

Abstract
Bioorthogonal phosphines were introduced in the context of the Staudinger ligation over twenty years ago. Since that time, phosphine probes have been used in myriad applications to tag azide-functionalized biomolecules. The Staudinger ligation also paved the way for the development of other phosphorus-based chemistries, many of which are widely employed in biological experiments. Several reviews have highlighted early achievements in the design and application of bioorthogonal phosphines. This review summarizes more recent advances in the field. We discuss innovations in classic Staudinger-like transformations that have enabled new biological pursuits. We also highlight relative newcomers to the bioorthogonal stage, including the cyclopropenone-phosphine ligation and the phospha-Michael reaction. The review concludes with chemoselective reactions involving phosphite and phosphonite ligations. For each transformation, we describe the overall mechanism and scope. We also showcase efforts to fine-tune the reagents for specific functions. We further describe recent applications of the chemistries in biological settings. Collectively, these examples underscore the versatility and breadth of bioorthogonal phosphine reagents.
Graphical Abstract

1. INTRODUCTION
Triarylphosphines have found widespread use in organic chemistry over the last century.1 These reagents are mild nucleophiles subject to react with a variety of electrophiles. Triarylphosphines can also be used in a variety of solvents, including water, and are tolerant of many functional groups. These features enabled the successful transition of phosphine reagents from round bottom flasks to biological samples.2–5 In 2000, Bertozzi reported the first bioorthogonal reaction between triarylphosphines and organic azides—a transformation inspired by the classic Staudinger reaction. Azides, like their triarylphosphine counterparts harbor several desirable characteristics for bioorthogonal reaction—they are bioinert and can be used in aqueous settings. The remarkable chemoselectivity between azides and triarylphosphines enabled selective biomolecule tagging in cells and even live organisms.6
Since Bertozzi’s seminal report, azides and phosphines have been routinely employed as chemical tools to both probe and control biological processes. Common examples include metabolite profiling, imaging, and biomolecule uncaging. Triarylphosphines and azides have also found extensive use in biomolecule synthesis, including polypeptide construction and modification, nucleic acid templated reactions, and biomaterial fabrication. These and other developments (up to 2011) have been extensively reviewed. For more information, please consult the reviews of Bertozzi, Köhn and Breinbauer, van Delft, van Hest, Bräse and Schepers, Waldmann, Hackenberger, and Raines.7–16
The past decade has seen a steady increase in bioorthogonal phosphine development and application.17–31 Many examples feature classic phosphine reagents applied in new settings. Others showcase entirely new bioorthogonal reactions. This review will cover both the refinement of classic transformations and the development of new chemistries over the last decade (Table 1). We will discuss recent developments involving the various Staudinger reactions: the reduction reaction (section 2.1), the Staudinger ligation (section 2.2), and the traceless Staudinger ligation (section 2.3). We will also cover more recent work involving phosphine reactions with alternate electrophiles, such as cyclopropenones and related analogs (section 2.4) and α,β-unsaturated carbonyl groups (section 2.5). The final section of the review will feature bioorthogonal reactions of phosphites and phosphonites (section 2.6). These reactions feature phosphorus probes with different oxidation states that react with a similarly high degree of chemoselectivity. Each section introduces the mechanism and theory behind the bioorthogonal reaction, then showcases several examples of their application in biological settings. The sections conclude with an analysis of the current scope of the reagents and opportunities for further exploration.
Table 1.
Bioorthogonal chemistries of phosphines and related analogs.

|
2. BIOORTHOGONAL REACTIONS OF TRIARYLPHOSPHINES AND RELATED DERIVATIVES
2.1. Staudinger reduction
The Staudinger reduction was reported over 100 years ago.1 This reaction employs triarylphosphines and organic azides to generate amines. The mild nature of the transformation makes this reaction an attractive method to introduce amines in complex products (Figure 1).32–33 From its foundation in synthetic chemistry, the Staudinger reduction has evolved into a broadly useful platform in chemical biology. Organic azides are small, stable motifs that can be readily installed into many biological targets. Their reaction with phosphine nucleophiles is highly chemoselective and compatible with a range of complex environments. The selectivity of the Staudinger reduction, along with its ability to be executed in aqueous solvents, has been leveraged by chemical biologists for biomolecule modification. The reaction has also been used to selectively liberate key amino groups, and thereby activate a variety of biomolecules.34–36
Figure 1.

The Staudinger reduction of organic azides with phosphines yields amine products.
The biocompatibility of the Staudinger reduction has been leveraged for a number of pursuits. Early applications included deoxyribonucleic acid (DNA)-templated organic synthesis. DNA strands bearing aryl azides were selectively reduced by phosphines appended to complementary strands.37 The close positioning of the reagents upon hybridization drove the Staudinger reduction. The liberated amines were then subsequently modified with various probes. Other early uses of the reaction involved oligonucleotide detection via the templated activation and release of optical reporters.38–40 Amino fluorophores caged with azides were selectively reduced by triarylphosphines, releasing viable light-emitting probes.38–39
While the Staudinger reduction has long been prized for its biological breadth and high chemoselectivity, the reaction is not without limitation. A historical drawback involves its sluggish kinetic profile (k2 = ~ 10−3 M−1 s−1). Recent efforts to improve the reaction rate have resulted in new phosphine and azide reagents.41–42 Many of these tools have been leveraged for biomolecule synthesis, imaging, and probe activation in cells.36, 43–44 In this section, we will detail efforts to improve the rate and other features of the Staudinger reduction. We will also showcase applications in biological systems over the past ten years.
2.1.1. Reaction basics
The Staudinger reduction converts an organic azide to an amine (Scheme 1). The reduction begins with a nucleophilic attack by the phosphine onto the terminal nitrogen of the azide, yielding a phosphazide intermediate. Subsequent electrocyclization provides a four-membered ring. Nitrogen is extruded in an irreversible retrocycloaddition. The resulting iminophosphorane 1 is ultimately hydrolyzed to amine 2 with concomitant formation of phosphine oxide 3. Under certain conditions, the iminophosphorane can be stable to hydrolysis. This feature has been exploited to develop a new bioorthogonal transformation termed the nonhydrolysis Staudinger ligation (see section 2.2.1). Overall, the Staudinger reaction tolerates a wide variety of functional groups, including amines and carboxylic acids, and can be applied in a variety biological settings. Indeed, it is one of the few organic reactions that can be successfully employed in both test tubes and living cells.
Scheme 1.

The mechanism of the Staudinger reduction reaction.1
While highly chemoselective, the Staudinger reduction is relatively slow compared to most bioorthogonal reactions. The rate-determining step depends on the nature of the reactants. For alkyl azides, the initial phosphine addition is rate determining. For some aryl azides, the unimolecular decomposition of the phosphazide is rate-limiting.45–46 Reasonable rates have been achieved in both cases using large boluses of phosphine to drive the reaction.35 More nucleophilic phosphine probes can also accelerate the reaction, but these reagents are typically more prone to degradation by molecular oxygen or cellular enzymes. Such phosphines can also reduce disulfide bonds, which are critical structural elements in many proteins.
Several efforts have focused on phosphine tuning to boost reaction rates and yields. A balance must be struck between phosphine nucleophilicity and iminophosphorane stability. Nucleophilic triarylphosphines afford faster rates, but also stabilize the iminophosphorane intermediates, preventing amine release. Electron-rich iminophosphoranes thus require either long reaction times or an acidic or basic workup to liberate the desired amines. By contrast, less nucleophilic triarylphosphines react more sluggishly with azides, but hydrolysis of the iminophosphorane intermediates is faster.41
One strategy to promote rapid amine production relies on neighboring group participation. For example, appending an ortho-carboxamide moiety to the triarylphosphine core activates the iminophosphorane for hydrolysis (Figure 2A).41–42 The carboxamide adds into the iminophosphorane (4), promoting rapid hydrolysis and amine release. In one example, methyl 4-azidobenzoate (5) was readily reduced by triphenylphosphinecarboxamide 6b to the corresponding amine (7a, Figure 2B). In the absence of the carboxamide, no amine was observed.42 The reaction stalled at the iminophosphorane intermediate (7b). The ortho-substituted carboxamide phosphine 6b also efficiently reduced protein-bound aryl azides in cellular conditions.41
Figure 2.

Neighboring group participation increases the efficiency of the Staudinger reduction.42 (A) Phosphines bearing ortho-carbonyl or thiocarbonyl groups enable faster cyclization and hydrolysis. (B) Triphenylphosphinecarboxamide 6b reacted with aryl azide 5 to produce amine 7a. The same phosphine reacted with aryl azide 5 to form a hydrolytically stable iminophosphorane product 7b (THF/H2O 9:1 v/v).
Azide reagents have also been modified to achieve more broadly useful Staudinger reductions. Designer azides now exist that promote rapid amine (or alcohol) release via self-immolation (Figure 3). Such probes can be leveraged to selectively release (or “uncage”) biomolecules of interest. One example includes α-azido ethers (8).47 These motifs undergo Staudinger reduction reactions, releasing alcohol (9) and aldehyde (10) products (Figure 3A). Rate enhancements can be achieved via reagent proximity.40, 48–49
Figure 3.

Self-immolative Staudinger reductions. (A) α-Azido ethers (8a) react with bioorthogonal phosphines to form hemiaminal products (8b).47 Subsequent hydrolysis provides alcohols 9 and aldehydes 10. (B) Staudinger reduction products can undergo intramolecular cyclization reactions to form lactams (13).51 (C) Azide-functionalized benzyl carbamates can be reduced to anilines (14b). Subsequent elimination of CO2 can unmask amines (15).36
Other self-immolative Staudinger reductions feature alkyl azides with pendant esters (11, Figure 3B). These groups react with phosphines to release amines via intramolecular cyclization. Alkyl azides tend to react more sluggishly with phosphines than aryl azides and require excess phosphine to achieve reasonable rates.43 Upon reduction, cyclization releases an alcohol (12) while forming a five- or six-membered ring product (13).50 The rate of cyclization can be increased using more electrophilic esters.51
Amine release via Staudinger reduction can also be triggered with carbamate-linked aryl azides (14). These reagents undergo spontaneous elimination upon phosphine treatment (Figure 3C). The resulting products comprise amine (15) and alcohol (16) groups. Probes with methyl groups at the benzylic position react more rapidly than their non-methylated counterparts.36, 41 This substituent stabilizes positive charge that builds up at the benzylic position during the elimination. The Deiters lab also found that para-azido substituents afford faster rates of amine liberation than ortho-substituted variants (60-80% increase).36 These and other self-immolative probes have been leveraged for “turning on” biomolecule activity (see section 2.1.2).
2.1.2. Biological applications
The Staudinger reduction’s mild and chemoselective nature is well suited for biological use. Initial applications focused on biomolecule modification, enabling selective chemical transformations in complex environments.37, 52 Azide reduction has also been leveraged to liberate bioactive amines in cellular settings. Such chemically triggered release platforms have been heavily employed for oligonucleotide sensing and selective delivery of small molecule drugs and other therapeutics. In this section, we review these and other applications of the Staudinger reduction.
Several recent applications of the Staudinger reduction involve optical detection of nucleic acids in living cells. When exocyclic amines are replaced with azides on canonical fluorophore architectures, light emission is typically dampened due to interrupted push-pull systems. Phosphine treatment reduces the azides to amines, restoring fluorescence.38–39 In an early example, azide-caged profluorophores and triarylphosphines were conjugated to separate strands of peptide nucleic acids (PNA). Hybridization of the two strands to a template brought the reactants into proximity. The ensuing Staudinger reduction liberated the amino fluorophore and activated fluorescence, enabling facile detection of the template oligonucleotide. The template strand also served as a catalyst, promoting multiple Staudinger reductions among the probe strands. Sufficient amplification was achieved to detect 5 nM template in vitro (500 nM PNA probes, 1% DNA template, 50% formamide in 500 mM PBS, pH 7.0, 37 °C).38 Importantly, the “turn-on” readout did not require strand isolation or significant washing steps.
More recently, a doubly caged azido rhodamine probe was developed for oligonucleotide detection.53 This probe provided a four-fold higher signal-to-noise ratio than singly caged azido rhodamines.53 Reduction of both azides was required for fluorescence turn-on. The probe was affixed to PNA and used in conjunction with tris(2-carboxyethyl)phosphine (TCEP)-PNA to detect template strands. Fluorescent outputs were significantly increased compared to the singly modified azido rhodamine (30 fold–120 fold).53 The enhanced signal enabled miRNA templates to be detected at low nM concentrations. Staudinger reduction with the doubly caged probe was ultimately used to quantify miRNA levels in cancer cell lines.
The Staudinger reduction has also been used in combination with a profluorescent version of hemicyanine (HC-N3). HC is a near-infrared fluorescent reporter. Azide replacement of a key amine resulted in quenched emission. Fluorescence was restored upon Staudinger reduction. HC-N3 was developed for quantitative detection of TCEP in bacterial cultures. The infrared reporter could detect nanomolar levels of TCEP in aqueous buffered solution (PBS, pH 7.4).54 HC-N3 reduction was also used to detect and quantify E. coli in cell culture.
Fluorogenic assays have been similarly developed using α-azido ether probes. Many feature azide-linked quenchers in proximity to light-emitting scaffolds. Treatment with phosphines results in quencher release and fluorescence turn-on. A large number of such quenched Staudinger triggered α-azido ether release (Q-STAR) probes have been developed for oligonucleotide detection.40, 47–49 Early versions comprised a fluorophore and quencher attached to a single strand (17), with the quencher linked via an α-azido ether unit. Hybridization of this strand and triphenylphosphine-modified DNA (TPP-DNA, 18) to a template strand positioned the azide and phosphine units close to one another. The ensuing Staudinger reduction (Figure 4) drove quencher release and signal production. Quencher release was minimized in the absence of template, due to the slow rate of the Staudinger reaction. Hybridization was required to boost the local concentration of the reactants.
Figure 4.

General scheme for Q-STAR probe reactivity.40 The fluorophore (green circle) is quenched (gray circle) until templated reduction with TPP-DNA 18. Staudinger reduction results in quencher removal and fluorescence turn-on.
Q-STAR probes enable fast, selective detection of target sequences even when they are in low abundance. To showcase this method, the Kool lab designed a synthetic DNA strand complementary to E. coli 16S rRNA. Fluorescein or cyanine dyes were attached to the strand, along with an appropriate quencher.49 In the presence of TPP-DNA, rapid fluorescence turn-on was observed in buffer (90% activation in 32 minutes, 200 nM Q-STAR probe, 200 nM template, 600 nM TPP-DNA). The Q-STAR method was further applied to a co-culture of E. coli and S. enterica. Probes were designed to target the 16S rRNA sequences from both bacterial strains. These sequences differed by only one nucleotide. When mixed bacterial cultures were treated with the Q-STAR probes and TPP-DNA, selective and robust fluorescence was observed within 3 hours.49 Additional sequence specificity and fluorescence turn-on was obtained using Q-STAR probes comprising two quenchers and TPP-DNA strands comprising two triphenyl phosphine units.47
Fluorogenic Q-STAR chemistry has also been used to detect abasic sites, the products of base-excision repair.55 Q-STAR and TPP-DNA probes were designed to selectively bind a DNA duplex containing a defined abasic site. In addition to a fluorophore/quencher pair, the Q-STAR probe comprised a bulky pyrene or imidazophenanthrene moiety in place of a normal purine. These large bases could bind across from the abasic site, but were sterically occluded from other positions. In the presence of TPP-DNA, Staudinger reduction resulted in quencher release and fluorescence turn-on.
Q-STAR probes have proven useful for detecting a range of oligonucleotides, including miRNA. The small sizes, low abundance, and similar sequences of miRNAs often complicate their detection. To address these issues, the Kool lab combined Q-STAR probes with rolling circle amplification (RCA) for miRNA detection.56 RCA enables target sequences (in this case, miRNA) to be ligated into a circular backbone for efficient replication by DNA polymerase. The amplified DNA can then be more readily detected. Q-STAR and TPP probes used in combination with RCA detected as little as 200 pM of miRNA in solution. Only a single reaction tube and a simple heating block were required for the assay.
Q-STAR is compatible with other assays for nucleic acid detection. In one example, the Kool lab used Q-STAR templates with quenched autoligation (QUAL) probes.57 QUAL probes bind target sequences and undergo SN2-type reactions, displacing fluorescent quenchers and “lighting up” upon ligation. When QUAL and Q-STAR were combined, 10 pM of DNA was detected with single nucleotide resolution. The hybrid method was also able to detect a point mutation correlating with tetracycline-resistant Helicobacter pylori. The probes could be used directly in cell lysate, eliminating the need for cumbersome sample handling steps and improving the overall accuracy of detection.
Related fluorogenic probes have enabled single-molecule imaging of oligonucleotides and other targets in cells. In one approach, the Guo lab used bioorthogonal cleavable fluorescent oligonucleotides (BoCFO) and antibodies to iteratively detect biomolecules (Figure 5).58 Fluorophores were appended to oligonucleotide sequences via α-azido ether linkages. The fluorescent probes hybridized with their target biomolecules in cells, and images were acquired. Fluorophores were removed via Staudinger reduction with TCEP. Signal was cleared within 30 minutes without deleterious effects. Repeated labeling and probe removal enabled biomolecules to be tracked in a single sample. Ten unique transcripts were imaged in mammalian cells via iterative imaging-cleavage cycles. The approach was also used to image and quantify DNA, RNA, and protein targets in the same cell. Additional multiplexing could potentially enable hundreds of different biomolecule targets to be tracked in single cells.
Figure 5.

Multiplexed fluorescence imaging by Staudinger reduction.58 Fluorescent oligonucleotides hybridize with their intracellular targets. Following imaging, TCEP-mediated reduction liberates the fluorescent probe. Additional probes targeting the same or different sequences can then be administered.
The Staudinger reduction has been leveraged to detect collections of miRNAs via a logic-gating approach.59 Oligonucleotide logic gates require multiple inputs to achieve a defined output (Figure 6A). Such approaches are useful for reporting on arrays of oligonucleotides versus individual sequences, and using this information for downstream outcomes. For example, logic gates can program the selective release of therapeutics within a cell upon recognition of a particular set of miRNAs. Multiple inputs increase the stringency of release. The Deiters lab designed a series of nucleic acid logic gates using Staudinger reduction and DNA-templated synthesis. DNAs outfitted with azido-caged fluorophores or TPP were used. The overall strategy leveraged toe-hold-mediated strand exchange to selectively release oligonucleotides bearing TPP or azidocaged fluorophores in the presence of displacing miRNAs (Figure 6). The liberated strands hybridized, enabling proximity-driven Staudinger reduction and fluorescence turn-on. OR logic gates were achieved using two different toe-holds on the TPP-DNA strand that could be displaced by one of two distinct miRNAs. The liberated TPP-DNA probe could then hybridize the aryl azide strand for subsequent fluorogenic reaction. AND logic gates required two defined miRNA strands to be present to displace toe-holds on both phosphine and fluorophore strands. More complex AND/OR logic gates were also developed enabling distinct outputs from various combinations of six miRNA inputs.
Figure 6.
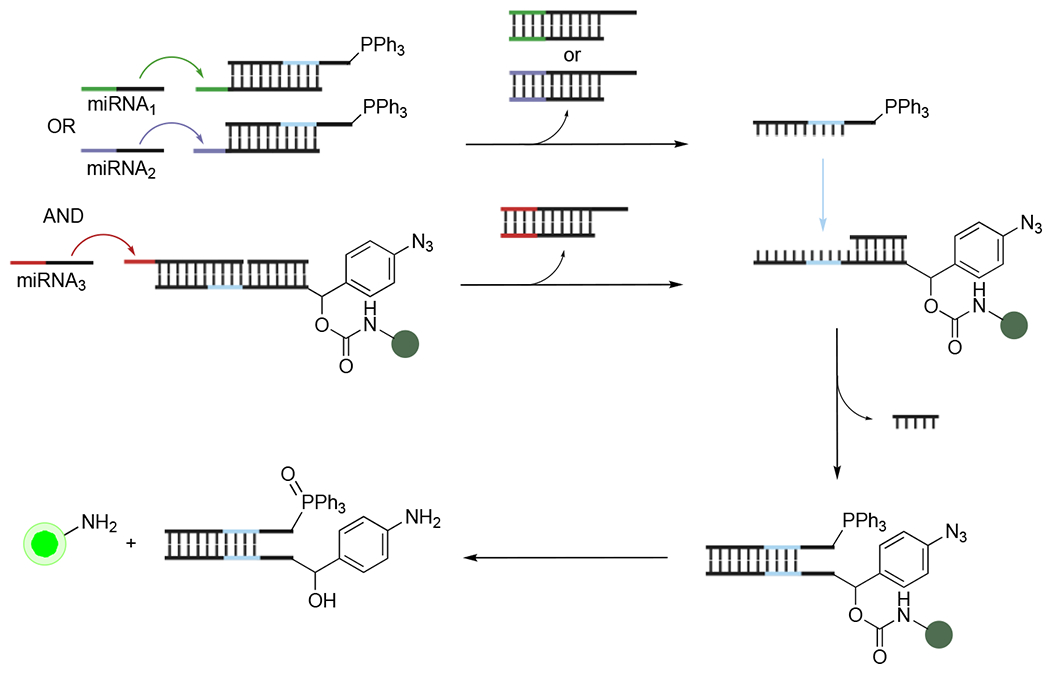
DNA logic gating via Staudinger reduction.59 Multiple miRNA inputs were required to displace sequences comprising fluorogenic azides or triarylphosphines. The liberated sequences could then hybridize, resulting in turn-on fluorescence following Staudinger reduction.
RNA-templated Staudinger reductions have been similarly employed to trigger the release of bioactive molecules. In a recent example, the Abe lab linked isopropyl-β-D-1-thiogalactopyranoside (IPTG) to a DNA strand via an α-azido ether linkage.34 This strand was designed to hybridize with a complementary RNA target in cells. In the presence of TPP-DNA (engineered to bind the same RNA strand), IPTG was released via templated Staudinger reduction and protein expression was induced. As a proof of concept, the caged IPTG strand was used to target the 23S rRNA sequence in E. coli. Upon binding the target in the presence of TPP-DNA, IPTG was released and eGFP expression was induced.
In addition to releasing fluorescent or bioactive molecules, the Staudinger reduction can directly modulate the activity of RNA targets. Several examples involve “cloaked” RNA. The cloaks comprise azido cages that block binding, folding, or other functions. These features are recovered upon cloak removal via Staudinger reduction.51, 60 To modify RNA targets with azides, the Kool lab used an azidoalkanoyl imidazole acylating reagent (19). This reagent non-specifically modifies 2′-OH groups on RNA (Figure 7A), disrupting various functions.43, 51 Phosphine treatment removes the azido cloaks. Amine (20) formation followed by intermolecular cyclization liberates the 2′-OH groups and restores RNA function. In one example, a fluorogenic aptamer (Spinach) was treated with the azido acylating agent. The cloaked RNA conjugate was less fluorescent than Spinach, due to perturbed folding of the RNA aptamer. Uncloaking with 2-(diphenylphosphino)ethylamine treatment rapidly restored Spinach fluorescence (5 mM phosphine, 1 h, 80% recovery of fluorescence).51
Figure 7.

RNA cloaking and activation with phosphines.43 (A) RNA sequences were non-specifically acylated with azide handles. Removal of these cloaking groups via Staudinger reduction restored the native RNA structures. (B) Chemical control of CRISPR-Cas9 gene editing in live human cells. Cloaked sgRNA could not engage Cas9. Upon reduction with intracellular phosphine probes, CRISPR-Cas9 editing was restored.
Staudinger-mediated RNA uncloaking has been further used to control CRISPR-Cas9 gene editing in vitro and in cells.43 CRISPR editing relies on single guide RNA (sgRNA) strands to target DNA sequences. Cloaked sgRNA strands inhibit Cas9 activity (Figure 7B). Triggered release of these probes could potentially reduce off-target effects and regulate gene editing. Toward this end, sgRNA was non-specifically cloaked with the azidoalkanoyl acylating reagent (19) (NAI-N3, 0.2 M, pH 7.0, 20 min). The resulting conjugate inhibited Cas9 activity to <1%. The cloak was removed upon treatment with tris(hydroxpropyl)phosphine (THPP) or diphenylphosphinobenzene-3-sulfonate (TPPMS), restoring CRISPR-Cas9 activity in vitro (1-5 mM phosphine, 1 h, PBS buffer pH 7.4). The strategy was also effective in cellulo. GFP-positive HeLa cells were transfected with Cas9 and cloaked sgRNA targeting GFP. Gene editing was disrupted in cells containing cloaked sgRNA. Upon removal of the azido cloaks with TPPMS (1 mM, 17 h) active sgRNA was liberated and gene editing (assayed via reduction in GFP fluorescence) was restored.
Aside from oligonucleotides, the Staudinger reduction has been used to control peptide and protein function.35, 61 Conditional control over a protein’s activity is a powerful method for studying biological systems in a gain-of-function manner. Towards this end, Deiters developed a series of azidobenzyloxycarbonyl caged lysine analogs.35 Masked Lys residues block protein function when installed at critical sites. Activity is restored upon phosphine treatment, with the Staudinger reduction removing the cage and liberating the amine. Both ortho- and para-azidobenzyloxycarbonyl residues (20–21) were spite-specifically incorporated into protein targets via genetic code expansion (Figure 8A). This technology enables non-natural amino acids to be installed in response to amber stop codons. A previously established orthogonal pyrrolysyl-tRNA synthetase (PylRS)-tRNAPyl pair was used to install the azido amino acids.35–36,62 Staudinger reduction with cell-permeable phosphines (22–23, Figure 8B) liberated the Lys residues. 2-(Diphenylphosphino)benzamide (23) was found to be particularly effective at azide removal. This phosphine was used to “uncage” eGFP in HEK293 cells. When K85 in eGFP was replaced with ortho-21, chromophore maturation and fluorescence were blocked. Fluorescence was restored upon treatment of cells with 23. Staudinger reduction was also used to regulate enzyme activity. Firefly luciferase (FLuc) was deactivated by replacing a key lysine residue with ortho-21 (Figure 8B). Enzyme function was restored upon phosphine-mediated decaging. Bioorthogonal Staudinger reduction was similarly used to control Cre recombinase and Cas9 activity, along with the localization of transcription factor SATB1. In this latter example, the azide-bearing amino acid was incorporated into the nuclear localization sequence of a SATB1-mCherry fusion (Figure 8C). Translocation into the nucleus was blocked until application of the phosphine probe.
Figure 8.

Staudinger reduction for protein activation. (A) Azidobenzyloxycarbonyl lysine derivatives were site-specifically installed into proteins using an engineered PylRS/tRNAPyl pair.35–36 The caged lysines disrupted protein function. Phosphine treatment restored native lysine residues and protein activity. (B) Caged firefly luciferase (FLuc) was activated by a triarylphosphine trigger. Upon liberation of the key lysine residue, FLuc reacts with d-luciferin to emit light.35 (C) Phosphine-controlled protein translocation in live cells.35 Azidobenzyloxycarbonyl lysine residues were installed in the nuclear localization sequence (NLS) of transcription factor SATB1, preventing nuclear translocation. Removal of the azido cage enables nuclear import of SATB1. Scale bar, 10 μm. Reprinted with permission from ref 35. Copyright 2016 Nature Publishing Group.
Uncaging experiments with the para-substituted azido lysine analog proved to be more efficient. In a comparative study, Deiters showed that phosphine-treatment liberates amines >3.5 fold faster than the analogous ortho-substituted probe.36 The para-substituted analog was then used to activate Ran GTPase-activating protein 1 (RanGAP1). RanGAP1 translocates to the nucleus upon SUMOylation of key lysine residues. Caging RanGAP1 with the azide-modified amino acid prevented SUMOylation and nuclear translocation in NIH3T3 cells. Treatment with phosphine 23 removed the cage, triggering SUMOylation and eventual nuclear import.
The Staudinger reduction, like other bioorthogonal chemistries, has been used to access homogenous samples of modified proteins. Such samples are key to deciphering the roles of defined post-translational modifications (PTMs), including ubiquitination. Many cellular processes, including the cell cycle, are regulated by ubiquitination.63 Ubiquitin-like modifications or multiple ubiquitin (Ub) molecules are added to a variety of protein targets, but the downstream functions of the modified proteins remain unknown in many cases. The complete repertoire of enzymes involved in adding and removing Ub are also not known. Synthesizing defined protein-Ub conjugates in cellulo would provide a powerful method for studying the role of ubiquitination. Toward this end, the Lang lab developed a bioorthogonal approach to site-specifically ubiquitinate protein targets.44 This method relies on selective installation of a sortase recognition motif (GGK). This sequence can be selectively modified in a second step, using engineered sortases to covalently append ubiquitin to the protein scaffold. To enable the modification “on demand”, azide-caged GGK sequences were installed into proteins of interest (Figure 9). Staudinger reduction with a phosphine trigger (22, 0.4 mM) liberated the sortase recognition motif (24). Proteins were then subject to ubiquitination in sortase-expressing cells.
Figure 9.

Site-specific ubquitination via Staudinger reduction and sortase-mediated conjugation in live cells.44 Azides were installed as part of latent GG motifs in a protein of interest (POI). Staudinger reduction revealed a GG sequence competent for transpeptidation. An engineered sortase (mut SrtA) was used to covalently attach ubiquitin to the proteins of interest.
Staudinger reduction methods have also been used to access proteins bearing methylated lysine residues.62 Lysine methylation is a post-translational modification commonly associated with histones and other proteins involved in transcription. Accessing pure variants is critical to unraveling the roles of lysine methylation in cells, but few methods exist to precisely introduce this motif. To address this limitation, a strategy was developed using non-natural amino acid, Nε-(4-azidobenzoxycarbonyl)-δ,ε-dehydrolysine. Staudinger reduction of this probe with TCEP (PBS, pH 7) yields a dehydrolysine residue.64 Dehydrolysines rapidly hydrolyze to aldehydes, which can be readily converted to methylated amines via reductive amination with NaBH3CN. This strategy was used to construct methylated variants of histone H3. An orthogonal PylRS-tRNAPyl pair was first used to install the azido precursor in the histone target. Subsequent reduction, hydrolysis, and reductive amination provided a series of dimethyl-histone H3 variants. Follow-up assays were performed with histone demethylases to confirm that the synthetic proteins were functional. The same synthetic strategy was used to prepare methylated lysine variants of the tumor suppressor protein p53 and investigate the role of methylation at residue K372.
2.1.3. Chemical and biological scope
The Staudinger reduction has developed a foothold in bioorthogonal chemistry as a reliable method to sense and activate bioactive molecules.30 The reaction is selective and mild, and compatible with a multitude of cellular functional groups. Organic azides and phosphines are mostly stable in cells and can be used for intracellular applications. Many other popular bioorthogonal reagents are susceptible to thiol degradation.65
Compared to other bioorthogonal reactions, the Staudinger reaction is fairly slow. For example, the ligations of tetrazine (Tz) and trans-cyclooctene (TCO) analogs are orders of magnitude faster (k2 > 103 M−1s−1) than phosphine-azide reduction.20 Tz-TCO reactions have thus attracted much attention for rapid uncaging experiments. In many cases, though, the release rates are only on par with the overall Staudinger reduction. The initial Tz-TCO cycloaddition is fast, but subsequent elimination steps to release bioactive molecules can occur on much slower timescales.66–67 Recent efforts to tune phosphine and azide reagents are also enabling more rapid reactions and expanding the scope of the bioorthogonal release. In some cases, cellular imaging and biomolecule activation studies can be completed within one hour, using biologically relevant concentrations.36, 49
2.2. Staudinger ligation
The Staudinger ligation remains one of the most widely recognized bioorthogonal chemistries. The reaction was inspired by the classic Staudinger reduction of organic azides and triphenylphosphine – functional groups that are quite bioinert and biocompatible (see section 2.1).6 As described in the previous section, the Staudinger reaction proceeds via an iminophosphorane intermediate. Subsequent hydrolysis yields separate amine and phosphine oxide products. To instead form a single ligated adduct, Bertozzi reasoned that the iminophosphorane could be captured with an electrophilic trap (Figure 10). Proximal ester groups were appended to the phosphine for this purpose. The resulting modified phosphine reagents reacted with azides to afford stable amide products. This version of the Staudinger reaction (termed the Staudinger ligation or Staudinger-Bertozzi ligation) has been used to selectively tag azide-functionalized biomolecules in a range of cellular contexts.
Figure 10.

The Staudinger ligation of organic azides and phosphines yields amide products.
The Staudinger ligation was initially used to tag cell surface glycans but has since been applied to numerous other pursuits. Glycosylation is a common post-translational modification, but the heterogeneity and non-templated biosynthesis of glycans complicates their study.68–70 Bioorthogonal chemistries offered a unique platform to examine glycoconjugates. Azide-modified sugars could be metabolically introduced into glycan structures and subsequently tagged with phosphine probes.71 This combination of metabolic engineering and covalent labeling has been applied to studies of sialoglycans, N-linked glycoproteins, mucin type O-linked glycoconjugates, cytosolic O-GlcNAc, and fucosylated glycoproteins, among others.6, 72–75 The Staudinger ligation of azide-modified glycans was also the first bioorthogonal reaction to be performed in living animals.73, 76–77 The remarkable biorthogonality of the reaction propelled its use in other contexts, such as tagging proteins, nucleic acids, and lipids in live cells.78–84 The Staudinger ligation has also found application in materials chemistry for microarray preparation and liposome functionalization.85, 86 These and other early applications of the Staudinger ligation have been extensively reviewed. 9–10, 12–13, 16, 87
While the Staudinger ligation helped to launch the field of bioorthogonal chemistry, some drawbacks have limited its utility. The Staudinger ligation is slower than most other bioorthogonal reactions.20 To compensate, phosphine probes are typically employed in large excess to drive reactions to completion on reasonable timescales. Still, the Staudinger ligation can be too slow to observe dynamic biological events, especially in vivo.83, 88–90 In addition to exhibiting slow reaction kinetics, the requisite phosphine reagents can be poorly soluble in aqueous solution and prone to oxidation.
The past decade has seen many efforts to improve the Staudinger ligation in terms of its rate and scope. New azide and phosphine reagents have been developed that provide new modes of reactivity and enhanced kinetic profiles (see section 2.2.1). In particular, electron-deficient aryl azides have emerged as rapid reaction partners for nucleophilic phosphines. In some cases, 50,000-fold rate enhancements over the canonical reaction have been observed. Improved phosphine probes have also been constructed that are even more compatible with biological samples. Many have enabled new applications in cells and live animals (see section 2.2.2). The following sections summarize recent achievements in Staudinger ligation probe development that have broadened the utility of this now iconic bioorthogonal transformation.
2.2.1. Reaction basics
The Staudinger ligation uses organic azides and phosphines equipped with electrophilic traps to afford amide linked products.91 The ligation begins identically to other Staudinger reactions: nucleophilic attack of the phosphine on the terminal nitrogen of the azide, forming phosphazide intermediate 26 (Scheme 2). Intramolecular rearrangement proceeds via a four-membered transition state to yield iminophosphorane 27. Nitrogen is extruded in this step. The iminophosphorane is hydrolyzed to amine products in the classic Staudinger reduction (see section 2.1.1). In the Staudinger ligation, the iminophosphorane is trapped by proximal electrophiles on the phosphine core. The resulting adducts (28) are ultimately hydrolyzed to provide the amide-linked product 29. The last step proceeds via the phosphorane intermediate shown in Scheme 2.91 For additional details, Bräse and coworkers recently summarized the mechanistic details of the Staudinger ligation.17
Scheme 2.

The thermodynamic and kinetic parameters of the Staudinger ligation are well characterized.91 The reaction is driven by nitrogen extrusion, phosphine oxidation, and amide bond formation. The ligation is first or second-order overall depending on the reactants. For aliphatic azides, the rate-determining step involves initial phosphine attack on the terminal nitrogen atom (k2 = 10−3 M−1 s−1).91 In reactions with aryl azides, iminophosphorane cyclization is rate-determining. In these cases, the iminophosphorane intermediate (27) is stabilized via resonance donation from the arene. Larger activation energy barriers must thus be overcome for subsequent cyclization.92
Several factors influence the rate of the Staudinger ligation. Faster reactions are achieved with more nucleophilic phosphines.91 Unfortunately, the fastest-reacting phosphines are also generally the most prone to oxidation under ambient conditions. The Staudinger ligation is also sensitive to solvent polarity.91 Faster reaction rates are observed in polar solvents, like water, due to stabilization of charged transition states (~3-fold rate enhancement in DMSO versus THF). The identity of the ortho-substituted ester does not significantly influence the overall rate, but it can influence the distribution of products (ligated or reduced) observed.91 The reaction rate further depends on the electronic properties of the azide and resulting iminophosphorane stability. Electron-deficient azides generate more stable iminophosphorane intermediates, leading to slower overall ligation reactions. For example, p-nitrophenylazide reacts ~6-fold slower with canonical Staudinger ligation phosphine probes compared to p-methoxyphenylazide.91
As noted earlier, slow reaction rates limited early applications of the Staudinger ligation (see section 2.2.3).73 Efforts to improve reaction speed initially focused on enhancing phosphine nucleophilicity. However, many of the resulting probes were more susceptible to oxidation or readily reduced disulfide bonds in protein structures.9,93 Alternative tuning strategies, including phosphine protecting groups, were explored to balance probe reactivity and stability.94–98 The limited aqueous solubility of phosphine probes also hindered early applications.6,83 Aqueous solubilities were increased via functionalization to peptides (e.g., FLAG peptide). However, less sterically demanding water-soluble probes were desired to broaden the scope of the Staudinger ligation.
Over the past ten years, several efforts have addressed the historical limitations of the Staudinger ligation. For example, electron-deficient aryl azides have been introduced as rapid ligation partners for triarylphosphines (k2 = 0.63–139 M−1 s−1).92, 99 The solubilities of phosphine probes have also been targeted for improvement, and mechanisms to trigger their reactivity have been explored. New water-soluble phosphine reagents enable more facile applications of the Staudinger ligation in aqueous solution and biological settings.100 Phosphines bearing photoremovable protecting groups were also recently developed to prevent oxidation and enable spatiotemporal activation in situ.101 More details on these efforts are provided in the following paragraphs.
Halogenated aryl azide reagents have been developed as Staudinger ligation reagents. Such electron-poor aryl azides (30) react with phosphine probes to provide iminophosphoranes (31, Figure 11).102 Unlike the intermediates formed in the classic Staudinger ligation, though, these electron-deficient iminophosphoranes are stable and do not further react to provide amide products.91, 45 The stability of the iminophosphorane depends on the nature of the ester trap. If phosphines with alkyl esters or no proximal electrophile are used, the reaction stops at the iminophosphorane (31, Figure 12).103–104 This version of the reaction has since been termed the “nonhydrolysis Staudinger ligation”.105 Phosphines bearing more electrophilic aryl esters still favor cyclization and subsequent hydrolysis (via classic Staudinger ligation).92
Figure 11.

Azides bearings electron-deficient arenes react with triarylphosphines to yield stable iminophosphorane adducts.92,99,103 In contrast to the canonical Staudinger ligation, these products are less prone to intramolecular cyclization.
Figure 12.

The product of the nonhydrolysis Staudinger ligation is influenced by the proximal ester on the phosphine core. Iminophosphorane intermediate 31 is stable in the presence of alkyl esters.99 With aryl esters, cyclization and ester cleavage predominate.92
Bis-halogenated aryl azides were originally used in the nonhydrolysis Staudinger ligation. 2,6-Dichloroaryl azide analogs (32) were found to moderately boost the overall Staudinger ligation rate (Figure 13, k2 = 0.63 M−1 s−1 in CD3CN) compared to conventional alkyl azides.99, 106 Analogous fluorine-substituted aryl azides (33) also resulted in accelerated reactions (kobs = 0.86 s−1 in PBS, pH 7.4). Both classes of bis-halogenated aryl azides provided iminophosphoranes. The products were stable for extended time periods in solution (bis-chlorinated aryl azide: 1 d in MeOH, bis-fluorinated aryl azide: 2 d in PBS).105, 99
Figure 13.

Electrophilic aryl azides react rapidly with triarylphosphines.92, 99, 103, 106 Second-order rate constants for reactions between triarylphosphines and a range of azides are shown. Perfluorinated aryl azides exhibit the fastest Staudinger ligation rates measured to date.
Building on the success of bis-halogenated aryl azides, perfluorinated probes were investigated for fast bioorthogonal ligation.107 Perfluoroaryl azide groups are widely used in chemical biology for modulating amino acid polarities, cycloaddition reactions, and photocrosslinking.108, 109 These perfluorinated reagents also react readily with triarylphosphines to form iminophosphorane products. The adducts are stable in some solvents (5 weeks in 10% D2O/CD3CN), and have been characterized by a variety of methods, including X-ray crystallography.103,52 The fluorine substituents greatly lower the LUMO energy of the aryl azide, resulting in rapid reactions with phosphines (Figure 13, 139 M−1 s−1 in 2% DMF/PBS).108, 92, 110–111 Perfluoroaryl azides (34) currently boast the fastest ligation speeds with triarylphosphines. While such electrophilic species run the risk of being reduced by other cellular nucleophiles, they appear to be sufficiently stable for some applications cells and rodents (see section 2.2.2).105, 103, 92
The nonhydrolysis Staudinger ligation is also compatible with other bioorthogonal reactions.112 Mutually orthogonal bioorthogonal chemistries are desirable for multi-component labeling studies. However, only a handful of such reactions can be used in tandem. Identifying combinations of compatible bioorthogonal chemistries is currently an active area of research.23, 113 The nonhydrolysis Staudinger ligation was recently applied alongside the classic Staudinger ligation. The two reactions share similar mechanisms, but exhibit vastly different rates, depending on the azide used. This kinetic preference was exploited to ligate electron-deficient azides in the presence of alkyl azides.99 The nonhydrolysis Staudinger ligation is also compatible with strain-promoted azide–alkyne cycloaddition (SPAAC), another common class of bioorthogonal transformations.110 Cyclooctyne and triarylphosphine reagents were mixed together to trigger either SPAAC or Staudinger ligation, depending on the electronics of the azide. Alkyl azides engaged preferentially in SPAAC ligations with cyclooctyne, while the electrophilic aryl azide reacted with the phosphine reagent.
In addition to tuning azide reagents, modifications to phosphine probes have resulted in enhanced Staudinger ligation capabilities. Photoactivable phosphine probes have recently been developed to provide spatiotemporal control over the covalent labeling reaction.101 Photocages are commonly used to activate reagents in situ due to their biological stability and orthogonal mode of cleavage.114–115 Phosphine probes were functionalized with 4,5-dimethoxy-2-nitrobenzyl photocleavable groups (35, Figure 14).101 The caged reagents were refractory to oxidation. Irradiation with UV light removed the cage, restoring a viable phosphine nucleophile for azide reaction. The photoactivated Staudinger ligation was used to tag azide-functionalized glycoproteins in cells and zebrafish embroys.101 Toxicity issues limited broader applications of the caged phosphines. Future work will likely address the need for more biocompatible probes that can be activated with longer (more biofriendly) wavelengths of light.116
Figure 14.

A photoactivatable phosphine reagent for Staudinger ligation.101 Phosphines were functionalized with photocages to prevent oxidation. UV irradiation rapidly removed the photocage to enable ligation with an azido-biomolecule.
Additional bioorthogonal labeling applications have been enabled by more water-soluble phosphine reagents. Included in this group are phosphines functionalized with proximal fluorosulfate groups instead of the typical ester trap (36, Figure 15).100 Fluorosulfate and fluorosulfonate moieties are polar motifs and common electrophiles in substitution reactions via sulfur (VI) fluoride exchange (SuFEx) chemistry.117 Fluorosulfates can similarly trap aza-ylide intermediates (37) formed upon azide treatment with phosphines. Subsequent hydrolysis provides aryl sulfamate ester products (38, >80% yields in 0.5–4 h). To favor fluoride departure in the reaction (versus the phenoxide-linked phosphine), an electron rich triarylphosphine must be used. Fluorosulfate-functionalized phosphines are more soluble in buffer (500 μM in PBS, pH 7.4) compared to other Staudinger ligation probes. Additionally, the sulfamate ester products can be useful proxies for phosphate linkages in biomolecule targets.
Figure 15.
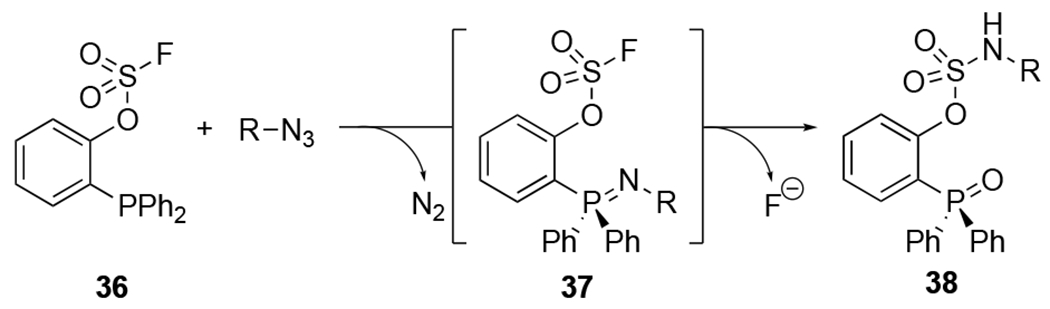
Fluorosulfate -functionalized phosphines react with azides to provide aryl sulfamate ester products.100
2.2.2. Biological applications
A suite of reagents is now available to facilitate biological applications of the Staudinger ligation. Electron-deficient aryl azides enable rapid ligations with phosphines (see section 2.2.1). The Staudinger ligation has also been combined with different imaging modalities, including bioluminescence, to track dynamic biological events in vivo. New slates of cell permeable phosphines are enabling intracellular applications of the Staudinger ligation.35–36, 43–44, 118 Some of these phosphines have also been used for biomolecule release via Staudinger reduction inside of living cells (see section 2.1.2). In this section, we will discuss recent applications in biological settings.
Similar to early applications of the Staudinger ligation, many azide and phosphine probes have been used to track biomolecules via metabolic engineering and covalent reaction.119 The Staudinger ligation is essentially background free, and is a reliable method to detect azide-modified biomolecules. Azide groups can be biosynthetically incorporated into a range of biomolecules, including glycans, proteins, lipids, and nucleic acids.78–84 The Staudinger ligation can then be used to attach a variety tags to the azide-modified biomolecules for enrichment, imaging, or other applications. Biotin-modified phosphines have been commonly used to image and profile azide-labeled proteins.120–124 Mammalian tissue- and cell-specific glycosylation patterns have been similarly characterized using azide-labeled metabolic precursors and the Staudinger ligation.125–126 Phosphine-biotin probes have also enabled probing of crosstalk between glycan metabolic pathways.127 In another study, the Staudinger ligation was used to characterize the role of O-GlcNAc in blocking ubiquitination of nascent polypeptide chains.128–129
Metabolic engineering and the Staudinger ligation were recently used to examined microbial glycosylation.130 Microbes harbor unique glycans that are absent in host glycomes. Such structures, and the enzymes that produce them, are thus attractive therapeutic targets.130 The Dube lab capitalized on a metabolic labeling strategy to probe and disrupt the pathogenesis of several microbes.131–133 Microbial biosynthetic pathways can process some azide-functionalized sugars and append them to underlying proteins. The modified glycoconjugates can then be detected via Staudinger ligation. Metabolic labeling followed by covalent detection was recently used to examine small molecule inhibitors of bacterial glycosylation.133 In this strategy, metabolic inhibitors were administered to various microbes along with azidosugars. Disruptions in glycan biosynthesis were read out via Staudinger ligation with triarylphosphine probes. Different degrees of inhibition were observed depending on metabolic inhibitor and bacterial species.
In another study, the metabolic incorporation of azidosugars was used to identify glycosyltansferases in Heliobacter pylori (H. pylori), a common gastric pathogen.134 A third strategy used the Staudinger ligation to append immune stimulants to the exterior of H. pylori (Figure 16).131 In this approach, bacterial cell surfaces were first decorated with azides via metabolism of an azide-containing GlcNAc sugar (39). Subsequent incubation with an adjuvant-functionalized phosphine (40) targeted the pathogen for immune detection and killing in vitro.
Figure 16.

Immune system activation via metabolic labeling and Staudinger ligation.131 An azido GlcNAc analog (39) was metabolized by H. pylori. Subsequent ligation with phosphines bearing 2,4-dinitrophenol (40) triggered an immune reaction. 2,4-Dinitrophenol (DNP) recruited host antibodies to the H. pylori surface to promote microbial killing.
The Staudinger ligation was among the first bioorthogonal chemistries to be applied in rodent models.73, 134 The reaction continues to be useful in this environment and has enabled dynamic biological processes to be visualized in vivo.135 Due to its slow rate, the Staudinger ligation must be used in concert with ultrasensitive or amplifiable imaging modalities. In a recent example, positron-emission tomography (PET) was used in combination with Staudinger ligation to monitor cancer growth in vivo.135 PET with radioactive probes is among the most sensitive imaging platforms. Brindle and coworkers used this modality to detect sialic acid residues in living mice.135 Sialylation is upregulated in certain forms of cancer.136–138 Sialic acid is therefore an attractive biomarker of disease progression. To target these residues, Brindle used an established approach to outfit sialic acid residues with chemical reporters in mice.73, 76 Briefly, an azide-modified sialic acid precursor (ManNaz) was administered to cancer-bearing mice. The unnatural sugar was metabolized and displayed on cells in vivo. The azido sugars were then then ligated with phosphine-biotin probes. Subsequent labeling with streptavidin-PET probes provided a visualize readout on tumor growth.135 Although this work represents the first demonstration of the Staudinger ligation for imaging in living mice, several steps were required to provide adequate signal-to noise levels.
The Staudinger ligation and bioluminescence have also been paired to visualize biological events in living systems. Bioluminescence imaging (BLI) uses luciferase enzymes and luciferin small molecules to produce light.139–141 BLI is a sensitive imaging technique, as it is “background free”. Mammals do not naturally produce large numbers of photons. Bertozzi and coworkers used bioluminescence in conjunction with the Staudinger ligation to visualize azido-sialosides on mammalian cells.142 Due to the intrinsic sensitivity of BLI, robust light emission was observed even with low reagent concentrations (~5 nM, 5 min). The Staudinger ligation and BLI have also been used to track more dynamic biological features in vivo.92, 143–144 In one example, Goun and coworkers applied bioluminescence and bioorthogonal chemistry to image mitochondrial membrane potential (ΔΨm).143 ΔΨm reports on mitochondrial function and cell health, and several diseases correlate with abnormal ΔΨm.values.145 To visualize ΔΨm, phosphine-caged luciferin and azide substrates were directed to the mitochondria via triphenylphosphonium groups.146 Upon localization and subsequent Staudinger ligation, functional luciferin was released. The luciferin was then processed by luciferase enzymes to produce photons. Light emission was dependent on metabolite trafficking, and ΔΨm was inferred from the photon outputs. Dynamic ΔΨm changes were observed in response to drug treatment and tumor growth in vivo.
The Staudinger ligation has been used in combination with genetic code expansion to target G protein-coupled receptors (GPCRs).147–148 GPCRs are a superfamily of transmembrane proteins that regulate a variety of biological processes.149 Over 25% of clinically used drugs target GPCRs.150 Despite their biological and pharmacological importance, a precise understanding of GPCR conformational dynamics and signal transduction is lacking in many cases.149, 151 The Staudinger ligation has been used to gain insight into GPCR activity. Non-natural amino acids bearing aryl azides were installed into the protein targets. Subsequent Staudinger ligation enabled the attachment of various biophysical tags.19, 152–159 In one example, azido amino acids (41) were incorporated into rhodopsin and CCR5 using an established orthogonal tyrosyl-tRNA synthetase (TyrRS)-tRNA pair (Figure 17).160–164 The GPCRs were then labeled with fluorophore-phosphine conjugates (42). Non-natural amino acid installation and Staudinger ligation efficiencies were found to be highly context-dependent, suggesting that a range of locations should be explored for targeting related receptors.161 The studies further revealed limitations of the Staudinger ligation for GPCR labeling. In some cases, excess phosphine non-covalently associated with cellular membranes and was difficult to remove without damaging the receptors.163 Incomplete ligations were also observed due to competitive Staudinger reduction pathways (only 30% ligation observed at 12 h, in some contexts).160 Cyclooctynes and other azide-targeting reagents have been used to circumvent some of these issues.147–148
Figure 17.

GPCR labeling via genetic code expansion and Staudinger ligation.161–164 Azidophenylalanine (41) was site-specifically installed in response to an amber stop codon (TAG) by an engineered aminoacyl-tRNA synthetase (AARS)/tRNA pair. Azide-modified GPCRs were subsequently tagged using functionalized phosphine probes (42).
In addition to GPCRs, the Staudinger ligation has been used to outfit a wide range of proteins and peptides with detectable probes and drugs165–170 Antibodies, in particular, have been the targets of chemoselective modification. Outfitting these reagents with toxic drugs can greatly increase their therapeutic potential171–172 In one example, antibodies labeled with azides were localized to tumor surfaces and ultimately conjugated with radiolabeled phosphines via Staudinger ligation.173–174 The antibodies targeted cell surface receptors overexpressed on neck cancer cells. Following antibody injection and localization to tumor sites, phosphine probes bearing radiolabeled agents were injected to tag the cancer cells for imaging and therapy. Poor labeling efficiencies were observed in vivo, likely due to the slow kinetics of the Staudinger ligation.174
Nucleic acids have been successfully outfitted with detectable probes via Staudinger ligation.175–177 Azide-functionalized oligonucleotides can be readily generated via enzymatic or chemical synthesis. The Staudinger ligation can then be used to attach a variety of detectable probes. Azide reporters have been introduced into a variety of cellular nucleic acid polymers, including DNA and RNA.175, 178–181 In some cases, azide-labeled nucleotide triphosphates were metabolically incorporated into the target. For example, azido-uridine triphosphate analogs were used to label RNA via T7 RNA polymerase activity.175, 177–178, 182 Labeled RNA products were then visualized via covalent attachment of fluorophore-phosphine conjugates. In another approach, Kurinomaru and coworkers used the Staudinger ligation to immobilize genomic DNA in detection assays for 5-hydroxymethylcytosine.180 Guanosine residues were chemically modified with 4-azidophenylglyoxyl. Subsequent Staudinger ligation with phosphine-biotin provided a handle for immobilization.
In addition to biological pursuits, the Staudinger ligation has enabled advances in synthetic chemistry, materials science, and supramolecular chemistry. The high chemoselectivity and mild reaction conditions enable covalent adducts to be forged in a variety of contexts. In materials chemistry, the Staudinger ligation has been routinely employed to attach molecules to solid supports for biosensor development and biomolecule delivery systems.183–184 In one example, photoswitchable azobenzenes were ligated to azide-functionalized surfaces via Staudinger ligation.185–186 Azobenzene is a common motif used to modulate photoresponsive materials, proteins, and therapeutics with external light.187–188 In addition to azobenzene groups, oligosaccharides and proteins have been attached to surfaces using Staudinger ligation.183, 189 The reaction has also been used to functionalize nanoparticles with peptides and carbohydrates.25, 190 The resulting conjugates were used for targeted drug delivery. Gold nanoparticles have also been functionalized with oligopeptide, fluorogenic dyes, and targeting elements via phosphine-azide chemistry.184, 191–192 One recent example combined the Staudinger ligation with other bioorthogonal reactions to attach multiple probes to the nanoparticles surface.104 Organic azides and phosphine reagents have further been used to craft oligosaccharide drug delivery vectors and other polymers. 193–196
The development of the nonhydrolysis Staudinger ligation has enabled new biological experiments. Electrophilic aryl azides have been ligated with phosphines in a range of settings, including live animals. For successful application, the reagents and iminophosphorane products must be stable in biological media. Bis-chlorinated aryl azide analogs were found to be stable in eukaryotic cell lysate (~80% intact after 24 h).99 Perfluorinated aryl azides were also found to tolerate aqueous conditions (no degradation observed after 60 min in 5% DMSO/H2O). The perfluorinated reagents were successfully applied in mammalian cells and living mice.92, 103 Importantly, the iminophosphorane adducts for both classes of aryl azides were found to be stable in a range of biological environments.99
Glycoconjugates, proteins, and nucleic acid targets have been probed using the nonhydrolysis Staudinger ligation. In an early report from Yan and coworkers, electron-deficient aryl azides were appended to monosaccharide precursors (GalNAc and ManNAc).103 The unnatural sugars were metabolized and displayed on mammalian cell surfaces. Subsequent incubation with a fluorescently labeled phosphine probe rapidly illuminated the azido metabolites. Fluorescent products were observed in as little as 1 minute. It should be noted that aryl azides are larger than their alkyl counterparts and may not be as generally tolerated in metabolic pathways. Electron deficient aryl azide reagents have further been used in conjunction with self-labeling tags, including HaloTag, for protein targeting99, 197 Nucleic acids are also amenable to visualization with the nonhydrolysis Staudinger reaction.110 Model DNA and RNA strands were synthesized with a tetrafluorinated azide. Subsequent labeling with a triarylphosphine dye was observed by gel electrophoresis.
The nonhydrolysis Staudinger ligation has recently been applied to visualize dynamic biological events in live animals.92, 144 The Goun lab used the bioorthogonal reaction in conjunction with bioluminescence imaging (BLI) to visualize glucose flux in vivo (Figure 18A).92 Aberrant glucose metabolism is a hallmark for several diseases, notably cancer and autoimmune disorders.198–199 To image glucose flux, phosphine-caged luciferin probes (43) were delivered into cells. Glucose analogs bearing perfluorinated azide groups (44) were then used to trigger luciferin release via nonhydrolysis Staudinger ligation. Light emission correlated with glucose import via the GLUT1 transporter (Figure 18B). Glucose transport was further visualized in tumor-bearing mice (Figure 18C).
Figure 18.

Visualizing glucose transport via Staudinger ligation and bioluminescence imaging.92 (A) A phosphine-caged luciferin 43 reacts with perfluorinated azido glucose analog 44 to release d-luciferin. d-Luciferin (45) is processed by firefly luciferase (FLuc) to emit light, providing a real-time readout of glucose transport. (B) Glucose transport was tracked over time in luciferase-expressing transgenic mice. The bioluminescent signal was diminished in the presence of the natural substrate d-glucose, suggesting that the azido analog uses the same transporter (GLUT1). (C) Bioluminescence imaging of glucose uptake in FLuc-expressing 4T1 tumor cells with (left) or without (right) GLUT1. Removal of the transporter decreases bioluminescent signal by 38%. Reprinted with permission from ref 92. Copyright 2019 Nature Publishing Group.
Similar to the canonical Staudinger ligation, the nonhydrolysis Staudinger ligation of perfluorinated aryl azides and phosphines has found utility in materials chemistry.104, 200–202 The reaction has been used to rapidly functionalize nanoparticles and prepare glycopolymers.104, 201 Glycopolymers are useful for understanding glycan-biomolecule interactions, but methods to access these scaffolds without heavy metal catalysts are limited. Perfluorinated aryl azide polymers were used to provide maltoheptaose- and mannose-functionalized polymers via nonhydrolysis Staudinger ligation. Gold nanoparticles have also been outfitted with various payloads and targeting groups via nonhydrolysis Staudinger ligation. For example, phosphine-functionalized nanoparticles were conjugated with perfluorinated aryl azide rhodamine dyes.104 The resulting conjugates were used to image human fibroblast cell surfaces. In addition to derivatizing particles, the nonhydrolysis Staudinger ligation has been leveraged to synthesize polyphosphazene polymers.200 Polyphosphazenes are attractive vaccine delivery agents and tissue engineering platforms.203–204 In one study, polyphosphazenes were synthesized using bis-functionalized perfluoroazide and phosphine reagents.200 The polymerization was complete within 30 minutes (20 mM azide, 20 mM phosphine, MeCN, 23 °C). The resulting polyphosphazenes exhibited molecular weights larger >59,000 Da with narrow dispersity (Ð = 1.1–1.2).
As the above examples highlight, the Staudinger ligation continues to be a useful reaction in the bioorthogonal toolkit. Azide and phosphine reagents were the first truly abiotic probes to transition from manifold to mouse (see section 2.2.1). The ligation also inspired several new classes of bioorthogonal reactions, including a “traceless” variant (see section 2.3) and other chemistries described in the latter sections. Historically, the Staudinger ligation has been primarily used for probing biomolecules in extracellular environments in vitro and in vivo. Newly developed probes are enabling more applications inside cells, though, and broadening the scope of the reaction.
2.2.3. Chemical and biological scope
The Staudinger ligation was one of the first transformations to be broadly used for bioorthogonal labeling. Since its introduction, several new chemistries have appeared that span a spectrum of reactivities.205–206 Many of these transformations have drawn inspiration from the Staudinger ligation mechanism, which features the formation of an unstable intermediate and subsequent intramolecular capture. Bioorthogonal ligations that exploit a similar strategy include the traceless Staudinger ligation (see section 2.3), the cyclopropenone-phosphine ligation (see section 2.4), the Pictet-Spengler ligation, and the boron-stabilized oxime and hydrazone ligations.207–213
Traditionally, the Staudinger ligation ranks among the slower bioorthogonal reactions (Figure 19, k2 = 10−3 M−1 s−1).9 Efforts to increase the speed of the reaction with electron-deficient aryl azides have been successful. Perfluorinated reporters currently undergo the fastest Staudinger ligations (k2 = 139 M−1 s−1).92 The rate of this non-hydrolysis variant compares favorably with other common bioorthogonal reactions of organic azides, including SPAAC ligations (k2 = ~1 M−1 s−1) and CuI-catalyzed azide–alkyne cycloaddition reactions (CuAAC) (k2 = 102 M−1 s−1).20 However, the fastest Staudinger ligations are still orders of magnitude slower than the fastest inverse-electron demand Diels–Alder (IEDDA) cycloadditions, where second-order rate constants can exceed 106 M−1 s−1.19, 214–215 Fast ligations are especially useful for in vivo work, where high reagent concentrations often cannot be used to drive covalent reactions.21, 154
Figure 19.

New variants of the Staudinger ligation exhibit faster reaction kinetics.20 The rates of the nonhydrolysis Staudinger ligations, in particular, are on par with several common bioorthogonal reactions. Adapted with permission from ref 20. Copyright 2014 American Chemical Society.
The Staudinger ligation is generally best applied in situations that require minimal off-target reactivity and mild conditions.22 Azide and phosphine probes are remarkably chemoselective and afford little background signal.9, 216–217 Improved ligation rates are now enabling more facile imaging experiments on live cell surfaces.103 Staudinger ligation applications were also traditionally limited to use on cellular exteriors, but suitable phosphine probes for intracellular labeling have been identified and such applications are becoming more routine (see section 2.1.2).
2.3. Traceless Staudinger ligation
The Staudinger ligation set the stage for the development of numerous other chemoselective phosphine-based transformations. Included in this group are reactions with phosphines bearing cleavable electrophilic traps (Figure 20). Such phosphine reagents enable the formation of amide bonds upon reaction with azides via acyl group transfer and phosphine expulsion. This “traceless” variant was developed contemporaneously by Raines and Bertozzi and reported in 2000.207–208 The reaction is chemoselective, versatile, and occurs under mild conditions.14–15, 25–26, 96, 218 It is also one of the few ligations that provides a native biological linkage.
Figure 20.

The “traceless” Staudinger ligation links two biomolecules via a native amide bond.
The traceless Staudinger ligation has been routinely applied to polypeptide synthesis and modification.15, 218 Early work leveraged the reaction for natural and unnatural oligopeptide synthesis, macrocyclization, and glycopeptide preparation.95, 98, 142, 219–221 The reaction has also been used for whole protein synthesis in buffered aqueous solution.96, 222–224 For protein and peptide construction, the traceless Staudinger ligation has typically been achieved via an intramolecular acyl group transfer. Intermolecular variants are possible, but less common.225–226 The traceless Staudinger ligation has also been used to attach azido-protein or -peptide cargo to solid supports for biomaterial applications.223, 227
While the traceless Staudinger ligation can, in theory, be used to forge amide linkages at any point along a peptide backbone, the reactions are less efficient when bulky amino acids flank the ligation site.228–229 Reactions with sterically occluded reagents often result in formation of amine products via Staudinger reduction.226 Early versions of the traceless ligation were more limited in scope, and the lack of generality precluded access to some polypeptides.
More recent advances have addressed some of the historic limitations of the traceless Staudinger ligation.26, 230 Reagents were identified to minimize off-target pathways and afford higher yields of amide products, even at sterically occluded junctures.231 Methods to trigger phosphine reactivity have also been identified, enabling a broader scope of applications.232–233 This section will detail the recent development of traceless Staudinger ligation reagents and their applications in biology.
2.3.1. Reaction basics
The mechanism of the traceless Staudinger ligation is similar to other Staudinger-type transformations (see sections 2.1.1 and 2.2.1).207–208, 222, 226 The reaction begins with nucleophilic attack of a phosphine on an organic azide to form phosphazide 46 (Scheme 3). This intermediate undergoes a retro-cycloaddition reaction to extrude nitrogen and form an iminophosphorane (47). Iminophosphoranes can participate in intra- and intermolecular acyl transfer reactions.234–239 In the case of the traceless Staudinger ligation, the iminophosphorane attacks the proximal acyl group to form a tetrahedral intermediate (48). This intermediate subsequently collapses to afford an amidophosphonium salt (49). Hydrolysis ultimately yields a nascent amide linkage between the azide and the acyl group. The oxidized phosphine and cleaved linker (50) are released as byproducts. Because the phosphine component is not retained in the final product, amide formation is “traceless”. Successful phosphine release requires the presence of scissile linkers, typically ester or thioester moieties (Scheme 3, X = O, S, respectively). Thioester groups undergo traceless Staudinger ligation faster and in higher yield than the ester analogs.226 Therefore, phosphinothioesters are often employed in the reaction.
Scheme 3.
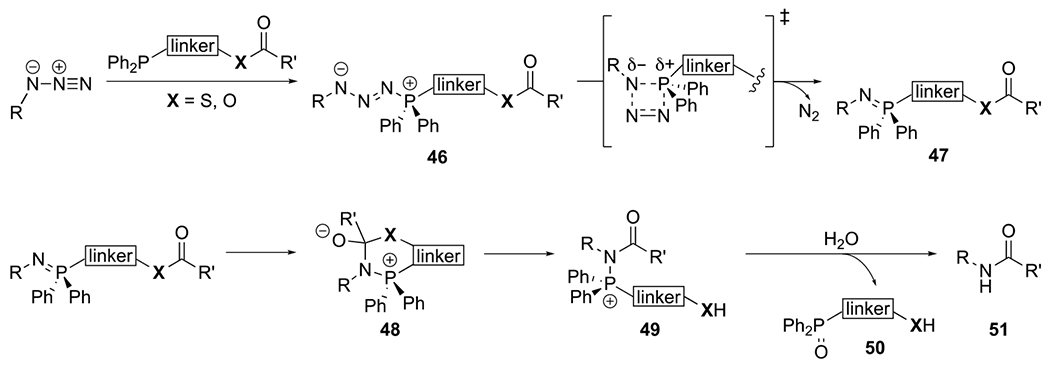
The mechanism of the traceless Staudinger ligation.26, 226, 231
The thermodynamic and kinetic parameters of the traceless Staudinger ligation are well established. The reaction is thermodynamically favored due to dinitrogen release, amide bond formation, and phosphine oxidation. The traceless Staudinger ligation typically proceeds with second-order rate constants of ~10−3 M−1 s−1.226 The rate-determining step is the initial phosphine attack on the azide, similar to the majority of Staudinger reduction and ligation reactions (see sections 2.1.1 and 2.2.1).
Several factors influence the efficiency of the reaction. Faster ligations occur in more polar solvents.226 Initial reports featured reactions in DMF and THF, but conditions for ligations in aqueous environments were later identified.222, 224 Ligation yields are also dependent on the pH of the solution.222, 224 The traceless reaction is most efficient at pH 8.0. More acidic or basic conditions result in poorer yields due to iminophosphorane protonation or thioester hydrolysis, respectively.
Off-target pathways can compete with amide formation in the traceless Staudinger ligation.26, 226, 231 Following initial formation of phosphazide intermediate 53 (Figure 21, pathway A), the desired pathway proceeds via iminophosphorane (54) formation, nitrogen expulsion, and intramolecular acyl transfer. Intramolecular acyl transfer can occur prior to nitrogen expulsion, though, yielding linear acyl triazine 53a (Figure 21, pathway B). Hydrolysis of this intermediate ultimately provides primary amide 53b and deacylated phosphine oxide 53c. A second off-target pathway diverts iminophosphorane 54. Protonation and hydrolysis of this intermediate yields amine 54a and acylated phosphine oxide 54b (Figure 21, pathway C). In a third competitive process, tetrahedral intermediate 55 can rearrange via an aza-Wittig reaction to yield oxazaphosphatane 55a (Figure 21, pathway D). Subsequent hydrolysis produces phosphonamide 55b.
Figure 21.

Side reactions can compete with formation of traceless Staudinger ligation adducts.26, 226, 231
Depending on the reactants, these off-target pathways can predominate and limit the utility of the traceless ligation. Productive amide bond formation is generally most favored with less sterically hindered reactants. In the case of polypeptide synthesis, ligations with amino acids lacking α-substitution, namely glycine residues, are most efficient (95% yield for Gly-Gly, Table 2). With bulkier reagents, the traceless ligation yields drop (e.g., 27% yield for Gly-Ala).221, 226, 240–241 The acyl transfer reaction becomes sluggish, and iminophosphorane hydrolysis outcompetes formation of the desired amide (Figure 21, pathway C). To avoid this side reaction in oligopeptide synthesis, the traceless Staudinger ligation is typically employed at sterically accessible junctures comprising glycine residues.
Table 2.
Dipeptide synthesis using the traceless Staudinger ligation.224, 226, 228–229, 241 Adapted with permission from ref 229. Copyright 2020 American Chemical Society.

|
Phosphine reagents have also been extensively modified to promote amide bond formation. More nucleophilic phosphines comprising alkyl substituents (instead of the traditional aryl groups) were found to favor amide products.226 The alkyl groups were hypothesized to stabilize the iminophosphorane 54 and limit the competing hydrolysis step.208 The electron-rich phosphines also boosted the overall reaction rate. However, the probes were more prone to oxidation and not further pursued.208 An alternative strategy explored phosphines comprising electron-donating arenes. These substituents were used to minimize the competitive aza-Wittig reaction (Figure 21, pathway D).228 Phosphines with p-methoxylphenyl or p-diethylamino substituents were found to disfavor oxazaphosphatane formation (Figure 21, pathway D), providing improved ligation yields in some cases (80% yield for Phe-Ala coupling).224, 226 In other cases, reaction yields decreased (59% yield for Ala-Gly coupling) likely due to the increased basicity of iminophosphorane 54, leading to faster protonation and Staudinger reduction (Figure 21, pathway C).224, 228
The acyl donor and linker components of the phosphine probes also influence product composition. If acyl transfer to iminophosphorane 54 is sluggish, off-target Staudinger reduction can compete for product formation (Figure 21, pathway C). Productive amide bond formation (pathway A) is thus favored with more electrophilic acyl groups and short, flexible linkers. Both esters and thioesters are sufficiently electrophilic to promote on-target reactivity. Phosphines with thioester groups were found to react faster and provide higher yields, compared to ester analogs.15, 226 Thus, phosphines bearing short alkyl thioester acyl donors (Figure 21, phosphinothioester 52) are among the most popular reagents for traceless Staudinger ligation.
A series of phosphinoesters was also recently developed to promote amide bond formation by limiting off-target pathways.229, 231, 242–243 These esters (57a–d, Figure 22) enabled efficient ligations even at sterically congested sites. The reagents were used to synthesize the natural product yaku’amide B. This non-ribosomal peptide features several non-proteinogenic modifications.231 The β,β-dialkylated α,β-dehydro amino acid linkages were forged via traceless Staudinger ligation. Initial coupling attempts with azide-bearing amino acid 58 and conventional phosphine probes (57a, Figure 22) were low yielding. Instead of the desired ligation product 59, the undesired primary amide 60 was formed (pathway B, Figure 21). To disfavor this pathway and promote iminophosphorane formation (pathway A), the phosphine was functionalized with electron-withdrawing groups (57b). The electron-deficient arenes boosted ligation yields (41%, Figure 22), but off-target primary amide products were still observed. To further minimize premature acyl transfer to phosphazide intermediate 61, phosphines with less electrophilic esters were used (57c). However, overall ligation yields decreased (22%, Figure 22). The “sweet spot” of reactivity was found when the electron-donating and withdrawing substituents were merged in a single scaffold. Phosphine 58d provided excellent ligation yields (76%, Figure 22). Traceless Staudinger ligations with these reagent afforded the three native β,β-dialkylated α,β-dehydro linkages in yaku’amide B. Alkene stereochemistry was also largely retained with either stereoisomer. Other peptide natural products have been accessed using similar phosphine reagents, suggesting that they are versatile probes.242–243
Figure 22.

The traceless Staudinger ligation was used to construct amide bonds in the total synthesis of natural product yaku’amide B.231 Triarylphosphine phosphinoester 57d was developed to efficiently access the required amide linkage based on mechanistic guidance.
Intermolecular versions of the traceless Staudinger ligation have been developed over the past decade. The canonical traceless ligation features an intramolecular acyl transfer between azide and phosphine moieties. The intermolecular version uses phosphine reagents to couple azides to carboxylic acids. The azide and phosphine undergo an initial Staudinger reaction to form an iminophosphorane (Figure 23). This intermediate then reacts with the carboxylic acid reagent to yield an esterified aza-ylide (61). Subsequent intramolecular rearrangement yields the desired amide and expels the phosphine byproduct. Both triphenylphosphine (Figure 23A) and chlorophosphite reagents (Figure 23B) have been used for intermolecular traceless Staudinger ligation.244–245 Phosphite-azide reactions are covered in more detail in section 2.6.
Figure 23.

Amide bond coupling with triphenyl phosphine and chlorophosphite reagents.244 (A) Triphenylphosphines activate carboxylates and azides to yield phosphonium carboxylate intermediates. 1,3-Acyl migration provides the amide linkage.245 (B) Chlorophosphites activate both carboxylates and azides to yield iminophosphorane ester intermediates. Acyl transfer yields the amide linkage.
Some traceless Staudinger ligation variants can be used to access diazo groups, versatile building blocks in organic synthesis and chemical biology.246–248 Diazo groups can be challenging to install in richly functionalized molecules.246, 249 Thus, the traceless Staudinger ligation is an attractive approach given its mild and biocompatible features. Similar to the canonical Staudinger reaction, this transformation begins with nucleophilic attack of phosphine 62 onto the azide to yield phosphazide 63 (Figure 24). Intermediate 63 can proceed via two possible pathways to form either an amide or diazo product. To encourage diazo formation, phosphine reagents were outfitted with electrophilic esters. These electrophiles rapidly transfer acyl groups to phosphazide 63 to form acyl triazine 64. Subsequent hydrolysis yields separate products comprising diazo 65 and a primary amide. Phosphines functionalized with 4-nitrophenol groups were found to be particularly effective for diazo formation in physiological buffer (70–92% yield).247 Furthermore, the reaction was shown to be compatible with protein side chains, suggesting its potential biological utility.
Figure 24.

The mechanism of organic azide conversion to diazo groups using phosphine 62.247
Phosphines used in traceless Staudinger ligations are susceptible to oxidation in aerobic environments.9, 15, 101 To mitigate this issue, borane protection is frequently employed. Borane adducts prevent undesirable reactions between the phosphine core and molecular oxygen. The complexes can be liberated on demand, releasing active phosphine for desired transformations.95, 220 Recently, a complimentary protecting group strategy emerged that masks phosphines with a photocleavable group (see section 2.2.1).101 Upon photocage removal via UV irradiation, phosphines are liberated and available for ligation. In one example, photolabile anthracenylmethyl groups were attached to diphenylphosphonium thioester analogs (66).232 No reaction between the phosphine reagent and azido probes was observed in the absence of light. UV irradiation triggered photocage removal and subsequent traceless Staudinger ligation (Figure 25A, λ= 360–400 nm). While the ligation could be controlled with light, lengthy irradiation times were necessary (Φ= 0.07, 500 W Hg lamp, 1.5 h). More biocompatible photocages were developed using anthracenyl groups with extended Π-conjugation. These scaffolds absorb more visible wavelengths of light.233 Photocages comprising 3,3-dimethylbutynyl groups provided the largest shift in decaging wavelength (λ > 420 nm). Phosphines outfitted with these modified photocages (67) could be activated with more biofriendly blue light (Figure 25B, λ > 420 nm, Φ= 0.025). The released phosphines reacted with organic azides via traceless Staudinger ligation (yields 31–41%). While the wavelength of activation was successfully red-shifted, phosphine activation still required intense irradiation (500 W Hg lamp, 2 h).
Figure 25.

Photoactivatable traceless Staudinger ligations with anthracene-caged phosphinothioesters. (A) A phosphinothioester was released upon UV illumination of phosphonium 66 (500 W Hg lamp).232 (B) Photocaged phosphine 67 was activated upon exposure to blue visible light (500 W Hg lamp).233 The released phosphine probes were used in subsequent traceless Staudinger ligations.
Although the traceless Staudinger ligation was introduced over two decades ago, it remains a useful reaction to forge native amide bonds. The traceless Staudinger ligation gained prominence due to its high chemoselectivity and versatility. However, initial reaction variants had some drawbacks. Ligation yields were poor with sterically congested reactants and many phosphines were not generally applicable. The last decade has provided an expanded set of probes for traceless Staudinger ligation, many of which provide reliable yields for a broad range of substrates. New variants of the reaction are also enabling access to other desirable functional groups. Finally, methods to control the ligation chemistry have been afforded by phoactivatable phosphine reagents.
2.3.2. Biological applications
The traceless Staudinger ligation has been used for a number of biological applications. Among the most popular include modification of azido peptides and proteins. As methods to outfit proteins with non-natural amino acids have improved, the traceless Staudinger ligation has found application in preparing designer proteins. A notable feature is that the reaction produces a native amide linkage. In addition to peptides and proteins, nucleic acids have been targeted for functionalization. Other applications have leveraged the reaction to detect rare oxidized biological metabolites and fabricate biomaterials. These and other applications of the traceless Staudinger ligation will be covered.
The traceless Staudinger ligation is useful to ligate probes onto azide-containing oligopeptide and small molecule targets. The high chemoselectivity of the reaction permits its use in the presence of native, unprotected biomolecules. In one example, the traceless Staudinger ligation was used to attach position emission tomography (PET) tags to small molecules.28, 251 Phosphines comprising radioactive 18F- or 131I-labeled acyl chains were incubated with azide-bearing pharmaceutical agents.252–256 The PET-tagged products could be used immediately for in vivo imaging. The products did not require purification owing to the mild nature of the ligation and lack of toxic byproducts. In another application, the traceless Staudinger ligation was used to synthesize a ruthenium (II) organometallic prodrug.257 The prodrug comprised a CO-releasing ruthenium center conjugated to a BODIPY dye. The conjugate was delivered into mammalian cells, and upon UV irradiation, carbon monoxide was liberated and cell death was observed.257
In addition to small molecule modification, the traceless Staudinger ligation is a reliable reaction to functionalize entire proteins.258 Azido amino acids can be non-specifically appended to protein targets or site-specifically installed using genetic code expansion techniques.19, 152–159 The azide handles can be subsequently conjugated with cargo via the traceless Staudinger ligation. The products ultimately contain native amide bonds. Fluorophores, cytotoxic drugs, and other moieties have been attached to various proteins with the traceless Staudinger ligation.259–260 In one example, the Jewett lab functionalized azide-bearing proteins with photoactivatable crosslinkers.261 Phosphine probes were synthesized with transferrable acyl groups bearing diazirines. Incubation with azides led to robust amide bond formation and diazirine transfer. The photocrosslinkers enabled capture of local protein interactions.
In addition to unnatural cargos, a variety of natural post-translational modifications have been installed onto biomolecules via traceless Staudinger ligation. Glycosylation is a common post-translational modification (PTM) that regulates biomolecule function, stability, and solubility.262 The heterogeneous nature of this modification complicates the study of glycosylated proteins. Homogenous samples could help to elucidate specific biological activities, but methods to install user-defined glycans on peptide backbones are rare.263–264 Several groups have used the traceless Staudinger ligation to address this issue and append specific glycans on peptide and protein substrates.240 In one example, the Bernardi group developed synthetic methods to append N-glycans onto peptide building blocks.265–266 In another study, Guo and coworkers prepared an analog of the glycopeptide CD52 using the traceless Staudinger ligation.267 The peptide was synthesized via SPPS. After cleavage from solid support, the C-terminus was converted to a thioester by coupling with a borane-protected phosphinothiol. The peptide sidechains and phosphine were deprotected under acidic conditions to reveal the CD52 peptide phosphinothioester. The thioester served as a substrate for traceless Staudinger ligation with an azido trisaccharide to provide the CD52 analog in high yield (66%).
The Davis lab has also applied phosphine reagents to glycosylate proteins.240 In their strategy, phosphines were linked to the anomeric position of monosaccharides via labile ester bonds. Upon incubation with azide-labeled proteins, traceless Staudinger ligation forged native glycopeptide bonds. Variable yields were obtained depending on the glycan unit transferred and the local environment surrounding the azide. In some cases, a competitive Staudinger reduction limited the efficiency of amide bond formation.
Beyond glycosylation, other protein modifications have been synthesized using the traceless Staudinger ligation. Lysine acylation (e.g., acetylation, succinylation) encompasses a common family of PTMs with diverse cellular roles.268–269 Acylated lysine residues can be installed in proteins using the traceless Staudinger ligation, providing homogeneous samples. Acetylated lysine residues were recently synthesized using modified phosphines to target ε-azidonorleucine in pentapeptides.270 Entire proteins have also been site-specifically modified via traceless Staudinger ligation. Depending on the modification, some lysine PTMs may be directly installed into protein targets using genetic code expansion. For example, N-ε-acetyl lysine was readily incorporated into defined protein positions using this method.271–272 Larger acyl modifications, though, have been more challenging to genetically encode. The Liu group recently developed a strategy to synthesize proteins bearing succinylated lysine residues.273 In their approach, ε-azidonorleucine (AznL) was installed into proteins using a mutated pyrrolysine amino acyl tRNA synthetase/tRNAPyl pair. Subsequent conjugation with a succinylated phosphinothioester conjugate (68) yielded native succinyl modifications (Figure 26A) on protein targets. Unfortunately, the reaction conditions also non-specifically succinylated endogenous protein residues. This background reactivity was hypothesized to originate from succinic anhydride (69) released via an intramolecular cyclization (Figure 26B). To prevent this side reaction, an o-nitrobenzyl photocage was installed on the phosphinothioester (dPPMT-NB-Su, Figure 26C). The caged phosphine reacted with azide-functionalized protein targets to afford conjugated products, with no off-target labeling. Subsequent photocleavage provided proteins bearing native lysine succinylated residues (Figure 26D).
Figure 26.

Site-specific succinylation via traceless Staudinger ligation.272 (A) Azidonorleucine (AznL) was installed in ubiquitin using a mutated PylRS/tRNAPyl pair. Phosphinothioester 68 non-specifically succinylates both native lysine and AznL residues. (B) Non-specific reactivity was attributed to release of succinic anhydride (69). (C) A photocaged phosphinothioester (dPPMT-NB-Su) was prepared to mitigate against succinic anhydride formation. (D) Site-specifically modified ubiquitin (Ub-K48AznL) was prepared by installing AznL at residue 48, then treating the conjugate with dPPMT-NB-Su. Following traceless Staudinger ligation, the native succinyl group was liberated via photocleavage of the o-nitrobenzyl group. Proteins were analyzed using SDS-PAGE and Western blot. Reprinted with permission from reference 272. Copyright Wiley Publishing Group.
In addition to polypeptide functionalization, the traceless Staudinger ligation has recently been applied to cyclize biomolecule substrates. Cyclic peptides are attractive drug candidates due to their diverse pharmaceutical activities and high metabolic stability.230 Peptide cyclization is often difficult to achieve with conventional coupling reactions, though, due to off-target intermolecular polymerization or loss of stereochemical information. One common solution uses native chemical ligation to couple N-terminal cysteine and C-terminal thioester residues.274 If necessary, the cysteine side chain can be removed using desulfurization chemistries.275 To directly cyclize peptides without the need for cysteine residues or subsequent modification, several groups have applied the traceless Staudinger ligation.
Recent work has focused on syntheses of small and medium-sized macrocycles.276–278 In these cases, linear peptide precursors were functionalized with N-terminal azide groups using standard solid-phase peptide synthesis (Figure 27). Following resin cleavage, the required C-terminal thioester was generated via incubation with a thiol-functionalized phosphine and activation agent. The phosphine reagent was protected with a borane group (70) to prevent premature reaction with the azide and/or oxidation. The borane can be removed under either acidic or basic conditions.95 Acidic deprotection was used in this case to both liberate the phosphine and simultaneously cleave side chain protecting groups. Borane removal ultimately triggered a head-to-tail cyclization reaction between the phosphinothioester and azide. In addition to peptide cyclization, the traceless Staudinger ligation is also useful to synthesize macrocyclic cyclic dinucleotide biomolecules.279
Figure 27.

Peptide macrocyclization using the traceless Staudinger ligation.277 An oligopeptide was prepared and functionalized with an N-terminal azido amino acid. A borane-protected phosphine (70) was installed at the C-terminus via a thioesterification reaction. Following deprotection, the traceless Staudinger ligation afforded the macrocycle.
The traceless Staudinger ligation has also been used in combination with other synthetic methods to prepare whole proteins with defined functionality. Solid-phase-peptide synthesis, expressed protein ligation, and unnatural amino acid mutagenesis have all been used in combination with the reaction.258 The overall strategy involves joining two functionalized protein halves (Figure 28). One fragment is substituted with a C-terminal phosphinothioester, typically produced via expressed protein ligation as an intein fusion.280–281 This piece comprises a transient C-terminal thioester (Figure 28A). Transthioesterification with a phosphine thiol probe releases the intein and installs the requisite phosphine for biomolecule assembly. To produce the other protein fragment, genetic code expansion is often used to install the requisite azide handle. ε-Azidonorleucine (71) is a common choice due to its ease of installation with known methionine amino acyl tRNA synthetase/tRNA pairs (Figure 28B).282 When the ε-azidonorleucine fragment is combined with the phosphine fragment, traceless Staudinger ligation links the fragments via an isopeptide bond (if the azide is not directly at the N-terminus, Figure 28C). Isopeptides mimic natural linkages observed in post-translational modifications with ubiquitin and ubiquitin-like modifications.283–284 Future work may apply this strategy to site-specifically modify proteins with other post-translational modifications to elucidate their downstream effects.258
Figure 28.

Convergent protein synthesis using expressed protein ligation, unnatural amino acid mutagenesis, and the traceless Staudinger ligation.15,26 (A) Expressed protein ligation can provide C-terminal thioester fragments. Transthioesterification with a water-soluble phosphinothiol probe yields the requisite phosphinothioester. (B) Azidonorleucine (71) can be installed in protein targets using an engineered MetRS/tRNAMet pair.281 (C) The two protein halves can be ligated via traceless Staudinger ligation.
Outside of biomolecule synthesis and modifications, phosphines have been applied to detect rare oxidized metabolites, including azanone (HNO). HNO is an endogenously produced metabolite that affects diverse biological processes.285–286 A platform for HNO detection was recently developed using a traceless Staudinger-like reaction.287–288 The method involves nitroxyl group reduction by phosphine 72 to form a transient iminophosphorane (Figure 29). The iminophosphorane cyclizes in a second step, releasing a detectable probe (e.g., 4-nitrophenol or fluorescein). The signal provides a means to quantify HNO in biological samples. Phosphine probes were also developed to detect nitrosothiol (R-SNO) species via a similar reaction pathway.289–290
Figure 29.

Phosphinoester reagents can detect nitroxyl metabolites.286 Phosphine 72 reduces nitroxyl species to form iminophosphorane intermediates. Subsequent cyclization forms urea products and releases p-nitrophenol, enabling a colorimetric readout.
The traceless Staudinger ligation has been leveraged to prepare biomaterials via native amide bond linkages.25, 190, 194 Polymers have been outfitted with biomolecules or synthetic modifications using the highly chemoselective reaction. For example, polysaccharide biomaterials were recently functionalized with synthetic cargo.291–293 In this strategy, azide groups were appended to carbohydrate units via activation of reactive alcohols and subsequent displacement with sodium azide. Traceless Staudinger ligation with PPh3 and carboxylic anhydrides enabled regioselective acylation. The traceless Staudinger ligation was also used to prepare functionalized probes for self-assembled monolayer (SAM) construction294 The reaction is further compatible with other polymer synthesis methods. In one example, reversible addition-fragmentation chain transfer (RAFT) and the traceless Staudinger ligation were used in tandem to assemble macromolecules.295 The synthesized polymers exhibited a high degree of uniformity and the molecular weights were controllable.
2.3.3. Chemical and biological scope
Over the last ten years, the traceless Staudinger ligation has been routinely employed to modify biomolecules with natural and synthetic cargo via native amide bonds.26 In its original form, the traceless Staudinger ligation suffered from a limited substrate scope.226, 228 Improved phosphine reagents now enable applications at sterically occluded junctures (see section 2.3.1). Other recent developments have provided azide and phosphinothioesters to functionalize biomolecules ranging from small oligopeptides to whole proteins (see section 2.3.2). The improved phosphine reagents complement other protein synthesis strategies to provide a suite of biomolecule assembly techniques.258
The traceless Staudinger ligation is one of few biocompatible reactions to yield native biological linkages.218, 296 Another well-known example is native chemical ligation (NCL).297 Early variants of NCL required an N-terminal cysteine residue.298 In many cases, cysteine residues were installed at ligation sites in place of the native amino acids. Such non-native cysteine residues can perturb polypeptide folding or activity following assembly299 The requirement for cysteine has been addressed with the development of desulfurization reactions to convert cysteine to more common residues post-ligation.275, 300–301 Newer reaction variants have also been developed that use alternative nucleophiles, eliminating the need for cysteine altogether.297, 302–303 Compared to NCL, the traceless Staudinger ligation suffers from a more limited substrate scope.218 The appropriate strategy likely depends on the application and requirement. Both reactions are complementary, though, and have been employed in tandem to assemble complex synthetic proteins.258, 277
In the future, the traceless Staudinger ligation will likely continue to be a valuable tool to modify peptide substrates in a variety of applications. For example, oligopeptide therapeutics are becoming increasingly common in clinical settings.304–305 Current and future drug development efforts will be aided by chemoselective reactions to synthesize and modify peptides. For additional information, Bräse and coworkers recently published a review on the traceless Staudinger ligation.17
2.4. Cyclopropenone-phosphine reactions
The cyclopropenone-phosphine reaction is a recent addition to the bioorthogonal toolkit. Cyclopropenone (CpO) motifs are small and stable in cellular environments, suggesting broad biological compatibility.306–308 CpO scaffolds react with phosphine probes to reveal ketene-ylide intermediates (Figure 30).209 These electrophiles can trap proximal nucleophiles, providing ring opened products.309–310 The electrophilic trap has been used for proximity-based biomolecule labeling and crosslinking applications.209, 309–310 CpO analogs also exhibit a range of reactivities with phosphines.311–312
Figure 30.

Cyclopropenone-phosphine ligations provide α,β-unsaturated carbonyl products.
This section will explore the development and application of bioorthogonal CpO-phosphine reactions. Tuning of the CpO scaffold afforded reagents that were stable to cellular thiols and other biological nucleophiles.309, 311 Heteroatom replacement further expanded the breadth of CpO reactions. For example, cyclopropenethione (CpS) variants exhibit markedly faster reactions with triarylphoshines than CpOs.4 Cyclopropeniminium (CpN+) ions also react with triarylphosphines, although they form cyclopropane-phosphonium adducts via a unique mechanism.311–312 Complementary phosphines have been developed for each class of CpO analog.209, 309–312 Excitingly, many of the probes are mutually compatible and can be used in tandem for multi-component labeling. CpOs can also be metabolically incorporated into various biomolecules, setting the stage for imaging and tagging studies.309
2.4.1. Reaction basics
The structures and reactivities of CpO motifs have been extensively characterized. One of the earliest reports involved CpO as a synthetic target to examine ring systems with two-electron aromatic character.313 The cyclopropenone scaffold is highly polarized (μ > 4.5 D), exhibits some C–O single bond character, and has significant ring strain ( > 75 kcal/mol).314–317 These potential liabilities in terms of stability are offset by the semi-aromatic character of CpO.314, 318–319 Charge separated resonance structures, cyclopropenylium ions, have two π-electrons delocalized over three 2p orbitals and are the smallest of the Hückel aromatic systems. Later reports established CpOs as versatile synthons. These motifs can react with electrophiles, nucleophiles, dipolar reagents, and other π systems.320
Most pertinent to bioorthogonal chemistry, CpOs react with bioorthogonal, triarylphosphines to afford ring opened carbonyl products (Scheme 4).321 The reaction is thought to proceed via conjugate addition of a functionalized-phosphine (e.g., 73 in Scheme 4) into the cyclopropenone (74) unit.209, 321 Subsequent ring opening reveals a ketene-ylide intermediate (75). Ketene-ylides are isolable under air- and moisture-free conditions.322 However, several nucleophiles (including O-, N-, and S-based groups) can trap the ketene both intermolecularly and intramolecularly.209, 309–310 Elimination of the phosphine results in an α,β-unsaturated carbonyl product (e.g., 76).
Scheme 4.

When the trapping reaction occurs intermolecularly, three components are required for a successful ligation: the CpO, the phosphine, and an exogenous nucleophile.209, 310 Early examples showcased hydroxy- and amine-functionalized nucleophiles as trapping reagents. Selective ligation was achieved using excess nucleophile in the reaction medium or high local concentrations of the desired nucleophile afforded by proximity. The reaction provided unsaturated, linear carbonyl products. The three-component reaction also releases the phosphine reagent, which can go on to react with additional CpO equivalents. For example, initial reports featured mono-substituted CpOs, amine nucleophiles, and catalytic water-soluble, triarylphosphines. The reaction was complete within thirty minutes (10 mol% phosphine, 10 equiv. nucleophile).209 This version of the CpO-phosphine ligation is one of the few examples of organocatalytic reactions in bioorthogonal chemistry.
When the trapping nucleophile is present on the phosphine itself, a bimolecular reaction ensues. A variety of ortho-substituted nucleophiles have been installed on triarylphosphine probes to act as intramolecular traps.209, 309 The ligation products in these cases are unsaturated carbonyl adducts (e.g., 76) bearing a phosphine tether. Initial bimolecular ligation attempts featured ortho-substituted phosphines and mono-substituted CpOs (77). These reactions were remarkably fast (Figure 31, k2 >20 M−1 s−1).309 Fast reactivity came at the expense of probe stability, though. While monoalkyl CpO reporters exhibited robust reactivity with bioorthogonal phosphines, they also reacted efficiently with cysteine and other biological nucleophiles, limiting their use in some cellular contexts.309
Figure 31.

Mono-substituted CpOs (77) react with phosphines to enable intermolecular or intramolecular trapping with nucleophiles.209
Striking the right balance of reactivity and stability required further reagent tuning. A panel of CpOs with varying alkyl or phenyl substituents was synthesized. The compounds were incubated with equimolar cysteine and monitored by NMR spectroscopy. Only dialkyl-substituted CpOs were inert to cysteine over seven days.309 Improved stability came at the expense of reactivity, though. The more stable dialkyl-substituted CpOs reacted ~100-1000 fold slower than their mono-substituted counterparts. Addition of the second alkyl group (i.e., methyl substituent) reduced the rate by multiple orders of magnitude (102-103 fold decrease in k2).309
To recoup reactivity, dialkyl-substituted CpO reagents were reacted with more nucleophilic phosphines. These reagents (79a–e) comprised triarylphosphines with ortho-substituted nucleophiles, along with cyclohexyl-substituted phosphines bearing vicinal nucleophiles. A model disubstituted CpO probe (78) was used to measure reaction rates with the phosphine panel. Replacing a phenyl group with a cyclohexyl unit resulted in enhanced rates for phosphines bearing hydroxy (79a–79b) or amine (79c–79d) traps (Figure 32, ~18-fold and ~137 fold increases in k2, respectively).309 Despite the increased nucleophilicity of cyclohexyl phosphines 79b and 79d, negligible oxidation was observed. The hydroxy derivatives also provided the fastest rates, likely due to their hydrogen bonding capability. Hydrogen bonds can activate CpO scaffolds for nucleophilic attack. Alcohols have lower pKa values compared to analogous amines and are thus better hydrogen bond donors.
Figure 32.

Kinetic experiments to analyze cyclopropenone and phosphine reactivity.308 Reactions between CpO 78 and a panel of phosphines were monitored via NMR in benzene–d6. Second-order rate constants of selected phosphine reactions with CpO 78.
The products of the CpO-phosphine ligation are ripe for additional modification. For example, when CpO scaffolds were treated with 79a, the resulting phenol ester product (80) was subjected to traceless Staudinger ligation with azide probes. The two-step sequence provided amide products (81, Figure 33).309 Thioesters formed from CpO reactions with thiophenol-substituted phosphines are also prime for further modification via native chemical ligation.297 Additionally, CpO ligation products comprise α,β-unsaturated carbonyl units. Such motifs are subject to conjugate addition reactions. This mode of reactivity could enable additional probe attachments or target enrichment.323–324
Figure 33.

CpO-phosphine ligation products (80) undergo traceless Staudinger ligation with organic azides.308
The CpO-phosphine reaction is compatible with and orthogonal to several common bioorthogonal transformations. Mutually orthogonal reactions are desirable for simultaneous biomolecule tagging or activation.113, 325 To date, several pairs of compatible bioorthogonal reactions have been identified.23, 113 However, very few examples of simultaneous triple-component labeling exist.113, 326–329 The CpO-phosphine ligation is amenable to triple labeling studies, as it has a unique reaction mechanism. The majority of orthogonal bioorthogonal reactions proceed via cycloaddition.113, 330 Many of the reagents are also inert to both CpOs and phosphines. For example, cyclopropenone (82a) and phosphine (82b) reactants are orthogonal to both triazine (83a) and teteramethylthiacycloheptyne (TMTH, 83b) groups. All of these reagents can also be used in tandem with cyclopropene (84a) and tetrazine (84b) reporters (Figure 34) for simultaneous labeling329
Figure 34.

Triple, one-pot bioorthogonal ligations.328 The CpO-phosphine ligation (red reagents) is compatible with cyclopropene-tetrazine cycloadditions (green reagents) and triazine-TMTH reactions (blue reagents).
Thus far, we have discussed the development of CpO scaffolds and their reactions with bioorthogonal phosphines to provide unsaturated carbonyl products. Additional tuning of the CpO scaffold was pursued for improved ligation rates and stability. In one case, the CpO carbonyl was substituted with a thiocarbonyl to provide cyclopropenethione (CpS) reagents.311 CpS motifs have been previously shown to react with triarylphosphines.329–330 The reaction follows a similar mechanism as CpO probes (Figure 35), proceeding through a thioketene intermediate (85). The thioketene can then be trapped by a proximal nucleophile to afford a ligated adduct. Following phosphine protonation and elimination, thioamide products (86) are formed. DFT calculations suggested that CpS had consistently lower LUMO energies compared to their CpO counterparts.311 Thus, it was surmised that CpSs could provide faster ligation rates.
Figure 35.

Cyclopropenethione-phosphine ligation.310 Cyclopropenethione (CpS) reagents react with phosphines via thioketene-ylide intermediates. Thioketene trapping with tethered nucleophiles yields ring-opened thiocarbonyl products.
A model CpS probe (87) was synthesized and incubated with various phosphines (88a–d) (Figure 36). The reaction of phosphine 88a and CpS probe 87 proceeded two orders of magnitude faster than the corresponding reaction with CpO 78. Interestingly, the most nucleophilic phosphines did not provide the fastest rates. Triarylphosphine 88a ligated CpS within five minutes, while the more nucleophilic cyclohexyl derivative 88b required 1 hour at similar concentrations (Figure 36).311 This rate differential was attributed to the enhanced hydrogen bonding capability of the phenol group compared to the cyclohexyl alcohol. Despite being a more reactive motif, the CpS scaffold maintained stability to cellular thiols. Disubstituted CpS motifs comprising at least one alkyl group were stable to glutathione over seven days.
Figure 36.

Kinetic experiments to analyze cyclopropenone and phosphine reactivity.310 Dialkyl cyclopropenonethione 87 was treated with a panel of functionalized phosphines 88a–d to afford thiocarbonyl products.
The nitrogenous equivalent of CpO has also been pursued for bioorthogonal labeling. Cyclopropeniminium ion scaffolds (CpN+, 89) were synthesized from the corresponding CpO scaffolds via carbonyl activation and amine displacement.312 CpN+ probes ligated phosphines (90a–b) to yield bicyclic phosphonium adducts (Figure 37).312 The reaction begins with phosphine conjugate addition into CpN+ probe 89, followed by intramolecular proton transfer to reform the iminium ion 91.312 Nucleophilic attack by the ortho-substituent on the phosphine probe results in the final adduct (92). The rates of the CpN+ ligation (~10−3 M−1s−1) are slower than analogous CpO and CpS reactions. Interestingly, only phosphines with thiophenol and phenol pendant nucleophiles provided covalent adducts. This result suggested that the intramolecular proton transfer requires a proton with pKa <16. Similar to CpOs, the dialkylated CpN+ scaffold (89) was refractory to hydrolysis for extended periods (t1/2 = ~13 d). CpN+ 89 showed some reactivity with glutathione, albeit over a relatively long time scale (t1/2 = ~41 h).
Figure 37.

Mechanism of CpN+-phosphine ligation.311 ortho-Substituted phosphines (90a–b) undergo conjugate addition reactions with CpN+ 89. Subsequent intramolecular proton transfer and nucleophilic attack yield bicyclic adducts 92a–b.
The unique mechanism of the CpN+-phosphine ligation enabled its immediate pairing with the classic CpO ligation. Cyclohexyl-substituted phosphine 88b selectively ligates CpOs in the presence of CpN+ scaffolds. This selectivity enabled sequential ligation of CpN+ and CpO reports with phosphine derivatives.312 Incubation of CpO, CpN+, and 88b resulted in complete, selective ligation of the CpO. Subsequent administration of a thiophenol-substituted phosphine tagged the remaining CpN+ motif. This result is notable, as the two cyclpropenone derivatives are quite similar in structure.
As the above examples highlight, CpO-phosphine reactivity can be tuned for selective bioorthogonal ligation and diverse applications. Cyclopropenone modifications can substantially alter reactivities with phosphine nucleophiles.309 Monosubstituted cyclopropenones enabled fast phosphine ligations, but were susceptible to thiol addition. Substituting a methyl group at the C3 position enhanced stability, but significantly reduced the rate of the phosphine ligation.309 Heteroatom substitution also impacted ligation rates. CpS-phosphine reactions were >100-fold faster than the corresponding CpO ligations with certain phosphines.311 Additionally, CpS probes were stable to biological nucleophiles, even with an aryl substituent. CpN+ probes are orthogonally reactive to CpO and CpS motifs.312 CpN+ analogs react with phosphines to form cyclopropane-phosphonium adducts. The mechanism relies on a crucial intramolecular protein transfer. This step limits the phosphines that can be used, but it also sets the stage for orthogonal labeling methods.
2.4.2. Biological applications
CpOs have historically been used as masking groups for strained cyclooctynes in bioorthogonal chemistry applications.331–332 Upon UV irradiation, CO is released to reveal a strained alkyne. The liberated alkyne can then participate in subsequent strain-promoted azide-alkyne cycloaddition (SPAAC) reactions or inverse electron-demand Diels–Alder cycloadditions (IEDDAC).104, 332–337 CpO decarbonylation has been applied to label and immobilize biomolecules in vitro and in mammalian cells.332, 337–338
More recently, CpO probes have been leveraged to tag biomolecules. The reaction proceeds via activation with phosphines followed by nucleophilic trapping of the ketene-ylide intermediate. As an initial demonstration, lysozyme was nonspecifically functionalized with mono-substituted CpO handles (Lys-CpO, 93). CpO-phosphine ligation was then performed in a three-component fashion by incubating Lys-CpO, water-soluble phosphine 94, and rhodamine 95 as a labelling nucleophile (Figure 38A, 100 μM P(3-SO3NaPh)3, 100 μM rhodamine, 1% DMSO/PBS).209 The reaction was monitored via in-gel fluorescence. The ligation was complete within 45 min. Three-component ligation enabled fast labeling in vitro, but was not well suited for live cell labeling, as all three components must converge in a single spot.
Figure 38.

Protein labeling via bioorthogonal ligation with CpOs and functionalized phosphines.209, 308 (A, B) Monosubstituted CpOs were non-specifically conjugated to a model protein (POI, 93).209 Subsequent reactions with phosphine probes (94, 96) could be performed in either a two-or three-component fashion. (C) CpO motifs are amenable to recombinant protein production and subsequent bioorthogonal labeling.308 An unnatural CpO amino acid 97 was site-specifically installed in GFP using genetic code expansion techniques. Subsequent ligation was performed with a phosphine-biotin probe (98).
Bimolecular reactions with CpO and ortho-substituted phosphines enabled more efficient labeling in cellular environments (Figure 38B).209 Model reactions were performed using Lys-CpO 93 with an excess of phosphine bearing pendant nucleophiles (96, 1 mM phosphine, 50% DMSO/buffer, 20 min). Mass spectrometry analysis confirmed that ligated adducts were formed. No hydrolysis products or adducts formed from non-specific ketene trapping were observed. Lys-CpO was further incubated with a rhodamine-functionalized phosphine with or without cellular lysate (0.25–2 μg CpO, 250 μM phosphine, 20 μg HEK293 lysate, 10% DMSO/PBS, 45 min). The reaction was monitored by in-gel fluorescence. Ligation adducts were detected in the presence of lysate, on par with reactions performed in buffer only.
CpS probes have also been used in model protein labeling studies. In an initial example, lysozyme (Lys) was non-specifically functionalized with CpS probes.311 Lys was also labeled with CpO for comparison. CpS-functionalized lysozyme (Lys-CpS) was treated with phosphine probes and labeling was visible by gel electrophoresis within 5 minutes. The reaction was complete within 30 minutes (20 μM Lys-CpS, 50 μM phosphine, PBS) Under similar conditions, Lys-CpO required 30 minutes for detection (20 μM Lys-CpO, 50 μM phosphine, PBS). Thus, CpS enabled a 300-fold faster reaction. Robust labeling of Lys-CpS was also achieved in cell lysate in one hour with 1 mM phosphine. Off-target labeling was not observed, suggesting that the CpS-phosphine ligation can be applied in cellular contexts.
CpO reporters have been site-specifically installed in proteins using genetic code expansion techniques. Precise placement of CpO handles in proteins of interest is useful for a number of imaging and tagging applications. Toward this end, a mutant PylRS/tRNA pair was used to site-specifically install CpO-pyrrolysine derivative 97 into GFP as a model protein (Figure 38C).309 Successful incorporation was confirmed by mass spectrometry. The conjugate was successfully ligated with an ortho-substituted phosphine biotin probe (1–2 μg GFP-CpO, 30 μg lysate, 100 μM phosphine).309 Robust labeling was detected by Western blot within an hour.
In addition to biomolecular labeling, ketene ylide formation can promote biomolecule crosslinking. As noted earlier, high concentrations of exogenous amines can trap ketene-ylide intermediates formed by CpO reactions with phosphines. The reaction can also proceed under catalytic conditions in buffer (75–81%, 50 mM CpO, 5 mM amine, 0.5 mM P(3-SO3NaPh)3, 20% MeCN/PBS).310 To investigate trapping by potential protein sidechains, a model CpO scaffold was incubated with various Fmoc-protected amino acids and a water-soluble triarylphosphine (250 μM–2 mM CpO, 2 equiv. amino acid, 1 equiv. P(3-SO3NaPh)3, PBS, pH 7.3). Only amino acids with nucleophilic side chains (Ser, Cys, Lys, and Tyr) were able to form crosslinks, albeit in low yields (<10%, 4 mM CpO). Most of the products comprised α,β-unsaturated carboxylates, resulting from addition of water to the ketene intermediate.
These results suggested that CpOs could be used as chemically triggered crosslinkers. If positioned on a biomolecule interface, CpOs could form covalent adducts with nucleophiles present on neighboring biomolecules (i.e., those with the highest local concentration). CpO-based chemical crosslinking was demonstrated using a split NanoLuc enzyme comprising a C-terminal peptide (SmBiT-CpO) and a larger fragment (LgBiT).310 In aqueous buffers, the two fragments hybridize to form a functional enzyme. A CpO amino acid was installed on the SmBiT peptide via standard solid-phase peptide synthesis. Docking studies suggested that the CpO amino acid was in proximity to trapping nucleophiles when bound to LgBiT (Figure 39A). When the two BiTs were mixed together, crosslinking was triggered by water-soluble phosphine 94. The resulting ketene-ylide was trapped by LgBiT nucleophiles and detected by SDS-PAGE within 60 min (40 μM LgBiT, 120 μM SmBiT, 1 mM 94, PBS, pH 7.4). Further experiments revealed that Tyr16 was the primary residue responsible for the covalent link. Faster crosslinking was also achieved using more nucleophilic phosphine probes. For example, cyclohexyldiphenylphosphine 3,3′-disulfonic acid (99) afforded detectable crosslinking within 10 min (40 μM LgBiT, 120 μM SmBiT, 1 mM 99, PBS, pH 7.4).
Figure 39.

Chemically triggered crosslinking via the CpO-phosphine reaction.309 (A) Crosslinking of split luciferase fragments (LgBit and a SmBit). SmBit was outfitted with a CpO amino acid (SmBit-CpO). The interaction between LgBit and SmBit-CpO can be captured upon phosphine triggering. (B) Luciferase crosslinking was observed in cell lysate, upon treatment with phosphine 99. SDS-PAGE analysis reveals the presence of a higher molecular weight crosslinked band in both the presence and absence of cellular lysate (13% and 5% crosslinking, respectively.)
The effect of probe affinity on crosslinking yields was further investigated. Variants of CpO-SmBiT were synthesized and crosslinked to LgBiT (40 μM LgBiT, 120 μM SmBiT, 500 μM 94, PBS, pH 7.4.) SmBiT fragments with greater binding affinities resulted in more crosslinking, while maintaining specificity for LgBiT-SmBiT crosslinking was also evaluated in bacterial lysate to simulate crosslinking in cellular contexts (40 μM LgBit, 120 μM SmBiT, 1 mM 99, 30 μg lysate, 30 min, Figure 39B). More crosslinking was detected in lysate than buffer alone. Macromolecular crowding was hypothesized to promote more productive binding interactions.
Phosphine-triggered reactivity was recently used to construct butenolides on demand. Butenolides comprise bioactive molecules and are found in a number of natural products.339–341 CpOs can be mapped onto these target structures, with pendant alcohols providing an intramolecular trap upon phosphine treatment. To explore whether hydroxymethyl-substituted CpOs could serve as general synthons for butenolides, a variety of substrates were produced from propargyl alcohol precursors.342 A panel of CpO probes were reacted with various phosphine triggers, and butenolide yields were measured. The CpO-butenolide formation reaction was tolerant of α-alkyl and aryl substituents on the CpO core as well as γ-methyl groups. The reactions could be performed with catalytic amounts of phosphine to furnish butenolide products (5-20% catalyst loading) in reasonable yields. The mild conditions are attractive for general synthetic use. Other adaptations of this approach could potentially provide bioactive drugs via bioorthogonal chemistry.
In summary, CpOs and phosphines are emerging bioorthogonal reagents with a number of potential applications. CpOs can label proteins in a two- or three-component fashion. Genetic code expansion has enabled site-specific installation of CpO amino acids in protein targets. Both nonspecifically and site-specifically CpO modified proteins were successfully labeled in mammalian cell lysate. Improved biomolecule labeling was achieved with CpS-functionalized proteins. In addition to biomolecule labeling, CpO scaffolds can be used as chemically triggered crosslinkers. Finally, the CpO-phosphine reaction has been used as a synthetic method to access butenolides with a variety of structures.
2.4.3. Chemical and biological scope
The CpO-phosphine ligation is a relatively new member to the bioorthogonal reaction toolbox, so its applicability, limitations, and scope are still being characterized. The availabilities of the reactants could limit general use. CpOs are not currently sold by commercial vendors. Among bioorthogonal transformations, the CpO-phosphine ligation is unique in its overall mechanism. CpOs have enabled different pursuits as triggerable, masked electrophiles. The selective liberation of an electrophilic intermediate is useful for both covalent labeling and crosslinking applications. Compared to other bioorthogonal reactions, such as IEDDA ligations with strained alkenes, the CpO-phosphine reaction is quite slow. CpOs are generally smaller and less perturbing to biomolecules, though, than many of the fastest reacting bioorthogonal reporters. They are also stable in complex biological media for extended time periods.
2.5. Phospha-Michael addition
The phospha-Michael addition is a relatively new reaction to the chemical biology toolkit. This ligation features tertiary phosphine nucleophiles and α,β-unsaturated carbonyl groups that react to provide phosphonium adducts (Figure 40).343 The phospha-Michael reaction is both mild and chemoselective, and can be performed in a variety of solvents. The overall 1,4 addition is similar to thiol-ene344–346 and radical additions with electrophilic olefins.263–264, 347 Compared to thiol nucleophiles, though, the phosphine equivalents generally react faster and have a wider substrate scope.
Figure 40.

Phosphines react with α,β-unsaturated carbonyls to yield phosphonium adducts via a phospha-Michael addition.
Phospha-Michael-type reactivity has been exploited in a variety of contexts. Phosphine addition to electrophilic alkenes is central to Morita-Baylis-Hilman and Rahut-Currier reactions. These transformations are widely used in organic synthesis and exploit the mild nature of phosphines for carbon-carbon bond construction.2, 348–349 Phospha-Michael-type additions have been more recently employed to tag activated alkenes in biological contexts. Examples include profiling α,β-unsaturated carbonyl modifications present on some endogenous proteins. A variety of alkenes comprise common lysine post-translational modifications found on histone proteins (e.g., dehydroalanine, acrylamide, dehydrobutyrine, and crotonyl groups). These modifications can alter protein activity gene expression and serve as epigenetic markers.350–352 The phospha-Michael addition has enabled chemical retrieval of α,β-unsaturated carbonyl modifications and numerous profiling experiments.323
Electron-deficient alkenes are relatively rare elsewhere in the cell. Thus, biomolecule targets purposefully outfitted with α,β-unsaturated carbonyl groups can be selectively ligated with detectable probes via phopha-Michael addition.353 In this section, we will summarize these and other applications of the phospha-Michael reaction (see section 2.5.2), following a brief discussion on reagent tuning (section 2.5.1). We will also compare and contrast the phospha-Michael reaction with other bioorthogonal chemistries (see section 2.5.3).
2.5.1. Reaction basics
The mechanism of the phospha-Michael reaction entails an initial 1,4-addition of a phosphine to an electrophilic α,β-unsaturated carbonyl (Scheme 5).343 Subsequent protonation of the resulting enolate provides a carbonyl adduct bearing a β-phosphonium substituent (100). The addition reaction is reversible, and the phosphonium adduct is subject to E1cB elimination.354–355 The reversibility of the phospha-Michael reaction is problematic for conventional covalent labeling applications. To circumvent this issue, high concentrations of the phosphine probe are used to drive product formation.323–324, 343 More nucleophilic trialkylphosphine probes can also favor product formation.323–324, 343, 353 For example, tris(2-carboxyethyl)phosphine (TCEP) ligates α,β-unsaturated carbonyls to yield stable phosphonium adducts (100, Scheme 5).
Scheme 5.

The mechanism of the phospha-Michael addition.343
The rate of the phospha-Michael reaction is dependent on phosphine nucleophilicity and the steric environment of the alkene. Initial conjugate addition is the rate-determining step, and bimolecular rate constants vary widely depending on the reactants (6.0×10−4–2.6×10−1 M−1 s−1).323–324, 353 Triarylphosphines are desirable labeling reagents based on their broad biocompatibility, but they are less reactive than trialkylphosphines with unsaturated carbonyls.356 Thus, these probes are used to ligate only highly reactive α,β-unsaturated carbonyls.343 Trialkylphosphines can label a broader set of alkenes, due to their increased nucleophilicity. Among the most popular reagents in this regard is tris(2-carboxylethyl)phosphine (TCEP). TCEP analogs can label both di- and tri-substituted α,β-unsaturated carbonyl group at reasonable rates in a variety of buffered aqueous solutions.324
Phospha-Michael reactions with TCEP analogs were recently employed for protein bioconjugation. The TCEP probes were incubated with protein or peptide substrates bearing 1,1-disubstituted α,β-unsaturated carbonyl groups (101a).324, 353 Stable phosphonium adducts (102a) were formed (Figure 41A). Notably, the phosha-Michael reaction was 50-fold faster (k2 = 0.26 M−1 s−1 in aqueous buffer pH 7.4, 23 °C) compared to analogous conjugate addition with a thiol probe.353 The phospha-Michael reaction was used to ligate dehydroalanine residues. Proteins were first functionalized with acrylamide groups using site-specific non-natural amino acid installation.353 The modified proteins were then incubated with TCEP analogs bearing biotin or fluorophore tags. Robust labeling was observed via Western blot and in-gel fluorescence.
Figure 41.

Phosphines were found to react more readily and have a wider substrate scope than thiol nucleophiles in Michael addition reactions.323 (A) 1,1-Disubstituted Michael acceptors ligated both thiols and phosphines via conjugate addition reactions. (B)1,1,2-Trisubstituted olefins reacted only with phosphine nucleophiles.
The scope of the phospha-Michael reaction was recently expanded to 1,1,2-trisubstituted alkenes. Tri-substituted alkenes comprise a number of post-translational modifications.323–324 These motifs (e.g., 101b, Figure 41) are significantly less electrophilic than their disubstituted counterparts. Thus, they react more slowly with nucleophilic phosphine and thiol probes (Figure 41B, k2 = 2.5 × 10−3 M−1 s−1).324 In many cases, such highly substituted olefins are refractory to ligation with thiols.324, 357 In their quest to identify crotonylated modifications (comprising 1,1,2-trisubstituted alkenes), the Muir group screened a panel of phosphines.323 TCEP was the only reagent that successfully ligated the substrates (k2 = 6 × 10−4 M−1 s−1 in pH 8 buffer).323 The addition product 103a was stable in buffer and was also unaffected by prolonged heating (pH 2.5–10, 80 °C over 2 d), suggesting that the phospha-Michael adduct was sufficiently stable for downstream purification and analysis.323 Related trisubstitued alkenes, dehydrobutyrine residues, could be similarly ligated upon incubation with TCEP (k2 = 2.5 × 10−3 M−1 s−1).324 These two examples suggest that phospha-Michael reactions with TCEP probes are generally useful for profiling substituted α,β-unsaturated carbonyls in biological systems.
Trialkylphosphines can also react with electrophilic alkene moieties present in several common fluorophores, forming photoactivatable complexes (Figure 42).358–359 Such phosphonium-fluorophore conjugates exhibit quenched emission. Light irradiation can photochemically release the phosphine and restore fluorescence, with the optical signature providing a readout on phosphine availability. The released phosphine can ligate the fluorophore again, rendering the overall process reversible. Photochemical control of fluorophore emission is useful in several biological applications. In one example, subcellular structures were visualized by leveraging photoactivatable trialkyphosphine-cyanine dyes (104).14 Trialkylphosphines (e.g. TCEP) quench the fluorescence of cyanine dyes upon conjugate addition to the polymethine bridge (104, Figure 42A). Irradiation with UV light released the phosphine and resulted in cyanine fluorescence (λ = 350 nm). The photoreversible process was used to control fluorophore emission and image cellular microtubules via super-resolution microscopy.359–360 In another approach, photoactivatable fluorophore-phosphine adducts were used to trigger reductive stress in cells.358 Trialkylphosphine nucleophiles reacted with coumarin dyes via conjugate addition to quench fluorophore emission (106, Figure 42B). Under blue light illumination, the phosphine groups were photochemically released from the coumarin scaffold (107). The liberated phosphines then reduced disulfide bonds in endogenous proteins to simulate reductive stress. Furthermore, reductive species could be generated in a single cell via spatially controlled administration of light. These photoactivable phosphines enable spatiotemporally bioorthogonal reactions, similar to other photocaged phosphine probes (see sections 2.2.1, 2.3.1, and 2.6.1).
Figure 42.

Photoactivatable phosphine-fluorophore conjugates enable spatiotemporal control of reactivity. (A) Cyanine dyes are reversibly quenched by the conjugate addition of TCEP. Irradiation with UV light liberates the phosphine.359 (B) Carbocyanine-phosphonium adducts release tributylphosphine upon illumination with visible light.358
2.5.2. Biological applications
Phosphines ligate α,β-unsaturated carbonyls with high chemoselectivity, enabling their use in complex biological environments. In one area, the phospha-Michael reaction has been employed to profile crotonyl groups in histone proteins.323 Lysine crotonylation is an epigenetic marker of transcriptional activation, and all histone proteins contain these post-translational modifications.352 The entire scope and downstream functions of these modifications, though, remains undefined. To gain insight, the phospha-Michael reaction was used to selectively tag proteins bearing electrophilic olefins. Trialkylphosphine reagents were incubated with synthetic mononucleosomes and histones bearing crotonyl groups (Figure 43A). Endogenous crotonyl groups were readily detected with TCEP-biotin conjugate 108 in mammalian cell lysate. In the absence of crotonyl modification, signal was diminished. The TCEP-biotin probe was further used to probe the effect of crotonylation on the activity of the transcriptional coactivator p300.361
Figure 43.

Trialkylphosphine-biotin conjugates can label tri-substituted alkenes in biological systems. (A) Crotonylation modifications on histones were detected in cell lysate with TCEP-biotin probe 108.322 (B) Phospholyase OspF deactivates ERK1/2 activity by converting a critical phosphothreonine modification to a dehydrobutyrine residue. The resultant α,β-unsaturated carbonyl groups were detected using TCEP-biotin probe 108.323
Trialkylphosphines were also used to label dehydrobutyrine residues in host proteins.324 In some bacterial infections, pathogenic phospholyases convert endogenous phosphoryl groups in host proteins to dehydrobutyrine units (via β-elimination).351 The resulting α,β-unsaturated carbonyl groups can deactivate key host proteins, modulating host metabolism and immune responses.351 The complete inventory of such modifications, along with their downstream effects, remain unknown. Until recently, chemical methods to track dehydrobutyrine formation were rare. The Scheck group developed biocompatible phospha-Micahel reagents to purify dehydrobutyrine modified targets. They focused specifically on substrates of the phospholyase OspF from the pathogen Shigella flexneri.324 OspF dephosphorylates ERK1/2 to attenuate cell proliferation.362 Upon incubation of OspF with mammalian cell lysate, phosphoryl groups on target residues were removed, leaving behind dehydrobutyrine units. TCEP-biotin probe 108 was used to ligate and retrieve the dehydrobutyrine modifications. Subsequent mass spectrometry and kinetic analyses ultimately identified the substrates of OspF (Figure 43B). Both known OspF targets and unknown protein substrates were identified. Notably, the trialkylphosphine probe was found to be more reactive and selective for dehydrobutyrine detection than the corresponding thiol reagent.
Beyond profiling endogenous olefins, trialkylphosphine reagents can be used to ligate non-natural proteins bearing α,β-unsaturated carbonyls.353 In one example, amino acids bearing acrylamide groups were installed in proteins using genetic code expansion. Lysine analogs were synthesized with ε-acrylamide substitutes and incorporated into protein targets using a mutated PylRS/tRNAPyl pair.363 The unnatural proteins readily underwent conjugate addition reactions with TCEP analogs in vitro. To determine if the phospha-Michael reaction could label proteins on bacterial membranes, the acrylamide-lysine analog was installed in the extracellular face of a prokaryotic membrane protein. Acrylamide groups were selectively detected by incubation with fluorophore-TCEP conjugates on the surface of living bacteria. In addition to protein visualization, the phospha-Michael reaction was used to retrieve acrylamide-labeled glycoproteins from cell lysate.353
2.5.3. Chemical and biological scope
Over the last four years, the phospha-Michael reaction has emerged as a new method to selectively label α,β-unsaturated carbonyls in biomolecules. Acrylamide, dehydroalanine, dehydrobutyrine, and crotonyl groups readily undergo covalent reactions with trialkylphosphine nucleophiles. These same nucleophiles can potentiate cell toxicity via disulfide bond reduction and subsequent protein denaturation93. Thus, the phospha-Michael reaction is typically used in vitro or in cell lysate to avoid undesirable phosphine toxicity.93 Trialkylphosphines are also more prone to non-specific oxidation than their triaryl counterparts. A notable exception is TCEP, which is stable to oxidation in aqueous solutions in pH ranges common for protein conjugation reactions.93
The phospha-Michael reaction is one of several bioconjugation reactions that can modify electron-deficient alkenes. Thiol nucleophiles can also ligate 1,1-disubstituted Michael acceptors in thiol-ene reactions. Compared to thiols, though, phosphines offer faster, more selective reactions and have a wider substrate scope. Electron-deficient alkenes can also be targeted via radical reactions. Many of these chemistries have recently been used to modify substituted olefins in proteins or oligonucleotides substrates.263, 346–347, 364–366 Some rely on cytotoxic metal catalysts or harsh irradiation conditions that may lead to protein degradation or are otherwise incompatible with cells. By comparison, the phospha-Michael reaction occurs under relatively mild conditions without the need for catalyst additives.
2.6. Staudinger-phosphite and -phosphonite ligations
The Staudinger-phosphite and -phosphonite reactions are mild, chemoselective methods to access phosphoramidates and phosphonamidates, respectively (Figure 44). While both reactions feature phosphines with different oxidation states than those featured in the preceding sections, they share similar reactivity features. The Staudinger-phosphite reaction was originally developed and coined by Hackenberger.367,368 The chemoselectivity of phosphite-azide reactions enabled facile access to phosphoramidates, in the presence of Lewis acids. Phosphoramidates have been used in a variety of settings as catalyst ligands and amine protecting groups.367, 369 Staudinger-phosphonite reactivity was first explored to access phosphonamidates in organic solvents.370 Recently, Hackenberger modified the reaction to access a range of biologically relevant phosphonamidates in aqueous conditions.371–372
Figure 44.

The Staudinger (A) phosphite and (B) phosphonite ligations provide phosphoramidate and phosphonamadite products, respectively.
Similar to other Staudinger-type bioorthogonal chemistries, the phosphite ligation has enabled numerous pursuits. The reaction can be performed under a variety of conditions, including in organic solvents, aqueous solutions, and even in cell lysate.373 The reaction is also chemoselective and tolerant of many types of functionality. For this reason, phosphite reactions with organic azides have been used to install a variety of substituents, including glycans, onto peptide and protein backbones.374–375 This reaction has further been used to synthesize mimics of post-translational modifications (PTMs) and to outfit biomolecules with various probes. An early variant of the Staudinger-phosphite reaction was used to prepare phosphorylated Tyr mimics on proteins, even in cell lysate.368, 376 Phosphite reactions with azides have also been used to install polyethylene glycol (PEG) groups onto protein biologics to improve their stability. Other early applications focused on site-specific modification of synthetic peptides, including the preparation of phosphoglycopeptides.377 These peptides mimic rare examples of phosphoglycosylation in cells and serve as useful synthetic standards.374
Since 2011, the scope of the Staudinger-like reactions with P(III) probes has expanded. New phosphite probes have enabled access to additional protein mimics and other biomolecules.378–381 Optimized phosphites were successfully used to functionalize a number of proteins and antibodies.382–384 Phosphonites were also identified as effective ligation partners with organic azides.371 Although phosphonites are prone to rapid hydrolysis in aqueous conditions, sufficiently stable probes have been identified,371, 385 enabling protein modification in water.386 These and other applications will be discussed in the following sections, along with the basic parameters of the Staudinger-phosphite and -phosphonite reactions.
2.6.1. Reaction basics
The mechanism of the Staudinger-phosphite reaction is similar to other Staudinger-type reactions (Scheme 6). The phosphite initially attacks the terminal nitrogen of an azide to yield a phosphazide intermediate (109). Subsequent cyclization and retro-cycloaddition releases nitrogen to yield a phosphorimidate intermediate (110). Hydrolysis of the phosphorimidate releases an alcohol and provides the final phosphoramidate (111). Phosphorimidate hydrolysis can also result in amine displacement when electron poor azides are used. However, this reaction is uncommon, typically accounting for <7% of products when aryl azides are used.368
Scheme 6.

Mechanism of the Staudinger-phosphite reaction.368
Phosphites are air stable, making them less prone to autooxidation than other bioorthogonal phosphines. A variety of phosphite probes can be prepared from chlorine and dimethylamino precursors via substitution at the phosphorus atom.373 Excess nucleophile is typically used to drive such reactions.376 Phosphites are prone to hydrolyze to the corresponding phosphonates as well as other products, especially at low pH.368, 376 Phosphite stability in aqueous solution is dependent on the substituents themselves. For example, trimethyl phosphite was found to hydrolyze at pH 7.4 over 30 min (57% phosphonate observed). A phosphite bearing PEG units, though, was more stable, with 17% decomposition observed under similar conditions.376 Phosphonate formation was less pronounced at pH 8.2. Interestingly, a bis(4-oligo-ethyleneglycol-2-nitrobenzyl) phosphite was stable for over two days at pH 7.4–8.2.
Phosphite substituents also influence the final phosphoramidate structure. The last step of the Staudinger phosphite reaction involves release of a substituent. For symmetrical phosphites, the same group is released, and a single product is formed. With unsymmetrical phosphites, phosphoramidates with varying substitution patterns can be formed. The propensity for different substituents to be released in the reaction was examined by Hackenberger.373, 380 Unsymmetrical phosphites were synthesized as model compounds and subjected to Staudinger-phosphite reaction with an aryl azide (112, Figure 45). Product formation was monitored by HPLC to identify the leaving group in each case. Benzyloxy (113a) and phenoxy (113b) groups proved to be the most labile. By contrast, aliphatic groups were mostly retained in the final products. Ethoxy substituents (113c) were more retained than methoxy groups, and even longer alkyl chains (e.g., decyl group 113d) improved retention380 Collectively, these results implied that aliphatic substituents on the phosphite core are more readily preserved. Biomolecule tagging applications thus typically append probes to the phosphite core via alkyl linkages. The remaining substituents comprise benzyl or phenyl groups, which serve as sacrificial leaving groups in the labeling reaction.
Figure 45.

The hydrolyses of unsymmetrical phosphites were analyzed.374 An aryl azide substrate 112 was reacted with a panel of phosphites bearing various substituents (113a–d) to form phosphoramidate 114. Hydrolysis was dependent on the nature of the phosphite substituents. Benzyl and phenyl groups were found to hydrolyze more readily than methyl substituents. Longer carbon chains were less likely to hydrolyze.
Phosphoramidate products formed from Staudinger-phosphite ligations are generally stable under physiological conditions (pH 7.4–8.2, multiple days).368 More basic conditions can promote β-elimination of the phosphoramidate.374 The end result is C–O bond cleavage and release of an alcohol. Phosphoramidate stability at more acidic pH values depends on the amine substituent. Phosphoramidates with aryl or sterically encumbered amines (115) are stable at low pH values, and some can even survive TFA cleavage cocktails common to solid-phase peptide synthesis (Figure 46A).387 Primary amines are more susceptible to P–N bond cleavage, though, under acidic conditions.378–379
Figure 46.

Phosphoramidate adducts can be further modified. (A) Aryl-linked phosphoramidate 115 was stable to acid-mediated cleavage of protecting groups in oligopeptide substrates.374 (B) Phosphoramidate 116 was stable to basic ester cleavage conditions.374 (C) Photocaged phosphoramidate 117 was efficiently deprotected with UV light, affording phosphorylated residues.368
In addition to solvents, phosphoramidates are compatible with a range of useful synthetic transformations. For example, acetyl protecting groups on sugars (116) and other biomolecules can be removed under mildly basic conditions, without detriment to phosphoramidate linkages (Figure 46B).374 This feature enabled facile access to phosphorylated molecules via Staudinger-phosphite chemistry.368,378–379 Phosphoramidates are also compatible with light-triggered reactions. In one example, Staudinger-phosphite ligation was performed with a phosphite bearing a photolabile group (117). Following the reaction, the phosphoramidate product was illuminated with UV light to release the photocaging group and reveal a free phosphate mimic (Figure 46C). This reaction sequence has been used to prepare mimics of biological linkages and PTMs.
The Staudinger-phosphonite reaction leverages more reactive phosphonite probes for chemoselective ligation. Similar to phosphites, phosphonites (P(OR′)2R′′, 118) react with organic azides to provide covalent adducts. The reaction proceeds identically to the Staudinger-phosphite reaction (Scheme 7) with initial nucleophilic attack of the azide. In the hydrolysis step, the C-P linkage is preserved in the phosphonamidate product (119, Scheme 7) while one of the O-linked substituents is removed.
Scheme 7.

Mechanism of the Staudinger-phosphonite reaction.386
Phosphonites (particularly alkyl-substituted variants) are highly prone to air oxidation and hydrolysis in aqueous solution.371, 386 Thus, Staudinger-phosphonite reactions have historically been performed in dry organic solvents.370 More stable phosphonite probes have been developed in recent years, enabling Staudinger-phosphonite reactions to proceed in the presence of water. These probes have even been leveraged to synthesize phosphonamidates on intact biomolecules. The most common method to stabilize phosphonites involves borane protection.371,385 Borane-protected phosphonites (both sp2- and sp-hybridized variants) were shown to be stable at 4 °C for >1 year. Protected phosphonites used in aqueous conditions comprised a phenyl substituent and two O-linked PEG groups.371 The borane group was removed with DABCO, and the liberated phosphonite was dissolved in buffer (Tris-HCl buffer, pH 8.2). Phosphonite degradation was observed after 24 minutes by 31P NMR spectroscopy. The probe was also not fully soluble. Despite these drawbacks, the phosphonite was able to selectively ligate azides on model peptides when excess probe was used (100 μM azido peptide, 500 equiv. phosphonite, 10% DMSO/Tris/HCl, 95%).
Borane protection has enabled broad classes of phosphonites to be accessed. For example, borane protected ethynyl phosphonites (120) served as a modular platform for phosphonite production (Figure 47). The ethynyl probe was ligated with different azides via CuAAC to yield borane-protected triazole adducts (121).372 Fluorophores, carbohydrates, and other groups have all been conjugated to phosphonites using this strategy.385 The resulting conjugates (122) were amenable to Staudinger-phosphonite ligations following borane deprotection. Triazole phosphonites bearing PEG or electron-deficient benzyl substituents were found to be completely soluble in buffer containing 10% DMSO. They were also stable for up to 3 hours (Tris-HCl, pH 8.2).372 These phosphonites were able to ligate model azido peptides in good yield (50 μM azidopeptide, 100–500 equiv. phosphonite, 86–100% yields).372
Figure 47.

Synthesis of triazole phosphonites.372, 385 Borane-protected ethynyl phosphonites were converted to triazoles via CuAAC. Subsequent borane deprotection by DABCO yielded active triazole phosphonite for Staudinger-phosphonite ligation.
Staudinger-phosphonite ligations have also been performed with vinyl and ethynylphosphonite probes (123a–b, Figure 48). The resulting phosphonamidate products (124a–b) comprise Michael-type acceptors that are subject to additional selective reactions.383–384, 388 One of the most common reactions involves cysteine addition. This mode of reactivity was exploited for protein and antibody bioconjugation. The resulting thioether conjugates (125a–b) displayed excellent stability in neutral and basic conditions over several days. Also, thiol exchange was not observed in the presence of thiols. The ability to perform sequential chemoselective reactions with unsaturated phosphonites expands the scope of the Staudinger-phosphonite reaction.
Figure 48.

Phosphonamidate groups ligate thiols via Michael-type additions.385, 389 Electron-rich vinyl and ethynyl phosphonites (123a–b) react with azide handles to yield electrophilic phosphonamidate products (124a–b). Thiols selectively add into the unsaturated carbon substituent to provide thioether adducts 125a–b.
2.6.2. Biological applications
Over the past ten years, Staudinger-type reactions with phosphite and phosphonite probes have been developed for biological application. The propensity of phosphites and phosphonites to oxidize under ambient conditions necessitated the development of more stable derivatives. As noted in the preceding section, improved reagents can now efficiently ligate azido biomolecules in biological buffers, even at low concentrations. The Staudinger-phosphite ligation was initially used to synthesize mimics of native biological linkages and functionalize biomolecules.368, 374, 376–377 In recent years, the reaction has been used to synthesize additional phosphorylated peptide and protein mimics that have been historically difficult to access. The Staudinger-phosphonite reaction has also been used for bioconjugation reactions to modify biological therapeutics for enhanced efficacy.378–379, 389 Modular phosphonite synthesis has enabled a wider scope of phosphonamidates to be accessed, including glycoconjugates and antibody-drug conjugates.375, 381 Recent applications of Staudinger-phosphite and -phosphonite ligations are discussed in more detail below.
The Staudinger-phosphite reaction has been used to produce site-specifically phosphorylated oligopeptide targets. Protein phosphorylation is a common PTM with important roles in biological processes, but a complete picture is lacking due to limited methods to prepare pure samples.390–391 Early work from the Hackenberger group leveraged photocleavable phosphites and azide-modified peptides and proteins to examine tyrosine phosphorylation.368 Following conjugation, the photocleavable group was removed via irradiation to yield phosphorylated residues. Recently, this strategy was extended to prepare more delicate phosphorylated lysine residues. Lysine phosphorylation is known to occur naturally, but has not been easy to profile due to the acid labile P–N bond. Phosphorylated Lys (pLys) groups are also not tolerant of the acidic deprotection conditions commonly employed in solid-phase peptide synthesis. The Staudinger-phosphite reaction provided a means to circumvent these issues and access pLys at defined positions in peptides (Figure 49).378–379 Toward this end, azidonorleucine was substituted for lysine in the target peptide. Phosphites bearing nitrobenzyl (126a) or base-cleavable cyanoethyl substituents (126b) were synthesized and reacted with the azido peptides. Phosphoramidate conjugates were formed in reasonable yields (50 μM peptide, 5 mM 126a–b, 45 °C, 500 mM Tris-HCl, pH 8.2, 30–50%).378 The use of acid-cleavable resins necessitated that the peptide was first removed from the solid support and deprotected prior to phosphoramidate formation. The phoshoramidate was then subsequently cleaved by UV irradiation or basic conditions to provide the desired pLys conjugate. When base-cleavable resins were used, the Staudinger-phosphite reaction could be performed directly on the resin.378 Subsequent removal from the solid support and cleavage of the base-labile phosphoramidate substituents yielded the pLys-containing peptide. The model pLys peptides were used in a variety of cellular experiments. Importantly, the pLys modification was found to be stable in cellular environments.
Figure 49.

The Staudinger-phosphite reaction can provide phosphorylated lysine residues.378, 379 Peptide or proteins labeled with azidonorleucine were ligated with phosphite reagents bearing cleavable groups (126a–b). Phosphorimate products can be subsequently cleaved via photoirradiation (top pathway) or base-mediated elimination (bottom pathway) to yield mimics of lysine phosphorylation.
The Staudinger-phosphite reaction has also been applied as a synthetic tool to access phosphoramidate-linked glycoconjugates. Pure, homogenous glycoconjugates are useful for studying numerous cell and molecular recognition events, but they have been difficult to synthesize or isolate from natural sources.392 The Staudinger-phosphite ligation provides a convenient method to prepare pure glycopeptide mimics. Early on, the Hackenberger lab showed how to access such conjugates using glycosyl azides and peptidyl phosphite residues. The resulting structures mimic naturally occurring phosphate-linked glycopeptides.374 This strategy has been expanded to include other classes of glycosidic linkages including glyco-phosphodiesters. These molecules are present in pathogen virulence factors, and their multivalent display is crucial for binding and recognition events.393–394 To synthesize multivalent mimics, symmetrical glycosyl phosphites were used to conjugate azidopeptides and polymers.375 Phosphoramidate products comprised two carbohydrates conjugated to peptides or polyglycerols.375
In addition to synthesizing biomolecule mimics and PTMs, the Staudinger-phosphite reaction has been used to access biomolecules with improved biological stability. For example, the Staudinger-phosphite reaction is well suited to ligate PEG groups onto biological proteins. PEG groups provide enhanced solubility and stability for protein therapeutics.381, 395 As a proof of concept, an apoptosis-inducing peptide, BH3, was modified using a PEG-functionalized phosphite reagent 127 (Figure 50A, TRIS buffer, pH 8.2).395 The resulting BH3-PEG2000 hybrid displayed a 57-fold increase in proteolytic stability in Jurkat cell lysate. When the conjugate was introduced in cells, a dose-dependent increase in apoptotic activity was observed. The same PEG-phosphite 127 was used to site-specifically PEGylate erythropoietin (EPO, Figure 50B).381 Numerous synthetic and semi-synthetic methods have been explored to produce bioactive EPO. EPO requires specific glycosylation patterns for bioavailability and biological activity.396 Current methods to produce EPO often fail to recapitulate these patterns. Traditional expression methods provide mixtures of isoforms, some of which can be immunogenic.397 Efforts to produce more homogenous, efficacious, and bioavailable EPO derivatives have proven difficult. 398 The Staudinger-phosphite reaction was used to examine whether PEG groups could be substituted for glycans at defined residues, simplifying the overall conjugate. Toward this end, the unnatural amino acid azido phenylalanine (AzF) was site-specifically installed at three positions (K24, K38, or K83) in EPO. The Staudinger-phosphite reaction was performed on the modified proteins using PEG-phosphite 127 (40 μM EPO-AzF, 8 mM phosphite, 3 M guanidinium hydrochloride, 20 mM tris-HCl, pH 8, 40 °C, 3 days). The PEGylated proteins were found to be more soluble and stable to proteolysis, and less prone to aggregation. Importantly, in vitro bioactivity was maintained with and, in some cases enhanced by, PEGylation.
Figure 50.

The Staudinger-phosphite reaction can modify protein targets. (A) BH3, an apoptosis-inducing peptide, was outfitted with an azide handle. The peptide was conjugated to two PEG groups via Staudinger-phosphite reaction. The resulting conjugates displayed improved proteolytic stability compared to unmodified BH3.395 (B) Genetic code expansion was used to site-specifically introduce azidophenylalanine (AzF) into erythropoietin (EPO, PDB: 1BUY).381 Subsequent Staudinger-phosphite reaction was used to PEGylate EPO, improving its stability.
In addition to introducing PEG groups for biological stability, the Staudinger-phosphite reaction has been used to append lipids and other substituents to proteins. Such groups can enhance intracellular delivery of peptide reagents. Peptides functionalized with a single hydrophobic carbon chain can form micelle-like structures that can permeate mammalian cell membranes.380 In one example, a model peptide was synthesized with an N-terminal aryl azide. The Staudinger-phosphite reaction was used to attach a C18 alkyl chain to the peptide (2.3 mM peptide, 34 mM phosphite, DMSO, 16 h, 30 °C, 31%). The phosphite probe comprised the C18 chain and two picolyl substituents. Picolyl groups, similar to benzyl substituents, are readily hydrolyzed from phosphorimidate intermediates to selectively transfer alkyl chains to the desired target.381 To confirm that the resulting lipidated peptides could form micelles, dynamic light scattering measurements were performed. Cell permeability was also examined using a fluorescent version of the peptide. Only lipidated peptides effectively penetrated HeLa cells, as determined by fluorescence microscopy.
Bioconjugation reactions can also be performed with the Staudinger-phosphonite ligation. In an initial example, polyglycerol azide was functionalized with a triazole-lactose-phosphonite.385 More recently, the Staudinger-phosphonite reaction was applied to immobilize protein targets.372 One protein examined was Rasa1. Rasa1 binds a phosphotyrosine residue on the human T-cell protein ADAP. Rasa1 was engineered to display AzF at Tyr2. Incubation with biotinylated phosphonite in Tris-HCl (pH 8.0) provided functionalized Rasa1. Streptavidin-immobilized Rasa1 retained its affinity for ADAP and could be further purified via affinity chromatography.
Additional tuning of the Staudinger-phosphonite reaction enabled temporal attachment and cleavage of phosphonamidate-linked bioconjugates for protein purification. For example, a labile biotin was conjugated via 2-nitrobenzyl phosphonamidate to a model peptide. This linkage was leveraged for temporal release of the native protein from streptavidin beads.382 After binding, irradiation of the immobilized phosphonamidate conjugate provided a phosphonamidate acid. This intermediate was subjected to acidic conditions (pH 2) to cleave the P–N bond and release the native amine.
As described earlier, electron-deficient ethynyl or vinyl phosphonamidates (the products of Staudinger-phosphonite ligation) can react further with thiols via Michael-type addition.383–384, 388, 399 This layer of reactivity has been leveraged for attaching various moieties to antibodies to form antibody-drug conjugates (ADC). Common ADC synthesis methods rely on maleimide-cysteine addition. These methods can be somewhat limited due to maleimide exchange with reactive thiols in albumin, glutathione, and other cysteines, leading to premature release of cytotoxic payloads and potential off-target effects.400 For example, one clinically used conjugate, Adcetris, comprises trastuzumab conjugated to monomethylauristatin E (MMAE), a potent chemotherapeutic. The efficacy of Adcetris is limited by maleimide exchange in circulation. 383 Phosphonamidate-linked MMAE could provide additional stability. Toward this end, an antibody-drug conjugate was synthesized (Figure 51). First, an ethynylphosphonamidate-MMAE conjugate 128 was formed via the Staudinger-phosphonite reaction. Next, reagent 128 was conjugated to trastuzumab via Michael-type addition. Prior treatment with dithiothreitol (DTT) was necessary (Figure 51, 1000 equiv. DTT, then 80 equiv. 128).388 Mass spectrometry analysis revealed an average drug-to-antibody ratio (DAR) of 3.8, similar to the DAR of Adcetris.383 Importantly, the phosphonamidate ADC was more stable in rat serum, retaining 90% of its payload after seven days. By comparison, Adcetris retained only 30% of the drug under similar conditions. The phosphonamidate ADC also exhibited comparable efficacy to a clinically used therapy in cell proliferation assays, in addition to cell selectivity.388 The phosphonamidite ADC was also more effective at a lower dose in vivo.
Figure 51.

Alkyne-modified Staudinger-phosphonite products react with cysteine residues.383,388 Antibodies are reduced with dithiothreitol (DTT) to unveil cysteine residues. The cysteine residues can then be ligated with ethynyl phosphonamidate 128 to attach payloads to the antibody target (DAR = n = 3.8). The conjugated group comprised a short protease-sensitive linker and cytotoxic drug, monomethylauristatin E (MMAE). The resulting thioether adduct demonstrated improved stability and efficacy in vivo.
In addition to ADCs, vinyl phosphonamidates have been used in cysteine selective peptide stapling (Figure 52).384 Peptide staples are an attractive means to synthesize macrocyclic drugs, molecules that often exhibit improved proteolytic stability.401–403 Staudinger-phosphonite ligations have been leveraged for stapling peptides via phosphonamidate linkages. In a recent example, a peptide comprising flanking azide and cysteine motifs (129) was cyclized through a two-step process. First, peptide 129 was reacted with vinyl phosphonite 130 via a Staudinger-phosphonite reaction. Second, a conjugate addition reaction between the vinyl group and terminal cysteine formed cyclic peptide 131. This strategy was applied as a synthetic technique to access inhibitors for helical protein-protein interactions.384
Figure 52.

The Staudinger-phosphonite reaction was used to form peptide macrocycles.384 Linear oligopeptide 129 was functionalized with terminal azide and thiol groups using SPPS. Azide-phosphonite ligation followed by thiol conjugate addition yielded the cyclized adduct 131.
2.6.3. Chemical and biological scope
Staudinger-phosphite and -phosphonite reactions are mild, chemoselective techniques to access many biologically relevant motifs. These reactions have enabled syntheses of a variety of bioconjugates with peptides, proteins, and other biomolecules, even in aqueous conditions. Ethynyl and vinyl phosphonite probes have further enabled quick and modular ‘click’ ligations for additional functionalization. The activated alkynes and alkenes have since been used for antibody conjugation and peptide stapling. An array of Staudinger-phosphite and -phosphonite bioconjugation techniques has been developed for selective biomolecule synthesis.
Other cysteine-selective reactions have been developed as complimentary bioconjugation methods.404 For example, the Hackenberger lab exploited phosphonthiolates as alternatives to phosphonamidate electrophiles for synthesizing ADCs and other conjugates.405 Some phosphites and phosphonites have also been used to ligate electrophiles other than azides, including disulfides.405 Such reactions can site-selectively install phosphorylate groups on peptides. Another cysteine selective bioconjugation strategy involves thiol-ene reactivity with α,β-unsaturated carbonyl probes.406, 407 Each reaction has its strengths and weaknesses in terms of speed and accessibility. The best choice for a given application is context-dependent.
3. Conclusions and future directions
Since Bertozzi’s seminal report on the Staudinger ligation in 2000, a family of phosphine-based bioorthogonal transformations has emerged. New variants of the Staudinger ligation are enabling rapid reactions inside cells and other complex environments. Many have been leveraged to tag biomolecules with visual probes. In addition to biomolecule labeling, the traceless Staudinger ligation remains a viable method to synthesize biomolecules via native amide bonds. Recent phosphine reactants now provide a broadened substrate scope (see section 2.3). Alongside the classic azide, cyclopropenone and α,β-unsaturated carbonyl groups have been added to the portfolio of chemical reporters (see sections 2.4. and 2.5., respectively). Phosphites and phosphonites have also been introduced as chemoselective and robust agents for protein modification (see section 2.6).
The past decade has seen both new and old phosphine reagents used to construct and probe biomolecules. Cell-permeable triarylphosphines have been introduced as powerful tools to control and interrogate biological processes in living cells (see sections 2.1 and 2.2). When used in conjunction with aryl azide reagents, such probes have enabled visualization of dynamic biological processes in vivo via Staudinger ligation (see section 2.2). As phosphines transition to applications in intracellular environments, the bioorthogonality of phosphines may warrant more rigorous characterization. Phosphines have long been considered among the most biocompatible and chemoselective probes in bioorthogonal chemistry. Indeed, they are among a handful of reactants that can be applied in living animals. However, highly nucleophilic phosphines are known reducing agents for disulfide bond in proteins. A more rigorous characterization of phosphine bioorthogonality may be useful to demarcate reagent utility and identify potential avenues for further reagent tuning.
The widespread adoption of the original Staudinger ligation was due, in part, to the commercial availability of the reagents and easily understandable protocols. While reagents remain accessible for this reaction, several of the reactants for newer transformations are not yet available from commercial sources. We anticipate that the increased availability of these recent transformations would encourage their adoption by a broad audience of chemical biologists. Indeed, reagent and protocol accessibility are key concepts to transition these reactions from the “tool-maker” to the “tool-user”.
Along similar lines, more careful comparisons of bioorthogonal reactions under defined conditions would help to identify the most effective chemistry for a given application. Such comparisons are useful to determine each reaction’s advantages and disadvantages. Furthermore, such studies would guide both experts and non-experts seeking to apply bioorthogonal reactions in novel pursuits. The broadened and improved spectrum of phosphine reactivity, combined with improved availability, should enable new and diverse biological investigations in the coming years. We anticipate that the collection of phosphines and related analogs will be key chemical tools in the modern bioorthogonal reaction toolbox.
Acknowledgments
J.A.P. is a Cottrell Scholar, Alfred P. Sloan Fellow, and Dreyfus Scholar. This work was supported by the U.S. National Institutes of Health (award no. R01 GM126226 to J.A.P.). We thank members of the Prescher laboratory for manuscript edits and helpful discussions. BioRender (https://biorender.com/) was used to prepare some figures in this text.
Abbreviations used
- AARS
aminoacyl-tRNA synthease
- ADC
antibody-drug conjugate
- AzF
4-azido-l-phenylalanine
- AznL
ε-azidonorleucine
- BoCFO
bioorthogonal cleavable fluorescent oligonucleotides
- BLI
bioluminescence imaging
- CD3CN
acetonitrile-d3
- CH3CN
acetonitrile
- CpO
cyclopropenone
- CpS
cyclopropenethione
- CpN+
cyclopropeniminium
- CRISPR
clustered regularly interspaced short palindromic repeats
- CuAAC
CuI-catalyzed azide–alkyne cycloaddition
- Da
dalton
- DABCO
1,4-diazabicyclo[2.2.2]octane
- DAR
drug-to-antibody ratio
- DFT
density-functional theory
- DNA
deoxyribonucleic acid
- DNP
2,4-dinitrophenol
- DMF
dimethylformamide
- DMSO
dimethylsulfoxide
- eGFP
enhanced green fluorescent protein
- EPO
erythropoietin
- E1cB
elimination unimolecular conjugate base
- GPCR
G protein-coupled receptor
- FLuc
firefly luciferase
- GalNAc
N-acetylgalactosamine
- GlcNAc
N-acetylglucosamine
- GFP
green fluorescent protein
- GlcNAz
N-azidoacetylglucosamine
- GLUT1
glucose transporter 1
- GPCR
G protein-coupled receptor
- HC
hemicyanine
- H. pylori
Heliobacter pylori
- IEDDA
inverse-electron demand Diels–Alder
- IPTG
isopropyl-β-d-1-thiogalactopyranoside
- IR
infrared
- LUMO
lowest unoccupied molecular orbital
- MeOH
methanol
- miRNA
microRNA
- ManNAc
N-acetylmannosamine
- ManNAz
N-azidoacetylmannosamine
- MetRS
methionyl-tRNA synthetase
- MMAE
monomethylauristatin E
- NCL
native chemical ligation
- NLS
nuclear localization sequence
- NMR
nuclear magnetic resonance
- PEG
polyethylene glycol
- PET
positron-emission tomography
- PBS
phosphate-buffered saline
- PNA
peptide nucleic acid
- POI
protein of interest
- PTM
post-translational modification
- PylRS
pyrrolysyl-tRNA synthetase
- Q-STAR
quenched Staudinger-triggered α-azidoether release
- QUAL
quenched autoligation
- RanGAP1
Ran GTPase-activating protein 1
- RAFT
reversible addition-fragmentation chain transfer
- RCA
rolling circle amplification
- RNA
ribonucleic acid
- rRNA
ribosomal RNA
- SDS-PAGE
sodium dodecyl-sulfate polyacrylamide gel electrophoresis
- SAM
self-assembled monolayer
- sgRNA
single guide RNA
- SN2
biomolecular nucleophilic substitution
- SPAAC
strain-promoted azide–alkyne cycloaddition
- SPPS
solid-phase peptide synthesis
- SuFEx
sulfur(VI) fluoride exchange
- SrtA
sortase A
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TCEP
tris(2-carboxyethyl)phosphine
- TCO
trans-cyclooctene
- THPP
tris(hydroxypropyl)phosphine
- TMTH
tetramethylthiacycloheptyne
- TPP-DNA
triphenylphosphine-modified DNA
- TPPMS
diphenylphosphinobenzene-3-sulfonate
- TyrRS
tyrosyl-tRNA synthetase
- Tz
tetrazine
- tRNA
transfer RNA
- UAA
unnatural amino acid
Biographies
Tyler K. Heiss received his undergraduate degree in Biochemistry from the University of Delaware in 2016. He worked with Professor Catherine Leimkuhler-Grimes to study bacterial cell wall biosynthesis with unnatural metabolic precursors. In 2016, he enrolled in graduate school at the University of California, Irvine. Under the guidance of Professor Jennifer Prescher, his research focuses on developing new bioorthogonal reactions to probe biological systems.
Robert S. Dorn received his undergraduate degree in Chemistry from Whitman College. Working in the lab of Professor Marion Götz, he synthesized non-covalent macrocyclic peptide proteasomal inhibitors to treat multiple myeloma. In 2018, he enrolled in graduate school at the University of California, Irvine. In the laboratory of Professor Jennifer Prescher, his research focuses on developing new bioorthogonal reactions and biological tools to probe cellular systems.
Jennifer A. Prescher is a Professor of Chemistry, Molecular Biology & Biochemistry, and Pharmaceutical Sciences at UC Irvine. She earned her B.S. degree at the University of Wisconsin–La Crosse in 2001 and obtained her Ph.D. in Chemistry at UC Berkeley in 2006 with Prof. Carolyn Bertozzi. She conducted postdoctoral research with Prof. Christopher Contag at Stanford University before joining the UC Irvine faculty in 2010.
Footnotes
The authors declare no competing financial interests.
REFERENCES
- 1.Staudinger H; Meyer J Über Neue Organische Phosphorverbindungen III. Phosphinmethylenderivate und Phosphinimine. Helv. Chim. Acta 1919, 2, 635–646. [Google Scholar]
- 2.Guo H; Fan YC; Sun Z; Wu Y; Kwon O Phosphine Organocatalysis. Chem. Rev 2018, 118, 10049–10293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tolman CA Steric Effects of Phosphorus Ligands in Organometallic Chemistry and Homogeneous Catalysis. Chem. Rev 1977, 77, 313–348. [Google Scholar]
- 4.Surry DS; Buchwald SL Biaryl Phosphane Ligands in Palladium-Catalyzed Amination. Angew. Chem., Int. Ed 2008, 47, 6338–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Surry DS; Buchwald SL Dialkylbiaryl Phosphines in Pd-Catalyzed Amination: A User’s Guide. Chem. Sci 2011, 2, 27–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saxon E; Bertozzi CR Cell Surface Engineering by a Modified Staudinger Reaction. Science 2000, 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- 7.Prescher JA; Bertozzi CR Chemistry in Living Systems. Nat. Chem. Biol 2005, 1, 13–21. [DOI] [PubMed] [Google Scholar]
- 8.Sletten EM; Bertozzi CR Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem., Int. Ed 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sletten EM; Bertozzi CR From Mechanism to Mouse: a Tale of Two Bioorthogonal Reactions. Acc. Chem. Res 2011, 44, 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Köhn M; Breinbauer R The Staudinger Ligation-a Gift to Chemical Biology. Angew. Chem., Int. Ed 2004, 43, 3106–3116. [DOI] [PubMed] [Google Scholar]
- 11.Debets MF; van der Doelen CW; Rutjes FP; van Delft FL Azide: A Unique Dipole for Metal-Free Bioorthogonal Ligations. ChemBioChem 2010, 11, 1168–1184. [DOI] [PubMed] [Google Scholar]
- 12.van Berkel SS; van Eldijk MB; van Hest JCM Staudinger Ligation as a Method for Biocojugation. Angew. Chem., Int. Ed 2011, 50, 8806–8827. [DOI] [PubMed] [Google Scholar]
- 13.Schilling CI; Jung N; Biskup M; Schepers U; Bräse S Bioconjugation via Azide–Staudinger Ligation: an Overview. Chem. Soc. Rev 2011, 40, 4840–4871. [DOI] [PubMed] [Google Scholar]
- 14.Hackenberger CPR; Schwarzer D Chemoselective Ligation and Modification Strategies for Peptides and Proteins. Angew. Chem., Int. Ed 2008, 47, 10030–10074. [DOI] [PubMed] [Google Scholar]
- 15.McGrath NA; Raines RT Chemoselectivity in Chemical Biology: Acyl Transfer Reactions with Sulfur and Selenium. Acc. Chem. Res 2011, 44, 752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y-X; Triola G; Waldmann H Bioorthogonal Chemistry for Site-Specific Labeling and Surface Immobilization of Proteins. Acc. Chem. Res 2011, 44, 762–773. [DOI] [PubMed] [Google Scholar]
- 17.Bednarek CW, I.; Jung N; Schepers U; Bräse S The Staudinger Ligation. Chem. Rev 2020, 120, 4301–4354. [DOI] [PubMed] [Google Scholar]
- 18.Spicer CD; Davis BG Selective Chemical Protein Modification. Nat. Commun 2014, 5, 4740. [DOI] [PubMed] [Google Scholar]
- 19.Lang K; Chin JW Cellular Incorporation of Unnatural Amino Acids and Bioorthogonal Labeling of Proteins. Chem. Rev 2014, 114, 4764–4806. [DOI] [PubMed] [Google Scholar]
- 20.Lang K; Chin JW Bioorthogonal Reactions for Labeling Proteins. ACS Chem. Biol 2014, 9, 16–20. [DOI] [PubMed] [Google Scholar]
- 21.Shih H-W; Kamber DN; Prescher JA Building Better Bioorthogonal Reactions. Curr. Opin. Chem. Biol 2014, 21, 103–111. [DOI] [PubMed] [Google Scholar]
- 22.Patterson DM; Nazarova LA; Prescher JA Finding the Right (Bioorthogonal) Chemistry. ACS Chem. Biol 2014, 9, 592–605. [DOI] [PubMed] [Google Scholar]
- 23.Row RD; Prescher JA Constructing New Bioorthogonal Reagents and Reactions. Acc. Chem. Res 2018, 51, 1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKay CS; Finn MG Click Chemistry in Complex Mixtures: Bioorthogonal Bioconjugation. Cell Chem. Biol 2014, 21, 1075–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramakers BEI; van Hest JCM; Löwik DWPM Molecular Tools for the Construction of Peptide-Based Materials. Chem. Soc. Rev 2014, 43, 2743–2756. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z-PA; Tian C-L; Zheng J-S The Recent Developments and Applications of the Traceless-Staudinger Reaction in Chemical Biology Study. RSC Adv. 2015, 5, 107192–107199. [Google Scholar]
- 27.Boutureira O; Bernardes GJ Advances in Chemical Protein Modification. Chem. Rev 2015, 115, 2174–2195. [DOI] [PubMed] [Google Scholar]
- 28.Mamat C; Gott M; Steinbach J Recent Progress Using the Staudinger Ligation for Radiolabeling Applications. J. Label Compd. Radiopharm 2017, 61, 165–178. [DOI] [PubMed] [Google Scholar]
- 29.Schäfer RJB; Aronoff MR; Wennemers H Recent Advances in Bioorthogonal Reactions. Chimia 2019, 73, 308–312. [DOI] [PubMed] [Google Scholar]
- 30.Li J; Chen PR Development and Application of Bond Cleavage Reactions in Bioorthogonal Chemistry. Nat. Chem. Biol 2016, 12, 129–137. [DOI] [PubMed] [Google Scholar]
- 31.Tu J; Xu M; Franzini RM Dissociative Bioorthogonal Reactions. ChemBioChem 2019, 20, 1615–1627. [DOI] [PubMed] [Google Scholar]
- 32.Tanimoto H; Kakiuchi K Recent Applications And Developments of Organic Azides in Total Synthesis of Natural Products. Nat. Prod. Commun 2013, 8, 1021–1034. [PubMed] [Google Scholar]
- 33.Nie L-D; Shi X-X; Ko KH; Lu W-D A Short And Practical Synthesis of Oseltamivir Phosphate (Tamiflu) From (−)-Shikimic Acid. J. Org. Chem 2009, 74, 3970–3973. [DOI] [PubMed] [Google Scholar]
- 34.Shibata A; Ito Y; Abe H RNA-Templated Molecule Release Induced Protein Expression in Bacterial Cells. Chem. Commun 2013, 49, 270–272. [DOI] [PubMed] [Google Scholar]
- 35.Luo J; Liu Q; Morihiro K; Deiters A Small-Molecule Control of Protein Function through Staudinger Reduction. Nat. Chem 2016, 8, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wesalo JS; Luo J; Morihiro K; Liu J; Deiters A Phosphine-Activated Lysine Analogues for Fast Chemical Control of Protein Subcellular Localization and Protein SUMOylation. ChemBioChem 2020, 21, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakurai K; Snyder TM; Liu DR DNA-Templated Functional Group Transformations Enable Sequence-Programmed Synthesis Using Small-Molecule Reagents. J. Am. Chem. Soc 2005, 127, 1660–1661. [DOI] [PubMed] [Google Scholar]
- 38.Pianowski ZL; Winssinger N Fluorescence-Based Detection of Single Nucleotide Permutation in DNA Via Catalytically Templated Reaction. Chem. Commun 2007, 3820–3822. [DOI] [PubMed] [Google Scholar]
- 39.Pianowski Z; Gorska K; Oswald L; Merten CA; Winssinger N Imaging of mRNA in Live Cells Using Nucleic Acid-Templated Reduction of Azidorhodamine Probes. J. Am. Chem. Soc 2009, 131, 6492–6497. [DOI] [PubMed] [Google Scholar]
- 40.Franzini RM; Kool ET Efficient Nucleic Acid Detection by Templated Reductive Quencher Release. J. Am. Chem. Soc 2009, 131, 16021–16023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lukasak B; Morihiro K; Deiters A Aryl Azides as Phosphine-Activated Switches for Small Molecule Function. Sci. Rep 2019, 9, 1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saneyoshi H; Ochikubo T; Mashimo T; Hatano K; Ito Y; Abe H Triphenylphosphinecarboxamide: an Effective Reagent for the Reduction of Azides and its Application to Nucleic Acid Detection. Org. Lett 2014, 16, 30–33. [DOI] [PubMed] [Google Scholar]
- 43.Habibian M; McKinlay C; Blake TR; Kietrys AM; Waymouth RM; Wender PA; Kool ET Reversible RNA Acylation for Control of CRISPR–Cas9 Gene Editing. Chem. Sci 2020, 11, 1011–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fottner M; Brunner A-D; Bittl V; Horn-Ghetko D; Jussupow A; Kaila VRI; Bremm A; Lang K Site-Specific Ubiquitylation and SUMOylation Using Genetic-Code Expansion and Sortase. Nat. Chem. Biol 2019, 15, 276–284. [DOI] [PubMed] [Google Scholar]
- 45.Leffler JE; Temple RD Staudinger Reaction Between Triarylphosphines and Azides. A Study of the Mechanism. J. Am. Chem. Soc 1967, 89, 5235–5246. [Google Scholar]
- 46.Leffler JE; Tsuno Y Some Decomposition Reactions of Acid Azides. J. Org. Chem 1963, 28, 902–906. [Google Scholar]
- 47.Franzini RM; Kool ET Two Successive Reactions on a DNA Template: a Strategy for Improving Background Fluorescence and Specificity in Nucleic Acid Detection. Chem. Eur. J 2011, 17, 2168–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H; Franzini RM; Bruner C; Kool ET Templated Chemistry for Sequence-Specific Fluorogenic Detection of Duplex DNA. ChemBioChem 2010, 11, 2132–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franzini RM; Kool ET Improved Templated Fluorogenic Probes Enhance the Analysis of Closely Related Pathogenic Bacteria by Microscopy and Flow Cytometry. Bioconjugate Chem. 2011, 22, 1869–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alouane A; Labruère R; Le Saux T; Schmidt F; Jullien L Self-Immolative Spacers: Kinetic Aspects, Structure–Property Relationships, and Applications. Angew. Chem., Int. Ed 2015, 54, 7492–7509. [DOI] [PubMed] [Google Scholar]
- 51.Park HS; Kietrys AM; Kool ET Simple Alkanoyl Acylating Agents for Reversible RNA Functionalization and Control. Chem. Commun 2019, 55, 5135–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cimecioglu AL; Ball DH; Kaplan DL; Huang SH Preparation of Amylose Derivatives Selectively Modified at C-6. 6-Amino-6-Deoxyamylose. Macromolecules 1994, 27, 2917–2922. [Google Scholar]
- 53.Gorska K; Keklikoglou I; Tschulena U; Winssinger N Rapid Fluorescence Imaging of miRNAs in Human Cells Using Templated Staudinger Reaction. Chem. Sci 2011, 2, 1969–1975. [Google Scholar]
- 54.Guo H; Tang W; Duan X A Selective and Sensitive Azido Near-Infrared Fluorescent Probe for Tris(2-carboxyethyl)phosphine Quantitative Detection and its application for E. coli determination. Anal. Methods 2018, 10, 5823–5826. [Google Scholar]
- 55.Lee SH; Wang S; Kool ET Templated Chemistry for Monitoring Damage and Repair Directly in Duplex DNA. Chem. Commun 2012, 48, 8069–8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harcourt EM; Kool ET Amplified microRNA Detection by Templated Chemistry. Nucleic Acids Res. 2012, 40, e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Velema WA; Kool ET Fluorogenic Templated Reaction Cascades for RNA Detection. J. Am. Chem. Soc 2017, 139, 5405–5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mondal M; Liao R; Nazaroff CD; Samuel AD; Guo J Highly Multiplexed Single-Cell in Situ RNA and DNA Analysis With Bioorthogonal Cleavable Fluorescent Oligonucleotides. Chem. Sci 2018, 9, 2909–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morihiro K; Ankenbruck N; Lukasak B; Deiters A Small Molecule Release and Activation through DNA Computing. J. Am. Chem. Soc 2017, 139, 13909–13915. [DOI] [PubMed] [Google Scholar]
- 60.Kadina A; Kietrys AM; Kool ET RNA cloaking by Reversible Acylation. Angew. Chem., Int. Ed 2018, 57, 3059–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pawlak JB; Gential GPP; Ruckwardt TJ; Bremmers JS; Meeuwenoord NJ; Ossendorp FA; Overkleeft HS; Filippov DV; van Kasteren SI Bioorthogonal Deprotection on the Dendritic Cell Surface for Chemical Control of Antigen Cross-Presentation. Angew. Chem., Int. Ed 2013, 54, 5628–5631. [DOI] [PubMed] [Google Scholar]
- 62.Yanagisawa T; Ishii R; Fukunaga R; Kobayashi T; Sakamoto K; Yokoyama S Multistep Engineering Of Pyrrolysyl-tRNA Synthetase to Genetically Encode Nɛ-(o-azidobenzyloxycarbonyl) Lysine for Site-Specific Protein Modification. Chem. Biol 2008, 15, 1187–1197. [DOI] [PubMed] [Google Scholar]
- 63.Rape M Ubiquitylation At The Crossroads of Development and Disease. Nat. Rev. Mol. Cell Biol 2018, 19, 59–70. [DOI] [PubMed] [Google Scholar]
- 64.Wang ZA; Zeng Y; Kurra Y; Wang X; Tharp JM; Vatansever EC; Hsu WW; Dai S; Fang X, et al. A Genetically Encoded Allysine for the Synthesis of Proteins with Site-Specific Lysine Dimethylation. Angew. Chem., Int. Ed 2017, 56, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Davies S; Qiao L; Oliveira BL; Navo CD; Jiménez-Osés G; Bernardes GJL Tetrazine-Triggered Release of Carboxylic-Acid-Containing Molecules for Activation of an Anti-Inflammatory Drug. ChemBioChem 2019, 20, 1541–1546. [DOI] [PubMed] [Google Scholar]
- 66.Wang X; Liu Y; Fan X; Wang J; Ngai WSC; Zhang H; Li J; Zhang G; Lin J; Chen PR Copper-Triggered Bioorthogonal Cleavage Reactions for Reversible Protein and Cell Surface Modifications. J. Am. Chem. Soc 2019, 141, 17133–17141. [DOI] [PubMed] [Google Scholar]
- 67.Zhang G; Li J; Xie R; Fan X; Liu Y; Zheng S; Ge Y; Chen PR Bioorthogonal Chemical Activation of Kinases in Living Systems. ACS Cent. Sci 2016, 2, 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khoury GA; Baliban RC; Floudas CA Proteome-Wide Post-Translational Modification Statistics: Frequency Analysis and Curation of the Swiss-Prot Database. Sci. Rep 2011, 1, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Solís DB, N. V; Davis AP; Jiménez-Barbero J; Romero A; Roy R; Smetana K Jr.; Gabius H-J A Guide into Glycosciences: How Chemistry, Biochemistry And Biology Cooperate to Crack the Sugar Code. Biochim. Biophys. Acta 2015, 1850, 186–235. [DOI] [PubMed] [Google Scholar]
- 70.Moremen KW; Tiemeyer M; Nairn AV Vertebrate Protein Glycosylation: Diversity, Synthesis and Function. Nat. Rev. Mol. Cell. Biol 2012, 13, 448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mahal LK; Yarema KJ; Bertozzi CR Engineering Chemical Reactivity on Cell Surfaces through Oligosaccharide Biosynthesis. Science 1997, 276, 1125–1128. [DOI] [PubMed] [Google Scholar]
- 72.Saxon E; Luchansky SJ; Hang HC; Yu C; Lee SC; Bertozzi CR Investigating Cellular Metabolism of Synthetic Azidosugars with the Staudinger Ligation. J. Am. Chem. Soc 2002, 124, 14893–14902. [DOI] [PubMed] [Google Scholar]
- 73.Prescher JA; Dube DH; Bertozzi CR Chemical Remodelling of Cell Surfaces in Living Animals. Nature 2004, 30, 873–877. [DOI] [PubMed] [Google Scholar]
- 74.Vocadlo DJ; Hang HC; Kim E-J; Hanover JA; Bertozzi CR A Chemical Approach for Identifying O-GlcNAc-Modified Proteins in Cells. Proc. Natl. Acad. Sci. U.S.A 2003, 100, 9116–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rabuka D; Hubbard SC; Laughlin ST; Argade SP; Bertozzi CR A Chemical Reporter Strategy to Probe Glycoprotein Fucosylation. J. Am. Chem. Soc 2006, 128, 12078–12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dube DH; Prescher JA; Quang CN; Bertozzi CR Probing Mucin-Type O-Linked Glycosylation in Living Animals. Proc. Natl. Acad. Sci. U.S.A 2006, 103, 4819–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baskin JM; Prescher JA; Laughlin ST; Agard NJ; Chang PV; Miller IA; Lo A; Codelli JA; Bertozzi CR Copper-Free Click Chemistry for Dynamic in Vivo Imaging. Proc. Natl. Acad. Sci. U.S.A 2007, 104, 16793–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kiick KL; Saxon E; Tirrell DA; Bertozzi CR Incorporation of Azides into Recombinant Proteins for Chemoselective Modification by the Staudinger Ligation. Proc. Natl. Acad. Sci. U.S.A 2002, 99, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tsao M-L; Tian F; Schultz PG Selective Staudinger Modification of Proteins Containing p-Azidophenylalanine. ChemBioChem 2005, 6, 2147–2149. [DOI] [PubMed] [Google Scholar]
- 80.Slavoff SA; Chen I; Choi Y-A; Ting AY Expanding the Substrate Tolerance of Biotin Ligase through Exploration of Enzymes from Diverse Species. J. Am. Chem. Soc 2008, 130, 1160–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang CCY; Seo TS; Li Z; Ruparel H; Ju J Site-Specific Fluorescent Labeling of DNA Using Staudinger Ligation. Bioconjugate Chem. 2003, 14, 697–701. [DOI] [PubMed] [Google Scholar]
- 82.Weisbrod SH; Marx A A Nucleoside Triphosphate for Site-Specific Labelling of DNA by the Staudinger Ligation. Chem. Commun 2007, 18, 1828–1830. [DOI] [PubMed] [Google Scholar]
- 83.Chang PV; Prescher JA; Hangauer MJ; Bertozzi CR Imaging Cell Surface Glycans with Bioorthogonal Chemical Reporters. J. Am. Chem. Soc 2007, 129, 8400–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chandra RA; Douglas ES; Mathies RA; Bertozzi CR; Francis MB Programmable Cell Adhesion Encoded by DNA Hybridization. Angew. Chem., Int. Ed 2006, 45, 896–901. [DOI] [PubMed] [Google Scholar]
- 85.Köhn M; Wacker R; Peters C; Schröder H; Soulére L; Breinbauer R; Niemeyer CM; Waldmann H Staudinger Ligation: a New Immobilization Strategy for the Preparation of Small-Molecule Arrays. Angew. Chem., Int. Ed 2003, 42, 5830–5834. [DOI] [PubMed] [Google Scholar]
- 86.Zhang H; Ma Y; Sun X-L Chemically-Selective Surface Glyco-Functionalization of Liposomes through Staudinger Ligation. Chem. Commun 2009, 21, 3032–3034. [DOI] [PubMed] [Google Scholar]
- 87.Köhn M Immobilization Strategies for Small Molecule, Peptide and Protein Microarrays. J. Pept. Sci 2009, 15, 393–397. [DOI] [PubMed] [Google Scholar]
- 88.Lemieux GA; de Graffenried CL; Bertozzi CR A Fluorogenic Dye Activated by the Staudinger Ligation. J. Am. Chem. Soc 2003, 125, 4708–4709. [DOI] [PubMed] [Google Scholar]
- 89.Hangauer MJ; Bertozzi CR A FRET-Based Fluorogenic Phosphine for Live-Cell Imaging with the Staudinger Ligation. Angew. Chem., Int. Ed 2008, 47, 2394–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shieh P; Bertozzi CR Design Strategies for Bioorthogonal Smart Probes. Org. Biomol. Chem 2014, 12, 9307–9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lin FL; Hoyt HM; van Halbeek H; Bergman RG; Bertozzi CR Mechanistic Investigation of the Staudinger Ligation. J. Am. Chem. Soc 2005, 127, 2686–2695. [DOI] [PubMed] [Google Scholar]
- 92.Maric T; Mikhaylov G; Khodakivskyi P; Bazhin A; Sinisi R; Bonhoure N; Yevtodiyenko A; Jones A; Muhunthan V; Abdelhady G, et al. Bioluminescent-Based Imaging and Quantification of Glucose Uptake in Vivo. Nat. Methods 2019, 16, 526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Burns JA; Butler JC; Moran J; Whitesides GM Selective Reduction of Disulfides by Tris(2-carboxyethyl)phosphine. J. Org. Chem 1991, 56, 2648–2650. [Google Scholar]
- 94.Soellner MB; Nilsson BL; Raines RT Staudinger Ligation of α-Azido Acids Retains Stereochemistry. J. Org. Chem 2002, 67, 4993–4996. [DOI] [PubMed] [Google Scholar]
- 95.Mühlberg M; Jaradat DM; Kleineweischede R; Papp I; Dechtrirat D; Muth S; Broncel M; Hackenberger CP Acidic and Basic Deprotection Strategies of Borane-Protected Phosphinothioesters for the Traceless Staudinger Ligation. Bioorg. Med. Chem 2010, 18, 3679–3686. [DOI] [PubMed] [Google Scholar]
- 96.Tam A; Raines RT Protein Engineering with the Traceless Staudinger Ligation. Methods Enzymol. 2009, 462, 25–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Grandjean C; Boutonnier A; Guerreiro C; Fournier JM; Mulard LA On the Preparation of Carbohydrate-Protein Conjugates Using the Traceless Staudinger Ligation. J. Org. Chem 2005, 70, 7123–7132. [DOI] [PubMed] [Google Scholar]
- 98.He Y; Hinklin RJ; Chang J; Kiessling LL Stereoselective N-Glycosylation by Staudinger Ligation. Org. Lett 2004, 6, 4479–4782. [DOI] [PubMed] [Google Scholar]
- 99.Meguro T; Terashima N; Ito H; Koike Y; Kii I; Yoshida S; Hosoya T Staudinger Reaction Using 2,6-Dichlorophenyl Azide Derivatives for Robust Aza-Ylide Formation Applicable to Bioconjugation in Living Cells. Chem. Commun 2018, 54, 7904–7907. [DOI] [PubMed] [Google Scholar]
- 100.Ren G; Zheng Q; Wang H Aryl Fluorosulfate Trapped Staudinger Reduction. Org. Lett 2017, 19, 1582–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shah L; Laughlin ST; Carrico IS Light-Activated Staudinger–Bertozzi Ligation within Living Animals. J. Am. Chem. Soc 2016, 138, 5186–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hammett LP The Effect of Structure upon the Reactions of Organic Compounds. Benzene Derivatives. J. Am. Chem. Soc 1937, 59, 96–103. [Google Scholar]
- 103.Sundhoro M; Jeon S; Park J; Ramström O; Yan M Perfluoroaryl Azide-Staudinger Reaction: a Fast and Bioorthogonal Reaction. Angew. Chem., Int. Ed 2017, 56, 12117–12121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Luo W; Luo J; Popik VV; Workentin MS Dual-Bioorthogonal Molecular Tool: “Click-To-Release” and “Double-Click” Reactivity on Small Molecules and Material Surfaces. Bioconjugate Chem. 2019, 30, 1140–1149. [DOI] [PubMed] [Google Scholar]
- 105.Cheng L; Kang X; Wang D; Gao Y; Yi L; Xi Z The One-Pot Nonhydrolysis Staudinger Reaction and Staudinger or SPAAC Ligation. Org. Biomol. Chem 2019, 17, 5675–5679. [DOI] [PubMed] [Google Scholar]
- 106.Zhang J; Gao Y; Kang X; Zhu Z; Wang Z; Xi Z; Yi L o,o-Difluorination of Aromatic Azide Yields a Fast-Response Fluorescent Probe for H2S Detection and for Improved Bioorthogonal Reactions. Org. Biomol. Chem 2017, 15, 4212–4217. [DOI] [PubMed] [Google Scholar]
- 107.Kang X; Cai X; Yi L; Xi Z Multifluorinated Aryl Azides for the Development of Improved H2S Probes, and Fast Strain-Promoted Azide–Alkyne Cycloaddition and Staudinger Reactions. Chem. Asian. J 2020, 15, 1420–1429. [DOI] [PubMed] [Google Scholar]
- 108.Xie S; Sundhoro M; Houk KN; Yan M Electrophilic Azides for Materials Synthesis and Chemical Biology. Acc. Chem. Res 2020, 53, 937–948. [DOI] [PubMed] [Google Scholar]
- 109.Nguyen T-A; Cigler M; Lang K Expanding the Genetic Code to Study Protein-Protein Interactions. Angew. Chem., Int. Ed 2018, 57, 14350–14361. [DOI] [PubMed] [Google Scholar]
- 110.Xie Y; Cheng L; Gao Y; Cai X; Yang X; Yi L; Xi Z Tetrafluorination of Aromatic Azide Yields a Highly Efficient Staudinger Reaction: Kinetics and Biolabeling. Chem. Asian. J 2018, 13, 1791–1796. [DOI] [PubMed] [Google Scholar]
- 111.Xie S; Lopez SA; Ramström O; Yan M; Houk KN 1,3-Dipolar Cycloaddition Reactivities of Perfluorinated Aryl Azides with Enamines and Strained Dipolarophiles. J. Am. Chem. Soc 2015, 137, 2958–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yoshida S Sequential Conjugation Methods Based on Triazole Formation and Related Reactions Using Azides. Org. Biomol. Chem 2020, 18, 1550–1562. [DOI] [PubMed] [Google Scholar]
- 113.Patterson DM; Prescher JA Orthogonal Bioorthogonal Chemistries. Curr. Opin. Chem. Biol 2015, 28, 141–149. [DOI] [PubMed] [Google Scholar]
- 114.Aubert S; Bezagu M; Spivey AC; Arseniyadis S Spatial and Temporal Control of Chemical Processes. Nat. Rev. Chem 2019, 3, 706–722. [Google Scholar]
- 115.Li J; Kong H; Zhu C; Zhang Y Photo-Controllable Bioorthogonal Chemistry for Spatiotemporal Control of Bio-Targets in Living Systems. Chem. Sci 2020, 11, 3390–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Klán P; Šolomek T; Bochet CG; Blanc A; Givens R; Rubina M; Popik V; Kostikov A; Wirz J Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy. Chem. Rev 2013, 113, 119–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dong J; Krasnova L; Finn MG; Sharpless KB Sulfur(VI) Fluoride Exchange (Sufex): Another Good Reaction for Click Chemistry. Angew. Chem., Int. Ed 2014, 53, 9430–9448. [DOI] [PubMed] [Google Scholar]
- 118.Pham D; Deter CJ; Reinard MC; Gibson GA; Kiselyov K; Yu W; Sandulache VC; St. Croix CM; Koide K Using Ligand-Accelerated Catalysis to Repurpose Fluorogenic Reactions for Platinum or Copper. ACS Cent. Sci 2020, 6, 1772–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gilormini P-A; Batt AR; Pratt MR; Biot C Asking More from Metabolic Oligosaccharide Engineering. Chem. Sci 2018, 9, 7585–7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Belardi B; de la Zerda A; Spiciarich DR; Maund SL; Peehl DM; Bertozzi CR Imaging the Glycosylation State of Cell Surface Glycoproteins by Two-Photon Fluorescence Lifetime Imaging Microscopy. Angew. Chem., Int. Ed 2013, 52, 14045–14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nazarova LA; Ochoa RJ; Jones KA; Morrissette NS; Prescher JA Extracellular Toxoplasma gondii Tachyzoites Metabolize and Incorporate Unnatural Sugars into Cellular Proteins. Microbes Infect. 2016, 18, 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Darabedian N; Gao J; Chuh KN; Woo CM; Pratt MR The Metabolic Chemical Reporter 6-Azido-6-Deoxy-Glucose Further Reveals the Substrate Promiscuity Of O-GlcNAc Transferase and Catalyzes the Discovery of Intracellular Protein Modification by O-Glucose. J. Am. Chem. Soc 2018, 140, 7092–7100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang H; Wang R; Cai K; He H; Liu Y; Yen J; Wang Z; Xu M; Sun Y; Zhou X, et al. Selective in Vivo Metabolic Cell-Labeling-Mediated Cancer Targeting. Nat. Chem. Biol 2017, 13, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hoegl A; Nodwell MB; Kirsch VC; Bach NC; Pfanzelt M; Stahl M; Schneider S; Sieber SA Mining the Cellular Inventory of Pyridoxal Phosphate-Dependent Enzymes with Functionalized Cofactor Mimics. Nat. Chem 2018, 10, 1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hubbard SC; Boyce M; McVaugh CT; Peehl DM; Bertozzi CR Cell Surface Glycoproteomic Analysis of Prostate Cancer-Derived PC-3 Cells. Bioorg. Med. Chem. Lett 2011, 21, 4945–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tian Y; Almaraz RT; Choi CH; Li QK; Saeui C; Li D; Shah P; Bhattacharya R; Yarema KJ; Zhang H Identification of Sialylated Glycoproteins from Metabolically Oligosaccharide Engineered Pancreatic Cells. Clin. Proteom 2015, 12, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Boyce M; Carrico IS; Ganguli AS; Yu SH; Hangauer MJ; Hubbard SC; Kohler JJ; Bertozzi CR Metabolic Cross-Talk Allows Labeling of O-linked β-N-Acetylglucosamine-Modified Proteins via the N-Acetylgalactosamine Salvage Pathway. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 3141–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhu Y; Liu TW; Cecioni S; Eskandari R; Zandberg WF; Vocadlo DJ O-GlcNAc Occurs Cotranslationally to Stabilize Nascent Polypeptide Chains. Nat. Chem. Biol 2015, 11, 319–325. [DOI] [PubMed] [Google Scholar]
- 129.Zhu Y; Willems LI; Salas D; Cecioni S; Wu WB; Foster LJ; Vocadlo DJ Tandem Bioorthogonal Labeling Uncovers Endogenous Cotranslationally O-GlcNAc Modified Nascent Proteins. J. Am. Chem. Soc 2020, 142, 15729–15739. [DOI] [PubMed] [Google Scholar]
- 130.Longwell SA; Dube DH Deciphering The Bacterial Glycocode: Recent Advances in Bacterial Glycoproteomics. Curr. Opin. Chem. Biol 2013, 17, 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kaewsapsak P; Esonu O; Dube DH Recruiting the Host’s Immune System to Target Helicobacter pylori’s Surface Glycans. ChemBioChem 2013, 14, 721–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Champasa K; Longwell SA; Eldridge AM; Stemmler EA; Dube DH Targeted Identification of Glycosylated Proteins in the Gastric Pathogen Helicobacter pylori (Hp). Mol. Cell Proteom 2013, 12, 2568–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Williams DA; Pradhan K; Paul A; Olin IR; Tuck OT; Moulton KD; Kulkarni SS; Dube DH Metabolic Inhibitors of Bacterial Glycan Biosynthesis. Chem. Sci 2020, 11, 1761–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Porte K; Riberaud M; Châtre R; Audisio D; Papot S; Taran F Bioorthogonal Reactions in Animals. ChemBioChem 2020, 22, 100–113. [DOI] [PubMed] [Google Scholar]
- 135.Neeves AA; Stöckman H; Hamston RR; Pryor HJ; Alam IS; Ireland-Zecchini H; Lewis DY; Lyons SK; Leeper FJ; Brindle KM Imaging Sialylated Tumor Cell Glycans in Vivo. FASEB J. 2011, 25, 2528–2537. [DOI] [PubMed] [Google Scholar]
- 136.Zhou X; Yang G; Guan F Biological Functions and Analytical Strategies of Sialic Acids in Tumor. Cells 2020, 9, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Büll C; Stoel MA; den Brok MH; Adema GJ Sialic Acids Sweeten a Tumor’s Life. Cancer Res. 2014, 74, 3199–3204. [DOI] [PubMed] [Google Scholar]
- 138.Pearce OM; Läubli H Sialic Acids in Cancer Biology And Immunity. Glycobiology 2016, 26, 111–128. [DOI] [PubMed] [Google Scholar]
- 139.Contag CH; Bachman MH Advances in in Vivo Bioluminescence Imaging of Gene Expression. Annu. Rev. Biomed. Eng 2002, 4, 235–260. [DOI] [PubMed] [Google Scholar]
- 140.Mezzanotte L; van ‘t Root M; Karatas H; Goun EA; Löwick CWGM In Vivo Molecular Bioluminescence Imaging: New Tools and Applications. Trends Biotechnol. 2017, 35, 640–652. [DOI] [PubMed] [Google Scholar]
- 141.Love AC; Prescher JA Seeing (and Using) the Light: Recent Developments in Bioluminescence Technology. Cell Chem. Biol 2020, 27, 904–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cohen AS; Dubikovskaya EA; Rush JS; Bertozzi CR Real-Time Bioluminescence Imaging of Glycans on Live Cells. J. Am. Chem. Soc 2010, 132, 8563–8565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bazhin AA; Sinisi R; De Marchi U; Hermant A; Sambiagio N; Maric T; Budin G; Goun EA A Bioluminescent Probe for Longitudinal Monitoring of Mitochondrial Membrane Potential. Nat. Chem. Biol 2020, 1385–1393. [DOI] [PubMed] [Google Scholar]
- 144.Godinat A; Bazhin AA; Goun EA Bioorthogonal Chemistry in Bioluminescence Imaging. Drug Discov. Today. 2018, 23, 1584–1590. [DOI] [PubMed] [Google Scholar]
- 145.Zorova LD; Popkov VA; Plotnikov EY; Silachev DN; Pevzner IB; Jankauskas SS; Babenko VA; Zorov SD; Balakireva AV; Juhaszova M, et al. Mitochondrial Membrane Potential. Anal. Biochem 2018, 552, 50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Murphy MP Targeting Lipophilic Cations to Mitochondria. Biochim. Biophys. Acta 2008, 1777, 1028–1031. [DOI] [PubMed] [Google Scholar]
- 147.Huber T; Sakmar TP Chemical Biology Methods for Investigating G Protein-Coupled Receptor Signaling. Cell Chem. Biol 2014, 21, 1224–1237. [DOI] [PubMed] [Google Scholar]
- 148.Tian H; Fürstenberg A; Huber T Labeling and Single-Molecule Methods to Monitor G Protein-Coupled Receptor Dynamics. Chem. Rev 2017, 117, 186–245. [DOI] [PubMed] [Google Scholar]
- 149.Weis WI; Kobilka BK The Molecular Basis of G Protein-Coupled Receptor Activation. Annu. Rev. Biochem 2018, 87, 897–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hauser AS; Attwood MM; Rask-Andersen M; Schiöth HB; Gloriam DE Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discov 2017, 16, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gregorio GG; Masureel M; Hilger D; Terry DS; Juette M; Zhao H; Zhou Z; Perez-Aguilar JM; Hauge M; Mathiasen S, et al. Single-Molecule Analysis of Ligand Efficacy in β2AR–G-Protein Activation. Nature 2017, 547, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chin JW Expanding and Reprogramming the Genetic Code of Cells and Animals. Annu. Rev. Biochem 2014, 83, 379–408. [DOI] [PubMed] [Google Scholar]
- 153.Brown W; Liu J; Deiters A Genetic Code Expansion in Animals. ACS Chem. Biol 2018, 13, 2375–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hoyt EA; Cal PMSD; Oliveira BL; Bernardes GJ Contemporary Approaches to Site-Selective Protein Modification. Nat. Rev. Chem 2019, 3, 147–171. [Google Scholar]
- 155.Wan W; Tharp JM; Liu WR Pyrrolysyl-tRNA Synthetase: an Ordinary Enzyme but an Outstanding Genetic Code Expansion Tool. Biochim. Biophys. Acta 2014, 1844, 1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liu CC; Schultz PG Adding New Chemistries to the Genetic Code. Annu. Rev. Biochem 2010, 79, 413–444. [DOI] [PubMed] [Google Scholar]
- 157.Vargas-Rodriguez O; Sevostyanova A; Söll D; Crnković A Upgrading Aminoacyl-tRNA Synthetases for Genetic Code Expansion. Curr. Opin. Chem. Biol 2018, 46, 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ye S; Huber T; Vogel R; Sakmar TP FTIR Analysis of GPCR Activation Using Azido Probes. Nat. Chem. Biol 2009, 5, 397–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ye S; Zaitseva E; Caltabiano G; Schertler GF; Sakmar TP; Deupi X; Vogel R Tracking G-protein-Coupled Receptor Activation Using Genetically Encoded Infrared Probes. Nature 2010, 464, 1386–1389. [DOI] [PubMed] [Google Scholar]
- 160.Tian H; Naganathan S; Kazmi MA; Schwartz TW; Sakmar TP; Huber T Bioorthogonal Fluorescent Labeling of Functional G-Protein-Coupled Receptors. ChemBioChem 2014, 15, 1820–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Naganathan S; Ray-Saha S; Park M; Tian H; Sakmar TP; Huber T Multiplex Detection of Functional G Protein-Coupled Receptors Harboring Site-Specifically Modified Unnatural Amino Acids. Biochemistry 2015, 54, 776–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Naganathan S; Grunbeck A; Tian H; Huber T; Sakmar TP Genetically-Encoded Molecular Probes to Study G Protein-Coupled Receptors. J. Vis. Exp 2013, 79, e50588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Huber T; Naganathan S; Tian H; Ye S; Sakmar TP Unnatural Amino Acid Mutagenesis of GPCRs Using Amber Codon Suppression and Bioorthogonal Labeling. Methods Enzymol. 2013, 520, 281–305. [DOI] [PubMed] [Google Scholar]
- 164.Naganathan S; Ye S; Sakmar TP; Huber T Site-Specific Epitope Tagging of G Protein-Coupled Receptors by Bioorthogonal Modification of a Genetically Encoded Unnatural Amino Acid. Biochemistry 2013, 52, 1028–1036. [DOI] [PubMed] [Google Scholar]
- 165.Rochefort MM; Girgis MD; Ankeny JS; Tomlinson JS Metabolic Exploitation of the Sialic Acid Biosynthetic Pathway to Generate Site-Specifically Labeled Antibodies. Glycobiology 2014, 24, 62–69. [DOI] [PubMed] [Google Scholar]
- 166.Rampoldi F; Sandhoff R; Owen RW; Gröne HJ; Porubsky S A New, Robust, and Nonradioactive Approach for Exploring N-Myristoylation. J. Lipid Res 2012, 53, 2459–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chen X; Henschke L; Wu Q; Muthoosamy K; Neumann B; Weil T Site-Selective Azide Incorporation into Endogenous RNase A via a “Chemistry” Approach. Org. Biomol. Chem 2013, 11, 353–361. [DOI] [PubMed] [Google Scholar]
- 168.Van Dyke AR; Etemad LS; Vessicchio MJ; Naclerio GA; Jedson V Capture-Tag-Release: a Strategy for Small Molecule Labeling of Native Enzymes. ChemBiochem 2016, 17, 1602–1605. [DOI] [PubMed] [Google Scholar]
- 169.Banerjee PS; Ostapchuk P; Hearing P; Carrico IS Unnatural Amino Acid Incorporation onto Adenoviral (Ad) Coat Proteins Facilitates Chemoselective Modification and Retargeting of Ad Type 5 Vectors. J. Virol 2011, 85, 7546–7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Banerjee PS; Carrico IS Chemoselective Modification of Viral Proteins Bearing Metabolically Introduced “Clickable” Amino Acids and Sugars. Bioconjugation Protocols 2011, 751, 55–66. [DOI] [PubMed] [Google Scholar]
- 171.Perez HL; Cardarelli PM; Deshpande S; Gangwar S; Schroeder GM; Vite GD; Borzilleri RM Antibody-Drug Conjugates: Current Status and Future Directions. Drug Discov. Today 2014, 19, 869–881. [DOI] [PubMed] [Google Scholar]
- 172.Beck A; Goetsch L; Dumontet C; Corvaïa N Strategies and Challenges for the Next Generation of Antibody-Drug Conjugates. Nat. Rev. Drug Discov 2017, 16, 315–337. [DOI] [PubMed] [Google Scholar]
- 173.Rondon A; Degoul F Antibody Pretargeting Based on Bioorthogonal Click Chemistry for Cancer Imaging and Targeted Radionuclide Therapy. Bioconjugate Chem. 2020, 31, 159–173. [DOI] [PubMed] [Google Scholar]
- 174.Vugts DJ; Vervoort A; Stigter-van Walsum M; Visser GWM; Robillard MS; Versteegen RM; Vulders RCM; Herscheid JDM; van Dongen GAMS Synthesis of Phosphine and Antibody–Azide Probes for in Vivo Staudinger Ligation in a Pretargeted Imaging and Therapy Approach. Bioconjugate Chem. 2011, 22, 2072–2081. [DOI] [PubMed] [Google Scholar]
- 175.George JT; Srivatsan SG Posttranscriptional Chemical Labeling of RNA by Using Bioorthogonal Chemistry. Methods 2017, 120, 28–38. [DOI] [PubMed] [Google Scholar]
- 176.Ivancová I; Leone DL; Hocek M Reactive Modifications of DNA Nucleobases for Labelling, Bioconjugations, and Cross-Linking. Curr. Opin. Chem. Biol 2019, 52, 136–144. [DOI] [PubMed] [Google Scholar]
- 177.George JT; Srivatsan SG Bioorthogonal Chemistry-Based RNA Labeling Technologies: Evolution and Current State. Chem. Commun 2020, 56, 12307–12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rao H; Tanpure AA; Sawant AA; Srivatsan SG Enzymatic Incorporation of an Azide-Modified UTP Analog into Oligoribonucleotides for Post-Transcriptional Chemical Functionalization. Nat. Protoc 2012, 7, 1097–1112. [DOI] [PubMed] [Google Scholar]
- 179.Sabale PM; Ambi UB; Srivatsan SG Clickable PNA Probes for Imaging Human Telomeres and Poly(A) RNAs. ACS Omega 2018, 3, 15343–15352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wu C; Kurinomaru T Development of the Bioluminescent Immunoassay for the Detection of 5-Hydroxymethylcytosine in Dinoflagellate. Anal. Sci 2019, 35, 301–305. [DOI] [PubMed] [Google Scholar]
- 181.Rao H; Sawant AA; Tanpure AA; Srivatsan SG Posttranscriptional Chemical Functionalization of Azide-Modified Oligoribonucleotides by Bioorthogonal Click and Staudinger Reactions. Chem. Commun 2012, 48, 498–500. [DOI] [PubMed] [Google Scholar]
- 182.Gopalakrishna S; Gusti V; Nair S; Sahar S; Gaur RK Template-Dependent Incorporation of 8-N3AMP into RNA With Bacteriophage T7 RNA Polymerase. RNA 2004, 10, 1820–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yoshimura SH; Khan S; Ohno S; Yokogawa T; Nishikawa K; Hosoya T; Maruyama H; Nakayama Y; Takeyasu K Site-Specific Attachment of a Protein to a Carbon Nanotube End Without Loss of Protein Function. Bioconjugugate Chem. 2012, 23, 1488–1493. [DOI] [PubMed] [Google Scholar]
- 184.Luo W; Gobbo P; Gunawardene PN; Workentin MS Fluorogenic Gold Nanoparticle (AuNP) Substrate: a Model For The Controlled Release of Molecules from AuNP Nanocarriers via Interfacial Staudinger-Bertozzi Ligation. Langmuir 2017, 33, 1908–1913. [DOI] [PubMed] [Google Scholar]
- 185.Szymánski W; Wu B; Janssen DB; Feringa BL Azobenzene Photoswitches for Staudinger-Bertozzi Ligation. Angew. Chem., Int. Ed 2013, 52, 2068–2072. [DOI] [PubMed] [Google Scholar]
- 186.Poloni C; Szymánski W; Hou L; Browne WR; Feringa BL A Fast, Visible-Light Sensitive Azobenzene for Bioorthogonal Ligation. Chem. Eur. J 2014, 20, 946–951. [DOI] [PubMed] [Google Scholar]
- 187.Velema WA; Szymanski W; Feringa BL Photopharmacology: Beyond Proof of Principle. J. Am. Chem. Soc 2014, 136, 2178–2191. [DOI] [PubMed] [Google Scholar]
- 188.Mart RJ; Allemann RK Azobenzene Photocontrol of Peptides and Proteins. Chem. Commun 2016, 52, 12262–12277. [DOI] [PubMed] [Google Scholar]
- 189.Freichel T; Eierhoff S; Snyder NL; Hartmann L Toward Orthogonal Preparation of Sequence-Defined Monodisperse Heteromultivalent Glycomacromolecules on Solid Support Using Staudinger Ligation and Copper-Catalyzed Click Reactions. J. Org. Chem 2017, 82, 9400–9409. [DOI] [PubMed] [Google Scholar]
- 190.Algar WR; Prasuhn DE; Stewart MH; Jennings TL; Blanco-Canosa JB; Dawson PE; Medintz IL The Controlled Display of Biomolecules on Nanoparticles: a Challenge Suited to Bioorthogonal Chemistry. Bioconjugate Chem. 2011, 22, 825–858. [DOI] [PubMed] [Google Scholar]
- 191.Gobbo P; Luo W; Cho SJ; Wang X; Biesinger MC; Hudson RH; Workentin MS Small Gold Nanoparticles for Interfacial Staudinger-Bertozzi Ligation. Org. Biomol. Chem 2015, 13, 4605–4612. [DOI] [PubMed] [Google Scholar]
- 192.Zhang P; Zhang X; Li C; Zhou S; Wu W; Jiang X Target-Amplified Drug Delivery of Polymer Micelles Bearing Staudinger Ligation. ACS Appl. Mater. Interfaces 2019, 11, 32697–32705. [DOI] [PubMed] [Google Scholar]
- 193.Manouilidou MD; Lazarou YG; Mavridis IM; Yannakopoulou K Staudinger Ligation Towards Cyclodextrin Dimers in Aqueous/Organic Media. Synthesis, Conformations and Guest-Encapsulation Ability. Beilstein J. Org. Chem 2014, 10, 774–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Liu S; Edgar KJ Staudinger Reactions for Selective Functionalization of Polysaccharides: a Review. Biomacromolecules 2015, 16, 2556–2571. [DOI] [PubMed] [Google Scholar]
- 195.Gattás-Asfura KM; Stabler CL Bioorthogonal Layer-By-Layer Encapsulation of Pancreatic Islets via Hyperbranched Polymers. ACS Appl. Mater. Interfaces 2013, 5, 9964–9974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Rengifo HR; Giraldo JA; Labrada I; Stabler CL Long-Term Survival of Allograft Murine Islets Coated via Covalently Stabilized Polymers. Adv. Healthc. Mater 2014, 3, 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.England CG; Luo H; Cai W HaloTag technology: a Versatile Platform for Biomedical Applications. Bioconjugate Chem. 2015, 26, 975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hay N Reprogramming Glucose Metabolism in Cancer: Can It Be Exploited for Cancer Therapy? Nat. Rev. Cancer 2016, 16, 635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Chang CH; Pearce EL Emerging Concepts of T Cell Metabolism as a Target of Immunotherapy. Nat. Immunol 2016, 17, 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Sundhoro M; Park J; Wu B; Yan M Synthesis of Polyphosphazenes by a Fast Perfluoroaryl Azide-Mediated Staudinger Reaction. Macromolecules 2018, 51, 4532–4540. [Google Scholar]
- 201.Nduguire W; Wu B; Yan M Synthesis of Carbohydrate-Grafted Glycopolymers Using a Catalyst-Free, Perfluoroarylazide-Mediated Fast Staudinger Reaction. Molecules 2019, 24, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Noy J-M; Li Y; Smolan W; Roth PJ Azide–para-Fluoro Substitution on Polymers: Multipurpose Precursors for Efficient Sequential Postpolymerization Modification. Macromolecules 2019, 52, 3083–3091. [Google Scholar]
- 203.Allcock HR; Morozowich NL Bioerodible Polyphosphazenes and Their Medical Potential. Polym. Chem 2012, 3, 578–590. [Google Scholar]
- 204.Nukavarapu SP; Kumbar SG; Brown JL; Krogman NR; Weikel AL; Hindenlang MD; Nair LS; Allcock HR; Laurencin CT Polyphosphazene/Nano-Hydroxyapatite Composite Microsphere Scaffolds for Bone Tissue Engineering. Biomacromolecules 2008, 9, 1818–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Bertozzi CR; Wu P In Vivo Chemistry. Curr. Opin. Chem. Biol 2013, 17, 717–718. [DOI] [PubMed] [Google Scholar]
- 206.Devaraj NK The Future of Bioorthogonal Chemistry. ACS Cent. Sci 2018, 4, 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Nilsson BL; Kiessling LL; Raines RT Staudinger Ligation: a Peptide from a Thioester and Azide. Org. Lett 2000, 2, 1939–1941. [DOI] [PubMed] [Google Scholar]
- 208.Saxon E; Armstrong JI; Bertozzi CRA “Traceless” Staudinger Ligation for the Chemoselective Synthesis of Amide Bonds. Org. Lett 2000, 2, 2141–2143. [DOI] [PubMed] [Google Scholar]
- 209.Shih H-W; Prescher JA A Bioorthogonal Ligation of Cyclopropenones Mediated by Triarylphosphines. J. Am. Chem. Soc 2015, 137, 10036–10039. [DOI] [PubMed] [Google Scholar]
- 210.Agarwal P; van der Weijden J; Sletten EM; Rabuka D; Bertozzi CR A Pictet-Spengler Ligation for Protein Chemical Modification. Proc. Natl. Acad. Sci. U.S.A 2013, 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Agarwal P; Kudirka R; Albers AE; Barfield RM; de Hart GW; Drake PM; Jones LC; Rabuka D Hydrazino-Pictet-Spengler Ligation as a Biocompatible Method for the Generation of Stable Protein Conjugates. Bioconjugate Chem. 2013, 24, 846–851. [DOI] [PubMed] [Google Scholar]
- 212.Bandyopadhyay A; Cambray S; Gao J Fast Diazaborine Formation of Semicarbazide Enables Facile Labeling of Bacterial Pathogens. J. Am. Chem. Soc 2017, 139, 871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Meadows MK; Roesner EK; Lynch VM; James TD; Anslyn EV Boronic Acid Mediated Coupling of Catechols And N-Hydroxylamines: a Bioorthogonal Reaction to Label Peptides. Org. Lett 2017, 19, 3179–3182. [DOI] [PubMed] [Google Scholar]
- 214.Oliveira BL; Guo Z; Bernardes GJL Inverse Electron Demand Diels–Alder Reactions in Chemical Biology. Chem. Soc. Rev 2017, 46, 4895–4950. [DOI] [PubMed] [Google Scholar]
- 215.Fang Y; Zhang H; Huang Z; Scinto SL; Yang JC; am Ende C; Dmitrenko O; Johnson DS; Fox JM Photochemical Syntheses, Transformations, and Bioorthogonal Chemistry of Trans-Cycloheptene and Sila Trans-Cycloheptene Ag(I) Complexes. Chem. Sci 2018, 9, 1953–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Agard NJ; Baskin JM; Prescher JA; Lo A; Bertozzi CR A Comparative Study of Bioorthogonal Reactions with Azides. ACS Chem. Biol 2006, 1, 644–648. [DOI] [PubMed] [Google Scholar]
- 217.Chang PV; Prescher JA; Sletten EM; Baskin JM; Miller IA; Agard NJ; Lo A; Bertozzi CR Copper-Free Click Chemistry in Living Animals. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 1821–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Pattabiraman VR; Bode JW Rethinking Amide Bond Synthesis. Nature 2011, 480, 471–479. [DOI] [PubMed] [Google Scholar]
- 219.David O; Meester WJ; Bieräugel H; Schoemaker HE; Hiemstra H; van Maarseveen JH Intramolecular Staudinger Ligation: a Powerful Ring-Closure Method to Form Medium-Sized Lactams. Angew. Chem., Int. Ed 2003, 42, 4373–4375. [DOI] [PubMed] [Google Scholar]
- 220.Kleineweischede R; Hackenberger CP Chemoselective Peptide Cyclization by Traceless Staudinger Ligation. Angew. Chem., Int. Ed 2008, 47, 5984–5988. [DOI] [PubMed] [Google Scholar]
- 221.Liu L; Hong ZY; Wong CH Convergent Glycopeptide Synthesis by Traceless Staudinger Ligation and Enzymatic Coupling. ChemBioChem 2006, 7, 429–432. [DOI] [PubMed] [Google Scholar]
- 222.Tam A; Soellner MB; Raines RT Water-Soluble Phosphinothiols for Traceless Staudinger Ligation and Integration with Expressed Protein Ligation. J. Am. Chem. Soc 2007, 129, 11421–11430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Nilsson BL; Hondal RJ; Soellner MB; Raines RT Protein Assembly by Orthogonal Chemical Ligation Methods. J. Am. Chem. Soc 2003, 125, 5268–5269. [DOI] [PubMed] [Google Scholar]
- 224.Tam A; Raines RT Coulombic Effects on the Traceless Staudinger Ligation in Water. Bioorg. Med. Chem 2009, 17, 1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kosiova I; Janicova A; Kois P Synthesis of Coumarin or Ferrocene Labeled Nucleosides via Staudinger Ligation. Beilstein. J. Org. Chem 2006, 2, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Soellner MB; Nilsson BL; Raines RT Reaction Mechanism and Kinetics of the Traceless Staudinger Ligation. J. Am. Chem. Soc 2006, 128, 8820–8828. [DOI] [PubMed] [Google Scholar]
- 227.Azoulay M; Tuffin G; Sallem W; Florent JC A New Drug-Release Method Using the Staudinger Ligation. Bioorg. Med. Chem. Lett 2006, 16, 3147–3149. [DOI] [PubMed] [Google Scholar]
- 228.Soellner MB; Tam A; Raines RT Staudinger Ligation of Peptides at Non-Glycyl Residues. J. Org. Chem 2006, 71, 9824–9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Bajaj K; Pillai GG; Sakhuja R; Kumar D Expansion of Phosphane Treasure Box for Staudinger Peptide Ligation. J. Org. Chem 2020, 85, 12147–12159. [DOI] [PubMed] [Google Scholar]
- 230.Chow HY; Zhang Y; Matheson E; Li X Ligation Technologies for the Synthesis of Cyclic Peptides. Chem. Rev 2019, 119, 9971–10001. [DOI] [PubMed] [Google Scholar]
- 231.Itoh H; Miura K; Kamiya K; Yamashita T; Inoue M Solid-Phase Total Synthesis Of Yaku’amide B Enabled by Traceless Staudinger Ligation. Angew. Chem., Int. Ed 2020, 59, 4564–4571. [DOI] [PubMed] [Google Scholar]
- 232.Hu P; Feng T; Yeung CC; Koo CK; Lau KC; Lam MH A Photo-Triggered Traceless Staudinger-Bertozzi Ligation Reaction. Chem. Eur. J 2016, 22, 11537–11542. [DOI] [PubMed] [Google Scholar]
- 233.Hu P; Berning K; Lam YW; Ng IH; Yeung CC; Lam MH Development of a Visible Light Triggerable Traceless Staudinger Ligation Reagent. J. Org. Chem 2018, 83, 12998–13010. [DOI] [PubMed] [Google Scholar]
- 234.Garcia J; Urpí F; Vilarrasa J New Synthetic “Tricks”. Triphenylphosphine-Mediated Amide Formation from Carboxylic Acids and Azides. Tetrahedron Lett. 1984, 25, 4841–4844. [Google Scholar]
- 235.Bosch I; Romea P; Urpí F; Vilarrasa J Alternative Procedures for the Macrolactamisation of ω-Amino Acids. Tetrahedron Lett. 1993, 34, 4671–4674. [Google Scholar]
- 236.Bosch I; Urpí F; Vilarrasa J Epimerisation-Free Peptide Formation from Carboxylic Acid Anhydrides and Azido Derivatives. J. Chem. Soc., Chem. Commun 1995, 1, 91–92. [Google Scholar]
- 237.Ariza X; Viladomat C; Vilarrasa J One-Pot Conversion of Azides to Boc-Protected Amines with Trimethylphosphine and Boc-ON. Tetrahedron Lett. 1998, 39, 9101–9102. [Google Scholar]
- 238.Ariza X; Pineda O; Urpí F; Vilarrasa J From Vicinal Azido Alcohols to Boc-Amino Alcohols or Oxazolidinones, with Trimethylphosphine and Boc2O or CO2. Tetrahedron Lett. 2001, 42, 4995–4999. [Google Scholar]
- 239.Garcia J; Vilarrasa J; Bordas X; Banaszek A New Synthetic “Tricks”. One-Pot Preparation of N-Substituted Phthalimides from Azides and Phthalic Anhydride. Tetrahedron Lett. 1986, 27, 639–640. [Google Scholar]
- 240.Bernardes GJ; Linderoth L; Doores KJ; Boutureira O; Davis BG Site-Selective Traceless Staudinger Ligation for Glycoprotein Synthesis Reveals Scope and Limitations. ChemBioChem 2011, 12, 1383–1386. [DOI] [PubMed] [Google Scholar]
- 241.Tam A; Soellner MB; Raines RT Electronic and Steric Effects on the Rate of the Traceless Staudinger Ligation. Org. Biomol. Chem 2008, 6, 1173–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Yamashita T; Kuranaga T; Inoue M Solid-Phase Total Synthesis of Bogorol A: Stereocontrolled Construction of Thermodynamically Unfavored (E)-2-Amino-2-Butenamide. Org. Lett 2015, 17, 2170–2173. [DOI] [PubMed] [Google Scholar]
- 243.Yamashita T; Matoba H; Kuranaga T; Inoue M Total Synthesis of Nobilamides B and D: Application of Traceless Staudinger Ligation. Tetrahedron 2014, 70, 7746–7752. [Google Scholar]
- 244.Kosal AD; Wilson EE; Ashfeld BL Direct Acyl Substitution of Carboxylic Acids: a Chemoselective O- to N-Acyl Migration in the Traceless Staudinger Ligation. Chem. Eur. J 2012, 18, 14444–14453. [DOI] [PubMed] [Google Scholar]
- 245.Kosal AD; Wilson EE; Ashfeld BL Phosphine-Based Redox Catalysis in the Direct Traceless Staudinger Ligation of Carboxylic Acids and Azides. Angew. Chem., Int. Ed 2012, 51, 12036–12040. [DOI] [PubMed] [Google Scholar]
- 246.Myers EL; Raines RT A Phosphine-Mediated Conversion of Azides into Diazo Compounds. Angew. Chem., Int. Ed 2009, 48, 2359–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Chou H-H; Raines RT Conversion of Azides into Diazo Compounds in Water. J. Am. Chem. Soc 2013, 135, 14936–14939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Mix KA; Aronoff MR; Raines RT Diazo Compounds: Versatile Tools for Chemical Biology. ACS Chem. Biol 2016, 11, 3233–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Xiao Q; Zhang Y; Wang J Diazo Compounds and N-Tosylhydrazones: Novel Cross-Coupling Partners in Transition-Metal-Catalyzed Reactions. Acc. Chem. Res 2013, 46, 236–247. [DOI] [PubMed] [Google Scholar]
- 250.Pretze M; Pietzsch D; Mamat C Recent Trends in Bioorthogonal Click-Radiolabeling Reactions Using Fluorine-18. Molecules 2013, 18, 8618–8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Mamat C; Franke M; Peppel T; Köckerling M; Steinbach J Synthesis, Structure Determination, And (Radio-) Fluorination of Novel Functionalized Phosphanes Suitable for the Traceless Staudinger Ligation. Tetrahedron 2011, 67, 4521–4529. [Google Scholar]
- 252.Carroll L; Boldon S; Bejot R; Moore JE; Declerck J; Gouverneur V The Traceless Staudinger Ligation for Indirect 18F-Radiolabelling. Org. Biomol. Chem 2011, 9, 136–140. [DOI] [PubMed] [Google Scholar]
- 253.Mamat C; Köckerling M Preparation Of 4-Halobenzoate-Containing Phosphane-Based Building Blocks for Labeling Reactions Using the Traceless Staudinger Ligation. Synthesis 2015, 47, 387–394. [Google Scholar]
- 254.Köckerling M; Mamat C Structural and Kinetic Considerations for the Application of the Traceless Staudinger Ligation to Future 18F Radiolabeling Using XRD and 19F NMR. Int. J. Chem 2017, 50, 31–40. [Google Scholar]
- 255.Vaidyanathan G; White B; Affleck DJ; McDougald D; Zalutsky MR Radioiodinated O(6)-Benzylguanine Derivatives Containing an Azido Function. Nucl. Med. Biol 2011, 38, 77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Geri S; Krunclova T; Janouskova O; Panek J; Hruby M; Hernández-Valdés D; Probst B; Alberto RA; Mamat C; Kubeil M, et al. Light-Activated Carbon Monoxide Prodrugs Based on Bipyridyl Dicarbonyl Ruthenium(II) Complexes. Chem. Eur. J 2020, 26, 10992–11006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Andersen KA; Raines RT Creating Site-Specific Isopeptide Linkages between Proteins with the Traceless Staudinger Ligation. Methods Mol. Biol 2015, 1248, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Heldt J-M; Kerzendörfer O; Mamat C; Starke F; Pietzsch H-J; Steinbach J Synthesis of Short and Versatile Heterobifunctional Linkers for Conjugation of Bioactive Molecules with (Radio-)Labels. Synlett 2013, 24, 432–436. [Google Scholar]
- 259.Wodtke R; König J; Pigorsch A; Köckerling M; Mamat C Evaluation of Novel Fluorescence Probes for Conjugation Purposes Using the Traceless Staudinger Ligation. Dyes Pigm. 2015, 113, 263–273. [Google Scholar]
- 260.Ahad AM; Jensen SM; Jewett JC A Traceless Staudinger Reagent to Deliver Diazirines. Org. Lett 2013, 15, 5060–5063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Varki A Biological Roles of Glycans. Glycobiology 2017, 1, 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Wright TH; Bower BJ; Chalker JM; Bernardes GJL; Wiewiora R; Ng W-L; Raj R; Faulkner S; Vallée MRJ; Phanumartwiwath A, et al. Posttranslational Mutagenesis: a Chemical Strategy for Exploring Protein Side-Chain Diversity. Science 2016, 354, aag1465. [DOI] [PubMed] [Google Scholar]
- 263.Wright TH; Davis BG Post-Translational Mutagenesis for Installation of Natural and Unnatural Amino Acid Side Chains into Recombinant Proteins. Nat. Protoc 2017, 12, 2243–2250. [DOI] [PubMed] [Google Scholar]
- 264.Colombo C; Bernardi A Synthesis of α-N-Linked Glycopeptides. Eur. J. Org. Chem 2011, 2011, 3911–3919. [Google Scholar]
- 265.Speciale G; Bernardi A; Nisic F A Facile Synthesis of α-N-Ribosyl-Asparagine and α-N-Ribosyl-Glutamine Building Blocks. Molecules 2013, 18, 8779–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Zhu S; Guo Z Chemical Synthesis of GPI Glycan–Peptide Conjugates by Traceless Staudinger Ligation. Org. Lett 2017, 19, 3063–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Drazic A; Myklebust LM; Ree R; Arnesen T The World of Protein Acetylation. Biochim. Biophys. Acta 2016, 1864, 1372–1401. [DOI] [PubMed] [Google Scholar]
- 268.Choudhary C; Weinert BT; Nishida Y; Verdin E; Mann M The Growing Landscape of Lysine Acetylation Links Metabolism and Cell Signaling. Nat. Rev. Mol. Cell. Biol 2014, 15, 536–550. [DOI] [PubMed] [Google Scholar]
- 269.Sowa S; Mühlberg M; Pietrusiewicz KM; Hackenberger CP Traceless Staudinger Acetylation of Azides in Aqueous Buffers. Bioorg. Med. Chem 2013, 21, 3465–3472. [DOI] [PubMed] [Google Scholar]
- 270.Neumann H; Peak-Chew SY; Chin JW Genetically Encoding Nε-Acetyllysine in Recombinant Proteins. Nat. Chem. Biol 2008, 4, 232–234. [DOI] [PubMed] [Google Scholar]
- 271.Elsässer SJ; Ernst RJ; Walker OS; Chin JW Genetic Code Expansion in Stable Cell Lines Enables Encoded Chromatin Modification. Nat. Methods 2016, 13, 158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Wang ZA; Kurra Y; Wang X; Zeng Y; Lee YJ; Sharma V; Lin H; Dai SY; Liu WR A Versatile Approach for Site-Specific Lysine Acylation in Proteins. Angew. Chem., Int. Ed 2017, 56, 1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.White CJ; Yudin AK Contemporary Strategies for Peptide Macrocyclization. Nat. Chem 2011, 3, 509–524. [DOI] [PubMed] [Google Scholar]
- 274.Wan Q; Danishefsky SJ Free-Radical-Based, Specific Desulfurization of Cysteine: a Powerful Advance in the Synthesis of Polypeptides and Glycopolypeptides. Angew. Chem., Int. Ed 2007, 46, 9248–9252. [DOI] [PubMed] [Google Scholar]
- 275.Beagle LC; Hansen FK; Monbaliu J-CM; DesRosiers MP; Phillips AM; Stevens CV; Katritzky AR Efficient Synthesis of 2,5-Diketopiperazines by Staudinger-Mediated Cyclization. Synlett 2012, 23, 2337–2340. [Google Scholar]
- 276.Panda SS; Jones RA; C. DH; Katritzky AR, Applications of Chemical Ligation in Peptide Synthesis via Acyl Transfer. In Protein Ligation and Total Synthesis I, Springer International Publishing: Cham, 2015; pp 229–265. [DOI] [PubMed] [Google Scholar]
- 277.Ha K; Monbaliu J-CM; Williams BC; Pillai GG; Ocampo CE; Zeller M; Stevens CV; Katritzky AR A Convenient Synthesis of Difficult Medium-Sized Cyclic Peptides by Staudinger Mediated Ring-Closure. Org. Biomol. Chem 2012, 10, 8055–8058. [DOI] [PubMed] [Google Scholar]
- 278.Fletcher MH; Burns-Lynch CE; Knouse KW; Abraham LT; DeBrosse CW; Wuest WM A Novel Application of the Staudinger Ligation to Access Neutral Cyclic Di-Nucleotide Analog Precursors via a Divergent Method. RSC Adv. 2017, 7, 29835–29838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Muir TW Semisynthesis of Proteins by Expressed Protein Ligation. Annu. Rev. Biochem 2003, 72, 249–289. [DOI] [PubMed] [Google Scholar]
- 280.Berrade L; Camarero JA Expressed Protein Ligation: a Resourceful Tool to Study Protein Structure and Function. Cell Mol. Life Sci 2009, 66, 3909–3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Tanrikulu IC; Schmitt E; Mechulam Y; Goddard WA; Tirrell DA Discovery of Escherichia Coli Methionyl-tRNA Synthetase Mutants for Efficient Labeling of Proteins with Azidonorleucine in Vivo. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 15285–15290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Komander D; Rape M The Ubiquitin Code. Annu. Rev. Biochem 2012, 81, 203–229. [DOI] [PubMed] [Google Scholar]
- 283.van der Veen AG; Ploegh HL Ubiquitin-Like Proteins. Annu. Rev. Biochem 2012, 81, 323–357. [DOI] [PubMed] [Google Scholar]
- 284.Smith BC; Marletta MA Mechanisms of S-Nitrosothiol Formation and Selectivity in Nitric Oxide Signaling. Curr. Opin. Chem. Biol 2012, 16, 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Dedon PC; Tannenbaum SR Reactive Nitrogen Species in the Chemical Biology of Inflammation. Arch. Biochem. Biophys 2004, 423, 12–22. [DOI] [PubMed] [Google Scholar]
- 286.Reisz JA; Zink CN; King SB Rapid and Selective Nitroxyl (HNO) Trapping by Phosphines: Kinetics and New Aqueous Ligations for HNO Detection and Quantitation. J. Am. Chem. Soc 2011, 133, 11675–11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Miao Z; Reisz JA; Mitroka SM; Pan J; Xian M; King SB A Selective Phosphine-Based Fluorescent Probe for Nitroxyl in Living Cells. Bioorg. Med. Chem. Lett 2015, 25, 16–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Bechtold E; King SB Chemical Methods for the Direct Detection and Labeling of S-Nitrosothiols. Antioxid. Redox Signal 2012, 17, 981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Seneviratne U; Godoy LC; Wishnok JS; Wogan GN; Tannenbaum SR Mechanism-Based Triarylphosphine-Ester Probes for Capture of Endogenous RSNOs. J. Am. Chem. Soc 2013, 135, 7693–7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Pereira JM; Edgar KJ Regioselective Synthesis of 6-Amino- and 6-Amido-6-Deoxypullulans. Cellulose 2014, 21, 2379–2396. [Google Scholar]
- 291.Zhang R; Edgar KJ Synthesis of Curdlan Derivatives Regioselectively Modified at C-6: O-(N)-Acylated 6-Amino-6-Deoxycurdlan. Carbohydr. Polym 2014, 105, 161–168. [DOI] [PubMed] [Google Scholar]
- 292.Fox SC; Edgar KJ Staudinger Reduction Chemistry of Cellulose: Synthesis of Selectively O-Acylated 6-Amino-6-Deoxy-Cellulose. Biomacromolecules 2012, 13, 992–1001. [DOI] [PubMed] [Google Scholar]
- 293.Senapati S; Biswas S; Zhang P Traceless Staudinger Ligation for Biotinylation Of Acetylated Thiol-Azido Heterobifunctional Linker and its Attachment to Gold Surface. Curr. Org. Chem 2018, 22, 411–415. [Google Scholar]
- 294.Pötzsch R; Fleischmann S; Tock C; Komber H; Voit BI Combining RAFT and Staudinger Ligation: a Potentially New Synthetic Tool for Bioconjugate Formation. Macromolecules 2011, 44, 3260–3269. [Google Scholar]
- 295.de Figueiredo RM; Suppo JS; Campagne JM Nonclassical Routes for Amide Bond Formation. Chem. Rev 2016, 116, 12029–12122. [DOI] [PubMed] [Google Scholar]
- 296.Agouridas V; El Mahdi O; Diemer V; Cargoët M; Monbaliu J-CM; Melnyk O Native Chemical Ligation and Extended Methods: Mechanisms, Catalysis, Scope, and Limitations. Chem. Rev 2019, 119, 7328–7443. [DOI] [PubMed] [Google Scholar]
- 297.Dawson PE; Muir TW; Clark-Lewis I; Kent SB Synthesis of Proteins by Native Chemical Ligation. Science 1994, 266, 776–779. [DOI] [PubMed] [Google Scholar]
- 298.Moura A; Savageau MA; Alves R Relative Amino Acid Composition Signatures of Organisms and Environments. PLoS One 2013, 8, e77319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Chisholm TS; Clayton D; Dowman LJ; Sayers J; Payne RJ Native Chemical Ligation–Photodesulfurization in Flow. J. Am. Chem. Soc 2018, 140, 9020–9024. [DOI] [PubMed] [Google Scholar]
- 300.Jin K; Li X Advances In Native Chemical Ligation-Desulfurization: a Powerful Strategy for Peptide and Protein Synthesis. Chemistry 2018, 24, 17397–17404. [DOI] [PubMed] [Google Scholar]
- 301.Conibear AC; Watson EE; Payne RJ; Becker CFW Native Chemical Ligation in Protein Synthesis and Semi-Synthesis. Chem. Soc. Rev 2018, 47, 9046–9068. [DOI] [PubMed] [Google Scholar]
- 302.Kulkarni SS; Sayers J; Premdjee B; Payne RJ Rapid and Efficent Synthesis through Expansion of the Native Chemical Ligation Concepts. Nat. Rev. Chem 2018, 2, 0122. [Google Scholar]
- 303.Fosgerau K; Hoffmann T Peptide Therapeutics: Current Status and Future Directions. Drug Discov. Today 2015, 20, 122–128. [DOI] [PubMed] [Google Scholar]
- 304.Usmani SS; Bedi G; Samuel JS; Singh S; Kalra S; Kumar P; Ahuja AA; Sharma M; Gautam A; Raghava GPS THPdb: Database of FDA-Approved Peptide and Protein Therapeutics. PLoS One 2017, 12, e0181748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Reber KP; Gilbert IW; Strassfeld DA; Sorensen EJ Synthesis Of (+)-Lineariifolianone and Related Cyclopropenone-Containing Sesquiterpenoids. J. Org. Chem 2019, 84, 5524–5534. [DOI] [PubMed] [Google Scholar]
- 306.Kogen H; Kiho T; Tago K; Miyamoto S; Fujioka T; Otsuka N; Suzuki-Konagai K; Ogita T Alutacenoic Acids A and B, Rare Naturally Occurring Cyclopropenone Derivatives Isolated from Fungi: Potent Non-Peptide Factor XIIIa Inhibitors. J. Am. Chem. Soc 2000, 122, 1842–1843. [Google Scholar]
- 307.Cohen M; Bretler U; Albeck A Peptidyl Cyclopropenones: Reversible Inhibitors, Irreversible Inhibitors, or Substrates of Cysteine Proteases? Protein Sci. 2013, 22, 788–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Row RD; Shih H-W; Alexander AT; Mehl RA; Prescher JA Cyclopropenones for Metabolic Targeting and Sequential Bioorthogonal Labeling. J. Am. Chem. Soc 2017, 139, 7370–7375. [DOI] [PubMed] [Google Scholar]
- 309.Row RD; Nguyen SS; Ferreira AJ; Prescher JA Chemically Triggered Crosslinking with Bioorthogonal Cyclopropenones. Chem. Commun 2020, 56, 10883–10886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Row RD; Prescher JA A Cyclopropenethione-Phosphine Ligation for Rapid Biomolecule Labeling. Org. Lett 2018, 20, 5614–5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Heiss TK; Prescher JA Cyclopropeniminium Ions Exhibit Unique Reactivity Profiles with Bioorthogonal Phosphines. J. Org. Chem 2019, 84, 7443–7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Breslow R; Haynie R; Mirra J The Synthesis of Diphenylcyclopropenone J. Am. Chem. Soc 1959, 81, 247–248. [Google Scholar]
- 313.Breslow R; Altman LJ; Krebs A; Mohacsi E; Murata I; Peterson RA; Posner J Substituted Cyclopropenones. J. Am. Chem. Soc 1965, 87, 1326–1331. [Google Scholar]
- 314.Greenberg A; Tomkins RPT; Dobrovolny M; Liebman JF The Strain Energy of Diphenylcyclopropenone: A Reexamination. J. Am. Chem. Soc 1983, 105, 6855–6858. [Google Scholar]
- 315.Hopkins HP; Bostwick D; Alexander CJ The Thermochemistry of Diphenylcyclopropenone. Strain Vs. Delocalization Energy. J. Am. Chem. Soc 1976, 98, 1355–1357. [Google Scholar]
- 316.Benson RC; Flygare WH; Oda M; Breslow R Microwave Spectrum, Substitutional Structure, and Stark and Zeeman Effects in Cyclopropenone. J. Am. Chem. Soc 1973, 95, 2772–2777. [Google Scholar]
- 317.Potts KT; Baum JS Chemistry of Cyclopropenones. Chem. Rev 1974, 74, 189–213. [Google Scholar]
- 318.Breslow R; Eicher T; Krebs A; Peterson RA; Posner J Diphenylcyclopropenone. J. Am. Chem. Soc 1965, 87, 1320–1325. [Google Scholar]
- 319.Komatsu K; Kitagawa T Cyclopropenylium Cations, Cyclopropenones, and Heteroanalogues–Recent Advances. Chem. Rev 2003, 103, 1371–1428. [DOI] [PubMed] [Google Scholar]
- 320.Hamada A; Takizawa T Reactions of Diphenylcyclopropenone with Trivalent Phosphorus Compounds. Chem. Pharm. Bull 1975, 23, 2933–2938. [Google Scholar]
- 321.Bestmann HJ; Sandmeier D Simple Synthesis of Ketenylidenetriphenylphosphorane and its Thioanalogs. Angew. Chem., Int. Ed 1975, 14, 634. [Google Scholar]
- 322.Bos J; Muir TW A Chemical Probe for Protein Crotonylation. J. Am. Chem. Soc 2018, 140, 4757–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Chambers KA; Abularrage NS; Hill CJ; Khan IH; Scheck RA A Chemical Probe for Dehydrobutyrine. Angew. Chem., Int. Ed 2020, 59, 7350–7355. [DOI] [PubMed] [Google Scholar]
- 324.Shao Z; Liu W; Tao H; Liu F; Zeng R; Champagne PA; Cao Y; Houk KN; Liang Y Bioorthogonal Release of Sulfonamides and Mutually Orthogonal Liberation of Two Drugs. Chem. Commun 2018, 54, 14089–14092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Sletten EM; Bertozzi CR A Bioorthogonal Quadricyclane Ligation. J. Am. Chem. Soc 2011, 133, 17570–17573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Tu J; Svatunek D; Parvez S; Liu AC; Levandowski BJ; Eckvahl HJ; Peterson RT; Houk KN; Franzini RM Stable, Reactive and Orthogonal Tetrazines: Dispersion Forces Promote the Cycloaddition with Isonitriles. Angew. Chem., Int. Ed 2019, 58, 9043–9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Italia JS; Addy PS; Erickson SB; Peeler JC; Weerapana E; Chatterjee A Mutually Orthogonal Nonsense-Suppression Systems and Conjugation Chemistries for Precise Protein Labeling at up to Three Distinct Sites. J. Am. Chem. Soc 2019, 141, 6204–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Kamber DN; Nguyen SS; Liang Y; Liu F; Briggs JS; Shih H-W; Houk KN; Prescher JA Isomeric 1,2,4-Triazines Exhibit Distinct Profiles of Bioorthogonal Reactivity. Chem. Sci 2019, 10, 9109–9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Liu F; Liang Y; Houk KN Bioorthogonal Cycloadditions: Computational Analysis with the Distortion/Interaction Model and Predictions of Reactivities. Acc. Chem. Res 2017, 50, 2297–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.McNitt CD; Popik VV Photochemical Generation of Oxa-Dibenzocyclooctyne (ODIBO) for Metal-Free Click Ligations. Org. Biomol. Chem 2012, 10, 8200–8202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Arumugam S; Popik VV Sequential “Click” – “Photo-Click” Cross-Linker for Catalyst-Free Ligation of Azide-Tagged Substrates. J. Org. Chem 2014, 79, 2702–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Mayer SV; Murnauer A; von Wrisberg M-K; Jokisch M-L; Lang K Photo-Induced and Rapid Labeling of Tetrazine-Bearing Proteins via Cyclopropenone-Caged Bicyclononynes. Angew. Chem., Int. Ed 2019, 58, 15876–15882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Sutton DA; Yu S-H; Steet R; Popik VV Cyclopropenone-Caged Sondheimer Diyne (Dibenzo[a,e]cyclooctadiyne): a Photoactivatable Linchpin for Efficient SPAAC Crosslinking. Chem. Commun 2016, 52, 553–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Sutton DA; Popik VV Sequential Photochemistry of Dibenzo[a,e]dicyclopropa[c,g][8]annulene-1,6-dione: Selective Formation of Didehydrodibenzo[a,e][8]annulenes with Ultrafast SPAAC Reactivity. J. Org. Chem 2016, 81, 8850–8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.McNitt CD; Cheng H; Ullrich S; Popik VV; Bjerknes M Multiphoton Activation of Photo-Strain-Promoted Azide Alkyne Cycloaddition “Click” Reagents Enables in Situ Labeling with Submicrometer Resolution. J. Am. Chem. Soc 2017, 139, 14029–14032. [DOI] [PubMed] [Google Scholar]
- 336.Luo W; Legge SM; Luo J; Lagugné-Labarthet F; Workentin MS Investigation of Au SAMs Photoclick Derivatization by PM-IRRAS. Langmuir 2020, 36, 1014–1022. [DOI] [PubMed] [Google Scholar]
- 337.Nainar S; Kubota M; McNitt C; Tran C; Popik VV; Spitale RC Temporal Labeling of Nascent RNA Using Photoclick Chemistry in Live Cells. J. Am. Chem. Soc 2017, 139, 8090–8093. [DOI] [PubMed] [Google Scholar]
- 338.Krenske EH; Petter RC; Houk KN Kinetics and Thermodynamics of Reversible Thiol Additions to Mono- and Diactivated Michael Acceptors: Implications for the Design of Drugs that Bind Covalently to Cysteines. J. Org. Chem 2016, 81, 11726–11733. [DOI] [PubMed] [Google Scholar]
- 339.Gan L-S; Zheng Y-L; Mo J-X; Liu X; Li X-H; Zhou C-X Sesquiterpene Lactones from the Root Tubers of Lindera aggregata. J. Nat. Prod 2009, 72, 1497–1501. [DOI] [PubMed] [Google Scholar]
- 340.Lu A; Wang J; Liu T; Han J; Li Y; Su M; Chen J; Zhang H; Wang L; Wang Q Small Changes Result in Large Differences: Discovery of (−)-Incrustoporin Derivatives as Novel Antiviral and Antifungal Agents. J. Agric. Food Chem 2014, 62, 8799–8807. [DOI] [PubMed] [Google Scholar]
- 341.Hou Y; Shi T; Yang Y; Fan X; Chen J; Cao F; Wang Z Asymmetric Total Syntheses and Biological Studies of Tuberostemoamide and Sessilifoliamide A. Org. Lett 2019, 21, 2952–2956. [DOI] [PubMed] [Google Scholar]
- 342.Nguyen SS; Ferreira AJ; Long ZG; Heiss TK; Dorn RS; Row RD; Prescher JA Butenolide Synthesis from Functionalized Cyclopropenones. Org. Lett 2019, 21, 8695–8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Lenzen A; Lannou M-I; Saint-Dizier A; Enders D The Phospha-Michael Addition in Organic Synthesis. Eur. J. Org. Chem 2005, 2006, 29–49. [Google Scholar]
- 344.Liu Y; Hou W; Sun H; Cui C; Zhang L; Jiang Y; Wu Y; Wang Y; Li J; Sumerlin BS; Liu Q; Tan W Thiol-Ene Click Chemistry: a Biocompatible Way for Orthogonal Bioconjugation of Colloidal Nanoparticles. Chem. Sci 2017, 8, 6182–6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Cramer NB; Bowman CN Thiol-Ene Chemistry. Chemoselective and Bioorthogonal Ligation Reactions: Concepts and Applications. Algar WR Dawson PE; Medintz IL 2017, 1, 117–145. [Google Scholar]
- 346.Galan SRG; Wickens JR; Dadova J; Ng W-L; Zhang X; Simion RA; Quinlan R; Pires E; Paton RS; Caddick S, et al. Post-Translational Site-Selective Protein Backbone α-Deuteration. Nat. Chem. Biol 2018, 14, 955–963. [DOI] [PubMed] [Google Scholar]
- 347.Dadová J; Galan SR; Davis BG Synthesis of Modified Proteins via Functionalization of Dehydroalanine. Curr. Opin. Chem. Biol 2018, 46, 71–81. [DOI] [PubMed] [Google Scholar]
- 348.Aroyan CE; Dermenci A; Miller SJ The Rahut-Currier Reaction: a History and its Synthetic Applications. Tetrahedron 2009, 65, 4069–4084. [Google Scholar]
- 349.Singh N; Pandey J Advances in Henry Reaction: a Versatile Method in Organic Synthesis. Mini-Rev. Org. Chem 2020, 17, 297–308. [Google Scholar]
- 350.Huang H; Lin S; Garcia BA; Zhao Y Quantitative Proteomic Analysis of Histone Modifications. Chem. Rev 2015, 115, 2376–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Chambers KA; Scheck RA Bacterial Virulence Mediated by Orthogonal Post-Translational Modification. Nat. Chem. Biol 2020, 16, 1043–1051. [DOI] [PubMed] [Google Scholar]
- 352.Tan M; Luo H; Lee S; Jin F; Yang JS; Montellier E; Buchou T; Cheng Z; Rousseaux S; Rajagopal N, et al. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell 2011, 146, 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Lee Y-L; Kurra Y; Liu WR Phospha-Michael Addition as a New Click Reaction for Protein Functionalization. ChemBioChem 2016, 17, 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Chen J; Jiang X; Carroll SL; Huang J; Wang J Theoretical and Experimental Investigation of Thermodynamics and Kinetics of Thiol-Michael Addition Reactions: a Case Study of Reversible Fluorescent Probes for Glutathione Imaging in Single Cells. Org. Lett 2015, 17, 5978–5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Heo CKM; Bunting JW Rate-Determining Steps in Michael-Type Additions and E1cb Reactions in Aqueous Solution. J. Org. Chem 1992, 57, 3570–3578. [Google Scholar]
- 356.Stewart IC; Bergman RG; Toste FD Phosphine-Catalyzed Hydration and Hydroalkoxylation of Activated Olefins: Use of a Strong Nucleophile to Generate a Strong Base. J. Am. Chem. Soc 2003, 125, 8696–8697. [DOI] [PubMed] [Google Scholar]
- 357.Jin K; Li T; Chow HY; Liu H; Li X P-B Desulfurization: an Enabling Method for Protein Chemical Synthesis and Site-Specific Deuteration. Angew. Chem., Int. Ed 2017, 56, 14607–14611. [DOI] [PubMed] [Google Scholar]
- 358.Tirla A; Rivera-Fuentes P Development of a Photoactivatable Phosphine Probe for Induction of Intracellular Reductive Stress with Single-Cell Precision. Angew. Chem., Int. Ed 2016, 55, 14709–14712. [DOI] [PubMed] [Google Scholar]
- 359.Vaughan JC; Dempsey GT; Sun E; Zhuang X Phosphine Quenching of Cyanine Dyes as a Versatile Tool for Fluorescence Microscopy. J. Am. Chem. Soc 2013, 135, 1197–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Vogelsang J; Steinhauer C; Forthmann C; Stein IH; Person-Skegro B; Cordes T; Tinnefeld P Make Them Blink: Probes for Super-Resolution Microscopy. ChemPhysChem 2010, 11, 2475–2490. [DOI] [PubMed] [Google Scholar]
- 361.Sabari BR; Tang Z; Huang H; Yong-Gonzalez V; Molina H; Kong HE; Dai L; Shimada M; Cross JR; Zhao Y, et al. Intracellular Crotonyl-CoA Stimulates Transcription through P300-Catalyzed Histone Crotonylation. Mol. Cell 2015, 58, 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Chambers KA; Abularrage NS; Scheck RA Selectivity within a Family of Bacterial Phosphothreonine Lyases. Biochemistry 2018, 57, 3790–3796. [DOI] [PubMed] [Google Scholar]
- 363.Lee Y-J; Wu B; Raymond JE; Zeng Y; Fang X; Wooley KL; Liu WR A Genetically Encoded Acrylamide Functionality. ACS Chem. Biol 2013, 8, 1664–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Josephson B; Fehl C; Isenegger PG; Nadal S; Wright TH; Poh AWJ; Bower BJ; Giltrap AM; Chen L; Batchelor-McAuley C, et al. Light-Driven Post-Translational Installation of Reactive Protein Side Chains. Nature 2020, 585, 530–537. [DOI] [PubMed] [Google Scholar]
- 365.Wang J; Lundberg H; Asai S; Martín-Acosta P; Chen JS; Brown S; Farrell W; Dushin RG; O’Donnell CJ; Ratnayake AS, et al. Kinetically Guided Radical-Based Synthesis of C(sp3)-C(sp3) Linkages on DNA. Proc. Natl. Acad. Sci. U.S.A 2018, 115, E6404–E6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Phelan JP; Lang SB; Sim J; Berritt S; Peat AJ; Billings K; Fan L; Molander GA Open-Air Alkylation Reactions in Photoredox-Catalyzed DNA-Encoded Library Synthesis. J. Am. Chem. Soc 2019, 141, 3723–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Wilkening I; del Signore G; Hackenberger CPR Synthesis of N,N-Disubstituted Phosphoramidates via a Lewis Acid-Catalyzed Phosphorimidate Rearrangement. Chem. Commun 2008, 25, 2932–2934. [DOI] [PubMed] [Google Scholar]
- 368.Serwa R; Wilkening I; Del Signore G; Mühlberg M; Claußnitzer I; Weise C; Gerrits M; Hackenberger CPR Chemoselective Staudinger-Phosphite Reaction of Azides for the Phosphorylation of Proteins. Angew. Chem., Int. Ed 2009, 48, 8234–8239. [DOI] [PubMed] [Google Scholar]
- 369.Wilkening I; del Signore G; Ahlbrecht W; Hackenberger CPR Lewis Acid or Alkyl Halide Promoted Rearrangements of the Phosphor- and Phosphinimidates to N,N-disubstituted Phoshpor- and Phosphinimidates. Synthesis 2011, 17, 2709–2729. [Google Scholar]
- 370.Mastryukova TA; Maschckenko NV; Odinets IL; Petrovskii PV; Kabachnik MI Imide-Amide Rearrangement of β-Chloroethylesters of N-Phenylimides of Phosphoric-Acids. Zh. Obshch. Khim 1988, 58, 1967–1973. [Google Scholar]
- 371.Vallée MRJ; Majkut P; Wilkening I; Weise C; Müller G; Hackenberger CPR Staudinger-Phosphonite Reactions for the Chemoselective Transformation of Azido-Containing Peptides and Proteins. Org. Lett 2011, 13, 5440–5443. [DOI] [PubMed] [Google Scholar]
- 372.Vallée MRJ; Majkut P; Krause D; Gerrits M; Hackenberger CPR Chemoselective Bioconjugation of Triazole Phosphonites in Aqueous Media. Chem. Eur. J 2015, 21, 970–974. [DOI] [PubMed] [Google Scholar]
- 373.Böhrsch V; Serwa R; Majkut P; Krause E; Hackenberger CPR Site-Specific Functionalisation of Proteins by a Staudinger-Type Reaction Using Unsymmetrical Phosphites. Chem. Commun 2010, 46, 3176–3178. [DOI] [PubMed] [Google Scholar]
- 374.Jaradat DMM; Hamouda H; Hackenberger CPR Solid-phase Synthesis of Phosphoramidate-Linked Glycopeptides. Eur. J. Org. Chem 2010, 2010, 5004–5009. [Google Scholar]
- 375.Böhrsch V; Mathew T; Zieringer M; Vallée MRJ; Artner LM; Dernedde J; Haag R; Hackenberger CPR Chemoselective Staudinger-Phosphite Reaction of Symmetrical Glycosyl-Phosphites with Azido-Peptides and Polygycerols. Org. Biomol. Chem 2012, 10, 6211–6216. [DOI] [PubMed] [Google Scholar]
- 376.Serwa R; Majkut P; Horstmann B; Swiecicki J-M; Gerrits M; Krause E; Hackenberger CPR Site-Specific PEGylation of Proteins by a Staudinger-Phosphite Reaction. Chem. Sci 2010, 1, 596–602. [Google Scholar]
- 377.Serwa RA; Swiecicki J-M; Homann D; Hackenberger CPR Phosphoramidate-Peptide Synthesis by Solution- and Solid-Phase Staudinger-Phosphite Reactions. J. Pept. Sci 2010, 16, 563–567. [DOI] [PubMed] [Google Scholar]
- 378.Bertran-Vicente J; Serwa RA; Schümann M; Schmieder P; Krause E; Hackenberger CPR Site-Specifically Phosphorylated Lysine Peptides. J. Am. Chem. Soc 2014, 136, 13622–13628. [DOI] [PubMed] [Google Scholar]
- 379.Bertran-Vicente J; Schümann M; Schmieder P; Krause E; Hackenberger CPR Direct Access To Site-Specifically Phosphorylated-Lysine Peptides from a Solid-Support. Org. Biomol. Chem 2015, 13, 6839–6843. [DOI] [PubMed] [Google Scholar]
- 380.Nischan N; Kasper M-A; Mathew T; Hackenberger CPR Bis(Arylmethyl)-Substituted Unsymmetrical Phosphites for the Synthesis of Lipidated Peptides via Staudinger-Phosphite Reactions. Org. Biomol. Chem 2016, 14, 7500–7508. [DOI] [PubMed] [Google Scholar]
- 381.Hoffmann E; Streichert K; Nischan N; Seitz C; Brunner T; Schwagerus S; Hackenberger CPR; Rubini M Stabilization of Bacterially Expressed Erythropoietin by Single Site-Specific Introduction of Short Branched PEG Chains at Naturally Occurring Glycosylation Sites. Mol. Biosyst 2016, 12, 1750–1755. [DOI] [PubMed] [Google Scholar]
- 382.Siebertz KD; Hackenberger CPR Chemoselective Triazole-Phosphonamidate Conjugates Suitable for Photorelease. Chem. Commun 2018, 54, 763–766. [DOI] [PubMed] [Google Scholar]
- 383.Kasper M-A; Stengl A; Ochtrop P; Gerlach M; Stoschek T; Schumacher D; Helma J; Penkert M; Krause E; Leonhardt H, et al. Ethynylphosphonamidates for the Rapid and Cysteine-Selective Generation of Efficacious Antibody–Drug Conjugates. Angew. Chem., Int. Ed 2019, 58, 11631–11636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Kasper M-A; Glanz M; Oder A; Schmieder P; Von Kries JP; Hackenberger CPR Vinylphosphonites for Staudinger-Induced Chemoselective Peptide Cyclization and Functionalization. Chem. Sci 2019, 10, 6322–6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Vallée MRJ; Artner LM; Dernedde J; Hackenberger CPR Alkyne Phosphonites for Sequential Azide–Azide Couplings. Angew. Chem., Int. Ed 2013, 52, 9504–9508. [DOI] [PubMed] [Google Scholar]
- 386.Wilkening I; Signore G. d.; Hackenberger CPR Synthesis of Phosphonamidate Peptides by Staudinger Reactions of Silylated Phosphinic Acids and Esters. Chem. Commun 2011, 47, 349–351. [DOI] [PubMed] [Google Scholar]
- 387.Bourguet CB; Proulx C; Klocek S; Sabatino D; Lubell WD Solution-Phase Submonomer Diversification of Aza-Dipeptide Building Blocks and Their Application in Aza-Peptide and Aza-DKP Synthesis. J. Pept. Sci 2010, 16, 284–296. [DOI] [PubMed] [Google Scholar]
- 388.Kasper M-A; Glanz M; Stengl A; Penkert M; Klenk S; Sauer T; Schumacher D; Helma J; Krause E; Cardoso MC, et al. Cysteine-Selective Phosphonamidate Electrophiles for Modular Protein Bioconjugations. Angew. Chem., Int. Ed 2019, 58, 11625–11630. [DOI] [PubMed] [Google Scholar]
- 389.Riva E; Wilkening I; Gazzola S; Li WMA; Smith L; Leadlay PF; Tosin M Chemical Probes for the Functionalization of Polyketide Intermediates. Angew. Chem., Int. Ed 2014, 53, 11944–11949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Bertran-Vicente J; Penkert M; Nieto-Garcia O; Jeckelmann J-M; Schmieder P; Krause E; Hackenberger CPR Chemoselective Synthesis and Analysis of Naturally Occurring Phosphorylated Cysteine Peptides. Nat. Commun 2016, 7, 12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Kee J-M; Villani B; Carpenter LR; Muir TW Development of Stable Phosphohistidine Analogues. J. Am. Chem. Soc 2010, 132, 14327–14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Bertozzi CR; Kiessling; Laura L Chemical Glycobiology. Science 2001, 291, 2357–2364. [DOI] [PubMed] [Google Scholar]
- 393.Hansson J; Oscarson S Complex Bacterial Carbohydrate Surface Antigen Structures: Syntheses of Kdo- and Heptose-Containing Lipopolysaccharide Core Structures and Anomerically Phosphodiester-Linked Oligosaccharide Structures. Curr. Org. Chem 2000, 4, 535–564. [Google Scholar]
- 394.Kanistanon D; Hajjar AM; Pelletier MR; Gallagher LA; Kalhorn T; Shaffer SA; Goodlett DR; Rohmer L; Brittnacher MJ; Skerrett SJ, et al. A Francisella Mutant in Lipid A Carbohydrate Modification Elicits Protective Immunity. PLOS Pathog. 2008, 4, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Nischan N; Chakrabarti A; Serwa RA; Bovee-Geurts PHM; Brock R; Hackenberger CPR Stabilization of Peptides for Intracellular Applications by Phosphoramidate-Linked Polyethylene Glycol Chains. Angew. Chem., Int. Ed 2013, 52, 11920–11924. [DOI] [PubMed] [Google Scholar]
- 396.Wasley LC; Timony G; Murtha P; Stoudemire J; Dorner AJ; Caro J; Krieger M; Kaufman RJ The Importance of N- and O-Linked Oligosaccharides for the Biosynthesis and in Vitro and in Vivo Biologic Activities of Erythropoietin. Blood 1991, 77, 2624–2632. [PubMed] [Google Scholar]
- 397.Gerngross TU Advances in the Production of Human Therapeutic Proteins in Yeasts and Filamentous Fungi. Nat. Biotechnol 2004, 22, 1409–1414. [DOI] [PubMed] [Google Scholar]
- 398.Leist M; Ghezzi P; Grasso G; Bianchi R; Villa P; Fratelli M; Savino C; Bianchi M; Nielsen J; Gerwien J, et al. Derivatives of Erythropoietin That Are Tissue Protective But Not Erythropoietic. Science 2004, 305, 239–242. [DOI] [PubMed] [Google Scholar]
- 399.Ochtrop P; Hackenberger CPR Recent Advances of Thiol-Selective Bioconjugation Reactions. Curr. Opin. Chem. Biol 2020, 58, 28–36. [DOI] [PubMed] [Google Scholar]
- 400.Shen B-Q; Xu K; Liu L; Raab H; Bhakta S; Kenrick M; Parsons-Reponte KL; Tien J; Yu S-F; Mai E, et al. Conjugation Site Modulates the in Vivo Stability and Therapeutic Activity of Antibody-Drug Conjugates. Nat. Biotechnol 2012, 30, 184–189. [DOI] [PubMed] [Google Scholar]
- 401.Otero-Ramirez ME; Passioura T; Suga H Structural Features and Binding Modes of Thioether-Cyclized Peptide Ligands. Biomedicines 2018, 6, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Celine A; Claudio S Converting a Peptide into a Drug: Strategies to Improve Stability and Bioavailability. Curr. Med. Chem 2002, 9, 963–978. [DOI] [PubMed] [Google Scholar]
- 403.Walensky LD; Kung AL; Escher I; Malia TJ; Barbuto S; Wright RD; Wagner G; Verdine GL; Korsmeyer SJ Activation of Apoptosis in Vivo by a Hydrocarbon-Stapled BH3 Helix. Science 2004, 305, 1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Gunnoo SB; Madder A Chemical Protein Modification through Cysteine. ChemBioChem 2016, 17, 529–553. [DOI] [PubMed] [Google Scholar]
- 405.Baumann AL; Schwagerus S; Broi K; Kemnitz-Hassanin K; Stieger CE; Trieloff N; Schmieder P; Hackenberger CPR Chemically Induced Vinylphosphonothiolate Electrophiles for Thiol–Thiol Bioconjugations. J. Am. Chem. Soc 2020, 142, 9544–9552. [DOI] [PubMed] [Google Scholar]
- 406.Bernardim B; Cal PMSD; Matos MJ; Oliveira BL; Martínez-Sáez N; Albuquerque IS; Perkins E; Corzana F; Burtoloso ACB; Jiménez-Osés G, et al. Stoichiometric and Irreversible Cysteine-Selective Protein Modification Using Carbonylacrylic Reagents. Nat. Commun 2016, 7, 13128. [DOI] [PMC free article] [PubMed] [Google Scholar]