

Abstract
Background and Objectives
Sporadic inclusion body myositis (IBM) is the most common acquired myopathy in individuals older than 50 years. The disorder is slowly progressive, and although many therapies have been investigated, response has generally been poor. Clinical heterogeneity may influence treatment responsiveness; however, data regarding heterogeneity in IBM are limited and often conflicting. We aim to identify clinically distinct subgroups within a large IBM cohort and prognostic factors for disease progression.
Methods
Clinical, histologic, radiologic, and electrophysiologic data were analyzed for all patients with IBM and other forms of myositis enrolled in a longitudinal cohort from The Johns Hopkins Myositis Center from 2003 to 2018. Patients with IBM were included if they met at least one of the following criteria: Griggs possible, European Neuromuscular Centre 2011 probable, or Lloyd-Greenberg data-derived criteria for IBM. Univariate, multivariate, and graphical analyses were used to identify prognostic factors in patients with IBM. Thus, linear and logistic regressions were used to adjust for potential confounding variables. The evolution of creatine kinase and muscle strength was studied using multilevel linear regression models. Nonmodifiable risk factors (sex, race, disease duration, and age at the onset of first symptoms) were used as adjusting covariates for the regression analyses.
Results
Among the 335 patients meeting the inclusion criteria for IBM, 64% were male with an average age of disease onset of 58.7 years and delay to diagnosis of 5.2 years. Initial misdiagnosis (52%) and immunosuppressant treatment (42%) were common. Less than half (43%) of muscle biopsies demonstrated all 3 pathologic hallmarks: endomysial inflammation, mononuclear cell invasion, and rimmed vacuoles. Black patients had significantly weaker arm abductors, hip flexors, and knee flexors compared with non-Black patients. Female patients had stronger finger flexors and knee extensors compared with their male counterparts. Younger age (<50 years) at onset was not associated with increased weakness.
Discussion
Our study demonstrates that female and Black patients have distinct clinical phenotypes and trajectories within the overarching IBM clinical phenotype. These subgroups may have different responses to therapies, which may influence the design of future clinical trials in IBM.
Sporadic inclusion body myositis (IBM) is the most common acquired myopathy among individuals older than 50 years. Gradual progression of muscle weakness and atrophy, particularly of the finger flexors and quadriceps muscles, and muscle biopsies showing endomysial inflammation with auto-invasive T-cells and rimmed vacuoles are hallmarks of the disease. Although there is evidence for dysregulation of the immune system in IBM, multiple trials of immunosuppressive agents have failed to show a benefit.1-5 Both degenerative and neoplastic processes have been suggested as alternative mechanisms.6,7
Since the initial description of IBM in 1971, multiple cohort studies have examined the clinical symptoms and course of this disorder.8-16 Although these studies have described a consistent pattern of weakness with prominent involvement of the finger flexors and knee extensors, there is evidence of substantial variability both within and between studies with regards to presentation, clinical progression, and accumulation of disability. Although the average age at onset across these studies is 59 years, onset as young as 28 years has been reported,8 and some studies have reported that 17%–20% of patients have symptom onset at <50 years.8,10 Onset of weakness in the lower extremities was most common; however, onset in the upper extremities was reported in 10%–20% of patients and onset in the bulbar muscles in 5%–10% of patients. About half of patients developed dysphagia during their disease course, and there was considerable variability in the progression of weakness in other skeletal muscles both between muscle groups and among individual patients.
Most patients eventually required the use of an assistive device for ambulation; however, there was a wide range in the time from diagnosis to the need for an assistive device (range 6–16 years). Data on mortality were mixed, with some studies showing increased mortality in patients with IBM, often related to respiratory causes,16-18 whereas others demonstrated no effect of IBM on mortality11,12
Data on prognostic factors have likewise been limited and often conflicting. Several studies have shown a more rapid functional decline in patients with older age at onset.9-11,14 However, others found mixed results13,19 or no correlation between age at onset and progression of weakness,12 and 1 study reported a more rapid decline in pinch strength among individuals with a younger age at onset.15 A more rapid decline in pinch and knee extensor strength has been reported in males; however, sex was not found to have an association with strength decline in other studies.12,13 The presence of anti-NT5c1A autoantibodies has also been reported as a poor prognostic factor in some studies,15,20 although in others, no correlation was seen between seropositivity and prognosis.21,22
There are also limited data on the effect of racial or ethnic background in IBM. Non-White patients have been underrepresented in IBM cohort studies. Of 17 IBM cohort studies,8-16,23-30 only 5 report on the racial or ethnic makeup of their patient population.13,14,16,29,30 Of these, only a single study specifically reported the inclusion of Black patients.13 One additional study reported that 4.5% of patients were non-White but did not specify further.30 Studies assessing the effect of racial or ethnic background in other forms of autoimmune myositis have demonstrated that Black race is associated with increased muscle weakness,31 increased severity of interstitial lung disease,32 and increased morbidity and poor overall outcomes, including survival.33,34 We hypothesize that racial/ethnic background may also influence clinical phenotypes and progression in IBM.
These studies suggest that IBM is a much more heterogeneous condition than has previously been considered. A larger study of IBM is important to identify distinct subgroups of patients with IBM and the prognostic factors that may influence their clinical trajectory. Such information can better inform patients about their expected disease course and can influence the design of clinical trials. We conducted a prospective longitudinal cohort study of 335 patients with IBM followed at the Johns Hopkins (JH) Myositis Center to identify IBM subgroups and their associated prognostic indicators.
Methods
Patients
The JH Myositis Center is a tertiary care referral center with a catchment area inclusive of the United States but concentrated in the mid-Atlantic region. All patients who provided informed consent to be enrolled in the JH Myositis Center Registry between 2003 and 2018 with a diagnosis of sporadic IBM were reviewed. All patients were evaluated by a JH Myositis Center physician. Those meeting at least one of the following criteria were included for analysis: Griggs possible, European Neuromuscular Centre (ENMC) 2011 probable, or Lloyd-Greenberg data-derived criteria (DDC) for IBM.35-37 We included as comparators all those myositis patients that were positive for autoantibodies recognizing Mi-2, NXP-2, TIF1-γ, MDA-5, Jo-1, PL-7, PL-12, SRP, or HMGCR by at least 2 different techniques from among the following: ELISA, immunoprecipitation of in vitro transcribed and translated protein, line blotting (EUROLINE myositis profile), and immunoprecipitation from S35-labeled HeLa cell lysates (S35 immunoprecipitation). Comparators were included in the dermatomyositis (DM) group if they had autoantibodies recognizing Mi-2, NXP-2, TIF1-γ, or MDA-5. Alternatively, patients were classified as having antisynthetase syndrome (AS) if they had autoantibodies against Jo-1, PL-7, or PL-12. Patients were included in the immune-mediated necrotizing myopathy (IMNM) group if they tested positive for anti-SRP or anti-HMGCR autoantibodies.38 Patient sex and race/ethnicity were self-reported.
Investigations
All clinical assessments were performed by either a JH Myositis Center physician or another neuromuscular specialist at The JH University. Disease onset was defined as the time of onset of the initial symptom (presenting symptom) leading to a diagnosis of IBM. Date of diagnosis was defined as the date the diagnosis of IBM was made by a treating physician. Dates were derived as patient reported or as otherwise noted in the medical record. If a specific date (MM/DD/YYYY) of onset or diagnosis was not available, the midpoint of the specified month or year was selected for the purposes of statistical analysis. Time to diagnosis was defined as the time interval between disease onset and IBM diagnosis date. Disease duration to first visit was defined as the time interval between disease onset and first visit to the JH Myositis Center. Initial symptoms and initial diagnoses other than IBM were derived as patient reported or as otherwise noted in the medical record.
Manual muscle testing was recorded using the Medical Research Council scale and converted to a 10-point Kendall scale for analysis. Muscle strength for each muscle group was reported as the average of the right and left measurements for each patient at each time point. The presence of dysphagia was assessed throughout the study based on patient report of symptoms. Sera from a subset of patients with IBM were tested for anti-NT5c1A antibodies by ELISA (ELISA MUP-44 [NT5c1A] Catalog #1675-4801 G). Only electrodiagnostic studies performed at JH by an experienced electromyographer were included in this study. Conventional techniques were used, and JH electrodiagnostic laboratory normal values were used for analysis. The presence of sensory peripheral neuropathy was defined as a sural sensory nerve action potential (SNAP) amplitude of <9 μV. Muscle biopsy reports from both JH and outside institutions were included. Reports were reviewed by JH Myositis Center physicians and considered sufficient for diagnostic purposes if cardinal IBM histopathologic features were assessed. A subset of reports containing more detailed histopathologic descriptions was included for further analysis.
Statistics
Dichotomous variables were expressed as percentages and absolute frequencies, and continuous features were reported as means and SD. Pairwise comparisons for categorical variables between groups were made using the χ2 test or Fisher exact test, as appropriate. The Student t test was used to compare continuous variables among groups. Creatine kinase (CK), a highly positively skewed variable, was expressed as median, first, and third quartile for descriptive purposes. Linear and logistic regressions were used to adjust for potential confounding variables.
Indirect standardization was used to compare mortality in patients with IBM compared with the general population as described elsewhere.31,32 In short, mortality incidence by age and sex groups was compared with the general population using indirect standardization based on the 1999–2014 Compressed Mortality File (wonder.cdc.gov/).
To account for the different number of visits per patient, the evolution of CK levels and muscle strength was studied using multilevel linear regression models with random slopes and random intercepts. We analyzed 2 models for each dependent variable, a simplified model excluding the interaction with time, and a more complex model including the interaction with time. Nonmodifiable risk factors (sex, race, disease duration, and age at the onset of first symptoms) were used as adjusting covariates for the multivariate analysis. Generalized additive models were applied to graphically analyze the longitudinal changes in strength and CK levels. All statistical analyses were performed using Stata/MP 14.1, and R v.4.0.3. A 2-sided p-value of 0.05 or less was considered significant with no correction for multiple comparisons.
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by an institutional review board at The JH Hospital. Written informed consent was obtained from all participants at the time of their enrollment in the JH Myositis Center Registry.
Data Availability
Deidentified data, study protocol, and statistical analysis plan will be made available in the case of a reasonable request, providing that the proposed use of the data has been approved by an independent review committee and that investigators meet The JH University policies for data sharing and management.
Results
Patient Characteristics
Clinical Criteria
Within the JH Myositis Center Registry, a total of 387 patients were identified with a listed diagnosis of IBM. Of these, 335 met at least Griggs possible, ENMC probable, or DDC for IBM and were included in the subsequent analysis. Only a small percentage of patients with IBM met either Griggs definite (2%) or ENMC clinicopathologically defined (1%) criteria (eTable 1, links.lww.com/WNL/C609). This was primarily due to the lack of electron microscopy and evaluation for protein accumulations other than amyloid in routine muscle biopsy assessments. Nearly half of patients (46%) met the criteria for Griggs, ENMC, and DDC, with 16% meeting only ENMC criteria, 15% meeting only DDC, and 16% meeting both ENMC and DDC (Figure 1).
Figure 1. Overlap of Diagnostic Criteria in a Large IBM Cohort.
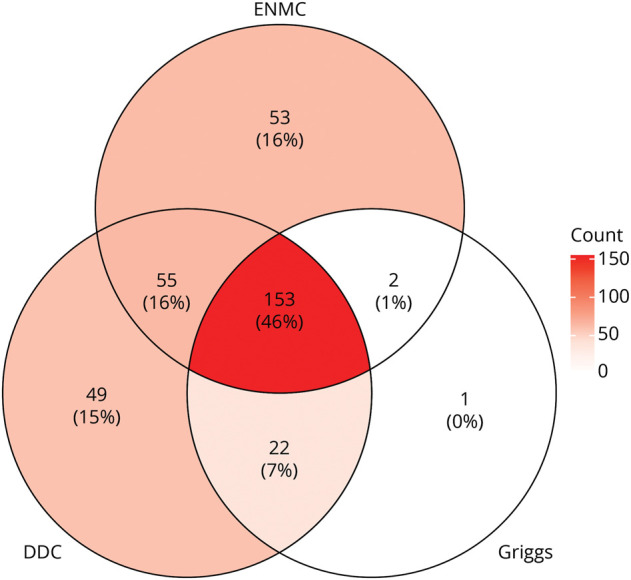
Venn diagram showing the overlap of diagnostic criteria met by each of the 335 patients with IBM studied in this cohort. DDC = data-derived criteria (also referred to as Lloyd-Greenberg criteria); ENMC = European Neuromuscular Centre 2011 probable IBM diagnostic criteria; GRIGGS = Griggs possible diagnostic criteria; IBM = inclusion body myositis.
An additional 524 patients with other forms of autoimmune myositis were identified as a comparator group. These included 165 patients with IMNM, 199 patients with DM, and 160 patients with AS.
IBM Clinical Characteristics
Baseline clinical information for the overall cohort is summarized in eTable 2, links.lww.com/WNL/C609. Patients with IBM had a mean follow-up period of 2.3 years, with a mean of 4.2 visits per patient. Sixty-five percent of patients with IBM had more than 1 visit. Compared with other types of myositis, IBM was more common in male (64%, p < 0.001), White (84%, p < 0.001), and older patients (median age at onset 58.7, p < 0.001). The average time to diagnosis for IBM was longer than for other myositis groups (5.2 years, p < 0.001). Only 4% of patients with IBM reported dysphagia as a presenting symptom, which was less than IMNM (19%, p < 0.001) or DM (19%, p < 0.001). However, 60% developed symptoms of dysphagia during the course of their illness, which was significantly higher compared with patients with IMNM (45%, p = 0.007) or the AS (38%, p < 0.001) but not DM (53%, p = 0.99). A considerable number of patients (42%) were treated with immunosuppressive agents, although this was lower than any other type of myositis (p < 0.001) in our study. Corticosteroids and methotrexate were the most common agents used. Compared with the other clinical groups, the overall mortality rate was higher in IBM than in IMNM; however, there were no significant differences in mortality between IBM and the general population (standardized mortality ratio 1.15, 95% CI 0.71–1.75).
Compared with patients with other types of myositis, those with IBM had more severe finger flexor and knee extensor weakness (eTable 3, links.lww.com/WNL/C609), showing a steady decline (about 0.33 Kendall scale strength points per year) in these 2 muscle groups during the evolution of their disease (Figure 2). In contrast, hip flexors and arm abductors tended to be stronger in IBM than in IMNM during the first 10 years after the onset of the disease (Figure 2).
Figure 2. Evolution of Strength in IBM Compared With Other Types of Autoimmune Myositis Using Generalized Additive Models.

Generalized additive models (GAMs) were applied to graphically analyze the longitudinal changes in strength observed for different subtypes of idiopathic inflammatory myopathy. Only measurements after 2 years from onset are shown due to the low number of datapoints for patients with IBM. IBM = inclusion body myositis.
Over half of patients with IBM received an initial diagnosis other than IBM (eTable 4, links.lww.com/WNL/C609). The most common misdiagnoses were polymyositis (22%) and unspecified myopathy (13%). Patients with initial misdiagnoses were more likely to be female and to have a younger age at disease onset. Finger flexor weakness and rimmed vacuoles on muscle biopsy were less common in this group. Those with an initial diagnosis of polymyositis were more likely to be ANA positive compared with those initially diagnosed with IBM (50% vs 31%, p = 0.04).
IBM Ancillary Testing
The average duration of disease at the time of muscle biopsy was 5.5 years. The combination of endomysial inflammation, invasion of non-necrotic fibers, and rimmed vacuoles was seen in 43% of biopsies. An additional 41% contained at least 2 of these cardinal features, and 34% did not show rimmed vacuoles (Figure 3). The quadriceps muscle was the most common muscle selected for biopsy. The location of the muscle biopsy did not influence the number of pathologic features present, except for the more common presence of cytochrome oxidase-negative fibers in the biceps (82%) or deltoid (83%) muscles compared with the quadriceps (47%).
Figure 3. Cardinal IBM Muscle Biopsy Features.

Venn diagram showing the distribution of pathologic features seen among patients with IBM in this cohort. COXNEG = cytochrome oxidase-negative fibers; ENDOMYS = endomysial inflammation; IBM = inclusion body myositis; MONONUCL = mononuclear cell invasion of non-necrotic myocytes; RIMMED = rimmed vacuoles.
Electrodiagnostic data were available for 285 (85%) patients with IBM (eTable 5, links.lww.com/WNL/C609). Myopathic motor unit action potentials (small duration and small amplitude) were seen in nearly all patients, with 44% also showing neurogenic units (large duration and large amplitude). Abnormal sural SNAP amplitudes (<9 μV) were present in 31% of patients with IBM compared with 24% of patients with other forms of myositis. Multivariate analysis suggested that this difference may be accounted for by the older age of patients with IBM at the time of EMG. We replicated the analysis decreasing the cutoff to sural <4 μV as recommended in practice guidelines,39 and this conclusion remained unchanged. The median CK in our IBM cohort was 426 IU/L with a Q1–Q3 range of 230–707 IU/L. A positive ANA was found in 40% of patients tested (n = 199). Testing for the anti-NT5c1A autoantibody was performed by ELISA for 320 patients with IBM. Forty-seven percent were positive for this autoantibody.
IBM Heterogeneity
Race
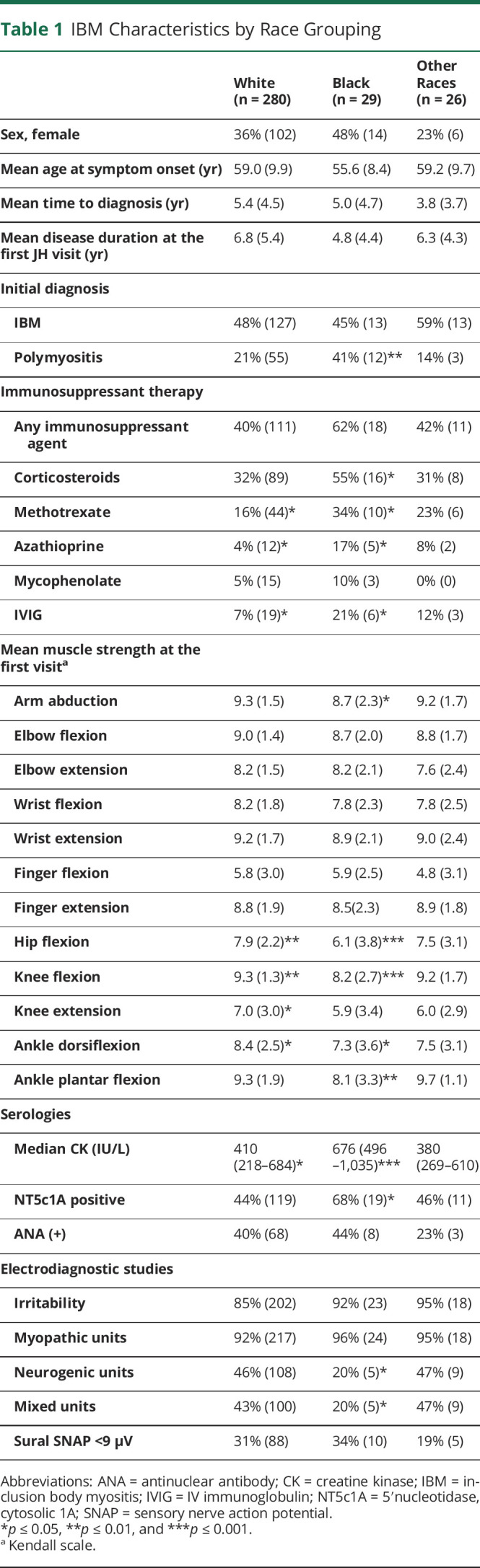
Compared with other races, Black patients with IBM had a similar age at symptom onset and duration of symptoms at the first visit. Black patients had significantly weaker arm abductors, hip flexors, and knee flexors (Table 1, eFigure 1, links.lww.com/WNL/C609) at the first JH Myositis Center visit. They were less likely to report dysphagia during their disease course (45%) compared with non-Black patients (61%, p = 0.05). Black patients were more likely to have been misdiagnosed with polymyositis and treated with immunosuppressive agents. The median CK was higher in this group (676 IU/L, Q1–Q3 range 496–1,035 IU/L), and neuropathic motor units were less commonly observed in electrodiagnostic studies (20%). Anti-NT5c1A autoantibodies were more commonly detected among Black patients (68%, p = 0.02). There was no difference in muscle biopsy features across races.
Table 1.
IBM Characteristics by Race Grouping
Multilevel regression models (independent of all the above-mentioned potential confounding factors) confirmed that, independent of the time from onset, Black patients had more severe weakness of hip flexors (−2.1 strength points, p < 0.001), arm abductors (−0.6 strength points, p = 0.02), and knee extensors (−1.3 strength points, p = 0.02) and had a trend toward weaker finger flexor muscles (−1.1 strength points, p = 0.06). Analyzing the slope and the intercept of the multilevel regression model separately, hip flexor strength declined at a similar rate in Black and non-Black patients (p = 0.7), and there was a more rapid decline in arm abduction (p = 0.05) and knee extensor (p = 0.03) strength in Black patients (eTable 6, links.lww.com/WNL/C609). It also confirmed that Black patients with IBM have higher CK levels (p = 0.001) compared with non-Black patients with IBM; however, this difference was not greater than that between Black and non-Black patients with other types of myositis (p > 0.05). Logistic regression showed that Black patients had lower odds of developing dysphagia (OR 0.4, p = 0.05).
Female Sex
Females had a longer time to diagnosis and were more likely to have been misdiagnosed with polymyositis than males (Table 2). Corticosteroid use was more common in females, although overall use of immunosuppressants did not differ between the sexes. Strength profiles showed that females had stronger knee extensors (p = 0.003) and a trend toward having stronger finger flexors (p = 0.06) (Figure 4). Females were more likely to develop dysphagia during their disease course (69%) compared with their male counterparts (55%, p = 0.02). Myopathic units were equally common between the sexes, although females tended to have less abnormal spontaneous activity on EMG.
Table 2.
IBM Characteristics by Sex Grouping
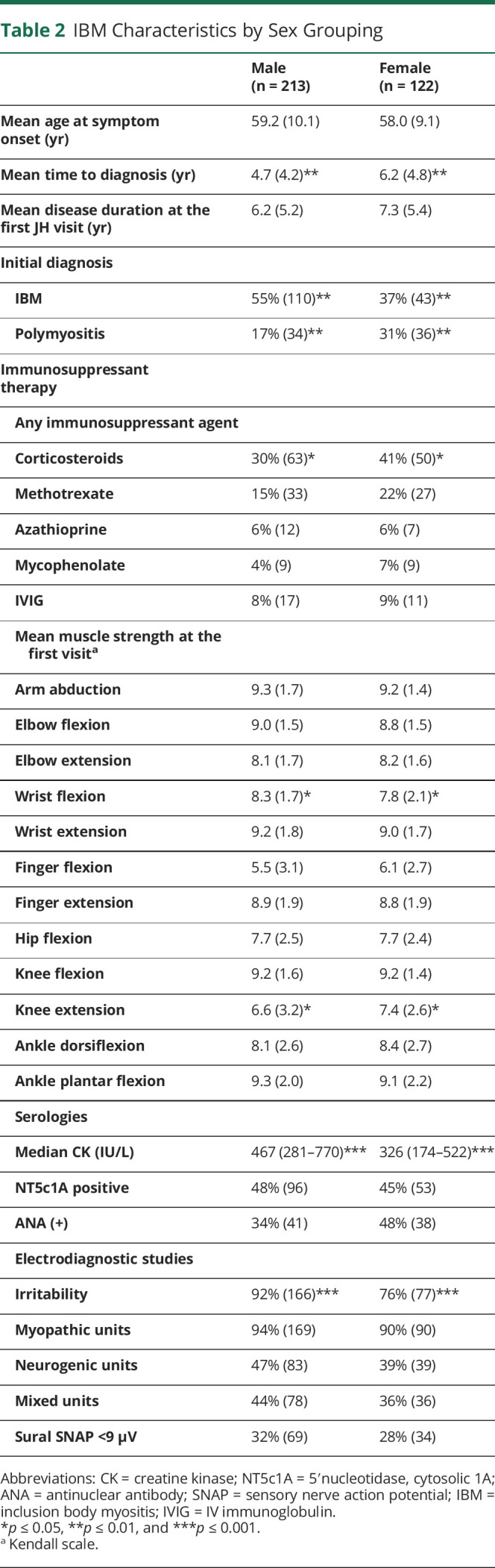
Figure 4. Evolution of Strength in IBM According to Sex Using Generalized Additive Models.

Generalized additive models (GAMs) were applied to graphically analyze the longitudinal changes in strength observed for males vs females. IBM = inclusion body myositis.
Multilevel regression models confirmed that, independent of the time from onset, females had stronger finger flexors (0.8 strength points, p = 0.02) and knee extensor muscles (0.9 strength points, p = 0.009) with no significant difference in hip flexor or arm abductor strength (both p > 0.05). Analyzing the slope and the intercept of the multilevel regression model separately, finger flexor (p = 0.003) and knee extensor (p = 0.05) strength declined more slowly in females (eTable 6, links.lww.com/WNL/C609). CK was lower in female patients with IBM compared with male patients with IBM (p = 0.02); however, this difference was not greater than that between female and male patients with other types of myositis (p > 0.05). Logistic regression showed that females developed dysphagia more frequently than males (OR 1.8, p = 0.02).
Age at Onset
Patients with a younger age at onset had a longer time to diagnosis and time to referral to the JH Myositis Center. Those with onset at <50 years of age were also more likely to be initially misdiagnosed with polymyositis and treated with immunosuppressive therapy, especially methotrexate, mycophenolate, and IV immunoglobulin. Muscle biopsy features did not differ between the age groups, although patients with a younger age at onset had a significantly longer disease duration at the time of biopsy, 7.3 years for the <50 age group vs 2.9 years for the >70 age group.
On univariate analysis, younger-onset patients had a more severe and diffuse pattern of weakness at the first visit, including more profound proximal weakness (Table 3, eFigure 2, links.lww.com/WNL/C609). However, multivariate regression models demonstrated that this difference was driven by the longer disease duration at the first visit in the younger-onset age group. Older age at onset was associated with lower CK levels than expected for the 2 independent effects (having IBM and being older) (p = 0.01). Logistic regression showed that there was no association between the prevalence of dysphagia and the age at onset, but there was a strong association between older age at onset and abnormal sural SNAP amplitudes (p < 0.001).
Table 3.
IBM Characteristics by Age at Onset Grouping

NT5c1A Status
Anti–NT5c1A-positive patients had higher median CK levels, but antibody status did not otherwise influence clinical characteristics, the pattern of weakness, muscle biopsy features, or electrodiagnostic findings.
Discussion
This study demonstrates that Black and female patients with IBM differ from their White, male counterparts with respect to both pattern and progression of weakness. Although both Black and female patients demonstrated an overall pattern of weakness consistent with that typically seen in IBM, Black patients had significantly weaker proximal muscles, and female patients had less prominent knee extension and finger flexion weakness. Black patients exhibited more rapid development of weakness in arm abductors and knee extensors. In contrast, female patients demonstrated a slower decline in finger flexor and knee extensor strength compared with male patients, and females were more likely to develop dysphagia. These characteristics may explain why both Black and female patient populations were more likely to be initially misdiagnosed with polymyositis and treated with corticosteroids.
Patients with an age of symptom onset <50 years had more diffuse and severe weakness at the initial JH visit compared with their older onset counterparts. However, this effect was confounded by a longer disease duration at the time of the first visit in the younger-onset group. Adjusting by this factor, we found that patients with IBM with onset at an older age had more severe weakness in finger flexors, whereas other muscles showed similar levels of weakness. Other studies have reported more rapid progression in patients with an older age at onset, although this was not observed in our cohort.9-11,14
These findings suggest that Black and female patients represent clinically distinct subgroups within IBM with unique disease trajectories and, potentially, different responses to therapeutic interventions. The origin of these differences in clinical phenotype and disease progression are unclear.
In accordance with prior studies, a large percentage of our patients experienced a prolonged time to diagnosis, during which period many were initially misdiagnosed with polymyositis and treated with ineffective immunosuppressive therapies. We suspect that overreliance on muscle biopsy features, in particular the presence of rimmed vacuoles, and underreliance on physical examination (e.g., presence of finger flexor weakness and quadriceps weakness) are major factors in the long delay to diagnosis that is common for patients with IBM. Only 43% of muscle biopsies in our study contained all 3 cardinal features of IBM (endomysial inflammation, mononuclear cell invasion, and rimmed vacuoles). In particular, rimmed vacuoles were less common in our study (66%) compared with prior studies (75%–100%)8,11,14 and were only seen in the absence of endomysial inflammation or mononuclear invasion in 3% of patients. This likely reflects the increased reliance of Griggs and ENMC diagnostic criteria for inclusion in prior published reports, both of which require the presence of rimmed vacuoles to meet definite or clinicopathologically defined IBM, respectively. This highlights the need for more accurate and less invasive diagnostic methods to identify patients with IBM at earlier stages of disease when they may be more responsive to therapeutics.
Many of our patients with IBM also had electrodiagnostic evidence of sensory peripheral neuropathy as defined by decreased sural SNAP amplitudes. Although a few small studies have also reported evidence of peripheral nerve dysfunction in up to 30% of patients with IBM, the involvement of peripheral nerves has not been extensively studied.40,41 The results of our study suggest that nerve dysfunction is primarily driven by older age rather than by the presence of IBM. However detailed studies of electrodiagnostic data in IBM are needed to explore this further.
Our study has several limitations. Data from clinical assessments were constrained to visits at the JH Myositis Center at which point patients had had symptoms for an average of 6.6 years. Although female and Black patients with IBM did have different patterns of weakness by this time, their initial symptoms may have differed more dramatically. In addition, younger-onset patients may have had a distinct phenotype at presentation which became obscured by more advanced disease.
In conclusion, sporadic IBM has long been considered a disease predominately affecting older White males. Our cohort included a significant number of female, Black, and younger-onset patients and demonstrated a unique clinical profile in both female and Black patients that may explain the propensity for initial misdiagnosis and ineffective immunosuppressant agent use in these populations. Further longitudinal studies are needed to confirm and further characterize these findings; however, it is important to recognize the presence of these subgroups as they may have different responses to therapies, and their presence may influence the design of future clinical trials in IBM. More sensitive and accessible diagnostics are needed to identify patients with IBM at earlier stages and within these less well-recognized subpopulations to further our understanding of clinical heterogeneity within this disorder.
Glossary
- AS
antisynthetase syndrome
- CK
creatine kinase
- DDC
data-derived criteria
- DM
dermatomyositis
- ENMC
European Neuromuscular Centre
- IBM
inclusion body myositis
- IMNM
immune-mediated necrotizing myopathy
- JH
Johns Hopkins
- SNAP
sensory nerve action potential
Appendix. Authors
Study Funding
This study was funded by The Sandra and Malcolm Berman Brain & Spine Institute, The Peter and Carmen Lucia Buck Foundation, The Huayi and Siuling Zhang Discovery Fund, and the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH. The authors thank these individuals and institutions for their contribution. J.J. Paik is supported by K23AR073927 from NIAMS/NIH. T.E. Lloyd is supported by R01 AR076390 from NIAMS/NIH and MDA630399 from the Muscular Dystrophy Association.
Disclosure
E.H. Michelle reports funding from The Sandra and Malcolm Berman Brain & Spine Institute. I. Pinal-Fernandez received support from The Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH. M. Casal-Dominguez received support from The Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH. J. Albayda was supported by contributions from The Peter and Carmen Lucia Buck Foundation and The Huayi and Siuling Zhang Discovery Fund. J.J. Paik was supported by funding from The National Institute of Arthritis and Musculoskeletal and Skin Diseases of the NIH under award number K23AR073927 and contributions from The Peter and Carmen Lucia Buck Foundation and The Huayi and Siuling Zhang Discovery Fund. E. Tiniakou was supported by contributions from The Peter and Carmen Lucia Buck Foundation and The Huayi and Siuling Zhang Discovery Fund. B. Adler was supported by contributions from The Peter and Carmen Lucia Buck Foundation and The Huayi and Siuling Zhang Discovery Fund. C.A. Mecoli was supported by contributions from The Peter and Carmen Lucia Buck Foundation and The Huayi and Siuling Zhang Discovery Fund. S.K. Danoff was supported by contributions from The Peter and Carmen Lucia Buck Foundation and The Huayi and Siuling Zhang Discovery Fund. L. Christopher-Stine was supported by contributions from The Peter and Carmen Lucia Buck Foundation and The Huayi and Siuling Zhang Discovery Fund. A.L. Mammen was supported by The Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH. T.E. Lloyd was supported by R01 AR076390 from NIAMS/NIH and MDA630399 from the Muscular Dystrophy Association and by contributions from The Peter and Carmen Lucia Buck Foundation and The Huayi and Siuling Zhang Discovery Fund. Go to Neurology.org/N for full disclosures.
References
- 1.Dalakas MC, Koffman B, Fujii M, Spector S, Sivakumar K, Cupler E. A controlled study of intravenous immunoglobulin combined with prednisone in the treatment of IBM. Neurology. 2001;56(3):323-327. doi: 10.1212/wnl.56.3.323. [DOI] [PubMed] [Google Scholar]
- 2.Badrising UA, Maat-Schieman MLC, Ferrari MD, et al. . Comparison of weakness progression in inclusion body myositis during treatment with methotrexate or placebo. Ann Neurol. 2002;51(3):369-372. doi: 10.1002/ana.10121. [DOI] [PubMed] [Google Scholar]
- 3.The Muscle Study Group. Randomized pilot trial of high-dose betaINF-1a in patients with inclusion body myositis. Neurology. 2004;63(4):718-720. doi: 10.1212/01.wnl.0000134675.98525.79. [DOI] [PubMed] [Google Scholar]
- 4.Barohn RJ, Herbelin L, Kissel JT, et al. . Pilot trial of etanercept in the treatment of inclusion-body myositis. Neurology. 2006;66(Issue 1, Suppl1):S123-S124. doi: 10.1212/01.wnl.0000192258.32408.54. [DOI] [PubMed] [Google Scholar]
- 5.Dalakas MC, Rakocevic G, Schmidt J, et al. . Effect of alemtuzumab (CAMPATH 1-H) in patients with inclusion-body myositis. Brain. 2009;132(6):1536-1544. doi: 10.1093/brain/awp104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benveniste O, Stenzel W, Hilton-Jones D, Sandri M, Boyer O, van Engelen BGM. Amyloid deposits and inflammatory infiltrates in sporadic inclusion body myositis: the inflammatory egg comes before the degenerative chicken. Acta Neuropathol. 2015;129(5):611-624. doi: 10.1007/s00401-015-1384-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenberg SA, Pinkus JL, Amato AA, Kristensen T, Dorfman DM. Association of inclusion body myositis with T cell large granular lymphocytic leukaemia. Brain. 2016;139(5):1348-1360. doi: 10.1093/brain/aww024. [DOI] [PubMed] [Google Scholar]
- 8.Lotz BP, Engel AG, Nishino H, Stevens JC, Litchy WJ. Inclusion body myositis: observations in 40 patients. Brain. 1989;112(3):727-747. doi: 10.1093/brain/112.3.727. [DOI] [PubMed] [Google Scholar]
- 9.Peng A, Koffman BM, Malley JD, Dalakas MC. Disease progression in sporadic inclusion body myositis: observations in 78 patients. Neurology. 2000;55(2):296-298. doi: 10.1212/wnl.55.2.296. [DOI] [PubMed] [Google Scholar]
- 10.Badrising UA, Maat-Schieman MLC, Houwelingen JC, et al. . Inclusion body myositis: clinical features and clinical course of the disease in 64 patients. J Neurol. 2005;252(12):1448-1454. doi: 10.1007/s00415-005-0884-y. [DOI] [PubMed] [Google Scholar]
- 11.Benveniste O, Guiguet M, Freebody J, et al. . Long-term observational study of sporadic inclusion body myositis. Brain. 2011;134(11):3176-3184. doi: 10.1093/brain/awr213. [DOI] [PubMed] [Google Scholar]
- 12.Cox FM, Titulaer MJ, Sont JK, Wintzen AR, Verschuuren JJGM, Badrising UA. A 12-year follow-up in sporadic inclusion body myositis: an end stage with major disabilities. Brain. 2011;134(11):3167-3175. doi: 10.1093/brain/awr217. [DOI] [PubMed] [Google Scholar]
- 13.Cortese A, Machado P, Morrow J, et al. . Longitudinal observational study of sporadic inclusion body myositis: implications for clinical trials. Neuromuscul Disord. 2013;23(5):404-412. doi: 10.1016/j.nmd.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 14.Hori H, Yamashita S, Tawara N, et al. . Clinical features of Japanese patients with inclusion body myositis. J Neurol Sci. 2014;346(1-2):133-137. doi: 10.1016/j.jns.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 15.Oldroyd AGS, Lilleker JB, Williams J, Chinoy H, Miller JAL. Long-term strength and functional status in inclusion body myositis and identification of trajectory subgroups. Muscle Nerve. 2020;62(1):76-82. doi: 10.1002/mus.26859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shelly S, Mielke MM, Mandrekar J, et al. . Epidemiology and natural history of inclusion body myositis. Neurology. 2021;96(21):e2653-e2661. doi: 10.1212/wnl.0000000000012004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naddaf E, Shelly S, Mandrekar J, et al. . Survival and associated comorbidities in inclusion body myositis. Rheumatology. 2021;61(5):2016-2024. doi: 10.1093/rheumatology/keab716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price MA, Barghout V, Benveniste O, et al. . Mortality and causes of death in patients with sporadic inclusion body myositis: survey study based on the clinical experience of specialists in Australia, Europe and the USA. J Neuromuscul Dis. 2016;3(1):67-75. doi: 10.3233/jnd-150138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sangha G, Yao B, Lunn D, et al. . Longitudinal observational study investigating outcome measures for clinical trials in inclusion body myositis. J Neurol Neurosurg Psychiatry. 2021;92(8):854-862. doi: 10.1136/jnnp-2020-325141. [DOI] [PubMed] [Google Scholar]
- 20.Lilleker JB, Rietveld A, Pye SR, et al. . Cytosolic 5'-nucleotidase 1A autoantibody profile and clinical characteristics in inclusion body myositis. Ann Rheum Dis. 2017;76(5):862-868. doi: 10.1136/annrheumdis-2016-210282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ikenaga C, Findlay AR, Goyal NA, et al. . Clinical utility of anti-cytosolic 5'-nucleotidase 1A antibody in idiopathic inflammatory myopathies. Ann Clin Transl Neurol. 2021;8(3):571-578. doi: 10.1002/acn3.51294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paul P, Liewluck T, Ernste FC, Mandrekar J, Milone M. Anti-cN1A antibodies do not correlate with specific clinical, electromyographic, or pathological findings in sporadic inclusion body myositis. Muscle Nerve. 2021;63(4):490-496. doi: 10.1002/mus.27157. [DOI] [PubMed] [Google Scholar]
- 23.Ringel SP, Kenny CE, Neville HE, Giorno R, Carry MR. Spectrum of inclusion body myositis. Arch Neurol. 1987;44(11):1154-1157. doi: 10.1001/archneur.1987.00520230042011. [DOI] [PubMed] [Google Scholar]
- 24.Beyenburg S, Zierz S, Jerusalem F. Inclusion body myositis: clinical and histopathological features of 36 patients. Clin Investig. 1993;71(5):351-361. doi: 10.1007/BF00186623. [DOI] [PubMed] [Google Scholar]
- 25.Lindberg C, Persson LI, Björkander J, Oldfors A. Inclusion body myositis: clinical, morphological, physiological and laboratory findings in 18 cases. Acta Neurol Scand. 2009;89(2):123-131. doi: 10.1111/j.1600-0404.1994.tb01647.x. [DOI] [PubMed] [Google Scholar]
- 26.Amato AA, Gronseth GS, Jackson CE, et al. . Inclusion body myositis: clinical and pathological boundaries. Ann Neurol. 1996;40(4):581-586. doi: 10.1002/ana.410400407. [DOI] [PubMed] [Google Scholar]
- 27.Rose MR, McDermott MP, Thornton CA, Palenski C, Martens WB, Griggs RC. A prospective natural history study of inclusion body myositis: implications for clinical trials. Neurology. 2001;57(3):548-550. doi: 10.1212/wnl.57.3.548. [DOI] [PubMed] [Google Scholar]
- 28.Felice KJ, North WA. Inclusion body myositis in Connecticut: observations in 35 patients during an 8-year period. Medicine. 2001;80(5):320-327. doi: 10.1097/00005792-200109000-00006. [DOI] [PubMed] [Google Scholar]
- 29.Dimachkie MM, Barohn RJ. Inclusion body myositis. Neurol Clin. 2014;32(3):629-646. doi: 10.1016/j.ncl.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paltiel AD, Ingvarsson E, Lee DKK, et al. . Demographic and clinical features of inclusion body myositis in North America. Muscle Nerve. 2015;52(4):527-533. doi: 10.1002/mus.24562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tiniakou E, Pinal-Fernandez I, Lloyd TE, et al. . More severe disease and slower recovery in younger patients with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Rheumatology. 2017;56(5):787-794. doi: 10.1093/rheumatology/kew470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pinal-Fernandez I, Casal-Dominguez M, Huapaya JA, et al. . A longitudinal cohort study of the anti-synthetase syndrome: increased severity of interstitial lung disease in black patients and patients with anti-PL7 and anti-PL12 autoantibodies. Rheumatology. 2017;56(6):999-1007. doi: 10.1093/rheumatology/kex021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phillippi K, Hoeltzel M, Byun Robinson A, Kim S, Childhood Arthritis and Rheumatology Research Alliance CARRA Legacy Registry Investigators. Race, income and disease outcomes in juvenile dermatomyositis. J Pediatr. 2017;184:38-44.e1. doi: 10.1016/j.jpeds.2017.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schiopu E, Phillips K, MacDonald PM, Crofford LJ, Somers EC. Predictors of survival in a cohort of patients with polymyositis and dermatomyositis: effect of corticosteroids, methotrexate and azathioprine. Arthritis Res Ther. 2012;14(1):R22. doi: 10.1186/ar3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griggs RC, Askanas V, DiMauro S, et al. . Inclusion body myositis and myopathies. Ann Neurol. 1995;38(5):705-713. doi: 10.1002/ana.410380504. [DOI] [PubMed] [Google Scholar]
- 36.Rose MR, ENMC IBM Working Group. 188th ENMC international workshop: inclusion body myositis, 2-4 December 2011, Naarden, The Netherlands. Neuromuscul Disord. 2013;23(12):1044-1055. doi: 10.1016/j.nmd.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 37.Lloyd TE, Mammen AL, Amato AA, Weiss MD, Needham M, Greenberg SA. Evaluation and construction of diagnostic criteria for inclusion body myositis. Neurology. 2014;83(5):426-433. doi: 10.1212/wnl.0000000000000642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Casal-Dominguez M, Pinal-Fernandez I, Pak K, et al. . Performance of the 2017 EULAR/ACR classification criteria for inflammatory myopathies in patients with myositis-specific autoantibodies. Arthritis Rheumatol. 2021;74(3):508-517.doi: 10.1002/art.41964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen S, Andary M, Buschbacher R, et al. . Electrodiagnostic reference values for upper and lower limb nerve conduction studies in adult populations. Muscle Nerve. 2016;54(3):371-377. doi: 10.1002/mus.25203. [DOI] [PubMed] [Google Scholar]
- 40.Lee JH, Boland-Freitas R, Liang C, Howells J, Ng K. Neuropathy in sporadic inclusion body myositis: a multi-modality neurophysiological study. Clin Neurophysiol. 2020;131(11):2766-2776. doi: 10.1016/j.clinph.2020.07.025. [DOI] [PubMed] [Google Scholar]
- 41.Joy JL, Oh SJ, Baysal AI. Electrophysiological spectrum of inclusion body myositis. Muscle Nerve. 1990;13(10):949-951. doi: 10.1002/mus.880131010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Deidentified data, study protocol, and statistical analysis plan will be made available in the case of a reasonable request, providing that the proposed use of the data has been approved by an independent review committee and that investigators meet The JH University policies for data sharing and management.