

Abstract
Post Tuberculosis Lung Disease (PTLD) affects millions of tuberculosis survivors and is a global health burden. The immune mechanisms that drive PTLD are complex and have historically been under investigated. Here, we discuss two immune-mediated paradigms that could drive human PTLD. We review the characteristics of a fibrotic granuloma that favors the development of PTLD via an abundance of T-helper-2 and T-regulatory cells and an upregulation of TGF-β mediated collagen deposition. Next, we discuss the post-primary tuberculosis paradigm and the complex mixture of caseous pneumonia, cavity formation and fibrosis that can also lead to PTLD. We review the delicate balance between cellular subsets and cytokines of the innate and adaptive immune system in conjunction with host-derived proteases that can perpetuate the parenchymal lung damage seen in PTLD. Next, we discuss the role of novel host directed therapies (HDT) to limit the development of PTLD and in particular, the recent repurposing of established medications such as statins, metformin and doxycycline. Finally, we review the emerging role of novel imaging techniques as a non-invasive modality for the early recognition of PTLD. While access to computed tomography imaging is unlikely to be available widely in countries with a high TB burden, its use in research settings can help phenotype PTLD. Due to a lack of disease-specific biomarkers and controlled clinical trials, there are currently no evidence-based recommendations for the management of PTLD. It is likely that an integrated antifibrotic strategy that could simultaneously target inflammatory and pro-fibrotic pathways will probably emerge as a successful way to treat this complex condition. In a disease spectrum as wide as PTLD, a single immunologic or radiographic marker may not be sufficient and a combination is more likely to be a successful surrogate that could aid in the development of successful HDTs.
Keywords: PTLD, Granuloma, Host directed therapy, Biomarker, Imaging, Radiographic signature
1. Introduction
Tuberculosis (TB) remains a leading cause of death from a single infectious agent, killing more than a million people annually [1]. TB programs have historically emphasized the need for rapid diagnosis and treatment of active TB, but as the prevalence of successful TB treatments has increased, the global numbers of TB survivors has also gone up exponentially, with an estimated 155 million TB survivors in 2020 [1]. With the help of a modeling study, Dodd et al., [2], estimated that the number of TB survivors alive in 2020 was ten times more than the estimated annual TB incidence. In another study that used a hypothetical cohort of individuals with TB and disability-adjusted life-years (DALY) to summarize health losses due to TB, 58 million DALYs were attributable to post TB sequelae [3]. Such astronomical case numbers are undisputedly concerning for healthcare sectors and institutions around the globe. Additionally, despite successful treatment and microbiological cure, post TB patients experience high morbidity and mortality [4], with a standardized mortality ratio 3 times higher than that of the control population [5]. Despite these concerning data, post tuberculosis lung disease (PTLD) is a term sparingly used by the international TB research community, thus impacting its recognition and treatment [6]. A systematic review of over 200 TB guidelines revealed that only 3 international TB guidelines mentioned PTLD, with little guidance on how to identify or treat it [6]. For example, the World Health Organization (WHO) End TB strategy makes no mention of PTLD and current TB registries only capture morbidity and mortality during treatment.
PTLD is marked by tissue necrosis and aberrant healing that leads to permanent destruction and distortion of the lung architecture [7]. Pathological features of PTLD include but are not limited to cavitation, fibrosis (both limited and extensive), bronchiectasis and small-airway disease (sometimes termed as tuberculosis-associated obstructive pulmonary disease), airway stenosis, fibrosing mediastinitis, fibrothorax and bronchopleural fistulas [8,9]. The heterogeneous pattern of disease in human PTLD suggests that risk factors are an interplay between the host, the pathogen and the environment. These include modifiable risk factors such as smoking, biomass fuel exposure, occupational exposures and concurrent respiratory tract infections and less modifiable risk factors such as a pre-existing lung disease [10]. For the purpose of this review, we have used the definition of PTLD as described at the First International Symposium on post TB disease as “evidence of chronic respiratory abnormality, with or without symptoms attributable at least in part to previous pulmonary TB” [11]. Our review has focused on the cellular subsets, cytokines and host-derived proteases of the innate and adaptive immune system that drive the parenchymal lung damage seen in PTLD. This includes host-pathogen interactions and immune dynamics that trigger and maintain the tuberculous granuloma as well as the various stages of post-primary/cavitary TB. We also discuss the role of novel host directed therapies (HDTs) to limit the development of PTLD. Finally, we review recent advances in imaging techniques such as computed tomography (CT) and positron emission tomography (PET)-CT that are being increasingly used as surrogates for the identification of PTLD.
2. Burden of PTLD
Studies have shown that subjects with residual lung damage/cavitation after successful completion of anti-TB therapy (ATT) can have an up to fourfold higher risk of re-infection, relapse and progressive tissue damage [12,13]. It has been estimated that up to 50% of individuals treated for TB have some form of persistent pulmonary dysfunction, despite successful bacteriologic cure [14]. Evidence of chronic airway obstruction is commonly found in subjects who have previously been treated for TB [15] and several systematic reviews have shown that a past history of TB can increase the risk of developing chronic obstructive pulmonary disease (COPD) [16]. Baez-Saldana et al. [17], showed a significant inverse correlation between both FEV1 and FVC and the extent of radiographic damage in patients who were at a median of approximately one year post ATT. Problems are perpetuated by the Xpert nucleic amplification tests that have a 1 in 7 false positive rate and [18] and if patients return with symptoms after the completion of their regimen, ATT is often re-commenced even in the absence of positive smears or cultures with the the possibility of PTLD commonly overlooked.
But there have been some promising developments in recent years. First, a multi-center TB cohort study called TB Sequel has been launched to assess predictors of PTLD and will give valuable information on the clinical, microbiological, immunological and socio-economic factors that drive this disease [19]. The primary outcome in the TB Sequel study is functional impairment but it will also collect biological samples in a longitudinal manner to assess risk in a nested case-control fashion and factor in the influence of comorbidities such as HIV, COPD and diabetes [19]. Secondly, a universal standard was recently defined with special considerations to identifying and counseling PTLD patients for pulmonary rehabilitation, followed by guidelines for evaluating the efficacy of pulmonary rehabilitation programs [20]. These standards recommend that every patient who completes TB therapy should be clinically evaluated for PTLD with a clinical examination, nutritional status assessment, chest radiology, pulmonary function test, 6-min walk test and functional scores. In addition, co-morbid medical conditions such as HIV, diabetes and cardiovascular conditions would be noted and follow up plans would include counseling/health education for patients as well as their household contacts. Patients at risk of developing PTLD would be identified as early as possible as they are the most likely to benefit from smoking cessation, airway clearance and bronchodilator therapy. The presence of persistent radiological features of past or healed TB is one of the best criteria for identifying such high-risk individuals PTLD and therefore the presence of such features should trigger active surveillance for the development of PTLD.
3. Pathogenesis of PTLD: immune mechanisms and current paradigms
The immune mechanisms that drive PTLD are complex and subtle permutations in the cytokine and cellular milieu can lead to major shifts in the phenotype. The vast majority of data in this field are from pre-clinical studies and animal models and the pathways that trigger PTLD in human TB survivors are largely unknown. The lack of an animal model that could be a good representation of the heterogeneous and dynamic nature of human PTLD has contributed to the under-investigation of PTLD [21]. Given the complete lack of studies with lower airway samples and a paucity of clinical trials in human PTLD subjects, it is challenging to draw “causal” inferences between the immune mechanisms and the development of PTLD. Here, we discuss two plausible immune mediated paradigms that could drive human PTLD.
3.1. The fibrotic tuberculous granuloma paradigm
The tuberculous granuloma is ground zero of the immune response to Mtb and in its simplest form is a collection of macrophages and leukocytic cells that organize after Mtb encounters the alveolar macrophage [22]. The granuloma has been traditionally believed to be protective by preventing the dissemination of Mtb but this has been challenged in recent years after it was shown that they can be conducive to the spread of Mtb [23]. In its classic form, the granuloma has a central focus of acellular necrotic caseum surrounded by epithelioid macrophages and multi-nucleated giant cells, that are then encapsulated by a rim of fibroblasts and lymphocytes. The caseous granuloma can become calcified and threby fall on the more successful end of the spectrum of disease outcomes [24]. Conversely, as depicted in Fig. 1, pro-inflammatory/permissive granulomas can also evolve into fibrotic types. Marakala et al. [25], generated a molecular map of granuloma progression in tissue obtained from surgical specimens by utilizing laser-capture micro-dissection, mass spectrometry and confocal microscopy. They identified over 3000 proteins that were differentially expressed between different regions and whilst the proteomic profiles of caseous or cavitary centers were composed of pro-inflammatory pathways, the periphery displayed a more anti-inflammatory signature. Recent research in non-human primates (NHP) has shown that the evolution of granuloma is less linear than previously believed [26] and the local balance of host pro- and anti-inflammatory cytokines play a critical role in determining their evolution [27].
Fig. 1.
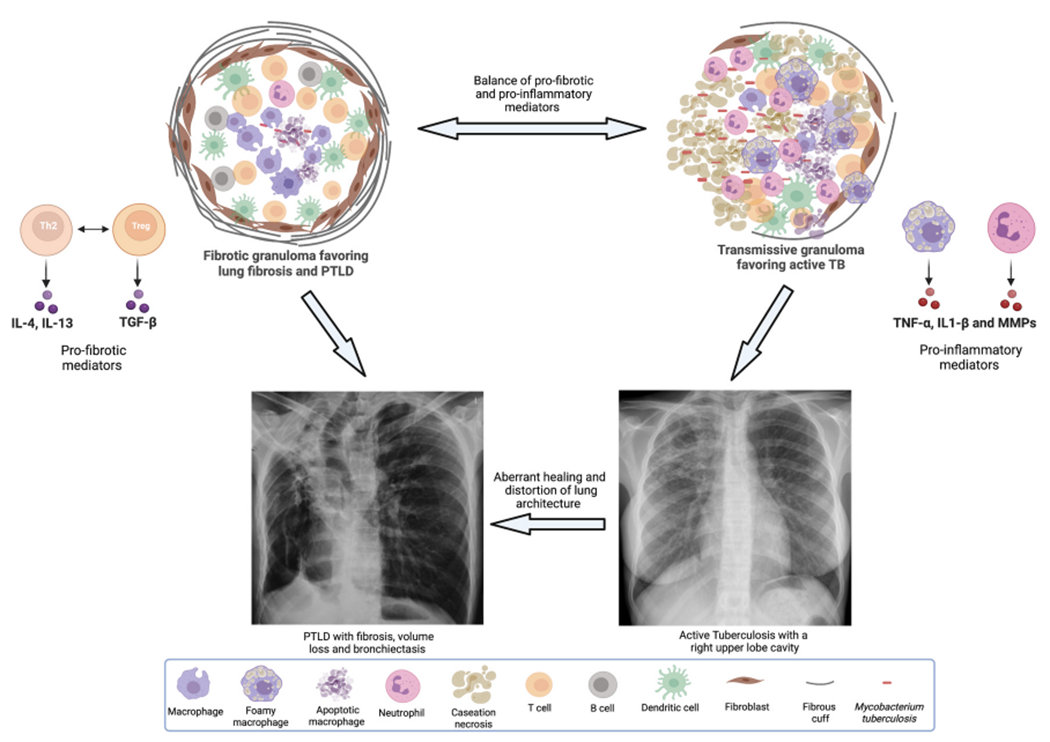
Depicts the characteristics of a fibrotic granuloma (left) and a transmissive granuloma (right). Fibrotic granulomas have an abundance of Th2 and Treg cells that drive TGF-β mediated collagen deposition, thick capsule consisting of myofibroblasts and an abnormal fibrous cuff. Transmissive granulomas are rich in classically activated and foamy macrophages as well as neutrophils. These cells produce inflammatory mediators such as tumor necrosis factor (TNF)-α and matrix metalloproteinases (MMPs) and depending on other host modulating factors, can result in the development of active cavitary TB disease. As the figure depicts, fibrotic granulomas favor the development of PTLD and transmissive granulomas favor the development of active TB, but granulomas can undergo changes driven by host factors and surrounding milieu such that they can morph into one or the other kind. Finally, the figure also depicts that active cavitary TB can lead to PTLD in the event of aberrant healing and distortion of lung architecture.
Several immune mediators and cell subtypes that can activate transforming growth factor (TGF)-β, the prototypic fibrogenic cytokine, have been observed in human fibrotic TB granulomas [28]. Using a hybrid multi-scale model of fibrotic granuloma formation, GranSim, Warsinke et al. [29], combined molecular, cellular and tissue scale models to characterize peripherally fibrotic and centrally fibrotic granulomas. They found that fibroblasts and myofibroblasts drive wound healing in the granuloma via the secretion of extracellular matrix (ECM) proteins, TGF-β and IL-10. In an NHP study that combined computational modeling with wet lab methodologies, sequential stimulation of alveolar macrophages drove a macrophage-to-myofibroblast transformation (MMT), followed by the development of peripheral fibrosis in TB granulomas [30]. Through complementary immunohistochemical staining of TB granulomas, a population of CD11c+ and α-smooth-muscle actin (αSMA)+ cells were identified in the fibrotic region suggesting the involvement of MMTs. Fibrosis occurs in and around inflamed or damaged tissue and fibrotic granulomas can help isolate infected tissue, but excess collagen can also lead to scar formation and lay the foundation for the development of PTLD. The identification of host immune response markers in accessible lung samples can help identify the interplay between fibrotic pathways that can trigger such lung damage in PTLD [31].
3.2. The post-primary/cavitary TB paradigm
As the name implies, post-primary TB follows primary TB and starts as an obstructive lobular bronchogenic pneumonia [32]. Post-primary TB can regress or undergo necrosis to become a caseous pneumonia that softens and then dissolves to form cavities. Alternatively, it can also progress to form either post-primary granulomas or fibrocaseous disease [33,34]. Over a period of months, secreted mycobacterial antigens and host lipids accumulate in foamy alveolar macrophages that then serve as a nutrient-rich reservoir for Mtb, thus increasing the risk for cavitation [35]. In a study that investigated regulatory markers within early infiltrates of fibrocaseous TB [36], foamy alveolar macrophages over-expressed mTORC1, programed death-1 ligand (PD-L1) and COX-2, suggesting that these alveolar macrophages facilitated disease progression to fibrocaseous disease. Different stages such as fibrosis, caseation and aberrant healing can co-exist and lesions in the same lung may evolve in radically different directions [37–39].
Small areas of tuberculous pneumonia may heal to leave only fibrous scars but large areas of caseous pneumonia are often surrounded by multiple concentric rings of fibrous tissue suggesting that lesions progressed before they subsequently healed; on multiple occasions [40]. Often, caseous pneumonia continues after the formation of cavities and produces a complex mixture of new cavity formation, fibrosis and healing. The fibrosis tends to worsen over time and is the leading cause of pulmonary fibrosis, lung collapse and atelectasis seen in PTLD. Fig. 1 depicts the characteristics of a fibrotic granuloma (left) and a transmissive granuloma (right). Fibrotic granulomas have an abundance of Th2 and Treg cells that drive TGF-β mediated collagen deposition and a thick capsule consisting of myofibroblasts and an abnormal fibrous cuff. Transmissive granulomas are rich in classically activated and foamy macrophages as well as neutrophils. These cells produce inflammatory mediators such as tumor necrosis factor (TNF)-α and matrix metal-loproteinases (MMPs) and depending on other host modulating factors can result in the development of active cavitary TB disease. As the figure depicts, fibrotic granulomas favor the development of PTLD and transmissive granulomas favor the development of active TB, but granulomas can undergo changes driven by host factors and surrounding milieu such that they can morph into one or the other kind. Finally, the figure also depicts that active cavitary TB can lead to PTLD in the event of aberrant healing and distortion of lung architecture.
4. Immunologic mediators of PTLD
The immunologic mediators that drive PTLD can be broadly categorized as belonging to the innate or the adaptive arms of immunity and are discussed below. Delineating the cellular and cytokine pathways involved can help discover adjunctive therapies with the potential to limit immunologically driven lung damage.
4.1. Innate immune response
4.1.1. Macrophages and neutrophils
Although there are several leukocytic cells that drive pathology in TB, the macrophage has a central role. Macrophages harbor the majority of Mtb and have various effector functions in the production of cytokines and chemokines, in the evolution of granulomas and in tissue remodeling [24]. Mtb infection skews the transcriptional and functional phenotype of macrophages towards the classically activated macrophage (CAM) that drives a Th1- response and up-regulates antimicrobial pathways [41]. This is counter-balanced by the alternatively activated macrophages (AAM) that are activated by pro-fibrotic cytokines such as IL-4 and IL-13 and have an immuno-suppressive and wound healing phenotype; driving weakly antibacterial and anti-inflammatory pathways, and ultimately contributing to damage control. In a study by Huang et al., [42] using lung tissues from human TB patients in addition to an in vitro tuberculous granuloma model, it was demonstrated that CAM type macrophages dominated during the initial stages of granuloma formation and macrophage bactericidal activity, whereas AAM type macrophages inhibited these effects. Macrophages can therefore have a dual, temporally-driven effect on the development of PTLD, with a pro-inflammatory role in the early stages via mediators that drive tissue injury, such as Interleukin (IL)-1β, TNF-α, reactive oxygen species and reactive nitrogen species. In later stages, they may acquire a more pro-fibrotic role via anti-inflammatory mediators such as IL-10 and programmed death ligand-2 [43].
The vast influx and subsequent quick demise of neutrophils after initial infection with Mtb are central to the development of necrotic and caseous cores of the granulomas [44]. Neutrophils are the only cells that store MMPs without co-existent tissue inhibitors of metalloproteinases (TIMPs), and thus have the capability to drive unrestrained matrix degradation in TB [45]. An excessive neutrophilic response is a sign of TB severity and lung destruction [46]. Barry et al. [47], have shown that tuberculous cavities contain more neutrophils and less lymphocytes compared to non-cavitary pulmonary infiltrates in TB. As a part of their role in TB pathogenesis, neutrophils have been shown to release neutrophil extracellular traps (NET), that are composed of condensed chromatin fibers coated with antimicrobial proteins [48]. NETs can entrap and prevent the dissemination of Mtb [49] but their lack of killing ability favors lung destruction and subsequent development of PTLD in TB. A study from the Rede-TB Study group [50] investigated the role of NETs in the occurrence of tissue damage as assessed on a chest radiograph and found a direct association between tissue damage and the various inflammatory proteins involved in the NET pathways.
4.1.2. Cytokines
The production of TGF-β correlates with fibrosis and blocking it has been demonstrated to reduce fibrosis in experimental animal models [51]. TGF-β is secreted in its latent form at the site of disease in TB from cells such as macrophages, fibroblasts and epithelial cells and then activated by various elements of the granuloma such as reactive oxygen and nitrogen species, low pH and hypoxia [52,53]. TGF-β induces the differentiation of fibroblasts into collagen-producing myofibroblasts and affects numerous other biological pathways such as cellular proliferation and migration. Elevated levels of TGF-β in serum and bronchoalveolar lavage fluid (BALF) correlated with an increase in fibrosis seen on high-resolution CT scans 6 months into TB treatment [54]. In a group of 51 patients from Indonesia who had completed ATT, there was a significant correlation between blood TGF-β levels and the presence of fibrosis on chest x-rays [55]. Higher pleural fluid TGF-β levels were also observed in patients with greater pleural thickening as observed on CT imaging [56]. Other studies have confirmed that the induction of inflammatory molecules such as TNF-α after Mtb infection correlates with clinical deterioration and severe tissue destruction [57,58]. In a seminal mouse study, neutralization of TNF-α resulted in rapid reactivation of TB, with a striking loss of granuloma structure and a substantially worsened pathologic process [59]. These studies therefore imply that TNF-α facilitates the structural integrity of the TB granuloma and drives necrosis and irreversible tissue damage [60], with a positive correlation with larger cavity size [61]. IL-1β is another innate inflammatory cytokine that can be a potent driver of fibrosis [62] and blood and BALF TNF-α and IL-1β levels correlated with the extent of disease and size of cavities [61,63]. Both TNF-α and IL-1β affect the production of MMPs [64] and IL-1β is also directly linked with the recruitment of neutrophils and lung damage.
4.2. Adaptive immune response
CD4+ T cells are well-established key players in TB immunopathology. T helper (Th)-1 subsets are defined by the production of IFN-γ [65] and Th2 subsets are defined by the production of pro-fibrotic cytokines IL-4 and IL-13 [66]. Via the activation of fibroblasts, Th2 cytokines drive fibrosis and collagen deposition, a phenomenon that is reciprocally inhibited by the Th1 cytokines [67,68]. In humans, BALF from patients with cavitary TB had increased IL-4-producing Th2 lymphocytes whereas those with non-cavitary disease had a predominantly Th1 profile [69]. Observations from these studies have led to the ‘Th2 hypothesis of fibrosis’, mediated by IL-4, IL-5 and IL-13 [70], via the activation of TGF-β. In the context of TB, an initial Th1 response followed by a Th2 response seeks to isolate the bacterium and drives a pro-fibrotic process [71]. Mechanistically, IFN-γ inhibits fibrosis by antagonizing TGF-β [72] and, in addition to inhibiting direct fibroblast proliferation, IFN-γ also inhibits TGF-β mediated pro-collagen and collagen synthesis in myofibroblasts. In experimental models, up-regulating Th1 cytokines has been shown to inhibit immune-mediated fibrosis but clinical studies investigating the therapeutic potential of IFN-γ in the limitation of fibrosis in fibrotic lung diseases have been disappointing [73]. A study by Harari et al. [74], showed that at the end of successful treatment, patients shifted from an Mtb-specific TNF-α+ CD4+ T cell profile towards a polyfunctional IL-2+ TNF-α+ IFN-γ+ CD4+ T cell profile. It is surmisable that single cytokine positive T cells correlate with a higher mycobacterial load and therefore assaying them early during treatment can help identify patients at high risk for PTLD as patients with high bacterial loads are more likely to have extensive inflammation and subsequent development of PTLD. In recent years, the Th17 subset producing Interleukin-17 (IL-17) has been recognized as an important driver of fibrosis in the lungs [75]. IL-17 expression is often associated with neutrophilia, which directly contributes to tissue damage and fibrosis [76]. IL-17 is expressed around TB granulomas in patients and drives the production of epithelial- and fibroblast-derived MMP-3 (a stromelysin) in cellular models of TB [77], another driver of tissue destruction. Finally, TREG cells are also an important source of pro-fibrotic cytokines such as TGF-β [78] and their preferential recruitment can have a decisive role as they exacerbate the effects of TGF-β, leading to an increased risk of PTLD. Fig. 1 depicts the delicate balance between various cellular subsets and cytokines of the innate and adaptive immune system and host-derived proteases that can drive the parenchymal lung damage seen in PTLD or the cavitary phenotype in active TB.
5. Matrix metalloproteinases in PTLD
MMPs are a family of host-derived zinc and calcium dependent proteolytic enzymes with the primary function of ECM degradation and tissue homeostasis [79]. In the context of TB, they have been implicated in tissue damage and cavitation [80]. In a study from Taiwan, MMP-1 (1607G) polymorphism was associated with more extensive lung fibrosis one year after the completion of ATT [81]. Elevated MMP levels in respiratory secretions are distinctly associated with more extensive disease and cavitation [82,83], with mice transgenic for MMP-1 exhibiting increased tissue damage and collagen destruction after Mtb infection [84]. Using a combination of an in vitro model, a cohort of 108 patients and patient lung biopsies, it was shown that neutrophil-derived MMP-8 (a collagenase) was up-regulated in TB and led to matrix destruction [45]. Sputum MMP-8 correlated with TB severity score and cavitation and neutrophils containing MMP-8 were found at the center of cavities and in areas of caseous necrosis.
There have been numerous studies to investigate the modulation of MMPs to reduce tissue damage and inflammation in TB. Specific MMP inhibitors (batimastat, cipemastat, marimastat) have been studied in animal models [85] but they generated variable host responses. More recently, the combined use of MMP inhibitors (marimastat) and a first-line ATT (isoniazid) have shown a synergistic effect in reducing Mtb burden in the mouse [86]. Marimastat also reduced vascular leakage surrounding TB granulomas and increased the concentration of isoniazid in the lungs. In a similar study, co-administration of an MMP-9 antibody with ATT reduced pulmonary bacterial burden and relapse rates [87]. These studies illustrate that MMP inhibition in conjunction with antibiotic therapy may help limit PTLD, whilst emphasizing that a better understanding of MMP driven pathology is required to develop more targeted therapies.
6. Host directed therapy in PTLD
The general mainstays of treatment for PTLD are the same as those for the majority of chronic lung conditions. These include pulmonary toilet and muco-ciliary clearance with inhaled bronchodilators. Studies have reported modest improvement in pulmonary function and dyspnea scores with these conservative measures [88,89], but there remains a paucity of treatment strategies that would specifically target the disease process in PTLD. The Host-Directed Therapies Consortium Network was launched in April 2015 to commence trials of TB HDTs [90] and approved several drugs already in clinical use to be repurposed for phase 2 trials in TB. But there were few trials targeting PTLD. In this section, we specifically focus on and review adjunctive HDTs that could have a potential role in limiting pathology and accelerating cure in PTLD. The existing evidence points towards a few pathways/avenues that could be exploited safely. For example, anti-fibrotic drugs could minimize the distortion of normal tissue architecture associated with PTLD. Given the central role that macrophages play in fibrosis, several experimental antifibrotic strategies have been designed to regulate the activation and recruitment of distinct macrophage populations. For example, ligands that engage peroxisome proliferator-activated receptor-α (PPAR-α) and PPAR-γ can inhibit the development of fibrosis by decreasing the production of inflammatory cytokines from CAM’s and by repressing TGF-β mediated fibrosis [91]. There are multiple pathways through which neutrophils drive extracellular matrix degradation and the formation of necrotic and hypoxic granulomas in TB. The detrimental effect of excessive NET release is particularly important in TB as NETs can expand in the pulmonary alveoli and cause extensive lung damage. Recent therapies targeting NETs include DNA disintegration with recombinant human DNase and the neutralization of NET proteins with anti-histone antibodies and protease inhibitors could potentially be utilized for PTLD [48].
Corticosteroids have long been used as adjunctive therapy in TB meningitis and pericarditis but recent reviews and meta-analyses have concluded that there is a lack of convincing evidence that steroids can limit long term sequela in pulmonary TB [92,93]. In a mouse study, Ibuprofen, a non-steroidal anti-inflammatory drug that inhibits cyclooxygenases, reduced murine lung pathology and mycobacterial load at 3–4 weeks after Mtb infection, resulting in smaller lesions [94]. But human studies have raised concerns against the use of non-steroidal agents due to an increased risk of active TB [95]. There has been a longstanding interest in identifying mechanisms to reduce TNF-α production to limit inflammation and improve treatment outcomes in TB. But since TNF-α is also essential for a protective immune response against Mtb, caution must be exercised when blocking this cytokine. Using a rabbit model of TB, Subbian et al. [96], demonstrated that the use of a phophodiesterase-4 (PDE4) inhibitor resulted in an enhanced response to isoniazid therapy and was associated with a striking resorption of granulomas, with limitation of necrosis and tissue damage. The PDE4 inhibitor, CC-3052, achieved suppression of mRNA levels of TNF-α, IL-4, IL-8 and several MMPs without a generalized immune suppression, thereby facilitating microbial killing and clearance via improved penetration of isoniazid into the lesions. Blocking TNF-α with etanercept in the presence of 1st line anti-TB drugs was also noted to be beneficial by reducing the bacterial load and limiting tissue pathology in a murine model [97]. Finally, IL-17 targeted therapy has the potential to limit PTLD related tissue damage, especially via modulation of neutrophilic responses.
Various medications already in clinical use can be repurposed for PTLD. Studies have reported that the widely used anti-diabetic medication metformin dampens immunopathology and enhances the efficacy of anti-TB drugs in mouse models of TB [98]. The lungs of metformin-treated mice had increased lymphocyte infiltration with fewer coalescent lesions, ascompared to the untreated control mice.. The protective effect of metformin is mediated via host cell production of reactive oxygen species and acidification of the mycobacterial phagosomes. Patients who were taking metformin had fewer pulmonary cavities and reduced progression to severe TB disease when compared to patients who were taking sulfonylureas [99], but several drug interactions and pharmacokinetic aspects need to be studied before mteformin can be used as an HDT to limit PTLD. Parihar et al. [100], demonstrated that statins arrest bacterial growth in Mtb-infected macrophages by enhancing phagosome maturation and autophagy and the treatment of Mtb-infected mice with statins increased survival, reduced the size and inflammation of lung lesions and shortened the duration of 1st line TB therapy. Doxycycline, the only licensed MMP inhibitor in clinical use, is known to non-specifically inhibit multiple MMPs, independent of its antimicrobial activity [83]. Walker et al. [83], first reported that doxycycline modulated MMP expression in Mtb-infected cells and reduced mycobacterial growth in the guinea pig model. In a phase 2 randomized controlled trial, the use of Doxycycline within 7 days of starting ATT led to significant changes in the host transcriptome and the suppression of systemic and respiratory markers of tissue destruction [101]. Whole blood RNA-sequencing demonstrated that doxycycline restored the dysregulated gene expression in TB towards normalcy and these effects persisted for 6 weeks. Doxycycline was also associated with concurrent suppression of plasma and respiratory MMPs and reduction of pulmonary cavity volume.
RNA interference is a rapidly evolving field that is yet to be exploited for PTLD. MicroRNAs (miRNAs) include a broad group of evolutionarily conserved non-coding RNAs that play an important role in pathophysiological processes by blocking translation or promoting degradation of complementary target messenger RNAs. A subset of miRNA’s constitutively expressed in healthy tissues are down-regulated as fibrosis develops, suggesting that they might have an anti-fibrotic role [102]. Identifying such specific miRNAs that can promote tissue healing could be an adjunctive treatment modality in PTLD [103]. Such miRNAs could be targeted via small molecule inhibitors or via miRNA mimics; via the use of techniques that encapsulate and deliver miRNAs to specific cells. Direct delivery of miRNAs by targeting macrophages and dendritic cells is one approach that can be exploited to aim for an anti-inflammatory phenotype and the prevention of PTLD [104]. Another approach is to deliver small-interfering RNAs (siRNA) such as let-7e and miR-29a in order to dampen the expression of cytokines such as TNF-α [105] or modulate the expression of MMP-9 [106]. A limitation is that most miRNAs target multiple mRNAs and their exogenous administration can therefore have unwanted off-target effects. Fig. 2 gives an overview of various HDTs that are undergoing clinical trials, such as PDE4 inhibitors, repurposed drugs and RNA interference technology.
Fig. 2.
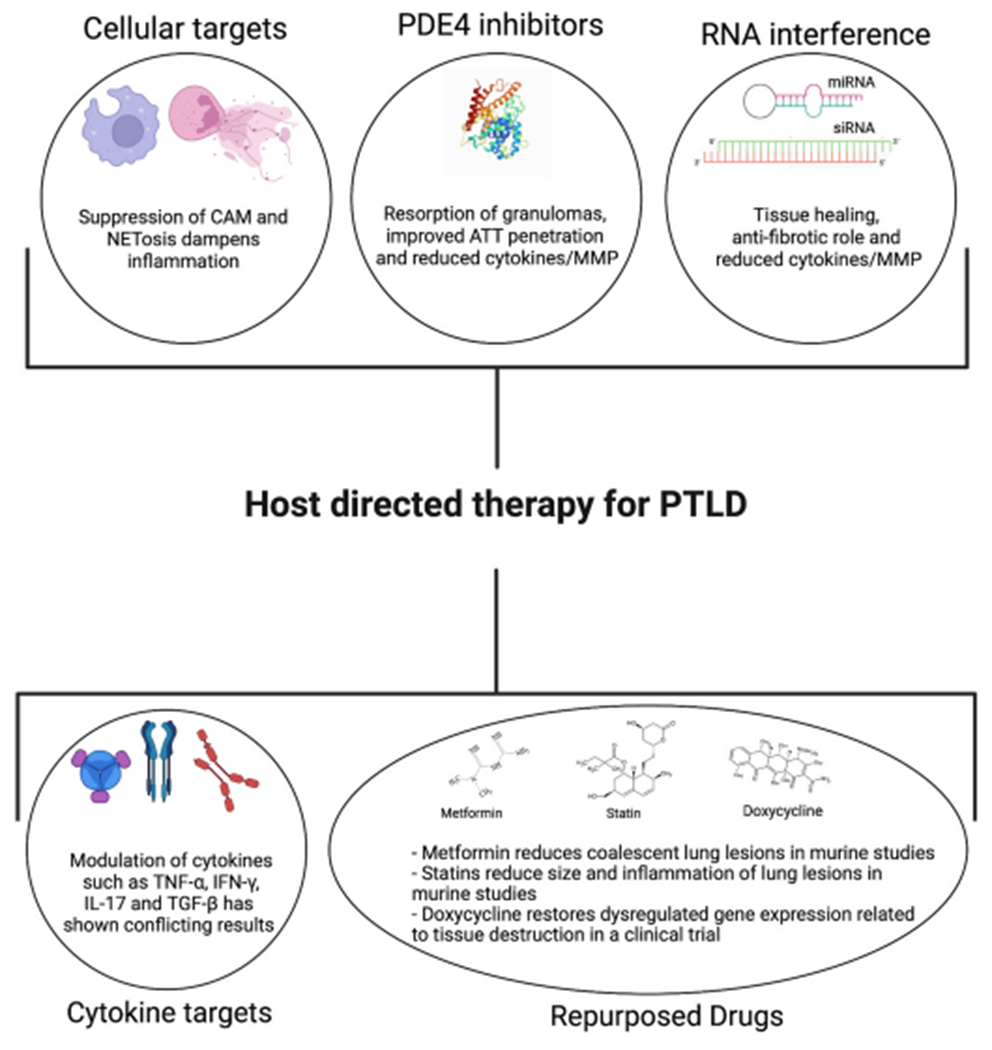
An overview of the various HDTs that are undergoing clinical trials, such as PDE4 inhibitors, cellular and cytokine targets, repurposed drugs and RNA interference technology. Here we focus on HDTs that can potentially be used for individuals that are at high risk of developing PTLD.
7. Imaging in PTLD
PTLD has moderate correlations with imaging, as patients without lung function loss or symptoms may have abnormal imaging post cure. Thus, while imaging is an important tool in evaluating PTLD characteristics, it can not be used in isolation to indicate the presence of clinically meaningful PTLD. Although the CXR is widely used and ubiquitously available, chest CT provides a much higher level of detail on the true nature of underlying lung damage after TB treatment completion. Persistence of TB cavities is associated with poor treatment outcomes and being able to identify the lesions that respond slowly can help optimize regimens early and thus limit the development of PTLD. Unfortunately, there is no uniform scoring system for chest radiographs or CT scans that would comprehensively define PTLD and this has been a hindrance for screening high-risk individuals. Additionally, most of the systems in place for classifying radiographic abnormalities associated with TB were designed for active TB, but such systems have the potential to be quickly adapted for PTLD. While access to CT imaging is unlikely to be available widely in the developing world where the burden of TB is the highest, its use within research settings can help phenotype PTLD. Finally, molecular imaging has gained prominence as a biomarker for various lung conditions. Positron emission tomography (PET) using 18F-fluorodeoxyglucose (18FDG) as a tracer to reflect glucose metabolism in combination with CT imaging constitutes a powerful imaging tool that can give valuable structural information [107].
In a recent review, Meghji et al. [108], examined several cohort studies, cross-sectional studies and randomized controlled trials on imaging-defined PTLD and its co-relation with symptoms and functional impairment. The total number of patients was nearly 5000 and the CT studies reported a higher prevalence rate of both fibrosis (70–92%) and bronchiectasis (35–86%) compared to the CXR. A more diverse range of pathologies were noted on CT imaging such as pleural thickening, nodules, consolidation and mosaicism, hence making a strong point about its utility in the diagnosis of PTLD. In a prospective cohort study in Malawi, the prevalence and pattern of residual lung damage at the completion of ATT was investigated using high resolution CT scans and spirometry [109]. In total, 385 scans were completed: moderate to severe bronchiectasis was seen in ≥1 lobe in 44% of participants. Atelectasis, banding and mosaicism were the most common patterns seen, and 10% of the participants had ≥1 destroyed lung lobe. On average, the majority of airways and parenchymal pathologies were significantly more extensive in HIV-negative compared to HIV-positive participants, with lower FEV1 and FVC z-scores and more abnormal lung parenchyma on HRCT imaging. This could be a reflection of the protective effects of a low CD4 count and reduced MMP production in the lungs of HIV -TB co-infected patients and may also be associated with a reduced clinical severity of PTLD [83]. In another study from India, a semi-quantitative analysis was done for all CT abnormalities and the scores were added to obtain a total morphological score (TMS) [110]. The lungs were then divided into three zones and scores added to obtain the total lung score (TLS). There was a significant difference in TMS and TLS between the dyspnea and non-dyspnea groups and significant correlations were obtained between the grades of dyspnea and the scores (nodule scores, TMS and TLS). The overall extent of radiological abnormalities did not however, correlate with the severity of ventilatory defect. Since the subjects in the study were all symptomatic, the correlations may not be applied accurately to PTLD, but can definitely be modified by factoring in spirometric changes and dyspnea scores that persist in patients with PTLD.
An international study involving 113 HIV-negative patients was conducted with PET-CT at different time points before, during and after ATT [111]. On completion of therapy, patients who had achieved a clinical cure had different patterns of 18FDG uptake compared to baseline. In some patients, there was complete resolution of metabolic activity; in others, most of the lesions had resolved; and in the rest, some lesions had become more intense or new lesions had developed. These new TB lesions may have developed due to a differential evolution of the various TB lesions and microevolution of different subpopulations of Mtb. Thus, tracking of individual lesions, including size and isotope uptake, can be of particular importance when investigating the treatment response and predicting the risk of PTLD. There are several limitations and challenges however and worth mentioning would be the concerns around safety due to repeated and high radiation exposures associated with PET- CT. It is nonetheless surmisable that PET-CT can help delineate the various stages of PTLD and the changes in glycolytic activity within TB lesions as measured by FDG uptake correlate well with the evolution of individual TB lesions [112].
8. Future research directions
The exact mechanisms that lead to PTLD have been difficult to study as human lungs cannot be sampled easily and no small animal model has been able to recapitulate the full spectrum of human TB disease . Computational models that integrate pharmacokinetics, pharmacodynamics and specific morphologic features of the granuloma highlight the fact that reducing granuloma associated fibrosis could improve lung function and therefore possibly limit the development of PTLD [113]. One of the major obstacles in this field has been the lack of disease-specific biomarkers that can be used to endotype patients who could benefit from a specific therapy. One way to achieve this would be the incorporation of genetic phenotyping in the clinical workup of patients diagnosed with PTLD. With the advent of more sophisticated DNA sequencing and gene expression technologies, coordinated human and animal studies can finally address the long-standing questions about PTLD. De Groote et al. [114], conducted a targeted proteomic analysis employing modified DNA aptamers to identify and quantify 1030 protein markers that were associated with active TB and that changed in response to treatment. They discovered changes in many proteins that were involved in lung healing and extracellular matrix (ECM) remodeling such as MMPs, TIMPs, plasminogen and thrombospondins. Using a blood 12-gene signature (AIRE, B2M, CD19, CXCL10, CXCL13, NCAM1, NLRC4, NLRP1, NLRP2, NOD2, TLR6 and TLR8) in combination with some basic clinical characteristics such as gender, body mass index, smoking history and previous history of TB, Sivakumaran et al. [115], identified signatures for treatment failure in TB patients after completion of standard ATT. These studies highlight that in-roads into the use of blood/site of disease multiomics could help predict the progression to PTLD. There is now increasing evidence that in addition to the lung microbiome (the larger local microbial environment beyond the pathogen, in this case Mtb) [116,117], the gut microbiome also plays a significant role in determining the lung immune tone and different aspects of lung immunity [118]. Thus, it is possible that the balance between the inflammatory and fibrotic processes determining PTLD is in part affected by a complex set of microbial and host interactions beyond those related to Mtb alone. It is likely that an integrated antifibrotic strategy that could simultaneously target inflammatory and pro-fibrotic pathways will probably emerge as the most successful way to treat the highly complex pathologies of PTLD.
9. Conclusions
PTLD significantly adds to the global burden of chronic lung diseases. Due to a lack of controlled clinical trials, there are no evidence-based recommendations for the management of PTLD and the condition has been largely unrecognized within TB management pathways [119]. Chest imaging is a non-invasive modality that can quickly characterize the changes in PTLD, and offers a rapid, non-invasive, point-of-care test. But the lack of a uniform and quantitative scoring system has limited its widespread application. This review highlights that our understanding of the burden of PTLD and of the immune mechanisms associated with it are limited. The key to preventing PTLD is early diagnosis, as there are millions of subjects worldwide who are at risk of developing PTLD [6]. Investigations of the interplay between the host and the pathogen are required to identify modifiable risk factors in such individuals. A better understanding of the underlying immunological mechanisms driving PTLD could also facilitate therapies to reduce the burden of PTLD and make an impact on quality of life measures. Transcriptomic and mulitomic profiling has the potential to expeditiously further research on HDTs but the variability in individual immune profiles and co-existent morbidities need to be factored in. In a disease spectrum as wide as PTLD, a single immunologic or radiographic marker cannot be sufficient and a combination is more likely to be a successful surrogate that could aid in the development of HDTs.
Acknowledgements
SS acknowledges grant support from the American Thoracic Society (RP- 2021-03), the Doris Duke Foundation-FRCS (2020140) and the Stony Wold Herbert Inc., Foundation. GT acknowledges funding from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI136894, European and Developing Countries Clinical Trials Partnership (EDCTP) program supported by the European Union (TMA-2015-SF-1041) and the South African Medical Research Council (SAMRC) Intramural Flagship Project. CCN is partially supported by funding from the EDCTP (TMA2017CDF-1914-MOSAIC), L’Oréal-UNESCO for Women in Science South African National Young Talents Program, and the SAMRC through its Division of Research Capacity Development under the SAMRC Intramural Post-doctoral program from funding received from the South African National Treasury. The content hereof is the sole responsibility of the authors and do not necessarily represent the official views of the SAMRC or the funders. SM is supported by the EDCTP (TMA2019CDF-2738-ESKAPE-TB) and the Faculty of Medicine and Health Sciences (FMHS), Stellenbosch University. TLC acknowledges funding from Deutscher Akademischer Austauschdienst (ST32-PKZ-91770486), the FMHS and Harry Crossley Foundation. LNS is supported by the National Institutes of Health under award number R37 CA244775.We acknowledge the use of biorender for making the figures (license numbers OW23JR19FG and RJ23JR1MQO).
References
- [1].Organisation WH. Global tuberculosis report. 2021.
- [2].Dodd PJ, Yuen CM, Jayasooriya SM, van der Zalm MM, Seddon JA. Quantifying the global number of tuberculosis survivors: a modelling study. Lancet Infect Dis 2021;21(7):984–92. [DOI] [PubMed] [Google Scholar]
- [3].Menzies NA, Quaife M, Allwood BW, Byrne AL, Coussens AK, Harries AD, et al. Lifetime burden of disease due to incident tuberculosis: a global reappraisal including post-tuberculosis sequelae. Lancet Global Health 2021;9(12):e1679–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lambert ML, Hasker E, Van Deun A, Roberfroid D, Boelaert M, Van der Stuyft P. Recurrence in tuberculosis: relapse or reinfection? Lancet Infect Dis 2003;3(5):282–7. [DOI] [PubMed] [Google Scholar]
- [5].Romanowski K, Baumann B, Basham CA, Ahmad Khan F, Fox GJ, Johnston JC. Long-term all-cause mortality in people treated for tuberculosis: a systematic review and meta-analysis. Lancet Infect Dis 2019;19(10):1129–37. [DOI] [PubMed] [Google Scholar]
- [6].van Kampen SC, Wanner A, Edwards M, Harries AD, Kirenga BJ, Chakaya J, et al. International research and guidelines on post-tuberculosis chronic lung disorders: a systematic scoping review. BMJ global health 2018;3(4):e000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bobrowitz ID, Rodescu D, Marcus H, Abeles H. The destroyed tuberculous lung. Scand J Respir Dis 1974;55(1):82–8. [PubMed] [Google Scholar]
- [8].Kim HY, Song KS, Goo JM, Lee JS, Lee KS, Lim TH. Thoracic sequelae and complications of tuberculosis. Radiographics 2001;21(4):839–58.; discussion 59-60. [DOI] [PubMed] [Google Scholar]
- [9].Hicks A, Muthukumarasamy S, Maxwell D, Howlett D. Chronic inactive pulmonary tuberculosis and treatment sequelae: chest radiographic features. Int J Tubercul Lung Dis : Off. J. Int. Union against Tuberc. Lung Dis 2014;18(2):128–33. [DOI] [PubMed] [Google Scholar]
- [10].Dickson RP, Martinez FJ, Huffnagle GB. The role of the microbiome in exacerbations of chronic lung diseases. Lancet 2014;384(9944):691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Allwood BW, van der Zalm MM, Amaral AFS, Byrne A, Datta S, Egere U, et al. Post-tuberculosis lung health: perspectives from the first international Symposium. Int J Tubercul Lung Dis : Off. J. Int. Union against Tuberc. Lung Dis 2020;24(8):820–8. [DOI] [PubMed] [Google Scholar]
- [12].Verver S, Warren RM, Beyers N, Richardson M, van der Spuy GD, Borgdorff MW, et al. Rate of reinfection tuberculosis after successful treatment is higher than rate of new tuberculosis. Am J Respir Crit Care Med 2005;171(12):1430–5. [DOI] [PubMed] [Google Scholar]
- [13].Hamilton CD, Stout JE, Goodman PC, Mosher A, Menzies R, Schluger NW, et al. The value of end-of-treatment chest radiograph in predicting pulmonary tuberculosis relapse. Int J Tubercul Lung Dis : Off. J. Int. Union against Tuberc. Lung Dis 2008;12(9):1059–64. [PMC free article] [PubMed] [Google Scholar]
- [14].Pasipanodya JG, Miller TL, Vecino M, Munguia G, Garmon R, Bae S, et al. Pulmonary impairment after tuberculosis. Chest 2007;131(6):1817–24. [DOI] [PubMed] [Google Scholar]
- [15].Willcox PA, Ferguson AD. Chronic obstructive airways disease following treated pulmonary tuberculosis. Respir Med 1989;83(3):195–8. [DOI] [PubMed] [Google Scholar]
- [16].Sarkar M, Srinivasa, Madabhavi I, Kumar K. Tuberculosis associated chronic obstructive pulmonary disease. Clin. Respir. J 2017;11(3):285–95. [DOI] [PubMed] [Google Scholar]
- [17].Báez-Saldaña R, López-Arteaga Y, Bizarrón-Muro A, Ferreira-Guerrero E, Ferreyra-Reyes L, Delgado-Sánchez G, et al. A novel scoring system to measure radiographic abnormalities and related spirometric values in cured pulmonary tuberculosis. PLoS One 2013;8(11):e78926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Theron G, Venter R, Calligaro G, Smith L, Limberis J, Meldau R, et al. Xpert MTB/RIF results in patients with previous tuberculosis: can we distinguish true from false positive results? Clin Infect Dis : Off. Publ. Infect. Dis. Soc. Am 2016;62(8):995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rachow A, Ivanova O, Wallis R, Charalambous S, Jani I, Bhatt N, et al. TB sequel: incidence, pathogenesis and risk factors of long-term medical and social sequelae of pulmonary TB - a study protocol. BMC Pulm Med 2019;19(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Migliori GB, Marx FM, Ambrosino N, Zampogna E, Schaaf HS, van der Zalm MM, et al. Clinical standards for the assessment, management and rehabilitation of post-TB lung disease. Int J Tubercul Lung Dis : Off. J. Int. Union against Tuberc. Lung Dis 2021;25(10):797–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Basaraba RJ, Hunter RL. Pathology of tuberculosis: how the pathology of human tuberculosis informs and directs animal models. Microbiol Spectr 2017;5(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Martin CJ, Carey AF, Fortune SM. A bug’s life in the granuloma. Semin Immunopathol 2016;38(2):213–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Davis JM, Ramakrishnan L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 2009;136(1):37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Flynn JL, Chan J, Lin PL. Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunol 2011;4(3):271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Marakalala MJ, Raju RM, Sharma K, Zhang YJ, Eugenin EA, Prideaux B, et al. Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat Med 2016;22(5):531–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lin PL, Ford CB, Coleman MT, Myers AJ, Gawande R, Ioerger T, et al. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med 2014;20(1):75–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gideon HP, Phuah J, Myers AJ, Bryson BD, Rodgers MA, Coleman MT, et al. Variability in tuberculosis granuloma T cell responses exists, but a balance of pro- and anti-inflammatory cytokines is associated with sterilization. PLoS Pathog 2015;11(1):e1004603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Aung H, Toossi Z, McKenna SM, Gogate P, Sierra J, Sada E, et al. Expression of transforming growth factor-beta but not tumor necrosis factor-alpha, interferon-gamma, and interleukin-4 in granulomatous lung lesions in tuberculosis. Tuber Lung Dis 2000;80(2):61–7. [DOI] [PubMed] [Google Scholar]
- [29].Warsinske HC, DiFazio RM, Linderman JJ, Flynn JL, Kirschner DE. Identifying mechanisms driving formation of granuloma-associated fibrosis during Mycobacterium tuberculosis infection. J Theor Biol 2017;429:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Evans S, Butler JR, Mattila JT, Kirschner DE. Systems biology predicts that fibrosis in tuberculous granulomas may arise through macrophage-to-myofibroblast transformation. PLoS Comput Biol 2020;16(12):e1008520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol 2008;214(2):199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hunter RL, Actor JK, Hwang SA, Khan A, Urbanowski ME, Kaushal D, et al. Pathogenesis and animal models of post-primary (bronchogenic) tuberculosis, A review. Pathogens 2018;7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hunter RL. The pathogenesis of tuberculosis: the early infiltrate of post-primary (adult pulmonary) tuberculosis: a distinct disease entity. Front Immunol 2018;9:2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol 2012;12(5):352–66. [DOI] [PubMed] [Google Scholar]
- [35].Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog 2008;4(11):e1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Brown RE, Hunter RL, Hwang SA. Morphoproteomic-guided host-directed therapy for tuberculosis. Front Immunol 2017;8:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hunter RL. Pathology of post primary tuberculosis of the lung: an illustrated critical review. Tuberculosis 2011;91(6):497–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Barry CE 3rd, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol 2009;7(12):845–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rook GA, Dheda K, Zumla A. Immune responses to tuberculosis in developing countries: implications for new vaccines. Nat Rev Immunol 2005;5(8):661–7. [DOI] [PubMed] [Google Scholar]
- [40].North RJ, Jung YJ. Immunity to tuberculosis. Annu Rev Immunol 2004;22:599–623. [DOI] [PubMed] [Google Scholar]
- [41].Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008;8(12):958–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Huang Z, Luo Q, Guo Y, Chen J, Xiong G, Peng Y, et al. Mycobacterium tuberculosis-induced polarization of human macrophage orchestrates the formation and development of tuberculous granulomas in vitro. PLoS One 2015;10(6):e0129744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 2011;11(11):723–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dallenga T, Schaible UE. Neutrophils in tuberculosis–first line of defence or booster of disease and targets for host-directed therapy? Pathog Dis 2016;74(3). [DOI] [PubMed] [Google Scholar]
- [45].Ong CW, Elkington PT, Brilha S, Ugarte-Gil C, Tome-Esteban MT, Tezera LB, et al. Neutrophil-derived MMP-8 drives AMPK-dependent matrix destruction in human pulmonary tuberculosis. PLoS Pathog 2015,11(5):e1004917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lyadova IV. Neutrophils in tuberculosis: heterogeneity shapes the way? Mediat Inflamm 2017;2017:8619307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Barry S, Breen R, Lipman M, Johnson M, Janossy G. Impaired antigen-specific CD4(+) T lymphocyte responses in cavitary tuberculosis. Tuberculosis 2009;89(1):48–53. [DOI] [PubMed] [Google Scholar]
- [48].Porto BN, Stein RT. Neutrophil extracellular traps in pulmonary diseases: too much of a good thing? Front Immunol 2016;7:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ramos-Kichik V, Mondragón-Flores R, Mondragón-Castelán M, Gonzalez-Pozos S, Muñiz-Hernandez S, Rojas-Espinosa O, et al. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (Edinb). 2009;89(1):29–37. [DOI] [PubMed] [Google Scholar]
- [50].de Melo MGM, Mesquita EDD, Oliveira MM, da Silva-Monteiro C, Silveira AKA, Malaquias TS, et al. Imbalance of NET and alpha-1-antitrypsin in tuberculosis patients is related with hyper inflammation and severe lung tissue damage. Front Immunol 2018;9:3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Verrecchia F, Mauviel A. Transforming growth factor-beta and fibrosis. World J Gastroenterol 2007;13(22):3056–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Khalil N, Bereznay O, Sporn M, Greenberg AH. Macrophage production of transforming growth factor beta and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med 1989;170(3):727–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Limper AH, Colby TV, Sanders MS, Asakura S, Roche PC, DeRemee RA. Immunohistochemical localization of transforming growth factor-beta 1 in the nonnecrotizing granulomas of pulmonary sarcoidosis. Am J Respir Crit Care Med 1994;149(1):197–204. [DOI] [PubMed] [Google Scholar]
- [54].Ameglio F, Casarini M, Capoluongo E, Mattia P, Puglisi G, Giosue S. Post-treatment changes of six cytokines in active pulmonary tuberculosis: differences between patients with stable or increased fibrosis. Int J Tubercul Lung Dis : Off. J. Int. Union against Tuberc. Lung Dis 2005;9(1):98–104. [PubMed] [Google Scholar]
- [55].Christine T, Tarigan AP, Ananda FR. The correlation between levels of transforming growth factor-beta with pulmonary fibrosis in post pulmonary tuberculosis in medan, north sumatera - Indonesia. Open Access. Macedonian J. Med. Sci 2019;7(13):2075–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Seiscento M, Vargas FS, Antonangelo L, Acencio MM, Bombarda S, Capelozzi VL, et al. Transforming growth factor beta-1 as a predictor of fibrosis in tuberculous pleurisy. Respirology 2007;12(5):660–3. [DOI] [PubMed] [Google Scholar]
- [57].Lasco TM, Cassone L, Kamohara H, Yoshimura T, McMurray DN. Evaluating the role of tumor necrosis factor-alpha in experimental pulmonary tuberculosis in the Guinea pig. Tuberculosis 2005;85(4):245–58. [DOI] [PubMed] [Google Scholar]
- [58].Bekker LG, Maartens G, Steyn L, Kaplan G. Selective increase in plasma tumor necrosis factor-alpha and concomitant clinical deterioration after initiating therapy in patients with severe tuberculosis. J Infect Dis 1998;178(2):580–4. [DOI] [PubMed] [Google Scholar]
- [59].Mohan VP, Scanga CA, Yu K, Scott HM, Tanaka KE, Tsang E, et al. Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: possible role for limiting pathology. Infect Immun 2001;69(3):1847–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wolpe SD, Davatelis G, Sherry B, Beutler B, Hesse DG, Nguyen HT, et al. Macrophages secrete a novel heparin-binding protein with inflammatory and neutrophil chemokinetic properties. J Exp Med 1988;167(2):570–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tsao TC, Hong J, Li LF, Hsieh MJ, Liao SK, Chang KS. Imbalances between tumor necrosis factor-alpha and its soluble receptor forms, and interleukin-1beta and interleukin-1 receptor antagonist in BAL fluid of cavitary pulmonary tuberculosis. Chest 2000; 117(1):103–9. [DOI] [PubMed] [Google Scholar]
- [62].Fan JM, Huang XR, Ng YY, Nikolic-Paterson DJ, Mu W, Atkins RC, et al. Interleukin-1 induces tubular epithelial-myofibroblast transdifferentiation through a transforming growth factor-beta1-dependent mechanism in vitro. Am J Kidney Dis 2001;37(4):820–31. [DOI] [PubMed] [Google Scholar]
- [63].Sigal GB, Segal MR, Mathew A, Jarlsberg L, Wang M, Barbero S, et al. Biomarkers of tuberculosis severity and treatment effect: a directed screen of 70 host markers in a randomized clinical trial. EBioMedicine 2017;25:112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Elkington PT, Friedland JS. Matrix metalloproteinases in destructive pulmonary pathology. Thorax 2006;61(3):259–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Shao DD, Suresh R, Vakil V, Gomer RH, Pilling D. Pivotal Advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation. J Leukoc Biol 2008;83(6):1323–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wangoo A, Sparer T, Brown IN, Snewin VA, Janssen R, Thole J, et al. Contribution of Th1 and Th2 cells to protection and pathology in experimental models of granulomatous lung disease. J Immunol 2001;166(5):3432–9. Baltimore, Md : 1950. [DOI] [PubMed] [Google Scholar]
- [67].Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Investig 1999;103(6):779–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Muller KM, Jaunin F, Masouye I, Saurat JH, Hauser C. Th2 cells mediate IL-4-dependent local tissue inflammation. J Immunol 1993;150(12):5576–84. Baltimore, Md : 1950. [PubMed] [Google Scholar]
- [69].Mazzarella G, Bianco A, Perna F, D’Auria D, Grella E, Moscariello E, et al. T lymphocyte phenotypic profile in lung segments affected by cavitary and non-cavitary tuberculosis. Clin Exp Immunol 2003;132(2):283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol 2004;4(8):583–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Dheda K, Booth H, Huggett JF, Johnson MA, Zumla A, Rook GA. Lung remodeling in pulmonary tuberculosis. J Infect Dis 2005;192(7):1201–9. [DOI] [PubMed] [Google Scholar]
- [72].Ulloa L, Doody J, Massagué J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature 1999;397(6721):710–3. [DOI] [PubMed] [Google Scholar]
- [73].King TE Jr, Albera C, Bradford WZ, Costabel U, Hormel P, Lancaster L, et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet 2009;374(9685):222–8. [DOI] [PubMed] [Google Scholar]
- [74].Harari A, Rozot V, Bellutti Enders F, Perreau M, Stalder JM, Nicod LP, et al. Dominant TNF-alpha+ Mycobacterium tuberculosis-specific CD4+ T cell responses discriminate between latent infection and active disease. Nat Med 2011;17(3):372–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lorè NI, Bragonzi A, Cigana C. The IL-17A/IL-17RA axis in pulmonary defence and immunopathology. Cytokine Growth Factor Rev 2016;30:19–27. [DOI] [PubMed] [Google Scholar]
- [76].Lombard R, Doz E, Carreras F, Epardaud M, Le Vern Y, Buzoni-Gatel D, et al. IL-17RA in non-hematopoietic cells controls CXCL-1 and 5 critical to recruit neutrophils to the lung of mycobacteria-infected mice during the adaptive immune response. PLoS One 2016;11(2):e0149455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Singh S, Maniakis-Grivas G, Singh UK, Asher RM, Mauri F, Elkington PT, et al. Interleukin-17 regulates matrix metalloproteinase activity in human pulmonary tuberculosis. J Pathol 2018;244(3):311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Liu F, Liu J, Weng D, Chen Y, Song L, He Q, et al. CD4+CD25+Foxp3+ regulatory T cells depletion may attenuate the development of silica-induced lung fibrosis in mice. PLoS One 2010;5(11):e15404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Parks WC, Wilson CL, López-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol 2004;4(8):617–29. [DOI] [PubMed] [Google Scholar]
- [80].Ong CW, Elkington PT, Friedland JS. Tuberculosis, pulmonary cavitation, and matrix metalloproteinases. Am J Respir Crit Care Med 2014;190(1):9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Wang CH, Lin HC, Lin SM, Huang CD, Liu CY, Huang KH, et al. MMP-1(−1607G) polymorphism as a risk factor for fibrosis after pulmonary tuberculosis in Taiwan. Int J Tubercul Lung Dis : Off. J. Int. Union against Tuberc. Lung Dis 2010;14(5):627–34. [PubMed] [Google Scholar]
- [82].Singh S, Kubler A, Singh UK, Singh A, Gardiner H, Prasad R, et al. Antimycobacterial drugs modulate immunopathogenic matrix metalloproteinases in a cellular model of pulmonary tuberculosis. Antimicrob Agents Chemother 2014;58(8):4657–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Walker NF, Clark SO, Oni T, Andreu N, Tezera L, Singh S, et al. Doxycycline and HIV infection suppress tuberculosis-induced matrix metalloproteinases. Am J Respir Crit Care Med 2012;185(9):989–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Elkington P, Shiomi T, Breen R, Nuttall RK, Ugarte-Gil CA, Walker NF, et al. MMP-1 drives immunopathology in human tuberculosis and transgenic mice. J Clin Investig 2011;121(5):1827–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ordonez AA, Maiga M, Gupta S, Weinstein EA, Bishai WR, Jain SK. Novel adjunctive therapies for the treatment of tuberculosis. Curr Mol Med 2014;14(3):385–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Xu Y, Wang L, Zimmerman MD, Chen KY, Huang L, Fu DJ, et al. Matrix metalloproteinase inhibitors enhance the efficacy of frontline drugs against Mycobacterium tuberculosis. PLoS pathogens 2018;14(4):e1006974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Ordonez AA, Pokkali S, Kim S, Carr B, Klunk MH, Tong L, et al. Adjunct antibody administration with standard treatment reduces relapse rates in a murine tuberculosis model of necrotic granulomas. PLoS One 2018;13(5):e0197474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kim CJ, Yoon HK, Park MJ, Yoo KH, Jung KS, Park JW, et al. Inhaled indacaterol for the treatment of COPD patients with destroyed lung by tuberculosis and moderate-to-severe airflow limitation: results from the randomized INFINITY study. Int J Chronic Obstr Pulm Dis 2017;12:1589–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Yum HK, Park IN. Effect of inhaled tiotropium on spirometric parameters in patients with tuberculous destroyed lung. Tuberc Respir Dis 2014;77(4):167–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wallis RS, Maeurer M, Mwaba P, Chakaya J, Rustomjee R, Migliori GB, et al. Tuberculosis–advances in development of new drugs, treatment regimens, host-directed therapies, and biomarkers. Lancet Infect Dis 2016;16(4):e34–46. [DOI] [PubMed] [Google Scholar]
- [91].Kulkarni AA, Thatcher TH, Olsen KC, Maggirwar SB, Phipps RP, Sime PJ. PPAR-γ ligands repress TGFβ-induced myofibroblast differentiation by targeting the PI3K/Akt pathway: implications for therapy of fibrosis. PLoS One 2011;6(1):e15909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Critchley JA, Orton LC, Pearson F. Adjunctive steroid therapy for managing pulmonary tuberculosis. Cochrane Database Syst Rev 2014;2014(11). Cd011370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Schutz C, Davis AG, Sossen B, Lai RP, Ntsekhe M, Harley YX, et al. Corticosteroids as an adjunct to tuberculosis therapy. Expet Rev Respir Med 2018;12(10):881–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Vilaplana C, Marzo E, Tapia G, Diaz J, Garcia V, Cardona PJ. Ibuprofen therapy resulted in significantly decreased tissue bacillary loads and increased survival in a new murine experimental model of active tuberculosis. J Infect Dis 2013;208(2):199–202. [DOI] [PubMed] [Google Scholar]
- [95].Wu CW, Wu JY, Lee MG, Lai CC, Wu IL, Tsai YW, et al. Risk of incident active tuberculosis disease in patients treated with non-steroidal anti-inflammatory drugs: a population-based study. BMC Pulm Med 2017;17(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Subbian S, Tsenova L, O’Brien P, Yang G, Koo MS, Peixoto B, et al. Phosphodiesterase-4 inhibition combined with isoniazid treatment of rabbits with pulmonary tuberculosis reduces macrophage activation and lung pathology. Am J Pathol 2011,179(1):289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Skerry C, Harper J, Klunk M, Bishai WR, Jain SK. Adjunctive TNF inhibition with standard treatment enhances bacterial clearance in a murine model of necrotic TB granulomas. PLoS One 2012;7(6):e39680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Singhal A, Jie L, Kumar P, Hong GS, Leow MK, Paleja B, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med 2014;6(263). 263ra159. [DOI] [PubMed] [Google Scholar]
- [99].Pan SW, Yen YF, Kou YR, Chuang PH, Su VY, Feng JY, et al. The risk of TB in patients with type 2 diabetes initiating metformin vs sulfonylurea treatment. Chest 2018;153(6):1347–57. [DOI] [PubMed] [Google Scholar]
- [100].Parihar SP, Guler R, Khutlang R, Lang DM, Hurdayal R, Mhlanga MM, et al. Statin therapy reduces the mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis 2014;209(5):754–63. [DOI] [PubMed] [Google Scholar]
- [101].Miow QH, Vallejo AF, Wang Y, Hong JM, Bai C, Teo FS, et al. Doxycycline host-directed therapy in human pulmonary tuberculosis. J Clin Investig 2021;131(15):e141895.. 10.1172/JCI141895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Alipoor SD, Adcock IM, Garssen J, Mortaz E, Varahram M, Mirsaeidi M, et al. The roles of miRNAs as potential biomarkers in lung diseases. Eur J Pharmacol 2016;791:395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF, et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2010;182(2):220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Sabir N, Hussain T, Shah SZA, Peramo A, Zhao D, Zhou X. miRNAs in tuberculosis: new avenues for diagnosis and host-directed therapy. Front Microbiol 2018;9:602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Singh Y, Kaul V, Mehra A, Chatterjee S, Tousif S, Dwivedi VP, et al. Mycobacterium tuberculosis controls microRNA-99b (miR-99b) expression in infected murine dendritic cells to modulate host immunity. J Biol Chem 2013;288(7):5056–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Fu X, Zeng L, Liu Z, Ke X, Lei L, Li G. MicroRNA-206 regulates the secretion of inflammatory cytokines and MMP9 expression by targeting TIMP3 in Mycobacterium tuberculosis-infected THP-1 human macrophages. Biochem Biophys Res Commun 2016;477(2):167–73. [DOI] [PubMed] [Google Scholar]
- [107].Johnson DH, Via LE, Kim P, Laddy D, Lau CY, Weinstein EA, et al. Nuclear imaging: a powerful novel approach for tuberculosis. Nucl Med Biol 2014;41(10):777–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Meghji J, Simpson H, Squire SB, Mortimer K. A systematic review of the prevalence and pattern of imaging defined post-TB lung disease. PLoS One 2016;11(8):e0161176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Meghji J, Lesosky M, Joekes E, Banda P, Rylance J, Gordon S, et al. Patient outcomes associated with post-tuberculosis lung damage in Malawi: a prospective cohort study. Thorax 2020;75(3):269–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Panda A, Bhalla AS, Sharma R, Mohan A, Sreenivas V, Kalaimannan U, et al. Correlation of chest computed tomography findings with dyspnea and lung functions in post-tubercular sequelae. Lung India. Ogff. Organ. Indian. Chest. Soc 2016;33(6):592–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Heysell SK, Thomas TA, Sifri CD, Rehm PK, Houpt ER. 18-Fluorodeoxyglucose positron emission tomography for tuberculosis diagnosis and management: a case series. BMC Pulm Med 2013;13:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Ichiya Y, Kuwabara Y, Sasaki M, Yoshida T, Akashi Y, Murayama S, et al. FDG-PET in infectious lesions: the detection and assessment of lesion activity. Ann Nucl Med 1996;10(2):185–91. [DOI] [PubMed] [Google Scholar]
- [113].Pienaar E, Cilfone NA, Lin PL, Dartois V, Mattila JT, Butler JR, et al. A computational tool integrating host immunity with antibiotic dynamics to study tuberculosis treatment. J Theor Biol 2015;367:166–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].De Groote MA, Nahid P, Jarlsberg L, Johnson JL, Weiner M, Muzanyi G, et al. Elucidating novel serum biomarkers associated with pulmonary tuberculosis treatment. PLoS One 2013;8(4):e61002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Sivakumaran D, Jenum S, Vaz M, Selvam S, Ottenhoff THM, Haks MC, et al. Combining host-derived biomarkers with patient characteristics improves signature performance in predicting tuberculosis treatment outcomes. Commun Biol 2020;3(1):359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Segal LN, Clemente JC, Tsay JC, Koralov SB, Keller BC, Wu BG, et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat Microbiol 2016;1:16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Wu BG, Sulaiman I, Tsay JJ, Perez L, Franca B, Li Y, et al. Episodic aspiration with oral commensals induces a MyD88-dependent, pulmonary T-helper cell type 17 response that mitigates susceptibility to Streptococcus pneumoniae. Am J Respir Crit Care Med 2021;203(9):1099–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Dickson RP, Erb-Downward JR, Falkowski NR, Hunter EM, Ashley SL, Huffnagle GB. The lung microbiota of healthy mice are highly variable, cluster by environment, and reflect variation in baseline lung innate immunity. Am J Respir Crit Care Med 2018;198(4):497–508. 10.1164/rccm.201711-2180OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Allwood BW, Byrne A, Meghji J, Rachow A, van der Zalm MM, Schoch OD. Post-tuberculosis lung disease: clinical review of an under-recognised global challenge. Respiration 2021;100(8):751–63. [DOI] [PubMed] [Google Scholar]