics data and the kth module from another omics data module. The element in the attention matrix was devised as follows:
| (2) |
where for short and and are the lth module vector of ith omics data and the kth module vector of jth omics data. To highlight the important modules, the module vectors are multiplied by the attention matrices with the other omics data and concatenated. The updated module vector is
| (3) |
where for short. A toy example of the module attention mechanism is shown in Supplementary Figure S1.
2.3 Training
The fully connected layers are then applied, which flattens the multi-dimensional vectors and yields the final probabilities for each label. In the model, loss L is set to the cross-entropy error between the gold label and task-specific outputs:
| (4) |
where C represents the total number of classes, yc denotes a labeling of c, consists of multiple fully connected layers for jth omics data, θfc denotes the weights of ffc, W denotes the weights, and an L2-norm penalty with a regularization parameter λ was used for optimization to avoid overfitting the module encoders and the multiple connected layers. The time complexity of the proposed approach is , where I is the number of iterations, R is the number of samples, F is the number of features of omics data, N is the number of modules, J is the number of omics data types, and D is the dimension of the module vector. The MOMA models for two and three datasets are described in Supplementary Sections S2 and S3, respectively.
2.4 Module detection
The proposed model is an explainable model capable of not only predicting phenotypes, such as disease status, but also detecting modules containing genes related to phenotypes. The most relevant modules for a specific phenotype are identified using the module attention matrix. Each element in the module attention matrix represents the cosine similarity value between a pair of module vectors, each of which is obtained from a respective omics dataset. A pair of the modules having the largest cosine similarity paid the most attention in the training phenotype. Thus, they are selected as the most relevant modules.
From the most relevant modules identified, we next select the important features. Let be the value of feature a of jth omics data, be the weight vector between feature a and the b-th module, and be the bth module vector of jth omics data. Then, a feature vector of gene a is the product between and . The importance of the feature in the module is the inner product of the feature vector and the module vector, e.g. importance of feature a in module b is defined as follows:
| (5) |
Features with an importance greater than a predefined threshold are selected for each module.
3 Experiments
In this study, three experiments were performed: three different types of classification experiments, a turn-off and turn-on attention mechanism experiment, and a module detection experiment for the proposed MOMA validation method. TCGA datasets were used for the classification of 34 classes (33 types of cancer and normal) and for classification between the early and late stages of each cancer type. The Religious Orders Study (ROS) and Rush Memory and Aging Project (MAP) (ROSMAP) datasets were used for classification between Alzheimer’s disease (AD) and normal (NL) samples.
The classification performance of the proposed MOMA method was compared against that of eXtreme Gradient Boosting (XGBoost) and a DNN of single omics data. In a recent study (Ma et al., 2020), XGBoost displayed a remarkable performance in predicting early- and late-stage patients with kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma (KIRP), lung squamous cell carcinoma (LUSC), and head and neck squamous cell carcinoma (HNSC) in TCGA, when compared to the support vector machine, random forest, DNN, k-nearest neighbor, naive Bayes and elastic net models.
In addition, the MOMA model was compared with other multi-omics integration models based on an inductive learning approach. MOMA is an inductive learning approach, where the model is trained on a training dataset and then applied to a test dataset separated from the training dataset. Therefore, information related to the test dataset is not seen while creating the model. On the contrary, a transductive learning approach builds the model based on a training dataset and an unlabeled test dataset and then applies the model to the test dataset (Vapnik, 2000). For a fair comparison, we excluded the transductive supervised learning approach (Wang et al., 2017; Zhang et al., 2019). The compared methods include: (i) Multi-Omics gRaph cOnvolutional NETworks (MORONET) (Wang et al., 2020); (ii) Multi-Omics Factor Analysis (MOFA) (Argelaguet et al., 2018); (iii) Robust Multimodal Approach to Integrative Analysis of Multiomics Data (SMSPL) (Yang et al., 2020) and (iv) Pathway-aware multi-layered hierarchical NETwork (P-NET) (Elmarakeby et al., 2021). MORONET is a supervised multi-omics data classification model that uses graph convolutional networks incorporating patient associations for better performance. MORONET utilizes a view correlation discovery network to explore the cross-omics correlations at the label space. MOFA is a Bayesian model for unsupervised integration of multimodal data. For comparison of classification performance, we used a MOFA+SVM model that is composed of MOFA for feature extraction of multi-omics data and the SVM for classification. SMSPL is a self-paced learning model that interactively recommends high-confidence samples between multi-omics data. SMSPL demonstrates the advantage of a robust performance even in the presence of heavy noises and displays a good generalization performance. P-NET is a biologically informed neural network model and has been applied to predict the treatment-resistance state in prostate cancer patients using multi-omics datasets. It encodes different biological entities in a neural network with customized connections between consecutive layers of genes, pathways and biological processes.
In addition, for MOMA, in the attention mechanism turn-off and turn-on experiments, the ROSMAP data classification performance was compared through various parameter combinations. Module analysis was also performed to identify the effects of the multi-omics attention mechanism.
3.1 Datasets
3.1.1 ROSMAP datasets
Batch effect-normalized mRNA data, methylation data and clinical data were obtained from the ROSMAP cohort provided on the AMP-AD Knowledge Portal (https://adknowledgeportal.synapse.org/). Patients with gene expression and DNA methylation profiles were included. The clinical consensus diagnosis of cognitive status at the time of death (COGDX) scores 4 and 5 were labeled as AD, and the COGDX score 1 as NL. Quantile normalized fragments per kilobase of transcript per million mapped reads (FPKM) values of the gene expression profiles were log2-transformed. In the DNA methylation profile, β-values were measured using the Infinium HumanMethylation450 BeadChip and missing β-values were imputed using a k-nearest neighbor algorithm.
3.1.2 TCGA datasets
Batch effect-normalized mRNA data, methylation data and clinical data were obtained from the TCGA Pan-Cancer dataset provided on Xena (https://xenabrowser.net/). Patients with both gene expression and DNA methylation profiles and with available clinical information were included in the analyses. For classification of the 34 classes, ‘Primary Solid Tumor’, ‘Primary Blood-Derived Cancer Peripheral Blood’ and ‘Solid Tissue Normal’ sample types were included. For early- and late-stage classification, only primary tumors and samples with ‘pathologic stage’ information were included. Cancer types with at least 20 minor class patients were also included in experiments. The gene expression profile in this dataset has log2 transformed FPKM values. In the DNA methylation profile, CpG site levels were calculated as β-values measured using the Infinium HumanMethylation450 BeadChip.
3.2 Preprocessing
For DNA methylation data, CpGs located in promoter regions (TSS200 or TSS1500) were preprocessed into the corresponding gene. Next, all duplicated genes were averaged. For each dataset, the gene expression value and DNA methylation value were normalized to the range of 0–1. Finally, ROSMAP datasets, comprising 18 164 gene expression features and 19 353 DNA methylation features, and TCGA datasets, comprising 16 335 gene expression features and 17 016 DNA methylation features, were obtained. Table 1 shows the number of samples for each class in each experiment.
Table 1.
Summary of datasets used in this study
| Cohort | Experiment | Class (Number of samples) |
|---|---|---|
| ROSMAP Cohort | AD and NL classification | NL (171), AD (218) |
| TCGA Cohort | 34 classes classification | NL (394), ACC (78), BLCA (408), BRCA (777), CESC (303), CHOL (36), COAD (288), DLBC (48), ESCA (182), GBM (51), HNSC (515), KICH (65), KIRC (311), KIRP (270), LAML (170), LGG (514), LIHC (368), LUAD (452), LUSC (364), MESO (87), OV (9), PAAD (177), PCPG (178), PRAD (494), READ (94), SARC (255), SKCM (103), STAD (370), TGCT (149), THCA (501), THYM (120), UCEC (417), UCS (57), UVM (80) |
| Early- and late-stage classification | ACC (46/30), BLCA (132/274), BRCA (557/208), COAD (155/121), ESCA (94/65), HNSC (98/343), KICH (45/20), KIRC (130, 179), KIRP (183/63), LIHC (255/90), LUAD (355/92), LUSC (302/59), MESO (26/61), READ (40/46), SKCM (68/30), STAD (168/192), THCA (334/165), UVM (39/40) |
3.3 Experimental settings
The models were trained using nested cross-validation (CV) for unbiased performance estimates (Supplementary Fig. S2). Hyperparameters were estimated using inner CV loops nested in each outer CV loop. With an outer 5-fold CV, the classification performances were measured in terms of the accuracy (ACC), area under the receiver operating characteristic curve (AUC), F1 score, Matthews correlation coefficient (MCC) and average precision (AP), which is also known as a precision-recall AUC. Weighted AUCs, weighted F1 scores and weighted AP were used for multiclass classification.
The hyperparameters of each model were determined in the 3-fold inner CV on the training set. AUC was used as an evaluation metric. However, in the classification of 34 TCGA classes, because the AUC was ∼1, the ACC was used as an evaluation metric. For MOMA, the parameters of ‘the number of modules’ from the set {32, 64, 128}, ‘the learning rate’ from the set , ‘the weight decay’ from the set , and ‘the early stopping patience’ from the set {50, 100} were optimized. The number of fully connected layers was fixed according to the number of modules. The Adam optimizer (Kingma and Ba, 2014) was used in the MOMA model. Details on the algorithm for this experiment and the list of other model hyperparameters are described in Supplementary Section S2. The grid search results for MOMA on the ROSMAP validation sets are described in Supplementary Tables S1 and S2.
3.4 Experimental results
3.4.1 Classification performance
XGBoost based on gene expression datasets, XGBoost based on DNA methylation datasets, DNN based on gene expression datasets, DNN based on DNA methylation datasets and MOMA are compared in terms of their classification performance in the ROSMAP and TCGA cohorts in Table 2 and Supplementary Table S3. An average scoring metric of 5-fold CV was reported. MOMA demonstrated the best ACC, F1 score, AUC, MCC and AP in the ROSMAP cohort. For TCGA 34 classes classification, MOMA showed the best prediction performance in ACC, F1 and MCC, whereas XGBoost showed the best performance in the AUC and AP. In the TCGA early- and late-stage classification, MOMA showed the best ACC performance in 7 of 18 datasets, the best F1 score performance in 13 of 18 datasets, the best AUC performance in 9 of 18 datasets, the best MCC performance in 12 of 18 datasets, and the best AP performance in 8 of 18 datasets. The number of datasets showing the best performance in ACC was smaller than that for the F1 score; this might be because of the imbalanced numbers of positive and negative samples. MOMA showed the best ACC performance in 5 of 7 balanced datasets and 2 of 11 imbalanced data sets (the proportion of samples of minor class patients was <35%).
Table 2.
Classification performance of XGBoost, DNN, and MOMA in ROSMAP NL and AD classifications, TCGA 34 classes classification, and TCGA early- and late-stage classification
| XGBoost |
XGBoost |
DNN |
DNN |
MOMA |
|||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GE |
DM |
GE |
DM |
GEa |
DMa |
||||||||||||||||||||
| Task and datasets | ACC | F1 | AUC | MCC | ACC | F1 | AUC | MCC | ACC | F1 | AUC | MCC | ACC | F1 | AUC | MCC | ACC | F1 | AUC | MCC | ACC | F1 | AUC | MCC | |
| ROSMAP NL/AD | 0.686 | 0.723 | 0.757 | 0.363 | 0.596 | 0.664 | 0.632 | 0.168 | 0.653 | 0.740 | 0.764 | 0.284 | 0.589 | 0.581 | 0.616 | 0.180 | 0.737 | 0.740 | 0.812 | 0.488 | 0.720 | 0.753 | 0.807 | 0.432 | |
| TCGA 34 classes | 0.953 | 0.951 | 0.998 | 0.951 | 0.950 | 0.949 | 0.998 | 0.948 | 0.878 | 0.848 | 0.988 | 0.874 | 0.875 | 0.837 | 0.985 | 0.870 | 0.948 | 0.945 | 0.996 | 0.946 | 0.954 | 0.952 | 0.996 | 0.952 | |
| TCGA early- and late-stage | ACC | 0.633 | 0.455 | 0.681 | 0.191 | 0.672 | 0.435 | 0.673 | 0.241 | 0.618 | 0.386 | 0.671 | 0.157 | 0.578 | 0.305 | 0.710 | 0.024 | 0.723 | 0.654 | 0.725 | 0.429 | 0.710 | 0.611 | 0.720 | 0.393 |
| BLCA | 0.756 | 0.832 | 0.772 | 0.410 | 0.685 | 0.785 | 0.688 | 0.221 | 0.675 | 0.806 | 0.636 | 0.000 | 0.699 | 0.800 | 0.712 | 0.223 | 0.705 | 0.771 | 0.736 | 0.352 | 0.663 | 0.716 | 0.747 | 0.335 | |
| BRCA | 0.718 | 0.179 | 0.630 | 0.101 | 0.716 | 0.154 | 0.584 | 0.094 | 0.701 | 0.188 | 0.573 | 0.088 | 0.705 | 0.232 | 0.602 | 0.117 | 0.692 | 0.288 | 0.616 | 0.134 | 0.651 | 0.331 | 0.618 | 0.125 | |
| COAD | 0.634 | 0.539 | 0.664 | 0.247 | 0.605 | 0.499 | 0.627 | 0.185 | 0.579 | 0.513 | 0.626 | 0.178 | 0.609 | 0.367 | 0.644 | 0.179 | 0.630 | 0.624 | 0.703 | 0.310 | 0.667 | 0.605 | 0.726 | 0.322 | |
| ESCA | 0.547 | 0.369 | 0.508 | 0.033 | 0.548 | 0.319 | 0.457 | 0.013 | 0.479 | 0.347 | 0.472 | 0.000 | 0.566 | 0.218 | 0.542 | 0.000 | 0.509 | 0.467 | 0.526 | 0.026 | 0.510 | 0.417 | 0.441 | −0.001 | |
| HNSC | 0.780 | 0.874 | 0.607 | 0.131 | 0.605 | 0.499 | 0.627 | 0.185 | 0.778 | 0.875 | 0.542 | 0.000 | 0.778 | 0.875 | 0.501 | 0.000 | 0.728 | 0.832 | 0.631 | 0.079 | 0.648 | 0.727 | 0.643 | 0.196 | |
| KICH | 0.631 | 0.280 | 0.617 | 0.088 | 0.646 | 0.253 | 0.567 | 0.062 | 0.692 | 0.067 | 0.522 | 0.000 | 0.554 | 0.201 | 0.450 | 0.018 | 0.631 | 0.357 | 0.556 | 0.141 | 0.708 | 0.286 | 0.617 | 0.176 | |
| KIRC | 0.719 | 0.647 | 0.781 | 0.421 | 0.735 | 0.662 | 0.798 | 0.453 | 0.719 | 0.641 | 0.772 | 0.476 | 0.735 | 0.584 | 0.752 | 0.429 | 0.754 | 0.717 | 0.836 | 0.507 | 0.754 | 0.714 | 0.830 | 0.510 | |
| KIRP | 0.845 | 0.638 | 0.845 | 0.573 | 0.833 | 0.592 | 0.848 | 0.523 | 0.817 | 0.463 | 0.819 | 0.430 | 0.805 | 0.447 | 0.789 | 0.400 | 0.813 | 0.637 | 0.842 | 0.522 | 0.849 | 0.684 | 0.858 | 0.602 | |
| LIHC | 0.725 | 0.226 | 0.593 | 0.120 | 0.696 | 0.127 | 0.644 | −0.004 | 0.733 | 0.088 | 0.547 | 0.034 | 0.733 | 0.236 | 0.665 | 0.134 | 0.699 | 0.366 | 0.680 | 0.188 | 0.730 | 0.314 | 0.688 | 0.190 | |
| LUAD | 0.779 | 0.056 | 0.605 | 0.020 | 0.792 | 0.130 | 0.643 | 0.132 | 0.794 | 0.000 | 0.612 | 0.000 | 0.794 | 0.000 | 0.509 | 0.000 | 0.711 | 0.241 | 0.595 | 0.092 | 0.761 | 0.177 | 0.612 | 0.092 | |
| LUSC | 0.834 | 0.029 | 0.564 | 0.020 | 0.831 | 0.084 | 0.460 | 0.085 | 0.703 | 0.057 | 0.567 | 0.000 | 0.837 | 0.000 | 0.533 | 0.057 | 0.698 | 0.190 | 0.506 | 0.023 | 0.695 | 0.224 | 0.500 | 0.044 | |
| MESO | 0.758 | 0.837 | 0.682 | 0.401 | 0.610 | 0.740 | 0.511 | −0.027 | 0.701 | 0.822 | 0.622 | 0.033 | 0.690 | 0.816 | 0.421 | −0.032 | 0.643 | 0.773 | 0.572 | −0.064 | 0.609 | 0.719 | 0.607 | 0.019 | |
| READ | 0.571 | 0.624 | 0.519 | 0.138 | 0.536 | 0.560 | 0.582 | 0.063 | 0.523 | 0.620 | 0.605 | −0.007 | 0.501 | 0.415 | 0.521 | 0.000 | 0.410 | 0.473 | 0.461 | −0.203 | 0.465 | 0.516 | 0.479 | −0.121 | |
| SKCM | 0.622 | 0.160 | 0.457 | −0.059 | 0.623 | 0.237 | 0.590 | 0.027 | .614 | 0.092 | 0.528 | 0.000 | 0.684 | 0.000 | 0.382 | −0.030 | 0.654 | 0.292 | 0.490 | 0.101 | 0.644 | 0.153 | 0.446 | 0.001 | |
| STAD | 0.550 | 0.582 | 0.592 | 0.096 | 0.569 | 0.617 | 0.595 | 0.132 | 0.581 | 0.579 | 0.626 | 0.173 | 0.500 | 0.301 | 0.541 | 0.035 | 0.594 | 0.637 | 0.627 | 0.183 | 0.594 | 0.587 | 0.643 | 0.198 | |
| THCA | 0.719 | 0.460 | 0.718 | 0.307 | 0.731 | 0.487 | 0.732 | 0.343 | 0.705 | 0.350 | 0.694 | 0.262 | 0.735 | 0.434 | 0.708 | 0.322 | 0.687 | 0.595 | 0.761 | 0.378 | 0.727 | 0.618 | 0.769 | 0.418 | |
| UVM | 0.378 | 0.358 | 0.404 | −0.252 | 0.480 | 0.454 | 0.509 | −0.038 | 0.506 | 0.592 | 0.608 | 0.013 | 0.493 | 0.267 | 0.469 | 0.000 | 0.533 | 0.501 | 0.587 | 0.060 | 0.545 | 0.417 | 0.580 | 0.115 | |
Note: The performance of the methods was evaluated for gene expression (GE) and DNA methylation (DM) datasets. Bold texts indicate the best performance in each metric, ACC, F1-score, AUC and MCC.
The dataset used for the performance measurement (a task data set) is specified.
MORONET, MOFA+SVM, SMSPL, P-NET and Ensem-MOMA are compared in terms of their classification performance in the ROSMAP and TCGA cohorts in Table 3. Ensem-MOMA indicates an ensemble of the results of each MOMA task. The Ensem-MOMA model used logistic regression as a combiner. MORONET displayed a better performance than MOFA+SVM and SMSPL in TCGA 34 classes classification. Moreover, MOFA+SVM and SMSPL displayed a better performance than MORONET in a large number of binary classification tasks. These results might be attributed to the fact that MORONET is a complex multi-layered model, whereas MOFA, based on a Bayesian model, and SMSPL, based on a logistic regression model, are relatively uncomplicated. P-NET displays more strength in binary classification than in multiclass classification; this might be due to the smaller numbers of parameters in P-NET than in MORONET. Ensem-MOMA outperformed other methods in both binary classification and multiclass classification tasks. Supplementary Table S4 shows the results of the two-tailed paired t-test between other methods and MOMA for 5 CVs × 20 tasks (including ROSMAP NL/AD classification, TCGA 34 classes classification, and early- and late-stage classification of 18 cancer types). There were statistically significant differences in performance in all metrics except for ACC and an F1 score with P-NET. These results indicate that MOMA is a viable approach for improving performance in general classification tasks regardless of the datasets used.
Table 3.
Classification performance of MORONET, MOFA+SVM, SMSPL, P-NET and MOMA in ROSMAP NL and AD classification, TCGA 34 classes classification and TCGA early- and late-stage classification
| MORONET |
MOFA+SVM |
SMSPL |
P-NET |
Ensem-MOMA |
||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Task and datasets | ACC | F1 | AUC | MCC | AP | ACC | F1 | AUC | MCC | AP | ACC | F1 | AUCa | MCC | AP | ACC | F1 | AUC | MCC | AP | ACC | F1 | AUC | MCC | AP | |
| ROSMAP NL/AD | 0.612 | 0.647 | 0.648 | 0.222 | 0.661 | 0.663 | 0.699 | 0.769 | 0.320 | 0.826 | 0.689 | 0.732 | 0.678 | 0.368 | 0.757 | 0.663 | 0.708 | 0.777 | 0.313 | 0.843 | 0.732 | 0.750 | 0.815 | 0.466 | 0.861 | |
| TCGA 34 classes | 0.945 | 0.943 | 0.997 | 0.943 | 0.971 | 0.869 | 0.865 | 0.994 | 0.863 | 0.914 | 0.891 | 0.874 | 0.902 | 0.886 | 0.805 | 0.725 | 0.757 | 0.996 | 0.739 | 0.948 | 0.958 | 0.957 | 0.996 | 0.957 | 0.980 | |
| TCGA early- and late-stage | ACC | 0.712 | 0.508 | 0.747 | 0.351 | 0.663 | 0.553 | 0.084 | 0.670 | −0.087 | 0.541 | 0.592 | 0.413 | 0.557 | 0.115 | 0.458 | 0.659 | 0.465 | 0.777 | 0.228 | 0.706 | 0.723 | 0.663 | 0.730 | 0.438 | 0.596 |
| BLCA | 0.682 | 0.755 | 0.669 | 0.305 | 0.772 | 0.712 | 0.803 | 0.713 | 0.291 | 0.821 | 0.717 | 0.804 | 0.638 | 0.305 | 0.840 | 0.685 | 0.775 | 0.715 | 0.253 | 0.842 | 0.712 | 0.787 | 0.742 | 0.342 | 0.852 | |
| BRCA | 0.654 | 0.246 | 0.567 | 0.056 | 0.319 | 0.728 | 0.000 | 0.525 | 0.000 | 0.310 | 0.663 | 0.265 | 0.548 | 0.082 | 0.299 | 0.708 | 0.366 | 0.611 | 0.193 | 0.408 | 0.634 | 0.405 | 0.620 | 0.168 | 0.419 | |
| COAD | 0.623 | 0.580 | 0.633 | 0.244 | 0.568 | 0.649 | 0.575 | 0.681 | 0.279 | 0.615 | 0.645 | 0.613 | 0.645 | 0.292 | 0.584 | 0.656 | 0.605 | 0.738 | 0.302 | 0.700 | 0.641 | 0.660 | 0.713 | 0.335 | 0.665 | |
| ESCA | 0.534 | 0.255 | 0.477 | −0.032 | 0.454 | 0.567 | 0.188 | 0.434 | 0.018 | 0.411 | 0.585 | 0.395 | 0.547 | 0.110 | 0.416 | 0.554 | 0.393 | 0.488 | 0.055 | 0.462 | 0.484 | 0.459 | 0.489 | −0.007 | 0.467 | |
| HNSC | 0.748 | 0.852 | 0.555 | 0.028 | 0.819 | 0.733 | 0.838 | 0.617 | 0.051 | 0.857 | 0.757 | 0.853 | 0.562 | 0.142 | 0.819 | 0.753 | 0.856 | 0.609 | 0.060 | 0.855 | 0.726 | 0.834 | 0.632 | 0.035 | 0.859 | |
| KICH | 0.646 | 0.397 | 0.567 | 0.195 | 0.488 | 0.708 | 0.080 | 0.461 | 0.087 | 0.451 | 0.662 | 0.214 | 0.533 | 0.068 | 0.371 | 0.708 | 0.080 | 0.561 | 0.087 | 0.529 | 0.662 | 0.337 | 0.583 | 0.152 | 0.534 | |
| KIRC | 0.689 | 0.565 | 0.732 | 0.350 | 0.656 | 0.735 | 0.657 | 0.766 | 0.450 | 0.696 | 0.751 | 0.691 | 0.740 | 0.492 | 0.722 | 0.744 | 0.690 | 0.797 | 0.473 | 0.716 | 0.751 | 0.714 | 0.832 | 0.503 | 0.768 | |
| KIRP | 0.764 | 0.515 | 0.784 | 0.388 | 0.578 | 0.801 | 0.426 | 0.853 | 0.378 | 0.733 | 0.846 | 0.648 | 0.758 | 0.572 | 0.666 | 0.809 | 0.574 | 0.853 | 0.463 | 0.738 | 0.837 | 0.682 | 0.850 | 0.584 | 0.763 | |
| LIHC | 0.699 | 0.352 | 0.571 | 0.175 | 0.384 | 0.707 | 0.164 | 0.646 | 0.065 | 0.384 | 0.681 | 0.191 | 0.522 | 0.018 | 0.317 | 0.684 | 0.298 | 0.657 | 0.106 | 0.381 | 0.684 | 0.452 | 0.685 | 0.239 | 0.418 | |
| LUAD | 0.765 | 0.127 | 0.481 | 0.050 | 0.252 | 0.794 | 0.000 | 0.629 | 0.000 | 0.330 | 0.740 | 0.135 | 0.506 | 0.011 | 0.240 | 0.774 | 0.150 | 0.609 | 0.091 | 0.324 | 0.714 | 0.287 | 0.598 | 0.119 | 0.320 | |
| LUSC | 0.814 | 0.050 | 0.477 | 0.008 | 0.214 | 0.837 | 0.000 | 0.577 | 0.000 | 0.238 | 0.784 | 0.083 | 0.497 | −0.021 | 0.192 | 0.812 | 0.025 | 0.405 | −0.036 | 0.188 | 0.715 | 0.212 | 0.504 | 0.048 | 0.236 | |
| MESO | 0.608 | 0.715 | 0.584 | 0.004 | 0.790 | 0.690 | 0.816 | 0.481 | −0.032 | 0.731 | 0.622 | 0.736 | 0.521 | 0.036 | 0.750 | 0.642 | 0.768 | 0.562 | −0.056 | 0.792 | 0.666 | 0.790 | 0.621 | 0.044 | 0.813 | |
| READ | 0.534 | 0.633 | 0.486 | 0.062 | 0.562 | 0.443 | 0.448 | 0.614 | −0.121 | 0.673 | 0.594 | 0.578 | 0.598 | 0.210 | 0.574 | 0.522 | 0.593 | 0.488 | 0.024 | 0.592 | 0.523 | 0.592 | 0.473 | 0.046 | 0.606 | |
| SKCM | 0.602 | 0.194 | 0.565 | −0.015 | 0.445 | 0.694 | 0.000 | 0.625 | 0.000 | 0.487 | 0.592 | 0.150 | 0.463 | −0.095 | 0.262 | 0.603 | 0.057 | 0.433 | −0.138 | 0.335 | 0.644 | 0.260 | 0.456 | 0.070 | 0.391 | |
| STAD | 0.550 | 0.502 | 0.577 | 0.127 | 0.601 | 0.564 | 0.610 | 0.570 | 0.120 | 0.601 | 0.597 | 0.626 | 0.595 | 0.192 | 0.642 | 0.611 | 0.650 | 0.629 | 0.217 | 0.647 | 0.586 | 0.630 | 0.636 | 0.166 | 0.656 | |
| THCA | 0.669 | 0.469 | 0.690 | 0.237 | 0.535 | 0.701 | 0.337 | 0.698 | 0.222 | 0.582 | 0.693 | 0.546 | 0.667 | 0.329 | 0.579 | 0.723 | 0.515 | 0.745 | 0.338 | 0.648 | 0.703 | 0.602 | 0.765 | 0.385 | 0.701 | |
| UVM | 0.457 | 0.477 | 0.521 | −0.072 | 0.572 | 0.544 | 0.603 | 0.387 | 0.087 | 0.503 | 0.533 | 0.492 | 0.536 | 0.076 | 0.514 | 0.563 | 0.453 | 0.531 | 0.140 | 0.620 | 0.596 | 0.561 | 0.577 | 0.205 | 0.628 | |
Note: Bold texts indicate the best performance in each metric, accuracy, F1-score, AUC, MCC and AP.
In SMSPL, predictor is categorical. AUC is computed by categorical values.
Supplementary Table S5 shows the computational time, number of parameters, memory usage and graphics processing unit (GPU) memory usage of XGBoost, DNN, MORONET, MOFA, SMSPL, P-NET and MOMA. In multi-omics data, DNN requires a large number of parameters and consumes a large amount of memory resources. Although MORONET, P-NET and MOMA are neural network-based models, they have relatively few parameters. MORONET reduces the number of features through feature selection, and P-NET uses a biologically informed neural network with customized connections. MOMA maintains the strength of neural networks and reduces the number of parameters through interactions between input features through the modularization of features and the attention mechanism.
To further compare the classification performance on different numbers of tasks and different data types, we performed two additional experiments. First, we experimented with three datasets from the Alzheimer's Disease Neuroimaging Initiative (ADNI), where two bioimaging datasets [magnetic resonance imaging regions of interest (ROIs) and AV45 positron emission tomography ROIs] and gene expression data were included. Supplementary Table S6 shows that the proposed model outperformed the existing models. The MOMA algorithm and experimental details are shown in Supplementary Material. Second, we applied MOMA to single-cell multi-omics data (single-cell RNA-seq and DNA-methylation datasets) to classify 2i and serum conditions of mouse embryonic stem cells (Supplementary Material). Supplementary Table S7 shows that MOMA outperformed XGBoost, MORONET, MOFA+SVM and SMSPL when the single-cell datasets were used.
3.4.2 Comparison between turning the module attention on and off
The performance of turning the module attention on and off according to various hyperparameter sets was compared through inner CV results in every outer CV. Figure 2 shows the results of each inner CV in the ROSMAP cohort; each point shows a different set of hyperparameters. Classification of AD and NL samples with gene expression and DNA methylation data showed statistically significant differences between turning the module attention on and off (p-values = and with two-sided paired t-tests, respectively). We also found statistically significant differences between turning the module attention on and off in the test results of 5-fold CV (P-value = with a two-sided paired t-test). These results indicate that the module attention was helpful in learning the classification of AD and NL samples.
Fig. 2.

Comparison of turning the module attention on/off in the ROSMAP cohort. Every inner cross-validation result is reported along with the area under the receiver operating characteristic curve (AUC). Every point shows a different set of hyperparameters. (A) Results of gene expression classification. (B) Results of DNA methylation classification
To validate the generality of this experiment, the performance of turning the module attention on and off was compared in 18 cancer types of TCGA (Supplementary Figs S3–S7). In 5 of the 18 cancers, identifying related modules was not assisted by the attention mechanism. In esophageal carcinoma (ESCA) and LUSC, the attention mechanism negatively affected the identification of related modules (P-value < 0.05 with a t-test). In kidney chromophobe (KICH), rectum adenocarcinoma (READ) and skin cutaneous melanoma (SKCM), the attention mechanism had no role in identifying related modules. However, the classification performance on these data was low; thus, the attention mechanism might be difficult to use. Interestingly, there was a relationship between the classification performance and the impact of the module attention (Fig. 3). The higher the predictive performance, the more the attention mechanism helped. This result indicated that module attention was more effective for well-learned modules.
Fig. 3.
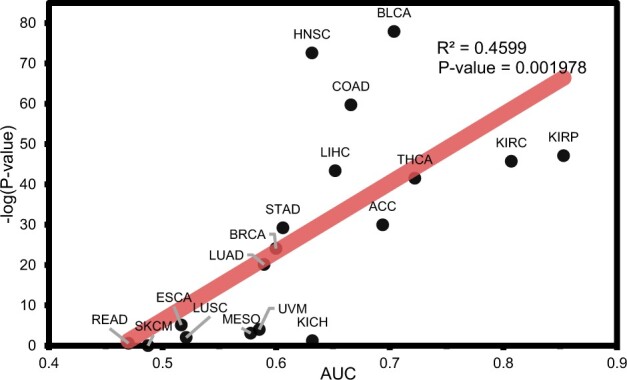
Relationship between the average area under the receiver operating characteristic curve (AUC) of inner cross-validation results in each TCGA cancer type and P-value in a paired t-test for the difference in turning the module attention on/off
3.4.3 MOMA module analysis
Figure 4 and Supplementary Figure S8 show the clustering results of the cosine similarity scores between gene expression and DNA methylation module vectors across the training and test datasets for each of the outer 5-fold CV results in the ROSMAP cohort. These results demonstrated that different module pairs of gene expression and DNA methylation were focused on while learning the AD and NL classification. Hierarchical clustering indicated that pairs that were important for each label during training were also maintained in the test.
Fig. 4.
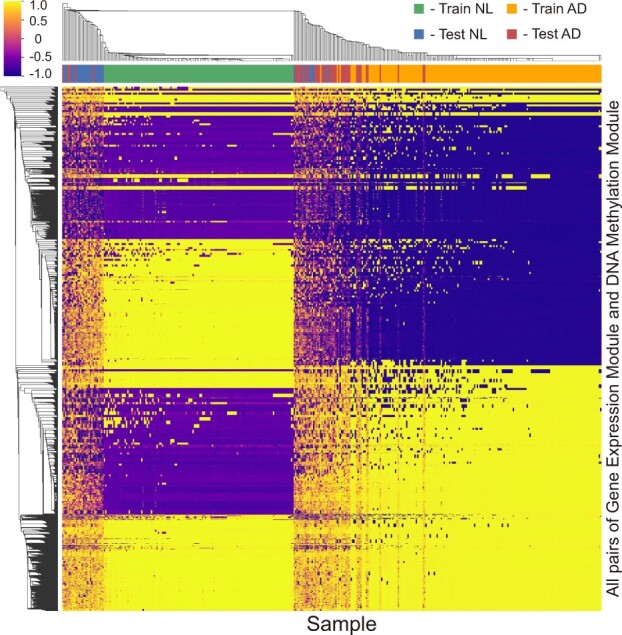
Heatmap of hierarchical clustered cosine similarity scores of the MOMA model in the ROSMAP cohort. Cosine similarity scores between the gene expression and DNA methylation modules of each sample are extracted
Next, we investigated the module attention to examine the genes contributing to the high classification performance in the ROSMAP cohort. A pair of the gene expression and DNA methylation module vectors that paid the most attention in training AD samples in each fold was selected. This pair had the largest cosine similarity score in the module attention matrix. In the selected modules, the importance of genes within each module was calculated using Equation (5) and their Z-scores were then calculated. Genes with greater than the Z-score threshold (empirically set to 99 percentile and 99.9 percentile for gene expression and DNA methylation, respectively) were selected. Pathway enrichment analysis was performed on these selected genes based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa and Goto, 2000) using Enrichr (Kuleshov et al., 2016).
Figure 5 shows the enriched KEGG pathways with P-values less than 0.05. The AD pathway was significantly enriched with q-values lesser than 0.05 in CV4 and CV5, and the AD pathway-related genes were also included in other CVs. Pathways associated with neurodegenerative disorders, including Parkinson’s disease and Huntington’s disease, were also significantly enriched (q-value < 0.05).
Fig. 5.
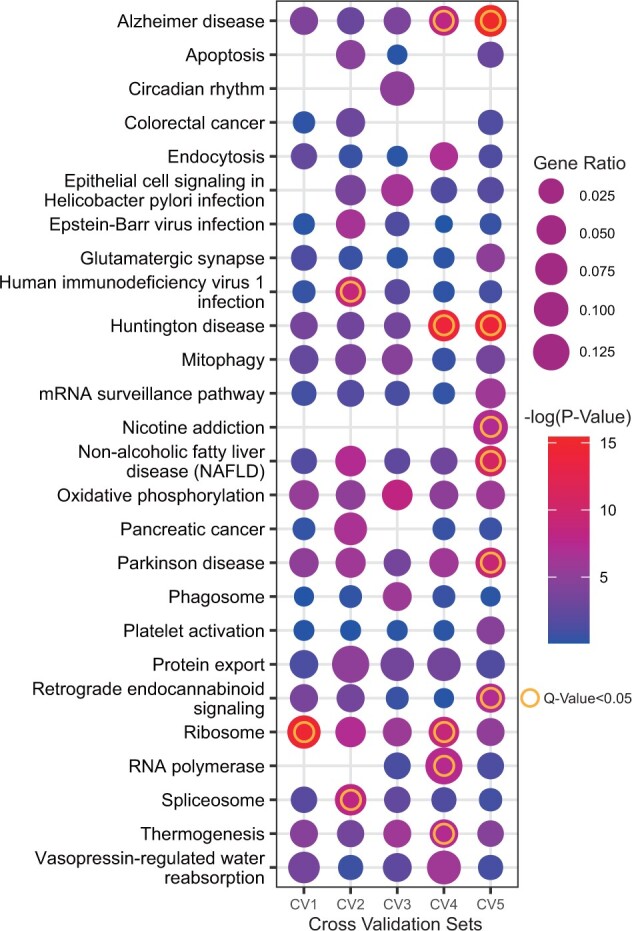
The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of each cross-validation (CV) model. The dot size indicates the gene ratio. The dot color indicates the P-value. The ring indicates a significant pathway (q-value < 0.05)
Previous studies have revealed that AD is closely related to enriched pathways, such as apoptosis (Shimohama, 2000), circadian rhythms (Wu and Swaab, 2007), glutamatergic synapses (Rudy et al., 2015), mitophagy (Chen and Chan, 2009), oxidative phosphorylation (Manczak et al., 2004), platelet activation (Sevush et al., 1998) and ribosome (Ding et al., 2005). This observation is an interesting example of effective learning, including biological meaning, as well as module disease detection.
Module analysis results on TCGA cohort are shown in Supplementary Figures S9–S11. Supplementary Figures S9–S11 show the well-separated clustering results of the similarity score across the training and test datasets for the 34 TCGA class classification and early- and late-stage classification on KIRC and KIRP. Supplementary Figures S10–S11 show the possibility of module analysis in various classification tasks with enrichment analysis of cancer-related functions and pathways.
We additionally performed an experiment to identify cancer-specific modules using 34 TCGA cancer types. The attention matrix was calculated based on the input values using samples of each cancer type. We averaged the cosine similarity score for each module of one omics dataset to all modules of another omics dataset. Next, we identified a module having the highest average similarity score for each cancer type. Supplementary Figure S12 shows the cancer-specific modules and NL-specific modules and demonstrates that different modules tend to be enriched in different pathways.
4 Conclusions
In summary, this study used DNNs to identify biologically important mechanisms from multi-biomedical data, outperforming the classification of disease-related phenotypes and module analysis. A MOMA interpretable supervised learning model was proposed. Using a geometric approach, a module encoder layer and module attention mechanism were designed that capture important related modules from multi-omics data. In the AD and NL classification tasks, 34 cancer type classification tasks, and early- and late-stage classification tasks, the proposed method outperformed the XGBoost and DNN models for each single dataset. The proposed model also outperformed the recent multi-omics integration models when compared. Furthermore, through internal comparison with turning the module attention on and off, the effectiveness of the module attention mechanism in MOMA was demonstrated. Through module analysis, it was demonstrated that modules enriched in disease-related biological pathways were focused on by the module attention mechanism.
However, some limitations remain, which should be addressed to further improve the performance of the model. The model involves simple multi-task learning with uniform loss weights. This may cause an optimization conflict for each task or unbalanced learning for each task. As in the inner CV1 of outer CV3 in Figure 2, negative transfer occurs. Although the performance improved on average over all tasks, the performance of the model that turned the attention mechanism off was worse than that of the model that turned the attention mechanism on for the gene expression classification task. Recently, multi-task learning with uncertainty (Kendall et al., 2018) has been shown to alleviate unbalanced learning for each task. Therefore, multi-task learning optimization has the potential to improve performance. In future studies, we plan to use various formats, such as learning each gene encoder as a common parameter with soft-parameter sharing and learning with an adaptive sharing approach (Sun et al., 2019), which learns what to share for efficient multi-task learning. Another direction for improving performance is to consider training on a single output as well. In this study, our model had multiple outputs for prediction, the number of which was equal to the number of input datasets. Ensemble running was then performed with these outputs. We expect that the end-to-end training on one output together with multimodal learning may further improve the performance of the model. Furthermore, a bootstrapping technique (Li et al., 2019) and an accurate estimation of the density of models (Liu et al., 2021) can be incorporated to overcome the instability of models and improve performances of the model.
Supplementary Material
Acknowledgements
The authors thank Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago for making their data available. Data collection was supported through funding by NIA (Grants P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36042 and R01AG36836); the Illinois Department of Public Health; and the Translational Genomics Research Institute.
Data availability
The data underlying this article are publicly available as described in the Datasets section.
Funding
This work was supported by the Bio & Medical Technology Development Program of National Research Foundation of Korea (NRF) funded by the Korean government (MSIT) [NRF-2018M3C7A1054935], an NRF grant funded by the Korea government (MEST) [2021M3A9E4021818], and a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea [HI18C0460].
Conflict of Interest: none declared.
References
- Argelaguet R. et al. (2018) Multi-omics factor analysis—a framework for unsupervised integration of multi-omics data sets. Mol. Syst. Biol., 14, e8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahdanau D. et al. (2014) Neural machine translation by jointly learning to align and translate. arXiv, preprint arXiv:1409.0473.
- Bennett D. et al. (2018) Religious orders study and rush memory and aging project. J. Alzheimer’s Dis., 64, S161–S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary K. et al. (2018) Deep learning-based multi-omics integration robustly predicts survival in liver cancer . Clin. Cancer Res., 24, 1248–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Chan D.C. (2009) Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum. Mol. Genetics, 18, R169–R176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.-I.H. et al. (2018) GSAE: an autoencoder with embedded gene-set nodes for genomics functional characterization. BMC Syst. Biol., 12, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Zhang S. (2018) Discovery of two-level modular organization from matched genomic data via joint matrix tri-factorization. Nucleic Acids Res., 46, 5967–5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. et al. (2021) Openannotate: a web server to annotate the chromatin accessibility of genomic regions. Nucleic Acids Res., 49, W483–W490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q. et al. (2005) Ribosome dysfunction is an early event in Alzheimer’s disease. J. Neurosci., 25, 9171–9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi S.K. et al. (2020) Deriving disease modules from the compressed transcriptional space embedded in a deep autoencoder. Nat. Commun., 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmarakeby H.A. et al. (2021) Biologically informed deep neural network for prostate cancer discovery. Nature, 598, 348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L., Medico E. (2007) Flame, a novel fuzzy clustering method for the analysis of dna microarray data. BMC Bioinformatics, 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanczar B. et al. (2020) Biological interpretation of deep neural network for phenotype prediction based on gene expression. BMC Bioinformatics, 21, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasin Y. et al. (2017) Multi-omics approaches to disease. Genome Biol., 18, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L. et al. (2019a) Driver network as a biomarker: systematic integration and network modeling of multi-omics data to derive driver signaling pathways for drug combination prediction. Bioinformatics, 35, 3709–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z. et al. (2019b) SALMON: survival analysis learning with multi-omics neural networks on breast cancer. Front. Genetics, 10, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter C., Zenklusen J.C. (2018) The Cancer Genome Atlas: creating lasting value beyond its data. J. Alzheimer’s Dis., 173, 283–285. [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res., 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall A. et al. (2018). Multi-task learning using uncertainty to weigh losses for scene geometry and semantics. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Salt Lake City, Utah, United States, IEEE, pp. 7482–7491.
- Kingma D.P., Ba J. (2014) Adam: a method for stochastic optimization. arXiv, preprint arXiv:1412.6980.
- Koh H.W. et al. (2019) iOmicsPASS: network-based integration of multiomics data for predictive subnetwork discovery. NPJ Syst. Biol. Appl., 5, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M.V. et al. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res., 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. et al. (2019) DeepTACT: predicting 3d chromatin contacts via bootstrapping deep learning. Nucleic Acids Res., 47, e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q. et al. (2021) Density estimation using deep generative neural networks. Proc. Natl. Acad. Sci. USA, 118, e2101344118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock E.F. et al. (2013) Joint and individual variation explained (jive) for integrated analysis of multiple data types. Ann. Appl. Stat., 7, 523–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B. et al. (2020) Diagnostic classification of cancers using extreme gradient boosting algorithm and multi-omics data. Comput. Biol. Med., 121, 103761. [DOI] [PubMed] [Google Scholar]
- Manczak M. et al. (2004) Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease. Neuromol. Med., 5, 147–162. [DOI] [PubMed] [Google Scholar]
- Min W. et al. (2021) TSCCA: a tensor sparse CCA method for detecting microRNA-gene patterns from multiple cancers. PLoS Comput. Biol., 17, e1009044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappoport N., Shamir R. (2018) Multi-omic and multi-view clustering algorithms: review and cancer benchmark. Nucleic Acids Res., 46, 10546–10562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy C.C. et al. (2015) The role of the tripartite glutamatergic synapse in the pathophysiology of Alzheimer’s disease. Aging Dis., 6, 131–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevush S. et al. (1998) Platelet activation in Alzheimer disease. Arch. Neurol., 55, 530–536. [DOI] [PubMed] [Google Scholar]
- Sharifi-Noghabi H. et al. (2019) Moli: multi-omics late integration with deep neural networks for drug response prediction. Bioinformatics, 35, i501–i509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimohama S. (2000) Apoptosis in Alzheimer’s disease—an update. Apoptosis, 5, 9–16. [DOI] [PubMed] [Google Scholar]
- Subramanian I. et al. (2020) Multi-omics data integration, interpretation, and its application. Bioinformatics Biol. Insights, 14, 1177932219899051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X. et al. (2019) Adashare: learning what to share for efficient deep multi-task learning. arXiv, preprint arXiv:1911.12423.
- Vapnik V. (2000). The Nature of Statistical Learning Theory. New York: Springer. [Google Scholar]
- Wang Q. et al. (2017). Multi-modality disease modeling via collective deep matrix factorization. In Proceedings of the 23rd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, pp. 1155–1164.
- Wang T. et al. (2020) MORONET: multi-omics integration via graph convolutional networks for biomedical data classification. bioRxiv.
- Wu Y.-H., Swaab D.F. (2007) Disturbance and strategies for reactivation of the circadian rhythm system in aging and Alzheimer’s disease. Sleep Med., 8, 623–636. [DOI] [PubMed] [Google Scholar]
- Yang Z., Michailidis G. (2016) A non-negative matrix factorization method for detecting modules in heterogeneous omics multi-modal data. Bioinformatics, 32, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. et al. (2020) SMSPL: robust multimodal approach to integrative analysis of multiomics data. IEEE Trans. Cybern., 1. [DOI] [PubMed] [Google Scholar]
- Zeng W. et al. (2021) Silencerdb: a comprehensive database of silencers. Nucleic Acids Res., 49, D221–D228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Zhang S. (2019) Learning common and specific patterns from data of multiple interrelated biological scenarios with matrix factorization. Nucleic Acids Res., 47, 6606–6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. et al. (2011) A novel computational framework for simultaneous integration of multiple types of genomic data to identify microRNA-gene regulatory modules. Bioinformatics, 27, i401–i409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. et al. (2012) Discovery of multi-dimensional modules by integrative analysis of cancer genomic data. Nucleic Acids Res., 40, 9379–9391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. et al. (2019). Integrated multi-omics analysis using variational autoencoders: application to pan-cancer classification. In: 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), SanDiego, CA, USA, IEEE, pp. 765–769.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are publicly available as described in the Datasets section.