

Abstract
Summary
Non-coding RNAs are often neglected during genome annotation due to their difficulty of detection relative to protein coding genes. FindNonCoding takes a pattern mining approach to capture the essential sequence motifs and hairpin loops representing a non-coding RNA family and quickly identify matches in genomes. FindNonCoding was designed for ease of use and accurately finds non-coding RNAs with a low false discovery rate.
Availability and implementation
FindNonCoding is implemented within the DECIPHER package (v2.19.3) for R (v4.1) available from Bioconductor. Pre-trained models of common non-coding RNA families are included for bacteria, archaea and eukarya.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Non-coding RNAs play central roles in the information flow from DNA to protein and many other cellular processes (Fremin and Bhatt, 2021). While it is common to analyze the protein content of a genome, non-coding RNAs are often ignored in bioinformatic analyses. This is partly because it remains challenging to annotate the wide variety of non-coding RNAs relative to the ease with which proteins can be annotated. Protein coding genes are readily identifiable by their distinct composition and the start/stop codons delimiting their boundaries. In contrast, non-coding RNAs are difficult to identify ab initio because their distinguishing characteristics—elevated GC-content and density of folding—are common throughout the genome. Therefore, detection of non-coding RNAs often requires prior knowledge of their sequence, complicating their identification. Simple software for identifying known non-coding RNAs would facilitate their analysis and inclusion in bioinformatic pipelines.
Many of the existing tools to identify non-coding RNAs are family-specific. For example, there are methods designed to detect only transfer RNAs (tRNAs), such as tRNAscan-SE (Lowe and Eddy, 1997). Analogous tools exist for many other families of RNAs that leverage specific features of the non-coding RNA for detection (Stadler, 2014). General purpose tools based on searches for sequence homology have low sensitivity because non-coding RNA families are often highly divergent (Freyhult et al., 2007). High accuracy approaches, such as Infernal (Nawrocki and Eddy, 2013), apply models of covariation that search for a combination of sequence and structure consensus. Since these models tend to be difficult to apply, user-friendly packages have been developed to facilitate their adoption [e.g. StructRNAfinder (Arias-Carrasco et al., 2018)]. Nevertheless, ease of use remains a major impediment to the routine annotation of non-coding RNAs in genomes, partly because existing tools lack the simplicity of protein coding gene callers where annotation is often a one step process.
Here, I set out to take an alternative approach to the annotation of non-coding RNAs that was designed specifically for ease of use. The method begins with a sequence alignment and learns a compact representation of sequence and structure patterns specific to the RNA family. Multiple families are combined into a single object that allows for the rapid and accurate detection of many non-coding RNAs in genomes. Care was taken to avoid complex training procedures or poor scalability for long RNAs. Using a new validation approach, I show the merits of FindNonCoding for the quick and practical annotation of common non-coding RNAs.
2 Materials and methods
The training process begins with a multiple alignment of sequences belonging to a non-coding RNA family (Fig. 1A). The LearnNonCoding function automatically extracts four features: (i) sequence motifs in the form of a position weight matrix, (ii) conserved hairpin loops and pseudoknots in the consensus secondary structure predicted by DECIPHER (Wright, 2020), (iii) a k-mer usage profile and (iv) the distribution of sequence lengths. These signals are used to construct a log-odds model of the sequences, where the score is obtained by summing the log-odds of features representing the RNA family relative to a background of random nucleotides (see Supplementary Methods for a complete description). Matches to sequence motifs and hairpin loops are separated into multiple bins based on their score or predicted free energy of pairing (Yilmaz et al., 2012), respectively. Patterns are ranked by their log-odds scores, and up to 20 (by default) are selected for the model. A collection of models can then be given along with a genome sequence to the FindNonCoding function, which returns any hits and their associated scores (Fig. 1A).
Fig. 1.
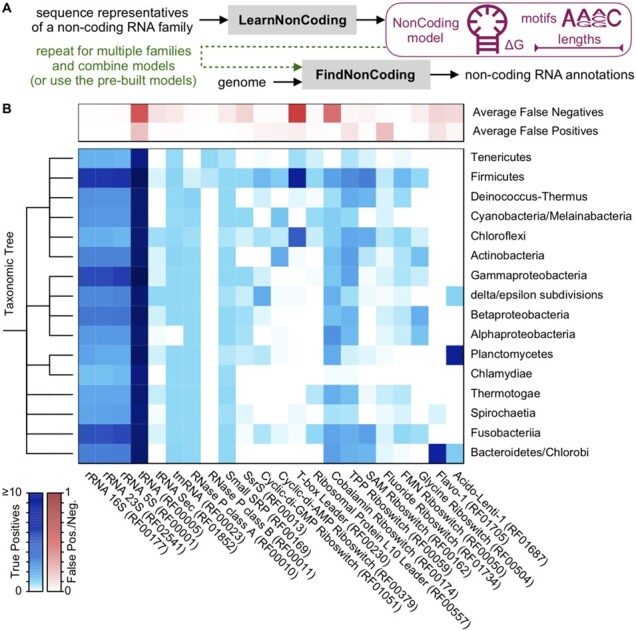
Application of FindNonCoding to detect non-coding RNA families. (A) The process begins by training a set of NonCoding models using LearnNonCoding or loading the pre-built models that come with the program. Each model contains a compact description of features characteristic of sequences belonging to a family. Models are then provided with a genome to FindNonCoding, which returns the location and score of any hits. (B) The distribution of true and false positives among bacteria for common non-coding RNA families. Non-coding RNAs were correctly identified within the intergenic space, with relatively few false positives substantially overlapping with protein coding genes. Ubiquitous non-coding RNAs were identified in an average of one or more copies among almost all genomes with few false negatives found by Infernal but not FindNonCoding. Only phyla with at least 10 representative genomes are shown
Previous attempts to benchmark tools for identifying non-coding RNAs have relied on embedding distant homologs in a background of random DNA representing a genome (Nawrocki, 2014). To quantify accuracy in a more realistic fashion, 2774 NCBI Refseq representative genomes were searched for a set of RNA families conserved among bacteria. RNA families were chosen that were found by Rfam (Kalvari et al., 2018) (via Infernal) in more than half the genomes belonging to at least 10% of bacterial phyla. Up to 1000 representatives of each family were obtained from Rfam and aligned with DECIPHER (Wright, 2020). Hits to each non-coding RNA family were classified as false positives if they substantially overlapped (>50% of RNA sites) with predicted open reading frames or true positives if they did not (<10%). Since this approach only provides information about true and false positives, Infernal's predictions were used to identify false negatives absent from FindNonCoding's predictions under the assumption that Infernal had a 0% false-positive rate. To this end, covariance models for each non-coding RNA family were downloaded from Rfam and combined with cmpress. Genomes were searched for matches using the cmscan command with non-default parameters ‘–cpu 1 -oskip –fmt 2 –tblout –notrunc’. Per the developer's suggestion, I subset hits to those spanning 90% of the model's length (CLEN), rather than using the ‘-g’ parameter, to obtain global hits for equivalence with FindNonCoding.
3 Results
FindNonCoding is conceptually different from other non-coding RNA detection tools in that it searches for patterns relative to the ends of the non-coding RNA rather than searching for the entire sequence or structure. Since the number of patterns is fixed, FindNonCoding scales independently of query sequence length, making it amenable to the quick detection of longer RNAs such as ribosomal RNA genes. This approach also facilitates the handling of large insertions and deletions within the non-coding RNA, so long as conserved patterns remain uninterrupted. FindNonCoding takes about one second to detect each RNA family (query) per million base pairs searched (Supplementary Fig. S1). FindNonCoding was able to detect almost all non-coding RNAs embedded in random sequence when the training set did not contain any sequences within 40% sequence identity of a test sequence (Supplementary Fig. S2). This benchmarking approach is similar to that used by most previous analyses of non-coding RNA detection programs but lacks realism because genomic sequence is non-random.
Bacterial genomes are around 90% protein coding and gene calling is relatively high accuracy (Korandla et al., 2020), which permits the quantification of true and false positives under the assumption that most non-coding RNAs should fall largely within intergenic spaces between protein coding genes. Figure 1B shows the breakdown of non-coding RNAs found by FindNonCoding across genomes spanning bacterial phyla in Refseq. FindNonCoding correctly detected non-coding RNAs across most genomes, with a false discovery rate far less than one per genome on average. The ubiquitous non-coding RNAs, such as tRNAs, ribosomal RNAs, transfer-messenger RNA (tmRNA), RNase P and the signal recognition particle, were identified in almost every bacterial group. The main exception being the tmRNA of Alphaproteobacteria, which is two-piece and has its own Rfam family (RF01849). The false-negative rate, i.e. non-coding RNAs identified by Infernal but not by FindNonCoding, was generally low with the exception of some families with scores near the threshold of detection (Supplementary Fig. S3). Notably, FindNonCoding identified some RNAs that were not found by Infernal and did not overlap protein coding genes (Supplementary Fig. S4), suggesting Infernal occasionally misses some non-coding RNAs or FindNonCoding misclassifies some non-coding RNAs as belonging to the wrong RNA family.
FindNonCoding works well to annotate non-coding RNAs in practice, and has several attributes that make it straightforward to use. First, a secondary structure annotation is not required with the input multiple sequence alignment, although LearnNonCoding is dependent on alignment quality and sequence diversity to correctly predict conserved secondary structure (Wright, 2020). To further simplify use, pre-trained models are provided based on alignments of common non-coding RNAs belonging to archaea, bacteria and eukarya. Second, FindNonCoding selects the top scoring model in each region when there are multiple hits. This facilitates greater resolution when multiple models are expected to match the same region, although it is also possible to obtain all hits by allowing overlaps. Third, the use of LearnNonCoding and FindNonCoding is well documented and requires few commands, making the software relatively user-friendly. A direct comparison of default outputs from StructRNAfinder with an E-value cutoff ≤ 1e-4 are shown in Supplementary Table S1 for the genome of the bacterium Chlamydia trachomatis (NC_000117). FindNonCoding provides information on cognate amino acids for tRNAs, does not report any hits to eukaryotic Rfam families, and avoids multiple hits of related RNA families (e.g. rRNAs) matching the same region. It is anticipated that these features will facilitate non-coding RNA genome annotation with FindNonCoding.
Funding
This work was supported by the National Institutes of Health (NIAID grant 1DP2AI145058-01).
Data availability
The data underlying this article are publicly available in Rfam at http://rfam.xfam.org and RefSeq at https://www.ncbi.nlm.nih.gov/refseq/.
Conflict of Interest: none declared.
Supplementary Material
References
- Arias-Carrasco R. et al. (2018) StructRNAfinder: an automated pipeline and web server for RNA families prediction. BMC Bioinformatics, 19, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremin B.J., Bhatt A.S. (2021) Comparative genomics identifies thousands of candidate structured RNAs in human microbiomes. Genome Biol., 22, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyhult E.K. et al. (2007) Exploring genomic dark matter: a critical assessment of the performance of homology search methods on noncoding RNA. Genome Res., 17, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvari I. et al. (2018) Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res., 46, D335–D342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korandla D.R. et al. (2020) AssessORF: combining evolutionary conservation and proteomics to assess prokaryotic gene predictions. Bioinformatics, 36, 1022–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe T.M., Eddy S.R. (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res., 25, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki E.P. (2014) Annotating functional RNAs in genomes using infernal. In: Gorodkin J., Ruzzo W.L. (eds) RNA Sequence, Structure, and Function: Computational and Bioinformatic Methods. Humana Press, Totowa, NJ, pp. 163–197. [DOI] [PubMed] [Google Scholar]
- Nawrocki E.P., Eddy S.R. (2013) Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics, 29, 2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler P.F. (2014) Class-Specific Prediction of ncRNAs. In: Gorodkin J., Ruzzo W.L. (eds) RNA Sequence, Structure, and Function: Computational and Bioinformatic Methods. Humana Press, Totowa, NJ, pp. 199–213. [Google Scholar]
- Wright E.S. (2020) RNAconTest: comparing tools for noncoding RNA multiple sequence alignment based on structural consistency. RNA, 26, 531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz L.S. et al. (2012) Modeling formamide denaturation of probe-target hybrids for improved microarray probe design in microbial diagnostics. PLoS One, 7, e43862. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are publicly available in Rfam at http://rfam.xfam.org and RefSeq at https://www.ncbi.nlm.nih.gov/refseq/.
Conflict of Interest: none declared.