

Abstract
Soft polymer nanoparticles designed to disassemble and release an antagonist of the neurokinin 1 receptor (NK1R) in endosomes provide efficacious yet transient relief from chronic pain. These micellar nanoparticles are unstable and rapidly release cargo, which may limit the duration of analgesia. We examined the efficacy of stable star polymer nanostars containing the NK1R antagonist aprepitant-amine for the treatment of chronic pain in mice. Nanostars continually released cargo for 24 h, trafficked through the endosomal system, and disrupted NK1R endosomal signaling. After intrathecal injection, nanostars accumulated in endosomes of spinal neurons. Nanostar-aprepitant reversed mechanical, thermal and cold allodynia and normalized nociceptive behavior more efficaciously than free aprepitant in preclinical models of neuropathic and inflammatory pain. Analgesia was maintained for >10 h. The sustained endosomal delivery of antagonists from slow-release nanostars provides effective and long-lasting reversal of chronic pain.
Keywords: Endosomes, Pain, Nociception, Receptors, Signal transduction
1. Introduction
Chronic pain is common, debilitating and poorly treated by available drugs. Whereas acute pain provides awareness and avoidance of injury and is thus necessary for survival, chronic pain can persist after healing and is maladaptive [1]. The factors that underlie the transition from acute (physiological) to chronic (pathological) pain are poorly understood. In consequence, the treatments for chronic pain are often ineffective or have unacceptable side effects. Although opioids are commonly used to treat acute pain, their analgesic properties wane with continued use (i.e., tolerance), while their on-target side effects of respiratory depression, constipation and sedation remain sustained, are life-threatening and are exacerbated by opioid addiction [2]. The opioid crisis highlights the need to develop improved treatments for chronic pain that are devoid of the side-effects of opioids.
G protein-coupled receptors (GPCRs) control most steps of pain transmission and are major therapeutic targets of treatments for pain [3]. GPCRs at the peripheral terminals of primary sensory neurons detect peptides, proteases, prostaglandins, amines and purines that are generated at sites of injury and disease. GPCRs expressed by second order spinal neurons detect substance P (SP), calcitonin gene-related peptide (CGRP) and glutamate that are released from the central projections of primary sensory neurons. GPCRs regulate the activity of ion channels that control neuronal activity and pain transmission, including members of the transient receptor potential family of cation channels [4].
GPCRs initiate signaling at the plasma membrane, where receptors interact with extracellular agonists and couple to intracellular heterotrimeric G proteins. However, plasma membrane GPCR signaling is transient. GPCR kinases phosphorylate activated receptors, increasing their affinity for β-arrestins (βARRs). βARRs uncouple GPCRs from G proteins, which desensitizes signaling, and couple GPCRs to clathrin and adaptor protein-2, which mediate receptor endocytosis [5]. Desensitization and endocytosis limit GPCR signaling at the surface of cells. Endosomes were formerly considered to be conduits for GPCR trafficking to degradatory or recycling pathways. Recent evidence suggests that GPCRs can form multiprotein signaling complexes within endosomes, including GPCR, Gα and βARRs “megaplexuses”, where Gα subunits convey signals and βARRs act as molecular scaffolds for signaling enzymes, including mitogen-activated protein kinases [6–10]. Evidence derived from use of inhibitors of clathrin-mediated endocytosis, and biosensors that detect signaling events with subcellular resolution, suggests that GPCRs in endosomes control a subset of signals in defined subcellular compartments. The SP neurokinin 1 receptor (NK1R), CGRP calcitonin-like receptor (CLR), protease-activated receptor-2 (PAR2) and the δ-opioid receptor signal from endosomes of primary sensory or spinal neurons to control neuronal excitation and pain transmission [11–15].
Since endosomal signals control pain transmission, GPCRs in endosomes might be the optimal therapeutic target for pain [16]. The inadequacy of conventional GPCR antagonists in clinical trials for chronic pain, where activated receptors may redistribute to endosomes due to chronic agonist production [11], might be related to their inability to engage GPCRs in endosomes. Whether conventional GPCR antagonists can penetrate the plasma and endosomal membrane and interact with GPCR conformations within multiprotein complexes in an acidic endosomal environment is seldom studied. Antagonists of the NK1R, CLR and PAR2 conjugated to a flexible PEG linker and the lipid cholestanol accumulate into plasma and endosomal membranes, block endosomal signals and provide more effective and long-lasting relief from pain than conventional antagonists [11–15]. The propensity of nanoparticles to accumulate in endosomes offers the prospect of bypassing the plasma membrane and direct delivery of antagonists to endosomes. When encapsulated into soft polymer nanoparticles designed to bypass cell surface receptors and release cargo in acidic endosomes, an antagonist of the NK1R, aprepitant (AP), provides potent and sustained analgesia in preclinical models of inflammatory and neuropathic pain [17,18]. These micellar nanoparticles rapidly disassemble and release cargo, are inherently unstable and are prone to dilution effects, which might limit the duration of action of analgesics. To surmount these limitations in the present study, we developed an analogue of AP functionalized with PEG-amine for conjugation to nanostars. Nanostars are dilution-stable and can be designed for tunable and sustained cargo release, which could provide a longer lasting analgesia. Core-crosslinked and functional polymeric nanostars are versatile soft matter nanoparticles engineered to be analogs of dendrimers [19]. We previously investigated the pharmacokinetic properties, uptake and association of nanostars with tumors, cancer cells and blood components [20–22]. We also explored the use of nanostars as platforms for the delivery of cancer therapeutics, antioxidants and imaging agents [23–25].
The current study compared the therapeutic efficacy of AP-nanostars and micellar AP-nanoparticles in preclinical models of neuropathic and inflammatory pain by studying evoked and non-evoked nociceptive behavior in mice. The analgesic properties of AP-nanostars with different degrees of hydrolytic stability were explored. The results show that the slow release of AP from nanostars in endosomes of spinal neurons provides a more complete and long-lasting analgesia than attained by free AP or AP encapsulated into micellar nanoparticles.
2. Materials and methods
2.1. Synthesis and characterization of micellar nanoparticles
DIPMA nanoparticles were generated and loaded as described [18]. In brief, diblock copolymers were synthesized with a hydrophilic shell of P(PEGMA-co-DMAEMA) and a hydrophobic core of P (DIPMA-co-DEGMA) to form pH-responsive DIPMA nanoparticles. Nanoparticles were self-assembled with AP (MK-869 [26]), a hydrophobic NK1R antagonist, forming DIPMA-aprepitant (DIPMA-AP). Empty nanoparticles (DIPMA-Ø) served as a control. The physicochemical properties of nanoparticles have been described [18,27]. DIPMA nanoparticles enter cells by clathrin- and dynamin-mediated endocytosis and release cargo under acidified conditions of endosomes (>50% cargo release, pH < 6.0).
2.2. Synthesis of nanostars
2.2.1. Materials
Oligo(ethylene glycol) methyl ether acrylate (OEGA, Mn ~480 g/mol, Merck) was deinhibited before use by percolation over basic aluminum oxide. Azobis(isobutyronitrile) (AIBN, Merck) was recrystallized from methanol before use. Sources of other reagents were: N,N′-methylenebis(acrylamide) (MBAA, Merck, 98%), 3-vinylbenzaldehyde (VBA, Sigma Aldrich, 97%), pentafluorophenyl acrylate (PFPA, Sigma Aldrich, 98%), trifluoroethanol (TFE, Sigma Aldrich, ≥ =99%), 4-benzoylphenyl acrylate (Benzo, BOC Biosciences), cyanine5 amine (Cy5 amine, Lumiprobe), tetrahydrofuran (THF, LiChrosolv). Chain transfer agent (3-benzylsulfanylthiocarbonylsulfanyl) propionic acid (BSPA) was synthesized as described [18]. Aprepitant amine (AP-NH2, MIPS-0020668) was synthesized in-house (Fig. S1). Petroleum benzine (b.p 60–80 °C), toluene, chloroform, diethyl ether, acetone and methanol were from Merck Millipore.
2.2.2. Synthesis, purification and activity of aprepitant-NH2 (AP-NH2)
AP-NH2 was synthesized for incorporation into nanostars. AP was from Combi blocks (CA, USA) and BocNH-PEG2-NH2 was from PurePEG (CA, USA). The synthesis scheme is shown in Fig. S1. The reaction was monitored by liquid chromatography-mass spectrometry (LC-MS) using an Agilent 6100 Series Single Quad LC-MS coupled to an Agilent 1200 Series HPLC. AP (Fig. S1, step 1; 600 mg, 1.123 mmol), 4-(3-(((2S,3R)-2-((S)-1-(3,5-bis(trifluoromethyl)phenyl)ethoxy)-3-(4-fluorophenyl)morpholino)methyl)-5-oxo-4,5-dihydro-1H-1,2,4-triazol-1-yl)butanoic acid, potassium iodide (37 mg, 0.245 mmol), potassium carbonate (310 mg, 2.245 mmol) and ethyl-4-bromobutyrate (162 μL, 1.123 mmol) were stirred in DMF (6 mL) at 60 °C for 6 h then at room temperature for 18 h. The mixture was diluted in EtOAc and washed with water, dried (MgSO4), filtered and concentrated to a resin that was chromatographed on silica gel with 60% EtOAc-pet ether to provide the intermediate ester as a clear colorless resin (62% yield, LCMS m/z 649.0 [M+H]+). The resin was taken up in MeOH (6.74 mL) and 1 M NaOH solution (3.37 mL) and stirred at room temperature overnight. The mixture was concentrated to remove volatiles and then diluted with water and acidified to pH 7 with 1 M HCl and then extracted with DCM. The combined DCM layers were dried (MgSO4), filtered and concentrated to provide (Fig. S1, step 2) as a resin/foam (400 mg. LCMS: m/z 621.0 [M+H]+). N-(2-(2-(2-aminoethoxy)ethoxy)ethyl)-4-(3-(((2S,3R)-2-((S)-1-(3,5-bis(trifluoromethyl)phenyl)ethoxy)-3-(4-fluorophenyl)morpholino)methyl)-5-oxo-4,5-dihydro-1H-1,2,4-triazol-1-yl)butanamide (Fig. 1, step 3) was prepared by dissolving the acid (Fig. 1, step 2) (105 mg, 0.169 mmol), HATU (64 mg, 0.169 mmol) and BocNH-PEG2-NH2 (42 mg, 0.169 mmol) in DMF (2 mL) and DIPEA (88 μL, 0.507 mmol) and stirred at room temperature for 3 h. The mixture was partitioned between EtOAc and water. The organic layer was washed with water, dried (MgSO4), filtered and concentrated to a resin. The resin was taken up in 3 mL of 1% cHCl in HFIP and left to stand for 4 h and then concentrated. The crude was purified by HPLC to provide 40 mg of resin (32% yield, LCMS: m/z 649.0 [M+H]+). 19F NMR (400 MHz, DMSO) δ −61.20 (s, 6F), δ −114.35 (s, 3F); 1H NMR (400 MHz, DMSO) δ 8.11–7.70 (m, 6H), 7.57–7.49 (m, 2H), 7.45 (s, 2H), 7.30–6.99 (m, 2H), 4.96 (q, J = 6.4 Hz, 1H), 4.31 (d, J = 2.3 Hz, 1H), 4.12 (t, J = 10.7 Hz, 1H), 3.69–3.49 (m, 12H), 3.20 (dd, J = 11.7, 5.9 Hz, 2H), 2.95 (dt, J = 11.2, 5.6 Hz, 2H), 2.79 (dd, J = 31.5, 13.3 Hz, 2H), 2.44–2.34 (m, 1H), 2.13 (t, J = 7.7 Hz, 2H), 1.98–1.74 (m, 2H), 1.39 (d, J = 6.5 Hz, 3H). To assess antagonistic activity, HEK293Tcells expressing human (h) NK1R were preincubated with graded concentrations of AP and AP-NH2 for 30 min, and were then challenged with 10 nM SP. Changes in [Ca2+]i were monitored using Fura-2/AM [14].
Fig. 1. DIPMA nanoparticles, mechanical and cold allodynia in neuropathic pain.
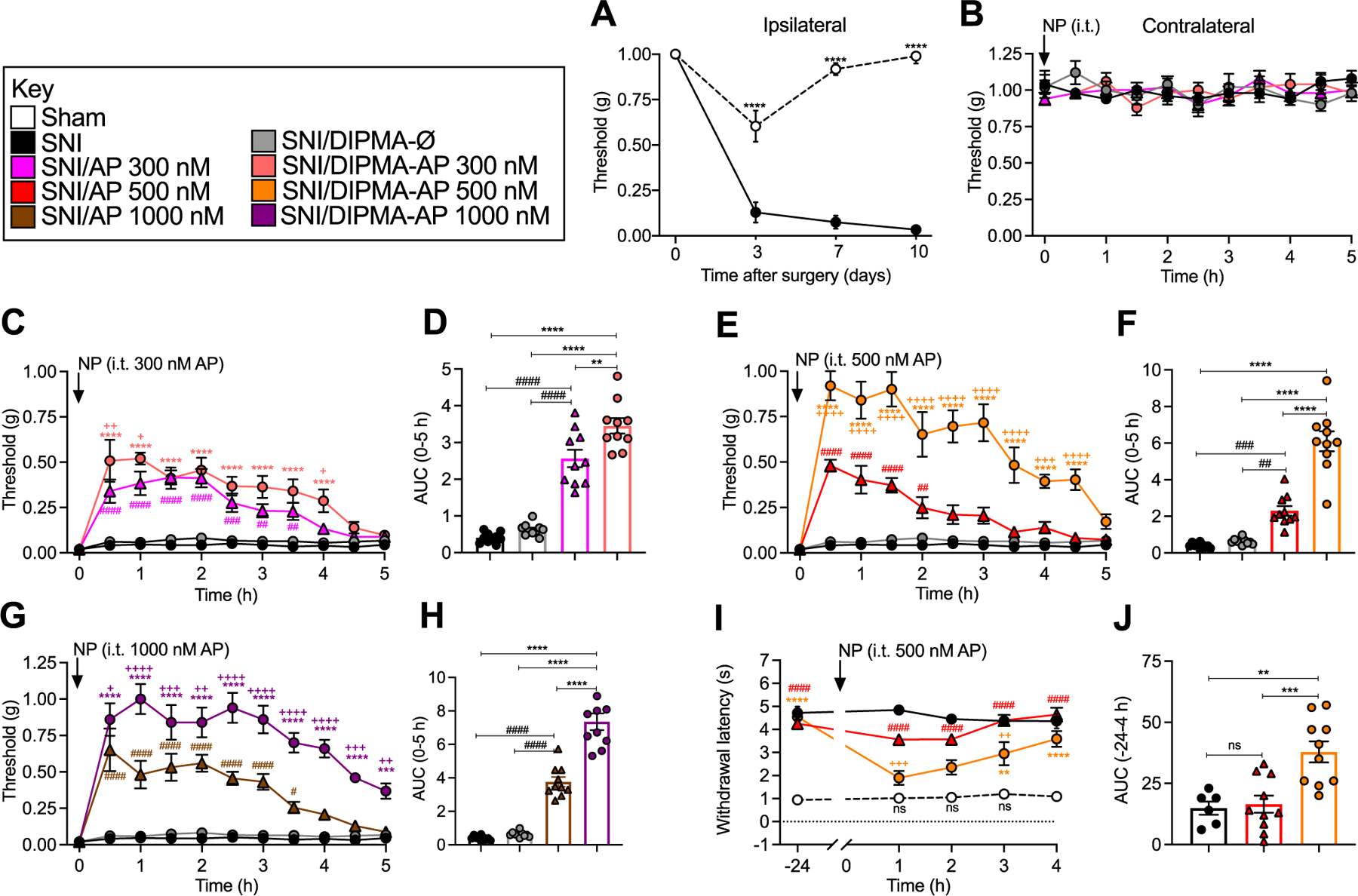
A. Time course of development of VFF withdrawal responses (mechanical allodynia) of ipsilateral paw in SNI and sham control mice. All subsequent studies were at 10 days post SNI surgery. B. Effects of DIPMA-AP, AP or DIPMA-Ø on VFF withdrawal responses of contralateral paw. C–H. Effects of DIPMA-AP, AP or DIPMA-Ø on VFF withdrawal responses of ipsilateral paw. C, E, G show time course and D, F, H show area under curve (AUC) of effects of 300, 500 or 1000 nM DIPMA-AP or free AP. I, J. Effects of DIPMA-AP, AP or DIPMA-Ø on acetone withdrawal responses (cold allodynia) of ipsilateral paw in SNI and sham mice. I shows time course and J shows of effects of 500 nM DIPMA-AP or AP. Mean ± SEM, N = 10 mice. *, +, #P < 0.05; **, ++, ##, P < 0.01; ***, +++, ###P < 0.005; ****, ++++, ####P < 0.001. * DIPMA-AP to SNI, DIPMA-Ø and AP; + DIPMA-AP to AP; # AP to SNI. Two-way ANOVA (mechanical and cold allodynia), one-way ANOVA (AUC) with Tukey’s multiple comparisons test.
2.2.3. Synthesis of nanostar POEGA arm
A macromolecular chain transfer agent (macro-CTA, POEGA) was synthesized by the reversible addition fragmentation chain transfer (RAFT) polymerization method using the RAFT agent benzylsulfanylthiocarbonylsufanylpropionic acid (BSPA, 0.051 g, 0.19 mmol, 1 eq.), the initiator azobisisobutyronitrile (AIBN, 0.003 g, 0.02 mol, 0.1 eq.), and oligo(ethylene glycol) methyl ether acrylate Mn ~480 g/mol (OEGA, 3.0 g, 6.25 mmol, 32 eq.). The mixture was dissolved in 9 mL toluene and deoxygenated by sparging with nitrogen for 30 min in an ice/water bath. The reaction proceeded at 70 °C, 400 RPM for 4 h. The polymerization was stopped by cooling in an ice/water bath and exposure to air. The resulting arm polymer (BSPA-POEGA) was purified by repeated precipitations in cold petroleum benzine 60–80 °C and then dried for 24 h under vacuum at 37 °C and 1 mbar.
2.2.4. Synthesis of core-crosslinked nanostar polymers
A series of core-crosslinked nanostar polymers was synthesized with the functional monomers 3-vinylbenzaldehyde (VBA), 4-benzoylphenyl acrylate (Benzo) or pentafluorophenyl acrylate (PFPA) incorporated into the core. All reactions were carried out in 5 mL of toluene and deoxygenated by sparging with nitrogen for 30 min in an ice/water bath. Polymerizations proceeded in an oil bath at 70 °C for 24 h and were stopped by cooling on ice/water and exposure to air. Nanostar polymers were synthesized with the following stoichiometries:
Nanostar-VBA:
[POEGA]0:[MBAA]0:[VBA]0:[AIBN]0 = 1:8:10:0.3. BSPA-POEGA linear arm (500 mg, 0.0532 mmol), N,N′-methylenebisacrylamide (65.7 mg, 0.426 mmol), VBA (70.4 mg, 0.532 mmol), AIBN (2.6 mg, 0.0160 mmol).
Nanostar-Benzo:
[POEGA]0:[MBAA]0:[Benzo]0:[AIBN]0 = 1:8:10:0.3. BSPA-POEGA linear arm (500 mg, 0.0532 mmol), N,N′-methylenebisacrylamide (65.7 mg, 0.426 mmol), 4-benzophenyl acrylate (134.3 mg, 0.532 mmol), AIBN (2.6 mg, 0.0160 mmol).
Nanostar-PFPA:
[POEGA]0:[MBAA]0:[PFPA]0:[AIBN]0 = 1:8:10:0.3. BSPA-POEGA linear arm (500 mg, 0.0532 mmol), N,N′-methylenebisacrylamide (65.7 mg, 0.426 mmol), PFPA (126.8 mg, 0.532 mmol), AIBN (2.6 mg, 0.0160 mmol).
2.2.5. Purification of nanostar polymers
Nanostars were purified by five successive precipitation and centrifugation steps using chloroform into diethyl ether (2% v/v) to remove unreacted cross-linker, arm polymer and monomers. Nanostar polymers were characterized by 1H NMR and gel permeation chromatography (GPC).
2.2.6. Synthesis of nanostar-Cy5-NH2
Nanostar-Cy5 conjugates (designated PFPA-Cy5, VBA-Cy5, Benzo-Cy5) were polymerized in DMF (1 mL) in dark at room temperature for 72 h with the following stoichiometries:
VBA-Cy5:
Nanostar-VBA (0.02 g), Cy5-NH3⋅Cl (1.3 mg, 2.1 × 10−6 mol) and triethylamine (5.0 μL, 3.59 × 10−5 mol).
Benzo-Cy5:
Nanostar-Benzo (0.02 g), Cy5-NH3⋅Cl (1.2 mg, 2.0 × 10−6 mol) and triethylamine (5.0 μL, 3.59 × 10−5 mol).
PFPA-Cy5:
Nanostar-PFPA (0.02 g), Cy5-NH3⋅Cl (1.9 mg, 3.23 × 10−6 mol) and triethylamine (5.0 μL, 3.59 × 10−5 mol).
2.2.7. Synthesis of nanostar-AP-NH2
AP-NH3 was synthesized and purified as described in supplementary methods. Nanostar-AP conjugates were polymerized in DMSO‑d6 (0.6 mL) in dark at room temperature for 155 h for nanostar-PFPA-AP (designated PFPA-AP), 155 h for nanostar-VBA-AP (VBA-AP) and 84 h for nanostar-Benzo-AP (Benzo-AP). The state of the reaction was periodically assessed using 19F NMR. Stoichiometries of the reactions were as follows:
VBA-AP:
Nanostar-VBA (0.02 g), AP-NH3⋅Cl (1.5 mg, 2.0 × 10−6 mol) and triethylamine (5.0 μL, 3.59 × 10−5 mol).
Benzo-AP:
Nanostar-Benzo (0.02 g), AP-NH3⋅Cl (1.6 mg, 2.1 × 10−6 mol) and triethylamine (5.0 μL, 3.59 × 10−5 mol).
PFPA-AP:
Nanostar-PFPA (0.02 g), AP-NH3⋅Cl (2.5 mg, 3.3 × 10−6 mol) and triethylamine (5.0 μL, 3.59 × 10−5 mol).
2.2.8. Purification of nanostars
Nanostar-Cy5 or nanostar-AP was purified by preparative size exclusion chromatography using Bio-Beads S-X1 support and chromatographic grade THF as the mobile phase. Fractions containing nanostar-Cy5 or nanostar-AP conjugates were combined and stored at −20 °C. Successful Cy5 conjugation and the removal of free Cy5 was confirmed by GPC using refractive index (RI) and UV-VIS detection (λ = 646). AP-NH2 content (conjugated or free contaminant) was determined by 19F NMR using a trifluoroethanol internal standard, while 1H NMR confirmed macromolecular structure. The expected increase in apparent molecular weight of the nanostars on conjugation with AP-NH2 was confirmed by GPC. Nanostar-AP conjugates were stored in DMSO‑d6 at −20 °C.
2.3. Characterization of nanostars
2.3.1. Gel permeation chromatography (GPC)
Nanostars were characterized using a Shimadzu GPC system comprising three KF-805L columns (300 × 8 mm, bead size: 10 mm, pore size maximum: 5000 Å) and SPD-20A UV/Vis and RID-10A differential refractive-index detectors. The temperature of columns was maintained at 40 °C. The eluent was N,N-dimethylacetamide (CHROMASOLV Plus for HPLC) with 0.03% w/v LiBr at a flow rate of 1.0 mL/min. A molecular weight calibration curve was produced using polystyrene standards (500–2 × 106 g mol−1). Polymer solutions (~2 mg/mL) were prepared and filtered through 0.45 μm PTFE filters before injection.
2.3.2. Proton-nuclear magnetic resonance (1H NMR) and fluorine-19 nuclear magnetic resonance (19F NMR)
NMR spectra were recorded on a Bruker Advance III (400 MHz for 1H NMR and 376 MHz for 19F NMR) spectrometer using an external lock. The 1H NMR spectra were referenced to the residual nondeuterated solvent and 19F NMR spectra were referenced to trifluoroethanol (TFE). Chemical shifts (δH) are reported in parts per million (ppm). CDCl3 and DMSO‑d6 (Cambridge Isotope Laboratories, Inc) were used as NMR solvents. Conversion and degree of polymerization (DP) were calculated by 1H NMR using peak integrals (I) where the subscript number indicates the location of the peak in ppm (Ix). The DP for BSPA-POEGA was calculated using the 1H NMR spectra with normalized to peaks a-c = 5 (I7.32). The molecular weight of the polymer (Mn) was calculated using the 1H NMR spectra with .
2.3.3. Dynamic light scattering (DLS) and ζ-potential analysis
Dynamic light scattering and ζ-potential analysis were determined at 25 °C using a Malvern Zetasizer ZS series equipped with a 4 mW laser at λ = 633 nm and a detector angle 173° running DTS software. Viscosity and refractive index were assumed to be 0.899 cP and 1.338 as for PBS and PEG particles: the DH number, hydrodynamic diameter, and ZETA potential are reported as determined by the DTS software.
2.3.4. Transmission electron microscopy (TEM)
The morphology of nanostars was determined by TEM imaging using a Thermo Scientific L120C electron microscope at an accelerating voltage of 120 kV at room temperature. Formvar coated copper grids (GSCu100F-50, Proscitech) were pretreated in a plasma discharger for 35 s. Nanostars (3 μL, 3 μg/mL) were deposited on grids, immediately blotted, and dried for 1 h followed by negative staining with Ammonium molybdate (5 μL, 5 wt %, 25 s).
2.3.5. pH-dependent release of Cy5-NH2. Cy5-NH2 release from nanostars was examined at pH 7.4, 6.0 and 5.0 by UV–visible spectrophotometry
Nanostar-Cy5 conjugate was dried, reconstituted in PBS (pH 7.4, 6.0, 5.0), and placed into the inner sample tube of an individual Spectra-Por Float-A-Lyser G2 Dialysis Device, 3.5–5k MWCO (1.0 mL). Cy5 release study was examined under sink conditions in 1 L PBS buffer (pH 7.4 or 6.0). At defined times, an aliquot (0.5 mL) of solution within the sample tube was removed, analyzed by UV–Vis (λ = 646 nm), and returned to the sample compartment. Changes in the absorbance at λ = 646 nm are reported relative to the starting solution.
2.3.6. Release of AP-NH2
AP-NH2 release from nanostars was examined at pH 7.4 by HPLC. Nanostar-AP conjugates were suspended in PBS containing 20% DMSO, adjusted to pH 7.4. Nanostar-AP conjugates (500 μL, 200 μM) were placed into a dialysis device (Spectra-Por Float-A-Lyser G2, 3.5–5k MWCO) and dialyzed against 15 mL of PBS/DMSO. At defined time points, 300 μL of the release solution was collected for HPLC analysis. The same volume of PBS/DMSO was added immediately into the release solution to maintain constant volume. Release was studied under stirring at room temperature. AP-NH2 release was quantified by HPLC using an ultra-fast liquid chromatograph (UFLC, Shimadzu) with a C18 column (150 × 4.6 mm, 5 μm) using a detection wavelength of 215 nm. The mobile phase was 0.01% phosphoric acid in water and acetonitrile. Samples (300 μL) for analysis were concentrated to a final volume of 100 μL using an ISSI110 SpeedVac Concentrator (Thermo Fisher). AP-NH2 release was quantified against standards (6.25–200 μM), and compounds were eluted under gradient conditions at a flow rate of 1.5 mL/min.
2.4. Studies of nanostars in cell lines
2.4.1. Cell lines
HEK293T (#CRL-3216™, American Type Culture Collection) cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with heat inactivated fetal bovine serum (fbs, 10%), L-glutamine (2 mM), penicillin (100 U/ml) and streptomycin (100 mg/mL) at 37 °C in 5% CO2 and 95% O2. cDNA encoding the hNK1R with an extracellular N-terminal FLAG-tag was transfected into HEK293Tcells to generate a stable cell line expressing hNK1R (HEK-hNK1R) as described [18].
2.4.2. Cell viability assays
HEK293Tcells were plated (25,000 cells/well) into poly-D-lysine-coated 96 well CulturPlates (PerkinElmer, USA). Cells were incubated with vehicle (HBSS), VBA-Ø, Benzo-Ø, PFPA-Ø (1–100 μg/mL) or the positive control Triton X-100 (1%) for 24 h and 48 h. Viability was assessed using AlamarBlue (In Vitro Technologies), propidium iodide (Sigma Aldrich) or Thiazolyl Blue Tetrazolium Bromide (MTT, Sigma Aldrich), according to the manufacturers’ instructions. Viability was calculated by subtracting the values of cells treated with the positive control Triton X-100 and expressing the values as a percentage of the vehicle control.
2.4.3. Endocytosis assays
HEK293T cells were plated into 12 mm glass coverslips and cultured for 24 h. Cells were incubated with PFPA-Cy5 nanostars (20 μg/mL) in Hank’s balanced salt solution (HBSS) for 30, 60 or 120 min at 37 °C. Cells were washed and fixed in 4% paraformaldehyde (10 min on ice). Cells were incubated in permeabilizing/blocking buffer (5% normal horse serum in PBS containing and 0.3% saponin) for 1 h at room temperature. Cells were incubated with a polyclonal anti-EEA1 antibody (rabbit, #ab2900, Abcam, 1:1000) or polyclonal anti-Rab7 antibody (rabbit, #9367, Cell Signaling, 1:1000) in blocking buffer overnight at 4 °C. Cells were washed and incubated with secondary antibody (donkey anti-rabbit Alexa488, ThermoFisher, 1:1000). Coverslips were mounted using Prolong diamond antifade (ThermoFisher) and images were captured on a Leica TCSSP8 confocal using an HCX PL APO Å~63 NA 2.0 oil objective. Images were analyzed using Fiji [28] with the toolbox plugin JACoP for Manders’ overlap coefficient analysis [29].
2.4.4. Enhanced bystander bioluminescence resonance energy transfer (EbBRET) assays
Mini-G proteins coupled to Venus were from N. A. Lambert (Augusta University). Constructs were modified to replace Venus with Renilla (R) luciferase 8 (Rluc8). Renilla GFP-CAAX (prenylation CAAX box of KRas), tdRGFP-Rab5a and Rluc2-βARR2 were from M. Bouvier (Université de Montréal). HEK293T cells were transfected using PEI with hNK1R (0.2 μg) and Rluc8-mGαsi/s/sq (0.1 μg) [30,31]and either RGFP-CAAX (0.2 μg) for cell surface activation or tdRGFP-Rab5a (0.15 μg) for activation in early endosomes [32]. For EbBRET assays of recruitment of βARR2 to the plasma membrane or early endosomes, HEK293T cells were transfected with PEI with hNK1R (0.2 μg) and Rluc2-βARR2 (0.1 μg) and RGFP-CAAX (0.2 μg) for plasma membrane recruitment or tdRGFP-Rab5a (0.15 μg) for early endosome recruitment [32]. For EbBRET assays, cells were washed with HBSS containing 10 mM Hepes (pH 7.4). Cells were incubated with Prolume Purple Coelenterazine (2.5 μM, 5 min, 37 °C; NanoLight Technology). EbBRET was recorded for 22.5 min in a Synergy Neo2 Microplate reader (BioTek) (acceptor filter: 515 ± 30 nm; donor filter: 410 ± 80 nm). Baseline was measured for 2.5 min and cells were challenged with SP (10 nM) or vehicle. ΔBRET represents the EbBRET signal in the presence of agonist subtracted by the EbBRET signal in the presence of vehicle.
2.5. Studies of nanostars in mice
2.5.1. Animals
Adult male C57BL/6 mice (6–10 weeks) were from the Monash Animal Research Platform and Jackson Laboratories (#000664) USA. Mice were maintained in a light-controlled (12-h light/dark cycle) temperature-controlled (22 ± 4 °C) environment with at libitum access to food and water. Studies on mice were in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and adhered to the ethical guidelines of the International Association for the Study of Pain [33]. Studies were approved by the animal ethics committees of the Monash Institute of Pharmaceutical Sciences, Monash University and of New York University Langone Medical Center. At the end of the studies, animals were euthanized by anesthetic overdose (5% isoflurane) and thoracotomy.
2.5.2. Nanostar biodistribution in animals
PFPA-Cy5 nanostars (1 mg/mL, 5 μL) were administered to mice (i.t., L4/L5) under sedation (2% isoflurane). For assessment of biodistribution in whole animals, mice were sedated again (2% isoflurane) and placed in an in vivo imaging system (IVIS spectrum, PerkinElmer, USA). Posterior images were obtained before and at 1 min, 2 h and 8 h post PFPA-Cy5 administration using PerkinElmer Living Image software v4.3.1.
2.5.3. Nanostar imaging in spinal cord sections
At 4 h after administration of PFPA-Cy5 nanostars, mice were transcardially with 4% paraformaldehyde in PBS and frozen sections of spinal cord were prepared as described [18]. Sections were incubated with mouse anti-Hu (1:2000, AB_221448), goat anti-GFAP (1:1000, Abcam, AB_880202) or rat anti-CD11b (1:1000, AB_312785) in PBS containing 0.2% Triton X-100 and 5% NHS (overnight, 4 °C). Sections were washed 3× in PBS for 10 min and incubated with donkey anti-rat Alexa 488 (1:500, ThermoFisher Scientific, AB_2535794), anti-mouse Alexa 568 (1:500, ThermoFisher Scientific AB_2534013) and anti-sheep DyLight™ 405 (1:250, Jackson Immunoresearch, AB_2340740) (2 h, room temperature). Sections were counter-stained with DAPI (5 μg/mL, 5 min), washed and mounted with ProLong Glass mounting medium (ThermoFisher Scientific). Sections were imaged on TCS-SP8 Lightning confocal system (20× HC PL APO NA 0.88 water immersion objective) and images were processed using FIJI.
2.5.4. Drug administration
The following drugs were administered by intrathecal (i.t.) injection (5 μL) into the intervertebral space (L4/L5) of conscious mice: AP (300–1000 nM), AP-NH2 (1000 nM), DIPMA-AP delivering an equivalent dose of AP (300–1000 nM AP), or nanostars delivering an equivalent dose of AP (1000 nM). Controls included empty nanoparticles (DIPMA-Ø, 100 μg/mL; VBA-Ø, 17 μg/mL; Benzo-Ø, 13 μg/mL; PFPA-Ø, 8.33 μg/mL), or vehicle (artificial cerebrospinal fluid, aCSF). Drugs were injected 10 days after induction of neuropathic pain or 2 days after induction of inflammatory pain.
2.5.5. Neuropathic pain
The spared nerve injury (SNI) procedure [34] with modifications was used to induce neuropathic pain. Under anesthesia (5% isoflurane), the left thigh was shaved and cleaned with isopropyl alcohol and iodide. The skin was incised (1 cm) on the lateral surface of the left thigh and the muscle was bluntly dissected to identify the sural, common peroneal and tibial branches of the sciatic nerve. The common peroneal and tibial nerves were ligated with 6–0 silk adjacent to the trifurcation, and a 1 mm segment of the two nerves was removed immediately distal to the ligature. For sham controls, the trifurcation was identified, but the nerves were untouched. The muscle layer was closed using 5–0 vicryl and the skin was closed with 5–0 surgipro (Ethicon, USA).
2.5.6. Inflammatory pain
An emulsion of Complete Freund’s Adjuvant (CFA, 0.5 mg/mL) and saline (0.9% NaCl) was prepared by vortexing for 1 min. CFA or vehicle (0.9% NaCl) was administered by intraplantar (i.pl.) injection (10 μL) into the left hindpaw of sedated mice (2% isoflurane) [11,35].
2.5.7. Assessment of nociception
Investigators were blinded to the procedure (CFA or vehicle, SNI or sham) and treatment. Mice were randomly assigned to treatments (www.randomization.com). Mice were studied during the light cycle (8:00 a.m. to 2:00 p.m.). Prior to experiments, mice were acclimatized to the experimental apparatus and environment for >1 h on 2 successive days.
Mechanical allodynia.
Mechanical nociception was assessed by measuring withdrawal thresholds to stimulation of the plantar surfaces of the hindpaws with calibrated von Frey filaments (VFFs) [13]. Mice were placed on a metal grid. VFF withdrawal thresholds were measured in triplicate to establish a baseline for each mouse. VFF responses were then measured at various times after drug treatment. Results are expressed as nociceptive threshold in grams, as percentage of baseline or as area under curve during the experimental period.
Cold allodynia.
Cold nociception was assessed by measuring the acute nociceptive response to evaporative cooling induced by topical application of acetone to the plantar surface of the hindpaw [36]. Mice were placed on a metal grid. Acetone (50 μL) was applied to the plantar surface of the hindpaw using a pipette. The time spent in flicking or licking of the plantar region over 60 s was measured.
Behavioral spectrometer.
Non-evoked nociception was assessed using a behavioral spectrometer, which eliminates operator bias (Behavior Sequencer, Behavioral Instruments, NJ; BiObserve, DE) [37,38]. The spectrometer comprised a 40 cm2 arena with a CCD camera mounted in the center of the ceiling and a door aperture in the front area of the arena. Mouse movement was assessed by a floor mounted vibration sensor and 32 wall mounted infrared transmitter and receiver pairs. Mice were individually placed in the center of the behavioral spectrometer and their behavior was recorded, tracked, evaluated and analyzed using a computerized video tracking system (Viewer3, BiObserve, DE) for 30 min. Total distance traveled in the open field, average velocity of locomotion, wall distance, ambulation and grooming were recorded and analyzed.
2.6. Statistical analyses
Data are presented as mean ± SEM unless noted otherwise. Student’s t-test was used for two comparisons. For multiple comparisons, results were compared using one- or two-way ANOVA followed by Tukey’s or Dunnett’s multiple comparison tests. A P < 0.05 was considered significant.
3. Results
3.1. pH-tunable DIPMA-AP nanoparticles provide efficacious relief from neuropathic pain
We have previously determined the efficacy of intrathecal DIPMA-AP for reversal of mechanical allodynia in preclinical models of inflammatory and neuropathic pain in mice and rats [18]; other indices of evoked and non-evoked nociception were not examined. Herein, we compared the effects of graded doses of DIPMA-AP and free AP on mechanical allodynia, cold allodynia and non-evoked nociceptive behavior in a preclinical model of neuropathic pain in mice. The SNI model of neuropathic pain was studied in which the common peroneal and tibial branches of the sciatic nerve of the left hindpaw were transected, leaving the sural branch intact [34]. Control mice underwent sham surgery. To assess mechanical allodynia, withdrawal responses to stimulation of the planar surfaces of the lesioned (left, ipsilateral) and unaffected (right, contralateral) hindpaws with graded VFFs were measured. Cold allodynia was assessed by measuring nociceptive responses (paw flicking and licking over 60 s) to evaporative cooling induced by topical application of acetone to the plantar surface of the ipsilateral hindpaw. Non-evoked nociception was assessed using a behavioral spectrometer, which recorded multiple behaviors and eliminated operator bias [37, 38].
Mechanical allodynia.
The VFF response measured in the ipsilateral paw of SNI mice declined at 3 days post-surgery and was sustained for at least 10 days, indicating development of sustained mechanical allodynia (Fig. 1A). In sham mice, the VFF response declined at 3 days, suggesting post-incisional pain, and recovered to pre-surgical within 7–10 days. In all subsequent experiments, mice were studied 10 days after SNI. Graded doses of DIPMA-AP, an equivalent dose of free AP (300, 500, 1000 nM AP) or DIPMA-Ø was injected intrathecally (all 5 μL), and mechanical allodynia was measured every 30 min for 5 h. These treatments had no effect on VFF withdrawal responses of the contralateral paw (Fig. 1B). Free AP caused a dose-related and partial reversal of VFF withdrawal responses of the ipsilateral paw for up to 3.5 h (Fig. 1C–H). At all doses, the magnitude and duration of the anti-nociceptive effects of DIPMA-AP significantly exceeded that of free AP and was sustained for at least 5 h (Fig. 1C–H). The VFF response (expressed as a percentage of baseline values) at 1 h was 40.3 ± 7.6% and 48.2 ± 9.5% basal after free AP 500 and 1000 nM respectively, but 84 ± 10.2% and 100 ± 10.3% basal after DIPMA-AP 500 and 1000 nM (P < 0.0001) respectively. DIPMA-Ø did not affect mechanical allodynia.
Cold allodynia.
At 10 days surgery, application of acetone to the paw caused a nocifensive response that was more sustained for SNI than sham mice, indicating cold allodynia (Fig. 1I and J). DIPMA-AP (500 nM AP) almost normalized cold allodynia at 1 and 2 h in SNI mice, whereas AP was less efficacious at all time points. DIPMA-Ø did not affect cold allodynia.
Non-evoked nociceptive behavior.
At 90 min after intrathecal drug administration, non-evoked nociceptive behavior was measured over 30 min. Compared to sham operated controls, SNI mice exhibited significant decreases in average distance from the wall (Fig. 2A), grooming time (Fig. 2B) and ambulation time (Fig. 2C), whereas track length (Fig. 2D), average velocity (Fig. 2E) and locomotor activity (Fig. 2F) were unaffected. The number of times that the SNI mice crossed a central area of the recording chamber was also significantly diminished in SNI mice (Fig. 2G, H, L). Intrathecal injection of gabapentin (1000 nM), an effective treatment for neuropathic pain, normalized affected behaviors in SNI mice. Free AP (500 nM) failed to normalize distance from wall, grooming and ambulation (Fig. 2A–C) but partially normalized center area crossing (Fig. 2J, L). In contrast, DIPMA-AP (500 nM) fully restored distance from wall (Fig. 2A), grooming time (Fig. 2B), ambulation time (Fig. 2C) and center crossing (Fig. 2L, L) in SNI mice to levels observed in sham mice. Neither AP nor DIPMA-AP affected track length, ambulation velocity or locomotor activity (Fig. 2D–F). DIPMA-Ø did not affect any measured parameter.
Fig. 2. DIPMA nanoparticles, non-evoked behavior in neuropathic pain.
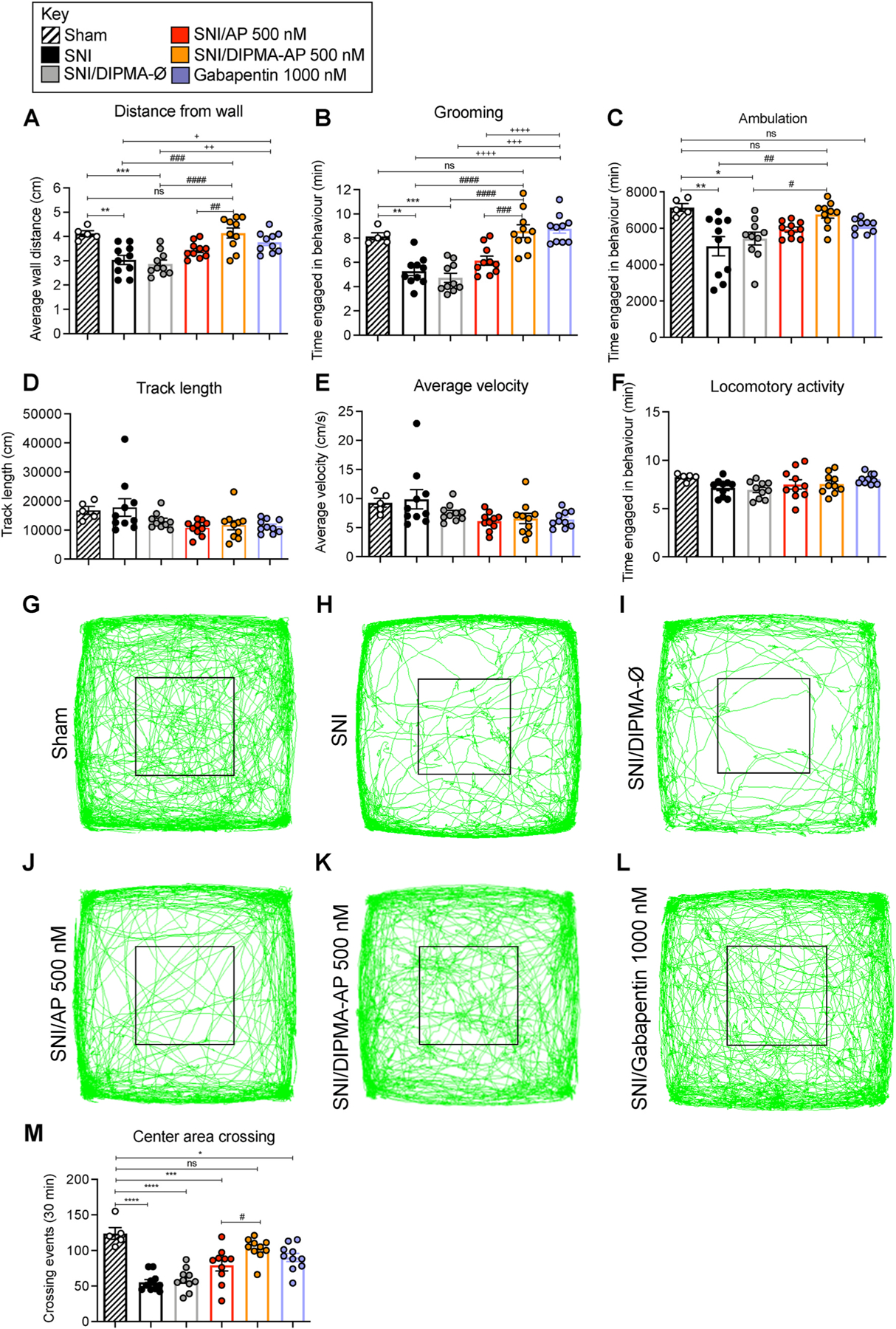
Non-evoked nociceptive behavior in SNI and sham control mice recorded for 30 min at 90 min after intrathecal injection of DIPMA-AP, AP (500 nM AP), DIPMA-Ø or gabapentin (1000 nM). Distance from wall (A), grooming (B), ambulation (C), track length (D), average velocity, (E) and locomotor activity (F) were recorded. G-L are records of tracks of mice. M shows crossing events in area marked by black square. Mean ± SEM, N = 10 mice. *, +, #P < 0.05; **, ++, ##P < 0.01; ***, +++, ###P < 0.005; ****, ++++, ####P < 0.001. * Sham to all others groups; + gabapentin to sham, SNI and DIPMA-Ø; # DIPMA-AP to sham, SNI, DIPMA-Ø and AP. Two-way ANOVA, Tukey’s multiple comparisons test.
These results show that encapsulation into pH-responsive DIPMA nanoparticles enhances the efficacy and duration of effect of AP for reversal of multiple measures of evoked and non-evoked nociception in a preclinical model of chronic neuropathic pain. Since the analgesic properties of DIPMA-AP waned after 5 h, we sought to generate and characterize nanostars as a sustained delivery vehicle for AP.
3.2. Synthesis and characterization of nanostars
Nanostars were synthesized by an arm-first methodology (Fig. 3A). Initially, a macromolecular chain transfer agent (BSPA-POEGA, incorporating a polyacrylate backbone with pendent oligo(ethylene glycol) side chains) was synthesized by polymerizing oligo(ethylene glycol) methyl ether acrylate (OEGA480) in the presence of 3-(benzyl sulfanyl thiocarbonyl sulfanyl)-propanoic acid (BSPA) in toluene. The resulting POEGA-BSPA, which includes a non-reactive benzyl group at one chain end and a reactive trithiocarbonate group at the other, was purified by precipitation into cold petroleum ether and drying in vacuo. Characterization by GPC (Mn 8600 g/mol, Ð 1.24) and 1H NMR demonstrated suitable molecular weight and functional groups for use in the subsequent synthetic steps (Fig. S2).
Fig. 3. Synthesis and characterization of nanostars.
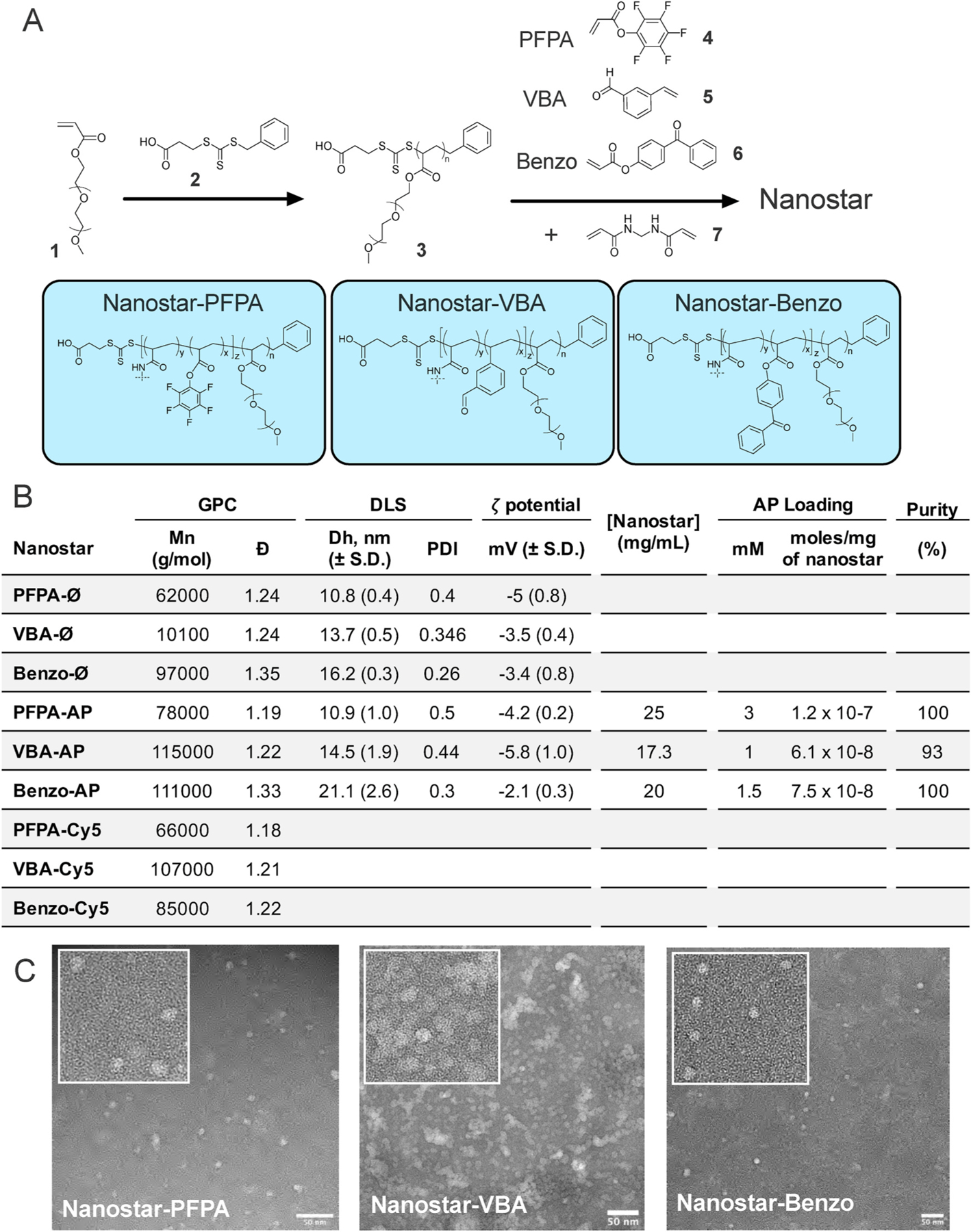
A. Scheme for the arm-first synthesis of core functional nanostars for the preparation of nanostar-AP and nanostar-Cy5 conjugates. B. Molecular weight and distribution data, hydrodynamic diameter and ζ-potential (DLS), AP loading and final purity as determined by 19F NMR. C. TEM images of nanostars. Scale bar = 50 nm.
To form nanostars, purified BSPA-POEGA was chain extended with one of three distinct functional co-monomers, VBA, Benzo or PFPA, in the presence of N,N′-methylenebis(acrylamide) (MBAA) cross-linker (Fig. 3A). The nanostars (each denoted by the functional monomer) were purified by fractional precipitation into 2% (v/v) chloroform in diethyl ether. The molecular weights, molecular weight distributions and shape and uniformity of the nanostars were determined by GPC (Fig. 3B, Fig. S2), 1H NMR (Fig. S2), DLS (Fig. 3B) and TEM (Fig. 3C). Each nanostar exhibited unimodal molecular weight distribution with narrow dispersity and quantitative removal of BSP0A-POEGA arm precursor, with the measured number average molecular weight of 61.9 K–100.9 K. Number-average hydrodynamic diameters determined by DLS were 10.8–16.2 nm, and ζ-potential was close to neutrality (< −5 mV) for each material in PBS. TEM revealed nanostars to be uniformly spherical and well dispersed, although nanostar-VBA images revealed some aggregated material.
3.3. Nanostars demonstrate slow and sustained release of Cy5-NH2 cargo
To examine the release kinetics, PFPA, VBA and Benzo nanostars were fluorescently labelled with Cy5-NH2 in the presence of triethylamine (TEA) as base (Fig. S3). Cy5 nanostars were purified by preparative size exclusion chromatography. Successful Cy5 conjugation and removal of residual Cy5 was confirmed by GPC using both RI and UV-VIS detection (λ = 646) (Fig. S3). Narrow molecular weight distribution was preserved throughout this procedure. Measured molecular weights were comparable to those determined for the non-labelled materials, notwithstanding a small decrease in apparent molecular weight for the Benzo nanostar (Fig. 3B). Release kinetics were determined by spectrophotometry at pH 7.4, 6.0 and 5.0. VBA-Cy5 and Benzo-Cy5 released most Cy5 cargo within 10 h, with continued release for at least 24 h (Fig. S4A, B). PFPA-Cy5 released minimal amounts of cargo for up to 48 h (Fig. S4C). The release rate was VBA > Benzo > PFPA. The kinetics of Cy5 release from VBA or Benzo nanostars was similar at pH 7.4, 6.0 and 5.0.
3.4. Nanostars can be conjugated with amine-functionalized AP
AP was alkylated to facilitate functionalization with a PEG2-amine (AP-NH2) for conjugation to nanostars. AP-NH2 inhibited SP-evoked Ca2+ signaling in HEK293T cells expressing hNK1R with similar potency and efficacy to AP, and is thus fully functional (Fig. S5). AP-conjugated nanostars, VBA-AP, Benzo-AP and PFPA-AP, were prepared by reacting AP-NH2 with VBA, Benzo and PFPA, respectively, in the presence of a suitable base (TEA) (Fig. S6). Conjugation of the AP-NH2 to the nanostar was followed by 19F NMR, with the 19F resonance arising from the 2 × (CF3) groups of the AP moiety at δ = −61.21 ppm undergoing a well resolved shift of ~ −0.1 ppm upon conjugation, accompanied by significant broadening due to the tethering of the AP-NH2 to the high molecular weight nanostar. Conjugation reactions were continued over several days until no further incorporation of the AP-NH2 into the nanostar was observed. AP-conjugated nanostars were purified by preparative size exclusion chromatography using Bio-Beads with THF as the mobile phase. GPC analysis of purified nanostars revealed a slight shift to higher molecular weight upon conjugation, with preservation of unimodal distribution and narrow MWD (Fig. S6). 1H NMR spectroscopic analysis of purified nanostars revealed peaks corresponding to the incorporated aprepitant at 4.9–5.0 ppm (Fig. S7A–C). 19F NMR spectra of the purified AP-conjugated nanostars were recorded in the presence of an internal standard (trifluoroethanol, 0.63 mM), enabling determination of the loaded drug concentration. Physicochemical properties and loading of AP-NH2 into nanostars are shown in Fig. 3B. TEM revealed the expected nanostar morphology (Fig. S8).
3.5. Nanostars demonstrate slow and sustained release of AP-NH2 cargo
Time-dependent release of AP-NH2 from VBA-AP and Benzo-AP was examined at pH 7.4, using HPLC to quantify free AP-NH2. VBA-AP and Benzo-AP released AP-NH2 at a similar rate (Fig. S4D–F). Release was more rapid over 10 h and was sustained for at least 24 h. AP-NH2 release from PFPA-AP nanostars could not be quantified because AP-NH2 levels were below the sensitivity of detection by HPLC.
3.6. Nanostars are not toxic to HEK293Tcells
To assess potential cytotoxic effects of nanostars, HEK293Tcells were incubated with VBA-Ø, Benzo-Ø and PFPA-Ø (1–100 μg/mL) for 2, 6 and 24 h. Cells were incubated with Triton X-100 as a positive control. Plasma membrane integrity (propidium iodide assay), redox and metabolic activity (Alamar Blue assay) and mitochondrial activity (thiazolyl blue tetrazolium bromide, MTT assay) were assessed. VBA-Ø, Benzo-Ø and PFPA-Ø showed no detectable effects on plasma membrane integrity, redox activity or mitochondrial activity for any tested concentration and studied time point (Fig. S9). Thus, nanostars do not have toxic actions in HEK293Tcells under these conditions.
3.7. Nanostars rapidly traffic to endosomes
The uptake of PFPA-Cy5 nanostars into HEK293Tcells was examined by immunofluorescence and confocal microscopy. Due to the negligible release of Cy5 from the PFPA linker (Fig. S4C), Cy5 fluorescence is likely to denote localization of nanostar material during the period of assay (30–120 min). HEK293Twere incubated with PFPA-Cy5 nanostars for 30, 60 or 120 min, washed and fixed. Early and late endosomes were detected by immunofluorescence staining for early endosomal antigen 1 and Rab7a, respectively. At 30 and 60 min, PFPA-Cy5 nanostars were detected in early endosomes, where colocalization was verified by measurement of the Manders’ overlap coefficient (>0.6) (Fig. 4A and B). After 120 min colocalization with early endosomes declined (Manders’ overlap coefficient <0.2). PFPA-Cy5 nanostars were most prominently colocalized with late endosomes after 60 min (Manders’ overlap coefficient 0.59) and still detected in late endosomes after 120 min (Manders’ overlap coefficient 0.45) (Fig. 4C and D). Thus, nanostars internalize into early endosomes and then late endosomes of HEK293Tcells.
Fig. 4. Endocytosis of Cy5-nanostars in HEK293Tcells.
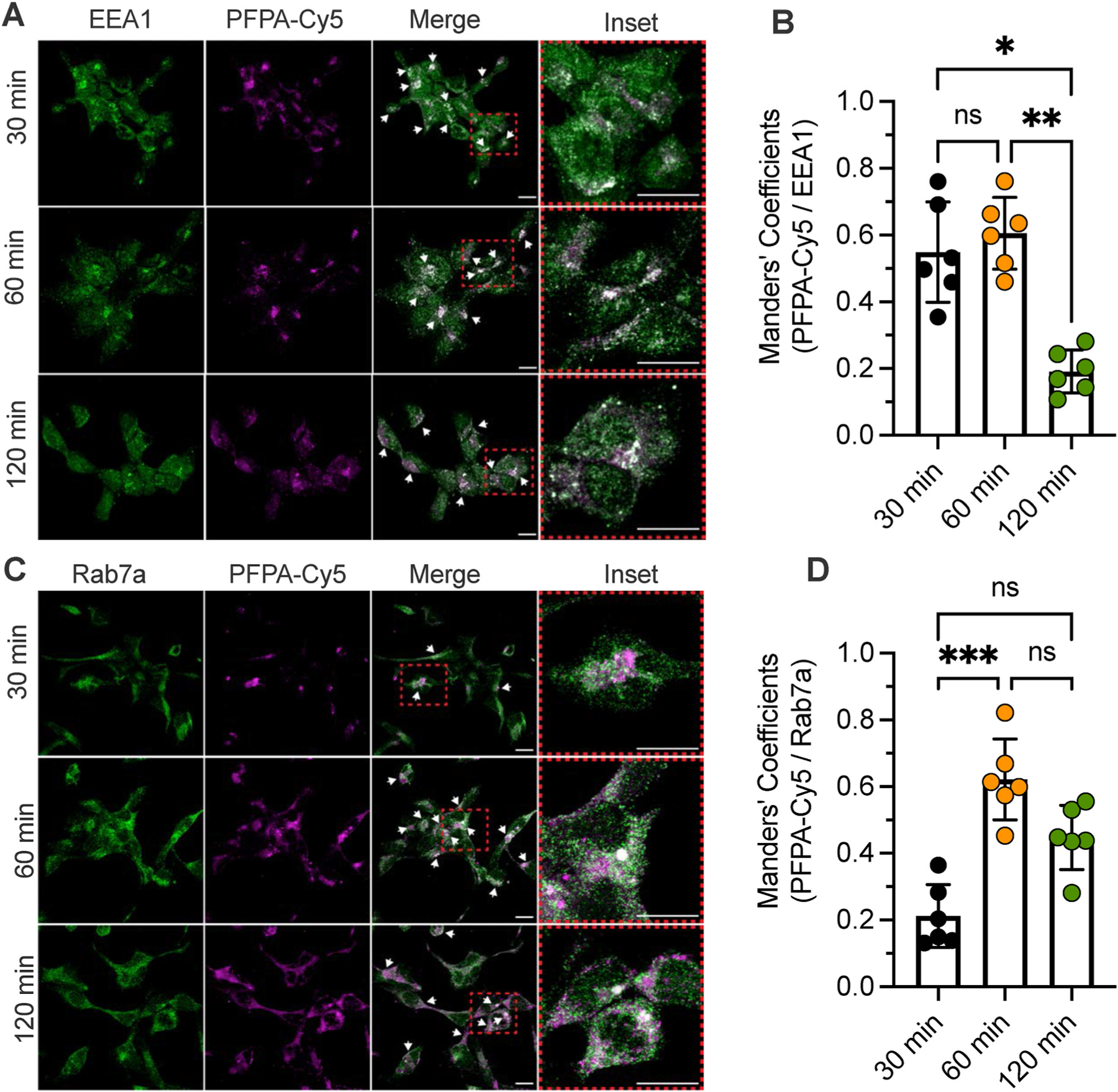
The uptake and intracellular trafficking of PFPA-Cy5 nanostars in HEK293T cells was examined by immunofluorescence and confocal microscopy. Antibodies to early endosomal antigen 1 (EEA1) and Rab7a were used to detect early and late endosomes, respectively. A. Localization of PFPA-Cy5 nanostars and early endosomal antigen 1 at 30, 60 and 120 min. B. Manders’ overlap coefficient for PFPA-Cy5 nanostars and early endosomal antigen 1. C. Localization of PFPA-Cy5 nanostars and Rab7a at 30, 60 and 120 min. D. Manders’ overlap coefficient for PFPA-Cy5 nanostars and Rab7a. N = 6 independent experiments. *P < 0.05, **P < 0.01. 1-way ANOVA, Duns’s multiple comparison test. Scale bar, 10 μm.
3.8. Nanostars antagonize endosomal and plasma membrane signaling of the NK1R in HEK293Tcells
Once activated by SP, the NK1R couples to heterotrimeric G proteins at the plasma membrane and recruits βARRs, which translocate from the cytosol to the plasma membrane. βARRs couple the NK1R to clathrin and adaptor protein-2, which mediate endocytosis. SP/NK1R continues to signal from endosomes of spinal neurons to regulate excitation and pain transmission in the spinal cord [11]. By antagonizing endosomal SP/NK1R signaling, DIPMA-AP inhibits pain transmission in the spinal cord [18]. We examined whether nanostar-AP similarly inhibits SP-evoked activation of the NK1R at the plasma membrane and in endosomes. EbBRET was used to study the activation of Gα and recruitment of βARR2 to the plasma membrane and early endosomes of HEK293T cells. SP-dependent activation of Gαi and Gαq was assessed using an EbBRET assay that detects recruitment of mini (m) Gαsi and mGαsq coupled to Renilla (R)luc8 to the plasma membrane marker CAAX coupled to RGFP or the early endosome marker Rab5a coupled to tandem (td)RGFP. Translocation of mGα to receptors located at the plasma membrane or in endosomes reflects the activation of full length Gα at these locations [30,31]. Recruitment of βARR2 was assessed by measuring EbBRET between Rluc2-βARR2 and RGFP-CAAX or tdRGFP-Rab5a [32].
HEK293T cells expressing hNK1R and EbBRET sensors were incubated with vehicle (control), VBA-AP, Benzo-AP, PFPA-AP (500 or 1000 nM AP), VBA-Ø, Benzo-Ø or PFPA-Ø for 4 h and were then challenged with SP (10 nM). EbBRET was measured for 1400 s. In vehicle-treated cells, SP stimulated an increase in EbBRET between Rluc8-mGαsi, Rluc8-mGαsq and Rluc2-βARR2 with RGFP-CAAX (Fig. S10A, D, G) and tdRGFP-Rab5a (Fig. 5A, D, G). VBA-AP (500, 1000 nM) prevented SP-induced recruitment of Rluc8-mGαsi, Rluc8-mGαsq and Rluc2-βARR2 to RGFP-CAAX (Fig. S10A–I) and tdRGFP-Rab5a (Fig. 5A–I). Benzo-AP (500 nM) inhibited recruitment of Rluc8-mGαsi to RGFP-CAAX and tdRGFP-Rab5a, but did not affect recruitment of Rluc8-mGαsq or Rluc2-βARR2. At a higher concentration (1000 nM), Benzo-AP inhibited recruitment of Rluc8-mGαsi, Rluc8-mGαsq and Rluc2-βARR2 to RGFP-CAAX, but did not inhibit recruitment of Rluc8-mGαsq and Rluc2-βARR2 to tdRGFP-Rab5a. PFPA-AP, VBA-Ø, Benzo-Ø or PFPA-Ø did not inhibit SP-stimulated recruitment of Rluc8-mGαsi, Rluc8-mGαsq and Rluc2-βARR2 to RGFP-CAAX or tdRGFP-Rab5a (Fig. S11). Thus, when encapsulated into VBA or Benzo nanostars, AP can inhibit SP-evoked activation of G proteins and βARRs at the plasma membrane and in endosomes. The activation of Gαi is more susceptible to inhibition than the activation of Gαq or βARR2. Effective antagonism requires the release of AP from nanostars because PFPA-AP, which does not release AP, had no effect.
Fig. 5. Nanoparticles and endosomal NK1R signaling in HEK293T cells.
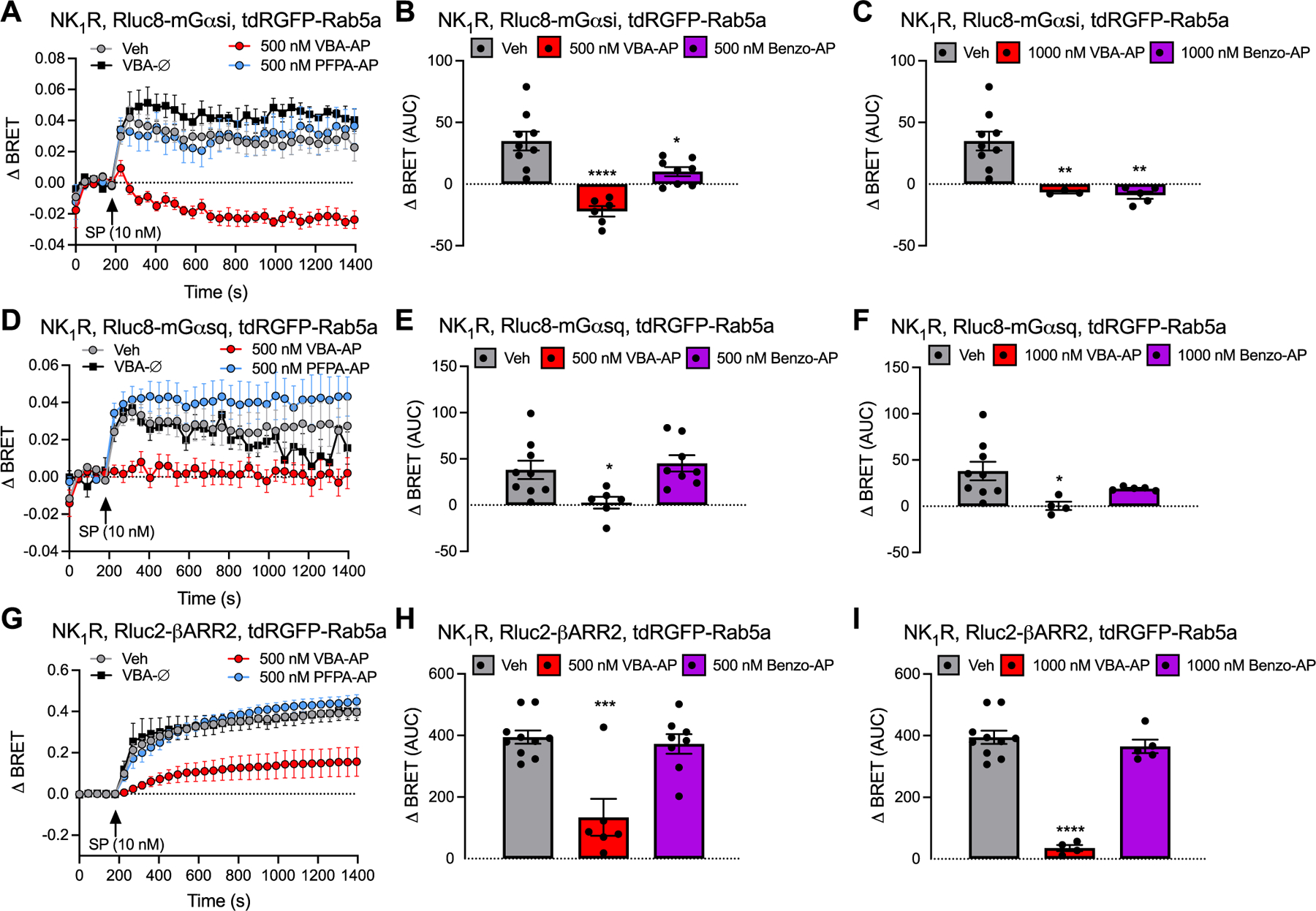
Effects of SP (10 nM) on EbBRET between Rluc8-mGαsi (A, B, C), Rluc8-mGαsq (D, E, F) and Rlu2-βARR2 (G, H, I) with tdRGFP-Rab5a. Cells were preincubated with vehicle (Veh, control), VBA-AP, Benzo-AP, PFPA-AP (500 nM AP loaded in nanoparticles: A, B, D, E, G, H; 1000 nM AP loaded in nanoparticles: C, F, I) or VBA-Ø for 4 h before stimulation with SP and measurement of EbBRET. A, D, G: time course of EbBRET prior and after SP (10 nM) stimulation. B, C, E, F, H, I: area under the curves (AUC) of corresponding time course of EbBRET. Mean ± SEM, N = 3–10 independent experiments. *P < 0.05, **P < 0.001, ***P < 0.001, ****P < 0.0001 compared to vehicle. One-way ANOVA, Dunnett’s multiple comparisons test.
3.9. Nanostars accumulate in intracellular membranes of spinal neurons
To investigate the biodistribution of nanostars, Cy5-PFPA nanostars were administered to mice by intrathecal injection. The potential redistribution for nanostar Cy5 fluorescence away from the injection site was evaluated by non-invasive whole body near-infrared imaging. Within 1 min of injection, Cy5 was intensely concentrated at the site of injection (Fig. 6A and B). The signal was retained within the region of the spinal cord for at least 8 h, with minimal reduction in signal intensity between 2 and 8 h post-injection. To examine uptake of nanostars into the spinal cord, tissues were collected from mice 4 h after intrathecal injection of PFPA-Cy5 nanostars. Spinal cord sections were processed to localize markers of neurons (anti-Hu), glial cells (anti-Cd11b) and astrocytes (anti-GFAP) by immunofluorescence and confocal microscopy. Representative images of spinal cord sections (L4/L5) demonstrated that the PFPA-Cy5 remained in the ipsilateral region (Fig. 6B, D, E) and predominantly accumulated in a perinuclear region of spinal neurons identified by the pan-neuronal marker Hu, and showed a punctate distribution consistent with endosomal accumulation (Fig. 6B). In contrast, minimal Cy5 fluorescence distribution was observed in GFAP-positive astrocytes, or Cd11b-positive microglial cells (Fig. 6E).
Fig. 6. Spinal cord biodistribution and neuronal uptake of nanostars.
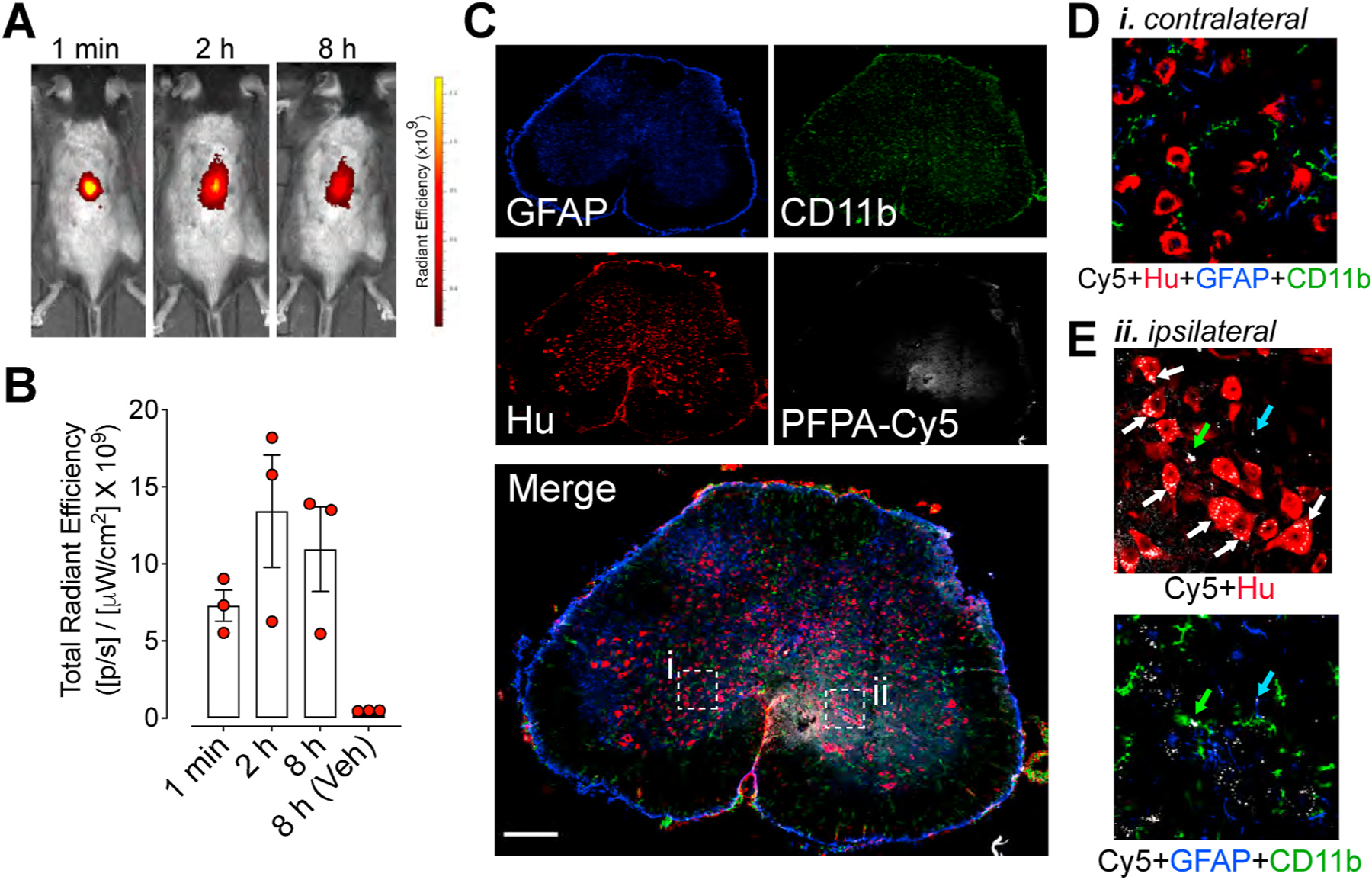
A. Distribution and intensity of PFPA-Cy5 at 1 min, 2 h and 8 h after intrathecal injection of mice. Representative images of experiments on N = 3 mice are shown. Color Scale, Cy5 fluorescence intensity measured as radiant efficiency (units reported as 109 × ps−1μW−1cm−2; p, photons; s, seconds; W, watts). B. Quantification of PFPA-Cy5 fluorescence in spinal cord region after intrathecal injection of mice, assessed as the radiant efficiency of the images. Mean ± SEM, N = 3 mice. C. Localization of PFPA-Cy5 in the spinal cord 4 h after intrathecal injection, showing single channels followed by merged image of a whole spinal cord section following labelling with anti-GFAP (reactive astrocytes, blue), anti-CD11b (microglia, green), anti-Hu (neurons, red) and PFPA-Cy5 nanostars (white). D. Inset (i) shows magnified region and lack of Cy5 fluorescence on contralateral side of spinal cord. E. Inset (ii) shows magnified region and accumulation of Cy5 nanostars predominantly in neurons in ipsilateral region of spinal cord. Representative images 12 sections each from N = 2 mice. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.10. VBA-AP and Benzo-AP nanostars provide sustained relief from neuropathic pain
To assess whether nanostar incorporation enhanced and extended the therapeutic efficacy of AP, we compared the effects of VBA-AP, Benzo-AP and free AP on mechanical allodynia, cold allodynia and non-evoked nociceptive behavior in mice with neuropathic pain.
Mechanical allodynia.
VBA-AP, Benzo-AP, PFPA-AP, an equivalent dose of free AP or AP-NH2 (1000 nM AP) or VBA-Ø, Benzo-Ø or PFPA-Ø was injected intrathecally, and mechanical allodynia was measured. These treatments had no effect on VFF withdrawal responses of the contralateral paw (Fig. 7A). Free AP caused a partial reversal of VFF withdrawal responses of the ipsilateral paw at 2 h (Fig. 7B and C). AP and AP-NH2 suppressed mechanical allodynia to the same degree (Fig. 7D). The magnitude and duration of the anti-nociceptive effects of VBA-AP and Benzo-AP significantly exceeded that of free AP and were apparent from 2 to at least 10 h (Fig. 7B and C). The VFF response at 6 h was 15.8 ± 6% of baseline values after free AP but 72 ± 9% basal after VBA-AP and 64 ± 7.8% basal after Benzo-AP (P < 0.0001). PFPA-AP, which did not release AP, had no effects on mechanical allodynia (Fig. 7B and C). VBA-Ø, Benzo-Ø or PFPA-Ø did not affect mechanical allodynia.
Fig. 7. Nanostars, mechanical and cold allodynia in neuropathic pain.
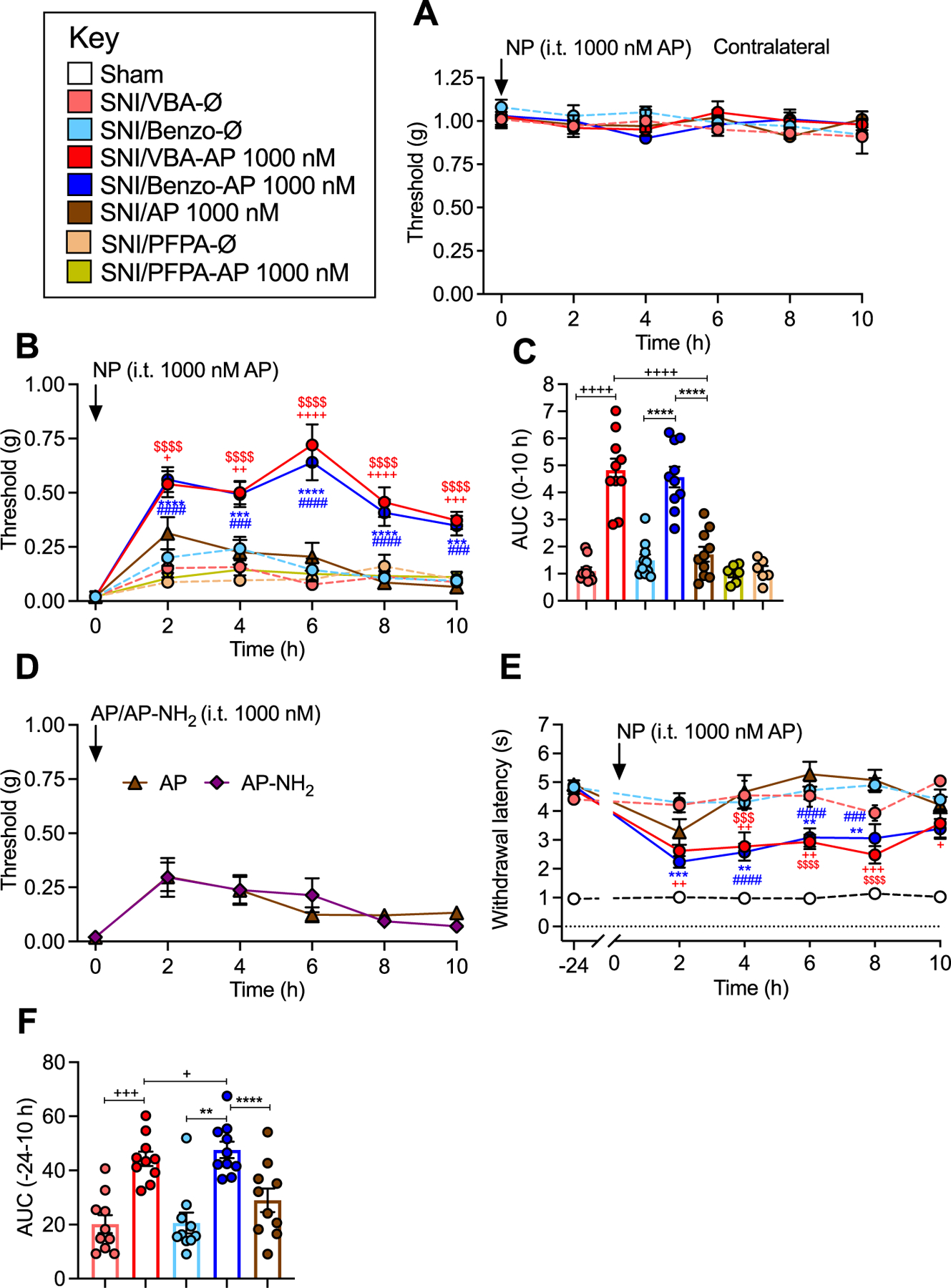
A. Effects of VBA-AP, Benzo-AP or AP (1000 nM AP), VBA-Ø or Benzo-Ø on VFF withdrawal responses (mechanical allodynia) of contralateral paw. B, C. Effects of VBA-AP, Benzo-AP, PFPA-AP, AP (1000 nM AP), VBA-Ø, Benzo-Ø or PFPA-Ø on VFF withdrawal responses of ipsilateral paw. B shows time course and C shows AUC. D. Comparison between AP and AP-NH2 in SNI mice. E,F. Effects of VBA-AP, Benzo-AP, AP (1000 nM AP), VBA-Ø or Benzo-Ø on acetone withdrawal responses (cold allodynia) of ipsilateral paw in SNI mice. E shows time course and F shows AUC. Mean ± SEM, N = 10 mice. *, +, #, $ P < 0.05; **, ++, ##, $ $ P < 0.01; ***, +++, ###, $ $ $ P < 0.005; ****, ++++, ####, $ $ $ $ P < 0.001. * Benzo-AP to AP; + VBA-AP to AP; # Benzo-AP to Benzo-Ø; $ VBA-AP to VBA-Ø. Two-way ANOVA (mechanical and cold allodynia), one-way ANOVA (AUC) with Tukey’s multiple comparison test.
Cold allodynia.
Whereas free AP had a small but insignificant effect on cold allodynia at 2 h, VBA-AP and Benzo-AP reversed cold allodynia from 2 to at least 10 h (Fig. 7E and F). VBA-Ø and Benzo-Ø did not affect cold allodynia.
Non-evoked nociceptive behavior.
Non-evoked nociceptive behavior was measured for 30 min 90 min after intrathecal drug administration. SNI mice exhibited significant decreases in average distance from the wall (Fig. 8A), grooming time (Fig. 8B) and ambulation time (Fig. 8C) compared to sham controls, whereas track length (Fig. 8D), average velocity (Fig. 8E) and locomotor activity (Fig. 8F) were unaffected. Central area crossing was also significantly diminished in SNI mice (Fig. 8G, H, I, L). VBA-AP and Benzo-AP (1000 nM) fully normalized distance from wall (Fig. 8A), grooming time (Fig. 8B), ambulation time (Fig. 8C) and center crossing (Fig. 8J–L) in SNI mice to levels observed in sham mice. VBA-Ø and Benzo-Ø did not affect any measured parameter.
Fig. 8. Nanostars, non-evoked behavior in neuropathic pain.
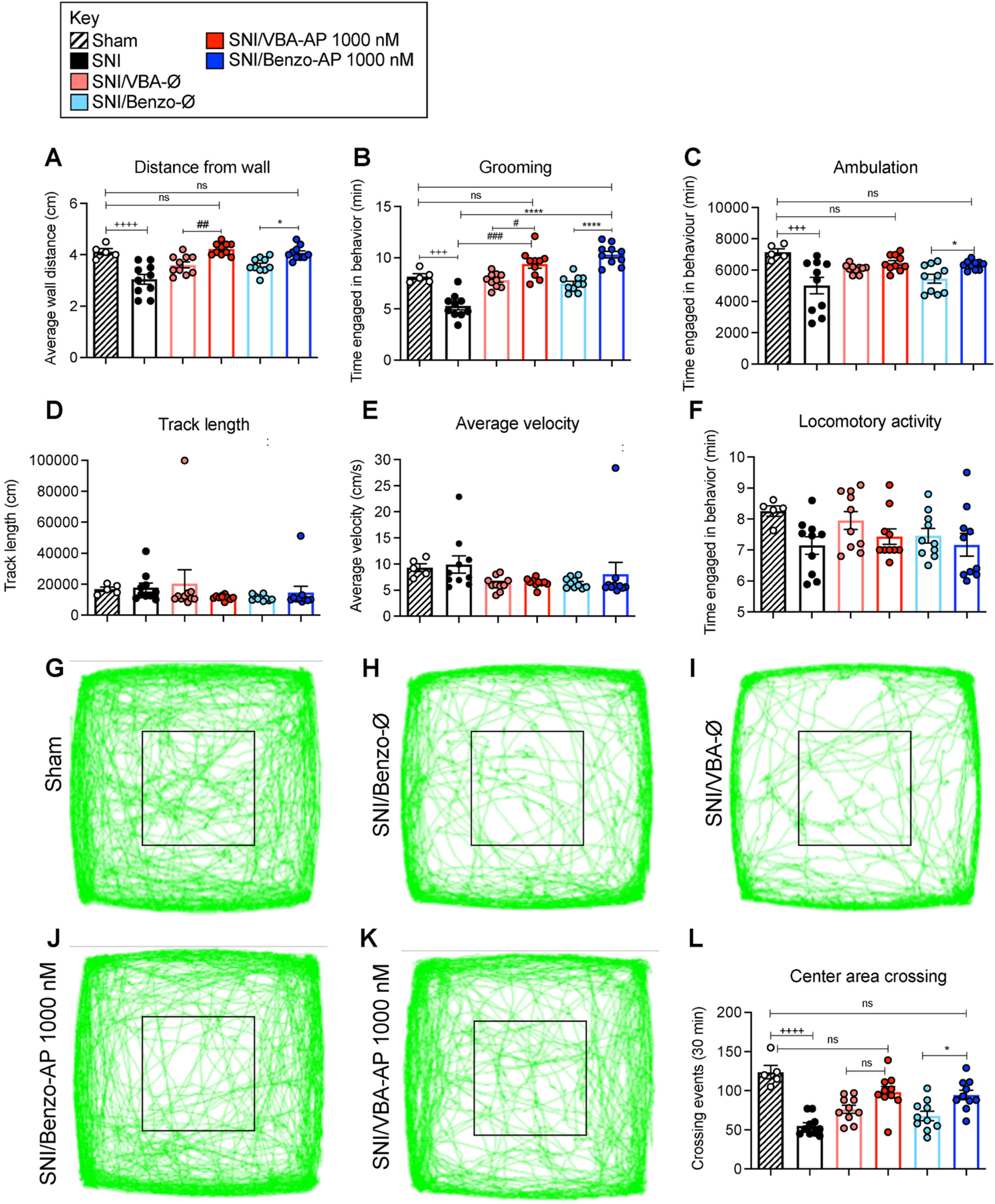
Non-evoked nociceptive behavior in SNI and sham control mice recorded for 30 min at 90 min after intrathecal injection of VBA-AP, Benzo-AP, AP (1000 nM AP), VBA-Ø or Benzo-Ø. Distance from wall (A), grooming (B), ambulation (C), track length (D), average velocity, (E) and locomotor activity (F) were recorded. G-K are records of tracks of mice. L shows crossing events in area marked by black square. Mean ± SEM, N = 10 mice. *, +, #P < 0.05; **, ++, ##P < 0.01; ***, +++, ###P < 0.005; ****, ++++, ####P < 0.001. * Benzo-AP to Benzo-Ø and, SNI; + Sham to SNI; # VBA-AP to VBA-Ø and SNI. Two-way ANOVA, Tukey’s multiple comparisons test.
3.11. VBA-AP and Benzo-AP nanostars provide sustained relief from inflammatory pain
To confirm the superior efficacy and duration of analgesia of nanostar AP in a second preclinical model of pain, the effects of VBA-AP, Benzo-AP and free AP on mechanical allodynia, in mice with inflammatory pain was examined. VBA-AP, Benzo-AP or free AP (1000 nmol) was administered by intrathecal injection 48 h after intraplanar injection of CFA. Whereas free AP had a minor inhibitory effect on mechanical allodynia at 2 h, VBA-AP and Benzo-AP inhibited mechanical allodynia to a similar degree for 10 h, although the effects were lost after 24 h (Fig. 9A and B).
Fig. 9. Nanostars, mechanical allodynia in inflammatory pain.
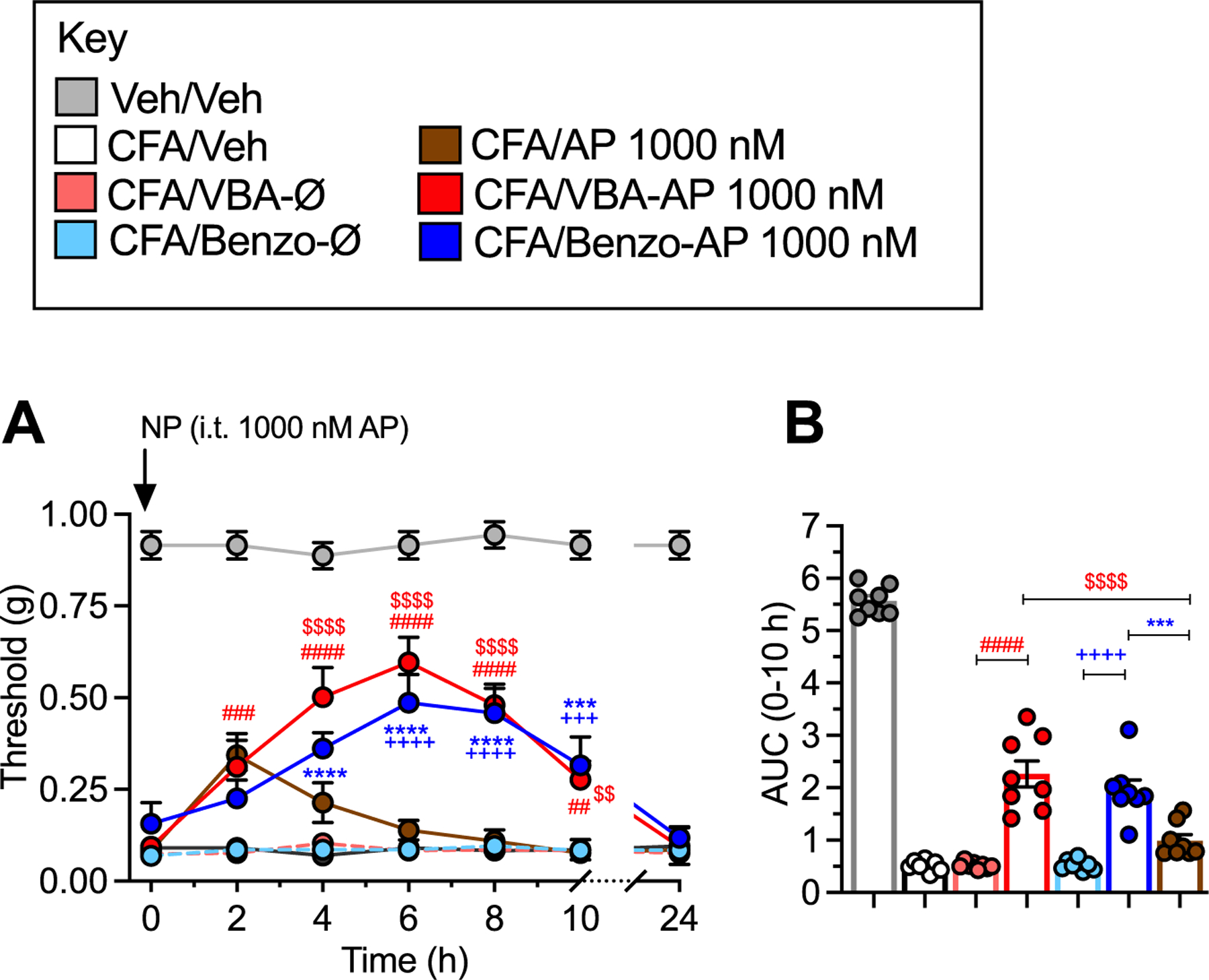
A-B. . Effects of VBA-AP, Benzo-AP, AP (1000 nM AP), VBA-Ø or Benzo-Ø on VFF withdrawal responses of ipsilateral paw. A shows time course and B shows AUC. Mean ± SEM, N = 8 mice. *, +, #, $ P < 0.05; **, ++, ##, $ $ P < 0.01; ***, +++, ###, $ $ $ P < 0.005: ****, ++++, ####, $ $ $ $ P < 0.001. * Benzo-AP to Benzo-AP; + Benzo-Ø to AP; # cVBA-AP to VBA-Ø; $ VBA-AP to AP. X-way ANOVA, Dunnett’s multiple comparisons test.
4. Discussion
Although opioids that activate the μ-opioid receptor are widely prescribed for the treatment of acute pain, their efficacy wanes with continued use and they can evoke paradoxical hyperalgesia. The over-prescription of opioids, coupled to their addictive properties and severe on-target side effects of respiratory depression, sedation and constipation, underlie the opioid crisis; drug overdose, chiefly opioids, is a leading cause of death world-wide that has worsened during the COVID-19 pandemic. The realization that certain non-μ-opioid GPCRs continue to signal from endosomes to control neuronal activity and pain transmission [11–15], and the propensity of nanoparticles to accumulate in endosomes [12,17,18], raises the possibility of harnessing endosomal acidity to trigger nanoparticle disassembly and GPCR antagonist release for the treatment of pain. Our results show that AP encapsulated into soft polymer DIPMA nanoparticles effectively reverses evoked and non-evoked nociception in a preclinical mouse model of chronic neuropathic pain. The assessment of non-evoked nociception obviates observational and operator bias that plagues studies of evoked nociception in rodents. The demonstration that gabapentin, which is clinically approved for the treatment of neuropathic pain, reverted all measures of non-evoked nociception similar to DIPMA-AP, validates the usefulness of measures of spontaneous nociception. We hypothesize that nanoparticle encapsulation enhances efficacy by delivery of AP to the key therapeutic target, the NK1R in endosomes of spinal neurons, and that the limited therapeutic efficacy of free AP relates to its inability to effectively engage with conformations of the activated NK1R within multiprotein signalosomes in the acidified endosomal environment [16]. An advantage of soft polymer DIPMA nanoparticles is that they permit encapsulation of unmodified drugs. Limitations include their inherent instability, which is exacerbated by dilution that exceeds the critical micellar concentration, and immediate disassembly and short-term cargo release at pH < 6 [18]. Although administration to confined volumes such as the intrathecal space avoids dilution-related instability, DIPMA nanoparticles are unsuitable for systemic administration because dilution alone would evoke disassembly. Moreover, immediate disassembly in acidic compartments would be expected to limit the duration of therapeutic efficacy of analgesic cargos.
To surmount these limitations, we synthesized nanostars with three distinct reactive groups for conjugation to cargo by RAFT polymerization using an arm first methodology. Nanostars incorporating pentafluorophenyl (PFPA), benzaldehyde (VBA) and benzophenone (Benzo) groups were prepared (Fig. 3). Upon exposure to amine functionalized cargo molecules, these incorporated groups would be expected to form amide, aldimine and ketimine linked nanostar-cargos, respectively, and thus exhibit differential hydrolytic stability. Each nanostar synthesized in this study incorporated a hydrophilic mPEG brush corona, which has the dual function of maintaining drug solubility in an aqueous environment and evading clearance. Nanostars were small (10–20 nm), uniformly spherical and largely unaggregated, and with a small negative charge (−5 mV), properties that would be expected to enhance cellular uptake. The molecular weight, hydrodynamic diameter and ζ-potential of the synthesized nanostars are consistent with previous results on PEGylated nanostars [39–41].
To examine the capacity of the nanostars to undergo differential cargo release depending on the specific amine reactive moiety, the three nanostars (PFPA, VBA, Benzo) were conjugated to the amine functional dye, Cy5-NH2. The resulting nanostar-dye conjugates enabled examination of the release kinetics from each nanostar by spectrophotometry. Release studies under sink conditions allowed determination of the kinetics of cargo release and the pH-responsivity of the different linkers. In contrast to DIPMA-AP nanoparticles that immediately disassemble at pH < 6.0 [18], VBA and Benzo nanostars continued to release Cy5 cargo for at least 24 h and cargo release was not accelerated under acidic conditions. As anticipated, the aldimine-linked conjugate (VBA-Cy5) released the dye more readily than the ketimine-linked conjugate (Benzo-Cy5). The amide-linked variant, PFPA-Cy5, exhibited minimal dye release over the time course studied. The stable amide-bond linked PFPA-Cy5 also served as a convenient fluorescently-labelled reporter to investigate the trafficking and biodistribution of nanostars.
We synthesized amine functionalized AP for conjugation to nanostars. Structural studies indicate that the triazolinone substituent of AP is oriented toward the extracellular space and creates an extended binding pocket by promoting additional hydrogen bonding with E193 and W184 in the extracellular region of the NK1R [42]. Modifications to the triazolinone ring are well tolerated and have minimal impact on affinity of AP [43,44]. To enable functionalization of the nanostars with AP, we modified the triazolinone substituent with a 3-carbon linker and PEG2-amine for conjugation to the arm of the nanostar. AP-NH2 and AP showed similar potency and efficacy for antagonism of SP-evoked Ca2+ signaling in HEK293Tcells and reversal of neuropathic pain in mice. Thus, AP-NH2 retains full activity and was suitable for comparisons to the parent compound AP in subsequent experiments. We observed similar kinetics of AP-NH2 release from VBA-AP and Benzo-AP nanostars. Since the rate of cargo release likely depends on the nature of the amine reactive moieties, we had expected similar release kinetics of Cy5-NH2 and AP-NH2. The observed differences in the relative kinetics of release of AP-NH2 and Cy5-NH2 between VBA and Benzo nanostars is thus likely related to the nature of the cargo. A limitation of these in vitro measurements of cargo release is that they do not replicate the complex intracellular environment of spinal neurons, where release could be affected by multiple factors. However, these results are in line with the superior ability of VBA-AP and Benzo-AP to suppress NK1R endosomal signaling in HEK293T cell lines and inhibit nociception in mice when compared to PFPA-AP, which was inactive.
Uptake studies in HEK293Tcells found that PFPA-Cy5 nanostars accumulate in early and late endosomes; endosomes are thus a likely site of cargo release. Further studies will be required to ascertain the ultimate fate of the inert nanostar components in HEK293T cells and in the spinal cord after intrathecal injection. By using EbBRET to assess the recruitment of mGα proteins and βARR2 to the plasma membrane and early endosomes, it was possible to assess the effects of nanostar-AP on NK1R signaling in subcellular compartments of HEK293T cells. SP stimulated the expected sustained recruitment of mGαi and mGαq and βARR2 to the plasma membrane and early endosomes of HEK293T cells. Preincubation of cells with VBA-AP, to allow for nanostar uptake and AP-NH2 release, prevented SP-evoked activation of mGαi, mGαq and βARR2 at the plasma membrane and in endosomes. Since late endosomes were the site of nanostar accumulation and likely disassembly, we speculate that freed AP-NH2 diffuses and thereby antagonizes the NK1R in early endosomes and at the plasma membrane; the dynamic nature of the tubulovesicular endosomal network likely facilitates the spread of released cargo [45]. Benzo-AP antagonized NK1R signaling only at higher concentrations, which is consistent with the slower kinetics of Cy5 cargo release. PFPA-AP did not affect NK1R signaling at any tested concentration, in line with its inability to release cargo; empty nanostars were also inactive. We conclude that release of AP-NH2 cargo is necessary for antagonism of SP-stimulated NK1R signaling in HEK293T cells.
When injected intrathecally to mice, PFPA-Cy5 was retained at the site of injection for at least 8 h and was preferentially endocytosed by neurons throughout the spinal cord. Further studies are required to understand the reasons for preferential uptake by neurons rather than glial cells or astrocytes, although the extensive vesicular transport within neurons that is related to synaptic transmission might predispose uptake of nanomaterials. The mechanisms of nanostar uptake into neurons and the possibility of uptake into subtypes of neurons remain to be investigated. We speculate that nanostars enter cells by clathrin- and dynamin-mediated endocytosis since these mechanisms mediate entry of DIPMA nanoparticles to HEK293Tcells [18].
Intrathecal injection of VNA-AP and Benzo-AP provided far more efficacious reversal of evoked and non-evoked nociception in preclinical mouse models of neuropathic and inflammatory pain than free AP, which had minor and transient analgesic effect. Despite differences in the kinetics of Cy5 cargo release in vitro and some discrepancy in antagonism of NK1R signaling in HEK293T cells, the analgesic properties of the same dose of VBA-AP and Benzo-AP were identical. Full dose-response studies will be required to compare fully the analgesic properties of these nanomaterials. However, stable PFPA-AP and empty nanoparticles did not affect nociception, confirming the requirement of AP-NH2 release for analgesia. VBA-AP and Benzo-AP provided analgesia for at least 10 h, twice the duration of the same dose of DIPMA-AP. The prolonged analgesic efficacy of nanostar-AP is consistent with the slow kinetics of disassembly and cargo release in vitro. Sustained and efficacious analgesia of nanostar-AP was attained despite the pH-independence of disassembly in vitro. Thus, pH-dependent cargo release in endosomes is not required for analgesia, although it could further enhance pain relief, as is the case for DIPMA-AP nanoparticles. Future pharmacodynamic analyses will be necessary to relate the kinetics of drug release to analgesia.
A limitation of this study is that we did not study the possible toxic effects of nanostars in mice. However, nanostars did not affect several measures of cytotoxicity in HEK293Tcells, and AP-nanostars did not affect withdrawal responses of the contralateral paw, suggesting no effects on normal motor coordination. Moreover, empty nanostars did not affect withdrawal responses and had no effects on multiple measures of normal behavior, which argue against any neurotoxic actions under the conditions of these experiments. DIPMA nanoparticles also do not have detectable toxic actions in HEK293Tcells or after intrathecal injection to mice [18]. Potential long-term neurotoxicity of nanostars will be assessed in future studies. Another limitation is that we only assessed the efficacy of intrathecal administration of nanostars, which had the advantage of drug deliver to targets in spinal neurons. Whether systemically administered nanostars can be used to target peripheral GPCRs for the treatment of chronic pain remains to be studied.
The present investigation extends the use of nanostars beyond the delivery of chemotherapeutics, antioxidants and imaging agents to cancer cells and tumors [20,23–25] to the delivery of GPCR ligands to endosomes of neurons for the treatment of chronic pain. In addition to analgesia, nanoparticle encapsulation and consequent endosomal delivery may enhance other therapeutic actions of AP, including its actions on chemotherapy-induced nausea and vomiting and its antitumor actions. AP has antiproliferative and antimitogenic effects on a variety of cancers (reviewed in Ref. [46]), which may be enhanced by nanoparticle encapsulation. Thus, nanoparticle-AP might ameliorate cancer progression as well as associated pain, while reducing the side effects (nausea and vomiting of standard chemotherapeutic agents). Further studies are required to investigate these possibilities. Although rapid-release DIPMA nanoparticles enhance the therapeutic actions of AP, sustained-release nanostars provide additional benefit. Even longer lasting nanomedicines, such as hydrophobic ion pairing nano-formulations that allow cargo release over weeks [47], may provide even more long-lasting analgesia. Biodegradable nanomaterials have been proposed to be an effective strategy for slow, localized drug release to provide sustained relief of post-surgical pain [48]. Nanostars are an attractive platform for advanced drug delivery due to their modular nature and the potential to conjugate multiple drugs through various linkers, attach targeting moieties and readily adjust the hydrodynamic diameter. Further refinements that could improve the therapeutic benefits of analgesic nanostars could include incorporation of antagonists of multiple GPCRs that signal from endosomes, which could overcome the inherent redundancy of the pathways of pain transmission, and the specific targeting of nanoparticles to neurons that sense and transmit pain. Encapsulation of antagonists of SP, CGRP and glutamate receptors would be expected to strongly inhibit pain transmission in the dorsal horn of the spinal cord, which depends on endosomal signaling of these GPCRs [3,11,15]. Conjugation of an agonists of the ∂-opioid receptor to a liposome shell of mesoporous silica nanoparticles enhances targeting and improves analgesia, which illustrates the utility of nociceptor-targeted analgesics [12].
5. Conclusion
Nanostars that slowly release an antagonist of the NK1R within endosomes abrogate endosomal signaling and provide sustained antinociception in preclinical models of neuropathic and inflammatory pain. The enhanced efficacy and duration of analgesia of nanostar-aprepitant depends on persistent release of antagonist within endosomes of spinal neurons. The nanostar platform is amenable to co-loading multiple antagonists and targeting to neurons that sense and transmit pain, which would be expected to provide long-lasting and opioid-free analgesia.
Supplementary Material
Acknowledgements
Supported by grants from: National Institutes of Health (NS102722, DE026806, DK118971, DE029951, NWB, BLS) and Department of Defense (W81XWH1810431, W81XWH-22-1-0239, Expansion Award, NWB, BLS). We thank Eric Le for help with Ca2+ mobilization assays and Joseph B. Guttenplan and Shavonne Teng for help with the HPLC analysis of AP-NH2.
Footnotes
Credit author statement
Rocco Latorre: Methodology, Validation, Investigation, Writing. Paulina D. Ramírez-Garcia: Methodology, Validation, Investigation, Writing. Alan Hegron: Methodology, Validation, Investigation, Writing. James L. Grace: Methodology, Validation, Investigation. Jeffri S. Retamal: Methodology, Validation, Investigation. Priyank Shenoy: Methodology, Validation, Investigation. Mai Tran: Methodology, Validation, Investigation. Luigi Aurelio: Resources. Bernard Flynn: Resources. Daniel P. Poole: Methodology, Validation, Investigation. Dane D. Jensen: Methodology, Validation, Investigation. Thomas P. Davis: Conceptualization, Funding acquisition. Brian L. Schmidt: Conceptualization, Funding acquisition. John F. Quinn: Conceptualization, Methodology, Writing. Michael R. Whittaker: Conceptualization, Methodology, Validation, Investigation, Writing. Nicholas A. Veldhuis: Conceptualization, Methodology, Validation, Investigation, Writing. Nigel W. Bunnett: Conceptualization, Writing, Funding acquisition, Supervision.
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Nigel W. Bunnett reports financial support was provided by Endosome Therapeutics Inc and Takeda Pharmaceuticals International. Nigel W Bunnett reports a relationship with Endosome Therapeutics Inc and Takeda Pharmaceuticals International that includes: board membership, equity or stocks, funding grants, and travel reimbursement. Declaration of competing interest. NWB is a founding scientist of Endosome Therapeutics Inc. Research in NWB’s laboratory is funded, in part, by Takeda Pharmaceuticals International.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.biomaterials.2022.121536.
Data availability
The raw data required to reproduce these findings are available upon request. The scripts used for the image analyses are available upon request. The processed data required to reproduce these findings are available upon request.
References
- [1].Basbaum AI, Bautista DM, Scherrer G, Julius D, Cellular and molecular mechanisms of pain, Cell 139 (2) (2009) 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Shipton EA, Shipton EE, Shipton AJ, A review of the opioid epidemic: what do we do about it? Pain Ther 7 (1) (2018) 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Geppetti P, Veldhuis NA, Lieu T, Bunnett NW, G protein-coupled receptors: dynamic machines for signaling pain and itch, Neuron 88 (4) (2015) 635–649. [DOI] [PubMed] [Google Scholar]
- [4].Veldhuis NA, Poole DP, Grace M, McIntyre P, Bunnett NW, The G protein-coupled receptor-transient receptor potential channel axis: molecular insights for targeting disorders of sensation and inflammation, Pharmacol. Rev 67 (1) (2015) 36–73. [DOI] [PubMed] [Google Scholar]
- [5].Peterson YK, Luttrell LM, The diverse roles of arrestin scaffolds in G protein-coupled receptor signaling, Pharmacol. Rev 69 (3) (2017) 256–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C, Persani L, Lohse MJ, Persistent cAMP-signals triggered by internalized G-protein-coupled receptors, PLoS Biol 7 (8) (2009), e1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].DeFea KA, Vaughn ZD, O’Bryan EM, Nishijima D, Dery O, Bunnett NW, The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta -arrestin-dependent scaffolding complex, Proc. Natl. Acad. Sci. U. S. A 97 (20) (2000) 11086–11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SG, Sunahara RK, El-Samad H, Huang B, von Zastrow M, Conformational biosensors reveal GPCR signalling from endosomes, Nature 495 (7442) (2013) 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nguyen AH, Lefkowitz RJ, Signaling at the endosome: cryo-EM structure of a GPCR-G protein-beta-arrestin megacomplex, FEBS J 288 (8) (2021) 2562–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nguyen AH, Thomsen ARB, Cahill TJ 3rd, Huang R, Huang LY, Marcink T, Clarke OB, Heissel S, Masoudi A, Ben-Hail D, Samaan F, Dandey VP, Tan YZ, Hong C, Mahoney JP, Triest S, Little J.t., Chen X, Sunahara R, Steyaert J, Molina H, Yu Z, des Georges A, Lefkowitz RJ, Structure of an endosomal signaling GPCR-G protein-beta-arrestin megacomplex, Nat. Struct. Mol. Biol 26 (12) (2019) 1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jensen DD, Lieu T, Halls ML, Veldhuis NA, Imlach WL, Mai QN, Poole DP, Quach T, Aurelio L, Conner J, Herenbrink CK, Barlow N, Simpson JS, Scanlon MJ, Graham B, McCluskey A, Robinson PJ, Escriou V, Nassini R, Materazzi S, Geppetti P, Hicks GA, Christie MJ, Porter CJH, Canals M, Bunnett NW, Neurokinin 1 receptor signaling in endosomes mediates sustained nociception and is a viable therapeutic target for prolonged pain relief, Sci. Transl. Med 9 (392) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jimenez-Vargas NN, Gong J, Wisdom MJ, Jensen DD, Latorre R, Hegron A, Teng S, DiCello JJ, Rajasekhar P, Veldhuis NA, Carbone SE, Yu Y, Lopez-Lopez C, Jaramillo-Polanco J, Canals M, Reed DE, Lomax AE, Schmidt BL, Leong KW, Vanner SJ, Halls ML, Bunnett NW, Poole DP, Endosomal signaling of delta opioid receptors is an endogenous mechanism and therapeutic target for relief from inflammatory pain, Proc. Natl. Acad. Sci. U. S. A 117 (26) (2020) 15281–15292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jimenez-Vargas NN, Pattison LA, Zhao P, Lieu T, Latorre R, Jensen DD, Castro J, Aurelio L, Le GT, Flynn B, Herenbrink CK, Yeatman HR, Edgington-Mitchell L, Porter CJH, Halls ML, Canals M, Veldhuis NA, Poole DP, McLean P, Hicks GA, Scheff N, Chen E, Bhattacharya A, Schmidt BL, Brierley SM, Vanner SJ, Bunnett NW, Protease-activated receptor-2 in endosomes signals persistent pain of irritable bowel syndrome, Proc. Natl. Acad. Sci. U. S. A 115 (31) (2018) E7438–E7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mai QN, Shenoy P, Quach T, Retamal JS, Gondin AB, Yeatman HR, Aurelio L, Conner JW, Poole DP, Canals M, Nowell CJ, Graham B, Davis TP, Briddon SJ, Hill SJ, Porter CJH, Bunnett NW, Halls ML, Veldhuis NA, A lipid-anchored neurokinin 1 receptor antagonist prolongs pain relief by a three-pronged mechanism of action targeting the receptor at the plasma membrane and in endosomes, J. Biol. Chem 296 (2021), 100345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yarwood RE, Imlach WL, Lieu T, Veldhuis NA, Jensen DD, Klein Herenbrink C, Aurelio L, Cai Z, Christie MJ, Poole DP, Porter CJH, McLean P, Hicks GA, Geppetti P, Halls ML, Canals M, Bunnett NW, Endosomal signaling of the receptor for calcitonin gene-related peptide mediates pain transmission, Proc. Natl. Acad. Sci. U. S. A 114 (46) (2017) 12309–12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Thomsen ARB, Jensen DD, Hicks GA, Bunnett NW, Therapeutic targeting of endosomal G-protein-coupled receptors, Trends Pharmacol. Sci 39 (10) (2018) 879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bhansali D, Teng SL, Lee CS, Schmidt BL, Bunnett NW, Leong KW, Nanotechnology for pain management: current and future therapeutic interventions, Nano Today 39 (2021), 101223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ramirez-Garcia PD, Retamal JS, Shenoy P, Imlach W, Sykes M, Truong N, Constandil L, Pelissier T, Nowell CJ, Khor SY, Layani LM, Lumb C, Poole DP, Lieu T, Stewart GD, Mai QN, Jensen DD, Latorre R, Scheff NN, Schmidt BL, Quinn JF, Whittaker MR, Veldhuis NA, Davis TP, Bunnett NW, A pH-responsive nanoparticle targets the neurokinin 1 receptor in endosomes to prevent chronic pain, Nat. Nanotechnol 14 (12) (2019) 1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hu J, Qiao R, Whittaker MR, Quinn JF, Davis TP, Synthesis of star polymers by RAFT polymerization as versatile nanoparticles for biomedical Applications, Aust. J. Chem 70 (11) (2017) 1161–1170. [Google Scholar]
- [20].Bayat N, McOrist N, Ariotti N, Lai M, Sia KC, Li Y, Grace JL, Quinn JF, Whittaker MR, Kavallaris M, Davis TP, Lock RB, Thiol-reactive star polymers functionalized with short ethoxy-containing moieties exhibit enhanced uptake in acute lymphoblastic leukemia cells, Int. J. Nanomed 14 (2019) 9795–9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Glass JJ, Li Y, De Rose R, Johnston AP, Czuba EI, Khor SY, Quinn JF, Whittaker MR, Davis TP, Kent SJ, Thiol-reactive star polymers display enhanced association with distinct human blood components, ACS Appl. Mater. Interfaces 9 (14) (2017) 12182–12194. [DOI] [PubMed] [Google Scholar]
- [22].Khor SY, Hu J, McLeod VM, Quinn JF, Williamson M, Porter CJ, Whittaker MR, Kaminskas LM, Davis TP, Molecular weight (hydrodynamic volume) dictates the systemic pharmacokinetics and tumour disposition of PolyPEG star polymers, Nanomedicine 11 (8) (2015) 2099–2108. [DOI] [PubMed] [Google Scholar]
- [23].Esser L, Lengkeek NA, Moffat BA, Vu MN, Greguric I, Quinn JF, Davis TP, Whittaker MR, A tunable one-pot three-component synthesis of an125I and Gd-labelled star polymer nanoparticle for hybrid imaging with MRI and nuclear medicine, Polym. Chem 9 (25) (2018) 3528–3535. [Google Scholar]
- [24].Goos J, Davydova M, Dilling TR, Cho A, Cornejo MA, Gupta A, Price WS, Puttick S, Whittaker MR, Quinn JF, Davis TP, Lewis JS, Design and preclinical evaluation of nanostars for the passive pretargeting of tumor tissue, Nucl. Med. Biol 84–85 (2020) 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yu SH, Patra M, Ferrari S, Ramirez Garcia P, Veldhuis NA, Kaminskas LM, Graham B, Quinn JF, Whittaker MR, Gasser G, Davis TP, Linker chemistry dictates the delivery of a phototoxic organometallic rhenium(i) complex to human cervical cancer cells from core crosslinked star polymer nanoparticles, J. Mater. Chem. B 6 (47) (2018) 7805–7810. [DOI] [PubMed] [Google Scholar]
- [26].Kramer MS, Cutler N, Feighner J, Shrivastava R, Carman J, Sramek JJ, Reines SA, Liu G, Snavely D, Wyatt-Knowles E, Hale JJ, Mills SG, MacCoss M, Swain CJ, Harrison T, Hill RG, Hefti F, Scolnick EM, Cascieri MA, Chicchi GG, Sadowski S, Williams AR, Hewson L, Smith D, Carlson EJ, Hargreaves RJ, Rupniak NM, Distinct mechanism for antidepressant activity by blockade of central substance P receptors, Science 281 (5383) (1998) 1640–1645. [DOI] [PubMed] [Google Scholar]
- [27].Stenzel MH, Davis TP, Star polymer synthesis using trithiocarbonate functional ?-cyclodextrin cores (reversible addition-fragmentation chain-transfer polymerization), J. Polym. Sci. Polym. Chem 40 (24) (2002) 4498–4512. [Google Scholar]
- [28].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, Fiji: an open-source platform for biological-image analysis, Nat. Methods 9 (7) (2012) 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bolte S, Cordelieres FP, A guided tour into subcellular colocalization analysis in light microscopy, J. Microsc 224 (Pt 3) (2006) 213–232. [DOI] [PubMed] [Google Scholar]
- [30].Nehme R, Carpenter B, Singhal A, Strege A, Edwards PC, White CF, Du H, Grisshammer R, Tate CG, Mini-G proteins: novel tools for studying GPCRs in their active conformation, PLoS One 12 (4) (2017), e0175642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wan Q, Okashah N, Inoue A, Nehme R, Carpenter B, Tate CG, Lambert NA, Mini G protein probes for active G protein-coupled receptors (GPCRs) in live cells, J. Biol. Chem 293 (19) (2018) 7466–7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Namkung Y, Le Gouill C, Lukashova V, Kobayashi H, Hogue M, Khoury E, Song M, Bouvier M, Laporte SA, Monitoring G protein-coupled receptor and beta-arrestin trafficking in live cells using enhanced bystander BRET, Nat. Commun 7 (2016), 12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zimmermann M, Ethical guidelines for investigations of experimental pain in conscious animals, Pain 16 (2) (1983) 109–110. [DOI] [PubMed] [Google Scholar]
- [34].Cichon J, Sun L, Yang G, Spared nerve injury model of neuropathic pain in mice, Bio Protoc 8 (6) (2018), e2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Stein C, Millan MJ, Herz A, Unilateral inflammation of the hindpaw in rats as a model of prolonged noxious stimulation: alterations in behavior and nociceptive thresholds, Pharmacol. Biochem. Behav 31 (2) (1988) 445–451. [DOI] [PubMed] [Google Scholar]
- [36].Yoon C, Wook YY, Sik NH, Ho KS, Mo CJ, Behavioral signs of ongoing pain and cold allodynia in a rat model of neuropathic pain, Pain 59 (3) (1994) 369–376. [DOI] [PubMed] [Google Scholar]
- [37].Brodkin J, Frank D, Grippo R, Hausfater M, Gulinello M, Achterholt N, Gutzen C, Validation and implementation of a novel high-throughput behavioral phenotyping instrument for mice, J. Neurosci. Methods 224 (2014) 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Castro J, Harrington AM, Lieu T, Garcia-Caraballo S, Maddern J, Schober G, O’Donnell T, Grundy L, Lumsden AL, Miller P, Ghetti A, Steinhoff MS, Poole DP, Dong X, Chang L, Bunnett NW, Brierley SM, Activation of pruritogenic TGR5, MrgprA3, and MrgprC11 on colon-innervating afferents induces visceral hypersensitivity, JCI Insight 4 (20) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ferreira J, Syrett J, Whittaker M, Haddleton D, Davis TP, Boyer C, Optimizing the generation of narrow polydispersity ‘arm-first’ star polymers made using RAFT polymerization, Polym. Chem 2 (8) (2011) 1671–1677. [Google Scholar]
- [40].Liu J, Duong H, Whittaker MR, Davis TP, Boyer C, Synthesis of functional core, star polymers via RAFT polymerization for drug delivery applications, Macromol. Rapid Commun 33 (9) (2012) 760–766. [DOI] [PubMed] [Google Scholar]
- [41].Syrett JA, Haddleton DM, Whittaker MR, Davis TP, Boyer C, Functional, star polymeric molecular carriers, built from biodegradable microgel/nanogel cores, Chem. Commun 47 (5) (2011) 1449–1451. [DOI] [PubMed] [Google Scholar]
- [42].Schoppe J, Ehrenmann J, Klenk C, Rucktooa P, Schutz M, Dore AS, Pluckthun A, Crystal structures of the human neurokinin 1 receptor in complex with clinically used antagonists, Nat. Commun 10 (1) (2019) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Halik PK, Lipinski PFJ, Matalinska J, Kozminski P, Misicka A, Gniazdowska E, Radiochemical synthesis and evaluation of novel radioconjugates of neurokinin 1 receptor antagonist aprepitant dedicated for NK1R-positive tumors, Molecules 25 (16) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Harrison T, Owens AP, Williams BJ, Swain CJ, Williams A, Carlson EJ, Rycroft W, Tattersall FD, Cascieri MA, Chicchi GG, Sadowski S, Rupniak NM, Hargreaves RJ, An orally active, water-soluble neurokinin-1 receptor antagonist suitable for both intravenous and oral clinical administration, J. Med. Chem 44 (24) (2001) 4296–4299. [DOI] [PubMed] [Google Scholar]
- [45].Hopkins CR, Gibson A, Shipman M, Miller K, Movement of internalized ligand-receptor complexes along a continuous endosomal reticulum, Nature 346 (6282) (1990) 335–339. [DOI] [PubMed] [Google Scholar]
- [46].Munoz M, Covenas R, The neurokinin-1 receptor antagonist aprepitant: an intelligent bullet against cancer? Cancers 12 (9) (2020) 2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ashton S, Song YH, Nolan J, Cadogan E, Murray J, Odedra R, Foster J, Hall PA, Low S, Taylor P, Ellston R, Polanska UM, Wilson J, Howes C, Smith A, Goodwin RJ, Swales JG, Strittmatter N, Takats Z, Nilsson A, Andren P, Trueman D, Walker M, Reimer CL, Troiano G, Parsons D, De Witt D, Ashford M, Hrkach J, Zale S, Jewsbury PJ, Barry ST, Aurora kinase inhibitor nanoparticles target tumors with favorable therapeutic index in vivo, Sci. Transl. Med 8 (325) (2016), 325ra17. [DOI] [PubMed] [Google Scholar]
- [48].Brigham NC, Ji RR, Becker ML, Degradable polymeric vehicles for postoperative pain management, Nat. Commun 12 (1) (2021) 1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data required to reproduce these findings are available upon request. The scripts used for the image analyses are available upon request. The processed data required to reproduce these findings are available upon request.