

Abstract
The zinc finger protein ZFYVE21 is involved in immune signaling. Using humanized mouse models, primary human cells, and patient samples, we identified a T cell–autonomous role for ZFYVE21 in promoting chronic vascular inflammation associated with allograft vasculopathy. Ischemia-reperfusion injury (IRI) stimulated endothelial cells to produce Hedgehog (Hh) ligands, which in turn induced the production of ZFYVE21 in a population of T memory cells with high amounts of the Hh receptor PTCH1 (PTCHhi cells, CD3+CD4+CD45RO+PTCH1hiPD-1hi), vigorous recruitment to injured endothelia, and increased effector responses in vivo. After priming by interferon-γ (IFN-γ), Hh-induced ZFYVE21 activated NLRP3 inflammasome activity in T cells, which potentiated IFN-γ responses. Hh-induced NLRP3 inflammasomes and T cell–specific ZFYVE21 augmented the vascular sequelae of chronic inflammation in mice engrafted with human endothelial cells or coronary arteries that had been subjected to IRI before engraftment. Moreover, the population of PTCHhi T cells producing high amounts of ZFYVE21 was expanded in patients with renal transplant–associated IRI, and sera from these patients expanded this population in control T cells in a manner that depended on Hh signaling. We conclude that Hh-induced ZFYVE21 activates NLRP3 inflammasomes in T cells, thereby promoting chronic inflammation.
INTRODUCTION
We previously identified ZFYVE21 (zinc finger FYVE-type containing 21) as a Rab5 effector protein involved in allograft vasculopathy (AV), a sequela of chronic vascular inflammation in solid organ allografts (1). ZFYVE21 is a highly-conserved, zinc finger–containing protein that was originally identified as a component of the MT1-MMP1 (membrane type 1 matrix metalloproteinase) complex that promotes cell motility (2–6). Through proteomic analyses of sorted endosomes in endothelial cells (ECs), we identified ZFYVE21 as a Rab5 effector induced by complement membrane attack complexes (MAC, 1). Human ECs may function as semi-professional antigen-presenting cells (7), and ZFVYE21 modifies the lipid content of signaling endosomes to activate noncanonical (1,7–10) and canonical (11–12) nuclear factor κB (NF-κB) signaling to enhance the immunogenicity of ECs targeted by allogeneic CD4+ T cells. These processes potentiated type 1 immune responses, such as the production of interferon gamma (IFN-γ), in the T cells (1,7–12).
Herein, we investigated a T cell–autonomous role for ZFYVE21 in augmenting type 1 responses of CD4+ T cells. Circulating effector memory CD4+ T cells form immune synapse–like structures on abluminal surfaces of ECs as a prelude to tissue entry. Following formation of these immune synapse–like structures, the effector function of infiltrating T cells may be shaped by tissue-derived morphogens, including Wnt ligands, transforming growth factor β (TGF-β) ligands, retinoic acid, and Hedgehog (Hh) ligands. Tissue-derived morphogens qualitatively modulate the effector properties of tissue-infiltrating T cells and based on this we used humanized models and patient biospecimens to examine the role of ZFYVE21 in response to EC-derived Hh signals.
As morphogens, Hh ligands provides growth and spatial positioning information in developing and regenerating tissues. Hh family members, such as sonic hedgehog (SHH), engage the receptor patched (PTCH), leading to the activation smoothened (SMO), a G protein–coupled receptor. Through a phosphoinositide 3-kinase (PI3K)-Akt signaling axis, activated SMO promotes nuclear translocation of members of the GLI family of transcription factors, which induce Hh-responsive gene expression programs related to cell cycle entry and motility (13,14).
Although CD4+ T cells do not produce a primary cilium, an organelle involved in Hh signaling, (13), they do produce Hh signaling components and are capable of receiving and transmitting functional input from Hh agonists, (15) including non-lipidated Hh ligands (16–18). Hh signaling in T cells promotes lineage commitment of TH2 (20,21) and regulatory CD4+ T cells (22) and, in fully differentiated T cells, modifies CD4+ T cell receptor (TCR) signal strength (23) and provides positive costimulation (19). Through these processes, tissue-derived Hh ligands, similar to the other aforementioned vascular morphogens, modulate the effector function of CD4+ T cells within inflamed tissues (21).
We hypothesized that the proliferative and migratory programs conferred through Hh signaling could be pathologically subsumed by tissue-infiltrating T cells to promote inflammation. We found that EC-derived Hh signals selectively activated ZFYVE21 in T cells producing high amounts of PTCH (PTCHhi T cells) following ischemia-reperfusion injury (IRI). Hh-induced ZFYVE21 initiated signaling programs specifying tissue homing and triggered the assembly of NLRP3 inflammasomes in the T cells, which mediated interleukin 18 (IL-18)-dependent potentiation of type 1 responses.
RESULTS
Endothelial cells produce Hh ligands following IRI
Delayed graft function (DGF) is a clinical manifestation of severe IRI following renal transplantation. Due to peri-operative renal dysfunction, patients with DGF require dialysis prior to hospital discharge. Early graft dysfunction (EGD), where uni- or bi-ventricular dysfunction occurs peri-operatively, is believed to similarly reflect severe IRI in cardiac transplantation (25). We analyzed biopsies from patients with DGF and EGD to test associations of Hh signaling with IRI-related injury. In EGD samples, we detected SHH in glomerular and peritubular ECs displaying C6, a MAC component, (Fig. 1A) and E-selectin, a marker of endothelial inflammation (Fig. 1B). We did not observe significant C6 or SHH staining in ECs within control biopsies from routine surveillance. EGD coronary arteries co-stained with C6 and SHH, both of which were absent in controls (Fig. 1C), and SHH showed gradient-like staining occurring most strongly in the intima and progressively declining towards the adventitia. SHH concentrations in sera were increased in DGF patients vs controls (Fig. 1D), though this value did not reach statistical significance, likely reflecting Hh effects locally within tissues.
Fig. 1. Endothelial cells produce Hh ligands following IRI.
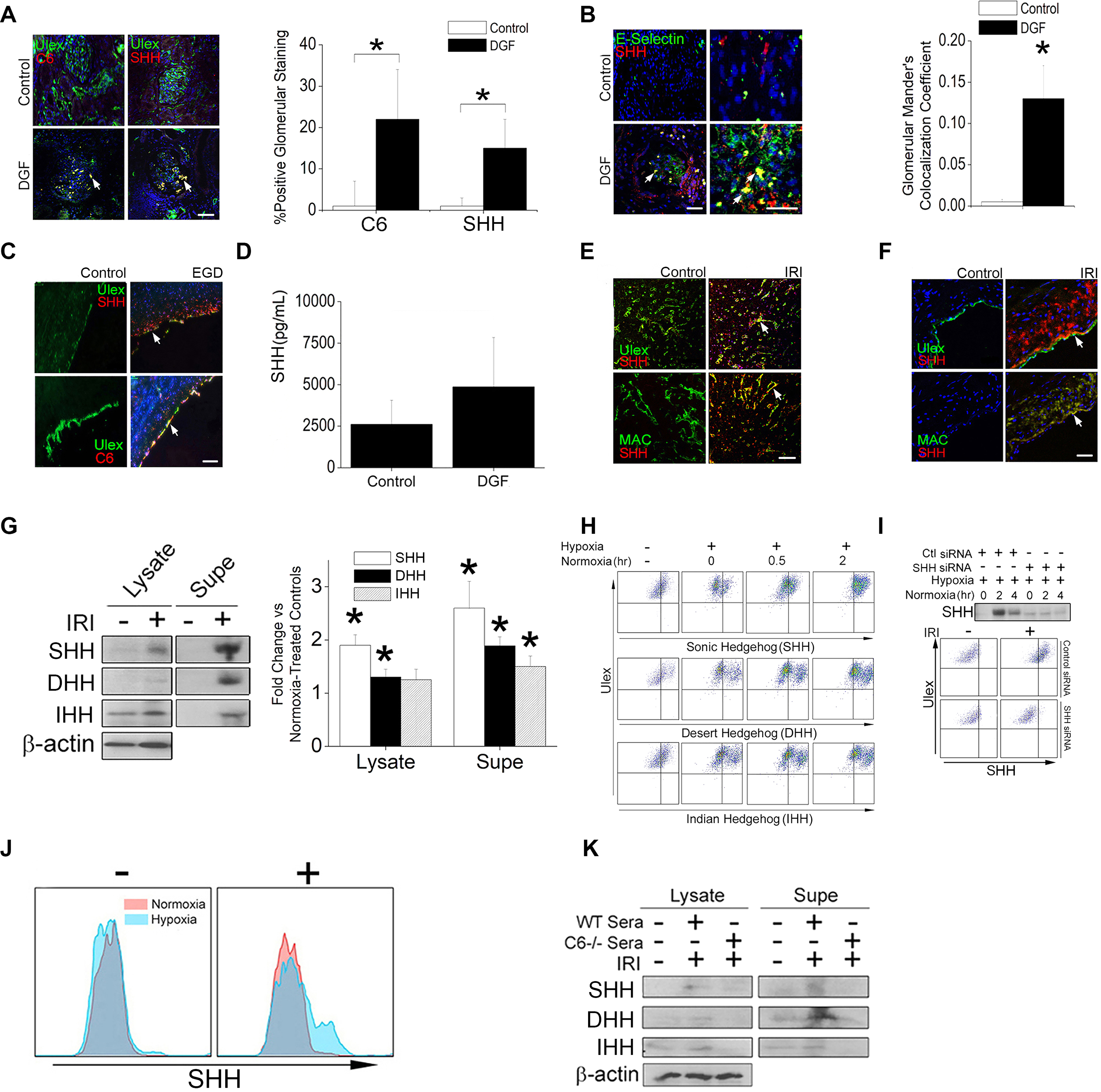
(A to C) Immunofluorescence (IF) staining for C6, SHH, the endothelial label Ulex, and the endothelial inflammation marker E-selectin in renal (A and B, n=3) biopsies from DGF patients and in coronary artery (C, n=4) biopsies from EGD patients. Cells showing C6 and SHH were quantified in (A), and colocalization of SHH and E-selectin was quantified in (B). (D) Quantification of SHH in sera from control (n=10), DGF (n=9), and AMR (n=2) patients by enzyme immunoassay. (E) IF staining for Ulex, MAC, and SHH in HUVECs embedded in collagen-fibronectin gels, implanted adjacent to proximal femoral arteries in SCID/bg mice, and subjected to IRI injury. Controls are implants from the sham-operated contralateral artery. n=3 mice. (F) IF staining for Ulex, MAC, and SHH in human coronary artery explants that were subjected to normoxia or IRI ex vivo prior to surgical implantation into the infrarenal aortae of SCID/bg mice. N=3 mice per treatment group. (G and H) Immunoblotting (G) and FACS analysis (H) for SHH, DHH, and IHH in cell lysates and supernatants of HUVECs subjected to normoxia or IRI-simulating conditions (11). β-actin is a loading control. (I) Immunoblotting for SHH in supernatants and FACS analysis for SHH in HUVECs treated with control or SHH-targeting siRNA and subjected to normoxia or IRI-simulating conditions. (J) FACS analysis of SHH in HUVECs cultured in WT serum (+) or C6-defiecient serum (–). (K) Immunoblotting for SHH, DHH, and IHH in HUVECs cultured with WT human serum or C6-deficient serum and subjected to IRI conditions as indicated. Data in (G to K) are representative of experiments using cells from 3 different human donors. All scale bars, 200μm. For quantifications in (A), 3–6 hpf fields per patient were analyzed in a blinded fashion by 3 independent reviewers. Data are presented as mean ± SD. For quantifications in A, B, and D Student’s t-test was used for statistical comparisons.
We sought to recapitulate these patient responses in ECs in mice. Human umbilical vein endothelial cells (HUVECs) embedded in collagen gel matrices (27) were implanted into mice and subjected to IRI following transient midfemoral artery occlusion. Harvested grafts showed costaining for C6 and SHH (Fig. 1E). In a second model, human coronary artery segments subjected to anoxia in organ culture (8,11,28) showed increased SHH in the tunica intima and tunica media (Fig. 1F). Finally, cultured HUVECs were subjected to an in vitro transient anoxia protocol that simulates IRI (11), which increased SHH in both cell lysates and culture supernatants (Fig. 1G). Surface-bound Hh ligands (SHH, DHH, and IHH) increased during post-hypoxic normoxia (Fig. 1H), a phase of the in vitro IRI simulation protocol when complement activation occurs (11). Small interfering RNA (siRNA)-mediated knockdown of SHH, the most abundant of the Hh ligands detected, prior to IRI simulation strongly reduced the abundances of both fully processed SHH in cell lysates and SHH on the cell surface (Fig. 1I), demonstrating an inducible, EC-derived source for this ligand. ECs lacking the ability to induce MAC formation because they were cultured without normal human serum as a source of complement (Fig. 1J) or with C6-deficient serum (Fig. 1K) failed to produce SHH when subjected to IRI conditions, indicating a MAC-dependent effect. These experiments demonstrate that endothelial SHH was detected in and on postnatal ECs subjected to IRI in patient samples, in vivo, and in vitro.
A subset of T cells produce high amounts of PTCH and are responsive to Hh agonism
With IRI, ECs were found to release Hh ligands (Fig 1A–F), and Hh ligands have been investigated for their positive costimulatory effects in CD4+ T cells (18,19). Because ECs do not produce the classical costimulatory molecules CD80 or CD86, which are found on professional antigen-presenting cells to engage the costimulatory receptor CD28 on T cells (APCs, 7), we embarked on studies to interrogate differential costimulatory effects between those mediated through EC-derived Hh agonism and those mediated by CD28 ligands on professional APCs. To do this, we first attempted to identify CD4+ T cell subset(s) responding to Hh agonism.
Hh signaling induces the production of PTCH1, SMO, and GLI1 (14), and the amounts of these proteins in various cell types proportionally correlate with Hh signal strength (26). Therefore, we reasoned that high abundance of Hh pathway component(s) might enable identification of Hh-responsive T cell population(s). Because we postulate this effect to occur within tissues, we focused upon CD4+CD45RO+ T memory populations (Tmem) from peripheral blood, which likely form interchangeable pools with populations recruited to tissues (57,58). T cell activation requires two signals, including the activation of TCR and a co-stimulatory activation. We stimulated human Tmem with an antibody targeting the TCR coreceptor CD3-stimulating antibody in the presence of either an CD28-stimulating antibody or the pharmacological reagent smoothened agonist (SAG), which was used to simulate the combined effects of Hh ligands. We then used FACS to separate activated HLA-DR+ T cells into 3 subsets based on Hh component abundance (lo, mid, hi). Compared to Tmem stimulated in the presence of CD28-stimulating antibody, cells treated with a CD3-stimulating antibody and concurrently exposed to SAG more strongly produced all Hh signaling mediators tested (Fig. 2A). High programmed cell death protein 1 (PD-1) production occurs among T cell subsets showing homing to inflamed peripheral tissues (29–32), and we found that Hh signal components (PTCH1, GLI1) showed very high and significant correlations with the abundance of PD-1 (Fig. 2B). We selected PTCH1 for downstream analyses due to its strong and reproducible surface staining. Exposure to Hh agonism alone (in the absence of an antibody that elicits CD3-mediated TCR stimulation) was insufficient for expanding HLA-DR+PTCH1hiPD-1hi Tmem but when combined with TCR stimulation, Hh agonism expanded PTCH1hiPD-1hi Tmem in a dose-dependent manner (Fig. 2, C and D). Concomitant exposure to Hh and CD28 agonism in the presence of TCR stimulation, a response that might occur following indirect allorecognition involving professional APCs positioned near ECs, did not affect frequencies of HLA-DR+PTCH1hiPD-1hi Tmem or the type 1 effector responses of these cells (fig. S1A). These data were compatible with the described costimulatory effects of Hh signaling (18,19).
Fig. 2. Identification of a PTCHhi T cell subset that responds to Hh agonism.
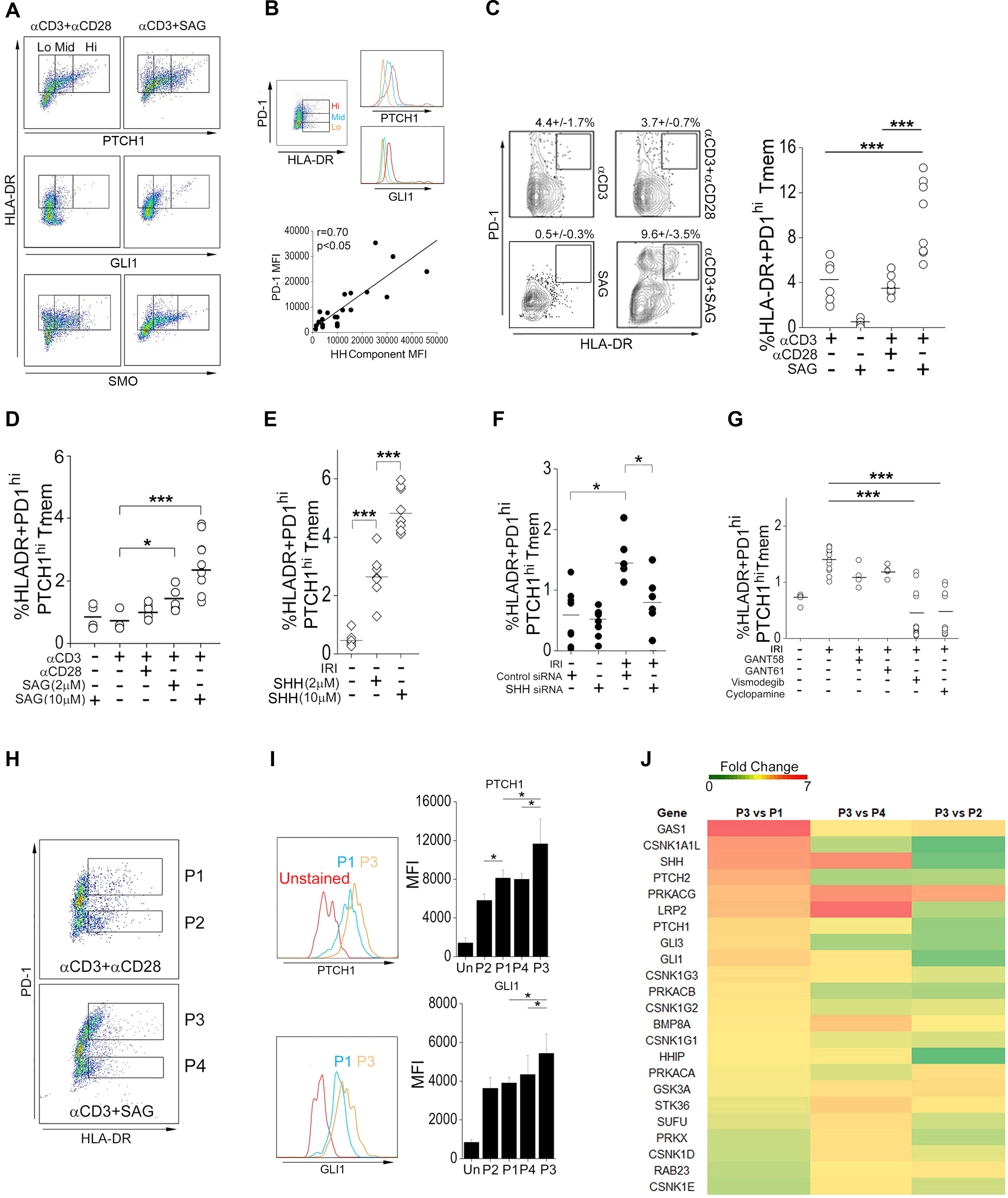
(A) FACS analysis of PTCH1, GLI1, and SMO in HLA-DR+CD4+CD45RO+ memory T cells (Tmem) pretreated with Smoothened agonist (SAG) or soluble antibody recognizing CD28 as indicated and stimulated with immobilized antibody specific for CD3. (B) Correlation of PD-1 abundance with that of Hh signaling components PTCH1 and GLI1 in Tmem activated with CD3-specfic antibody in the presence of SAG. MFI, mean fluorescence intensity. (C and D) FACS analysis (C) and HLA-DR+PD-1hi Tmem frequency (C and D) in Tmem treated with CD3-specific antibody, SAG, and CD28-sepecific antibody as indicated. (E) HLA-DR+PD1hiPTCHhi Tmem frequency in endothelial cell (EC):T cell cocultures with or without the indicated concentrations of SHH under normoxic conditions (no IRI). (F) HLA-DR+PD1hiPTCHhiTmem frequency in EC:T cell cocultures treated with control or SHH siRNA and subjected to IRI conditions as indicated. (G) HLA-DR+PD1hiPTCHhi Tmem frequency in EC:T cell cocultures treated with or without GANT58, GANT61, vismodegib, or cyclopamine and subjected to IRI conditions as indicated. (H to J) Tmem were pretreated with CD28-specific antibody or SAG before stimulation with immobilized CD3 antibody for 48 hrs. The P1–P4 populations defined according to PD-1 abundance in HLA-DR+ Tmem (H) were further classified as PTCHlo (P2), PTCHmid (P1,P4), and PTCHhi (P3) based on their abundance of PTCH1 and GLI1 (I). Heatmap of bulk RNA-seq analysis of the P1–P4 Tmem populations (J). Experimental points reflect technical replicates. Data shown are representative of 2–4 independent experiments using cells from N=3–6 separate leukopack donors. Data are presented as mean ± SD. * indicates p<0.05. Two-way ANOVA followed by Tukey’s pairwise comparison was used for statistical comparisons.
We proceeded to examine the effects of Hh agonism in modulating frequencies of HLA-DR+PTCH1hiPD-1hi Tmem in EC:T cell cocultures, which model direct allorecognition. In this assay, memory T cells, when cocultured with ECs from an allogeneic donor, show activation of 1–5% of the total T cell population based upon TCR recognition of class II MHC antigen differences (9). For this reason, the ECs must be pre-treated with IFN-γ to re-induce class II MHC expression to regain the in situ expression that is lost in cell culture. Expansion of HLA-DR+PTCH1hiPD-1hi Tmem was potentiated in a dose-dependent manner by lipidated Shh (Fig. 2E). Prior work has shown that ECs subjected to IRI conditions in vitro more strongly stimulate allogeneic T cells compared to control ECs in vitro and in vivo (8,11,28). Compared to co-cultures containing control ECs, HLA-DR+PTCH1hiPD-1hi Tmem were preferentially expanded by IRI-treated ECs as a source for Hh ligands, and this effect was abrogated by transfection of SHH siRNA to IRI-treated ECs prior to coculture (Fig. 2F). Pharmacological inhibitors of Smo (vismodegib and cyclopamine) abrogated HLA-DR+ PTCH1hiPD-1hi Tmem to a greater degree than did the GLI inhibitors GANT58 and GANT61 (Fig. 2G). These experiments indicated that ECs subjected to IRI could provide functional Hh agonism to T cells to preferentially expand the activated Ptch1hiPD-1hi Tmem population.
To confirm the use of PTCH1 and PD-1 as markers of Hh signal strength, we enriched for Tmem subsets with differential abundance of Hh signaling components and used gene expression profiling to determine relative amounts of Hh pathway–associated transcripts. To do this, Tmem from 4 donors were stimulated with an antibody against CD3 in the presence of either an antibody against CD28 or SAG, and the P1-P4 populations emerging from these treatments (Fig. 2H) were sorted by FACS for RNA sequencing (RNA-seq). Prior to testing and compared to unstained controls, we found that abundances of PTCH1 and GLI1 among these populations were P3>P1≈P4>P2 (Fig. 2I). After stimulation, in PCA plots we observed that the transcriptomic similarities among the P1-P4 transcriptomes correlated with Hh signal strength, with the P3 and P2 subsets showing highly dissimilar transcriptomes and with the P1 and P4 subsets showing comparatively similar transcriptomic profiles (fig. S1B). The P3 population was enriched in Hh signaling–associated gene products (Fig 2J), causing this subset to become separately clustered in an unsupervised hierarchical analysis of Hh pathway–associated genes (fig. S1B). Together, our data show that the abundance of Hh signaling components was a correlative marker for Hh signal strength in human Tmem. Based on this, we defined three CD4+ Tmem subsets, termed PTCHlo (P2), PTCHmid (P1,P4) and PTCHhi (P3) Tmem, showing differential Hh signal strengths.
PTCH hi CD4+ Tmem show enhanced immune effector functions
EC-dependent Hh signaling is expected to occur within peripheral tissues, and Hh ligands are well-described morphogens that induce gene expression programs specifying spatial positioning (13,14,16,26). We thus initially examined phenotypes linked to EC-mediated T cell recruitment of the PTCHhi (P3) subset, using PTCHlo (P2) and PTCHmid (P1) populations as controls. We chose the P1 subset for analysis because this subset showed equivalent Hh signal strength compared to the P4 subset and could be recovered at substantially higher cell numbers. PTCHhi Tmem (P3) were recruited under shear conditions by IRI-treated ECs at higher frequencies relative to the PTCHmid (P1) and PTCHlo (P2) subsets (Fig. 3A), and the observed enhanced recruitment of PTCHhi (P3) Tmem was abrogated by pretreatment of the ECs with vismodegib, indicating a SMO-dependent response (Fig. 3B). A re-analysis of our prior microarray data of MAC-treated ECs (9, GEO Dataset GSE50112) showed that MAC significantly increased the production of chemokines in ECs that were canonically associated with recruitment to peripheral tissues including CCL2, CCL5, and CXCL11, fig. S1C). Geneset analyses of PTCHhi (P3) Tmem revealed broad enrichment of chemokine receptors, some of which were cognate for the chemokines mentioned above (CCR2, CCR5, CXCR3), and additionally uncovered enrichment for α- and β-integrins (fig. S1D). Concurrently, this analysis revealed that PTCHhi Tmem reduced transcripts encoding proteins associated with tissue egress (S1PR1) and homing to secondary lymphoid organs (CCR7, CXCR4, CXCR5). In validation studies, we found that PTCHhi (P3) Tmem showed increased abundance of CCR2 and CD103 but not CXCR5 (fig. S1F), and in co-culture studies, expansion of HLA-DR+PTCHhi Tmem was strongly driven by PTCHhi Tmem coexpressing CCR2 (Fig 3C). In follow-up studies, we found that Hh agonism expanded PTCHhi Tmem coexpressing CCR2 but not CXCR5 (fig. S1G–I) and that Tmem recovered from 3 healthy donors in situ showed higher abundance of Hh signaling components in PD-1+ Tmem coexpressing CCR2 but not CXCR5. (fig. S1E). PD-1hiCXCR5+ Tmem showed increased expression of genes encoding Hh pathway inhibitors (Sufu, Hip1), suggesting that Hh signal signaling may differentially occur based on migratory homing phenotypes.
Fig. 3. Phenotypic analysis of PTCH hiCD4+ Tmem.
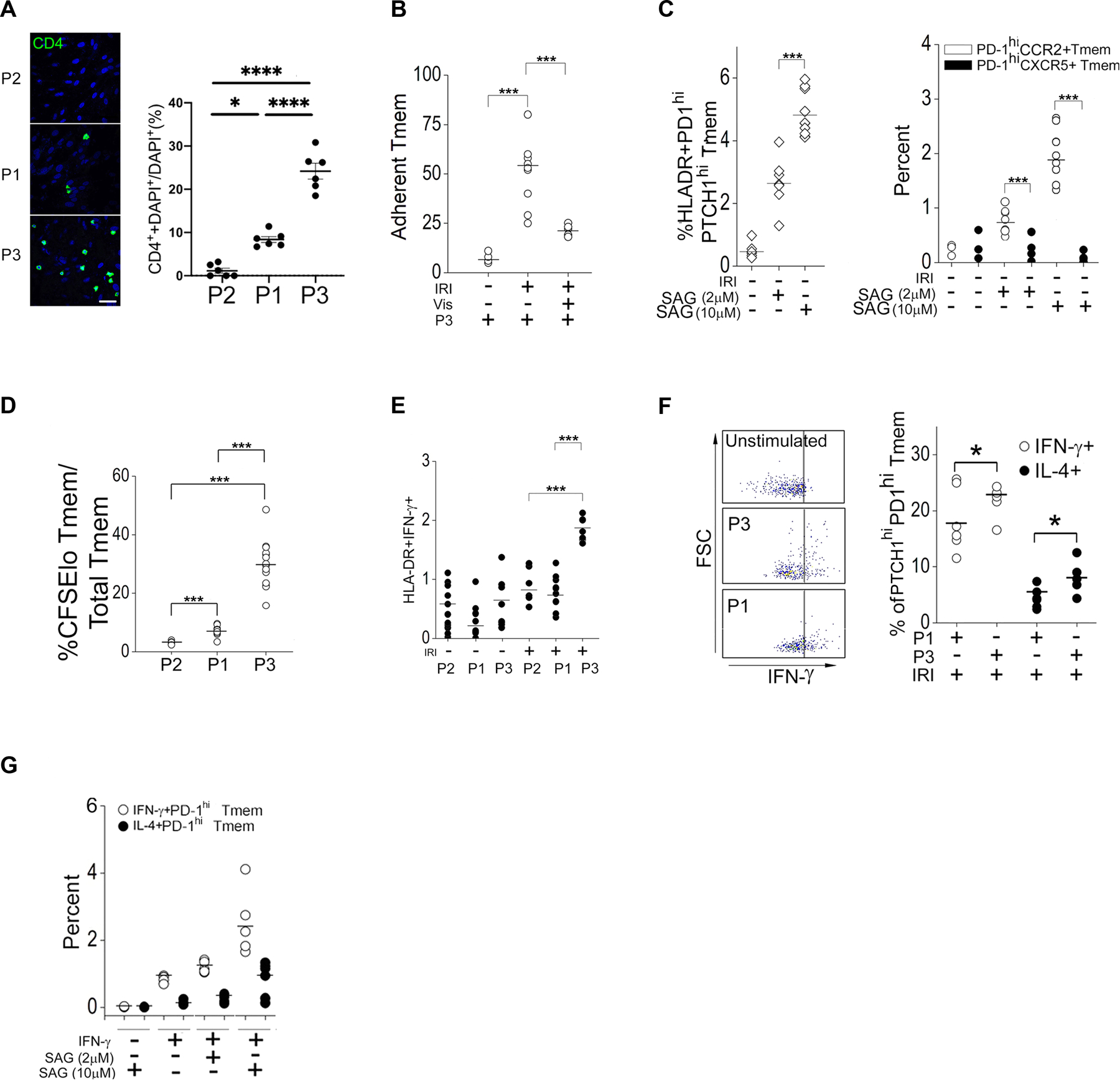
(A) Quantification of PTCHlo (P2), PTCHmid (P1), and PTCHhi (P3) Tmem adhesion to endothelial cells (ECs) in EC:T cell cocultures. Experiments were repeated 3 times. A total of 6 images per Tmem subset were quantified by 3 independent researchers. Scale bar, 200μm. (B) Quantification of the adhesion of PTCHhi (P3) Tmem in IRI-treated EC:T cell cocultures treated with vehicle or Vismodegib (Vis) as indicated. (C) Frequency of HLA-DR+PD1hiPTCHhi, PD1hiCCR2+, and PD1hiCXCR5+ Tmem frequency in IRI-treated EC:T cell cocultures treated with different concentrations of Smoothened agonist (SAG). (D) Frequency of CFSElo Tmem in PTCHlo (P2), PTCHmid (P1), or PTCHhi (P3) Tmem cocultured with IRI-treated ECs. (E) Frequency of HLA-DR+IFN-γ+ Tmem in PTCHlo (P2), PTCHmid (P1), or PTCHhi (P3) Tmem cocultured with IRI-treated ECs. (F) FACS analysis of IFN-γ production in unstimulated control, stimulated PTCHmid (P1), and PTCHhi (P3) Tmem and frequency of IL4+PTCHhiPD-1hiTmem and IFN-γ+PTCHhiPD-1hiTmem in IRI-treated EC:T cell cocultures. FSC, forward scatter. (G) Frequency of IL4+PD-1hi and IFN-γ+PD-1hi Tmem in IRI-treated EC:T cell cocultures treated with IFN-γ and different concentrations of SAG. Data in (A and B) are representative of 2–4 independent experiments using N=3 separate leukopack donors. Data in (C to G) are representative of 3 independent experiments using N=3 separate leukopack donors. Data are presented as mean ± SD. Two-way ANOVA followed by Tukey’s pairwise comparison was used for statistical comparisons.
As further confirmation of the ability of Hh to generate Tmem with putative tissue-homing phenotypes, we tested the effects of Hh agonism in Jurkat cells. Jurkat cells are amenable to genetic manipulations, and they produce functional PD-1 (33) and Hh signaling components including PTCH1 and GLI1 (34). We found that immobilized CD3 antibody plus SAG (fig. S2A) or lipidated SHH (fig. S2B) induced a CD69+ Jurkat cell population producing high amounts of PD-1, PTCH1, and CCR2 (fig. S2B). Due to shared surface phenotypes and dose-dependent expansion with SAG, we examined the CD69+PD-1hiPTCH1hiCCR2+ population as a surrogate for the P3 subset above. We found that expansion of CD69+PD-1hiPTCH1hiCCR2+ Jurkat cells, consistent with the P3 subset (fig. S2F and S2G), was more strongly blocked by SMO siRNA compared to GLI1 siRNA (fig. S2C) and by SMO-specific antagonists vs GLI-specific inhibitors (fig. S2D). Cumulatively, these data indicated that PTCHhi (P3) Tmem can be functionally recruited to sites of inflammation where EC-derived Hh morphogens are produced.
Alloantigens cause strong type 1 polarization of CD4+ T cells, and this response is potentiated through CD28-associated signaling. Because ECs lack abundance of CD80 and CD86, cognate ligands for CD28, we asked whether EC-derived Hh signals, in lieu of those associated with CD28, could modulate EC-induced type 1 responses in T cells. Analyses of PTCHhi CD4+ Tmem (P3) transcriptomes showed enrichment for a heterogeneous array of genes curated under the rubric of ‘Allograft Rejection’ in GSEA. Within this gene set of 196 genes, we uncovered significant enrichment for genes annotated for proliferation (fig. S1J) and IFN-γ signaling (fig. S1K).
We tested effects of Hh agonism on EC-induced type 1 T cell responses using EC:T cell cocultures. In cocultures containing ECs that had been subjected to IRI conditions, HLA-DR+ PTCHhi cells displayed stronger proliferative (Fig 3D) and type 1 effector (Fig 3, E and F) responses compared to PTCHmid (P1) and/or PTCHlo (P2) cells with lower Hh signal strengths. Failure to pretreat ECs with IFN-γ to restore in situ loss of the MHC class II alloantigen ablated type 1 responses despite the presence of high-dose SAG (Fig. 3G), consistent with our CD3 antibody experiments showing that Hh agonism alone in the absence of TCR stimulation was insufficient for expanding HLA-DR+PD-1hiPTCH1hi Tmem (Fig. 2C). These data indicate that Hh agonism did not incur mitogenic effects on CD4+ T cells. Moreover, Hh agonism appeared to proportionally enhance but not qualitatively alter the type 1 effector response (Fig. 3G). Dual Hh and CD28 agonism, when combined with T cell stimulation, did not appear to further potentiate type 1 responses (fig. S1A) and did not modify the abundances of CCR2 and CXCR5 compared to Hh agonism alone (fig. S1L). Tmem acquire migratory and proliferative phenotypes associated with Hh morphogenic signaling and display strong type 1 effector responses. We subsequently focused on defining a mechanism by which Hh signaling enhanced the EC-mediated type 1 response.
Hh signaling stimulates AV
We initially asked whether Hh signaling was required for the enhanced type 1 responses observed in PTCHhi Tmem. In coculture studies, IRI-treated ECs potentiated type 1 responses in PTCHhi Tmem (Fig 4A), and this response was significantly reduced by pretreatment of ECs with SHH siRNA, indicating a Hh-specific effect (Fig. 4A). We then extended this finding in vivo using a humanized mouse model of AV. Chronic exposure to IFN-γ appears to play a major role in the development of AV (7,25,35,36), characterized by vascular scarring, perivascular infiltrates, and obliterative lesions. We used these features as markers of chronic, type 1–mediated inflammation using a humanized mouse model for AV. In this model, human artery segments are surgically placed as interposition grafts in the infrarenal aorta of immunodeficient (SCID/bg) hosts engrafted with human lymphocytes (Fig. 4B). We and others have employed this system and adaptations thereof to model AV-like changes resulting from type 1–mediated inflammation (1,8–12,36,37). SCID/bg hosts engrafted with human lymphocytes subsequently received human artery segments that were subjected to transient anoxia ex vivo prior to implantation to simulate IRI and increase EC production of Hh ligands. At the time of implantation of IRI-treated arteries, SCID/bg hosts were concurrently implanted with osmotic pumps eluting vehicle or specific pharmacological inhibitors of the Hh signaling components SMO (vismodegib) and GLI (GANT61) (Fig. 4B). Subsequently, we monitored the appearance of PTCHhi Tmem in the 3 murine groups above, using a fourth group receiving no peripheral blood mononuclear cells (PBMCs), human artery, or osmotic pump implantations as negative controls. Hosts receiving IRI-treated human artery segments and vehicle-eluting pumps showed PTCHhi Tmem frequencies that were attenuated SMO or GLI inhibitors (Fig. 4C). Upon examination of human artery segments, compatible with in vitro results, we observed that the severity of AV (Fig. 4, D and E) as well as the strength of type 1 responses (Fig. 4F) were more strongly reduced in hosts treated with vismodegib than those treated with GANT61. Canonical Hh signaling involves cellular responses requiring both SMO and GLI effectors, whereas noncanonical Hh responses are exclusively dependent on either SMO or GLI (14,34). Our data showed dual contributions of SMO and GLI effectors with a predominant SMO-dependent response being required for type 1 responses in PTCHhi Tmem. Based on the minor contribution of GLI effectors, as-yet-unidentified pathway(s) likely intersect with proximal components of the Hh pathway to contribute to type 1 responses in PTCHhi Tmem.
Fig. 4. Hh signaling promotes AV pathology.
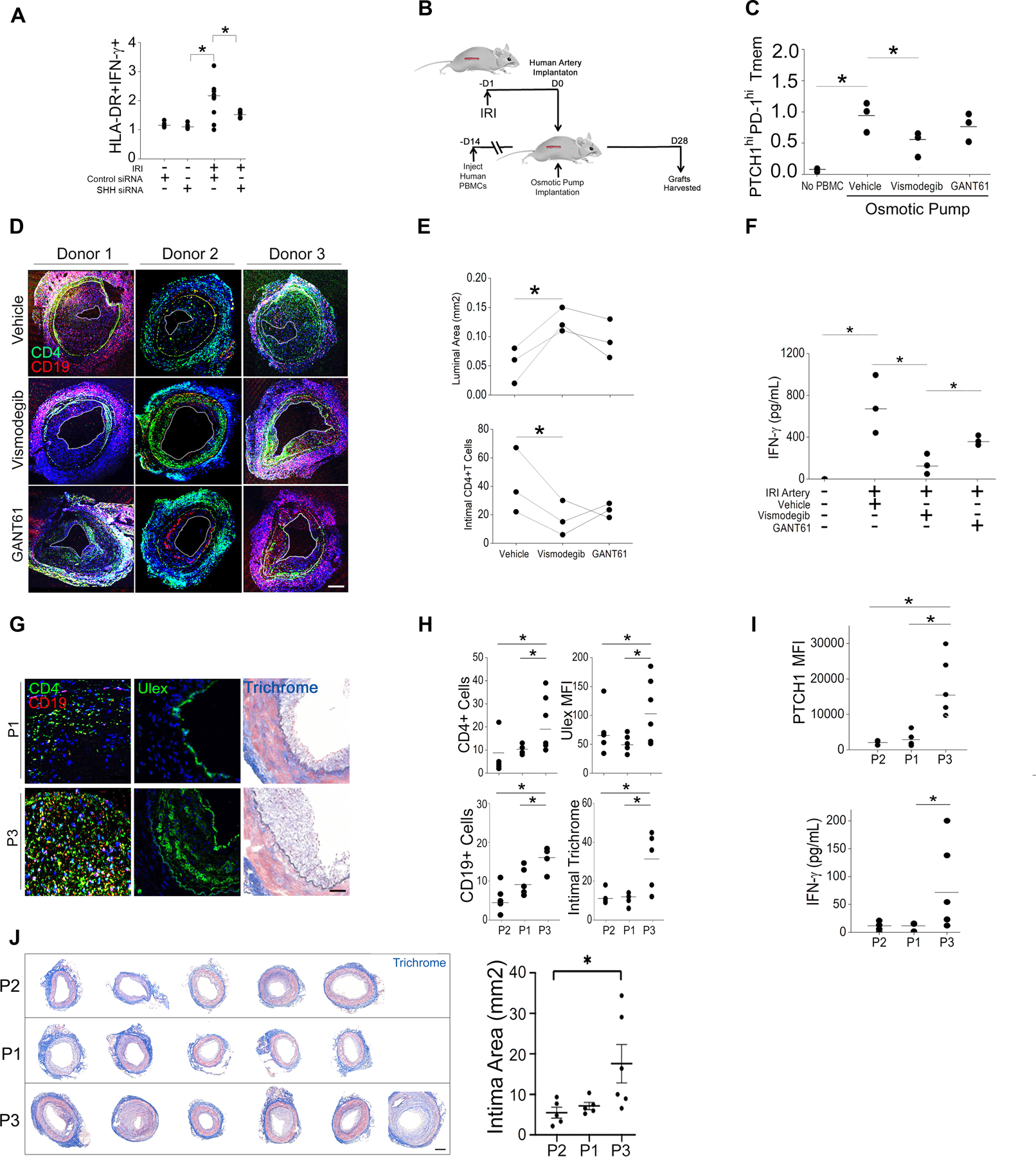
(A) Frequency of HLA-DR+IFN-γ+ Tmem in IRI-treated EC:T cell cocultures following EC transfection with control or SHH siRNA. 2–4 independent experiments were performed using N=3 separate leukopack donors (B) Schematic of artery xenograft experimental design. SCID/bg mice were engrafted with human arteries subjected ex vivo to IRI conditions or adoptively transferred with human PBMCs then implanted with osmotic pumps delivering the pharmacological inhibitors of SMO (vismodegib) or GLI (GANT61). (C) Circulating PTCH1hiPD-1hi Tmem frequency in xenografted mice. (D and E) Immunofluorescence staining for CD4+ and CD19+ cells (D) and quantitation of luminal area and intimal CD4+ T cells coverage area (E) in human artery xenografts harvested from mice. (F) IFN-γ concertation in the sera of the xenografted mice. (G and H) SCID/bg mice bearing human artery xenografts were adoptively transferred with FACS-sorted PTCHmid (P1), PTCHlo (P2), or PTCHhi (P3) Tmem. Representative images show immunofluorescence staining for CD4, CD19, and Ulex and Masson’s trichrome staining (G). CD4+ and CD19+ cell coverage, MFI of Ulex, and intimal Masson’s trichrome staining area were quantified (H). (I) FACS analysis of PTCH1 in circulating Tmem and serum IFN-γ concentration in SCID/bg mice that were treated with FACS-sorted PTCHmid (P1), PTCHlo (P2), or PTCHhi (P3) Tmem. (J) Masson’s trichrome staining and quantification of intimal area of human artery xenografts from SCID/bg mice that were treated with FACS-sorted PTCHmid (P1), PTCHlo (P2), or PTCHhi (P3) Tmem. Data in (C–F) represent N=3 mice receiving human arteries from 3 donors. Data in (G–J) represent N=5 mice receiving P2 and P1 Tmem, N=6 mice receiving P3 Tmem. Scale bars, 200μm. *indicates p<0.05, ** indicates p<0.005. Two-way ANOVA followed by Tukey’s pairwise comparison was used for statistical comparisons.
To more closely focus on the contribution of PTCHhi Tmem to AV, we passively transferred PTCHlo (P2), PTCHmid (P1), or Ptchhi Tmem (P3), along with autologous B cells into SCID/bg hosts bearing human artery xenografts (Fig. 4G). Compared to hosts adoptively receiving PTCHlo and PTCHmid subsets, hosts receiving PTCHhi Tmem showed significantly exacerbated readouts of AV, including increased neointimal infiltrates, neovessel formation, and fibrous deposition compared to hosts receiving Tmem of lesser Hh signal strengths (Fig. 4, G and H). Phenotypes of PTCHhi Tmem (P3) observed prior to adoptive transfer, including higher Hh signal strength and stronger type 1 responses (Fig. 4I), were retained in recipients. Congruent with increased IFN-γ amounts, hosts receiving PTCHhi Tmem showed significantly exacerbated intimal hyperplasia (Fig. 4J). Tmem were recovered at comparable frequencies across groups (fig. S1M), indicating that the observed phenotypes were not due to differential engraftment of the passively transferred subsets. Thus, a pathologic PTCHhi Tmem population showing strong type 1 responses was expanded by EC-dependent Hh agonism in vivo.
NLRP3 inflammasome activity in Ptchhi Tmem promotes AV
We sought to define a mechanism by which Hh agonism enhanced type 1 responses in PTCHhi Tmem. Because constitutive Hh activation is implicated in tumorigenesis, we retrieved public transcriptomes examining constitutive Hh activation in various cell types including a study focusing on CD4+ T cells (21). We observed moderate, significant correlations between genes annotated for Hh signaling (GLI1, GLI2, SMO) and for inflammasome activation (CASP1, IL1b, GSDMD, Fig. 5A). This prompted a re-examination of our RNA-seq data of PTCHhi (P3) Tmem, which showed, relative to PTCHmid (P2) and PTCHlo (P1) subsets, enrichment in genes related to assembly and/or activation of inflammasomes (fig. S3A) and their principal effector pathway, NF-κB (fig. S3B). NLRP3 inflammasome activity has been detected in CD4+ T cells, where it contributes to IFN-γ production (38–40), and we thus tested whether Hh signaling enhanced type 1 responses of PTCH1hi Tmem by inducing inflammasome activation.
Fig. 5. NLRP3 inflammasome activity in PTCHhi Tmem promotes AV.
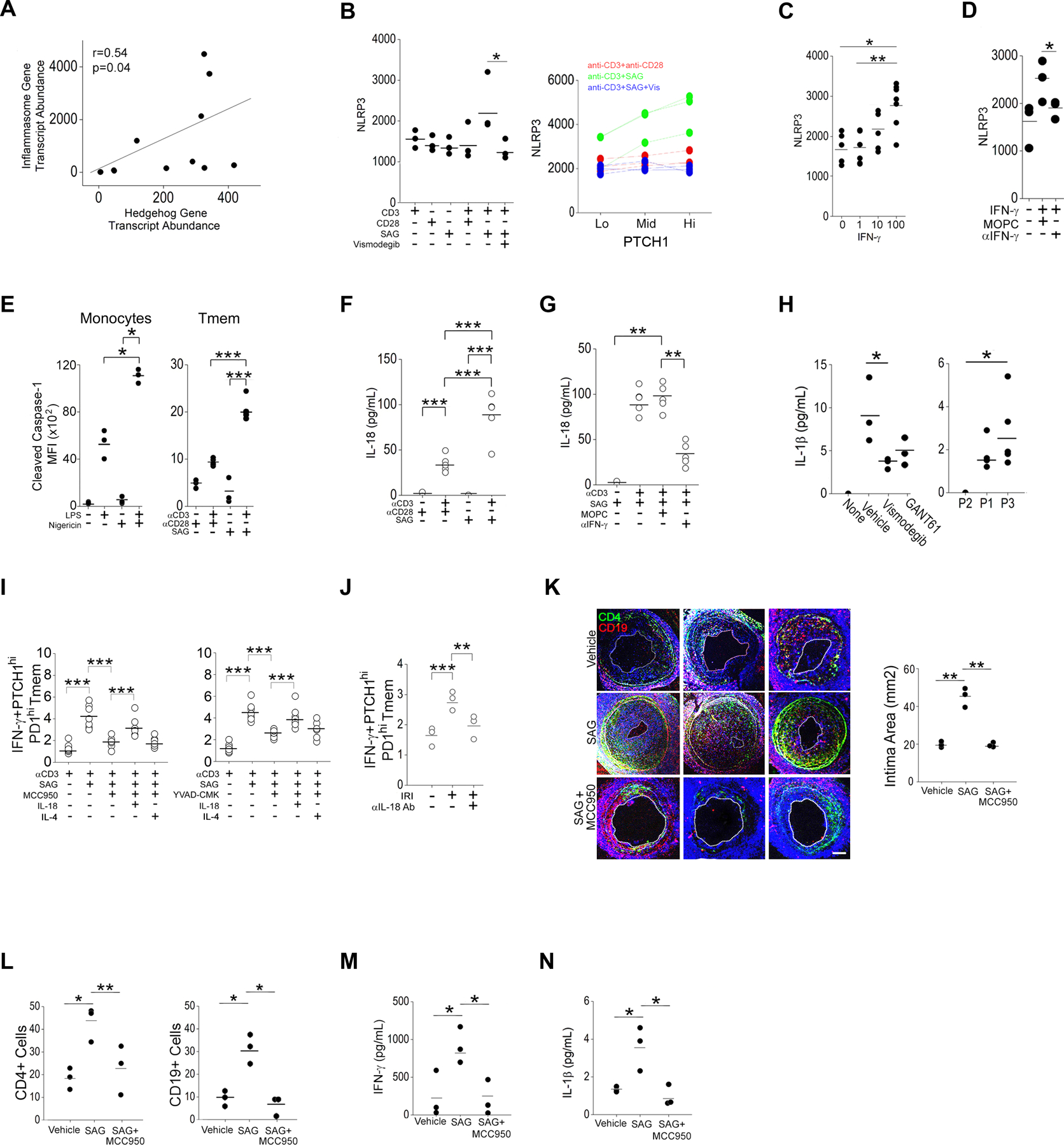
(A) Correlation coefficients (Pearson’s r) between hedgehog (Hh)- and inflammasome-associated transcripts in publicly available RNA-seq data. (B) MFI of NLRP3 in bulk Tmem and in PTCHlo, PTCHmid, and PTCHhi Tmem that were pretreated with CD28-specific antibody, Smoothened agonist (SAG), and vismodegib and stimulated with immobilized CD3 antibody as indicated. (C) MFI of NLRP3 in Tmem treated with varying doses of IFN-γ. (D) MFI of NLRP3 in Tmem treated with IFN-γ in the presence of isotype control MOPC-21 (MOPC) or IFN-γ–neutralizing antibody. (E) MFI of cleaved caspase-1in monocytes treated with LPS or nigericin and Tmem treated with CD28-specific antibody and SAG before stimualtion of CD3. (F and G) IL-18 concertation in culture supernatants from Tmem that were pretreated with CD28-specific antibody, SAG, MOPC, and IFN-γ neutralization antibody before stimulation of CD3 antibody as indicated. (H) IL-1β concentration in the sera from mice bearing artery xenografts and treated with the indicated drugs (Fig. 4D) or adoptively transferred with FACS-sorted PTCHmid (P1), PTCHlo (P2), and PTCHhi (P3) Tmem (Fig. 4G). (I and J) Frequency of IFN-γ+PTCHhiPD-1hi Tmem in T cells pretreated with MCC950 or Ac-YVAD-CMK, SAG, IL-18, and IL-4 as indicated before CD3 stimulation (I), or pretreated with IL-18 neutralization antibody prior to CD3 stimulation in EC:T cell cocultures under IRI conditions. (K to N) SCID/bg mice were engrafted with human arteries, adoptively transferred with PBMCs, and implanted with osmotic pumps eluting SAG or SAG + MCC950. Immunofluorescence staining for CD4+ and CD19+ cells (K) was used to quantify the intima area (K) and intima CD4+ and CD19+ cell coverage (L) in the human artery xenografts. IFN-γ+ (M) and IL-1β (N) serum concentrations were quantified. Data in (A to J) are representative of 2–4 experiments using N=3 separate leukopack donors. Data in (K to N) represent N=3 mice implanted with human artery from 3 donors per group. Scale bars, 200μm. * indicates p<0.05, ** indicates p<0.01. Two-way ANOVA followed by Tukey’s pairwise comparison was used for statistical comparisons.
We first tested abundance of NLRP3 inflammasome–related proteins for which an increase in abundance driven by a priming stimulus is required for subsequent inflammasome activity and inflammasome-mediated maturation of IL-1 family cytokines. Among inflammasome-related proteins, we focused on NLRP3, which showed strong, reproducible effects. NLRP3 siRNA reduced NLRP3 mean fluorescence intensity (MFI) in Jurkat cells, confirming the specificity of staining (fig. S2F). In CD4+CD45+ Tmem, we found that Tmem exposed to SAG combined with an CD3-stimulating antibody showed the highest amount of NLRP3 protein at the 48 hr timepoint, and this increase in NLRP3 was blocked by vismodegib (Fig 5B). Upon further gating of these cells, in 3 separate donors we found that the observed induction of NLRP3 by SAG had occurred mainly in PTCHhi Tmem (Fig. 5B). To test whether NLRP3 priming occurred through contact- and/or humoral-dependent mechanism(s), we overlaid conditioned media from cultures treated with an antibody specific for CD3, antibodies specific for CD3 and CD28, or SAG plus an antibody specific for CD3 onto autologous Tmem. We observed that conditioned media from cultures treated with the CD3 antibody plus SAG significantly induced NLRP3 (fig. S3C) in a concentration-dependent manner (fig. S3D), indicating a humoral effect. To test whether the observed humoral effects occurred through a direct or indirect effect on Hh agonism, we added an SHH-neutralizing antibody to the conditioned media from cells treated with the CD3 antibody plus SAG (Fig. S3E). The SHH-neutralizing antibody had no effect on NLRP3 abundance (fig. S3E), and the addition of SAG (fig. S3F) or lipidated SHH (fig. S3G) to unstimulated Tmem failed to increase NLRP3, excluding inflammasome priming in Tmem through direct Hh agonism. We next tested whether IFN-γ, the production of which was potentiated by SAG plus TCR stimulation, could induce inflammasome components in T cells, an effect that occurs in IFN-γ–treated human ECs (12) and macrophages (41). Addition of IFN-γ to unstimulated Tmem increased the abundance of NLRP3 in a concentration-dependent manner (Fig. 5C), and NLRP3 production was attenuated by the addition of an IFN-γ–neutralizing antibody (Fig. 5D). Similar but weaker increases in caspase-1, proIL-18, and proIL-1β occurred with high-dose IFN-γ (100ng/mL)) and was similarly reversed with IFN-γ–neutralizing antibody (fig. S3H). From these data we conclude that Hh agonism indirectly primes NLRP3 inflammasome components through IFN-γ, the production of which occurs most highly in PTCHhi Tmem.
To assess inflammasome activity, we analyzed staining for cleaved caspase-1, using adherent human monocytes in validation studies. Following treatment with canonical inflammasome activators, lipopolysaccharide (LPS) or LPS plus nigericin (67) we detected increased cleaved caspase-1 MFIs in human monocytes at high signal-to-noise ratios (Fig 5E). In Tmem, Hh agonism in conjunction with T cell activation increased cleaved caspase-1 (Fig. 5E), and culture supernatants showed increased IL-18 (Fig. 5F). Amounts of IL-18 were not substantially altered by SAG treatment alone (Fig. 5F) but were strongly reduced by IFN-γ–neutralizing antibody (Fig. 5G). Upon re-analysis of in vivo experiments (Fig 4C), we found that circulating IL-1β amounts were relatively decreased in hosts implanted with vismodegib-eluting pumps that depleted PTCHhi Tmem and increased in hosts passively receiving PTCHhi Tmem (Fig. 5H), indicating that inflammasome activity occurred in direct proportion to frequencies of PTCHhi Tmem in vivo. In EC:T cell cocultures, pharmacological inhibition of NLRP3 (MCC950) or caspase-1 (Ac-YVAD-CMK) reduced the frequencies of IFN-γ+ PTCHhi Tmem, and these attenuated responses were rescued by exogenous IL-18 but not IL-4 (Fig. 5I). In these studies, MCC950 did not show significant effects on cell viability (fig. S3I). Further, IFN-γ responses of PTCHhi cells were blocked by an IL-18 antibody (Fig. 5J). We further confirmed inflammasome assembly using Jurkat cells. We stably transduced Jurkat cells with ASC-green fluorescent protein (GFP) constructs and observed an increase in speck formation following TCR stimulation when the cells were pretreated with SAG (fig. S2E). Moreover, frequencies of CD69+PD-1hiCCR2+ Jurkat cells were decreased by siRNA-mediated knockdown of NLRP3 (fig. S2F). These results are consistent with IRI-induced, EC-dependent Hh agonism potentiating type 1 responses in PTCHhi cells by promoting T cell–intrinsic NLRP3 inflammasome activation.
To test the role of Hh-induced inflammasome activation in vivo, we implanted osmotic pumps loaded with vehicle, SAG, or SAG plus MCC950 into hosts bearing normoxia-treated human artery xenografts and engrafted with human PBMCs. We observed that Hh agonism by SAG strongly potentiated readouts for AV compared to controls (Fig. 5, K and L), and these pathologies were significantly reduced in hosts dually implanted with pumps eluting SAG plus MCC950 (Fig. 5K, L). Compatible with its role in exacerbating AV, SAG induced higher circulating amounts of IFN-γ (Fig 5M) and IL-1β (Fig. 5N) compared to vehicle-treated controls, and the abundances of these cytokines were significantly attenuated by MCC950 (Fig. 5, M and N). Thus, Hh-induced NLRP3 inflammasome activity potentiated AV, a sequela of chronic type 1–mediated inflammation in vivo.
Hh signaling induces ZFYVE21 in Ptchhi Tmem
Signaling responses implicated in Hh-induced T cell activation include CD28-associated cell cycle genes (18), profibrotic TGF-β signaling (42), and immune synapse assembly (15). These pathways, including Hh signaling itself (43), may be propagated intracellularly in a Rab5-dependent manner. We therefore tested whether Hh signaling might also involve ZFYV21 which we previously identified as a Rab5 effector protein (1). Re-analysis of public transcriptomes (Fig. 5A) uncovered a significant correlation between ZFYVE21 and expression of bona fide Hh target genes, including Ptch1, Smo, and Gli1 (Fig. S4A). Among the studies analyzed, we found that amounts of ZFYVE21 increased in proportion to Gli2 cleavage in murine CD4+ T cells (21), suggesting a link between Hh signaling and ZFYVE21 abundance in T cells. We analyzed ZFYVE21 staining in T cells in biopsies obtained from renal transplant patients with chronic antibody-mediated rejection (CAMR), and heart transplant patients with cardiac allograft vasculopathy (CAV). CAMR and CAV are sequelae of DGF and EGD, respectively, and both are characterized by chronic, type 1–mediated vascular inflammation and AV. In all tissues examined, including renal and cardiac tissues, we observed staining for ZFYVE21 in vascular structures that separately stained with Ulex, a plant lectin that binds to glycoproteins on the surface of ECs (Fig 1, A to F), consistent with publicly available gene expression data [Human Protein Atlas (www.proteinatlas.org), Gene Expression Database (informatics.jax.org/expression.shtml)] and our prior findings examining endothelial ZFYVE21 production (1). Control renal transplant subjects without CAMR showed low colocalization of ZFYVE21 with CD3 in lymph nodes (LN), spleen, and peripheral renal tissues (Fig. 6A). In CAMR patients, ZFYVE21 colocalization with CD3 was modestly increased in draining LN compared with non-draining peri-aortic LN and spleen but strongly increased in perivascular areas of inflamed peripheral kidney tissues (Fig. 6B). These data indicated that ZFVYE21 production in T cells was nominal under basal conditions, similar to EC-derived SHH (Fig. 1A–C), but became strongly increased in inflamed peripheral tissues but not in secondary lymphoid organs. We re-analyzed coronary artery biopsies from heart transplant patients with CAMR to better isolate region(s) of ZFYVE21 production within vascular structures. We found that ZFYVE21 colocalization with CD3 occurred most strongly in the intima, progressively declined towards the media, and was absent in the adventitia (Fig. 6C). This gradient-like staining pattern phenocopied that of SHH in EGD patients (Fig. 1C) and did not qualitatively differ with regards to cardiac AV severity. We were unable to obtain specific staining for ZFYVE21 in murine tissues following testing of 4 commercially available antibodies.
Fig. 6. Hh agonism induces ZFYVE21 in PTCHhi Tmem.
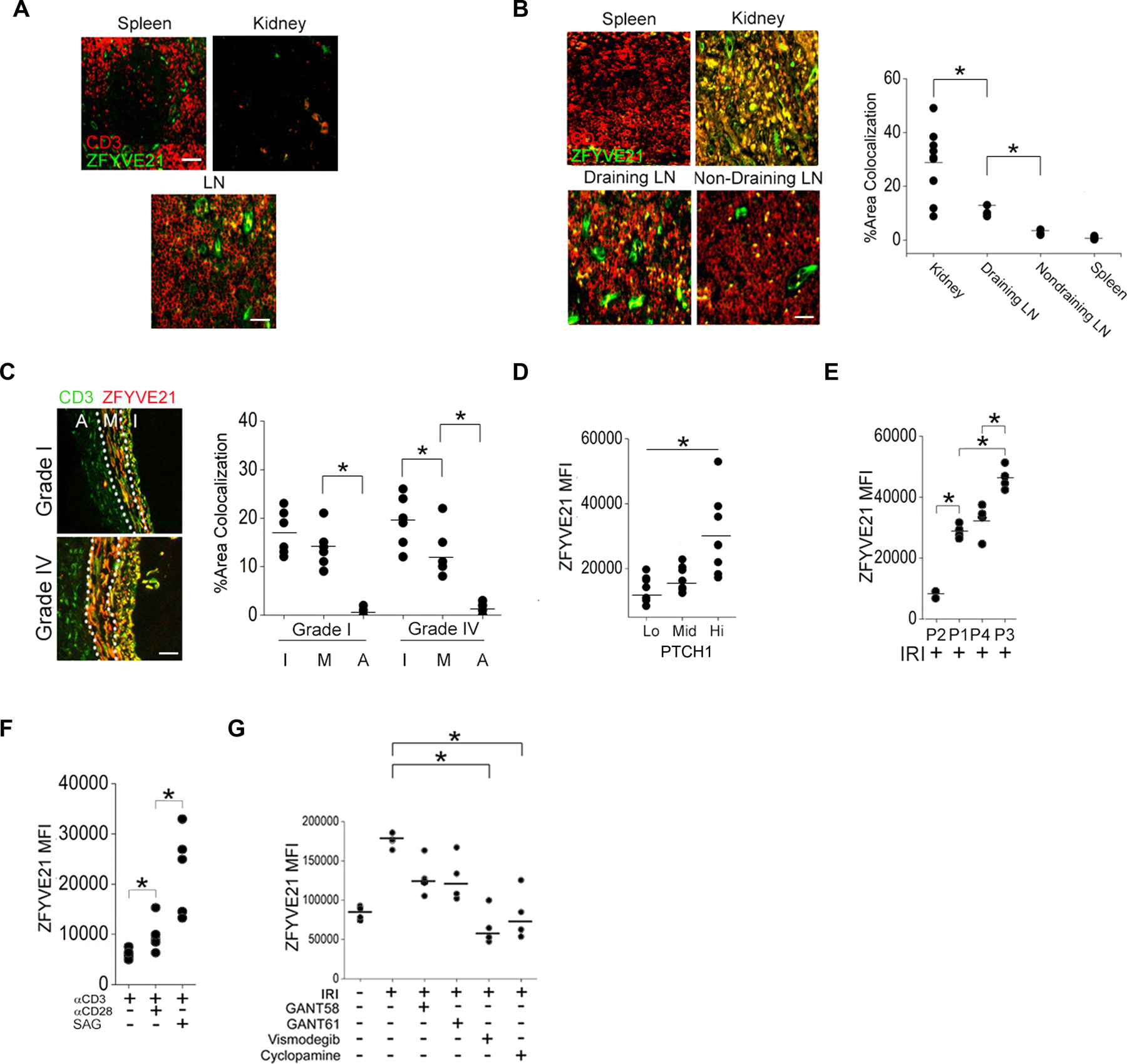
(A to C) CD3 and ZFYVE21 staining in tissues from control renal transplant patients (A, N=3), renal transplant patients with CAMR (B, N=3), or heart transplant patients with AV (C, N=4). ZFYVE21 and CD3 colocalization was quantified in (B) and (C). (D) Abundance of ZFYVE21 in PTCHlo, PTCHmid, and PTCHhi Tmem treated with SAG and stimulated with CD3-specific antibody. (E) Abundance of of ZFYVE21 in PTCHlo (P2), PTCHmid (P1,P4), and PTCHhi (P3) Tmem in IRI-treated EC:T cell cocultures. (F) Abundance of ZFYVE21 in PTCH1hi Tmem following stimulation with antibodies against CD3 and CD28 in the absence or presence of SAG as indicated. (G) Abundance of ZFYVE21 in PTCH1hi Tmem in IRI-treated EC:T cell cocultures in the presence of GANT58, GANT61, vismodegib, or cyclopamine as indicated. Experimental points reflect technical replicates. Data in (D–G) are representative of 4–5 experiments using N=3 separate leukopack donors. Scale bars, 200μm. * indicates p<0.05, ** indicates p<0.01. Two-way ANOVA followed by Tukey’s pairwise comparison was used for statistical comparisons.
In CD4+CD45RO+ Tmem, ZFVYE21 transcripts were minimally increased by treatment with a CD3-stimulating antibody plus SAG (fig. S4C), but ZFYVE21 transcripts were strongly induced in PTCHhi (P3) Tmem at the 72 hr timepoint compared to PTCHlo (P2) Tmem (fig. S4C). ZFYVE21 amounts increased in proportion to Hh signal strength (Fig. 6D) and were greatest in PTCHhi (P3) Tmem relative to comparator subsets showing lesser Hh signal strengths (Fig 6E). Combined treatment with the CD3-stimulating antibody and low-dose SAG (0.4μM) to improve segregation of PD-1 populations showed that Hh agonism induced ZFVYE21 in PTCHhi cells to a greater degree than did a CD28-stimulating antibody (Fig. 6F). Similar to what was observed in IFN-γ+PTCHhi Tmem (Fig 4B), the increase of ZFYVE21 production in cocultures containing ECs subjected to IRI conditions (Fig 6G) was more strongly reduced by pharmacological inhibitors of SMO (vismodegib, cyclopamine) than with inhibitors of GLI (GANT58, GANT61). ZFYVE21 production in Jurkat cells was reduced after knocking down SMO or GLI1 (fig. S2G). Thus, Hh agonism occurring in peripheral tissues selectively induced ZFYVE21 in PTCHhi Tmem.
Hh-induced ZFYVE21 activates NLRP3 inflammasomes in T Cells
We subsequently assessed a role for ZFYVE21 in expanding Ptchhi Tmem. We transduced PBMCs from different human donors with control short hairpin RNA (shRNA) or ZFYVE21 shRNA constructs also encoding GFP and tested their effects on Hh signal strength. Due to variability in transduction efficiencies between donors, analyses were performed following gating of total GFP+ cells for normalization prior to downstream analyses. Cells transduced with ZFYV21 shRNA showed low amounts of PD-1 (Fig. 7A) and relatively decreased ability to induce PTCH1 production in response to SAG (Fig. 7B). In ZFYVE21 shRNA Tmem, PTCH1 amounts were significantly reduced in GFP+ (PD-1lo) populations containing high amounts of the ZFYVE21 shRNA construct compared to PD-1hi cells lacking GFP which in contrast showed higher PTCH1 expression (fig. S5A), indicating a cell autonomous effect of ZFYVE21. In contrast, separate cells from these same donors, when transduced with ZFYVE21 expression vectors, produced high amounts of PD-1 (P1, Fig. 7C) and PTCH1 (Fig. 7D) compared to empty vector controls despite the absence of exogenous Hh agonism. These data together indicated that ZFYVE21 was sufficient for expanding Tmem producing high amounts of both PTCH1 and PD-1 (PTCH1hiPD-1hi), a surface phenotype compatible with PTCHhi Tmem.
Fig. 7. Hh-induced ZFYVE21 activates NLRP3 inflammasomes in T cells.
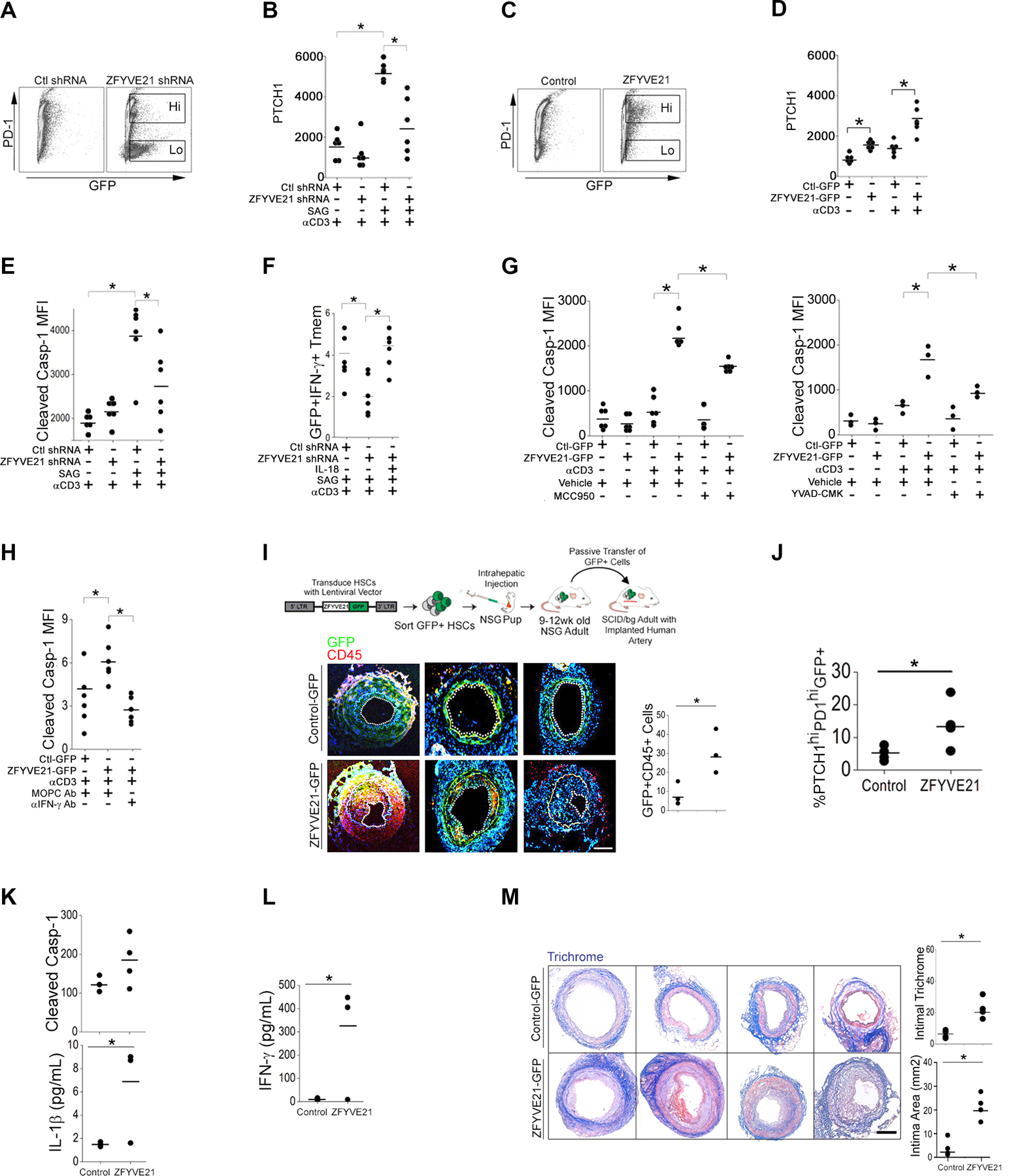
(A and B) Abundance of PD-1 (A) in Tmem expressing control (Ctl) or ZFYVE21 shRNA GFP-marked constructs and abundance of PTCH1 (B) when these cells were treated with SAG and stimulated with immobilized CD3-specific antibody. (C and D) Abundance of PD-1 (C) in Tmem overexpressing GFP-tagged ZFYVE21 or empty vector (Ctl-GFP) and abundance of PTCH1 (D) when these cells were stimulated with CD3-specific antibody. (E and F) Quantification of cleaved casp-1 (E) and frequency of GFP+IFNγ+ Tmem (F) in Tmem expressing control or ZFYVE21 shRNA, treated with SAG and IL-18 as indicated, and stimulated with immobilized CD3-specific antibody. (G and H) Quantification of cleaved casp-1 in Tmem overexpressing ZFYVE21-GFP or GFP alone following stimulation with immobilized CD3-specific antibody. Cells were treated with MCC950 or YVAD-CMK (G) or with IFN-γ neutralization antibody (H) as indicated. MOPC is an isotype-specific control for IFN-γ antibody. Data in (A to H) represent experiments from N=6 separate leukopack donors. (I to M) Human CD34 stem cells (HSCs) transduced with control GFP (N=4) or ZFYVE21-GFP expression (N=4) constructs were intrahepatically injected into NSG pups. PBMCs and splenocytes from these adult animals were passively transferred into SCID/bg hosts bearing human artery xenografts. Immunofluorescence images show CD45 and GFP in tissue grafts and were used to quantify GFP+CD45+ cells (I). PTCHhiPDhiGFP+ PBMC frequency (J), cleaved casp-1 and IL-1β concentration in PBMCs (K), and IFN-γ concentration in sera (L) were quantified in the mice receiving xenografts and donor PBMCs. Xenografts were harvested and stained with Masson’s trichrome (M). N=3–4 mice implanted with human arteries from 3 donors were used in each group. Scale bars, 200μm. *indicates p<0.05. Two-way ANOVA followed by Tukey’s pairwise comparison (B, D, E, F, G, H) and Student’s t-test (I-M) were used for statistical comparisons.
We next tested effects of ZFYVE21 on functional phenotypes of this PTCH1hiPD-1hi population. ZFYVE21 production was sufficient for increasing molecules prototypically conferring homing to peripheral (CCR2, CXCR3) but not secondary (CXCR5, CCR7) lymphoid tissues (fig. S5B), and these effects were amplified following TCR stimulation (fig. S4B). ZFYVE21 production, although sufficient for inducing PTCH1 production and the chemokine receptors above with or without TCR stimulation (Fig. 7D), was insufficient for increasing NLRP3 or for augmenting NLRP3 priming following IFN-γ exposure (fig. S5C), indicating that ZFYVE21 did not directly prime NLRP3 inflammasomes in Tmem. ZFYVE21 shRNA attenuated the ability of SAG to increase inflammasome activity (Fig. 7E), thereby reducing frequencies of GFP+IFN-γ+ Tmem (Fig. 7F). The deficit in type 1 responses in ZFYVE21 shRNA Tmem was rescued by IL-18 (Fig. 7F). Despite the absence of exogenous SAG, upon TCR stimulation, GFP+ Tmem carrying ZFYVE21 expression constructs showed strongly increased inflammasome activity (Fig. 7G) compared to empty vector controls, and this effect was significantly reduced by inhibition of NLRP3 with MCC950, caspase-1 with YVAD-CMK (Fig. 7G), or IFN-γ with IFN-γ neutralization antibody (Fig 7, G and H). These data collectively indicated that ZFYVE21 production was sufficient to expand a cell population with surface and functional phenotypes consistent with PTCHhi Tmem.
To assess a role for this ZFYVE21-induced population in vivo, we expanded Tmem containing control-GFP or ZFYVE21-GFP expression vectors according to a workflow adapted from a prior report (Fig. 7I) (44). We transduced human CD34+ hematopoietic stem cells with control or ZFYVE21 expression constructs and engrafted NSG pups with transduced GFP+ stem cells. Nine- to twelve- weeks later, human T cells constituted 40–56% of circulating murine CD45+ leukocytes (fig. S5D). Among treatment groups, we did not observe differences in human T cell engraftment, of which ~80–90% were GFP+ (fig. S5E). Of GFP+ cells, CD19+ cells showed comparatively lower engraftment percentages vs CD3+ cells (fig. S5E). Following engraftment, 2–3×106 GFP+ cells, including ZFYVE21+ T cells and B cells, were sorted and passively transferred into SCID/bg mice bearing human artery segments. We employed this passive transfer strategy because NSG mice showed thromboses at sites of surgical anastomoses and were thus not amenable to surgical implantation of human artery segments. Compared to controls, hosts receiving ZFYVE21+ lymphocytes showed increased GFP+ neointimal infiltrates (Fig. 7, I and J) and expanded percentages of PTCH1hiPD-1hi Tmem (Fig. 7J), similar to what was observed in vitro (Fig 7D). ZFVYVE21-GFP Tmem upon ex vivo recovery coexpressed tissue homing molecules, including CCR2 and CD103 (fig. S5F) and displayed potentiated markers of inflammasome activity (Fig 7K). Moreover, hosts receiving ZFYVE21-GFP lymphocytes showed increased type 1 responses (Fig 7L) and exacerbated arteriosclerotic lesions (Fig 7M), thereby phenocopying the effects of passively transferred PTCHhi Tmem (Fig. 4G). These studies indicated that lymphocyte production of ZFYVE21 functionally recapitulated pathologic features of PTCHhi Tmem to induce sequelae of chronic, type 1–mediated inflammation in vivo.
Hh-activated ZFYVE21-Akt-Casp1 signaling in Tmem occurs in DGF patients
The kinase AKT has been shown to potentiate Hh signaling (13,43) and we previously found that AKT activation was regulated by ZFYVE21 (1). On this basis we again analyzed RNA-seq data from PTCHlo (P2), PTCHmid (P1,P4), and PTCHhi (P3) subsets and uncovered enrichment of genes annotated for PI3K-AKT signaling in the PTCHhi subset (Fig. S5G), suggesting that ZFYVE21 could modulate AKT activity in Tmem. To test this, we performed phosphoFlow studies and found that SAG increased AKT phosphorylated on Ser473 (pAKTSer473) similarly to that in CD28 antibody–treated Tmem (Fig. 8A), an effect most pronounced in PTCH1hi cells (Fig. 8B). AktVIII, a selective AKT inhibitor, partially blocked SAG-dependent inflammasome activity in activated PCTHhi Tmem (Fig. 8C), suggesting the existence of additional pathways through which ZFYVE21 may modulate caspase-1 activity. ZFYVE21 overexpression or ZFYVE21 shRNA significantly increased or decreased pAKTSer473, respectively (Fig. 8, D and E). In Jurkat cells, isoform-specific knockdowns of AKT, in particular AKT2, blocked caspase-1 cleavage (fig. S2H). Together, these data support a ZFYVE21-AKT-Caspase-1 signaling axis wherein Hh-induced ZFYVE21 promotes AKT activation in PTCHhi T cells to induce T cell–intrinsic cleavage of caspase-1.
Fig. 8. Hh-induced ZFYVE21-Akt-Casp1 signaling in Tmem from DGF patients.
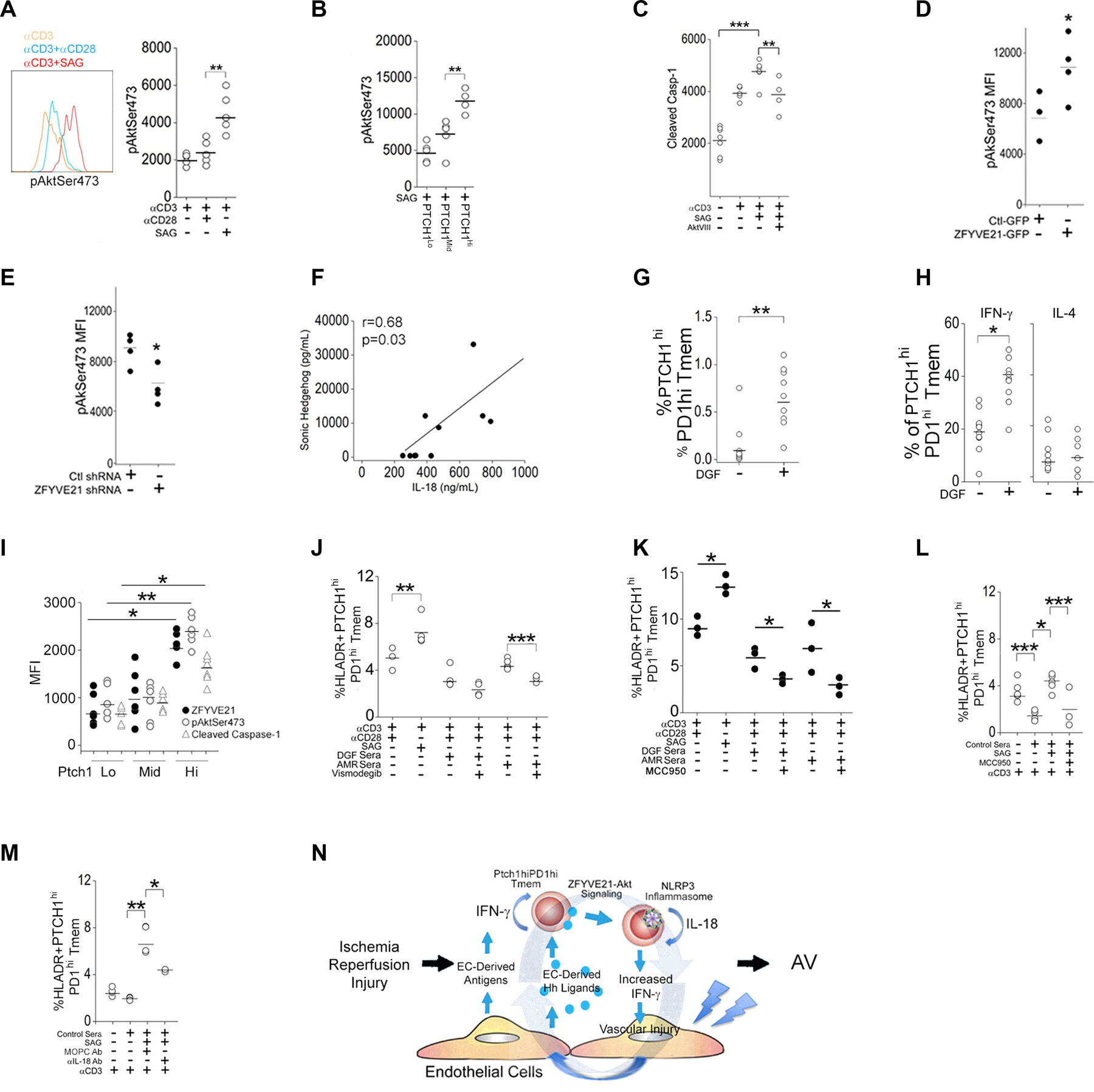
(A) Quantification of pAktSer473 in Tmem pretreated with antibody against CD28 or Smoothened agonist (SAG) prior to stimulation with antibody against CD3. (B) Quantification of pAktSer473 in FACS-sorted PTCHlo, PTCHmid, and PTCHhi Tmem. (C) Quantification of cleaved Casp-1 in Tmem pretreated with SAG or AktVIII prior to CD3 stimulation. (D and E) Quantification of pAktSer473 in Tmem overexpressing ZFYVE21-GFP (D) or expressing ZFYVE21 shRNA (E) and stimulated with antibody against CD3. (F) Pearson correlation between SHH and IL-18 in DGF patient sera (N=7). (G) PTCHhiPD-1hi Tmem frequency in control (–) and DGF (+) PBMCs. (H) IFN-γ and IL-4+ PTCHhiPD-1hi Tmem frequency in control and DGF PBMCs. (I) Quantification of ZFYVE21, pAktSer473, and cleaved casp-1 in PTCHlo, PTCHmid, and PTCHhi Tmem. (J to L) HLA-DR+PTCHhiPD-1hi Tmem frequency in non-autologous Tmem from a healthy donor stimulated with an antibody against CD3 in the presence of CD28-specific antibody, SAG, MCC950, or vismodegib as indicated. DGF or AMR sera (J and K) or control sera (L) were used as indicated. (M) HLA-DR+PTCHhiPD-1hi Tmem frequency in non-autologous Tmem from a healthy donor stimulated with an antibody specific for CD3 in control serum in the presence of SAG or antibodies specific for MOPC or IL-18 as indicated. (N) Working model for an Hh-induced ZFYVE21-Akt-Casp-1 signaling axis in Tmem. Experimental points in (A–E) and (G–M) reflect technical replicates and were repeated 2–3 times using N=6 separate leukopack donors. * indicates p<0.05, ** indicates p<0.01. Two-way ANOVA followed by Tukey’s pairwise comparison (A-C, I-L) and Student’s t-test (D, E, G, H) were used for statistical comparisons.
Finally, we tested the ZFYVE21-AKT-Caspase-1 signaling axis in biospecimens from patients with DGF, a subset of which developed antibody-mediated rejection (AMR). The inclusion criteria and baseline characteristics of this cohort along with control patients were previously described (11). Prior analyses of patient sera showed that DGF patients had higher circulating amounts of IL-18 (11) and SHH (Fig. 1D) compared to controls. In light of the new findings herein, we re-analyzed these sera and detected a significant correlation between the amounts of Shh and IL-18 (Fig. 8F). Moreover, compared to controls, DGF patients showed increased frequencies of PCTHhi Tmem (Fig. 8G) showing comparatively stronger type 1 effector responses upon ex vivo stimulation (Fig. 8H). Abundances of ZFYVE21, pAKTSer473, and cleaved casp-1 increased in proportion to PTCH1 amounts in Tmem from DGF patients (Fig. 8I). To specifically test effects of Hh signaling, we overlaid sera from DGF or AMR patients containing high amounts of SHH onto non-autologous Tmem. In all cultures tested, including those from controls, we observed that addition of either control or DGF sera caused a generalized reduction in percentages of HLA-DR+ Tmem, an observation made previously (11) that we surmise was likely due to the presence of immunosuppressives in the sera. Because strong stimulation could not be achieved in the presence of patient sera, assessment of inhibitory drug effects in our studies were likely underestimated. Samples containing DGF sera showed significant expansion of PCTHhi Tmem (Fig. 8J). Addition of vismodegib or MCC950 to these samples reduced measured frequencies of PTCHhi Tmem (Fig. 8J–K). Conversely, addition of Hh agonist to autologous Tmem and sera from control patients containing low levels of SHH boosted frequencies of PTCHhi Tmem, and this potentiated effect was reduced by MCC950 (Fig. 8L) and by IL-18 neutralization antibody (Fig. 8M). These data support a role for Hh signaling, NLRP3 inflammasome activity, and IL-18 in modulating the frequencies of PTCHhi Tmem in patients with DGF, a clinical manifestation of IRI, where Hh ligands are released by ECs.
DISCUSSION
Here, we describe a previously unrecognized role for ZFYVE21, a Rab5 effector, in T cell–mediated inflammation. (Fig. 8N). We showed that Hh morphogens released by damaged tissues expand a T cell subset, PTCHhi Tmem, that showed strong homing and type 1 effector responses. Through a ZFYVE21-AKT signaling axis, Hh agonism promoted NLRP3 inflammasome activity in PTCHhi Tmem and potentiated IFN-γ responses, a process that furthers Hh ligand release from injured tissues to create a positive feedback loop promoting chronic inflammation.
Divergent effects of SHH have been observed with regards to lung vs skin, with SHH worsening allergic lung disease through the potentiation of TH2 cells (21,) the polarization of which was subsequently found to be dependent on Hh signaling (20), but decreasing T cell activation in atopic dermatitis through the potentiation of Tregs in a TGF-β–dependent manner (23). In the context of its role as a morphogen in wound healing, Hh signaling contributes to parenchymal quiescence in some studies (45–49) but not in others (50–55). Based on this, Hh signaling effects on adaptive immunity may incorporate additional, yet undefined factors that may occur in a tissue- or context-specific fashion (56). Hh-induced NLRP3 inflammasomes, in addition to occurring in CD4+ T cells, may occur in other cell types, particularly passenger myeloid cells contained within transplanted artery grafts as well as ECs (11,12), which may influence systemic inflammasome activity as well as in vivo phenotypes following pharmacological inhibition of NLRP3. Our data show that, in addition to provision of positioning cues, EC-derived morphogens like Hh might iteratively heighten the effector response of certain tissue-localized immune cell population(s).
Our study results differ from prior reports showing anti-inflammatory effects of Hh signaling (57,58). We have specifically attempted to model human EC interactions with T cells through direct allorecognition, a response fundamentally lacking in murine models where indirect allorecognition predominates due to the relative inability of murine ECs to directly prime murine CD4+ T cells. We used HUVECs to simulate IRI-induced signaling responses. HUVECs are a widely used model system whose immune responses in direct allorecognition have been well-characterized and that model immune responses at the microvascular level. HUVECs were used to define core signaling responses which most likely are present in HUAECs based on experiments in vivo. We have identified and interrogated a specific subset of PTCH1hi CD4+ T cells that show strong Hh responses, and the effector qualities of this specific subset differ from responses seen in bulk CD4+ T cell populations examined in other studies.
Our study is a proof of principle to show that EC-derived alloantigens, which strongly mediate type 1 responses, may utilize a damage-related morphogen, Hh, to incur vascular inflammation. Our humanized models, in addition to skewing towards direct allorecognition responses as above, lack human myeloid cells, which do not significantly engraft in SCID/bg mice. These fundamental immune differences along with differences in experimental model systems as below might explain the proinflammatory effects of Hh agonism in our study when compared to prior reports in the literature (59,60).
In murine models, nonlipidated Hh agonists (22) and genetic deficiency of various Hh signaling components (61,62) are routinely used to study Hh effects on T cells. We have used a chemical agonist, SAG, with a different half-life from exogenous non-lipidated Hh ligands and which elicits strong Hh signal activation. The dose and duration of Hh agonism with SAG may further explain the inflammatory effects of PTCHhi Tmem observed herein when compared to prior reports. Additionally, to explore EC:T cell interactions, we have used ECs treated with conditions that simulate IRI, which release all 3 Hh ligands (SHH, DHH, and IHH). Because DHH (63) and IHH (63) may affect myelopoiesis and thymopoiesis, respectively, the effects of these Hh ligands when combined with SHH may exert differential immune effects on T cells. Also, we have not used germline deletions of SMO or other Hh signaling components, which, due to Hh effects on thymic selection (23), may alter the pathogenicity of the adult T cell repertoire.
We have observed a gradient effect of SHH release in IRI-treated human arteries where SHH production appears strongest in the intima containing ECs and dissipates towards the adventitia (Fig. 1C). This gradient of SHH is phenocopied by the relative abundance of infiltrating ZFVYE21+ T cells (Fig. 6C). SHH is well known to exert gradient effects during development (65), but the effects of such gradients in adult tissues in vivo are not as well studied. It is possible that PTCHhi T cells, in addition to showing high Hh responsiveness, may sequester Hh ligands to spatially constrain SMO-dependent signaling. We have observed differential amounts of Hh signaling components that correlate with chemokine receptor expression (fig. S1D). Examination of Hh signaling components as well as the other downstream pathways they influence, like NF-κB (62), and transcriptional targets, including Rab5b (66) and IL-1β (62), in tissues is of future interest.
MATERIALS AND METHODS
Cell cultures
All protocols were approved by the Yale Institutional Review Board. HUVEC were isolated as healthy, de-identified tissues from the Dept of Obstetrics and Gynecology at Yale New Haven Hospital as previously described. HUVEC were pooled from 3 human donors. For “IRI” treatment, HUVEC were pre-treated with IFN-γ (50ng/mL, Invitrogen) for 48–72h prior to placement in gelatin veronal buffer (Sigma) containing 50% v/v normal human sera (Innovative Research) or C6-deficient human sera (Sigma). HUVEC were then placed in a humidified incubator at 5% CO2 and 37°C with 0% FiO2 for 4h to simulate ischemia followed by 2h in a humidified incubator at 5% CO2 and 37°C with 21% FiO2 for 2h to simulate reperfusion. Normoxia controls were pretreated with IFN-γ and placed in gelatin veronal buffer containing 50% v/v normal human sera under normoxic conditions with 21% FiO2 for 6h.
HUVEC were then subjected to “IRI” prior to addition of human CD4+CD45RO+ T cells which were added at 0.5–1×106 cells/well at a volume of 200μL in RPMI (Gibco) supplemented with 5% FBS, 1.5% L-glutamine, and 1% penicillin/ streptomycin. T cells were harvested 10–14 days after co-culture in a humidified incubator at 5% CO2 and 37°C with 21% FiO2 and analyzed by flow cytometry (Becton Dickinson, LSRII). Human Jurkat cells (clone E6–1) were purchased from ATCC (#TIB152).
siRNA-mediated knockdown of EC proteins
Small interfering (si) RNA (Dharmacon, Waltham, MA) targeting Sonic Hedgehog (SmartPool #L-006036–00-0005, target sequences: GUG CAC AGC GUG ACC CUA A, GGU GUA AGG ACA AGU UGA A, CAG CUG CUC UAC CAA AUA G, GAA ACU CCG AGC GAU UUA A), or non-targeting siRNA (target sequence UAA CGA CGC GAC GAC GUA A) were transfected into HUVEC in 24-well plates (BD Falcon) at 20nM concentrations. To do so, siRNA was mixed at stated concentrations with RNAiMax transfection reagent (Invitrogen) for 45 minutes at room temperature, then added to cultures for 6 hours prior to washing and buffer exchange with complete EGM2 media. IFN-γ was added to transfected cultures the following day for 48 hours followed by treatment with the “IRI” protocol. Knockdown efficiency was confirmed by Western blotting and consistently showed 60–90% decrement in protein expression relative to HUVEC treated with control siRNA.
Human CD4+ T cell isolation, RNA-seq, and EC:T cell coculture
All protocols were approved by the Yale Institutional Review Board (Protocol #0601000969). PBMCs were isolated from leukopacks using density centrifugation as described previously and cryopreserved in liquid nitrogen.13 CD4+CD45RO+ T cells were isolated from thawed cryovials using magnetic bead separation kits (Miltenyi) with HLA-DR Ab (clone L243, Novus #NB100–77855) and CD45RA Ab negative depletion (10μL per cryovial, eBiosciences, 14–0458-82). Where indicated, flat-bottom 96-well microtiter plates were coated with anti-CD3 Ab (1μg/mL, eBiosciences, #16003785) in sterile PBS at 4°C overnight, and the following day isolated CD4+CD45RO+ T cells (Tmem) were pre-treated with soluble anti-CD28 Ab (1μg/mL, eBiosciences, #16–0281-82) or SAG (5μM) for 1 hr prior to addition to anti-CD3 Ab-coated plates. Tmem were stimulated for 72 hrs prior to use in downstream applications including RNA sequencing, EC:T cell cocultures, T cell adhesion assays, and passive transfer into humanized mice.
For RNA sequencing, Tmem were homogenized via QiaShredder spin columns (Qiagen, #79656) for 2 min at room temperature, and total RNA was isolated using RNeasy spin columns (Qiagen, #74004). 200ng-1ug of purified total RNA was suspended in nuclease-free H2O (30 μL) and submitted for RNA expression profiling at the Yale Center for Genomic Analysis. Quality control checks for submitted samples included OD260/280 ratio of 1.8–2 (Agilent Technologies 2100 Bioanalyzer) and a 2:1 intensity ratio of intact 28S:18S rRNA bands on 1% agarose gel electrophoresis. The sequencing library was prepared from extracted RNA using the Illumina platform for cDNA synthesis, amplicon generation, tagmentation, and cleaning. The pooled and cleaned library was sequenced on an Illumina NovaSeq platform with each sample given at least 1 million reads.
For EC:T cell co-cultures, HUVEC isolated from a single donor were grown in U-bottom 96-well microtiter plates, pretreated with human IFN-γ (50ng/mL, Invitrogen) for 48–72hr. Human T cells were incubated for 1 hr with Ac-YVAD-CMK (20μM), cyclopamine (5μM), GANT58 (5μM), GANT61 (2.5μM), MCC950 (5μM), SAG, or vismodegib (2.5μM) prior to addition at 5×105-1×106 Tmem per well for 7–10 days prior to analysis. For EC:T cell coculture studies, Tmem were initially enriched from total human PBMCs by magnetic bead separation as per the manufacturer’s instructions above (Miltenyi). Cells were then stained with the appropriate fluorophore-labeled antibodies and sorted using the FACSAria Cell Sorter (Becton Dickinson).
T cell adhesion assays
T cell adhesion to EC monolayers was assessed under conditions of venular flow as described previously (9). In brief, confluent HUVEC monolayers on a human plasma fibronectin-coated coverglass were pre-treated with “IRI” to induce Hh ligand production. EC monolayers were then assembled into a parallel plate flow chamber apparatus (Glycotech, #31,001, Gaithersburg, MD), and T cells were injected over the EC monolayer at a rate imparting 0.75 dyne/cm2 of shear stress using a syringe pump device (New Era Pump Systems, Farmingdale, NY) over a period of ~15 min. Adherent T cells were fixed in 4% formaldehyde, stained with anti-human CD45-FITC antibody (Beckman Coulter, #A07782, San Diego, CA), and manually quantified in ten high-powered fields per sample. The data were calculated as CD4+DAPI+ cells per total DAPI+ cells in ≥5 high power fields.
Collagen-fibronectin gel model
HUVEC were grown to confluency in T75 flasks, harvested, pelleted, and resuspended in 500μL of a solution mixture containing 50μL 10X M199 (Sigma), 50μL of 1mg/mL fibronectin (Millipore), 12.5μL 1M HEPES, 188μL of 10mg/mL NaHCO3, 300μL of 3.66mg/mL Collagen (Corning), and 6μL 1M NaOH. Gels were solidified at 37°C for 1 hour prior to implantation to the distal iliac artery/proximal femoral artery. Four weeks after implantation to allow anastomosis formation, the proximal iliac artery was then surgically ligated for 24 hrs prior to removal of the surgical ligature. Twenty-four hours after removal of the surgical ligature, grafts were harvested, flash frozen in OCT, sectioned, stained, and analyzed by I.F. as indicated. Staining Abs were used at 1:200 dilution including Ulex (Vector) and Shh (Novus, #NBP2–2212655).
FACS analysis
T cell surface staining used the following primary antibodies at 1:100 for 1 hr at 4°C in PBS containing 3% BSA and 0.01% NaN3: CD4+ (Biolegend), HLA-DR FITC (Biolegend, #307604), PD-1 PE (eBiosciences, #12–2799-42), CCR2 APC (Biolegend, #257208), CXCR5 Pacific Blue (Biolegend, #356918), and Ptch1 (Cell Signal, clone C53A3, #2468T). For intracellular staining, T cells were fixed and permeabilized using a FOXP3 Staining Kit according to the manufacturer’s specifications (R&D Systems). Prior to fixation and permeabilization, cells were stained using the following antibodies at 1:100 for 1 hr at 4°C: Gli1 (Cell Signal, clone C68H3, #3538T), NLRP3 (Cell Signal, clone DRD8T, #15101) and cleaved caspase-1 (Cell Signal, cat #4199).
For cellular staining, antibodies included ZFYVE21 (Biorbyt, #221973), IFN-γ PE (Biolegend), IL-4 PE/Cy7, Biolegend, #500824), IL-1β (Cell Signaling, clone D3U3E, #12703), IL-18 (Proteintech, #10663–1-ap) cleaved caspase-1 (Cell Signaling, clone D57A2, #4199). T cells were stimulated for 2 h with PMA/ionomycin prior to addition of GolgiStop (Becton Dickinson) for 4h prior to harvest and FACS analysis as previously described.13 T cells were stained with CFSE (Molecular Probes) as previously described.15,17–18 T cells were gated and analyzed using an LSR II flow cytometer (Becton Dickinson) and sorted using a FACSAria cell sorter (Becton Dickinson). Blocking anti-IL-18 (Invivogen, #mabg-hil18), anti-IFN-γ, clone B27, #554699), and isotype control (Biolegend, clone MOPC-21, #400101) antibodies were used at 5μg/mL. MOPC-21 (MOPC) is a monoclonal mouse IgG1 Ab that is commonly used as an isotype control. Human IL-18 (Sinobiologicals) was added at either 0.5μg or 5μg as indicated.
Western blot analysis
For Western blot analysis, antibodies against SHH (Novus, #NBP2–2212655), DHH (BosterBio, #A03731), IHH (BosterBio, #PA2225), and β-actin (Sigma, #A5441) were added at a concentration of 1:1000 at 4°C overnight with gentle agitation. Secondary HRP-conjugated anti-mouse (ThermoFisher) and anti-rabbit (ThermoFisher) secondary antibodies were added at 1:5000 for 1h at room temperature with gentle agitation.
Immunofluorescence analysis
All protocols were approved Yale Institutional Review Board (Protocol #1212011221). Human artery xenografts upon harvest were flash frozen in OCT, sectioned, stained, and analyzed by I.F. For I.F. analysis, antibodies against C6 (R&D), SHH (Novus, NBP2–22126SS), Ulex (VectorLabs), Formalin-fixed paraffin embedded coronary biopsy sections (n=4) were obtained as archival samples the Yale Dept of Pathology. Following deparaffinization and hydration, antigen retrieval was performed at 98°C for 1hr in Antigen Unmasking Solution (VectorLabs) and stained using antibodies against SHH (Novus, #NBP2–22126SS), Ulex (VectorLabs), and MAC (Agilent, clone AE11, #M0777)) at 1:200 dilution at 4°C overnight prior to addition of secondary Abs (1:500 as above for 1 hour at room temperature. Fresh tonsillar (n=3) and appendiceal tissues (n=3) were obtained as de-identified samples from the Yale Dept of Pathology. Tissues were mechanically disrupted through a cell strainer, placed into single cell suspension in PBS, and washed three times prior to FACS analysis.
I.F. images were analyzed by 3 blinded reviewers for colocalization using ImageJ software using the JaCop plugin. Merged images were deconvoluted into red, green, and, in the case of triple colocalization studies, blue color channels. Pixels ≤1500 were selected for analysis in the case of CD4, CD19, TPH, and TFH cell quantification and sequentially analyzed for colocalization with colocalized pixels being defined as < 50 diameters.
Animal studies
All protocols were approved by the Yale Institutional Animal Care and Use Committee (Protocol #: 2021–20175) and were performed in accordance with institutional guidelines. Human coronary arteries were interposed into the infrarenal abdominal aortae of adult 6–12-week-old female C.B-17 SCID/beige mice (Taconic, #CBSCBG-F) for ~30 days to logistically hold and quiesce vessel grafts prior to use. Prior for inclusion into the study, SCID/beige mice were tested for ‘leakiness’ or presence of murine CD3+ and CD19+ cells in PBMCs. Mice showing CD3 and/or CD19 percentages >5% of CD45+ cells were classified as ‘leaky’ and excluded from the study. Prior to artery implantation, mice were tested for presence of murine T cells via FACS analysis of sera. In studies where mice were engrafted with human lymphocytes, 1×108 allogeneic PBMCs were injected ~2 weeks prior to artery implantation. CD3+ T cell engraftment was tested every 3–4 days via FACS for human lymphocyte engraftment, and hosts were used for artery implantation experiments when human CD3+ T cell engraftment was ≥3% of total murine CD45+ PBMCs. For passive transfer studies, CD4+ T cells isolated from human PBMCs by magnetic bead separation were stimulated with plate-bound CD3 specific antibody in the presence of either soluble CD28-specific antibody (1μg/mL) or SAG (50μM) for 48 hr. Tmem were then FACS-sorted and injected i.p. at 5×106 cells/host into SCID/bg mice bearing human artery segments along with 10×106 autologous B cells. Sample sizes for all studies were ≥3 hosts per group to enable calculation of standard deviations. Artery graft segments from the same human donor were randomized among treatment groups. Grafts and sera were harvested 8 weeks later for analysis.
To induce IRI, human artery segments were placed at 0% FiO2 5% CO2 for 37°C for 12 hours in DMEM in organ culture prior to surgical reimplantation into the infrarenal position of a second set of SCID/bg hosts previously injected with 1×108 allogeneic PBMCs i.p. ~2 weeks prior, a procedure which typically engrafts ~0.5–5.0% human T cells and B cells.11,13,14,15,18 Human arteries and host sera were harvested 4 weeks after artery implantation. Where indicated, micro-osmotic pumps (Alzet model 1004, #9922) were concurrently implanted subcutaneously into the flank of SCID/bg hosts. Prior to implantation, pumps were primed according to the manufacturer’s specifications to elute vismodegib (12mg/kg/dy), GANT61 (6mg/kg/dy), MCC950 (4mg/kg/dy), or SAG (15mg/kg/dy) over a 28-day period.
Viral transduction of human T Cells and CD34+ stem cells
Lentiviral constructs encoding control shRNA-GFP (SHC016V0718127MN) or ZFYVE21 shRNA-GFP (TRCN0000436194) constructs were purchased from Sigma-Aldrich. Lentiviral constructs encoding ZFYVE21-GFP (pLenti-GIII-CMV-GFP-2A-Puro) were purchased from Applied Biological Materials. CD4+CD45RO+ T cells (5×104 cells per well) were electroporated with 2μg of the plasmids above according to the manufacturer’s specifications (Amaxa, #VPA-1002) using the Nucleofector II device (Amaxa). Seventy-two hours later, for a total of 4 d in culture, T cells were washed and restimulated with plate-bound anti-CD3 (1μg/mL) and treated as indicated in the text. Lentivirus containing the constructs above were used to infect 1×105 human CD34+ stem cells for 8 hrs. The next day, transduced GFP+ stem cells were FACS-sorted and intrahepatically injected into sublethally irradiated (180 cGy) 1–3dy old female NSG (Jackson, #00557) pups. Nine-to-twelve weeks later, mice were bled and human Tmem were analyzed as indicated in the text. In certain experiments, 8×106 PBMCs from the mice above were passively transferred i.p. into adult 6–12-week-old SCID/bg hosts containing human artery segments implanted into the infrarenal position of the descending aorta. Four weeks later, grafts and host sera were harvested for analysis.
DGF patient study
All protocols were approved Yale Institutional Review Board (Protocol #2000020032), and participants gave informed consent. Eligible patients were prospectively recruited from a single center (Yale New Haven Hospital) between the dates of 2016–2018 and included renal transplant recipients receiving deceased donor kidney transplants with either an uncomplicated post-operative hospital course (controls, n=10) or a post-operative hospital course requiring one or more sessions of dialysis (DGF, n=7) prior to discharge. Patients with DGF who subsequently developed a clinical diagnosis of AMR were also recruited. Patients with a pre-existing complement-mediated condition, e.g., systemic lupus erythematosus or rheumatoid arthritis, were excluded as were patients having peri-operative infection(s). Sera and PBMCs used in the study were collected 2–4 weeks post-transplantation. Analysis of Patient Sera. Patient sera were analyzed using EIA for human SHH (R&D Systems, #DSHH00), human IL-1β (ThermoFisher), and human IFN-γ (ThermoFisher). The recruitment details and baseline characteristics of these patients were previously reported.11
Data availability
All data and methods are available from the authors upon reasonable request. RNA-seq data sets were deposited in the Gene Expression Omnibus (GSE208349). Publicly available transciptomic data (Fig. 5A) were retrieved from the Gene Expression Omnibus analyzing inflammasome-related transcript abundance for casp1, ILb, and GSDMD and Hh signaling genes Ptch1, Gli1, and Smo for the following datasets in the Gene Expression Omnibus: GSE110932, GSE11987, GSE20065, GSE103171, GSE29316, GSE81963, and GSE40359.
Statistical methods
Comparisons between two groups were performed using two-sample t-test, and multiple comparisons were performed using a one-way or two-way ANOVA followed by Tukey’s pairwise comparison test using Origin computer software. Pearson’s r was used to calculate correlation coefficients. RNA gene expression profiling data were analyzed using Rstudio gene software. p-values <0.05 were considered statistically significant. Standard deviations are reported throughout the text.
Supplementary Material
ACKNOWLEDGEMENTS:
We would like to thank Ricarda Tomlin, Summer Guo, and Sanjay Kulkarni for assistance with collection of clinical samples. We wish to thank Tassos Kyriakides and Dejian Zhao for independent statistical reviews of the manuscript.
FUNDING:
D.J. was supported by grants from the NIH (R01HL141137-01, R01HL141137-04S2), the Veterans Hospital Administration (I01 BX005117), and the American Lung Association (ETRA 736563). J.S.P. was supported by grants from the NIH (R01HL-51014).
Footnotes
COMPETING INTERESTS: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
Figs. S1 to S5.
DATA AND MATERIALS AVAILABILITY:
RNA-seq data have been deposited into the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) with the dataset identifier GSE208349. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Uekita T, Gotoh I, Kinoshita T, Itoh Y, Sato H, Shiomi T, Okada Y, Seiki M. Membrane-type 1 matrix metalloproteinase cytoplasmic tail-binding protein-1 is a new member of the Cupin superfamily. A possible multifunctional protein acting as an invasion suppressor down-regulated in tumors. J Biol Chem. 2004. 279:12734–43. [DOI] [PubMed] [Google Scholar]
- 2.Hoshino D, Nagano M, Saitoh A, Koshikawa N, Suzuki T, Seiki M. The phosphoinositide-binding protein ZF21 regulates ECM degradation by invadopodia. PLoS One. 2013. 8:e50825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagano M, Hoshino D, Sakamoto T, Akizawa T, Koshikawa N, Seiki M. ZF21 is a new regulator of focal adhesion disassembly and a potential member of the spreading initiation center. Cell Adh Migr. 2011. 5:23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagano M, Hoshino D, Toshima J, Seiki M, Koshikawa N. NH2 -terminal fragment of ZF21 protein suppresses tumor invasion via inhibiting the interaction of ZF21 with FAK. Cancer Sci. 2020. 111:4393–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagano M, Hoshino D, Koshiba S, Shuo T, Koshikawa N, Tomizawa T, Hayashi F, Tochio N, Harada T, Akizawa T, Watanabe S, Handa N, Shirouzu M, Kigawa T, Yokoyama S, Seiki M. ZF21 protein, a regulator of the disassembly of focal adhesions and cancer metastasis, contains a novel noncanonical pleckstrin homology domain. J Biol Chem. 2011. 286:31598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang C, Manes TD, Liu L, Liu K, Qin L, Liu R, Tietjen GT, Nigrovic PA, Tellides G, Pober JS, Jane-Wit D. ZFYVE21 is a complement-induced Rab5 effector that activates non-canonical NF-κB via phosphoinosotide remodeling of endosomes. Nat Commun. 2019. 10:2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pober JS, Jane-wit D, Qin L, Tellides G. Interacting mechanisms in the pathogenesis of cardiac allograft vasculopathy. Arterioscler Thromb Vasc Biol. 2014. 34:1609–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qin L, Li G, Kirkiles-Smith N, Clark P, Fang C, Wang Y, Yu ZX, Devore D, Tellides G, Pober JS, Jane-Wit D. Complement C5 Inhibition Reduces T Cell-Mediated Allograft Vasculopathy Caused by Both Alloantibody and Ischemia Reperfusion Injury in Humanized Mice. Am J Transplant. 2016. 16:2865–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jane-Wit D, Manes TD, Yi T, Qin L, Clark P, Kirkiles-Smith NC, Abrahimi P, Devalliere J, Moeckel G, Kulkarni S, Tellides G, Pober JS. Alloantibody and complement promote T cell-mediated cardiac allograft vasculopathy through noncanonical nuclear factor-κB signaling in endothelial cells. Circulation. 2013. 128:2504–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jane-wit D, Surovtseva YV, Qin L, Li G, Liu R, Clark P, Manes TD, Wang C, Kashgarian M, Kirkiles-Smith NC, Tellides G, Pober JS. Complement membrane attack complexes activate noncanonical NF-κB by forming an Akt+ NIK+ signalosome on Rab5+ endosomes. Proc Natl Acad Sci U S A. 2015. 112:9686–9691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu L, Fang C, Fu W, Jiang B, Li G, Qin L, Rosenbluth J, Gong G, Xie CB, Yoo P, Tellides G, Pober JS, Jane-Wit D. Endothelial Cell-Derived Interleukin-18 Released During Ischemia Reperfusion Injury Selectively Expands T Peripheral Helper Cells to Promote Alloantibody Production. Circulation. 2020. 141:464–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie CB, Qin L, Li G, Fang C, Kirkiles-Smith NC, Tellides G, Pober JS, Jane-Wit D. Complement Membrane Attack Complexes Assemble NLRP3 Inflammasomes Triggering IL-1 Activation of IFN-γ-Primed Human Endothelium. Circ Res. 2019. 124:1747–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramsbottom S, Pownall ME. Regulation of Hedgehog Signaling Inside and Outside the Cell. J Dev Biol. 2016. 4:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang L, Xie G, Fan Q, Xie J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene. 2010. 29:469–481.15. [DOI] [PubMed] [Google Scholar]
- 15.de la Roche M, Ritter AT, Angus KL, Dinsmore C, Earnshaw CH, Reiter JF, Griffiths GM. Hedgehog Signaling Controls T Cell Killing at the Immunological Synapse. Science. 2013. 342:1247–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crompton T, Outram SV, Hager-Theodorides AL. Sonic hedgehog signaling in T-cell development and activation. Nat Rev Immunol. 2007. 7:726–735. [DOI] [PubMed] [Google Scholar]
- 17.Stewart GA, Lowrey JA, Wakelin SJ, Fitch PM, Lindey S, Dallman MJ, Lamb JR, Howie SE. Sonic hedgehog signaling modulates activation and cytokine production by human peripheral CD4+ T cells. J Immunol. 2002. 169:5451–5457. [DOI] [PubMed] [Google Scholar]
- 18.Lowrey JA, Stewart GA, Lindey S, Hoyne GF, Dallman MJ, Howie SE, Lamb JR. Sonic hedgehog promotes cell cycle progression in activated peripheral CD4(+) T lymphocytes. J Immunol. 2002. 169:1869–1875. [DOI] [PubMed] [Google Scholar]
- 19.Chan VS, Chau SY, Tian L, Chen Y, Kwong SK, Quackenbush J, Dallman M, Lamb J, Tam PK. Sonic hedgehog promotes CD4+ T lymphocyte proliferation and modulates the expression of a subset of CD28-targeted genes. Int Immunol. 2006. 18:1627–1636. [DOI] [PubMed] [Google Scholar]
- 20.Yánez DC, Lau CI, Chawda MM, Ross S, Furmanski AL, Crompton T. Hedgehog signaling promotes TH2 differentiation in naïve human CD4 T cells. J Allergy Clin Immunol. 2019. 144:1419–1423.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furmanski AL, Saldana JI, Ono M, Sahni H, Paschalidis N, D’Acquisto F, Crompton T. Tissue-derived hedgehog proteins modulate Th differentiation and disease. J Immunol. 2013. 190:2641–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papaioannou E, Yánez DC, Ross S, Lau CI, Solanki A, Chawda MM, Virasami A, Ranz I, Ono M, O’Shaughnessy RFL, Crompton T. Sonic Hedgehog signaling limits atopic dermatitis via Gli2-driven immune regulation. J Clin Invest. 2019. 129:3153–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rowbotham NJ, Hager-Theodorides AL, Cebecauer M, Shah DK, Drakopoulou E, Dyson J, Outram SV, Crompton T. Activation of the Hedgehog signaling pathway in T-lineage cells inhibits TCR repertoire selection in the thymus and peripheral T-cell activation. Blood. 2007. 109:3757–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jane-Wit D, Fang C, Goldstein DR. Innate immune mechanisms in transplant allograft vasculopathy. Curr Opin Organ Transplant. 2016. 21:253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valenzuela NM, Reed EF. Antibody-mediated rejection across solid organ transplants: manifestations, mechanisms, and therapies. J Clin Invest. 2017. 127:2492–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hall ET, Cleverdon ER, Ogden SK. Dispatching Sonic Hedgehog: Molecular Mechanisms Controlling Deployment. Trends Cell Biol. 2019. 29:385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jay SM, Shepherd BR, Bertram JP, Pober JS, Saltzman WM. Engineering of multifunctional gels integrating highly efficient growth factor delivery with endothelial cell transplantation. FASEB J. 2008. 22:2949–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yi T, Fogal B, Hao Z, et al. Reperfusion injury intensifies the adaptive human T cell alloresponse in a human-mouse chimeric artery model. Arterioscler Thromb Vasc Biol. 2012. 32:353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, Donlin LT, Henderson LA, Wei K, Mizoguchi F, Teslovich NC, Weinblatt ME, Massarotti EM, Coblyn JS, Helfgott SM, Lee YC, Todd DJ, Bykerk VP, Goodman SM, Pernis AB, Ivashkiv LB, Karlson EW, Nigrovic PA, Filer A, Buckley CD, Lederer JA, Raychaudhuri S, Brenner MB. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. 2017. 542:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noël G, Langouo Fontsa M, Garaud S, De Silva P, de Wind A, Van den Eynden GG, Salgado R, Boisson A, Locy H, Thomas N, Solinas C, Migliori E, Naveaux C, Duvillier H, 51. Lucas S, Craciun L, Thielemans K, Larsimont D, Willard-Gallo K. Functional Th1-oriented T follicular helper cells that infiltrate human breast cancer promote effective adaptive immunity. J Clin Invest. 2021. doi: 10.1172/JCI139905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abou-Daya KI, Tieu R, Zhao D, Rammal R, Sacirbegovic F, Williams AL, Shlomchik WD, Oberbarnscheidt MH, Lakkis FG. Resident memory T cells form during persistent antigen exposure leading to allograft rejection. Sci Immunol. 2021. 6:eabc8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szabo PA, Miron M, Farber DL. Location, location, location: Tissue resident memory T cells in mice and humans. Sci Immunol. 2019. 4(34): eaas9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, Vale RD. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017. 355:1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chai JY, Sugumar V, Alshawsh MA, Wong WF, Arya A, Chong PP, Looi CY. The Role of Smoothened-Dependent and -Independent Hedgehog Signaling Pathway in Tumorigenesis. Biomedicines. 2021. 9:1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tellides G, Tereb DA, Kirkiles-Smith NC, Kim RW, Wilson JH, Schechner JS, Lorber MI, Pober JS. Interferon-gamma elicits arteriosclerosis in the absence of leukocytes. Nature. 2000, 403:207–211. [DOI] [PubMed] [Google Scholar]
- 36.Stolp J, Zaitsu M, Wood KJ. Immune Tolerance and Rejection in Organ Transplantation. Methods Mol Biol. 2019. 1899:159–180. [DOI] [PubMed] [Google Scholar]
- 37.Akiyama M, Ohtsuki S, Berry GJ, Liang DH, Goronzy JJ, Weyand CM. Innate and Adaptive Immunity in Giant Cell Arteritis. Front Immunol. 2021. 11:621098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arbore G, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science. 2016. 352: aad1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uchimura T, et al. The Innate Immune Sensor NLRC3 Acts as a Rheostat that Fine-Tunes T Cell Responses in Infection and Autoimmunity. Immunity. 2018. 49: 1049–1061.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin BN, et al. T cell-intrinsic ASC critically promotes TH17-mediated experimental autoimmune encephalomyelitis. Nat Immunol. 2016. 17: 583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, MacMicking JD. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science. 2012. 336:481–485. [DOI] [PubMed] [Google Scholar]
- 42.Tan RJ, Zhou D, Liu Y. Signaling Crosstalk between Tubular Epithelial Cells and Interstitial Fibroblasts after Kidney Injury. Kidney Dis. 2016, 2:136–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D’Angelo G, Matusek T, Pizette S, Thérond PP. Endocytosis of Hedgehog through dispatched regulates long-range signaling. Dev Cell. 2015. 32:290–303. [DOI] [PubMed] [Google Scholar]
- 44.Schickel JN, Kuhny M, Baldo A, Bannock JM, Massad C, Wang H, Katz N, Oe T, Menard L, Soulas-Sprauel P, Strowig T, Flavell R, Meffre E. PTPN22 inhibition resets defective human central B cell tolerance. Sci Immunol. 2016. 1:aaf7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Asai J, Takenaka H, Kusano KF, Ii M, Luedemann C, Curry C, Eaton E, Iwakura A, Tsutsumi Y, Hamada H, Kishimoto S, Thorne T, Kishore R, Losordo DW. Topical Sonic Hedgehog Gene Therapy Accelerates Wound Healing in Diabetes by Enhancing Endothelial Progenitor Cell-Mediated Microvascular Remodeling. Circulation 2006, 113:2413–2424. [DOI] [PubMed] [Google Scholar]
- 46.Peng T, Frank DB, Kadzik RS, Morley MP, Rathi KS, Wang T, Zhou S, Cheng L, Lu MM, Morrisey EE. Hedgehog actively maintains adult lung quiescence and regulates repair and regeneration. Nature. 2015. 526:578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paulis L, Fauconnier J, Cazorla O Activation of Sonic hedgehog signaling in ventricular cardiomyocytes exerts cardioprotection against ischemia reperfusion injuries. Sci Rep. 2015. 5: 7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petrova R, Joyner AL. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development. 2014, 141:3445–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Le H, Kleinerman R, Lerman OZ, Brown D, Galiano R, Gurtner GC, Warren SM, Levine JP, Saadeh PB. Hedgehog signaling is essential for normal wound healing. Wound Repair Regen. 2008. 16:768–773. [DOI] [PubMed] [Google Scholar]
- 50.Chen Y, Li X, Tian L, Lui VC, Dallman MJ, Lamb JR, Tam PK. Inhibition of sonic hedgehog signaling reduces chronic rejection and prolongs allograft survival in a rat orthotopic small bowel transplantation model. Transplantation. 2007. 83:1351–1357. [DOI] [PubMed] [Google Scholar]
- 51.Pratap A, Panakanti R, Yang N, Lakshmi R, Modanlou KA, Eason JD, Mahato RI. Cyclopamine attenuates acute warm ischemia reperfusion injury in cholestatic rat liver: hope for marginal livers. Mol Pharm. 2011, 8:958–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang F, Hao M, Jin H, Yao Z, Lian N, Wu L, Shao J, Chen A, Zheng S. Canonical hedgehog signalling regulates hepatic stellate cell-mediated angiogenesis in liver fibrosis. Br J Pharmacol. 2017. 174:409–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bijlsma MF, Leenders PJ, Janssen BJ, Peppelenbosch MP, Ten Cate H, Spek CA. Endogenous hedgehog expression contributes to myocardial ischemia-reperfusion-induced injury. Exp Biol Med. 2008. 233:989–96. [DOI] [PubMed] [Google Scholar]
- 54.Bolaños AL1, Milla CM, Lira JC, Ramírez R, Checa M, Barrera L, García-Alvarez J, Carbajal V, Becerril C, Gaxiola M, Pardo A, Selman M. Role of Sonic Hedgehog in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012. 303:L978–90. [DOI] [PubMed] [Google Scholar]
- 55.Krause A1, Xu Y, Joh J, Hubner R, Gess A, Ilic T, Worgall S. Overexpression of sonic Hedgehog in the lung mimics the effect of lung injury and compensatory lung growth on pulmonary Sca-1 and CD34 positive cells. Mol Ther. 2010. 18:404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smelkinson MG. The Hedgehog Signaling Pathway Emerges as a Pathogenic Target. J Dev Biol. 2017. 5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klicznik MM, Morawski PA, Höllbacher B, et al. Human CD4+CD103+ cutaneous resident memory T cells are found in the circulation of healthy individuals. Sci Immunol. 2019. 4(37):eaav8995. doi: 10.1126/sciimmunol.aav8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Collins N, Jiang X, Zaid A, Macleod BL, Li J, Park CO, Haque A, Bedoui S, Heath WR, Mueller SN, Kupper TS, Gebhardt T, Carbone FR, Skin CD4+ memory T cells exhibit combined cluster-mediated retention and equilibration with the circulation, Nat Commu. 2016, 7:11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Caradu C, Guy A, James C, Reynaud A, Gadeau AP, Renault MA. Endogenous Sonic Hedgehog limits inflammation and angiogenesis in the ischaemic skeletal muscle of mice. Cardiovasc Res. 2018. 114:759–770. [DOI] [PubMed] [Google Scholar]
- 60.Otsuka A, Dreier J, Cheng PF, Nägeli M, Lehmann H, Felderer L, Frew IJ, Matsushita S, Levesque MP, Dummer R. Hedgehog pathway inhibitors promote adaptive immune responses in basal cell carcinoma. Clin Cancer Res. 2015. 21:1289–97. [DOI] [PubMed] [Google Scholar]
- 61.Rowbotham NJ, Furmanski AL, Hager-Theodorides AL, Ross SE, Drakopoulou E, Koufaris C, Outram SV, Crompton T. Repression of hedgehog signal transduction in T-lineage cells increases TCR-induced activation and proliferation. Cell Cycle. 2008. 7:904–8. [DOI] [PubMed] [Google Scholar]
- 62.Furmanski AL, Barbarulo A, Solanki A, Lau CI, Sahni H, Saldana JI, D’Acquisto F, Crompton T. The transcriptional activator Gli2 modulates T-cell receptor signalling through attenuation of AP-1 and NFκB activity. J Cell Sci. 2015. 128:2085–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Outram SV, Hager-Theodorides AL, Shah DK, Rowbotham NJ, Drakopoulou E, Ross SE, Lanske B, Dessens JT, Crompton T. Indian hedgehog (Ihh) both promotes and restricts thymocyte differentiation. Blood. 2009. 113:2217–28. [DOI] [PubMed] [Google Scholar]
- 64.Lau CI, Outram SV, Saldaña JI, Furmanski AL, Dessens JT, Crompton T. Regulation of murine normal and stress-induced erythropoiesis by Desert Hedgehog. Blood. 2012. 119:4741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goodrich LV, Jung D, Higgins KM, Scott MP. Overexpression of ptc1 inhibits induction of Shh target genes and prevents normal patterning in the neural tube. Dev Biol. 1999. 211:323–34. [DOI] [PubMed] [Google Scholar]
- 66.Mengrelis K, Lau CI, Rowell J, et al. Sonic Hedgehog Is a Determinant of γδ T-Cell Differentiation in the Thymus. Front Immunol. 2019. 10:1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gritsenko A, Yu S, Martin-Sanchez F, Diaz-Del-Olmo I, Nichols EM, Davis DM, Brough D, Lopez-Castejon G. Priming Is Dispensable for NLRP3 Inflammasome Activation in Human Monocytes In Vitro. Front Immunol. 2020. 11:565924. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and methods are available from the authors upon reasonable request. RNA-seq data sets were deposited in the Gene Expression Omnibus (GSE208349). Publicly available transciptomic data (Fig. 5A) were retrieved from the Gene Expression Omnibus analyzing inflammasome-related transcript abundance for casp1, ILb, and GSDMD and Hh signaling genes Ptch1, Gli1, and Smo for the following datasets in the Gene Expression Omnibus: GSE110932, GSE11987, GSE20065, GSE103171, GSE29316, GSE81963, and GSE40359.
RNA-seq data have been deposited into the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) with the dataset identifier GSE208349. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.